Machine learning for accelerated prediction of lattice thermal conductivity at arbitrary temperature
Received
5th September 2024
, Accepted 25th November 2024
First published on 27th November 2024
Abstract
Efficient evaluation of lattice thermal conductivity (κL) is critical for applications ranging from thermal management to energy conversion. In this work, we propose a neural network (NN) model that allows ready and accurate prediction of the κL of crystalline materials at arbitrary temperature. It is found that the data-driven model exhibits a high coefficient of determination between the real and predicted κL. Beyond the initial dataset, the strong predictive power of the NN model is further demonstrated by checking several systems randomly selected from previous first-principles studies. Most importantly, our model can realize high-throughput screening on countless systems either inside or beyond the existing databases, which is very beneficial for accelerated discovery or design of new materials with desired κL.
1. Introduction
The lattice thermal conductivity (κL) plays an essential role in various application scenarios. For example, thermoelectric materials require low κL to enhance energy conversion efficiency,1–4 while high κL is needed to dissipate excessive thermal energy in electronic devices.5–7 It is therefore extremely important to discover or design specific systems with desired κL. Theoretically speaking, the κL can be accurately calculated by solving the phonon Boltzmann transport equation (BTE)8,9 within the framework of density functional theory (DFT), which is however limited by high computational cost, especially for systems with large unit cells and/or low symmetry. On the other hand, although classic molecular dynamics (MD) simulations10,11 can deal with large-scale systems, their accuracy is highly dependent on the choice of interatomic potentials.
Very recently, machine learning (ML) methods have attracted considerable attention in predicting the κL of given systems since they can deal with a huge search space at extremely low computational cost.12–25 For instance, Wang et al.14 developed various nonlinear regression ML models based on the κL of 5486 materials, which were computed by using the Automatic GIBBS Library (AGL) method. They found that the eXtreme Gradient Boosting (XGBoost) model exhibits the best prediction performance, which is utilized to screen candidate thermoelectric materials with ultra-low κL. By combining graph neural networks and random forest algorithms, Zhu et al.17 predicted the room temperature κL of numerous inorganic compounds directly from their atomic structures, and a set of rare-earth chalcogenides were identified as a new class of promising thermoelectric materials. After a thorough algorithm comparison, Yang et al.19 found that Bayesian optimization20 using the Gaussian process allows for fast and accurate measurement of κL over a wide temperature range. In addition, Qin et al.24 constructed fifteen ML models for accurate prediction of κL, where the dataset consists of experimentally measured κL of 350 different materials and the input features include 8 basic properties of the compounds obtained from first-principles calculations. It should be noted that most of these studies were focused on the κL at 300 K, which is not beneficial for the discovery of systems with desired κL in a wide temperature range and large search space. Besides, some of the involved datasets contain κL calculated using semi-empirical models, which may lead to insufficient accuracy of the derived ML model. Moreover, to predict κL in a high-throughput style, it is necessary to adopt input features that can be readily obtained, which is however less considered in previous studies.
In this work, using a dataset completely obtained from first-principles calculations, we propose a neural network (NN) model by which the κL can be readily obtained at arbitrary temperature. The strong predictive power of our model is demonstrated by good agreement between the predicted and real κL, both inside and beyond the initial dataset. By leveraging the established NN model, we give a high-throughput prediction of the κL of 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 252 compounds from the Inorganic Crystal Structure Database (ICSD)26 in a wide temperature range from 100 to 1000 K, where many promising candidates are quickly identified for effective thermoelectric conversion or heat dissipation.
252 compounds from the Inorganic Crystal Structure Database (ICSD)26 in a wide temperature range from 100 to 1000 K, where many promising candidates are quickly identified for effective thermoelectric conversion or heat dissipation.
2. Methods
Our ML approach has been performed by adopting a NN algorithm, where the fully connected pyramid architecture contains an input unit, hidden layers, and the quantitative labels of κL as the output unit.27 To obtain an accurate NN model, the initial dataset is randomly divided into training (80%), validation (10%), and testing (10%) sets. The input features consist of 290 compositional descriptors derived from 58 elemental properties of the constituent atoms, which can be readily generated by the XENONPY package.28–30 As generally used in many ML methods, all the feature values are scaled to a range of 0–1. During the training process, the input unit receives the feature data, which is then manipulated by three hidden layers, each containing 150 neurons. When the data are moved from one neuron to another, their value will be multiplied by a particular weight parameter, and all the neurons are activated with an Exponential Linear Unit (ELU).31 It should be noted that the NN algorithm allows back propagation, which means that the weight and bias parameters of each neuron are updated so that the loss function can be minimized. To achieve the best prediction accuracy, the hyperparameters are fine-tuned by adopting the Adam optimizer with a learning rate of 0.005. Meanwhile, the batch size is set as 128 and a dropout of 0.2 is used for each hidden layer.
3. Results and discussion
To construct a reliable dataset, we have collected the κL of 103 crystalline systems at various temperatures (84–1200 K) from the literature,32,33 which in turn leads to 1795 entries for the NN training. It should be noted that all the κL values were obtained from accurate first-principles calculations by solving the linearized phonon BTE using an iterative process. Fig. 1(a) shows the distribution of the 1795 samples, where we find that the κL value spans several orders of magnitude. For example, the κL of CsK2Sb (space group no. 225) is only 0.03 W m−1 K−1 at 1012 K. In contrast, the κL of SiC (space group no. 216) exceeds 5000 W m−1 K−1 at 148 K. Besides, we see from the inset that most κL are located in a narrow range of 0–10 W m−1 K−1. Such an uneven distribution of input data may adversely affect the training process and thus the prediction accuracy of the NN model. Therefore, instead of directly using the original κL values, we adopt their natural logarithms so that an approximate normal distribution can be obtained, as shown in the histogram in Fig. 1(b).
 |
| | Fig. 1 The distribution of 1795 entries in the initial dataset according to (a) their lattice thermal conductivities and (b) the natural logarithmic values. The inset of (a) shows detailed distribution for those with small κL. | |
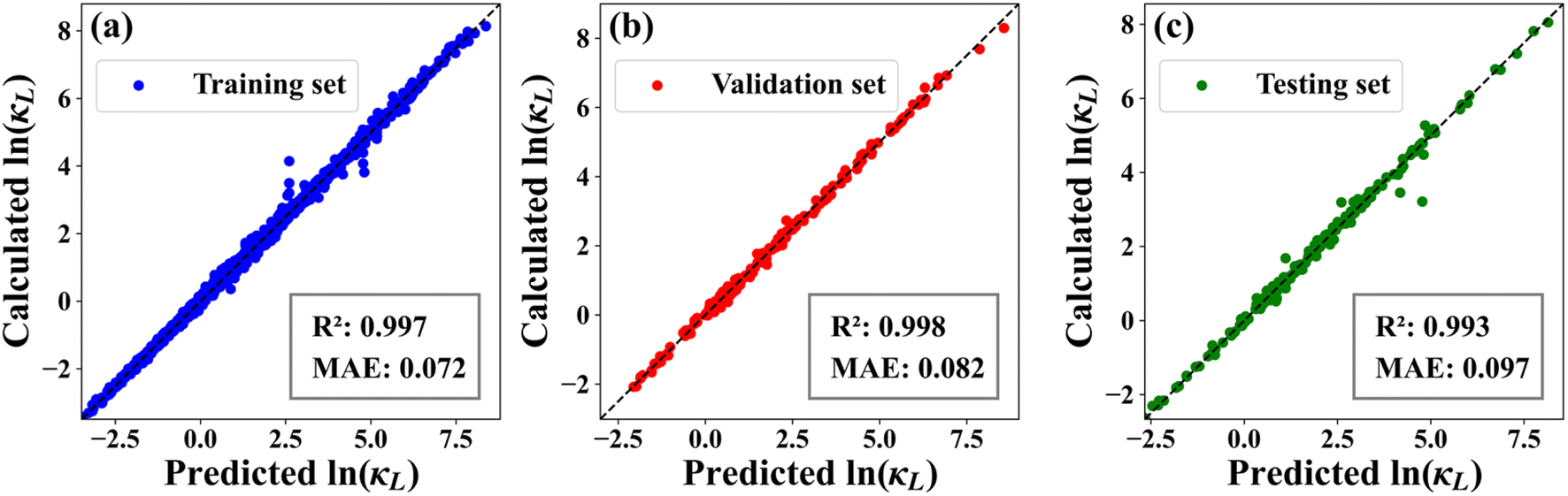
It is well known that the input features play a crucial role in determining the predictive power of the ML model. In the present work, we adopt 290 compositional descriptors generated from 58 elemental properties of the constituent atoms. In addition, the feature vector includes space groups of the systems and temperature by default. By utilizing the training and validation sets as benchmarks for hyperparameter tuning, we establish a well-optimized NN model to rapidly predict the κL of any given system at arbitrary temperature. Fig. 2(a) and (b) respectively show the intuitive linear correlation between the NN-predicted and real κL (on the natural logarithmic scale) for the training (1436 entries) and validation sets (179 entries), where we see that all the data points are located around the dashed line representing equality. Besides, the coefficient of determination (R2) between the predicted and real ln(κL) is found to be 0.997 and 0.998 for the training and validation sets, respectively. Meanwhile, the corresponding mean absolute errors (MAEs) are as small as 0.072 and 0.082 (note that the involved ln(κL) varies from −4 to 8). Even for the testing set (180 entries) that is not used during the training process, the NN model can still give strong prediction accuracy. As illustrated in Fig. 2(c), the R2 between the predicted and real ln(κL) is as high as 0.993 with a small MAE of 0.097. All these findings suggest that the data-driven NN model is highly reliable and can be used to effectively predict the κL of crystalline materials.
 |
| | Fig. 2 The intuitive linear correlation between the real and NN-predicted lattice thermal conductivities (natural logarithmic values) for the (a) training, (b) validation, and (c) testing sets. | |
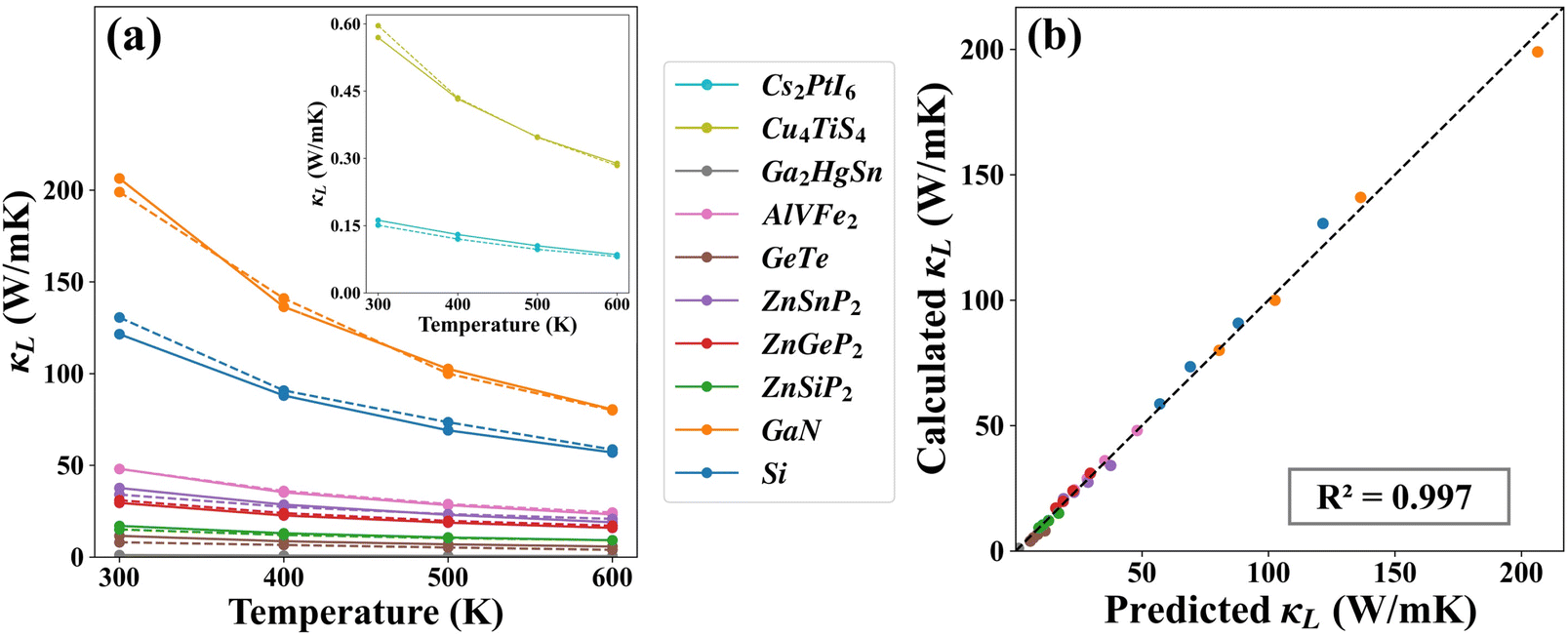
Beyond the initial dataset, we have employed the NN model to predict the κL of 10 compounds that are randomly selected from the literature, as shown in Fig. 3(a) in a temperature range from 300 to 600 K. Although the values (40 entries in total) span over several orders of magnitude, the NN-predicted κL are in good agreement with those obtained from first-principles calculations.34–41 For example, the room temperature κL of AlVFe2 (space group no. 216) is calculated to be 48.0 W m−1 K−1,39 which almost coincides with our NN-predicted result of 48.1 W m−1 K−1. Besides, the κL of GaN (space group no. 186) is predicted to be 102.5 W m−1 K−1 at 500 K, which is close to the calculated value of 100.0 W m−1 K−1.37 At a higher temperature of 600 K, the NN-predicted κL of Cu4TiS4 (space group no. 219) is found to be 0.29 W m−1 K−1, which is almost identical to the first-principles result of 0.28 W m−1 K−1.41 To have a statistical analysis, Fig. 3(b) shows the intuitive linear correlation between the real and predicted values of κL for these 10 compounds at different temperatures. We see that all the data points are distributed around the dashed line with a slope of 1, and the R2 between the real and predicted κL is as high as 0.997. All these observations indeed substantiate the strong predictive power of our NN model in evaluating the κL at various temperatures.
 |
| | Fig. 3 (a) The NN-predicted κL (solid line) of ten randomly selected compounds beyond the initial dataset, plotted as a function of temperature. For comparison, the results from first-principles calculations (dashed line) are also shown. The inset shows an enlarged view of those with ultra-small κL. (b) The intuitive linear correlation between the NN-predicted κL and those from first-principles calculations. | |
As mentioned above, the input features for our NN model can be readily obtained from 58 elemental properties of the constituent atoms, which is very beneficial to predict κL of any crystalline system at negligible computational cost. For instance, the κL of 32252 systems in the ICSD can be quickly obtained in a wide temperature range from 100 to 1000 K. Fig. 4 plots the distribution of these systems according to their predicted ln(κL), where we see that the quantity of systems possessing lower κL becomes increased while that with higher κL is decreased at elevated temperature. As a consequence, the average ln(κL) decreases with increasing temperature. Such an observation is consistent with the general understanding that the κL is usually inversely proportional to the temperature for most systems. More importantly, our high-throughput prediction provides a good opportunity to discover new materials with desired κL that are suitable for different application scenarios. For example, it is well-known that good thermoelectric materials require low κL to enhance the energy conversion efficiency. If we focus on room temperature, we find that 22050 compounds have ultra-small κL in the range of 0.1–5 W m−1 K−1. Among them, 4957 systems exhibit moderate band gaps (0.1–2.0 eV), which implies that they could be possible high-performance thermoelectric candidates. In particular, it is found that 582 compounds are composed of non-toxic and earth-abundant elements,42 which is strongly desirable and highly competitive for thermoelectric applications. Table 1 summarizes some of these candidate systems, where the room temperature κL is further restricted to be lower than 0.15 W m−1 K−1. On the other hand, we see from Table 2 that there are 50 systems with very high κL (exceeding 300 W m−1 K−1) at 300 K, which are suggested to be very promising candidates for heat dissipation. A typical example is the diamond with a NN-predicted κL as high as 2091.76 W m−1 K−1, which is consistent with that measured experimentally43 and further confirms the reliability of our ML approach. It should be noted that we focus on the systems with finite band gaps and the effect of electron–phonon interactions is not considered. Besides, it is surprising to find that some systems exhibit κL even larger than that of the diamond, such as C4Os2 (2148.42 W m−1 K−1, space group no. 194) and COs (3797.02 W m−1 K−1, space group no. 216), which deserve further theoretical and experimental investigations. It should be emphasized that although we are dealing with room temperature, a similar picture can also be found at other temperatures, as already implied in Fig. 4.
 |
| | Fig. 4 The distribution of 32252 systems in the ICSD according to their predicted lattice thermal conductivities (natural logarithmic values) at different temperatures. The blue circles refer to the average values. | |
Table 1 The 40 compounds screened from the ICSD with moderate band gaps (in units of eV) and ultra-small κL (in units of W m−1 K−1) at 300 K, which are desirable for thermoelectric conversion. The corresponding space group is also shown
| Compound |
Space group |
κ
L
|
Gap |
| Cs16O16Zn8 |
14 |
0.103 |
1.94 |
| Fe4K12O8 |
92 |
0.105 |
1.29 |
| I10K2Sn4 |
140 |
0.106 |
1.50 |
| Ca4Sn4Sr4 |
62 |
0.110 |
0.34 |
| Cs8I24Sn4 |
225 |
0.113 |
0.12 |
| Fe4Rb12S12 |
64 |
0.113 |
1.34 |
| Cs8Cu2K4O16Si4 |
136 |
0.113 |
1.65 |
| C12Cs4Fe2K2N12 |
14 |
0.114 |
0.14 |
| Cr8Fe4O32Rb4 |
62 |
0.116 |
1.89 |
| Cs2Mo6O18 |
12 |
0.120 |
0.77 |
| Fe4Rb8S10 |
2 |
0.120 |
0.93 |
| Cs4S6Ti2 |
36 |
0.121 |
1.70 |
| Fe4Rb12S12 |
14 |
0.121 |
1.31 |
| Cs6S27Ti6 |
146 |
0.124 |
1.45 |
| Ba6Cr4O18W2 |
194 |
0.125 |
1.75 |
| Ba6Cr2O10 |
140 |
0.125 |
1.47 |
| Cs4Li2Mn2O8 |
36 |
0.126 |
1.84 |
| Fe10K14S20 |
15 |
0.126 |
1.02 |
| Ba4Fe4Li2N6 |
15 |
0.127 |
0.10 |
| Fe6Na14O16 |
2 |
0.128 |
1.99 |
| Ba6Cr4Mo2O18 |
194 |
0.129 |
1.12 |
| Cr5CsS8 |
12 |
0.132 |
0.71 |
| Fe4Rb2S6 |
63 |
0.134 |
0.25 |
| Ba8Cr4O16 |
62 |
0.135 |
1.75 |
| Fe2K6O5 |
8 |
0.135 |
1.27 |
| Ba8Cr4Nb4O24 |
194 |
0.136 |
1.78 |
| Fe4K4Na8O12 |
62 |
0.138 |
1.98 |
| Fe4K8O10 |
14 |
0.138 |
2.00 |
| Fe4K12O16 |
62 |
0.139 |
0.29 |
| Fe2K4Na2O6 |
67 |
0.139 |
1.99 |
| I6Sn3 |
12 |
0.142 |
1.70 |
| Fe2K6O6 |
12 |
0.142 |
1.80 |
| Fe4K8O16 |
62 |
0.143 |
1.54 |
| Fe2Na12S8 |
36 |
0.144 |
1.87 |
| Fe2Na8O6 |
9 |
0.145 |
1.10 |
| Fe10Na6O18 |
15 |
0.146 |
1.54 |
| Fe2K3NaO8 |
164 |
0.147 |
1.40 |
| Fe4K12S12 |
14 |
0.147 |
1.25 |
| Cs4Cu4S16 |
19 |
0.148 |
1.87 |
| Cu4K4O36Ta12 |
53 |
0.149 |
1.44 |
Table 2 The 50 systems screened from the ICSD with finite band gaps and very high κL (in units of W m−1 K−1) at 300 K, which are desirable for efficient heat dissipation. The corresponding space group is also shown
| Compound |
Space group |
κ
L
|
| B4N4 |
62 |
327.14 |
| CSn |
216 |
338.96 |
| He |
191 |
351.72 |
| B2P2 |
186 |
359.27 |
| C2N4 |
36 |
373.29 |
| B4N4 |
9 |
378.98 |
| B4N4 |
8 |
379.82 |
| CHN |
44 |
388.99 |
| B3N3 |
160 |
413.05 |
| SiSn |
216 |
414.30 |
| C2Si2 |
186 |
421.59 |
| He |
225 |
424.36 |
| He |
229 |
433.51 |
| He2 |
194 |
438.02 |
| B12 |
166 |
449.77 |
| B6Si |
221 |
473.05 |
| CN2 |
119 |
492.50 |
| AsB2P |
115 |
514.13 |
| B12C3 |
166 |
519.33 |
| BP |
216 |
537.37 |
| C16 |
194 |
547.26 |
| CHN |
107 |
550.65 |
| C16 |
62 |
603.44 |
| C16 |
67 |
605.57 |
| B2N2 |
194 |
607.09 |
| CSi |
216 |
608.69 |
| B2N2 |
187 |
618.17 |
| B2N2 |
186 |
619.76 |
| BSb |
216 |
620.35 |
| C14 |
166 |
687.27 |
| C8 |
12 |
690.93 |
| C4Os4 |
198 |
749.25 |
| C12 |
194 |
795.39 |
| BN |
216 |
816.54 |
| C8 |
65 |
896.92 |
| B2C4N2 |
17 |
902.67 |
| CRu |
216 |
904.49 |
| AsB |
216 |
916.91 |
| C10 |
166 |
969.22 |
| CGe |
216 |
981.78 |
| C8 |
194 |
1176.85 |
| C8 |
206 |
1185.38 |
| C8 |
229 |
1192.31 |
| C4 |
139 |
1208.59 |
| C2 |
166 |
1495.80 |
| C4 |
194 |
1635.08 |
| BC2N |
25 |
1912.43 |
| C2 |
227 |
2091.76 |
| C4Os2 |
194 |
2148.42 |
| COs |
216 |
3797.02 |
It is important to note that the above-mentioned 32252 systems all feature integer stoichiometry. Beyond the ICSD or other materials databases, it is possible to construct countless samples with fractional stoichiometry by alloying or doping, which provides additional degrees of freedom to tune the κL. Within the framework of DFT, it is rather time-consuming or even prohibitive to calculate the κL of alloyed or doped systems because very large supercells are usually involved. This is especially the case for high-entropy materials, which hold promise for various applications by selecting specific elements and altering stoichiometry. Fortunately, such a challenging task can be readily fulfilled by using our NN model since the required 290 compositional descriptors are directly derived from the 58 elemental properties of the constituent atoms. Taking a binary system AwABwB as an example, where the stoichiometry wA and wB could be an integer or a fractional, if the elemental properties of A and B atoms are respectively denoted by fA,i and fB,i (i = 1, 2, …, 58), the 290 compositional descriptors can be calculated using:
| | | fmax,i = max(fA,i,fB,i) (max-pooling) | (1) |
| | | fmin,i = min(fA,i,fB,i) (min-pooling) | (2) |
| | | fsum,i = wAfA,i + wBfB,i (weighted sum) | (3) |
| |  | (4) |
| |  | (5) |
where

and

refer to the normalized composition summing up to one. In other words, the NN model is applicable for any compound with either integer or fractional stoichiometry, which allows us to discover or design new materials with target
κL in an even larger search space.
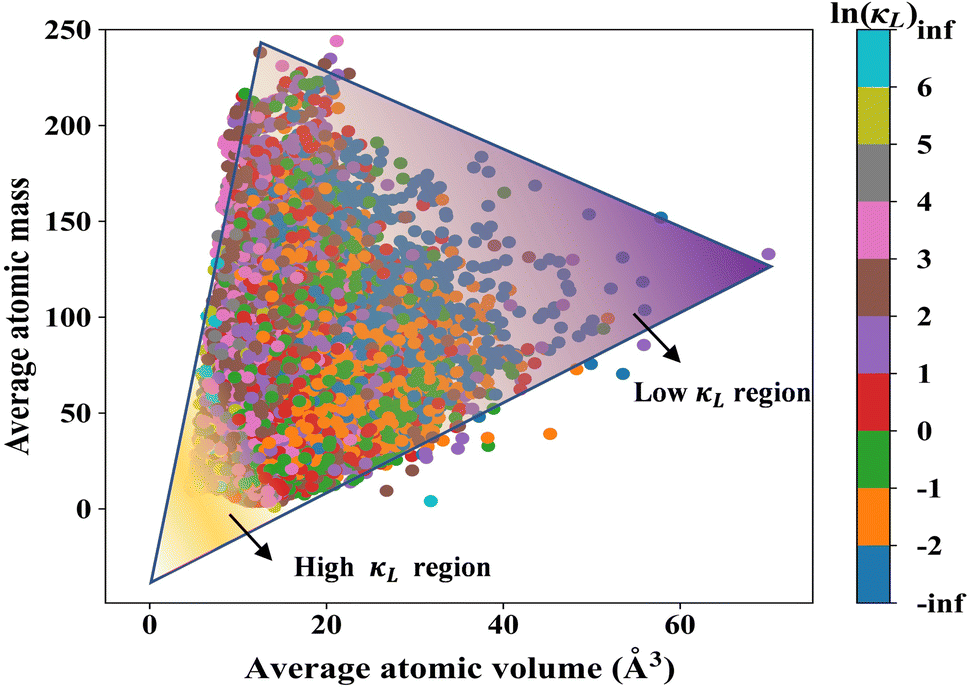
Although our NN model can be used to accurately predict the κL of any crystalline system at arbitrary temperature, it is somewhat similar to a “black box” which is not beneficial to understand the inherent physical mechanism. To address this issue, we become aware that materials with lower κL usually have weaker chemical bonds, lower phonon frequencies, and complex unit cells.44–46 In principle, such characteristics can be described by two simple structural parameters, namely, the average atomic volume (Vave) and the average atomic mass (mave). By respectively using Vave and mave as the horizontal and vertical coordinates, we plot in Fig. 5 the distribution of the above-mentioned 32252 compounds, where the corresponding room temperature κL (natural logarithmic value) is indicated by a color scale. It is interesting to note that the distribution can be approximately viewed as a triangle, where systems with low κL tend to be distributed in the upper right corner and those with high κL are more likely to be found in the lower left corner. The physical origin is that a larger Vave usually indicates longer distances between atoms in a given system and thus weaker bond strength, while heavier mave of the constituent atoms in general corresponds to lower phonon frequency. It should be mentioned that such kinds of systems also tend to have complex unit cells. As a consequence, the systems with simultaneously large Vave and mave would exhibit small κL and appear in the upper right corner of the triangle. All these findings demonstrate that our NN model has effectively captured and learned the inherent connection between the κL and the fundamental structural properties of crystalline materials. Accordingly, the predicted results are highly reliable and very beneficial for accelerated discovery of promising systems with desired κL in a large exploration space.
 |
| | Fig. 5 The distribution of 32252 compounds in the two-dimensional space defined by the average atomic volume and average atomic mass, where the color of the data points represents the NN-predicted lattice thermal conductivities (natural logarithmic values) at 300 K. | |
4. Summary
In summary, using the NN algorithm, we propose a machine learning model for rapid and accurate prediction of the κL of any crystalline system at arbitrary temperature. It is found that the NN model shows strong predictive power in the training, validating, and testing sets, as demonstrated by the high coefficient of determination and low mean absolute error. By leveraging such a data-driven model with physical intuition, we give a high-throughput prediction of the κL of 32252 compounds in a wide temperature range from 100 K to 1000 K, where many systems with ultra-small or extremely high κL are quickly identified. Our work not only enables accelerated discovery of candidate materials with desired κL, but also highlights their diverse applications such as thermoelectric conversion and heat dissipation.
Data availability
This study was carried out using publicly available data from ref. 32 and 33.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (Grant No. 62074114 and 12474019). The numerical calculations in this work have been done on the platform in the Supercomputing Center of Wuhan University.
References
-
D. M. Rowe, CRC Handbook of Thermoelectric, CRC Press, 1995 Search PubMed
 .
.
- K. F. Hsu, S. Loo, F. Guo, W. Chen, J. S. Dyck, C. Uher, T. Hogan, E. K. Polychroniadis and M. G. Kanatzidis, Cubic AgPbmSbTe2+m: Bulk Thermoelectric Materials with High Figure of Merit, Science, 2004, 303, 818–821 CrossRef CAS PubMed .
- K. Biswas, J. He, I. D. Blum, C. I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, High-performance bulk thermoelectrics with all-scale hierarchical architectures, Nature, 2012, 489, 414–418 CrossRef CAS PubMed .
- L. D. Zhao, S. H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals, Nature, 2014, 508, 373–377 CrossRef CAS PubMed .
- A. L. Moore and L. Shi, Emerging challenges and materials for thermal management of electronics, Mater, Today, 2014, 17, 163–174 CAS .
- X. Qian, J. Zhou and G. Chen, Phonon-engineered extreme thermal conductivity materials, Nat. Mater., 2021, 20, 1188–1202 CrossRef CAS .
- Z. He, Y. Yan and Z. Zhang, Thermal management and temperature uniformity enhancement of electronic devices by micro heat sinks: A review, Energy, 2021, 216, 119223 CrossRef .
- D. A. Broido, M. Malorny, G. Birner, N. Mingo and D. A. Stewart, Intrinsic lattice thermal conductivity of semiconductors from first principles, Appl. Phys. Lett., 2007, 91, 231922 CrossRef .
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, ShengBTE: A solver of the Boltzmann transport equation for phonons, Comput. Phys. Commun., 2014, 185, 1747–1758 CrossRef CAS .
- P. K. Schelling, S. R. Phillpot and P. Keblinski, Comparison of atomic-level simulation methods for computing thermal conductivity, Phys. Rev. B:Condens. Matter Mater. Phys., 2002, 65, 144306 CrossRef .
- Z. Fan, L. F. C. Pereira, H. Q. Wang, J. C. Zheng, D. Donadio and A. Harju, Force and heat current formulas for many-body potentials in molecular dynamics simulations with applications to thermal conductivity calculations, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 92, 094301 CrossRef .
- A. Seko, H. Hayashi, K. Nakayama, A. Takahashi and I. Tanaka, Representation of compounds for machine-learning prediction of physical properties, Phys. Rev. B, 2017, 95, 144110 CrossRef .
- L. Chen, H. Tran, R. Batra, C. Kim and R. Ramprasad, Machine learning models for the lattice thermal conductivity prediction of inorganic materials, Comput. Mater. Sci., 2019, 170, 109155 CrossRef CAS .
- X. Wang, S. Zeng, Z. Wang and J. Ni, Identification of Crystalline Materials with Ultra-Low Thermal Conductivity Based on Machine Learning Study, J. Phys. Chem. C, 2020, 124, 8488–8495 CrossRef CAS .
- C. Loftis, K. Yuan, Y. Zhao, M. Hu and J. Hu, Lattice Thermal Conductivity Prediction Using Symbolic Regression and Machine Learning, J. Phys. Chem. A, 2021, 125, 435–450 CrossRef CAS PubMed .
- H. Miyazaki, T. Tamura, M. Mikami, K. Watanabe, N. Ide, O. M. Ozkendir and Y. Nishino, Machine learning based prediction of lattice thermal conductivity for half-Heusler compounds using atomic information, Sci. Rep., 2021, 11, 13410 CrossRef CAS PubMed .
- T. Zhu, R. He, S. Gong, T. Xie, P. Gorai, K. Nielschb and J. C. Grossman, Charting lattice thermal conductivity for inorganic crystals and discovering rare earth chalcogenides for thermoelectrics, Energy Environ. Sci., 2021, 14, 3559 RSC .
- P. R. Chowdhury and X. Ruan, Unexpected thermal conductivity enhancement in aperiodic superlattices discovered using active machine learning, npj Comput. Mater., 2022, 8, 12 CrossRef .
- L. Yang, A. Gil, P. S. H. Leong, J. O. Khor, B. Akhmetov, W. L. Tan, S. Rajoo, L. F. Cabeza and A. Romagnoli, Bayesian optimization for effective thermal conductivity measurement of thermal energy storage: An experimental and numerical approach, J. Energy Storage, 2022, 52, 104795 CrossRef .
- M. F. Lazin, C. R. Shelton, S. N. Sandhofer and B. M. Wong, High-dimensional multi-fidelity Bayesian optimization for quantum control, Mach. Learn.: Sci. Technol., 2023, 4, 045014 Search PubMed .
- Z. Guo, P. R. Chowdhury, Z. Han, Y. Sun, D. Feng, G. Lin and X. Ruan, Fast and accurate machine learning prediction of phonon scattering rates and lattice thermal conductivity, npj Comput. Mater., 2023, 9, 95 CrossRef .
- Y. Srivastava and A. Jain, End-to-end material thermal conductivity prediction through machine learning, J. Appl. Phys., 2023, 134, 225101 CrossRef CAS .
- Z. Wang, J. Ma, R. Hu and X. Luo, Predicting lattice thermal conductivity of semiconductors from atomic-information-enhanced CGCNN combined with transfer learning, Appl. Phys. Lett., 2023, 122, 152106 CrossRef CAS .
- G. Qin, Y. Wei, L. Yu, J. Xu, A. D. Rodriguez, H. Wang, Z. Qin and M. Hu, Predicting lattice thermal conductivity from fundamental material properties using machine learning techniques, J. Mater. Chem. A, 2023, 11, 5801 RSC .
- Y. Luo, M. Li, H. Yuan, H. Liu and Y. Fang, Predicting lattice thermal conductivity via machine learning: a mini review, npj Comput. Mater., 2023, 9, 4 CrossRef .
-
ICSD, https://icsd.products.fiz-karlsruhe.de, accessed June 2024.
-
M. Anthony and P. L. Bartlett, Neural Network Learning: Theoretical Foundations, Cambridge University Press, 1999 Search PubMed .
- S. Ju, R. Yoshida, C. Liu, S. Wu, K. Hongo, T. Tadano and J. Shiomi, Exploring diamondlike lattice thermal conductivity crystals via feature-based transfer learning, Phys. Rev. Mater., 2021, 5, 053801 CrossRef CAS .
- C. Liu, E. Fujita, Y. Katsura, Y. Inada, A. Ishikawa, R. Tamura, K. Kimura and R. Yoshida, Machine Learning to Predict Quasicrystals from Chemical Compositions, Adv. Mater., 2021, 33, 2102507 CrossRef CAS PubMed .
-
XenonPy, https://github.com/yoshida-lab/XenonPy, accessed June 2024.
-
D. A. Clevert, T. Unterthiner, and S. Hochreiter, Fast and Accurate Deep Network Learning by Exponential Linear Units (ELUs), arXiv, 2016, preprint, arXiv:1511.07289, DOI:10.48550/arXiv.1511.07289.
- R. Juneja, G. Yumnam, S. Satsangi and A. K. Singh, Coupling the High-Throughput Property Map to Machine Learning for Predicting Lattice Thermal Conductivity, Chem. Mater., 2019, 31, 5145–5151 CrossRef CAS .
- R. Jaafreh, Y. S. Kang and K. Hamad, Lattice Thermal Conductivity: An Accelerated Discovery Guided by Machine Learning, ACS Appl. Mater. Interfaces, 2021, 13, 57204–57213 CrossRef CAS PubMed .
- J. Garg, N. Bonini and N. Marzari, High Thermal Conductivity in Short-Period Superlattices, Nano Lett., 2021, 11, 5135–5141 CrossRef PubMed .
- J. He, M. Amsler, Y. Xia, S. S. Naghavi, V. I. Hegde, S. Hao, S. Goedecker, V. Ozoliņš and C. Wolverton, Ultralow Thermal Conductivity in Full Heusler Semiconductors, Phys. Rev. Lett., 2016, 117, 046602 CrossRef PubMed .
- L. Wei, X. Lv, Y. Yang, J. Xu, H. Yu, H. Zhang, X. Wang, B. Liu, C. Zhang and J. Zhou, Theoretical Investigation on the Microscopic Mechanism of Lattice Thermal Conductivity of ZnXP2 (X = Si, Ge, and Sn), Inorg. Chem., 2019, 58, 4320–4327 CrossRef CAS PubMed .
- Q. Zheng, C. Li, A. Rai, J. H. Leach, D. A. Broido and D. G. Cahill, Thermal conductivity of GaN, 71GaN, and SiC from 150 K to 850 K, Phys. Rev. Mater., 2019, 3, 014601 CrossRef CAS .
- M. Sajjad, Q. Mahmood, N. Singh and J. A. Larsson, Ultralow Lattice Thermal Conductivity in Double Perovskite Cs2PtI6: A Promising Thermoelectric Material, ACS Appl. Energy Mater., 2020, 3, 11293–11299 CrossRef CAS .
- K. Berland, O. M. Løvvik and R. Tranås, Discarded gems: Thermoelectric performance of materials with band gap emerging at the hybrid-functional level, Appl. Phys. Lett., 2021, 119, 081902 CrossRef CAS .
- T. Xing, C. Zhu, Q. Song, H. Huang, J. Xiao, D. Ren, M. Shi, P. Qiu, X. Shi, F. Xu and L. Chen, Ultralow Lattice Thermal Conductivity and Superhigh Thermoelectric Figure-of-Merit in (Mg, Bi) Co-Doped GeTe, Adv. Mater., 2021, 33, 2008773 CrossRef CAS .
- T. Zhang, T. Yu, S. Ning, Z. Zhang, N. Qi, M. Jiang and Z. Chen, Extremely Low Lattice Thermal Conductivity Leading to Superior Thermoelectric Performance in Cu4TiSe4, ACS Appl. Mater. Interfaces, 2023, 15, 32453–32462 CrossRef CAS .
- O. Caballero-Calero, J. R. Ares and M. Martín-González, Environmentally Friendly Thermoelectric Materials: High Performance from Inorganic Components with Low Toxicity and Abundance in the Earth, Adv. Sustainable Syst., 2021, 5, 2100095 CrossRef CAS .
- S. Li, Q. Zheng, Y. Lv, X. Liu, X. Wang, P. Y. Huang, D. G. Cahill and B. Lv, High thermal conductivity in cubic boron arsenide crystals, Science, 2018, 361, 579–581 CrossRef CAS PubMed .
- E. S. Toberer, A. Zevalkink and G. J. Snyder, Phonon engineering through crystal chemistry, J. Mater. Chem., 2021, 21, 15843 RSC .
- W. G. Zeier, A. Zevalkink, Z. M. Gibbs, G. Hautier, M. G. Kanatzidis and G. J. Snyder, Thinking Like a Chemist: Intuition in Thermoelectric Materials, Angew. Chem., Int. Ed., 2016, 55, 6826–6841 CrossRef CAS PubMed .
- T. Zhu, Y. Liu, C. Fu, J. P. Heremans, J. G. Snyder and X. Zhao, Compromise and Synergy in High-Efficiency Thermoelectric Materials, Adv. Mater., 2017, 29, 1605884 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Ying
Fang
*b
*a and
Ying
Fang
*b




 and
and  refer to the normalized composition summing up to one. In other words, the NN model is applicable for any compound with either integer or fractional stoichiometry, which allows us to discover or design new materials with target κL in an even larger search space.
refer to the normalized composition summing up to one. In other words, the NN model is applicable for any compound with either integer or fractional stoichiometry, which allows us to discover or design new materials with target κL in an even larger search space.