
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge chemical language models for property prediction and high-throughput screening of ionic liquids†
Yuxin
Qiu
a,
Zhen
Song
 *ac,
Guzhong
Chen
b,
Wenyao
Chen
a,
Long
Chen
a,
Kake
Zhu
*a,
Zhiwen
Qi
a,
Xuezhi
Duan
*a and
De
Chen
a
*ac,
Guzhong
Chen
b,
Wenyao
Chen
a,
Long
Chen
a,
Kake
Zhu
*a,
Zhiwen
Qi
a,
Xuezhi
Duan
*a and
De
Chen
a
aState Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: songz@ecust.edu.cn; kakezhu@ecust.edu.cn; xzduan@ecust.edu.cn
bDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA
cEngineering Research Center of Resource Utilization of Carbon-Containing Waste with Carbon Neutrality (Ministry of Education), East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
First published on 28th April 2025
Abstract
Ionic liquids (ILs) possess unique physicochemical properties and exceptional tunability, making them versatile materials for a wide range of applications. However, their immense design flexibility also poses significant challenges in efficiently identifying outstanding ILs for specific tasks within the vast chemical space. In this study, we introduce ILBERT, a large-scale chemical language model designed to predict twelve key physicochemical and thermodynamic properties of ILs. By leveraging pre-training on over 31 million unlabeled IL-like molecules and employing data augmentation techniques, ILBERT achieves superior performance compared to existing machine learning methods across all twelve benchmark datasets. As a case study, we highlight ILBERT's ability to screen ILs as potential electrolytes from a database of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333096 synthetically feasible ILs, demonstrating its reliability and computational efficiency. With its robust performance, ILBERT serves as a powerful tool for guiding the rational discovery of ILs, driving innovation in their practical applications.
333096 synthetically feasible ILs, demonstrating its reliability and computational efficiency. With its robust performance, ILBERT serves as a powerful tool for guiding the rational discovery of ILs, driving innovation in their practical applications.
1 Introduction
Ionic liquids (ILs) are typically defined as compounds consisting entirely of ions with melting points below 100 °C.1,2 Their distinctive properties, including nonvolatility, wide liquidus range, high thermal stability, and high ionic conductivity, have facilitated their applications across a wide range of fields.3–5 The diverse combinations of cations and anions provide significant design flexibility, enabling the tailoring of ILs to meet specific applications.6 However, this diversity also necessitates considerable time and costs for experimental evaluation of various combinations of cations and anions. Consequently, efficient and accurate tools for predicting the properties of ILs are highly desirable.7,8As a result of continuous efforts over the last decades, researchers have developed a variety of computational methods to predict the properties of ILs, including but not limited to equation of state (EoS) methods, group contribution (GC) methods, quantum chemistry (QC) calculations, and conductor-like screening model (COSMO) based methods.6,9–14 EoS methods possess a solid theoretical foundation in thermodynamics, while their application is hindered by complexity for estimating the required model parameters.15,16 GC methods assume that the contributions of functional groups to a specific target property are additive, which has been shown to perform well in estimating certain properties (such as density and heat capacity); nevertheless, not all properties adhere to the simple additivity rule.17–19 QC calculations can provide in-depth insights into the characteristics and behaviors of ILs at the microscopic scale, while the high computational costs restrict their application in large-scale screening.20 The COSMO-RS and COSMO-SAC models are versatile predictive methods for thermodynamic properties of fluids and their mixtures, including ILs.21,22 However, COSMO-based models necessitate prior availability of the σ-profiles of all involved molecules and in some cases provide qualitative rather than quantitative prediction.8,23,24
Apart from the methods mentioned above, quantitative structure–property relationship (QSPR) models that correlate molecular properties with their corresponding chemical structures have gained significant popularity driven by advancements in machine learning (ML).25–31 These methods can utilize various molecular representations, such as groups, descriptors and fingerprints, demonstrating considerable flexibility and accuracy.20,32–34 However, these molecular representations are essentially manually engineered based on expert knowledge, which requires feature engineering tailored to specific types of ILs or target properties. This dependence may limit their scalability to other IL property prediction tasks.7 In recent years, deep learning, as a subset of ML, has achieved remarkable success in various fields.35 One key principle of deep learning is to design suitable deep neural networks and train them on large amounts of raw data, which allows models to automatically learn feature representations and reduces the need for manual feature engineering.36 Nevertheless, when it comes to the task of IL property prediction based on deep learning, databases such as ILThermo that have even been elaborately accumulated for years are still far from sufficient compared to the vast potential chemical space.37
The challenge of data scarcity faced by IL property prediction tasks is essentially also encountered in natural language processing (NLP), that is, unlimited unlabeled datasets versus limited labeled datasets. As a significant advancement in the NLP field, transformer architecture proposed by Vaswani et al.38 laid the foundation for subsequent research, particularly with the emergence of pre-trained large language models such as BERT (Bidirectional Encoder Representations from Transformers) and GPT (Generative Pre-trained Transformer).39,40 These models operate within a pre-training and fine-tuning framework, leveraging large-scale unlabeled text data during pre-training, resulting in impressive performance across diverse downstream tasks. The success of NLP has enlightened molecular property prediction based on chemical languages, such as the Simplified Molecular Input Line Entry System (SMILES).41 For instance, Chithrananda et al.42 collected 77 million SMILES strings from PubChem and constructed a chemical language model named ChemBERTa, which demonstrates a competitive performance against the best models on MoleculeNet. Kuenneth and Ramprasad43 introduced a model based on chemical languages, termed polyBERT, which is capable of predicting a wide range of polymer properties and identifying suitable candidates with exceptional speed and accuracy. As for ILs, Chen et al.7 proposed ILtransR that can predict IL properties from SMILES by combining transformer and convolutional neural network (CNN) architectures, which well manifests the potential of chemical languages for IL representation. However, these efforts are predominantly constrained by their reliance on SMILES representations and purely NLP methods, which limits the exploration of alternative chemical languages and the domain-specific characteristics inherent to chemical structures.
In this work, building upon the aforementioned content, we introduce ILBERT, a BERT-based chemical language model for predicting twelve properties of ILs. ILBERT follows the framework of pre-training and fine-tuning based on the collection of 31 million unlabeled IL-like molecules and twelve IL property datasets. By comprehensively evaluating twelve IL property prediction tasks, ILBERT exhibits superior performance compared to other ML-based methods of corresponding literature. Moreover, the comparative analyses of how different chemical languages and tokenization methods affect model performance are conducted, and the impacts of pre-training dataset size and the number of model parameters are investigated. Apart from model performance, the attention mechanism is utilized to analyze the learned representation from ILBERT to provide the interpretability of the model. As an exemplary application, ILBERT successfully identified electrolyte candidates with high electrical conductivity and low viscosity from 8333096 synthetically feasible ILs. To facilitate the widespread use of ILBERT for assisting researchers in designing ILs for specific processes, a web server thereon is developed at https://ai4solvents.com/prediction, and source codes of ILBERT and data are also provided in the GitHub repository at https://github.com/Yu-Xin-Qiu/ILBERT.
2 Methodology
2.1 Workflow
The workflow of ILBERT proposed herein is illustrated in Fig. 1, encompassing three stages of pre-training, fine-tuning and high-throughput screening. During the pre-training stage, the masked language model (MLM) is utilized to learn the implicit context information of chemical languages based on 31 million unlabeled molecules. During the fine-tuning stage, twelve benchmark datasets are compiled to evaluate the performance of the model, covering various physicochemical and thermodynamic properties of ILs. Comprehensive experiments are carried out to determine the optimal chemical language and tokenization methods for IL property prediction. Subsequently, we investigate the impacts of pre-training data quantity and the number of model parameters on model performance and conduct ablation studies to evaluate the efficacy of pre-training and data augmentation. Finally, a high-throughput screening case study is exemplified to screen superior IL electrolytes from 8333096 synthetically feasible ILs.
 | ||
| Fig. 1 Workflow of the proposed ILBERT. (A and B) Pre-training and fine-tuning framework. (C) High-throughput IL screening case study for electrolytes. | ||
2.2 Chemical language and tokenization
To explore which chemical language is more suitable for IL property prediction, prominent chemical languages, including SMILES,51 DeepSMILES,52 InChI53 and SELFIES,54 are used for comparative analysis. It is noteworthy that the default configuration of SELFIES is unable to encode all anions and cations, such as PF6−. To overcome this limitation, the constraints for phosphorus in the hypervalence constraints are relaxed to enable the encoding of all ILs. Additionally, we also investigate various tokenization methods: character-level (CL), atom-level (AL), SMILES pair encoding (SPE), and atom-in-SMILES (AIS), with AIS being applicable only to SMILES.55,56 The vocabularies are constructed based on the entire fine-tuning dataset for IL property prediction. Tables S1 and S2† show the different tokenization results for the same IL across various chemical languages. Fig. S1† illustrates the length distribution of the fine-tuning dataset under different combinations of chemical languages and tokenization methods.2.3 Data augmentation
To address the challenge of relatively scarce labeled data of ILs, data augmentation (DA) is employed based on SMILES enumeration. Specifically, for each canonical SMILES input, 9 additional non-canonical SMILES strings are generated. During the training period, all SMILES strings are utilized to enable the model to recognize molecular structures from various “perspectives”. During testing or validation periods, the final prediction value is obtained by averaging the predictions from all 10 SMILES strings. It should be noted that different SMILES strings for the same IL do not appear in the training and test sets at the same time to avoid data leakage.2.4 Dataset collection
A large unlabeled database is compiled by collecting 1 billion SMILES strings from ZINC (https://zinc.docking.org/) and 117 million SMILES strings from PubChem (https://pubchem.ncbi.nlm.nih.gov/). As this study focuses solely on predicting the properties of ILs, two specific filtering criteria are employed to screen molecules that are closely related to ILs. The first criterion is that SMILES strings must contain either ionic bonds represented by “.” or both “+” and “-” charges, while the second criterion specifies that sequence lengths must be less than 100. Following data cleaning and standardization, approximately 31 million distinct SMILES strings are retained for pre-training. In the fine-tuning stage, twelve datasets of physicochemical and thermodynamic properties are collected from literature, which can be divided into three types. The first type of property is independent of temperature and pressure (melting point Tm, glass transition temperature Tg, thermal decomposition temperature Td, and cytotoxicity towards the leukemia rat cell line IPC-81 (log10EC50), the second type of property is dependent on temperature (electrical conductivity lnκ, viscosity lnη, surface tension γ, refractive index nD and heat capacity Cp), and the third type of property is associated with both temperature and pressure (thermal conductivity λ, density ρ and CO2 solubility xCO2). It should be noted that while viscosity and electrical conductivity exhibit both temperature and pressure dependence, the majority of available experimental data correspond to ambient pressure conditions. In this work, we focus specifically on the temperature-dependent behavior of viscosity and electrical conductivity. Detailed information about these twelve datasets is presented in Table 1, and the distribution of each dataset is depicted in Fig. S2.† The total number of SMILES strings involved in the pre-training and fine-tuning datasets are 30526093 and 64226, respectively, with their length distribution shown in Fig. S3.†
| Property | Number of data points | Number of ILs | Units | Data source |
|---|---|---|---|---|
| Melting point Tm | 2673 | 2673 | K | Makarov et al.44 |
| Glass transition temperature Tg | 798 | 798 | K | Makarov et al.45 |
| Thermal decomposition temperature Td | 2780 | 2780 | K | Makarov et al.45 |
| Cytotoxicity towards the leukemia rat cell line IPC-81 log10EC50 | 355 | 355 | 1 | Wang et al.46 |
| Electrical conductivity lnκ |
2168 | 242 | S m−1 | Chen et al.33 |
| Viscosity lnη |
15368 |
1964 | mPa s | Chen et al.7 |
| Surface tension γ | 6051 | 542 | mN m−1 | Baran and Kloskowski47 |
| Refractive index nD | 2963 | 350 | 1 | Cao et al.32 |
| Heat capacity Cp | 11521 |
256 | J mol−1 K−1 | Liaqat et al.18 |
| Thermal conductivity λ | 606 | 44 | W m−1 K−1 | Wan et al.48 |
| Density ρ | 31167 |
2257 | kg m−3 | Paduszyński49 |
| CO2 solubility xCO2 | 10116 |
124 | mol% | Song et al.50 |
2.5 Implementation details and model construction
During the data preprocessing stage, RDKit (https://www.rdkit.org) is employed to process SMILES, including filtering invalid SMILES, SMILES standardization, and SMILES enumeration. Additionally, the deepsmiles, RDKit, and selfies packages are utilized to convert canonical SMILES into DeepSMILES, InChI, and SELFIES, respectively.In the pre-training stage, 31 million canonical SMILES strings are utilized as inputs for our model. After tokenization, 15% of the tokens are randomly masked before being fed into the BERT model. The objective of the pre-training task is to predict the masked tokens and minimize the cross-entropy loss associated with the MLM. Compared to the original BERT model, the data volume and complexity of the IL property prediction task are relatively smaller. Consequently, the number of transformer encoder layers, heads in multi-head attention, and embedding dimensions in BERT-base are modified to construct three pre-trained models with varying model parameters, as detailed in Table S3.† The Hugging Face library (https://huggingface.co/) is employed to build the pre-trained models, training with the Adam optimizer for five epochs, an initial learning rate of 1 × 10−4, and other settings consistent with BERT-base. To independently evaluate the performance of pre-trained models, 1% of the pre-training dataset was randomly selected as the validation set.
In the fine-tuning stage, both transfer learning (TL) and fine-tuning (FT) approaches are employed for IL property prediction tasks. For the TL approach, the weights of the pre-trained transformer encoder are frozen and a convolutional neural network (CNN) model is added, followed by the inclusion of conditional variables such as temperature and pressure, before finally inputting these into Multilayer Perceptron (MLP) for IL property prediction. For the FT approach, not only the weights of the CNN and MLP but also the pre-trained models are updated to better adapt to the target task. Mean Squared Error (MSE) is developed as the loss function and hyperparameter search is performed for each task. Table S4† summarizes the optimal hyperparameters for each IL property prediction task. To avoid overfitting, the early stopping strategy is adopted, and the training is suspended if no loss reduction was observed within 15 epochs.
In our previous work, we highlighted the distinction between two dataset split strategies: data point-based and IL-based.7,32,33 When handling tasks related to temperature/pressure, the data point-based dataset split strategy allows the same IL (with only a difference in temperature/pressure) to appear in both the training and test sets, leading to data leakage and overestimation of model performance. In contrast, the IL-based split strategy mitigates this problem by ensuring that the same IL does not appear in both sets, thus providing more rigorous evaluation. Unless specifically noted, this study follows the rigorous IL-based dataset split strategy, and five-fold cross-validations (CVs) are repeated five times to report the final results. The final model is integrated with five individual models that are obtained from five-fold cross-validations. The average prediction across these models is used as the final result, and the standard deviation serves as the estimate of uncertainty.
2.6 Experiment details
Based on the results of high-throughput screening, two ILs, 1-ethyl-3-methylimidazolium dicyanamide ([EMIM][DCA]) and 1,3-diethylimidazolium dicyanamide ([DEIM][DCA]), were selected to measure their melting point, electrical conductivity, viscosity and density. [EMIM][DCA] (CAS: 370865-89-7, ≥98%, wwater = 0.4884%) was purchased from Adamas-beta. Additionally, [DEIM][DCA] (≥98%, wwater = 0.09466%) was first synthesized in this study, with the synthesis process shown in Fig. S4.† The chemical structure and composition of both ILs were confirmed using 1H NMR and 13C NMR spectroscopy. NMR spectra were recorded on a NMR spectrometer (AV-400 MHz, Bruker, Switzerland) using DMSO-d6 as the solvent. Water content was measured by Karl-Fischer volumetric titration (AQV-300, Hiranuma, Japan). The 1H NMR and 13C NMR spectra of [EMIM][DCA] and [DEIM][DCA] are presented in Fig. S5–S8.†The melting point was determined by differential scanning calorimetry (DSC 25, TA Instruments, USA) and the DSC curves are provided in Fig. S9 and S10.† Electrical conductivity was measured with a conductivity meter (SD30, Mettler Toledo, Switzerland) and a conductivity sensor (InLab731, Mettler Toledo, Switzerland) inside a glove box (MKUS2-2309-0069, Mikrouna, China) that maintained water and oxygen levels below 0.01 ppm. Viscosity and density were measured using an automated falling ball viscometer (Lovis 2000 ME, Anton Paar, Austria). All experiments were carried out at temperatures ranging from 293.15 K to 323.15 K.
3 Results and discussion
In this section, comprehensive experiments of the proposed ILBERT are conducted to answer these six questions: (1) which chemical language and tokenization method are the most appropriate for IL property prediction tasks? (2) how do the amount of pre-training data and the number of model parameters affect the performance of ILBERT? (3) how do transfer learning, fine-tuning and data augmentation strategies influence the performance of ILBERT? (4) how does ILBERT perform in different IL property prediction tasks compared with other ML-based methods? (5) what insights could we acquire from the representations learned by ILBERT? (6) could ILBERT efficiently and accurately identify promising candidates from the vast chemical space for specific tasks?3.1 Chemical language and tokenization (Q1)
To investigate the most effective chemical languages and tokenization methods for predicting IL properties, various combinations of chemical languages and tokenization methods are compared across three IL property prediction tasks (Tm, lnη and ρ, each as an example of the three types of IL properties) in Fig. 2. It can be found that SMILES, DeepSMILES, and SELFIES provide approximate prediction results for all three tasks. However, InChI performs relatively poorly across all tokenization methods, indicating that InChI may not be suitable for predicting IL properties. This is probably due to more complicated syntax and arithmetic rules, which are challenging for language models. Moreover, SELFIES + CL results in notably poor performance due to excessively long sequence lengths (see Fig. S1D†) and erroneous splitting of multicharacter entities such as “[Ring1]” and “[Branch1]”. It should be noted that increasing the maximum sequence lengths of SMILES in the fine-tuning stage does not increase the performance of the model, but leads to a redundant increase in computational cost (see Table S5†). Among all combinations, AIS + SMILES consistently achieves the best prediction performance, suggesting that the classic SMILES representation is highly effective for IL property modeling. Compared with other tokenization methods, the AIS tokenization method not only eliminates ambiguities inherent in SMILES tokens but also better reflects the chemical environment around the corresponding atoms, resulting in superior modeling performance. Consequently, we recommend utilizing AIS + SMILES for IL property prediction.
 | ||
| Fig. 2 Impact of chemical language and tokenization on model performance in property prediction tasks. (A) Melting point. (B) Electrical conductivity. (C) Density. | ||
3.2 Pre-training dataset size and the number of model parameters (Q2)
To thoroughly examine the impacts of pre-training dataset size and the number of model parameters on model performance, three pre-trained models with varying parameters (see Table S3†) are constructed to evaluate their performance in the pre-training task and four representative downstream tasks (Tm, lnκ, ρ and λ). As illustrated in Fig. 3, the performance of the pre-training task and the first three downstream tasks (Tm, lnκ, and ρ) generally improves with an increase in pre-training dataset size. However, the performance increment gradually diminishes once the dataset size exceeds millions. Similarly, the same trend can be observed for the number of model parameters (see Fig. 3A–D). In contrast, for the specific task of λ, the above trend does not always remain consistent (see Fig. 3E), which is mainly because of the highest sparsity of data points (606) and IL types (44) among all properties. It is important to note that continuously increasing the number of model parameters and the pre-training dataset size also brings about higher and unaffordable computational costs. In this context, considering the trade-off between computational costs and performance, the pre-trained model with a moderate number of parameters (14 M) is selected for further modeling.
 | ||
| Fig. 3 Impact of pre-training dataset size and the number of model parameters on model performance in property prediction tasks. (A) Pre-training task. (B) Melting point. (C) Electrical conductivity. (D) Viscosity. (E) Thermal conductivity. | ||
3.3 Transfer learning, fine-tuning and data augmentation (Q3)
To demonstrate the effectiveness of pre-training, we compared the performance of three strategies: training from scratch, transfer learning, and fine-tuning of the twelve IL property prediction tasks, as illustrated in Fig. 4A. In comparison with training from scratch, transfer learning achieves an average reduction of 12.08% in MAE across all twelve tasks, while fine-tuning achieves a slightly greater reduction of 13.74%, with specific results detailed in Table S6.† The pre-training strategy proves to be beneficial for ten out of the twelve tasks, while making only trivial changes for the heat capacity and CO2 solubility prediction tasks. Possible reasons for the latter findings can be attributed to: (1) the data distribution of the heat capacity dataset is highly imbalanced, where a single IL accounts for 15% and the top three ILs make up 27.2% of the data points (see Fig. S11†); (2) CO2 solubility is strongly influenced by the interaction between ILs and CO2, which pre-trained models may not capture as they primarily focus on general IL features without considering such specific interactions. | ||
| Fig. 4 Overview of model performance and ablation study for IL property prediction. (A) Comparison of training from scratch, transfer learning, and fine-tuning approaches for modeling twelve IL properties. (B) Ablation study of data augmentation (DA). | ||
The impact of data augmentation on model performance is further analyzed for the tasks with fewer than 10000 data points. The results of ablation study (see Fig. 4B and Table S7†) indicate that both data augmentation and fine-tuning independently enhance the performance of the model, respectively. Furthermore, when applied together, they lead to additional improvements in the model performance, achieving an average reduction of 20.87% in MAE. Fig. 5 illustrates the results of five-fold cross-validation across all twelve IL property prediction tasks, verifying that most of the data points are concentrated along the diagonal region in the parity plot. In conclusion, the combination of fine-tuning and data augmentation is highly effective for IL property prediction and successfully mitigates the challenge of data scarcity.
 | ||
| Fig. 5 Results of five-fold cross-validation across all twelve IL property prediction tasks. (A) Melting point. (B) Glass transition temperature. (C) Thermal decomposition temperature. (D) Cytotoxicity towards the leukemia rat cell line IPC-81. (E) Electrical conductivity. (F) Viscosity. (G) Surface tension. (H) Refractive index. (I) Heat capacity. (J) Thermal conductivity. (K) Density. (L) CO2 solubility. | ||
3.4 Model performance of ILBERT (Q4)
To comprehensively evaluate the performance of ILBERT, the final models in the corresponding literature from which we collected the twelve IL property datasets are compared as benchmarks, respectively, including various ML-based prediction methods such as group contribution (GC) + ML,18,32 COSMO-RS derived descriptors + ML,33,48 RDKit descriptors + ML,46 graph convolutional networks (GCNs),47 transformer-CNN,44 ILTransR,7 and the consensus model.45 To ensure a fair comparison, we maintain consistency in the dataset and dataset split strategies during the evaluation. The detailed results are presented in Table 2.| Properties | Models | Split by | MAE | RMSE | R 2 | Source |
|---|---|---|---|---|---|---|
| T m | This work | IL | 26.03 ± 0.17 | 35.04 ± 0.25 | 0.782 ± 0.003 | Makarov et al.44 |
| Transformer-CNN | — | 36 | 0.77 | |||
| T g | This work | IL | 9.88 ± 0.23 | 15.96 ± 0.23 | 0.708 ± 0.009 | Makarov et al.45 |
| Consensus modela | 10.4 | 17 | 0.67 | |||
| T d | This work | IL | 21.88 ± 0.20 | 32.63 ± 0.32 | 0.816 ± 0.004 | Makarov et al.45 |
| Consensus modela | 24.6 | 33.6 | 0.81 | |||
| log10EC50 | This work | IL | 0.2007 ± 0.0039 | 0.2777 ± 0.0026 | 0.9400 ± 0.0011 | Wang et al.46 |
| SVM | 0.2628 | 0.3204 | 0.9202 | |||
| lnκ |
This work | IL | 0.350 ± 0.010 | 0.530 ± 0.012 | 0.888 ± 0.005 | Chen et al.33 |
| ML boosting COSMO-RS | 0.396 | — | 0.870 | |||
| lnη |
This work | IL | 0.326 ± 0.002 | 0.555 ± 0.006 | 0.883 ± 0.002 | Chen et al.7 |
| ILTransR | 0.35 | — | — | |||
| γ | This work | IL | 2.34 ± 0.15 | 3.65 ± 0.24 | 0.835 ± 0.021 | Baran and Kloskowski47 |
| GCNb | 2.71 ± 0.12b | 4.09 ± 0.11b | 0.794 ± 0.011b | |||
| n D | This work | IL | 0.0055 ± 0.0001 | 0.0086 ± 0.0002 | 0.9538 ± 0.0018 | Cao et al.32 |
| GC + XGBoost | — | 0.0149 | 0.863 | |||
| C p | This work | Random | 15.89 ± 3.18 | 24.30 ± 3.38 | 0.990 ± 0.003 | Liaqat et al.18 |
| GC | — | — | 0.987 | |||
| λ | This work | Random | 0.0021 ± 0.0001 | 0.0029 ± 0.0001 | 0.9880 ± 0.0010 | Wan et al.48 |
| COSMO-RS+MLR | — | 0.004281 | 0.9733 | |||
| ρ | This work | IL | 13.24 ± 0.26 | 26.20 ± 0.43 | 0.979 ± 0.001 | Chen et al.7 |
| ILTransR | 16.46 | — | — | |||
| xCO2 | This work | IL | 0.0343 ± 0.0004 | 0.0595 ± 0.0014 | 0.937 ± 0.003 | Chen et al.7 |
| ILTransR | 0.057 | — | — |
As seen, the models proposed in this work demonstrate superior performance across all twelve IL properties compared with the reference models in the corresponding literature. For properties related only to the molecular structure (namely Tm, Tg, Td, and log10EC50), the root mean square error (RMSE) decreases by 3.00%, 6.12%, 2.89%, and 13.33%, respectively. Notably, for the Tg and Td tasks, our model outperforms the ensemble results of four descriptor-less deep learning models, demonstrating its strong predictive capability. As for the second and third types of IL properties that are dependent on temperature and/or pressure, ILBERT also surpasses all the reference models, decreasing the MAE ranging from 6.85% for lnη to 39.82% for xCO2. To further assess whether our model could effectively capture temperature and/or pressure dependence, an IL namely 1-methyl-1-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide ([C3MPr][NTf2]) that appears in all twelve datasets is chosen as an example. The results shown in Fig. S12† indicate that the model accurately captures the temperature and/or pressure dependence of these properties in a wide range. Even in some cases of the lnη, γ, Cp, and ρ datasets, ILBERT still demonstrates robustness against data of uncertain quality. To further illustrate the differences between the two data split strategies (data point-based and IL-based), their impacts on the performance of 5-fold cross-validation evaluation are compared using conductivity, viscosity, and surface tension datasets as examples (see Table S9†). This demonstrates that the prediction metrics following the IL-based split strategy are significantly decreased, as the splitting ensures that the same IL does not appear simultaneously in both the training and testing sets. This approach enables a more rigorous assessment of model performance on unseen ILs, thereby providing a more reliable evaluation of the model's generalization capability. Furthermore, Table S10† presents a more extensive comparison of ILBERT's performance with that of models from other literature, further confirming its exceptional predictive capabilities.
3.5 Interpretability of ILBERT learned representations (Q5)
In addition to modeling performance, the interpretability of the model is another critical aspect that warrants further attention. To reveal the intrinsic knowledge learned by ILBERT, the attention mechanism of the transformer model is leveraged to visualize and interpret the attention scores from ILBERT using [C3MPr][NTf2] as an example (see Fig. 6A). It is noteworthy that the breaking and flattening of rings at specific atoms can result in non-adjacent positions in SMILES for atoms that are actually bonded in the IL structure. For instance, the tokens “[N+]1” and “C1”, as well as the brown-colored “O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S(O)” and yellow-colored “C(F)(F)F”, are not directly adjacent or explicitly related in the SMILES string, while these components are adjacent in the actual IL structure. ILBERT successfully differentiates the information of substructures and the connectivity of atoms from SMILES directly. Moreover, two visualization tools, Attention Visualizer and BertViz,57,58 are employed to interpret the ILBERT model from different perspectives. Attention Visualizer provides an intuitive illustration of token importance in transformer-based encoder models. Using the melting point prediction task as an example, we analyzed the contributions of SMILES tokens from four representative ILs (belonging to pyrrolidinium, phosphonium, imidazolium and pyridinium, respectively) to melting point prediction, with contribution magnitude represented by color intensity (see Fig. S13†). The results showed that the model primarily focused on the positive and negative charge centers of the ILs, which are inherently determined by their ionic composition, and assigned higher attention scores to specific functional groups, such as hydroxyl, ether, and tertiary amine groups. Additionally, BertViz, an interactive tool, is used to visualize the attention mechanisms in transformer-based language models. Fig. 6B and C illustrate the attention scores between input tokens in the first and sixth (final) layers, aggregated across four attention heads. Fig. 6D depicts the visualization effects of the first heads in the sixth layer. BertViz provided a comprehensive view of the implicit relationships learned by the ILBERT model in the chemical language, with certain heads focusing on functional groups and charge centers (e.g., Fig. 6D). However, given the complexity of deep learning models, we acknowledge that this interpretation represents only a preliminary understanding, and further research is required to fully explain such complex models.
S(O)” and yellow-colored “C(F)(F)F”, are not directly adjacent or explicitly related in the SMILES string, while these components are adjacent in the actual IL structure. ILBERT successfully differentiates the information of substructures and the connectivity of atoms from SMILES directly. Moreover, two visualization tools, Attention Visualizer and BertViz,57,58 are employed to interpret the ILBERT model from different perspectives. Attention Visualizer provides an intuitive illustration of token importance in transformer-based encoder models. Using the melting point prediction task as an example, we analyzed the contributions of SMILES tokens from four representative ILs (belonging to pyrrolidinium, phosphonium, imidazolium and pyridinium, respectively) to melting point prediction, with contribution magnitude represented by color intensity (see Fig. S13†). The results showed that the model primarily focused on the positive and negative charge centers of the ILs, which are inherently determined by their ionic composition, and assigned higher attention scores to specific functional groups, such as hydroxyl, ether, and tertiary amine groups. Additionally, BertViz, an interactive tool, is used to visualize the attention mechanisms in transformer-based language models. Fig. 6B and C illustrate the attention scores between input tokens in the first and sixth (final) layers, aggregated across four attention heads. Fig. 6D depicts the visualization effects of the first heads in the sixth layer. BertViz provided a comprehensive view of the implicit relationships learned by the ILBERT model in the chemical language, with certain heads focusing on functional groups and charge centers (e.g., Fig. 6D). However, given the complexity of deep learning models, we acknowledge that this interpretation represents only a preliminary understanding, and further research is required to fully explain such complex models.
 | ||
| Fig. 6 Visualization of model interpretability, taking [C3MPr][NTf2] as an example. (A) Attention scores within SMILES strings, and the higher scores indicate higher correlation of tokens. Attention visualization of SMILES tokens in ILBERT provided by BertViz. (B) Layer 1 (all head). (C) Layer 6 (all head). (D) The first head in Layer 6. | ||
To further analyze the learned representations, t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis is employed for dimensionality reduction. Specifically, we collect all distinct cations from the density dataset (that includes 31167 data points, 2257 types of ILs, and 763 types of cations). After extracting features using ILBERT, each cation is represented by a 512-dimensional feature vector. The t-SNE visualization of learned representations is shown in Fig. 7A, while Fig. S14A† displays the visualization of extended connectivity fingerprints (ECFPs) as comparisons. Even without further fine-tuning, ILBERT effectively separates almost all cation types, demonstrating its ability to capture rich structural information from chemical languages during pre-training. Additionally, the features fine-tuned for specific tasks (such as the melting point) are also visualized in Fig. 7B, with S14B† showing the corresponding results of ECFPs. The ILs with high melting points (depicted in light colors) are primarily composed of smaller halide anions. This is because smaller anions increase the melting point by promoting crystal effective accumulation as their volume decreases. It is evident that the clustering performance of the learned representations before and after fine-tuning surpasses that of the ECFPs, which partially explains the superior predictive performance of ILBERT.
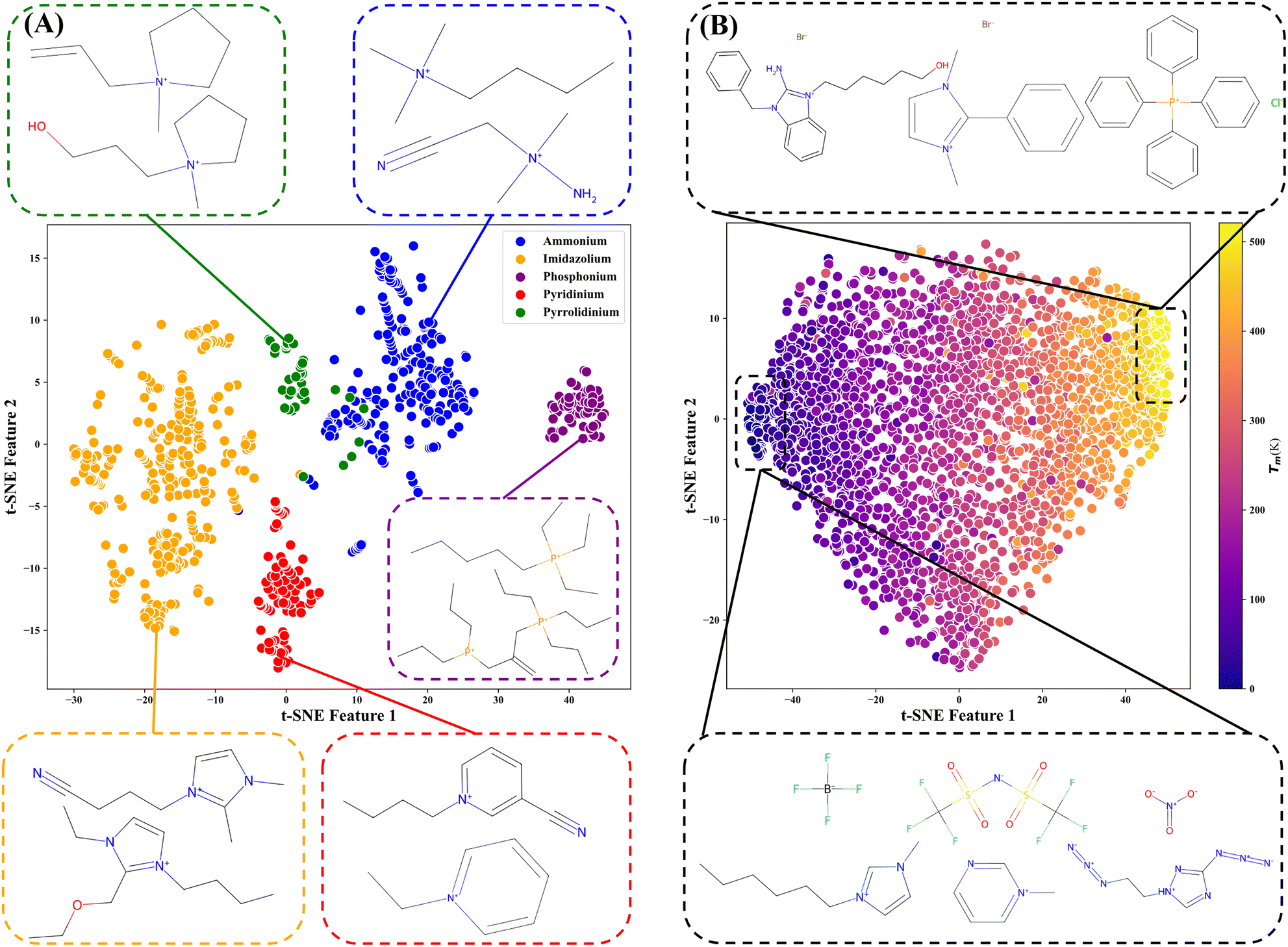 | ||
| Fig. 7 t-SNE analysis of learned representations before and after fine-tuning for specific tasks (melting point). (A) Learned representation (before fine-tuning) for cation classification. (B) Learned representation (after fine-tuning) for melting point prediction. | ||
3.6 Model application case study: high-throughput screening of IL electrolyte (Q6)
Using the proposed ILBERT model, the twelve physicochemical and thermodynamic properties of ILs can be predicted efficiently and accurately, enabling the design of task-specific ILs. In this study, a large-scale virtual screening database of 8333096 possible combinations of ILs established by Venkatraman et al.59 is utilized to demonstrate high-throughput screening. Thanks to the efficiency of ILBERT, predicting each property takes only about 2.5 hours on a single RTX A6000 GPU. The prediction results for all 12 predicted properties are available at https://github.com/Yu-Xin-Qiu/ILBERT.
As a case study, IL electrolytes are screened for their potential suitability in lithium-ion batteries. ILs are increasingly recognized as promising electrolyte materials due to their unique properties, such as high ionic conductivity at room temperature, excellent thermal stability, and improved safety compared to traditional organic solvents.60 Key characteristics include high ionic conductivity and low viscosity, while other factors such as the melting point, thermal decomposition temperature, and toxicity are also crucial for practical applications.61 Based on the review of literature and practical application requirements, the screening criteria in this study are as follows: Tm < 298 K, Td > 473 K, log10EC50 > 3.4; κ > 1.2 S m−1 and η < 100 mPa s at T = 298.15 K and P = 1 bar. Followed by these criteria, 50 candidates are retained, and their predicted viscosity and electrical conductivity are shown in Fig. 8A. From these, five ILs with the highest electrical conductivity are selected for further analysis. It can be observed that all five ILs share dicyanamide anions paired with imidazolium, pyrrolidinium and pyridinium cations (see Fig. 8B). To further illustrate ILBERT's ability to predict ILs not included in the training set, we select the first two ILs ([EMIM][DCA] and [DEIM][DCA]) with the highest conductivity for experimental validation, which do not appear in the conductivity training set. To be specific, [EMIM][DCA] is commercially available while [DEIM][DCA] is synthesized for the first time in this work.
 | ||
| Fig. 8 High-throughput screening results and experimental validation for ILs as electrolytes. (A) Viscosity and electrical conductivity of 50 candidate ILs. The top five ILs with the highest electrical conductivity are highlighted with red stars. (B) Structures of the five screened ILs with the highest electrical conductivity. (C–E) Electrical conductivity, viscosity and density of [EMIM][DCA]. (F–H) Electrical conductivity, viscosity and density of [DEIM][DCA]. | ||
Experimental data on melting points, electrical conductivity, viscosity, and density for the two ILs are shown in Fig. 8C–H and provided in Tables S11 and S12.† The results demonstrate that ILBERT maintains high predictive accuracy for novel ILs not included in the training set, with maximum errors within the cross-validation margin. In summary, ILBERT offers a reliable and efficient tool for predicting IL properties and enables large-scale high-throughput screening, paving the way for the design of advanced IL-based materials.
4 Conclusion
In this work, we present ILBERT, a flexible and powerful model for predicting IL properties based on chemical language. To address the challenge of limited data in IL property prediction, ILBERT is pre-trained on 31 million unlabeled molecular structures, enabling it to capture the inherent contextual information in chemical language. This pre-trained model is then fine-tuned on specific IL property prediction datasets. Benchmark tests across twelve representative IL properties (including both physicochemical and thermodynamic properties) demonstrate that ILBERT consistently outperforms other ML methods, proving its reliability and versatility.Our analysis of different chemical languages and tokenization combinations reveals that using SMILES with the AIS method is the most effective strategy for IL property prediction. In general, enlarging the pre-training dataset and the number of model parameters gradually improves prediction accuracy, but varies to a large extent and even does not hold depending on the size and distribution of the downstream database. Rigorous ablation studies confirm the benefits of transfer learning, fine-tuning and data augmentation. Notably, fine-tuning reduces the MAE by an average of 13.74% across all twelve prediction tasks compared to training from scratch. For tasks with fewer than 10000 data points, combining data augmentation with fine-tuning achieves an average MAE reduction of 20.87%.
Finally, we demonstrate ILBERT's capability in high-throughput screening of a large chemical space. In this case study, ILBERT is applied to identify promising IL candidates as electrolytes from a large dataset of 8333096 potential ILs. Two of the top candidates are experimentally validated and found to exhibit excellent electrochemical properties. We believe that ILBERT will serve as a valuable tool for the rational design of task-specific ILs, advancing their applications in diverse fields. Moving beyond, this study underscores that large chemical language models combining advanced natural language processing techniques with chemical informatics hold the power to transform the paradigms in computational chemistry and materials discovery.
While ILBERT represents a significant advancement in predicting physicochemical properties of ILs, we acknowledge that future research directions will focus on addressing its limitations, including high computational demands, challenges in interpretability, and overdependence on training data. To further move forward, given the widespread presence of data imbalance in IL datasets, one of the key focuses of future work will be to develop effective solutions through in-depth research to address this challenge. Additionally, other advanced deep learning models, such as Graphormer that has demonstrated effectiveness in various applications, have the potential to improve the accuracy of IL property predictions. Besides, future studies are highly worthwhile to investigate advanced modeling approaches for even more complex mixture systems such as deep eutectic solvents (DESs), aiming to guide the rational discovery of mixture systems and unlock their diverse applications.
Data availability
The datasets, code and trained models for this work have been made publicly available at Github https://github.com/Yu-Xin-Qiu/ILBERT and with DOI – 10.5281/zenodo.14601047. The version of the code employed for this study is version v1.0.0.Author contributions
Yuxin Qiu: conceptualization, methodology, writing – original draft. Zhen Song: conceptualization, validation, formal analysis, writing – review & editing, project administration. Guzhong Chen: data curation, writing – review & editing. Wenyao Chen: investigation, resources. Long Chen: formal analysis, resources. Kake Zhu: conceptualization, supervision, resources, project administration. Zhiwen Qi: resources, supervision. Xuezhi Duan: resources, supervision, project administration, funding acquisition. De Chen: supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China under the grant of 2024YFA1510302 and the National Natural Science Foundation of China (NSFC) under the grants of 22208098 and 22278134. Z. S. also achknowledges the support from the Shanghai Municipal Commission of Education.References
- Z. Lei, C. Dai, J. Hallett and M. Shiflett, Chem. Rev., 2024, 124, 7533–7535 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef CAS PubMed.
- H. Struebing, Z. Ganase, P. G. Karamertzanis, E. Siougkrou, P. Haycock, P. M. Piccione, A. Armstrong, A. Galindo and C. S. Adjiman, Nat. Chem., 2013, 5, 952–957 CrossRef CAS PubMed.
- S. Koutsoukos, F. Philippi, F. Malaret and T. Welton, Chem. Sci., 2021, 12, 6820–6843 RSC.
- G. Chen, Z. Song, Z. Qi and K. Sundmacher, Digital Discovery, 2023, 2, 591–601 RSC.
- Z. Song, J. Chen, J. Cheng, G. Chen and Z. Qi, Chem. Rev., 2024, 124, 248–317 CrossRef CAS PubMed.
- R. L. Gardas and J. A. P. Coutinho, AIChE J., 2009, 55, 1274–1290 CrossRef CAS.
- M. L. Alcantara, G. L. Bressan, P. V. A. Santos, M. F. V. Nobre, J. A. P. Coutinho, C. A. O. Nascimento and L. A. Follegatti-Romero, J. Mol. Liq., 2025, 417, 126616 CrossRef CAS.
- A. K. Halder, R. Haghbakhsh, E. S. C. Ferreira, A. R. C. Duarte and M. N. D. S. Cordeiro, J. Mol. Liq., 2025, 418, 126707 CrossRef CAS.
- A. Roosta, R. Haghbakhsh, A. R. C. Duarte and S. Raeissi, J. Mol. Liq., 2023, 388, 122747 CrossRef CAS.
- N. Hayer, T. Wendel, S. Mandt, H. Hasse and F. Jirasek, Chem. Eng. J., 2024, 158667, DOI:10.1016/j.cej.2024.158667.
- F. Jirasek and H. Hasse, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 31–51 CrossRef CAS PubMed.
- J. A. P. Coutinho, P. J. Carvalho and N. M. C. Oliveira, RSC Adv., 2012, 2, 7322–7346 RSC.
- R. Zhu, H. Kang, Q. Liu, M. Song, C. Gui, G. Li and Z. Lei, Ind. Eng. Chem. Res., 2024, 63, 1670–1679 CrossRef CAS.
- D. K. Mital, P. Nancarrow, T. H. Ibrahim, N. Abdel Jabbar and M. I. Khamis, Ind. Eng. Chem. Res., 2022, 61, 4683–4706 CrossRef CAS.
- S. Liaqat, M. d. B. Shahin, P. Nancarrow, S. Zeinab, T. Ibrahim, N. Abdel Jabbar, M. Khamis and S. McCormack, Ind. Eng. Chem. Res., 2023, 62, 16093–16112 CrossRef CAS.
- A. Roosta, R. Haghbakhsh, A. R. C. Duarte and S. Raeissi, Fluid Phase Equilib., 2023, 565, 113672 CrossRef CAS.
- E. I. Izgorodina, Z. L. Seeger, D. L. A. Scarborough and S. Y. S. Tan, Chem. Rev., 2017, 117, 6696–6754 CrossRef CAS PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- I. H. Bell, E. Mickoleit, C.-M. Hsieh, S.-T. Lin, J. Vrabec, C. Breitkopf and A. Jäger, J. Chem. Theory Comput., 2020, 16, 2635–2646 CrossRef CAS PubMed.
- M. G. Freire, L. M. Santos, I. M. Marrucho and J. A. P. Coutinho, Fluid Phase Equilib., 2007, 255, 167–178 CrossRef CAS.
- K. Paduszyński and M. Królikowska, Ind. Eng. Chem. Res., 2020, 59, 11851–11863 CrossRef.
- K. Klimenko and G. V. S. M. Carrera, J. Cheminf., 2021, 13, 83 Search PubMed.
- J. Jiang, W. Duan, Q. Wei, X. Zhao, L. Ni, Y. Pan and C.-M. Shu, J. Mol. Liq., 2020, 301, 112471 CrossRef CAS.
- B. Winter, C. Winter, T. Esper, J. Schilling and A. Bardow, Fluid Phase Equilib., 2023, 568, 113731 CrossRef CAS.
- B. Winter, J. Schilling and A. Bardow, Chem. Ing. Tech., 2022, 94, 1320 CrossRef.
- L. Fleitmann, P. Ackermann, J. Schilling, J. Kleinekorte, J. G. Rittig, F. vom Lehn, A. M. Schweidtmann, H. Pitsch, K. Leonhard, A. Mitsos, A. Bardow and M. Dahmen, Energy Fuels, 2023, 37, 2213–2229 CrossRef CAS.
- F. Jirasek and H. Hasse, Fluid Phase Equilib., 2021, 549, 113206 CrossRef CAS.
- R. Gurnani, S. Shukla, D. Kamal, C. Wu, J. Hao, C. Kuenneth, P. Aklujkar, A. Khomane, R. Daniels, A. A. Deshmukh, Y. Cao, G. Sotzing and R. Ramprasad, Nat. Commun., 2024, 15, 6107 CrossRef CAS PubMed.
- P. Cao, J. Chen, G. Chen, Z. Qi and Z. Song, Chem. Eng. Sci., 2024, 298, 120395 CrossRef CAS.
- Z. Chen, J. Chen, Y. Qiu, J. Cheng, L. Chen, Z. Qi and Z. Song, ACS Sustain. Chem. Eng., 2024, 12, 6648–6658 CrossRef CAS.
- H. Tran, R. Gurnani, C. Kim, G. Pilania, H.-K. Kwon, R. P. Lively and R. Ramprasad, Nat. Rev. Mater., 2024, 9, 866–886 CrossRef CAS.
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444 CrossRef CAS PubMed.
- X.-C. Zhang, C.-K. Wu, Z.-J. Yang, Z.-X. Wu, J.-C. Yi, C.-Y. Hsieh, T.-J. Hou and D.-S. Cao, Briefings Bioinf., 2021, 22, bbab152 CrossRef PubMed.
- Q. Dong, C. D. Muzny, A. Kazakov, V. Diky, J. W. Magee, J. A. Widegren, R. D. Chirico, K. N. Marsh and M. Frenkel, J. Chem. Eng. Data, 2007, 52, 1151–1159 CrossRef CAS.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, arXiv, 2017, preprint, arXiv:1706.03762, DOI:10.48550/arXiv.1706.03762.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, arXiv, 2018, preprint, arXiv:1810.04805, DOI:10.48550/arXiv.1810.04805.
- A. Radford, K. Narasimhan, T. Salimans and I. Sutskever, Improving language understanding by generative pre-training, 2018 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2020, preprint, arXiv:2010.09885, DOI:10.48550/arXiv.2010.09885.
- C. Kuenneth and R. Ramprasad, Nat. Commun., 2023, 14, 4099 CrossRef CAS PubMed.
- D. M. Makarov, Y. A. Fadeeva, L. E. Shmukler and I. V. Tetko, J. Mol. Liq., 2021, 344, 117722 CrossRef CAS.
- D. M. Makarov, Y. A. Fadeeva, L. E. Shmukler and I. V. Tetko, J. Mol. Liq., 2022, 366, 120247 CrossRef CAS.
- Z. Wang, Z. Song and T. Zhou, Processes, 2020, 9, 65 CrossRef.
- K. Baran and A. Kloskowski, J. Phys. Chem. B, 2023, 127, 10542–10555 CrossRef CAS PubMed.
- R. Wan, M. Li, F. Song, Y. Xiao, F. Zeng, C. Peng and H. Liu, Ind. Eng. Chem. Res., 2022, 61, 12032–12039 CrossRef CAS.
- K. Paduszyński, Ind. Eng. Chem. Res., 2019, 58, 5322–5338 CrossRef.
- Z. Song, H. Shi, X. Zhang and T. Zhou, Chem. Eng. Sci., 2020, 223, 115752 CrossRef CAS.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- N. O'Boyle and A. Dalke, DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures, 2018 Search PubMed.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, J. Cheminf., 2015, 7, 23 Search PubMed.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn.: Sci. Technol., 2020, 1, 045024 Search PubMed.
- X. Li and D. Fourches, J. Chem. Inf. Model., 2021, 61, 1560–1569 CrossRef CAS PubMed.
- U. V. Ucak, I. Ashyrmamatov and J. Lee, J. Cheminf., 2023, 15, 55 Search PubMed.
- A. Alam Falaki and R. Gras, arXiv, 2023, preprint, arXiv:2308.14850, DOI:10.48550/arXiv.2308.14850.
- J. Vig, arXiv, 2019, preprint, arXiv:1904.02679, DOI:10.48550/arXiv.1904.02679.
- V. Venkatraman, S. Evjen and K. Chellappan Lethesh, Data, 2019, 4, 88 CrossRef.
- H. Niu, L. Wang, P. Guan, N. Zhang, C. Yan, M. Ding, X. Guo, T. Huang and X. Hu, J. Energy Storage, 2021, 40, 102659 CrossRef.
- D. M. Makarov, Y. A. Fadeeva and L. E. Shmukler, J. Mol. Liq., 2023, 391, 123323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dd00035a |
| This journal is © The Royal Society of Chemistry 2025 |