
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple similarity metric for comparing synthetic routes†
Samuel
Genheden
 *a and
Jason D.
Shields
*b
*a and
Jason D.
Shields
*b
aMolecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Pepparedsleden 1, SE-431 83 Mölndal, Sweden. E-mail: samuel.genheden@astrazeneca.com
bMedicinal Chemistry, Research and Early Development, Oncology R&D, AstraZeneca, 35 Gatehouse Drive, Waltham, MA 02451, USA. E-mail: jason.shields@astrazeneca.com
First published on 6th November 2024
Abstract
Experimentally validated routes to synthetic compounds can be compared to each other by quantitative metrics (step count, yield, atom economy), or by qualitative assessments (strategy, novelty). AI-predicted routes are typically compared to experimental syntheses to check for an exact match among the top-ranked predictions (top-N accuracy). This method is ideal for the evaluation of retrosynthetic algorithms on large datasets (>106 routes), but it cannot assess a degree of similarity between routes, which would be desirable for small datasets (<102 routes). Here, we present a simple method to calculate a similarity score between any two synthetic routes to a given molecule. The score is based on two concepts: which bonds are formed during the synthesis; and how the atoms of the final compound are grouped together throughout the synthesis. As a result, the similarity score overlaps well with chemists' intuition and provides a finer assessment of prediction accuracy.
Introduction
In the field of organic synthesis, molecules are synthesized in a stepwise fashion, starting with simple and commercially available building blocks and then carrying out chemical reactions to forge the desired bonds (and break undesired bonds) until the target molecule is in hand. The set of steps used to construct a molecule is known as the synthetic route. Due to the immense size of synthetically accessible chemical space (estimated to encompass >1020 compounds for molecules with ≤36 heavy atoms)1 and the large number of known chemical reactions,2 a given target molecule could theoretically have tens of thousands of plausible routes. Indeed, retrosynthesis algorithms, which predict synthetic routes by iterating backwards from the target molecule until commercial starting materials are achieved, are quite capable of generating >104 routes and are usually explicitly programmed to output a more human-manageable number. In actual practice, even the most intensively studied molecules have on the order of a hundred reported routes.‡How does a chemist compare one route to another? If both routes in question have been experimentally validated, then a set of practical concerns dominate the comparison: overall yield; cost of goods; and the safety and environmental sustainability of the overall process are all common metrics. This method may establish which route is the most efficient, but it is not well suited to addressing theoretical routes. When assessing theoretical routes, step count, complexity scores, and feasibility predictions can provide rank ordering. This approach is used by AI tools like AiZynthFinder and ASKCOS to present a small set of prioritised routes to the chemist for expert assessment.3,4 Routes can also be clustered by Tree Edit Distance (TED), which we currently use for AiZynthFinder output.5,6 This method is best suited to avoiding degeneracy, which arises from the algorithm selecting different versions of the same synthon. Another method is the Retro-BLEU score, which was introduced to estimate the overlap of the predicted sequences of reactions in a route with known sequences of reactions. However, such an approach is naturally limited by the availability of known sequences of reactions.7 Finally, when comparing one experimental route with multiple predicted routes, top-N accuracy is routinely used, with the experimental route considered as ground truth.8 A relaxed version of this has been suggested, where only the starting material overlap between the predicted route and the experimental route is taken into account.9
A more qualitative method of route comparison can be found in the field of total synthesis. Here, complex natural products are obtained in laboratory syntheses, often necessitating numerous steps and the development of new chemistry. When comparing routes to natural products, it is common to consider the “key step(s)” or “strategy” of the routes: which bond-forming step(s) generated the most complexity or provided the most novelty?10 Cernak et al. have recently described a graph edit distance method to visualize and select the most direct retrosynthetic routes to target molecules; setting up the calculation requires some manual definitions beforehand.11
We found the methods described above to be wanting for several desirable applications in drug discovery. In particular, we sought a similarity metric to compare the predicted routes of a given compound at point-of-design with its subsequent experimental route. Monitoring this score in aggregate for newly synthesized molecules would help to continuously assess the performance of AiZynthFinder. Furthermore, it could be a step on the way to “closing the loop,” on AI-proposed syntheses, making future predictions more accurate. Finally, it could replace our current clustering algorithm to ensure that chemists can quickly access a diverse selection of routes for their expert appraisal. An additional, more challenging goal was to approximate “key step” analysis such that the output concorded with chemist intuition.
Results and discussion
Defining similarity
| mR,i = {a1, a2,…,an} |

The atom similarity, Satom is then computed by summing the maximum overlap for each molecule in a route, doing this for both route X and Y, and then normalizing by the total number of molecules (N) in both routes:

It should be noted that the target compound is excluded from these calculations as by definition we ensure that both instances of the target compound are atom-mapped identically.
The bond similarity of a route is based on an analysis of which bonds in the target compound are formed over the course of the route. In particular, we define a reaction as a set of bonds, bij:
| rR,i = {b1,2, b3,4,…,bn,m} |
| ρR = {rR,i|∀i} |
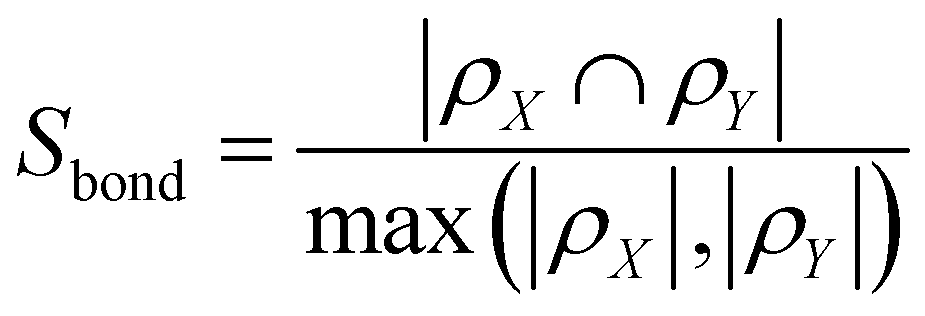
The total similarity is then computed as the geometric mean of the atom and bond similarity:

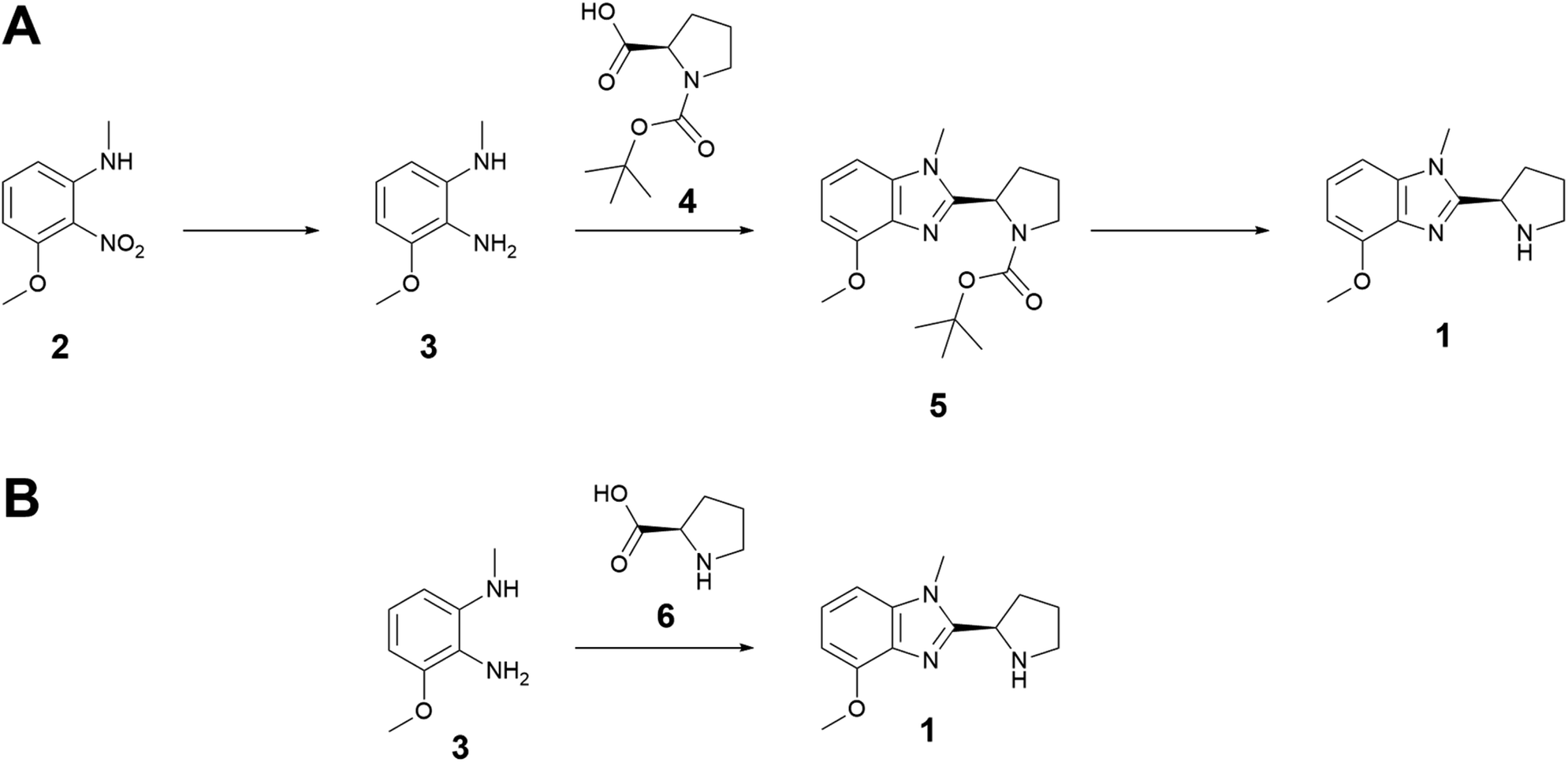 | ||
| Fig. 1 (A) Experimental route to benzimidazole 1, using nitroaniline 2 as a cheaper starting material and Boc-protected proline 4 to prevent side reactivity. (B) AiZynthFinder-predicted route to 1. The key cyclization step is the same and SA,B = 0.97. TEDA,B = 5.25. | ||
The combination of bond and atom similarity is a further advantage. Sbond is rooted in human practice, as numbering atoms and then keeping track of which bonds are formed over the course of a synthesis is a common exercise for students of organic chemistry. Satom addresses step sequence, another fundamental consideration in the practice of organic synthesis. For example, routes A and B (Fig. 2) differ only in the order of steps. The bond forming events are the same, but intermediates 9 and 12 have different atoms, leading to an Satom score of 0.90 and an overall SA,B of 0.95.
 | ||
| Fig. 2 Three syntheses to make hypothetical pyrimidine 11 starting from 7. (A) Buchwald–Hartwig, SNAr sequence. (B) SNAr, Buchwald–Hartwig sequence. (C) Chan–Lam, SNAr sequence. SA,B = SB,C = 0.95; SA,C = 1.0. TEDA,B = 4.62; TEDB,C = 4.63; TEDA,C = 1.04. | ||
There are several limitations that must be considered when using this similarity score. First, if rxnmapper fails to assign atom numberings correctly in any given reaction, then the score will be inaccurate.§ In the case of simple errors like failure to recognize hydroxide as the source of oxygen in an ester hydrolysis, this will not perturb the score greatly, but for larger errors like incorrectly mapped rearrangement reactions the effect is greater. Second, the Satom component is calculated based on atom groupings but ignores connectivity. Third, stereochemistry is ignored altogether. A more subtle limitation is shared with other methods of comparing synthetic routes: namely, the concept of a “step” is underdefined. Multiple reactions are often carried out in a one-pot or telescoped fashion. Two routes that share the same fundamental transformations in the same sequence but report the individual steps differently will return a score less than unity (see ESI† for an example).
Beyond these “hard” limitations there are also limitations by design. As written, the similarity algorithm does not consider atoms that are absent in the final product. This approach is well suited to our specific interests in AI retrosynthesis and medicinal chemistry, in which synthons and overall strategy are more important than (for example) atom economy or choice of protecting group. Thus, routes A and C in Fig. 2 return SA,C = 1, because the Buchwald–Hartwig and Chan–Lam couplings—although they would have different conditions, reagents, byproducts, and side products—form the same C–N bond at the same point in the synthesis. Modifying the algorithm to include atoms that are not present in the target could prove useful if comparing many, highly similar routes. Furthermore, the current code can only compare routes that terminate in the same final product; comparing routes to different final compounds for similarity of overall strategy will be the focus of future work.
Case studies
 | ||
| Fig. 3 Select key steps and intermediates of the medicinal chemistry routes (A and B), process route (C), and AiZynthFinder route (D) to atorvastatin 14. | ||
As a further test, we used AiZynthFinder to predict a retrosynthesis of atorvastatin.¶ The highest ranked predicted route was compared to the three experimental routes. Simple visual analysis of the key steps suggests high similarity to the process chemistry route: both routes employ a Knoevenagel condensation, a Stetter reaction, and a Paal–Knorr pyrrole cyclization, all in the same sequence (see ESI† for full routes). The similarity algorithm concurs, with SC,D = 0.74 compared to scores less than 0.5 between both medicinal chemistry routes and the predicted route. It is worth noting that none of the AiZynthFinder results match any of the published routes exactly, demonstrating again that top-N analysis is not fit for small datasets.
In the ESI,† we compare the route similarities to TED. There is a good correlation for these four routes, but TED is unbounded and therefore harder to interpret than the similarity metric.
Before running the analysis, we predicted that the routes would split into two clusters based on their penultimate intermediate (Fig. 4). Beyond that, it seemed likely that the two Kuehne syntheses would give the highest similarity scores among all pairwise comparisons, as they intercept complex intermediate 26; and that the Woodward route, being the only exclusively linear sequence, would have the lowest similarity to all other routes. The Martin route is explicitly aimed at mimicking the biosynthesis of strychnos alkaloids via a geissoschizine-like intermediate (29), and we predicted that it would be most similar to the proposed biosynthetic pathway as a result.
 | ||
| Fig. 4 (A) The two penultimate intermediates used to make strychnine 21, isostrychnine 22 and Wieland-Gumlich aldehyde 23. (B) Kuehne's racemic synthesis begins with tryptamine 25 while the enantioselective synthesis begins with methyl L-tryptophanate 27. Both sequences intercept complex intermediate 26, then diverge again. (C) Biosynthetic intermediate geissoschizine 28 and Martin's similar intermediate 29. | ||
These predictions were largely borne out. As expected, the Kuehne enantioselective and racemic syntheses provided the highest pairwise similarity score (0.91). The Woodward route was noticeably dissimilar to the others even by visual inspection using a heat map (Fig. 5), with all SWoodward,route <0.6. The proposed biosynthesis of strychnine was indeed most similar to the Martin synthesis (0.74), although several other syntheses came close. One result highlighting limitations of the similarity algorithm is that the Vanderwal synthesis gave mostly high scores with all other routes, presumably as a consequence of its much lower step count than the others, as Satom is normalized by number of molecules in both routes. Finally, our prediction that the twelve routes would cluster into two overall groups based on penultimate intermediate was inaccurate (see ESI†); presumably the interception of a very late-stage common intermediate is insignificant compared to the highly variable earlier intermediates across a small dataset.
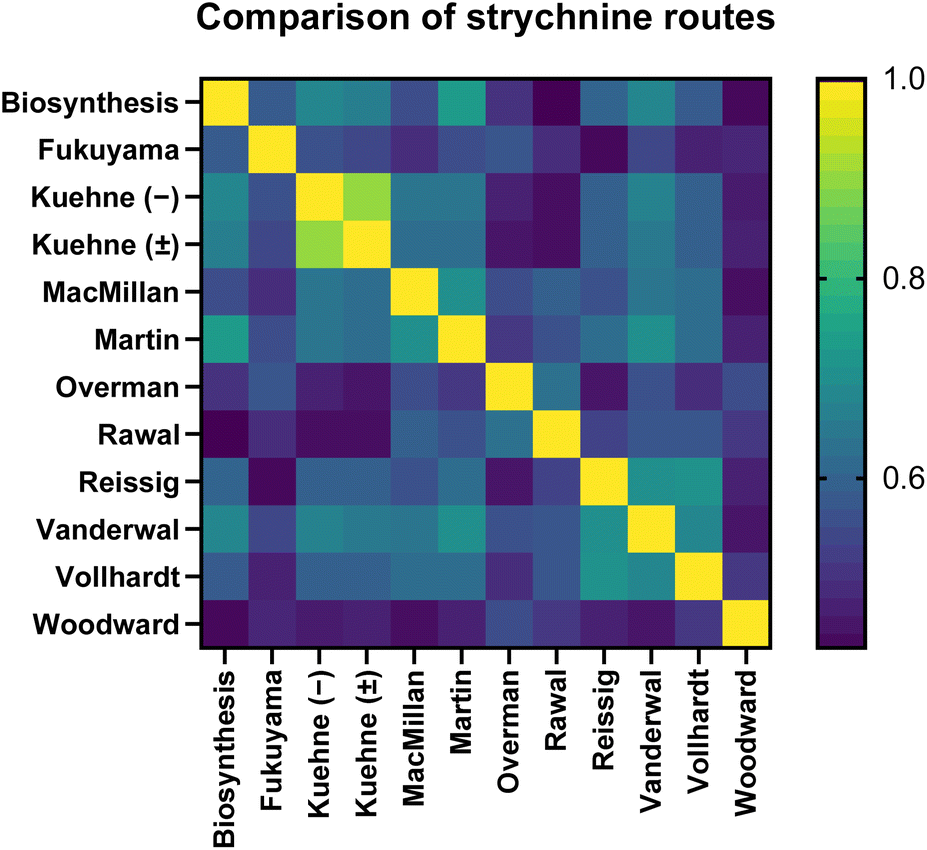 | ||
| Fig. 5 Heat map comparing similarity of the 12 routes to strychnine 21. See ESI† for numerical results. | ||
The TED calculations for the twelve strychnine routes return distances that are consistently larger than 30, due to the longer routes. The correlation between the similarity metric and TED is also considerably weaker than in the case of atorvastatin, especially for more dissimilar routes (see ESI†).
| Model | Success rate | Accuracya | Similarity | |
|---|---|---|---|---|
| Top-1 | Top-10 | |||
| a The fraction of targets for which we find the reference route as the first ranked prediction or among the top-10 ranked predictions. | ||||
| USPTO-PaRoutes32 | 97.2% | 0.24 | 0.54 | 0.91 |
| AZ-2019 (ref. 33) | 91.6% | 0.10 | 0.38 | 0.85 |
| AZ-retrained31 | 97.8% | 0.11 | 0.44 | 0.91 |
We then performed a similar benchmark on 4934 routes extracted from the Journal of Medicinal Chemistry, which is likely a more relevant benchmark set for pharmaceutical applications than PaRoutes. In Table 2, we show the results when employing a stock consisting of only the starting materials in the experimental reference routes. For this benchmark set, the success rate ranges from 69% for USPTO-PaRoutes to 86% for AZ-retrained, although the top-N accuracies are all relatively low. When it comes to similarity, the AZ-retrained model outperforms the other models with an average of 0.84, which is significantly higher than 0.71 for the older AZ-2019 model and 0.75 for USPTO-PaRoutes. Thus we observe a clear effect of training a retrosynthesis model on a combination of literature, patent, and internal data when predicting routes for medicinal chemistry targets. Looking at the distribution of the maximum similarity (see ESI†), we see that for a majority of the targets, the similarity is very high, with only a few targets for which the predictions are far from the reference routes. In the ESI† we outline additional benchmarks of retrosynthesis predictions.
Conclusions
We have presented a simple method for pairwise comparison of any set of routes to a given compound. For small datasets such as might be viewed by a synthetic chemist, it provides a suitably granular metric for route comparison. For large datasets of importance to data scientists, it provides an additional metric that supplements success rate and top-N accuracy. It is important to stress that the resulting similarity score is unrelated to feasibility, although we do speculate that if a theoretical route is similar to a proven experimental route, it is more likely to be feasible. Future research in this area will be to extend similarity comparison to routes for similar compounds, e.g. compounds that share a substructure but vary one portion of the molecule.Data availability
Routes and processed versions of them for atorvastatin and strychnine, Reaxys IDs for reactions in J. Med. Chem. routes, as well as PaRoutes reference routes for targets also in J. Med. Chem. are available as ESI.† PaRoutes data and code can be downloaded from Github: https://github.com/MolecularAI/paroutes. Code for the route similarity metric is available on Github: https://github.com/MolecularAI/reaction_utils.Author contributions
Conceptualization, investigation, writing – original draft, writing – review & editing: SG and JDS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to Matthew Burns, Mikhail Kabeshov, Christos Kannas, and Per-Ola Norrby for helpful discussions.Notes and references
- P. Ertl, J. Chem. Inf. Comput. Sci., 2003, 43, 374–380 CrossRef CAS.
- S. Szymkuć, T. Badowski and B. A. Grzybowski, Angew. Chem., Int. Ed., 2021, 60, 26226–26232 CrossRef PubMed.
- L. Saigiridharan, A. K. Hassen, H. Lai, P. Torren-Peraire, O. Engkvist and S. Genheden, J. Cheminf., 2024, 16, 57 Search PubMed.
- Y. Mo, Y. Guan, P. Verma, J. Guo, M. E. Fortunato, Z. Lu, C. W. Coley and K. F. Jensen, Chem. Sci., 2021, 12, 1469–1478 RSC.
- S. Genheden, O. Engkvist and E. Bjerrum, J. Chem. Inf. Model., 2021, 61, 3899–3907 CrossRef CAS.
- S. Genheden, O. Engkvist and E. Bjerrum, Mach. Learn.: Sci. Technol., 2022, 3, 015018 Search PubMed.
- J. Li, L. Fang and J.-G. Lou, Digital Discovery, 2024, 3, 482–490 RSC.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Chem. Sci., 2020, 11, 3316–3325 RSC.
- P. Torren-Peraire, A. K. Hassen, S. Genheden, J. Verhoeven, D.-A. Clevert, M. Preuss and I. V. Tetko, Digital Discovery, 2024, 3, 558–572 RSC.
- E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, Wiley, 1989 Search PubMed.
- Y. Lin, R. Zhang, D. Wang and T. Cernak, Science, 2023, 379, 453–457 CrossRef CAS.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Sci. Adv., 2021, 7(15) DOI:10.1126/sciadv.abe4166.
- J. D. Shields, R. Howells, G. Lamont, Y. Leilei, A. Madin, C. E. Reimann, H. Rezaei, T. Reuillon, B. Smith, C. Thomson, Y. Zheng and R. E. Ziegler, RSC Med. Chem., 2024, 15, 1085–1095 RSC.
- S. P. Kane, ClinCalc DrugStats Database, Version 2024.01, https://clincalc.com/DrugStats, updated January 1, 2024, accessed July 23, 2024 Search PubMed.
- B. D. Roth, D. F. Ortwine, M. L. Hoefle, C. D. Stratton, D. R. Sliskovic, M. W. Wilson and R. S. Newton, J. Med. Chem., 1990, 33, 21–31 CrossRef CAS PubMed.
- D. E. Butler, T. V. Le and T. N. Nanninga, US Pat., US5298627A, 1993 Search PubMed.
- J. S. Cannon and L. E. Overman, Angew Chem. Int. Ed. Engl., 2012, 51, 4288–4311 CrossRef CAS PubMed.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, J. Am. Chem. Soc., 1954, 76, 4749–4751 CrossRef CAS.
- S. D. Knight, L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1993, 115, 9293–9294 CrossRef CAS.
- V. H. Rawal, C. Michoud and R. Monestel, J. Am. Chem. Soc., 1993, 115, 3030–3031 CrossRef CAS.
- V. H. Rawal and S. Iwasa, J. Org. Chem., 1994, 59, 2685–2686 CrossRef CAS.
- M. E. Kuehne and F. Xu, J. Org. Chem., 1997, 62, 7950–7960 CrossRef CAS PubMed.
- M. J. Eichberg, R. L. Dorta, K. Lamottke and K. P. Vollhardt, Org. Lett., 2000, 2, 2479–2481 CrossRef CAS PubMed.
- M. Ito, C. W. Clark, M. Mortimore, J. B. Goh and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 8003–8010 CrossRef CAS.
- Y. Kaburagi, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2004, 126, 10246–10247 CrossRef CAS PubMed.
- C. Beemelmanns and H. U. Reissig, Angew Chem. Int. Ed. Engl., 2010, 49, 8021–8025 CrossRef CAS PubMed.
- D. B. C. Martin and C. D. Vanderwal, Chem. Sci., 2011, 2, 649–651 RSC.
- S. B. Jones, B. Simmons, A. Mastracchio and D. W. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS PubMed.
- M. E. Kuehne and F. Xu, J. Org. Chem., 1993, 58, 7490–7497 CrossRef CAS.
- B. Hong, D. Grzech, L. Caputi, P. Sonawane, C. E. R. Lopez, M. O. Kamileen, N. J. Hernandez Lozada, V. Grabe and S. E. O'Connor, Nature, 2022, 607, 617–622 CrossRef CAS PubMed.
- S. Genheden, P.-O. Norrby and O. Engkvist, J. Chem. Inf. Model., 2023, 63, 1841–1846 CrossRef CAS PubMed.
- S. Genheden and E. Bjerrum, Digital Discovery, 2022, 1, 527–539 RSC.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, Chem. Sci., 2020, 11, 154–168 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00292j |
| ‡ A Reaxys search for “ibuprofen” conducted on July 23, 2024 provided ∼600 published preparations of ibuprofen, many of which are degenerate; “taxol” ∼400; “aspirin” and “morphine” ∼100. The vast majority of laboratory-synthesized molecules have only one reported route, simply because they were made to test a specific hypothesis and then not pursued. Furthermore, the route in question is often only “reported” in that it exists in a notebook. |
| § To ensure correct atom mapping in our case studies, all results with our small datasets were checked by eye. In four cases this led to re-mapping, typically involving either one-atom reactants (e.g. a sulfur ylide in Kuehne's racemic strychnine synthesis) or carbon-centered leaving groups (e.g. Woodward's formal homologation of an acid to a vinyl acetate, which involves a decarboxylation). The more complex the route, the more likely rxnmapper is to misassign atoms and thus propagate errors; strychnine is an extreme example and we do not anticipate the need for manual re-mapping outside of natural products. |
| ¶ Initially AiZynthFinder predicted a one-step synthesis from commercially available atorvastatin lactone. In order to prevent AiZynth from utilizing any commercially available late-stage intermediates, we imposed a 250 Da weight limit on the starting materials, and restricted it to eMolecules stock. |
| || Some of these syntheses are “formal” syntheses; that is, they were only carried out experimentally up to a known late-stage intermediate, typically isostrychnine or the Wieland-Gumlich aldehyde. Because the formal syntheses terminate in strychnine itself they are all directly comparable using our algorithm. |
| This journal is © The Royal Society of Chemistry 2025 |