
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell-free synthetic biology for natural product biosynthesis and discovery
Andrew J.
Rice
 a,
Tien T.
Sword
b,
Kameshwari
Chengan
c,
Douglas A.
Mitchell
ad,
Nigel J.
Mouncey
e,
Simon J.
Moore
*f and
Constance B.
Bailey
*g
a,
Tien T.
Sword
b,
Kameshwari
Chengan
c,
Douglas A.
Mitchell
ad,
Nigel J.
Mouncey
e,
Simon J.
Moore
*f and
Constance B.
Bailey
*g
aDepartment of Biochemistry, School of Medicine – Basic Sciences, Vanderbilt University Medical Research Building-IV, Nashville, Tennessee 37232, USA
bDepartment of Chemistry, University of Tennessee-Knoxville, Knoxville, TN, USA
cSchool of Biosciences, University of Kent, Canterbury, CT2 7NZ, UK
dDepartment of Chemistry, Vanderbilt University, Medical Research Building-IV, Nashville, Tennessee, 37232, USA
eDOE Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
fDepartment of Life Sciences, Imperial College London, London, SW7 2AZ, UK. E-mail: simon.moore@imperial.ac.uk
gSchool of Chemistry, University of Sydney, Camperdown, NSW 2001, Australia. E-mail: constance.bailey@sydney.edu.au
First published on 19th March 2025
Abstract
Natural products have applications as biopharmaceuticals, agrochemicals, and other high-value chemicals. However, there are challenges in isolating natural products from their native producers (e.g. bacteria, fungi, plants). In many cases, synthetic chemistry or heterologous expression must be used to access these important molecules. The biosynthetic machinery to generate these compounds is found within biosynthetic gene clusters, primarily consisting of the enzymes that biosynthesise a range of natural product classes (including, but not limited to ribosomal and nonribosomal peptides, polyketides, and terpenoids). Cell-free synthetic biology has emerged in recent years as a bottom-up technology applied towards both prototyping pathways and producing molecules. Recently, it has been applied to natural products, both to characterise biosynthetic pathways and produce new metabolites. This review discusses the core biochemistry of cell-free synthetic biology applied to metabolite production and critiques its advantages and disadvantages compared to whole cell and/or chemical production routes. Specifically, we review the advances in cell-free biosynthesis of ribosomal peptides, analyse the rapid prototyping of natural product biosynthetic enzymes and pathways, highlight advances in novel antimicrobial discovery, and discuss the rising use of cell-free technologies in industrial biotechnology and synthetic biology.
 Andrew J. Rice | Andrew J. Rice received his BSc in Biochemistry with Honors in Chemistry at Muhlenberg College. In 2019, he started his PhD in the Mitchell group at the University of Illinois at Urbana-Champaign where his research spanned a breadth of enzymes generating a variety of complex post-translational modifications on peptides and proteins. He received his PhD in 2024. He has been supported by an NSF Graduate Research Fellowship and an NIH-sponsored Chemistry–Biology Interface Training Program. |
 Tien T. Sword | Tien T. Sword received her BS in Chemistry from the University of Tennesee-Knoxville in 2019 and in 2020 started her PhD in the Bailey group at the University of Tennessee-Knoxville where her research focused on developing strategies to optimise biosynthetic protein expression in lysate-based cell-free expressions including nonribosomal peptide synthetases and polyketide synthases. She received her PhD in 2024. |
 Kameshwari Chengan | Kameshwari Chengan received her BS in Biomedical Sciences with Honours from the University of Kent in 2019. In 2020, she started her PhD in the Moore group at the University of Kent where her research was focused on developing a Klebsiella pneumoniae cell-free gene expression tool for antibiotic discovery, screening, and resistance studies. She joined the start-up company Evolutor Ltd in 2024, where she took a strain development role producing precision evolved microbial chassis and building adaptive laboratory evolution platform technologies. |
 Douglas A. Mitchell | Douglas A. Mitchell received a BSc in Chemistry from Carnegie Mellon University in 2002, a PhD from the University of California, Berkeley in 2006, followed by postdoctoral studies at the University of California, San Diego. Mitchell joined the Department of Chemistry at the University of Illinois at Urbana-Champaign in 2009 and held the John and Margaret Witt Professorship. In 2025, Mitchell moved to the School of Medicine at Vanderbilt University and is appointed as the William Kelly Warren S. Chair in Biochemistry and Director of the Vanderbilt Institute for Chemical Biology. The Mitchell lab employs various theoretical and experimental approaches to identify, characterise, and engineer natural product biosynthetic pathways. |
 Nigel J. Mouncey | Nigel J. Mouncey received his BSc in Microbiology in 1992 from the University of Glasgow and his DPhil in Biochemistry in 1995 at the University of Sussex where he studied molybdenum uptake and regulation. Following postdoctoral fellowships at Harvard Medical School and the University of Texas Medical School Houston, he joined Roche Vitamins, Inc. as a Senior Research Scientist. Subsequently he became Senior Scientist at DSM Nutritional Products and Bioengineering and Bioprocessing R&D Director and Leader at Dow AgroSciences. During his time in industry, Dr Mouncey directed R&D teams that focused on the discovery, development and commercialization of novel production organisms and fermentation processes for vitamins, insecticides, fungicides, platform chemicals, cosmetics and new crop traits. Currently, Dr Mouncey serves as the Director of the DOE Joint Genome Institute in Berkeley and also as Head of the JGI Secondary Metabolites Science Program. His research interests span regulation of secondary metabolites biosynthesis, synthetic biology and microbiome research. |
 Simon J. Moore | Simon J. Moore received his BSc in Biochemistry in 2007 and his PhD in 2011 from the University of Kent under the supervision of Professor Martin Warren, where he studied tetrapyrrole biosynthesis, elucidating the anaerobic biosynthesis of vitamin B12 and coenzyme F430 pathways. In 2014, he joined Imperial College London with Professor Paul Freemont to explore synthetic biology for natural product discovery. He began an early-career Wellcome Trust-Imperial College London ISSF fellowship in 2017 to study natural product biosynthesis, including the development of Streptomyces cell-free gene expression. In 2018, Simon started as a Lecturer in Biochemistry at the University of Kent, before moving to Queen Mary University of London in 2023. In 2025, Moore transferred to the Department of Life Sciences, Imperial College London. His group has a specialist interest in cell-free synthetic biology, as well as broader expertise in studying and engineering biosynthetic pathways. |
 Constance B. Bailey | Constance B. Bailey received her BA in Biochemistry and Molecular Biology from Reed College in 2010 and PhD from the University of Texas-Austin in Chemistry in 2015 where she investigated the enzymology and biocatalytic applications of type I polyketide synthases under the mentorship of Prof. Adrian Keatinge-Clay. Following her PhD she pursued an NIH Postdoctoral Fellowship with Prof. Jay Keasling at UC Berkeley and Lawrence Berkeley National Laboratory in synthetic biology generating bioproducts from engineered polyketide synthase assembly lines. She started her independent career at the University of Tennessee-Knoxville as an Assistant Professor from 2018 to 2023 and in 2023 moved to the University of Sydney as a Senior Lecturer and independent group leader. Her laboratory focuses on developing heterologous expression strategies for antibiotic discovery and development, including developing cell-free strategies for prototyping engineered secondary metabolites. |
1. Introduction
Over the last 20 years, there has been a resurgence in using cell-free technologies – broadly referred to as cell-free synthetic biology – for a range of applications including the production of commodity and specialty chemicals, proteins, therapeutics, prototyping of gene expression, and diagnostics.1,2 However, only more recently have cell-free technologies been applied to complex and commercially relevant natural products. Historically, natural product biosynthesis has been investigated in an interdisciplinary fashion that draws upon synthetic chemistry, enzymology, genome mining, and cellular microbiology. As our ability to tackle complexity increases in cell-free synthetic biology, its applicability to natural product biosynthesis expands. This review summarises efforts to date and the strengths and limitations in applying cell-free transcription/translation systems to elucidating natural product biosynthesis and generating engineered secondary metabolites. This includes bioactivity-guided drug discovery, antimicrobial resistance (AMR) studies, and the growing role of cell-free innovation in industrial applications.2 Natural products have widespread applications and their chemical scaffolds are found in about one-third of U.S. Food and Drug Administration (FDA)-approved new molecular entities.3 They are an immensely important source of therapeutics, including but not limited to antimicrobial, anti-tumour, and anti-parasitic compounds. Many natural products have complex molecular architectures, are often greater than >500 Da, and thus do not always meet Lipinski's rule of five for drug-like chemical properties.4 In the past, the pharmaceutical industry discovered most of the antibiotic chemical scaffolds through Selman Waksman's approach of using high-throughput screening of extracts from a range of natural sources including environmental bacteria, fungi, and plants.5 In particular, the Streptomyces genus was a dominant source of antibiotics – e.g., streptomycin, kanamycin, griseomycin.6 This period, known as the “Golden Age of Antibiotics,” has long since declined due to the rediscovery of known compounds, and the shift of the pharmaceutical industry to synthetic chemistry.Together with traditional chemistry approaches, the emergence of both DNA recombinant technologies and enzymology studies aided several chemistry and chemical biology groups to decipher the biosynthesis of some of the most prominent natural products, successfully correlating genes to molecules. The release of the model Streptomyces coelicolor A3(2) genome sequence in the early 2000s provided a significant advance.7 Its genome contained clusters of genes spread throughout its linear chromosome that were linked to the biosynthesis of known natural products as biosynthetic gene clusters (BGCs). However, S. coelicolor A3(2) possessed 27 BGCs, which was a much higher number than the known natural products identified under standard laboratory culture conditions.8 With further research, 17 out of these 27 BGCs now have been assigned to known metabolites.8 As genome sequencing became more widely available, this pioneering work inspired a new era of using genomics to guide natural product discovery. Now, there are about 1.2 million bacterial genomes fully sequenced and approximately half a million sequenced metagenomes.9 Combined with advances in freely available genome mining tools such as antiSMASH, BAGEL, RODEO, ARTS and many others,10–15 this rich pool of genomic data suggests that the true chemical diversity of natural products vastly exceeds the number of known natural products,16–18 which have mostly been discovered through traditional bioactivity-guided isolations from crude extract and fractionation methods.19–21 Many of these advances have been driven by improved capacity in high-throughput screening, heterologous expression and genome and pathway engineering8,22–26 across a range of organisms.27
2. Cell approaches to natural product discovery.
Microbes generate natural products in their environment to help them survive, often triggered in response to specific chemical or physical cues, or other stress factors leading to genomic changes and adaptive evolution triggering BGC gene expression. Those BGCs that cannot be correlated to known natural products are often called “silent” or “cryptic”.28 “Silent” BGCs are transcriptionally or translationally dormant under standard laboratory conditions. “cryptic” BGCs refer to lesser characterised BGCs, where knowledge of the regulation or biosynthetic genes is incomplete, missing or unknown (i.e., abundance of hypothetical genes, domains of unknown function), or spread throughout the genome rather than clustered. In addition, some natural products and pathway intermediates are labile. Therefore, in general, isolated natural products tend to be highly abundant and chemically stable in metabolite extracts. Considerable efforts have been put forth to elicit BGC activation in natural hosts, such as the one-strain many compounds (OSMAC) approach. In OSMAC, microbial isolates are cultured under an array of conditions to activate different BGCs.29 Other screening efforts include small molecule libraries to elicit BGC activation30,31 and co-culturing to simulate environmental competition and collusion between microbes.32–34 Genetic manipulations have been performed to express BGCs, such as over expressing or deleting regulators, replacing promoters or using heterologous expression systems. However, in general, discovery of molecules appropriate as therapeutics from natural sources often requires further modification of molecular structure beyond those found in naturally occurring molecules. Frequently, there is a need to manipulate enzymes and biosynthetic pathways with precise control to alter the molecular structure of natural products and improve bioactive properties. This can be to improve target affinity, but also to improve drug properties. In many cases, natural products are not optimised for pharmacological interactions with human physiology as they evolved in a distinct environment within the producing organism. These molecules typically must be further altered for mammalian cellular penetrance, reduced cytochrome P450 metabolism, and other properties that affect pharmacokinetics and toxicity.20,35 Given the immense molecular complexity of many natural products, analogue generation presents a significant barrier to their development as therapeutics.363. Why cell-free synthetic biology?
Synthetic biology holds promise in generating new natural products by enabling activation of “silent” or “cryptic” BGCs or allowing precise control over molecular structures. However, there are challenges associated with cell-based synthetic biology. It is often necessary to screen multiple genetic designs through iterative and resource-intense experiments that can be challenging, especially if a less genetically tractable or slower growing heterologous host is used, or if the intermediate/product is toxic.Cell-free technologies have emerged as an alternative tool to accelerate the discovery and development of natural products and their derivatives. We focus here on describing the emerging uses of cell-free gene expression (CFE) for engineering natural product biosynthesis. By removing the cell wall, cell membrane and genomic DNA, cell-free extracts provide a quasi-chemical bioreactor platform, which can be modularly controlled to both make1,37,38 and detect39–42 RNA, peptides, proteins, and small molecules. Cell-free enzymes and ribozymes (i.e., ribosomes) provide a powerful catalytic unit with the dexterity to study biological chemistry, and flexibility to work at different levels of scale from microfluidics43,44 through to 100 L reactions,45–47 and with evidence of linearity and low variability across these scales.45 Depending on the analysis type, CFE experiments can take a few minutes to hours,48–50 whereas depending on the context, a cell-based approach can take several days to weeks, potentially even longer if one performs genetic modification. Thus, CFE enables rapid cycling between experimental design and analysis51 of mRNA and protein synthesis.44,52,53 The starting concentration of the substrates and proteins can be determined,52 or controlled (i.e., addition of purified proteins and chemicals), aiding standardisation and predictive modelling.51,54 CFE reactions can be extended by replenishment of the reaction substrates and removal of metabolic end-products (e.g., lactate, acetate), through dialysis or microfluidics devices, extending steady-state protein synthesis for up to 30 hours.44 To increase the speed, or directly engineer the DNA templates, CFE reactions can work with linear DNA products.55 In addition, since cell-free extracts are non-living, they are safer to explore with low biocontainment facilities and expertise. CFE can also be freeze-dried to facilitate transport at room temperature, and later use upon rehydration.39,56 These collective advantages make CFE increasingly attractive to chemists, chemical engineers, and synthetic biologists. However, there are also significant challenges in using CFE for natural product discovery, which we will discuss at specific points through this review (Fig. 1). We will next discuss the historical context of CFE, what constitutes the molecular machinery and reactions of CFE, and different CFE methods currently established. Where possible, we will also highlight similarities between CFE and synthetic chemistry.
 | ||
| Fig. 1 Overview of the advantages and disadvantages of (a) whole cell and (b) cell-free approaches to natural product biosynthesis and discovery. | ||
3.1 Historical context of cell-free expression
CFE has played a major role in the fields of genetics and biochemistry. The pioneering efforts of the Nobel laureate Eduard Buchner (Nobel Prize in Chemistry in 1907) led to the discovery that yeast cell extracts ferment glucose into carbon dioxide and water.57 In addition, a major discovery of the 20th century was the unravelling of the genetic code by Marshall Nirenberg, Har Khorana and Robert Holley, which led to a shared Nobel Prize in Physiology or Medicine in 1968.58–60 Critical to these experiments were the use of Escherichia coli cell-free extracts containing active ribosomes, combined with poly-RNA and single 14C-labeled amino acids to initiate protein synthesis. This was remarkable since, at this time, the biochemistry of protein synthesis was unknown. To determine the genetic code, radioactivity was used to assign which DNA triplet (i.e., 64 codons) codes for which of the 20 canonical amino acids. This research was an important stepping stone in the development of modern molecular biology and highlights a key strength of cell-free extracts to study and manipulate gene expression. In another example, Alfred Goldberg and Tom Maniatis determined the mechanism for the ubiquitin-proteasome pathway using yeast CFE.61,62 Closer to natural product biosynthesis, Ian A. Scott initially developed the concept of total enzyme biosynthesis using cell-free extracts and purified enzymes to perform complex chemistry, including the biosyntheses of S-adenosyl-L-methionine,63 vitamin B1264–66 and taxol.67 More recently, this approach has been applied for the biosynthesis of a variety of complex natural products.68–733.2 What is a cell extract?
At their simplest level, cell-free experiments are composed of a cell extract, devoid of a genome, cell wall, and cell membrane. Typically, the extract contains all the proteins, RNA and small molecules required to generate new proteins through transcription–translation, as well as many metabolic enzymes present at the point of cell lysis. Focusing on CFE, this is similar to synthetic chemistry in a broad sense. CFE reactions start from known substrates, and through ribozyme and enzyme-catalysed reactions – some dependent on energy, metals or specialised cofactors to aid catalysis – generate products, often proteins, and sometimes at high yields and efficiency. In contrast to chemistry reactions that can require elevated temperatures, typical CFE models derived from cells grown at mesophilic conditions function best between ∼20–40 °C, although there is a Thermus thermophilus CFE model that works at thermophilic temperatures.74,75 Finally, in contrast to most synthetic chemistry reactions, CFE does not need high concentrations of expensive or unsustainable metal catalysts, protection/deprotection steps, or toxic organic solvents.
E. coli is the dominant CFE model because it is extensively characterised in terms of physiology and multi-omics (i.e., DNA, RNA, protein and metabolite) level characterisation. E. coli cells double approximately every 20 minutes when supplemented with rich organic media and saturating oxygen at 37 °C.76 Under these conditions, E. coli requires less energy for anabolic processes such as making amino acids77 or cofactors. About 55% of an E. coli cell's biomass is protein,78 with ribosome biogenesis requiring up to one-third of a cell's volume or mass.79,80 The total number of ribosomes is proportional to cell growth. Fast dividing cells (TD = 20 min) contain approximately 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ribosomes per cell.80 Slow dividing cells (TD > 1 hour) contain less than 8000 ribosomes per cell.80 While ribosomes and translational machinery from stationary cells are active,81 the most active E. coli cell extracts for CFE are typically derived from rapidly dividing cells.
000 ribosomes per cell.80 Slow dividing cells (TD > 1 hour) contain less than 8000 ribosomes per cell.80 While ribosomes and translational machinery from stationary cells are active,81 the most active E. coli cell extracts for CFE are typically derived from rapidly dividing cells.
The cell wall and cell membrane are physical barriers that contain a highly crowded molecular mixture of proteins, RNA, and small molecules inside a set volumetric unit – the cell. After cell lysis, the cell extract is less crowded – by between one to two orders of magnitude52,82–84 – compared to the inside of a cell. Dividing cells are also continuously making and recycling RNA/proteins. For an E. coli cell (∼1 μm3) this results in 3–4 million proteins (average size 35 kDa) per cell.85 In the cytoplasm there is an estimated total protein concentration of 200–300 mg mL−1.86,87 Up to one-third of this content is ribosomes, which catalyse their own biogenesis, as well as the cell's proteome. The rest of the cytoplasmic proteins are dedicated to gene expression, metabolism and other processes essential to a cell including structural proteins involved in controlling cell division and cell trafficking.88,89 While some of these interactions are well mapped in model organisms such as E. coli, their role in molecular crowding and pathway functions remains relatively uncharacterised yet can influence the function of key cell-free enzymatic processes. Examples include reconstituted ribosomes90 and enzyme RNA polymerase, whose activity is enhanced by artificial molecular crowding agents in CFE studies.91 Other biocatalytic processes can also be stimulated by molecular crowding chemical mimics, including polymers such as polyethylene glycol.92–95 However, the rates of activity are less in CFE than within a cell. For example, the ribosomes in CFE translate proteins approximately an order of magnitude slower than inside the cell.52,87 This is an important limitation in considering the potential applications of CFE.
Last, engineering living cells to make chemicals can be a challenge. The concentration of metabolites and chemicals spans several orders of magnitude inside a cell.96,97 Within any cell type, the optimal balance of single-molecule concentrations is achieved through homeostasis, a process regulated by genetic changes, metabolic flux, and the import and export of chemicals across the cell membrane. This intricate process relies on individually evolved catalytic efficiencies and molecular interactions inherent to each cell type and their associated microenvironment.97 Frequently, challenges arise in synthetic biology and metabolic engineering studies when engineering cells to accommodate heterologous enzymes and chemicals. This can lead to disruptions in homeostasis and impairments in cell growth. Each protein has an evolved function within a specific cell type, including how it interacts with other biomolecules (nucleic acids, lipids, proteins and small molecules) and how its own distinct physiochemical properties (i.e., thermostability, solubility limits) control function. Therefore, when placing a heterologous gene/protein into a cell, its expression can lead to various metabolic and genetic imbalances – often generically referred to as “burden”98–100 – as well as solubility issues,101 leading to stress responses to restore order – i.e., DNA recombination and mutations. The above factors are not relevant for CFE, providing an advantage to this approach.
3.3 Cell-free gene expression using E. coli
A crude cell-free extract has all the essential proteins and RNA required for catalysing gene expression, as well as a variety of anabolic and catabolic metabolic pathways (Fig. 2). There are about 90 proteins, three ribosomal RNA (rRNA) and 33 transfer RNA (tRNA) species required for catalysing CFE.102,103 Gene expression begins through expression of DNA by a multienzyme RNA polymerase (RNAP) complex (αβ1β2ω), which synthesises messenger RNA (mRNA) from the nucleotide bases adenosine triphosphate (ATP), cytosine triphosphate (CTP), guanosine triphosphate (GTP) and thymidine triphosphate (TTP). Specifically, a Sigma factor (σ) guides the RNAP holoenzyme to the so-called promoter region of the DNA helix to initiate mRNA transcription, while several other enzymes conduct DNA winding/unwinding. Instead of using the native RNA polymerase, CFE models typically use the heterologous T7 RNA polymerase (derived from T7 bacteriophage – a virus that infects and replicates within E. coli) as a high powered and orthogonal alternative. Translation in E. coli requires ribosomes, 33 translation factors, 22 aminoacyl-tRNA ligases, 33 tRNA species, 20 amino acids and energy as ATP equivalents. The ribosome is an RNA–protein complex composed of a small 30S (Svedberg units) and large 50S subunit. The 30S subunit requires ribosomal proteins (labelled S1–S12) and both 5S and 16S ribosomal RNA (rRNA). In contrast, the 50S subunit is composed of ribosomal proteins (L1–L21) and 23 rRNA. When total RNA is extracted from actively growing bacterial cells, rRNA and tRNA are the most abundant and stable RNA species, comprising ∼95% of total RNA.104 Translation factors are critical for ribosome activity including three initiation factors (IF1–3), three elongation factors (EF-Tu, EF-Ts, and EF-G), three release factors (RF1–3), a ribosome recycling factor (RRF), and the 20 canonical amino acids for coupling to a cognate tRNA. One essential precursory step occurs via an aminoacylation reaction catalysed by an aminoacyl-tRNA ligase. Beyond the transcription–translation machinery, there are endo- and exo-nucleases (RNAses) that recycle bulk RNA, as well as peptidases and proteases (∼74 in E. coli), chaperones, transcription factors and metabolic enzymes. | ||
| Fig. 2 Components and metabolic pathways required for (a) PURExpress® and (b) crude cell extract-based CFE. Metabolic pathways related to cell-free energy regeneration are shown with black solid arrows. Cofactors and primary energy metabolites involved in primary and secondary energy regeneration are coloured orange and green respectively. | ||
Compared to peptide chemical synthesis,105E. coli ribosomes catalyse amino acid elongation at rates of ∼10–20 amino acids per second.52 Atom efficiency and success rate is close to 100%, while incomplete translation products and mRNA transcripts are degraded and recycled back into amino acids and nucleotides.106,107 In comparison, solid phase peptide synthesis (SPPS) can be performed at rates of near 10–15 residues per hour108 (at most 2.5 residues per minute). SPPS can also make up to 50–100 amino acid polymers with yields approaching 99% for each addition. In terms of limitations, CFE is sensitive to many physical and chemical variables (e.g., pH, temperature, ionic strength). However, there are clear advantages for biological peptide/protein synthesis over SPPS, especially as many proteins are much larger than 100 amino acids in length. In addition, peptides and proteins can be modified post-translationally, and non-canonical amino acids can be incorporated, which is comparable to SPPS. By leveraging transcription/translation however, vast libraries of variable peptide and protein sequences can be generated from DNA templates. Here, CFE provides a unique advantage for making and modifying ribosomal peptides, as discussed specifically in Section 4.1.
In terms of concentration and diversity of proteins in cell extracts, early proteomics studies found about 500–800 different proteins in E. coli CFE,109,110 while a more recent study (with increased sensitivity) found up to 1892111 different proteins – or 43% of what is encoded by the E. coli genome. In terms of protein concentration, gene expression proteins are dominant, with elongation factor Tu (EF-Tu) being the most abundant, followed by the ribosomal proteins.112 In complement, quantitative mass spectrometry methods have been used to estimate that the ribosome concentration (at 10 mg mL−1 total protein) is ∼2.2 μM,113 with E. coli estimates between 1.6 to 2.3 μM.52,114 Individual levels of intracellular proteins were also shown to be sensitive to several physical conditions during cell extract preparation,111 including heating and pH/salts. Such information is valuable towards understanding the limitations of CFE and to inform future efforts to improve variability and performance.
Currently, E. coli is the most popular cell type for making cell extracts, with the B strains specifically developed as a base for strong heterologous recombinant protein production. During the early 2000s, an E. coli derived ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) rotein synthesis
rotein synthesis ![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) sing
sing ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ecombinant
ecombinant ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) lements (PURE) system was developed from purified components.115 This includes purified ribosomes (54 ribosomal proteins) and translation factors, which when combined with T7 RNAP, tRNA, energy regeneration enzymes, substrates (i.e., amino acids, creatine phosphate) and synthetic DNA, reconstitutes transcription–translation within a test-tube.103,116 This remarkable engineering feat is commercially available as the PURExpress® kit (New England Biolabs), while another group recently made available a step-by-step protocol and bacterial strains (His6-tagged translation factors) for manual preparation of the PURE cell-free reaction.117,118 An advantage of PURE is its high efficiency because of an absence of competing side-reactions such as non-specific phosphatases, nucleases and peptidases/proteases, which degrade the energy source, DNA/RNA and proteins. However, limitations of PURE include its high cost and limited potential for scaling up. Several groups have used PURE systems for a variety of applications,119 including natural product biosynthesis.120
lements (PURE) system was developed from purified components.115 This includes purified ribosomes (54 ribosomal proteins) and translation factors, which when combined with T7 RNAP, tRNA, energy regeneration enzymes, substrates (i.e., amino acids, creatine phosphate) and synthetic DNA, reconstitutes transcription–translation within a test-tube.103,116 This remarkable engineering feat is commercially available as the PURExpress® kit (New England Biolabs), while another group recently made available a step-by-step protocol and bacterial strains (His6-tagged translation factors) for manual preparation of the PURE cell-free reaction.117,118 An advantage of PURE is its high efficiency because of an absence of competing side-reactions such as non-specific phosphatases, nucleases and peptidases/proteases, which degrade the energy source, DNA/RNA and proteins. However, limitations of PURE include its high cost and limited potential for scaling up. Several groups have used PURE systems for a variety of applications,119 including natural product biosynthesis.120
3.4 Alternative CFE methods
To provide alternative CFE methods, there is a rising interest in exploiting alternative microbes that offer unique benefits, such as resilience to harsh manufacturing conditions or specific functional advantages – e.g., photosynthesis, minimal cells, natural product hosts. As such, alternative hosts are expected to play an increasingly significant role in synthetic biology.121 However, there are several limitations (e.g., genetic tools, characterisation) that hinder wider use. Here, CFE provides an enabling technology to explore such non-model hosts to study gene expression and metabolic reactions. Recent CFE tools from microbes include Bacillus subtilis,122Priestia megaterium,113Pseudomonas putida,123Vibrio natriegens,124–126Clostridium autoethanogenum,127 and multiple Streptomyces spp.128–133 In addition, a related study showed the general flexibility of cell-free methods across several bacterial species and even multi-extract CFE.134The Bacillus Gram-positive low G + C genus are traditionally popular in industry because of their strong potential for enzyme and protein secretion, and natural product discovery.135 There are two well-established systems for this potential application. First, a B. subtilis CFE,122 which is derived from a major industrial microbiology model organism. Second, a Priesta megaterium CFE (formerly Bacillus megaterium) tool, which demonstrated stronger levels of protein synthesis than B. subtilis CFE and has minimal protease activity.136P. megaterium is notable for its industrial use including amylase and vitamin B12 production.136,137 For future progress, tools that accelerate the development of novel molecular tools are needed for non-model microbes. To meet this need, the P. megaterium CFE was used as a rapid prototyping platform for developing gene expression tools, an approach assisted by automation.136
Another promising candidate is Vibrio natriegens, which has the fastest doubling time of any known cultured microbe at TD < 10 min.138V. natriegens is also compatible with E. coli plasmids and genetic systems.138–140 Three groups reported the development of V. natriegens CFE tools, providing high protein yields (up to 1.6 mg mL−1) equivalent to E. coli CFE.124–126 The V. natriegens CFE was also active with linear DNA templates with protection from degradation by the Cro DNA-binding protein.141
Finally, the high G + C Actinomycetota phylum, which includes the Streptomyces genus, remain the most studied in terms of natural products isolated and characterised. Over 3000 strains have been catalogued, and many are slow-growing and/or challenging to genetically engineer. For this, Streptomyces CFE from different models has been developed for studying gene expression or pilot expression studies.128–133 Examples of their initial uses for natural product discovery will be highlighted within the review, while a more focused review on Streptomyces CFE is available.37
3.5 Cell-free metabolism
Early CFE studies were based on in vitro translation (IVT) reactions, when an RNA template was added to a cell extract with ATP as the primary energy source. However, the native energy pool and extract associated ATP consuming phosphorylases are a key limiting factor for extended mRNA and protein synthesis in these early IVT/CFE methods. Crude cell extracts are dynamic in terms of their ability to catalyse anabolic and catabolic metabolic reactions.113,142,143 There are hundreds of metabolic enzymes present in the cell extract dependent on the growth condition and processing steps used for extraction,111 suggesting that cell-free extracts have the unrealised potential to be engineered to catalyse more complex chemical reactions. CFE can make energy as ATP equivalents from a secondary energy source. Through catabolism of a high-energy carbon substrate, CFE will generate reducing equivalents via primary metabolism, which have the potential to couple to other biosynthetic pathways via redox active proteins (e.g., Fe–S) and protein-bound cofactors, such as nicotinamide adenine dinucleotide (NAD), flavin adenine dinucleotide (FAD) and mononucleotide (FMN). In synthetic chemistry, rare metal catalysts such as palladium are often used to perform equivalent reduction reactions. Another advantage that CFE provides over living cells is the facile integration of synthetic enzyme pathways through the addition of purified enzymes or mixing of different cell-free extracts and enzymes, also referred to a cell-free metabolic engineering.144–147 This enables rapid prototyping, as well as the use of non-natural precursors (i.e., non-canonical amino acids) and sacrificial substrates such as polyphosphate or phosphite for energy regeneration such as ATP148 and NADH/NADPH149,150 recycling via kinase and dehydrogenase enzymes, respectively.Focusing on energy sources, several CFE studies initially used creatine phosphate/kinase-based systems for energy regeneration.41,151,152 Recent research has since identified a range of high energy carbon substrates as an alternative energy source to regenerate ATP within the cell extracts.113,143,153–155 These energy sources are categorised as primary or secondary. The primary energy sources (∼0.5–3 mM) are the nucleotides ATP, GTP, CTP and TTP (or diphosphate and monophosphate equivalents), which are utilised as the building blocks for mRNA synthesis in transcription. ATP/GTP then has an additional role as energy for translation: 1 molar equivalent of ATP is required to initiate translation, 2 for activation of the amino acids to form a single aminoacyl-tRNA, 2–3 for peptide elongation, and 1 for translation termination by a release factor protein (RF1–3).156,157 The secondary energy source (∼10–50 mM) is required to regenerate the primary energy source (∼1–2 mM) that initially powers mRNA synthesis, while some ATP/GTP is used to begin protein synthesis and in charging the aminoacylated-tRNA species. Therefore, most CFE reactions use a working concentration of ATP/GTP higher than CTP and TTP.158 The secondary energy source then generates ATP equivalents through catabolism of a high-energy carbon substrate. This occurs because the extracts contain the metabolic enzymes and complete pathways for these reactions. While glucose can be used as an energy source in CFE,142,143,159 other high-energy substrates from the glycolysis pathway such as pyruvate,160 phosphoenolpyruvate,161 3-phosphoglycerate,162 and L-glutamate163 provide more direct sources of energy (Fig. 2). Interestingly, E. coli cell-free extracts also contain inverted vesicles. Early studies established that CFE synthesis of chloramphenicol acetyltransferase is oxygen-dependent and disrupted by known electron transport chain (ETC) inhibitors.164 These vesicles are an artefact of cell lysis and contain parts of the ETC and ATP synthase, also observed by proteomics.111,165
A limitation of cell-free extracts is the presence of many non-specific phosphorylase enzymes, which degrade the bulk nucleotide triphosphate (NTP) pool and therefore contribute to overall energy loss in CFE. Calculations highlight up to 98% of total energy is wasted in cell extract CFE.113 Here, advancements in machine learning have assisted with modelling and prediction of “winning” combinations of this multi-parameter space in CFE.166 In addition, there are background metabolic enzymes and pathways that can be activated to contribute to energy production. For example, L-glutamate, an amino acid and nitrogen donor, can provide extra ATP. L-Glutamate enters the Krebs cycle via a transaminase reaction to generate α-ketoglutarate. Uniquely, P. megaterium CFE was active with succinate as a secondary energy source,113 presumably catabolised via the succinate dehydrogenase complex to feed into to the ETC.
Cell-free extracts also contain a range of amino acid, nucleotide, fatty acid, and sugar metabolites, whereas in PURE all starting substrates and cofactors are controlled for. In crude cell-free extracts, the addition of exogenous NAD+ and coenzyme A can improve CFE productivity,52,158 suggesting these cofactors are rate-limiting in cell-free extracts. Together with energy and reducing equivalents, NAD and coenzyme A are important cofactors or coenzymes for secondary metabolism. For example, acetyl-CoA, malonyl-CoA and related analogues are substrates for polyketide biosynthesis.
4. CFE for biosynthesis
In previous sections, we explored the distinct features of CFE and its potential uses for biosynthesis. Now, we will delve into specific types of natural products and other chemical products engineered using CFE. We will refer to this broader approach as cell-free biosynthesis (CFB), or cell-free transcription–translation and biosynthesis. For instances where there are gaps in the literature, we will highlight similar approaches using cell-free extracts or purified enzymes. We will also highlight key advantages and limitations of CFE/CFB for specific chemical classes or related biomolecules where applicable.4.1 CFB of RiPPs
The ribosomally synthesised and post-translationally modified peptides (RiPPs) family are a well-suited class of natural products for CFB (Fig. 3 and Table 1). This is because the structure and function of RiPPs are directly amendable at the level of the DNA sequence. RiPPs are translated as an inactive precursor peptide, often consisting of an N-terminal leader region involved in substrate binding and a C-terminal core region modified by one or several enzymes.167 Proteolysis of the leader peptide often results in the mature RiPP, although further tailoring modifications can occur. By separating the sites primarily responsible for recognition and modification, Nature exploits this as a strategy for library generation. As RiPPs are DNA encoded, variants of the final product can be readily produced by mutagenesis of the precursor peptide gene. However, generating extensive libraries of precursor peptides is laborious, and there is no guarantee that any given modifying enzyme will accommodate these variations in substrate. Furthermore, variants produced by heterologous expression are limited to the 20 standard amino acids unless amber stop codon suppression, chemical modification, or another strategy is used.168,169 SPPS is an option, but this is a relatively low-throughput strategy, and most precursor peptides are long enough that cost, peptide solubility, and secondary structure can be problematic. | ||
| Fig. 3 Summary of RiPP biosynthesis. (a) A generalised schematic of RiPP biosynthesis, in which one or several enzymes act on a precursor peptide to form a mature RiPP. (b) Several experiments in which CFB can be leveraged are depicted. | ||
| Year(s) | RiPP class | Biosynthetic enzyme(s) | RiPP(s) generated | Cell-free strategy | Metabolite yield(s) | Ref. |
|---|---|---|---|---|---|---|
| 2022 | Pyritide | MroB, MroC, MroD | Pyritide A1 and analogs | PURExpress | Not generated | (Nguyen et al., 2022)170 |
| 2020 | Pyritide, thiopeptide | LazB, LazC, LazD, LazE, LazF | Lactazole and analogs | FIT-Laz | Not generated | (Vinogradov et al., 2020)171 |
| 2021, 2023, 2025 | Lasso peptide | FusB, FusC, FusE | Fusilassin and analogs | E. coli BL21 (DE3) lysate | Not generated | (Si et al., 2021),172 (Kretsch et al. 2023),173 (Barrett et al. 2025)174 |
| 2021 | Lasso peptide | BurB, BurC, BurD | Burhizin | E. coli BL21 (DE3) lysate | Not generated | (Si et al., 2021)172 |
| 2021 | Lasso peptide | CapB, CapC, CapD | Capistruin | E. coli BL21 (DE3) lysate | ∼40 μg mL−1 | (Si et al., 2021)172 |
| 2021 | Lasso peptide | CelB, CelC, CelE | Celulassin | E. coli BL21 (DE3) lysate | Not generated | (Si et al., 2021)172 |
| 2021 | Lasso peptide | FusB, FusC, FusE | Halolassin | E. coli BL21 (DE3) lysate | Not generated | (Si et al., 2021)172 |
| 2007 | Lanthipeptide | NisB, NisC | Nisin | In vitro rapid translation system, supplemented with ZnSO4 | ∼200 IU per mL | (Cheng et al., 2007)175 |
| 2020 | Lanthipeptide | NisB, NisC, NisP | Nisin and analogs | E. coli BL21 (DE3) lysate, supplemented with ZnCl2 | ∼180 IU per mL | (Liu et al., 2020)176 |
| 2024 | Lanthipeptide | SboM, SboT | Salivaricin B and analogs | UniBioCat, derived from E. coli BL21 Star (DE3) ΔdegP ΔpepN lysate | Not generated | (Liu et al., 2024)177 |
| 2022 | Circular bacteriocin | N/A | Garvicin ML | PURExpress, SICLOPPS | Not generated | (Peña et al., 2022)178 |
| 2020 | Pantocins | PaaA | Pantocin A and analogs | FIT system | Not generated | (Fleming et al., 2020)179 |
| 2021 | Pyrroloquinoline quinones | N/A | PQQ | Gluconobacter oxydans 621H cell lysate | Not generated | (Wang et al., 2021)180 |
| 2019 | Proteusin | N/A | N-Methylated Nhis-AerA | Microvirgula aerodenitrificans cell lysate | Not generated | (Bhushan et al., 2019)181 |
CFB enables the rapid assessment of each residue's potential contributions to substrate recognition and modification status (Fig. 3). Additionally, core peptides can often be swapped out to generate novel natural products, and the production of vast libraries of precursor peptides has been readily achieved. Variants can be generated in a high-throughput manner by several methods, including error-prone polymerase chain reaction (PCR) and site saturation mutagenesis.182,183 This library can then be assayed against the modifying enzyme(s), often without additional purification steps. In some examples, both the precursor peptide and modifying enzymes have been expressed through CFB, resulting in the de novo cell-free production of RiPP natural products directly from DNA.172 Orthologous enzymes could be assayed this way without expressing and purifying each individually. However, modifying enzymes are primarily purified separately via affinity chromatography and added to the reaction to conserve resources for CFB of the precursor peptide. For several uses of CFB in studying the bioactivity of RiPPs, we direct the reader to a recent review on RiPP mechanisms of action.184
As an example of the flexizyme system's usage, a platform for assessing the percentage of peptide natural products accessible by CFB was devised.189 By parsing data from several online repositories, the authors developed a chemoinformatic resource for data on both linear and macrocyclic peptide natural products. Leveraging this data, they chose 43 peptides to express in either PURExpress® or PUREfrex, most of which (36, ∼84%) were successfully produced. Cyclisation of 14 of 31 head-to-tail macrocyclised candidates was achieved using either the macrocyclase PatGmac from the patellamide biosynthetic pathway or PCY1 from the orbitide biosynthetic pathway. Flexizymes were successfully utilised to incorporate a highly represented piperazic acid motif and N-methyl, D- and β-amino acids, although deficiencies are often observed during their incorporation.190 Overall, this work provides a wealth of peptide natural product data and highlights potential areas of prioritisation in the CFB production of peptide natural products, including RiPPs.
In a separate example, a macrocycle was generated that was inspired by amphotericin B, an antifungal polyketide metabolite that has activity that depends on both a hydrophobic polyol region as well as a lipophilic polyene region191 (Fig. 4). Importantly, amphotericin, despite being an incredibly valuable antifungal, displays high levels of renal-toxicity because of sterol sequestering in humans and in fungal pathogens.192 Thus, developing compounds that retain bioactivity, but decrease its toxicity are desired. Despite amphotericin lacking an overall peptide scaffold, the researchers devised a peptide with general mimicry of amphotericin B's physiochemical features.191
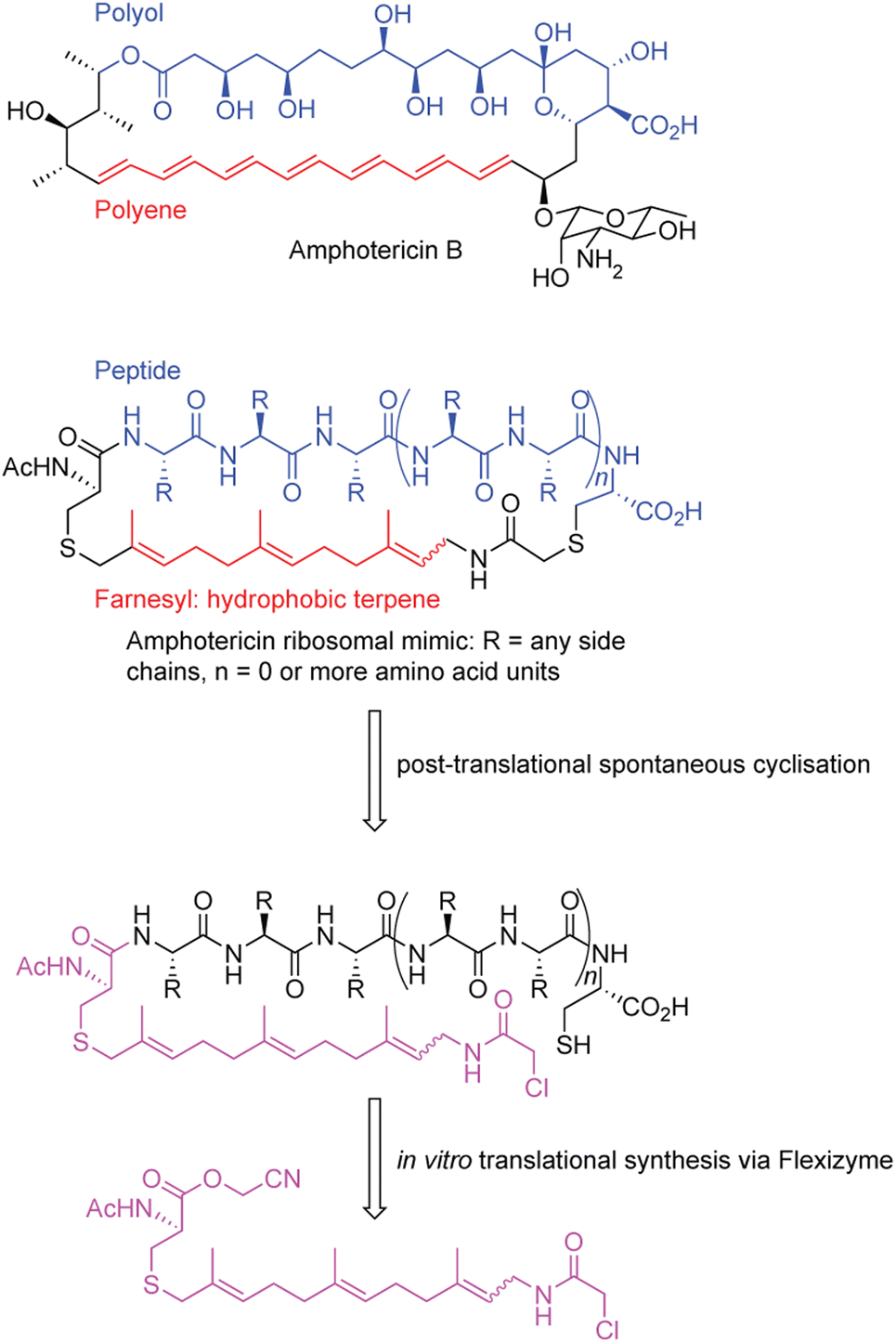 | ||
| Fig. 4 Generation of amphotericin-like peptide analogues with the flexizyme system.191 | ||
To construct a molecule resembling amphotericin, the authors used a peptide backbone to mimic the polar polyol portion and a terpene farnesyl region to mimic the polyene. To generate their structure, an SN2 reaction is leveraged between an N-terminal chloroacetamido moiety at the end of the farnesyl group, incorporated via FIT, and a cysteine residue in the peptidic portion.193 While structurally impressive, the activity of this compound is unexplored. Overall, the above examples show the utility of FIT to create natural products and natural product inspired molecules via synthetic biology.
CFB has been used to assess the substrate scope of the MroBCD enzymes involved in the biosyntheses of pyritide A1 and A2 from Micromonospora rosaria. An array of substrates was rapidly evaluated using commercially available PURExpress®. MroBCD exhibited a wide substrate tolerance, including macrocycle expansion and contraction, suggesting that pyritide enzymes may be suitable for future engineering.170 Notably, recognition of both an N-terminal leader sequence and the C-terminal tripeptide “WLI” on MroA2 by MroB and MroD explain the broad substrate promiscuity shown between these regions of the precursor peptide.
Almost all pyritides are also thiopeptides, which feature additional modification and frequently possess potent antibacterial activity.198,199 Besides a pyridine or dehydropiperdine, thiopeptides contain azol(in)e heterocycles formed from Ser, Thr, and/or Cys residues. Core biosynthetic steps were first elucidated using the enzymes from the thiomuracin BGC, found in Thermobispora bispora.72,195 An ATP-dependent YcaO cyclodehydratase (TbtFG) phosphorylates the peptide backbone and uses the amino acid residue side chain for nucleophilic attack to generate a 5-membered heterocycle, which is often dehydrogenated by a FMN-dependent enzyme (TbtE).200 Azole formation typically precedes the installation of additional PTMs in biosynthesis.72,195
CFB has been extensively leveraged in work on the thiopeptide lactazole to develop pseudo-natural products that could serve as drug candidates201,202 (Fig. 5a and b). While thiomuracin biosynthesis had been studied previously,72,195 it remained to be seen if the proposed order of biosynthetic events was conserved in other thiopeptide BGCs. CFB was successfully leveraged to explore the substrate scope of most enzymes in the lactazole BGC171 (LazA-F) (Fig. 5a). LazA was produced through a flexible in vitro translation (FIT) system (FIT-Laz),203 consisting of purified ribosomes and necessary components for transcription-coupled translation in vitro204 (Fig. 5c). The FIT system has previously been used for CFB of azoline-containing peptides and dissection of goadsporin biosynthesis, all of which are linear.205,206 Various combinations of purified biosynthetic enzymes were added to assess activity on LazA and elucidate any cooperativity between the enzymes.171 While thiomuracin biosynthesis can be cleanly delineated into several steps, LazA-F act in a more complex and nuanced manner. Leveraging the capacity for unnatural amino acid installation, the authors incorporated cycloleucine and several backbone N-methylated amino acids into a mature thiopeptide and assessed which regions of LazA are amenable to insertion, deletion, and substitution. The FIT-Laz system circumvented the requirement for intensive cloning, heterologous expression, and purification of the assessed substrates.
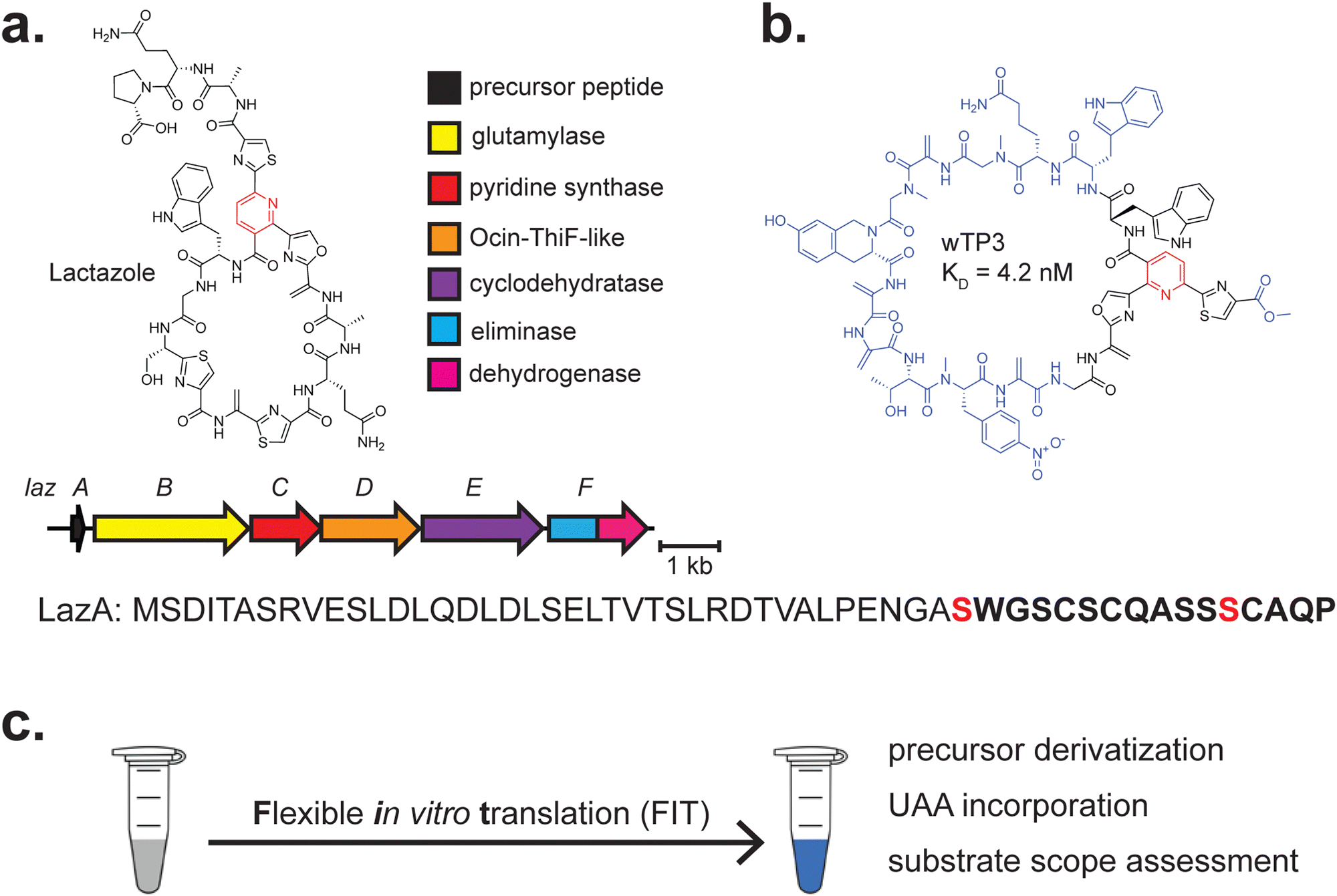 | ||
| Fig. 5 Thiopeptides. (a) BGC and structures of lactazole. The class-defining PTM is highlighted in red. The core peptide is bolded, and the residue(s) on which class-defining PTMs occur are red. (b) The structure of wTP3, a pseudo-natural product developed through years of extensive work using LazB-F. The regions of the compound that differ from lactazole are highlighted in blue. (c) Stylised workflow visualisation for the Flexible in vitro Translation (FIT) system, in which the components for transcription/translation, DNA, flexizyme(s), and unnatural amino acid(s) are combined to achieve the goals stated to the right. | ||
This work proved foundational for future engineering efforts with lactazole fueled by CFB. A site saturation mutagenesis library was generated for LazA and assessed using mRNA display, in which a translated peptide is joined to its reverse-transcribed coding sequence via puromycin to covalently link phenotype with genotype.207,208 The authors used a precursor peptide library with N-terminal biotinylation generated using FIT, as well as a C-terminal human influenza hemagglutinin (HA) affinity tag and puromycin linker. After modification by LazB-F (Fig. 5a), HA affinity purification removes cleaved leader peptide from the reaction mixture. An additional streptavidin purification separates modified products from unmodified products in which the leader peptide and biotin groups are attached.209
After using mRNA display to assess the full scope of LazB-F modification, the authors evaluated the precise substrate requirements for each modification.210 Using numerous LazA variants, they determined the order of biosynthetic events and found that, rather than formation of azoles first and Dhas second as in thiomuracin biosynthesis, lactazole biosynthesis intertwines these steps. Again, the use of CFB to generate these precursor peptides streamlined this process. The FIT-Laz system was then leveraged to explore the substrate promiscuity of glutamate elimination domains such as LazC, finding the enzymes to be highly substrate promiscuous, as was suspected previously.211 CFB and mRNA display have also been coupled with machine learning to assess substrate preferences for LazBF and LazDEF.212 In this approach, next-generation sequencing was used to generate vast quantities of processing data on substrates. A relatively simple machine learning approach was employed in which peptides were represented as extended-connectivity fingerprints.213,214 These data were used to train a deep convolutional neural network215 in assessing the potential suitability of substrates. Despite utilizing ∼107 samples for training this model, the preferences for both groups of enzymes remain complex, highlighting the need for high-throughput testing of variable precursor peptides.
Most recently, LazB-F have been used to generate de novo thiopeptide pseudo-natural products with nanomolar affinity to Traf2- and NCK-interacting kinase (TNIK), an established target in several cancers.201,216 This was achieved by using a similar approach to previous mRNA display work209 but including an additional step in which counter-selection against immobilised TNIK enriches for binders to the targeted kinase. Leveraging of a minimised genetic code resulted in the selection of another TNIK binder with a similar affinity to their previous compound (nM) but improved serum stability and incorporation of several unnatural amino acids201 (Fig. 5b). This strategy could, in principle, be applied to generate thiopeptides with affinity to any desired immobilised target. This has recently been achieved to evolve binders to both interleukin-1 receptor-associated kinase 4 (IRAK4), an important component of Toll-like receptor signaling,217 and the TLR10 cell surface receptor, which exhibits unique anti-inflammatory activity.218,219 Overall, the ability to assess extensive libraries of variants (>1012) in a proven directed-evolution workflow has enabled an unprecedented understanding of this biosynthetic pathway's potential and streamlined the selection of designer pseudo-natural products.
Alternative strategies for thiopeptide intermediate generation have also been employed. In the production of thiocillin and lactazole, flexizymes have been used to incorporate phenylselenocysteine, which undergoes oxidative elimination with H2O2 to generate Dha.220 This circumvented the need to use a glutamyl-tRNA-dependent dehydratase (such as TbtB or LazB) in producing Dha, which is required for thiopeptide production. While other chemical methods to generate Dhas exist, such as dehydrothiolation of Cys,221 this method often conflicts with the activity of azole-forming YcaO enzymes (such as TbtG and LazE), which typically act first in biosynthesis and may cyclise the target Cys residues.195
Streptolysin S (SLS) belongs to the linear azole-containing peptide class of RiPPs. SLS is a critical virulence factor in the human pathogen Streptococcus pyogenes.222 In the biosynthesis of SLS, S. pyogenes and other organisms use a putative protease SagE. To assess if SagE truly is a leader peptidase, in vitro transcribed and translated precursor peptide SagA, featuring 35S labeling, was supplied to S. pyogenes lysate.223 The proteolysis of SagA supported the putative role of SagE, and additional experiments established this protease is critical for SLS production and showed inhibition by FDA-approved HIV protease inhibitors.
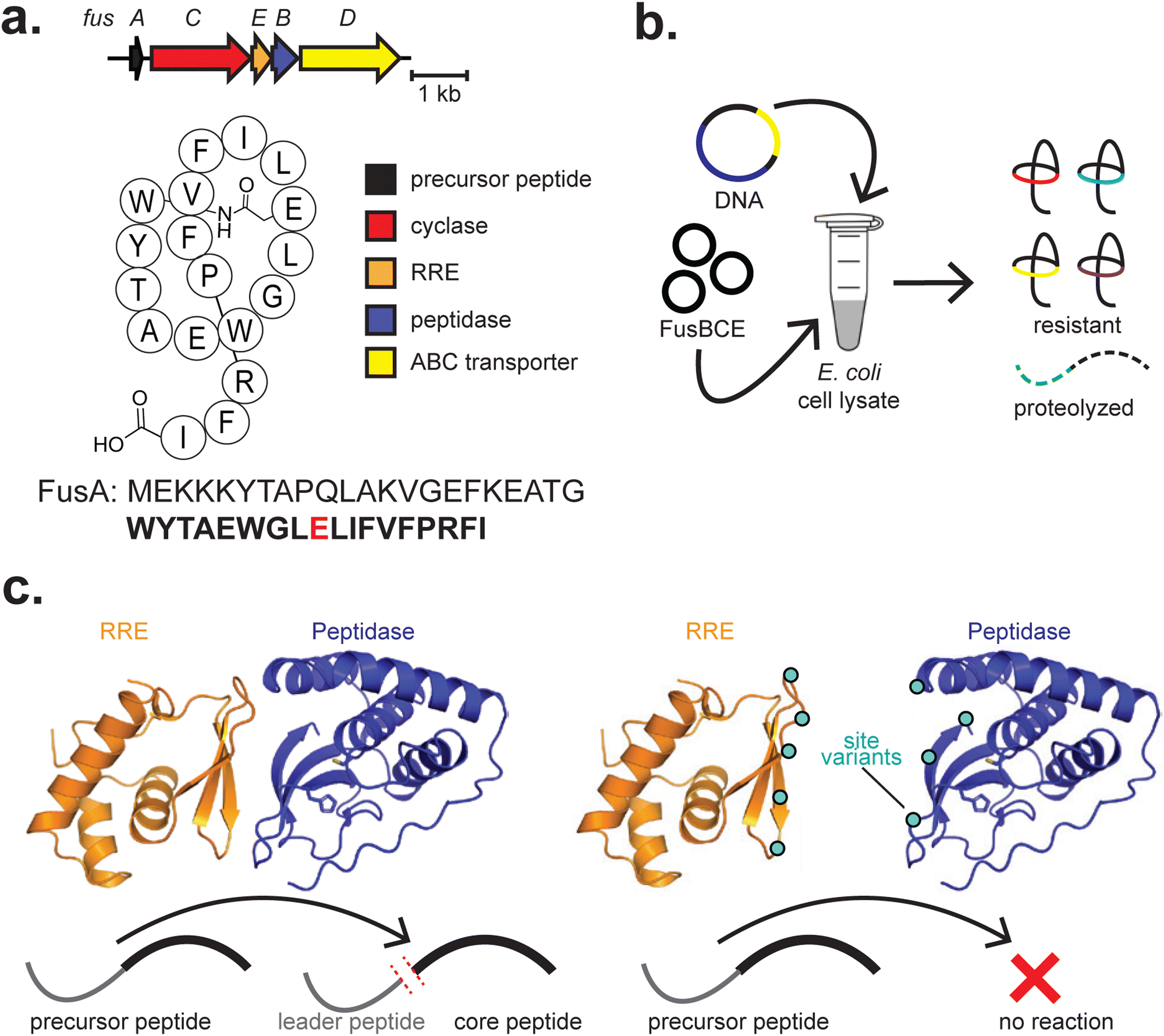 | ||
| Fig. 6 Lasso peptides. (a) BGCs and structures of fusilassin. The class-defining PTM is highlighted in red. The core peptide is bolded, and the residue(s) on which class-defining PTMs occur are red. (b) Stylised workflow visualisation for lasso peptide derivatisation by CFB. The necessary components (DNA and biosynthetic enzymes) are added to clarified E. coli lysate to produce lasso peptides and native proteases digest unmodified precursors. (c) An allosteric interaction between an RRE (FusE) and a leader peptidase (FusB) leads to successful leader peptide cleavage in fusilassin biosynthesis. Site variants of the RRE–peptidase interface, depicted as cyan circles, can disrupt the protein–protein interaction, preventing leader peptide cleavage. | ||
In subsequent work on lasso peptides, site-directed mutagenesis and cell-free production of the fusilassin protease (FusB) and precursor recognition (FusE) enzymes were critical in revealing an allosteric interaction between the two173,180 (Fig. 6c). While FusE variants could be readily produced by PURExpress, robust production of FusB variants was only achieved in E. coli cell extracts, highlighting the complementary use of different CFE platforms.173 In the extract, crowding effects and chaperones may have potentially contributed to successfully producing FusB variants, though any precise mechanism has not yet been elucidated. Cell-free production of numerous protein variants expedited testing, and additional experiments corroborated the results. Both studies highlight the potential for cell-free production of enzyme site variants in assessing complex biosynthetic pathways and protein–protein interactions.
Recently, interactions between the FusA precursor peptide and FusC have also been assessed.174 Cell-free generation of both FusA and FusC variants was utilized, in combination with molecular dynamics simulations and bioinformatic analysis, to explore how FusC interacts with FusA. This work further highlights the utility of CFB in generating variants of both a RiPP enzyme and its peptide substrate.
In a separate example, a Zn2+ supplemented E. coli cell extract was used to produce nisin and several genome-mined lanthipeptides.176 All 18 uncharacterised nisin analogs in the National Center for Biotechnology Information (NCBI) database were identified. Genes encoding the core peptides of these RiPPs were then fused C-terminally to the nisin Z leader peptide gene sequence, and the resulting chimeric precursors were subjected to NisBC modification. Six peptides showed dehydration, and four were shown to have antibacterial activity. This work further emphasises the breadth at which predicted RiPPs can be generated with CFB. Saturation mutagenesis was used to generate a nisin variant, M5, with improved antibacterial activity against E. coli. CFB mixture was co-incubated with a growing E. coli culture to assess antibacterial activity. This approach circumvents potential cytotoxicity issues from the production of fully modified nisin in vivo and exemplifies the utility of CFB in producing cytotoxic RiPPs.
Recently, a unified biocatalysis (UniBioCat) system for the rapid engineering of lanthipeptides was achieved.177 This strategy differs from previous work in that all components are generated via CFB, including the precursor peptide, modification enzyme(s), requisite protease(s), and transporter(s). Using this UniBioCat system, they first biosynthesized the lanthipeptide, salivaricin B from Streptococcus salivaris K12 in an E. coli-derived lysate. Further, they utilized a modified E. coli lysate with several proteases knocked out to screen both salivaricin B and 46 analogues. After establishing proof of concept, the UniBioCat system was used to express uncharacterized lanthipeptides identified via genome mining. Similarly to previous work on lasso peptides,172 the core peptide genes of uncharacterized lanthipeptides were fused to the salivaricin precursor peptide (SboA) leader for modification by its cognate lanthipeptide cyclase SboM. This represents further advancement of the ability to use RiPPs for CFE and highlights the potential to generate all requisite components for biosynthesis in a single CFB reaction (Table 1).
Display technologies have also been frequently used with CFB. The biosynthetic scope of PaaA, an enzyme catalysing two dehydration/decarboxylation reactions on two Glu residues of the precursor peptide PaaP to form pantocin A (Fig. 7a), has been explored through the FIT system coupled with mRNA display.179 The authors use endoproteinase GluC cleavage to assess substrate modification, rapidly sorting viable substrates from non-substrates (Fig. 7a). To validate hits from their screening, they produced several precursor variants using PURExpress and then assessed in vitro activity. In a single set of experiments, they glean insight into the core recognition and enzyme promiscuity of PaaA.
 | ||
| Fig. 7 Additional examples of cell-free biosynthesis in RiPPs. (a) The structure of the modified precursor peptide PaaP is derived from two glutamate residues. Once modified by the enzyme PaaA, PaaP is resistant to GluC cleavage, providing a selection mechanism for modified variants of PaaP. (b) Supplying M. aerodenitrificans lysate with pure Nhis-AerA co-expressed with AerD in E. coli led to five side-chain N-methylations, represented by the red outlines and CH3 groups. The dashed outline on Asn43 indicates that methylation was not directly observed but proposed based on additional MS fragmentation data. | ||
Several other examples of CFB use lysates from the native producers, including production of the RiPP pyrroloquinoline quinone (PQQ). PQQ is a redox cofactor used by several bacterial dehydrogenases.230 PQQ biosynthesis occurs mostly in Pseudomonodota, although other microbes incapable of its production can scavenge exogenous PQQ.231,232 Precursor peptide PqqA was heterologously expressed and purified, then modified in cell lysate from a strain that naturally produces PQQ, Gluconobacter oxydans 621H.233 This strategy may prove useful for scaling up the production of RiPPs with robust biosynthetic enzymes.
A unique strategy was employed while screening for the production of a predicted proteusin product, aeronamide.181 Proteusins are a heavily modified class of RiPPs defined by an unusually long leader peptide composed of a bundle of alpha-helices, and were initially bioinformatically predicted before their isolation and structural elucidation.167,234,235 These compounds feature extensive epimerisation and methylation, exhibiting antibacterial activity through the formation of ion channels.236–239 To confirm the expression and activity of the remaining enzymes in the aeronamide BGC, researchers spiked epimerised, partially modified precursor peptide AerA into lysate (Fig. 7b). They observed a mass shift indicative of five expected substrate methylations.181 Because lysate from the native producer Microvirgula aerodenitrificans was used for this experiment, the authors confirmed active enzymes in their culturing conditions before scaling up production. As RiPP precursor peptides and their expected mass shifts due to modification are often predictable, we envision this being a beneficial strategy.
While most uses feature the addition of DNA to generate proteins of interest, several examples feature the use of in vitro transcription to generate biosynthetically crucial tRNA. Reconstitution of NisB and TbtB, dehydratase enzymes involved in lanthipeptide and thiopeptide biosynthesis, respectively, has required glutamyl-tRNAGlu.72,240 In these cases, the tRNA was generated first as a DNA template and then transcribed using RNA polymerase. tRNAGlu was glutamylated either separately or concurrently with NisB/TbtB activity in vitro. Further in vitro characterisation of tRNA-dependent RiPP enzymes will likely benefit from a similar strategy for tRNA generation.
A similar enzyme is responsible for pearlin biosynthesis. In pearlins, a peptide aminoacyl-tRNA-ligase (PEARL) enzyme elongates the C-terminus of a precursor peptide by a single amino acid residue in a tRNA-dependent manner.241 PEARLs are structurally related to tRNAGlu-dependent dehydratases. In studying the biosynthesis of 3-thiaglutamate by the PEARL TglB, modified TglA-derived substrates were generated by PURExpress.242 This enabled further characterisation of downstream modifying enzymes TglHI.
While broadly applicable for a substrate scope assessment, CFB is often performed qualitatively. Frequently, a significant excess of enzyme relative to substrate is provided in each reaction, and reaction times may be long. When assayed non-quantitatively, potentially poor substrates can be more effectively modified. Recent work on the lactazole BGC supports this, with a more narrow window of preferred substrates than modified substrates.218 It should, therefore, be stressed that while some precursor peptides may be tolerated in CFB, they may be poor substrates when assayed under more stringent conditions. In short, while any given enzyme may modify a substrate, this method does not inform whether this modification is necessarily efficient.
Most RiPP CFB studies use PURExpress. Reasons for this include ease of use and much higher replicability when compared to preparing E. coli cell lysate for CFE in-house. Endogenous proteases in cellular extract pose a significant issue in RiPP biosynthesis. Several classes of RiPPs, including pyritides, lasso peptides, and lanthipeptides, are highly resistant to proteases because of their macrocyclic structures. This resistance can provide a selection mechanism for modified precursor peptides.172 For protease-susceptible classes of RiPPs, additional measures must be employed to use cell lysates from E. coli and other bacteria fully. Unidentified components in these lysates may assist in the folding and stability of biosynthetic enzymes,44 but a direct mechanism has yet to be identified.
CFB will also expedite RiPP engineering, which may involve assaying millions of substrate variants for processing and activity. Designer substrates can readily interrogate complex pathways' biosynthetic order of events.171 Individual components can be included, excluded, or altered to assess interactions between RiPP-modifying enzymes. Numerous classes of RiPPs have not yet been extensively characterised, and could be, in an approach like that taken with the lactazole pathway. As has been discussed, both RiPPs and other ribosomal peptides can also be tailored using the FIT system to develop pseudo-natural products that improve upon Nature's designs. As cell-free methods are optimised and lysate sources beyond E. coli are more widely used, we predict the study of RiPPs will benefit greatly.
4.2 CFB of non-ribosomal peptides
Although both generate peptides, the biosynthetic logic behind RiPPs and nonribosomal peptides (NRPS) is distinct, though there is an overlap in their overall scaffolds via convergent evolution to form bioactive moieties. For example, both non-ribosomal peptides and RiPPs frequently have numerous N-methyl and D-amino acids, and heavily oxidised and/or cross-linked side chains.167 Recently, acylated RiPPs, including lipoavitides245 and selidamides,246 have been discovered that have similarities to non-ribosomal lipopeptides such as the polymyxins,247 and daptomycin.248 This is suggestive that regardless of biosynthetic machinery, using predicted and known structures from peptide natural products is a fruitful area for bioactive discovery. Applications of CFB to investigate non-ribosomal peptides (Table 2) have been pursued both using the PURE system and crude lysates.249 NRPS are composed of a series of modular domains that select building blocks (with NRPS, amino acids) that are elongated on a phosphopantetheinylated carrier protein in a colinear fashion.250–252 Briefly, NRPS consist of an adenylation (A) domain that recognises the amino acid and activates the amino acid monomer at the C-terminus as an adenylate followed by transfer to a peptidyl carrier protein (PCP) sometimes known as a thiolation (T) domain. The activated, loaded amino acid is condensed via the C-domain. Optional domains such as epimerase (E), cyclisation (Cy), methyltransferase (MT) and oxidase (Ox) domains generate structural variety. Chain release typically occurs via hydrolysis or macrocyclisation by a thioesterase (TE) domain (Fig. 8),253–255 though in some systems a reductase (R) domain reduces the product to a primary alcohol256,257 (Fig. 8). Additional tailoring may occur on the carrier protein and the released metabolite. One of the attractive features of NRPS is that non-proteinogenic amino acids can be incorporated without the use of flexizymes, genetic reprogramming, or further tailoring (e.g. as occurs with modifying enzymes in RiPPs). This modularity is attractive from a synthetic biology perspective, and determining the best junctions for chimeric and selectivity swapped pathways has been an area of considerable attention recently.258–262| Year(s) | NRPS gene(s) | Non-ribosomal peptide metabolite(s) | Cell-free strategy | Metabolite yield(s) | Protein yield(s) | Ref. |
|---|---|---|---|---|---|---|
| 2017 | GrsA and GrsB1 from Brevibacillus brevis | D-Phe-L-Pro diketopiperazine (DKP), shunt product of gramicidin S | E. coli BL21 Star (DE3) lysate | ∼12 mg L−1 | GrsA ∼106 μg mL−1 | (Goering et al., 2017)263 |
| GrsB1 ∼77 μg mL−1 | ||||||
| 2020 | GrsA from Brevibacillus brevis | Gramicidin S | PURExpress | Not generated | Not quantiated | (Siebels et al., 2020b)264 |
| 2020 | Vlm1 and Vlm2 from Streptomyces tsusimaensis | Valinomycin | E. coli BL21 Star (DE3) lysate and cell-free protein synthesis-metabolic engineering (CFPS-ME) | ∼30 mg L−1 | Not quantitated | (Zhuang et al., 2020)265 |
| 2020 | BpsA from Streptomyces lavenduale | Indigiodine | PURExpress | ∼62 μg mL−1 | BpsA: ∼28.7 μg mL−1 | (Siebels et al., 2020b)264 |
| 2023 | BpsA from Streptomyces lavenduale | Indigiodine | E. coli BL21 Star (DE3) lysate | ∼223 μg mL−1 | BpsA: ∼95 μg mL−1 | (Dinglasan et al., 2023)266 |
| 2020 | KJ12A, KJ12B, KJ12C from Xenorhabdus KJ12·A | RXP (rhabdopeptide-like peptides) | PURExpress | Not quantified | Not quantified | (Siebels et al., 2020b)264 |
| 2020 | IndC from Photorhabdus luminescens | Indigiodine | PURExpress | Not generated | Not quantified | (Siebels et al., 2020b)264 |
| 2020 | TycB from Bacillus brevis | Tyrocidine | PURExpress | Not generated | Not quantified | (Siebels et al., 2020b)264 |
| 2023 | TycA from Bacillus brevis | Tyrocidine | E. coli BL21 Star (DE3) lysate | Not generated | Not quantified | (Dinglasan et al., 2023)266 |
| 2023 | Pys from Pseudomonas entomophilia | Pyreudione | E. coli BL21 Star (DE3) lysate | Not generated | Not quantified | (Dinglasan et al., 2023)266 |
| 2021 | TxtA and TxtB from Streptomyces scabiei | Thaxtomin A | Streptomyces venezuelae ATCC 10712 lysate | Not generated | Not quantified | (Moore et al., 2021)132 |
| 2021 | NH08_RS0107360 from Streptomyces rimosus | Uncharacterized | Streptomyces venezuelae ATCC 10712 lysate | Not generated | Not quantified | (Moore et al., 2021)132 |
 | ||
| Fig. 8 Overview of CFB of NRPS products (a) GrsA and GrsB1, the first couple modules from the gramicidin S NRPS were used to generate a D-Phe-L-Pro diketopiperize product.263 (b) Valinomycin NRPS, the largest NRPS enzyme and metabolite produced by CFE to date.267 (c) Blue pigment synthetase A (BpsA) which generates the blue pigment indigiodine.264 (d) Rhabdopeptide-like peptide (RXP) from Xenorhabdus KJ12.1.264 Abbreviations: A = adenylation, T = thiolation, C= condensation, Ox = oxidase, TE = thioesterase, MT = methyltransferase, KR = ketoreductase, E = epimerase. | ||
:1 (cell lysate-Vlm1:cell lysate-Vlm2). To increase valinomycin production, TE-II was added using a two-step approach. TEII was first produced by CFE and then added to the CFB reaction of Vlm1 and Vlm2. This single change increased valinomycin production 375-fold to 29 mg L−1. As glucose is the key reaction substrate, further optimisation of glucose concentrations gave the highest valinomycin titre at ∼30 mg L−1 (200 mM glucose). Taken together, this is the first example of a whole natural product NRPS gene cluster of valinomycin (>19 kb) made using CFB. The yield of valinomycin improved with the addition of an associated editing enzyme (TE-II).
:1.5:1 (KJ12A:B:C): mV-V-mV-mV-PEA, V-V-V-mV-PEA, and V-mV-V-mV-PEA (N-methylated valine (mV), valine (V), and phenylethylamine (PEA)) (Fig. 8d). The yields of rhabdopetides were not quantitated, and only relative ratios of proteins were reported. This work shows the utility of applying the PURExpress® system directly to generate natural products from genomic DNA.
Ultimately, the reporter-based system determined reaction conditions that were more optimal for BpsA than initial conditions optimised on GFP. These optimised reaction conditions can be used for other monomodular NRPSs with similar architectures to BpsA: TycA (124 kDa), and Pys (142 kDa). The result demonstrated that with these two enzymes, the improvement in expression compared to ‘standard CFE conditions’ was even more dramatic than the improvement in BpsA expression (∼1 order of magnitude). Further, this work reveals the importance of optimizing beyond simple fluorescent protein reporters when using CFE approaches.
4.3 CFB of polyketides
To our knowledge, there is a single example of a detectable amount of a polyketide metabolite being produced by CFE.288 However, there have been several examples of polyketide synthases (PKSs) being generated in cell-free studies, which along with in vitro studies utilising purified proteins, are suggestive of future promise for applying CFE to generate polyketide metabolites (Table 3).| Year(s) | PKS gene(s) | PKS type | Associated polyketide metabolite | Cell-free strategy | Metabolite generated? | Protein yield(s) | Ref. |
|---|---|---|---|---|---|---|---|
| 2017 | DEBS1-TE (first gene from the 6-deoxyerythronlide B fused to the thioesterase) | Type I | Erythromycin | E. coli BL32 Star (DE3) lysate E. coli TB3 lysate | Not generated | Not quantified | (Hurst et al., 2017)276 |
| 2020 | PikAIII (5th module from the pikromycin PKS) | Type I | Pikromycin | PURExpress | Not generated | Not quantified | (Siebels et al., 2020b)264 |
| 2020 | DEBS module 4 | Type I | Erythromycin | PURExpress | Not generated | Not quantified | (Siebels et al., 2020b)264 |
| 2024 | RppA | Type III | Flaviolin | E. coli BL32 Star (DE3) lysate | Flaviolin generated, not quantified | Not quantified | (Sword et al., 2024)288 |
Polyketides are a family of diverse and complex natural products that possess chemical architectures with broad-ranging bioactivity. Polyketide biosynthesis consists of sequential condensations of malonyl-CoA or malonyl-CoA derivatives in a fashion analogous to fatty acid biosynthesis, albeit with a greater variance of reductive processing of the β-keto moiety. Canonical PKSs consist of the following enzymatic domains: a ketosynthase (KS) that is responsible for condensation of malonyl-CoA and derivatives thereof in a Claisen-like fashion, and an acyltransferase (AT) which selects an extender unit and condenses it to an acyl carrier protein (ACP) (Fig. 9). Scaffolds are additionally modified by optional beta carbon processing domains, including a ketoreductase (KR), which reduces the keto group stereoselectively to a hydroxy group, a dehydrase (DH), which dehydrates to form an olefin, and an enoylreductase (ER), which forms a saturated carbon backbone. Often, the polyketide is terminated by a thioesterase which typically either hydrolyses from a linear product or macrolactonises.289,290
 | ||
| Fig. 9 CFB of PKS products. (a) Enzymatic reactions catalysed by PKS domains. (b) Type I PKSs consist of all domains fused as one “megasynthase” and either can be modular in the case of bacteria (one round of extension and β-carbon processing per module) or iterative (multiple rounds of β-carbon processing per module) in the case of fungi. β-Carbon processing domains (KR, DH, and ER) are optional. (c) Type II PKSs consist of the domains as discrete enzymes. A KS-chain length factor (CLF) is responsible for condensation, a malonyl acyl transferase (MAT) extends and a cyclase TE (CYC-TE) is responsible for cyclisation of the aromatic products. β-Carbon processing domains (KR and DH) are optional. (d) Type III PKS acts upon a CoA substrate rather than an acyl carrier protein in an interative fashion. (e) DEBS-1 TE, the first gene in the 6-deoxyerythronolide B synthase (DEBS) fused to the thioesterase, consisting of a loading module and two extension modules, 275 KDa which was generated in an E. coli cell-free lysate. (f) RppA, the type III PKS which generates the red brown polyketide synthase, flaviolin which was generated in an E. coli cell-free lysate. Abbreviations: ACP = acyl carrier protein, AT = acyltransferase, KS = ketosynthase, KR = ketoreductase, DH = dehydratase, ER = enoylreductase, TE = thioesterase. | ||
Polyketide synthases are divided into three sub-classes: type I, type II, and type III. Type I PKSs have a multi-domain architecture that is similar to the mammalian fatty acid synthase, wherein domains with discrete enzymatic activity are covalently linked and are the prototypical megasynthases.291–295 Type I PKSs from bacteria are typically modular, where there is one elongation per module (set of condensations followed by subsequent reduction) and are further categorised as cis-AT and trans-AT PKSs depending on whether the acyltransferase domain is embedded or present on a separate domain.296–298 The type I modular PKSs are found in Actinomycetota and Myxobacteria298 whereas trans-AT PKS are found in a broad range of bacterial phyla.299 Type I fungal PKSs296,300–303 and some other eukaryotic PKSs304 have a similar multi-domain architecture, but catalyse the elongation of the polyketide chain in an iterative fashion. Type II PKSs are almost exclusively found in soil and marine bacteria, traditionally Actinomycetota, but have recently been shown to occur in other phyla. Type II PKSs have domains organised into discrete proteins that dock to a KS/chain length factor (KS/CLF) complex, and traditionally form non-highly reductive products, such as oxytetracyclines.305–308 Type III PKSs are found in plants,309,310 fungi,300 and bacteria311 and function on standalone CoA substrates and typically form chalcone-like scaffolds.312,313 Many iterations of polyketide pathways exist outside of these paradigms, including fusions with other natural product classes314,315 (PKS/NRPS316 or PKS/terpene hybrids317), unusual domains318–324 and irregular substrate selectivity,325 rendering PKSs one of the most fascinating targets for construction of diverse carbon scaffolds via bioengineering.
PURExpress® has also been explored for generating type I PKSs, however, only protein synthesis has been achieved. A selection of type I PKS proteins including the 14th module from the rapamycin PKS (RAPS module 14), the 5th module from the pikromycin PKS (PikAIII), and DEBS module 4, have been generated.264 Besides PKS proteins, four variations of the murine fatty acid synthase (FAS) were generated, which shares architectural similarity to type I PKSs. In all cases, successful labelling with a fluorescent mimic of CoA (CoA-647) was observed, demonstrating they were likely correctly folded as the ACP domain could be modified, suggesting promise for in vitro catalytic activity.264
4.4 Terpenes
Unlike PKS or NRPS biosyntheses, terpene biosynthesis consists of discrete terpene synthases that polymerise and isomerise isoprene units (C5H8)n as dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP), two structural isomers. These isoprene units are produced by two distinct metabolic pathways: the mevalonate (MVA) pathway and the non-mevalonate (MEP) pathway. After these units are polymerised, they are further rearranged, oxidized, and reduced. Terpenes form an immense amount of the biodiversity observed in microbial and plant biosynthesis and are bioactive compounds (e.g. the anticancer agent taxol, the antimalarial artemisinin), lubricants, flavourants, and fragrances. Both the biosynthesis of monomers and the chemistry done to the isoprenoid scaffold require substantial quantities of cofactors. The titres of terpenes are typically low to moderate depending on application (<0.5 g L−1 up to ∼1.5 g L−1) with a few exceptions such as amphoadiene which can be produced at 40 g L−1 in Saccharomyces cerevisiae (Table 4).| Year(s) | Terpene | Cell-free strategy | Metabolite yield | Ref. |
|---|---|---|---|---|
| 2016 | Limonene | E. coli BL32 Star (DE3) lysate | 60 μg L−1 | (Dudley et al., 201 6)146 |
| 2020 | Limonene | E. coli BL21 Star (DE3) lysate, iPROBE | 610 mg L−1 | (Dudley et al., 2020)145 |
| 2020 | Bisabolene | E. coli BL21 Star (DE3) lysate, iPROBE | 1010 mg L−1 | (Dudley et al., 2020)145 |
| 2020 | Pinene (mixture of α- and β-pinene) | E. coli BL21 Star (DE3) lysate, iPROBE | Not reported | (Dudley et al., 2020)145 |
 | ||
| Fig. 10 Cell-free biosynthesis of limonene, α/β-pinene and bisabolene. The authors used cell-free protein synthesis for rapid testing of different enzyme combinations to optimise and modularise the pathway.142 Abbreviations: AA-CoA = acetoacetyl-CoA, IPP = isopentylpyrophosphate, DMAPP = dimethylallyl pyrophosphate, GPP = geranyl pyrophosphate. | ||
After initial efforts established that generating limonene in CFE was feasible, a cell-free approach called in vitro Prototyping and Rapid Optimisation of Biosynthetic Enzymes (or iPROBE) was developed. When using iPROBE, many pathway combinations can be rapidly built and optimised via separation into discrete reactions that are then mixed to assemble distinct biosynthetic pathways. The limonene pathway with nine biochemical steps served as an excellent test case to prototype. 150 unique sets of enzymes (particularly distinct homologs of enzymes) in 580 pathway conditions were screened using iPROBE to prototype the best metabolic pathway. This included distinct homologs of enzymes used to design the pathway and fine tuning of the cofactors, which turned out to be a critical parameter. This approach resulted in an increase in limonene production of 25-fold (from ∼23 to 610 mg L−1). To show the modularity and broader applicability of this approach, the terpene synthase enzymes were replaced and just the isoprenoid module was kept, allowing for the generation of the terpenes pinene and bisabolene.145
5. CFE for antimicrobials and resistance
AMR is a pressing global challenge359 causing approximately 700000 annual global deaths, and is predicted to rise to 10 million by 2050.360 Importantly, no new chemical classes of antibiotics have been approved for clinical treatment of Gram-negative bacteria since the synthetic fluoroquinolones developed during the 1970s.361 While there are several economic and scientific challenges to antibiotic discovery,6,362 there is a renewed desire to find novel chemical scaffolds and targets, as well as the use of non-standard antibiotics (i.e., peptides, phages, nanomaterials).
In considering the challenge of AMR, antibiotics are one of the most significant discoveries of the 20th century, and a highly successful form of chemotherapy in terms of lives saved.362–364 While less studied, we will discuss the application of CFE for the initial drug screening process, and its potential for structure–activity relationship (SAR), antibiotic mechanism of action (MOA) and resistance studies.
5.1 Traditional antimicrobial screening
Antimicrobial discovery typically requires phenotypic whole cell bioactivity assays, selecting for compounds that either target the cell envelope or are favourably transported inside the cells to bind intracellular targets. Therefore, primary screening typically obtains low hit rates of discovery and is reliant on growth-inhibition (e.g., agar diffusion, broth dilution) assays. The development of bioactive leads specifically for Gram-negative bacteria is a priority, including extended-spectrum beta-lactamases (ESBLs) and carbapenemase-resistant strains.365,366 Gram-negative bacteria are particularly insidious since they are protected from many antibiotics due to cell envelope barriers. This includes an inner membrane, peptidoglycan and outer membrane, as well as extracellular decorations, including sugar capsules (e.g., Klebsiella pneumoniae) and lipids/polysaccharides. Together these components form a permeability barrier restricting access of antibiotics across the inner and outer membrane.367 Because of the differences in cell envelope physiology, bioactive compounds are easier to obtain for most Gram-positive bacteria. For antibiotic screening, most studies have prioritised bioactive compounds that permeate the cell envelope or target the cell membrane (e.g., pore forming). However, previous screening of synthetic compound libraries suggest that finding compounds with structural properties that satisfy both bioactivity and cell permeability is challenging.368 In contrast, while antimicrobial natural products typically do not obey Lipinski's rule of five for drug-like properties,4 they have evolved to exploit specific transporters, such as TolC-BtuB369 and OmpA,370 to penetrate the cell envelope defences and engage their target(s).The Nobel laureate Frances Arnold notably said, “you get what you screen for”.371 Systematic industrial antibiotic screening used growth inhibition, which led to the discovery of many important antibiotics from natural product libraries5,372 often containing complex mixtures of metabolites, and of variable concentration, quality, and chemical stability. However, considering that transport is limiting for many small molecules, whole cell screening only selects for the most abundant bioactive molecules or those with potent and favourable import into cells. While this is a drawback, cell-based growth inhibition screening assays remain the gold standard due to their success.373 In contrast to natural products, HTS screens of synthetic small molecule libraries has had limited success, in part due to non-specific activity of pan-assay interference compounds.374 In addition, with the rise of genomics technologies, antimicrobial discovery has also attempted target-based screening assays as a direct method for MOA elucidation. This approach investigates effects on a single purified protein or complex target, although there have been limited leads despite significant investment, largely due to poor translation of target binding into cellular potency.368 From 1995 to 2001, GSK evaluated over 300 target genes and 70 target-based HTS campaigns against their collection of 260000–530000 compounds, which resulted in five hits-to-leads.375 Although target-based assays are less successful than whole cell primary screening platforms for antimicrobial discovery, MOA studies such as topoisomerase inhibitor development have shown promise.376,377 Alternatively, screening against targets from pathogenic bacteria, instead of model organisms, increases the potential to identify narrow spectrum leads.378,379
5.2 Antibacterial mechanisms of action
Before considering the application of CFE for antimicrobial screening, we will briefly discuss four major targets in bacterial cells that antibacterial compounds act on, ultimately leading to cell death (bactericidal) or growth inhibition (bacteriostatic). The primary targets include (1) protein synthesis, (2) nucleic acid synthesis, (3) primary metabolism, and (4) cell wall synthesis and/or cell membrane disruption. Within the context of this review, protein synthesis is the major target, however, inhibitors have been demonstrated for DNA gyrase165 and energy regeneration46,380 targets. Several antibiotic classes target protein synthesis by binding to ribosomes at different locations. Aminoglycosides and tetracyclines interact with the 16S rRNA of the 30S subunit near the A-site, causing premature termination of mRNA translation.381,382 Kasugamycin, derived from Streptomyces kasugaensis, is an aminoglycoside that acts as mRNA mimic disrupting mRNA–tRNA codon-anticodon interaction.383 Macrolides, chloramphenicol derived from Streptomyces venezuelae, and synthetic oxazolidinones bind to the 23S rRNA of the 50S subunit at the polypeptide exit tunnel. Specifically, bulky macrolides bind the nascent peptide exit tunnel resulting in peptidyl-tRNA drop off and translation abortion while the other two have a π-stacking interaction that bind at the A-site of the peptidyl transferase centre and prevent binding of incoming charged-tRNA.384,385 The third major type of antibiotic targets includes nucleic acid synthesis and DNA repair mechanisms. Rifamycin sourced from Amycolatopsis mediterranei, and its semi-synthetic derivatives, including rifampicin, inhibit RNA synthesis by targeting DNA-dependent RNA polymerase at the DNA/RNA channel of the RNA polymerase β-subunit.386 This interaction blocks further RNA elongation. Quinolones (synthetic) target DNA gyrase and topoisomerase IV, which are involved in relieving torsional stress during DNA replication and supercoiling of bacterial chromosome.387,388 Alterations in supercoiling control leads to the formation of cleaved complexes (drug–enzyme–DNA), chromosomal fragmentation and ultimately cell death.3895.3 CFE-based antimicrobial screening
In contrast to cell-based screening, CFE provides an alternative approach to antimicrobial discovery, with advantages and disadvantages (Table 5). First, a cell-free extract provides a low cost (<$0.10 per drug) screening assay for detecting antimicrobial compounds from natural product and synthetic sources. For comparison, most HTS assays cost $0.10–0.50 per drug for single target enzyme assays, or $1–10 for whole cell targets.390 Historically, IVT assays have been used to validate the MOAs of antibiotics and toxins, using E. coli, Bacillus sp. and cyanobacteria extracts.151,391–396 For example, a dipeptide antibiotic TAN1057 A/B (GS7128), derived from the soil bacterium Flexibacter, was discovered by a E. coli IVT system and determined to disrupt the peptidyl transferase reaction.395 Interestingly, GS7128 displayed whole cell activity, possibly through import by a dipeptide transporter. In addition, gene reporter systems have been developed for antibiotic interference such as translation initiation.21 Therefore, broadly, IVT assays have provided a tool for characterising novel antimicrobial activity.397–400 In the early 2000s, a dual E. coli and S. cerevisiae IVT assay based on a single mRNA transcript21 was developed to verify target specificity and HTS of 25000 microbial natural product extracts (Fig. 11). This led to the discovery of GE81112, a tetrapeptide inhibitor of prokaryotic translation initiation and first in class hit.401 GE81112 was later established to be a non-ribosomal peptide derived from a 61 kb BGC from Streptomyces sp. L-49973.402 Besides GE81112, the cyclic peptide GE82832 was also discovered through IVT and shown to inhibit translocation by binding to the 30S subunit.403 However, despite demonstrating novelty in terms of MOA and chemical structures, IVT screening encountered issues such as a high hit rate, which is unfavourable for industrial screening. In addition, the assay enriched for novel compounds that showed high activity in vitro but unfortunately had limited activity in whole cell assays. However, while GE81112 had poor transport into bacterial cells, structural derivatives were later developed with improved membrane permeability.42,404–407 This evidence supports that CFE approaches have the potential to deliver hit novelty in terms of MOA, whereas activity in cells can later be achieved through rational drug design to reach a lead. Alternatively, by focusing on natural product extracts displaying both in vitro and whole cell activity to reduce the hit rate, additional work led to the discovery of orthoformimycin from Streptomyces sp. ID107558. This compound inhibited EF-G dependent movement of the mRNA on the ribosome and Pi release from EF-G without affecting EF-G dependent aminoacyl-tRNA translocation.21,408 Employing reporter systems in CFE also aids the detection of novel gene expression inhibitors.409
| Assay | Whole-cell assay | Target-based assay | CFE |
|---|---|---|---|
| Screening format | Growth inhibition | Effect on pre-determined target | Inhibition of mRNA and protein synthesis |
| Advantages | Standardised method | HTS | HTS |
| Translation into clinic | Mechanism of action | Mechanism of action | |
| Structure–activity relationship | |||
| Physiological effect | Structure–activity relationship | Mimics native cytoplasmic environment | |
| Multiple intracellular targets | |||
| Limitations | Mechanism of action unknown | Characterised target | Hits may not be cell permeable |
| Scalability to HTS | Purified target required | Low permeability – limited to cellular activity | |
| Permeability barrier – false negatives | Low hit rate |
 | ||
| Fig. 11 Outline of HTS of a 25000 member natural product library using an E. coli IVT assay to discover GE81112 and GE82832, the former, a first in class antimicrobial disrupting the formation of the pre-initiation translation complex.21 The stereochemistry of GE82832 is not shown, as this was not reported. | ||
The ribosome and the core transcription and translation machinery are both highly conserved within, and evolutionarily divergent between, prokaryotes and eukaryotes, which enables selectivity in terms of antibiotic drug design. However, there are substantial differences between individual microbial cell types across all levels of genetics, biochemistry and physiology that could be exploited for the development of narrow-spectrum antimicrobials. While antimicrobial screening has previously focused on broad spectrum approaches (i.e., Gram-negative and Gram-positive bioactivity), there is more scope for species-specific targeted development of antimicrobials as measure to minimise the risk of AMR.361,410 For this purpose, some cell-free studies have developed species-specific CFE tools. For example, a Staphylococcus aureus CFE assay was developed to evaluate transcription–translation inhibitors from a cytoplasmic extract and purified ribosomes.411 Then, a HTS platform was developed for a clinically relevant Streptococcus pneumoniae strain and used to screen 220000 compounds using a dual translation-activity assay with luciferase as reporter.152 Identified hits were screened in a panel of species-specific CFE assays to evaluate specificity. A limitation of this study was the high rate of off-target inhibition of the luciferase enzyme, leading to false-positives. More recently, Klebsiella pneumoniae CFE was developed from laboratory and clinical strains and used for the study of AMR genetic determinants, as well as for targeted antimicrobial characterisation to explore differences between K. pneumoniae and an E. coli CFE model.165 This showed that the minimum concentration of antibiotic to inhibit 50% of K. pneumoniae cells (MIC50) ranged between one to two an orders of magnitude higher (with the exception of chloramphenicol and tetracycline) than the equivalent 50% inhibitory concentration (IC50) value determined with K. pneumoniae CFE for most antibiotics tested.165 This was insightful, since past industrial-led antimicrobial screening with whole cell assays was typically performed with complex natural product extracts and fractionated libraries, derived from laboratory grown microbial isolates, and diversified through temporal and geographic sampling from biologically diverse regions.21,412,413 This is important because natural products are typically only produced at low concentrations by laboratory grown microbes. Potentially, CFE could offer an opportunity to detect low abundance bioactive compounds from complex natural product libraries otherwise missed by whole cell assays.21,42,152,401
5.4 CFE as a tool to study antibiotic resistance
AMR arises through adaptive chromosomal or plasmid mutations in response to antibiotic-induced cell stress, or through acquisition of AMR genes (ARGs) from the environment.414 Resistance mechanisms are grouped into six categories: (1) reduced permeability, (2) increased efflux of antibiotics from the periplasm and cytoplasm, (3) modification of antibiotic target, (4) target mimicry, where an evolved ortholog bypasses an inhibited metabolic step, (5) antibiotic inactivation by modification, sequestration, or degradation, or (6) biofilm formation.414For profiling of antibiotic resistance, there are two predominant approaches: genotyping and phenotyping. Genotyping uses whole-genome next-generation sequencing (NGS) and bioinformatics tools, such as RESfinder,415,416 to detect known genetic determinants associated with AMR (i.e., resistance genes, single-nucleotide polymorphisms in AMR determinants). Uniquely, NGS can study single strains and mixed populations. However, both genotyping and bioinformatics analysis still requires laboratory-based phenotypic experiments, especially for emerging resistance determinants. Phenotyping requires cell culture, broth dilution, time kill, and disc diffusion assays, following the recommended standards from the U.S. Food and Drug Administration (FDA), World Health Organization, National Committee for Clinical Laboratory Standards and EUCAST. Critically, the MIC50/90 value is a complex measure of overall antibiotic activity, a combination of antibiotic import, efflux, metabolism, selectivity, and potency, all overlaid by mutations associated with the resistome.417 Additionally, mutations associated with a specific AMR phenotype can confer collateral resistance or sensitivity to structurally or functionally unrelated antibiotics.418–420 Similarities occur in cancer drug resistance.421
As an extra tool for phenotypic analysis of AMR, several studies have reported the use of IVT and CFE tools for mechanistic studies of resistance, using either crude extracts or purified ribosomes. For example, purified ribosomes from a mutant E. coli strain containing single or double 16S rRNA mutations (U1060A and U1052G) in the head domain of 30S ribosomal subunit were investigated.422 U1052 16S rRNA mutants increased resistance to negamycin and susceptibility to tetracycline resulting from an unexpected tetracycline-binding site.422 Ribosomal resistance from antibiotic-producing Streptomyces strains in CFE also provide insights into the protective mechanism against pactamycin (a ribosome inhibitor) specific to this host.423 Additionally, a small pentapeptide encoded within the E. coli 23S rRNA, if overexpressed, confers resistance to erythromycin.424 However, while the RNA encoding the pentapeptide conferred erythromycin resistance in an E. coli translation system, the corresponding synthetic peptide did not, suggesting the peptide has a short half-life and requires continuous expression.424 In related studies where AMR and drug discovery were not the focus, the use of CFE to study ribosomal mutants was established. Seven isolated ribosomal mutants from streptomycin-resistant E. coli BL21 were used to quantify CFE of chloramphenicol acetyltransferase.425 Most mutants showed reduced ribosomal activity and more interestingly increased translation accuracy measured by mis-incorporation of Leu in a poly(U) encoded poly-Phe sequence. A yeast CFE carrying ribosomal mutations in the S28 and S3 proteins (S12, S5 homologs in E. coli) was used to contrast translational accuracy and sensitivity/resistance to paromomycin.426 In addition, clindamycin-resistance was used as a selection approach to validate an in vitro ribosome synthesis and evolution via ribosome display technology.427 A fungal IVT system resistant to the translational inhibitor trichothecin428 was also investigated. Collectively, these studies reveal the potential of CFE to reconstitute mutant ribosomes and study the differential effects on antibiotic resistance, ribosomal processivity and translational accuracy.
CFE is a viable tool to profile ARGs. However, except for chloramphenicol acetyltransferase, which historically was used as a gene expression reporter with CFE,429 only a handful of studies, mostly during the 1980s, have used IVT/CFE to study ARGs. For example, both Streptomyces and E. coli CFE were used to express and identify resistance gene products against novobiocin,430 hygromycin B,431 and thiostrepton.285 More recently, a K. pneumoniae CFE tool was combined with adaptive laboratory evolution to study chloramphenicol (50S ribosome), valnemulin (50S ribosome), and rifampicin (RNA polymerase) resistance.165 Genomic sequencing of a selection of these mutants was performed to determine the genotype, followed by phenotypic characterisation of the extracts derived from these strains, through measuring protein synthesis and resistance to these set gene expression inhibitors. Specifically, an RNA polymeraseβ-subunit (rpoB) H526L mutant displayed 40-fold increased resistance to rifampicin, while moderate resistance was observed for chloramphenicol (Fig. 12). Outside of academia, Biobits® has made a cell-free educational kit for expressing an aminoglycoside kinase for inactivating the translational inhibitor kanamycin.432
 | ||
| Fig. 12 The application of a K. pneumoniae CFE tool to study antibiotic resistance associated mutations and their effects on protein synthesis for (a) rifampicin resistance, and (b) chloramphenicol resistance. Figure reproduced and adapted with permission.165 | ||
5.5 Prospects for CFE for antimicrobials and resistance
Overall, CFE technologies can serve a range of uses within antimicrobial discovery, and the study of antimicrobial resistance. Key advantages include the speed, cost, and potential for automation, while natural products are notoriously produced at limited quantities, whereas cell-free bioassays are shown to have orders of magnitude higher sensitivity than conventional whole cell antimicrobial assays. We suggest CFE provides a rapid and flexible research tool to support the characterisation of new ARGs and resistance profiles that will become more prevalent with the rise of AMR and the use of new antibiotics. There is also potential to employ CFE to mimic the biochemistry of a cell and study structure–activity relationship studies for antimicrobial development. In addition, there is evidence CFE is beneficial for identifying novel modes of action, such as peptide-based antimicrobials.6. Beyond the lab: cell-free technology in industry
Finally, we shall consider the wider use of CFE and CFB within industry (Fig. 13). Cell-free (both CFE and extract) approaches are attractive for industrial biomanufacturing because of separation of biomass growth (for lysate production) and product formation (performing biosynthetic reactions using the lysate), increased biosynthesis rates due to the abolishment of the need for transport of substrates and products across membranes and/or cell walls, and substrate and product tolerance. Owing to technology advances and scale-up capabilities, cell-free is emerging as an attractive and alternative platform for development and commercialisation of next-generation pharmaceuticals, cosmetic and food ingredients and specialty and commodity industrial chemicals. A recent analysis showed that cell-free patent filings increased five-fold over the last decade (compared to just a 1.5-fold increase in peer-reviewed publications) with an increase in company filed patent applications by seven-fold between 2015 and 2020.2 Here we briefly describe some companies and applications of cell-free biomanufacturing they are employing and note whilst these are largely not-focused on natural products per se, are illustrative of commercial applications that could be employed for natural products. | ||
| Fig. 13 Overview of industrial strategies to apply CFE as a prototyping and production platform. | ||
LanzaTech, a company that uses gas fermentation with Clostridium autoethanogenum for the production of commodity chemicals, has developed a cell-free prototyping platform that enabled them to identify enzymes causing unwanted side reactions which they then eliminated from the genome, resulting in increased product titers of acetone and isopropanol.433 This platform, termed iPROBE (discussed specifically in Section 4.4.1) allows for the rapid prototyping of biosynthetic pathways, combinatorial pathway design and testing, and pathway optimisation.434 This two-step system is initiated with CFE synthesis of desired enzymes and is followed by the reaction phase upon addition of substrates and cofactors. They also achieved increased production of 3-hydroxybutyrate, 2,3-butanediol and butanol, showing the versatility and usefulness of this platform.434
Other companies are using cell-free technology to produce vaccines and therapeutics. Nature's Toolbox Inc.435 has developed a continuous flow CFE using hollow fibre bioreactors for production of RNA molecules, especially those mRNA for therapeutics, that provides flexibility and accelerated production compared with traditional cell-based or synthesis systems. Such an approach could be highly valuable when rapid responses are required (e.g. such as in pandemics). They also offer a cell-free protein expression platform. Vaxcyte436 is using their CFE platform, licensed from Sutro Biopharma,437 to engineer, optimise and manufacture vaccines, focusing on vaccines for invasive pneumococcal disease, Group A Strep, periodontitis and Shigella. They couple CFE with conjugate chemistry to add functionality. Sutro Biopharma has developed CFE that incorporate non-natural amino acids (nnAAs) using orthogonal translation systems consisting of non-native tRNAs and aminoacyl-tRNA synthetases that lead to single-species antibody–drug conjugates for antitumor applications. Ginkgo Bioworks recently entered into an agreement with DARPA to deliver therapeutic proteins such as antibodies, cytokines and clotting factors using cell-free methods in conjunction with their biofoundry. Resilience acquired Swiftscale Biologics438 CFE platform and now uses that for production of various biologics.
Several companies have developed CFB catalytic processes for products across multiple applications. FabricNano439 uses enzyme immobilisation to conduct biocatalysis for a range of applications. They have built and offer a platform for predictive immobilisation (Predictive Immobilisation Plates) which allows for optimisation of enzyme and immobilisation methods in a combinatorial manner that reduces the need for mass experimentation. They have partnered with several companies. EnginZyme has developed a patented process for pseudouridine biosynthesis using immobilised enzymes, that leads to a purer version of pseudouridine compared to conventional synthesis approaches. N1-Methylpseudouridine-5′-triphosphate, which is derived from pseudouridine, helps stabilise and reduce the immunogenicity of mRNA vaccines, such as that for COVID-19.
Besides the animal cell and microbial CFE platforms described above, plant CFE have also been developed and commercialised. Most notably is the development of tobacco BY-2 cell suspension cultures that have achieved protein yields of 3 mg mL−1 in a coupled CFE reaction and have been used for the production of lycopene, indigoidine, and betalains.440 This system has been commercialised by LenioBio GmbH as ALiCE® (Almost Living Cell-free extract) and further been shown to produce virus-like particles, membrane receptors and a monoclonal antibody.441
The advent and application of artificial intelligence has been applied to cell-free technology. While various academic efforts that combine automation, artificial intelligence and cell-free technology, the commercial sector is also seeing these combined technologies being employed. Debut442 has a scalable cell-free platform that spans discovery to commercialisation and who is focusing on products for the personal care industry. Tierra Biosciences443 is combining cell-free protein expression with AI for on-demand and custom protein synthesis.
Broader adoption of cell-free technology in industrial biomanufacturing has been hampered by the costs and scale of lysate preparation, cofactor provision, and the ability to operate in a continuous mode. It has been calculated that it costs approximately $5000 per L for a standard cell-free reaction formulation with most of the cost coming from the phosphorylated energy substrate, phosphoenolpyruvate (a detailed breakdown of these costs can be found in the Supporting reference of a comprehensive review by Silverman et al.).1 As described above, the commercial applications are predominately in the therapeutic space, where the economics can sustain these higher costs, and where scales are diminished compared to commodity chemicals. With further optimisation and development work, alternative approaches to mitigate such economic and scale issues could be possible which will open new avenues for cell-free biomanufacturing.
7. Future outlook
The ability to use CFE and cell-extract based technologies for a variety of applications has exploded in recent years. However, applying the wealth of options to generate complex proteins, pathways, and ultimately small molecule metabolites has only recently been directed towards applications relevant to the expression of biosynthetic gene clusters and their corresponding natural products. To date, preliminary efforts to generate peptide, polyketide, and terpene natural products have been performed both in lysate-based and PURExpress® systems. These initial studies can be directed in multiple ways to further natural products research. Generating uncharacterised natural products in CFE can be used to access compounds that are not readily biosynthesised in their producing organisms under laboratory conditions and can produce molecules that present toxic effects when overexpressed in heterologous hosts. CFE can also be used to rapidly evaluate modes of activity, such as antibiotic resistance.Prototyping design for bioengineering applications remains a huge unrealised application of CFE. Due to the complexity of these metabolic pathways, there are numerous avenues for optimisation both of precursor flux and of pathway design. For the so-called “megasynthase” enzymes (PKS and NRPS enzymes), using this iterative “design-build-test-learn” cycle has promise for designing chimeric assembly lines.444–446 Efforts to generate soluble, full-length proteins present a major step towards this application.266
For RiPPs, as the precursor is a proteinogenic amino acid, there are many opportunities for pathway elucidation and engineering.167,172,447 One of several challenges in CFE for RiPPs natural products is establishing appropriate cofactor recycling for redox sensitive enzymes, such as radical SAM enzymes. Recent progress in strategies for complex cofactor recycling, such as efforts to generate iron sulphur clusters under aerobic conditions,448 may further expand the application of CFE in RiPPs. Terpenes have been shown to be highly modular for bioengineering applications in vivo449 as evidenced by efforts to produce complex terpene natural products (a key example being artemisinin450) in tractable heterologous hosts. Metabolic engineering approaches such as iPROBE demonstrates that similar engineering approaches can be employed in cell-free environments.145 Furthermore, once generated, CFE can be used to rapidly evaluate bioactivity of these compounds.
In summary, advances in technology continue to push the horizons of what is achievable in CFE. Whether it is democritising the PURE® system,117 improving co-factor recycling, or adapting novel lysates to cell-free applications with natural products, these advances highlight continued development of these robust platforms. Ultimately, this will allow biomanufacturing to extend to a broader range of metabolic pathways and expand the synthetic biology of natural products.
Author contributions
AJR, TTS, KC, DAM, NJM, SJM, and CBM wrote the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AJR acknowledges funding support from a Chemical-Biology Interface Training Grant (Grant T32-GM136629) and a National Science Foundation Graduate Research Fellowship (Grant DGE 21-46756). SJM acknowledges funding support from the Leverhulme Trust (RPG-2021-018), Royal Society (RGS\R1\231113) and Biotechnology and Biological Sciences Research Council (BB/Y005074/1). KC acknowledges support by the Global Challenges Doctoral Centre at the University of Kent. CBB acknowledges support from the University of Sydney School of Chemistry and the Sydney Anti-Biotics Accelerator from the University of Sydney Centre of Drug Discovery Innovation and is a member the University of Sydney Infectious Diseases Institute, and the University of Sydney Nanoscience Institute. TTS acknowledges support from the University of Tennessee-Knoxville Department of Chemistry, the University of Tennessee-Oak Ridge Innovation Institute, and a University of Tennessee-Oak Ridge Innovation Institute Graduate Advancement, Training, and Education (GATE) fellowship. DAM acknowledges support from NIH/NIGMS (GM123998). The work of NJM at the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231.Notes and references
- A. D. Silverman, A. S. Karim and M. C. Jewett, Nat. Rev. Genet., 2020, 21, 151–170 Search PubMed.
- C. Meyer, Y. Nakamura, B. J. Rasor, A. S. Karim, M. C. Jewett and C. Tan, Life, 2021, 11, 551 Search PubMed.
- E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discovery Today, 2016, 21, 204–207 Search PubMed.
- A. Ganesan, Curr. Opin. Chem. Biol., 2008, 12, 306–317 Search PubMed.
- H. B. Woodruff, Appl. Environ. Microbiol., 2014, 80, 2–8 Search PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 Search PubMed.
- S. D. Bentley, K. F. Chater, A.-M. Cerdeño-Tárraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C.-H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O’Neil, E. Rabbinowitsch, M.-A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 Search PubMed.
- G. L. Challis, J. Ind. Microbiol. Biotechnol., 2014, 41, 219–232 Search PubMed.
- D. A. Benson, M. Cavanaugh, K. Clark, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and E. W. Sayers, Nucleic Acids Res., 2013, 41, D36–42 Search PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–46 Search PubMed.
- K. Blin, S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann, M. Alanjary, A. Fetter, B. R. Terlouw, W. W. Metcalf, E. J. N. Helfrich, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 51, W46–W50 Search PubMed.
- A. J. van Heel, A. de Jong, M. Montalbán-López, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2013, 41, W448–53 Search PubMed.
- M. A. Georgiou, S. R. Dommaraju, X. Guo, D. H. Mast and D. A. Mitchell, ACS Chem. Biol., 2020, 15, 2976–2985 Search PubMed.
- H. Ren, C. Shi and H. Zhao, iScience, 2020, 23, 100795 Search PubMed.
- M. Alanjary, B. Kronmiller, M. Adamek, K. Blin, T. Weber, D. Huson, B. Philmus and N. Ziemert, Nucleic Acids Res., 2017, 45, W42–W48 Search PubMed.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 Search PubMed.
- C. Cano-Prieto, A. Undabarrena, A. C. de Carvalho, J. D. Keasling and P. Cruz-Morales, Annu. Rev. Biochem., 2024, 93(1), 411–445 Search PubMed.
- F. Hemmerling and J. Piel, Nat. Rev. Drug Discovery, 2022, 21, 359–378 Search PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 Search PubMed.
- K. Lewis, Cell, 2020, 181, 29–45 Search PubMed.
- P. Monciardini, M. Iorio, S. Maffioli, M. Sosio and S. Donadio, Microb. Biotechnol., 2014, 7, 209–220 Search PubMed.
- M. Myronovskyi, B. Rosenkränzer, S. Nadmid, P. Pujic, P. Normand and A. Luzhetskyy, Metab. Eng., 2018, 49, 316–324 Search PubMed.
- J. P. Gomez-Escribano and M. J. Bibb, Microb. Biotechnol., 2011, 4, 207–215 Search PubMed.
- A. Thanapipatsiri, J. Claesen, J.-P. Gomez-Escribano, M. Bibb and A. Thamchaipenet, Microb. Cell Fact., 2015, 14, 145 Search PubMed.
- R. E. Cobb, Y. Wang and H. Zhao, ACS Synth. Biol., 2015, 4, 723–728 Search PubMed.
- L. Li, G. Zheng, J. Chen, M. Ge, W. Jiang and Y. Lu, Metab. Eng., 2017, 40, 80–92 Search PubMed.
- G. Wang, Z. Zhao, J. Ke, Y. Engel, Y.-M. Shi, D. Robinson, K. Bingol, Z. Zhang, B. Bowen, K. Louie, B. Wang, R. Evans, Y. Miyamoto, K. Cheng, S. Kosina, M. De Raad, L. Silva, A. Luhrs, A. Lubbe, D. W. Hoyt, C. Francavilla, H. Otani, S. Deutsch, N. M. Washton, E. M. Rubin, N. J. Mouncey, A. Visel, T. Northen, J.-F. Cheng, H. B. Bode and Y. Yoshikuni, Nat. Microbiol., 2019, 4, 2498–2510 Search PubMed.
- P. A. Hoskisson and R. F. Seipke, mBio, 2020, 11(5), e02642–20 Search PubMed.
- J. Schwarz, G. Hubmann, K. Rosenthal and S. Lütz, Biomolecules, 2021, 11, 193 Search PubMed.
- F. Xu, B. Nazari, K. Moon, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2017, 139, 9203–9212 Search PubMed.
- K. Moon, F. Xu, C. Zhang and M. R. Seyedsayamdost, ACS Chem. Biol., 2019, 14, 767–774 Search PubMed.
- T. Chen, Y. Zhou, Y. Lu and H. Zhang, Biotechnol. Lett., 2019, 41, 27–34 Search PubMed.
- R. Wang, S. Zhao, Z. Wang and M. A. Koffas, Curr. Opin. Biotechnol, 2020, 62, 65–71 Search PubMed.
- M. Wong, A. Badri, C. Gasparis, G. Belfort and M. Koffas, Crit. Rev. Biochem. Mol. Biol., 2021, 56, 587–602 Search PubMed.
- M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 Search PubMed.
- Z.-C. Wu and D. L. Boger, Nat. Prod. Rep., 2020, 37, 1511–1531 Search PubMed.
- S. J. Moore, H.-E. Lai, J. Li and P. S. Freemont, Nat. Prod. Rep., 2023, 40, 228–236 Search PubMed.
- M. Bujara, M. Schümperli, S. Billerbeck, M. Heinemann and S. Panke, Biotechnol. Bioeng., 2010, 106, 376–389 Search PubMed.
- J. K. Jung, K. K. Alam, M. S. Verosloff, D. A. Capdevila, M. Desmau, P. R. Clauer, J. W. Lee, P. Q. Nguyen, P. A. Pastén, S. J. Matiasek, J.-F. Gaillard, D. P. Giedroc, J. J. Collins and J. B. Lucks, Nat. Biotechnol., 2020, 38, 1451–1459 Search PubMed.
- M. K. Takahashi, J. Chappell, C. A. Hayes, Z. Z. Sun, J. Kim, V. Singhal, K. J. Spring, S. Al-Khabouri, C. P. Fall, V. Noireaux, R. M. Murray and J. B. Lucks, ACS Synth. Biol., 2015, 4, 503–515 Search PubMed.
- B. Rasmussen, H. F. Noller, G. Daubresse, B. Oliva, Z. Misulovin, D. M. Rothstein, G. A. Ellestad, Y. Gluzman, F. P. Tally and I. Chopra, Antimicrob. Agents Chemother., 1991, 35, 2306–2311 Search PubMed.
- L. Brandi, A. Fabbretti, A. La Teana, M. Abbondi, D. Losi, S. Donadio and C. O. Gualerzi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 39–44 Search PubMed.
- Y. Hori, C. Kantak, R. M. Murray and A. R. Abate, Lab Chip, 2017, 17, 3037–3042 Search PubMed.
- H. Niederholtmeyer, V. Stepanova and S. J. Maerkl, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15985–15990 Search PubMed.
- J. F. Zawada, G. Yin, A. R. Steiner, J. Yang, A. Naresh, S. M. Roy, D. S. Gold, H. G. Heinsohn and C. J. Murray, Biotechnol. Bioeng., 2011, 108, 1570–1578 Search PubMed.
- A. M. Voloshin and J. R. Swartz, Biotechnol. Bioeng., 2005, 91, 516–521 Search PubMed.
- K. M. Wilding, S.-M. Schinn, E. A. Long and B. C. Bundy, Curr. Opin. Biotechnol, 2018, 53, 115–121 Search PubMed.
- Z. Z. Sun, E. Yeung, C. A. Hayes, V. Noireaux and R. M. Murray, ACS Synth. Biol., 2014, 3, 387–397 Search PubMed.
- S. J. Moore, H.-E. Lai, R. J. R. Kelwick, S. M. Chee, D. J. Bell, K. M. Polizzi and P. S. Freemont, ACS Synth. Biol., 2016, 5, 1059–1069 Search PubMed.
- J. Chappell, K. Jensen and P. S. Freemont, Nucleic Acids Res., 2013, 41, 3471–3481 Search PubMed.
- D. Siegal-Gaskins, Z. A. Tuza, J. Kim, V. Noireaux and R. M. Murray, ACS Synth. Biol., 2014, 3, 416–425 Search PubMed.
- J. Garamella, R. Marshall, M. Rustad and V. Noireaux, ACS Synth. Biol., 2016, 5, 344–355 Search PubMed.
- J. Kim, I. Khetarpal, S. Sen and R. M. Murray, Nucleic Acids Res., 2014, 42, 6078–6089 Search PubMed.
- E. Karzbrun, J. Shin, R. H. Bar-Ziv and V. Noireaux, Phys. Rev. Lett., 2011, 106, 048104 Search PubMed.
- H. Niederholtmeyer, Z. Z. Sun, Y. Hori, E. Yeung, A. Verpoorte, R. M. Murray and S. J. Maerkl, eLife, 2015, 4, e09771 Search PubMed.
- K. Pardee, S. Slomovic, P. Q. Nguyen, J. W. Lee, N. Donghia, D. Burrill, T. Ferrante, F. R. McSorley, Y. Furuta, A. Vernet, M. Lewandowski, C. N. Boddy, N. S. Joshi and J. J. Collins, Cell, 2016, 167, 248–259.e12 Search PubMed.
- A. Harden and F. R. Henley, Biochem. J., 1927, 21, 196–197 Search PubMed.
- P. Leder, B. F. Clark, W. S. Sly, S. Pestka and M. W. Nirenberg, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 1135–1143 Search PubMed.
- M. W. Nirenberg, J. H. Matthaei, O. W. Jones, R. G. Martin and S. H. Barondes, Fed. Proc., 1963, 22, 55–61 Search PubMed.
- S. H. Barondes and M. W. Nirenberg, Science, 1962, 138, 813–817 Search PubMed.
- V. J. Palombella, O. J. Rando, A. L. Goldberg and T. Maniatis, Cell, 1994, 78, 773–785 Search PubMed.
- A. Orian, S. Whiteside, A. Israël, I. Stancovski, A. L. Schwartz and A. Ciechanover, J. Biol. Chem., 1995, 270, 21707–21714 Search PubMed.
- J. Park, J. Tai, C. A. Roessner and A. I. Scott, Bioorg. Med. Chem., 1996, 4, 2179–2185 Search PubMed.
- A. I. Scott, N. E. Mackenzie, P. J. Santander, P. E. Fagerness, G. Müller, E. Schneider, R. Sedlmeier and G. Wörner, Bioorg. Chem., 1984, 12, 356–362 Search PubMed.
- P. J. Santander, C. A. Roessner, N. J. Stolowich, M. T. Holderman and A. I. Scott, Chem. Biol., 1997, 4, 659–666 Search PubMed.
- A. I. Scott, N. J. Stolowich, J. Wang, O. Gawatz, E. Fridrich and G. Müller, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14316–14319 Search PubMed.
- Q. Huang, C. A. Roessner, R. Croteau and A. I. Scott, Bioorg. Med. Chem., 2001, 9, 2237–2242 Search PubMed.
- C. Greunke, A. Glöckle, J. Antosch and T. A. M. Gulder, Angew. Chem., Int. Ed., 2017, 56, 4351–4355 Search PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 Search PubMed.
- J. A. Kalaitzis, Q. Cheng, P. M. Thomas, N. L. Kelleher and B. S. Moore, J. Nat. Prod., 2009, 72, 469–472 Search PubMed.
- Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi and B. S. Moore, Nat. Chem. Biol., 2007, 3, 557–558 Search PubMed.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 16012–16015 Search PubMed.
- S. J. Moore, A. D. Lawrence, R. Biedendieck, E. Deery, S. Frank, M. J. Howard, S. E. J. Rigby and M. J. Warren, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14906–14911 Search PubMed.
- A. L. J. L. Ribeiro, P. Pérez-Arnaiz, M. Sánchez-Costa, L. Pérez, M. Almendros, L. van Vliet, F. Gielen, J. Lim, S. Charnock, F. Hollfelder, J. E. González-Pastor, J. Berenguer and A. Hidalgo, Microb. Cell Fact., 2024, 23, 169 Search PubMed.
- Y. Ohno-Iwashita, T. Oshima and K. Imahori, J. Biochem., 1976, 79, 1245–1252 Search PubMed.
- G. Reshes, S. Vanounou, I. Fishov and M. Feingold, Phys. Biol., 2008, 5, 046001 Search PubMed.
- C. Kaleta, S. Schäuble, U. Rinas and S. Schuster, Biotechnol. J., 2013, 8, 1105–1114 Search PubMed.
- H. Taymaz-Nikerel, A. E. Borujeni, P. J. T. Verheijen, J. J. Heijnen and W. M. van Gulik, Biotechnol. Bioeng., 2010, 107, 369–381 Search PubMed.
- H. Brunschede, T. L. Dove and H. Bremer, J. Bacteriol., 1977, 129, 1020–1033 Search PubMed.
- M. Nilsson, L. Bülow and K. G. Wahlund, Biotechnol. Bioeng., 1997, 54, 461–467 Search PubMed.
- J. Failmezger, M. Rauter, R. Nitschel, M. Kraml and M. Siemann-Herzberg, Sci. Rep., 2017, 7, 16524 Search PubMed.
- M. B. Elowitz, M. G. Surette, P. E. Wolf, J. B. Stock and S. Leibler, J. Bacteriol., 1999, 181, 197–203 Search PubMed.
- S. B. Zimmerman and S. O. Trach, J. Mol. Biol., 1991, 222, 599–620 Search PubMed.
- S. Cayley, B. A. Lewis, H. J. Guttman and M. T. Record Jr, J. Mol. Biol., 1991, 222, 281–300 Search PubMed.
- R. Milo, BioEssays, 2013, 35, 1050–1055 Search PubMed.
- R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21465–21471 Search PubMed.
- R. Milo, P. Jorgensen, U. Moran, G. Weber and M. Springer, Nucleic Acids Res., 2010, 38, D750–3 Search PubMed.
- A. Vendeville, D. Larivière and E. Fourmentin, FEMS Microbiol. Rev., 2011, 35, 395–414 Search PubMed.
- R. J. Ellis, Curr. Opin. Struct. Biol., 2001, 11, 114–119 Search PubMed.
- B. R. Fritz, O. K. Jamil and M. C. Jewett, Nucleic Acids Res., 2015, 43, 4774–4784 Search PubMed.
- S. Chung, E. Lerner, Y. Jin, S. Kim, Y. Alhadid, L. W. Grimaud, I. X. Zhang, C. M. Knobler, W. M. Gelbart and S. Weiss, Nucleic Acids Res., 2019, 47, 1440–1450 Search PubMed.
- N. Kozer, Y. Y. Kuttner, G. Haran and G. Schreiber, Biophys. J., 2007, 92, 2139–2149 Search PubMed.
- X. Ge, D. Luo and J. Xu, PLoS One, 2011, 6, e28707 Search PubMed.
- G. E. Vezeau and H. M. Salis, ACS Synth. Biol., 2021, 10, 2508–2519 Search PubMed.
- L. Kai, V. Dötsch, R. Kaldenhoff and F. Bernhard, PLoS One, 2013, 8, e56637 Search PubMed.
- B. D. Bennett, J. Yuan, E. H. Kimball and J. D. Rabinowitz, Nat. Protoc., 2008, 3, 1299–1311 Search PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593–599 Search PubMed.
- F. Ceroni, R. Algar, G.-B. Stan and T. Ellis, Nat. Methods, 2015, 12, 415–418 Search PubMed.
- C. Barajas, H.-H. Huang, J. Gibson, L. Sandoval and D. Del Vecchio, Nat. Commun., 2022, 13, 7054 Search PubMed.
- Z.-K. Wang, J.-S. Gong, J. Qin, H. Li, Z.-M. Lu, J.-S. Shi and Z.-H. Xu, ACS Synth. Biol., 2021, 10, 2796–2807 Search PubMed.
- A. Ramón, M. Señorale-Pose and M. Marín, Front. Microbiol., 2014, 5, 56 Search PubMed.
- A. Doerr, D. Foschepoth, A. C. Forster and C. Danelon, Sci. Rep., 2021, 11, 1898 Search PubMed.
- Y. Shimizu, T. Kanamori and T. Ueda, Methods, 2005, 36, 299–304 Search PubMed.
- Z. Li and M. P. Deutscher, Methods Enzymol., 2008, 447, 31–45 Search PubMed.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 Search PubMed.
- T. D. Loan, C. J. Easton and A. Alissandratos, New Biotechnol., 2018, 49, 104–111 Search PubMed.
- L. Herzel, J. A. Stanley, C.-C. Yao and G.-W. Li, Nucleic Acids Res., 2022, 50, 5029–5046 Search PubMed.
- L. P. Miranda and P. F. Alewood, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1181–1186 Search PubMed.
- D. Foshag, E. Henrich, E. Hiller, M. Schäfer, C. Kerger, A. Burger-Kentischer, I. Diaz-Moreno, S. M. García-Mauriño, V. Dötsch, S. Rupp and F. Bernhard, New Biotechnol., 2018, 40, 245–260 Search PubMed.
- D. Garenne, C. L. Beisel and V. Noireaux, Rapid Commun. Mass Spectrom., 2019, 33, 1036–1048 Search PubMed.
- B. J. Rasor, P. Chirania, G. A. Rybnicky, R. J. Giannone, N. L. Engle, T. J. Tschaplinski, A. S. Karim, R. L. Hettich and M. C. Jewett, ACS Synth. Biol., 2023, 12(2), 405–418 Search PubMed.
- H. Bremer and P. P. Dennis, EcoSal Plus, 2008, 3(1) DOI:10.1128/ecosal.5.2.3.
- S. J. Moore, J. T. MacDonald, S. Wienecke, A. Ishwarbhai, A. Tsipa, R. Aw, N. Kylilis, D. J. Bell, D. W. McClymont, K. Jensen, K. M. Polizzi, R. Biedendieck and P. S. Freemont, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4340–E4349 Search PubMed.
- J. Li, L. Gu, J. Aach and G. M. Church, PLoS One, 2014, 9, e106232 Search PubMed.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 Search PubMed.
- H. Ohashi, T. Kanamori, E. Osada, B. K. Akbar and T. Ueda, Methods Mol. Biol., 2012, 805, 251–259 Search PubMed.
- B. Lavickova and S. J. Maerkl, ACS Synth. Biol., 2019, 8, 455–462 Search PubMed.
- L. Grasemann, B. Lavickova, M. C. Elizondo-Cantú and S. J. Maerkl, J. Visualized Exp., 2021, 23(172), 62625 Search PubMed.
- Y. Cui, X. Chen, Z. Wang and Y. Lu, Biodes. Res., 2022, 2022, 9847014 Search PubMed.
- Y. Khatri, R. M. Hohlman, J. Mendoza, S. Li, A. N. Lowell, H. Asahara and D. H. Sherman, ACS Synth. Biol., 2020, 9, 1349–1360 Search PubMed.
- M. Eisenstein, Nature, 2016, 531, 401–403 Search PubMed.
- R. Kelwick, A. J. Webb, J. T. MacDonald and P. S. Freemont, Metab. Eng., 2016, 38, 370–381 Search PubMed.
- H. Wang, J. Li and M. C. Jewett, Synth. Biol., 2018, 3, ysy003 Search PubMed.
- J. Failmezger, S. Scholz, B. Blombach and M. Siemann-Herzberg, Front. Microbiol., 2018, 9, 1146 Search PubMed.
- B. J. Des Soye, S. R. Davidson, M. T. Weinstock, D. G. Gibson and M. C. Jewett, ACS Synth. Biol., 2018, 7, 2245–2255 Search PubMed.
- D. J. Wiegand, H. H. Lee, N. Ostrov and G. M. Church, ACS Synth. Biol., 2018, 7, 2475–2479 Search PubMed.
- A. Krüger, A. P. Mueller, G. A. Rybnicky, N. L. Engle, Z. K. Yang, T. J. Tschaplinski, S. D. Simpson, M. Köpke and M. C. Jewett, Metab. Eng., 2020, 62, 95–105 Search PubMed.
- H. Xu, C. Yang, X. Tian, Y. Chen, W.-Q. Liu and J. Li, ACS Synth. Biol., 2022, 11, 570–578 Search PubMed.
- H. Xu, W.-Q. Liu and J. Li, Methods Mol. Biol., 2022, 2433, 89–103 Search PubMed.
- H. Xu, W.-Q. Liu and J. Li, ACS Synth. Biol., 2020, 9, 1221–1224 Search PubMed.
- J. Li, H. Wang, Y.-C. Kwon and M. C. Jewett, Biotechnol. Bioeng., 2017, 114, 1343–1353 Search PubMed.
- S. J. Moore, H.-E. Lai, S.-M. Chee, M. Toh, S. Coode, K. Chengan, P. Capel, C. Corre, E. L. de Los Santos and P. S. Freemont, ACS Synth. Biol., 2021, 10, 402–411 Search PubMed.
- S. J. Moore, H.-E. Lai, H. Needham, K. M. Polizzi and P. S. Freemont, Biotechnol. J., 2017, 12, 1600678 Search PubMed.
- S. S. Yim, N. I. Johns, J. Park, A. L. Gomes, R. M. McBee, M. Richardson, C. Ronda, S. P. Chen, D. Garenne, V. Noireaux and H. H. Wang, Mol. Syst. Biol., 2019, 15, e8875 Search PubMed.
- T. Clements-Decker, M. Kode, S. Khan and W. Khan, Front. Chem., 2022, 10, 1025979 Search PubMed.
- R. Biedendieck, T. Knuuti, S. J. Moore and D. Jahn, Appl. Microbiol. Biotechnol., 2021, 105, 5719–5737 Search PubMed.
- R. Biedendieck, M. Malten, H. Barg, B. Bunk, J.-H. Martens, E. Deery, H. Leech, M. J. Warren and D. Jahn, Microb. Biotechnol., 2010, 3, 24–37 Search PubMed.
- M. T. Weinstock, E. D. Hesek, C. M. Wilson and D. G. Gibson, Nat. Methods, 2016, 13, 849–851 Search PubMed.
- I. Maida, E. Bosi, E. Perrin, M. C. Papaleo, V. Orlandini, M. Fondi, R. Fani, J. Wiegel, G. Bianconi and F. Canganella, Genome Announc., 2013, 1(4) DOI:10.1128/genomeA.00648-13.
- Z. Wang, B. Lin, W. J. Hervey 4th and G. J. Vora, Genome Announc., 2013, 1(4), e00589-13 Search PubMed.
- B. Zhu, R. Gan, M. D. Cabezas, T. Kojima, R. Nicol, M. C. Jewett and H. Nakano, Biotechnol. Bioeng., 2020, 117, 3849–3857 Search PubMed.
- F. Caschera and V. Noireaux, Biochimie, 2014, 99, 162–168 Search PubMed.
- M. J. Anderson, J. C. Stark, C. E. Hodgman and M. C. Jewett, FEBS Lett., 2015, 589, 1723–1727 Search PubMed.
- X. Yi, B. J. Rasor, N. Boadi, K. Louie, T. R. Northen, A. S. Karim, M. C. Jewett and H. S. Alper, Metab. Eng., 2023, 80, 241–253 Search PubMed.
- Q. M. Dudley, A. S. Karim, C. J. Nash and M. C. Jewett, Metab. Eng., 2020, 61, 251–260 Search PubMed.
- Q. M. Dudley, K. C. Anderson and M. C. Jewett, ACS Synth. Biol., 2016, 5, 1578–1588 Search PubMed.
- A. S. Karim and M. C. Jewett, Metab. Eng., 2016, 36, 116–126 Search PubMed.
- P.-H. Wang, K. Fujishima, S. Berhanu, Y. Kuruma, T. Z. Jia, A. N. Khusnutdinova, A. F. Yakunin and S. E. McGlynn, ACS Synth. Biol., 2020, 9, 36–42 Search PubMed.
- L. Zhang, E. King, W. B. Black, C. M. Heckmann, A. Wolder, Y. Cui, F. Nicklen, J. B. Siegel, R. Luo, C. E. Paul and H. Li, Nat. Commun., 2022, 13, 5021 Search PubMed.
- J. M. Vrtis, A. K. White, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2002, 41, 3257–3259 Search PubMed.
- D. L. Shinabarger, K. R. Marotti, R. W. Murray, A. H. Lin, E. P. Melchior, S. M. Swaney, D. S. Dunyak, W. F. Demyan and J. M. Buysse, Antimicrob. Agents Chemother., 1997, 41, 2132–2136 Search PubMed.
- S. D. Pratt, C. A. David, C. Black-Schaefer, P. J. Dandliker, X. Xuei, U. Warrior, D. J. Burns, P. Zhong, Z. Cao, A. Y. C. Saiki, C. G. Lerner, L. E. Chovan, N. B. Soni, A. M. Nilius, F. L. Wagenaar, P. J. Merta, L. M. Traphagen and B. A. Beutel, J. Biomol. Screen., 2004, 9, 3–11 Search PubMed.
- F. Caschera and V. Noireaux, Metab. Eng., 2015, 27, 29–37 Search PubMed.
- Y. Wang and Y.-H. P. Zhang, BMC Biotechnol., 2009, 9, 58 Search PubMed.
- A. S. Karim, J. T. Heggestad, S. A. Crowe and M. C. Jewett, Metab. Eng., 2018, 45, 86–94 Search PubMed.
- X.-P. Hu, H. Dourado, P. Schubert and M. J. Lercher, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- K. A. Calhoun and J. R. Swartz, in In Vitro Transcription and Translation Protocols, ed. G. Grandi, Humana Press, Totowa, NJ, 2007, vol. 375, pp. 3–17 Search PubMed.
- N. E. Gregorio, M. Z. Levine and J. P. Oza, Methods Protoc., 2019, 2(1), 24 Search PubMed.
- K. A. Calhoun and J. R. Swartz, Biotechnol. Bioeng., 2005, 90, 606–613 Search PubMed.
- D. M. Kim and J. R. Swartz, Biotechnol. Bioeng., 1999, 66, 180–188 Search PubMed.
- D. M. Kim and J. R. Swartz, Biotechnol. Bioeng., 2001, 74, 309–316 Search PubMed.
- Z. Z. Sun, C. A. Hayes, J. Shin, F. Caschera, R. M. Murray and V. Noireaux, J. Visualized Exp., 2013, 50762, e50762 Search PubMed.
- Q. Cai, J. A. Hanson, A. R. Steiner, C. Tran, M. R. Masikat, R. Chen, J. F. Zawada, A. K. Sato, T. J. Hallam and G. Yin, Biotechnol. Prog., 2015, 31, 823–831 Search PubMed.
- M. C. Jewett, K. A. Calhoun, A. Voloshin, J. J. Wuu and J. R. Swartz, Mol. Syst. Biol., 2008, 4, 220 Search PubMed.
- K. Chengan, C. Hind, M. Stanley, M. E. Wand, L. K. Nagappa, K. Howland, T. Hanson, R. Martín-Escolano, A. D. Tsaousis, J. A. Bengoechea, J. Mark Sutton, C. M. Smales and S. J. Moore, npj Antimicrob. Resist., 2023, 1, 16 Search PubMed.
- O. Borkowski, M. Koch, A. Zettor, A. Pandi, A. C. Batista, P. Soudier and J.-L. Faulon, Nat. Commun., 2020, 11, 1872 Search PubMed.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 Search PubMed.
- M. A. Shandell, Z. Tan and V. W. Cornish, Biochemistry, 2021, 60, 3455–3469 Search PubMed.
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 Search PubMed.
- D. T. Nguyen, T. T. Le, A. J. Rice, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2022, 144, 11263–11269 Search PubMed.
- A. A. Vinogradov, M. Shimomura, Y. Goto, T. Ozaki, S. Asamizu, Y. Sugai, H. Suga and H. Onaka, Nat. Commun., 2020, 11, 2272 Search PubMed.
- Y. Si, A. M. Kretsch, L. M. Daigh, M. J. Burk and D. A. Mitchell, J. Am. Chem. Soc., 2021, 143, 5917–5927 Search PubMed.
- A. M. Kretsch, M. G. Gadgil, A. J. DiCaprio, S. E. Barrett, B. L. Kille, Y. Si, L. Zhu and D. A. Mitchell, Biochemistry, 2023, 62, 956–967 Search PubMed.
- S. E. Barrett, S. Yin, P. Jordan, J. K. Brunson, J. Gordon-Nunez, G. Costa Machado da Cruz, C. Rosario, B. K. Okada, K. Anderson, T. A. Pires, R. Wang, D. Shukla, M. J. Burk and D. A. Mitchell, Nat. Chem. Biol., 2025, 21, 412–419 Search PubMed.
- F. Cheng, T. M. Takala and P. E. J. Saris, J. Mol. Microbiol. Biotechnol., 2007, 13, 248–254 Search PubMed.
- R. Liu, Y. Zhang, G. Zhai, S. Fu, Y. Xia, B. Hu, X. Cai, Y. Zhang, Y. Li, Z. Deng and T. Liu, Adv. Sci., 2020, 7, 2001616 Search PubMed.
- W.-Q. Liu, X. Ji, F. Ba, Y. Zhang, H. Xu, S. Huang, X. Zheng, Y. Liu, S. Ling, M. C. Jewett and J. Li, Nat. Commun., 2024, 15, 4336 Search PubMed.
- N. Peña, M. J. Bland, E. Sevillano, E. Muñoz-Atienza, I. Lafuente, M. E. Bakkoury, L. M. Cintas, P. E. Hernández, P. Gabant and J. Borrero, Front. Microbiol., 2022, 13, 1052686 Search PubMed.
- S. R. Fleming, P. M. Himes, S. V. Ghodge, Y. Goto, H. Suga and A. A. Bowers, J. Am. Chem. Soc., 2020, 142, 5024–5028 Search PubMed.
- A. Alfi, A. Popov, A. Kumar, K. Y. J. Zhang, S. Dubiley, K. Severinov and S. Tagami, ACS Synth. Biol., 2022, 11, 2022–2028 Search PubMed.
- A. Bhushan, P. J. Egli, E. E. Peters, M. F. Freeman and J. Piel, Nat. Chem., 2019, 11, 931–939 Search PubMed.
- D. S. Wilson and A. D. Keefe, Current Protocols in Molecular Biology, 2001, ch. 8, Unit8.3 Search PubMed.
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 Search PubMed.
- C. Ongpipattanakul, E. K. Desormeaux, A. DiCaprio, W. A. van der Donk, D. A. Mitchell and S. K. Nair, Chem. Rev., 2022, 122, 14722–14814 Search PubMed.
- M. Ohuchi, H. Murakami and H. Suga, Curr. Opin. Chem. Biol., 2007, 11, 537–542 Search PubMed.
- H. Murakami, D. Kourouklis and H. Suga, Chem. Biol., 2003, 10, 1077–1084 Search PubMed.
- H. Murakami, A. Ohta, H. Ashigai and H. Suga, Nat. Methods, 2006, 3, 357–359 Search PubMed.
- T. Kawakami, H. Murakami and H. Suga, J. Am. Chem. Soc., 2008, 130, 16861–16863 Search PubMed.
- J. M. Pelton, J. E. Hochuli, P. W. Sadecki, T. Katoh, H. Suga, L. M. Hicks, E. N. Muratov, A. Tropsha and A. A. Bowers, J. Am. Chem. Soc., 2024, 146, 8016–8030 Search PubMed.
- R. Takatsuji, K. Shinbara, T. Katoh, Y. Goto, T. Passioura, R. Yajima, Y. Komatsu and H. Suga, J. Am. Chem. Soc., 2019, 141, 2279–2287 Search PubMed.
- K. Torikai and H. Suga, J. Am. Chem. Soc., 2014, 136, 17359–17361 Search PubMed.
- R. Laniado-Laborín and M. N. Cabrales-Vargas, Rev. Iberoam. Micol., 2009, 26, 223–227 Search PubMed.
- J. Lee, K. E. Schwieter, A. M. Watkins, D. S. Kim, H. Yu, K. J. Schwarz, J. Lim, J. Coronado, M. Byrom, E. V. Anslyn, A. D. Ellington, J. S. Moore and M. C. Jewett, Nat. Commun., 2019, 10, 5097 Search PubMed.
- G. A. Hudson, A. R. Hooper, A. J. DiCaprio, D. Sarlah and D. A. Mitchell, Org. Lett., 2021, 23, 253–256 Search PubMed.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2016, 138, 15511–15514 Search PubMed.
- W. J. Wever, J. W. Bogart, J. A. Baccile, A. N. Chan, F. C. Schroeder and A. A. Bowers, J. Am. Chem. Soc., 2015, 137, 3494–3497 Search PubMed.
- A. J. Rice, J. M. Pelton, N. J. Kramer, D. S. Catlin, S. K. Nair, T. V. Pogorelov, D. A. Mitchell and A. A. Bowers, J. Am. Chem. Soc., 2022, 144, 21116–21124 Search PubMed.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 Search PubMed.
- A. A. Vinogradov and H. Suga, Cell Chem. Biol., 2020, 27, 1032–1051 Search PubMed.
- K. L. Dunbar, J. O. Melby and D. A. Mitchell, Nat. Chem. Biol., 2012, 8, 569–575 Search PubMed.
- A. A. Vinogradov, Y. Zhang, K. Hamada, S. Kobayashi, K. Ogata, T. Sengoku, Y. Goto and H. Suga, J. Am. Chem. Soc., 2024, 146, 8058–8070 Search PubMed.
- S. Hayashi, T. Ozaki, S. Asamizu, H. Ikeda, S. Ōmura, N. Oku, Y. Igarashi, H. Tomoda and H. Onaka, Chem. Biol., 2014, 21, 679–688 Search PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 Search PubMed.
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 Search PubMed.
- Y. Goto, Y. Ito, Y. Kato, S. Tsunoda and H. Suga, Chem. Biol., 2014, 21, 766–774 Search PubMed.
- T. Ozaki, K. Yamashita, Y. Goto, M. Shimomura, S. Hayashi, S. Asamizu, Y. Sugai, H. Ikeda, H. Suga and H. Onaka, Nat. Commun., 2017, 8, 14207 Search PubMed.
- C. Helmling and C. N. Cunningham, ACS Symposium Series, American Chemical Society, Washington, DC, 2022, pp. 27–53 Search PubMed.
- M. S. Newton, Y. Cabezas-Perusse, C. L. Tong and B. Seelig, ACS Synth. Biol., 2020, 9, 181–190 Search PubMed.
- A. A. Vinogradov, E. Nagai, J. S. Chang, K. Narumi, H. Onaka, Y. Goto and H. Suga, J. Am. Chem. Soc., 2020, 142, 20329–20334 Search PubMed.
- A. A. Vinogradov, M. Shimomura, N. Kano, Y. Goto, H. Onaka and H. Suga, J. Am. Chem. Soc., 2020, 142, 13886–13897 Search PubMed.
- A. A. Vinogradov, M. Nagano, Y. Goto and H. Suga, J. Am. Chem. Soc., 2021, 143, 13358–13369 Search PubMed.
- A. A. Vinogradov, J. S. Chang, H. Onaka, Y. Goto and H. Suga, ACS Cent. Sci., 2022, 8, 814–824 Search PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 Search PubMed.
- S. Mohapatra, N. Hartrampf, M. Poskus, A. Loas, R. Gómez-Bombarelli and B. L. Pentelute, ACS Cent. Sci., 2020, 6, 2277–2286 Search PubMed.
- L. Alzubaidi, J. Zhang, A. J. Humaidi, A. Al-Dujaili, Y. Duan, O. Al-Shamma, J. Santamaría, M. A. Fadhel, M. Al-Amidie and L. Farhan, J. Big Data, 2021, 8, 53 Search PubMed.
- A. A. Vinogradov, Y. Zhang, K. Hamada, J. S. Chang, C. Okada, H. Nishimura, N. Terasaka, Y. Goto, K. Ogata, T. Sengoku, H. Onaka and H. Suga, J. Am. Chem. Soc., 2022, 144, 20332–20341 Search PubMed.
- S. Li, A. Strelow, E. J. Fontana and H. Wesche, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5567–5572 Search PubMed.
- J. S. Chang, A. A. Vinogradov, Y. Zhang, Y. Goto and H. Suga, ACS Cent. Sci., 2023, 9, 2150–2160 Search PubMed.
- F. Fore, C. Indriputri, J. Mamutse and J. Nugraha, Immune Netw., 2020, 20, e21 Search PubMed.
- S. R. Fleming, T. E. Bartges, A. A. Vinogradov, C. L. Kirkpatrick, Y. Goto, H. Suga, L. M. Hicks and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 758–762 Search PubMed.
- P. M. Morrison, P. J. Foley, S. L. Warriner and M. E. Webb, Chem. Commun., 2015, 51, 13470–13473 Search PubMed.
- E. M. Molloy, P. D. Cotter, C. Hill, D. A. Mitchell and R. P. Ross, Nat. Rev. Microbiol., 2011, 9, 670–681 Search PubMed.
- T. Maxson, C. D. Deane, E. M. Molloy, C. L. Cox, A. L. Markley, S. W. Lee and D. A. Mitchell, ACS Chem. Biol., 2015, 10, 1217–1226 Search PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 Search PubMed.
- G. C. A. da Hora, M. Oh, M. C. Mifflin, L. Digal, A. G. Roberts and J. M. J. Swanson, J. Am. Chem. Soc., 2024, 146, 4444–4454 Search PubMed.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 Search PubMed.
- B. Li, J. P. J. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk and S. K. Nair, Science, 2006, 311, 1464–1467 Search PubMed.
- R. H. Perez, T. Zendo and K. Sonomoto, Front. Microbiol., 2018, 9, 2085 Search PubMed.
- C. P. Scott, E. Abel-Santos, M. Wall, D. C. Wahnon and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13638–13643 Search PubMed.
- N. Goosen, R. G. Huinen and P. van de Putte, J. Bacteriol., 1992, 174, 1426–1427 Search PubMed.
- D. H. Haft, BMC Genomics, 2011, 12, 21 Search PubMed.
- W. Zhu and J. P. Klinman, Curr. Opin. Chem. Biol., 2020, 59, 93–103 Search PubMed.
- G. Wang, Y. Zhou, K. Ma, F. Zhang, J. Ye, G. Zhong and X. Yang, Protein Expression Purif., 2021, 178, 105777 Search PubMed.
- N. A. Nguyen, F. N. U. Vidya, N. H. Yennawar, H. Wu, A. C. McShan and V. Agarwal, Nat. Commun., 2024, 15, 1265 Search PubMed.
- D. H. Haft, M. K. Basu and D. A. Mitchell, BMC Biol., 2010, 8, 70 Search PubMed.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H.-G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387–390 Search PubMed.
- T. Hamada, S. Matsunaga, G. Yano and N. Fusetani, J. Am. Chem. Soc., 2005, 127, 110–118 Search PubMed.
- S. Matsuoka, N. Shinohara, T. Takahashi, M. Iida and M. Inoue, Angew. Chem., Int. Ed., 2011, 50, 4879–4883 Search PubMed.
- A. Hayata, H. Itoh and M. Inoue, J. Am. Chem. Soc., 2018, 140, 10602–10611 Search PubMed.
- M. A. Ortega, Y. Hao, Q. Zhang, M. C. Walker, W. A. van der Donk and S. K. Nair, Nature, 2015, 517, 509–512 Search PubMed.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 Search PubMed.
- M. I. McLaughlin, Y. Yu and W. A. van der Donk, ACS Chem. Biol., 2022, 17, 930–940 Search PubMed.
- R. S. Ayikpoe, C. Shi, A. J. Battiste, S. M. Eslami, S. Ramesh, M. A. Simon, I. R. Bothwell, H. Lee, A. J. Rice, H. Ren, Q. Tian, L. A. Harris, R. Sarksian, L. Zhu, A. M. Frerk, T. W. Precord, W. A. van der Donk, D. A. Mitchell and H. Zhao, Nat. Commun., 2022, 13, 6135 Search PubMed.
- A. M. King, Z. Zhang, E. Glassey, P. Siuti, J. Clardy and C. A. Voigt, Nat. Microbiol., 2023, 8, 2420–2434 Search PubMed.
- H. Ren, C. Huang, Y. Pan, S. R. Dommaraju, H. Cui, M. Li, M. G. Gadgil, D. A. Mitchell and H. Zhao, Nat. Chem., 2024, 16, 1320–1329 Search PubMed.
- F. Hubrich, N. M. Bösch, C. Chepkirui, B. I. Morinaka, M. Rust, M. Gugger, S. L. Robinson, A. L. Vagstad and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2113120119 Search PubMed.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898–1916 Search PubMed.
- R. H. Baltz, Curr. Opin. Chem. Biol., 2009, 13, 144–151 Search PubMed.
- T. T. Sword, G. S. K. Abbas and C. B. Bailey, Front. Nat. Prod., 2024, 3, 1353362 Search PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y.-N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 Search PubMed.
- C. Beck, J. F. G. Garzón and T. Weber, Biotechnol. Bioprocess Eng., 2020, 25, 886–894 Search PubMed.
- D. Xu, Z. Zhang, L. Yao, L. Wu, Y. Zhu, M. Zhao and H. Xu, Appl. Microbiol. Biotechnol., 2023, 107, 4187–4197 Search PubMed.
- E. A. Felnagle, E. E. Jackson, Y. A. Chan, A. M. Podevels, A. D. Berti, M. D. McMahon and M. G. Thomas, Mol. Pharm., 2008, 5, 191–211 Search PubMed.
- B. R. Miller and A. M. Gulick, Methods Mol. Biol., 2016, 1401, 3–29 Search PubMed.
- S. L. Wenski, S. Thiengmag and E. J. N. Helfrich, Synth. Syst. Biotechnol., 2022, 7, 631–647 Search PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S.-C. Tsai, Chem. Biol., 2015, 22, 1018–1029 Search PubMed.
- H. Heinemann, H. Zhang and R. J. Cox, Chemistry, 2024, 30, e202302590 Search PubMed.
- C. Khosla and R. J. Zawada, Trends Biotechnol., 1996, 14, 335–341 Search PubMed.
- H. Kries, D. L. Niquille and D. Hilvert, Chem. Biol., 2015, 22, 640–648 Search PubMed.
- H. Sun, Z. Liu, H. Zhao and E. L. Ang, Drug Des., Dev. Ther., 2015, 9, 823–833 Search PubMed.
- J. Zhang, Y.-J. Yan, J. An, S.-X. Huang, X.-J. Wang and W.-S. Xiang, Microb. Cell Fact., 2015, 14, 152 Search PubMed.
- K. A. J. Bozhüyük, L. Präve, C. Kegler, L. Schenk, S. Kaiser, C. Schelhas, Y.-N. Shi, W. Kuttenlochner, M. Schreiber, J. Kandler, M. Alanjary, T. M. Mohiuddin, M. Groll, G. K. A. Hochberg and H. B. Bode, Science, 2024, 383, eadg4320 Search PubMed.
- A. W. Goering, J. Li, R. A. McClure, R. J. Thomson, M. C. Jewett and N. L. Kelleher, ACS Synth. Biol., 2017, 6, 39–44 Search PubMed.
- I. Siebels, S. Nowak, C. S. Heil, P. Tufar, N. S. Cortina, H. B. Bode and M. Grininger, ACS Synth. Biol., 2020, 9, 2418–2426 Search PubMed.
- L. Zhuang, S. Huang, W.-Q. Liu, A. S. Karim, M. C. Jewett and J. Li, Metab. Eng., 2020, 60, 37–44 Search PubMed.
- J. L. N. Dinglasan, T. T. Sword, J. W. Barker, M. J. Doktycz and C. B. Bailey, ACS Synth. Biol., 2023, 12, 1447–1460 Search PubMed.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 Search PubMed.
- S. Gruenewald, H. D. Mootz, P. Stehmeier and T. Stachelhaus, Appl. Environ. Microbiol., 2004, 70, 3282–3291 Search PubMed.
- A. Stanišić, A. Hüsken, P. Stephan, D. L. Niquille, J. Reinstein and H. Kries, ACS Catal., 2021, 11, 8692–8700 Search PubMed.
- A. M. Matter, S. B. Hoot, P. D. Anderson, S. S. Neves and Y.-Q. Cheng, PLoS One, 2009, 4, e7194 Search PubMed.
- Y.-Q. Cheng, ChemBioChem, 2006, 7, 471–477 Search PubMed.
- N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 10698–10699 Search PubMed.
- H. Kries, J. Pept. Sci., 2016, 22, 564–570 Search PubMed.
- J. Li, J. Jaitzig, L. Theuer, O. E. Legala, R. D. Süssmuth and P. Neubauer, J. Biotechnol., 2015, 193, 16–22 Search PubMed.
- M. C. Jewett and J. R. Swartz, Biotechnol. Bioeng., 2004, 86, 19–26 Search PubMed.
- G. B. Hurst, K. G. Asano, C. J. Doktycz, E. J. Consoli, W. L. Doktycz, C. M. Foster, J. L. Morrell-Falvey, R. F. Standaert and M. J. Doktycz, Anal. Chem., 2017, 89, 11443–11451 Search PubMed.
- M. Vilkhovoy, N. Horvath, C.-H. Shih, J. A. Wayman, K. Calhoun, J. Swartz and J. D. Varner, ACS Synth. Biol., 2018, 7, 1844–1857 Search PubMed.
- S. Wick, D. I. Walsh 3rd, J. Bobrow, K. Hamad-Schifferli, D. S. Kong, T. Thorsen, K. Mroszczyk and P. A. Carr, ACS Synth. Biol., 2019, 8, 1010–1025 Search PubMed.
- B. Shen, L. Du, C. Sanchez, D. J. Edwards, M. Chen and J. M. Murrell, J. Nat. Prod., 2002, 65, 422–431 Search PubMed.
- O. W. Choroba, D. H. Williams and J. B. Spencer, J. Am. Chem. Soc., 2000, 122, 5389–5390 Search PubMed.
- A. M. van Wageningen, P. N. Kirkpatrick, D. H. Williams, B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. Jones and P. J. Solenberg, Chem. Biol., 1998, 5, 155–162 Search PubMed.
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105–1118 Search PubMed.
- J.-Q. Gu, K. T. Nguyen, C. Gandhi, V. Rajgarhia, R. H. Baltz, P. Brian and M. Chu, J. Nat. Prod., 2007, 70, 233–240 Search PubMed.
- M. Debono, B. J. Abbott, R. M. Molloy, D. S. Fukuda, A. H. Hunt, V. M. Daupert, F. T. Counter, J. L. Ott, C. B. Carrell and L. C. Howard, J. Antibiot., 1988, 41, 1093–1105 Search PubMed.
- J. Thompson, S. Rae and E. Cundliffe, Mol. Gen. Genet., 1984, 195, 39–43 Search PubMed.
- X. Tian, W.-Q. Liu, H. Xu, X. Ji, Y. Liu and J. Li, Synth. Syst. Biotechnol., 2022, 7, 775–783 Search PubMed.
- X. Ji, W.-Q. Liu and J. Li, Curr. Opin. Microbiol., 2022, 67, 102142 Search PubMed.
- T. T. Sword, J. L. N. Dinglasan, G. S. K. Abbas, J. W. Barker, M. E. Spradley, E. R. Greene, D. S. Gooden, S. J. Emrich, M. A. Gilchrist, M. J. Doktycz and C. B. Bailey, Sci. Rep., 2024, 14, 12983 Search PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 Search PubMed.
- A. M. Hill, Nat. Prod. Rep., 2006, 23, 256–320 Search PubMed.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 Search PubMed.
- J. W. Giraldes, D. L. Akey, J. D. Kittendorf, D. H. Sherman, J. L. Smith and R. A. Fecik, Nat. Chem. Biol., 2006, 2, 531–536 Search PubMed.
- L. Katz, Methods Enzymol., 2009, 459, 113–142 Search PubMed.
- Y. Katsuyama and A. Miyanaga, Curr. Opin. Chem. Biol., 2022, 71, 102223 Search PubMed.
- M. Grininger, Nat. Chem. Biol., 2023, 19, 401–415 Search PubMed.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 Search PubMed.
- H. Chen and L. Du, Appl. Microbiol. Biotechnol., 2016, 100, 541–557 Search PubMed.
- H. Jenke-Kodama, A. Sandmann, R. Müller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 2027–2039 Search PubMed.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 Search PubMed.
- D. Yu, F. Xu, J. Zeng and J. Zhan, IUBMB Life, 2012, 64, 285–295 Search PubMed.
- C. R. Hutchinson, J. Kennedy, C. Park, S. Kendrew, K. Auclair and J. Vederas, Antonie van Leeuwenhoek, 2000, 78, 287–295 Search PubMed.
- J. M. Crawford and C. A. Townsend, Nat. Rev. Microbiol., 2010, 8, 879–889 Search PubMed.
- X.-L. Yang, S. Friedrich, S. Yin, O. Piech, K. Williams, T. J. Simpson and R. J. Cox, Chem. Sci., 2019, 10, 8478–8489 Search PubMed.
- J. P. Torres, Z. Lin, J. M. Winter, P. J. Krug and E. W. Schmidt, Nat. Commun., 2020, 11, 2882 Search PubMed.
- C. M. McBride, E. L. Miller and L. K. Charkoudian, Microb. Genomes, 2023, 9(3), 000965 Search PubMed.
- J. Kim and G.-S. Yi, BMC Microbiol., 2012, 12, 169 Search PubMed.
- A. Chen, R. N. Re and M. D. Burkart, Nat. Prod. Rep., 2018, 35, 1029–1045 Search PubMed.
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 Search PubMed.
- R. Bisht, A. Bhattacharyya, A. Shrivastava and P. Saxena, Front. Plant Sci., 2021, 12, 746908 Search PubMed.
- C. Stewart Jr, C. R. Vickery, M. D. Burkart and J. P. Noel, Curr. Opin. Plant Biol., 2013, 16, 365–372 Search PubMed.
- Y. Katsuyama and Y. Ohnishi, Methods Enzymol., 2012, 515, 359–377 Search PubMed.
- M. B. Austin and J. P. Noel, ChemInform, 2003, 34(17), 284 Search PubMed.
- Y. Shimizu, H. Ogata and S. Goto, ChemBioChem, 2017, 18, 50–65 Search PubMed.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 Search PubMed.
- A. T. Keatinge-Clay, Chem. Rev., 2017, 117, 5334–5366 Search PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 Search PubMed.
- D. Yan and Y. Matsuda, Chem. Sci., 2024, 15, 3011–3017 Search PubMed.
- D. C. Gay, P. J. Spear and A. T. Keatinge-Clay, ACS Chem. Biol., 2014, 9, 2374–2381 Search PubMed.
- G. Berkhan, C. Merten, C. Holec and F. Hahn, Angew. Chem., Int. Ed., 2016, 55, 13589–13592 Search PubMed.
- D. T. Wagner, Z. Zhang, R. A. Meoded, A. J. Cepeda, J. Piel and A. T. Keatinge-Clay, ACS Chem. Biol., 2018, 13, 975–983 Search PubMed.
- G. Berkhan and F. Hahn, Angew. Chem., Int. Ed., 2014, 53, 14240–14244 Search PubMed.
- K. H. Sung, G. Berkhan, T. Hollmann, L. Wagner, W. Blankenfeldt and F. Hahn, Angew. Chem., Int. Ed., 2018, 57, 343–347 Search PubMed.
- Y. Sugimoto, L. Ding, K. Ishida and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 1560–1564 Search PubMed.
- H.-Y. He, H. Yuan, M.-C. Tang and G.-L. Tang, Angew. Chem., Int. Ed., 2014, 53, 11315–11319 Search PubMed.
- L. Ray and B. S. Moore, Nat. Prod. Rep., 2016, 33, 150–161 Search PubMed.
- A. Hagen, S. Poust, T. de Rond, J. L. Fortman, L. Katz, C. J. Petzold and J. D. Keasling, ACS Synth. Biol., 2016, 5, 21–27 Search PubMed.
- S. Yuzawa, C. H. Eng, L. Katz and J. D. Keasling, Biochemistry, 2013, 52, 3791–3793 Search PubMed.
- S. Yuzawa, C. B. Bailey, T. Fujii, R. Jocic, J. F. Barajas, V. T. Benites, E. E. K. Baidoo, Y. Chen, C. J. Petzold, L. Katz and J. D. Keasling, ACS Chem. Biol., 2017, 12, 2725–2729 Search PubMed.
- A. Zargar, R. Lal, L. Valencia, J. Wang, T. W. H. Backman, P. Cruz-Morales, A. Kothari, M. Werts, A. R. Wong, C. B. Bailey, A. Loubat, Y. Liu, Y. Chen, S. Chang, V. T. Benites, A. C. Hernández, J. F. Barajas, M. G. Thompson, C. Barcelos, R. Anayah, H. G. Martin, A. Mukhopadhyay, C. J. Petzold, E. E. K. Baidoo, L. Katz and J. D. Keasling, J. Am. Chem. Soc., 2020, 142, 9896–9901 Search PubMed.
- S. Yuzawa, M. Mirsiaghi, R. Jocic, T. Fujii, F. Masson, V. T. Benites, E. E. K. Baidoo, E. Sundstrom, D. Tanjore, T. R. Pray, A. George, R. W. Davis, J. M. Gladden, B. A. Simmons, L. Katz and J. D. Keasling, Nat. Commun., 2018, 9, 4569 Search PubMed.
- A. Zargar, L. Valencia, J. Wang, R. Lal, S. Chang, M. Werts, A. R. Wong, A. C. Hernández, V. Benites, E. E. K. Baidoo, L. Katz and J. D. Keasling, Metab. Eng., 2020, 61, 389–396 Search PubMed.
- M. F. J. Mabesoone, S. Leopold-Messer, H. A. Minas, C. Chepkirui, P. Chawengrum, S. Reiter, R. A. Meoded, S. Wolf, F. Genz, N. Magnus, B. Piechulla, A. S. Walker and J. Piel, Science, 2024, 383, 1312–1317 Search PubMed.
- K. A. Ray, J. D. Lutgens, R. Bista, J. Zhang, R. R. Desai, M. Hirsch, T. Miyazawa, A. Cordova and A. T. Keatinge-Clay, Nat. Commun., 2024, 15, 6585 Search PubMed.
- T. Miyazawa, M. Hirsch, Z. Zhang and A. T. Keatinge-Clay, Nat. Commun., 2020, 11, 80 Search PubMed.
- S. Yuzawa, J. D. Keasling and L. Katz, J. Antibiot., 2016, 69, 494–499 Search PubMed.
- A. A. Nava, J. Roberts, R. W. Haushalter, Z. Wang and J. D. Keasling, ACS Synth. Biol., 2023, 12, 3148–3155 Search PubMed.
- B. Pang, L. E. Valencia, J. Wang, Y. Wan, R. Lal, A. Zargar and J. D. Keasling, Biotechnol. Bioprocess Eng., 2019, 24, 413–423 Search PubMed.
- R. Pieper, G. Luo, D. E. Cane and C. Khosla, Nature, 1995, 378, 263–266 Search PubMed.
- A. D. Harper, C. B. Bailey, A. D. Edwards, J. F. Detelich and A. T. Keatinge-Clay, ChemBioChem, 2012, 13, 2200–2203 Search PubMed.
- A. J. Hughes, J. F. Detelich and A. T. Keatinge-Clay, MedChemComm, 2012, 3, 956 Search PubMed.
- K. P. Yuet, C. W. Liu, S. R. Lynch, J. Kuo, W. Michaels, R. B. Lee, A. E. McShane, B. L. Zhong, C. R. Fischer and C. Khosla, J. Am. Chem. Soc., 2020, 142, 5952–5957 Search PubMed.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 Search PubMed.
- J. Yan, C. Hazzard, S. A. Bonnett and K. A. Reynolds, Biochemistry, 2012, 51, 9333–9341 Search PubMed.
- M. Oliynyk, M. J. Brown, J. Cortés, J. Staunton and P. F. Leadlay, Chem. Biol., 1996, 3, 833–839 Search PubMed.
- L. Kellenberger, I. S. Galloway, G. Sauter, G. Böhm, U. Hanefeld, J. Cortés, J. Staunton and P. F. Leadlay, ChemBioChem, 2008, 9, 2740–2749 Search PubMed.
- A. Ranganathan, M. Timoney, M. Bycroft, J. Cortés, I. P. Thomas, B. Wilkinson, L. Kellenberger, U. Hanefeld, I. S. Galloway, J. Staunton and P. F. Leadlay, Chem. Biol., 1999, 6, 731–741 Search PubMed.
- J. Li, H. Wang and M. C. Jewett, Biochem. Eng. J., 2018, 130, 29–33 Search PubMed.
- J. G. Klein, Y. Wu, B. Kokona and L. K. Charkoudian, Bioorg. Med. Chem., 2020, 28, 115686 Search PubMed.
- M. E. Hillenmeyer, G. A. Vandova, E. E. Berlew and L. K. Charkoudian, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13952–13957 Search PubMed.
- X. Liu, K. Hua, D. Liu, Z.-L. Wu, Y. Wang, H. Zhang, Z. Deng, B. A. Pfeifer and M. Jiang, ACS Chem. Biol., 2020, 15, 1177–1183 Search PubMed.
- B. S. Moore and J. Piel, Antonie van Leeuwenhoek, 2000, 78, 391–398 Search PubMed.
- X. Yan, K. Probst, A. Linnenbrink, M. Arnold, T. Paululat, A. Zeeck and A. Bechthold, ChemBioChem, 2012, 13, 224–230 Search PubMed.
- W. Zhang, K. Watanabe, X. Cai, M. E. Jung, Y. Tang and J. Zhan, J. Am. Chem. Soc., 2008, 130, 6068–6069 Search PubMed.
- L. E. Day, J. Bacteriol., 1966, 91, 1917–1923 Search PubMed.
- S. J. Moore, T. Tosi, D. Bell, Y. B. Hleba, K. M. Polizzi and P. S. Freemont, Synth. Biol., 2021, 6(1), ysab021 Search PubMed.
- S. J. Moore, Y. B. Hleba, S. Bischoff, D. Bell, K. M. Polizzi and P. S. Freemont, Microb. Cell Fact., 2021, 20, 116 Search PubMed.
- T. P. Korman, P. H. Opgenorth and J. U. Bowie, Nat. Commun., 2017, 8, 15526 Search PubMed.
- J. Misa, J. M. Billingsley, K. Niwa, R. K. Yu and Y. Tang, ACS Synth. Biol., 2022, 11, 1639–1649 Search PubMed.
- A. R. Mahoney, M. M. Safaee, W. M. Wuest and A. L. Furst, iScience, 2021, 24, 102304 Search PubMed.
- M. E. A. de Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184 Search PubMed.
- S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415–12474 Search PubMed.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 Search PubMed.
- S. J. Projan, Curr. Opin. Pharmacol., 2002, 2, 513–522 Search PubMed.
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 Search PubMed.
- J. J. Rahal, Clin. Infect. Dis., 2009, 49(Suppl 1), S4–S10 Search PubMed.
- H. S. Fraimow and C. Tsigrelis, Crit. Care Clin., 2011, 27, 163–205 Search PubMed.
- H. I. Zgurskaya and V. V. Rybenkov, Ann. N. Y. Acad. Sci., 2020, 1459, 5–18 Search PubMed.
- R. Tommasi, D. G. Brown, G. K. Walkup, J. I. Manchester and A. A. Miller, Nat. Rev. Drug Discovery, 2015, 14, 529–542 Search PubMed.
- N. G. Housden, M. N. Webby, E. D. Lowe, T. J. El-Baba, R. Kaminska, C. Redfield, C. V. Robinson and C. Kleanthous, Nat. Commun., 2021, 12, 4625 Search PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 Search PubMed.
- C. Schmidt-Dannert and F. H. Arnold, Trends Biotechnol., 1999, 17, 135–136 Search PubMed.
- S. A. Waksman, A. Schatz and D. M. Reynolds, Ann. N. Y. Acad. Sci., 2010, 1213, 112–124 Search PubMed.
- Z. A. Khan, M. F. Siddiqui and S. Park, Diagnostics, 2019, 9, 49 Search PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 Search PubMed.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29–40 Search PubMed.
- R. H. Flatman, A. J. Howells, L. Heide, H.-P. Fiedler and A. Maxwell, Antimicrob. Agents Chemother., 2005, 49, 1093–1100 Search PubMed.
- M. J. Buttner, M. Schäfer, D. M. Lawson and A. Maxwell, FEMS Microbiol. Rev., 2018, 42(1), fux005 Search PubMed.
- K.-J. Choi, Y. G. Yu, H. G. Hahn, J.-D. Choi and M.-Y. Yoon, FEBS Lett., 2005, 579, 4903–4910 Search PubMed.
- F. Arhin, O. Bélanger, S. Ciblat, M. Dehbi, D. Delorme, E. Dietrich, D. Dixit, Y. Lafontaine, D. Lehoux, J. Liu, G. A. McKay, G. Moeck, R. Reddy, Y. Rose, R. Srikumar, K. S. E. Tanaka, D. M. Williams, P. Gros, J. Pelletier, T. R. Parr Jr and A. R. Far, Bioorg. Med. Chem., 2006, 14, 5812–5832 Search PubMed.
- K. A. Calhoun and J. R. Swartz, Methods Mol. Biol., 2007, 375, 3–17 Search PubMed.
- C. U. Chukwudi, Antimicrob. Agents Chemother., 2016, 60, 4433–4441 Search PubMed.
- Y. Zhou, V. E. Gregor, Z. Sun, B. K. Ayida, G. C. Winters, D. Murphy, K. B. Simonsen, D. Vourloumis, S. Fish, J. M. Froelich, D. Wall and T. Hermann, Antimicrob. Agents Chemother., 2005, 49, 4942–4949 Search PubMed.
- F. Schluenzen, C. Takemoto, D. N. Wilson, T. Kaminishi, J. M. Harms, K. Hanawa-Suetsugu, W. Szaflarski, M. Kawazoe, M. Shirouzu, K. H. Nierhaus, S. Yokoyama and P. Fucini, Nat. Struct. Mol. Biol., 2006, 13, 871–878 Search PubMed.
- P. Vannuffel and C. Cocito, Drugs, 1996, 51(Suppl 1), 20–30 Search PubMed.
- B. Bozdogan and P. C. Appelbaum, Int. J. Antimicrob. Agents, 2004, 23, 113–119 Search PubMed.
- W. Wehrli, Rev. Infect. Dis., 1983, 5(Suppl 3), S407–S411 Search PubMed.
- K. Drlica, Curr. Opin. Microbiol., 1999, 2, 504–508 Search PubMed.
- K. Drlica, H. Hiasa, R. Kerns, M. Malik, A. Mustaev and X. Zhao, Curr. Top. Med. Chem., 2009, 9, 981–998 Search PubMed.
- K. Drlica, M. Malik, R. J. Kerns and X. Zhao, Antimicrob. Agents Chemother., 2008, 52, 385–392 Search PubMed.
- A. S. Verkman, Am. J. Physiol.: Cell Physiol., 2004, 286, C465–74 Search PubMed.
- H. Yamaguchi, C. Yamamoto and N. Tanaka, J. Biochem., 1965, 57, 667–677 Search PubMed.
- H. L. Ennis, Antimicrob. Agents Chemother., 1972, 1, 204–209 Search PubMed.
- S. Perzynski, M. Cannon, E. Cundliffe, S. B. Chahwala and J. Davies, Eur. J. Biochem., 1979, 99, 623–628 Search PubMed.
- B. Rasmussen, H. F. Noller, G. Daubresse, B. Oliva, Z. Misulovin, D. M. Rothstein, G. A. Ellestad, Y. Gluzman, F. P. Tally and I. Chopra, Antimicrob. Agents Chemother., 1991, 35, 2306–2311 Search PubMed.
- N. Böddeker, G. Bahador, C. Gibbs, E. Mabery, J. Wolf, L. Xu and J. Watson, RNA, 2002, 8, 1120–1128 Search PubMed.
- K. Kojima, M. Oshita, Y. Nanjo, K. Kasai, Y. Tozawa, H. Hayashi and Y. Nishiyama, Mol. Microbiol., 2007, 65, 936–947 Search PubMed.
- M. Weidlich, C. Klammt, F. Bernhard, M. Karas and T. Stein, Lett. Appl. Microbiol., 2008, 46, 155–159 Search PubMed.
- M. Taniguchi, A. Ochiai, S. Fukuda, T. Sato, E. Saitoh, T. Kato and T. Tanaka, J. Biosci. Bioeng., 2016, 122, 385–392 Search PubMed.
- M. Taniguchi, A. Ochiai, H. Kondo, S. Fukuda, Y. Ishiyama, E. Saitoh, T. Kato and T. Tanaka, J. Biosci. Bioeng., 2016, 121, 591–598 Search PubMed.
- A. Sonousi, J. C. K. Quirke, P. Waduge, T. Janusic, M. Gysin, K. Haldimann, S. Xu, S. N. Hobbie, S.-H. Sha, J. Schacht, C. S. Chow, A. Vasella, E. C. Böttger and D. Crich, ChemMedChem, 2021, 16, 335–339 Search PubMed.
- L. Brandi, A. Lazzarini, L. Cavaletti, M. Abbondi, E. Corti, I. Ciciliato, L. Gastaldo, A. Marazzi, M. Feroggio, A. Fabbretti, A. Maio, L. Colombo, S. Donadio, F. Marinelli, D. Losi, C. O. Gualerzi and E. Selva, Biochemistry, 2006, 45, 3692–3702 Search PubMed.
- T. M. Binz, S. I. Maffioli, M. Sosio, S. Donadio and R. Müller, J. Biol. Chem., 2010, 285, 32710–32719 Search PubMed.
- L. Brandi, A. Fabbretti, M. Di Stefano, A. Lazzarini, M. Abbondi and C. O. Gualerzi, RNA, 2006, 12, 1262–1270 Search PubMed.
- A. Maio, L. Brandi, S. Donadio and C. O. Gualerzi, Antibiotics, 2016, 5, 17 Search PubMed.
- C. R. Zwick 3rd, M. B. Sosa and H. Renata, J. Am. Chem. Soc., 2021, 143, 1673–1679 Search PubMed.
- S. M. M. Schuler, G. Jürjens, A. Marker, U. Hemmann, A. Rey, S. Yvon, M. Lagrevol, M. Hamiti, F. Nguyen, R. Hirsch, C. Pöverlein, A. Vilcinskas, P. Hammann, D. N. Wilson, M. Mourez, S. Coyne and A. Bauer, Microbiol. Spectrum, 2023, 11, e0224722 Search PubMed.
- J. P. López-Alonso, A. Fabbretti, T. Kaminishi, I. Iturrioz, L. Brandi, D. Gil-Carton, C. O. Gualerzi, P. Fucini and S. R. Connell, Nucleic Acids Res., 2017, 45, 2179–2187 Search PubMed.
- S. I. Maffioli, A. Fabbretti, L. Brandi, A. Savelsbergh, P. Monciardini, M. Abbondi, R. Rossi, S. Donadio and C. O. Gualerzi, ACS Chem. Biol., 2013, 8, 1939–1946 Search PubMed.
- E. S. Komarova Andreyanova, I. A. Osterman, P. I. Pletnev, Y. A. Ivanenkov, A. G. Majouga, A. A. Bogdanov and P. V. Sergiev, Biochimie, 2017, 133, 45–55 Search PubMed.
- D. Y. Travin, R. Jouan, A. Vigouroux, S. Inaba-Inoue, J. Lachat, F. Haq, T. Timchenko, D. Sutormin, S. Dubiley, K. Beis, S. Moréra, K. Severinov and P. Mergaert, mBio, 2023, 14, e0021723 Search PubMed.
- R. W. Murray, E. P. Melchior, J. C. Hagadorn and K. R. Marotti, Antimicrob. Agents Chemother., 2001, 45, 1900–1904 Search PubMed.
- E. Esquenazi, A. C. Jones, T. Byrum, P. C. Dorrestein and W. H. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5226–5231 Search PubMed.
- J. A. van Santen, G. Jacob, A. L. Singh, V. Aniebok, M. J. Balunas, D. Bunsko, F. C. Neto, L. Castaño-Espriu, C. Chang, T. N. Clark, J. L. Cleary Little, D. A. Delgadillo, P. C. Dorrestein, K. R. Duncan, J. M. Egan, M. M. Galey, F. P. J. Haeckl, A. Hua, A. H. Hughes, D. Iskakova, A. Khadilkar, J.-H. Lee, S. Lee, N. LeGrow, D. Y. Liu, J. M. Macho, C. S. McCaughey, M. H. Medema, R. P. Neupane, T. J. O’Donnell, J. S. Paula, L. M. Sanchez, A. F. Shaikh, S. Soldatou, B. R. Terlouw, T. A. Tran, M. Valentine, J. J. J. van der Hooft, D. A. Vo, M. Wang, D. Wilson, K. E. Zink and R. G. Linington, ACS Cent. Sci., 2019, 5, 1824–1833 Search PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 Search PubMed.
- V. Bortolaia, R. S. Kaas, E. Ruppe, M. C. Roberts, S. Schwarz, V. Cattoir, A. Philippon, R. L. Allesoe, A. R. Rebelo, A. F. Florensa, L. Fagelhauer, T. Chakraborty, B. Neumann, G. Werner, J. K. Bender, K. Stingl, M. Nguyen, J. Coppens, B. B. Xavier, S. Malhotra-Kumar, H. Westh, M. Pinholt, M. F. Anjum, N. A. Duggett, I. Kempf, S. Nykäsenoja, S. Olkkola, K. Wieczorek, A. Amaro, L. Clemente, J. Mossong, S. Losch, C. Ragimbeau, O. Lund and F. M. Aarestrup, J. Antimicrob. Chemother., 2020, 75, 3491–3500 Search PubMed.
- E. Zankari, H. Hasman, S. Cosentino, M. Vestergaard, S. Rasmussen, O. Lund, F. M. Aarestrup and M. V. Larsen, J. Antimicrob. Chemother., 2012, 67, 2640–2644 Search PubMed.
- G. D. Wright, Nat. Rev. Microbiol., 2007, 5, 175–186 Search PubMed.
- N. J. E. Waller, C.-Y. Cheung, G. M. Cook and M. B. McNeil, Nat. Commun., 2023, 14, 1517 Search PubMed.
- C. Pál, B. Papp and V. Lázár, Trends Microbiol., 2015, 23, 401–407 Search PubMed.
- F. Baquero, J. L. Martínez, Â. Novais, J. Rodríguez-Beltrán, L. Martínez-García, T. M. Coque and J. C. Galán, Front. Microbiol., 2021, 12, 757833 Search PubMed.
- B. Zhao, J. C. Sedlak, R. Srinivas, P. Creixell, J. R. Pritchard, B. Tidor, D. A. Lauffenburger and M. T. Hemann, Cell, 2016, 165, 234–246 Search PubMed.
- A. I. Cocozaki, R. B. Altman, J. Huang, E. T. Buurman, S. L. Kazmirski, P. Doig, D. B. Prince, S. C. Blanchard, J. H. D. Cate and A. D. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8188–8193 Search PubMed.
- M. J. Calcutt and E. Cundliffe, Microbiology, 1989, 135, 1071–1081 Search PubMed.
- T. Tenson, A. DeBlasio and A. Mankin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5641–5646 Search PubMed.
- N. Chumpolkulwong, C. Hori-Takemoto, T. Hosaka, T. Inaoka, T. Kigawa, M. Shirouzu, K. Ochi and S. Yokoyama, Eur. J. Biochem., 2004, 271, 1127–1134 Search PubMed.
- D. Synetos, C. P. Frantziou and L. E. Alksne, Biochim. Biophys. Acta, 1996, 1309, 156–166 Search PubMed.
- M. J. Hammerling, B. R. Fritz, D. J. Yoesep, D. S. Kim, E. D. Carlson and M. C. Jewett, Nat. Commun., 2020, 11, 1108 Search PubMed.
- M. Iglesias and J. P. G. Ballesta, Eur. J. Biochem., 1994, 223, 447–453 Search PubMed.
- W. C. Yang, K. G. Patel, H. E. Wong and J. R. Swartz, Biotechnol. Prog., 2012, 28, 413–420 Search PubMed.
- A. S. Thiara and E. Cundliffe, EMBO J., 1988, 7, 2255–2259 Search PubMed.
- M. Zalacain, F. Malpartida, D. Pulido and A. Jimenez, Eur. J. Biochem., 1987, 162, 413–418 Search PubMed.
- A. Huang, P. Q. Nguyen, J. C. Stark, M. K. Takahashi, N. Donghia, T. Ferrante, A. J. Dy, K. J. Hsu, R. S. Dubner, K. Pardee, M. C. Jewett and J. J. Collins, Sci. Adv., 2018, 4, eaat5105 Search PubMed.
- F. E. Liew, R. Nogle, T. Abdalla, B. J. Rasor, C. Canter, R. O. Jensen, L. Wang, J. Strutz, P. Chirania, S. De Tissera, A. P. Mueller, Z. Ruan, A. Gao, L. Tran, N. L. Engle, J. C. Bromley, J. Daniell, R. Conrado, T. J. Tschaplinski, R. J. Giannone, R. L. Hettich, A. S. Karim, S. D. Simpson, S. D. Brown, C. Leang, M. C. Jewett and M. Köpke, Nat. Biotechnol., 2022, 40, 335–344 Search PubMed.
- A. S. Karim, Q. M. Dudley, A. Juminaga, Y. Yuan, S. A. Crowe, J. T. Heggestad, S. Garg, T. Abdalla, W. S. Grubbe, B. J. Rasor, D. N. Coar, M. Torculas, M. Krein, F. E. Liew, A. Quattlebaum, R. O. Jensen, J. A. Stuart, S. D. Simpson, M. Köpke and M. C. Jewett, Nat. Chem. Biol., 2020, 16, 912–919 Search PubMed.
- Nature's toolbox, inc. (NTx), https://ntxbio.com/, (accessed July 4, 2024).
- Homepage, https://vaxcyte.com/, (accessed July 4, 2024).
- Sutro Biopharma, Inc. is a public biotechnology company focused on clinical stage drug discovery, development and manufacturing company, https://www.sutrobio.com/, (accessed July 4, 2024).
- Biopharmaceutical manufacturing company, https://resilience.com/explore/biologics/, (accessed July 4, 2024).
- Home, https://fabricnano.com/, (accessed July 4, 2024).
- M. Buntru, N. Hahnengress, A. Croon and S. Schillberg, Front. Plant Sci., 2021, 12, 794999 Search PubMed.
- M. D. Gupta, Y. Flaskamp, R. Roentgen, H. Juergens, J. Armero-Gimenez, F. Albrecht, J. Hemmerich, Z. A. Arfi, J. Neuser, H. Spiegel, S. Schillberg, A. Yeliseev, L. Song, J. Qiu, C. Williams and R. Finnern, Biotechnol. Bioeng., 2023, 120, 2890–2906 Search PubMed.
- Debut, https://www.debutbiotech.com/, (accessed July 4, 2024).
- Protein synthesis on-demand breaking the bounds of biology, https://www.tierrabiosciences.com/, (accessed July 4, 2024).
- K. J. Weissman, Nat. Chem. Biol., 2015, 11, 660–670 Search PubMed.
- A. M. Soohoo, D. P. Cogan, K. L. Brodsky and C. Khosla, Annu. Rev. Biochem., 2024, 93, 471–498 Search PubMed.
- G. D. Cook and N. M. Stasulli, SLAS Technol., 2024, 29, 100120 Search PubMed.
- H. Li, W. Ding and Q. Zhang, RSC Chem. Biol., 2024, 5, 90–108 Search PubMed.
- P.-H. Wang, S. Nishikawa, S. E. McGlynn and K. Fujishima, ACS Synth. Biol., 2023, 12, 2887–2896 Search PubMed.
- K. W. George, J. Alonso-Gutierrez, J. D. Keasling and T. S. Lee, Adv. Biochem. Eng. Biotechnol., 2015, 148, 355–389 Search PubMed.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |