
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the mechanism of 3d transition-metal-catalyzed directed C(sp3)–H bond functionalization reactions
Andrés
García-Viada
 a,
Juan C.
Carretero
abc,
Javier
Adrio
*abc and
Nuria
Rodríguez
*abc
a,
Juan C.
Carretero
abc,
Javier
Adrio
*abc and
Nuria
Rodríguez
*abc
aDpto. de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid (UAM), Cantoblanco, 28049, Madrid, Spain. E-mail: javier.adrio@uam.es; n.rodriguez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), UAM, 28049 Madrid, Spain
cCenter for Innovation in Advanced Chemistry (ORFEO-CINQA), Madrid, Spain
First published on 20th March 2025
Abstract
The growing interest in the catalytic activity of earth-abundant 3d transition-metals has led to the development of new and more sustainable methods for C–H bond functionalization reactions. However, this is an emerging field which involves considerable mechanistic complexity as the mode of action of 3d transition metals differs markedly from the well-studied mechanisms of precious metals. In this review, we present an overview of the research efforts in Ni-, Cu-, Fe- and Co-catalyzed directed C(sp3)–H bond functionalization reactions, covering design principles and mechanistic discussions, along with potential applications and limitations. To conclude, the unresolved challenges and future viewpoints are highlighted. We aspire for this review to serve as a relevant and valuable reference for researchers in this swiftly progressing field, helping to inspire the development of more original and innovative strategies.
 Andrés García-Viada | Andrés García-Viada obtained his BSc in Chemistry in 2019 from Universidad Autónoma de Madrid and his MSc in 2020 at the same university. He is currently doing his PhD at the Universidad Autónoma de Madrid under the supervision of Prof. Juan Carlos Carretero and Prof. Nuria Rodríguez. His research focuses on the selective functionalization of aliphatic C–H bonds using directing groups as structural tool for the control of the selectivity. |
 Juan C. Carretero | Juan Carlos Carretero received his PhD from the Universidad Autónoma de Madrid (UAM) in 1985 with Prof. José L. García Ruano. After postdoctoral studies at the Université Catholique de Louvain (Belgium) with Prof. Léon Ghosez, he joined the Department of Organic Chemistry of the UAM and became Associate Professor in 1988 and Professor of Organic Chemistry in 2000. His research interests are focused on developing new methodologies in asymmetric catalysis, C–H functionalization and stereocontrolled metal-catalyzed processes. |
 Javier Adrio | Javier Adrio completed his PhD in Organic Chemistry at the Universidad Autónoma de Madrid under the supervision of Prof. Juan Carlos Carretero. From 2001 to 2003 he worked as a senior scientist on PharmaMar. During this period, he also pursued postdoctoral studies at the University of Pennsylvania under the supervision of Professor Madeleine Joullié. He returned to UAM as a Ramón y Cajal researcher, a position he held until his appointment as Associate Professor in 2010 and subsequently as Professor in 2022. Throughout his career, he has undertaken several short research stays as a Visiting Scholar at the University of Pennsylvania, collaborating with Professor Patrick Walsh's lab. His research interest focus on metal-catalyzed reactions, C–H functionalization processes and catalytic asymmetric methodologies. |
 Nuria Rodríguez | Nuria Rodríguez was born in 1978 in Valencia (Spain). She received her D. Chem. in 2006 from the Department of Organic Chemistry at the University of Valencia (Spain) under the guidance of Professors G. Asensio and M. Medio-Simón. After spending about five years with Professor L. J. Gooßen at the Technische University of Kaiserslautern (Germany), in 2011 she took up a Ramón y Cajal researcher position at the University of Madrid (Spain), where she remains active as Associate Professor. Her major research goals now are the direct and selective functionalization of inert carbon–hydrogen bonds. |
1. Introduction
Transition-metal-catalyzed C–H bond functionalization has changed the mindset about the “inactivity” of these bonds, opening new retrosynthetic pathways that sidestep the need for pre-activated compounds.1–4 This breakthrough minimizes synthetic steps and reduces the production of undesired by-products in total synthesis.5–10 However, attaining site selectivity for the target C–H bond among numerous others in the substrate remains a significant challenge in C–H functionalization.11–25 Often, this problem is solved using directing groups (DGs). A DG is a basic functional group able to coordinate the metal catalyst, bringing it in proximity to the desired C–H bond, facilitating its regioselective activation and subsequent functionalization.26–35Directed C(sp3)–H functionalization is notably challenging due to the greater conformational flexibility and less favourable orbital interactions between the σ* orbitals of the C–H bonds and the metal's d orbitals, in contrast to the more favourable interactions observed with sp2 centers.36–43 However, direct functionalization of sp3 centers is of great interest in many areas, including the increase in the three-dimensional character of pharmacological compounds to improve their selectivity and efficiency.8–10,44–47
To date, this is an area in which catalysis with noble metal-based complexes has dominated, with palladium standing out among them mainly because of its low activation barrier during the change of redox state and its compatibility with many different ligands and oxidants.48–55 In the past decade, however, chemists have pursued sustainable methods for C(sp3)–H bond functionalization using abundant and cost-effective 3d transition metal catalysts (Scheme 1).29,40,56–60 This leap to the use of 3d transition-metals holds considerable mechanistic complexity, as the mode of action of 3d transition metals may differ from the well-studied mechanisms of precious metals. Therefore, innovation in this arena goes hand in hand with progress in the analysis and understanding of the mechanistic peculiarities that govern these processes.
 | ||
| Scheme 1 3d TM-catalyzed directed C(sp3)–H bond functionalization reactions. | ||
Recently published reviews have addressed specific aspects of the evolution of 3d transition-metal-catalyzed directed C(sp3)–H bond functionalization reactions.40,43,61–64 While these reports offer valuable insights, they do not provide broad mechanistic discussions. Covering this aspect, this article aims to present a thorough review of these methodologies, encompassing their scope, limitations and digging into the reaction mechanisms by highlighting key catalytic systems and integrating mechanistic insights. For example, a description of the oxidation states of the metal centers in the intermediate species, the role of additives and/or co-catalysts, isolated intermediates, deuterium scrambling experiments, KIE studies, and DFT calculations.
The content has been organized according to the metal employed (Ni, Cu, Fe and Co) and the nature of the bond formed (C–C, C–N, C–O and C–S). Although significant advances have been made in Mn catalysis,65–67 progress in Mn-catalyzed functionalization of inert C(sp3)–H bonds is beyond the scope of this review of directed transformations.
By understanding these recent advances and underlying principles, we hope that this review will be handy for researchers to further expand the potential of organic synthesis, ultimately contributing to the development of novel, sustainable, and efficient 3d-transition metal-catalyzed synthetic methodologies.
2. Ni-catalysis
Nickel, a group 10 metal located just above palladium, represents one of the most prominent alternatives for replacing noble metals.68 Inherent advantages of nickel-catalysis include a wide range of catalytically active oxidation states (ranging from 0 to +4), high abundance in the earth's crust and comparatively low cost.Its ability to activate C–H bonds was first demonstrated in the 1960s by Dubeck et al. in their study of the cyclometalation of azobenzene with Cp2Ni.69 However, it was not until the early 2000s that nickel-catalyzed chelation-assisted C–H functionalization began to emerge as a prominent alternative, especially with regard to C(sp2)–H bond functionalization.64,70–73 In contrast, the functionalization of unactivated aliphatic C–H bonds remains in its early stages. The following sections highlight key contributions, covering mechanistic discussions and potential applications.63
2.1. C–C bond formation
 | ||
| Scheme 2 β-C(sp3)–H arylation of amides with iodoarenes. | ||
Parallel to this work, You et al. published a similar reactivity using aryl iodides and bromides as coupling partners (Scheme 3).75 In this case, Ni(OTf)2 was also used as catalyst precursor, along with pivalic acid (PivOH) as additive, Na2CO3 as base and PPh3 as ligand, in DMSO/1,4-dioxane solvent mixture at 160 °C for 36 h. Remarkably, this methodology was suitable for the functionalization of both α-quaternary and α-tertiary propanamides. The system was selective for the β-CH3 functionalization, maintaining this selectivity even in the case of 2-methyl-3-phenylacrylamide derivative.
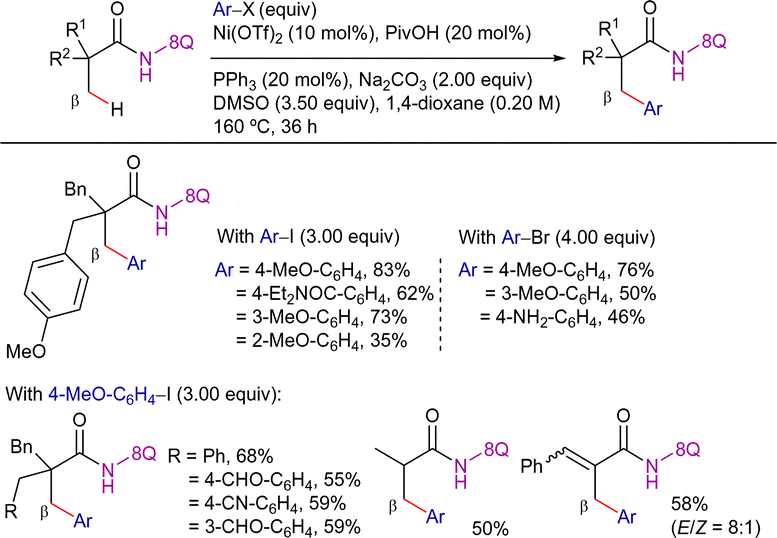 | ||
| Scheme 3 Ni-cat. β-C(sp3)–H arylation of aliphatic amides with aryl halides. | ||
Regarding the reaction mechanism, Chatani et al. conducted deuterium-labeling experiments and observed H/D exchange in both the product and the recovered amide (Scheme 4). Based on these findings, the C–H bond cleavage is presumably reversible and occurs rapidly, preceding the addition of the aryl halide. Furthermore, the addition of 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO) did not inhibit the reaction, suggesting that a single-electron transfer (SET) mechanism is not involved. On this basis, Chatani proposed the NiII/NiIV catalytic cycle depicted in Scheme 4. The process starts with the N,N-coordination of the amide derivative to the nickel(II)-catalyst followed by the reversible C(sp3)–H bond cleavage via a base-assisted concerted metalation-deprotonation mechanism (CMD), forming the nickel(II)-cyclometalated intermediate II. Subsequently, oxidative addition of the aryl halide leads to the highly oxidized nickel(IV)-complex III, from which the β-arylated product is formed through reductive elimination and protodemetalation, closing thus the catalytic cycle. This mechanistic pathway closely resembles those proposed for heavier metal catalysts.50,76
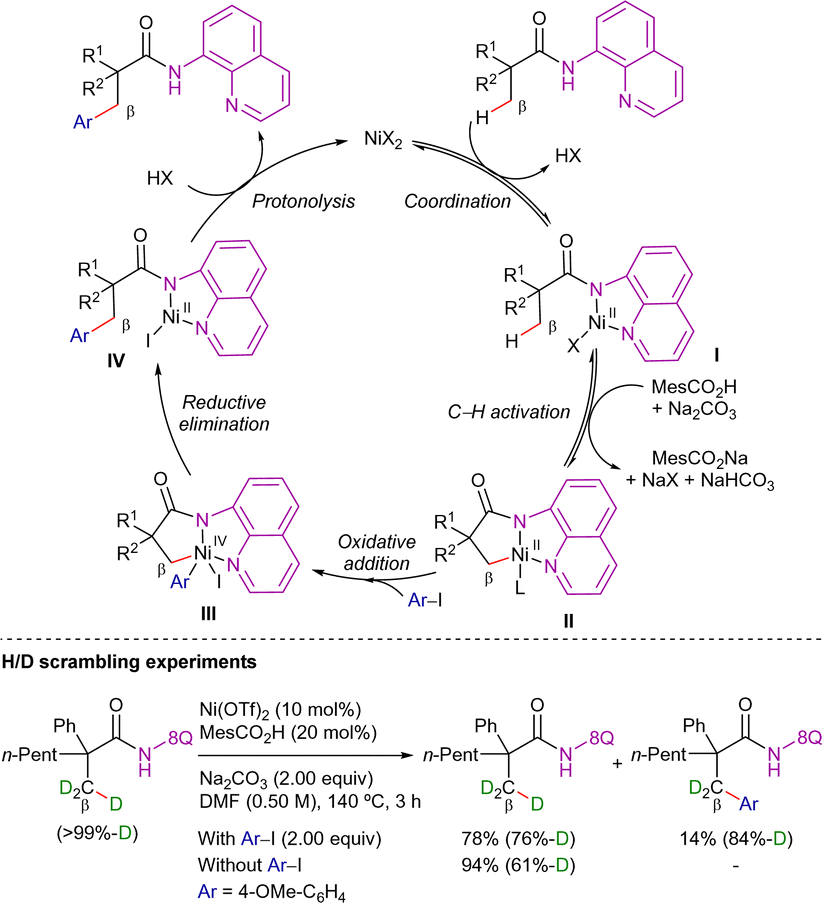 | ||
| Scheme 4 β-C(sp3)–H arylation with aryl halides. Mechanistic hypothesis. | ||
Moreover, this pathway was further supported by the computational studies conducted by Liu and Sunoj.77,78 Both authors discarded a radical pathway involving NiIII due to the inherent instability of phenyl radicals, highlighting certain distinctions from Pd chemistry. According to their computational studies, the formation of the nickelacycle II seems to be thermodynamically less favourable than the corresponding C–H metalation process with a palladium(II)-catalyst (with a difference of 1.6 kcal mol−1). The optimized transition state geometries suggest a later transition state in the metalation step with the nickel catalyst compared to palladium, which aligns with the predictions of the Hammond postulate. Furthermore, while the C–H metalation step with the palladium catalyst is most likely the r.d.s. step, in nickel arylation, the C–H metalation step is reversible and the subsequent oxidative addition step with the aryliodide is the r.d.s. Nonetheless, the barrier of the oxidative addition (∼14.3 kcal mol−1 for PhI) did not justify experimentally requiring high temperatures (140–160 °C).
Shedding light on this point, mechanistic studies reported by Johnson et al.79 showed that the intermediates prior to C–H activation and after functionalization were not diamagnetic but paramagnetic nickel(II)-species. Moreover, both the C–H activation and oxidative addition steps occurred stoichiometrically at 80 °C, a temperature much lower than the temperatures required for catalysis. On this basis, the authors postulated that under the original catalytic conditions employing Na2CO3 as a base, the deprotonation and binding of the substrate to the nickel-center was possibly the r.d.s. due to the insufficient basicity of Na2CO3. In fact, using NatBuO as base, the reaction occurred at only 100 °C rather than 140–160 °C required when Na2CO3 was used. Nevertheless, catalysis proceeded smoothly at higher temperatures, and the use of 10 equivalents of NatBuO resulted in a 94% conversion to the triarylated product (Scheme 5).
 | ||
| Scheme 5 β-C(sp3)–H tri-arylation of pivalamide derivative. | ||
Complementing the structural versatility of Chatani's and You's methodologies, Xu and Qiu disclosed the 8-AQ-assisted nickel(II)-catalyzed β-C(sp3)–H heteroarylation of aliphatic amides using heteroaryl bromides as coupling partners (Scheme 6).80 The reaction proceeded efficiently using NiBr2 as the catalyst precursor, MesCO2H and tetrabutylammonium iodide (TBAI) as additives, Na2CO3 as base, in DMF at 160 °C for 24 h. Overall, the reactivity pattern resembled that observed in previous couplings with aryl bromides,75 with electron-rich heteroarenes demonstrating better reactivity than their electron-deficient counterparts. Interestingly, in this work, the authors proposed a NiII/NiIV catalytic cycle involving diamagnetic mononuclear intermediates but pointing that in the coupling with heteroaryl bromides, the C–H bond cleavage seemed to be the r.d.s. (KIE = 6.4). The efficient removal of the 8-AQ moiety under basic conditions enhanced the practicability of the method (Scheme 7).
 | ||
| Scheme 6 Ni-cat. β-C(sp3)–H arylation with heteroarylbromides. | ||
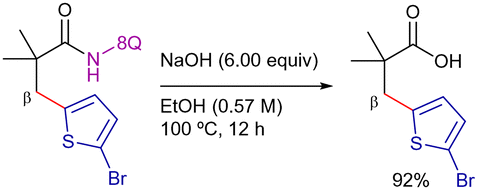 | ||
| Scheme 7 Removal of the 8-AQ with sodium hydroxide. | ||
Alternatively, Chatani et al. demonstrated the viability of the 8-AQ-assisted nickel-catalyzed β-C(sp3)–H arylation of aliphatic amides with diaryliodonium salts as electrophilic coupling partners (Scheme 8).81 In this case, the optimal reaction conditions were Ni(OTf)2 as catalyst precursor, Na2CO3 as base in 4-methyltetrahydro-2H-pyran (MTHP) at 140 °C for 24 h. Although diaryliodonium salts can decompose into aryl iodides and aryl triflates under these basic conditions at high temperatures, the authors showed that neither of them was suitable coupling partners in this process. Consequently, the diaryliodonium salts acted as both coupling partners and oxidants. Interestingly, the method was suitable for the functionalization of methyl and methylene positions in α,α- and α-substituted propanamide derivatives.
 | ||
| Scheme 8 β-C(sp3)–H arylation with diaryliodonium salts. | ||
In this work, H/D scrambling experiments showed deuterium incorporation at the β-position, suggesting that the C–H bond cleavage was reversible (Scheme 9). Moreover, as the H/D scrambling in the absence of the hypervalent iodide was higher than in its presence, the authors proposed that the oxidative addition step would take place after the C–H activation step. Furthermore, the addition of TEMPO to the reaction did not have much effect, suggesting that radical species were not involved in the reaction pathway.
 | ||
| Scheme 9 β-C(sp3)–H arylation with iodonium salts. Mechanistic hypothesis. | ||
Along these studies, the authors proposed the NiII/NiIV catalytic cycle disclosed in Scheme 9.82 This hypothesis was closely related to the mechanism suggested for the coupling with aryl halides. The cycle consists of (i) AQ-directed C–H activation leading to metallacycle II, (ii) oxidative addition of the diaryliodonium salt to form the cationic nickel(IV) intermediate V, and (iii) C–C bond-forming reductive elimination, followed by protonolysis to yield the functionalized product.
Taking this mechanism as reference, Sanford et al. disclosed a ligand design strategy to isolate a cyclometallated nickel(IV) complex similar to the key intermediate proposed in the aforementioned processes (Scheme 10).83 The reaction of the anionic nickel(II)-picolinate with the diaryliodonium salt resulted in the formation of a stable nickel(IV) σ-aryl complex at room temperature. This complex subsequently underwent C(sp3)–C(sp2) bond-coupling via reductive elimination in almost quantitative yield under mild conditions (70 °C, 120 min). The authors suggested that the high temperatures required for the catalytic reaction may be due to slow C(sp3)–H activation and/or to the formation of off-cycle Ni-species. Note that in these studies, no diamagnetic Ni-intermediates were detected by 1H-NMR spectroscopy under any conditions.
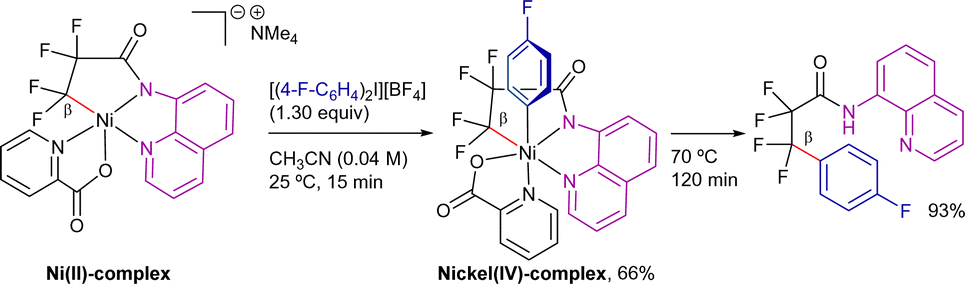 | ||
| Scheme 10 Nickel(IV)-intermediates in 8-AQ-directed C(sp3)–C(sp2) coupling. | ||
As an alternative strategy for C(sp3)–H arylation, Xia and Yin reported the Ni-catalyzed oxidative C–H/C–H cross-dehydrogenative coupling (CDC) reaction as an straightforward methodology to construct various highly functionalized alkyl (aryl)-substituted thiophenes (Scheme 11).84 Under optimized conditions (Ni(OTf)2 as catalyst source, Ag2CO3 as oxidant, MesCO2H and TBAB as additives, and KH2PO4 as base in DMSO at 160 °C for 24 h), a variety of alkyl-aryl substituted thiophenes were obtained in high yields via double C–H bond cleavage without affecting the C–Br or C–I bonds. Other heteroaryl groups, such as thiazole, benzofuran and furyl derivatives also participated in the reaction showing similar reactivity.
 | ||
| Scheme 11 β-C(sp3)–H arylation with heteroaryls through a CDC process. | ||
In this work, the KIE experiments suggested that the C(sp3)–H bond cleavage could be involved in the r.d.s (KIE value = 4.6) but not the activation of the aromatic position (KIE value = 1.1) (Scheme 12). Moreover, the addition of TEMPO or 1,4-benzoquinone (1,4-BQ) did not inhibit the reactivity, supporting a non-radical pathway. On the other hand, DFT calculations verified the importance of KH2PO4 as an additive for generating Ni(H2PO4)2 as the predominant active catalytic nickel(II)-species and promoting the C(sp3)–H bond cleavage. The mechanistic proposal is depicted in Scheme 12: (i) nickel(II)-mediated 8-AQ-directed C–H activation to form metallacycle II, (ii) Ag-mediated oxidation to nickel(III)-intermediate VII, (iii) metalation of the thiophene to form the nickel(III)-intermediate VIII, and finally, (iv) C–C bond-forming reductive elimination to release the functionalized product. Subsequently, Ag-mediated reoxidation of the generated nickel(I)-species would regenerate the active nickel(II)-catalyst.
 | ||
| Scheme 12 Proposed catalytic cycle for the Ni-catalyzed CDC. | ||
Following a similar strategy, You et al. reported the 8-AQ-assisted Ni-catalyzed oxidative heteroarylation of unactivated C(sp3)–H bonds with benzothiazoles (Scheme 13).85 In this case, the optimal conditions consisted on Ni(OAc)2·H2O as catalyst source, PPh3 as ligand, Ag2CO3 as oxidant, 1-adamantanecarboxylic acid (1-AdCO2H) as additive in ClPh at 160 °C for 24 h. The method was limited to aliphatic amides quaternized at the α-position. In this work, the elimination of the DG was achieved efficiently, accessing carboxylic acid under basic conditions or carboxylate ester using BF3·Et2O (Scheme 14).
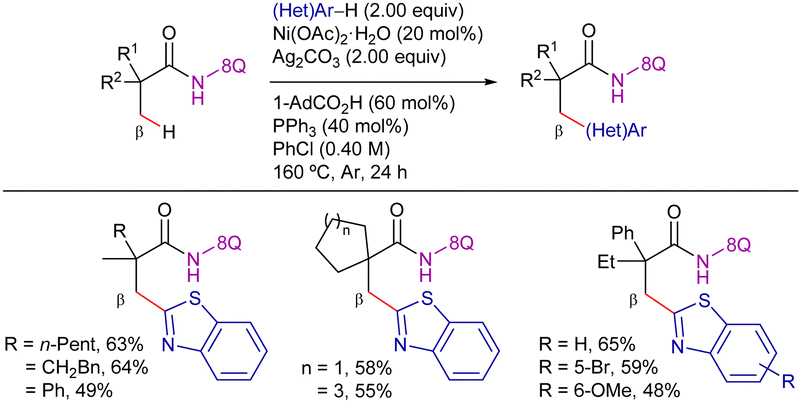 | ||
| Scheme 13 β-C(sp3)–H arylation with benzothiazoles through a CDC process. | ||
 | ||
| Scheme 14 Selective removal of the DG. | ||
 | ||
| Scheme 15 β-C(sp3)–H alkenylation with internal alkynes. | ||
 | ||
| Scheme 16 Synthesis of γ-butyrolactone scaffolds. | ||
In this work, deuterium labeling experiments suggested that the C–H activation step was reversible and not involved in the r.d.s. (KIE = 1.0), whereas the insertion of the alkyne was likely to occur a posteriori. The authors proposed the catalytic cycle depicted in Scheme 17, which involves: (i) reversible nickel(II)-mediated 8-AQ-directed C–H activation to form metallacycle II, (ii) coordination of the alkyne to form complex IX, and its subsequent migratory insertion to form the seven-membered metallacycle intermediate X, and (iii) final protonation of X, delivering the alkenylated product and the nickel(II) active species to close the catalytic cycle. In principle, the protonolysis step tends to generate the (Z)-isomer. However, under the reaction conditions, isomerization to the thermodynamically more stable (E)-isomer may occur, which accounts for the E/Z mixtures observed in the reaction.
 | ||
| Scheme 17 β-C(sp3)–H alkenylation with internal alkynes. Proposed mechanism. | ||
Interestingly, in the same year, Maiti et al. also published an improved methodology for this process, showing that by changing the solvent mixture of toluene/isopropanol to either DMF or NMP, the temperature could be decreased to 140 °C while maintaining similar reactivity (Scheme 18).87 The system was selective for the β-CH3 functionalization, observing this selectivity even in the case of methylacrylamide derivative. Moreover, the conditions were optimal for the decarboxylative coupling with phenyl propionic acid, yielding the alkenylated product in moderate yield and with complete regio- and stereoselectivity.
 | ||
| Scheme 18 Ni-catalyzed β-C(sp3)–H alkenylation with internal alkynes. | ||
Adding to these contributions, in 2017, Zhang et al. reported a new set of conditions for the reaction with terminal alkynes (Scheme 19).88 The reaction was performed in the presence of Ni(OAc)2 as precatalyst, MePh2P as ligand, and the appropriate combination of NaOAc with AcOH in DMF at 160 °C for 24 h. Under these conditions, the reaction displayed excellent functional group tolerance with respect to both aliphatic amides and terminal alkynes, being possible to apply less activated aliphatic alkynes. Remarkably, removal of the DG was achieved upon treatment with BF3·Et2O and sodium hydroxide (Scheme 20).
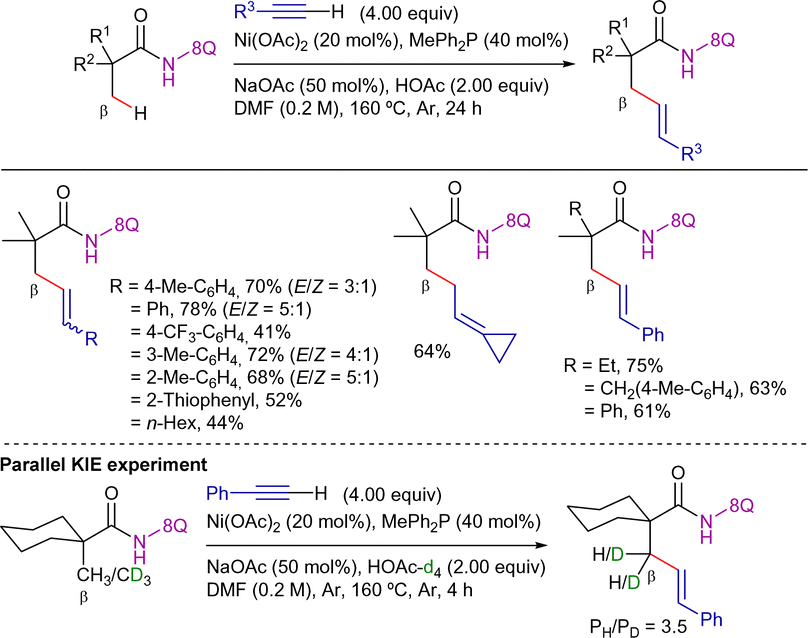 | ||
| Scheme 19 β-C(sp3)–H alkenylation with terminal alkynes. | ||
 | ||
| Scheme 20 Removal of the 8-AQ DG under basic conditions. | ||
Mechanistic experiments revealed that the reaction did not involve radical species and that the C–H activation step was presumably irreversible. Moreover, parallel KIE experiments provided a value of 3.5, while the intermolecular KIE value was 4.6, indicating that the C–H activation step was presumably involved in the r.d.s. The authors proposed a mechanistic hypothesis similar to that depicted in Scheme 17, which involves the following sequence: (i) nickel(II)-amide coordination, (ii) C–H bond cleavage, (iii) alkyne migratory insertion to generate a seven-membered nickelacycle, and (iv) protonolysis, yielding the alkenylated product.
Terminal alkynes have not only been employed in alkenylation processes but also in alkynylation/annulation sequences. Thus, in 2016, Zhang et al. described the 8-AQ-assisted nickel(II)-catalyzed synthesis of structurally diverse five-membered lactams from aliphatic amides and terminal acetylenes (Scheme 21).89 The optimized reaction conditions involved the use of (Cy3P)2NiCl2 as precatalyst, Ag2CO3 as oxidant, Na2CO3 as a base and TBAI as additive, in toluene at 150 °C for 24 h. Using this system, a broad range of terminal acetylenes and aliphatic amides were efficient coupling partners, furnishing the corresponding E-ethylidene-pyrrolidinones in good yields. Interestingly, amides with prochiral β-CH3 positions led to a mixture of diastereomers generated by the presence of the chiral C3 and the axial chirality of the lactam ring.
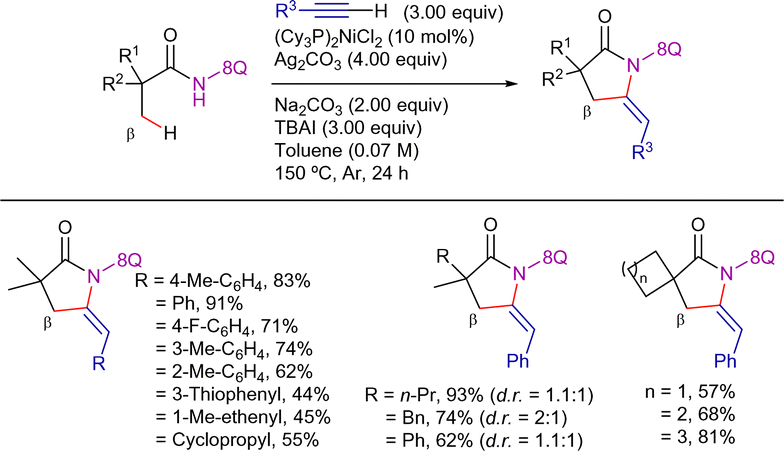 | ||
| Scheme 21 β-C(sp3)–H alkynylation/annulation with terminal alkynes. | ||
In this work, the authors suggested that the transformation presumably proceeds through an oxidative alkynylation process, followed by an Ag/TBAI-mediated intramolecular annulation reaction, ultimately leading to the thermodynamically favoured (E)-ethylidene-pyrrolidinones (Scheme 22). The addition of TEMPO or BHT (2,6-di-tert-butyl-4-methylphenol) did not influence the reactivity, and therefore the intervention of radical species in the process was ruled out. It is proposed that the catalytic cycle starts by the C–H bond cleavage for the formation of the nickel(II)-cyclometallated intermediate II. Then, silver-mediated single electron-oxidation and ligand exchange generate intermediate XI, which was detected by MALDI-TOF analysis. From here, reductive elimination leads to the formation of the β-alkynylated product, which, in the presence of Ag/TBAI, undergoes in situ cyclization to generate the γ-lactams. On the other hand, the nickel(I) complex generated in the reductive elimination step would be re-oxidized to nickel(II) species by the remaining Ag-salt.
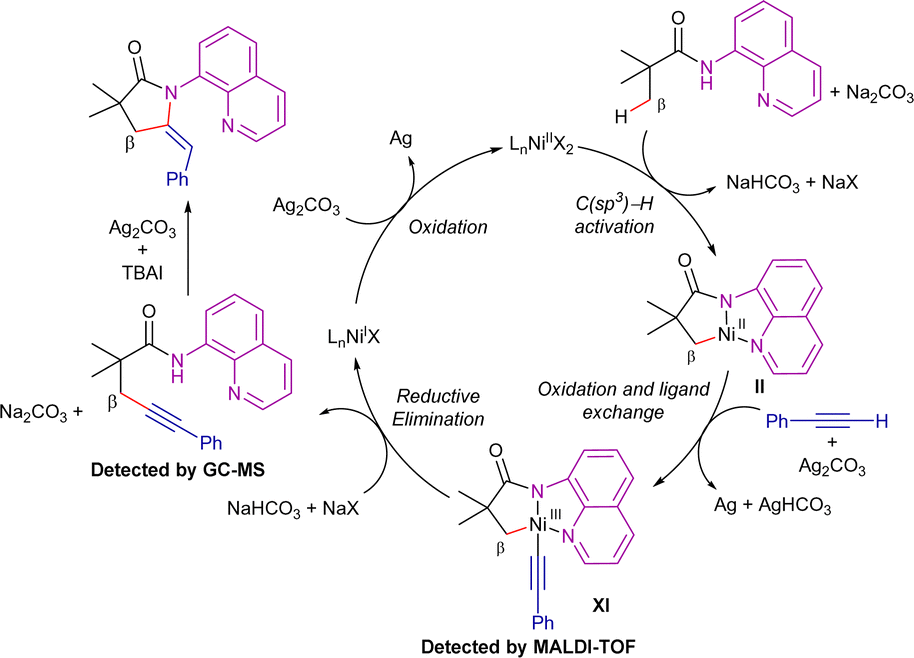 | ||
| Scheme 22 Sequential β-C(sp3)–H alkynylation/annulation with terminal alkynes. Proposed mechanism. | ||
In 2020, Shen et al. expanded this methodology to the nickel-catalyzed annulation of aliphatic amides with alkynylsilanes (Scheme 23).90 The method was effective in promoting the process with aryl, heteroaryl, and alkyl acetylenes, as well as with enynes. The authors suggested a mechanistic proposal like that of terminal alkynes (Scheme 22): the transformation is likely to occur via an oxidative alkynylation process, which is then followed by an intramolecular annulation reaction mediated by Ag/TBAI generating the thermodynamically favoured (E)-ethylidene-pyrrolidinones. The possibility of a mechanism involving radical intermediates was excluded since the addition of TEMPO did not affect the reaction.
 | ||
| Scheme 23 Sequential β-C(sp3)–H alkynylation/annulation with trimethylsilyl alkynes. | ||
On the other hand, Shi et al. disclosed the efficient 8-AQ-assisted nickel(II)-catalyzed alkenylation of β-methyl C(sp3)–H bonds of a broad range of α-quaternary aliphatic carboxamides using trans-vinyl iodides as coupling partners (Scheme 24).91 The catalytic system comprised the air-stable Ni(acac)2 as catalyst, BINOL as ligand, and the combination of KTFA and Li2CO3 as basic additives in DMSO at 140 °C for 8 h. The scope of carboxamides revealed that a wide variety of carboxamides bearing both linear and cyclic chains were compatible with this protocol, showing a preference for functionalization of aliphatic β-methyl than β-methylene and γ-C(sp2)–H bonds.
 | ||
| Scheme 24 β-C(sp3)–H alkenylation with aryl vinyl iodides. | ||
 | ||
| Scheme 25 Ni/Cu synergistic catalyzed β-C(sp3)–H carbonylation with DMF. | ||
 | ||
| Scheme 26 Post-synthetic modification to obtain 1,4-dicarbonyl compounds. | ||
As in previously reported examples, the authors proposed the nickel(II) mediated C(sp3)–H cleavage to afford the cyclometallated intermediate II (Scheme 27). Subsequent nucleophilic addition to the iminium ion intermediate a, generated in situ from DMF and copper species, forms intermediate XII that after oxidation, intramolecular nucleophilic addition and hydrolysis, liberates intermediate b. Finally, oxidation and hydrolysis of the resulting iminium intermediate generates the succinimide derivative. Agreeing with this mechanistic proposal, 13C-labeling studies confirmed that the carbonyl group inserted would not come directly from the carbonyl group of DMF. Instead, the incorporated carbonyl group mainly comes from the methyl group. Moreover, deuterium labeling and KIE experiments seemed to indicate that the C–H activation step was irreversible and involved in the r.d.s. (KIE = 2.1).
 | ||
| Scheme 27 Proposed catalytic cycle for the β-C(sp3)–H carbonylation with DMF. | ||
 | ||
| Scheme 28 Ni-cat. β-C(sp3)–H alkylation with alkyl halides. | ||
In this work, intermolecular competition KIE experiments provided a value of 4.1, indicating that the C–H bond cleavage was involved in the r.d.s. Furthermore, the reaction in the presence of TEMPO resulted in a great decrease in the yield, but not its inhibition, and a product derived from the coupling between TEMPO and alkyl halides was isolated. Therefore, radical alkyl species might be generated in the reaction and might be involved in the mechanism. The authors suggested two possible reaction pathways: (i) a NiII/NiIV catalytic cycle (path a) or (ii) a NiII/NiIII/NiI route (path b) (Scheme 29). Both routes have in common the initial formation of the nickel(II)-cyclometallated intermediate II. From this point, path a involves oxidative addition of the alkyl halide, generating the nickel(IV)-intermediate XIV, which, by subsequent reductive elimination and protonolysis, releases the alkylated product and nickel(II) species to accomplish the catalytic cycle. On the other hand, if path b is considered, the next step would be the alkyl radical addition to intermediate II, forming the nickel(III) intermediate XVI, followed by reductive elimination and nitrogen protonation to release the final product and nickel(I) species. Alkyl halide single electron oxidation of nickel(I) species would regenerate the active nickel(II) species, generating also an alkyl radical.
 | ||
| Scheme 29 Mechanistic proposals for β-C(sp3)–H alkylation with alkyl halides. | ||
In addition to these contributions, Chatani's work described one example of Ni-catalyzed 8-AQ-assisted β-C(sp3)–H methylation of aliphatic amides using phenyltrimethylammonium iodide as methyl source (Scheme 30).94 The reaction proceeds with high regiocontrol, providing the mono-functionalized product in 68% yield along with traces of the di-alkylated product.
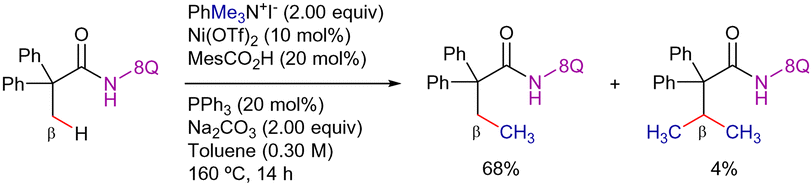 | ||
| Scheme 30 β-C(sp3)–H alkylation with phenyltrimethylammonium salts. | ||
As an alternative to the use of alkyl halides (or pseudo-halides), Maiti et al. disclosed the nickel-catalyzed insertion of electron-deficient olefins into unactivated C(sp3)–H bonds using also the 8-AQ as optimal DG (Scheme 31).87 The reaction conditions consisted on Ni(OAc)2·4H2O as catalyst precursor, PPh3 as ligand, in DMSO at 140 °C for 24 h. This system displayed excellent linear selectivity. The authors proposed a mechanistic hypothesis similar to alkynes (Scheme 17), involving the following sequence: (i) nickel(II)-amide coordination, (ii) C–H bond cleavage, (iii) alkene migratory insertion, leading to the formation of a seven-membered nickelacycle, and (iv) protonolysis, yielding the alkylated product. To the best of our understanding, the acetate counteranion facilitates the C–H activation step and acts as a proton shuttle, thereby contributing to the closure of the catalytic cycle. At no time do the authors mention having detected the alkenylation product, the formation of which could have been more favoured in the presence of basic additives.
 | ||
| Scheme 31 β-C(sp3)–H alkylation with activated olefins. | ||
The method was sensitive to the structure of both reagents, obtaining the best results with alkyl substitution at the amide substrate with alkyl acrylates and acrylonitrile.
 | ||
| Scheme 32 β-C(sp3)–H alkynylation with trimethylsilylacetylene. | ||
To gain mechanistic data about the transformation, the authors analysed the reactivity of 1-(methyl-d3)cyclohexanoic acid derivative under the standard conditions, in the absence of the alkyne, and adding 1.00 mL of H2O. The starting material was recovered unaltered and fully deuterated at the β-position. Moreover, the intermolecular competition KIE experiment provided a 2.03 value, while the parallel KIE experiment gave a 2.28 value. These results indicated, therefore, that the C–H activation step was irreversible and was involved in the r.d.s. Finally, the addition of TEMPO had a significant effect, suggesting the formation of radical species in the process.
Based on these results, the authors proposed the catalytic cycle depicted in Scheme 33. First coordination of the amide to the catalytically active nickel(II) species, followed by the β-C(sp3)–H bond cleavage would generate intermediate XIX. From this point, two pathways were proposed: (a) Cu-mediated single electron oxidation of nickel(II) to nickel(III), followed by transmetalation with the alkynyl cupper(I) species generated in situ by reaction between CuI and terminal alkynes, leading to intermediate XXI (path a); (b) single electron oxidation by alkynyl radical species generated by the homolytic cleavage of alkynyl cupper species to generate intermediate XXI (path b). From intermediate XXI, the subsequent reductive elimination step would generate the alkynylated product and nickel(I) species. Then Cu-mediated oxidation would further re-oxidize the nickel(I) species to the catalytically active nickel(II) species.
 | ||
| Scheme 33 Proposed catalytic cycle for β-C(sp3)–H alkynylation with trimethylsilylacetylene. | ||
2.2. C-Heteroatom bond formation
In a seminal contribution, Ge et al. reported the intramolecular dehydrogenative cyclization of aliphatic amides by a nickel-catalyzed C(sp3)–H bond functionalization process with the assistance of 8-AQ as a bidentate DG (Scheme 34).102 The reaction favoured the C–H bonds of β-methyl groups over the γ-methyl or β-methylene groups, as well as the β-methyl C–H bonds over aromatic C(sp2)–H bonds. Additionally, this process enabled the efficient functionalization of secondary benzylic C–H bonds with complete regioselectivity when there were no accessible β-methyl positions. The authors showed that the 8-amino-5-methoxyquinoline was as effective as the 8-AQ and could be removed under oxidative conditions to afford the desired NH-β-lactam product (Scheme 35).
 | ||
| Scheme 34 Ni-catalyzed β-C(sp3)–H intramolecular dehydrogenative cyclization. | ||
 | ||
| Scheme 35 Removal of the 8-amino-5-methoxyquinoline group. | ||
The proposed catalytic cycle is depicted in Scheme 36. The reaction starts with the coordination of the amide to nickel(II)-species and subsequent ligand exchange process under basic conditions, leading to the formation of the nickel complex XXII. Subsequent C(sp3)–H nickellation generates the nickel(II) complex XXIII, which is then oxidized to the nickel(III) species XXIV by TEMPO. Finally, reductive elimination from intermediate XXIV generates the desired product and a nickel(I)-species that would be re-oxidized by TEMPO closing the catalytic cycle.
 | ||
| Scheme 36 Proposed catalytic cycle for β-C(sp3)–H intramolecular amidation. | ||
Years later, Chatani et al. disclosed a methodology for the Ni-catalyzed synthesis of β-lactams from aliphatic amides employing the 8-amino-5-chloroquinoline as optimal DG (Scheme 37).103 The reaction occurred in the presence of Ni(OTf)2 as catalyst precursor, Na2CO3 as a base, catalytic amounts of Ag2CO3 and an excess of I2, in DMF at 140 °C for 24 h.
 | ||
| Scheme 37 β-C(sp3)–H cyclization in the presence of I2. | ||
In this work, the authors proposed that the reaction might occur through a direct pathway, in which the β-lactam would be formed by (i) nickel(II)-cyclometalation after the C–H activation step, and (ii) C–N bond reductive elimination from a nickel(III) or a nickel(IV) intermediate generated by iodine single-electron or oxidative addition oxidations, respectively. Nonetheless, their findings seemed to indicate that a stepwise pathway was also feasible, involving the direct β-iodination of the aliphatic amide and a facile subsequent intramolecular cyclization to form the β-lactam. Deuterium-labeling experiments revealed that the C(sp3)–H bond cleavage was irreversible and presumably the r.d.s. in this reaction. The addition of radical scavengers, such as TEMPO and BHT, completely inhibited the reaction, suggesting that radical intermediates may play a role in the process. However, the involvement of a background reaction in which radical scavengers react with I2 cannot be ruled out. Such a reaction could consume most of the I2, potentially influencing the observed inhibition.
Shedding light on this point, Sanford et al. synthesize a σ-alkyl nickelacycle complex that undergo intramolecular C(sp3)–N bond-forming at room temperature to form a β-lactam product (Scheme 38a).104 Moreover, this complex proved to be catalytically active under the previously reported reaction conditions by Chatani, which suggested its involvement in the reaction (Scheme 38b). In contrast, when a nickel(III)-complex was tested, no reactivity was observed, indicating that it is not a competent catalyst for the C–H amination process.
 | ||
| Scheme 38 Nickel(II)-intermediates in 8-AQ-directed C(sp3)–N coupling in the presence of I2. | ||
On the other hand, in 2021, Baik's and Chang's group reported a DG-assisted Ni-catalyzed intermolecular C(sp3)–H amidation using organic sulfonylazides as nitrene precursors (Scheme 39).105 In this transformation, 8-amino-5-trifluoromethyl quinoline was proved to be the optimal DG. The external oxidant-free conditions enabled the selective intermolecular amidation to outcompete an intramolecular counterpart. A complete chemoselectivity toward the β-methyl C(sp3)–H bond suggested that the inner-sphere mechanism was operative. The method provided amidated products with low to good yields, observing better reactivities with electron-rich aryl azides. In addition, alkyl azides were also compatible with the reaction conditions. Nevertheless, the method was less effective with α-substituted propanamide derivatives or for the functionalization of β-methylene positions.
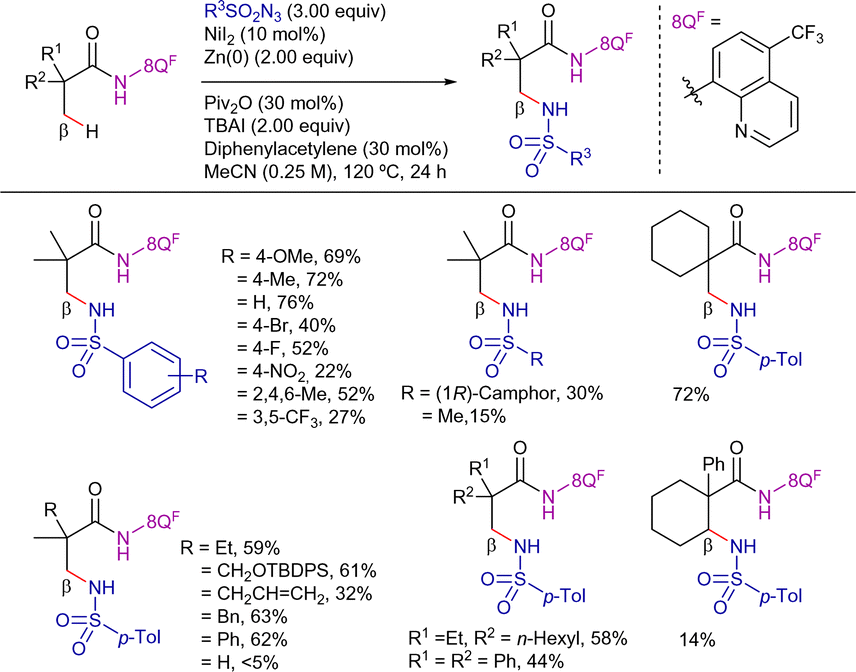 | ||
| Scheme 39 β-C(sp3)–H intermolecular amidation with sulfonylazides. | ||
Based on theoretical calculations, the authors suggested the catalytic cycle shown in Scheme 40. The reaction would start with the in situ reduction of nickel(II) species to nickel(0), followed by pivalic anhydride oxidation through a highly exothermic step to generate nickel(II) species.106,107 Subsequent amide coordination to generate intermediate XXV and irreversible C–H metalation (via CMD mechanism, intermediate XVI) followed by ligand exchange gives the azide-nickel(II) cyclometallated intermediate XXVII. Extrusion of N2 forms a nitrene-nickel(III) intermediate XXVII, that undergoes favourable inner-sphere C–N coupling to release the aminated product. Attending to the energy profile of the reaction, the azide coordination-N2 extrusion goes through the highest energy barrier (29.8 kcal mol−1), indicating that this step is the r.d.s. Moreover, DFT calculations showed that the substitution in the quinoline ring did not significantly affect the C–H activation step, but it influenced the efficiency of the aminated product release and regeneration of intermediate XXV. While this process was calculated to be only 13.4 kcal mol−1 uphill with an electron-withdrawing CF3 group at the quinoline core, the presence of an electron-donating methoxy functionality increases the energy requirement to 15.4 kcal mol−1.
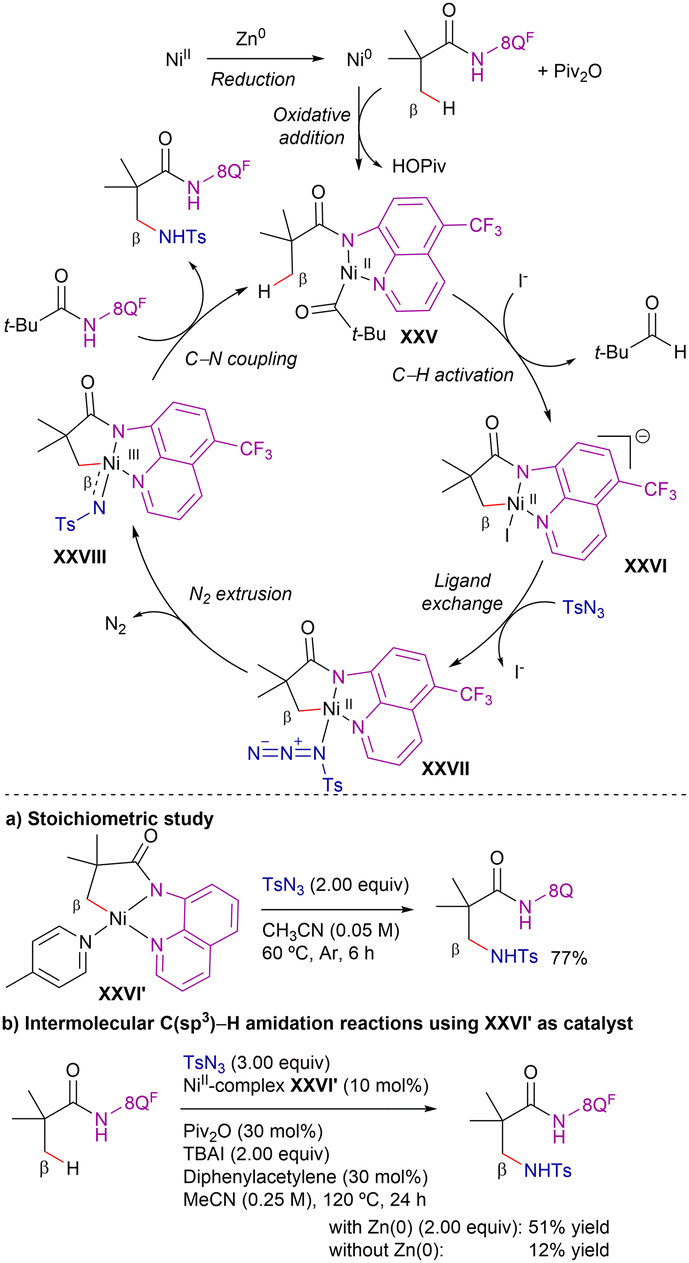 | ||
| Scheme 40 Proposed catalytic cycle for β-C(sp3)–H intermolecular amidation. | ||
To evaluate the feasibility of the above nickel(II/III) catalytic cycle proposed by computational studies, the authors performed stoichiometric amidation reactions using a C–H-activated nickel(II) complex (XXVI′, Scheme 40a), which exhibits structural similarity to the hypothesized Ni(II) intermediate XXVI. When the NiII–C complex XXVI′ was combined solely with TsN3 in acetonitrile at 60 °C, the desired intermolecular C(sp3)–H amidation proceeded efficiently (77% yield). This result indicates that nickel(II) serves as an active species in the amidation process and that the putative NiIII-nitrenoid species, responsible for C–N bond formation, originates from the nickel(II) intermediate.
To gain further mechanistic insights into the catalytic system, the authors employed the NiII–C complex XXVI′ as catalyst precursor under the amidation conditions (Scheme 40b). Under standard reaction conditions, the desired amidation product was obtained with a 51% yield; however, in the absence of Zn, the reaction efficiency significantly dropped (12%). Given that catalytic turnover was hindered by the absence of Zn, unlike in the stoichiometric amidation scenario, the authors inferred that while Zn does not participate in the key amidation step, it plays a crucial role in facilitating the regeneration of the active catalyst.
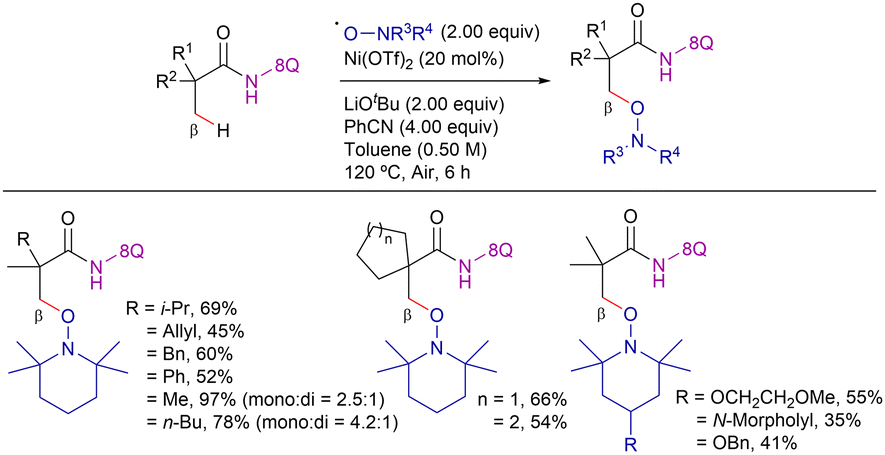 | ||
| Scheme 41 β-C(sp3)–H alkoxylation with TEMPO and analogues. | ||
 | ||
| Scheme 42 One-pot synthesis of α-formyl acid derivatives. | ||
The proposed catalytic cycle is depicted in Scheme 43. Initially, bidentate chelation of the amide to the nickel(II) species forms the nickel(II) complex I, accompanied by the generation of HX, which is captured by LitBuO. Subsequently, the β-C–H bond undergoes irreversible cleavage, resulting in the formation of the nickel(II) cyclometallated intermediate II. Next, oxidative addition of II with the nitroxyl radical leads to intermediate XXIX. Finally, reductive elimination and protonation deliver the desired product along with the generation of nickel(I) species. Oxidation to nickel(II) species by either the stable nitroxyl radical or oxygen in air furnishes the catalytic cycle. Mechanistic insights suggested that the C–H activation step was presumably irreversible and was involved in the r.d.s. (KIE = 2.4).
 | ||
| Scheme 43 Proposed catalytic cycle for the β-C(sp3)–H alkoxylation. | ||
In 2015, Zhang,112 Qiu, Xu and Yin,113 and Shi114 reported different protocols for the 8-AQ-assisted Ni-catalyzed β-C–H thioetherification of aliphatic amides using disulfides as coupling partners (Scheme 44). The three reaction conditions were rather similar, requiring the use of a nickel(II)-salt as catalyst precursor (Ni(OTf)2, NiBr2 or (dppp)NiCl2), a carboxylic acid or biaryl-type ligand (N-acetylglycine, MesCO2H or BINOL), a base (Na2CO3 or KTFA) and the use of a coordinating polar solvent (DMF or DMSO) at high reaction temperatures (140–160 °C). In addition, the work of Zhang required the use of TBAI as an extra additive (Scheme 44a), while the work of Shi used Ag2O (Scheme 44c). Analyzing the scope of these contributions, in general, electron-rich diaryl disulfides were more efficient than electron-withdrawing and the substituents at para-positions provided better yields than meta and ortho. Notably, the method of Qiu, Xu and Yin could be applied to challenging aliphatic disulfides, achieving the desired products in good yields (Scheme 44b).113
 | ||
| Scheme 44 β-C(sp3)–H thiolation with disulfides. | ||
Regarding the mechanistic studies disclosed in these papers, the addition of radical scavengers such as TEMPO or BHT did not affect the reactivity in any case. Therefore, radical species were presumably not involved in the reaction. Deuterium labeling experiments at short reaction times performed by Zhang indicated that the C–H bond cleavage was reversible while KIE experiments performed by Qiu, Xu and Yin, indicated that the C–H activation step was involved in the r.d.s. (KIE = 5.7).
With all the mechanistic data, a combined proposed catalytic cycle based on the ones proposed in each work is presented in Scheme 45. Firstly, coordination of nickel(II) species to the amide followed by base-assisted N–H deprotonation leads to intermediate I, which then undergoes C–H metalation to generate intermediate II. Then, oxidative addition of PhSSPh generates nickel(IV) intermediate XXX, which then evolves through a reductive elimination step to generate the new C–S bond and the nickel(II) intermediate XXXI. Finally, the presence of silver, sodium or potassium would capture the thiol ligand that has not been inserted in the activated carbon, facilitating the release of the final product and the regeneration of the catalytically active nickel(II) species.
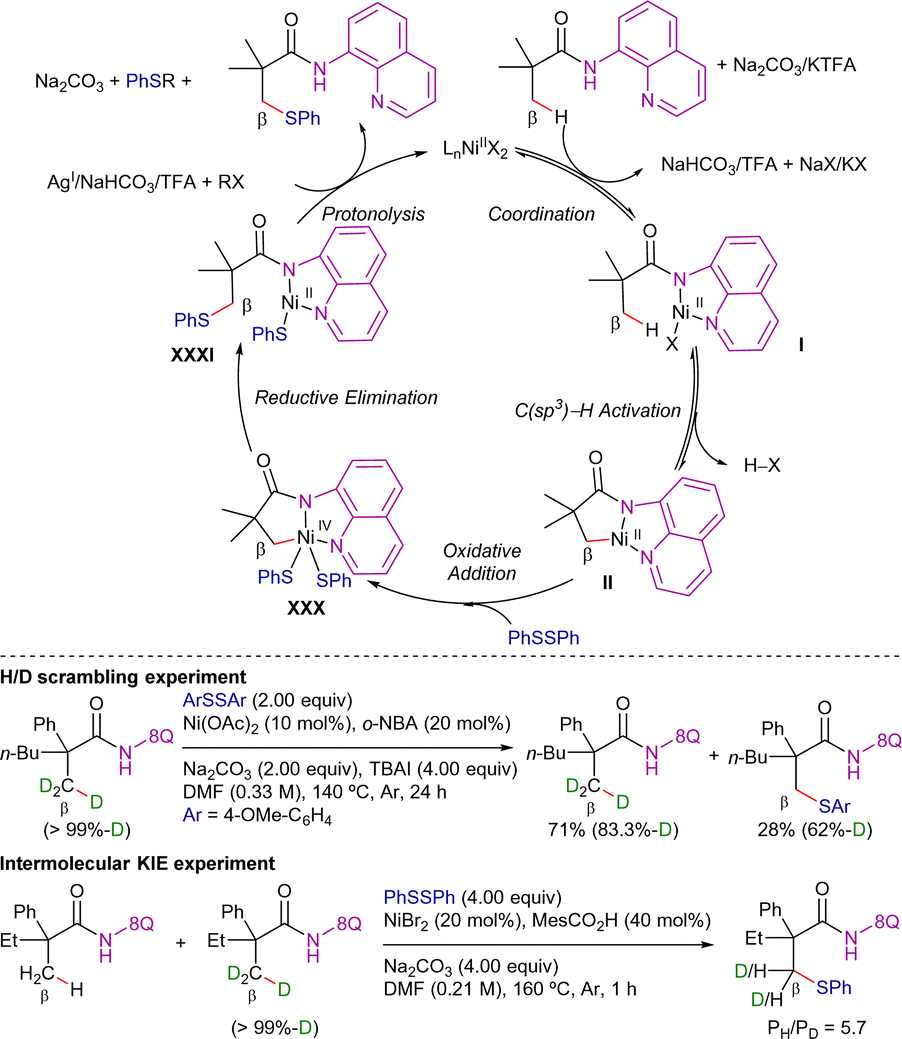 | ||
| Scheme 45 Proposed catalytic cycle for Ni-cat. β-C(sp3)–H thiolation with disulfides. | ||
On the other hand, Zhang (Scheme 46a),112 and Qiu, Xu and Yin (Scheme 46b)113 showed that diphenyl diselenide also reacted, rendering the β-phenylselenated product in 40% and 27% yield, respectively (Scheme 46).
 | ||
| Scheme 46 β-C(sp3)–H functionalization with diselenides. | ||
In parallel, Shi and co-workers reported a complementary method for the direct thiolation of β-C(sp3)–H bonds in α,α-disubstituted propanamide derivatives with disulfides and thiols as coupling partners (Scheme 47).115 The key to achieving good reactivities with thiols was to carry out the reaction under air. This could be due to the in situ generation of the disulfide from the thiol.116 The reaction was compatible with a wide range of aromatic thiols and lineal or cyclic carboxamides, providing the thiolated products in moderate to good yields.
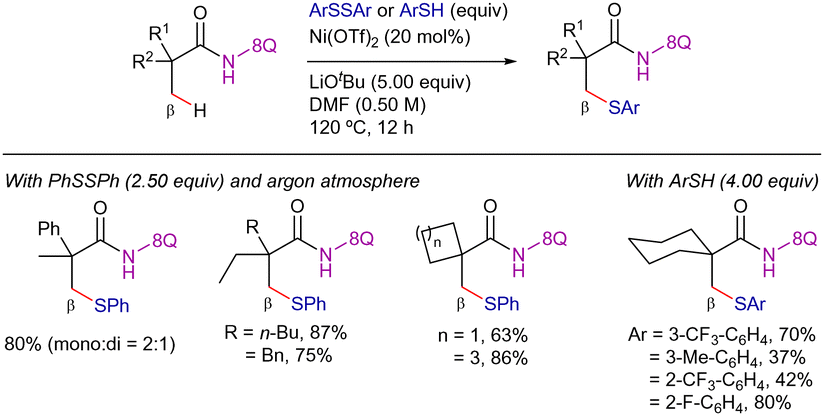 | ||
| Scheme 47 β-C(sp3)–H thiolation with disulfides and thiols. | ||
Later, Weng, Lu and co-workers reported a novel method for C–H thiolation with disulfides by using a 2-pyridyl hydrazine moiety as DG (Scheme 48).117 A mixture of mono- and di-thiolated products was isolated in 41% overall yield and 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
 | ||
| Scheme 48 Hydrazine-directed β-C(sp3)–H thiolation with disulfides. | ||
2.3. Exploring organometallic nickel intermediates
In all the contributions mentioned in the previous sections, the generally accepted mechanism would be that in which the activation of the C–H bond would take place at a low-valent NiII-center leading to the formation of a NiII-(σ-alkyl) intermediate. Its subsequent oxidative functionalization would release the desired product and regenerate the NiII-catalyst. In general, this mechanistic view has been shown to be valid for the functionalization of C(sp3)–H bonds proximal to AQ-type DGs.However, the substrate scope of Ni-catalyzed C–H functionalization is currently more limited compared to similar transformations using Pd. Additionally, most Ni-catalyzed C–H functionalization reactions necessitate harsh conditions (typically temperatures above 130 °C). These limitations are, at least in part, due to the C–H activation step, which is known to be significantly more challenging in NiII than in analogous PdII centers. Shedding light on this point, the group of Schafer and Love demonstrated that the C(sp3)–H activation in urea scaffolds took place in nickel(II) species under mild conditions, and was easier at β-methyl rather than β-methylene positions (Scheme 49).118 In fact, these studies showed that increasing the steric hindrance around the β-position with alkyl substituents resulted in lower reaction rates, not observing the formation of the nickelacycle with the substrate wearing a tertiary isopropyl group. These results agreed with the computational study reported by Omer and Lui.77 In contrast, with benzyl substitution, the reaction rates increased, favoured with electron-donating rather than with electron-withdrawing substitution. More recently, they also observed that the kinetic for the C–H activation step was modified by introducing substituents at the C5 position of the quinoline ring.119 Thus, with electron-rich substituents, the reaction rates were higher. However, despite these studies, no report has demonstrated the viability of functionalizing the β-position in urea moieties.
 | ||
| Scheme 49 Isolated thiourea nickel(II)-cyclometallated species. | ||
On the other hand, Sanford et al. have demonstrated that unactivated C(sp3)–H bonds could be cleaved and subsequently functionalized at high-valent Ni-centers with both heteroatom- and carbon-based nucleophiles under relatively mild conditions (Scheme 50).120 Moreover, these two steps were combined to demonstrate a proof-of-principle catalytic transformation for converting a C(sp3)–H bond into a C(sp3)–N bond. Unfortunately, the competing decomposition of the oxidant limited the catalyst turnover in this transformation. DFT studies revealed two possible mechanisms for the C–H activation process, involving triflate-assisted C–H activation at either a NiIV or NiIII intermediate. The former pathway was slightly favoured over the latter, with a difference of approximately 3 kcal mol−1.
 | ||
| Scheme 50 C(sp3)–H functionalization at high-valent Ni-centers. | ||
3. Cu-catalysis
The low price and toxicity of copper have awakened chemist interest in exploring its potential in organic synthesis.121 Since the discovery of the Ullmann and Golberg couplings,122 multiple radical relay transformations,123 cross-coupling reactions,124–126 Wacker-type oxidations127 or C–H oxidation processes,128–130 among others, have clearly shown the multidisciplinary of copper to promote reactions.131–133 Following the general trend, the directed C–H functionalization via in situ formation of Cu metallacyclic intermediates has predominantly been applied to the functionalization of C(sp2)–H bonds, facilitating the formation of new C–C and C–heteroatom bonds with the assistance of exogenous functional groups.134,135 In contrast, examples of C(sp3)–H functionalization remain relatively scarce. The primary challenge in using copper for C(sp3)–H bond functionalization lies in the difficulty of accessing copper(III) species, which is energetically demanding and often requires high reaction temperatures and elevated copper salt loadings.3.1. C–C bond formation
In 2015, Ge et al. reported the 8-AQ-assisted Cu-promoted β-C(sp3)–H bond functionalization of aliphatic amides with polyfluoroarenes through a CDC reaction (Scheme 51).136 In their study, the authors discovered that pyridine was a crucial additive and di-tert-butyl peroxide was the optimal oxidant. However, stoichiometric Cu(OAc)2 was required for this reaction. Under these conditions, the reaction exhibited good functional group tolerance and excellent site-selectivity. It favoured the functionalization of C(sp2)–H bonds at the ortho-position relative to two fluorine atoms, as well as the C–H bonds of β-CH3 groups over those of the β-CH2 or γ-CH3 groups in the amides.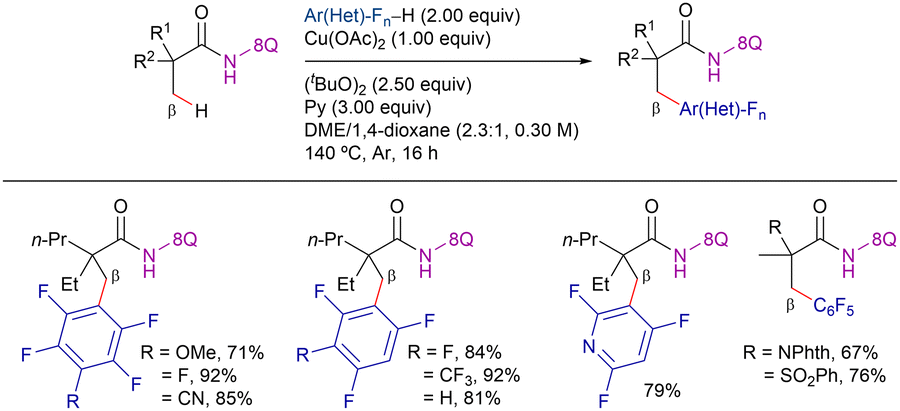 | ||
| Scheme 51 Cu-promoted β-C(sp3)–H arylation with polyfluorinated rings. | ||
Preliminary mechanistic studies indicated that C(sp3)–H bond cleavage was irreversible but not the r.d.s. (KIE = 1.1), and the C(sp2)–H activation of arenes appeared to be reversible, previous to the C(sp3)–H activation and could take place at copper(I) or copper(II) species. Based on these results, the authors proposed the reaction mechanism depicted in Scheme 52. Initially, in the presence of pyridine, a reversible C(sp2)–H bond cleavage mediated by copper(II) species leads to intermediate XXXII. Subsequently, N,N-coordination of the amide to the Cu–C(sp2) complex XXXII forms the chelate XXXIII, from which a copper(II) or (tBuO)2 single-electron oxidation generates copper(III) intermediate XXXIV. Following C(sp3)–H activation yields the five-membered metallacycle XXXV, from which final reductive elimination, along with nitrogen protonation releases the arylated product and copper(I) species. In principle, these copper(I) species would regenerate intermediate XXXIIvia two pathways: (a) initial (tBuO)2 oxidation followed by C(sp2)–H activation, or (b) aromatic C–H activation and further oxidation.
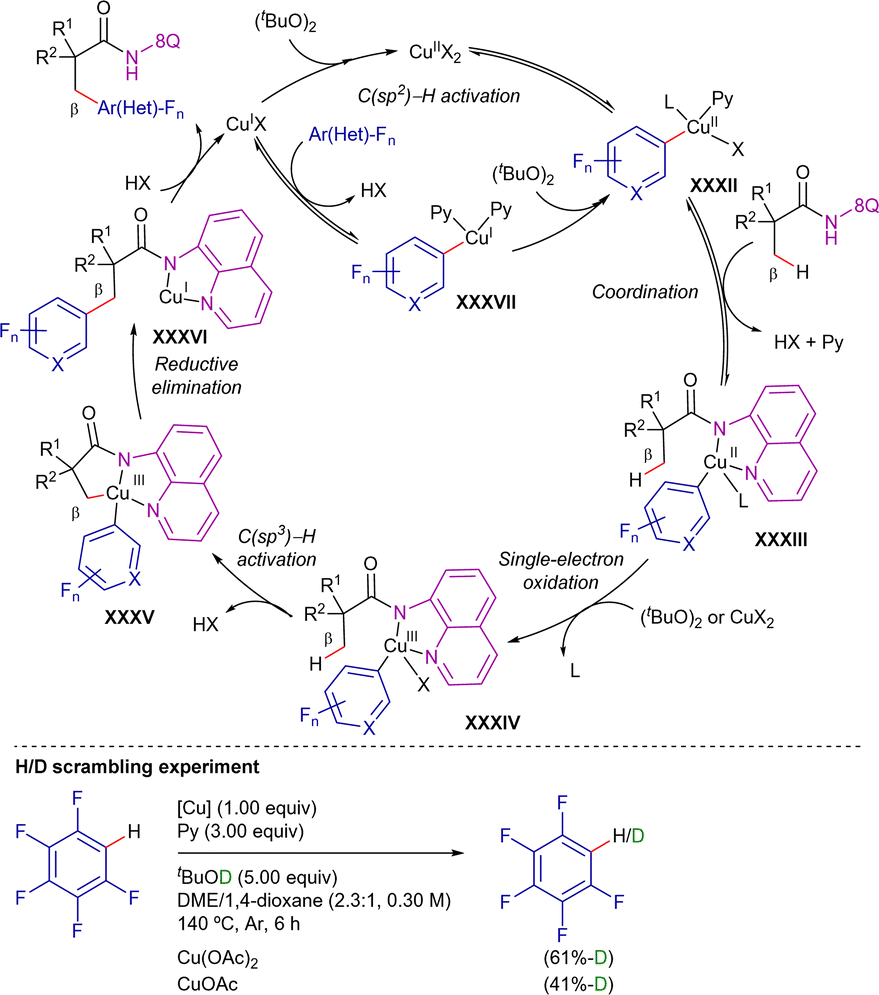 | ||
| Scheme 52 Proposed mechanism for Cu-promoted CDC with polyfluorinated rings. | ||
Two years later, You et al. reported a copper-mediated oxidative heteroarylation of unactivated C(sp3)–H bonds with azoles. The use of pyridine-N-oxide auxiliary facilitated the access to β-azolyl propanoic acid derivatives in good yields (Scheme 53).85 The reaction proceeded in the presence of super stoichiometric amounts of Cu(OAc)2·H2O as promoter, using PivOH and K2CO3 as additives in o-xylene at 140 °C under a N2 atmosphere for 24 h. In line with Ge's report, the reaction was limited to pivalamide derivatives with a quaternary α-carbon. Nonetheless, azoles such as oxazole, benzoxazole, thiazole, benzothiazoles, benzimidazole, purine, and even [1,2,4]triazolo[1,5-a]pyrimidine were well-tolerated under the optimized conditions. The practicability of the method was expanded by removing the auxiliary under basic conditions (Scheme 54).
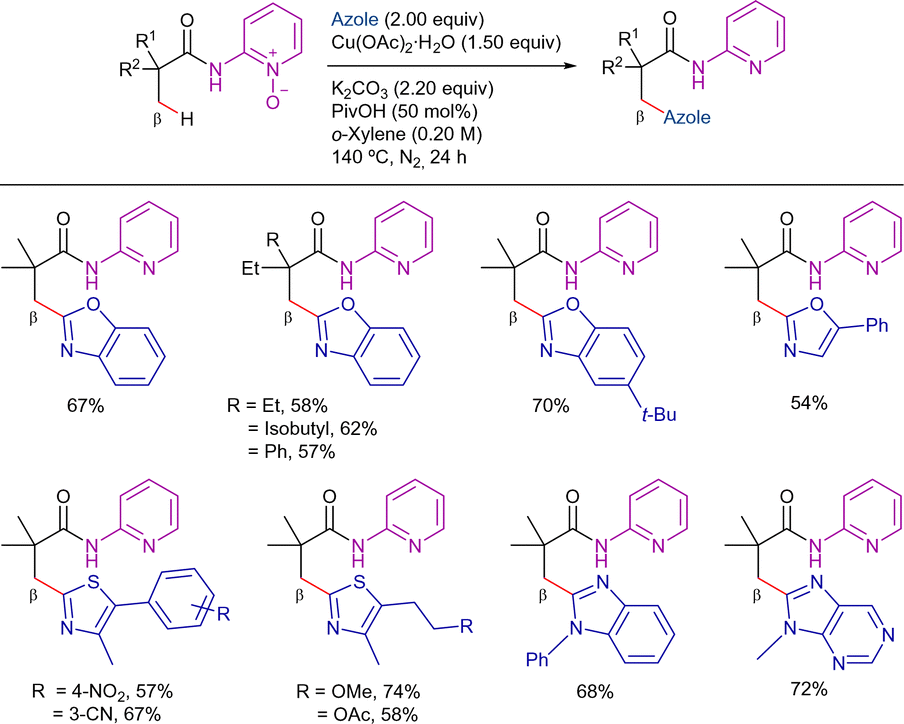 | ||
| Scheme 53 Cu-promoted β-C(sp3)–H arylation with azoles. | ||
 | ||
| Scheme 54 Removal of the pyridine-2-amine auxiliary. | ||
H/D scrambling experiments revealed that while the C(sp3)–H bond cleavage was an irreversible step, the C(sp2)–H bond activation was reversible. The proposed mechanism is depicted in Scheme 55. First, a ligand exchange of 2-pivalamidopyridine 1-oxide with Cu(OAc)2·H2O forms complex XXXVIII (synthesized and characterized by X-ray diffraction). Next, complex XXXVIII reacts with benzoxazole to produce intermediate XXXIX. Next, the C–H cupration of a primary C(sp3)–H bond forms the copper(III) complex XLI, with the involvement of additional Cu(OAc)2 through disproportionation. Subsequently, XLI undergoes reductive elimination to generate a copper(I)-coordinated intermediate XLII. Finally, the N–Oxide bond of intermediate XLII is reductively cleavaged by Cu(I), releasing the functionalized 2-pivalamidopyridine derivative and CuO (observed in the reaction mixture and tube wall). The formation of inert CuO would explain the requirement of super stoichiometric Cu(OAc)2·H2O for this transformation.
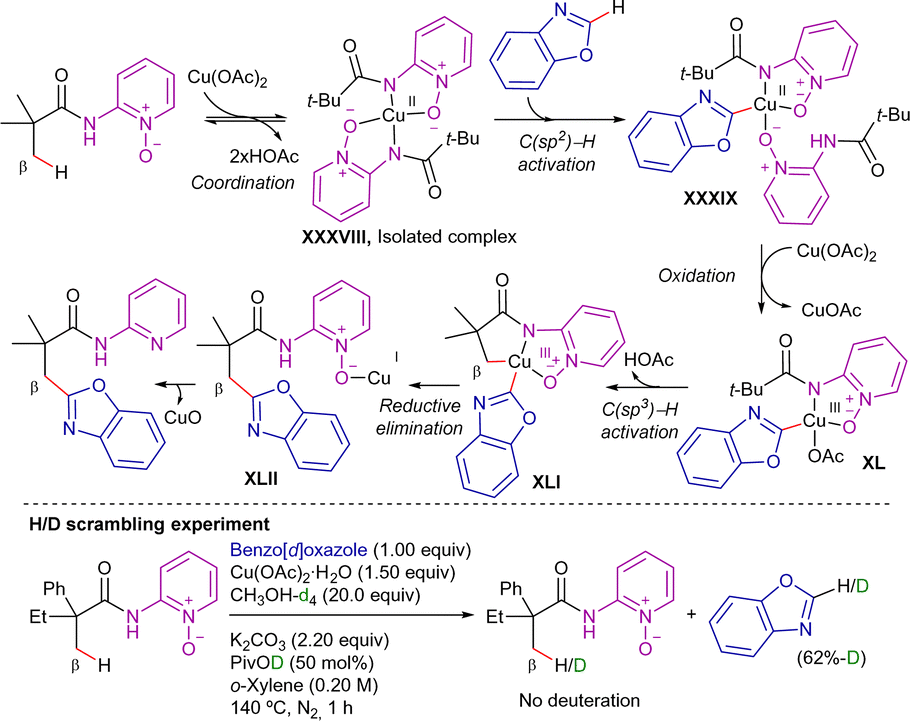 | ||
| Scheme 55 Proposed reaction mechanism for Cu-promoted β-C(sp3)-arylation with azoles. | ||
Ge et al. disclosed the 8-AQ-assisted Cu(OAc)2-promoted carbonylation of β-C(sp3)–H bonds with nitromethane as the carbonyl equivalent (Scheme 56).137 The optimized reaction conditions required the use of stoichiometric Cu(OAc)2, K2S2O8 as oxidant, PhCO2Na and Al2O3 as additives, along with DMPU in a solvent mixture 1,4-dioxane/isopropanol at 165 °C for 24 h. Using this system, the C(sp3)–H functionalization showed high site selectivity by favouring the C–H bonds of α-methyl groups. In this study, initial mechanistic analysis indicated that the substrate first undergoes a dehydrogenative coupling with nitromethane, followed by a Nef-type reaction, leading to the formation of the carbonylation product (Scheme 57).138 Deuterium labeling experiments unveiled that the C(sp3)–H activation was reversible.
 | ||
| Scheme 56 Cu-promoted β-C(sp3)–H carbonylative cyclization with nitromethane. | ||
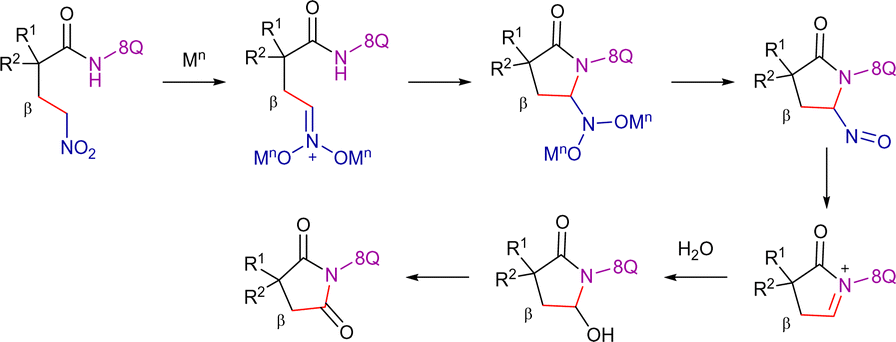 | ||
| Scheme 57 Proposed Nef-type reaction. | ||
In 2016, Zhang's group reported a copper-catalyzed C(sp3)–H alkynylation/annulation reaction between aliphatic amides and alkynyl carboxylic acids or terminal alkynes (Scheme 58).139 In this system, TBAI was used as a phase-transfer catalyst system and Ag2CO3 played a crucial role by: (i) generating the active silver acetylide species in situ,140–142 (ii) acting as oxidant, and (iii) promoting the cyclization of the alkynylated product, which leads to the thermodynamically favored (E)-ethylidene-pyrrolidinones. The reaction was carried out in DMF at 140–150 °C for 24 h. Interestingly, under the optimized conditions, terminal alkynes were less effective than the carboxylic acid analogues.
 | ||
| Scheme 58 Cu-cat. sequential β-C(sp3)–H activation/annulation with alkynes. | ||
Under these conditions, the reaction was limited to pivalamide derivatives with a quaternary α-carbon. The postulated mechanism would be analogous to the one depicted in Scheme 22. The catalytic cycle would involve (i) copper(II)-mediated DG-assisted C–H activation to form a copper(II)-cyclometalated intermediate, (ii) copper(II) disproportionation to generate an alkyl-Cu(III) metallacycle, (iii) transmetalation with the silver/copper acetylide generated in situ, and (iv) C–C bond reductive elimination to generate the alkynylated product and copper(I) species. The silver(I) would re-oxidize the copper(I) to the catalytically active copper(II) species, closing the catalytic cycle, whereas the alkynylated product would evolve to the β-lactam through a Ag/TBAI mediated cyclization reaction.
3.2. C–Heteroatom bond formation
In 2014, two protocols were independently reported for the synthesis of β-lactams through the 8-AQ-assisted copper-catalyzed intramolecular β-C(sp3)–H oxidative amidation of carboxamides. The protocol of Kuninobu' and Kanai's groups involved Cu(OAc)2 as catalyst and Ag2CO3 as oxidant in DCE at 140 °C (Scheme 59a),149 while Ge's group used CuCl as the catalyst and duroquinone as oxidant, PhCO2Na as base, ortho-xylene as solvent at 160 °C (Scheme 59b).150 In both protocols, similar reactivities and selectivities were observed. Additionally, both demonstrated that the 8-amino-5-methoxyquinoline DG was also efficient for this transformation and could be removed under oxidative conditions as shown in Scheme 35.
 | ||
| Scheme 59 Cu-cat. β-C(sp3)–H amidation. | ||
In both contributions, the initial C(sp3)–H bond activation occurs via cyclocupration, leading to the formation of an alkyl–copper(II) species. This species is then oxidized by either another Cu-salt or oxidant, resulting in the formation of an alkyl–copper(III) species. Subsequent C–N bond formation via reductive elimination leads to the final product with concomitant generation of a copper(I) species, which would be reoxidized to copper(II). However, in some cases, the copper(II) might first coordinate the DG before being oxidized to copper(III). Subsequent cyclocupration forms the alkyl–copper(III) species.
In 2015, Yang’ and You's groups described a more economical and practical protocol for the formation of β-lactam compounds using oxygen as the sole oxidant, observing complete β-CH3 selectivity in most cases (Scheme 59c).151 In this work, the authors proposed the plausible reaction mechanism shown in Scheme 60. Initially, copper(I) reacts with O2 to generate a copper(II)-superoxide radical, which undergoes electron transfer oxidation and H-abstraction to yield a copper(II)-hydroperoxo species. The resulting copper(II) species then coordinates with the amide, forming the N,N-chelated copper complex XLIII, followed by C(sp3)–H cupration to produce the cyclometallic intermediate XLIV. Subsequently, disproportionation generates copper(III) complex XLV. Finally, reductive elimination of the complex XLV releases the β-lactam and copper(I) species to close the catalytic cycle. It is worth noting that the direct oxidation of the copper(II) complex XLIV to the copper(III) species XLV by O2 was not excluded as a possibility.
 | ||
| Scheme 60 Proposed catalytic cycle for Cu-cat. β-C(sp3)–H amidation under O2 atmosphere. | ||
In 2016, two intermolecular versions for the coupling with secondary amines by using the 8-AQ as DG were disclosed independently by the groups of Qin and Yang. The method reported by the Qin's group, mediated by a Cu(OAc)2/O2 system at 130 °C, showed a useful substrate scope for α-quaternary carboxylic amides and cyclic secondary alkylamines (Scheme 61a).152 On the other hand, the protocol disclosed by Yang's group was carried out at 150 °C under air and required the presence of Na2HPO4 (Scheme 61b).153 This protocol was applied to a variety of cyclic and lineal secondary amines as well as to more challenging primary amines.
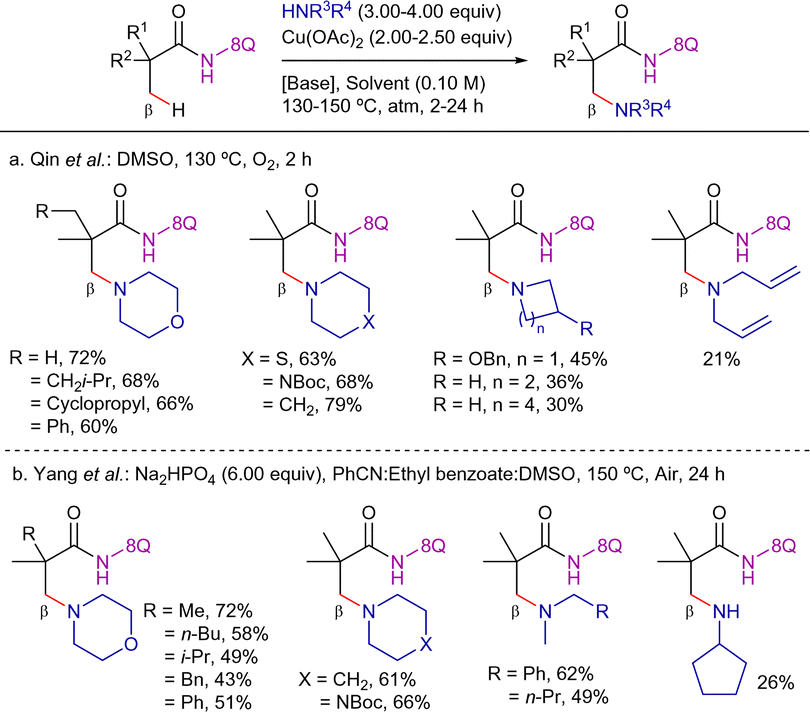 | ||
| Scheme 61 Cu-cat. β-C(sp3)–H intermolecular amination. | ||
Mechanistic studies indicated that the C–H activation step would be irreversible and involved in the r.d.s. Furthermore, the addition of TEMPO or BHT inhibited the reaction, suggesting the involvement of radical species. In fact, in the work of Yang et al., a coupling product between TEMPO and morpholine was isolated (Scheme 62).
 | ||
| Scheme 62 Proposed catalytic cycle for Cu-cat. β-C(sp3)–H intermolecular amination. | ||
On these bases, two reaction pathways have been proposed (Scheme 62): (a) copper(II) coordination to the carboxamide to generate intermediate XLVI, followed by the C–H activation step to generate a copper(II)-cyclometallated complex XLIV. Then, nitrogen radical oxidative addition, generated by copper(II)-mediated amine oxidation, forms intermediate XLVII (path a); or (b) intermediate XLVII could also be formed by first amine and carboxamine coordination to copper(II) species to generate intermediate XLVIII, followed by disproportionation to produce the copper(III)-intermediate XLIX, and subsequent C–H activation (path b). Finally, from intermediate XLVII, a reductive elimination step forms the final product and copper(I) species, that would be re-oxidized by oxygen.
 | ||
| Scheme 63 Cu-mediated β-C(sp3)–H acetoxylation. | ||
Additionally, in the same work, Ge's group demonstrated that other acyloxy groups can be introduced at β-C(sp3)–H bonds in the absence of acetoxy sources, under similar reaction conditions (Scheme 64).155 More recently, Zhang et al. improved these conditions by reporting a copper-catalyzed, TBAB/TBAC-accelerated, C(sp3)–H acyloxylation protocol (Scheme 65).156 In this work, heteroaryl and alkenyl carboxylic acids were also suitable coupling partners. Regarding the mechanism, the use of TEMPO as radical scavenger suppressed the reaction, suggesting the involvement of radical species. In addition, deuterium labeling and KIE experiments indicated that the C(sp3)–H activation was irreversible and involved in the r.d.s. (KIE = 2.8).
 | ||
| Scheme 64 Ge's et al. work on Cu-catalyzed β-C(sp3)–H acyloxylation. | ||
 | ||
| Scheme 65 Zhang's et al. work on Cu-catalyzed β-C(sp3)–H acyloxylation. | ||
Likewise, Zhang and co-workers reported a copper-mediated oxidation of β-C(sp3)–H bonds of propionamides with organosilanes (Scheme 66).157 Vinyl and arylsilanes were suitable for this transformation, providing the desired product from moderate to good yields. A pathway involving a C–O coupling of the C(sp3)–H bond with phenol generated from trimethoxy(phenyl)silane was ruled out since the phenol itself is not a suitable reaction partner. O18-labeling experiments showed that Cu(OAc)2 served as the source of the oxygen to generate the C(sp3)–O bond during the aryloxylation process.
 | ||
| Scheme 66 Cu-promoted β-C(sp3)–H aryl- and vinyloxylation with organosilanes. | ||
Based on these results, the proposed reaction pathway begins with the C(sp3)–H bond activation and oxidation to an alkyl–copper(III) species. This species then undergoes C–O reductive elimination between the alkyl group and an acetate ligand to provide the newly inserted ester group and a copper(I) species ligated to the DG (intermediate L in Scheme 67). Oxidation of copper(I) to copper(II), presumably with Ag2O and HOAc, generates intermediate LI, which allows the hydrolysis of the acetate inserted at the β-position, giving the copper(II) species LII. Oxidation via disproportionation and subsequent transmetalation gives intermediate LIV. Final reductive elimination between the alcohol and the aryl or alkenyl group provides the desired products.
 | ||
| Scheme 67 Reaction mechanism for the aryl- and vinyloxylation with organosilanes. | ||
An oxidative demethylation of α-quaternary propionamides was reported by the Fu's group using Cu(OAc)2 (2 equiv.), TMSOAc (4 equiv.) as additive, K2CO3 (2 equiv.) and Na2CO3 (1 equiv.) as bases in DMSO at 150 °C under O2 atmosphere, using 4Å molecular sieves (Scheme 68).158 The authors suggested that this transformation might proceed via similar mechanism of initial β-C(sp3)–H acetoxylation, next hydrolysis of the ester group and final oxidative decarboxylation. In fact, when a β-acetoxylated product was subjected to the reaction conditions, the formation of the demethylated product was observed.
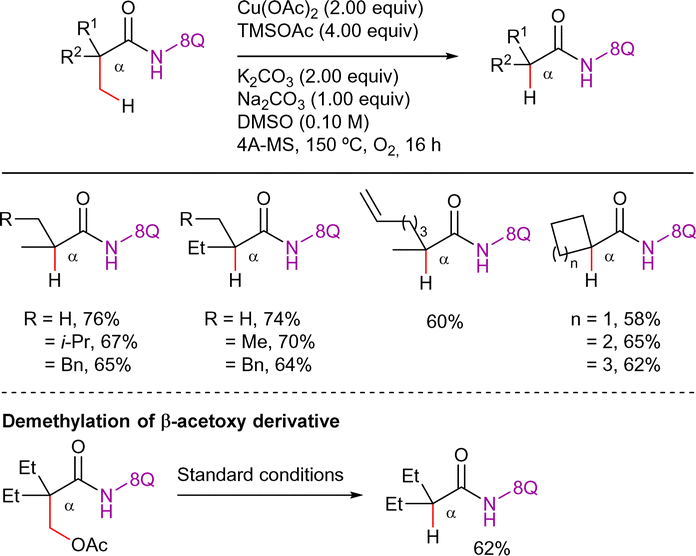 | ||
| Scheme 68 Cu-promoted β-C(sp3)–H demethylation. | ||
4. Fe-catalysis
Iron is the second-most abundant metal in Earth's crust, after aluminium, and is relatively inexpensive compared to the precious metals often used in catalysis.159 Moreover, iron can adopt oxidation states ranging from−2 to +5 (and in rare cases +6).160–162 High-oxidation state iron catalysts (+3, +4, or even +5) are most commonly employed in nonactivated C(sp3)–H bond functionalization reactions. Currently, iron-catalyzed C(sp3)–H bond functionalization is primarily limited to C–C bond-forming processes, such as arylation and alkylation.In 2013, Nakamura et al. reported the first 8-AQ-directed C(sp3)–H arylation of α,α-disubstituted propamide derivatives with diaryl zinc reagents employing Fe(acac)3 as catalyst. The use of 1,2-bis(diphenylphosphino)benzene (dppbz) as ligand and 1,2-dichloro-2-methylpropane (DCIB) as organic oxidant allowed the formation of arylated products in good yields (Scheme 69).163 The experimental procedure of this reaction involves the previous mixture of ArMgBr (7.00 equiv.) and ZnBr2·TMEDA (3 equiv.) to form in situ 3.00 equiv. of Ar2Zn which probably act as nucleophiles in the reaction. The remaining 1.00 equiv. of ArMgBr most likely deprotonates the amide proton.
 | ||
| Scheme 69 8-AQ-assisted β-C(sp3)–H arylation with Grignard reagents. | ||
The arylation reaction was susceptible to steric effects in the aryl magnesium bromide reagent. Whereas with para- and meta-tolyl magnesium bromides, the arylated products were isolated in good yields, with ortho-tolyl derivative, the starting carboxamide was recovered unaltered.
The significantly greater reactivity of a methyl group compared to a benzylic group rules out a radical mechanism, while the strong dependence of the yield on the substrate's structure and the ligand indicates the involvement of organoiron intermediates in key steps. The KIE experiments suggested that the C–H bond cleavage is the r.d.s. of the reaction, with a primary KIE of 2.4 observed in parallel reactions and an intermolecular KIE of 4.0.
Further mechanistic studies performed by Chen's group revealed that the C(sp3)–H activation step could be promoted by iron(II) or iron(III) species through a two-state reactivity (TSR) scenario. The low-spin iron(II) singlet state, initially an excited state, would transition to the quintet-spin ground state and facilitate the C–H bond cleavage.164 From this point, inspired by the mechanistic knowledge on iron-catalyzed C(sp2)–H arylation,62,165 the authors proposed that the next step involves an aryl transmetalation, followed by the oxidation of FeII to FeIII through a single-electron transfer (SET). The dichloroalkane would act as an oxidizing agent, facilitating the final C–C coupling and completing the C–H functionalization. FeI species would also be generated and reoxidized to the active FeII catalyst by another SET oxidation by DCIB. The ligand sphere of iron would thus be essential in the TSR mechanism by stabilization of the reactive low-spin state that mediates the C–H activation.
In 2014, Ackermann's group demonstrated that a triazole-based DG also afforded excellent results in the iron-catalyzed arylation of unactivated C(sp3)–H bonds with Ar2Zn (Scheme 70).166 The reaction conditions were very similar, with an increase in the catalyst loading and reaction temperature. Removal of the DG was achieved using concentrated HCl at 130 °C.
 | ||
| Scheme 70 β-C(sp3)–H arylation with Ar2Zn using a triazole type-DG. | ||
In 2015, Nakamura's group reported the direct methylation of a C(sp3)–H bond with a AlMe3/FeIII system by using the 8-AQ as DG (Scheme 71).167 In this work, AlMe3 was used as the methyl donor and 2,3-dichlorobutane (2,3-DCB) as oxidant, along with (Z)-1-phenyl-1,2-bis(diphenylphosphino)ethene (Ph-dppen) as ligand. The reaction was applied to a variety of α,α-disubstituted propanamide substrates.
 | ||
| Scheme 71 β-C(sp3)–H alkylation with AlMe3. | ||
Alternatively, Ackermann's group developed a complementary methodology for the β-C(sp3)–H alkylation with ZnCl2·TMEDA using TAM as DG (Scheme 72).168 In this work, optimal results were achieved at near ambient temperature with FeCl3 as catalyst and DCIB as a mild oxidant. The strong selectivity for functionalizing the methyl C–H bond over the weaker benzylic C–H bond suggested an organometallic C–H metalation mechanism.
 | ||
| Scheme 72 β-C(sp3)–H alkylation with MeMgBr/ZnBr2 using a triazole type-DG. | ||
On the other hand, in 2017, Nakamura's group expanded the methodology to aryl-, heteroaryl-, and alkenyl boron reagents as coupling partners (Scheme 73).169 The success of this reaction relied on the use of 8-AQ as DG, Fe(acac)3 as catalyst and a ZnII-salt as a co-catalyst, along with a diphosphine ligand bearing a conjugated backbone [(Z)-1,2-bis-(diphenylphosphino)ethene or its electron-rich congener, (Z)-1,2-bis[bis(4-methoxyphenyl)phosphine]ethene].
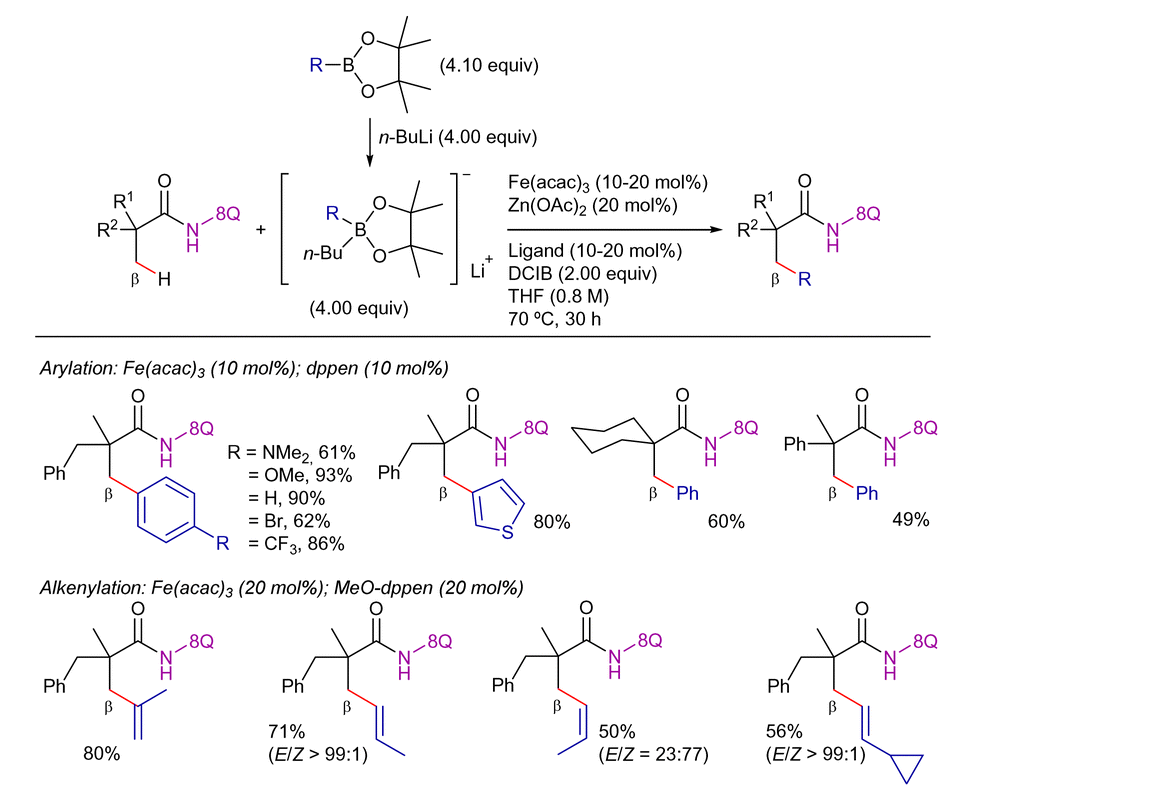 | ||
| Scheme 73 β-C(sp3)–H arylation and alkenylation with organoboron reagents. | ||
Based on experimental mechanistic studies, the authors postulated the mechanism shown in Scheme 74. Zinc-assisted deprotonation gives the zinc amide LV, and then boron/iron transmetalation gives the organoiron(III) intermediate LVI. Conversion of LVI to the β-arylated product via a C–H oxidative addition mechanism (via an iron(V) species LVII) would be unlikely. Alternatively, the C–H activation through σ-bond metathesis forming intermediate LVIII is postulated. Ferracycle LVIII then reacts with another phenyl group from phenylboronate and undergoes reductive elimination to form the new C–C bond product, generating the organoiron(I) intermediate LIX. This seemingly unstable iron(I) species may be stabilized through electron delocalization over the conjugated ligand backbone. Finally, the product is released by protonolysis, generating iron(I) species that can be easily oxidized in the presence of O2.
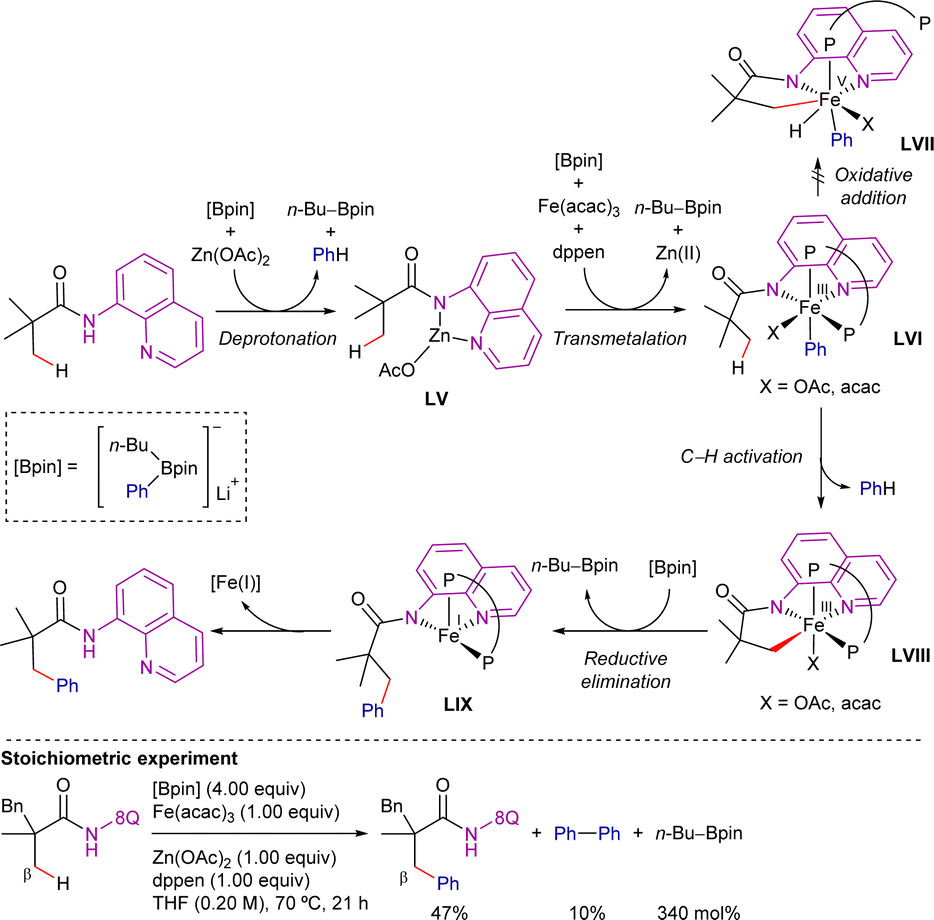 | ||
| Scheme 74 Proposed mechanism for C(sp3)–H arylation with organoboron reagents. | ||
5. Co-catalysis
Cobalt has achieved a privileged status as catalyst due to its low cost, high abundance and variable oxidation states (+1, +2, +3 and +4). Since the first cobalt-catalyzed work of Kharasch and Fields on the homocoupling of Grignard reagents,170 several efficient cross-couplings,171 hydroformylations,172 and many other reactions have been reported.56,173–175In the field of cobalt-catalyzed C–H functionalization reactions, two categories can be defined based on the oxidation state of the catalysts:176–179 a low-valent approach, where the active catalyst is a cobalt(0) or cobalt(I) species,180,181 and a high-valent approach, where the active catalysts typically feature a cobalt-center in the +3 oxidation state.182–186 Nonetheless, advancements in DG-assisted cobalt-catalyzed C(sp3)–H functionalization have predominantly been achieved through high-valent cobalt catalysis.61 These transformations primarily rely on bench-stable Cp*Co(III) complexes in combination with monodentate coordinating DGs.187,188 Alternatively, they can be facilitated via the in situ oxidation of cobalt(II) catalyst precursors, enabled by bidentate chelation assistance, particularly from 8-AQ, picolinamide, and pyridine-2-sulfonamide. In particular, when cobalt(II) precursors are employed, two plausible operative pathways can be considered. One option is that the catalytic cycle begins with the coordination of the substrate to the cobalt(II) salt, forming a cobalt(II)-substrate complex, which is subsequently oxidized to a cobalt(III) species that undergoes C–H activation. Alternatively, a second option is that the cobalt(II) catalyst is initially oxidized to a cobalt(III) salt, followed by substrate coordination. Literature evidence supports both pathways, and the most likely operative mechanism depends on the specific reaction conditions and/or the substrate used in the transformation.
The following are relevant contributions in this area grouped by the type of bond formed.
5.1. C–C bond formation
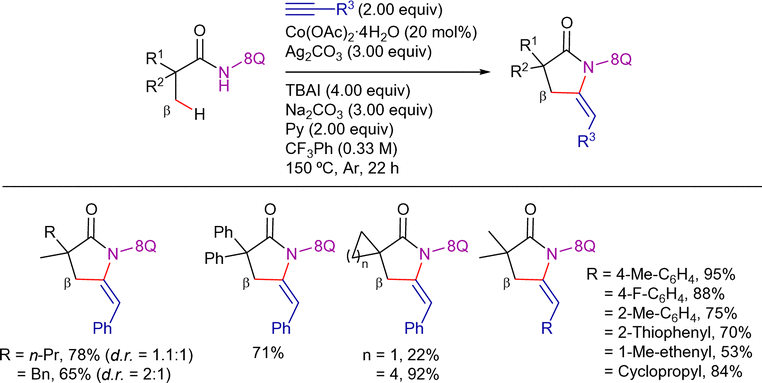 | ||
| Scheme 75 Sequential β-C(sp3)−H alkynylation/annulation with terminal alkynes. | ||
In line with the previously mentioned reports using nickel- and copper-catalysis,89,139 the authors suggested that this transformation goes through an oxidative alkynylation process and subsequent Ag/TBAI-mediated intramolecular annulation reaction (Scheme 76). The authors detected the alkynylated intermediate c in the reaction mixture through GC-MS analysis at short reaction times and demonstrated its conversion into the corresponding 5-(E)-ethylidene-pyrrolidin-2-one derivative under the reaction conditions. They proposed that an alkynyl–cobalt(IV) or alkynyl–cobalt(III) intermediate undergoes reductive elimination to generate intermediate c. Additionally, an alkynyl radical intermediate was suggested based on a control experiment using 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) as a radical inhibitor. However, since the reaction proceeded with moderate yield even in the presence of TEMPO and no TEMPO adduct was detected, the involvement of an alkynyl radical remains uncertain. Notably, radical-trapping experiments in transition-metal catalysis should be interpreted cautiously, as radical-trapping reagents may also interact with transition-metal species.190
 | ||
| Scheme 76 Sequential β-C(sp3)–H alkynylation/annulation process. Proposed mechanism. | ||
On the other hand, the use of aromatic solvents was essential for this transformation, and the solvated metallacycle intermediate LX′ was identified via MS (MALDI-TOF) analysis of the reaction solution. Therefore, both pyridine and PhCF3 may play a role in the formation of the metallacycle intermediate. Additionally, since no deuterium scrambling is observed, the C–H activation step is irreversible and presumably the r.d.s. (competitive KIE experiment with a value of PH/PD = 3.3).
Relying on related work, the authors suggest that TBAI may act as a phase-transfer catalyst.
Interestingly, Zhang et al. also reported one example demonstrating the viability of carrying the 8-AQ-assisted cobalt-catalyzed C(sp3)–H alkenylation of the pivalamide derivative with phenyl acetylene. The reaction was carried out using NaOAc (50 mol%) and HOAc (2 equiv.) as additives (Scheme 77).88 Although no mechanistic studies have been conducted on this outcome, the observed reactivity could be rationalized by a pathway involving C–H activation, followed by a plausible migratory insertion and subsequent protonolysis to yield the alkenylated product. Without a silver(I) salt in the medium, the alkynyl–cobalt(III) intermediate does not form, thereby preventing the formation of the alkynylated intermediate.
 | ||
| Scheme 77 β-C(sp3)–H alkenylation with terminal alkynes. | ||
Recently, Hamlin and Dixon et al. have described an efficient protocol for the enantioselective Co-catalyzed β-C(sp3)–H alkenylation of benzyl thioamide derivatives with but-2-ynoate ester coupling partners (Scheme 78).191 The mild reaction conditions were compatible with a broad range of α-dimethyl-substituted thioamide partners, delivering the alkenylated products as single regioisomers in excellent yields (up to 85%) and high enantiomeric excess [up to 91:9 enantiomeric ratio (er), or >99:1 er after a single recrystallization]. Post-synthetic modifications allowed the conversion of the thioamide group into a wide range of functional groups using MeOTf and different nucleophiles (Scheme 79).
 | ||
| Scheme 78 Enantioselective β-C(sp3)–H alkenylation with internal alkynes. | ||
 | ||
| Scheme 79 Conversion of the thioamide into different functional groups. | ||
As illustrated in Scheme 80, DFT calculations suggest that the active species, Cp*Co(O2CR)+, is generated through the interaction of Cp*Co(III) with the L-tert-leucine-derived chiral acid ligand under the reaction conditions. The catalytic cycle then begins with the coordination of the thioamide substrate to the proposed active catalyst, forming complex LXII. The C–H activation step is initiated via a κ2–κ1 displacement of the carboxylate by the β-hydrogen of the substrate (LXIII), proceeding through a CMD mechanism to afford intermediate LXIV. The steric bulk of the acid co-catalyst with the benzyl group is likely a key factor in governing stereoselectivity at this stage. Subsequent migratory insertion of the alkyne yields intermediate LXV, which, upon protonolysis, leads to the formation of the desired alkenylated product while regenerating the active Cp*Co(O2CR)+ species. The regioselectivity of the migratory insertion step is also influenced by steric repulsions with the benzyl group at the corresponding transition state.
 | ||
| Scheme 80 Enantioselective β-C(sp3)–H alkenylation with internal alkynes. Proposed mechanism. | ||
On the other hand, Sundararaju's group developed a highly regioselective and stereoselective method for the γ-alkenylation of 8-methylquinoline derivatives with alkynes under mild conditions.192 The reaction employed a Cp*Co(CO)I2 catalyst precursor (5 mol%), AgOTf and AdCO2H as additives (10 mol% each), and TFE as solvent at 80 °C (Scheme 81). Under these conditions, the cis-addition of the C(sp3)–H bond to the alkyne proceeded in good yields. The reaction tolerated a wide range of substituted quinolines and symmetrical alkyl internal alkynes. Unfortunately, aryl alkynes were less effective and mixtures of regioisomers were obtained using unsymmetrical alkynes. Terminal alkynes could also be used by changing to AgSbF6 and PivOH as additives, and DCE as solvent, albeit the products were isolated in low yields.
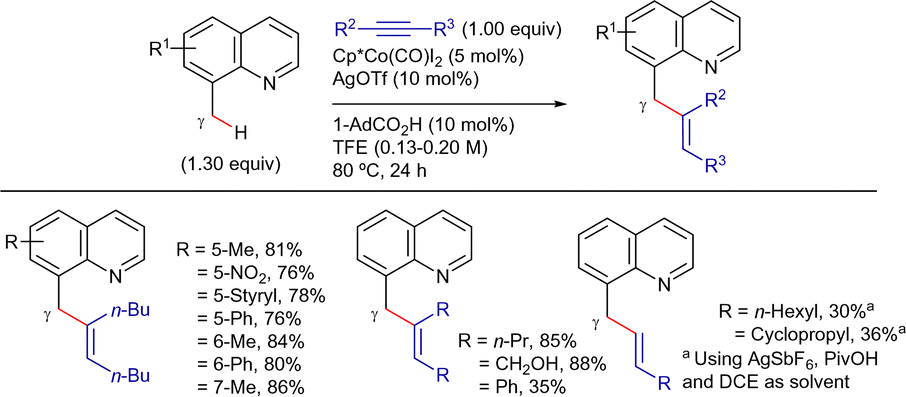 | ||
| Scheme 81 Cp*CoIII-cat. γ-C(sp3)–H alkenylation with alkynes. | ||
Intermolecular competitive experiments revealed that the reaction was favoured with electron-rich substituents at the quinoline ring and alkyl substitution at the internal alkyne. Deuterium labeling experiments suggested an irreversible cyclometalation pathway which, as determined by DFT calculations, appears to occur via an outer-sphere C–H activation rather than an inner-sphere pathway. The proposed reaction mechanism is depicted in Scheme 82. First, halogen abstraction from [Cp*Co(CO)I2] with AgOTf and ligand exchange generates the catalytically active cationic cobalt(III) species that then undergoes coordination to the 8-methylquinoline derivative to generate intermediate LXVI. Subsequent external base-assisted cyclometalation via a CMD mechanism leads to the cationic intermediate LXVII. From intermediate LXVII, coordination of the alkyne in a preorganized orientation followed by its insertion into the C(sp3)–Co bond leads to the cationic 7-membered intermediate LXIX. Finally, after a protodemetalation step, the final alkenylated product is released and the catalytically active cobalt(III)-species is regenerated.
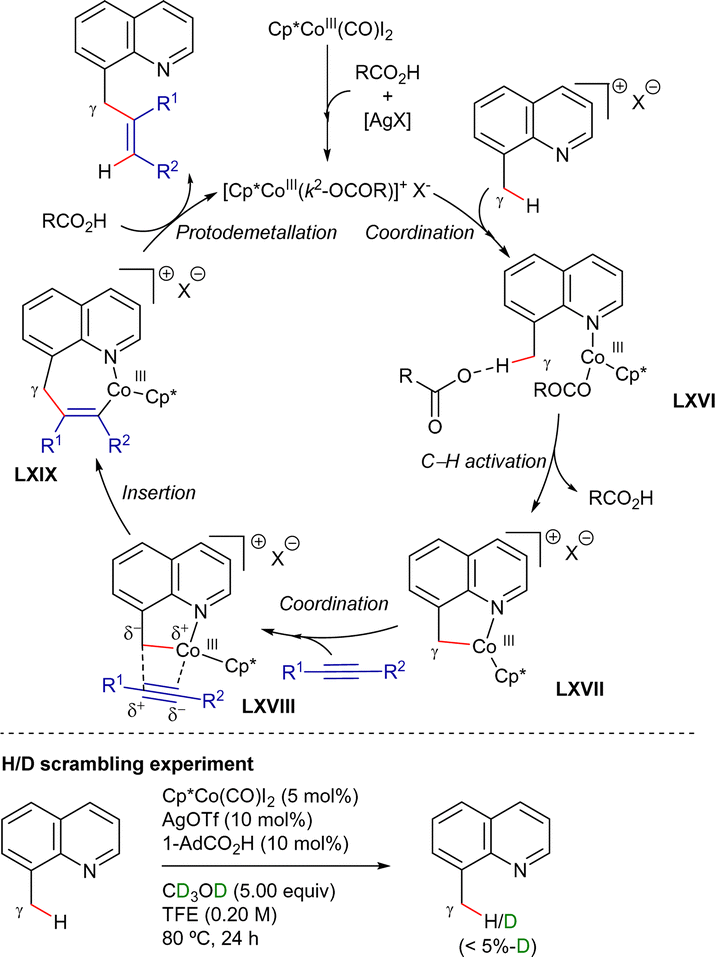 | ||
| Scheme 82 γ-C(sp3)–H alkenylation with alkynes. Proposed mechanism. | ||
 | ||
| Scheme 83 Sequential β-C(sp3)–H carbonylation/annulation. | ||
A parallel KIE experiment conducted by Sundararaju's group revealed a kH/kD value of 1.42, suggesting that C–H activation is not the r.d.s. Both Sundararaju's and Lei's groups reported that the addition of TEMPO significantly reduced reaction yields, implying the potential involvement of a SET pathway. However, no TEMPO adducts were identified, making the characterization of radical intermediates challenging, as previously mentioned. Furthermore, Lei's group conducted an X-ray absorption spectroscopy (XAS) analysis, which suggested that the oxidation of cobalt(II) to cobalt(III) is more likely to occur after cobalt(II) coordinates to the amide derivative. On these bases, although a plausible mechanism was still unclear at that stage, Lei and Sundararaju considered the possibilities shown in Scheme 84. Initial coordination of cobalt(II) to the propanamide substrate, followed by NH deprotonation (possibly by silver carbonate) and subsequent oxidation to cobalt(III) promoted by silver(I) leads to intermediate LXX. Subsequent carboxylate/carbonate assisted CMD provides cobaltacycle LXXI. Coordination of CO to the cobalt(III) intermediate LXXI, followed by its insertion into the Co–C bond generates intermediate LXXII. From here, the final product succinimide could be generated by means of: (i) a reductive elimination step, also generating cobalt(I) species which would be re-oxidized by silver (path a), or (ii) one electron oxidation to generate the cobalt(IV) intermediate LXXIII, which would then undergo the reductive elimination to give the final succinimide product, regenerating cobalt(II) species (path b). Besides these options, the authors also considered that intermediate LXXI could undergo one electron oxidation to the cobalt(IV) intermediate LXXIV, and then insertion of CO into the Co–C bond to lead to intermediate LXXIII (path c), which would subsequently undergo reductive elimination to generate the final product and cobalt(II) species.
 | ||
| Scheme 84 Proposed mechanism for the Co-cat. β-C(sp3)–H cyclative carbonylation. | ||
More recently, Sundararaju's group developed a complementary two-chamber method in which the CO atmosphere was created by CO2 reduction in the presence of disilanes, achieving similar yields to those of previous protocols (Scheme 85).196
 | ||
| Scheme 85 Sequential β-C(sp3)–H carbonylation/annulation with CO2 as CO-source. | ||
 | ||
| Scheme 86 β-C(sp3)–H methylation with trimethylaluminum. | ||
In the next year, Shi's group reported a Co(OAc)2·4H2O-catalyzed, 8-AQ-directed double C(sp3)–H activation strategy for the synthesis of bicyclo[4.1.0]heptane and bicyclo[3.1.0]hexane derivatives by using α-substituted 2-methylhept-6-enamide or 2-methylhex-5-enamide derivatives as starting materials (Scheme 87).198 The formation of larger bicyclo[5.1.0]octanes was not possible.
 | ||
| Scheme 87 Double β-C(sp3)–H bond functionalization. | ||
Since no deuterium scrambling is observed, the authors suggest that the C–H activation step is irreversible and presumably the r.d.s. As shown in Scheme 88, this reaction was proposed to start with the coordination of the cobalt(II)-species to the 8-AQ and the double bond of the substrate, followed by silver(I)-mediated single-electron oxidation to generate the cobalt(III)-species LXXV. Then, subsequent irreversible activation of the methyl C(sp3)–H bond generates intermediate LXXVI, which, after alkene migratory insertion, leads to intermediate LXXVII. From this point, methylene C(sp3)–H activation at the same β-position gives the formation of the four-membered cobaltacycle intermediate LXXVIII. Subsequent reductive elimination and decomplexation would release the bicyclo[n.1.0]alkane product and cobalt(I) species, that would be reoxidized by silver(I)-species to regenerate the active cobalt(II)-species, closing the catalytic cycle. To support this mechanism the authors analyzed the reactivity of a 2-methylhept-5-enamide derivative that presented two allylic hydrogens. In this case, a mixture of the desired bicyclo[3.1.0]hexane derivative along with a monocyclic vinylcyclopentane derivative was obtained. The formation of the later was explained via β-hydride elimination from intermediate LXXVII′ (analogue of intermediate LXXVII).
 | ||
| Scheme 88 Double β-C(sp3)–H functionalization. Proposed mechanism. | ||
Interestingly, the authors also reported the synthesis of a bicyclo[3.1.0]hexane from pivalamide and allyl methyl carbonate, under slightly modified reaction conditions. This reaction presumably proceeds through a C(sp3)–H allylation/cyclopropanation sequence, which involves the cleavage of three C(sp3)–H bonds (Scheme 89).
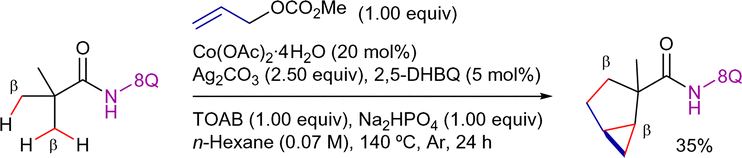 | ||
| Scheme 89 Triple β-C(sp3)–H functionalization. | ||
In parallel, Shi's group showed that the γ-alkylation of 8-methylquinolines was feasible by employing diazo compounds as coupling partners (Scheme 90).199 The reaction presumably proceeded through primary C(sp3)–H cobaltation/carbene insertion. C5, C6 and C7 substituted quinolines were equally reactive, tolerating the presence of electron donor and electron-withdrawing groups. Remarkably, halogen and Bpin substitution remained intact after the transformation, providing flexible handles for further orthogonal functionalization. Both the symmetrical and unsymmetrical diazomalonates were compatible. However, low yield was observed when di-tert-butyl diazomalonate was employed. The alkylated products served as precursors for the synthesis of tricyclic products under reductive conditions or as key intermediates for the synthesis of azatricyclic antibiotics (Scheme 91).200
 | ||
| Scheme 90 γ-C(sp3)–H alkylation with diazo compounds. | ||
 | ||
| Scheme 91 Synthetic applications of γ-alkylated 8-methylquinoline derivatives. | ||
Regarding the mechanism, deuterium labeling experiments indicated that the cyclometalation step was reversible (observing deuterium scrambling) but the subsequent carbene formation/insertion seemed faster than the back reaction. Furthermore, parallel and competitive KIE experiments provided values of 6.7 and 8.1, respectively, indicating that the cleavage of the C–H bond was likely involved in the r.d.s.
Later, Ackermann's group showed that the cobalt(III)-catalyzed γ-C(sp3)–H bond mono-methylation of 8-methylquinoline was viable using the commercially available trimethylboroxine as methyl source (Scheme 92).201
 | ||
| Scheme 92 γ-C(sp3)–H alkylation with trimethylboroxine. | ||
On the other hand, Sun et al. reported a novel way to introduce maleimides at the γ-CH3 position of 8-methylquinoline derivatives under cobalt(III)-catalysis using a combination of AgOTf and Zn(OAc)2 as additives (Scheme 93).202 The reaction mechanism, depicted in Scheme 94, involves the formation of the catalytically active cobalt(III)-species and coordination with the quinoline substrate to form cationic cobalt(III)-species. Next, C–H activation and subsequent migratory insertion of the maleimide into the Co–C bond generates a 7-membered cationic cyclometallated complex which by protodemetalation releases the final product, regenerating the active catalyst. Mechanistic studies showed that benzyl activation was reversible in the absence of maleimide. However, no deuteration at the benzyl position of quinoline was observed in its presence, indicating that functionalization of the C–Co bond formed after the C–H activation step is faster than its protodemetalation. A KIE value of 2.1 indicated that cleavage of the methyl C–H bond was potentially involved in the r.d.s.
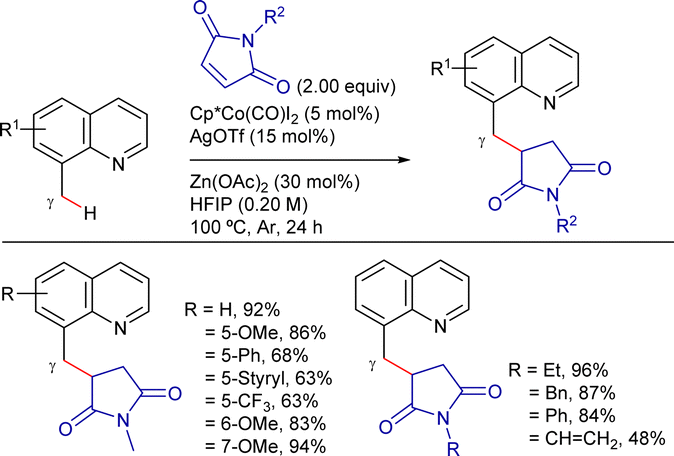 | ||
| Scheme 93 γ-C(sp3)–H alkylation with maleimides. | ||
 | ||
| Scheme 94 γ-C(sp3)–H alkylation with maleimides. Proposed mechanism. | ||
A complementary protocol for the coupling between 8-methylquinoline and maleimide derivatives was reported by Sharma et al.203 In this case, a carboxylic acid was used as additive and TFE as solvent (Scheme 95). Albeit the reaction yields were slightly lower, the scope was broad, being possible to apply the method to mono- and di-substituted quinoline substrates with diverse alkyl, unsaturated and halogen substitution at any position. Moreover, the method worked well with a wide range of N-protecting groups at the maleimide and more interestingly, the direct use of the NH free maleimide was also tolerated albeit provided the product in low yield. 8-Ethylquinoline was also compatible with the method, giving the alkylated products with moderate yields and diastereoselectivities. Since no deuterium scrambling is observed, the catalytic cycle proposed by the authors would be like the one previously proposed by Sun202 but, in this case the C–H activation step is irreversible and presumably the r.d.s. (kH/kD value of 2.1).
 | ||
| Scheme 95 γ-C(sp3)–H alkylation with maleimides. | ||
On the other hand, Kong, Chang, and Li described a method for the regio- and stereoselective benzylic allylation of 8-methylquinolines with (per)fluoroalkyl olefins, which involves benzylic C–H activation followed by C–F bond cleavage. (Scheme 96).204 Interestingly, a calcium additive was employed as halide scavenger to facilitate the formation of the active metal species. Regarding the organofluorine coupling partner, mono-substituted olefins were tolerated regardless of the length of the fluorine chain. The method was less effective with tetrasubstituted or less activated olefins.
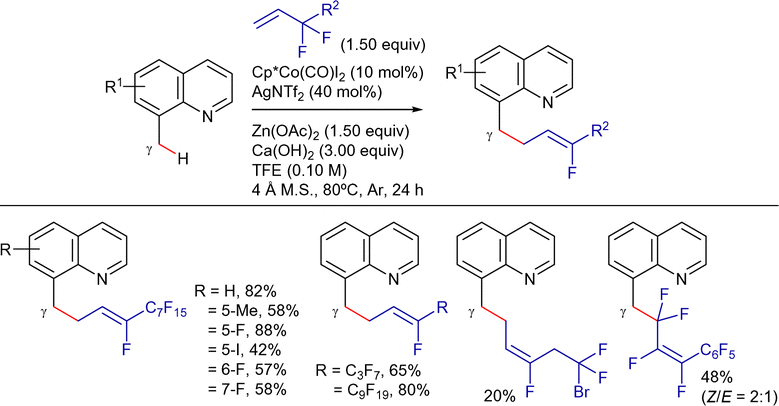 | ||
| Scheme 96 Cp*CoIII-cat. γ-C(sp3)–H allylation with polyfluorinated olefins. | ||
Based on previous works, the authors proposed that the catalytic cycle starts by first formation of the active Cp*CoX2 (X = OAc or NTf2) followed by coordination to the quinoline substrate and subsequent C–H activation to generate the five-membered metallacycle LXVII (Scheme 97). Next, olefin coordination and migratory insertion lead to the 7-membered cobaltacycle LXXXIII. Finally, β-F elimination via a syn-coplanar transition state delivers the final product with concomitant generation of a cobalt(III) fluoride species. Anion exchange would regenerate the active cobalt(III) species.
 | ||
| Scheme 97 γ-C(sp3)–H allylation with polyfluorinated olefins. Proposed mechanism. | ||
Later, Zhou, Fan and co-workers showed that azabenzonorbornadienes were compatible coupling partners for the benzylic functionalization of 8-methylquinolines under an Ag/Cp*CoIII catalytic system.205 Remarkably, the method proceeds at only 70 °C (Scheme 98). Electron-donating groups at the C5 position of the quinoline substrate provided the ring-opening products in better yields than electron-withdrawing substituents. However, the method was more sensitive to steric effects, observing a lower yield when a methyl group was at the C7 position rather than at the C6 or C5 ones. Interestingly, methylene benzylic positions were also reactive, providing the desired product in low yields. In contrast, the reaction did not proceed with any or N-protecting group at the azabenzonorbornadienes substrate, being not possible to use oxabenzonorbornadiene and benzonorbornadiene.
 | ||
| Scheme 98 γ-C(sp3)–H alkylation with azabenzonorbornadienes. | ||
Deuterium scrambling experiments indicated that the C–H bond cleavage was reversible (observing deuterium incorporation in the absence of the azabenzonorbornadiene). However, the evolution of the five-membered metallated intermediate to the final product was presumably faster than the back reaction. Nonetheless, a parallel KIE experiment revealed a kH/kD value of 7.9, suggesting that C–H activation is involved in the r.d.s.
4.2. C–N bond formation
In 2015, Ge et al. disclosed the cobalt-catalyzed site-selective dehydrogenative cyclization of propionamide and butyramide derivatives using the 8-AQ as DG (Scheme 99).206 The reaction favoured the functionalization of β-methyl groups over β-methylene and γ- or δ-methyl groups, providing the β-lactam derivatives with high regioselectivity. Interestingly, the authors also observed a preference for the functionalization of γ-benzylic over β-methyl and methylene C–H bonds, thus obtaining the γ-lactams as the major products.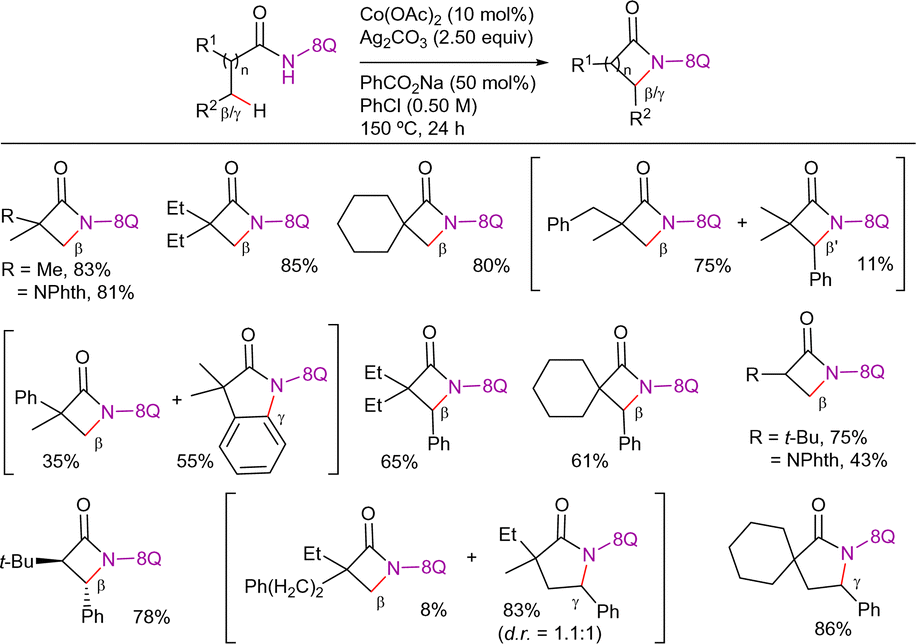 | ||
| Scheme 99 β/γ-C(sp3)–H intramolecular amidation. | ||
Based on mechanistic studies, two distinct pathways were proposed to explain the formation of the four- and five-membered ring products. On the one hand, it seemed unlikely that the formation of β-lactams was a radical-mediated process due to the high selectivity for β-methyl bonds over β-benzyl C–H bonds and the good yields obtained in the presence of TEMPO. Thus, the formation of β-lactams implies the formation of the cobalt(III) complex LXXXIV by coordination of the amide to the cobalt species and a subsequent ligand exchange under basic conditions (Scheme 100). Cyclometalation via a benzoate-assisted CMD mechanism produces intermediate LXXXV. Oxidation of LXXXV with Ag2CO3 gives the cobalt(IV) complex LXXXVI, which leads to the β-lactam compound upon reductive elimination. The newly generated cobalt(II) species could then be re-oxidized to cobalt(III) by silver(I) furnishing the catalytic cycle. Alternatively, the authors also considered that the cyclometalation of the amide can be at cobalt(II) species, forming intermediate LXXXVI by further oxidation. The KIE experiments revealed that the C–H cleavage was presumably irreversible (KIE value of 2.3).
 | ||
| Scheme 100 β-C(sp3)–H intramolecular amidation. Proposed catalytic cycle. | ||
In contrast, in the case of the formation of γ-lactam derivatives, a radical or cationic species seemed to be involved in the catalytic cycle due to the predominant preference for γ-benzylic over β-methyl C–H bonds. Moreover, in the case of γ-lactams the addition of TEMPO considerably reduced the yield, which suggested the participation of radical species or a SET pathway.
The applicability of the method was further enhanced by converting the 8-AQ into the 8-amino-5-methoxyquinoline group to access the free NH lactam products under oxidative conditions (Scheme 101).
 | ||
| Scheme 101 Removal of the DG under oxidative conditions. | ||
Additionally, in the above-mentioned contribution, Ge et al. also reported the cobalt(III)-catalyzed β-C(sp3)–H intermolecular amidation using electron-deficient amides as coupling partners (Scheme 102).206 In general, the desired products were obtained in modest yields, observing complete selectivity towards β-methyl positions over β-methylene and γ-methyl ones. In this case, the activation of γ-C(sp2)–H bonds competed, obtaining the corresponding γ-lactam as the major product.
 | ||
| Scheme 102 β-C(sp3)–H intermolecular amidation. | ||
It is interesting to enote that the synthesis of lactams has also been achieved via Pd-, Cu- and Ni-catalysis.207 However, Ni- (Section 2.2.1)102,103 and Cu-catalyzed (Section 3.2.1)149–151 processes were restricted to the synthesis of β-lactams substrates from α,α-disubstitued propanamide derivatives. On the other hand, Pd-catalyzed synthesis of β-lactams was restricted to α-unsubstituted and α,β-cyclic substrates, and the synthesis of γ-lactams was only feasible from β-substituted substrates.208 Therefore, the cobalt-catalyzed method provided a complementary approach to access monocyclic and spiro β- or γ-lactams.
In 2017, Seayad, Dixon, and their team developed the thioamide-directed Co-catalyzed amidation of C(sp3)–H bonds, utilizing (hetero)aryl- and alkyl-substituted dioxazolones (Scheme 103).209 The method was suitable for α-quaternary and tertiary substrates. Moreover, a broad range of thioamides containing N-heterocycles such as piperazine and morpholine were tolerated and provided the corresponding products in good yields. Notably, reduction of the thioamide group was achieved using a NiII/NaBH4 system or converted into an amide group using Ag2CO3 (Scheme 104).
 | ||
| Scheme 103 β-C(sp3)–H amidation with dioxazolones in carboxylic acid derivatives. | ||
 | ||
| Scheme 104 Post-synthetic modifications of the thioamide group. | ||
The authors proposed that the [Cp*Co(III)]-catalyzed thioamide-directed C–H amidation reaction follows the mechanism shown in Scheme 105. Notably, ab initio calculations suggest that the C–H activation step proceeds via an external concerted metalation-deprotonation (CMD) mechanism rather than an internal CMD pathway. The calculated energy difference between the external and internal CMD transition states was 35 kJ mol−1.
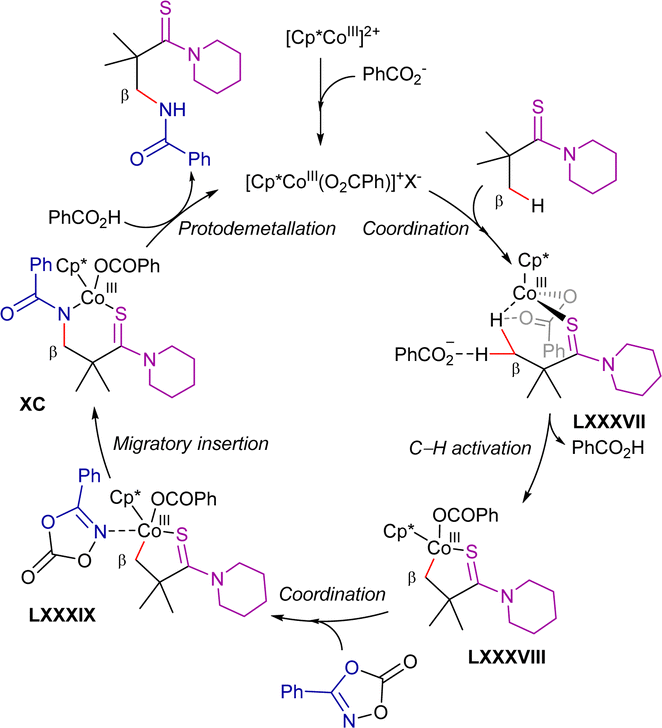 | ||
| Scheme 105 β-C(sp3)–H amidation with dioxazolones. Proposed mechanism. | ||
In 2019, Matsunaga's group reported the enantioselective C(sp3)–H amidation of thioamides with dioxazolones employing a Cp*Co/chiral carboxylic acid (CCA) system (Scheme 106a).210 In particular, N-protected tert-leucine derivatives induced high enantioselectivities. The reactivity and selectivity were both further improved by combining sterically hindered cobalt catalyst with the MS13X zeolite.
 | ||
| Scheme 106 Enantioselective β-C(sp3)–H amidation with dioxazolones. | ||
Later, Matsunaga' group introduced the 2-aryl ferrocene carboxylic acids as a new type of efficient chiral ligands for this transformation. Good reactivity and enantioselectivity were obtained for a wide range of structural diverse amino acids (Scheme 106b).211 Deuterium labeling and DFT calculations revealed that the C–H activation step was presumably irreversible, and it was involved in the enantio-determining step.
In 2019, Loh et al. reported the Cp*CoIII-catalyzed, thioamide-directed amination of C(sp3)–H bonds using anthranils as amine electrophiles (Scheme 107).212 Using 7.5% of [Cp*Co(CH3CN)3][SbF6]2 and 1 equiv. of 1-AdCO2H as additive, the method allowed the introduction of 2-formyl, -acetyl and -benzoyl anilines at the β-position of thioamide derivatives.
 | ||
| Scheme 107 β-C(sp3)–H amidation with anthranils. | ||
Disclosing a similar reactivity, more recently, Li et al. published a complementary method by using 20 mol% of [Cp*Co(CH3CN)3][SbF6]2 as catalyst and i-PrCO2Na as basic additive (Scheme 108).213
 | ||
| Scheme 108 β-C(sp3)–H amidation with anthranils. | ||
In 2016, Sundararaju et al. reported a methodology for the γ-amidation of 8-methylquinoline derivatives with (hetero)aryl dioxazolones under cobalt(III)-catalysis, employing AgSbF6 and NaOPiv as additives in DCE at 100 °C (Scheme 109).214 The reaction was selective and tolerated a variety of functional groups. However, the method was less effective with 3-nonyl-1,4,2-dioxazol-5-one, providing the γ-amidated product in low yield. Diversification of the amidated quinoline derivatives was achieved under a NiII/NaBH4 catalytic system (Scheme 110).
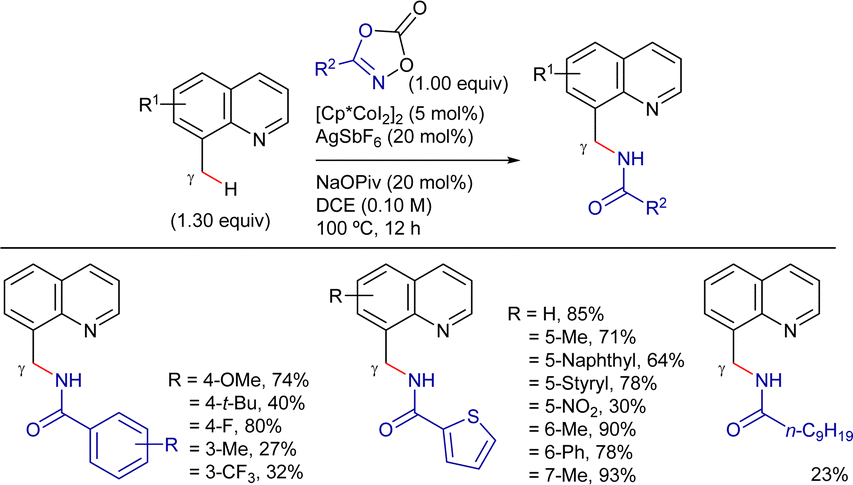 | ||
| Scheme 109 γ-C(sp3)–H intermolecular amidation. | ||
 | ||
| Scheme 110 Diversification of the amidated products. | ||
In 2022, Niu et al. developed two efficient methodologies for the synthesis of indoline and benzoxazine derivatives via cobalt(II)-catalyzed PA-directed selective C(sp3)–H functionalization processes (Schemes 111 and 112).215 A key feature in these protocols is the use of a hydroxylamine-based oxidant.
 | ||
| Scheme 111 δ-C(sp3)–H intramolecular amidation in aniline structures. | ||
 | ||
| Scheme 112 γ-C(sp3)–H intramolecular oxidation in aniline structures. | ||
On the one hand, as can be seen in Scheme 111, the synthesis of indolines required only 5 mol% of Co(OAc)2·4H2O, demonstrating the efficiency of this cobalt-system compared to Pd- or Cu- catalysis.216,217 A wide variety of para-substituted aniline and 4-methyl-2-(2-arylpropan-2-yl)aniline derivatives were well tolerated and afforded the corresponding indoline products in good yields by the direct functionalization of the δ-CH3 positions. Remarkably, substrates having alkenyl, alkynyl, alkyl, phenyl, naphthyl and heteroaryl groups were compatible with the reaction conditions.
On the other hand, as can be seen in Scheme 112, the synthesis of 4H-3,1-benzoxazine products occurred through direct activation of secondary and tertiary γ-C(sp3)–H bonds in ortho-substituted anilines. Interestingly, the reaction was effective with cycloalkyl framework substrates, leading to the desired spirocyclic products in good yields. Moreover, this method proved effective with para-substituted picolinamide derivatives and isoquinoline structures.
According to the authors, both procedures occur through the catalytic cycles depicted in Scheme 113. Both reactions begin with the oxidation of the cobalt(II)-salt to the cobalt(III)-species XCI (this complex was isolated when R1 = H, CCDC 2116720). Intermediate XCI is oxidized intramolecularly to afford cobalt(IV)-imido intermediate XCII. From this stage, the reaction proceeds via two distinct pathways, depending on the R1 group. If R1 is a methyl group, the cobalt(IV) species XCII undergoes intramolecular C–H insertion, followed by reductive elimination, yielding the indoline product and regenerating the cobalt(II) species (path a). While, if R1 group is H, the reaction proceeds via intramolecular hydrogen atom abstraction (HAA) from cobalt(IV) species XCII to deliver the radical intermediate XCIV, leading to the radical-adduct d. This adduct was isolated in some examples as by-product. Nonetheless, intermediate d cannot be transformed to product e by intramolecular nucleophilic substitution, ruling out this pathway. In contrast, isomerization of XCIV generates intermediate XCV, which undergoes radical substitution to deliver the target product e.
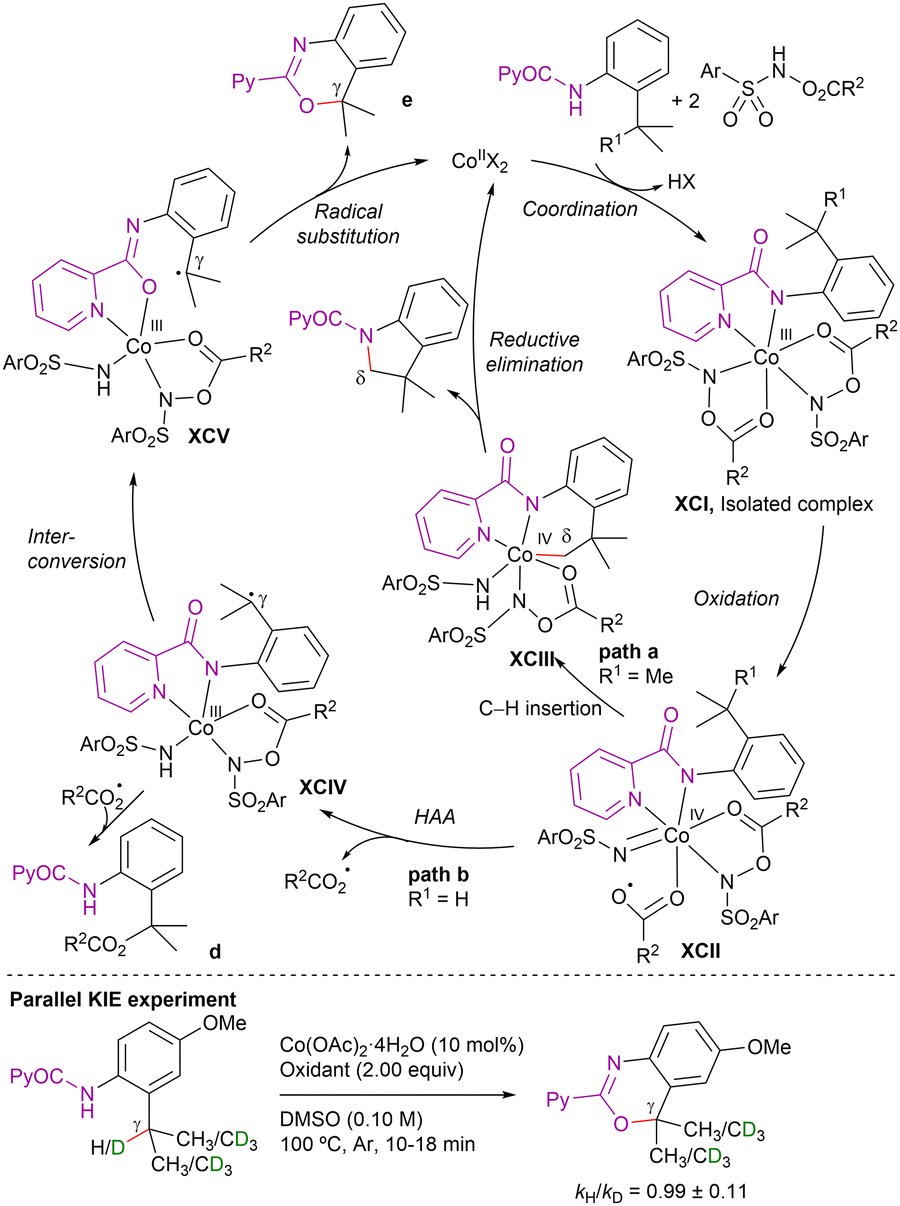 | ||
| Scheme 113 Formation of indoline and benzoxazine derivatives. Proposed mechanisms. | ||
In both the synthesis of indoline derivatives and benzoxazines, no deuterium scrambling was observed, leading the authors to suggest that the C–H activation step is irreversible. However, the parallel kinetic isotope effect (KIE) value of 0.99 ± 0.11 obtained for benzoxazine synthesis indicates that C–H bond cleavage is unlikely to be involved in the rate-limiting step in this case.
In 2022, Song et al. reported the synthesis of indole derivatives starting from (2-pyridyl)sulfonanilide derivatives (Scheme 114).218 This reaction also worked with a hydroxylamine-based compound (ArSO2NHOR) as oxidant. The method showed good functional group tolerance under the optimal conditions, tolerating the presence of halogen groups that could be used in post-synthetic modifications. Substrates with both electron-rich and electron-deficient substituents reacted effectively, yielding the desired products in moderate to good yields. Additionally, substrates containing secondary C–H bonds were also compatible with this protocol.
 | ||
| Scheme 114 SO2Py-directed β-C(sp3)–H intramolecular amidation. | ||
Regarding the mechanism, as no deuterium scrambling was observed, the authors proposed that the C–H activation step is irreversible. Furthermore, the reaction was inhibited upon the addition of TEMPO, and when BHT was introduced into the catalytic system, a TsNH-BHT adduct was detected. Based on these findings, the authors suggested that a single-electron transfer (SET) step is involved in the transformation. Based on these studies, the authors suggested the mechanism outlined in Scheme 115. First, in the presence of the (2-pyridyl)sulfonanilide derivative and TsNHOPiv, the cobalt(II) catalyst evolves into the cobalt(III) species XCVI, which by intramolecular one-electron oxidation would form the cobalt(IV) imide intermediate XCVII. Then, the intermediate XCVII undergoes a C(sp3)–H insertion forming the cobalt(IV) imide XCIX. However, there is an alternative pathway which would involve XCVI becoming XCIXvia the cobalt(III) intermediate XCVIII aminyl radical. This would involve a homogeneous intramolecular N–O bonding reaction followed by one-electron oxidation processes and hydrogen atom abstraction (HAA).
 | ||
| Scheme 115 Formation of indoline and benzoxazine derivatives. Proposed mechanisms. | ||
In any case, from the cobaltacycle, XCIX a β-H elimination step leads to intermediate C, and a reductive elimination results in intermediate CI and TsNH2. Subsequently, oxidation of CI to cobalt(IV) species by oxidative addition of TsNHOPiv, followed by C(sp2)–H activation produces intermediate CII. This intermediate CII undergoes reductive elimination to afford the desired product along with cobalt(II) species.
Alternatively, the authors also suggest that protonation of intermediate CI generates the dehydrogenation byproduct f (isolated product in some examples). Then, a Heck-type mechanism involving amino-metalation of the double bond and subsequent β-hydride elimination would lead to the indole product. Besides these options, the authors also considered an alternative path through a reductive elimination step from intermediate XCIX to generate indoline g and further oxidation to form product I. However, oxidation of indoline g was not observed under the optimized conditions.
Summary and outlook
The synthetic efficiency achieved in the directed functionalization of inactive C(sp3)–H bonds has established DG-assisted cyclometalation as one of the most optimal approaches to diversify organic molecules. To date, this area has mainly been governed by the use of 4d and 5d metals as catalysts. However, in recent decades, the impetus to move from precious metals to cost-effective, cheaper and earth-abundant first-row transition metals has driven new developments in nickel, iron, copper and cobalt catalysis.(A) Nickel catalysis has enabled the selective β-C(sp3)–H functionalization of carboxylic acid derivatives using 8-AQ and related DGs. Various oxidation states of Ni facilitate diverse bond-forming reactions (C–C, C–N, C–S, C–Se, and C–O) via NiII/NiII or NiII/NiIV pathways. However, Ni catalysts generally exhibit lower reactivity toward inert C(sp3)–H bonds compared to Pd and often require harsh conditions. Key challenges include oxidation state control, mechanistic complexity, and the development of milder catalytic systems. A unified mechanistic framework remains elusive, and advances in more versatile and traceless DGs and enantioselective transformations are needed for broader applicability.
(B) Copper(II)-salts, in conjunction with 8-AQ as a DG, have been employed in the β-functionalization of aliphatic carboxylic acid derivatives, enabling arylation, alkenylation, carbonylation, amidation, and acyl-, aryl-, and vinyloxylation reactions. However, these transformations often necessitate stoichiometric copper due to its multifaceted roles throughout the catalytic cycle and the challenges associated with stabilizing high-valent copper(III) species. The development of sustainable catalytic cycles, particularly those leveraging O2 or electrochemical approaches, represents a promising yet underexplored avenue. Additionally, the design of chiral ligands capable of achieving high enantioselectivity in these transformations remains a significant challenge.
(C) Iron catalysis remains relatively underexplored but has demonstrated remarkable versatility in β-arylation, alkenylation, and methylation of aliphatic carboxylic acids using various organometallic reagents. These transformations, facilitated by directing groups such as 8-aminoquinoline (8-AQ) or triazoles, often proceed under significantly milder conditions compared to other 3d transition metals. Nonetheless, the transient nature of iron intermediates complicates mechanistic elucidation, requiring a combination of computational studies, kinetic experiments, and spectroscopic techniques to unravel reaction pathways. Understanding and harnessing iron's multiple oxidation states, particularly FeII/FeIII and FeIII/FeV, is crucial for designing well-defined catalytic cycles. This knowledge will undoubtedly aid in advancing Fe-catalyzed enantioselective C(sp3)–H functionalization, a field that, like nickel and copper, remains largely unexplored.
(D) The d6-electron configuration of cobalt(III) complexes has been crucial in the recent development of numerous directed C–H transformations. Monodentate strongly coordinating DGs have been typically used in Cp*Co(III)-catalyzed reactions, while bidentate DGs (specially, 8-aminoquinoline and picolinamide) have commonly been exploited with cobalt(II)-salts under oxidative conditions. Most processes were directed at β-position in carboxylic acid derivatives, achieving also the functionalization at γ- and δ-position in aniline derivatives. In addition, a handful of enantioselective Cp*Co(III)-catalyzed C–H functionalization procedures have been reported using chiral carboxylic acids as chiral additives to promote the enantioselective C–H activation step. The field of enantioselective C–H functionalization holds immense potential in organic synthesis, drug discovery and medicinal chemistry.
(E) Overall, considering the sustainable and cost-effective nature of 3d metal-catalyzed C–H activation, substantial progress in this rapidly evolving field is expected. The primary challenges that remain include: (i) the widespread use of 8-AQ as a DG, which often requires harsh conditions for its removal, highlighting the need for more versatile or traceless alternatives, and (ii) the dependence on substantial steric activation, such as the Thorpe–Ingold effect, to facilitate C–H activation in most substrates. Undoubtedly, future progress will be driven by a deeper mechanistic understanding, particularly concerning oxidation state control, ligand effects, and reaction intermediates, ultimately leading to more efficient and widely applicable methodologies.
Furthermore, the integration of emerging technologies, such as photochemistry and electrochemistry, is expected to drive advancements in sustainable C–H functionalization by reducing reliance on stoichiometric metal oxidants. While these approaches have already shown significant impact in C(sp2)–H functionalization, their application to C(sp3)–H functionalization remains relatively underdeveloped, requiring further investigation.219
We hope that this review can provide new perspectives in the catalytic 3d-catalyzed C(sp3)–H functionalization to expand the practicability of these metals in chelation-assisted strategies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank FEDER/Ministerio de Ciencia, Innovación y Universidades–Agencia Estatal de Investigación (Grants PGC2018-098660-B-I00 and PID2021-1248553NB-100) for financial support. We also thank the MINECO for the financial support for the RED2022-134331-T.Notes and references
- J. A. M. Simoes and J. L. Beauchamp, Chem. Rev., 1990, 90, 629–688 CrossRef CAS.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 2002 Search PubMed.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 Search PubMed.
- K. M. Altus and J. A. Love, Commun. Chem., 2021, 4, 173 Search PubMed.
- J. Tsuji, Transitions Metal Reagents and Catalysts: Innovations in Organic Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- R. Jana, H. M. Begam and E. Dinda, Chem. Commun., 2021, 57, 10842–10866 RSC.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- J.-Q. Yu and Z. Shi, C–H Activation, Topics in Current Chemistry, Springer-Verlag, 2010, 292 Search PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed.
- Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed.
- M. Bietti, Angew. Chem., Int. Ed., 2018, 57, 16618–16637 CrossRef CAS PubMed.
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- D. Maiti, Handbook of C–H Functionalization of C–H Functionalization, John Wiley & Sons, Inc., 2022 Search PubMed.
- N. Holmberg and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef PubMed.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed.
- Q. Sun, X. Xu and X. Xu, ChemCatChem, 2022, 14, e202201083 CrossRef CAS.
- Y. He, Z. Huang, K. Wu, J. Ma, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2022, 51, 2759–2852 RSC.
- Y. Zhang, T. Zhang and S. Das, Chem, 2022, 8, 3175–3201 CAS.
- T. Punniyamurthy and A. Kumar, Transition-Metal-Catalyzed C–H Functionalization of Heterocycles, John Wiley & Sons, Inc., 2023 Search PubMed.
- J. Grover, A. T. Sebastian, S. Maiti, A. C. Bissember and D. Maiti, Chem. Soc. Rev., 2025, 54, 2006–2053 RSC.
- P. Belloti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef PubMed.
- J. J. Li, C–H Activation in Organic Synthesis, CRC Press, 1st edn, 2015 Search PubMed.
- I. J. S. Fairlamb and A. R. Kapdi, in Directed C–H Bond Functionalization Strategies for Synthesis, ed. A. R. Kapdi and D. Maiti, Elsevier Inc., 2017, pp. 9–48 Search PubMed.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- M. Kapoor, A. Singh, K. Sharma and M. H. Hsu, Adv. Synth. Catal., 2020, 362, 4513–4542 CrossRef CAS.
- K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. S. Júnior and D. Maiti, Chem. - Eur. J., 2021, 27, 12453–12508 CrossRef CAS PubMed.
- T. Sarkar, T. A. Shah, P. K. Maharana, K. Talukdar, B. K. Das and T. Punniyamurthy, Chem. Rec., 2021, 21, 3758–3778 CrossRef CAS PubMed.
- S. Rej, A. Das and N. Chatani, Coord. Chem. Rev., 2021, 431, 213683 CrossRef CAS.
- B. Desai, M. Patel, B. Z. Dholakiya, S. Rana and T. Naveen, Chem. Commun., 2021, 57, 8699–8725 RSC.
- J. Zhang, X. Lu, C. Shen, L. Xu, L. Ding and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263–3314 RSC.
- R. H. Crabtree, Chem. Rev., 1985, 85, 245–269 CrossRef CAS.
- A. A. Mishra, D. Subhedar and B. M. Bhanage, Chem. Rec., 2019, 19, 1829–1857 CrossRef CAS PubMed.
- P. W. Tan and J. Seayad, Tetrahedron Lett., 2019, 60, 151338 CrossRef.
- J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887–10909 RSC.
- B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed.
- B. Li, M. Elsaid and H. Ge, Chem, 2022, 8, 1254–1360 CAS.
- J. Das, W. Ali and D. Maiti, Trends Chem., 2023, 5, 551–560 CAS.
- F. Doraghi, M. M. A. Ashtiani, M. Ameli, B. Larijani and M. Mahdavi, Chem. Rec., 2024, 24, e202400116 CAS.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CAS.
- B.-B. Zhan, M.-X. Jiang and B.-F. Shi, Chem. Commun., 2020, 56, 13950–13958 CAS.
- S. Shabani, Y. Wu, H. G. Ryan and C. A. Hutton, Chem. Soc. Rev., 2021, 50, 9278–9343 RSC.
- I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CAS.
- H. Ha, J. Lee, M. H. Park, B. Jung and M. Kim, Bull. Korean Chem. Soc., 2020, 41, 582–587 CrossRef CAS.
- G. Saini and M. Kapur, Chem. Commun., 2021, 57, 1693–1714 RSC.
- A. Das and B. Maji, Chem. – Asian J., 2021, 16, 397–408 CrossRef CAS PubMed.
- S. A. Babu, Y. Aggarwal, P. Patel and R. Tomar, Chem. Commun., 2022, 58, 2612–2633 RSC.
- B.-B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–232 CrossRef CAS.
- W. Ali, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2024, 53, 9904–9953 RSC.
- M. Sadeghi, ACS Catal., 2024, 14, 15356–15373 CrossRef CAS.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- S. St John-Campbell and J. A. Bull, Adv. Synth. Catal., 2019, 361, 3662 CrossRef CAS.
- L. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef.
- J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- B. Desai, A. Uppuluru, A. Dey, N. Deshpande, B. Z. Dholakiya, A. Sivaramakrishna, T. Naveen and K. Padala, Org. Biomol. Chem., 2023, 21, 673–699 RSC.
- J. Mo, A. M. Messinis, J. Li, S. Warratz and L. Ackermann, Acc. Chem. Res., 2024, 57, 10–22 CrossRef CAS PubMed.
- C. Johnson, S. Li, R. Arora, B. Mirabi and M. Lautens, Synlett, 2024, 851–861 CAS.
- P. Xu and S. Zhao, ChemistrySelect, 2024, 9, e202305125 CrossRef CAS.
- J. Son, Beilstein J. Org. Chem., 2021, 17, 1733–1751 CrossRef CAS PubMed.
- T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, Catal. Sci. Technol., 2021, 11, 444–458 RSC.
- G. Anusree, P. S. Devi and G. Anilkumar, Adv. Synth. Catal., 2019, 366, 1–38 Search PubMed.
- V. M. Chernyshev and V. P. Ananikov, ACS Catal., 2022, 12, 1180–1200 CrossRef CAS.
- J. P. Kleiman and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 1544–1545 CrossRef CAS.
- S. M. Khake and N. Chatani, Trends Chem., 2019, 1, 524–539 CrossRef CAS.
- Y.-H. Liu, Y.-N. Xia and B.-F. Shi, Chin. J. Chem., 2020, 38, 635–662 CrossRef CAS.
- N. Chatani, Acc. Chem. Res., 2023, 56, 3053–3064 CrossRef CAS PubMed.
- F. Požgan, U. Grošelj, J. Svete, B. Štefane and H. H. Al Mamari, Molecules, 2024, 29, 1917 CrossRef PubMed.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed.
- M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944–3946 RSC.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909–9920 CrossRef CAS PubMed.
- S. Singh, K. Surya and R. B. Sunoj, J. Org. Chem., 2017, 82, 9619–9626 CrossRef CAS PubMed.
- J. Liu and S. A. Johnson, Organometallics, 2021, 40, 2970–2982 CrossRef CAS.
- X. Wang, L. Zhu, S. Chen, X. Xu, C.-T. Au and R. Qiu, Org. Lett., 2015, 17, 5228–5231 CrossRef CAS PubMed.
- M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11933–11939 CrossRef CAS PubMed.
- E. L. Nolan, I. M. Blythe, F. Qu, J. W. Kampf and M. S. Sanford, This proposal is comparable with the detailed organometallic studies conducted by M. Sanford on the aminoquinoline-directed Ni-catalyzed C−H functionalization of 2,3,4,5-tetrafluoro-N-(quinolin-8-yl)benzamide using diarylio-donium reagents. These studies implicated a NiII/III/IV catalytic cycle in which the diaryliodonium promotes a double single-electron oxidation process involving aryl radical intermediates in the transformation, J. Am. Chem. Soc., 2024, 146, 18128–18135 CrossRef CAS PubMed.
- M. W. Milbauer, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2022, 144, 21030–21034 CrossRef CAS PubMed.
- X. Wang, P. Xie, R. Qiu, L. Zhu, T. Liu, Y. Li, T. Iwasaki, C.-T. Au, X. Xu, Y. Xia, S.-F. Yin and N. Kambe, Chem. Commun., 2017, 53, 8316–8319 RSC.
- G. Tan, L. Zhang, X. Liao, Y. Shi, Y. Wu, Y. Yang and J. You, Org. Lett., 2017, 19, 4830–4833 CrossRef CAS PubMed.
- M. Li, Y. Yang, D. Zhou, D. Wan and J. You, Org. Lett., 2015, 17, 2546–2549 CrossRef CAS PubMed.
- S. Maity, S. Agasti, A. M. Earsad, A. Hazra and D. Maiti, Chem. – Eur. J., 2015, 21, 11320–11324 CrossRef CAS PubMed.
- C. Lin, Z. Chen, Z. Liu and Y. Zhang, Org. Lett., 2017, 19, 850–853 CrossRef CAS PubMed.
- C. Lin, J. Zhang, Z. Chen, Y. Liu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 1778–1793 CrossRef CAS.
- C. Lin, Y. Xu, Q. Teng, J. Lin, F. Gao and L. Shen, Synlett, 2020, 889–894 CrossRef CAS.
- Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899–7902 RSC.
- X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924–4927 CrossRef CAS PubMed.
- X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed.
- T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162–3165 CrossRef CAS PubMed.
- X. Li, J.-M. Lv, D. Hu and I. Abe, RSC Chem. Biol., 2021, 2, 166–180 RSC.
- P. T. Boeck, R. Yadav, B. S. Sumerlin and A. S. Veige, Macromolecules, 2024, 57, 71–77 CrossRef.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- A. S. Sarala, S. Bhowmick, R. L. de Carvalho, S. A. Al-Thabaiti, M. Mokhtar, E. N. da S. Júnior and D. Maiti, Adv. Synth. Catal., 2021, 363, 4994–5027 CrossRef.
- F.-X. Luo, Z.-C. Cao, H.-W. Zhao, D. Wang, Y.-F. Zhang, X. Xu and Z.-J. Shi, Organometallics, 2017, 36, 18–21 CrossRef CAS.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CAS.
- M. V. K. Rao, S. Kareem, S. R. Vali and V. S. Reddy, Org. Biomol. Chem., 2023, 21, 8426–8462 RSC.
- X. Wu, Y. Zhao and H. Ge, Chem. – Eur. J., 2014, 20, 9530–9533 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323–4329 CrossRef CAS.
- P. Roy, J. R. Bour, J. W. Kampf and M. L. Sanford, J. Am. Chem. Soc., 2019, 141, 17382–17387 CrossRef CAS PubMed.
- Y. B. Kim, J. Won, J. Lee, J. Kim, B. Zhou, J.-W. Park, M.-H. Baik and S. Chang, ACS Catal., 2021, 11, 3067–3072 CrossRef CAS.
- M. R. Ringenberg, D. L. Gray and T. B. Rauchfuss, Organometallics, 2011, 30, 2885–2888 CrossRef CAS.
- Q. Chen, X.-H. Fan, L.-P. Zhang and L.-M. Yang, RSC Adv., 2014, 4, 53885–53890 RSC.
- A. A. Antonov and K. P. Bryliakov, Appl. Organomet. Chem., 2022, 36, e6499 CrossRef CAS.
- C. Wang, L. Zhang and J. You, Org. Lett., 2017, 19, 1690–1693 CrossRef CAS PubMed.
- L. L. Hegedus and R. W. McCabe, Catalyst Poisoning, ed. M. Dekker, New York, 1984 Search PubMed.
- D. Rampon, D. Seckler, E. Q. da Luz, D. B. Paixão, A. M. Larroza, P. H. Schneider and D. Alves, Org. Biomol. Chem., 2022, 20, 6072–6177 RSC.
- C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 1340–1343 CrossRef CAS PubMed.
- X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970–1973 CrossRef CAS PubMed.
- S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341–7344 RSC.
- X. Ye, J. L. Pettersen and X. Shi, Chem. Commun., 2015, 51, 7863–7866 RSC.
- J. L. G. Ruano, A. Parra and J. Alemán, Green Chem., 2008, 10, 706–711 RSC.
- J.-M. Li, Y. Yu, J. Weng and G. Lu, Org. Biomol. Chem., 2018, 16, 6047–6056 RSC.
- D. D. Beattie, A. C. Grunwald, T. Perse, L. L. Schafer and J. A. Love, J. Am. Chem. Soc., 2018, 140, 12602–12610 CrossRef CAS PubMed.
- B. E. Nadeau, D. D. Beattie, E. K. J. Lui, M. Tewkesbury, J. A. Love and L. L. Schafer, Organometallics, 2023, 42, 2326–2334 CrossRef CAS.
- C. C. Roberts, E. Chong, J. W. Kampf, A. J. Canty, A. Ariafard and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 19513–19520 CrossRef CAS PubMed.
- G. Anilkumar and S. Saranya, Copper Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2020 Search PubMed.
- F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed.
- S. Thapa, B. Shrestha, S. K. Gurung and R. Giri, Org. Biomol. Chem., 2015, 13, 4816–4827 CAS.
- S. Radhika, N. A. Harry, M. Neetha and G. Anilkumar, Org. Biomol. Chem., 2019, 17, 9081–9094 RSC.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CAS.
- P. Rajeshwaran, J. Trouvé, K. Youssef and R. Gramage-Doria, Angew. Chem., Int. Ed., 2022, 61, e202211016 CrossRef CAS PubMed.
- T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, RSC Adv., 2020, 10, 34429–34458 CAS.
- H. Y. Kim and K. Oh, Org. Biomol. Chem., 2021, 19, 3569–3583 RSC.
- Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- R. Trammell, K. Rajabimoghadam and I. García-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed.
- L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- L. Marais, H. C. M. Vosloo and A. J. Swarts, Coord. Chem. Rev., 2021, 440, 213958 CrossRef CAS.
- J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174–1194 CrossRef CAS.
- I. M. Blythe, J. Xu, J. S. F. Odell, J. W. Kampf, M. A. Bowring and M. S. Sanford, Nowadays, there is little knowledge about the organometallic intermediates involved in directed Cu-catalyzed C–H functionalization reaction. Nonetheless, the group of Sanford described the isolation of key intermediates in the Cu-mediated 8-AQ-directed C(sp2)–H functionalization of polyfluorinated benzamide derivatives. This study showed that anionic copper complexes are involved in the transformation stabilize Cs+ counterion. Moreover, while the C–H activation step occurs at copper(II)-species via CMD mechanism assisted by carbonate basic additive, the subsequent steps involved copper(III) σ-aryl complexes, J. Am. Chem. Soc., 2023, 145, 18253–18259 CrossRef CAS PubMed.
- X. Wu, Y. Zhao and H. Ge, Chem. Sci., 2015, 6, 5978–5983 RSC.
- X. Wu, J. Miao, Y. Li, G. Li and H. Ge, Chem. Sci., 2016, 7, 5260–5264 RSC.
- W. E. Noland, Chem. Rev., 1955, 55, 137–155 CrossRef CAS.
- J. Zhang, D. Li, H. Chen, B. Wang, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 792–807 CrossRef CAS.
- U. Halbes-Letinois, J.-M. Weibel and P. Pale, Chem. Soc. Rev., 2007, 36, 759–769 RSC.
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- P. Sivaguru, S. Cao, K. R. Babu and X. Bi, Acc. Chem. Res., 2020, 53, 662–675 CrossRef CAS PubMed.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691–1692 CrossRef.
- X. Yan, X. Yang and C. Xi, Catal. Sci. Technol., 2014, 4, 4169–4177 RSC.
- A. Ribaucourt and J. Cossy, ACS Catal., 2020, 10, 10127–10148 CrossRef CAS.
- J.-P. Wan and Y. Jing, Beilstein J. Org. Chem., 2015, 11, 2209–2222 CrossRef CAS PubMed.
- J.-L. Ma, X.-M. Zhou, P.-H. Guo, H.-C. Cheng and H. B. Ji, Chin. J. Chem., 2022, 40, 1204–1223 CrossRef CAS.
- Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496–3499 CrossRef CAS PubMed.
- X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706–3710 CrossRef CAS PubMed.
- C. Wang, Y. Yang, D. Qin, Z. He and J. You, J. Org. Chem., 2015, 80, 8424–8429 CrossRef CAS PubMed.
- Q. Gou, Y.-W. Yang, Z.-N. Liu and J. Qin, Chem. – Eur. J., 2016, 22, 16057–16061 CrossRef CAS PubMed.
- C. Wang and Y. Yang, Tetrahedron Lett., 2017, 58, 935–940 CrossRef CAS.
- Z. Wang, Y. Koninobu and M. Kanai, Org. Lett., 2014, 16, 4790–4793 CrossRef CAS PubMed.
- X. Wu, Y. Zhao and H. Ge, Chem. – Asian J., 2014, 9, 2736–2739 CrossRef CAS PubMed.
- F. Wang, X. Li, Z. Li, S. Zhou and W. Zhang, ACS Omega, 2019, 4, 331–343 CrossRef CAS PubMed.
- J. Zhang, H. Chen, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2768–2771 CrossRef CAS PubMed.
- J.-H. Liu, M. Cui, X.-Y. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 1242–1245 RSC.
- N. E. Idoine, E. R. Raycraft, S. F. Hobbs, P. Everett, E. J. Evans, A. J. Mills, D. Currie, S. Horn and R. A. Shaw, World mineral production 2018-22, British Geological Survey, Keyworth, 2024.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- S. Cattani and G. Cera, Chem. – Asian J., 2024, 19, e202300897 CrossRef CAS PubMed.
- R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030–6032 CrossRef CAS PubMed.
- Y. Sun, H. Tang, K. Chen, L. Hu, J. Yao, S. Shaik and H. Chen, J. Am. Chem. Soc., 2016, 138, 3715–3730 CrossRef CAS PubMed.
- T. E. Boddie, S. H. Carpenter, T. M. Baker, J. C. DeMuth, G. Cera, W. W. Brennessel, L. Ackermann and M. L. Neidig, J. Am. Chem. Soc., 2019, 141, 12338–12345 CrossRef CAS PubMed.
- Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868–3871 CrossRef CAS PubMed.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 7660–7663 CrossRef CAS PubMed.
- K. Graczyk, T. Haven and L. Ackermann, Chem. – Eur. J., 2015, 21, 8812–8815 CrossRef CAS PubMed.
- L. Ilies, Y. Itabashi, R. Shang and E. Nakamura, ACS Catal., 2017, 7, 89–92 CrossRef CAS.
- M. S. Kharasch and E. K. Fields, J. Am. Chem. Soc., 1941, 63, 2316–2320 CrossRef CAS.
- G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed.
- M. M. Tohidi, B. Paymard, S. R. Vasquez-García and D. Fernández-Quiroz, Tetrahedron, 2023, 136, 133352 CrossRef CAS.
- P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed.
- H. Pellissier and H. Clavier, Chem. Rev., 2014, 114, 2775–2823 CrossRef CAS PubMed.
- M. Moselage and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- Y. Zheng, C. Zheng, Q. Gu and S.-L. You, Chem. Catal., 2022, 2, 2965–2985 CrossRef CAS.
- J. Bora, M. Dutta and B. Chetia, Tetrahedron, 2023, 132, 133248 CrossRef CAS.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed.
- D. Tilly, G. Dayaker and P. Bachu, Catal. Sci. Technol., 2014, 4, 2756–2777 RSC.
- R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, ChemSusChem, 2020, 13, 3306–3356 CrossRef CAS PubMed.
- L. Lukasevics, A. Cizikovs and L. Grigorjeva, Chem. Commun., 2021, 57, 10827–10841 RSC.
- S. Sunny and R. Karvembu, Adv. Synth. Catal., 2021, 363, 4309–4331 CrossRef CAS.
- D. Chandra, Manisha and U. Sharma, Chem. Rec., 2022, 22, e202100271 CrossRef CAS PubMed.
- E. L. Nolan, F. Qu and M. S. Sanford, To date, it remains unclear whether cobalt(II) centers are directly involved in the C–H activation step. Nonetheless, M. Sanford just reported that the dehydrogenative dimerization of 2,3,4,5-tetrafluoro-N-(quinolin-8-yl)benzamide occurs through a first cyclometalation with Co(OAc)2, forming an NNC cobalt(II) pincer complex. Single-electron oxidation with AgOAc generates a cobalt(III) intermediate, which undergoes a second directed C(sp2)−H activation, yielding a bis-cyclometalated octahedral cobalt(III) complex. This cobalt(III) species remains inert to thermal C–C bond-forming reductive elimination, even when heated at 160 °C for 48 hours. However, treatment with ferrocenium oxidants at room temperature induces oxidative carbon–carbon coupling, presumably via a cobalt(IV) intermediate, Organometallics, 2024, 43, 2165–2168 CrossRef CAS.
- T. Yoshino, S. Satake and S. Matsunaga, Chem. – Eur. J., 2020, 36, 7346–7357 CrossRef PubMed.
- T. Yoshino and S. Matsunaga, in C–H Functionalization Catalyzed by Cobalt(III)/Cp* and Related Complexes, ed. N. Yoshikai, Georg Thieme Verlag KG, 1st edn, 2023 Search PubMed.
- J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990–12996 CrossRef CAS PubMed.
- J.-H. Wu and H.-Q. Yu, Environ. Sci. Technol., 2024, 58, 18496–18507 CrossRef CAS PubMed.
- L. Staronova, K. Yamazaki, X. Xu, H. Shi, F. M. Bickelhaupt, T. A. Hamlin and D. J. Dixon, Angew. Chem., Int. Ed., 2024, 63, e202316021 CrossRef CAS PubMed.
- M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef CAS.
- N. Barsu, S. K. Bolli and B. Sundararaju, Chem. Sci., 2017, 8, 2431–2435 RSC.
- P. Williamson, A. Galván and M. J. Gaunt, Chem. Sci., 2017, 8, 2588–2591 RSC.
- L. Zeng, S. Tang, D. Wang, Y. Deng, J.-L. Chen, J.-F. Lee and A. Lei, Org. Lett., 2017, 19, 2170–2173 CAS.
- N. Barsu, D. Kalsi and B. Sundararaju, Catal. Sci. Technol., 2018, 8, 5963–5969 CAS.
- H. Wang, S. Zhang, Z. Wang, M. He and K. Xu, Org. Lett., 2016, 18, 5628–5631 CrossRef CAS PubMed.
- Z.-Z. Zhang, Y.-Q. Han, B.-B. Zhan, S. Wang and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 13145–13149 CrossRef CAS PubMed.
- S.-Y. Yan, P.-X. Ling and B.-F. Shi, Adv. Synth. Catal., 2017, 359, 2912–2917 CrossRef CAS.
- H. Christian, R. Georg, S. Jean-Philippe and Z. A. Cornelia, U.S. Pat., 2010/331318 A1, 2010 Search PubMed.
- S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed.
- X.-X. Chen, J.-T. Ren, J.-L. Xu, H. Xie, W. Sun, Y.-M. Li and M. Sun, Synlett, 2018, 1601–1606 CAS.
- R. Kumar, R. Kumar, D. Chandra and U. Sharma, J. Org. Chem., 2019, 84, 1542–1552 CrossRef CAS PubMed.
- N. Li, Y. Wang, L. Kong, J. Chang and X. Li, Adv. Synth. Catal., 2019, 361, 3880–3885 CrossRef CAS.
- H. Tan, R. Khan, D. Xu, Y. Zhou, X. Zhang, G. Shi and B. Fan, Chem. Commun., 2020, 56, 12570–12573 RSC.
- X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 6, 6462 CrossRef CAS PubMed.
- S. Dutta, S. Chatterjee, S. A. Al-Thabaiti, S. Bawaked, M. Mokhtar and D. Maiti, Chem. Catal., 2022, 2, 1046–1083 CrossRef CAS.
- Y. N. Timsina, B. F. Gupton and K. C. Ellis, ACS Catal., 2018, 8, 5732–5776 CrossRef CAS.
- P. W. Tan, A. M. Mak, M. B. Sullivan, D. J. Dixon and J. Seayad, Angew. Chem., Int. Ed., 2017, 56, 16550–16554 CAS.
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CAS.
- D. Sekine, K. Ikeda, S. Fukagawa, M. Kojima, T. Yoshino and S. Matsunaga, Organometallics, 2019, 38, 3921–3926 CrossRef CAS.
- R.-H. Liu, Q.-C. Shan, X.-H. Hu and T.-P. Loh, Chem. Commun., 2019, 55, 5519–5522 CAS.
- L. Liu, Z. Zhang, Y. Wang, Y. Zhang and J. Li, Synthesis, 2020, 3881–3890 CAS.
- N. Barsu, Md. A. Rahman, M. Sen and B. Sundararaju, Chem. – Eur. J., 2016, 22, 9135–9138 CAS.
- H. Zhang, M.-C. Sun, D. Yang, T. Li, M.-P. Song and J.-L. Niu, ACS Catal., 2022, 12, 1650–1656 CrossRef.
- J. J. Neumann, S. Rakshit, T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892–6895 CrossRef CAS PubMed.
- F. Pan, B. Wu and Z.-J. Shi, Chem. – Eur. J., 2016, 22, 6487–6490 CrossRef CAS PubMed.
- H. Zhang, D. Yang, X.-F. Zhao, J.-L. Niu and M.-P. Song, Org. Chem. Front., 2022, 9, 3723–3729 RSC.
- U. Dhawa, N. Kaplaneris and L. Ackermann, Org. Chem. Front., 2021, 8, 4886–4913 RSC.
| This journal is © The Royal Society of Chemistry 2025 |