
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-healing behavior of superhard covalent bond materials
Keliang
Qiu†
a,
Xiang
Li†
a,
Yanhong
Li
a,
Yonghai
Yue
*ab and
Lin
Guo
 *a
*a
aSchool of Chemistry, Beihang University, Beijing, China. E-mail: yueyonghai@buaa.edu.cn; guolin@buaa.edu.cn
bState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan, China
First published on 24th March 2025
Abstract
In recent years, superhard covalently bonded materials have drawn a great deal of attention due to their excellent mechanical properties and potential applications in various fields. This review focuses on the self-healing behavior of these materials, outlining state-of-the-art research results. In detail, we discuss current self-healing mechanisms of self-healing materials including extrinsic healing mechanisms (such as microencapsulation, oxidative healing, shape memory, etc.) and intrinsic healing (dynamic covalent bonding, supramolecular interactions, diffusion, defect-driven processes, etc.). We also provide an overview of the progress in the self-healing behavior of superhard covalently bonded materials and the mechanisms of permanent covalent bonding healing. Additionally, we analyze the factors that influence the healing properties of these materials. Finally, the main findings and an outlook on the future directions and challenges of this emerging field are summarized in the Conclusion section.
 Keliang Qiu | Keliang Qiu received his PhD from Beihang University under the supervision of Prof. Yonghai Yue. He is currently a postdoctoral researcher at Beihang University, focusing on multi-scale, multi-field coupled in situ studies of the structure–property relationships in structural materials. |
 Xiang Li | Xiang Li is a PhD student at Beihang University under the supervision of Professor Yue Yonghai. Xiang Li is dedicated to the study of failure mechanisms of mechanical behaviour of metallic materials under different external fields at the micro and nano scales. |
 Yonghai Yue | Yonghai Yue is a professor at the School of Chemistry, Beihang University. He received his PhD degree from the Institute of Microstructure and Properties of Advanced Materials, Beijing University of Technology in 2012. He was a visiting scholar at Arizona State University in 2014. His research area focuses on the design and preparation of lightweight and high-strength materials, the exploration of multi-field coupled in situ microscopic failure mechanisms and the exploration of potential applications. |
 Lin Guo | Guo Lin is a full-time professor at the School of Chemistry, Beihang University. he received his PhD degree from the School of Chemical Engineering and Materials, Beijing Institute of Technology in 1997. He was a visiting scholar at the Hong Kong University of Science and Technology in 1999 and a Humboldt Scholar at the Dresden University of Technology. His research interests focus on the design and preparation of amorphous micro- and nanomaterials and the exploration of potential performance applications. |
1. Introduction
Drawing inspiration from nature is a crucial strategy for developing new materials.1–5 Over billions of years of natural selection, biological organisms have evolved unique damage repair mechanisms, which is an essential survival feature of life forms. For instance, small wounds on human skin can heal after a sufficient period of time, as fractures can be gradually repaired. Even plants have a greater capacity for self-repair or regeneration compared to animals. In contrast, man-made materials often fail catastrophically due to natural or anthropogenic degradation or degradation over time, resulting in the formation of micro-cracks.6–8 Therefore, in order to improve the safety, reliability and service life of the materials, researchers have mimicked the self-healing properties of living organisms for applications in aerospace, energy, and biomedicine.9–13Currently, research on self-healing materials is only a few decades old. Their self-healing mechanisms have not yet attained the complexity found in the physical events and chemical reactions involved in the self-healing process of living systems. Nevertheless, significant progress has been made in the study of self-healing materials, mainly in the field of polymeric materials, which are generally soft and deformable and more likely to exhibit self-healing properties.14–16 In addition, some research progress have been made in self-healing metallic materials and self-healing ceramic materials likewise.17,18 In contrast, Research on self-healing superhard covalent materials remains preliminary and exploratory.
1.1 What is self-healing?
So, what is self-healing? Over time, the degradation of synthetic materials can cause damage or micro cracks, reducing the material's safety and reliability, potentially resulting in catastrophic failure within its effective service life.19,20 Therefore, “Removing” damage through damage management is an important solution, that explores the ability of materials to recover from physical damage, inspired by the self-healing capabilities of living organisms. The ability of materials to automatically and autonomously heal or repair damage without any external intervention is defined as self-healing.21 Once the self-healing behavior is triggered, the tendency of non-self-healing materials to progressively accumulate damage over time until failure (d(damage)/dt ≥ 0(0 < t < life time)) shifts to that of self-healing materials, which cyclically remove damage (d(damage)/dt < 0(ti < t < ti+Δt(healing))) to ensure sustainable service (Fig. 1a). The healing of material mechanical properties is paramount, as mechanical performance underpins the practical applications of materials. Once physical failures such as collapse and fracture occur in materials, they will directly trigger catastrophic failures. Therefore, in order to more accurately describe the self-healing ability of materials, the ratio of the healed stress (or strength) to the initial stress (or strength) is usually defined as the healing efficiency. Artificial self-healing materials can be divided into two categories based on whether their self-healing behavior needs to be triggered by the external environment: one category is active, i.e., it does not need to be triggered by the external environment to achieve active repair;22–26 the other category requires external intervention to trigger self-healing behavior.27–30 | ||
| Fig. 1 Diagram of self-healing material properties. (a) Diagram of relationship between damage and time of non-self-healing and self-healing materials. (b) The main factors affecting self-healing materials. | ||
It is important to ensure that the self-healing function of the material is realized, so the following aspects need to be fully understood (Fig. 1b): (1) the extent of damage. To clarify the location and scale of the damage of the material, it should be within the scope of the material's repairable capacity; (2) healing mechanism. Give full play to the self-healing ability of the material and improve the healing efficiency; (3) time dependence. Self-healing is not instantaneous but takes a certain amount of time, so the healing time should be reduced as much as possible; (4) long-term stability. This self-healing ability can remain effective until the material completely fails, allowing for multiple healing cycles. In addition, other factors must be considered, such as ensuring that the material's performance is not significantly reduced after healing, biocompatibility (biomaterials) and so on.
1.2 Types of self-healing materials
The self-healing phenomenon in non-living materials was discovered long ago, for example, mica in nature can heal itself after fracture.31,32 In addition, self-healing phenomena similar to biological self-healing have likewise been found in man-made materials, such as cements, polymer materials, and so on, which have been widely noticed.33–36 With the continuous research on self-healing materials, two main types of self-healing materials have been developed, i.e., extrinsic self-healing37–39 and intrinsic self-healing.40–42 Extrinsic self-healing refers to achieving self-healing by adding non matrix healing agents.43 Intrinsic self-healing materials achieve damage repair relying on their own dynamic chemical bonding or their own physical properties. A variety of self-healing mechanisms have been developed for both types of self-healing materials.1.3 Superhard covalently bonded materials
Superhard covalently bonded materials occupy a unique position within the broader spectrum of materials. These materials are mainly composed of covalent compounds. The covalent bonds within them are short and strong, endowing them with excellent resistance to elastic and plastic deformation. Generally, the (Vickers) hardness value of superhard materials exceeds 40 GPa, often far exceeding that of traditional metal materials and ordinary ceramic materials. Thanks to their exceptional hardness, superhard covalent bond materials exhibit remarkable wear resistance, making them highly effective in applications such as cutting tools, abrasives, and grinding tools. Furthermore, superhard materials typically showcase a range of other distinctive properties such as high compressive strength, remarkable shear resistance, a high bulk modulus, a high melting point, chemical inertness, and excellent thermal conductivity.44 These attributes further enhance their versatility and performance in demanding industrial applications.However, superhard materials are among the rarest of functional inorganic solids, representing less than 0.1% of the entire material system.45 Currently, the prevalent and commercially accessible superhard materials are predominantly diamond and cubic boron nitride. Over the past few decades, considerable efforts have been made to develop superhard materials that surpass the hardness and performance of cubic boron nitride and diamond. To this end, both quantitative hardness prediction techniques and experimental approaches have been advanced, aimed at significantly improving the properties of existing superhard materials.46,47 In addition, researchers design and discover novel superhard materials experimentally and theoretically by combining light elements like boron, carbon, nitrogen, and/or oxygen to form short covalent bonds or introducing elements with extremely high valence electron density.48 At present, the hardness of synthetic diamond blocks has been increased to 276 GPa.49 The hardness of cubic boron nitride has also exceeded 100 GPa.50 Even the hardness of silicon carbide has surpassed 40 GPa.51 However, in practical applications, superhard covalent bond materials are not without flaws. For example, diamond has serious limitations that severely restrict its industrial applications in the processing of ferrous metal materials: it has poor thermal stability and high chemical reactivity with ferroalloys. As for cubic boron nitride, although it is suitable for the processing of ferrous metal materials, the hardness and toughness of its single crystal are only half of those of single crystal diamond.52 It is important to note that when superhard covalent-bonded materials are exposed to demanding operational conditions—such as sustained mechanical stress, thermal cycling, and chemical corrosion—various forms of damage, including microcracks and other defects, inevitably develop within the materials. These defects continuously expand and accumulate during service, and ultimately are highly likely to trigger the catastrophic failure of the materials, posing serious potential threats to the safe operation of related engineering equipment.
Therefore, once the self-healing of superhard covalent bond materials is achieved, it will greatly expand their application scope, significantly enhance their service performance under extreme environments, and thus bring about a new revolution in the field of materials science and engineering. In view of this, in order to develop superhard covalent bond self-healing materials more effectively and provide guidance for the development of other self-healing materials, we have provided a comprehensive overview of the primary self-healing mechanisms currently in use. We have also highlighted the present state of development in self-healing superhard covalent-bond materials and, ultimately, outlined prospective directions for future advancements.
2. Mechanisms of self-healing
2.1 Extrinsic self-healing mechanisms
Extrinsic self-healing mechanism is an important branch in the study of self-healing materials, which refers to the self-healing of materials by externally-added restorative agents after damage. | ||
| Fig. 2 Schematic of a typical extrinsic self-healing materials. (a) Schematic diagram of microencapsulated self-healing. (b) Analysis of the effect of relative stiffness of microcapsules on proximity crack extension paths.37 Reproduced with permission from ref. 37, Copyright 2001, Springer Nature. (c) Schematic diagram of microvascular coating-substrate structure. (d) High-magnification cross-sectional image of the coating. (e) Healing efficiency for each successive loading.53 Reproduced with permission from ref. 53, Copyright 2007, Springer Nature. | ||
So far, in addition to polymer materials, a variety of self-healing materials based on extrinsic self-healing mechanisms have been developed in ceramics, metals and other materials. Mimicking the design of extrinsic self-healing mechanisms for polymers, self-healing can be achieved by embedding a capsule with a healing agent in the ceramic material. For example, the urea-formaldehyde microcapsules encapsulated with 1H,1H,2H,2H-perfluorodecyltriethoxysilane are homogeneously dispersed in a pure phosphate ceramic coating to obtain a phosphate ceramic coating with self-healing capability.56 Self-healing metal composites can be designed by applying microencapsulation design strategies to embed low-melting-point metals into high-melting-point metals that act as healing agents.57,58 Theoretically, this self-healing design model based on microcapsules demonstrates broad application potential and can endow the vast majority of materials with self-healing properties. However, in practical application scenarios, numerous factors have crucial impact on the initial mechanical properties of materials. These factors include the size and volume fraction of microcapsules, the interfacial bonding strength between microcapsules and the matrix, as well as the distribution state of microcapsules within the matrix. In matrix materials with relatively low mechanical properties, microcapsules may play a role in strengthening and toughening the material.
 | ||
| Fig. 3 Self-healing ceramics based on oxidizing mechanism. (a) Schematic of oxidative healing mechanisms. (b) Surface crack morphology of self-healing Al2O3 ceramic before and after healing.63 Reproduced with permission from ref. 63, Copyright 2023, Elsevier. (c) and (d) Fracture behavior of Ti2AlC before and after healing and EDS spectrum of the healed region.66 Reproduced with permission from ref. 66, Copyright 2022, Springer Nature. | ||
Oxidative healing mechanisms can be triggered at high temperatures and high oxygen partial pressure environments (air environments), however, when oxygen partial pressure is low, oxidative healing mechanisms are constrained and other healing mechanisms need to be considered. For example, Yao et al. found that Ti2AlC exhibits a new mechanism of decomposition-induced crack healing at a low oxygen partial pressure of about 1 Pa, i.e., microcracks are filled with decomposed TiCx as the main phase.66 The healed flexural strength exceeded the initial sample flexural strength after healing at 1400 °C for 2 h (Fig. 3c and d). From the energy dispersive spectrum (EDS) analysis, it can be seen that the Al2O3 grains fill the pores in the TiCx layer, resulting in a relatively dense healing zone. In low oxygen partial pressure environments of 5 Pa, another mechanism is the precipitation-induced crack healing mechanism, such as Sn-precipitation-induced crack healing in Ti2SnC.67
 | ||
| Fig. 4 Shape memory assists self-healing. (a) and (b) Healing process and mechanical properties of Al-3Si/SMA composites before and after healing.75 Reproduced with permission from ref. 66, Copyright 2018, Elsevier. (c) and (d) Schematic of SMP-assisted self-healing and mechanical properties of PPG-MPU0.7-PA0.3 before and after healing.74 Reproduced with permission from ref. 74, Copyright 2024, Elsevier. (e) and (f) Mechanical properties of TPU (MW ≈ 72 kDa) before and after healing, and optical images of TPU (MW ≈ 32 kDa) healing process.76 Reproduced with permission from ref. 76, Copyright 2020, Springer Nature. | ||
Shape memory-assisted self-healing methods have demonstrated effectiveness in promoting fracture closure; however, they generally require external conditions for activation. Hornatet et al. developed self-healing thermoplastic polyurethane (TPU) fibers and films assisted by autonomous shape memory under environmental conditions.76 The efficient storage and release of conformational entropy during damage and repair is achieved through shape memory effects and utilises surface energy/tension to drive the repair process, with lower molecular weight TPU (MW ≈ 32 kDa) requires more time to self-repair through a distinct mechanism driven by surface/surface energy (Fig. 4f). Higher molecular weight TPU (MW ≈ 72 kDa) heal faster and mechanical properties are fully restored (Fig. 4e).
The realization of extrinsic healing in materials via the incorporation of embedded microcapsules, 3D vascular networks, oxidative healing mechanism components, and shape memory components, etc. is relatively straightforward in terms of operation. Nevertheless, a series of pivotal issues that accompany this approach demand due attention. These encompass the interfacial bonding strength between the self-healing elements and the matrix, chemical stability, the efficacy and frequency of healing and so on. Specifically, for super-hard and hard materials, a significant disparity in hardness exists between the self-healing elements (e.g., embedded microcapsules) and the matrix, exacerbated by a weak interfacial bonding strength. Under external loading, the interface of such materials is highly susceptible to stress concentration, which in turn leads to the initiation interface cracking. Therefore, a trade-off between material properties and self-healing properties is needed to meet the demands of the working conditions.
2.2 Intrinsic self-healing mechanisms
Intrinsic self-healing relies on its own internal structure or physicochemical properties to achieve self-healing. In contrast to extrinsic self-healing, intrinsic self-healing usually repairs scratches or undetectable defects at the crack nucleation stage and can theoretically be repeated over and over again. In the case of superhard materials, due to their intrinsic brittleness, fracture failure occurs in the event of microcracking. Therefore, studying the intrinsic self-healing mechanism is highly significant for addressing the safety and reliability of superhard materials and their related applications. So far, some progress has been made in the study of intrinsic healing mechanisms, such as dynamic covalent bonding, supramolecular interactions, defect-driven self-healing, phase transitions, and so on. | ||
| Fig. 5 Research on self-healing materials based on dynamic covalent bonds. (a)–(d) The mechanism, healing process and self-healing tests of CG@CPA.83 Reproduced with permission from ref. 83, Copyright 2021, Springer Nature. (e)–(g) Self-healing mechanism diagram and self-healing tests of PUDS bulk material samples.84 Reproduced with permission from ref. 84, Copyright 2018, Wiley-VCH. (h)–(j) Schematic mechanism, optical image of the self-healing process of fractured CLCE and stress–strain curve with different healing time.85 Reproduced with permission from ref. 85, Copyright 2022, Wiley-VCH. | ||
In addition to DA reactions, dynamic covalent bonds used for the development of self-healing materials include disulfide bonds, boron ester bonds, imine bonds, etc.86–89 Disulfide bonds can be rapidly exchanged between disulfide or between disulfide and thiols, whose dynamic covalent properties can be stimulated by a variety of external stimuli such as sound, light, thermal and mechanical energy.90–92 Self-healing materials such as soft gels, elastomers, et al. with self-healing properties can be obtained by embedding disulfide bonds in polymer materials.93,94 For example, Lai et al. developed self-healing polyurethane elastomers with disulfide bonds (PUDS) by embedding and locking dynamic disulfide bonds in viscoelastic hard microphase regions, which healed surface scratches within 60 s at 70 °C, and achieved a healing efficiency of 85% at 70 °C for 6 h after complete fracture (Fig. 5e–g).84 Dynamic covalent bonding boron ester bonds can be obtained by hydrolysis reactions, ester exchange reactions with exogenous substances and exchange reactions between different boron ester compounds.88,95,96 The B–O bonds can be exchanged through both thermo-activated direct metathesis between different boronic ester compounds and water-assisted hydrolysis/dehydration. For example, Ma et al. endowed a cholesteric liquid crystal elastomer (CLCE) with programmable and self-healing capabilities by introducing boron ester bonds into CLCE polymer networks, which was attributed to the reversible B–O bond exchange carried out by water-assisted hydrolysis/dehydration (Fig. 5h).85 The fractures of completely broken CLCEs were joined together and a few drops of water were added to the damaged interface, followed by close contact at room temperature for 24 h (Fig. 5i). The samples self-healed with a maximum healing efficiency of 86.18% (Fig. 5j). The imine bond is capable of three dynamic transformations: transamination, decomposition of imine complexes, and hydrolysis, which can endow materials with the properties of self-healing, solderability, and degradability.97–99 Bhattacharya et al. endowed the gel with self-healing properties by generating imine bonds through Schiff base reactions between aldehyde units on the surface of the carbon dots and primary amine residues in the polyethyleneimine network.100 The addition of water or heating can both enhance the self-healing process.
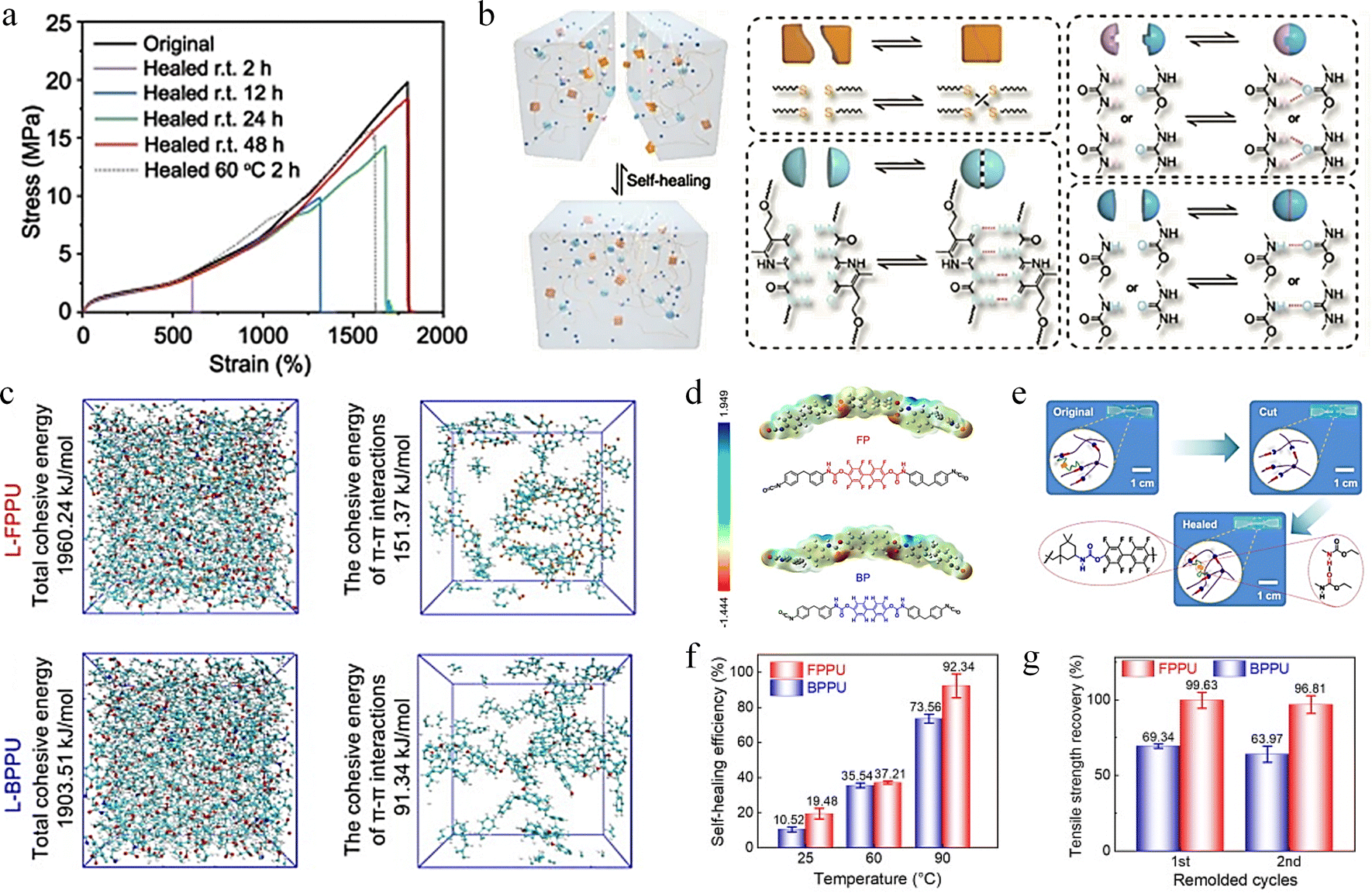 | ||
| Fig. 6 Research on self-healing behavior based on H-bonding and π–π stacking interaction. (a) and (b) Test of mechanical properties of DSE before and after healing, and schematic diagram of healing mechanism.108 Reproduced with permission from ref. 108, Copyright 2022, Springer Nature. (c) and (d) Molecular dynamics simulation and electrostatic potential calculation of L-FPPU and L-BPPU systems. (e) Schematic diagram of FPPU and BPPU healing mechanism. (f) and (g) Self-healing efficiency and recovery efficiency of tensile strength for FPPU and BPPU.109 Reproduced with permission from ref. 109, Copyright 2024, Elsevier. | ||
π–π stacking is a weak intermolecular interaction exhibited by π-electron conjugated systems under certain specific spatial arrangements. This type of interaction plays a significant role in molecular structure, reaction mechanisms, and optical properties. In aromatic compounds, π–π stacking is a special spatial arrangement that typically occurs between two molecules that are relatively electron-rich and electron-deficient, making it an important non-covalent interaction akin to hydrogen bonding. The interaction between the aromatic rings, the π–π stacking, helps to strengthen the non-covalent interaction of the polymer chains.110 According to Jia et al., fluorine atoms exhibit a stronger electron-pulling induction effect than hydrogen, and connecting fluorine atoms directly to the aromatic ring changes the π–π stacking pattern and enhances the π–π stacking interaction.109 They produced a fluorinated phenolic polyurethane (FPPU) elastomer based on octafluoro-4,4′-biphenol. The internal energy and electrostatic potential of linear fluorinated phenolic polyurethane (L-FPPU) and its non-fluorinated linear bisphenol phenolic polyurethane (L-BPPU) were compared by molecular dynamics (MD) simulations and electrostatic potential calculations (Fig. 6c and d), demonstrating that fluorine atoms further enhance the π–π stacking interaction of aromatic rings. The chopped FPPU material exhibits complete self-healing behavior when subjected to temperature and pressure. (Fig. 6e). FPPU has higher self-healing efficiency than BPPU (92.3% for FPPU and 73.56% for BPPU in Fig. 6f) and tensile strength recovery efficiency (96% for FPPU and 63% for BPPU in Fig. 6g).
Complex interfacial supramolecular interactions will occur at the interface of two different phases (such as solid and liquid or gas and liquid), including hydrogen bonding, π–π stacking, van der Waals forces, electrostatic interactions, hydrophobic interactions, and host–guest interactions. The characteristics of interfacial supramolecular interactions include high selectivity, reversibility and dynamics, which play a key role in regulating interfacial properties such as wettability, adhesion and mechanical properties.111–114 Xu et al. designed a non-covalent assembly of rigid 2D transition metal carbide/carbonitride (MXene) nanosheets based on the layered structure of biological tissues, and prepared a polyurea-carbamate (SHPU)/L-MXene composite (SHPULM).115 The mechanical properties of SHPULM composites are significantly improved due to the advantages of layered structure and dense interfacial supramolecular interactions. SHPULM is capable of self-healing autonomously and efficiently at room temperature and can be stretched to 5 times its original length after 5 minutes of healing (Fig. 7a). The fractured MXene skeleton can fully heal on a microscale after 30 hours of self-healing at 25 °C, as shown in Fig. 7b. The stress–strain curve in Fig. 7c indicates that the recombined SHPULM repaired at room temperature for 30 h almost overlapped with the original curve with self-healing efficiency of 97 ± 2%. In supramolecular chemistry, host and guest molecules interact through non-covalent bonds to form stable complexes. The strength and nature of the host–guest interaction can be modulated by changing the chemical structure of the subject or the guest, which provides a wide range of tunability for the design of materials.116–118 The Mo et al. fabricated a supramolecular hydrogel with host–guest complexes of two types of β-cyclodextrin (β-CD) using an entirely elastic and entirely polymeric approach, which possesses excellent self-healing characteristics.119Fig. 7d illustrates the self-healing mechanism of the supramolecular hydrogel: the residual amino acid molecules on the surface of the supramolecular hydrogel sheets interact with the grafted β-CD molecules, forming supramolecular bonds within the double-cross-linked hydrogel through host–guest interactions. Fig. 7e demonstrates that when cut hydrogel blocks are brought into contact under humid conditions, allowing the blocks to fully heal within 1 hour. In contrast, healing does not occur in a competitive guest solution because the competitive molecules occupy the β-CD, thereby inhibiting its self-repair characteristics. This demonstrates the intelligent response of supramolecular hydrogels to different environmental stimuli. Then, they tested the self-healing behavior of the supramolecular hydrogel integrated capacitor (Fig. 7f). After being cut and standing for 1 hour at ambient condition, it could still power an electronic watch. Even after being cut 30 times in a cycle, it could still support a weight of 500 g without breaking and maintain 95.8% of its capacitance, indicating that the supramolecular hydrogel capacitor has excellent self-healing characteristics.
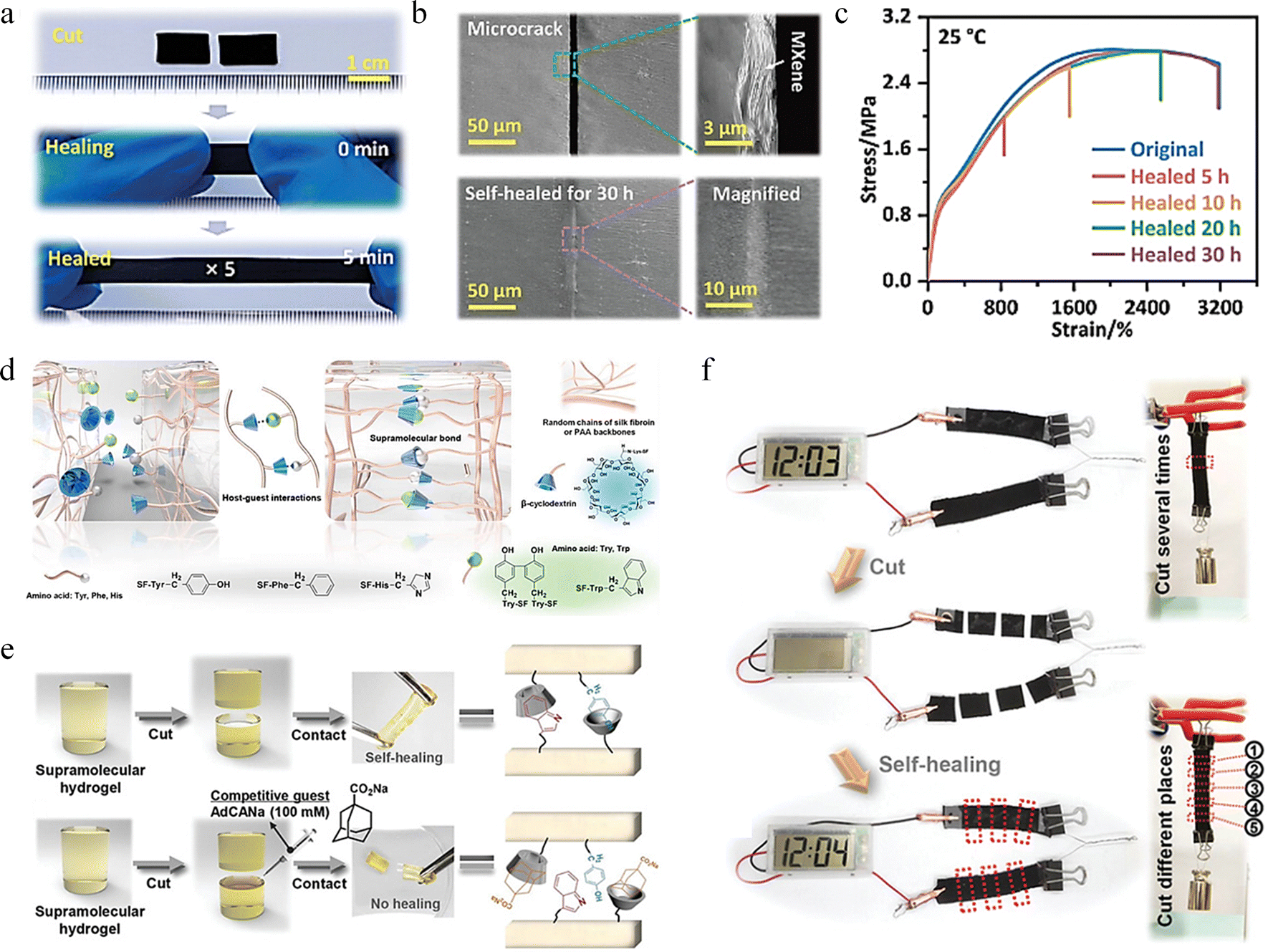 | ||
| Fig. 7 Research on self-healing behavior based on interfacial supramolecular interaction and host–guest interaction. (a) Photos of SHPULM-20 in instantaneous self-healing and stretching. (b) SEM images of cracks of SHPULM-20 composites before and after self-healing at 25 °C for 30 hours. (c) Stress–strain curves of original and healed SHPULM-20 under different healing time.115 Reproduced with permission from ref. 115, Copyright 2023, Wiley-VCH. (d) Self-healing mechanism of the supramolecular hydrogels. (e) Self-healing photos of supramolecular hydrogel in a wet and a competitive guest condition. (f) Self-healing behavior of the integrated supramolecular supercapacitor.119 Reproduced with permission from ref. 119, Copyright 2021, Wiley-VCH. | ||
 | ||
| Fig. 8 Structural self-healing behavior due to diffusion occurring in different types of materials. (a) Schematic representation of the method used for self-healing and mechanism of self-healing of MAPbI3.131 Reproduced with permission from ref. 131, Copyright 2022, Wiley-VCH. (b) Optical image of material surface damage when untreated and after heating at 100 °C for 1 h using different polymer mass percentage.133 Reproduced with permission from ref. 133, Copyright 2021, Elsevier. (c) Surface diffusion behaviour of gold nanowire.134 Reproduced with permission from ref. 134, Copyright 2018, Wiley-VCH. | ||
Finkenauer et al. doped thiourea-triglycol polymer molecules with mechanical toughness and self-healing into polycrystalline chalcogenide films, and synthesised chalcogenide films with glass transition temperatures close to room temperature and strong interactions with chalcogenide to form composites with bicontinuous interpenetrating networks. The TUEG3 polymer in the active layer was found to contribute to the reattachment of distant chalcogenide particles and repair of large cracks. This is due to the softening effect of the polymer addition, which promotes the mechanisms of synergistic grain growth and solid diffusion to repair the damage, as shown in Fig. 8b. The healing ability of the films exhibited differences with the addition of different contents of polymer molecules.133 Structural healing processes due to diffusion phenomena have also been of wide interest in the field of metallic materials, most of which are categorised as cold welding. Cold welding is a process of welding metals at near ambient temperatures (usually under atmospheric pressure) that requires clean and flat metal surfaces to be contacted under vacuum conditions to produce atomic diffusion leading to strong adhesion. Additional conditions generally need to be imposed, such as mechanical vibration, pressure, electrical impulses to induce the breakdown of the original stable atomic bonds on the metal surface. Consequently, atomic diffusion occurs at the separated interfaces on both sides, resulting in the formation of new bonds and leading to structural healing. Some experiments have been conducted by applying pressure to copper wires to eventually achieve the fusion of their ends at the point of contact.135 Nanomaterials such as nanoparticles, metal films or nanowires, have been found to exhibit cold welding behaviour. Rapid atomic rearrangements, surface diffusion, and atomic hopping are the fundamental modes for atomic transport on nanoscale metal surfaces.136 Wang et al. found that the cold welding process of gold nanowires can be assisted by simple mechanical vibration, and Fig. 8c shows that after using a tungsten probe to leave a crater at the fracture of a gold nanowire, the surface geometry of the end surface of the nanowire was reconstructed by a surface atomic diffusion process, which ultimately exhibits the pre-damage morphology. The surface atomic diffusion process can be accelerated with the addition of mechanical vibration assistance to achieve fusion of the nanowire fracture ends.134
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of cyclic loading. The presence of small-size twin boundaries is close to the crack healing region (GB34). Statistical graph shows the variation of crack length with emergence, extension, and self-healing behaviour during the complete cyclic loading process.143
000 of cyclic loading. The presence of small-size twin boundaries is close to the crack healing region (GB34). Statistical graph shows the variation of crack length with emergence, extension, and self-healing behaviour during the complete cyclic loading process.143
 | ||
| Fig. 9 Mechanisms of structural internal defects and microstructural effects on the self-healing behaviour of metals. (a) The self-healing behavior of ZnO nanobelt. D: dislocations, C: crack.142 Reproduced with permission from ref. 142, Copyright 2011, American Chemical Society. (b) Damage recovery from plastic deformation of gold particles.139 Reproduced with permission from ref. 139, Copyright 2017, Wiley-VCH. (c) Self-healing behaviour of polycrystalline Pt.143 Reproduced with permission from ref. 143, Copyright 2023, Springer Nature. | ||
In contrast to the extrinsic self-healing mechanism, the intrinsic self-healing mechanism operates on a distinct principle. It primarily relies on dynamic covalent bonds, supramolecular interactions, and atomic diffusion processes to exert its effectiveness. The relatively low bond energies involved in these processes allow the material to spontaneously undergo the cleavage and recombination of chemical bonds or the re-establishment of atomic/molecular interactions, either in the absence of external stimuli or with minimal energy input. Notably, the intrinsic self-healing mechanism effectively overcomes the limitation of the restricted number of healing cycles in the extrinsic mechanism. Moreover, it circumvents a series of issues arising from the interface mismatch between self-healing elements and the matrix. Consequently, by capitalizing on the advancements in the intrinsic self-healing mechanisms, the precise modulation of bond energies in super-hard covalent materials to align with the activation conditions of this mechanism— or the incorporation of chemical bonds capable of facilitating the autonomous re-bonding of fractured bonds—could play a pivotal role in the design of superhard covalent materials with self-healing properties.
3. Research on novel self-healing superhard covalent bond materials
3.1 Development of novel superhard materials
Hardness refers to the combined resistance of the chemical bonds in a crystal to indentation.46 The superhard properties derive from their unique microstructure and ultra-strong permanent covalent bonds. Following the successful development of polycrystalline diamond (110–140 GPa) (Fig. 10a), which exceeds the hardness of monocrystalline diamond,144 Tian et al. prepared nanotwinned diamond (200 GPa) (Fig. 10b) with hardness up to twice that of natural diamond, and nanotwinned cubic boron nitride (Fig. 10c) with hardness exceeding 100 GPa through the introduction of nanotwinned microstructural design.50,145 These superhard materials have a three-dimensional bond network with short and strong bonds, high bond density and high valence electron density.47 Subsequently, amorphous carbon materials (AM-III) (Fig. 10d) with a high content of sp3 hybridization (∼94%) that can scratch single crystal diamond were developed.146 In addition, polycrystalline diamond (Fig. 10e) and amorphous carbon (AC-3) (Fig. 10f), which also have a high proportion of sp3 hybridized carbon, have been reported.147,148 Without exception, these superhard materials are composed of ultra-strong permanent covalent bonds. However, a significant issue that is increasingly apparent is the intrinsic brittleness of the material, where even a small crack can lead to catastrophic fracture damage, resulting in structural material deterioration and posing serious risks to the safety and reliability of superhard materials and their associated devices. Yue et al. successfully prepared nanotwinned diamond composites (ntDC) assembled with coherently interfaced diamond polytypes, interwoven nanotwins and interlocked nanograins, and increased the fracture toughness to 26.6 MPa m1/2 without sacrificing hardness (∼200 GPa), which is five times higher than that of synthetic diamond.149 | ||
| Fig. 10 Superhard materials. (a) Polycrystalline diamond.144 Reproduced with permission from ref. 144, Copyright 2003, Springer Nature. (b) Nanotwinned diamond.145 Reproduced with permission from ref. 145, Copyright 2014, Springer Nature. (c) Nanotwinned cubic boron nitride.50 Reproduced with permission from ref. 138, Copyright 2013, Springer Nature. (d) AM-III.146 Reproduced with permission from ref. 146, Copyright 2021, Oxford University Press. (e) Paracrystalline diamond.147 Reproduced with permission from ref. 147, Copyright 2021, Springer Nature. (f) AC-3.148 Reproduced with permission from ref. 148, Copyright 2021, Springer Nature. | ||
To expand the application fields of superhard materials, researchers have been constantly searching for superhard materials other than diamond and cubic boron nitride. Recently, Tian et al. carried out research on grain refinement of cubic silicon carbide (SiC). They found that when the grain size of this material was refined to 10 nanometers, its hardness reached 41.5 GPa.51 This discovery implies that this category of silicon carbide meets the hardness criteria for superhard materials and can be classified as such. In fact, apart from silicon carbide, the hardness of certain diamond-like films, carbon nitrides, and other materials can also reach 40 GPa or even higher.150,151 In recent years, researchers have carried out predictions and explorations of potential superhard materials by means of theoretical calculations and other methods.152,153
In the research of superhard materials, researchers mainly focus on two key aspects. Firstly, through nanostructure design, such as nanotwins and gradient interfaces, the internal structure is optimized to increase hardness and improve toughness.52,154 Secondly, theoretical models of hardness are developed, alongside the exploration of new superhard phases, to push the boundaries of traditional materials and overcome their inherent limitations.47,155 Despite remarkable achievements, the proportion of superhard materials in practical applications is low. The inherent brittleness of high hardness materials makes them highly susceptible to the initiation and propagation of microcracks during long-term service due to external factors such as wear or impact. This, in turn, shortens the service life of the materials and makes it difficult to ensure their reliability. Meanwhile, the complexity of atomic bonding and phase-transformation kinetics hinders the optimization of the hardness of existing materials, and the synthesis of materials predicted by theory is also restricted by technical issues such as thermodynamic instability. Therefore, the coordinated regulation of hardness, toughness, and stability within the existing material system has become a central challenge in the field. The bionic self-healing mechanism, which draws on the self-healing characteristics of organisms, is expected to endow materials with self-healing capabilities and provide new ideas for solving the above-mentioned problems.
3.2 Study of self-healing superhard covalent bonding materials
In the previous chapter, we conducted a systematic and in-depth exploration of the existing self-healing mechanisms, covering both extrinsic and intrinsic self-healing mechanisms. These research findings provide ideas for the development of novel self-healing superhard covalent bond materials. However, numerous formidable challenges lie ahead. Common synthesis methods for superhard materials, such as high temperature and high pressure methods, chemical vapor deposition, etc.,44 encounter significant difficulties when incorporating self-healing elements (e.g., microcapsules and shape memory alloys). Under high temperature and high pressure synthesis conditions, the microcapsule wall materials may fail to withstand the extreme environment, resulting in the inability to effectively encapsulate the healing agents. Moreover, achieving uniform dispersion of microcapsules within the matrix remains a highly challenging task. When microcapsules are introduced using chemical vapor deposition, the surface quality of the coating may be compromised, leading to surface irregularities or defects. Additionally, due to the weak interfacial bonding force between the microcapsules and the coating, detachment is prone to occur. If shape memory alloys are incorporated into superhard materials, due to the obvious mismatch in coefficient of thermal expansion among these constituent phases,156,157 significant thermally-induced mismatch stress is likely to arise at the heterogeneous interface. This stress concentration effect tends to trigger interfacial delamination or microcracks, thus severely undermining the structural integrity and thermomechanical stability of the composite materials.Oxidation healing mechanisms rely on high temperatures and an air/oxygen environment, which is unfavorable for diamond. This is due to the poor thermal stability of diamond. Graphitization of diamond begins when the temperature exceeds approximately 780 °C.158 Moreover, neither diamond nor cubic boron nitride can form oxides to heal cracks. However, this oxidation healing mechanism appears to be applicable to SiC. When SiC undergoes oxidation at high temperatures (∼800 °C), it transforms into SiO2, which can fill the cracks in the material. Nevertheless, there is a substantial difference in hardness between the hardness of SiO2 (10.9 GPa) and that of pure SiC (33.3 GPa).159,160 More crucially, precisely controlling the high-temperature oxidation healing process exclusively at the sites where microcracks occur in SiC remains a significant challenge.
The defining feature of ultra-hard covalent bonds is their extremely high hardness. This property indicates a higher bond density/electron density, shorter bond lengths, and a greater degree of covalency.161 For instance, the super-strong C–C covalent bonds in diamond are among the most robust chemical bonds. They allow several carbon atom layers to withstand pressures up to 40 GPa.52,162 Unlike metallic materials, it is difficult for these bonds to heal through the atomic diffusion process. Moreover, such permanent covalent bonds have higher dissociation activation energies and binding energies compared to dynamic covalent bonds and supramolecular interactions. Under normal conditions, achieving reversibility is an arduous task. Consequently, the realization of the intrinsic self-healing of superhard covalent bond materials poses a significant challenge. Table 1 summarizes the damage mode, self-healing mechanism, self-healing condition and self-healing efficiency of superhard/hard covalent bond materials. In current research, hard covalent bond materials can achieve self-healing through processes such as oxidative healing, atomic diffusion, re-bonding of permanent covalent bonds, and grain growth. In contrast, given the unique properties of superhard covalent bond materials, we mainly focus on understanding the intrinsic mechanism of the self-healing process induced by the re-bonding of permanent covalent bonds.
| Superhard/hard covalent bond materials | Damage mode | Self-healing mechanism | Self-healing condition | Self-healing efficiency | Ref. | ||
|---|---|---|---|---|---|---|---|
| Nanotwinned diamond composite nanobeam | Width: 200nm |
Completely fractured | Formation of amorphous diamond osteoblasts hybridized by a mixture of sp2 and sp3 on the fracture surface, interatomic interaction forces change from repulsion to attraction, C–C re-bonding | Room temperature without pressure, high vacuum | Helaing time: 600 min | 34.1% | 163 |
| Width: 300nm |
Helaing time: 60 min | 24.2% | |||||
| Width: 400nm |
Helaing time: 60 min | 18.4% | |||||
| Width: 500nm |
Helaing time: 60 min | 18.3% | |||||
| Diamond single crystals nanobeam | Tensile direction: 〈100〉 | Completely fractured | Formation of amorphous diamond osteoblasts hybridized by a mixture of sp2 and sp3 on the fracture surface, interatomic interaction forces change from repulsion to attraction, C–C re-bonding | Room temperature without pressure, high vacuum | Healing time: 480 min | 6.7% | 163 |
| Tensile direction: 〈110〉 | Healing time: 60 min | 5.1% | |||||
| Tensile direction: 〈111〉 | Healing time: 480 min | 5.6% | |||||
| Single crystal SiC nanowires | Diameter: 87 nm | Completely fractured | Re-bonding and self-matching of stack faults | Room temperature, high vacuum | Healing time: 20 min | 12.9% | 164 |
| Amorphous SiC nanowires | Diameter: 102 nm | Completely fractured | Re-bonding of Si–C and Si–Si bonds, atomic diffusion | Room temperature, high vacuum | Healing time: 2 s | 63.6% | 164,165 |
| SiC with amorphous and crystalline composite structures nanowires | Diameter: 187 nm | Completely fractured | Re-bonding of Si–C, C–C and Si–Si bonds, atomic migration | Room temperature, high vacuum | Healing time: 20 min | 11.7% | 166 |
| Si with crystalline-amorphous composite structures nanowires | Diameter: 59 nm | Completely fractured | Spontaneous re-bonding, atomic rearrangement, and van der Waals attraction | Room temperature, high vacuum | Healing time: 20 min | 10.1% | 167 |
| Ti3AlC2 nanowires | Diameter: 93 nm | Completely fractured | The cleavage behavior, atomic migrations, and re-bonding on fracture surfaces | Room temperature, high vacuum | Healing time: 1 min | 27.6% | 168 |
| Healing time: 5 min | 36.2% | ||||||
| 2D Graphene | Plasma-induced damage | Atom migration and adatom-vacancy recombination | Plasma irradiation | Slower Damage at Grain Boundaries | 169 | ||
| SiBCN blocks | The cracks prepared using Vickers indentation | High-temperature oxidation generates liquid-like products to fill the crack | High temperature (1000 °C) air atmosphere | Cracks are eliminated | 168,170 | ||
| Polycrystalline Al2O3 blocks | Deep surface cracks | The growth of grains | High temperature (1700 °C) heat treatment for 1 hour | Cracks are eliminated | 171 | ||
| Spinel (MgAl2O4) ceramic | Pre-cracked | Abnormal grain growth and sintering phenomena | High temperature (1600 °C) heat treatment for 1 hour, air atmosphere | 91% | 172 | ||
nm, without external stimuli, can achieve up to 34% healing efficiency at room temperature simply by putting the fracture in contact (Fig. 11b). The self-healing performance is intricately linked not only to the healing time and frequency of healing but also to the maximum healing efficiency, which is profoundly influenced by the effective contact area when the two fractured surfaces make contact. In ntDC, as cracks initiate and expand, an amorphous carbon region forms at the crack tip. After the material fractures completely, this amorphous carbon distributes on the fracture surface (Fig. 11c). This amorphous carbon structure is composed of a mixture of sp2 and sp3 hybridized carbon which is named “Diamond Osteocytes” (DOs). It plays a crucial role in the self-healing process of the diamond and is the key factor for achieving self-healing. When fractures with DOs phases are close to each other, the interatomic interactions first show repulsive forces, and with further proximity to reach a critical distance the repulsive forces are changed to attractive forces, followed by the formation of new C–C bonds to achieve self-healing (Fig. 11d). This repulsive force is attributed to the fact that after the fracture of diamond, approximately half of the carbon atoms on the cracked surface undergo a transformation from sp3 hybridization to sp2 hybridization, and the half-occupied pz orbitals protruding outward from the surface form dangling bonds. Moreover, due to the insulating properties of diamond, charges accumulate on the surface of the fracture, thereby giving rise to the repulsive effect. When a critical distance is reached, it causes a sharp decrease in energy, thus changing from repulsive to attractive force. Benefiting from the advantages of the hierarchical structure of ntDC, such as diamond polytypes, nanotwins, stacking faults, etc., a large number of DOs can be generated at the fracture surface of ntDC, and these DOs promote the self-healing behavior. In addition, even the ntDC with width ∼500 nm showed room temperature self-healing ability, but the maximum healing efficiency was only 18.3%, which also indicates that the maximum healing efficiency is closely related to the cross-sectional size of the sample. This also explains why no room-temperature self-healing behavior was observed for diamond at the macroscopic size. Similarly, single crystal diamond exhibits a similar self-healing phenomenon. However, because only a small amount of DO forms on the diamond fracture surface, and the material at the fracture site disintegrates as a result of brittle fracture, the effective contact area is significantly reduced. As a result, the healing efficiency, which is approximately 6.7%, is much lower compared to that of nanocrystalline diamond (ntDC).
 | ||
| Fig. 11 Study of the self-healing behaviour of ntDC. (a) The SENB test of ntDC.149 Reproduced with permission from ref. 149, Copyright 2020, Springer Nature. (b) Testing of mechanical properties with different healing times of ntDC. (c) DOs formation process. (d) Atomic-scale healing processes of ntDC.163 Reproduced with permission from ref. 163, Copyright 2023, Springer Nature. | ||
Interestingly, even though the fracture surface was covered with amorphous DOs, Qiu et al. found no atomic diffusion behavior during the healing process.163 And DOs showed significant flexibility in tracing the atomic bonding self-healing process (Fig. 12a–f). Moreover, through first principles calculations, they further revealed the significance of amorphous DOs for the self-healing of diamond. The maximum repulsive force that needs to be overcome when two 3C diamond (111) facets are close to each other is about 12.82 GPa (Fig. 12g), whereas the maximum repulsive force that needs to be overcome when a DOs enriched with 34.7% of sp3 hybridized carbon is close to each other is only 5.73 GPa (Fig. 12h).163 This is due to the fact that the sp2 bonds in the DOs on the fracture surface can greatly increase the electrical conductivity, thereby reducing the charge accumulation on the surface, which effectively reduces the repulsive force and promotes the self-healing process. The relationship between the content of sp3 hybridized carbon in DOs and the maximum repulsive force also shows that the higher the content of sp3 bonding, the greater the maximum repulsive force (Fig. 12i), and the repulsive force also reaches a maximum of about 12.82 GPa when the sp3 content reaches 100%.
 | ||
| Fig. 12 Simulation of the self-healing process between two fractured ends of ntDC. (a)–(f) Molecular dynamics simulation of the ntDC crack healing process with DOs. (g) Force per area versus distance curve between two (111) surfaces of diamond. (h) Force per area versus distance curve between two amorphous DO structures with an sp3 ratio of 34.7%. (i) The maximum repulsive force per area versus sp3 content.163 Reproduced with permission from ref. 163, Copyright 2023, Springer Nature. | ||
Zeng et al. investigated the toughening and crack healing mechanism of diamond by molecular dynamics simulations.173 During the fracture processes of diamond single crystals, nanotwinned diamond, and 2H diamond polytypes, researchers have observed that the fracture surface of diamond single crystals is extremely flat and smooth (Fig. 13a). The fracture surface of nanotwinned diamond shows randomly distributed atomic clusters (Fig. 13b), while the fracture surface of 2H diamond polytypes exhibits an even larger number of disordered atomic clusters (Fig. 13c). These disordered atomic clusters can effectively impede crack propagation, thus significantly enhancing the fracture toughness of diamond materials. For diamond samples subjected to tensile fracture, once the applied load is removed, if the fracture surface exhibits exceptional smoothness, the fractured edges can align with remarkable precision, facilitating an almost flawless self-healing response (Fig. 13d–f). However, once there are disordered atomic clusters on the fracture surface, these atomic clusters will act as obstacles, making it difficult for the carbon atoms on the fracture surfaces to re-combine precisely and perfectly, thus severely affecting the degree of self-healing (Fig. 13g–i). Zeng et al. pointed out that the crack healing occurs during unloading, and its extent is associated with whether the fracture surfaces are clean.173 And a similar phenomenon occurs in 9R diamonds.
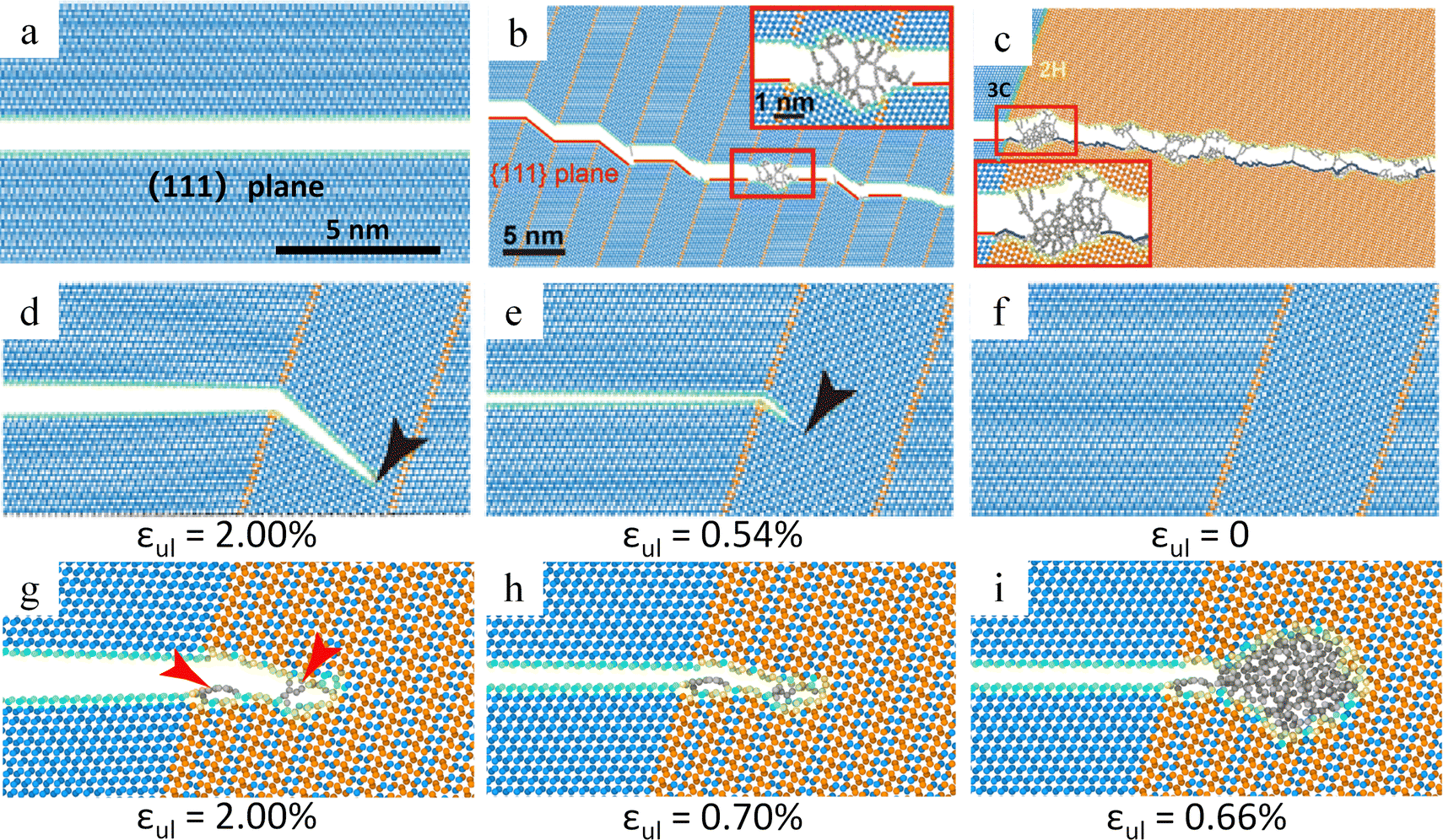 | ||
| Fig. 13 Theoretical study of self-healing behavior of diamonds. (a)–(c) Crack propagation path in diamond single crystal, nanotwinned diamond and 2H diamond. (d)–(f) Crack healing in nanotwinned diamond. (g)–(i) Crack healing in 4H diamond.173 Reproduced with permission from ref. 173, Copyright 2021, American Physical Society. | ||
Doping boron into diamond can transform it into p-type semiconductors suitable for microelectronics and photonic devices; however, the short and strong C–C bonds complicate the doping process. Ion implantation is one way of doping, but high-energy boron ions in the MeV range can cause damage to the diamond lattice.174 As detected by Raman spectroscopy, the one-phonon peak broadens and shifts to lower frequencies with a structured and intense background, indicating diamond lattice damage.174,175 Agulló-Rueda et al. found that after annealing at 1000 °C for 1 h, the position and width of the one-phonon peak were tilted towards the original values, while the background almost disappeared, indicating a partial recovery of the damaged diamond lattice.174 Further, Jiménez-Riobóo et al. found that carbon atoms are displaced from the boron atom injection path and location, eventually forming amorphous/crystalline hybridized regions.176 The Raman spectroscopic examination shows that the damaged area was repaired after vacuum annealing at 1000 °C (Fig. 14a). The recovery of the diamond lattice is achieved by the injection path of the C interstitial from the neighboring region to B and thermally activated diffusion at the B position. In addition, annealing at 1200 °C is detrimental to the diamond lattice and leads to traces of graphitization compared to 1000 °C. Focused ion beams of Ga ions, broad Ar ion beams, etc. cause damage to the structure of the diamond surface when processing diamond.177,178 The structural damage induced by ion implantation severely reduces the intrinsic thermal conductivity of diamond. Yuan et al. systematically investigated the evolution of corrugated morphology and chemical composition changes on the surface of the polycrystalline chemical vapor deposition diamond (PCD) under Ar ion beam bombardment with different incidence angles, ion energies and bombardment times.177 During ion bombardment, a large number of lattice damages or defects appeared on the PCD surface due to the conversion of sp3 hybridized C to sp2 hybridized C. After high temperature (1200 °C) annealing in vacuum (5.0 Pa), the physical phase did not change (Fig. 14b), while the intensity of the characteristic diamond peaks in the Raman spectra was restored, indicating that the damage was repaired (Fig. 14c), and that the resistance of the thin layer increased with annealing time under annealing conditions (Fig. 14d).
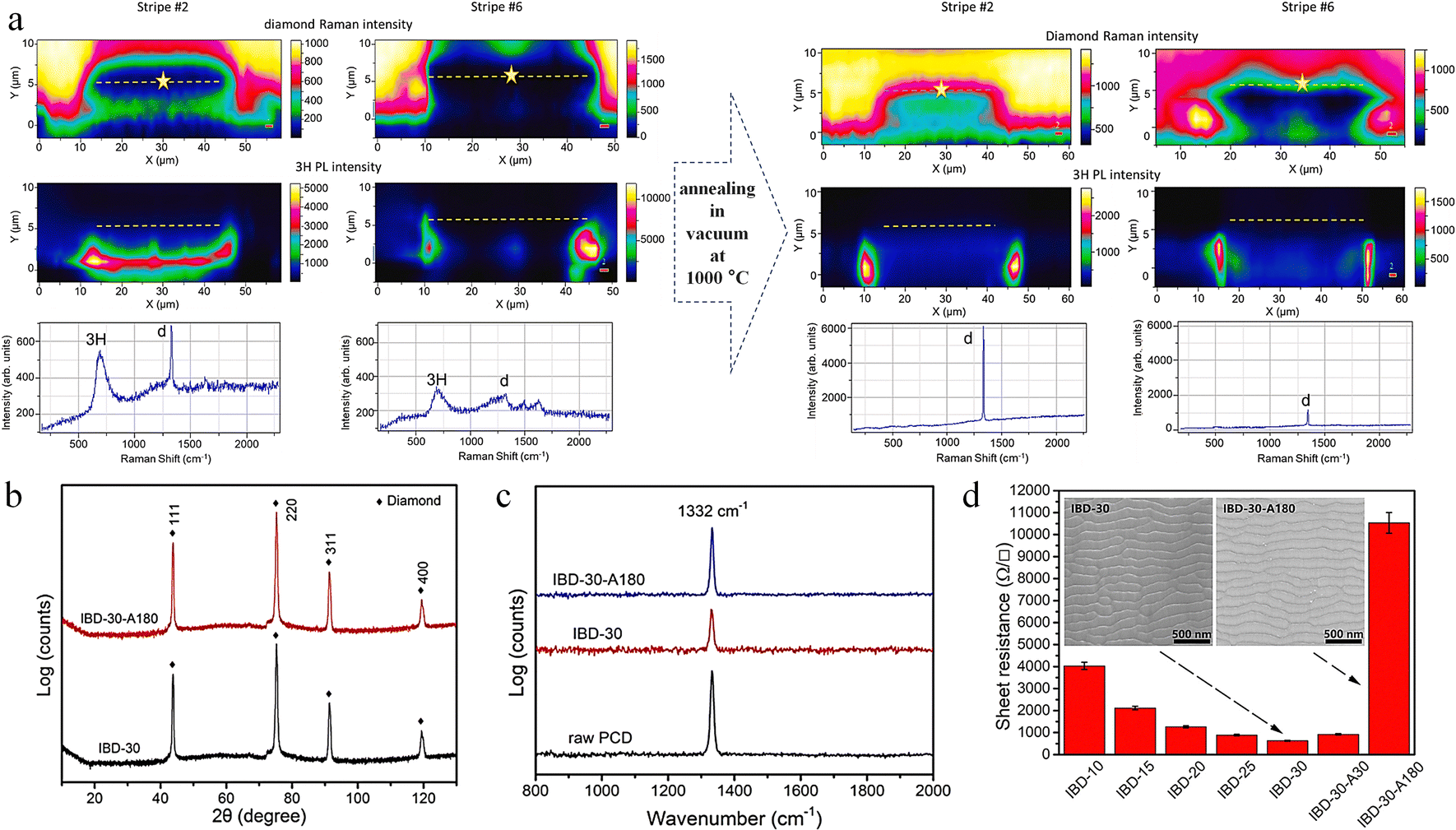 | ||
| Fig. 14 Annealing to repair diamond surface damage. (a) Repair of B-ion implantation-induced damage by annealing in vacuum at 1000 °C.176 Reproduced with permission from ref. 176, Copyright 2023, Elsevier. (b) X-ray diffraction spectrum of PCD before and after 30 keV Ar ion bombardment. (c) Raman spectra before and after the annealing in vacuum (5.0 Pa) at 1200 °C for 180 min. (d) The sheet resistance of the PCD.177 Reproduced with permission from ref. 177, Copyright 2021, Elsevier. | ||
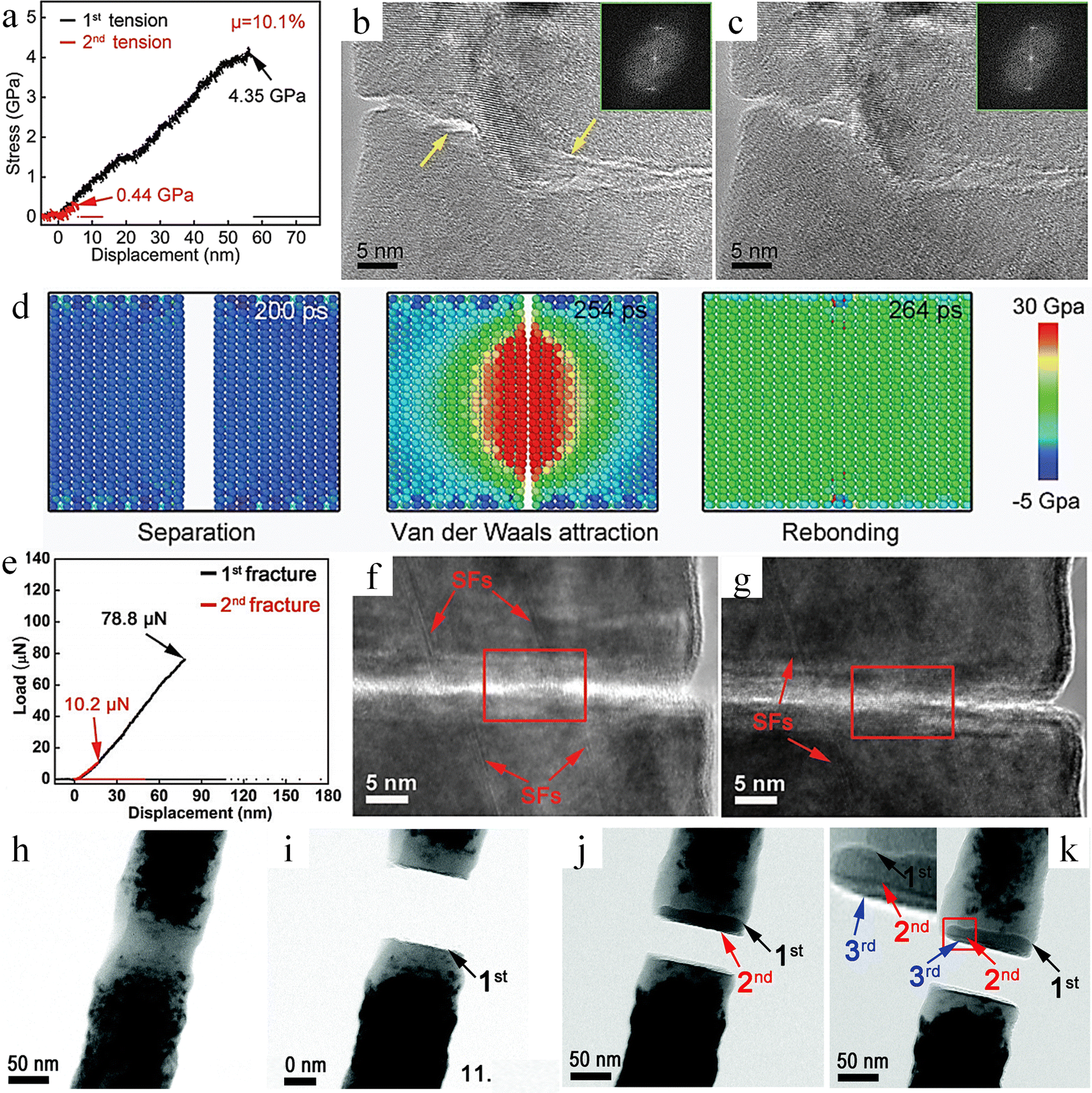 | ||
| Fig. 15 Study on self-healing behavior of single crystal Si and SiC. (a) Mechanical properties of Si nanowires before and after healing. (b) and (c) The HRTEM of Si nanowires before and after healing. (d) Molecular dynamics simulation of Si nanowire self-healing.167 Reproduced with permission from ref. 167, Copyright 2022, Wiley-VCH. (e) Mechanical properties of SiC nanowires before and after healing. (f) and (g) The HRTEM of SiC nanowires before and after healing. (h)–(k) TEM image at the fracture after multiple cycle fracture.164 Reproduced with permission from ref. 164, Copyright 2018, Royal Society of Chemistry. | ||
Different from non-polar covalent Si materials, polar covalent SiC exhibits different room temperature self-healing mechanisms. Zhang et al. investigated the self-healing behavior of SiC nanowires and revealed that single crystal SiC nanowires have a 12.9% self-healing efficiency (Fig. 15e).164 Each fracture occurred at the same position. According to the HRTEM results, the stacking faults were mismatched before healing (Fig. 15f), while the stacking faults were self-matched after self-healing (Fig. 15g). In addition, recrystallization and re-bonding occurred in the 〈111〉 direction during crack healing. Molecular dynamics simulations indicate that suspended bonds on the fracture surface do not oxidize under vacuum conditions, resulting in a high energy state. To reach a stable state, the system minimizes the potential energy of the suspended bonds, with the re-bonding of Si and C being the most effective method. After inducing the amorphization of single crystal SiC by electron beam irradiation and repeating the self-healing experiment, it was found that, different from single crystal SiC, amorphous SiC consistently fractured at distinct locations with each iteration (Fig. 15h–k). This indicates that the strength after healing is higher than that of the original amorphous phase, and the healing efficiency (67%) is much higher than that of single crystal SiC. This is due to the occurrence of recrystallization and re-bonding along the 〈111〉 orientation, forming a new interface composed of the amorphous phase and the crystalline phase. And atomic diffusion behavior was also found in this process. Single crystal SiC exhibits relatively ordered dangling bonds. In stark contrast, amorphous SiC not only has disordered dangling bonds but also has a greater number of dangling bonds per unit area. Disordered dangling bonds enables more dynamic movement of atoms on the fracture surface of amorphous SiC. Such atomic diffusion is conducive to eliminating the mismatched Si–C bonds at the healing sites and can relocate the atoms back to their original lattice structure in the 〈111〉 direction. Compared with single crystal SiC, amorphous SiC can more readily achieve the re-bonding of Si–C bonds. This process is beneficial for enhancing the fracture strength and elastic modulus of amorphous silicon carbide after crack healing.
Cui et al. also investigated the self-healing behavior of amorphous silicon carbide by in situ electron microscopy, and the results showed a self-healing efficiency of 63.6% (Fig. 16a).165 The HRTEM images taken before and after self-healing revealed that the cracks had disappeared, indicating effective healing (Fig. 16b). However, unlike the previous study, both fractures broke at the same location and no crystals were formed before and after self-healing.164 As a typical polar semiconductor, the fracture of SiC induces the generation of charges with opposite polarities on the fracture surfaces. These charges give rise to electrostatic forces, which play a pivotal role in facilitating the self-healing process of SiC nanowires.180 These charges then form electrostatic forces, which play a crucial role in the self-healing process of SiC nanowires. In addition, the disordered overhang bonds at the amorphous SiC fracture can be directly bonded without the need to reorient the bonds like crystals, and the atomic rearrangement is conducive to the self-healing of amorphous SiC. According to the results of molecular dynamics simulation, healing is achieved by reorganization of Si–C bonds forming Si–C and Si–Si bonds (Fig. 16c and d). Subsequently, Zhang et al. investigated the self-healing behavior on a fractured surface of SiC with an amorphous and crystalline (AAC) composite structure.166 Its diameter up to 187 nm can still show self-healing behavior, and the healing efficiency is about 11.7% (Fig. 16e).
 | ||
| Fig. 16 Study on self-healing behavior of amorphous SiC and AAC SiC. (a) Mechanical properties of amorphous SiC nanowires before and after healing. (b) The HRTEM of amorphous SiC nanowires before and after healing. (c) and (d) Molecular dynamics simulation of amorphous SiC nanowire self-healing.165 Reproduced with permission from ref. 165, Copyright 2019, American Chemical Society. (e) Mechanical properties of AAC SiC nanowires before and after healing. (f) The HRTEM of AAC SiC nanowires before and after healing.166 Reproduced with permission from ref. 166, Copyright 2020, Royal Society of Chemistry. | ||
Comparison of HRTEM images (Fig. 16f) before and after self-healing reveals that before self-healing, the mismatched fracture surface of the contact has a gap of 17 nm in length, and after self-healing, the mismatched fracture surface grows by 3 nm to fill up the gap, which is reduced to 14 nm. This is inconsistent with the phenomenon for pure single crystal SiC and pure amorphous SiC.164,165 Through molecular dynamics simulation, it is pointed out that the potential energy is reduced by re-bonding, while the potential energy is increased by debonding. The re-bonding of the C–Si, C–C and Si–Si pairs of atoms reduces the potential energy, respectively. Through coherent re-bonding and de-bonding, the potential energy of C–Si and Si–Si atoms is reduced respectively. Different from the self-healing mechanism of pure crystal and pure amorphous SiC, the healing process of AAC SiC achieves atomic migration through coherent re-bonding and debonding, thus filling the gap.
Unlike superhard/hard covalent bonding materials such as SiC, Si, and diamond, the MAX phase is a ternary compound due to the covalent bonding of M–X and the metal bonding of M–A.181 The Ti3AlC2 MAX phase is used as an example, where Ti–C is covalently bonded and Ti–Al is metallically bonded. Ti3AlC2 nanowires exhibit high room temperature self-healing efficiency, which can reach 36.2% when the diameter is 93 nm (Fig. 17a), and the maximum healing efficiency also exhibits a size dependence, with the larger the size, the lower the healing efficiency.168 After 1 min of self-healing in the fractured sample, the cracks on the fractured surface healed and atomic recombination was observed (Fig. 17b). Due to the lower cleavage energy of the Ti–Al bond compared to the Ti–C bond, fracture predominantly occurs along the Ti–Al interface, resulting in a smoother fracture surface and consequently enhancing the self-healing efficiency. Molecular dynamics simulation results indicate that initially, Al atoms on the opposing fracture surfaces form Al–Al bonds. This formation allows the nanowires to bear tensile stress. As the fracture surfaces continue to move closer, Al atoms re-bond with Ti atoms to form Al–Ti bonds, thereby releasing the tensile stress. This leads to a decrease in the potential energy and total energy of the nanowires, driving the self-healing process forward. During the healing process, surface relaxation is the primary factor affecting the self-healing efficiency. This is because after surface relaxation, some Al atoms move to the region between Al and Ti, impeding the direct bonding between Al and Ti. However, the atomic migration of Al atoms can effectively counteract the adverse effects caused by surface relaxation (Fig. 17c–h). Throughout the healing process, the Ti–C covalent bond did not directly participate in the breaking and re-bonding of chemical bonds.
 | ||
| Fig. 17 Study of the self-healing behavior of Ti3AlC2 MAX phase. (a) Stress–displacement curves obtained from healing experiments. (b) Characterization of fracture surfaces of Ti3AlC2. (c)–(h) The self-healing process between two fracture surfaces of the Ti3AlC2.168 Reproduced with permission from ref. 168, Copyright 2024, Wiley-VCH. | ||
1. Size effect: a strong and significant correlation exists between the healing efficiency of superhard and covalently bonded materials and the size of the samples. Generally speaking, the smaller the sample size, the higher the healing efficiency. This is mainly due to the fact that as the size decreases, the surface energy of the material increases accordingly. As a result, after fracture, the material is more likely to recombine to reduce the total potential energy of the system. In addition, the smaller the size, the smoother the fracture surface of the material, which undoubtedly creates favorable conditions for the self-healing process.
2. Degree of fracture mismatch: when two fractured surfaces approach each other, there is a substantial likelihood of experiencing relative misalignment during their contact. The impact of this phenomenon cannot be underestimated. It not only reduces the effective contact area, thereby leading to a decrease in strength and misassessment of stress, but also hinders the process of crystal-oriented bonding. The greater the degree of misalignment of the fractured surfaces, the lower the healing efficiency of the material.
3. Roughness of fracture surface: the research results indicate that the smoother and cleaner the fractured surfaces are, the higher the healing efficiency will be. When the fractured surfaces are rough, cavities will form during the closure of the fracture, leading to stress concentration in some areas. Such rough surfaces not only reduce the effective contact area but also prolong the healing time.
4. Healing time and healing frequency: healing time is a prerequisite for the self-healing of materials. When the self-healing process involves the re-formation of chemical bonds, extending the healing time is of vital importance. This can not only enable more chemical bonds to re-bond but also guarantee the high quality of these re-formed bonds, thus providing a solid foundation for the recovery of the material's properties. Meanwhile, sufficient time is also essential for the atomic diffusion process. An adequate amount of time ensures that the atomic diffusion occurs fully and perfectly, allowing atoms to migrate to appropriate positions and further facilitating the repair of the material's internal structure. Within the maximum achievable healing efficiency range of the material, there is an obvious positive correlation between the healing time and the healing efficiency. That is, the longer the healing time, the higher the healing efficiency. Research results on the self-healing behavior of diamond show that the healing frequency plays a crucial role in the self-healing performance of diamond. Increasing the healing frequency can promote the formation of DOs, which in turn drives a significant increase in the healing efficiency. Therefore, for those healing mechanisms involving the evolution of surface structure, the influence of healing frequency must be fully considered in practical research and application.
5. Degree of amorphization: current research results indicate that the amorphous structure, characterized by its disordered nature, offers significant advantages in terms of re-bonding. When a material undergoes an amorphization transformation, this change may potentially promote the self-healing behavior of the material. However, it is important to note that the amorphization process may also bring about some negative effects. One of these is the reduction in the surface cleanliness of the material, which in turn affects the fitting state of the fractured surfaces and ultimately leads to a decrease in the healing efficiency.
6. Environmental factors: the environment is crucial for a material's self-healing. Temperature, pressure, and oxygen content are key factors. Higher temperatures aid atomic diffusion by providing atoms with more energy to overcome lattice barriers, facilitating post-fracture re-bonding. However, excessive heat can cause thermal damage, altering the material's internal structure and impairing its performance and self-healing ability; pressure also matters. Appropriate pressure brings fractured surfaces closer, increasing atomic interaction and potentially enhancing bonding efficiency and the self-healing process; oxygen can also significantly impact self-healing. In an oxygen environment, dangling bonds on the fractured surface oxidize. Oxidation reduces their activity and the material's potential energy, making atomic re-bonding harder and hindering self-healing.
Research on self-healing superhard covalently bonded materials is still in its infancy. There may be a variety of potential factors in addition to those mentioned above. For example, the microstructural design of the material itself, as well as other factors that have not yet been identified. All these aspects urgently require more in-depth research. Especially at the atomic scale, in-depth dissection of the micro-mechanisms of self-healing is essential to further improve the healing efficiency of materials.
4. Conclusion and outlook
Currently, research on self-healing materials is being carried out vigorously in various fields. The concept of self-healing materials emerged as a significant innovative idea in the field of materials science. Its core objective is to endow materials with the function of autonomously repairing damage, thereby prolonging the service life of materials and enhancing their reliability. Among a wide variety of self-healing materials, new self-healing superhard covalent-bonded materials have stood out and attracted much attention. The unique covalent bonding in superhard covalently bonded materials endows them with a range of excellent mechanical properties, including very high hardness, excellent wear resistance, and strong strength, but also poses challenges for the design and development of self-healing superhard covalently bonded materials. Currently, the research on self-healing superhard covalent bonding materials is still in the early exploration stage. The covalent bonding system itself is extremely complex, and how to strike a delicate balance between maintaining superhard properties and realizing self-healing capability poses a great challenge to research efforts.This paper systematically and comprehensively reviews the extrinsic self-healing mechanisms, such as microencapsulation embedment, oxidative healing mechanism, and shape memory assisted healing, as well as the intrinsic self-healing mechanisms including dynamic covalent bonds, supramolecular interactions, diffusive behavior, and defect-driven processes. It elaborates on how these extrinsic and intrinsic healing mechanisms inspire the design of self-healing superhard covalently bonded materials. Meanwhile, it focuses on reviewing the research status of the self-healing behavior of superhard covalently bonded materials, deeply analyzes the key factors affecting their self-healing efficiency, aiming to assist researchers in better understanding and designing novel self-healing superhard covalently bonded materials.
For the study of the self-healing behavior of superhard covalent bonding materials, especially how to design and prepare superhard covalent bonding materials with high healing efficiency, the following key challenges must be addressed and overcome: (a) at present, a comprehensive and systematic theoretical model is lacking. This model should fully cover various influencing factors such as the type of inter atomic bonding, bond length/bond energy, crystal structure, grain size, dislocation density, etc. Through quantitative analysis, an accurate relationship between hardness and theoretical self-healing ability should be established, and then a practical theoretical guidance framework can be formed. (b) It is necessary to vigorously promote the continuous innovation of advanced characterization techniques, especially to focus on the development of direct bonding techniques for permanent covalent bonds. This not only plays a decisive role in the precise design of self-healing superhard covalent bond materials but also has equally important significance for the innovative development of wafer direct bonding technology. (c) When developing new self-healing superhard covalent bond materials, on the one hand, it is necessary to carefully design their microstructure at the micro-nano scale to ensure the maintenance of high hardness while significantly improving the comprehensive performance. On the other hand, starting from the atomic scale, chemical bonds should be precisely regulated to achieve high efficiency self-healing. (d) Achieving high precision positioning healing of the fracture surface, greatly improving the healing efficiency, and realizing controllable and rapid healing of permanent covalent bonds are crucial factors in overcoming the bottlenecks that hinder the practical application of materials. (e) Currently, the research on the self-healing behavior of superhard covalent bond materials mainly relies on transmission electron microscopes in a high vacuum environment to conduct analysis from the atomic scale. However, achieving reliable self-healing under complex working conditions (such as extreme conditions like high temperature, low temperature, high pressure, radiation, oxygen atmosphere, etc.) is the core and key to realizing the wide range practical application of this material. The development of self-healing superhard covalently bonded materials will have important potential applications in a number of industrial fields such as cutting tools, aerospace, electronics, etc. This progress could pave the way for the emergence of numerous innovative technologies in the near future, offering substantial benefits across various sectors.
Author contributions
K. Q., X. L. contributed equally to this work. Y. Y., L. G., proposed and supervised the project; K. Q., X. L., Y. L., Y. Y., L. G. wrote the review together.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2024YFA1209800), the National Natural Science Foundation of China (U23A20542, 52494930, 51972009, 51922017) and the State Key Laboratory of Silicate Materials for Architectures (Wuhan University of Technology) (SYSJJ2024-03).References
- M. Li, A. R. Mao, Q. W. Guan and E. Saiz, Chem. Soc. Rev., 2024, 53, 8240–8305 CAS.
- Y. Liu, B. Chen, Z. Liu, Z. Zhang and R. O. Ritchie, Prog. Mater. Sci., 2024, 144, 101281 CAS.
- S. Zhao and J. H. Ahn, Mater. Sci. Eng., R, 2022, 148, 100672 CrossRef.
- D. Nepal, S. Kang, K. M. Adstedt, K. Kanhaiya, M. R. Bockstaller, L. C. Brinson, M. J. Buehler, P. V. Coveney, K. Dayal, J. A. El-Awady, L. C. Henderson, D. L. Kaplan, S. Keten, N. A. Kotov, G. C. Schatz, S. Vignolini, F. Vollrath, Y. Wang, B. I. Yakobson, V. V. Tsukruk and H. Heinz, Nat. Mater., 2022, 22, 18–35 Search PubMed.
- S. An, B. Shi, M. Jiang, B. Fu, C. Song, P. Tao, W. Shang and T. Deng, Chem. Rev., 2023, 123, 7081–7118 CAS.
- U. H. Kim, E. J. Lee, C. S. Yoon, S. T. Myung and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1601417 Search PubMed.
- H. A. Bale, A. Haboub, A. A. MacDowell, J. R. Nasiatka, D. Y. Parkinson, B. N. Cox, D. B. Marshall and R. O. Ritchie, Nat. Mater., 2012, 12, 40–46 CrossRef PubMed.
- S. Lavenstein, Y. J. Gu, D. Madisetti and J. A. El-Awady, Science, 2020, 370, 190 CrossRef PubMed.
- G. Gao, F. Yang, F. Zhou, J. He, W. Lu, P. Xiao, H. Yan, C. Pan, T. Chen and Z. L. Wang, Adv. Mater., 2020, 32, 2004290 CrossRef CAS PubMed.
- F. Awaja, S. Zhang, M. Tripathi, A. Nikiforov and N. Pugno, Prog. Mater. Sci., 2016, 83, 536–573 CrossRef CAS.
- Y. Cao, Y. J. Tan, S. Li, W. W. Lee, H. Guo, Y. Cai, C. Wang and B. C. K. Tee, Nat. Electron., 2019, 2, 75–82 CrossRef.
- S. Bonardd, M. Nandi, J. I. Hernández García, B. Maiti, A. Abramov and D. Díaz Díaz, Chem. Rev., 2022, 123, 736–810 Search PubMed.
- A. Zarepour, S. Ahmadi, N. Rabiee, A. Zarrabi and S. Iravani, Nano-Micro Lett., 2023, 15, 100 CAS.
- J. Kang, J. B. H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 Search PubMed.
- I. L. Hia, V. Vahedi and P. Pasbakhsh, Polym. Rev., 2016, 56, 225–261 CAS.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CAS.
- P. Greil, J. Adv. Ceram., 2013, 1, 249–267 Search PubMed.
- S. Zhang, N. van Dijk and S. van der Zwaag, Acta Metall. Sin., 2020, 33, 1167–1179 Search PubMed.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CAS.
- J. W. C. Pang and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791–1799 CAS.
- S. K. Ghosh, Self-Healing Materials, 2008, pp. 1–28 DOI:10.1002/9783527625376.ch1.
- S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. M. Reddy, Science, 2021, 373, 321–327 CAS.
- J. Cui, Y. Bao, Q. Dai, F. Li, H. Wang, Y. Liu, F. Cao and J. Li, Adv. Funct. Mater., 2024, 2405737, DOI:10.1002/adfm.202405737.
- J. Xu, J. Chen, Y. Zhang, T. Liu and J. Fu, Angew. Chem., Int. Ed., 2021, 60, 7947–7955 CrossRef CAS PubMed.
- S. Zhang, Y. H. Cheng, L. Galuska, A. Roy, M. Lorenz, B. Chen, S. Luo, Y. T. Li, C. C. Hung, Z. Qian, P. B. J. St. Onge, G. T. Mason, L. Cowen, D. Zhou, S. I. Nazarenko, R. F. Storey, B. C. Schroeder, S. Rondeau-Gagné, Y. C. Chiu and X. Gu, Adv. Funct. Mater., 2020, 30, 2000663 CrossRef CAS.
- Y. Yao, Z. Xu, B. Liu, M. Xiao, J. Yang and W. Liu, Adv. Funct. Mater., 2020, 31, 2006944 CrossRef.
- E. Z. Alharissa, N. Nashrah, R. A. K. Putri, W. Al Zoubi and Y. G. Ko, Adv. Compos. Hybrid Mater., 2024, 7, 16 CrossRef CAS.
- C. Ye, D. Liu, X. Peng, Y. Jiang, R. Cheng, C. Ning, F. Sheng, Y. Zhang, K. Dong and Z. L. Wang, ACS Nano, 2021, 15, 18172–18181 CrossRef CAS PubMed.
- J. Liu, Z. Tong, F. Gao, J. Wang, J. Hu, L. Song, Y. Hou, J. Lu, X. Zhan and Q. Zhang, Adv. Mater., 2024, 36, 2401982 CrossRef CAS PubMed.
- Y. M. Li, Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Adv. Mater., 2023, 35, 2211009 CrossRef CAS PubMed.
- R. B. Leonesio, J. Am. Ceram. Soc., 2006, 55, 437–439 CrossRef.
- G. L. Gaines and D. Tabor, Nature, 1956, 178, 1304–1305 CrossRef CAS.
- Y. M. Malinskii, V. V. Prokopenko, N. A. Ivanova and V. A. Kargin, Polym. Mech., 1973, 6, 240–244 CrossRef.
- Y. M. Malinskii, V. V. Prokopenko, N. A. Ivanova and V. S. Kargin, Polym. Mech., 1973, 6, 382–384 CrossRef.
- Y. M. Malinskii, V. V. Prokopenko and V. A. Kargin, Polym. Mech., 1973, 6, 969–972 CrossRef.
- R. J. Gray, Cem. Concr. Res., 1984, 14, 315–317 CrossRef.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CAS.
- G. Williams, R. Trask and I. Bond, Composites, Part A, 2007, 38, 1525–1532 Search PubMed.
- K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399–1405 CAS.
- B. Aizenshtein and L. Etgar, Small, 2023, 20, 2305755 CrossRef PubMed.
- C. O. W. Trost, A. Lassnig, P. Kreiml, T. Jörg, V. L. Terziyska, C. Mitterer and M. J. Cordill, Adv. Mater., 2024, 36, 2401007 CrossRef CAS PubMed.
- A. K. Padhan, D. Sharma, T. S. Thomas, A. P. Sinha, A. N. Mallick and D. Mandal, J. Mater. Chem. A, 2024, 12, 9508–9517 RSC.
- J. Wang, J. Tang, D. Chen, S. Xing, X. Liu and J. Hao, Polym. Compos., 2023, 44, 6304–6323 CrossRef CAS.
- V. L. Solozhenko and E. Gregoryanz, Mater. Today, 2005, 8, 44–51 CrossRef CAS.
- Z. Zhang and J. Brgoch, J. Am. Chem. Soc., 2022, 144, 18075–18080 CrossRef CAS PubMed.
- B. Xu and Y. Tian, Sci. China: Mater., 2015, 58, 132–142 CAS.
- Y. Tian, B. Xu and Z. Zhao, Int. J. Refract. Met. Hard Mater., 2012, 33, 93–106 CrossRef CAS.
- R. B. Kaner, J. J. Gilman and S. H. Tolbert, Science, 2005, 308, 1268–1269 CrossRef CAS PubMed.
- P. Ying, B. Z. Li, M. D. Ma, Y. F. Gao, R. X. Sun, Z. H. Li, S. Chen, B. Zhang, H. F. Li, B. Liu, L. Sun, S. Zhao, K. Tong, W. T. Hu, Y. L. Pan, G. D. Tang, D. L. Yu, Z. S. Zhao, B. Xu and Y. J. Tian, Nat. Synth., 2025, 4, 391–398 CrossRef CAS.
- Y. Tian, B. Xu, D. Yu, Y. Ma, Y. Wang, Y. Jiang, W. Hu, C. Tang, Y. Gao, K. Luo, Z. Zhao, L.-M. Wang, B. Wen, J. He and Z. Liu, Nature, 2013, 493, 385–388 CrossRef CAS PubMed.
- R. X. Sun, X. D. Wei, W. T. Hu, P. Ying, Y. J. Wu, L. Y. Wang, S. Chen, X. Zhang, M. D. Ma, D. L. Yu, L. Wang, G. Y. Gao, B. Xu and Y. J. Tian, Small, 2022, 18, 2201212 CrossRef CAS PubMed.
- Z. Zhao, B. Xu and Y. Tian, Annu. Rev. Mater. Res., 2016, 46, 383–406 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS PubMed.
- S. Meure, D. Y. Wu and S. Furman, Acta Mater., 2009, 57, 4312–4320 CrossRef CAS.
- S. Gupta, Q. Zhang, T. Emrick, A. C. Balazs and T. P. Russell, Nat. Mater., 2006, 5, 229–233 CrossRef CAS.
- J. Wang, M. Wu, J. Huang, Y. Wang, X. Miao, Y. Chen, D. Bian and Y. Zhao, ACS Appl. Mater. Interfaces, 2024, 16, 26768–26786 CrossRef CAS PubMed.
- J. Martínez Lucci, R. S. Amano and P. K. Rohatgi, Heat Mass Transfer, 2016, 53, 825–848 CrossRef.
- K. K. Alaneme and M. O. Bodunrin, Appl. Mater. Today, 2017, 6, 9–15 Search PubMed.
- J.-J. Chen, H. Xie, L.-Z. Liu, H. Guan, Z. You, L. Zou and H.-J. Jin, Science, 2024, 385, 629–633 CrossRef CAS PubMed.
- X. Lu, S. Li, W. Zhang, B. Yao, W. Yu and Y. Zhou, J. Eur. Ceram. Soc., 2019, 39, 4023–4028 Search PubMed.
- K. Takahashi, M. Yokouchi, S. K. Lee and K. Ando, J. Am. Ceram. Soc., 2004, 86, 2143–2147 CrossRef.
- S. Li, L. Xiao, G. Song, X. Wu, W. G. Sloof, S. van der Zwaag and Y. Zhou, J. Am. Ceram. Soc., 2013, 96, 892–899 CrossRef CAS.
- Y. Liu, H. Liu, C. Huang, L. Ji, L. Wang, Y. Yuan, Q. Liu and Q. Han, Ceram. Int., 2023, 49, 13790–13798 Search PubMed.
- J. Lin, Y. Huang and H. Zhang, Ceram. Int., 2014, 40, 16811–16815 CrossRef CAS.
- G. M. Song, Y. T. Pei, W. G. Sloof, S. B. Li, J. T. M. De Hosson and S. van der Zwaag, Scr. Mater., 2008, 58, 13–16 CrossRef CAS.
- B. Yao, S. Li, W. Zhang, W. Yu, Y. Zhou, S. Fan and G. Bei, J. Adv. Ceram., 2022, 11, 1687–1695 CrossRef CAS.
- S. Li, L. Zhang, W. Yu and Y. Zhou, Ceram. Int., 2017, 43, 6963–6966 CrossRef CAS.
- W.-C. Kim, K.-R. Lim, W.-T. Kim, E.-S. Park and D.-H. Kim, Prog. Mater. Sci., 2022, 123, 100756 CrossRef.
- J. B. Ferguson, B. F. Schultz and P. K. Rohatgi, Mater. Sci. Eng., A, 2015, 620, 85–88 CrossRef.
- P. K. Rohatgi, Mater. Sci. Eng., A, 2014, 619, 73–76 CrossRef CAS.
- N. Salowitz, S. Misra, M. I. Haider, M. Povolo and P. Rohatgi, Materials, 2022, 15, 2970 CrossRef CAS PubMed.
- E. L. Kirkby, J. D. Rule, V. J. Michaud, N. R. Sottos, S. R. White and J. A. E. Månson, Adv. Funct. Mater., 2008, 18, 2253–2260 CrossRef CAS.
- J. Champagne, S.-S. Pang and G. Li, Composites, Part B, 2016, 97, 344–352 CrossRef CAS.
- Y. Shi, C. B. Cooper, T. Nogusa, J.-C. Lai, H. Lyu, M. Khatib, C. Xu, L. Michalek and Z. Bao, Matter, 2024, 7, 2108–2124 CrossRef CAS.
- C. R. Fisher, H. B. Henderson, M. S. Kesler, P. Zhu, G. E. Bean, M. C. Wright, J. A. Newman, L. C. Brinson, O. Figueroa and M. V. Manuel, Appl. Mater. Today, 2018, 13, 64–68 CrossRef.
- C. C. Hornat and M. W. Urban, Nat. Commun., 2020, 11, 1028 CrossRef CAS PubMed.
- S. Samanta, S. Kim, T. Saito and A. P. Sokolov, J. Phys. Chem. B, 2021, 125, 9389–9401 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- X. Liu, Y. Li, X. Fang, Z. Zhang, S. Li and J. Sun, ACS Mater. Lett., 2022, 4, 554–571 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- K. P. Faseela, A. P. Benny, Y. Kim and S. Baik, Small, 2021, 18, 2104764 CrossRef PubMed.
- T. Wang, W.-W. Kong, W.-C. Yu, J.-F. Gao, K. Dai, D.-X. Yan and Z.-M. Li, Nano-Micro Lett., 2021, 13, 162 CrossRef CAS PubMed.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef PubMed.
- J. Ma, Y. Yang, C. Valenzuela, X. Zhang, L. Wang and W. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116219 CrossRef CAS PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- G. P. Carden, M. L. Martins, G. Toleutay, S. Ge, B. Li, S. Zhao and A. P. Sokolov, Macromolecules, 2024, 57, 8621–8631 CrossRef CAS.
- S. Cho, S. Y. Hwang, D. X. Oh and J. Park, J. Mater. Chem. A, 2021, 9, 14630–14655 RSC.
- M. Irigoyen, A. Fernández, A. Ruiz, F. Ruipérez and J. M. Matxain, J. Org. Chem., 2019, 84, 4200–4210 CrossRef CAS PubMed.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS PubMed.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- M. O. Saed, A. Gablier and E. M. Terentejv, Adv. Funct. Mater., 2019, 30, 1906458 CrossRef.
- Y. Yuan, Y. Sun, S. Yan, J. Zhao, S. Liu, M. Zhang, X. Zheng and L. Jia, Nat. Commun., 2017, 8, 14657 CrossRef PubMed.
- H. Memon, H. Liu, M. A. Rashid, L. Chen, Q. Jiang, L. Zhang, Y. Wei, W. Liu and Y. Qiu, Macromolecules, 2020, 53, 621–630 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2018, 51, 9816–9824 CrossRef CAS.
- S. Bhattacharya, R. S. Phatake, S. Nabha Barnea, N. Zerby, J.-J. Zhu, R. Shikler, N. G. Lemcoff and R. Jelinek, ACS Nano, 2019, 13, 1433–1442 CrossRef CAS PubMed.
- D. Zhao, M. Feng, L. Zhang, B. He, X. Chen and J. Sun, Carbohydr. Polym., 2021, 256, 117580 CrossRef CAS PubMed.
- H. Feng, T. Wang, W. Wang, C. Ma, Y. Pu and S. Chen, J. Mater. Sci. Technol., 2024, 173, 121–136 CrossRef CAS.
- Y. Wang, M. Yang and Z. Zhao, Carbohydr. Polym., 2023, 310, 120723 CrossRef CAS PubMed.
- J.-H. Gao, B. Wan, M.-S. Zheng, L. Luo, H. Zhang, Q.-L. Zhao, G. Chen and J.-W. Zha, Mater. Horiz., 2024, 11, 1305–1314 RSC.
- C. Zhang, M. Wang, C. Jiang, P. Zhu, B. Sun, Q. Gao, C. Gao and R. Liu, Nano Energy, 2022, 95, 106991 CrossRef CAS.
- B. Zhou, D. He, J. Hu, Y. Ye, H. Peng, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2018, 6, 11725–11733 CAS.
- L. Wang, Z. L. Gong, S. Y. Li, W. Hong, Y. W. Zhong, D. Wang and L. J. Wan, Angew. Chem., Int. Ed., 2016, 55, 12393–12397 CAS.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CAS.
- Y. Jia, Q. Guan, C. Chu, L. Zhang, R. E. Neisiany, S. Gu, J. Sun and Z. You, Sci. Bull., 2024, 69, 1875–1886 CrossRef CAS PubMed.
- Y. Liu, X. Chen, S. Yin, X. Chang, C. Lv, J. Zang, X. Leng, T. Zhang and G. Zhao, ACS Nano, 2022, 16, 19472–19481 CAS.
- Z. Wang, J. Fan, D. He, L. Ren, Z. Hao, R. Sun and X. Zeng, J. Mater. Chem. A, 2022, 10, 21923–21932 CAS.
- X. Zhu, W. Zhang, G. Lu, H. Zhao and L. Wang, ACS Nano, 2022, 16, 16724–16735 CAS.
- R. Han, F. Zeng, Q. Xia, X. Pang and X. Wu, Carbohydr. Polym., 2024, 340, 122271 CrossRef CAS PubMed.
- Y. Han, K. Zhao, G. Chen, R. A. Li, C. Zhou, Z. Hua, H. Duan and M. He, J. Mater. Chem. A, 2023, 11, 19637–19644 RSC.
- J. Xu, Y. Li, T. Liu, D. Wang, F. Sun, P. Hu, L. Wang, J. Chen, X. Wang, B. Yao and J. Fu, Adv. Mater., 2023, 35, 2300937 CrossRef CAS PubMed.
- J.-B. Hou, X.-Q. Zhang, D. Wu, J.-F. Feng, D. Ke, B.-J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12105–12113 CrossRef CAS PubMed.
- M. Mohamadhoseini and Z. Mohamadnia, Coord. Chem. Rev., 2021, 432, 213711 CrossRef CAS.
- L. Xiao, W. Xu, L. Huang, J. Liu and G. Yang, Adv. Compos. Hybrid Mater., 2022, 6, 15 CrossRef.
- F. Mo, Q. Li, G. Liang, Y. Zhao, D. Wang, Y. Huang, J. Wei and C. Zhi, Adv. Sci., 2021, 8, 2100072 CrossRef CAS PubMed.
- H. Lv, X. Chu, Y. Zhang, Q. Liu, F. Wu and D. Mu, Mater. Today, 2024, 78, 181–208 CrossRef CAS.
- E. B. Stukalin, L.-H. Cai, N. A. Kumar, L. Leibler and M. Rubinstein, Macromolecules, 2013, 46, 7525–7541 CrossRef CAS PubMed.
- L. N. Neumann, E. Oveisi, A. Petzold, R. W. Style, T. Thurn-Albrecht, C. Weder and S. Schrettl, Sci. Adv., 2021, 7, eabe4154 CAS.
- Y. H. Kim and R. P. Wool, Macromolecules, 2002, 16, 1115–1120 CrossRef.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CAS.
- C. Chen, W. B. Ying, J. Li, Z. Kong, F. Li, H. Hu, Y. Tian, D. H. Kim, R. Zhang and J. Zhu, Adv. Funct. Mater., 2021, 32, 2106341 CrossRef.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 CAS.
- B. P. Finkenauer, Akriti, K. Ma and L. Dou, ACS Appl. Mater. Interfaces, 2022, 14, 24073–24088 CAS.
- Y. Cheng, X. Liu, Z. Guan, M. Li, Z. Zeng, H. W. Li, S. W. Tsang, A. G. Aberle and F. Lin, Adv. Mater., 2020, 33, 2006170 Search PubMed.
- Y. Rakita, I. Lubomirsky and D. Cahen, Mater. Horiz., 2019, 6, 1297–1305 CAS.
- Y. Rakita, S. R. Cohen, N. K. Kedem, G. Hodes and D. Cahen, MRS Commun., 2015, 5, 623–629 CAS.
- M. B. Al-Handawi, G. Dushaq, P. Commins, D. P. Karothu, M. Rasras, L. Catalano and P. Naumov, Adv. Mater., 2022, 34, 2109374 CAS.
- S. K. Yadavalli, Z. Dai, H. Zhou, Y. Zhou and N. P. Padture, Acta Mater., 2020, 187, 112–121 CAS.
- B. P. Finkenauer, Y. Gao, X. Wang, Y. Tian, Z. Wei, C. Zhu, D. J. Rokke, L. Jin, L. Meng, Y. Yang, L. Huang, K. Zhao and L. Dou, Cell Rep. Phys. Sci., 2021, 2, 100320 CAS.
- B. Wang, Y. Han, S. Xu, L. Qiu, F. Ding, J. Lou and Y. Lu, Small, 2018, 14, 1704085 Search PubMed.
- G. S. Ferguson, M. K. Chaudhury, G. B. Sigal and G. M. Whitesides, Science, 1991, 253, 776–778 CAS.
- D. V. Wagle and G. A. Baker, Mater. Horiz., 2015, 2, 157–167 RSC.
- S. Zhang, C. Kwakernaak, W. Sloof, E. Brück, S. van der Zwaag and N. van Dijk, Adv. Eng. Mater., 2015, 17, 598–603 CrossRef CAS.
- R. N. Lumley, A. J. Morton and I. J. Polmear, Acta Mater., 2002, 50, 3597–3608 CrossRef CAS.
- O. Kovalenko, C. Brandl, L. Klinger and E. Rabkin, Adv. Sci., 2017, 4, 1700159 CrossRef PubMed.
- C. Chen, S. Nagao, K. Suganuma, J. Jiu, H. Zhang, T. Sugahara, T. Iwashige, K. Sugiura and K. Tsuruta, Appl. Phys. Lett., 2016, 109, 093503 CrossRef.
- R. Xin, Q. Ma and W. Li, Mater. Sci. Eng., A, 2016, 662, 65–71 CrossRef CAS.
- J. Zang, Z.-H. Xu, R. A. Webb and X. Li, Nano Lett., 2010, 11, 241–244 CrossRef PubMed.
- C. M. Barr, T. Duong, D. C. Bufford, Z. Milne, A. Molkeri, N. M. Heckman, D. P. Adams, A. Srivastava, K. Hattar, M. J. Demkowicz and B. L. Boyce, Nature, 2023, 620, 552–556 CrossRef CAS PubMed.
- T. Irifune, A. Kurio, S. Sakamoto, T. Inoue and H. Sumiya, Nature, 2003, 421, 599–600 CAS.
- Q. Huang, D. Yu, B. Xu, W. Hu, Y. Ma, Y. Wang, Z. Zhao, B. Wen, J. He, Z. Liu and Y. Tian, Nature, 2014, 510, 250–253 CrossRef CAS PubMed.
- S. Zhang, Z. Li, K. Luo, J. He, Y. Gao, A. V. Soldatov, V. Benavides, K. Shi, A. Nie, B. Zhang, W. Hu, M. Ma, Y. Liu, B. Wen, G. Gao, B. Liu, Y. Zhang, Y. Shu, D. Yu, X.-F. Zhou, Z. Zhao, B. Xu, L. Su, G. Yang, O. P. Chernogorova and Y. Tian, Natl. Sci. Rev., 2022, 9, nwab140 CAS.
- H. Tang, X. Yuan, Y. Cheng, H. Fei, F. Liu, T. Liang, Z. Zeng, T. Ishii, M.-S. Wang, T. Katsura, H. Sheng and H. Gou, Nature, 2021, 599, 605–610 CAS.
- Y. Shang, Z. Liu, J. Dong, M. Yao, Z. Yang, Q. Li, C. Zhai, F. Shen, X. Hou, L. Wang, N. Zhang, W. Zhang, R. Fu, J. Ji, X. Zhang, H. Lin, Y. Fei, B. Sundqvist, W. Wang and B. Liu, Nature, 2021, 599, 599–604 CAS.
- Y. Yue, Y. Gao, W. Hu, B. Xu, J. Wang, X. Zhang, Q. Zhang, Y. Wang, B. Ge, Z. Yang, Z. Li, P. Ying, X. Liu, D. Yu, B. Wei, Z. Wang, X.-F. Zhou, L. Guo and Y. Tian, Nature, 2020, 582, 370–374 CAS.
- X. Zhao, X. He, Y. Sun, J. Yi and P. Xiao, Acta Mater., 2009, 57, 893–902 CAS.
- D. Laniel, F. Trybel, A. Aslandukov, S. Khandarkhaeva, T. Fedotenko, Y. Yin, N. Miyajima, F. Tasnádi, A. V. Ponomareva, N. Jena, F. I. Akbar, B. Winkler, A. Néri, S. Chariton, V. Prakapenka, V. Milman, W. Schnick, A. N. Rudenko, M. I. Katsnelson, I. A. Abrikosov, L. Dubrovinsky and N. Dubrovinskaia, Adv. Mater., 2023, 36, 2308030 Search PubMed.
- A. Mansouri Tehrani, A. O. Oliynyk, M. Parry, Z. Rizvi, S. Couper, F. Lin, L. Miyagi, T. D. Sparks and J. Brgoch, J. Am. Chem. Soc., 2018, 140, 9844–9853 CAS.
- A. L. Ivanovskii, Prog. Mater. Sci., 2012, 57, 184–228 CAS.
- Y. L. Pan, P. Ying, Y. F. Gao, P. Liu, K. Tong, D. L. Yu, K. L. Jiang, W. T. Hu, B. Z. Li, B. Liu, Z. S. Zhao, J. L. He, B. Xu, Z. Y. Liu and Y. J. Tian, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108340118 CAS.
- X. L. He, X. Dong, Q. S. Wu, Z. S. Zhao, Q. Zhu, A. R. Oganov, Y. J. Tian, D. L. Yu, X. F. Zhou and H. T. Wang, Phys. Rev. B, 2018, 97, 100102(R) Search PubMed.
- P. Jacobson and S. Stoupin, Diamond Relat. Mater., 2019, 97, 107469 CAS.
- X. M. Sun, D. Y. Cong, Y. Ren, D. E. Brown, R. G. Li, S. H. Li, Z. Yang, W. X. Xiong, Z. H. Nie, L. Wang and Y. D. Wang, Appl. Phys. Lett., 2018, 113, 041903 Search PubMed.
- S. Liu, L. Han, Y. Zou, P. Zhu and B. Liu, J. Mater. Sci. Technol., 2017, 33, 1386–1391 CAS.
- R. Sun, X. Zhang, X. Hao, W. Hu, X. Wei, X. Song, Z. Zhang, P. Ying, S. Zhao, Y. Wang, Y. Gao, D. Yu, B. Xu, G. Gao and Y. Tian, J. Eur. Ceram. Soc., 2025, 45, 116829 CrossRef CAS.
- E. Kulik, N. Nishiyama, Y. Higo, N. A. Gaida and T. Katsura, J. Am. Ceram. Soc., 2018, 102, 2251–2256 CrossRef.
- F. M. Gao, J. L. He, E. D. Wu, S. M. Liu, D. L. Yu, D. C. Li, S. Y. Zhang and Y. J. Tian, Phys. Rev. Lett., 2003, 91, 015502 CrossRef PubMed.
- Z. Zeng, J. Wen, H. Lou, X. Zhang, L. Yang, L. Tan, B. Cheng, X. Zuo, W. Yang, W. L. Mao, H.-K. Mao and Q. Zeng, Nature, 2022, 608, 513–517 CrossRef CAS PubMed.
- K. Qiu, J. Hou, S. Chen, X. Li, Y. Yue, B. Xu, Q. Hu, L. Liu, Z. Yang, A. Nie, Y. Gao, T. Jin, J. Wang, Y. Li, Y. Wang, Y. Tian and L. Guo, Nat. Mater., 2023, 22, 1317–1323 CrossRef CAS PubMed.
- Z. Zhang, J. Cui, B. Wang, H. Jiang, G. Chen, J. Yu, C. Lin, C. Tang, A. Hartmaier, J. Zhang, J. Luo, A. Rosenkranz, N. Jiang and D. Guo, Nanoscale, 2018, 10, 6261–6269 RSC.
- J. Cui, Z. Zhang, H. Jiang, D. Liu, L. Zou, X. Guo, Y. Lu, I. P. Parkin and D. Guo, ACS Nano, 2019, 13, 7483–7492 CrossRef CAS PubMed.
- Z. Zhang, D. Liu, F. Huo, S. Huang, J. Cui, Y. Lu, I. P. Parkin and D. Guo, Nanoscale, 2020, 12, 19617–19627 CAS.
- J. Cui, Y. Sun, H. Chen, Y. Yang, G. Chen, P. Ke, K. Nishimura, Y. Yang, C. Tang and N. Jiang, Adv. Funct. Mater., 2023, 33, 2210053 CrossRef CAS.
- J. Cui, X. Hu, L. Zhang, Y. Yang, Y. Li, G. Chen, C. Tang and P. Ke, Adv. Funct. Mater., 2024, 2422697 CrossRef.
- P. Vinchon, X. Glad, G. Robert Bigras, R. Martel and L. Stafford, Nat. Mater., 2020, 20, 49–54 CrossRef PubMed.
- F. Liu, J. Kong, C. Luo, F. Ye, X. Luan, N. Tian, Y. Liu, H. Zhang, J. Gu and Y. Tang, Adv. Compos. Hybrid Mater., 2018, 1, 506–517 CrossRef CAS.
- F. F. Lange and K. C. Radford, J. Am. Ceram. Soc., 1970, 53, 420–421 CrossRef CAS.
- F. Tavangarian and G. Li, Mater. Sci. Eng., A, 2015, 641, 201–209 CrossRef CAS.
- Y. P. Zeng, Q. Zhang, Y. J. Wang, J. X. Jiang, H. Z. Xing and X. Y. Li, Phys. Rev. Lett., 2021, 127, 066101 CrossRef CAS PubMed.
- F. Agulló-Rueda, M. D. Ynsa, N. Gordillo, A. Maira, D. Moreno-Cerrada and M. A. Ramos, Diamond Relat. Mater., 2017, 72, 94–98 CrossRef.
- J. O. Orwa, K. W. Nugent, D. N. Jamieson and S. Prawer, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 5461–5472 CrossRef CAS.
- R. J. Jiménez-Riobóo, N. Gordillo, A. de Andrés, A. Redondo-Cubero, M. Moratalla, M. A. Ramos and M. D. Ynsa, Carbon, 2023, 208, 421–431 CrossRef.
- Y. Yuan, W. Yang, J. Sang, J. Zhu, L. Fu, D. Li and L. Zhou, Diamond Relat. Mater., 2021, 120, 108670 CrossRef CAS.
- S. Kunuku, K. J. Sankaran, C.-Y. Tsai, W.-H. Chang, N.-H. Tai, K.-C. Leou and I. N. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 7439–7449 CrossRef CAS PubMed.
- T. Çagin, J. Che, M. N. Gardos, A. Fijany and W. A. Goddard, Nanotechnology, 1999, 10, 278–284 Search PubMed.
- R. Madar, Nature, 2004, 430, 974–975 CAS.
- H. Ding, Y. Li, M. Li, K. Chen, K. Liang, G. Chen, J. Lu, J. Palisaitis, P. O. Å. Persson, P. Eklund, L. Hultman, S. Du, Z. Chai, Y. Gogotsi and Q. Huang, Science, 2023, 379, 1130–1135 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |