
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel approach to protein chemical shift prediction from sequences using a protein language model†
He
Zhu
 ,
Lingyue
Hu
,
Yu
Yang
* and
Zhong
Chen
*
,
Lingyue
Hu
,
Yu
Yang
* and
Zhong
Chen
*
Department of Electronic Science, Fujian Provincial Key Laboratory of Plasma and Magnetic Resonance, State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen, China. E-mail: yuyang15@xmu.edu.cn; chenz@xmu.edu.cn
First published on 6th January 2025
Abstract
Chemical shifts are crucial parameters in protein Nuclear Magnetic Resonance (NMR) experiments. Specifically, the chemical shifts of backbone atoms are essential for determining the constraints in protein structure analysis. Despite their importance, protein NMR experiments are costly and spectral analysis presents challenges due to sample impurities, complex experimental environments, and spectral overlap. Here, we propose a chemical shift prediction method that requires only protein sequences as input. This low-cost chemical shift predictor provides a chemical shift corresponding to each backbone atom, offers valuable prior information for peak assignment, and can significantly aid protein NMR spectrum analysis. Our approach leverages recent advances in pre-trained protein language models (PLMs) and employs a deep learning model to obtain chemical shifts. Different from other chemical shift prediction programs, our method does not require protein structures as input, significantly reducing costs and enhancing robustness. Our method can achieve comparable accuracy to other existing programs that require protein structures as input. In summary, this work introduces a novel method for protein chemical shift prediction and demonstrates the potential of PLMs for diverse applications.
Introduction
Proteins are essential in biological systems, and understanding the structure of proteins is instrumental in comprehending their function. Nuclear Magnetic Resonance (NMR) spectroscopy serves as a complementary tool for studying proteins in their native environment, offering unique insights into their dynamics and interactions alongside the high-resolution structural information provided by X-ray crystallography and cryo-EM.1,2 Chemical shifts encode the local chemical environment around atoms and contain detailed information about protein structures. For instance, the chemical shifts of backbone atoms can be used to determine the internal restraints of a protein3 or serve as information for calculating the structural parameters.4,5 Despite these merits, NMR spectra of proteins are often intricate, and the spectral analysis is time-consuming, even for experienced researchers, owing to factors such as sample impurities and complex experimental conditions.To assist the process of protein structure determination via NMR, some chemical shift prediction programs based on machine learning have been proposed. These programs typically involve calculating expert-selected structural features such as amino acid type, ϕ/ψ/χ1 torsion angle, and other factors believed to influence chemical shifts. A model is then trained to map these features to chemical shifts. For example, PROSHIFT6 uses a comprehensive 350-dimensional feature set derived from protein structures, while SPARTA+7 and SHIFTX+8 utilize 113-dimensional and 97-dimensional feature sets, respectively. Some approaches do not rely on expert-selected features. Methods using graph neural networks, for instance, automatically extract features from protein structures.9 Additionally, sequence-based methods such as SPARTA10 and UCBShift-Y11 provide chemical shift predictions without requiring protein structures. These methods search databases for sequence fragments that match the local sequence of the target protein and use sequence homology assessments for predictions. However, these methods heavily rely on database search results and may fail when no suitable sequence match is found, reducing their robustness. Consequently, they are often combined with structure-based methods to create integrated models, exemplified by models like SHIFTX2 (ref. 8) and UCBShift.11 Beyond chemical shift prediction, some methods employ protein structure or statistical analysis to provide broader insights. These methods may offer rough chemical shift intervals to correct potential inaccuracies in chemical shift assignments. This holistic approach underscores the integration of various computational techniques to improve the reliability and accuracy of NMR-based protein studies.12,13
The majority of the aforementioned methods are effective only in the presence of high-quality protein structures, which are often unavailable as prior information in many NMR experiments. Notably, structure-based chemical shift prediction algorithms require high-quality protein structures, typically derived from crystallography, for their training sets. Herein lies a controversy: although the labels (chemical shifts) in these training sets originate from NMR experiments, the structures used for training are primarily from X-ray crystallography. This discrepancy raises concerns, as notable differences have been observed between crystal structures and solution-state NMR structures.14,15
Method
We introduce a method for predicting protein chemical shifts using only amino acid sequences, called PLM-CS (protein-language-model-based chemical shift prediction). This approach offers rapid inference speeds and remarkably simplifies the prediction process by eliminating the need for protein structure information. Central to inferring chemical shifts from protein sequences is the freeze fine-tuning16 applied to the protein language model. By treating the amino acid sequence like a language, the protein language model extracts semantic information from each amino acid sequence, encoding features that can be projected onto chemical shifts after training. This research not only presents a novel methodology for chemical shift prediction but also explores the potential applications of protein language models in a multitude of subsequent tasks.Protein language models
Protein language models initially gained prominence for their ability to infer the 3-dimensional structure of proteins from their sequences, addressing the longstanding protein folding problem.17 Understanding protein folding requires extracting co-evolutionary information from amino acid sequences. Unlike multi-sequence alignment (MSA) based methods, such as AlphaFold,18 protein language models eliminate the necessity for homologous sequences, enabling the extraction of evolutionary information from a single sequence and thus significantly reducing computational consumption. Protein language models treat protein sequences as sentences composed of 20 common amino acids as words. For instance, Evolutionary Scale Modeling (ESM)19 is a language model similar to BERT (Bidirectional Encoder Representations from Transformers)20 but tailored for proteins. It is trained to predict the types of amino acids that are randomly masked during the pre-training process, forcing it to learn the latent feature information embedded in the sequence. After pre-training, the model can transform each amino acid in the input protein sequence into a high-dimensional feature representation, encoding latent information in the sequence. This capability can be applied to various downstream tasks such as protein structure prediction,21–23 secondary structure prediction,24 and intrinsic protein disorder prediction.25By introducing the protein language model into chemical shift prediction, we eliminate the need for explicit protein structure information, unlike other chemical shift prediction programs. Our approach leverages the embedded structural information implicitly derived through the language model, which has proven effective in predicting chemical shifts through subsequent experiments.
Model
Our method combines a pre-trained ESM encoder (ESM2-650M21) with a transformer predictor, as illustrated in Fig. 1. The ESM2-650M model converts protein sequences into vector representations at the amino acid level. Using these vectors, the predictor calculates chemical shifts for each type of atom. Specifically, the ESM module treats each protein sequence as a sentence and functions as a semantic analyzer to extract intrinsic evolutionary information, which is then implicitly embedded in the output vector. In order to extract the information corresponding to chemical shifts in the embedding features obtained by ESM, we employ a predictor, which features two transformer encoders,26 with 8 attention heads and a 512-dimensional attention matrix. Before entering the encoder, the data are projected from 1280 to 512 dimensions via a linear layer, and positional encoding is added to capture sequential relationships among amino acids. We use the Gaussian Error Linear Unit (GELU)27 as the activation function in our model. The predictor takes these embeddings and outputs the chemical shifts for the backbone atoms of the corresponding amino acid. Detailed model structures of the multi-head attention module and feed-forward modules are shown in the ESI.†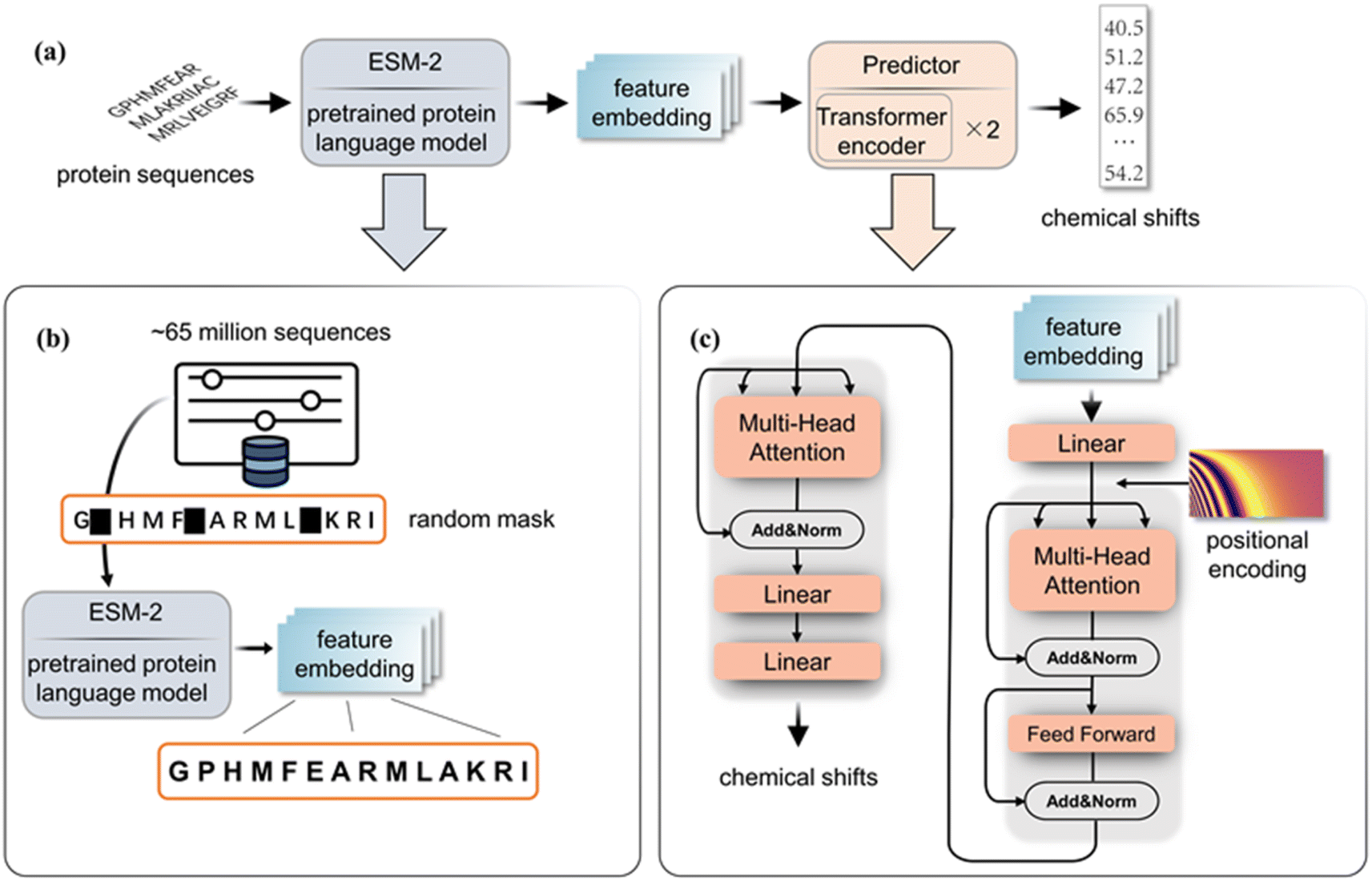 | ||
| Fig. 1 (a) The complete block diagram of PLM-CS. (b) The pre-training process of ESM2-650M. (c) The structure of the transformer predictor. | ||
Data preparation
To develop an accurate protein chemical shift predictor, a high-quality training set is essential. Advanced programs like SHIFTX2 and SPARTA+ achieve precise prediction results by carefully selecting the proteins with high-quality X-ray resolved crystal structures for their training sets. In contrast, our method does not require known high-quality protein structures, as it only uses protein sequences as input. Our training set includes 2429 re-referenced protein chemical shift files from RefDB.28 This dataset includes protein chemical shifts processed using the corresponding protein structure coordinate data via SHIFTX. This dataset has previously served as a sequence query database for SHIFTY8 and UCBShifty.11 While processing these data, we focus solely on the corrected Biological Magnetic Resonance Data Bank (BMRB)29 files from the RefDB dataset, as our method does not need protein structure information. For input sequences, we utilized those extracted from BMRB files to ensure that they correspond to actual protein sequences in NMR experiments, rather than relying on sequences from Protein Data Bank (PDB)30 files. This approach differs from structure-based chemical shift prediction methods that depend on structures and sequences in the PDB files. The sequences consist of the 20 common amino acid abbreviations, with other types of amino acids coded as ‘〈mask〉’ and excluded from training. The chemical shift distribution of each backbone atom for RefDB is illustrated in Fig. 2.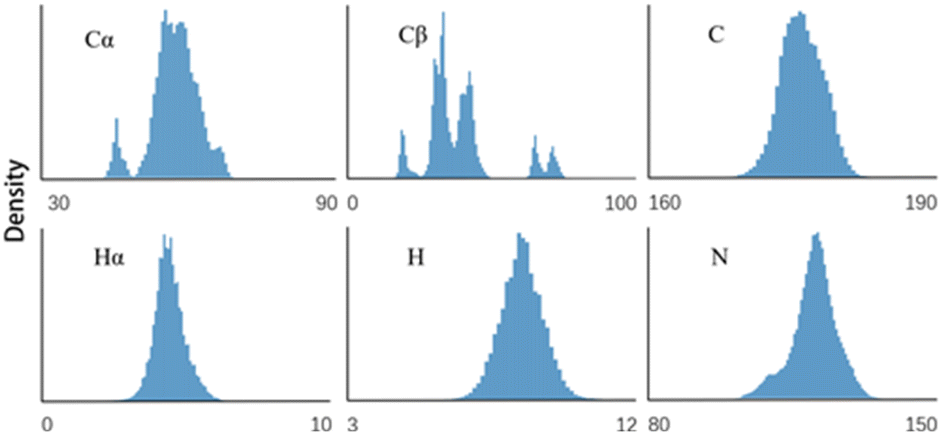 | ||
| Fig. 2 Chemical shift distributions of 6 backbone atoms in the RefDB database. | ||
To facilitate parallel training, sequences longer than 512 were removed, with only one protein (BMRB ID 5471) excluded from the dataset. Shorter sequences were padded to a uniform length of 512, resulting in a dataset of 2359 proteins. Each type of atom has its own label, necessitating the training of a separate model for each atom type. It should be noted that if longer sequences need to be processed, the network architecture may need to be modified, and a significant number of longer sequences (over 512 residues) should be included to improve performance.
Training
While training the model, we use Root Mean Square Error (RMSE) as the loss function. The ESM2-650M module is frozen during the training process, while parameters of the chemical shift predictor were initialized by the “Kaiming” initialization method31 and updated using the Adam optimization algorithm. It takes about 8 hours to train a single model on a GTX 3090 GPU. The hyperparameters for training the model of each atom type exhibit minor variations.Results
For consistency with other chemical shift prediction programs, we used the SHIFTX test dataset,8 which contains 61 proteins selected in consideration of whether they have a high-quality PDB structure solved by X-rays. These proteins were excluded from the training set to ensure unbiased test results. We refer to this as the SHIFTX test set.Fig. 3 illustrates the prediction results of our model on the SHIFTX test set. The Root Mean Square Error (RMSE) between the predicted values and actual labels is computed as a metric to evaluate the performance of the model, as shown in Table 1. The table also includes results from several state-of-the-art chemical shift prediction programs on the SHIFTX test set. The results of GNN are cited from ref. 9, while the results of the remaining programs are sourced from the SHIFTX2 website: https://www.shiftx2.ca/. The performance of UCBShift is not included because the SHIFTX test set was part of its training set.
 | ||
| Fig. 3 The predicted results of six models on their respective atoms after training. r stands for the correlation coefficient. | ||
| Method | Cα | Cβ | C | Hα | H | N |
|---|---|---|---|---|---|---|
| PLM-CS | 1.10 | 1.16 | 1.06 | 0.30 | 0.40 | 2.48 |
| GNN | 1.26 | 1.76 | 1.28 | 0.29 | 0.53 | 2.75 |
| SHIFTX+ | 0.77 | 0.86 | 0.87 | 0.20 | 0.38 | 2.09 |
| SHIFTX2 | 0.38 | 0.53 | 0.51 | 0.11 | 0.24 | 1.23 |
| PROSHIFT | 2.55 | 2.64 | 2.30 | 0.33 | 0.55 | 3.02 |
It is crucial to highlight that the performance comparison between our model and existing chemical shift prediction programs is not conducted under identical conditions. Unlike our model, which uses protein sequences as the only input, other programs rely on protein structures and some also require additional experimental parameters such as PH and temperature.
As evident from the table, SHIFTX+ and the integrated model SHIFTX2, which extract expert-selected features based on protein structures, have state-of-the-art performance on the SHIFTX test set, surpassing the GNN-based method that does not rely on expert-selected features. This indicates that features selected by experts still outperform those automatically extracted by the network in predicting protein chemical shifts. Our model, which only uses sequence input, achieves better performance than the GNN method but still does not reach the precision of SHIFTX2. This demonstrates the potential of the sequence-based protein chemical shift prediction method. However, this also highlights the limitations of PLM-CS: without sufficient prior knowledge, such as experimental conditions, the chemical shift information inferred from sequences alone still carries a certain degree of uncertainty.
Results on the custom dataset
In practical applications, protein crystal structures are not always available. Therefore, the structure-based approach needs to take into account the scenario when dealing with NMR-determined solution structures. Unlike methods like SHIFTX2 that rely on PDB structures for prediction, our approach does not require structure information, making it potentially more robust in cases where there are some differences between solution and solid-state protein structures. To verify this, we collected a dataset of 76 sequences, each with the corresponding chemical shifts from BMRB and structures from PDB. All structures in this dataset are solution structures determined by NMR. This custom test set allowed us to compare SHIFTX2's performance across different structural types and, moreover, to evaluate both SHIFTX2 and PLM-CS on previously unseen data, as none of these proteins are in RefDB. In selecting these sequences, our primary objective was to ensure a broad distribution of sequence diversity and corresponding protein structures, while also ensuring high-quality chemical shift assignments in BMRB. We refer to this as the solution-NMR test set. The PDB and BMRB information for the solution-NMR test set is all included in the ESI.†We tested the performance of SHIFTX2 and PLM-CS on the solution-NMR test set, as shown in Fig. 4 and Table 2. During evaluation, samples with RMSE values exceeding 3 times the mean for both SHIFTX2 and PLM-CS were treated as outliers and excluded from the results. During evaluation, samples with RMSE values exceeding 3 times the mean for both SHIFTX2 and PLM-CS were treated as outliers and excluded from the results. These samples contain a number of obvious or possible misassignments of chemical shifts, which are discussed in detail in the ESI.† Compared to its performance on the SHIFTX test set, the predictions of SHIFTX2 on the solution-NMR test set have a larger deviation from the reference values, resulting in higher RMSE values. This suggests that SHIFXT2, trained on crystal-structured proteins, gives a decreased prediction accuracy for proteins with solution structures, which are the focus of real solution NMR experiments. In contrast, our sequence-based PLM-CS demonstrates stable performance and robustness.
 | ||
| Fig. 4 Predicted results of SHIFTX2 and PLM-CS on the solution-NMR test set, presented as RMSE (ppm). | ||
| Method | Test set | Cα | Cβ | C | Hα | H | N |
|---|---|---|---|---|---|---|---|
| PLM-CS | SHIFTX | 1.10 | 1.16 | 1.06 | 0.30 | 0.40 | 2.48 |
| Solution-NMR | 1.32 | 1.39 | 1.28 | 0.31 | 0.41 | 2.71 | |
| SHIFTX2 | SHIFTX | 0.38 | 0.53 | 0.51 | 0.11 | 0.24 | 1.23 |
| Solution-NMR | 1.32 | 1.43 | 1.29 | 0.32 | 0.54 | 3.01 | |
| AF-SHIFTX2 | Solution-NMR | 1.36 | 1.40 | 1.37 | 0.27 | 0.46 | 2.92 |
We also consider scenarios where proteins lack experimentally determined crystal structures but can be modeled using tools like Alphafold (AF). To assess SHIFTX2's performance in such cases, we conducted tests using AF-predicted structures instead of the solution-NMR structures within the solution-NMR test set. As shown in Table 2, AF-SHIFTX2 outperforms SHIFTX2 with solution-NMR structures from PDB files, suggesting that Alphafold can supplement the protein structure information required by SHIFTX2. This is likely because AF-predicted structures are generally closer to crystal structures15 and thus align better with SHIFTX's training dataset. Compared to the proposed PLM-CS, AF-SHIFTX2 is slightly less accurate for most atoms but performs better on Hα atoms. Additionally, PLM-CS, which is based on a completely different framework, offers efficiency advantages, requiring only seconds to predict a protein's chemical shift, compared to the up to ten minutes Alphafold may take to predict a protein structure.
Ablation experiment
As aforementioned, the proposed method employs a dual-module design, utilizing the evolutionary information encapsulation capability of the ESM encoder and the advanced sequence-to-sequence transformation of the transformer, to provide a robust solution for protein chemical shift prediction. To assess the effects of these two modules, two ablation experiments were conducted as shown in Fig. 5. In the first ablation model, we replaced the transformer predictor with a linear layer, naming it ESM + Linear. Unlike the complete model, ESM + Transformer, ESM + Linear lacks crucial sequence-processing modules, such as the attention mechanism block, significantly limiting its ability to fully capture the internal correlations between the ESM output embeddings. As a result, the performance of this prediction model reflects the direct correlation between the ESM output embeddings and the chemical shifts. This ablation model enables us to contrastively verify the effectiveness of the transformer predictor in further extracting features from the ESM output embeddings. | ||
| Fig. 5 The structures for the ablation experiments. (a) ESM encoder combined with a linear layer as the predictor. (b) One-hot encoder with the linear-layer predictor. | ||
In the second ablation model, termed One-hot + Linear, the ESM module is replaced with a one-hot encoder that converts amino acids into distinct vectors. This model serves as a baseline, allowing us to assess how effective the features extracted by the pre-trained ESM models from amino acid sequences are for predicting chemical shifts. ESM + Linear and One-hot + Linear are optimized using LinearRegression in sklearn.32
Table 3 presents the results of the ablation experiment. The One-hot + Linear (baseline) model directly maps amino acid types to chemical shifts. A method can surpass this baseline only if it integrates feature representations beyond the amino acid level. As can be seen from the table, the prediction RMSE of the ESM + Linear model is better than the baseline on most atom types. This indicates that the amino-acid-level representation obtained by the ESM2-650M model not only contains sequence-level information but also includes higher-level features such as structure information, which can be used to make more accurate predictions of chemical shifts. Nonetheless, the prediction RMSE for the Cβ atom is worse than that of the One-hot + Linear model. One possible reason for this is that the Cβ atom is located within the side chain of the amino acid, causing its chemical environment to be more influenced by the specific type of amino acid, as opposed to the more uniform environment experienced by other backbone atoms. As we can see from Fig. 2, the distribution of Cβ is more heterogeneous, with multiple distinct clusters related to specific amino acid types.
| Method | Cα | Cβ | C | Hα | H | N |
|---|---|---|---|---|---|---|
| ESM + Predictor (PLM-CS) | 1.10 | 1.16 | 1.06 | 0.30 | 0.40 | 2.48 |
| ESM + Linear | 1.79 | 3.55 | 1.89 | 0.38 | 0.56 | 3.55 |
| One-hot + Linear (baseline) | 2.21 | 2.81 | 2.25 | 0.49 | 0.67 | 4.28 |
The results of ablation experiments also demonstrate that our model (PLM-CS) outperforms ESM vector projection (ESM + Linear), indicating the effectiveness of our model in further extracting protein sequence information for predicting chemical shifts based on ESM vector representations.
Potential applications of PLM-CS
Comparing experimental data with prediction results can provide valuable insights for spectral interpretation, experimental guidance, and protein property analysis. For instance, a large discrepancy between predicted values and experimental assignments may indicate misassignments or inherent sample flexibility that causes structural uncertainty (see S2 in the ESI†). Additionally, PLM-CS can assist in the peak assignment for raw experimental data by estimating the likelihood of assigning each experimental peak to specific residues based on predicted chemical shifts (see S3 in the ESI†). However, it should be noted that this peak assignment approach, based on predicted chemical shifts, may only address a limited subset of peaks in experimental data due to relatively high prediction uncertainties and spectral crowding. Exploring the applications of PLM-CS will also be a focus of our future research.Discussion
Overall, the results highlight the potential of the protein language model for predicting chemical shifts. To our knowledge, this is the first method for predicting chemical shifts of protein atoms that requires only sequence information as input. The findings demonstrate that the ESM vector from the pre-trained ESM module contains rich information about the correlations within the protein sequence, making it suitable for obtaining chemical shift values. Although our method may not be as accurate as SHIFTX2, it offers a promising approach to predicting chemical shifts using only sequence information with minimal loss in accuracy. Comparisons with other state-of-the-art methods indicate that our approach provides a viable alternative when structural information is not available. The ablation experiments confirm that the ESM module plays a crucial role in this prediction task. However, incorporating a well-designed predictor can further enhance accuracy.Conclusions
This PLM-CS prediction method could be useful in scenarios where structural data are incomplete or unavailable. For instance, it can be used in the early stages of protein structure determination or high-throughput protein analysis. This approach could also be integrated into the analysis or processing of complicated NMR data, where accurate prediction of chemical shifts can effectively guide spectrum reconstruction or peak assignment.Data availability
All the training and data processing code is available at: https://github.com/doorpro/predict-chemical-shifts-from-protein-sequence [https://doi.org/10.5281/zenodo.14546356]. The training set and the two test sets can be found at: https://github.com/doorpro/predict-chemical-shifts-from-protein-sequence/tree/main/dataset [https://doi.org/10.5281/zenodo.14546356].Author contributions
Zhu He: conceptualization, methodology, program, and writing – original draft. Lingyue Hu: validation and writing – original draft. Yu Yang: supervision and writing – review & editing. Zhong Chen: supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Fujian Provincial Natural Science Foundation of China (2024J01054), the Natural Science Foundation of Xiamen, China (3502Z202373005), and the National Natural Science Foundation of China (22161142024, 11975192, and 12175189).References
- P. Selenko, D. P. Frueh, S. J. Elsaesser, W. Haas, S. P. Gygi and G. Wagner, In situ observation of protein phosphorylation by high-resolution NMR spectroscopy, Nat. Struct. Mol. Biol., 2008, 15(3), 321–329 CrossRef CAS PubMed.
- K. Wüthrich, NMR with Proteins and Nucleic Acids, Europhys. News, 2017, 17(1), 11–13 CrossRef.
- N. J. Fowler, M. F. Albalwi, S. Lee, A. M. Hounslow and M. P. Williamson, Improved methodology for protein NMR structure calculation using hydrogen bond restraints and ANSURR validation: the SH2 domain of SH2B1, Structure, 2023, 31(8), 975–986 Search PubMed.
- Y. Shen and A. Bax, Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks, J. Biomol. NMR, 2013, 56, 227–241 CrossRef CAS PubMed.
- A. Cavalli, X. Salvatella, C. M. Dobson and M. Vendruscolo, Protein structure determination from NMR chemical shifts, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(23), 9615–9620 CrossRef CAS PubMed.
- J. Meiler, PROSHIFT: protein chemical shift prediction using artificial neural networks, J. Biomol. NMR, 2003, 26, 25–37 CrossRef CAS PubMed.
- Y. Shen and A. Bax, SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network, J. Biomol. NMR, 2010, 48, 13–22 CrossRef CAS PubMed.
- B. Han, Y. Liu, S. W. Ginzinger and D. S. Wishart, SHIFTX2: significantly improved protein chemical shift prediction, J. Biomol. NMR, 2011, 50(1), 43–57 CrossRef CAS PubMed.
- Z. Yang, M. Chakraborty and A. D. White, Predicting chemical shifts with graph neural networks, Chem. Sci., 2021, 12(32), 10802–10809 RSC.
- Y. Shen and A. Bax, Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology, J. Biomol. NMR, 2007, 38(4), 289–302 CrossRef CAS PubMed.
- J. Li, K. C. Bennett, Y. Liu, M. V. Martin and T. Head-Gordon, Accurate prediction of chemical shifts for aqueous protein structure on ‟Real World” data, Chem. Sci., 2020, 11(12), 3180–3191 RSC.
- B. Wang, Y. Wang and D. S. Wishart, A probabilistic approach for validating protein NMR chemical shift assignments, J. Biomol. NMR, 2010, 47(2), 85–99 CrossRef CAS PubMed.
- H. Dashti, M. Tonelli, W. Lee, W. M. Westler, G. Cornilescu and E. L. Ulrich, et al., Probabilistic validation of protein NMR chemical shift assignments, J. Biomol. NMR, 2016, 64(1), 17–25 CrossRef CAS PubMed.
- N. J. Fowler, A. Sljoka and M. P. Williamson, The accuracy of NMR protein structures in the Protein Data Bank, Structure, 2021, 29(12), 1430–1439 CrossRef CAS PubMed.
- N. J. Fowler and M. P. Williamson, The accuracy of protein structures in solution determined by AlphaFold and NMR, Structure, 2022, 30(7), 925–933 CrossRef CAS PubMed.
- Guo Y., Shi H., Kumar A., Grauman K., Rosing T. and Feris R., ed, Spottune: transfer learning through adaptive fine-tuning, Proceedings Of The IEEE/CVF Conference On Computer Vision And Pattern Recognition, 2019 Search PubMed.
- K. A. Dill and J. L. MacCallum, The protein-folding problem, 50 years on, Science, 2012, 338(6110), 1042–1046 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin and J. Liu, et al., Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(15) CrossRef CAS PubMed.
- J. Devlin, M.-W. Chang, K. Lee, and K. Toutanova, Bert: Pre-training of deep bidirectional transformers for language understanding, arXiv, 2018, preprint, arXiv:181004805, DOI:10.48550/arXiv.1810.04805.
- R. Chowdhury, N. Bouatta, S. Biswas, C. Floristean, A. Kharkar and K. Roy, et al., Single-sequence protein structure prediction using a language model and deep learning, Nat. Biotechnol., 2022, 40(11), 1617–1623 CrossRef CAS PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu and W. Lu, et al., Evolutionary-scale prediction of atomic-level protein structure with a language model, Science, 2023, 379(6637), 1123–1130 CrossRef CAS PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu and W. Lu, et al., Language models of protein sequences at the scale of evolution enable accurate structure prediction, bioRxiv, 2022, 2022, 500902 Search PubMed.
- M. H. Hoie, E. N. Kiehl, B. Petersen, M. Nielsen, O. Winther and H. Nielsen, et al., NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning, Nucleic Acids Res., 2022, 50(W1), W510–W515 CrossRef PubMed.
- I. Redl, C. Fisicaro, O. Dutton, F. Hoffmann, L. Henderson and B. M. J. Owens, et al., ADOPT: intrinsic protein disorder prediction through deep bidirectional transformers, NAR:Genomics Bioinf., 2023, 5(2), lqad041 CrossRef.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones and A. N. Gomez, et al., Attention is all you need, Adv. Neural Inf. Process. Syst., 2017, 30 Search PubMed.
- D. Hendrycks, and, K. Gimpel, Gaussian error linear units (gelus), arXiv, 2016, preprint, arXiv:160608415, DOI:10.48550/arXiv.1606.08415.
- H. Zhang, S. Neal and D. S. Wishart, RefDB: a database of uniformly referenced protein chemical shifts, J. Biomol. NMR, 2003, 25, 173–195 CrossRef CAS PubMed.
- E. L. Ulrich, H. Akutsu, J. F. Doreleijers, Y. Harano, Y. E. Ioannidis and J. Lin, et al., BioMagResBank, Nucleic Acids Res., 2007, 36(suppl_1), D402–D408 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat and H. Weissig, et al., The protein data bank, Nucleic Acids Res., 2000, 28(1), 235–242 CrossRef CAS PubMed.
- He K., Zhang X., Ren S., and, Sun J., ed, Delving deep into rectifiers: Surpassing human-level performance on imagenet classification, Proceedings Of The IEEE International Conference On Computer Vision, 2015 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion and O. Grisel, et al., Scikit-learn: Machine Learning in Python, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental results and descriptions of the network structure. S1: performance of PLM-CS and comparisons with SHIFTX2; S2: examples of using PLM-CS for validation of the chemical shift data; S3: examples of using PLM-CS for peak assignments; S4: detailed structures of the transformer predictor. See DOI: https://doi.org/10.1039/d4dd00367e |
| This journal is © The Royal Society of Chemistry 2025 |