
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in chiral analysis: from classical methods to emerging technologies
Roberto
Penasa
 ,
Giulia
Licini
and
Cristiano
Zonta
*
,
Giulia
Licini
and
Cristiano
Zonta
*
Department of Chemical Sciences, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: cristiano.zonta@unipd.it
First published on 20th October 2025
Abstract
The wide impact that chirality exerts in various fields of science has continuously pushed the development of methods and instruments capable of performing fast and reliable enantiomeric excess (ee) determination. From the historical optical polarimeter to modern instruments and supramolecular probes, this research field is continuously developing with the purpose of finding techniques of more general applicability and/or reducing the time and cost of analysis. The aim of this tutorial review is to provide the reader with a comprehensive overview of the available methodologies, referring to more specific reviews on the topic when available. Particular focus will be directed towards recent progress in this field, highlighting the advantages and disadvantages of the techniques and indicating recent trends in research.
 Roberto Penasa | Roberto Penasa received both his BSc and MSc degrees in Chemistry from the University of Padova, where he studied new supramolecular cages for the sensing of dicarboxylate species in complex media. He completed his PhD thesis under the supervision of Prof. Cristiano Zonta at the same university, focusing on the synthesis of new amine-based metal complexes and their applications in chiral sensing and reduction catalysis. He is currently a Researcher in the Traceability Unit at Fondazione Edmund Mach in San Michele all’Adige, Trentino. |
 Giulia Licini | Giulia Licini received her PhD from the University of Padova, working under the guidance of Prof. Giorgio Modena. She then spent a year as a postdoctoral fellow in the laboratory of Prof. Albert I. Meyers at Colorado State University, USA. Returning to the University of Padova, she began her academic career and is now a full professor. Her research explores innovative approaches to (stereo)selective homogeneous catalysis and reaction mechanism studies with a particular emphasis on small-molecule activation and the valorization of biomass into high-value products. |
 Cristiano Zonta | Cristiano Zonta received his PhD in 2003 from the University of Sheffield under the supervision of Prof. C. A. Hunter. After post-doctoral activities in Venice (with Prof. O. De Lucchi) and Padova (with Prof. G. Licini), he became a lecturer in 2006 at the University of Padova, where he is now a full professor. While his main research focus was initially directed towards the application of supramolecular cages in recognition and catalysis, the development of optical chiral probes, which started as a side project, has become central to his research. |
Key learning points(1) The concept of chirality at the molecular level, including key historical milestones that have shaped the modern understanding of this fundamental phenomenon across various scientific disciplines.(2) The principles of chiral discrimination, outlining the essential features that analytical methods must possess to successfully differentiate between enantiomers. (3) An overview of classical analytical techniques for chiral analysis, highlighting their basic operation, as well as their main strengths and limitations. (4) Recent advancements in chiral analytical methodologies, with a focus on current trends and the direction of modern research in the field. (5) Alternative analytical approaches and the specific advantages they offer, expanding the toolbox available for chiral analysis. |
Introduction
An elderly Jean-Baptiste Biot, overwhelmed with joy, exclaimed, “My son, all my life I have loved this science so deeply that I can now hear my heartbeat for joy.” These heartfelt words were in response to Louis Pasteur's groundbreaking separation of tartaric acid's chiral forms.1,2 Biot, the polarimeter's inventor, employed his instrument to verify that isolated crystals rotated linearly polarized light in opposing directions, a landmark observation that can be regarded as the inception of enantiomeric excess evaluation. In the wake of this seminal experiment, a multitude of “chiral assessment” techniques have flourished. While the principle of opposite light rotation continues to serve as an introductory concept in organic chemistry education, chromatography-based methods have emerged as the predominant analytical tools in practice.Chirality, a concept first articulated by Lord Kelvin,3 is a fundamental property of Nature. He defined a chiral object as one whose mirror image cannot be superimposed upon itself. Thus, this term covers a wide range of objects, from molecules and macromolecules to everyday items. The International Union of Pure and Applied Chemistry (IUPAC) defines a molecule's chirality based on “the geometric property of a rigid object (or spatial arrangement of points or atoms) of being nonsuperimposable on its mirror image; such an object has no symmetry elements of the second kind (a mirror plane, σ = S1, a centre of inversion, i = S2, a rotation-reflection axis, S2n). If the object is superposable on its mirror image, the object is described as being achiral”.4 Another correlated but independent concept is stereogenicity. While chirality originates from the structure and geometry of the molecules, stereogenicity refers to the capability of a molecule to generate stereoisomers (enantiomers or diastereomers) by group permutation.5 The tetrahedral geometry of an sp3 carbon atom bearing four different substituents was the first stereogenic element proposed by Jacobus Henricus van’t Hoff in 1874, but many others, such as geometrical atom arrangements, stereogenic axes or planes, have been described over the years.
The importance of chirality is crucial in biological-related fields where Nature's intrinsic homochirality has profound consequences for pharmaceutical and agricultural chemistry. For example, synthetic drugs are usually prepared in their enantiopure form, and controlling their enantiopurity is crucial. As a result, while synthetic chemists focus on novel and effective synthesis of enantiopure molecules, methods capable of performing fast and reliable chiral analysis are continuously being developed. The common “index” to measure their enantiopurity is defined as enantiomeric excess (ee). The ee is a measure of the purity of a chiral sample and it quantifies the relative difference in the amount of each enantiomer (excess of one enantiomer over the racemic part). Precisely, it is defined as:
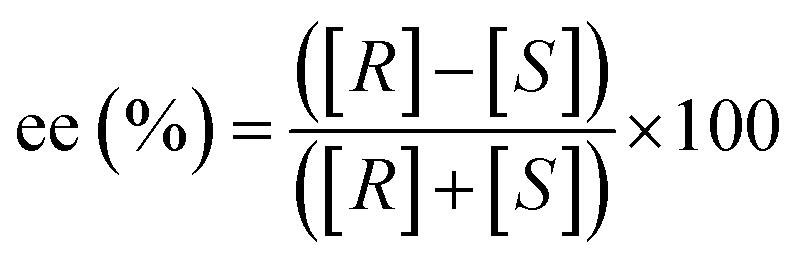 | (1) |
The aim of this tutorial review is to provide readers with a comprehensive overview of the available instrumental methods for analysing samples’ enantiopurity and absolute configuration. Focus will be on molecular probes, the impact of nanomaterials, and recent technological advancements in this field. Methods for investigating both absolute configuration and enantiopurity assessment will be discussed.
Principles for chiral discrimination
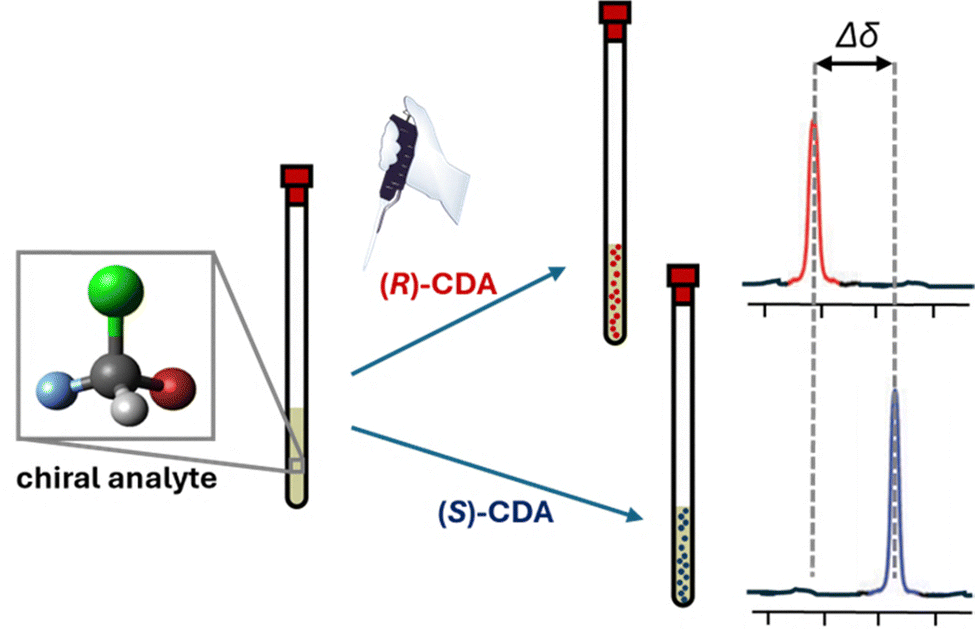
In the early 19th century, a pivotal moment in the field of stereochemistry unfolded when Kestner, an industrial chemist engaged in the purification of tartaric acid from wine barrels, made a serendipitous discovery. During the chemical purification and recrystallization processes of a particular batch of tartaric acid, Kestner observed the formation of an unexpected substance less soluble and optically inactive. Pasteur's subsequent investigations led to separation of the D and L enantiomers of tartaric acid from this mixture, a finding that would become the cornerstone of stereochemistry. This work provided fundamental insights into the principles of chiral discrimination and revealed that pure enantiomers and racemic mixtures crystallize differently. The difference in crystallization behavior is due to the presence of the opposite enantiomer in racemic mixtures, which acts as a chiral selector during the crystallization process, influencing the final crystal structure and properties.Differentiation of enantiomers requires determining their relative atomic coordinates (xyz) within a defined reference frame (x′y′z′). Possible approaches include: (i) exploiting incident or emitted polarized light, (ii) taking advantage of (non)covalent interactions with a separate chiral molecule of known enantiomeric composition, or (iii) creating a physical internal reference system within the analytical device (Fig. 1). Through the interactions with these stereodefined frames, diastereomeric non-isometric couples are formed and discrimination is possible. For example, most chromatographic methods for chiral analysis take advantage of chiral molecules in the stationary phase that interact with the analytes. Alternatively, some mass spectrometry methods are exploring the use of physical reference axes within the instrument to create a chiral environment.
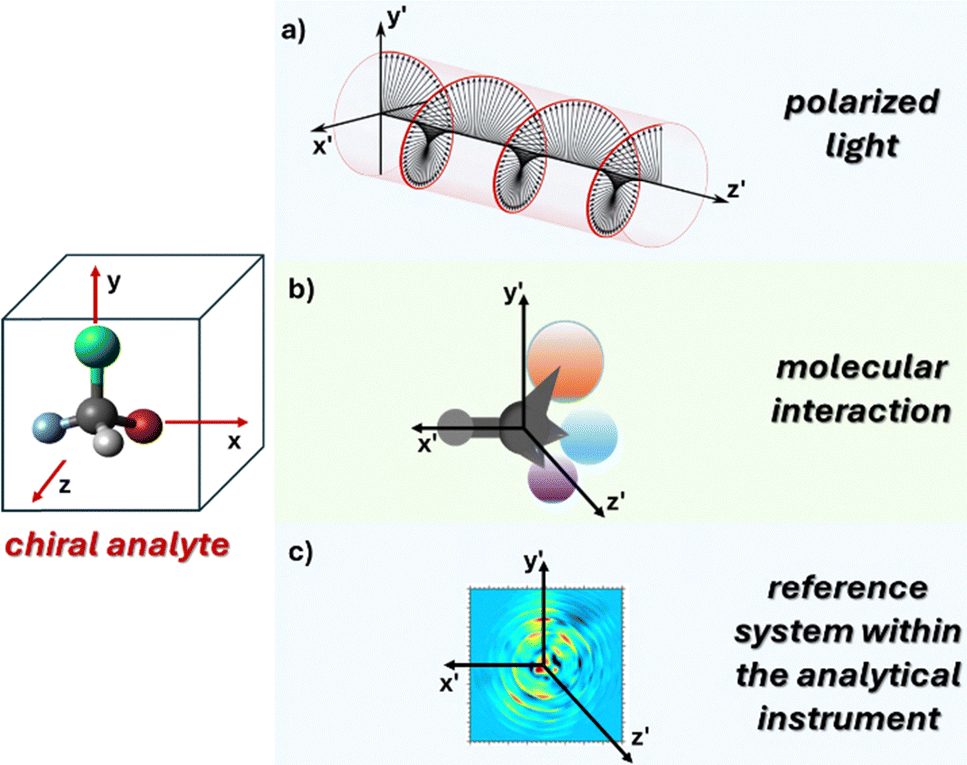 | ||
| Fig. 1 Distinguishing between enantiomers involves establishing the relative atom positions (xyz) using a specific reference frame (x′y′z′). Typical methods employ (a) interactions with or emission of polarized light, (b) (non)covalent interactions with another chiral molecule, and (c) the establishment of a reference system within the analytical instrument itself. | ||
In the following paragraphs, an overview of the recent advancements in different analytical techniques is presented. Besides introducing a brief historical overview of the reported analytical system, attention will be paid to the principle behind the technique, the chiral selector frame and the recent advancements to improve the efficiency of the system. Given the wide range of established and continuously emerging methods for the analysis of chiral molecules, the review is focused on the four most typical analytical techniques: (i) chromatography, (ii) chiroptical spectroscopies, (iii) NMR, and (iv) mass spectrometry. The last paragraph is dedicated to other emerging technologies in the field.
Within the subject of chiral discrimination, it should be pointed out that in some cases, as mentioned earlier, properties of racemic mixtures differ from those of their enantiopure or partially enriched mixtures, particularly in the solid state (e.g. melting point, solubility, etc.). This phenomenon can be rationalized by the difference between homochiral (narcissistic R with R or S with S) and heterochiral (anti-narcissistic R with S) interactions present in non-enantiopure samples. These differences have also been observed in mass spectrometry6 and NMR spectroscopy7 analyses. In other words, the opposite enantiomer acts as the x′y′z′ reference system for its mirror image. This phenomenon has also been identified as the cause of the non-linear correlation between optical rotation and enantiomeric excess, a behaviour known as the Horeau effect.8 Although the different properties of racemates represent a valuable proof of concept for ee determination, they are not broadly applicable for chiral analysis and, therefore, not discussed in the following sections.
Another alternative strategy for chiral analysis employs quasi-enantiomeric probes.9 These are structurally similar enantiomers that, through subtle differentiations like isotope labelling or minor functional group variations, maintain enantiomeric behaviour while remaining analytically distinguishable. When mixed with a chiral analyte, these quasi-enantiomers form distinguishable diastereomeric complexes, whose relative abundance reflects the analyte's enantiomeric composition. Isotope labelling is the most common method, enabling straightforward mass spectrometric differentiation. However, other detection methods, including NMR spectroscopy and optical techniques, have also proven effective. Despite its conceptual elegance and analytical potential, this approach faces significant practical limitations. The demanding synthetic effort required for probe preparation, coupled with challenges in calibration and accurate quantification, currently restricts its widespread use for ee determination.
This review includes also some relevant examples of enantiomer differentiation that involves kinetic resolution, since diastereomeric transition state differences have been extensively exploited. A prominent example is the use of lipases for the enantioselective hydrolysis of racemic esters, where the enzyme preferentially reacts with one enantiomer over the other due to steric and electronic differences in the transition state (Table 1).10
| Technique | Chiral selector | Unit of measurement | Method for distinguishing enantiomers (Fig. 1) | |
|---|---|---|---|---|
| α (°): degrees; θ (mdeg): millidegrees; I (a.u.): intensity; Abs: absorbance; tel (s): time of elution; f (ppm): chemical shift; tdr (s): drift time; tfl (s): time of flight; ρ (e− Å−3): electron density; ν (Hz): frequency; E (V): reduction (or oxidation) potential; [P] or [SM]: concentration of the product or the starting material. | ||||
| Chromatography | Chiral-GC | Chiral molecule (x′y′z′ within mobile and/or stationary phases) | t el (s) | (b) |
| Chiral-HPLC | (b) | |||
| Chiral-SFC | (b) | |||
| Chiral-CE | (b) | |||
| Optical methods | Optical rotatory dispersion (ORD) polarimeter | Linear polarised light (x′y′z′ = direction of propagation, magnetic and electric fields) | α (°) | (a) |
| Electronic circular dichroism (CD) | Difference in the absorption of opposite circularly polarised light (x′y′z′ = direction of propagation, magnetic and electric fields) | θ (mdeg) | (a) | |
| Vibrational circular dichroism (VCD) | (a) | |||
| Fluorescence detected circular dichroism (FDCD) | (a) | |||
| Circularly polarized luminescence (CPL) | Emission of circularly polarised light (x′y′z′ = direction of propagation, magnetic and electric fields) | I (a.u.) | (a) | |
| Raman optical activity (ROA) | Emission or absorption of circularly polarised light (x′y′z′ = direction of propagation, magnetic and electric fields) | (a) | ||
| UV-Vis | (Non)covalent functionalization with chiral molecules (x′y′z) | Abs | (b) | |
| Fluorescence | I (a.u.) | (b) | ||
| NMR | Chiral derivatizing agents | Covalent functionalization with chiral molecules (x′y′z′) | f (ppm) | (b) |
| Chiral solvating agents | Non-covalent interactions with chiral molecules (x′y′z′) | (b) | ||
| Chiral shift reagents | (b) | |||
| Mass | Chiral ion mobility | Chiral molecule or fragmentation detection on the x′y′z′ axis | t dr (s), tfl (s) | (b) |
| Coulomb explosion | (c) | |||
| X-ray | X-ray crystallography | Bijvoet pairs, crystal sponges | ρ (e− Å−3) | (c) |
| Electrochemical methods | Chiral CV | (Non)covalent functionalization of electrodes with chiral molecules | E (V) | (b) |
| Microwave | Chiral rotational spectroscopy (CRS) | Rotational transition frequency | ν (Hz) | (a) |
| Enzyme-based methods | Enzyme catalysis | Enzyme | [P] or [SM] | (b) |
Chiral analytical techniques
Chromatography11,12
Enantiomers can be separated using two main approaches in chromatography. The first involves covalently attaching a chiral auxiliary to the analyte and then using standard achiral columns. The second method, known as chiral chromatography, directly separates enantiomers exploiting their different affinities for a chiral stationary phase or a chiral mobile phase. After separation, analytes are typically detected via UV-Vis, CD,13 or MS and quantified by peak area integration. Originally developed in its achiral form by Mikhail Tsvet for plant pigment separation, chiral chromatography is now the leading technique for quantifying low-concentration analytes and determining enantiopurity. Its popularity stems from providing the highest accuracy in terms of ee determination compared to other commonly used techniques. Depending on the approach, analyses are routinely performed using gas chromatography (GC),14 high-performance liquid chromatography (HPLC),15 supercritical fluid chromatography (SFC),16 and capillary electrophoresis (CE).17The main advantage of chiral chromatography is its ability to perform both analysis and separation of enantiomers, offering an alternative to derivatization with enantiomerically pure reagents, followed by diastereomer separation. However, the technique's primary drawback lies in the need for extensive parameter optimization, typically involving the selection of a suitable stationary/mobile phase specific to the analyte, and generally long analysis times.
Chiral chromatography offers two analytical approaches for enantiomeric separation. One approach involves the addition of a chiral molecule in the mobile phase. These additives, usually cyclodextrins or chiral ionic liquids, avoid functionalisation with expensive chiral molecules. Nonetheless, the main approach involving the use of chiral stationary phases (CSPs) is most established, reliable and widely used. CSPs are made using chiral selectors that interact differently with the opposite enantiomers of the analyte, allowing for direct separation without the need for derivatization (Fig. 2). First introduced by Henderson and Rule in 1938 and later refined by Pirkle in the 1970s, CSPs have become the gold standard for ee determination.
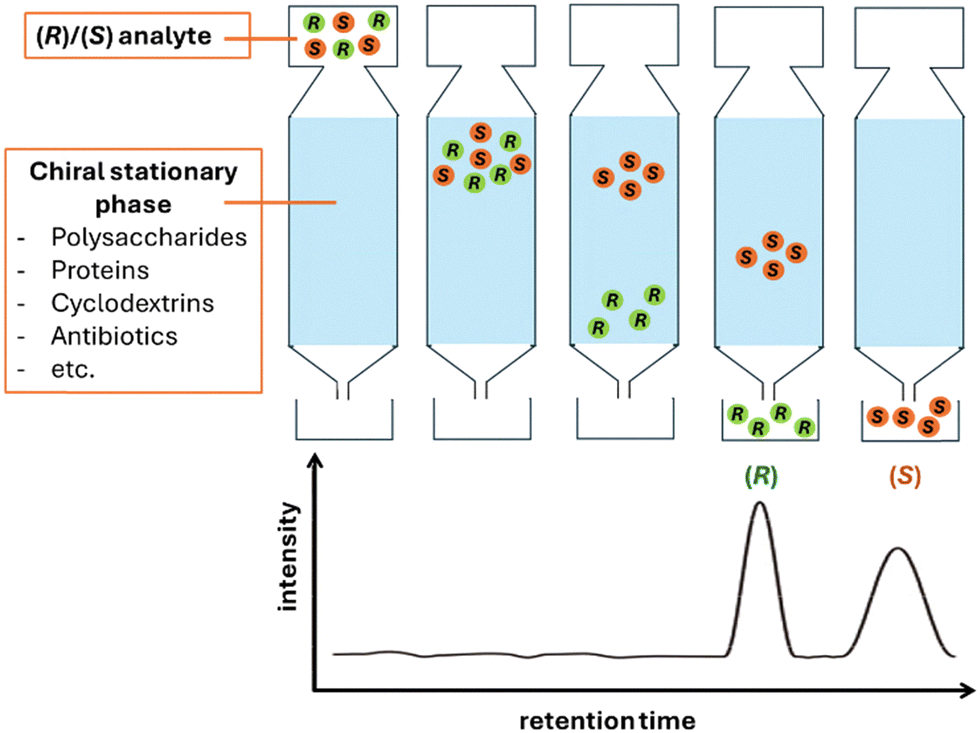 | ||
| Fig. 2 Schematic representation of chiral column chromatography. (R)- and (S)-enantiomers interact differently with the chiral stationary phase, resulting in distinct retention times during elution. | ||
Chromatography coupled with CSPs has exceptional efficacy not only in the separation and quantification of stereoisomers but also, in the case of liquid chromatography, in the preparative-scale isolation of enantiopure compounds.
In the pursuit of developing highly effective CSPs, researchers have explored different chiral selectors for many decades. These encompass modified cyclodextrins, crown ethers, Pirkle-type systems, polysaccharides, and antibiotics.12 Very recently, attention has also been directed toward porous materials such as covalent-organic frameworks (COFs), metal organic frameworks (MOFs), and porous organic cages (POCs), where interesting results have been obtained.18 The commercial availability of these varied CSPs provides analysts with a broad spectrum of options, each offering distinct enantioselective properties. However, economic viability together with limited substrate scope for each CSP and the need to develop specific separation conditions for each analyte are still issues that need to be solved. Consequently, there is ongoing research focused on the design of novel “universal” chiral stationary phases (CSPs).19
Another active area of research involves the development of ultrafast analytical methods, aiming to complete enantioseparations within a few minutes—or even under one minute—thus addressing one of the main limitations traditionally associated with chromatographic techniques. Fig. 3 shows, as an example, the reduction in separation times for three pharmaceuticals, achieving separations in under one minute for all.
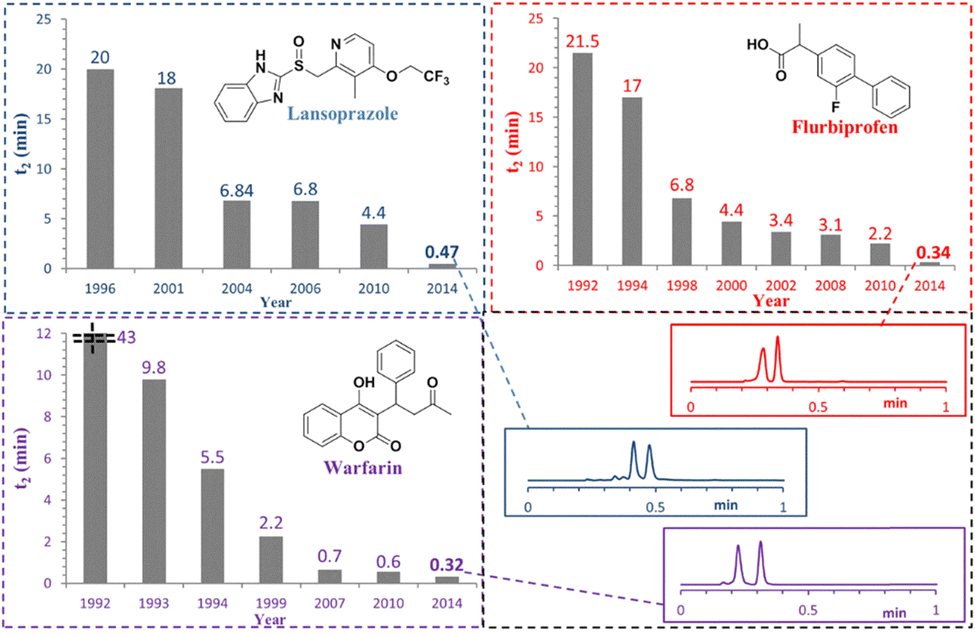 | ||
| Fig. 3 Evolution of the speeds for chromatographic separation of the enantiomers of lansoprazole, flurbiprofen and warfarin. Reproduced from ref. 20 with permission from John Wiley and Sons, copyright 2015. | ||
Chiroptical methods (ORD, CD, FDCD, CPL, VCD, and ROA)
Chiroptical analysis uses polarized electromagnetic radiation to distinguish chiral structures. This field encompasses various techniques across different spectral regions, measuring changes in refraction, absorption, luminescence, or scattering. These methods are categorized based on the nature of incident light and measured output. Techniques like optical rotatory dispersion (ORD), circular dichroism (CD), and vibrational circular dichroism (VCD) use polarized light, while others such as circularly polarized luminescence (CPL) employ unpolarized incident light and measure the polarization of emitted light. This diversity allows for comprehensive analysis of chiral molecules and their functional properties.Polarimetry and optical rotatory dispersion (ORD)21 are chiroptical techniques that measure the rotation of linearly polarized light as an indicator of enantiopurity. These methods rely on the small difference in velocities between right- and left-handed circular polarizations (circular birefringence) in solutions containing chiral molecules. Typically, a polarimeter operates at a single wavelength, usually 589 nm (sodium D line), while ORD instruments work across the UV-Vis spectrum, generally 170–800 nm (Fig. 4). Since absorption is not the primary phenomenon in these techniques, strong chromophores are not required, making it possible to observe this effect for virtually any chiral molecule. The rotated angle after passing through an optically active medium is related to the molecule's properties, the sample's enantiopurity, path length, and concentration (Biot's law). Polarimetry was the first method developed to determine enantiopurity, even before scientists understood the molecular basis of chirality. As a result, the terms ‘optical isomer’ and ‘optically pure’ are still used to describe enantiopure compounds.
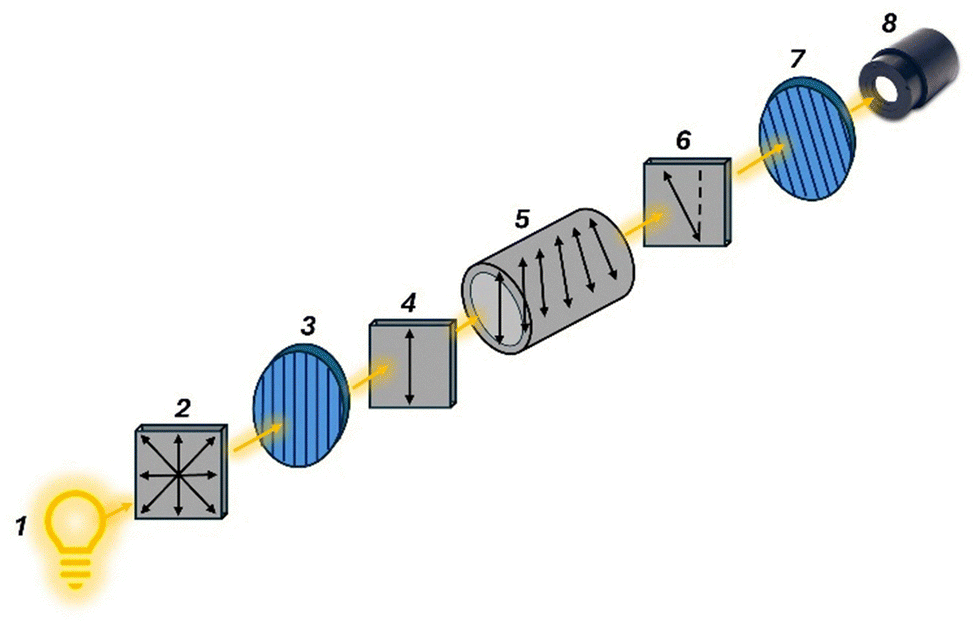 | ||
| Fig. 4 The light source (1), typically a sodium vapor lamp, emits unpolarized light (2). A linear polarizer (3) filters this light, producing linearly polarized light (4), which then passes through the sample (5) containing the chiral molecules under investigation. As the polarized light travels through the sample, it undergoes optical rotation (6) due to the presence of the chiral molecules. A rotatable linear analyzer (7) is used to determine the angle of rotation before the light reaches the detector (8). | ||
In this context, ‘optical purity’ serves as a parameter to evaluate the purity of scalemic mixtures—those containing unequal proportions of R and S enantiomers. It is defined as the ratio between the specific rotation of the mixture and that of the optically pure enantiomer, expressed as:

Given that the specific rotations of the two enantiomers have the same magnitude but opposite signs, knowing the specific rotation of one enantiomer allows for the straightforward determination of the ee through a simple optical purity measurement.
Although this methodology is simple and an in-house polarimeter can be built inexpensively,22 the technique presents some notable drawbacks compared to other chiroptical spectroscopies. In particular, it exhibits low sensitivity, lacks detailed structural information, and is significantly influenced by external factors such as temperature, solvents, and impurities. Recent advances in the field employ cavity-based spectroscopy, a technique that traps light between highly reflective mirrors. This approach creates an optical cavity, enabling light to pass through the sample multiple times, thus enhancing sensitivity and resolution.23
In the presence of chromophores, a dramatic change in rotation is observed near the absorption band (Cotton effect, Fig. 5). In a positive Cotton effect, the ORD signal increases as the wavelength decreases, crosses zero at the absorption maximum, and then decreases, forming a mirror-like band. Conversely, in a negative Cotton effect, the signal initially decreases, then increases after the absorption maximum. The Cotton effect is significant because it enhances the sensitivity and specificity of chiroptical techniques, and it is directly related to the ability of the molecule to absorb differently left and right circularly polarized light.
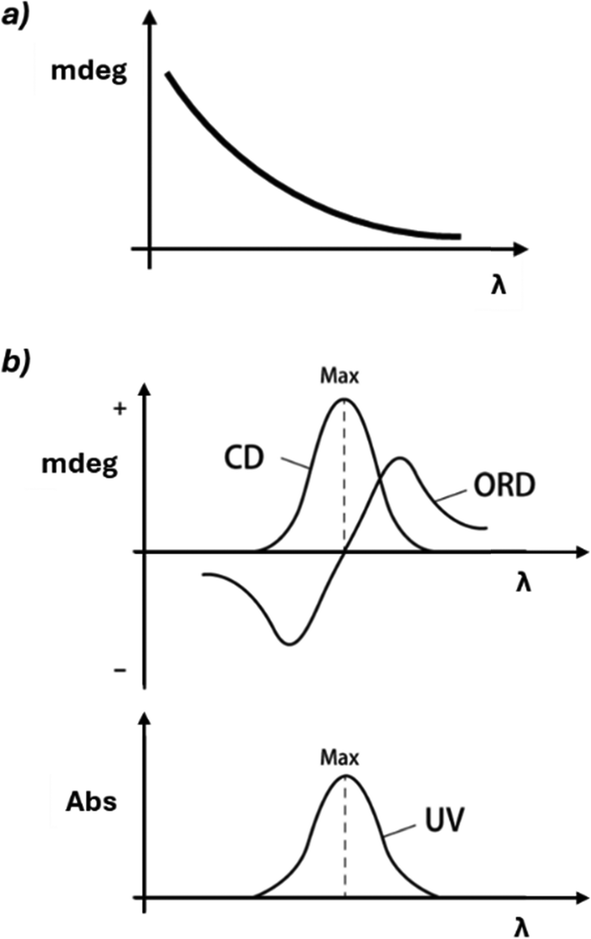 | ||
| Fig. 5 (a) In the absence of absorption, CD is not observed, and ORD exhibits ordinary dispersion, characterized by a monotonic change with increasing wavelength. (b) In the presence of absorption, both CD and ORD are detected. Typically, ORD displays anomalous dispersion (Cotton effect), which is characterized by a distinctive bisignate signal. | ||
Circular dichroism (CD)24 is one of the most widely used and established methods for chiral analysis. This technique exploits the unique property of chiral molecules containing chromophores to absorb left and right circularly polarized light to different extents. By measuring this differential absorption across a range of wavelengths, CD spectra are generated (Fig. 5 and 6). These spectra serve as characteristic fingerprints, allowing for the identification and quantification of chiral compounds, as well as the determination of their absolute configuration and conformational changes. For these reasons, CD is widely used in biochemistry and structural biology.25
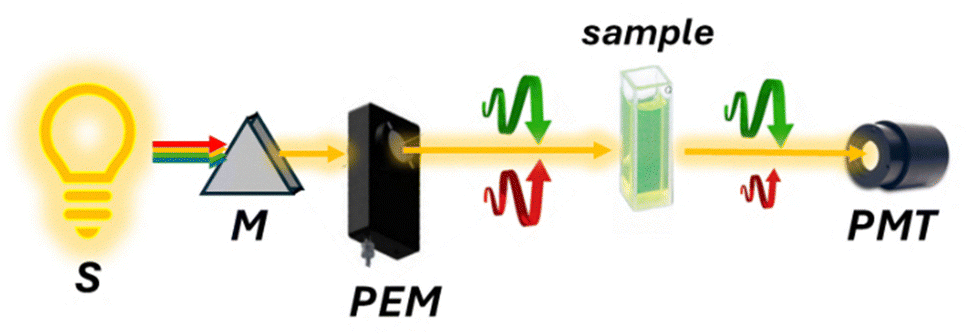 | ||
| Fig. 6 Schematic representation of an ECD spectropolarimeter. The light source (S), typically a xenon arc lamp, emits unpolarized light that passes through a monochromator (M) to select a specific wavelength. A photoelastic modulator (PEM) then alternates left- and right-circularly polarized light, which is directed through the sample. Differences in absorption between the two polarizations are detected by a photomultiplier tube (PMT). | ||
CD is generally preferred over ORD due to its ability to provide more resolved bands. For relatively simple molecules, CD spectra can be interpreted empirically using well-established theoretical descriptions such as the exciton chirality method.26
However, in modern practice, TD-DFT (time-dependent density functional theory) calculations have become more common in the interpretation of the spectra owing to their reliability and accuracy. In particular, TD-DFT sensitivity to conformations has made it an invaluable tool when combined with experimental data for structural characterization.13
CD has recently garnered significant attention for its potential implementation in high-throughput screening (HTS) instrumentation.27 The development of new methods for asymmetric synthesis, such as combinatorial chemistry and parallel synthesis, has led to the rapid production of chiral analytes, which in turn necessitates equally rapid methods of analysis. Optical methods are promising candidates due to their speed in ee analysis and their potential for automation.28 Even if CD instruments are widespread, they have low cost and short analysis times, and strong absorbing chromophores are required. For this reason, research interest in the last few years has been directed toward the development of molecules able to bind the analyte, either in a covalent or non-covalent fashion, and amplify the signals of “CD-silent” molecules. Current strategies are based on two main approaches: functionalizing analytes with strong chromophores or using rapidly interconverting enantiomeric conformers that bear strong chromophores (stereodynamic systems).29 In the latter approach, the interaction with chiral analytes generates diastereomeric complexes with differing thermodynamic stabilities. The resulting population imbalance gives rise to a CD signal proportional to the ee of the analyte.
From a practical standpoint, while computational tools can assist in assigning the absolute configuration, determining the enantiomeric composition typically relies on calibration curves. These are generated using either a pure reference standard of the target compound or, at minimum, a sample of known enantiopurity. Once the calibration curve is established, the ee of an unknown sample can be readily obtained by interpolating its CD response. However, because the CD response is compound and concentration specific, an individual calibration curve must be prepared for each analyte (Fig. 7). This requirement represents a major limitation of all chiroptical spectroscopies, as it is both time-consuming and dependent on the availability of reference materials with known enantiopurity. This represents a major drawback, particularly in high-throughput screening (HTS) experiments, where novel compounds are continuously generated and optically pure samples may not be readily available. In this context, interesting results have been obtained either by the use of concentration independent probes30 or by the use of classical physical organic chemistry tools with the aid of machine learning assistance and chemometric data processing.31,32
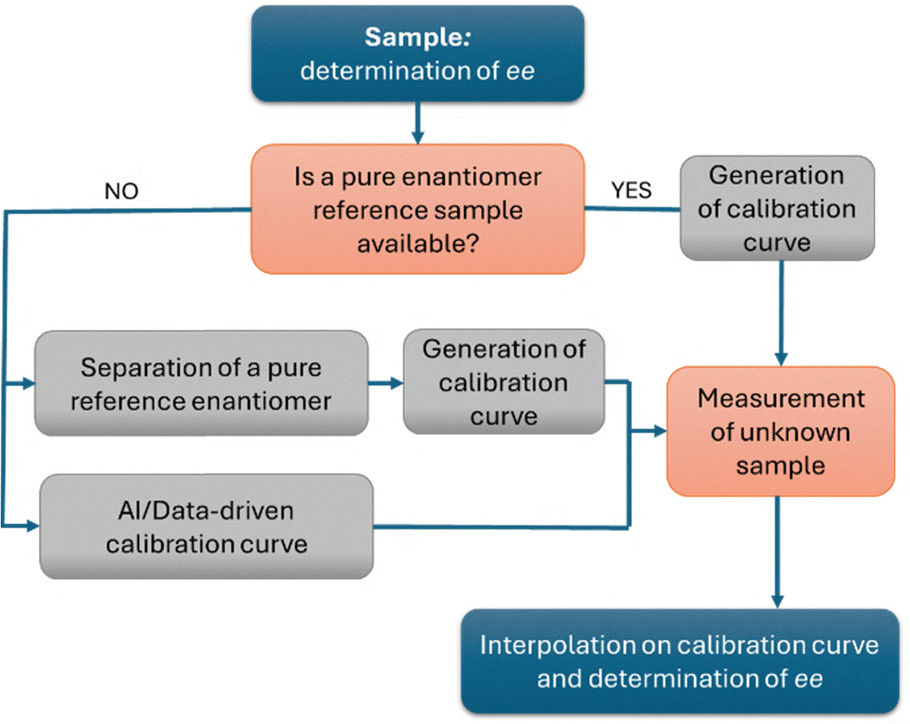 | ||
| Fig. 7 Schematic workflow for the determination of ee using chiroptical spectroscopies and other methods based on calibration curves. This is a simplified representation; additional steps (e.g., purification, dilution, temperature control) may be required depending on the specific method employed. | ||
More recent advances exploit photoelectron circular dichroism (PECD),33 a phenomenon observed during chiral molecule photoionization. PECD uses circularly polarized light to induce asymmetry in emitted photoelectron angular distribution, enabling rapid and sensitive enantiomeric composition measurement. Despite its advantages, complex instrumentation limits widespread adoption. A typical PECD setup records three-dimensional photoelectron angular distributions (PADs) using a multi-channel plate and CCD camera. Recent studies demonstrated femtosecond time-resolved PECD (TR-PECD) using ultrashort circularly polarized vacuum ultraviolet (VUV) pulses to monitor molecular chirality during reactions, enabling observation of chirality changes during photodissociation.34 Similarly, photoelectron elliptical dichroism (PEELD) uses elliptically polarized laser pulses to study chiral molecules in the gas phase. PEELD enables precise ee quantification (∼0.04% precision) across diverse compounds, including terpenes, amino acids, and iodoarenes. Its structural sensitivity highlights its potential as a valuable tool in chiral analysis.35
Circularly polarized luminescence (CPL)36 spectroscopy quantifies the difference in emission between left and right circularly polarized light from luminescent systems (Fig. 8a). This technique can be considered the “chiral version” of fluorescence emission spectroscopy. The popularity of CPL spectroscopy stems from its unique ability to probe excited-state chirality, complementing the CD technique. Its high sensitivity to structural changes, applicability to diverse systems, and non-destructive nature make it valuable across multiple fields. Recent technological advancements and growing interest in materials science and biology have further increased its relevance. CPL's role in developing new optoelectronic devices has also contributed to its rising prominence in research.37
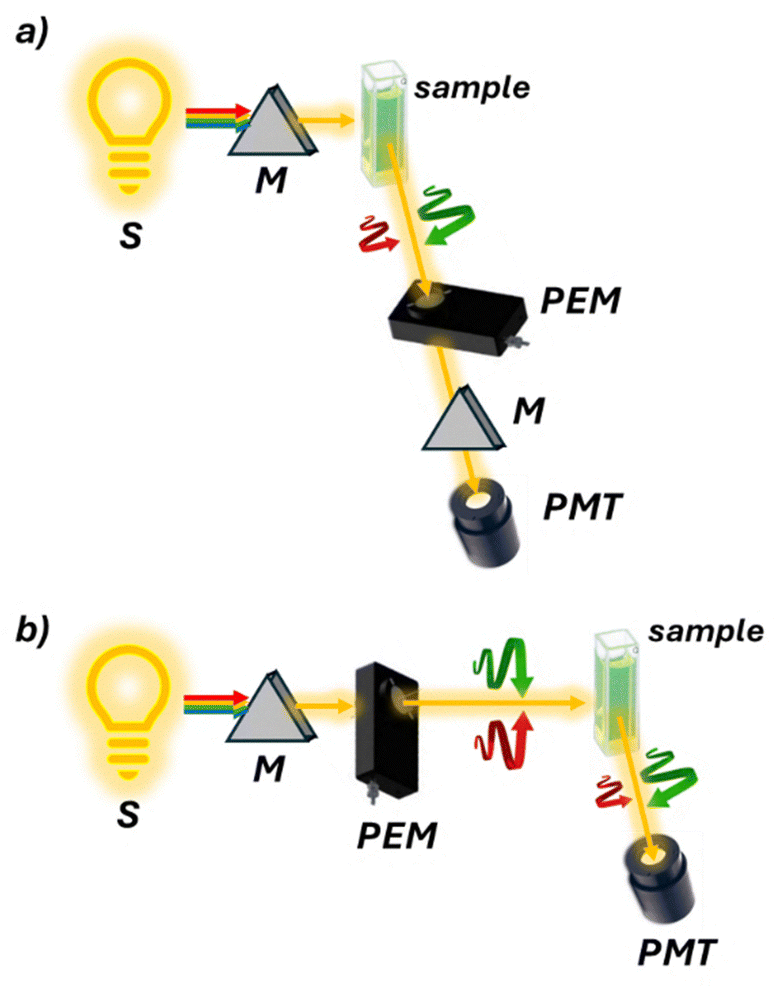 | ||
| Fig. 8 Schematic representation of (a) a CPL and (b) an FDCD spectropolarimeter. FDCD has the same optical components as CD with the photomultiplier tube (PMT) placed at 90° to collect the fluorescence. CPL instead has the photoelastic modulator (PME) after the sample to detect the differently emitted right and left circular polarizations. | ||
Chiral sensing has been extensively studied using lanthanide complexes due to their unique fluorescence properties with long excited state lifetimes and high luminescence dissymmetry factor, glum.38 As seen previously for CD, also in this case, the use of stereodynamic complexes where chirality is enabled by the helical twist of a macrocyclic ligand around the lanthanide ion is privileged. Sensing can be achieved either by perturbing a racemic system at equilibrium or by interacting with an enantiopure ligand. More recently, attention has also been directed toward the preparation of sensing materials with the use of small organic molecules, conjugated polymers, and organic frameworks.39 Similarly to CD, the determination of ee by fluorescence-based chiroptical spectroscopies also relies on the construction of calibration curves. Thanks to their inherently higher sensitivity, these techniques often enable a marked improvement in the detection limit for ee measurements.
Fluorescence detected CD (FDCD)40 is essentially the ‘chiral’ version of fluorescence excitation. Without any artefact effects, FDCD is related to CD in a similar way that excitation spectra are related to absorption spectra. In FDCD, the sample is exposed to alternating circularly polarized light, and the resulting fluorescence is measured (Fig. 8b). Despite its introduction in 1974, FDCD has seen limited applications in chiral sensing. This is surprising given its crucial advantages over absorption-based CD spectroscopy. Firstly, FDCD is inherently more sensitive, especially with technical precautions that enhance the signal-to-noise ratio. Secondly, it offers greater selectivity, allowing for the analysis of complex samples containing several absorbance interferents. Recent developments have highlighted FDCD's potential. Biderman et al. applied FDCD to supramolecular host–guest systems,41 finding that combining CD and FDCD provided valuable additional information, such as analyte chemical identity and hidden aggregation phenomena. Our group recently reported the first application of FDCD to a stereodynamic probe.42 This extended the probe's sensing performance to sub-micromolar concentrations. Importantly, the FDCD signal proved insensitive to chiroptical contaminants, opening possibilities for analyzing complex media with multiple interferences that typically hinder conventional absorption-based CD spectroscopy.
Vibrational circular dichroism (VCD)43 is a technique that utilizes circularly polarized light analysis in the infrared region. Using vibrational energy transitions, which are available in all organic structures, provides three key advantages: strong UV-Vis chromophores are not necessary, VCD spectra can be easily and confidentially calculated using DFT calculations, and the higher number of bands allows for a deeper and unique understanding of the analyte's molecular structure and conformation. For these reasons, it has gained significant interest, particularly for assigning the absolute configuration of challenging compounds and intermediates.44 Nevertheless, VCD's primary limitation lies in its inherently weak signal intensity. Measurements often require long acquisition times and large amounts of samples.
Despite this challenge, VCD is becoming more accessible and practical for chiral analysis, thanks to new, high-performance instrumentation. Similar to IR spectroscopy, but unlike conventional CD, typical VCD instrumentation operates using Fourier transform methods (FT-VCD). This requires the use of a Michelson interferometer, rather than a monochromator, to generate an interferogram that is subsequently converted into the conventional VCD spectrum via FT operation (Fig. 9).
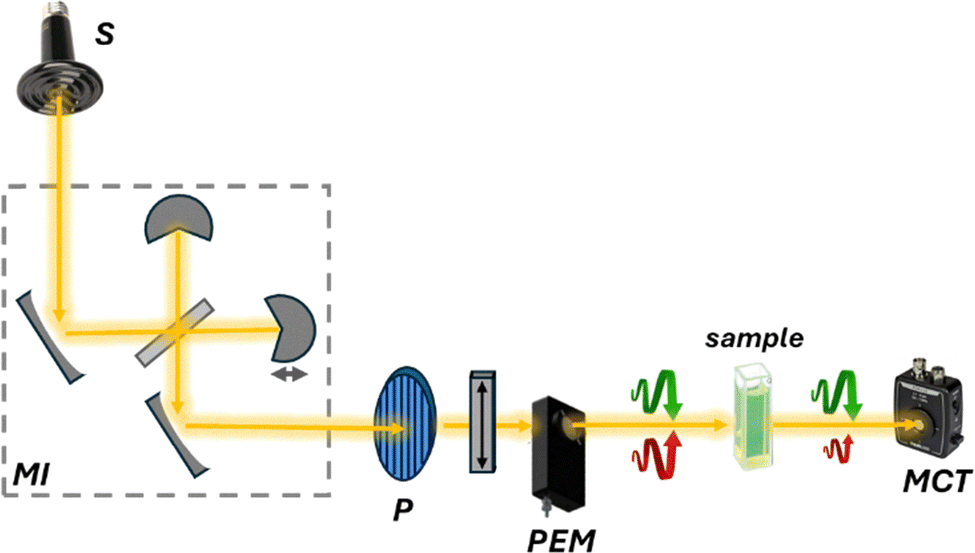 | ||
| Fig. 9 Schematic representation of an FT-VCD instrument. The source (S), typically a ceramic light emitter, emits unpolarized light that passes through a Michelson interferometer (MI) that modulates the optical length for the production of the interferogram. Then, similarly to CD, the polarizer (P) and the photoelastic modulator (PEM) alternate left- and right-circularly polarized light, which is directed through the sample. Differences in absorption between the two polarizations are detected by a mercury cadmium telluride (MCT) detector. | ||
While the application of VCD from small molecules to (bio)macromolecules has been extensively explored in recent years, including in the context of femtosecond time-resolved VCD,45 its use for ee determination remains limited due to the previously described limitations. To overcome these constraints, scientists are investigating signal enhancement arising from molecular aggregation or taking advantage of the presence of an open-shell metal center, which gives rise to low-lying electronic states (LLES) and electronic transitions at low energy.46
Raman optical activity (ROA)47,48 is a technique that exploits Raman scattered light signals in chiral analysis. ROA is the most recently developed technique among chiroptical spectroscopies, with the first experiments conducted by Barron, Bogaard, and Buckingham in 1973. Despite its relatively short history, ROA has evolved rapidly since the commercialization of its instrumentation. There are two main methods for measuring ROA: incident circular polarization (ICP) ROA, which measures the small difference in the intensity of vibrational Raman scattering from chiral molecules when exposed to right- and left-circularly polarized incident light (Fig. 10a), and scattered circular polarization (SCP) ROA, which measures the intensity of a small circularly polarized component in the scattered light using incident light of fixed polarization (Fig. 10b). These two methods offer different ways to analyze the chiral properties of molecules through their interaction with polarized light, providing valuable insights into molecular structure and behavior. Unlike VCD, ROA is better suited for analyzing samples in the presence of water. This makes it particularly valuable for studying biological molecules such as proteins and DNA.49,50 In recent years, the technique's performance has significantly improved. However, its applications remain somewhat restricted due to long acquisition times. Nevertheless, signal enhancement has been obtained by resonance ROA (R-ROA), by tuning the excitation wavelength near an electronic absorption band of the analyte, or by taking advantage of localized surface plasmon resonance of metallic nanostructures (SE-ROA).51 If the plasmonic structure presents an homochiral environment, either because of the ligand or because of the metallic arrangement, surface-enhanced Raman scattering (SERS) can be exploited.
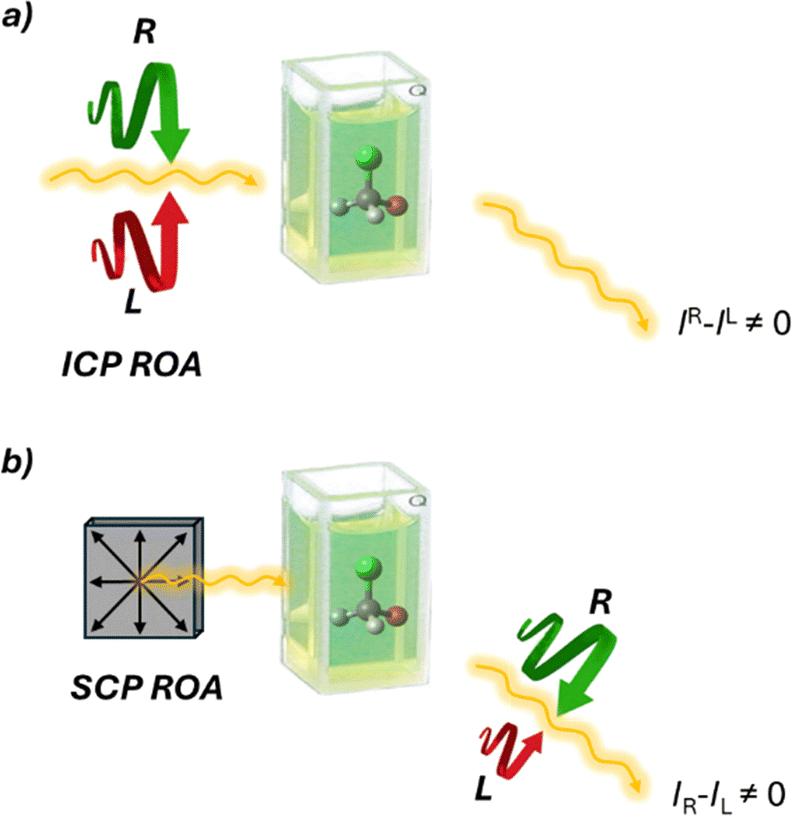 | ||
| Fig. 10 Schematic representation of the two ROA techniques. (a) ICP ROA measures (IR − IL), where IR and IL are the scattered intensities in right- and left-circularly polarized incident light, respectively. (b) SCP ROA measures (IR − IL), where IR and IL are the intensities of the right- and left circularly polarized components, respectively, of the scattered light using incident light of fixed polarization. | ||
Terahertz circular dichroism (TCD)52 expands the application of “classical” chiroptical spectroscopies to the far-IR and terahertz (THz) region of the electromagnetic spectrum. Unlike conventional UV-visible or infrared CD, which probes electronic or localized vibrational transitions, this low energy part of the electromagnetic spectrum carries a lot of information about different types of collective vibrational modes. For this reason, TCD is sensitive to intermolecular interactions, global conformational dynamics, and collective atomic vibrations in biomolecular assemblies, supramolecular systems, and chiral materials. While still in a developmental phase, advances in broadband THz generation, polarization control, and detection have made it possible to measure subtle chiroptical responses in both solid-state and solution-phase samples. THz-CD holds particular promise for investigating biomolecular hydration, protein collective motions, and chiral phononics, offering a complementary perspective to traditional chiral spectroscopies and expanding the frontiers of chiral analysis into the far-infrared domain.
Non-linear chiroptical spectroscopies53,54 are an emerging class of analytical techniques that exploit non-linear optical processes to probe molecular chirality with enhanced sensitivity and spatial selectivity. Indeed, one main limitation of linear spectroscopies relies on the small difference between the signal of the two enantiomers that causes low intensity signals. Techniques such as second harmonic generation circular dichroism (SHG-CD), sum frequency generation (SFG), and two-photon circular dichroism (TPCD) enable the investigation of chiral structures at surfaces, interfaces, and in complex heterogeneous environments that are often inaccessible to linear chiroptical methods. Their intrinsic surface specificity, combined with high spatial and temporal resolution, makes them particularly suited for studying chiral nanostructures, confined biomolecular systems, and engineered chiral metasurfaces. Moreover, the integration with nanophotonic architectures can lead to a significant enhancement of chiroptical signals through field confinement and resonance effects. These properties position non-linear chiroptical spectroscopies as powerful tools in the development of next-generation chiral analysis strategies, especially at the nanoscale.
Nanophotonic technologies55,56 manipulate light at the nanoscale using structures comparable to or smaller than the light's wavelength, enhancing light–matter interactions for applications like sensing and imaging. Recent studies have explored their potential in chiral sensing and ee quantification. These methods exploit metallic and dielectric nanostructures to generate superchiral light, characterized by an optical chirality density exceeding that of circularly polarized waves, enabling stronger interactions with chiral molecules and improving sensitivity beyond conventional techniques. A recent advancement in this area involves the use of gold-based nanostructured platforms to amplify the VCD signals of chiral molecules. This approach enabled enhancements of up to 13 orders of magnitude in detection sensitivity, allowing the simultaneous determination of both concentration and enantiomeric composition in complex mixtures (Fig. 11). These results highlight the exceptional potential of nanophotonic strategies in chiral analysis and point toward continued innovation in the field.
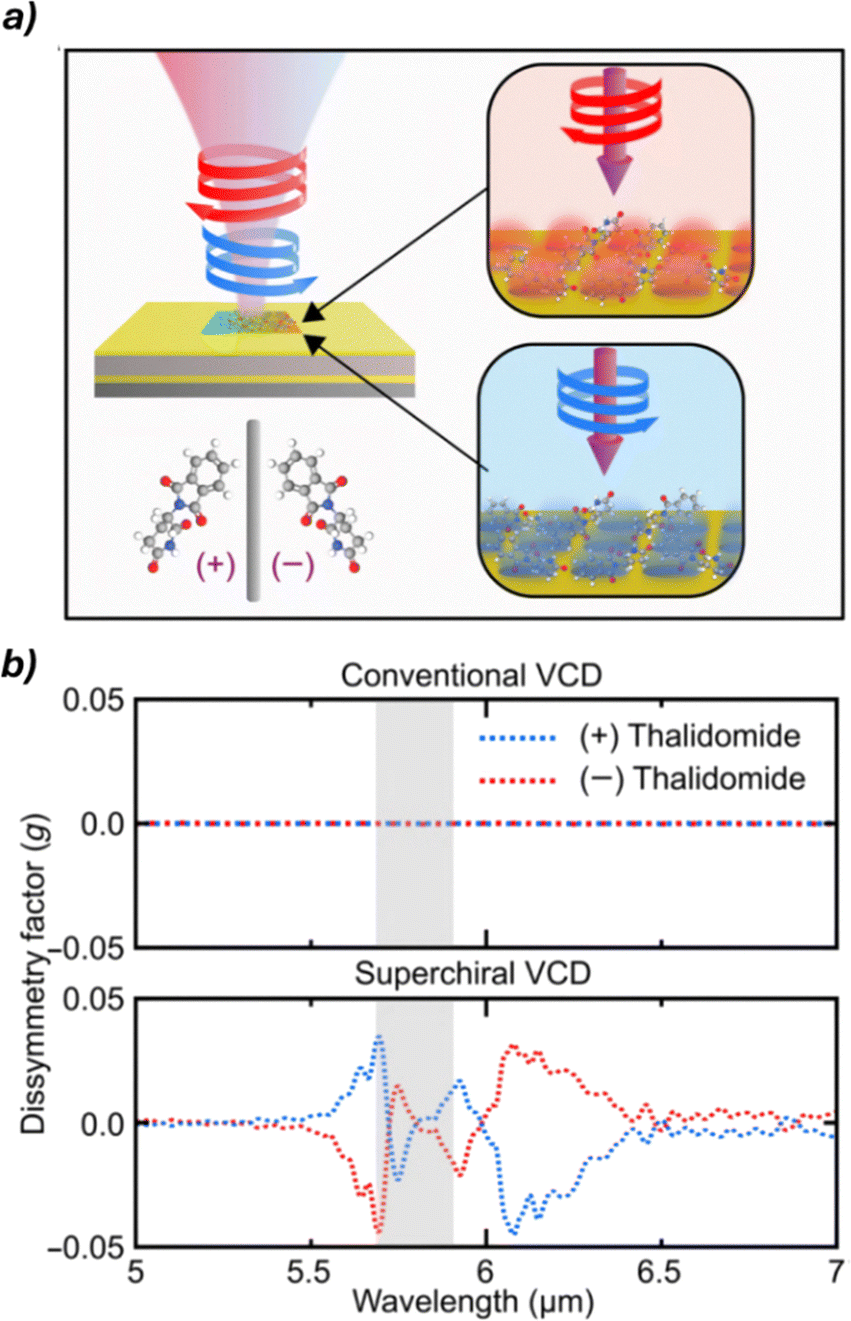 | ||
| Fig. 11 (a) Schematic representation of the chiral analysis of (+)/(−)-thalidomide using the gold-based plasmonic sensor. (b) Comparison of dissymmetry factors obtained with conventional VCD and superchiral light-enhanced VCD, highlighting the substantial signal enhancement achieved with the nanophotonic approach. Reproduced from ref. 54 with permission from The American Association for the Advancement of Science, copyright 2024. | ||
Nuclear magnetic resonance (NMR)57–59
Nuclear magnetic resonance (NMR) spectroscopy exploits the interaction of atomic nuclei under the influence of an external magnetic field with radiofrequency pulses to elucidate the structure and dynamics of organic molecules. The application of NMR spectroscopy for determining absolute configuration and assessing enantiomeric purity was first reported in the latter half of the twentieth century. Pioneering work in this field includes the studies by Raban and Mislow,60 who initially employed chiral derivatizing agents for chiral analysis via NMR. Enantiomers are typically indistinguishable by NMR because the resonances of enantiotropic nuclei are isochronous. However, chiral agents can be employed to render enantiomers distinguishable in NMR spectra. These optically pure molecules include chiral derivatizing agents (CDAs) that form covalent bonds and chiral solvating agents (CSAs) or chiral shift reagents (CSRs) that interact with the analyte through non-covalent interactions. These agents allow for the formation of diastereoisomers or diastereomeric complexes, which are distinguishable by NMR.CDAs are enantiopure compounds, which react with the analyte to form diastereomeric derivatives that can be distinguished by creating observable differences in chemical shifts and/or coupling constants in the NMR spectra (Fig. 12). To determine the enantiomeric composition of a sample, it is crucial to ensure that no racemization or kinetic resolution occurs during the derivatization process and, once this is confirmed, the enantiomeric excess can be easily determined by integrating the new diastereomeric peaks and applying eqn (1). In cases where the preferential conformation adopted by the CDA is known, it is also possible to determine the absolute configuration of an unknown analyte by comparison. A classical CDA is the Mosher α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) used for derivatizing chiral alcohols and amines.61 This CDA has been widely utilized due to several key features: the ability to use 1H and 19F NMR, characteristic OCH3 diagnostic singlet, and the presence of phenyl ring current effects. These properties make MTPA a powerful tool for analyzing chiral compounds, offering insights into both their composition and structure (Fig. 13).
 | ||
| Fig. 12 NMR analysis of a chiral analyte through derivatization with (R)- or (S)-CDA. The resulting diastereomeric complexes exhibit distinct chemical shifts, enabling enantiomer differentiation based on the CDA enantiomer employed. | ||
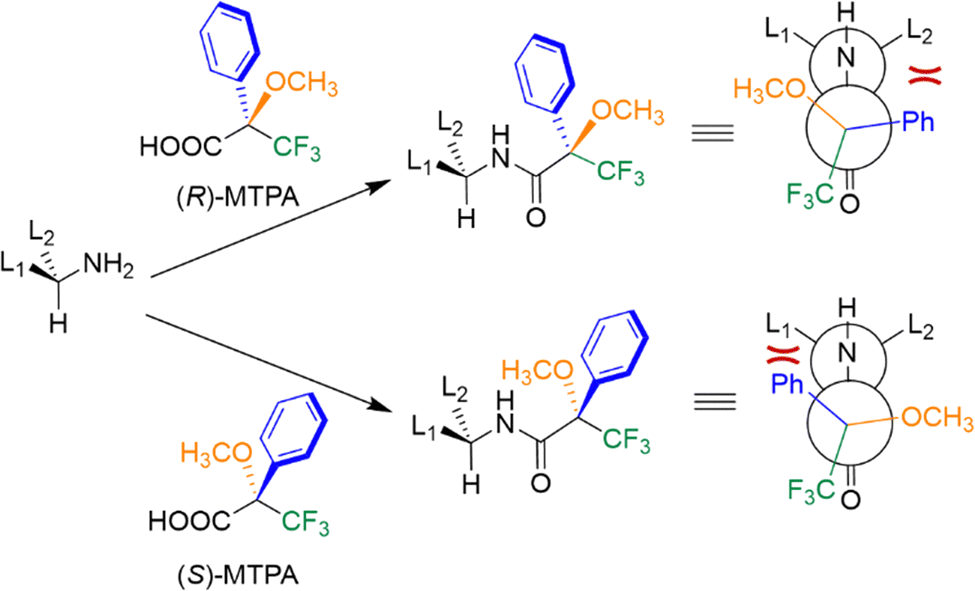 | ||
| Fig. 13 NMR analysis of a chiral amine through derivatization with Mosher acid. The different shielding of the L1 and L2 groups of the amine are highlighted in red. Depending on the enantiomer of MTPA acid employed, different shielding effects on the L1 and L2 groups of the amine are observed. | ||
CSRs are instead optically active, paramagnetic complexes, typically containing lanthanide metals such as europium or ytterbium complexed by a chiral ligand.62 These reagents form weak, reversible adducts with analyte molecules, inducing significant chemical shift changes and often signal broadening due to their paramagnetic nature. CSRs create distinctly different magnetic environments for each enantiomer, resulting in differentiated NMR signals, particularly for protons in close spatial proximity to the metal center.
On the other hand, CSAs are enantiopure diamagnetic organic compounds that form transient, non-covalent complexes with chiral analytes in solution through interactions such as hydrogen bonding, π–π stacking, or dipole–dipole interactions. CSAs create a chiral environment around the analyte, resulting in the formation of diastereomeric complexes that exhibit distinct chemical shifts in NMR spectra. CSRs and CSAs do not need to be covalently attached to the analyte for analysis, which simplifies the analytical procedure.
In recent years, research has focused on developing simpler chiral selectors that can be easily synthesized from commercially available reagents. These new selectors aim to be more versatile in terms of functional group applicability, faster in binding or functionalization processes, and capable of producing larger chemical shift separations in the resulting diastereomeric species. These include the use of chiral liquid crystals,63 chiral aluminium solvating agents,64 and densely fluorinated agents.65
It is important to highlight that research has demonstrated an interesting phenomenon in chiral molecules subjected to a strong magnetic field. Under these conditions, the precession of nuclear magnetic moments can induce a small electric polarization due to the nuclear magnetic shielding polarizability. Notably, this magnetoelectric response exhibits equal magnitude but opposite sign in a molecule and its mirror image, thus enabling the direct discrimination of enantiomers via NMR spectroscopy.66
Mass spectrometry (MS)67,68
Mass spectrometry (MS) involves the ionization of molecules or fragments and the measurement of their mass-to-charge ratio (m/z), providing information on the molecular weight and structural features of a target analyte. The MS technique offers some intrinsic fundamental advantages that make it an active research area for chiral analysis, particularly high sensitivity allowing the detection of analytes at parts per million (or billion) concentrations, the selectivity towards the desired analytes even in the presence of interfering molecules, the minimal sample preparation, the possibility to integrate automation in HTS methodology, and the versatility that allows coupling with various separation techniques. From a historical perspective, the use of MS for chiral analysis is more recent compared to other analytical techniques. As MS is an intrinsically achiral technique, achieving enantiodiscrimination requires the formation of diastereoisomers with an enantiopure molecule. This can be performed by (i) adding the chiral selector in the solution phase before ionisation and measuring the relative abundance in the gas phase (single-stage MS), (ii) fragmenting diastereomeric ions formed with a chiral selector in the gas phase to obtain information about their relative abundance (MS/MS), and (iii) using ion mobility in the presence of a chiral drift gas modifier able to form diastereomeric ions with different mobility (IM-MS).While single-stage MS is more affected by ionisation conditions (solvent, concentration, etc.), MS/MS has been shown to be more interesting in terms of reproducibility and sensibility. Chiral differentiation in these studies is based on different MS/MS fragmentation patterns of complexes formed by enantiomers and enantiopure chiral selectors, generally detected as proton or metal-bound cluster ions. Collision-induced dissociation (CID) is typically used to dissociate these complexes. Under identical CID conditions, complexes from different enantiomers can show varying dissociation rates, resulting in distinct spectral patterns. Chiral differentiation is determined by comparing the intensity ratios of product ions or product-to-precursor ions between enantiomers. Photodissociation (PD) is an alternative method in which ions absorb photons for dissociation. In PD, complexes from different enantiomers may produce significantly different fragmentation patterns, enabling chiral differentiation through direct spectral comparison or intensity ratio analysis. In ion mobility-mass spectrometry (IM-MS), gaseous ions are separated based on size and shape, measured as collision cross section (CCS). Diastereomeric complex ions of enantiomers can have different CCS values, allowing separation. Various IM-MS techniques have been applied to chiral analysis, Dwivedi demonstrated gas-phase separation of enantiomers by modifying drift gas with chiral (R)- or (S)-2-butanol vapor.69 This modification enabled selective interactions between enantiomers and the chiral modifier, resulting in different ion mobilities. While reducing the mobilities of both enantiomers, it did so to different extents, enabling distinction. The method successfully differentiated various chiral compounds, including drugs, amino acids, and sugars (Fig. 14). Despite limitations,70 this discovery opened possibilities for rapid chiral separation and measurement methods. Recent advances in this field exploit trapped ion mobility spectrometry (TIMS-MS).71 This technique uses an electric field opposite to the ion direction to enhance the resolution of ion separation. The power of this approach was used in a recent study to achieve the separation, after chiral derivatisation, of all the proteogenic amino acid enantiomer pairs in a single run. Application of IM-MS in HTS was also reported using the flow injection technique that significantly speeds up the time required for the analysis of eliminating the separation steps and the possibility of automation.
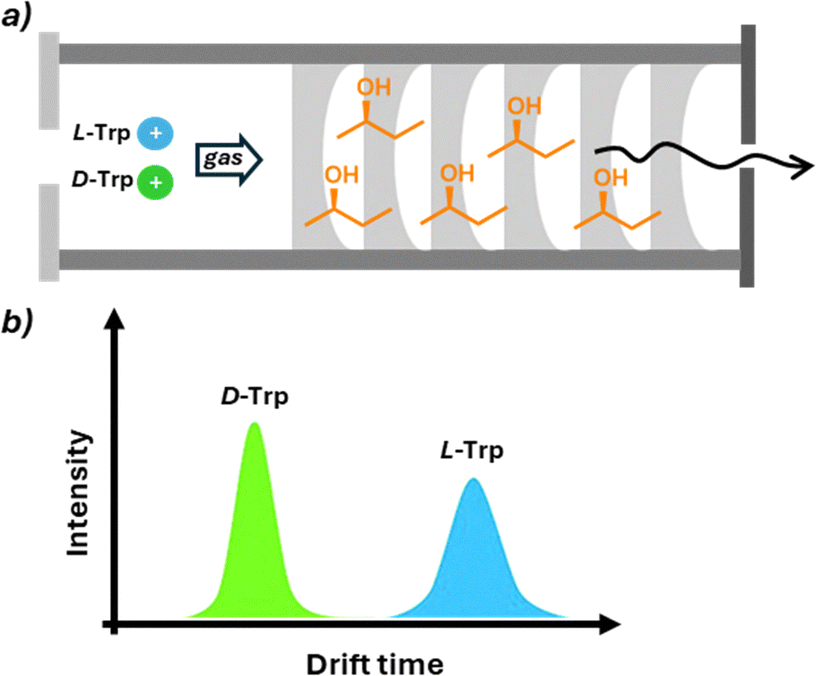 | ||
| Fig. 14 Separation of D- and L-tryptophan by IM-MS. (a) The presence of (R)-2-butanol as a chiral selector in the gas of the drift tube induces diverse drift times of the opposite enantiomers. (b) The intensity of peaks relative to the two enantiomers can be correlated to the ee of the sample. | ||
Due to the variety of outputs provided by mass-based techniques (e.g. drift time, intensity, etc.), specific calibration curves are generated by plotting the relevant instrument response against the enantiomeric composition. As with other techniques, the ee of an unknown sample is then obtained by interpolation on the corresponding curve.
A completely different approach called Coulomb explosion imaging (CEI) was introduced for the direct determination of AC in the gas phase.72 In summary, Coulomb explosions occur when multiple electrons are expelled from a molecule, leading to the repulsion of positively charged nuclei and resulting in fragmentation. Then, the use of a time- and position-sensitive detector allows the determination of the spatial disposition of the fragmented nuclei, which can be correlated with the AC of the molecule. Despite the significant advancements this technique has brought to molecular imaging, its application has been limited due to the high energy required for ionization and the difficulty in determining the AC of complex molecules.
Another interesting approach exploits the directional rotation of gas-phase ions.73 In more detail, a dual alternating current excitation was used to control the rotation of the trapped ions’ motion, including rotation around the centre of mass and macro movement around the trap centre. This motion induces differences in the enantiomers’ collision cross section, which allows their distinction.
Others
While chiral chromatography remains the gold standard, numerous alternative methods for chiral analysis have been developed over the years. These less frequently employed techniques, though not as widely adopted as chromatographic methods, or widespread such as optical and NMR technologies, offer unique advantages in specific applications. In the final section of this review, we will introduce the reader to emerging and specialized techniques in chiral sensing, highlighting their potential and current limitations in the field of enantiomeric analysis.X-ray crystallography74 is a reference methodology for determining AC. The anomalous scattering recorded in non-centrosymmetric space groups can be exploited for the determination of the configuration of crystalline molecules. This is achieved by measuring small differences in the intensity of Bijvoet pairs—reflections related by inversion—which arise due to the wavelength-dependent variation in atomic scattering factors of heavy atoms or other anomalous scatterers. Good crystal quality, especially for light-atom structures, is an essential condition to attribute the correct AC. The most important drawback of this technique is its limited applicability to molecules that can be obtained as crystals. To address the challenge of analyzing compounds that are difficult or impossible to crystallize, the Fujita group developed the innovative ‘crystal sponge methodology’.75 This structural analysis technique allows for the determination of molecular structure and absolute configuration without directly crystallizing the compound under examination. The method employs a porous host compound, aptly named the ‘crystal sponge’, typically a metal–organic complex with a well-defined three-dimensional structure. The analyte is diffused into this crystal sponge, and the resulting structure is analyzed using single-crystal X-ray diffraction, providing detailed structural information (Fig. 15). A key advantage of this technique is its ability to analyze liquids or difficult-to-crystallize compounds using only minute sample quantities, often just nanograms or micrograms. This makes it particularly valuable for elucidating the structures of natural products, metabolites, and other complex organic compounds that have traditionally challenged conventional crystallographic techniques.
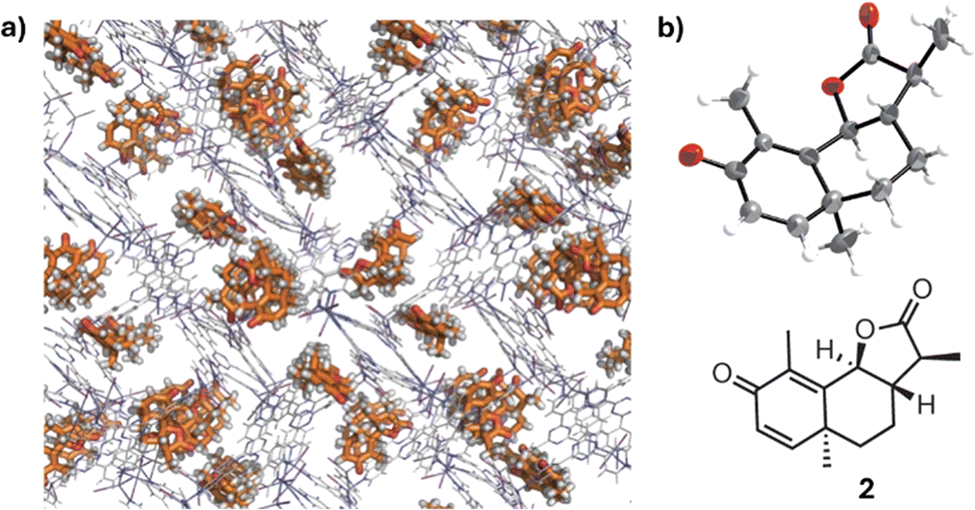 | ||
| Fig. 15 (a) The crystal structure of a chiral guest, santonin, trapped in a crystalline sponge. (b) ORTEP drawing (50% probability) of the santonin trapped in the pore and the chemical structure of santonin. Reproduced from ref. 72 with permission from Springer Nature BV, copyright 1995. | ||
Electrochemical methods76,77 typically involve electron transfer between a substance and an electrode to study molecular composition, structure, and reactivity. These techniques have gained interest for chiral sensing, where chiral recognition typically arises from the formation of transient diastereomeric complexes between a chiral selector, typically using modified electrodes coated with chiral molecules, and target enantiomers. Differences in Gibbs free energy between the diastereoisomers lead to distinct electrochemical signals such as shifts in oxidation or reduction peaks in cyclic voltammetry (Fig. 16). Despite being relatively new in this chiral analysis, electrochemical techniques offer notable advantages: high sensitivity and selectivity, low energy consumption, and operational simplicity. For instance, electroactive films based on chiral oligomers have shown separation of voltammogram peaks according to the enantiomer used for electrode coating, with peak shifts and intensities varying linearly with enantiomeric composition. This allowed the use of pure reference compounds for the construction of calibration curves (ee vs. ΔV) and the ee determination of unknown samples by curve interpolation. Recent studies suggest that the chiral-induced spin selectivity (CISS) effect can also be exploited in chiral electrochemical sensing.78
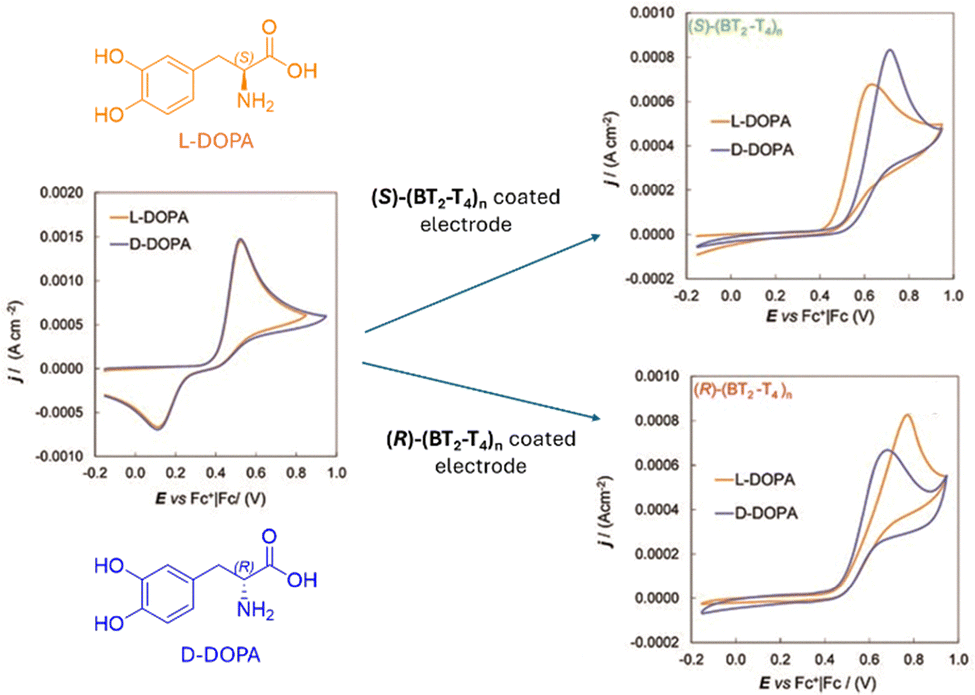 | ||
| Fig. 16 Cyclic voltammetry of DOPA enantiomers (4 mM H2O + 50 mM HCl) recorded with (left) a bare GC electrode, (right, up) a GC electrode coated with (S)-(BT2-T4)n, and (right, down) a GC electrode coated with (R)-(BT2-T4)n, the enantiomeric pair of bithianaphthene-based oligomers. Reproduced from ref. 79 with permission from The Royal Society of Chemistry, copyright 2010. | ||
Current research in this field is primarily focused on two main directions. On one hand, efforts are directed toward the development of chiral versions of various electrochemical techniques, including sweep and differential pulse voltammetry, electrochemical impedance spectroscopy, and others. On the other hand, significant attention is being devoted to the design of high-performance chiral selectors, leveraging recent advances in the synthesis of MOFs, COFs, and promising composite or hybrid materials.
Molecular rotational resonance (MRR) spectroscopy80,81 is an emerging tool in chiral analysis, which uses microwave radiation to probe pure rotational transitions and provide highly accurate structural information. It distinguishes isomeric species—such as regioisomers, stereoisomers, and isotopologues—with high resolution and sensitivity, often without prior separation. MRR has gained attention for its ability to analyze enantioisotopomers, which are difficult to resolve using conventional techniques.82
A major advancement emerged with microwave three-wave mixing (M3WM), which exploits the opposite signs of the product of dipole moment components (μαμβμγ) in enantiomers (Fig. 17). By exciting orthogonal transitions and monitoring the third, this method allows for precise chiral discrimination and ee determination with accuracies of up to 0.5%. Its resonant nature enables direct analysis of mixtures—as shown in the ee quantification of a conformational mixture of carvone, where each conformer's ee was resolved individually.
 | ||
| Fig. 17 The sign of the product between the dipole moment components (μαμβμγ) is enantiomer dependent and can be exploited in MRR spectroscopy for chiral analysis. Reproduced from ref. 81 with permission from Springer, copyright 2013. | ||
Enzyme-based methods83 represent a valuable and versatile approach for chiral sensing, particularly for biologically relevant molecules. These strategies exploit the inherent stereospecificity of enzymes, which typically convert only one enantiomer of a chiral substrate into a detectable product. The quantification of this product allows for the indirect determination of the sample's ee. One example involves monitoring the initial rate of enzymatic oxidation of a chiral alcohol, where the rate can be used for the construction of calibration curves, enabling accurate substrate ee analysis. The method also demonstrated robustness across various solvents, suggesting its good practical applicability. Another approach uses oxidoreductases, such as dehydrogenases, which produce NADH or NAD+-cofactors that are easily detected via UV-Vis spectroscopy due to their characteristic absorption at 340 nm. This principle has been applied in high-throughput methodologies for the rapid determination of ee in chiral secondary alcohols. These techniques are independent of substrate concentration and allow the simultaneous analysis of multiple samples in 96-well plates, with measurement times under five minutes, demonstrating excellent speed and suitability for high-throughput screening (HTS).
UV-Vis absorbance and fluorescence techniques,84 despite their achiral nature, have been extensively utilized in conjunction with chiral probes capable of forming diastereomeric pairs with different optical properties, owing to the widespread availability of these instruments. The chiral probe is typically designed to differentiate enantiomers either by forming diastereoisomers with distinct optical properties or by creating pseudo-racemic mixtures in which the two pseudo-enantiomers display different optical characteristics.
Pioneering within these techniques has been the work of Kubo,85 who employed indophenol dyes linked to a 1,1′-binaphthyl unit on a calix[4]crown platform that undergo different conformational changes depending on the specific enantiomer present in solution. The host exhibited colorimetric chiral recognition: one enantiomer showed a red-to-reddish-violet colour change, whereas the other led to almost no change in the absorption spectra. Diastereomeric optical differentiation has also been achieved using fluorescent probes, especially taking advantage of aggregate induced emission based luminophores and phosphorescence probes.86 Based on different principles are colour indicators based on doped liquid crystals.87 In these systems, the addition of a chiral analyte causes a variation in the liquid crystal's helical pitch. This variation results in light reflection at different wavelengths, leading to color changes.
Recent research in this field has taken two main directions: one focuses on employing chemometrics to develop more versatile probes capable of analyzing a wider range of substrates, while the other explores the use of nanomaterials such as carbon dots and gold nanoparticles, which exhibit enhanced optical properties for chiral sensing. As an example, Fig. 18 shows the functionalization of gold nanoparticles with D-gluconic acid and their application as a colorimetric sensor for various amino acids. Interestingly, the progressive colour shift depending on the ee of the sample enables a visual estimation of enantiopurity by simple naked-eye inspection.88
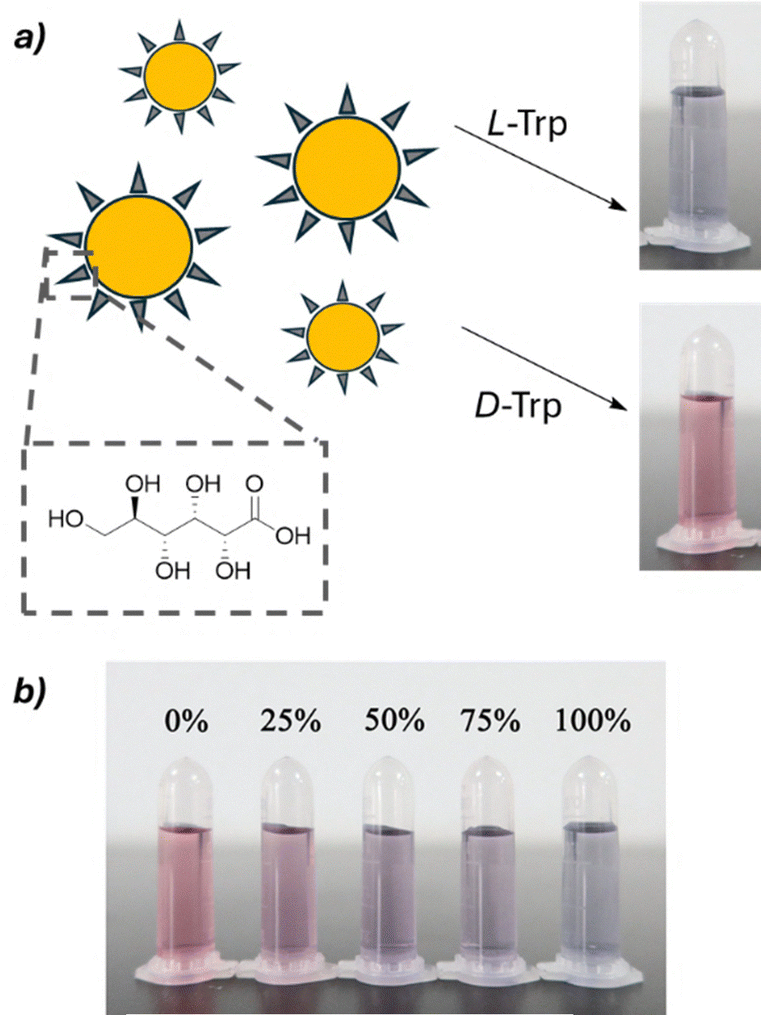 | ||
| Fig. 18 (a) Colorimetric shift induced by L- and D-tryptophan to a solution containing chiral gold nanoparticles functionalized with D-gluconic acid. (b) Different ee values furnish a diverse colorimetric output. Reproduced from ref. 88 with permission from Springer Nature, copyright 2021. | ||
Conclusions
The field of chiral analysis has witnessed remarkable advancements over the past few decades, evolving from classical polarimetry to sophisticated spectroscopic and chromatographic techniques. Besides classical techniques, the integration of cutting-edge technologies such as nanophotonics, machine learning, and automation is driving the development of increasingly sensitive, rapid, and high-throughput chiral analysis methods. Innovative techniques like Coulomb explosion imaging and crystal sponge methodologies are opening new avenues for direct determination of absolute configuration, addressing longstanding challenges in the field. Concurrently, the use of advanced materials such as metal–organic frameworks, covalent organic frameworks, and plasmonic nanostructures is enhancing the performance of established techniques like chromatography and chiroptical spectroscopies. The development of more universal methods applicable to a wide range of analytes remains a major challenge. While chiral chromatography remains the gold standard for many applications, the future of chiral analysis is likely to see further convergence of methods, with hybrid approaches combining the strengths of different techniques to achieve even more accurate and informative results.Continued innovation in this field not only enhances our ability to characterize and quantify enantiopurity but also has profound implications for drug development, materials science, and our fundamental understanding of biological and chemical processes at the molecular level. As we move forward, the advancements in chiral analysis will unlock new insights into the chiral world, fostering novel results across a wide spectrum of scientific disciplines and technological applications.
Author contributions
CZ conceived the idea of writing this review. All authors wrote the paper.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
The support from Fondazione Cassa di Risparmio di Padova e Rovigo (CHIRALSPACE), Italian Ministry of University and Research (MUR, PRIN grant: 2022CXHY3A) is acknowledged.References
- G. Vantomme and J. Crassous, Chirality, 2021, 33, 597–601 CrossRef CAS PubMed.
- J. Gal, Helv. Chim. Acta, 2013, 96, 1617–1657 CrossRef CAS.
- H. Gerlach, Chirality, 2013, 25, 684–685 CrossRef CAS PubMed.
- International Union of Pure and Applied Chemistry, ‘Chirality’ in IUPAC Compendium of Chemical Terminology, 2025, 5th edn, Online version 5.0.0, 2025 DOI:10.1351/goldbook.C01058.
- K. Mislow and J. Siegel, J. Am. Chem. Soc., 1984, 106, 3319–3328 CrossRef CAS.
- H. M. Fales and G. J. Wright, J. Am. Chem. Soc., 1977, 99, 2339 CrossRef CAS.
- T. Williams, R. G. Pitcher, P. Bommer, J. Gutzwiller and M. Uskokovic, J. Am. Chem. Soc., 1969, 91, 1871–1872, DOI:10.1021/ja01035a0601968–1969.
- P. L. Polavarapu, Org. Biomol. Chem., 2020, 18, 6801–6806 RSC.
- Q. Zhang and D. P. Curran, Chem. – Eur. J., 2005, 11, 4866–4880 CrossRef CAS PubMed.
- M. T. Reetz and K. E. Jaeger, Chem. – Eur. J., 2000, 6, 407–412 CrossRef CAS PubMed.
- K. Barman, M. M. Islam, K. S. Das, N. Singh, S. Priya, B. D. Kurmi and P. Patel, Biomed. Chromatogr., 2025, 39, e6073 CrossRef CAS PubMed.
- H. L. Qian, S. T. Xu and X. P. Yan, Anal. Chem., 2023, 95, 304–318 CrossRef CAS PubMed.
- P. Salvadori, C. Rosini and C. Bertucci, J. Org. Chem., 1984, 49, 5050–5054 CrossRef CAS.
- G. Betzenbichler, L. Huber, S. Kräh, M. L. K. Morkos, A. F. Siegle and O. Trapp, Chirality, 2022, 34, 732–759 CrossRef CAS PubMed.
- J. Teixeira, M. E. Tiritan, M. M. M. Pinto and C. Fernandes, Molecules, 2019, 24, 865, DOI:10.3390/molecules24050865.
- C. West, Trends Anal. Chem., 2019, 120, 115648 CrossRef CAS.
- Q. Zhang, S. Xue, A. Li and S. Ren, Coord. Chem. Rev., 2021, 445, 214108 CrossRef CAS.
- Z. Wang, W. Wang, A. Q. Luo and L. M. Yuan, J. Sep. Sci., 2024, 47, 2400073, DOI:10.1002/jssc.202400073.
- M. Lämmerhofer, J. Chromatogr. A, 2010, 1217, 814–856 CrossRef PubMed.
- E. L. Regalado and C. J. Welch, J. Sep. Sci., 2015, 38, 2826–2832 CrossRef CAS PubMed.
- E. Castiglioni, S. Abbate and L. Giovanna, Chirality, 2011, 23, 711–716 CrossRef CAS PubMed.
- L. Kvittingen and B. J. Sjursnes, J. Chem. Educ., 2020, 97, 2196–2202 CrossRef CAS PubMed.
- D. Sofikitis, L. Bougas, G. E. Katsoprinakis, A. K. Spiliotis, B. Loppinet and T. P. Rakitzis, Nature, 2014, 514, 76–79 CrossRef CAS PubMed.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- A. J. Miles, R. W. Janes and B. A. Wallace, Chem. Soc. Rev., 2021, 50, 8400–8413 RSC.
- G. Pescitelli, Chirality, 2022, 34, 333–363 CrossRef CAS PubMed.
- D. Leung, S. O. Kang and E. V. Anslyn, Chem. Soc. Rev., 2012, 41, 448–479 RSC.
- J. S. S. K. Formen, J. R. Howard, E. V. Anslyn and C. Wolf, Angew. Chem., Int. Ed., 2024, 63, e202400767, DOI:10.1002/anie.202400767.
- C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC.
- P. Zardi, K. Wurst, G. Licini and C. Zonta, J. Am. Chem. Soc., 2017, 139, 15616–15619 CrossRef CAS PubMed.
- J. R. Howard, J. R. Shuluk, A. Bhakare and E. V. Anslyn, Chem, 2024, 10, 2074–2088 CAS.
- J. J. Dotson, E. V. Anslyn and M. S. Sigman, J. Am. Chem. Soc., 2021, 143, 19187–19198 CrossRef CAS PubMed.
- C. Sparling and D. Townsend, Phys. Chem. Chem. Phys., 2025, 27, 2888–2907 RSC.
- V. Svoboda, N. B. Ram, D. Baykusheva, D. Zindel, M. D. J. Waters, B. Spenger, M. Ochsner, H. Herburger, J. Stohner and H. J. Wörner, Sci. Adv., 2022, 8, 1–8 Search PubMed.
- A. Comby, E. Bloch, C. M. M. Bond, D. Descamps, J. Miles, S. Petit, S. Rozen, J. B. Greenwood, V. Blanchet and Y. Mairesse, Nat. Commun., 2018, 9, 1–14 CrossRef PubMed.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1–32 Search PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- C. Zhang, S. Zhao, M. M. Zhang, B. Li, X. Y. Dong and S. Q. Zang, Coord. Chem. Rev., 2024, 514, 215859 CrossRef CAS.
- K. Tanaka, G. Pescitelli, K. Nakanishi and N. Berova, Monatsh. Chem., 2005, 136, 367–395 CrossRef CAS.
- A. Prabodh, Y. Wang, S. Sinn, P. Albertini, C. Spies, E. Spuling, L. P. Yang, W. Jiang, S. Bräse and F. Biedermann, Chem. Sci., 2021, 12, 9420–9431 RSC.
- R. Penasa, F. Begato, G. Licini, K. Wurst, S. Abbate, G. Longhi and C. Zonta, Chem. Commun., 2023, 59, 6714–6717 RSC.
- A. N. L. Batista, A. L. Valverde, L. A. Nafie and J. M. Batista, Chem. Commun., 2024, 60, 10439–10450 RSC.
- T. P. Golub, A. H. Abazid, B. J. Nachtsheim and C. Merten, Angew. Chem., Int. Ed., 2022, 61, e202204624, DOI:10.1002/anie.202204624.
- H. Rhee, Y. G. June, J. S. Lee, K. K. Lee, J. H. Ha, Z. H. Kim, S. J. Jeon and M. Cho, Nature, 2009, 458, 310–313 CrossRef CAS PubMed.
- G. Pescitelli and L. Di Bari, J. Phys. Chem. B, 2024, 128, 9043–9060 CrossRef CAS PubMed.
- C. R. Lightner, A. Kaczor and C. Johannessen, Vib. Spectrosc., 2024, 132, 103683 CrossRef CAS.
- Y. Tian, G. Fang, F. Wu, J. G. Kauno, H. Wei, H. Y. Hsu, F. Li, G. Xu and W. Niu, Coord. Chem. Rev., 2025, 526, 216375, DOI:10.1016/j.ccr.2024.216375.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- E. Er, T. H. Chow, L. M. Liz-Marzán and N. A. Kotov, ACS Nano, 2024, 18, 12589–12597 CrossRef CAS PubMed.
- F. Zhu, N. W. Isaacs, L. Hecht and L. D. Barron, Structure, 2005, 13, 1409–1419 CrossRef CAS PubMed.
- W. J. Choi, S. H. Lee, B. C. Park and N. A. Kotov, J. Am. Chem. Soc., 2022, 144, 22789–22804 CrossRef CAS PubMed.
- D. Verreault, K. Moreno, É. Merlet, F. Adamietz, B. Kauffmann, Y. Ferrand, C. Olivier and V. Rodriguez, J. Am. Chem. Soc., 2020, 142, 257–263 CrossRef CAS PubMed.
- P. Fischer and F. Hache, Chirality, 2005, 17, 421–437 CrossRef CAS PubMed.
- M. L. Solomon, A. A. E. Saleh, L. V. Poulikakos, J. M. Abendroth, L. F. Tadesse and J. A. Dionne, Acc. Chem. Res., 2020, 53, 588–598 CrossRef CAS PubMed.
- A. Biswas, P. Cencillo-Abad, M. W. Shabbir, M. Karmakar and D. Chanda, Sci. Adv., 2024, 10, eadk2560, DOI:10.1126/sciadv.adk2560.
- T. J. Wenzel and C. D. Chisholm, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 1–63 CrossRef CAS PubMed.
- D. Parker, Chem. Rev., 1991, 91, 1441–1457 CrossRef CAS.
- M. Seco, E. Quin and R. Riguera, Chem. Rev., 2004, 104, 17–117 CrossRef.
- M. Raban and K. Mislow, Tetrahedron Lett., 1965, 48, 4249–4253 CrossRef.
- J. A. Dale and H. S. Masher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS.
- M. D. Mccreary, D. W. Lewis, D. L. Wernick and G. M. Whitesides, J. Am. Chem. Soc., 1974, 96, 1038–1054 CrossRef CAS.
- P. Lesot, C. Aroulanda, P. Berdagué, A. Meddour, D. Merlet, J. Farjon, N. Giraud and O. Lafon, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 116, 85–154 CrossRef CAS PubMed.
- M. Seo, S. Jang and H. Kim, Chem. Commun., 2018, 54, 6804–6807 RSC.
- Y. Jia, L. Wen, W. Bao, Z. Xu, J. Wu and Y. Zhao, Anal. Chem., 2023, 95, 10362–10367 CrossRef CAS PubMed.
- T. Georgiou, J. L. Palma, V. Mujica, S. Varela, M. Galante, V. J. Santamaría-García, L. Mboning, R. N. Schwartz, G. Cuniberti and L. S. Bouchard, Nat. Commun., 2024, 15, 7367, DOI:10.1038/s41467-024-49966-8.
- X. Yu and Z. P. Yao, Anal. Chim. Acta, 2017, 968, 1–20 CrossRef CAS PubMed.
- D. Q. Han and Z. P. Yao, Trends Anal. Chem., 2020, 123, 115763 CrossRef CAS.
- P. Dwivedi, C. Wu, L. M. Matz, B. H. Clowers, W. F. Siems and H. H. Hill, Anal. Chem., 2006, 78, 8200–8206 CrossRef CAS PubMed.
- H. H. Hill, Anal. Chem., 2022, 94, 3020–3021 CrossRef PubMed.
- C. Xie, Y. Chen, X. Wang, Y. Song, Y. Shen, X. Diao, L. Zhu, J. Wang and Z. Cai, Chem. Sci., 2022, 13, 14114–14123 RSC.
- P. Martin, K. Maksim, J. Allan, S. J. Till, S. Hendrik, S. Felix, S. Lothar, S.-B. Horst, D. Reinhard, S. Jürgen, K. Julia, R. Michael, M. Sebastian, S. Alexander and S. Markus, Science, 2013, 341, 1096–1099 CrossRef PubMed.
- X. Zhou, Z. Wang, S. Li, X. Rong, J. Bu, Q. Liu and Z. Ouyang, Science, 2024, 383, 612–618 CrossRef CAS PubMed.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- J. Zou, G. Q. Zhao, G. L. Zhao and J. G. Yu, Coord. Chem. Rev., 2022, 471, 214732 CrossRef CAS.
- G. Zhu, O. J. Kingsford, Y. Yi and K. Wong, J. Electrochem. Soc., 2019, 166, H205–H217 CrossRef CAS.
- A. Stefani, T. Salzillo, P. R. Mussini, T. Benincori, M. Innocenti, L. Pasquali, A. C. Jones, S. Mishra and C. Fontanesi, Adv. Funct. Mater., 2024, 34, 1–11 CrossRef.
- F. S. Serena Arnaboldi, T. Benincori, R. Cirilli, W. Kutner, M. Magni, P. Romana Mussini and K. Noworytad, Chem. Sci., 2015, 6, 1706–1711 RSC.
- S. R. Domingos, C. Pérez, M. D. Marshall, H. O. Leung and M. Schnell, Chem. Sci., 2020, 11, 10863–10870 RSC.
- D. Patterson, M. Schnell and J. M. Doyle, Nature, 2013, 497, 475–477 CrossRef CAS PubMed.
- R. E. Sonstrom, Z. P. Vang, H. N. Scolati, J. L. Neill, B. H. Pate and J. R. Clark, Org. Process Res. Dev., 2023, 27, 1185–1197 CrossRef CAS PubMed.
- Z. Li, L. Bütikofer and B. Witholt, Angew. Chem., 2004, 116, 1730–1734 CrossRef.
- Y. Kubo, Molecules, 2025, 30(8), 1748 CrossRef CAS PubMed.
- Y. Kubo, S. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–524 CrossRef CAS.
- X. Chen, R. Zhu, B. Zhang, X. Zhang, A. Cheng, H. Liu, R. Gao, X. Zhang, B. Chen, S. Ye, J. Jiang and G. Zhang, Nat. Commun., 2024, 15, 1–11 CrossRef CAS PubMed.
- R. A. van Delden, Angew. Chem., Int. Ed., 2001, 40(17), 3918–3200 CrossRef.
- J. Yang, X. Li, Y. Du, M. Ma, L. Zhang, J. Zhang and P. Li, Amino Acids, 2021, 53, 195–204 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |