
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecarboxylative photocatalytic transformations
Francisco
Foubelo
 ab,
Carmen
Nájera
*b,
M. Gracia
Retamosa
ab,
José M.
Sansano
ab,
Ana
Sirvent
ab and
Miguel
Yus
b
ab,
Carmen
Nájera
*b,
M. Gracia
Retamosa
ab,
José M.
Sansano
ab,
Ana
Sirvent
ab and
Miguel
Yus
b
aDepartamento de Química Orgánica and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain
bCentro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain. E-mail: cnajera@ua.es
First published on 10th November 2025
Abstract
Photocatalytic decarboxylation of carboxylic acids or their redox active esters has become an important strategy in organic chemistry. Using catalytic amounts of metal-based or organic photocatalysts, normally under visible light irradiation, these substrates generate carbon centered radicals, which have been applied to a broad range of C–C and C–heteroatom bond forming reactions. Addition reaction to electron-deficient alkenes, hydroalkylation of unsaturated C–C bonds and addition to C–heteroatom multiple bonds have been extensively studied. Cross-coupling reactions such as arylation, alkylation, allylation, vinylation, alkynylation, acylation, cyanation and C–H functionalization reactions are also successfully performed. In the case of C–heteroatom bond forming reactions, C–halogen, C–oxygen, C–sulfur, C–nitrogen, C–phosphorus, C–boron and C–silicon are fundamental functionalization processes. Hydro- and deuterodecarboxylation reactions allow the substitution of the carboxylic group by a hydrogen or a deuterium atom regioselectively. Finally, decarboxylative elimination reactions, such as olefination reactions for the synthesis of alkenes and decarboxylative C–C bond cleavage of cyclic carboxylic acids, give 1,n-dicarbonyl compounds. These photoredox transformations, a renaissance in organic chemistry, starting from readily accessible carboxylic acids widely available in Nature and the pharma industry with a great structural diversity occur under mild and simple reaction conditions with excellent efficiency and clean energy input.
 Francisco Foubelo | Francisco Foubelo was born in 1961 and studied Chemistry at the University of Oviedo where he received his BS (1984), MS (1986) and PhD (1989) degrees, under the direction of Professors J. Barluenga, M. Yus and F. J. Fañanás. He then joined the laboratory of Professor M. F. Semmelhack at Princeton University as a Fulbright postdoctoral fellow, and, in 1991, the group of Professor M. Yus at the University of Alicante, where he became an associate professor in 1995 and a full professor in 2002. His current research interests are focused on the development of new synthetic methodologies involving chiral sulfinyl imines and on metal-promoted functionalization of alkenes and alkynes. |
 Carmen Nájera | Professor Carmen Nájera was born in Nájera (La Rioja, 1951) and studied chemistry at the University of Zaragoza (1973) and PhD at the University of Oviedo (1979). After postdoctoral stays at the ETH, Oxford, Harvard and Uppsala University, she became an Associate Professor (1985) at the University of Oviedo and a Full Professor (1993) at the University of Alicante. She is the coauthor of more than 400 papers (25 |
 M. Gracia Retamosa | Maria de Gracia Retamosa is a Senior Research fellow of the University of Alicante (CIDEGENT program of the G. Valenciana). She received her PhD in 2008 at the University of Alicante (Spain) under the guidance of Prof. Carmen Nájera and José Miguel Sansano. She is the coauthor of 45 articles and 5 patents. She did several postdoctoral stays [Prof. Michael Greaney at the University of Edinburgh (UK, 2009), Prof. Jesús M. Sanz at the University Miguel Hernández (Elche, Spain, 2009–2011) and Prof. Fernando P. Cossío at the University of the Basque Country and Donostia International Physics Center (Spain, 2012–2016)]. Recently, she has joined the group of Prof. Rosario Fernández and José M. Lassaletta as a postdoctoral researcher [CSIC (Sevilla, Spain)]. Her current research interests include asymmetric metal and organocatalysis and synthesis of compounds of pharmacological interest. |
 José M. Sansano | Professor José Miguel Sansano was born in Rojales (Alicante), studied chemistry at the University of Alicante, where he obtained his BSc and PhD degrees in 1988 and 1994, respectively. His thesis was supervised by Prof. C. Nájera and dealt about sulfone chemistry. After spending a two-year postdoctoral stay at the University of Leeds (UK) with Prof. R. Grigg, he joined the University of Alicante in 1996, where he was appointed Associate Professor in 2001. In 2010 he was promoted to Full Professor in the same University. He was an invited visiting Professor at Chuo University in 2014 and at the UFRJ (Brazil). He is the coauthor of more than 200 articles and he has supervised 13 PhD students. |
 Ana Sirvent | Ana Sirvent received her BSc (2015), MSc (2016) and PhD degrees (2021) from the University of Alicante (Spain) under the supervision of Prof. Francisco Foubelo and Prof. Miguel Yus. During her PhD studies, in 2019, she conducted a short research stay in the group of Prof. Jonathan A. Ellman at Yale University (USA). In 2021, she joined the group of Prof. Nuno Maulide at the University of Vienna (Austria) as a postdoctoral researcher. Since 2024, she has been a postdoctoral researcher at the University of Alicante. |
 Miguel Yus | Professor Miguel Yus was born in Zaragoza (1947) and received his BSc (1969), MSc (1971) and PhD (1973) degrees from the University of Zaragoza. After two years at Max Planck Institut für Kohlenforschung (Mülheim/Ruhr) he returned to the University of Oviedo, becoming associate professor (1977) and full professor (1987). In 1988 he went to the University of Alicante, being a visiting professor at ETH-Zentrum, Oxford, Harvard, Uppsala, Marseille, Tucson, Okayama, Paris, Strasbourg, Bolognia, Sassari, Tokyo and Kyoto. He has published 600+ papers, supervised 62 doctoral theses, getting 35 |
1. Introduction
Decarboxylative reactions performed under photoredox catalysis have gained much interest in the last decade as versatile strategies in organic synthesis.1–18 Carboxylic acids are suitable starting materials that can be mainly obtained from renewable feedstocks and possess a broad structural diversity, especially amino acids, fatty acids or sugar acids. The formation of an electrophilic radical occurs by single electron oxidation of a carboxylate ion using photocatalysts able to form excited state units as oxidants. On the other hand, under redox neutral decarboxylation conditions nucleophilic radicals are formed. These radicals can be used in a range of C–C and C–heteroatom bond formation reactions with appropriate acceptors. Alternatively, mainly N-(acyloxy)phthalimide (NHPI) esters can be used as redox-active esters (RAEs) in decarboxylative transformations under mild reaction conditions.1,14,19–22 In this case, the RAE (NHPI ester) is reduced by a photocatalyst, by accepting an electron, generating the corresponding radical after decarboxylation and formation of the phthalimidyl anion. This review covers the synthetic applications of decarboxylative transformations starting from carboxylic acids and their derivatives reported in the last seven years.2. C–C bond-forming reactions
In this section, addition reactions of electrophilic olefins (Giese reactions), unactivated alkenes, alkynes, carbonyl compounds and imines will be considered (Sections 2.1–2.3). Cross-coupling reactions by arylation, alkylation, allylation, alkenylation, alkynylation, acylation, cyanation, C–H bond functionalization and cyclization processes will be subsequently treated (Sections 2.4–2.12).2.1. Addition reactions of electron-deficient alkenes
Decarboxylative Giese reactions (DGRs) have been recently carried out under photocatalysis and have been covered by Kanegusuku and Roizen23 and by Lee and co-workers14 until 2021. Carbon-centered radicals, generated from NHPI esters or from carboxylic acids, added to Michael acceptors through a regioselective conjugate pathway under mild and environmentally benign photocatalytic conditions. Overman and co-workers24,25 employed NHPI esters for the synthesis of natural products under Ru(bpy)2(BF4)2 metallaphotocatalysis. König and co-workers26 carried out the DGR of N-Boc-protected α-amino acids (AAs) and fatty acids using the abundant dye Eosin Y as the photocatalyst. Oxidative decarboxylation of carboxylic acids was described by MacMillan and co-workers27,28 using K2HPO4 as base and an iridium photocatalyst. Carboxylic acids with a secondary or tertiary α-carbon, as well as primary α-substituted carboxylic acids bearing oxygen or nitrogen atoms at the α-position, were also used.29–43 Instead of iridium photocatalysts, organic dyes have been employed, such as 9-mesityl-10-methylacridinium perchlorate [Acr-Mes]ClO4 in aqueous acrylonitrile, allowing the use of a wide range of substrates.44 Other organic photocatalysts have been employed for DGRs.45–49 Titanium oxide was able to promote the DGRs by photoexcitation with 390 nm light.50Recent advances in photomediated decarboxylative Giese reactions of carboxylic acids are focused on metallaphotocatalysis15 and organocatalysis.51 Concerning metallaphotocatalysis, iridium, ruthenium, titania and iron have been mainly employed. In the case of carboxylic acids, the corresponding radicals are generated by a radical decarboxylation mechanism with extrusion of CO2. However, Yu and co-workers52 reported a carbon-economical and sustainable carbocarboxylation of alkenes. They employed carboxylic acids 1, including α-amino acids, peptides, terpenoids and alkyl carboxylic acids, for DGRs on activated alkenes 2 under Ir photocatalysis to obtain carbon radicals. Such radicals were reduced to carbanions that were able to react with the in situ generated CO2 affording γ-amino butyric acid derivatives (GABAs) 3 in the case of AAs (Scheme 1). This is the only example of carbocarboxylation of alkenes allowing the recycling of CO2. The most efficient metallaphotocatalyst was Ir[dF(CF3)(ppy)]2(dtbbpy)·PF6 (4), with CsF as a base in N,N-dimethylacetamide (DMA) under 1 atm N2 and 30 W blue LED irradiation at room temperature. The gram-scale reaction of 1,1-diphenylethylene with N-Cbz-Pro was carried out under sunlight irradiation, obtaining 3aa in 74% yield with only 0.1 mol% of the photocatalyst (PC). Mechanistic studies based on control and kinetic experiments indicate that the initial single-electron transfer (SET) between the photoexcited *[Ir]III and the carboxylate ion 1a′, generated by deprotonation of 1a with the base, formed the α-amino radical A and CO2. Radical addition of A to 2a produces radical B, which undergoes SET reduction by [Ir]II to provide the carbanion C. The final reaction of C with CO2 furnishes carboxylate ion D, which after protonation affords 3aa.
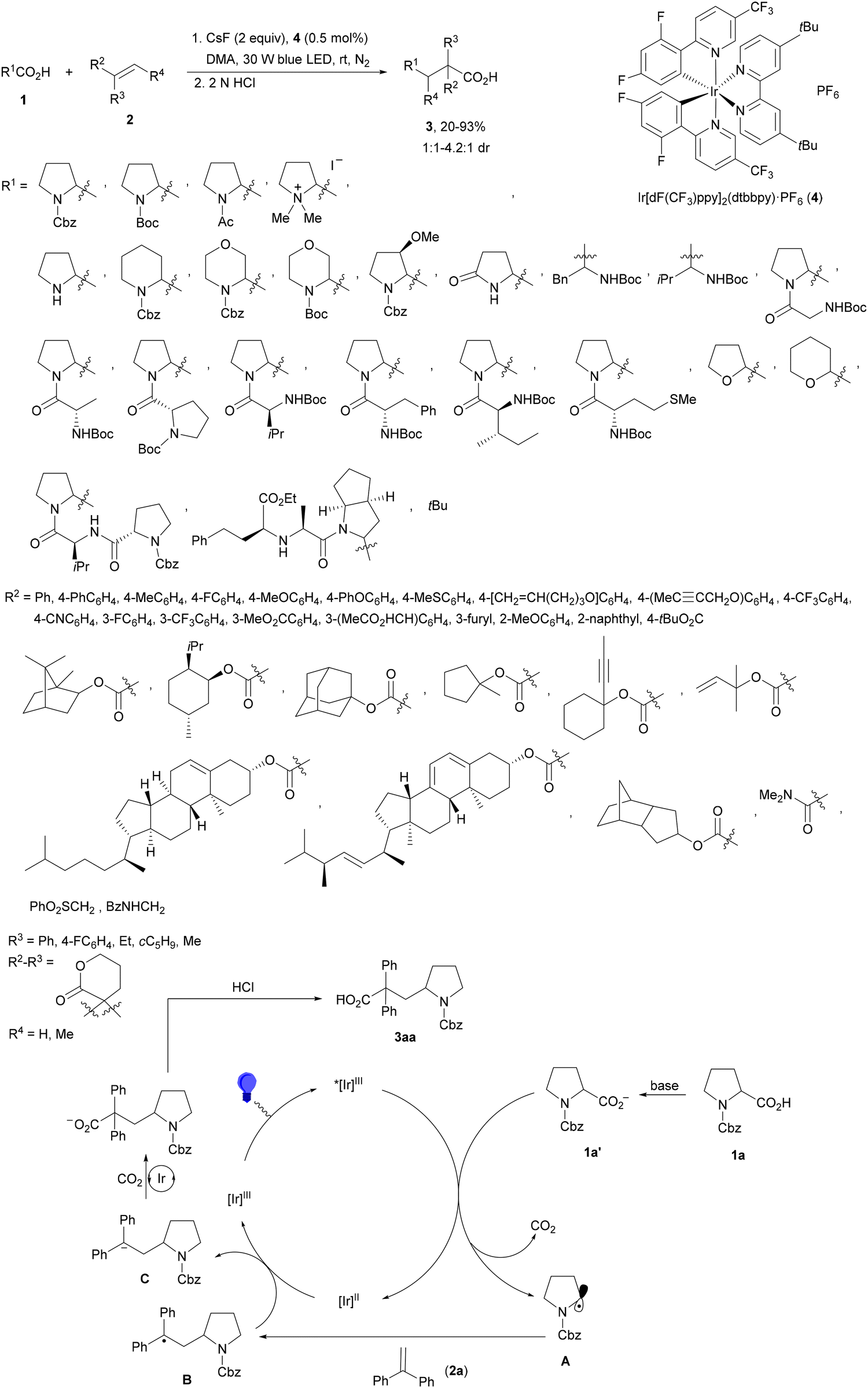 | ||
| Scheme 1 Carbocarboxylation of alkenes 2 with carboxylic acids 1 under Ir photocatalysis. | ||
A dual catalytic system developed by merging iron/copper catalysis has been employed by the Li and Zeng group53 for the decarboxylative alkylation of electron-deficient alkenes with alkyl carboxylic acids. An iron catalyst, tetrabutylammonium tetrachloroferrate(III) (TBAFeCl4), and Cu(MeCN)4PF6 as a cocatalyst and 1,4-diazabicyclo[2.2.2]octane (DABCO) as a base in dichloromethane (DCM) and a N2 atmosphere at room temperature produced the desired products 5 by irradiation with a 390 nm LED lamp (Scheme 2). Several carboxylic acids including various types of drugs or natural products, such as diclofenac, oxaprozin, ibuprofen, naproxen, linoleic acid, oleic acid, carbamic acid, gemfibrozil, indomethacin, aceclofenac, ketoprofen and stearic acid, were used. As olefinic counterparts, acrolein, acrylodinitrile, acrylonitrile, methyl acrylate, acrylamide, phenyl vinyl ketone and vinyl sulfone were employed. Mechanistic studies supported a ligand-to-metal change transfer (LMCT)54,55 pathway of the iron catalyst, whereas the copper catalyst here might act as a Lewis acid activating the electron-deficient olefin and inhibiting the potential polymerization. The carbon radical was formed through the LMCT of the iron catalyst followed by decarboxylation.
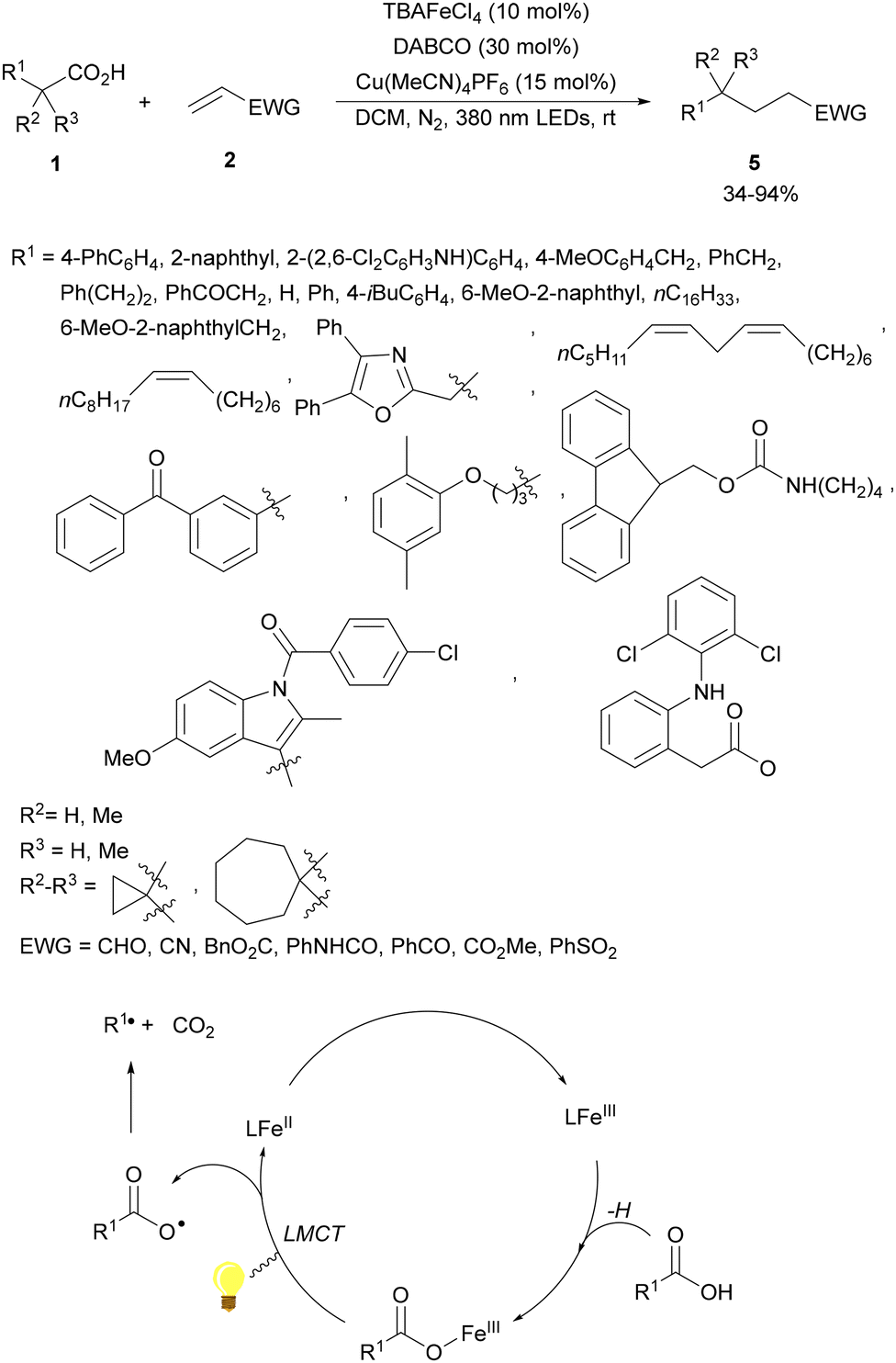 | ||
| Scheme 2 Decarboxylative Giese reaction of carboxylic acids and electron-deficient alkenes under Fe/Cu photocatalysis. | ||
Jung and co-workers56 performed the photoreductive β-aminoalkylation of an α,β-unsaturated carbasugar mimic 7 driven by blue LEDs light using 4 as an Ir photocatalyst (Scheme 3). Different N-Boc α-amino acids 6 reacted with the cyclopentenone derivative 7 in DMSO at room temperature to provide γ-amino ketones 8 with very good yields and moderate diastereoselectivity. The resulting products 8 were transformed into 5′-amino carbasugar nucleoside analogues for S-adenosyl methionine-based methyltransferase inhibitors.
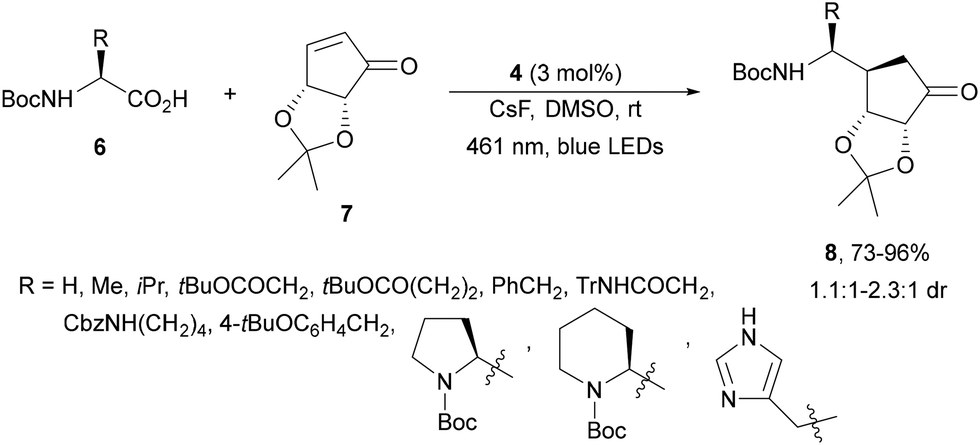 | ||
| Scheme 3 Decarboxylative Giese reaction of AAs with enone 7 under Ir photocatalysis. | ||
Four membered heterocyclic carboxylic acids 9 bearing an aryl group generate benzyl radicals using the Ir photocatalyst 4 (Scheme 4).57 Oxetane and azetidine carboxylic acids 9 reacted with electron-poor alkenes 2 in DMF at 36 °C to provide products 10 in moderate to good yields due to the formation of secondary products. Experimental and computational studies revealed that only oxetane and azetidine substrates favored the DGR pathway and minimized dimer formation.
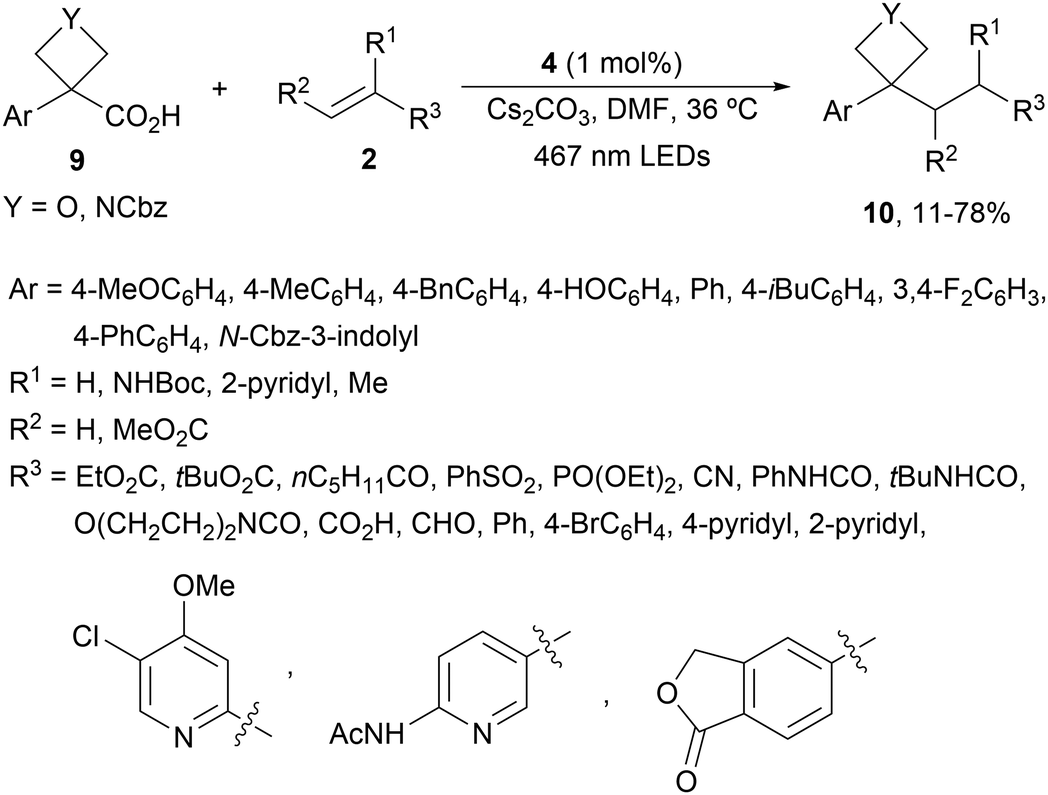 | ||
| Scheme 4 Decarboxylative Giese reaction of oxetane and azetidine carboxylic acids 9 and electron-poor alkenes 2 under Ir photocatalysis. | ||
Gómez-Suárez and co-workers58 have described a two-step procedure for the DGR generating in situ11 as electron-poor alkenes. Starting from malononitrile and cyclic ketones the corresponding alkenes 11 were generated by Knoevenagel condensation followed by a domino DGR/base-mediated cyclization. Intermediate 12 was formed using Ir complex 4 as a photocatalyst and after cyclization the resulting spirocyclic products 13 were isolated in modest to good yields (Scheme 5a). The same group59 recently reported a two-step procedure based on a domino DGR/oxidative functionalization to obtain β2- and α-quaternary β1,2-amino acid derivatives 14 in good yields (Scheme 5b). In this case, the aminoalkylation DGR was also carried out in the presence of Ir[dF(CF3)ppy2(dtbbpy)]PF6 (4). This strategy has been scaled up via continuous-flow technology.
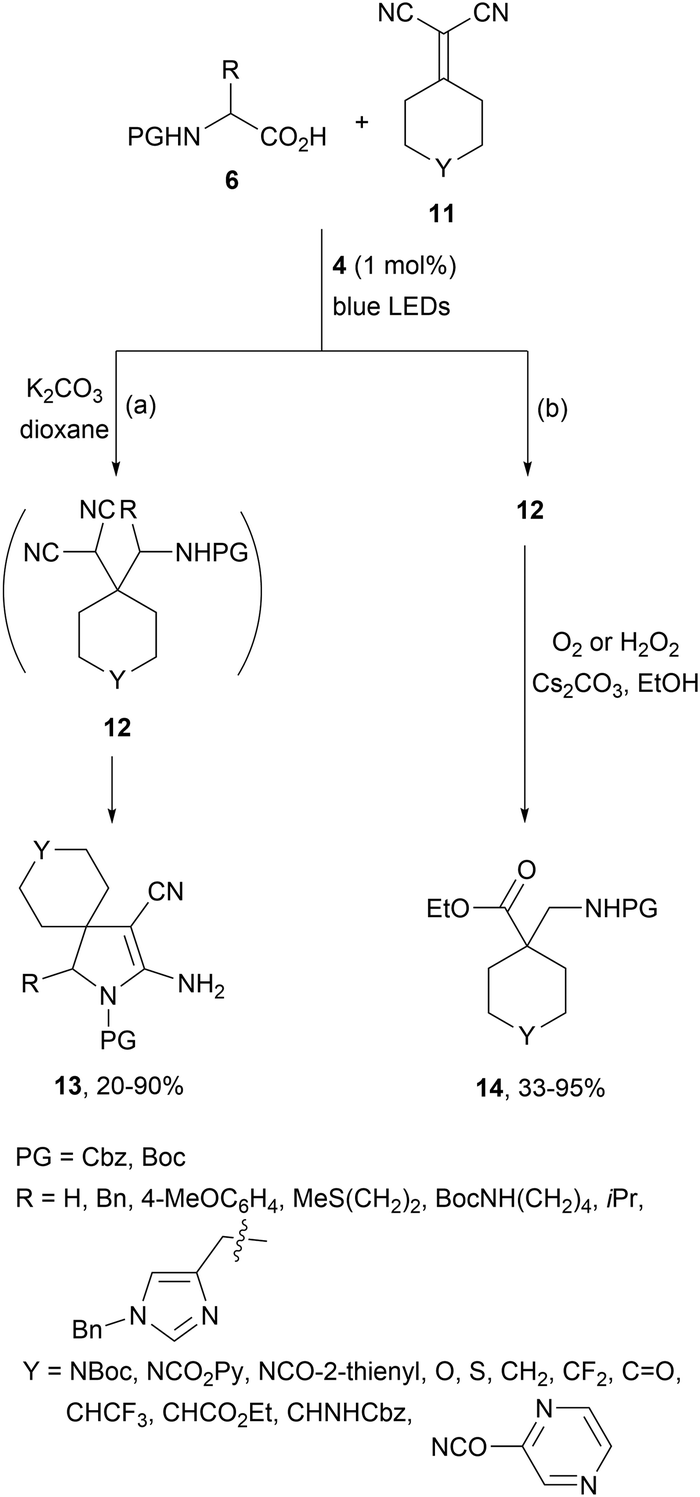 | ||
| Scheme 5 Two-step DGR/cyclization and DGR/oxidative functionalization between alkylidenemalononitriles and AAs under Ir photocatalysis (a, b). | ||
Decarboxylative 1,4-addition of α-oxo carboxylic acids to α,β-unsaturated carbonyl compounds gives the corresponding 1,4-dicarbonyl compound derivatives by intermediacy of acyl radicals. Ir-derived complexes have been mainly used as metallaphotocatalysts.60–62 Recently, Tunge and co-workers63 reported the aroylation of alkenes 2 with α-keto acids 15 using cobaloxime 16 and organophotocatalyst 17 (Mes-Acr-Ph+) as a cooperative catalyst. This method allowed access to chalcone cores 18 in moderate to good yields and diastereoselectivities (Scheme 6). In the proposed mechanism photocatalyst (PC) 17 gave upon irradiation Mes-Acr-Ph+* [E1/2 (PC*/PC) = +2.17 V]. A SET from the deprotonated α-keto acid would generate the carboxy radical intermediate A, which after decarboxylation afforded acyl radicals B. The reduced PC Mes-Acr-Ph˙ can be oxidized by the Co(III) catalyst to close the catalytic cycle. The generated Co(II) species would react with the acyl radical B to give intermediate C, which undergoes a radical addition with 2 forming intermediate D. Homolytic cleavage of the Co–C bond results in a Co(II) species E. Final hydrogen atom transfer provides product 18 and Co(III)–H, which can react with a proton from another molecule of α-keto acid 15 to evolve H2 gas and return Co to its catalytic cycle.
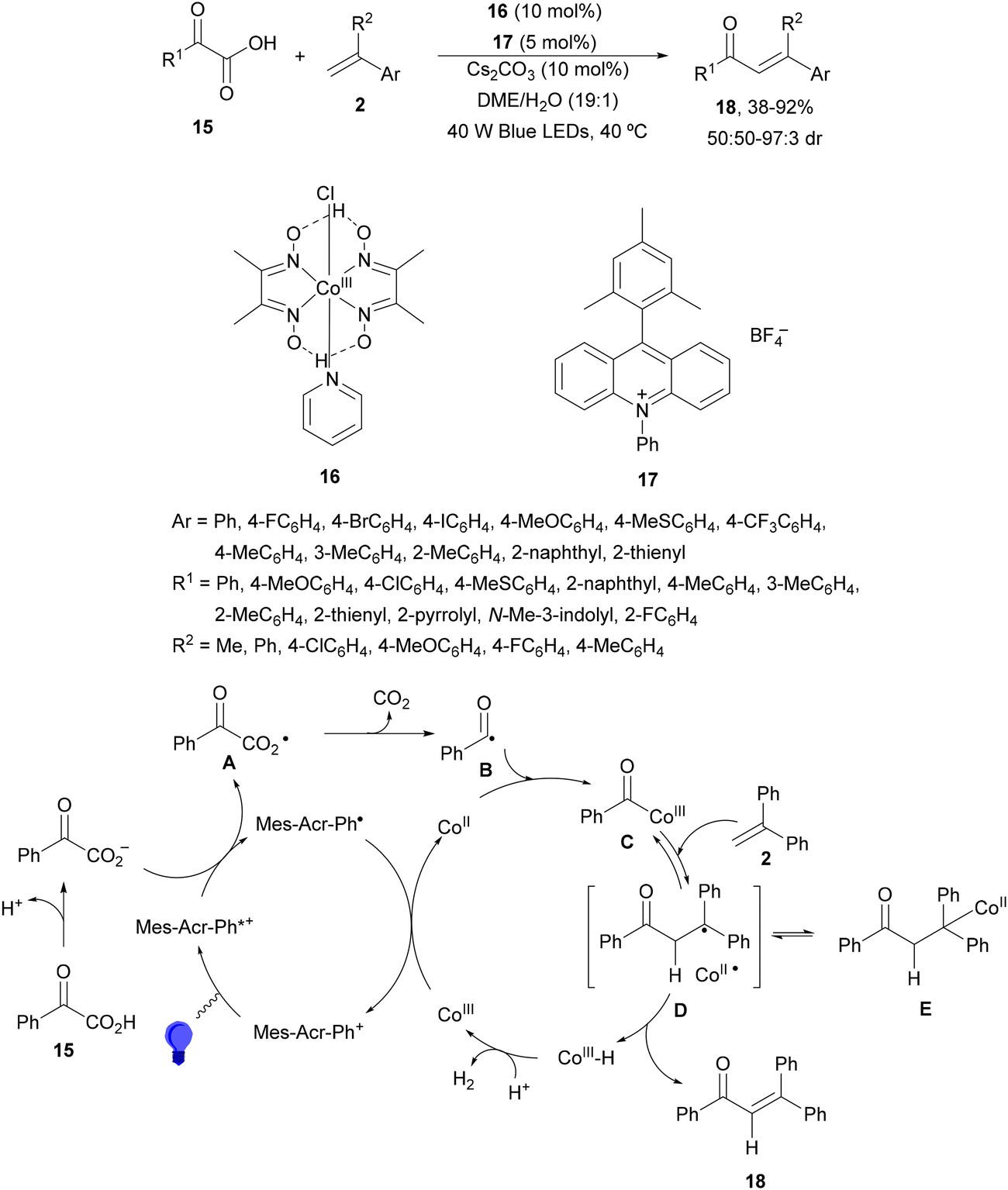 | ||
| Scheme 6 Decarboxylative Giese reaction of α-oxo acids 15 with styrenes 2 under cobaloxime 16 and Mes-Acr-Ph+ (17) dual photocatalysis. | ||
Organophotoredox catalysis has been expanded in the past few years as a cheaper alternative to Ir-based photocatalysis.15,51,64 The family of organophotoredox catalysts are composed of (a) arenes coupled with dicyanoarenes (DCAs), (b) carbazolyldicyanobenzene (CDCB) derivatives, (c) dicyanopyrazines (DPZs), (d) flavins (FLs), (e) thioxanthones (TXs), (f) pyrimidopteridine N-oxides (PPTNOs), (g) acridinium derivatives, (h) 2,2′-bipyridine and (i) potassium–carbon nitride (Fig. 1). All of them have been used as PCs in the DGR of carboxylic acids including α-keto acids.
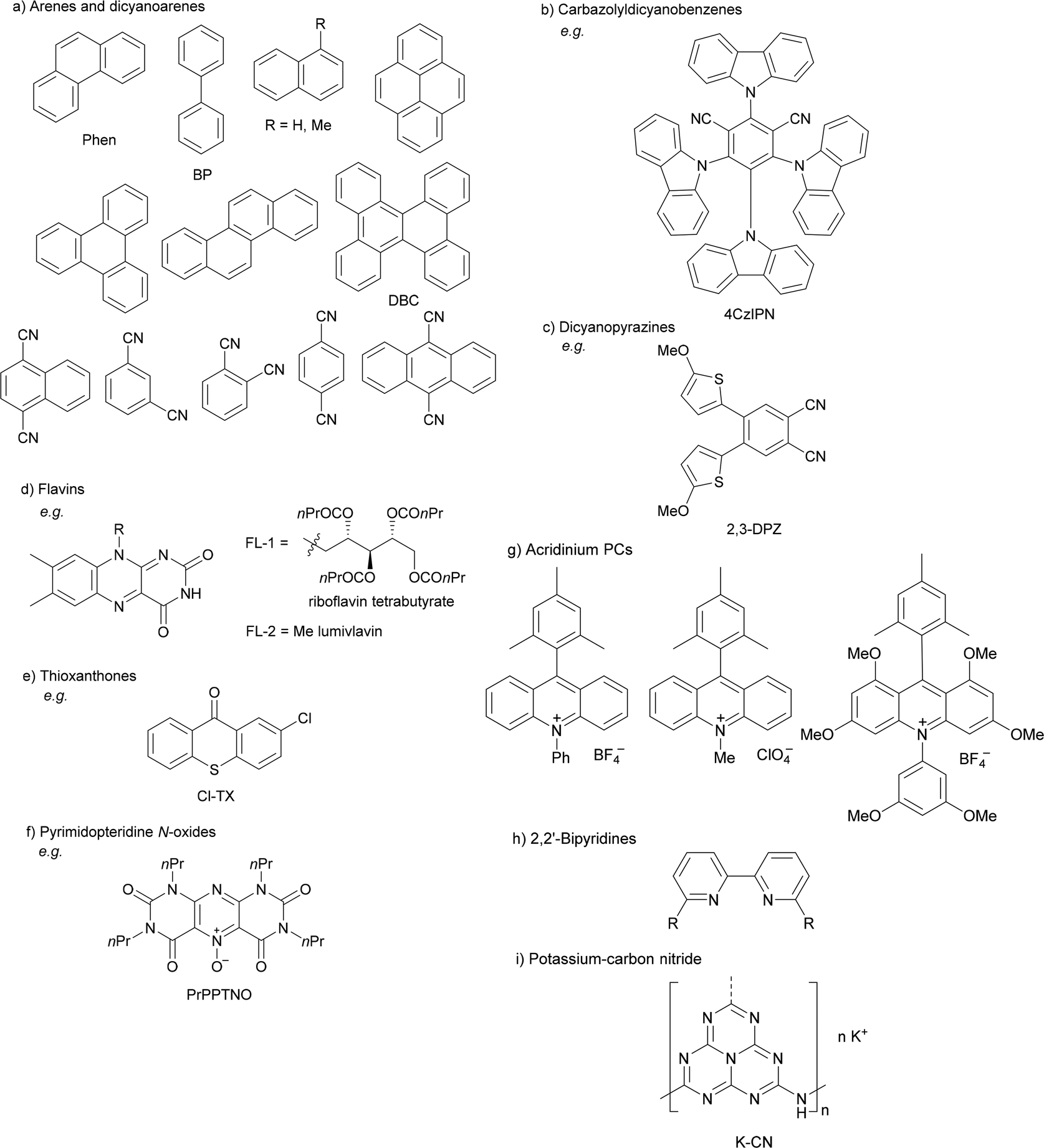 | ||
| Fig. 1 Representative organophotocatalysts (a–i) for decarboxylative Giese reaction of carboxylic acids. | ||
Recent applications of some of these organocatalysts will be considered. In the case of an arene as a photocatalyst and a dicyanoarene (DCA) as a redox mediator, Yoshimi and co-workers have reported the DGRs of aliphatic65,66 and aromatic67 carboxylic acids. In the first case, experiments conducted using phenanthrene (Phen) as the electron-donor (ED) and 1,4-dicyanobenzene as the electron-acceptor (EA) demonstrate photoinduced electron transfer (PET) from the ED to the EA, generating the radical cation of the ED, which oxidizes the alkyl carboxylate ion to form the carboxy radical able to generate the alkyl radical (Scheme 7A). When aromatic carboxylic acids are used, the EDs such as biphenyl (BP) and the EAs such as 1,4-dicyanonaphthalene (DCN) or 9,10-dicyanoanthracene (DCA) were used as photoredox catalysts (Scheme 7B). In the first case, the EA˙− undergoes back electron transfer (BET) to radical A generating anion B. In the second case, two BET processes occur in the oxidation of ED˙− to ED and of the carboxy radical to the anion.
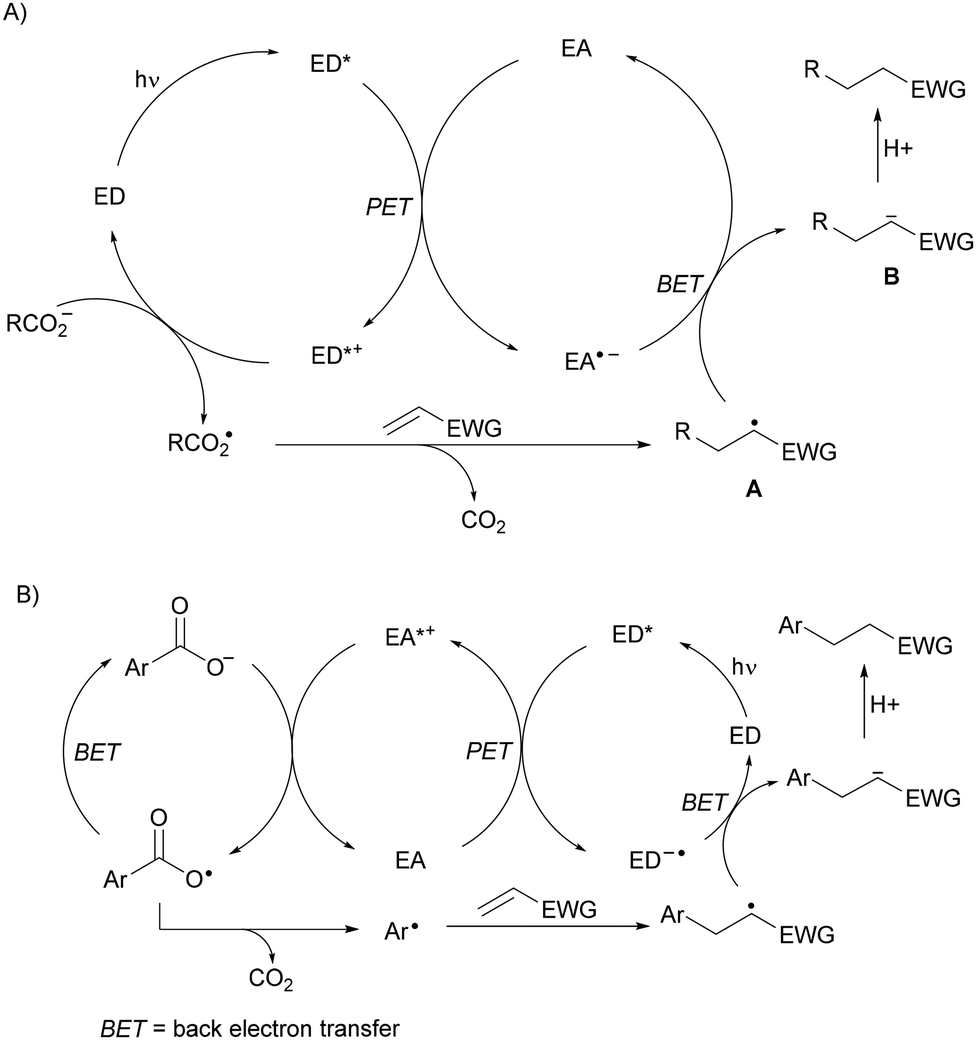 | ||
| Scheme 7 Plausible mechanisms (A, B) for decarboxylative Giese reactions using two organic photocatalysts. | ||
Yoshimi and co-workers68 recently reported the use of 9-cyano-10-methoxycarbonylanthracene 19 as the EA instead of 9,10-dicyanoanthracene (DCA), because of its better solubility, and BP in a two-molecule photoredox system for the DGR of aromatic and aliphatic carboxylic acids. For aromatic acids, BP/19 in aqueous MeCN as solvent and NaOH as a base were employed whereas for the aliphatic ones Phen/19 induced DGR under visible light (Scheme 8). The replacement of Phen by BP allowed a facile tuning of the oxidation potential of these electron donor molecules. Products 5 and 20 were obtained in moderate to good yields using visible light (405 nm) under an argon atmosphere at room temperature. The same group69 described that the EA 19 together with Phen or BP as the ED is slightly unstable and studied the modification of these EDs. Dibenzo[g,p]chrysene 21 absorbs blue LED light (405 nm) with a 0.10 × 103 molar extinction coefficient with an oxidation potential of 1.7 eV. Working with only 21/1,4-DCB (6.6 mol%) and KOH as a base in aqueous MeCN, aliphatic carboxylic acids gave DGR products 5 in higher yields (30–94%). In the case of aromatic carboxylic acids, the same group70 studied 19 as the EA and the effects of different EDs and of NaOH or KOH as bases. The influence of the counter-cation (Na+ or K+) was relatively minor. Additionally, the dependence of the oxidation ability of the radical cation on the ED allowed estimation of the oxidation potential of the benzoate ions.
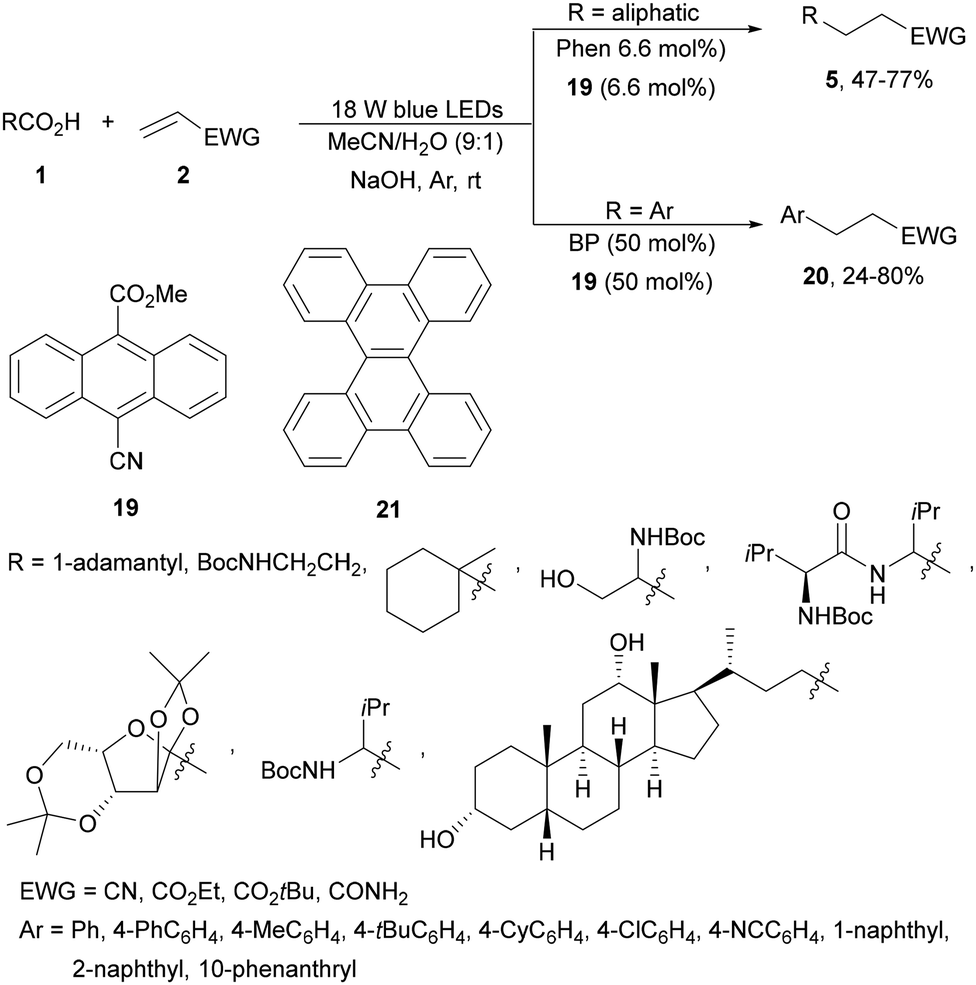 | ||
| Scheme 8 Decarboxylative Giese reaction of aliphatic and aromatic acids with electron-deficient alkenes under two organic photoredox systems. | ||
Primary carboxylic acids, such as L-glutamic acid and L-aspartic acid methyl esters 22, showed a low rate of decarboxylation as in the case of aromatic carboxylic acids due to the high oxidation potential of the carboxylate ion and the weak donating ability of the primary alkyl radical.71 The reaction of compounds 22 with acrylate and acrylamide derivatives 2 took place with BP/DCN and with Phen/CMA (19) under visible light (405 nm) or UV (313 nm) to give products 23 in moderate yields with complete retention of the configuration of the AA unit (Scheme 9). This method was used for the preparation of substrates with carbohydrates, amino acids and peptides.
 | ||
| Scheme 9 Decarboxylative Giese reaction of L-glutamic and L-aspartic acid methyl esters 22 with electron-deficient alkenes under two organic photoredox systems. | ||
Recently, this photoinduced decarboxylation of carboxylic acids was applied to the radical polymerization under visible light.72 Various carboxylic acids and electron-withdrawing alkenes afforded block copolymers 24 with number-average molecular weight (Mn) and polydispersity (Mw/Mn) depending on both the concentration of ED/EA and the carboxylic acid (Scheme 10).
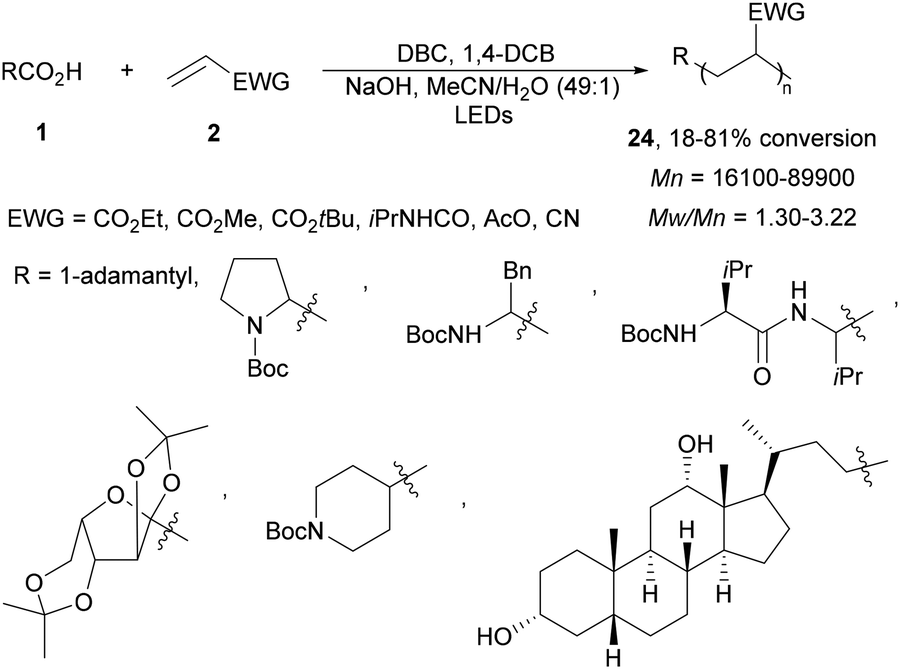 | ||
| Scheme 10 Decarboxylative co-polymerization of carboxylic acids and electron-poor alkenes under two organic photoredox systems. | ||
Carbazolyldicyanobenzenes (CDCBs) are structural derivatives of cyanoarenes with the phenyl core as an electron acceptor and the carbazole units as electron donor structures. They are characterized by a high singlet energy and therefore high oxidation potentials. The HOMO is mainly delocalized over the four carbazolyl moieties and the LUMO is centered on the dicyanobenzene ring. In addition, due to steric hindrance between the carbazolyl and nitrile groups, a large dihedral angle (about 60°) is observed, limiting torsional flexibility and reducing nonradiative decay. Therefore, these PCs exhibit bimodal redox properties both in their ground and excited states, which can be tuned by modification of both structural units.15,51,64 Generally, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN, 25) is the most used organocatalyst of this family.73 Zhang and Schubert74 demonstrated that 4CzIPN is a similar photocatalyst to Ir 475 for the DGR of carboxylic acids and dehydroalanine derivatives. In a recent publication, 2,2-diethoxy acetic acid 26 has been employed by Sun and co-workers76 as a formyl radical equivalent in the sequential formyl/carboxylation of alkylated alkenes 2 and CO2 using 4CzIPN 25 as the photocatalyst (Scheme 11). Adducts 27 were obtained after methylation of intermediate carboxylate ions in moderate to good yields. On the other hand, the acetal group can be further hydrolyzed to an aldehyde or transformed into diverse functionalized compounds.
 | ||
| Scheme 11 Formyl/carboxylation of activated alkenes 2 with diethyl glyoxylic acid acetal 26 and CO2. | ||
Wang and co-workers77 performed a DGR using carboxylic acids and imidazolidinone-derived dehydroalanines 28 as Michael acceptors. Under irradiation with blue LEDs using 4CzIPN (25) as a PC in the presence of Cs2CO3 as a base in DMF at room temperature, products 29 were isolated with good yields (Scheme 12). These compounds 29 were subjected to a Clayden rearrangement78,79 to deliver products 30 as precursors of chiral quaternary α-aryl amino acid derivatives with high diastereoselectivity via memory of chirality.
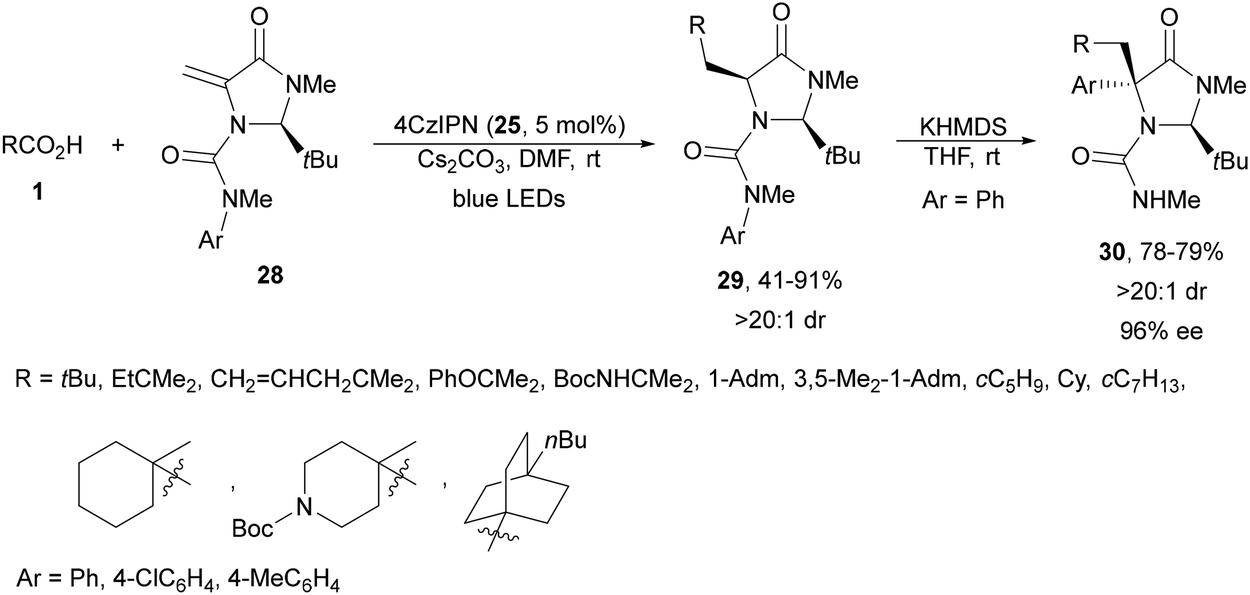 | ||
| Scheme 12 Decarboxylative Giese reaction of dehydroalanines 28 and aliphatic carboxylic acids under 4CzIPN (25) photocatalysis and subsequent Clayden rearrangement. | ||
Ionic photocatalysts such as acridinium salts15,51,64 are excellent alternatives to metal-based PCs, because of high oxidative and reducing power and more favorable electron-transfer between the ED and the PC excited state than metallacatalysts. A recent work of Tunge and co-workers80 employed 9-mesityl-10-phenylacridinium tetrafluoroborate (17) as a PC for the DGR of α-keto acids 15 with maleic anhydrides 31 as traceless synthons of acrylic acid (Scheme 13). A dual decarboxylative procedure resulted in γ-keto acids 32 in good yields working with 4-dimethylaminopyridine (DMAP) as a base in aqueous dimethoxyethane (DME) and 40 W blue LED irradiation at −40 °C. This process can be carried out for instance in aqueous methanol as solvent to obtain the corresponding methyl γ-keto esters. On the other hand, when acrylic acid or maleic acid 31a was used as a Michael acceptor the resulting products 32 were obtained in low yields. In the general proposed mechanism, Mes-Acr-Ph+ is excited to Mes-Acr-Ph+*, which after a SET from the carboxylate ion A generates the carboxy radical B intermediate. Subsequent decarboxylation of B gives the acyl radical C, which undergoes Giese addition into the maleic anhydride generating intermediate D. The reduced PC will return to its ground state via a SET process to D affording enolate E. Protonation of E by a new molecule of α-keto acid and subsequent hydrolysis of the maleic anhydride F unit gives intermediate G, which undergoes decarboxylation to furnish the γ-keto acid 32a.
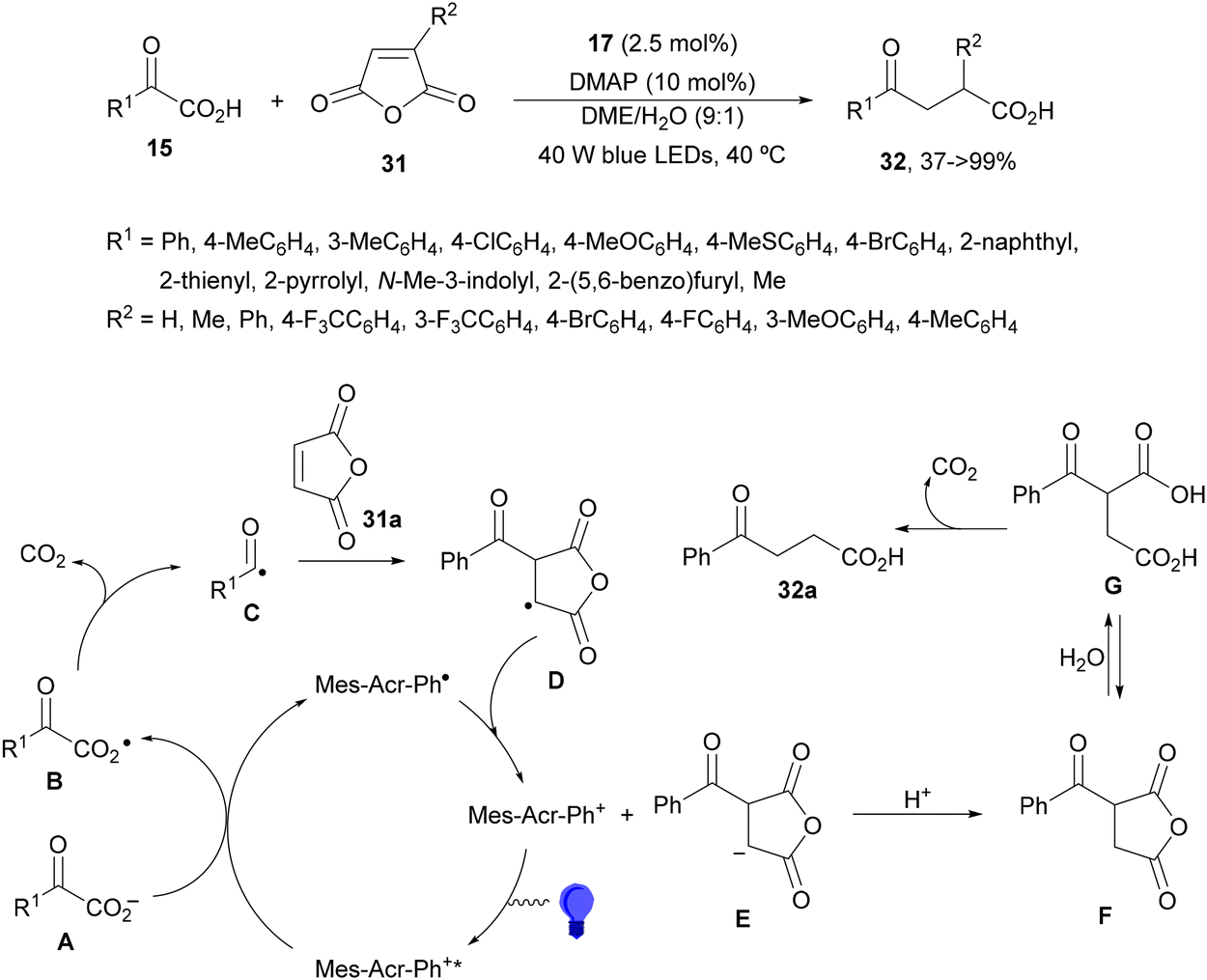 | ||
| Scheme 13 Decarboxylative Giese reaction of α-keto acids 15 and maleic anhydrides 31 using [Mes-Acr-Ph]+BF4− (17) as the photocatalyst. | ||
Radical polymerization by a reversible addition-fragmentation chain-transfer (RAFT) from carboxylic acids and acrylic derivatives has been recently performed using acridinium salts as PCs by Hooper and co-workers.81 In the presence of a thiocarbonyl disulfide 33 used as the RAFT agent under green light and the acridinium salt 34 polymers 35 were isolated with low dispersity (1.12–1.58) and an Mn of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 to 33300 g mol−1 determined by NMR (Scheme 14). In contrast, the use of Ir catalyst 4 gave poorer results. Different carboxylic acids such as AAs, secondary and tertiary ones were more effective than the corresponding primary ones.
900 to 33300 g mol−1 determined by NMR (Scheme 14). In contrast, the use of Ir catalyst 4 gave poorer results. Different carboxylic acids such as AAs, secondary and tertiary ones were more effective than the corresponding primary ones.
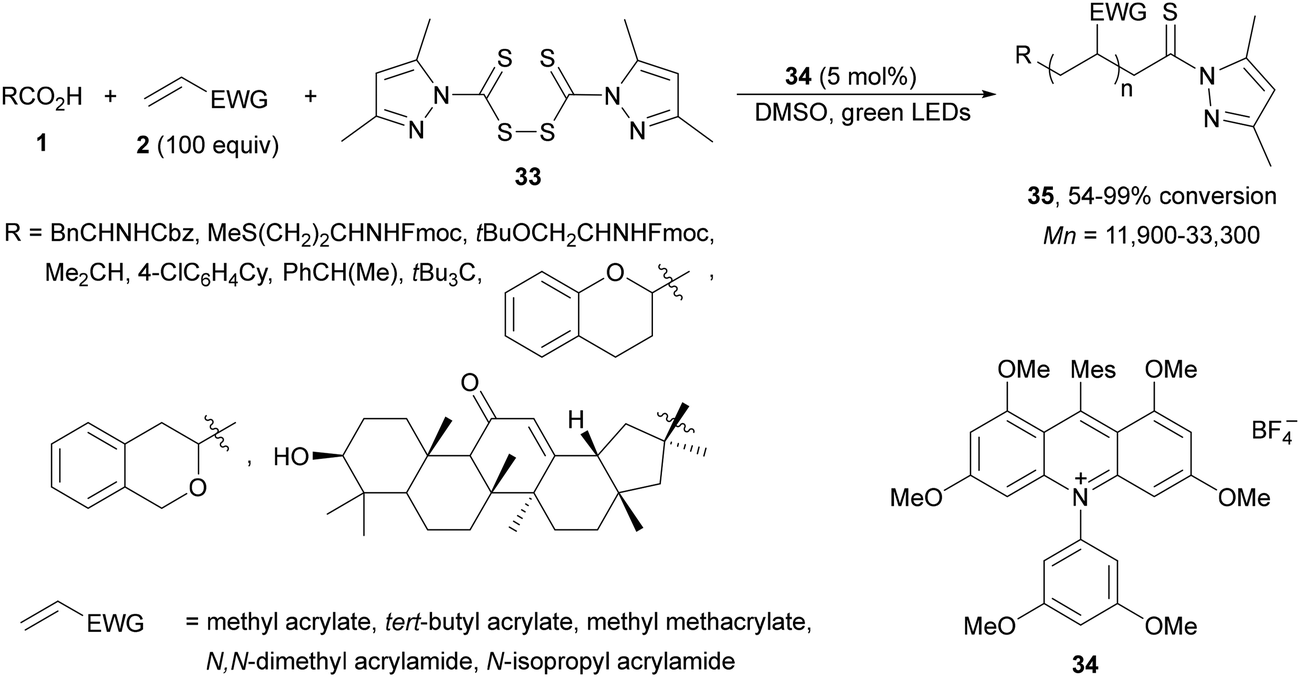 | ||
| Scheme 14 Decarboxylative RAFT polymerization of carboxylic acids and acrylates using acridinium salt 34 as a photocatalyst. | ||
Light-mediated DGR without a PC has been described by Lu and coworkers82 using α-keto acids 15 and Michael acceptors 2. This procedure was carried out under oxidative conditions in the presence of ammonium persulfate, γ-terpinene as a hydrogen atom transfer (HAT) source and 2,4,6-collidine as a base (Scheme 15). Products 36 were obtained in better yields under light-mediated than that under thermal (50 °C) reaction conditions. In the proposed mechanism, SO4˙− (C) is formed by photodecomposition of the combination of 2,4,6-collidine and S2O82− (B). Upon formation of C, SET between C and glyoxylate anion A can occur to provide radical D, which decarboxylates to the acyl radical E. Subsequent Giese addition to 2 forms radical F, which undergoes HAT from γ-terpinene to yield product 36.
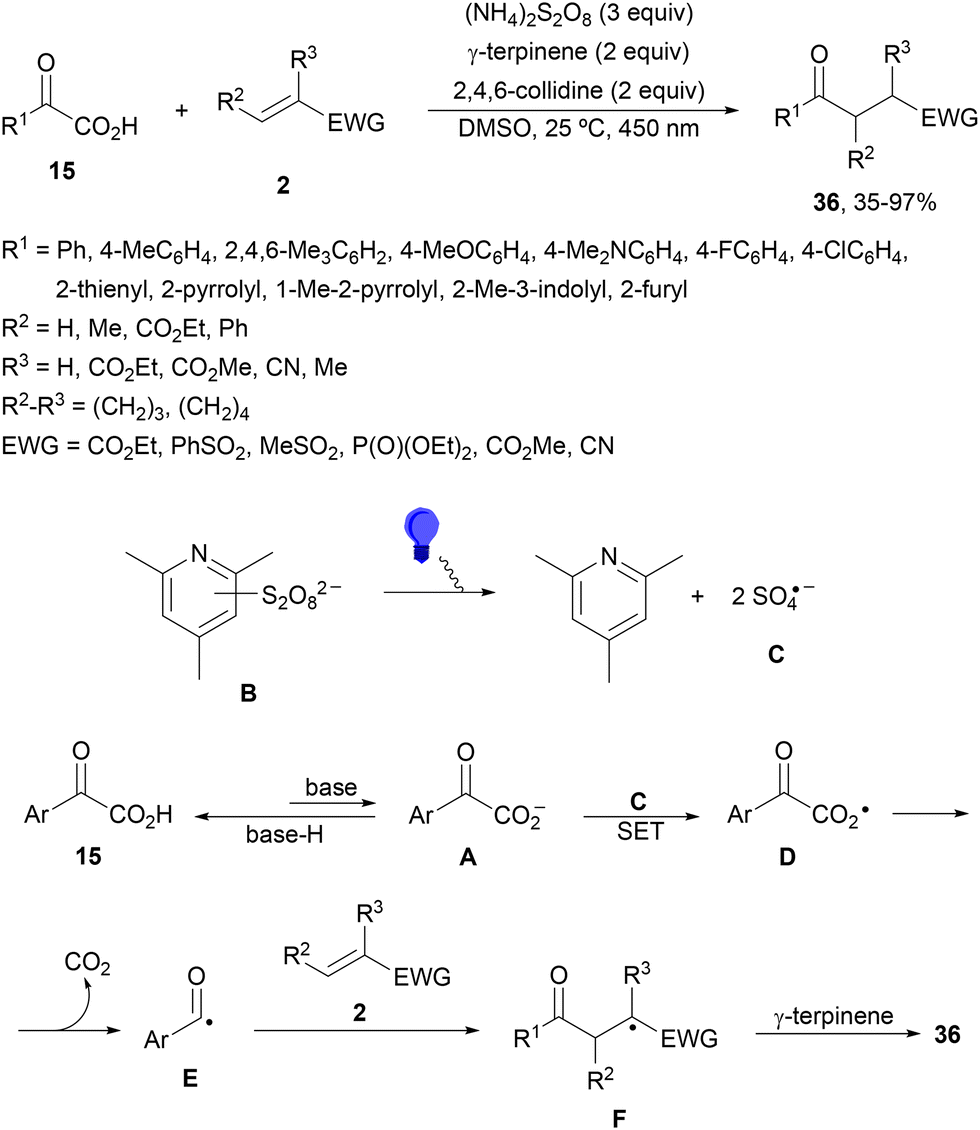 | ||
| Scheme 15 Light-mediated DGR of α-keto acids 15 and Michael acceptors under oxidative conditions without a photocatalyst. | ||
In conclusion, the two-molecule photoredox system employs inexpensive, stable and neutral organic molecules and allows the modulation of the back electron transfer from the radical anion of the EA and the replacement of the ED with the same EA facilitating a change in the oxidation potential maintaining the same EA. This modification is impossible for the one-molecule photoredox catalysis. However, 4CzIPN has shown very good photocatalytic properties being easily modified at the carbazole and dicyanobenzene units.
1,4-Dihydropyridine (DHP)-derived glycosyl esters 37 have been employed as redox-active ester (RAE) precursors of glycosyl radicals via anomeric C(sp3)–O bond homolysis and subsequent decarboxylation in DGRs.83 For instance, ester 37a reacted with oxazolidinone-derived dehydroalanine 38 and dehydroalanine peptides 39 using Ir complex 4 or 4CzIPN (25) as a photocatalyst under blue LED irradiation in MeCN or 1,4-dioxane at 85 °C to provide products 40 and 41, respectively (Scheme 16). A wide range of glycosyl DHP esters derived from monosaccharides and oligosaccharides were used to give products 40 in moderate to good yields (up to 87%) and good to excellent stereoselectivities (up to >20:1 dr). In the case of starting compounds 39, containing dipeptides and tripeptides, the corresponding mannofuranosyl peptides 41 were obtained in 34–65% yields with excellent anomeric selectivity. According to mechanistic studies a proposed mechanism is depicted in Scheme 16. The excited PC* promotes the SET oxidation of ester 37 to intermediate A followed by homolysis to the alkoxycarbonyl radical B and Hantzsch pyridine C. After decarboxylation of B, glycosyl radical D is formed, which adds to 38 giving radical E. Radical E undergoes SET with the reduced PC˙− to yield, after protonation, product 40.
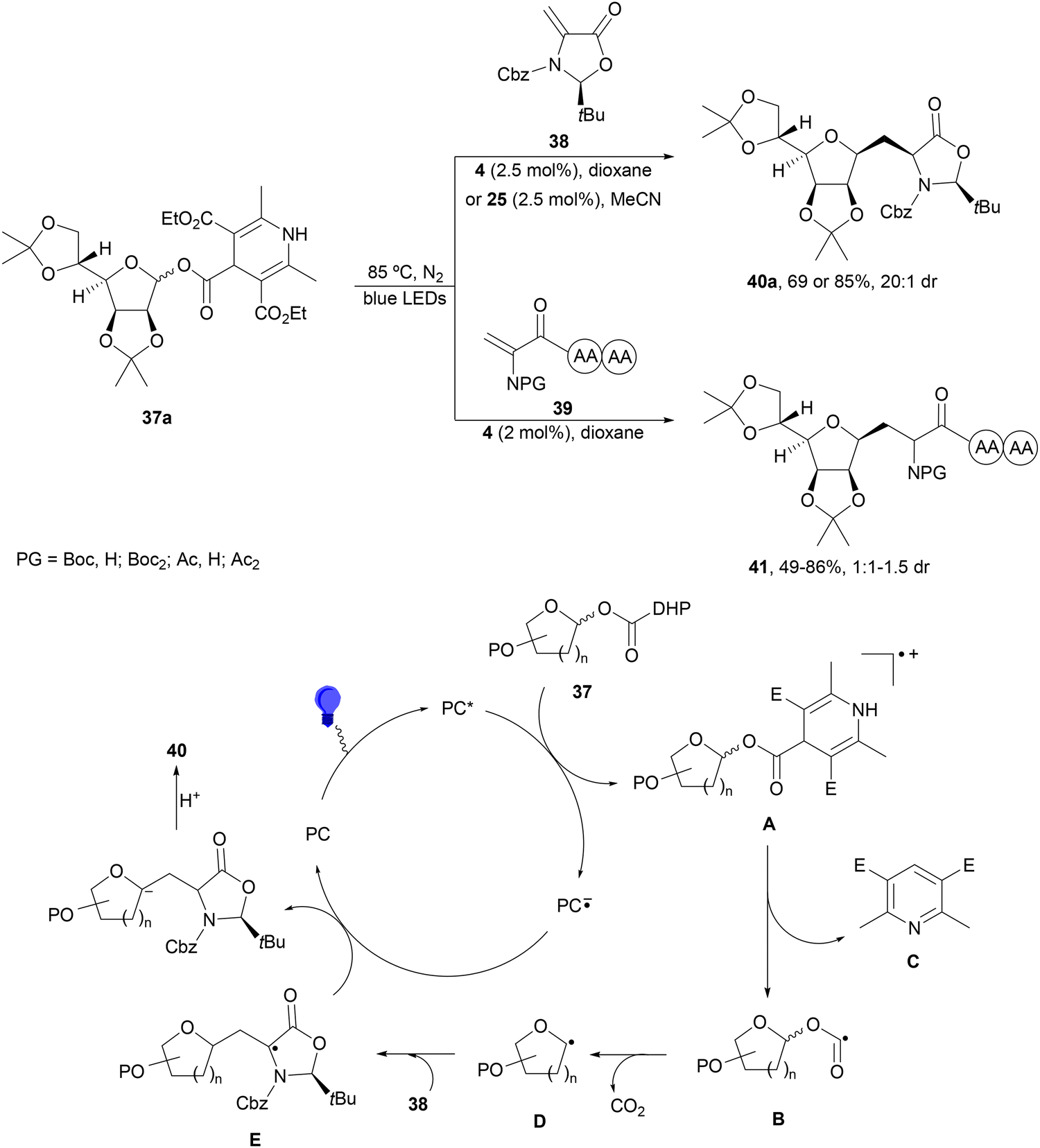 | ||
| Scheme 16 Decarboxylative Giese reaction of glycosyl DHP esters 37 with dehydroalanine-derivatives 38 and 39 under Ir 4 or 4CzIPN (25) photocatalysis. | ||
Recently, Fairbanks and co-workers84 described the DGR of unprotected glycosyl DHP esters 42 using 4CzIPN (25) as a PC in dry MeCN under blue LED irradiation at 85 °C (Scheme 17). Using acrylates as Michael acceptors and glucose DHP ester 42a products 43 were obtained in 51 to 62% yields. The reaction could be applied to mannose and galactose DHP esters, as well as to maltose and lactose DHP esters.
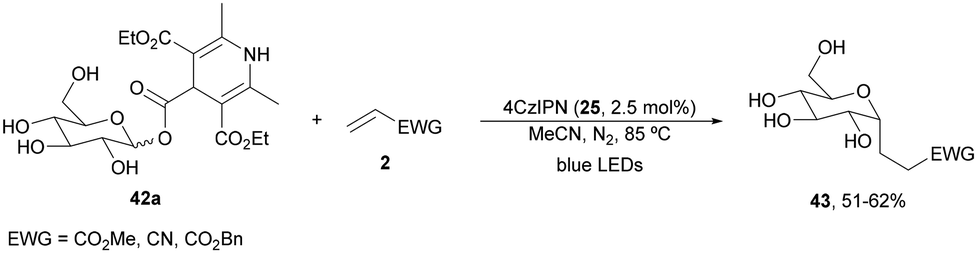 | ||
| Scheme 17 Decarboxylative Giese reaction of unprotected glycosyl DHP esters 42 and Michael acceptors under 4CzIPN (25) photocatalysis. | ||
2.2. Addition to electron-rich alkenes
Decarboxylative hydroalkylation of C–C double bonds has been performed with substituted naphthalene derivatives 44 and α-amino acids 1 using different PCs.85 This 1,2-hydroalkylation is a formal dearomatization carried out under redox-neutral conditions and using 4CzIPN (25) as a more efficient PC than Ir or Ru complexes. Substituted 1,2-dihydronaphthalenes 45 were obtained in good yields under visible light (Scheme 18). When an ester group was incorporated at the 2-position of naphthalene 44, tricyclic lactam-fused products 47 were also obtained. These compounds 47 were exclusively formed when the substituent at the 2-position of naphthalene was changed to N-acyl pyrazole or pentafluorophenyl ester with excellent diastereoselectivity. In the case of 3-hydroxy-2-methoxycarbonylnaphthalenes, products 46 were exclusively obtained with 63–99% yields. In the proposed mechanism, naphthalene 44 is transformed into the radical anion A by oxidation with the photoexcited species 4CzIPN* to deliver 4CzIPN+˙. Protonation of A gives radical B and N-arylglycine undergoes single-electron oxidation by 4CzIPN+˙ to provide after decarboxylation radical C. Final recombination of radical B and C gives products 45–47.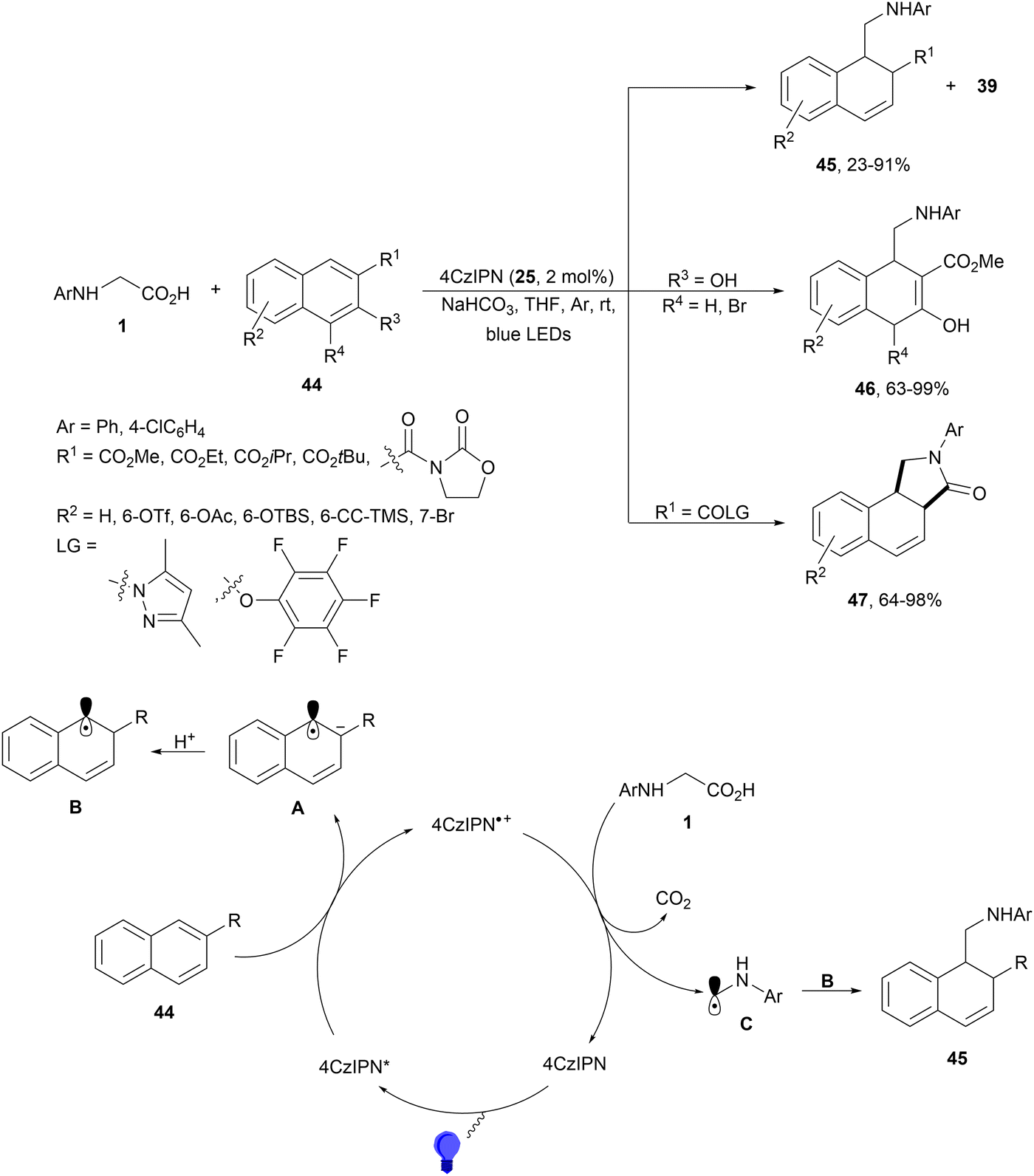 | ||
| Scheme 18 1,2-Hydroalkylation of naphthalene derivatives 44 with N-arylglycines under 4CzIPN (25) photocatalysis. | ||
Tandem aminomethylation/carboxylation of styrenes with sodium glycinates 48 has been developed under Ru(bpy)3·2PF6 photocatalysis using CO2 (1 atmosphere) in DMSO at room temperature (Scheme 19).86 This methodology afforded α,α-disubstituted γ-amino acids 49 in moderate to good yields. In some cases Ir complex 4 was also used in place of the Ru complex. When the resulting products 49 were treated with acetic anhydride the corresponding γ-lactams 50 were obtained in moderate to good yields. This procedure represents a step forward in the development of atom-economical decarboxylative reactions.
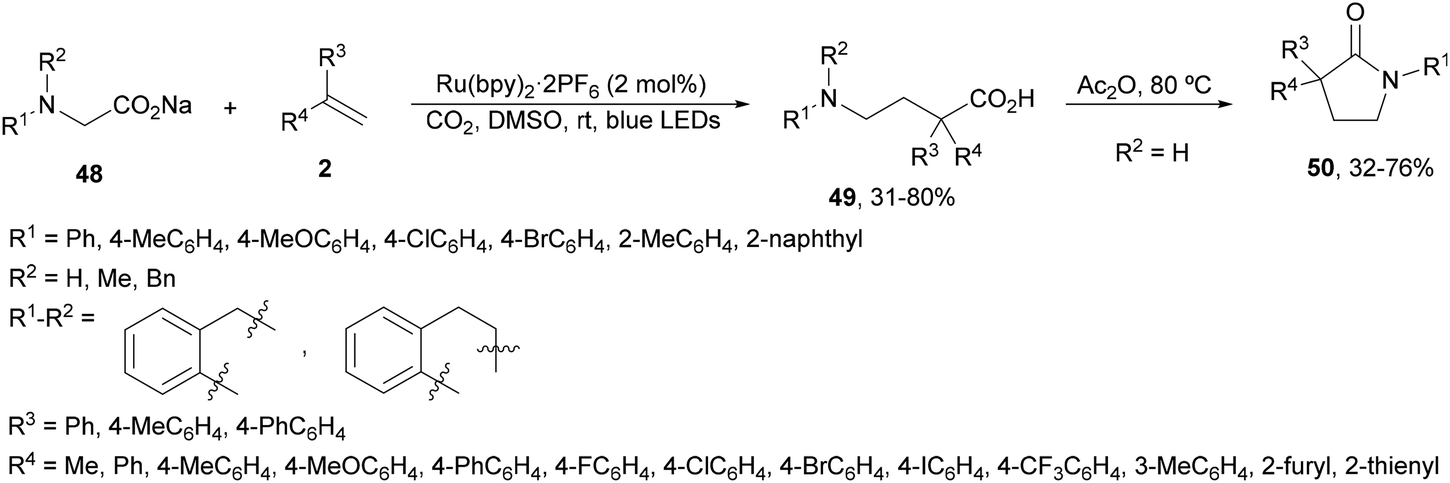 | ||
| Scheme 19 Aminomethylation/carboxylation of styrenes with sodium glycinates 48 and CO2 under Ru(bpy)2·2PF6 photocatalysis. | ||
In the case of allenes, a dual Pd/photoredox-catalyzed regio- and enantioselective decarboxylative hydroalkylation with N-aryl AAs has been recently reported by Breit and co-workers.87 When 1-alkoxyallenes 51 were hydroalkylated using Ir complexes 52 or 53 in the presence of 4-CF3C6H4CO2H and Feringa's phosphoramidite 54 as a chiral ligand syn-1,2-amino ethers 55 were obtained in good yields with good diastereo- and enantioselectivities under blue LED irradiation (Scheme 20a). The Ir catalyst gave better results than 4CzIPN. This transformation starts by oxidative addition of Pd(0) to the AA giving the palladium hydride species A. Migratory insertion of A into the allene forms the π-allyl palladium species B, which via a reductive quenching process of the excited Ir(III)* complex provides intermediate C. Subsequent SET from the carboxylate ion to Pd facilitates the decarboxylation step resulting in intermediate D. Final reductive elimination forms the product and the Pd(I) species, which is reduced by the Ir(II) complex. With monosubstituted allenes 56 linear decarboxylative hydroalkylation took place with N-aryl AAs using rac-BINAP (57) as a ligand for Pd to provide homoallyl amines 58 with good yields and regio- and E/Z-diastereoselectivities (Scheme 20b).
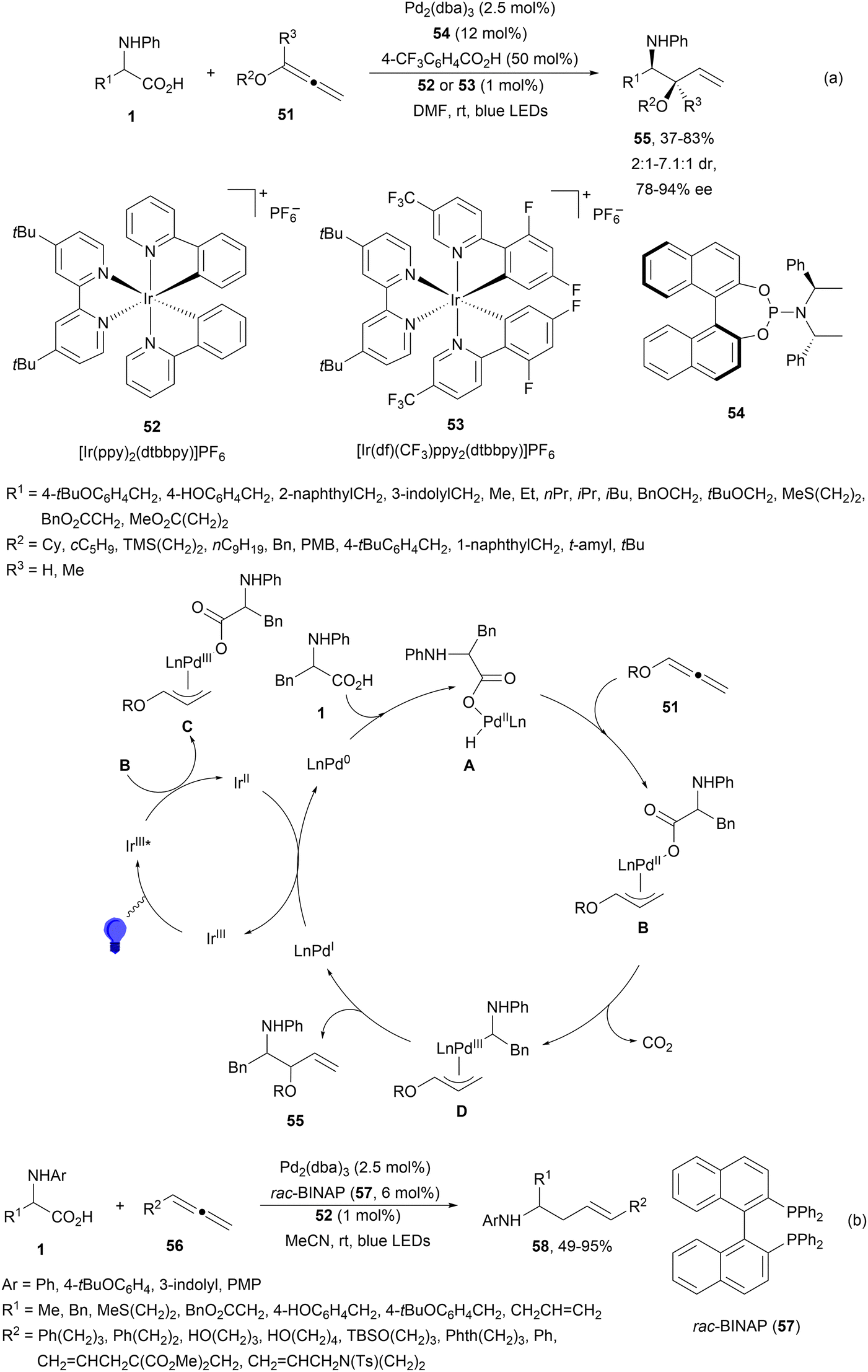 | ||
| Scheme 20 Decarboxylative hydrofluoroalkylation of allenes 51 (a) and 56 (b) with N-aryl AAs under Pd/Ir complex photocatalysis. | ||
Breit and co-workers88 performed the enantioselective hydroalkylation of dienol ethers 59 with AAs 6 using dual Pd/Ir photoredox catalysis. The decarboxylative 1,2-Markovnikov addition was carried out with an (η3-cinnamyl)PdCp complex (60) and the chiral phosphoramidite 61 and [Ir(ppy)2(dtbbpy)]PF6 (52) in acetone at room temperature under blue LED irradiation to provide products 62 up to >19:1 dr and up to >99% ee (Scheme 21). Mechanistic studies suggest a reversible hydropalladation as the key step.
 | ||
| Scheme 21 Decarboxylative hydroalkylation of dienol ethers 59 with N-aryl AAs under Pd/Ir complex photocatalysis. | ||
Zhang and co-workers89 have reported a three-component acetalation–pyridylation of alkenes using 4CzIPN (25) as an organophotocatalyst and Cs2CO3 as a base. Diethoxyacetic acids 26, cyanopyridines 63 and styrene derivatives 2 provided difunctional products 64 with very good yields and regioselectivities (Scheme 22). According to experimental results a plausible mechanism was proposed in which excited 4CzIPN* by irradiation of 25 with blue LED generates the formyl radical equivalent A and CO2. Hydroalkylation of styrene by intermediate A forms radical B, which reacts with 4-cyanopyridine 63 to generate radical C. Subsequent reduction of C by 4CzIPN−˙ forms anion D, which evolves to product 64a. Some products showed favorable in vitro antitumor activity.
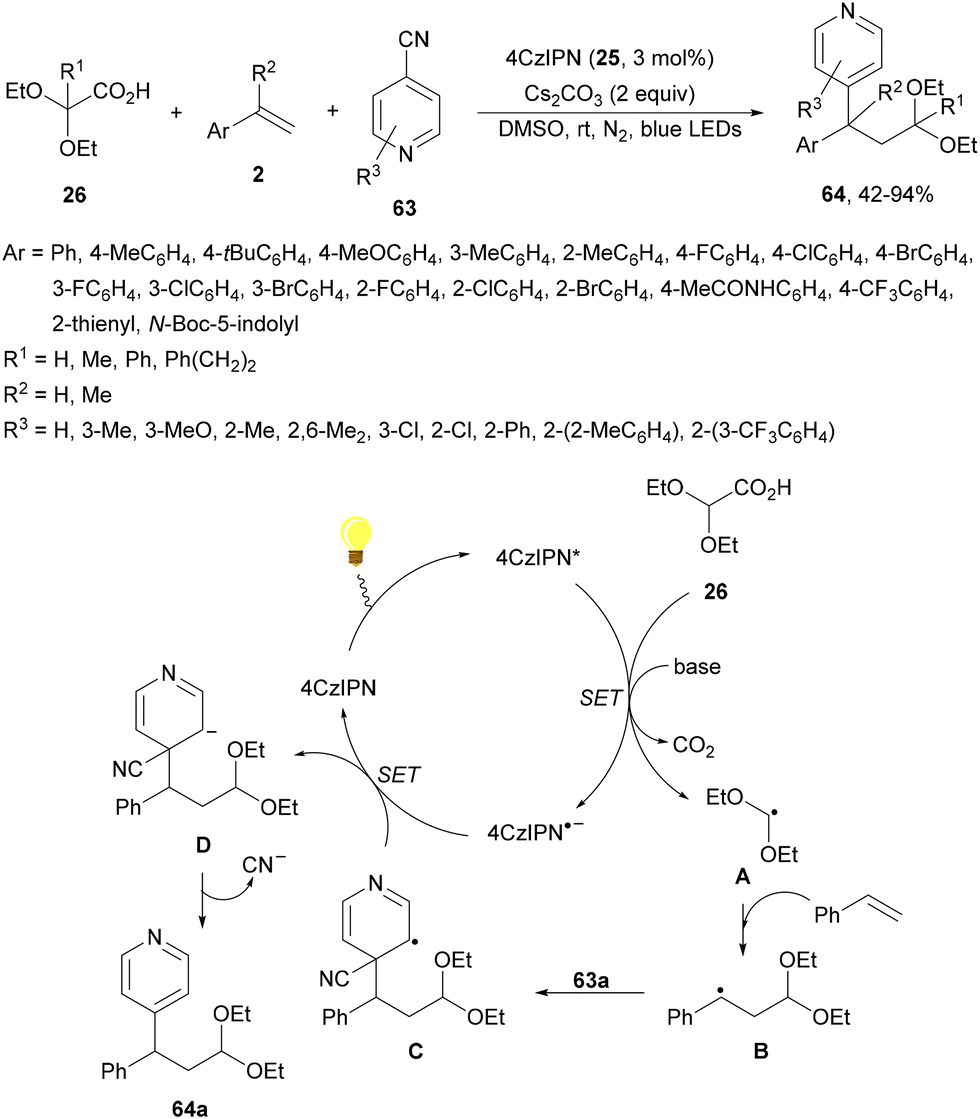 | ||
| Scheme 22 Three-component acetalation–pyridylation of styrene derivatives with diethoxyacetic acids (26) and cyanopyridines 63 under 4CzIPN (25) photocatalysis. | ||
Recently, a similar three-component decarboxylative coupling reaction of α-aryloxyacetic acids 65, cyanopyridines 63 and styrene derivatives 2 provided pyridines 66 with good yields (Scheme 23).90 In this case, Ir complex 4 was the best PC, with NaHCO3 as a base in DMSO at room temperature under blue LED irradiation. This process was carried out in a gram-scale.
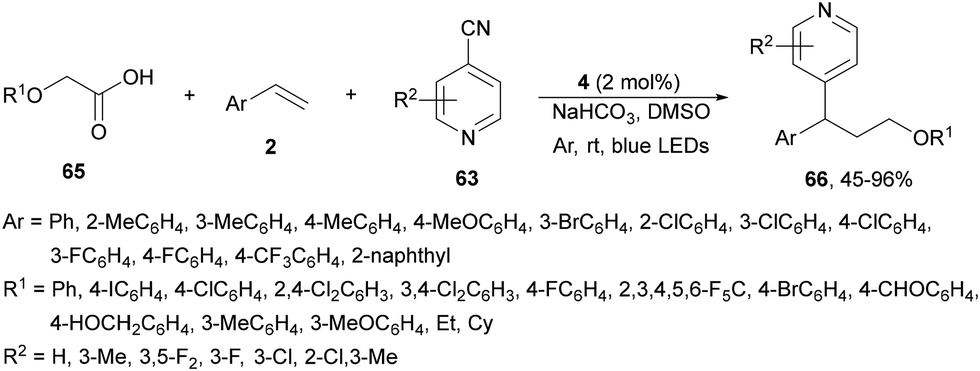 | ||
| Scheme 23 Three-component alkoxymethylation-pyridylation of styrene derivatives with α-alkoxy acetic acids 65 and cyanopyridines 63 under Ir photocatalysis. | ||
Aryl-alkylation of unactivated alkenes has been recently described by MacMillan and co-workers.91 This process took place by reaction of alkenes 67 with aryl bromides 68 and NHPI esters 69 under Ir complex 4 or 25 and Ni complex 70 photocatalysis in the presence of adamantylaminosupersilane (Admnsilane 71) and CsOAc as a base under blue LED irradiation (Scheme 24). By a triplet radical sorting mechanism of an aryl radical and a primary radical from the RAE 69 the corresponding products 72 resulted in good yields. Different unactivated alkenes 67 reacted with 4-bromo-2-chloro-pyridine 68a and NHPI 69a to provide products 72 in 59–79% yields (Scheme 24a). Different aryl bromides 68 were allowed to react with alkenes 67b and 67c and NHPI ester 69a giving products 72 in 41–72% yields (Scheme 24b). The scope of NHPI esters 69 was studied with N-vinylpyrrolidone 67b and 4-bromo-2-chloropyridine 68a to obtain products 72 in 47–83 yields using 4CzIPN (25) or Ir complex 4 as a photocatalyst (Scheme 24c). A proposed mechanism for the alkene aryl-alkylation is shown in Scheme 24: blue light excites the Ir photocatalyst 4 to the long-lived triplet excited state Ir(III)*, which is reductively quenched by Admnsilane (71). After aza-Brook rearrangement the silane radical A is formed, which undergoes a halogen atom transfer (XAT) with aryl bromide 68a to give an aryl radical B. Addition of B to alkene 67a results in the hindered alkyl radical C. To close the photoredox catalytic cycle Ir(II) reduces an NHPI ester 69a to provide the primary alkyl radical D after decarboxylation. This species is captured by the SH2 radical-sorting Ni catalyst 70 to afford complex E, which reacts with radical C providing product 72a.
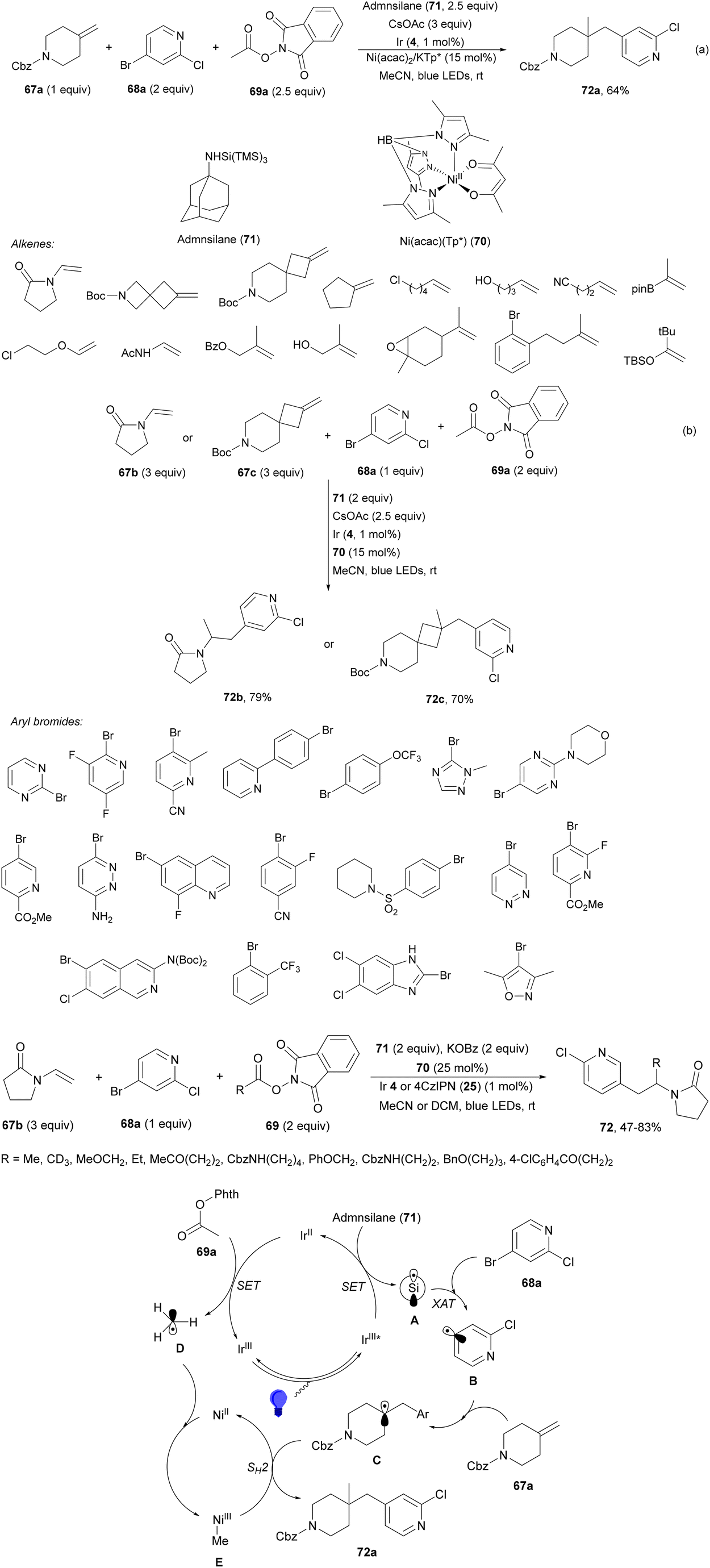 | ||
| Scheme 24 Aryl-alkylation of unactivated alkenes 67 with aryl bromides 68 and NHPI esters 69 using Admnsilane 71, Ir 4 and Ni (70) complexes as photoredox catalysts. | ||
Recently, several studies on iron-catalyzed hydrofluorination have been reported.92–94 West and co-workers92 published hydrofluoroalkylation of alkenes with fluoroalkylcarboxylic acids under mild conditions using Fe and redox-active thiol catalysis. Unactivated alkenes reacted with trifluoroacetic acid or difluoroacetic acid with Fe(OAc)2 and 2,4,6-triisopropylbenzenethiol (TRIP thiol) (or its disulfide) as catalysts, Na2CO3 as a base, aqueous MeCN at room temperature under 390 nm Kessil blue LED irradiation to furnish product 73 or 74, respectively, with good yields (Scheme 25a). These hydrotrifluoromethylation and hydrodifluoromethylation reactions have been applied to APIs and natural products. Non-steroidal and anti-inflammatory drugs such as ibuprofen, flurbiprofen and ioxoprofen provided the corresponding fluorinated products with 62–82% yields. Sulfonamide-containing probenecid and naproxen were trifluoromethylated in 89 and 43% yields, respectively. (−)-Borneol, L-menthol, oleic acid, monosaccharides, flavone and others were hydrofluoroalkylated with good yields. N-Boc-proline, estrone, 18β-glycyrrhetinic acid and pregnenolone derivatives gave useful conversions. Hydromonofluoroalkylation of styrene derivatives and hydro(polyfluoro)alkylation were performed under similar reaction conditions to provide products 75 and 76 (Scheme 25b). In the proposed mechanism, photochemical formation of a disulfide or thiyl radical enables oxidation of Fe(II) to Fe(III), which promotes the homolysis of the acids through an LMCT process54,55 of the Fe(III)-carboxylate. In the Fe photocatalysis cycle carboxylate ion A is transformed into B after LED irradiation, which evolves into Fe(II) and the carboxy radical C. Decarboxylation of C forms the fluorinated radical D. Subsequent radical addition onto the alkene provided the corresponding carbon centered radical, which reacts with the thiol as the HAT reagent to give the product, and the thiyl radical E will reoxidize the Fe(II) and receive a proton from the acid or from water closing both catalytic cycles.
 | ||
| Scheme 25 Decarboxylative hydrofluoroalkylation (a, b) of alkenes under Fe(OAc)2 or Fe(NO3)3 and TRIPSH photocatalysis. | ||
Guo, Xia and co-workers93 have reported photoinduced decarboxylative hydrodifluoromethylation of alkenes. In this case, terminal alkenes reacted with α,α-difluoroacetic acid using Fe(acac)3 and TRIPS as catalysts, with DMSO as solvent at room temperature under 390 nm LED irradiation to give products 74 with moderate to good yields (Scheme 26). These reaction conditions were also applied to APIs and natural products used in the pharmaceutical industry. In the presence of D2O, it was possible to perform deuteriodifluoromethylation of alkenes with >91% deuterium incorporation. Gram-scale experiments were carried out in continuous-flow in the synthesis of (+)-nootkalone to obtain 0.89 g of product 74a with 82% yield and 1:1 dr. In the proposed catalytic cycles, a LMCT process54,55 takes place through the iron complex A, providing similar intermediates to that shown in Scheme 25. Under similar reaction conditions, the Giese reaction of electron-deficient alkenes with aliphatic carboxylic acids using FeCl3 (10 mol%) and DABCO as a base in MeCN at room temperature under 390 nm LED irradiation occurs.
 | ||
| Scheme 26 Decarboxylative hydrofluoromethylation of alkenes with CF2HCO2H under Fe(acac)3 and TRIPSH photocatalysis. | ||
In the presence of a Brønsted acid, the formation of iron LMCT54,55 was facilitated for the activation of haloalkylcarboxylates (CnXmCO2−, X = F or Cl) to produce CnXm radicals.94 Unactivated terminal alkenes reacted with trifluoroacetic anhydride, Selectfluor, diphenyl ether and Fe(acac)3 as a catalyst in isopropanol and MeCN under a N2 atmosphere and blue light irradiation to form products 77 with moderate to good yields (Scheme 27). A wide variety of APIs were subjected to this Brønsted acid-unlocked iron LMCT photocatalysis. According to mechanistic studies, the photodecomposition of in situ-generated Fe(CF3CO2)3 occurred only under acidic conditions. In the proposed mechanism, the Fe(III) photoactive species, after irradiation through TS A, gives CF3CO2 radical B and Fe(II) intermediate C. After decarboxylation of B, the desired CF3 radical D is trapped by the alkene to form radical E, which is fluorinated by Selectfluor to give product 77. The generated N-radical cation F required the regulation by the redox buffer diphenyl ether to avoid the formation of C–N bonds.
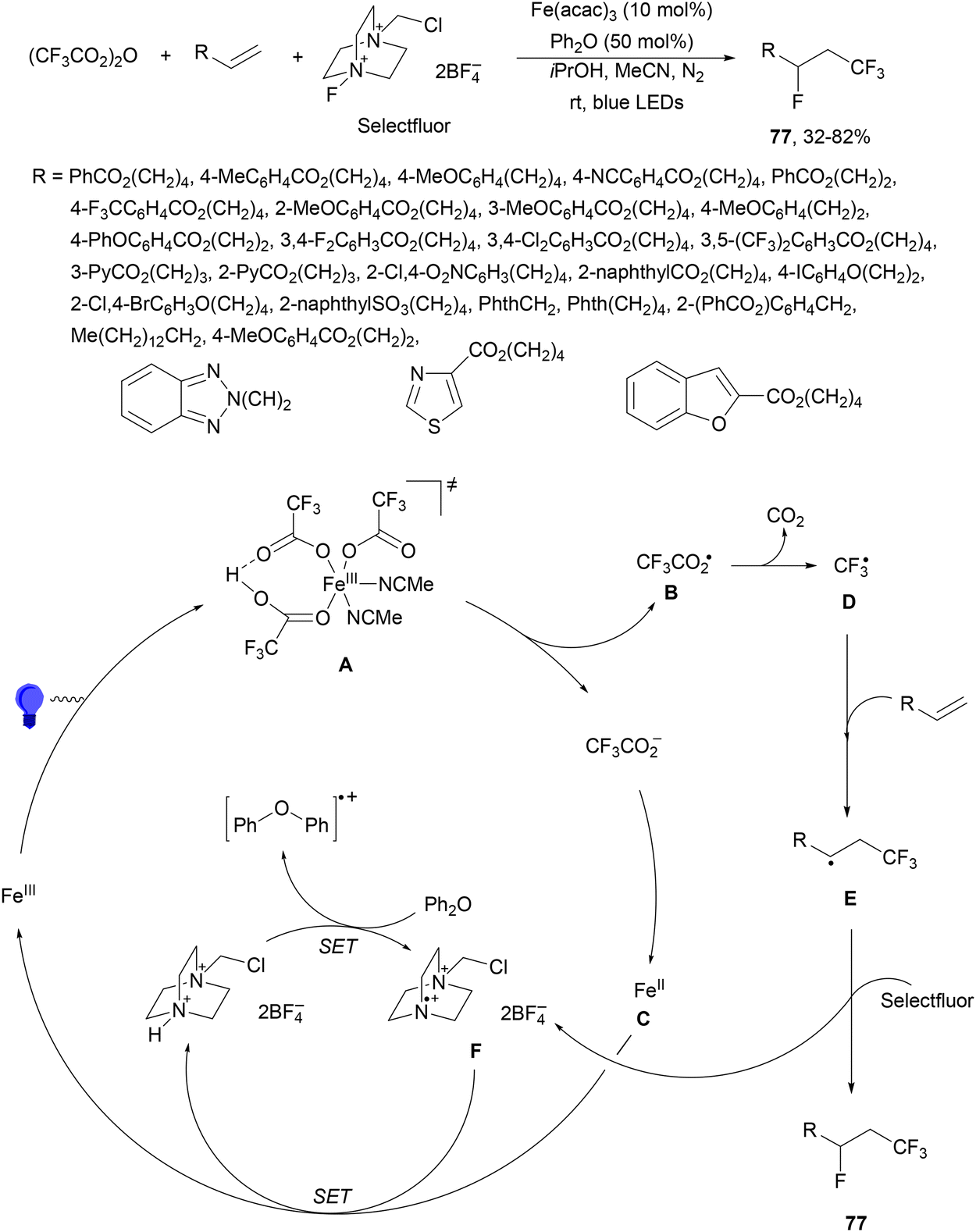 | ||
| Scheme 27 Decarboxylative fluorotrifluoromethylation of alkenes using Fe(acac)3, Selectfluor and Ph2O under photocatalysis. | ||
Radical polymerization of alkenes initiated by photocatalytic decarboxylation of carboxylic acids has been performed by Liao and Ni95 using titanium oxide nanoparticles (NPs) as a photocatalyst. The polymerization of vinyl acetate was achieved with carboxylic diacids 79 in aqueous media (Scheme 28). The rate of this polymerization depends strongly on the diacid structure and it was found that diacids with an even carbon number polymerized faster. This process was carried out in a reactor using a mercury vapor lamp at a frequency of 365 nm during 3 hours with 0.2% wt of TiO2. The number average molecular weight (Mn) of PVAc 80 ranges from 240 to 400 kDa and the molecular weight distribution ranges from 1.96 to 2.63.
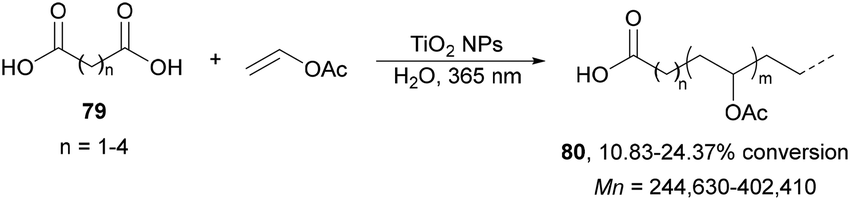 | ||
| Scheme 28 Photopolymerization of vinyl acetate initiated by carboxylic acids 79 under TiO2 NP photocatalysis. | ||
Naphthalene derivatives, styrenes, allenes and dienes can be hydroalkylated by photochemical decarboxylation of carboxylic acids under very different reaction conditions. Recently, iron-catalyzed hydrofluoroalkylation has been developed as an inexpensive process. In addition, three-component processes between carboxylic acids, styrenes, alkenes and cyanopyridines can be performed using 4CzIPN or Ir complexes as photocatalysts. Radical photopolymerization of alkenes has been efficiently carried out under TiO2 NP irradiation.
2.3. Addition reaction to alkynes
Decarboxylative hydroalkylation of unactivated alkynes was reported by MacMillan and co-workers96 using metallaphotoredox catalysis.15 This procedure involves photocatalytic decarboxylative radical generation from carboxylic acids and the Ir complex 4 and oxidative radical capture by low valent nickel species to form and alkyl-Ni(II) complex B, which undergoes a migratory insertion coupling step with the alkyne generating a vinyl-nickel complex Cvia cis-carbometallation. Terminal and internal alkynes 91 and carboxylic acids 1 reacted in the presence of Ir complex 4 and 5,5′-ditertbutylbipyridine (dtbbpy)·NiCl2 as dual catalysts and 1,1,3,3-teramethylguanidine (TMG) as a base in aqueous DMF under visible light to provide stereoselective products 92 in good yields (Scheme 29). In the case of terminal alkynes, branched products were regioselectively obtained instead of linear ones, indicating that the nickel-mediated pathway is exclusively operative. The proposed metallaphotoredox dual catalytic cycle supports the observed regio- and stereochemical outcomes.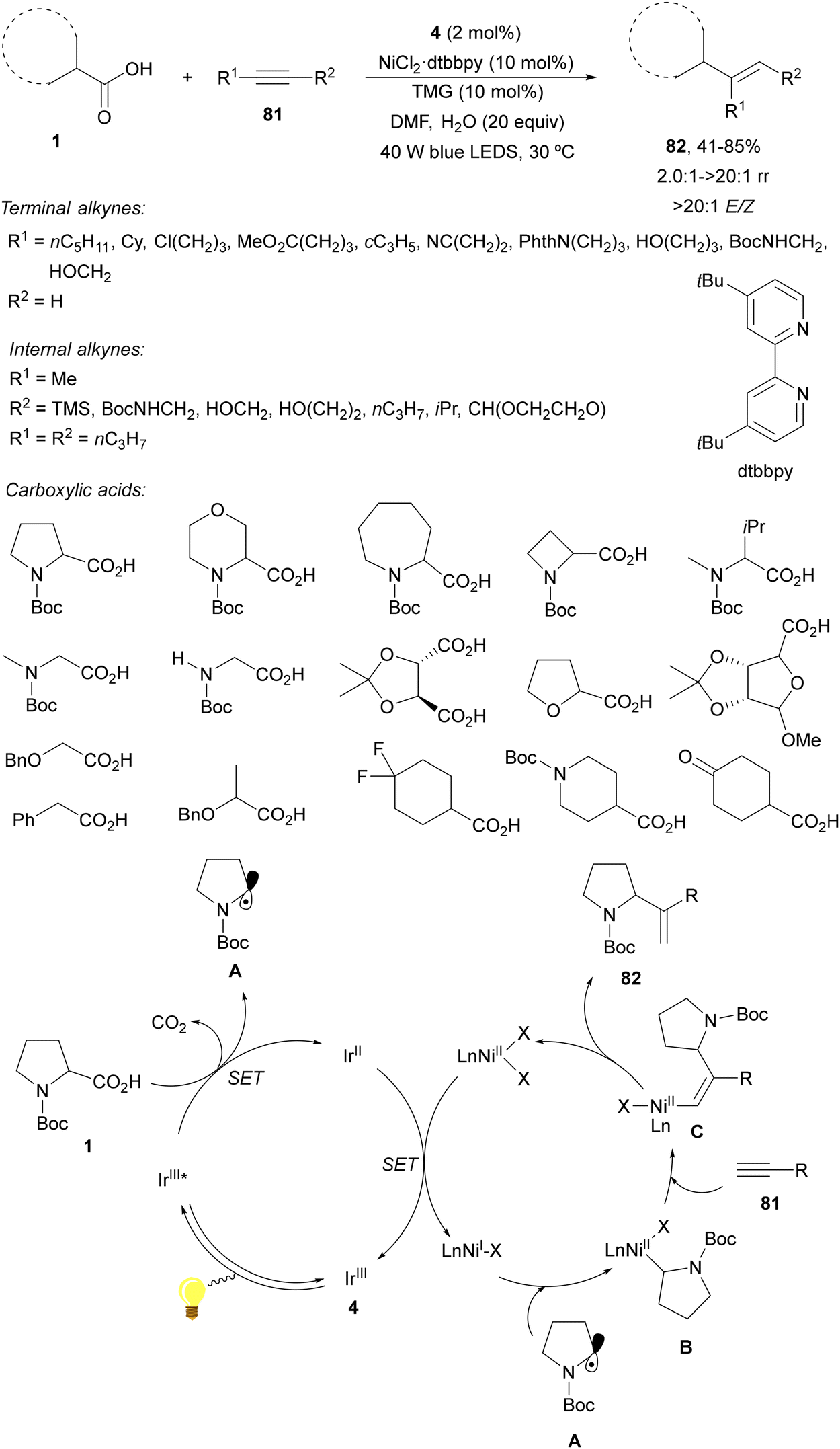 | ||
| Scheme 29 Decarboxylative hydroalkylation of unactivated alkynes 81 and carboxylic acids under Ir/Ni photoredox catalysis. | ||
Rueping and co-workers97 have reported the anti-Markovnikov dual catalyzed hydroalkylation of terminal alkynes 81 using N-protected AAs 6 and the Ir complex 83 and NiCl2·glyme as a catalyst under blue LED irradiation. Products 84 were obtained with moderate to complete Z/E diastereoselectivities and yields using Cs2CO3 as a base and a 1:1 mixture of MeCN and DMF at room temperature (Scheme 30a). In addition, arylalkylation of alkynes via a photoredox/nickel dual catalyzed three-component cross-coupling was achieved by changing the reaction conditions. Terminal alkynes 81, AAs 6 and aryl bromides were subjected to photoredox/nickel dual catalysis using Ir complex 4 and NiBr2·dtbbpy with Cs2CO3 as a base in DMF at room temperature to obtain products 85 with moderate diastereoselectivity (Scheme 30b). Control experiments for both processes and DFT calculations support the proposed mechanisms. In the case of hydroalkylation of alkynes, the Ir(III) complex 83 gives a triplet excited state Ir(III)* complex after absorption of visible light. Oxidation of the carboxylic acid and subsequent CO2 extrusion generates the alkyl radical intermediate A, which is trapped by the Ni(II) complex B to afford Ni(II) intermediate C. Reduction of C by an Ir(II) complex gives Ni(I) intermediate D, which after alkyne 1,2-migratory insertion forms the Ni(I) intermediate E. Final protonation of Evia intermediate F provides product (E)-84 and Ni complex B. The (Z)-isomer 84 is formed via intermediate G through an energy transfer pathway. In the second catalytic cycle, the proposed mechanism for the arylalkylation of alkynes is as follows: the Ni(II) intermediate Hvia a SET process gives Ni(I) intermediate I, which after addition to the alkyne and oxidative addition of an aryl bromide provides intermediates J and K, respectively. Final reductive elimination of K delivers the anti-addition three-component coupling product anti-85. The syn-addition product 85 is obtained via an energy transfer (ET) pathway.
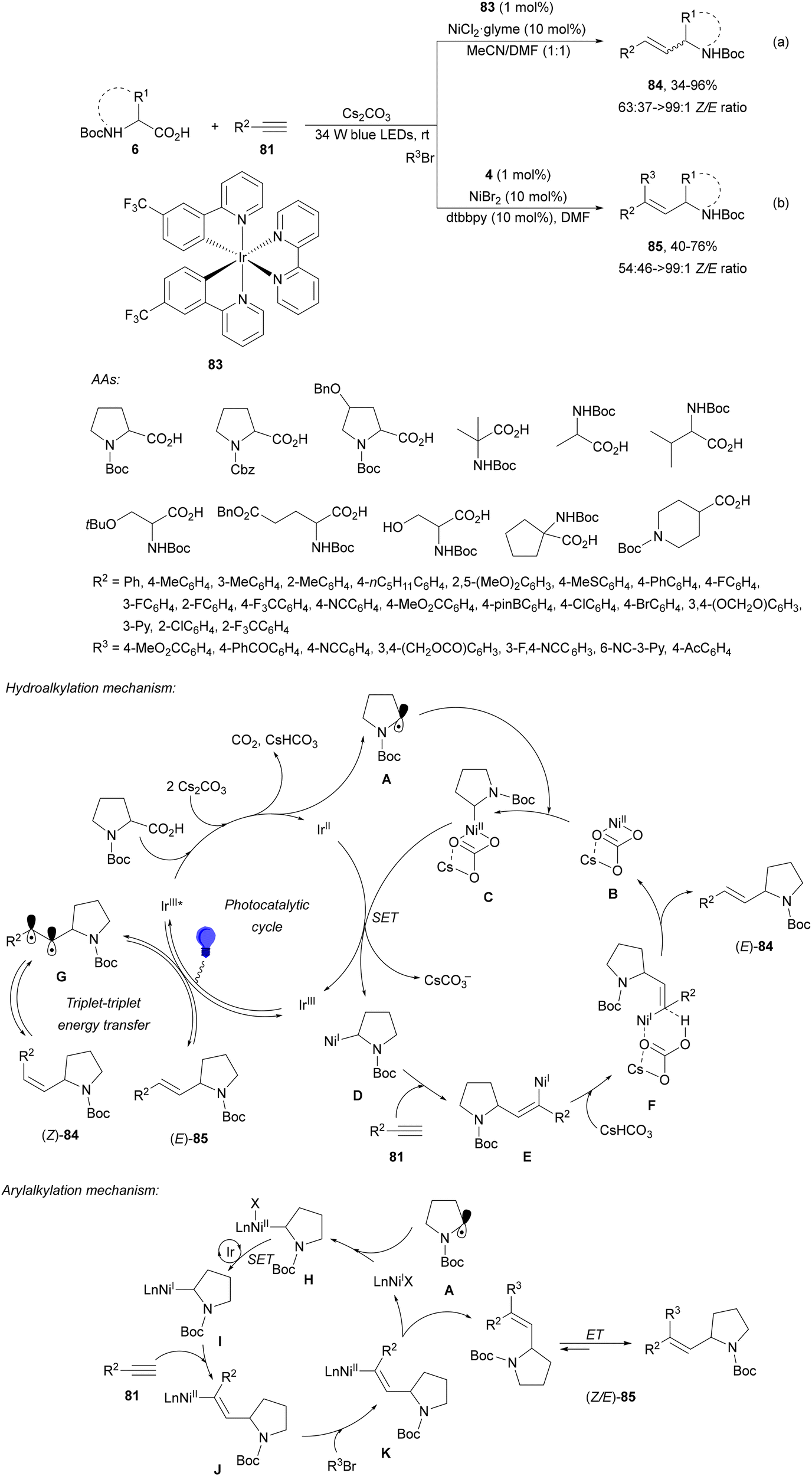 | ||
| Scheme 30 Decarboxylative hydroalkylation and arylalkylation of terminal alkynes 81 (a, b) with AAs under Ir/Ni dual photocatalysis. | ||
Pericàs and co-workers98 performed the decarboxylative stereodivergent99 hydroalkylation of alkynes using copper instead of nickel and 4CzIPN (25) instead of an Ir complex as a photoredox catalyst. By a multivariate high-throughput experimentation (HTE) approach different carboxylic acids, including AAs and α-oxy acids, reacted with terminal alkynes 81 using Cu(OAc)2, diamine 86 as a ligand, CsOAc as a base and DMA as solvent and blue LED at room temperature (Scheme 31). The resulting (Z)-alkenes 87 were obtained with moderate stereoselectivity. On the other hand, (E)-alkenes 87 were formed by changing ligand 86 to oleylamine (88) and using CsHCO3 as a base. In the proposed mechanism, upon irradiation of the PC the Cu(II) complexes were reduced to Cu(I) A and also the carboxylate ion was oxidized to radical C, which underwent decarboxylation. The Cu(I) complex A generates acetylide B, which after photoexcitation gives the species B* with a depletion of charge on the alkyne moiety through ligand-to-metal charge transfer (LMCT)54,55 accelerating the attack of the radical C to form the vinyl radical D. Subsequent oxidation of PC−˙ by D provides vinyl anion E, which after protonation and proto-demetallation of the Cu–C bond gives enriched (E)-87, generating the Cu(I) species. Isomerization of (E)-87via energy transfer (ET) mediated by the PC provides (Z)-87.
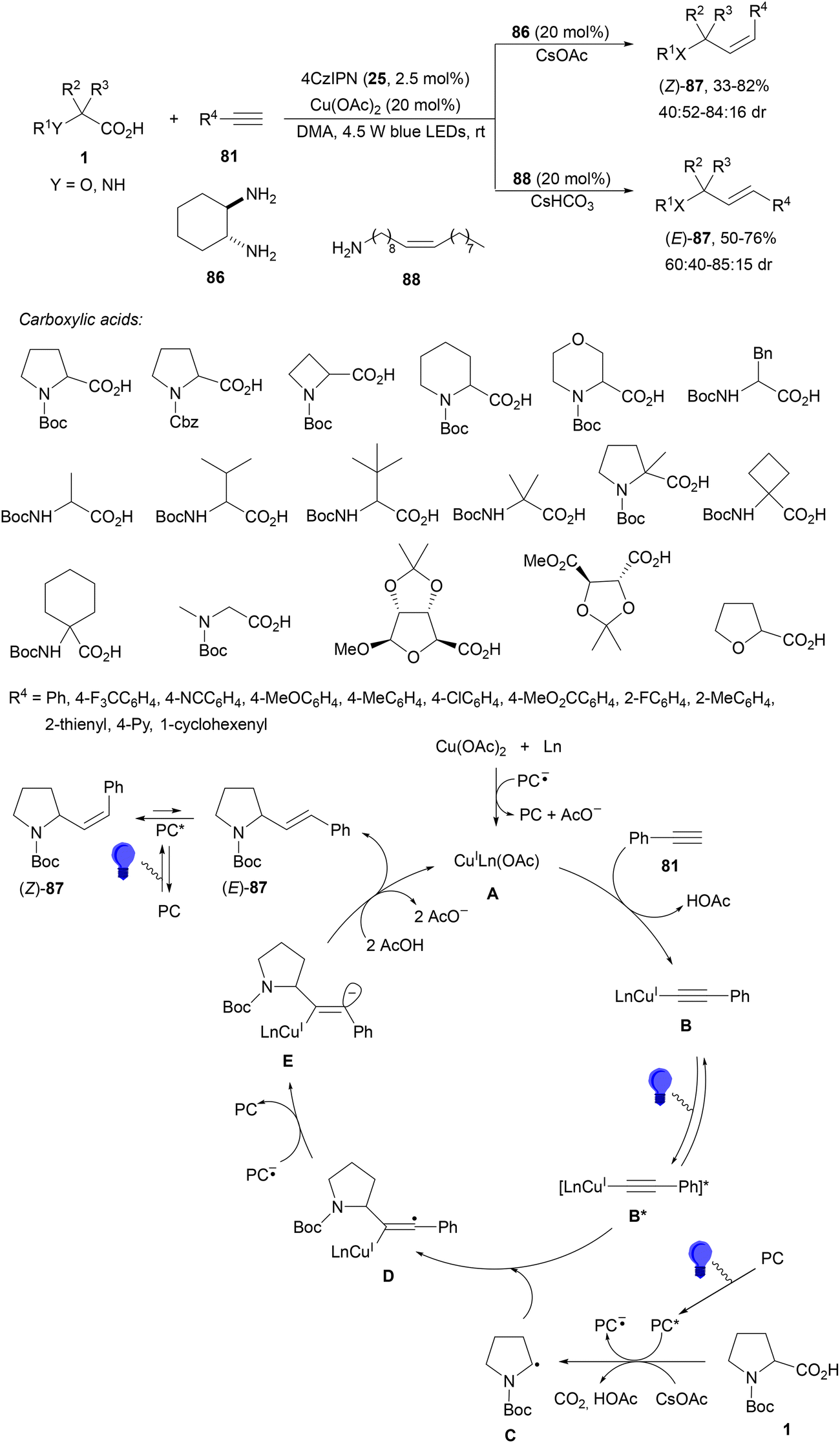 | ||
| Scheme 31 Decarboxylative hydroalkylation of terminal alkynes with carboxylic acids under Cu/4CzIPN (25) photocatalysis. | ||
Decarboxylative hydroalkylation of terminal acetylenes 81 using NHPI esters 69 has been performed by Luo, Tang and co-workers100 using an Ir(ppy)3 (89) complex as a photocatalyst and blue LED irradiation. A wide range of primary, secondary and tertiary carboxylate ions as well as α-amino and α-oxy acid-derived esters 69 reacted with terminal arylalkynes 81 to provide alkenes 88 with good yields and Z/E-diastereoselectivities (Scheme 32). This process took place at room temperature under an Ar atmosphere with DIPEA as a base and DMA as solvent without the requirement of metal activation.
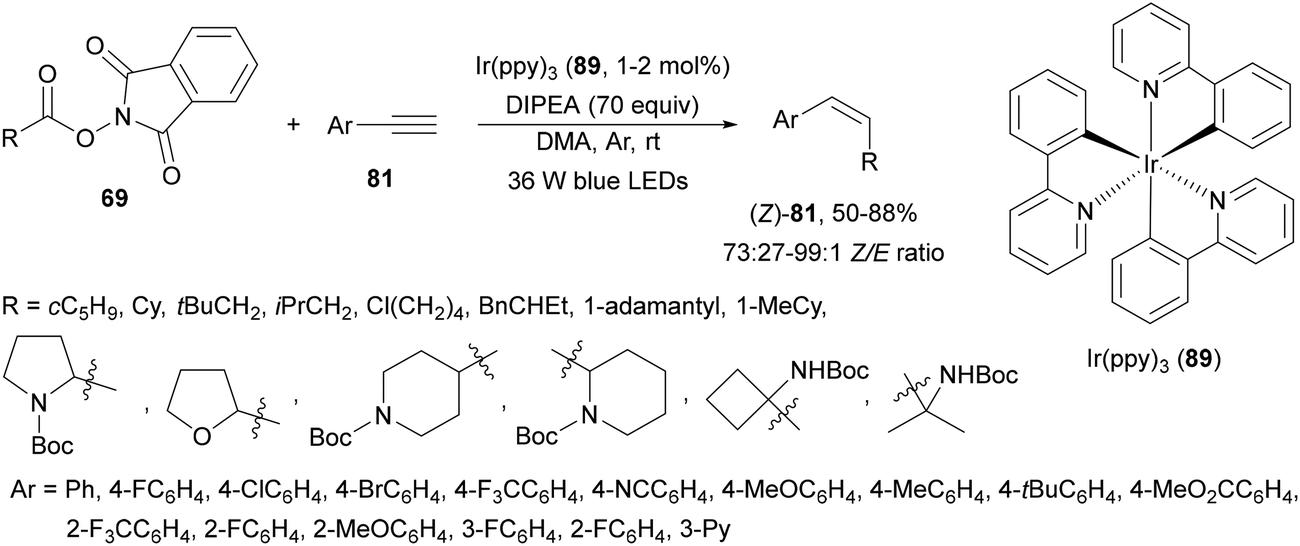 | ||
| Scheme 32 Decarboxylative alkylation of terminal arylalkynes 81 with NHPI esters 69 using Ir complex 89 as a photocatalyst. | ||
The photocatalytic hydroalkylation of alkynes with carboxylic acids must be carried out under Ni or Cu co-catalysis and under Ir photocatalysis in the case of NHPI esters with good diastereoselective control of the obtained alkenes.
2.4. Addition to C–heteroatom multiple bonds
Decarboxylative addition of benzylic radicals to carbonyl groups was initially carried out with arylacetic acids101 and 9-fluorenone102 or N-methylphthalimides103 under photocatalytic conditions. Jiang and co-workers104 reported the enantioselective radical addition to 1,2-diketones 90 and isatins 91 of N-acylglycines 6 using a visible light dual catalytic system dicyanopyrazine photocatalyst DPZ 92 and a chiral phosphoric acid (CPA), SPINOL 93a or 93b (Scheme 33). This reaction was performed in the presence of tetra-n-butylphosphonium bromide (TBPB) and Na2S2O4 in cyclopentyl methyl ether (CPME) at 10 °C to provide products 95 in good yields and up to 97% ee. In the case of isatins 94, SPINOL 93b was the best CPA and THF was used as solvent to obtain 3-hydroxy-3-aminomethylindolyn-2-ones 96 in good yields and ees. DPZ is able to generate intermediate A, which after decarboxylation forms the α-amino radical B. DPZ−˙ reduces the 1,2-diketone giving intermediate C. Both radicals B and C are activated by the CPA by hydrogen bonding favoring their coupling to provide 1,2-aminoalcohols 95.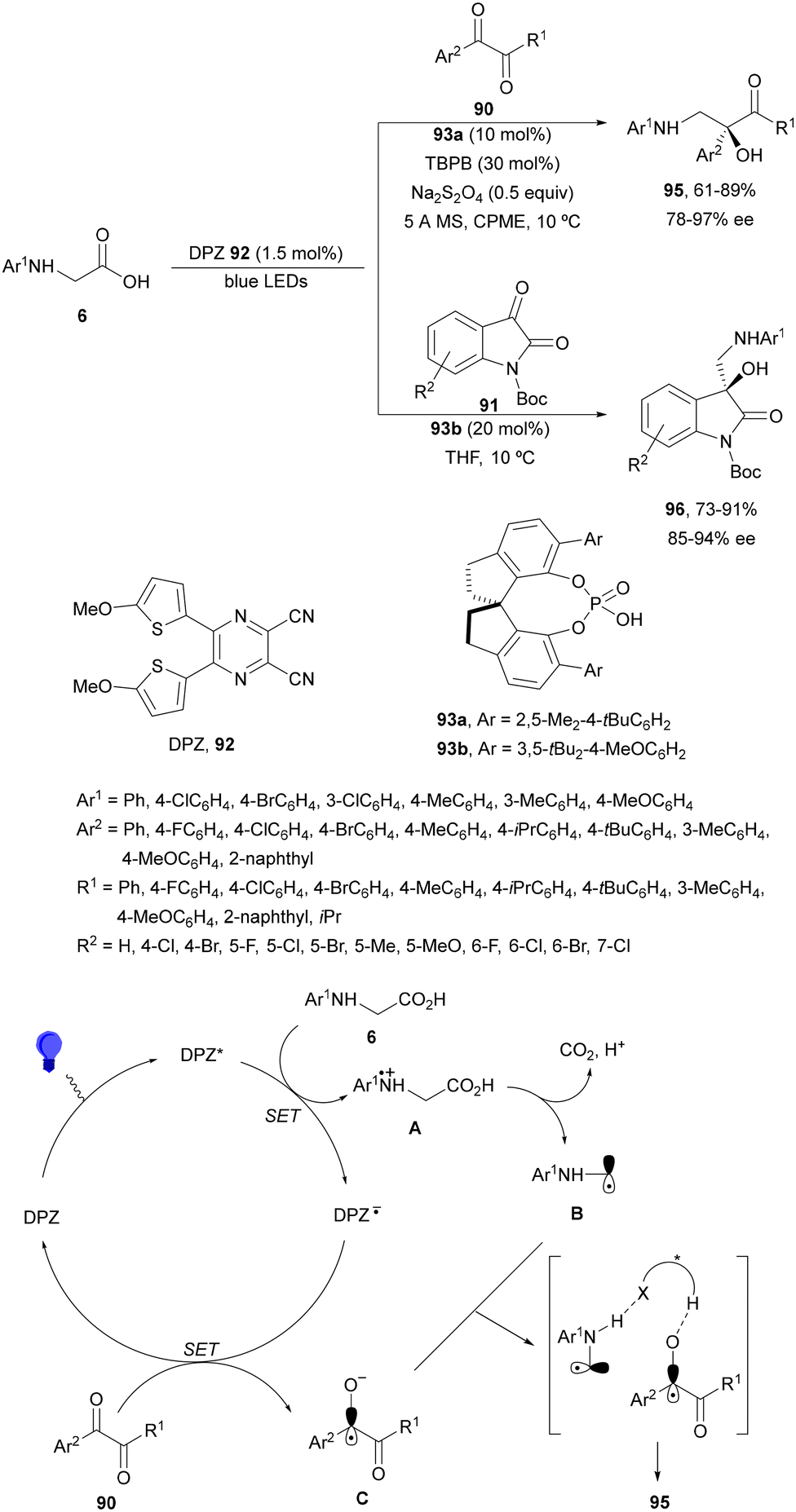 | ||
| Scheme 33 Enantioselective decarboxylative addition of N-arylglycines to 1,2-dicarbonyl compounds 90 or to isatins 91 under dual DPZ 92 and CPA 93 photocatalysis. | ||
Decarboxylative addition of N-aryl amino acids to aldehydes or ketones has been described under visible-light in water at room temperature using Ir complex 4 as a photocatalyst (Scheme 34).105 The corresponding 1,2-amino alcohols 97 were obtained in very good yields and the reaction of N-phenyl glycine and 4-MeOC6H4CHO was carried out on a gram scale. According to experimental studies the α-amino radical A reacts with the alcoholate radical B to provide the product.
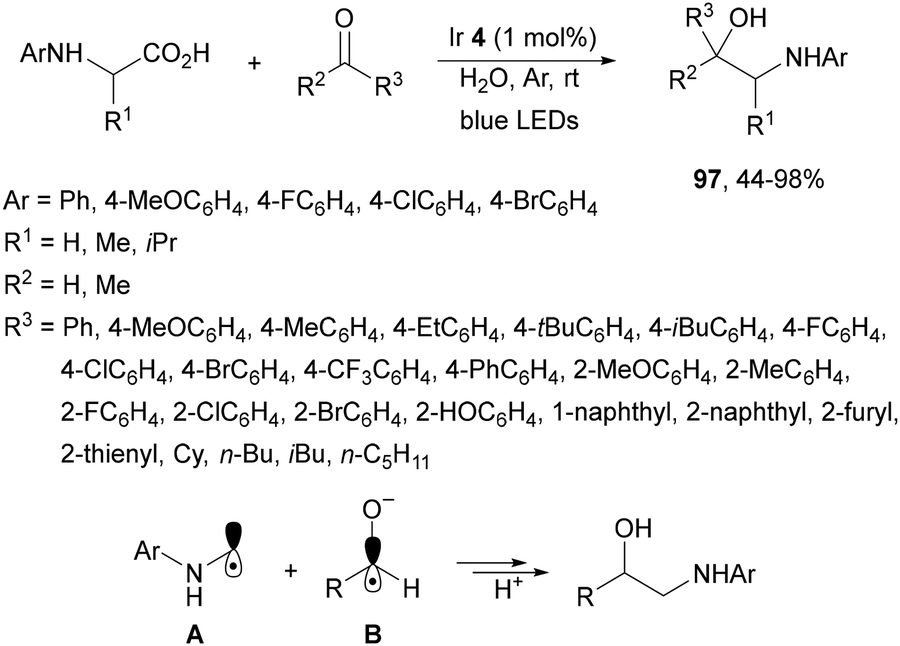 | ||
| Scheme 34 Decarboxylative radical addition of N-aryl amino acids to aldehydes or ketones in water under Ir 4 photocatalysis. | ||
In the case of DNA-conjugated aldehydes 98, the photoredox-mediated decarboxylative coupling with α-amino acids provided DNA-encoded 1,2-amino alcohols 99 (Scheme 35).106 This reaction proceeded for a wide range of aldehydes but also for ketones with conversions >50% using Ir complex 4 as a PC and K2HPO4 as a base in aqueous DMAc at room temperature in only 10 minutes irradiation time.
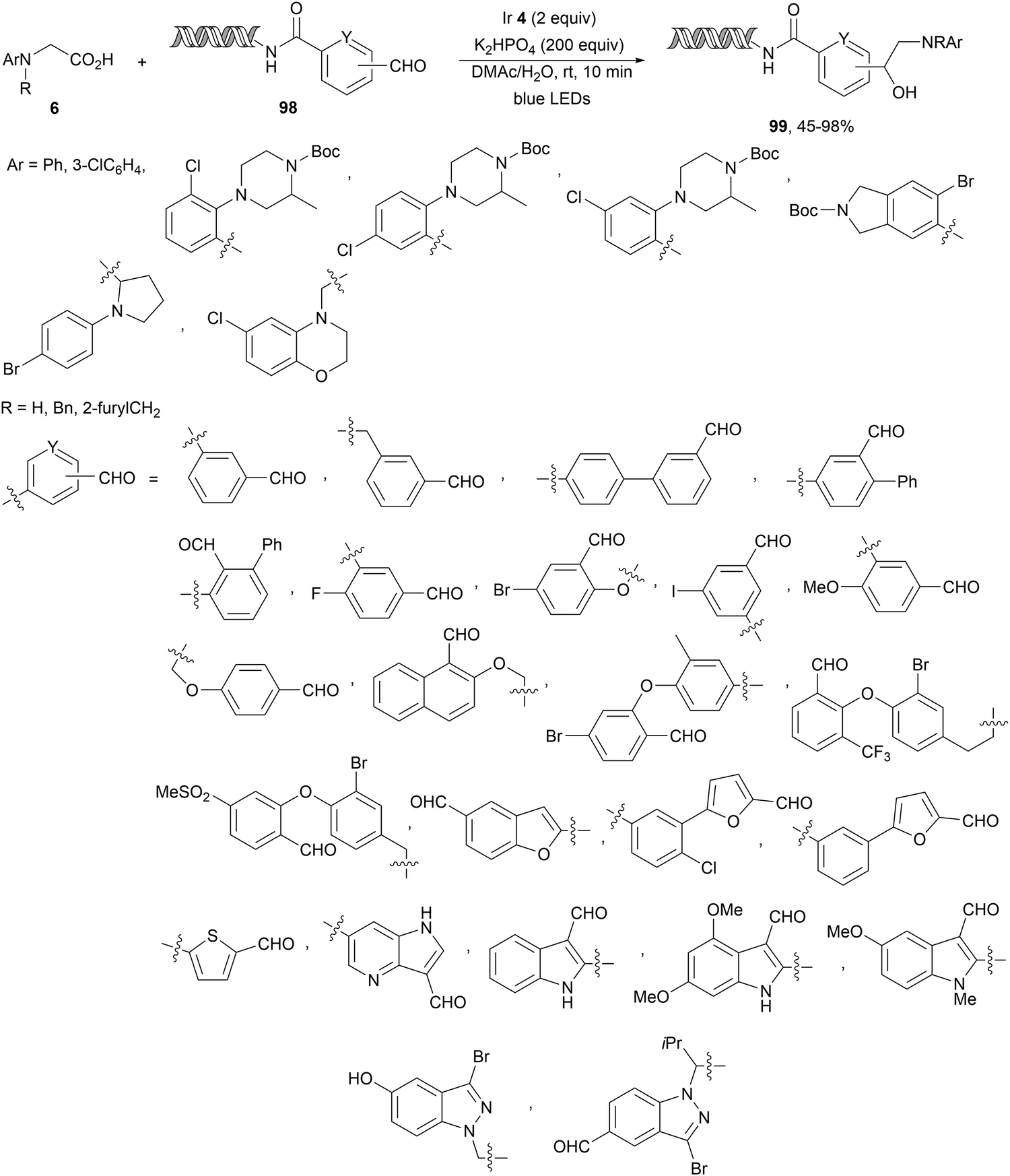 | ||
| Scheme 35 Decarboxylative coupling of α-amino acids with DNA-conjugated aldehydes under Ir 4 photocatalysis. | ||
For the synthesis of 1,2-amino alcohols by decarboxylative coupling of α-amino acids with aldehydes an urushiol derivative 102 has been used as a photocatalyst by Zhang and co-workers.107 This PC was prepared from urushiol 100 and 2,3,5,6-tetrafluorotherephthalonitrile (101) in the presence of K2CO3 (Scheme 36). Due to the photoredox properties of 102 it was used in MeCN as solvent with NaHCO3 as a base at room temperature under blue LED irradiation to obtain 1,2-amino alcohols 97 with modest to high yields (16–93%). This biomass-based PC exhibits absorption in the visible region (λmax = 426 nm) and its half-wave potentials were +1.17 and −1.24 V, which can be assigned to 102−˙/102 and 102/102−˙, respectively, according to cyclic voltammetry measurements.
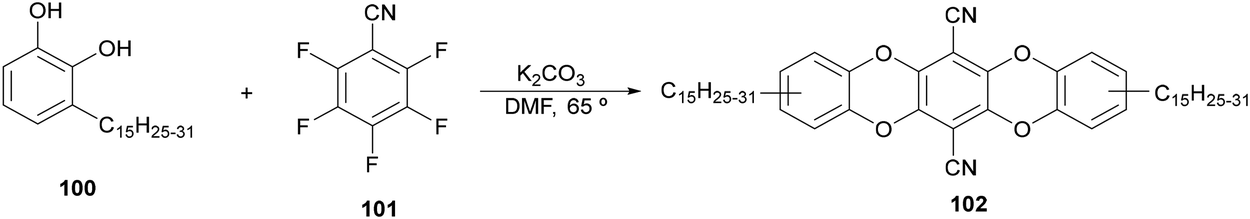 | ||
| Scheme 36 Synthesis of PC 102 derived from urushiol (100). | ||
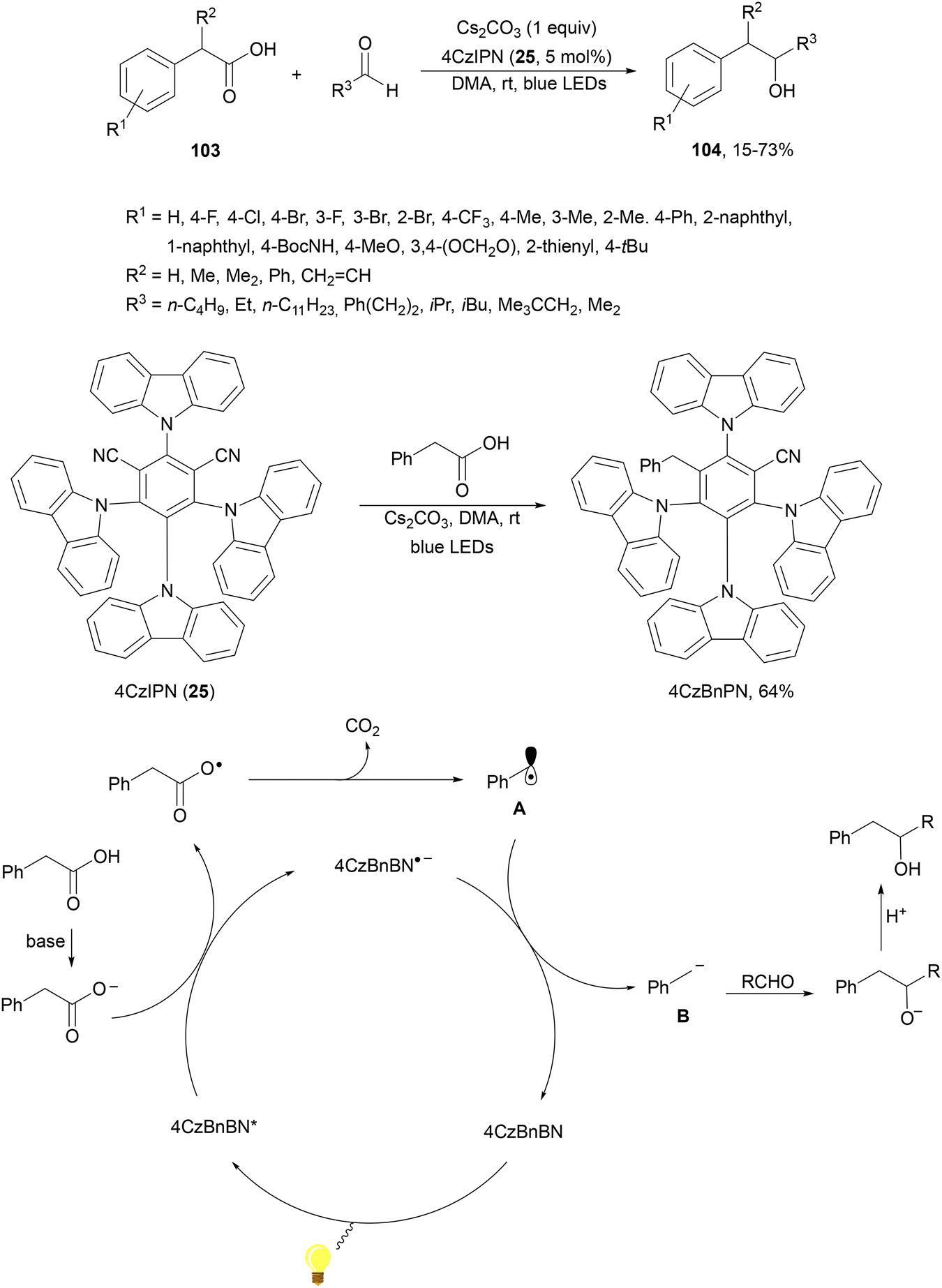
König and co-workers108 have reported a redox-neutral procedure for benzylate aliphatic aldehydes via the photocatalytic generation of benzyl carbanions109 from arylacetic acids 103. Different carboxylic acids 103 reacted with aldehydes using Cs2CO3 as a base, 4CzIPN (25) as a PC and DMA as solvent at room temperature to provide alcohols 104 in moderate to good yields (Scheme 37). Deuterium labeling experiments suggest the formation of an anion intermediate with D2O as an electrophile. Considering experimental and computational studies, the following mechanism is proposed: after deprotonation of the carboxylic acid the carboxylate ion is oxidized by the excited 4CzBnBN*. Actually, this organophotocatalyst 4CzBnBN has been formed by reaction of 4CzIPN with the benzyl anion B (generated from phenylacetic acid) through an E1cb mechanism. Radical A is converted into anion B by 4CzBnPN−˙, which adds to aldehyde forming the corresponding alcoholate, which suffers protonation to yield the final product 104.
 | ||
| Scheme 37 Decarboxylative addition of arylacetic acids 103 with aliphatic aldehydes under 4CzIPN photocatalysis. | ||
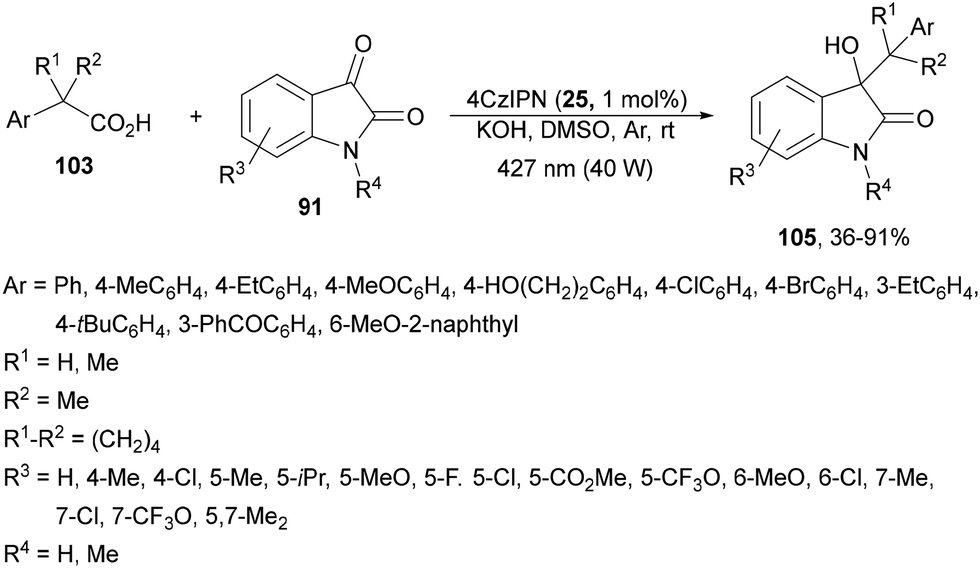
Recently, the addition of arylacetic acids to aromatic aldehydes has been described110 just changing Cs2CO3 by CsF as a base using DMF as solvent. The corresponding alcohols 104 (R3 = Ar, heteroaryl) were obtained with 50–96% yields and this process was performed on a gram-scale for the reaction of α,α-diphenylacetic acid with 4-fluorobenzaldehyde. Isatins 91 reacted with arylacetic acids 103 to provide 3-hydroxy-3-alkyloxindoles 105 using 4CzIPN as a PC, KOH as a base and DMSO as solvent at room temperature under two 40 W 427 nm Kessil tuna blue lamp irradiation (Scheme 38).111 The reaction of isatin (R3 = R4 = H) with α,α-dimethylphenylacetic acid was carried out on a gram-scale to obtain the corresponding oxindole 105 in 71% yield. In this case, mechanistic studies demonstrate that the process involves cross-coupling between a persistent ketyl radical and a transient alkyl radical from the carboxylic acid.
 | ||
| Scheme 38 Decarboxylative coupling of arylacetic acids 103 with isatins 91 under 4CzIPN photocatalysis. | ||
The reaction of carboxylic acids with carbonyl compounds under photocatalyzed decarboxylation takes place by cross-coupling of ketyl radicals and alkyl radicals from the carboxylic acid. However, in the case of arylacetic acids and aldehydes, the mechanism involves the addition of benzylic anions to the carbonyl group.
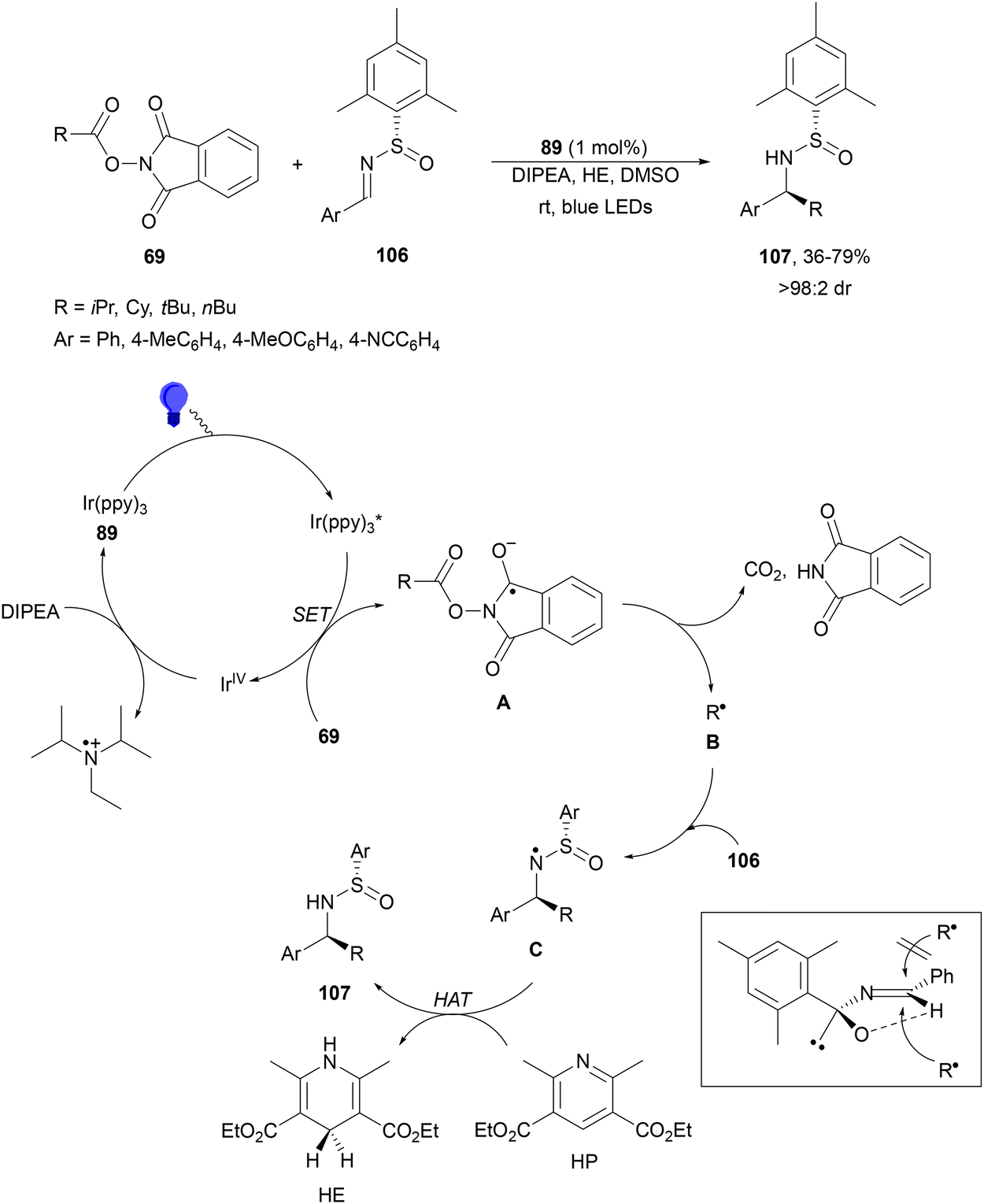
Decarboxylative radical addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds under photocatalytic conditions has been studied with imines, nitrones and isocyanates. Maestro, Alemán and co-workers112 described the enantioselective addition of alkyl radicals to chiral N-sulfinimines 106 (Scheme 39). They used NHPI esters 69, Ir(ppy)3 (89) as a PC, DIPEA as a base, visible light and DMSO as solvent in the presence of 1.5 equivalents of Hantzsch ester (HE) to provide products 107 with good diastereoselectivities. The best results were obtained with mesityl-substituted N-sulfinimines and secondary or tertiary alkyl groups. In the proposed mechanism, the photoexcited catalyst generates, through a SET process, the radical anion A from the NHPI ester by an oxidative pathway. Reductive elimination of the phthalimide group leads to decarboxylation and formation of radical B, which undergoes radical addition to N-sulfinimine to form radical C. Finally, HE donates hydrogen through hydrogen atom transfer (HAT) to C giving rise to product 107 and HP. The approach of the radical to 106 takes place by the less hindered Si-face of the imine explaining the S configuration of the new stereocenter.
N double bonds under photocatalytic conditions has been studied with imines, nitrones and isocyanates. Maestro, Alemán and co-workers112 described the enantioselective addition of alkyl radicals to chiral N-sulfinimines 106 (Scheme 39). They used NHPI esters 69, Ir(ppy)3 (89) as a PC, DIPEA as a base, visible light and DMSO as solvent in the presence of 1.5 equivalents of Hantzsch ester (HE) to provide products 107 with good diastereoselectivities. The best results were obtained with mesityl-substituted N-sulfinimines and secondary or tertiary alkyl groups. In the proposed mechanism, the photoexcited catalyst generates, through a SET process, the radical anion A from the NHPI ester by an oxidative pathway. Reductive elimination of the phthalimide group leads to decarboxylation and formation of radical B, which undergoes radical addition to N-sulfinimine to form radical C. Finally, HE donates hydrogen through hydrogen atom transfer (HAT) to C giving rise to product 107 and HP. The approach of the radical to 106 takes place by the less hindered Si-face of the imine explaining the S configuration of the new stereocenter.
 | ||
| Scheme 39 Decarboxylative asymmetric addition of NHPI esters 69 to mesityl-substituted N-sulfinimines 106 under Ir(ppy)3 (89) photocatalysis. | ||
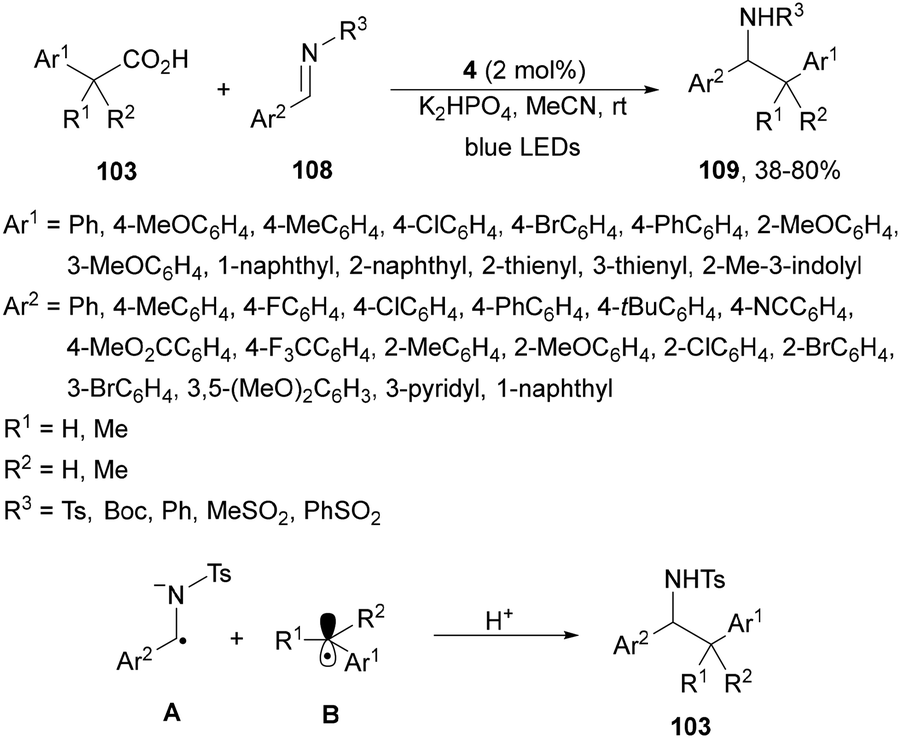
A visible-light-mediated decarboxylative benzylation of N-protected aldimines 108 with arylacetic acids 103101 has been reported by Weng, Lu and co-workers.113 This process took place using Ir complex 4 as PC, K2HPO3 as a base in MeCN at room temperature under blue LED irradiation to give rise to N-protected amines 109 with good yields (Scheme 40). In the proposed mechanism the excited Ir(III)* species forms the α-amino radical anion A, which combines with the benzylic radical B by a radical-radical coupling pathway giving after protonation products 109.
 | ||
| Scheme 40 Decarboxylative coupling of arylacetic acids 103 with aldimines 108 under Ir complex 4 photocatalysis. | ||
Zhong, Zeng and co-workers114 applied the decarboxylative radical coupling of N-aryl amino acids with aldehydes105 to imines. In this case, a three-component reaction between aromatic amines, aldehydes or acetophenone and N-aryl glycines 6 using the Ir complex 4 as a PC, DMA as solvent under Ar provided 1,2-diamines 110 in modest to high yields (Scheme 41). Under the same blue LED irradiation and using Co(dmgH2)PyCl (16) as an oxidant, imidazolines 111 were obtained in moderate to good yields. These reactions were scaled-up for products 110a and 111a, using benzaldehyde, anisidine and N-phenyl glycine, which were obtained on a gram-scale with 79 and 55% yields, respectively. According to control experiments two plausible mechanisms were proposed. Under the former reaction conditions, N-phenyl glycine is reduced by Ir(III)* to produce Ir(II) and the α-amino radical A after CO2 elimination. The intermediate imine is reduced by Ir(II) to generate Ir(III) and the radical B, which undergoes a radical coupling reaction with A to form the diamine 110a. In the case of product 111a, Co(III) can be reduced by Ir(III) to generate Co(II), which can be reduced by radical A to Co(I) and the iminium ion C. Subsequent oxidation of Co(I) to Co(III) can be performed by coordination with H+ followed by H2 elimination. Diamine 110a can be cyclized by formaldehyde, which was generated by hydrolysis of iminium ion C in the presence of water, and the corresponding imidazoline 111a is then produced.
 | ||
| Scheme 41 Decarboxylative radical coupling of aldehydes, amines and N-aryl glycines under Ir complex 4 photocatalysis. | ||
The same group achieved the asymmetric synthesis of imidazolidines by a three-component process involving a decarboxylative radical coupling/cyclization reaction of N-aryl glycines 6, aldehydes and hydrazones 112 under copper and visible light-induced photoredox catalysis (Scheme 42).115 This process took place using Cu(OTf)2 and chiral bisoxazoline 113 as a ligand, 4CzIPN (25) as a PC, and Cs2CO3 as a base in THF at 0 °C to room temperature to furnish imidazolidines 114 with good yields and up to 95% ee. When this reaction was performed in the absence of an aldehyde, chiral diamines116115 can be obtained with high yields and enantioselectivities. In the proposed reaction mechanism intermediate A is formed by the ligand exchange between the hydrazone 112a and the chiral copper catalyst [L-Cu(II)], which has been reduced to radical B by a SET of PC−˙. Subsequently, radical B reacts with the α-amino carbonyl radical C to produce D, which after protonation and ligand exchange gives rise to diamine 115 and regenerates intermediate A. Diamine 115a can be cyclized with the aldehyde to give imidazolidine 114a.
 | ||
| Scheme 42 Decarboxylative asymmetric coupling of N-aryl glycines and hydrazones 112 under Cu/4CzIPN photocatalysis. | ||
Recently, an iron catalyzed decarboxylative radical addition of N-indole acetic acid and related compounds 116 or carboxylic acids to chiral azomethine imines 117117 upon visible light irradiation was reported.118 In the presence of Fe2(SO4)3 (10 mol%) and Cs2CO3 in DMSO at 30 °C under blue LED irradiation the corresponding adducts 118 or 119 were obtained with good yields and diastereoselectivities (Scheme 43). In the proposed catalytic cycle, a LMCT process54,55 occurs upon irradiation of the in situ formed iron(III) carboxylate A with subsequent decarboxylation to provide radical R˙ (B), followed by addition of this radical B to the less shielded face of the chiral azomethine imine 117via model I. The generated Fe(II) would be re-oxidized into Fe(III) during the facile reduction of cation intermediate C, or the amidyl C′ derived thereof. Final protonation gives the addition product 118 or 119.
 | ||
| Scheme 43 Decarboxylative radical addition of carboxylic acids to chiral azomethine imines 117 under Fe2(SO4)3 photocatalysis. | ||
Spiro-imidazolidines 121 have been synthesized by a four-component method under carbon nitride photocatalysis.119 Starting from primary amines, cyclic ketones, N-aryl glycines 6 and aldehydes in dry dichloromethane at room temperature under visible light mediated photocatalysis, the corresponding products 121 were isolated with moderate to good yields (Scheme 44). The heterocatalyst carbon nitride 1.0 Ci–C3N4120 was prepared from melamine and glyoxal and can be recovered and recycled in multiple runs without loss of activity and was used in a gram-scale synthesis in a continuous photo flow fashion. In the proposed mechanism an α-amino radical intermediate A is formed from N-phenyl glycine. In the meantime, primary amine and ketone condensed to generate the iminium ion species B, which undergoes a free radical addition to give the radical adduct C. Subsequent reduction of C provides diamine D, which by reaction with formaldehyde gives rise to the corresponding spiro-imidazolidine 121a.
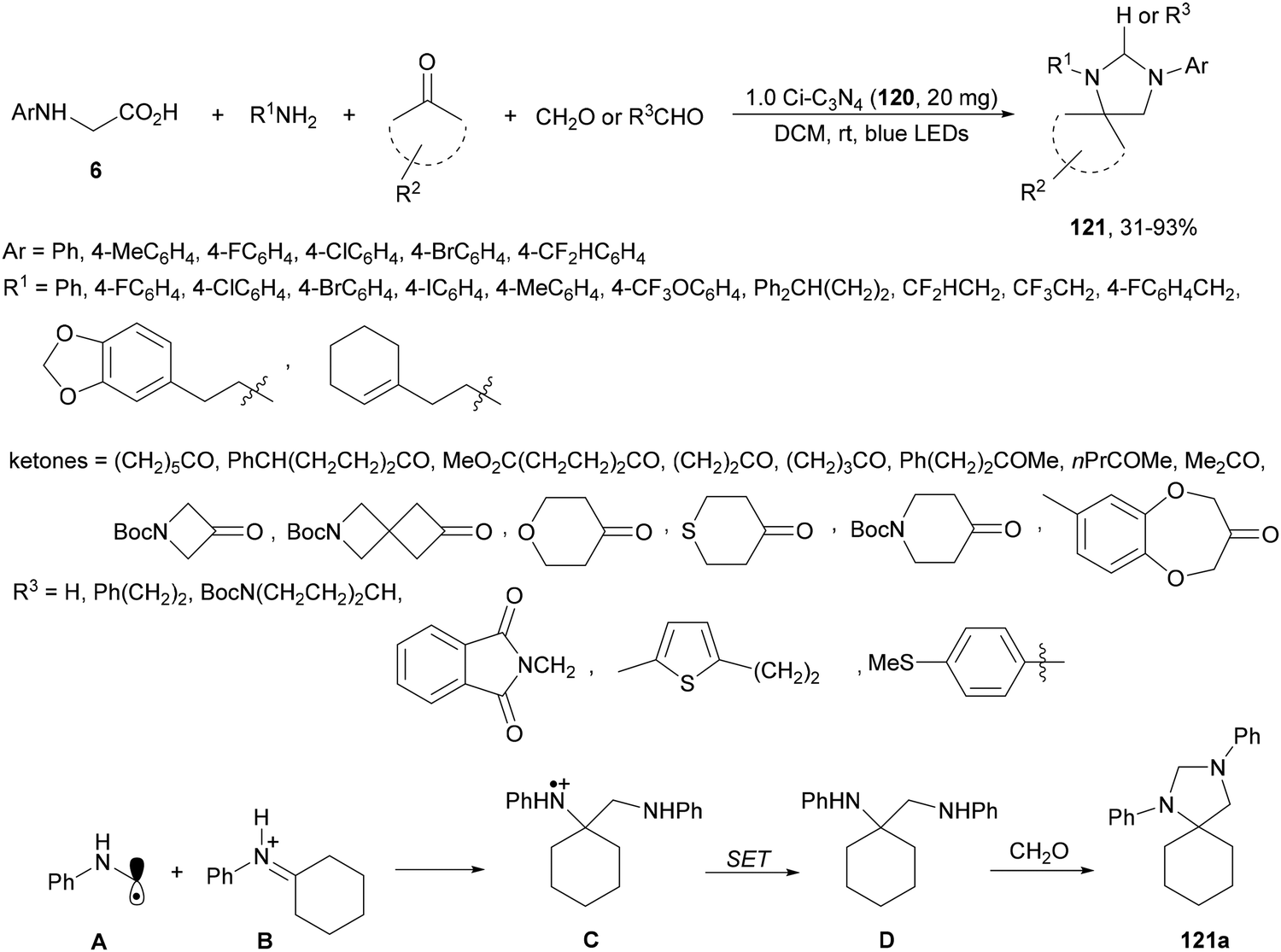 | ||
| Scheme 44 Decarboxylative four-component reaction of N-aryl glycines, amines, ketones and aldehydes under carbon nitride 120 photocatalysis. | ||
Hong and co-workers120 employed 4CzIPN (25) as an organic PC for the decarboxylative radical addition of the 2,2-diethoxyacetic acid 26 derived radical to imines 108 using (NH4)2S2O8 as an oxidant in catalytic amounts under visible-light irradiation (Scheme 45). α-Amino acetals 122 were obtained in the presence of Cs2CO3 as a base in DMSO at room temperature with modest to good yields. In the proposed mechanism, the excited state of 4CzIPN* undergoes reductive quenching by (EtO)2CHCO2− (A), which after decarboxylation forms a diethoxymethyl radical B. Addition of B to imine 108a gives the nitrogen centered radical C, which by reduction and protonation affords product 122a.
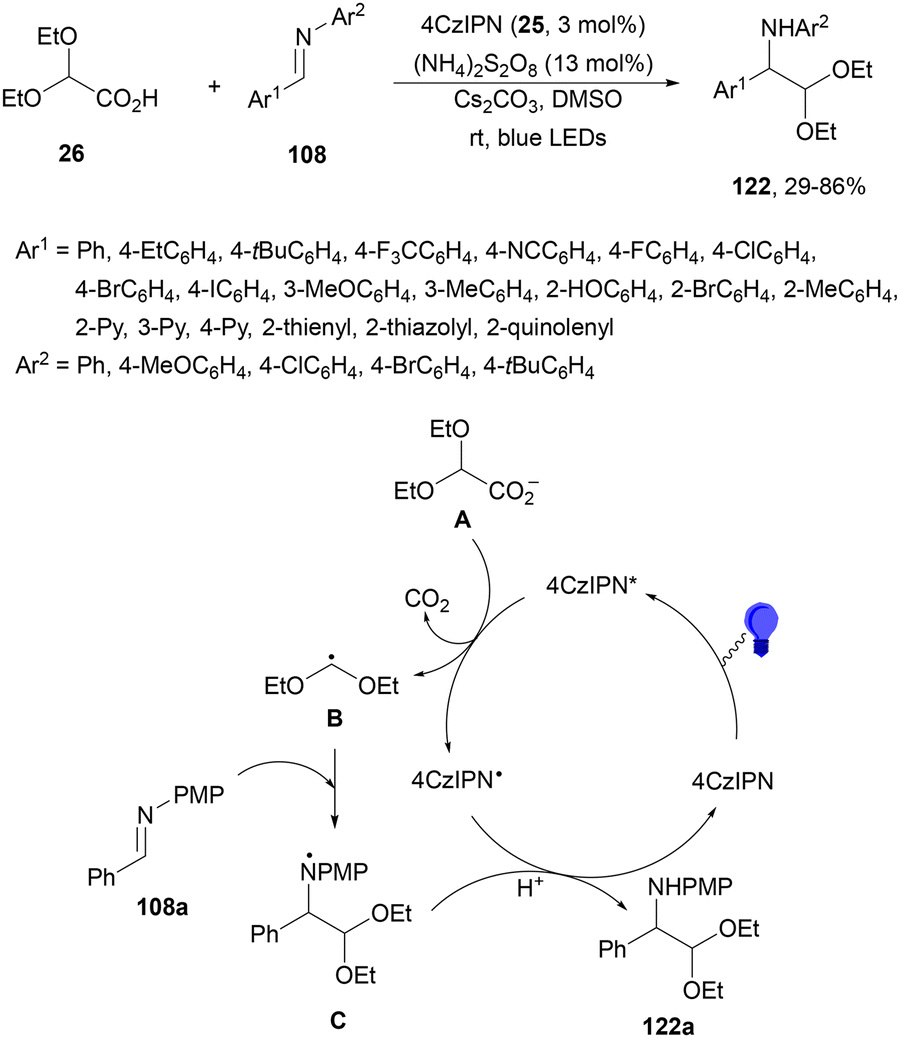 | ||
| Scheme 45 Decarboxylative radical addition of 2,2-diethoxyacetic acid 26 to aldimines 108 under 4CzIPN (25) photocatalysis. | ||
Dual acridine 123 and tetra-n-butylammonium decatungstate (TBADT) photocatalysis have been employed in the radical addition of alkyl carboxylic acids with in situ generated imines from aldehydes and aromatic amines.121 This three-component reaction allowed the preparation of amines 124 in the presence of methanesulfonic acid (MsOH) and MeCN under 400 nm LED irradiation (Scheme 46). The role of TBADT is to facilitate the turnover of acridine PC by means of hydrogen atom transfer. In the case of using a stoichiometric amount of MsOH, it is for the generation of iminium species, which are more susceptible to radical addition. Other azomethine substrates such as hydrazones and nitrones were successfully alkylated.
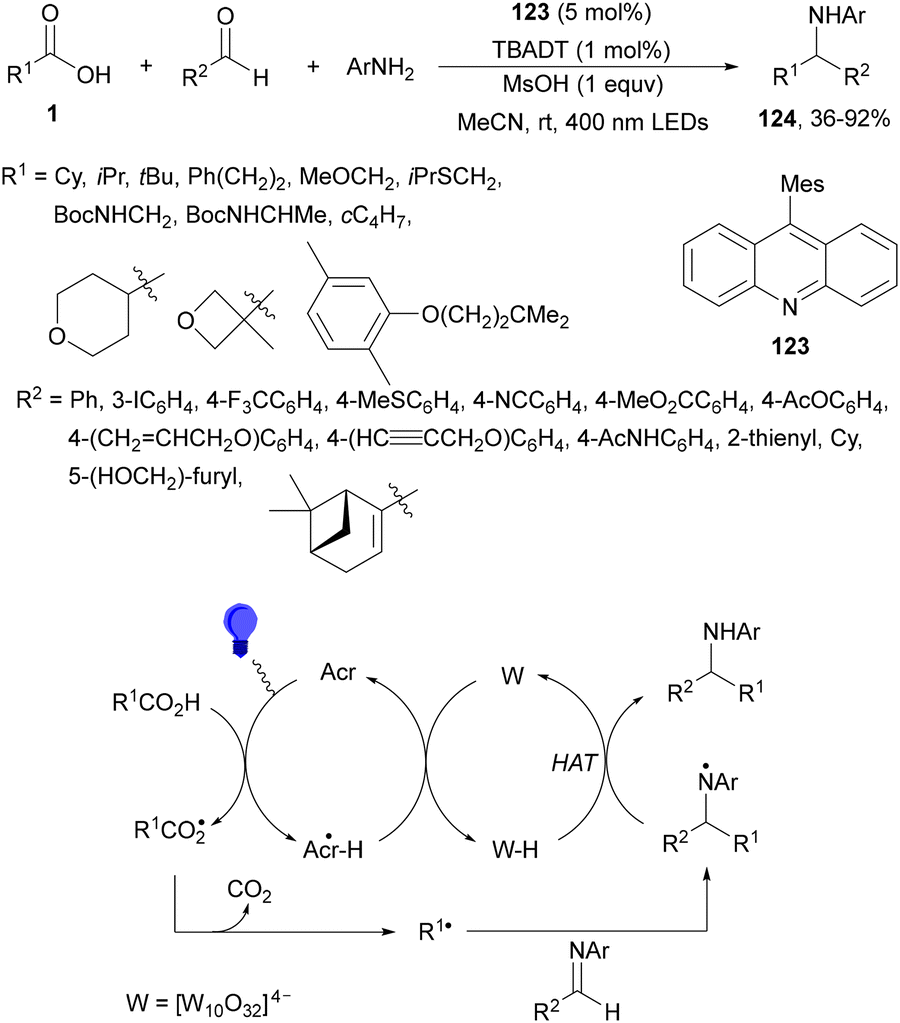 | ||
| Scheme 46 Decarboxylative radical addition of carboxylic acids to in situ generated imines under acridine 123/TBADT photocatalysis. | ||
A similar three-component transformation has been described by Larionov and co-workers122 using acridine 125 (8 mol%) (Fig. 2), Cu(MeCN)4BF4 (8 mol%), TsOH (8 mol%) and MeCN as solvent. The corresponding amines 124 were obtained in high yields (63–98%) employing secondary and tertiary carboxylic acids.
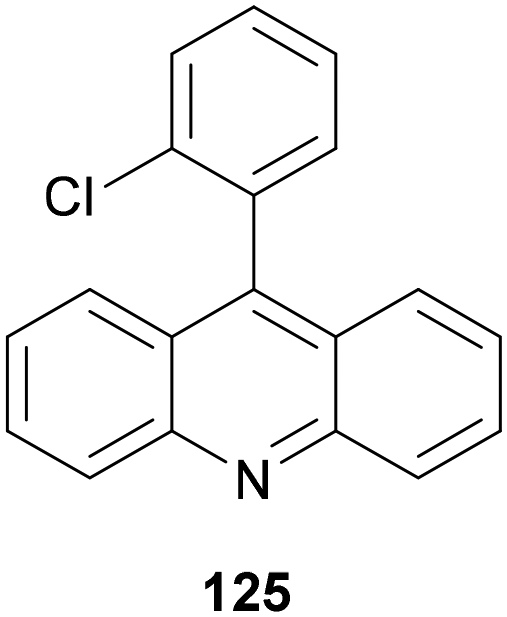 | ||
| Fig. 2 Acridine 125 used as a PC in the three-component reaction of carboxylic acids, aldehydes and amines. | ||
Reductive coupling of imines 108 with NHPI esters 69 has been achieved using rose Bengal (RB) as a photocatalyst by Rueping and co-workers.123 Working with Li2CO3 and DIPEA in aqueous EtOAc at room temperature under green LED irradiation, the corresponding amines 109 were obtained in good yields (Scheme 47).
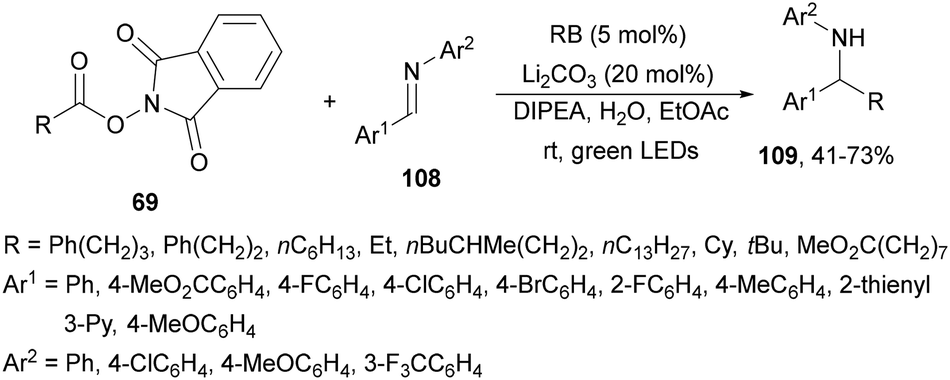 | ||
| Scheme 47 Decarboxylative coupling of NHPI esters 69 with aldimines 108 under rose Bengal photocatalysis. | ||
Dilman and co-workers124 reported the one-pot transformation of aldehydes to ketones by preliminary formation of aldimines, which reacted with alkyl radicals generated from NHPI esters 69 under photoredox conditions (Scheme 48). Starting from aldehydes in the presence of n-propylamine followed by addition of TMSCl in methanol, TEBACl and 4CzIPN (25) as a PC in DCM under blues LED irradiation, the corresponding ketones were obtained in 30–91% yields (Method A). On the other hand, starting from N-methyl imines under the same reaction conditions the resulting ketones were isolated in 64–86% yields (Method B). These procedures worked with NHPI esters to generate secondary and tertiary radicals. However, with esters 69 leading to less stable radicals such as primary or cyclopropyl, MeSO3H in acetonitrile was used instead of MeOH for the activation of imines to provide ketones in 32–96% yields (Method C). In the proposed mechanism, iminium ion A reacts with radical R1˙ giving radical cation B. Loss of proton in B leads to the formation of a carbon centered imine precursor of ketones after acidic work-up with 1 M HCl in EtOH.
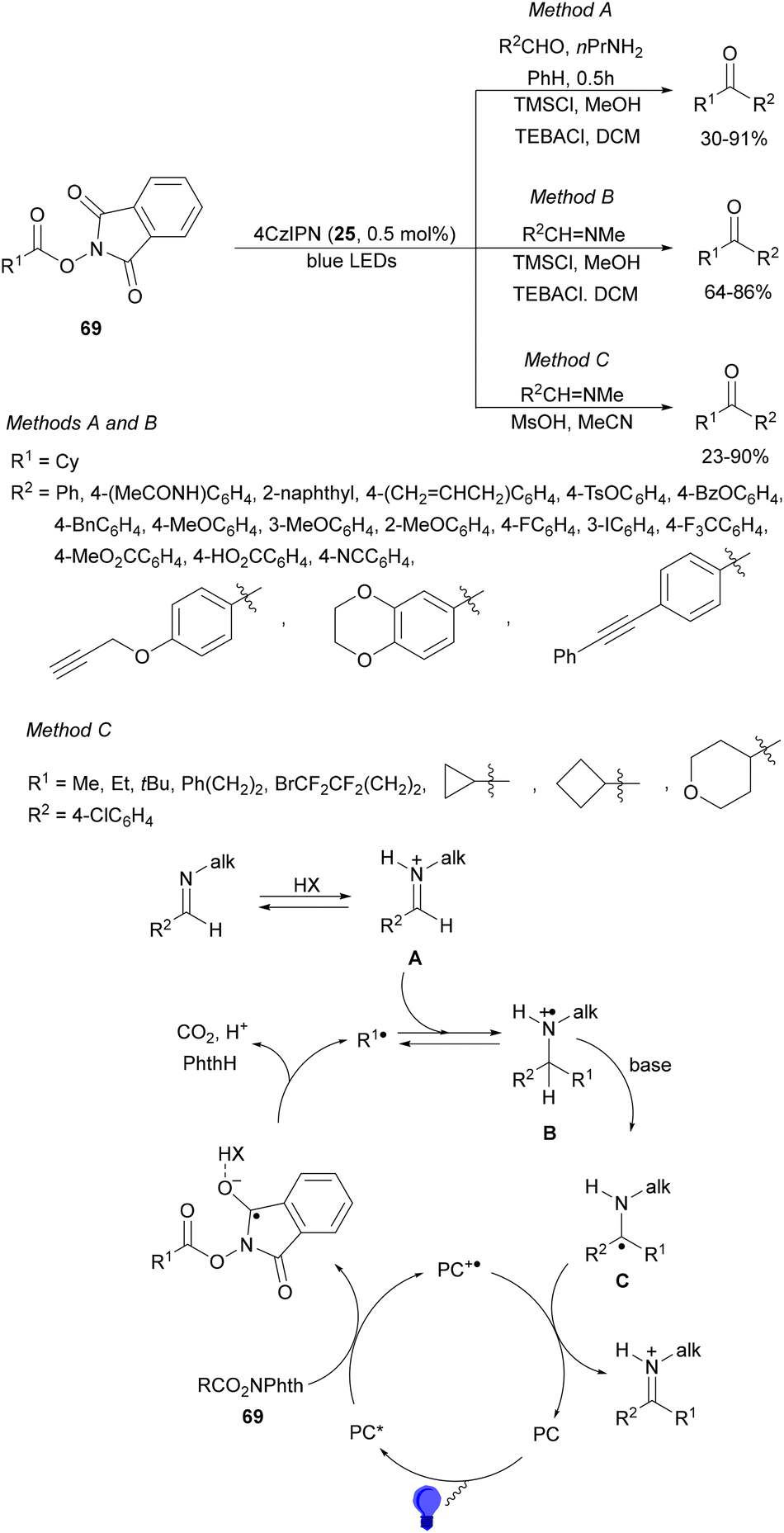 | ||
| Scheme 48 Decarboxylative radical addition of NHPI esters 69 to imines under 4CzIPN (25) photocatalysis. | ||
Glyoxylic oxime esters 126 underwent radical coupling with α-AA derivatives under photoredox decarboxylative conditions using acridinium salt 34 as a photocatalyst (Scheme 49).125 Cyclic and chain AAs, dipeptides and aliphatic acids were used as alkyl radical sources to provide α,β-diamino esters 127, multi-amino esters 128 and α-AA derivatives 129, respectively. Two possible mechanisms have been proposed: (a) the addition of radical A, formed from the acid after decarboxylation, to glyoxylic oxime ester 126 and (b) the coupling of radical A with B, which was reduced by Acr−˙* to give anion C, which after protonation afforded product 127a.
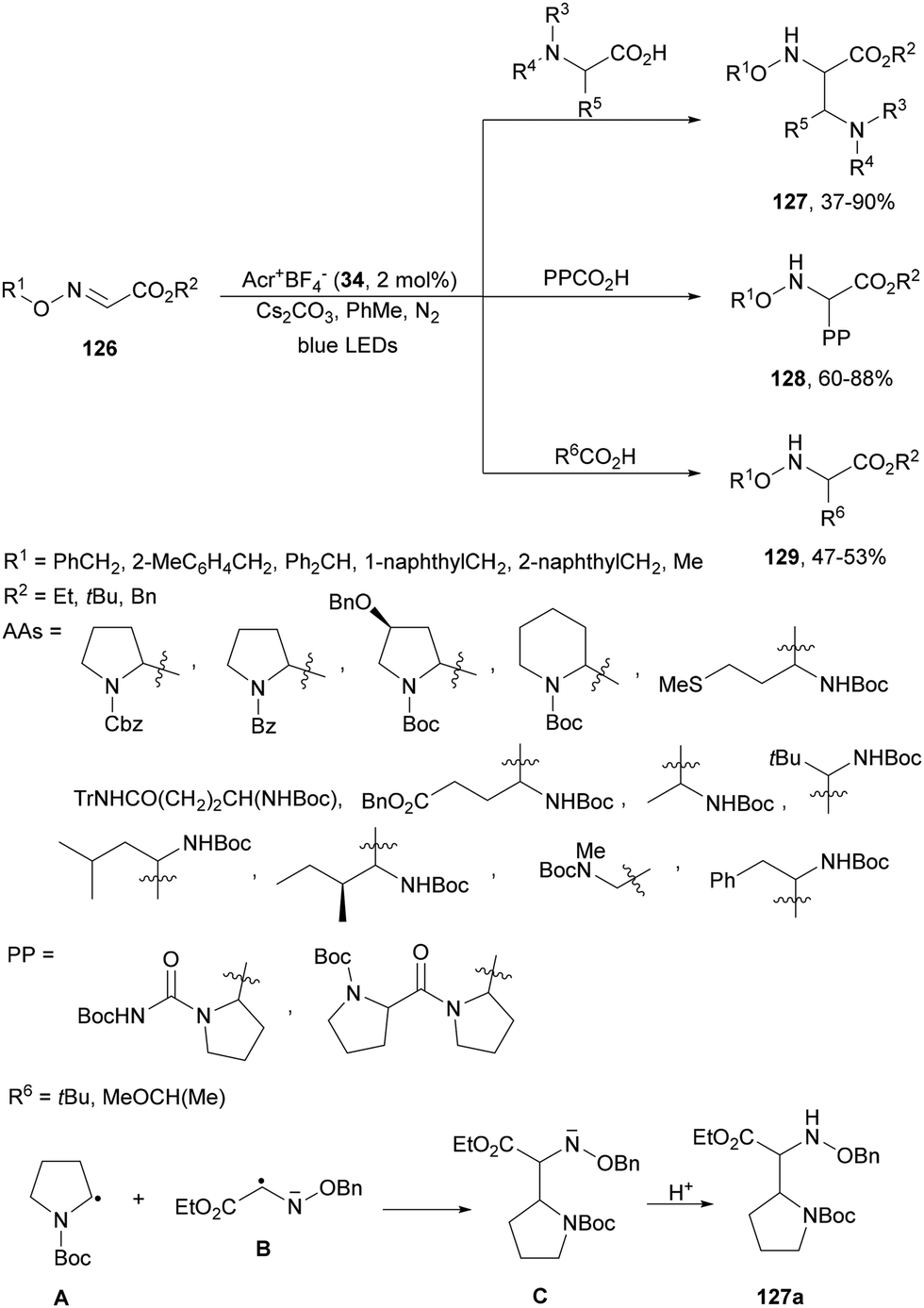 | ||
| Scheme 49 Decarboxylative coupling of glyoxylic oxime esters 126 with AAs, dipeptides and carboxylic acids under acridinium 34 photocatalysis. | ||
Mariano, Wang and co-workers126 performed the synthesis of C-glycosyl amino acids 132 by reaction of imines 108 and imino esters 131 with RAE of saccharides 130. This visible-light-promoted coupling took place in the absence of PC, using HE and DIPEA·HBF4 in acetonitrile at room temperature to form products 132 in moderate to good yields (Scheme 50). Pentoses and hexoses preserved the configuration of the anomeric carbon atom. In the proposed mechanism, HE in acetonitrile absorbs in the visible region to give excited HE*, which serves as a SET donor to glycosyl N-hydroxydichlorophthalimide (TCNHPI) ester 130a to provide after decarboxylation intermediate A and HE+˙. Radical A reacts with protonated imino ester (131H+BF4−) to form radical B and HBF4. Subsequent HAT of B by HE+˙ releases product 132 and the pyridine derivative (HP) of HE.
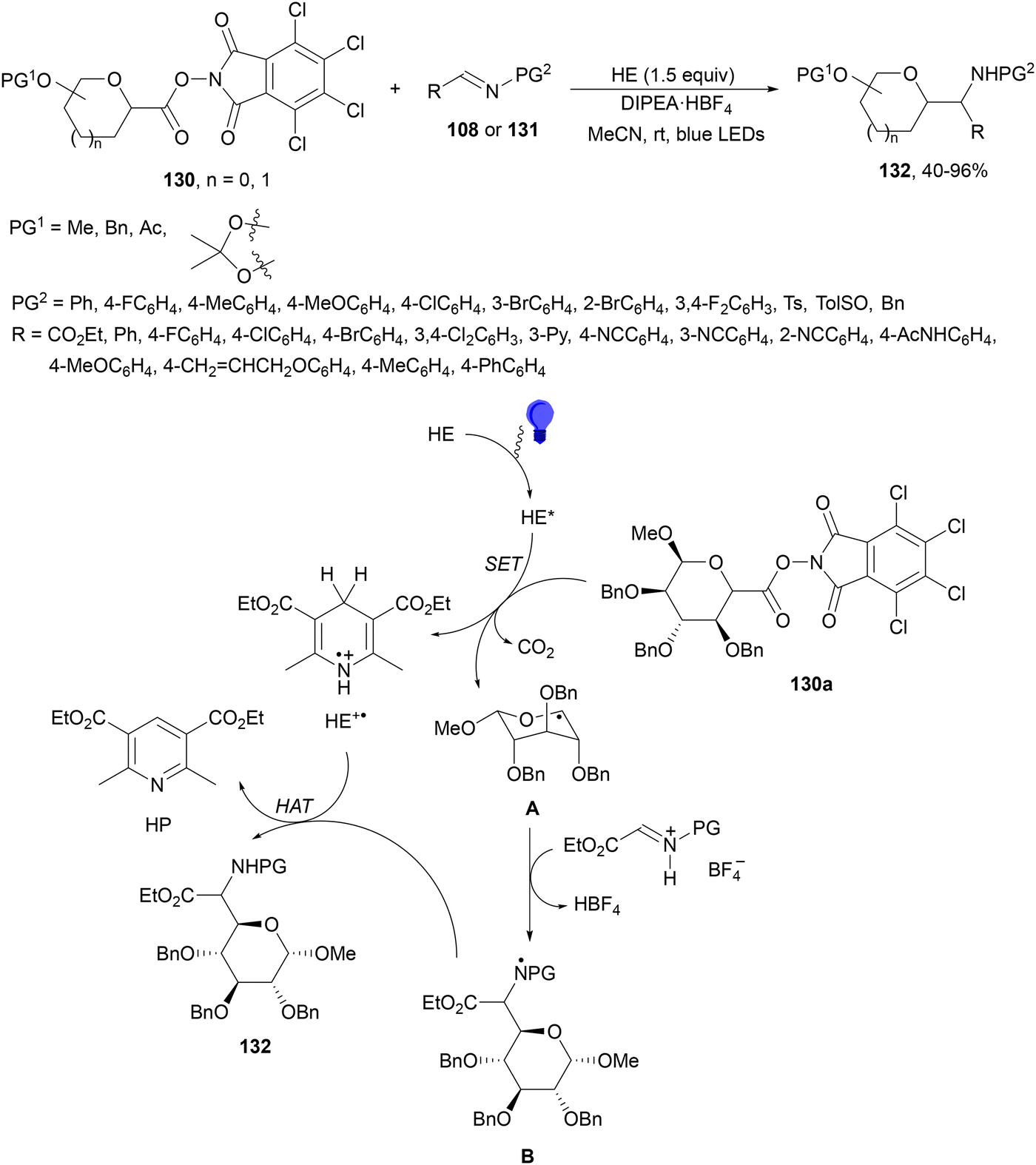 | ||
| Scheme 50 Decarboxylative radical addition of redox-active esters of saccharides 130 to imines 108 and imino esters 131 using Hantzsch ester under visible light. | ||
Photoredox radical alkylation of glyoxylic ester-derived hydrazones 133 with NHPI esters 69 has been carried out by Shen and co-workers.127 Under visible-light irradiation, in the presence of Ru(bpy)3Cl2 and HE using K2CO3 as a base in DCM at room temperature, α-amino ester derivatives 134 were obtained with high yields (Scheme 51). In addition to hydrazones 133, O-benzyl oximes and N-benzoyl hydrazones can be employed as radical acceptors. With respect to NHPI esters, the oleanolic acid derivative was obtained in 98% yield and the chenodeoxycholic acid derivative was obtained in 82% and 81% yields, respectively. Experimental studies suggest that the N-centered radical B after decarboxylation of radical A and addition to 133 would abstract a hydrogen atom from HE+˙ species. DFT calculations also suggest that K2CO3 has important beneficial functions in stabilizing HE intermediates facilitating HAT processes. Therefore, [HE+*–CO3]K has been postulated to produce product 134.
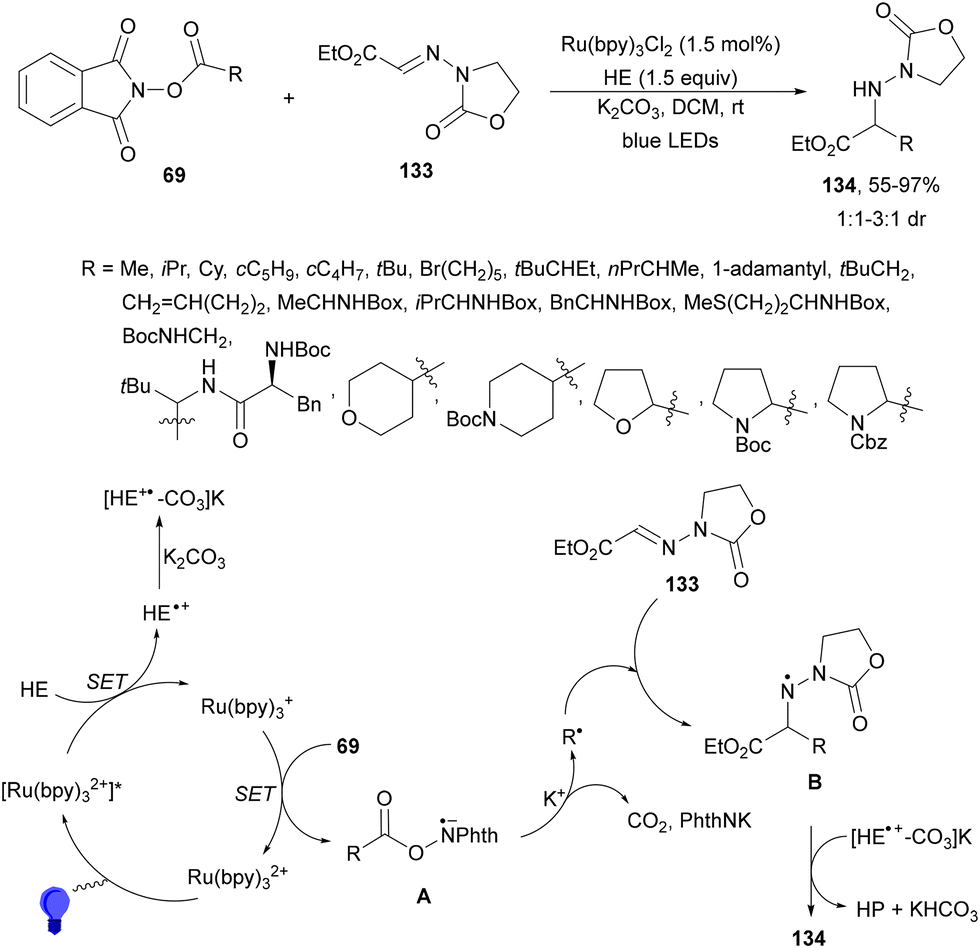 | ||
| Scheme 51 Decarboxylative addition of NHPI esters 69 to glyoxylic ester-derived hydrazones 133 under Ru(bpy)3Cl2 photocatalysis. | ||
Kärkäs and co-workers128 reported the synthesis of amino acid derivatives 137 by reaction of chiral glyoxylate derived sulfinyl imines 135 with carboxylic acids through photoredox catalysis with an acridinium-based PC 136 (Scheme 52). As in the case of Alemán's group112 (Scheme 39) this process allows the asymmetric synthesis of amines but as α-amino acid derivative 137 with modest to high yields and diastereoselectivities (>95:5 dr). In the proposed mechanism the radical from the carboxylic acid decarboxylation adds to the N-sulfinyl imine 135.
 | ||
| Scheme 52 Decarboxylative asymmetric radical addition of carboxylic acids to chiral N-sulfinyl imines 135 under [Mes-Me2Acr-Ph]BF4 (136) photocatalysis. | ||
Noël and co-workers129 reported recently a decarboxylative cross-coupling of NHPI esters 69 derived from α-AAs and sulfonyl hydrazones 138 (Scheme 53a). This process took place with ethyl glyoxylate-derived 4-trifluoromethylphenyl sulfonyl hydrazones 138 as the radical acceptor HE as a reductive quencher and disodium Eosin Y (EYNa2, 139) as a PC under blue LED irradiation. The obtained adducts were subjected to cleavage conditions in ethanol providing esters 140. This approach can be considered as a C1 homologation of carboxylic esters, a safer alternative to the traditional Arndt–Eistert reaction, and has been extended to peptides on a solid phase. In addition, aromatic and aliphatic N-sulfonyl hydrazones 141 were transformed into β-aryl ethylamines 142 with good yields (Scheme 53b). Mechanistic investigation suggests that excited EYNa2* is reductively quenched by the sacrificial electron donor HE to generate HE+˙. Subsequently, the NHPI ester is reduced by EYNa2−˙ giving the alkyl radical, after decarboxylation. Addition of R˙ to the sulfonyl hydrazone 141 provides the hydrazinyl radical A, which after HAT from HE+˙ or neutral HE generates the product and the pyridinium B.
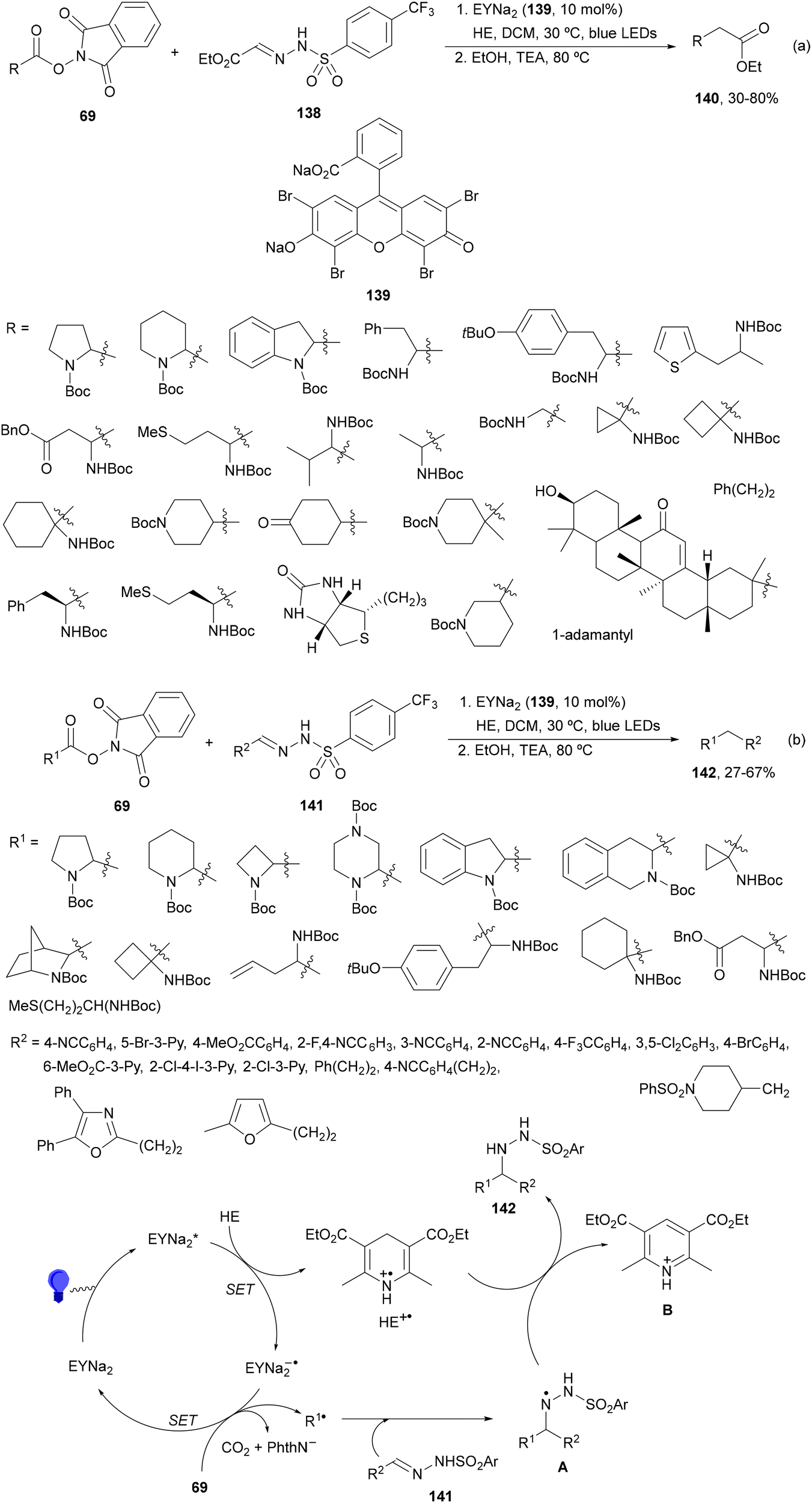 | ||
| Scheme 53 Decarboxylative radical addition of NHPI esters 69 to sulfonyl hydrazones 138 (a) and 141 (b) under Eosin Y (139) photocatalysis. | ||
N-Acyl imines have been generated in situ by decarboxylation of N-acyl α-amino acid derived NHPI esters 143 under visible-light photoredox conditions.130 By merging Ir complex 4 and chiral phosphate 145, NHPI esters 143 reacted with indoles 144 to form 3-substituted indoles 146 in up to 96% yield and up to 97% ee (Scheme 54). This asymmetric Friedel-Crafts reaction could take place by generation of an α-aminoalkyl radical A and Ir(III). Irradiation of Ir(III) with blue LEDs forms Ir(III)*, which oxidizes A to N-acyl imine B. The chiral phosphate acts as a bifunctional catalyst by activation of the N-acyl imine and hydrogen bonding with indole favoring indole attack on the Si face of the imine to afford 146 with R configuration. The removal of the N-acyl group from products 146 can be achieved by treatment with the Schwartz reagent (Cp2ZrHCl).131
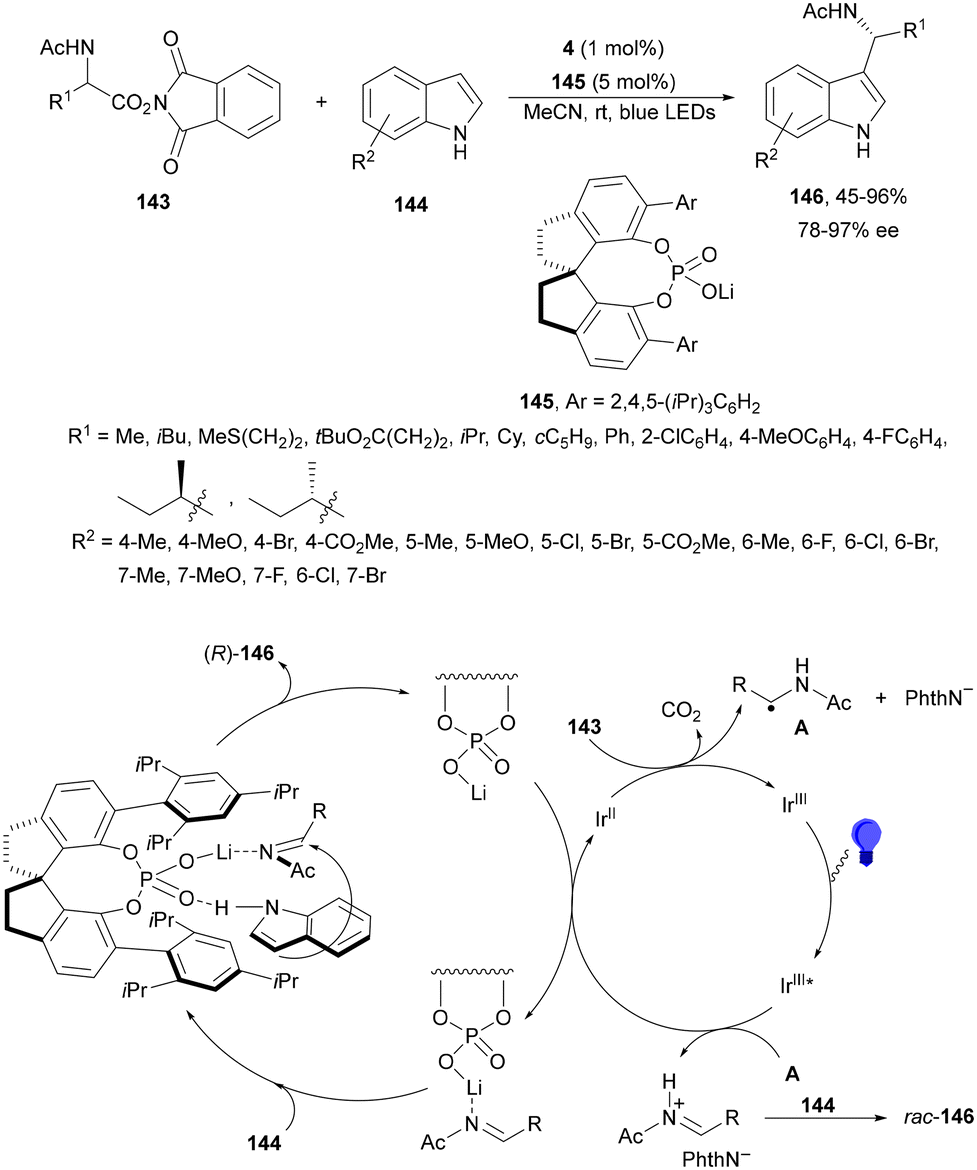 | ||
| Scheme 54 Decarboxylative generation of N-acyl imines from N-acyl α-amino acid derivatives NHPI esters 143 and enantioselective addition of indoles 144 under Ir 4 and chiral phosphate 145 photocatalysis. | ||
Maruoka and co-workers132 employed N-protected α-amino acid derived NHPI esters 143 as precursors of N-protected imines. In the presence of ester-stabilized phosphonium ylides 147, Ir(ppy)3 (89) as a PC and ZnCl2 as a Lewis acid under blue LED irradiation provided β-amino phosphonium ylides 148 with good yields (Scheme 55). These products 148 were further subjected to in situ photocatalytic transformations such as: (a) reduction with ascorbic acid or deuteration with trifluoroacetic acid in MeCN/D2O to form β-AAs 149 or 150 in high yields; (b) alkylation reaction with alkenes giving products 151 with modest diastereoselectivity; (c) (hetero)arylation by using electron-rich hetero(arenes) and HBF4 as Brønsted acid, resulting in product 152 with moderate dr; and (d) Wittig olefination without isolation of 148 with formaldehyde afforded products 153 with moderate to good yields. Concerning the first decarbonylative photoredox reaction, in the proposed mechanism the N-acyl iminium intermediate B is generated from 143via a radical-polar crossover process of the amino radical A. The role of ZnCl2 is the capture of the phthalimide anion by complexation and also accelerating the formation of a C–C bond by coordination of B, forming intermediate C, which after reaction with 147 facilitates the proton transfer from the α-position of the phosphonium ion to the nitrogen atom providing ylide 148.
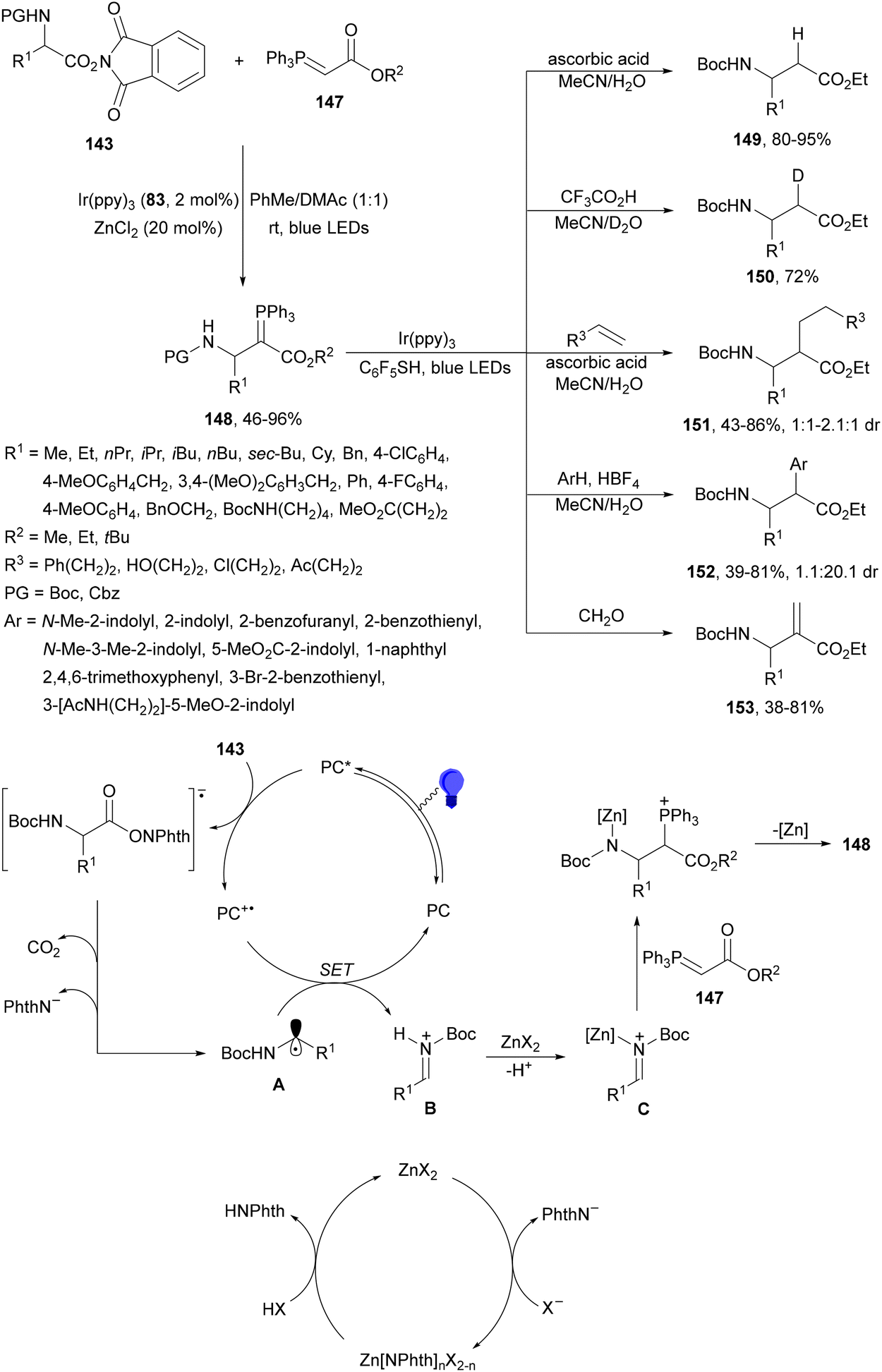 | ||
| Scheme 55 Decarboxylative generation of N-protected imines from NHPI esters 143 and addition of phosphonium ylides 147 under Ir(ppy)3 (89) photocatalysis. | ||
Seidel and co-workers133 recently reported the decarboxylative alkylation of cyclic imine-BF3154 with carboxylic acids under acridine (154)-copper dual catalysis to provide α-alkyl substituted azacycles 156 with good yields (Scheme 56a). This addition was carried out using Cu(OTf)2·toluene and acridine 155 in DCM at room temperature or in DCE at 70 °C when primary radicals are involved. In the proposed catalytic cycle, acridine 155 forms a photoactive H-bonded complex with carboxylic acid 1, which after excitation with purple light (395 nm) undergoes decarboxylation to the alkyl radical and acridinyl radical 155H˙. The alkyl radical A reacts with 154a in the presence of Cu(II) species to provide intermediate B. Acridinyl radical 155H˙ is oxidized to 155H+ by Cu(II) species LnCu(OTf)2. Protonation of B by 155H+ furnished complex 156H+-BF3 regenerating acridine 155. A three-component reaction of complexes 154, [1.1.1]propelane (157) and carboxylic acids under similar reaction conditions, in the absence of Cu(II), afforded products 158 in good yields (Scheme 56b). In this case, the initially formed radical A reacts with [1.1.1]propelane (157) to give radical C, which after addition to 154 leads to aminyl radical D. Acridinyl radical 155H˙ reduces aminyl radical D to generate intermediate E and acridinium 155H+. Finally, protonation of E provides product–BF3 complex 158H+–BF3.
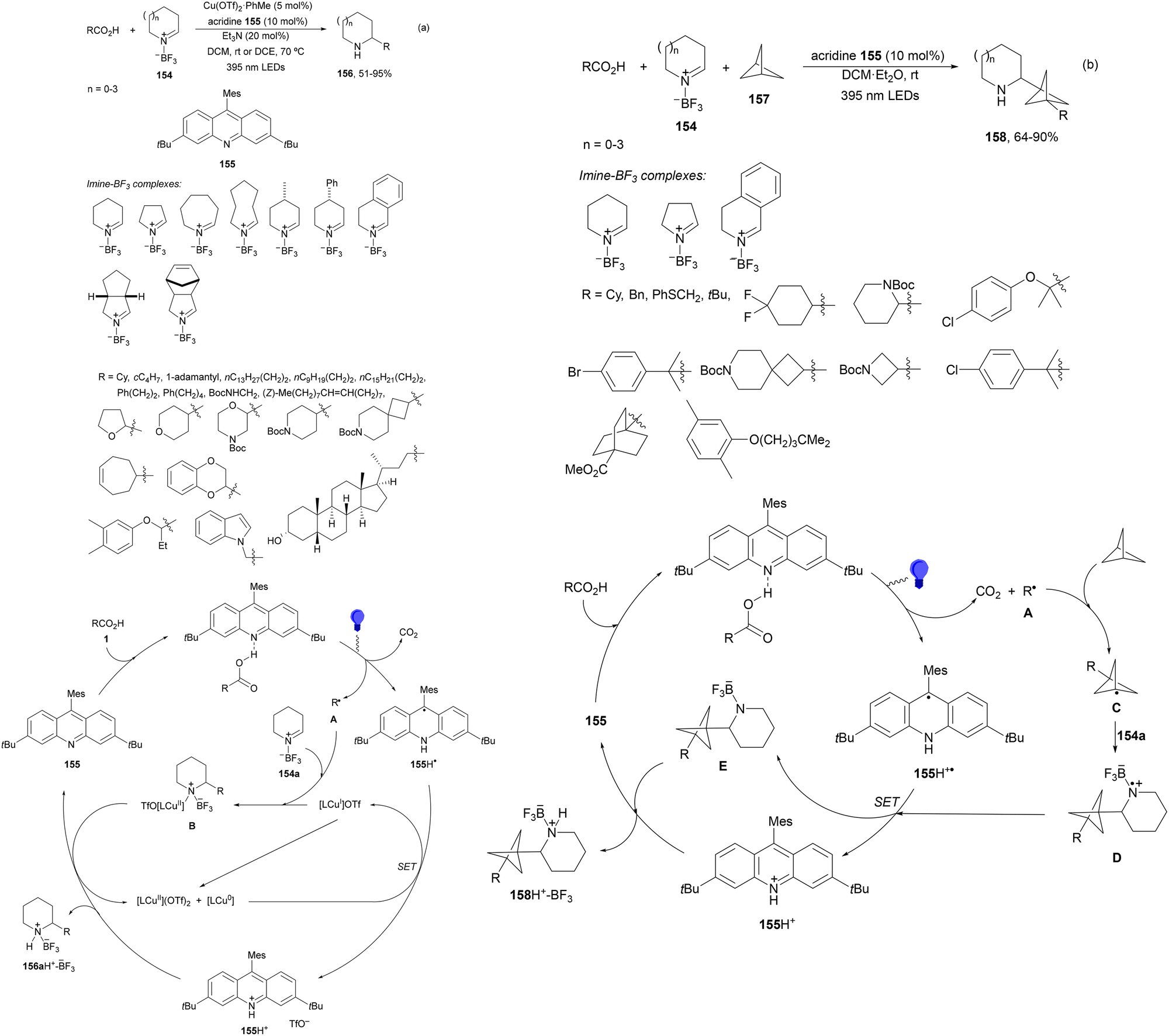 | ||
| Scheme 56 Decarboxylative radical alkylation of cyclic imine-BF3 complexes 154 with carboxylic acids (a) and propellane (b) 157 under acridine 155 photocatalysis. | ||
Decarboxylative addition of acid derived radicals to nitrones was previously described by Zheng, Huang and co-workers134 earlier than Dilman's group.121 In this case, α-AAs were used as α-aminoalkyl radical precursors and acyclic 159 and cyclic nitrones 160 as radical acceptors to provide acyclic 161 and cyclic 162 β-amino hydroxylamines with good yields and moderate to good diastereoselectivities (Scheme 57a). This process took place using Ir complex 4 as a PC and Li2CO3 as a base in DMF under blue LED irradiation at room temperature. The antihistamine drug mepyramine was prepared from nitrone 159a and N,N-dimethylglycine followed by N–O bond cleavage of 161a with Zn and Pd-catalyzed N-alkylation with 2-chloropyridine (Scheme 57b).
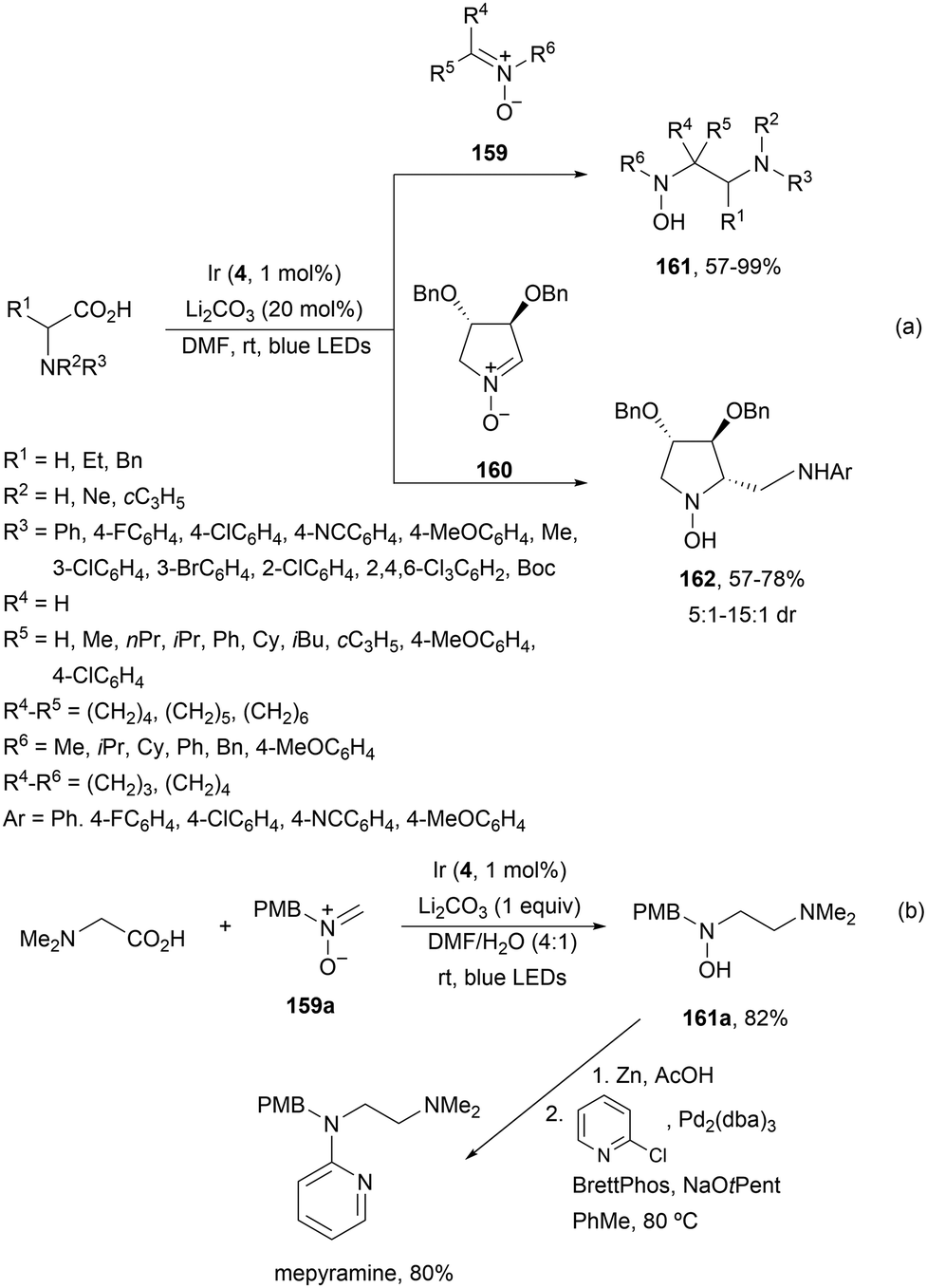 | ||
| Scheme 57 Decarboxylative radical addition to nitrones 159 and 160 of radicals from α-AAs under Ir photocatalysis (a). Synthesis of mepyramine (b). | ||
Aryl isocyanates 162 can be transformed into amides 163 by a Ce(III)-photocatalyzed addition of radicals from carboxylic acids. Primary, secondary and tertiary aliphatic carboxylic acids reacted with isocyanates 162 using catalytic amounts of CeCl3 and TBACl in MeCN at room temperature under blue LED irradiation (Scheme 58).135 A wide range of amides were obtained with up to 93% yield and on a gram scale. In the proposed mechanism, CeCl3 was oxidized by isocyanate 162 to a Ce(IV) complex, which underwent a photoinduced ligand-to-metal charge-transfer (LMCT)54,55 to generate the Ce(III) complex and Cl˙. The reaction of Cl˙ with the carboxylic acid forms radical A and HCl. Decarboxylation of A gives the alkyl radical B, which was then added to isocyanate 162 to provide radical C. After reduction of radical C by the Ce(III) complex, anion D and the Ce(III) complex are formed. Additionally, the coupling of radical anion E and radical A could also produce anion D. Finally, protonation of anion D gives product 163. Moreover, the HAT process between radical D and carboxylic acid 1 to give amide 163 cannot be ruled out. When the reaction was carried out in darkness at higher temperatures the yield increased smoothly with the temperature. Therefore, the thermal reaction pathway should coexist, but the radical addition pathway should be the main pathway.
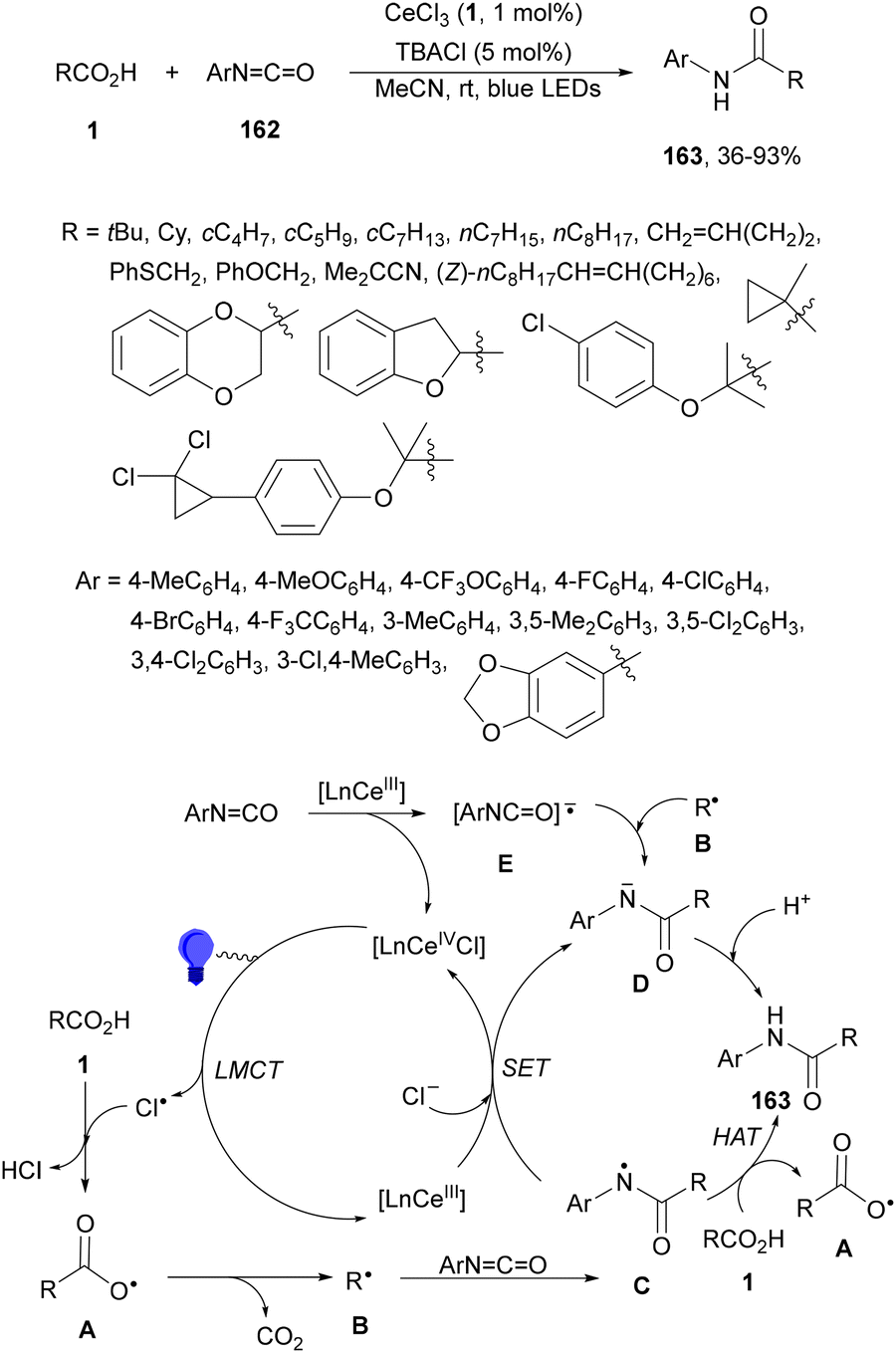 | ||
| Scheme 58 Decarboxylative radical addition of carboxylic acids to isocyanates 162 under Ce(III)-photocatalysis. | ||
Cross-coupling of arylacetic acids and α-AAs with imines or in situ generated imines takes place using Ir complex 4 as a PC to give amines and 1,2-diamines, respectively. When organic PCs, such as 4CzIPN or acridine, are used as PCs an additional oxidant must be used. However, active NHPI esters react with imines using rose Bengal or 4CzIPN through a radical addition mechanism. Imine derivatives of glycosylic esters react with carboxylic acids to give α-AA derivatives and with α-AAs to provide α,β-diamino acids using acridinium salts as PCs. In the case of NHPI esters α-AA derivatives are formed by addition reaction under Ru(bpy)3Cl2 catalysis and Hantzsch ester (HE) as a radical acceptor or with disodium Eosin Y as a PC. N-Protected α-amino NHPI esters generate N-protected imines under Ir catalysis, which are able to react with indoles in the presence of chiral phosphate to provide chiral 3-substituted indoles. When these NHPI esters are allowed to react with phosphonium ylides β-amino phosphonium ylides are formed, which can be further transformed in a one-pot process into β-amino ester derivatives. Other radical acceptors such as cyclic imine-BF3, nitrones and isocyanates react with carboxylic acids to provide cyclic amines, diamines and amides, respectively.
2.5. Arylation reactions
C(sp2)–C(sp3) cross-coupling of aromatic halides with aliphatic carboxylic acids or derivatives by photoredox decarboxylation processes is an excellent alternative to transition metal-catalyzed cross-coupling reactions. Metallaphotoredox decarboxylation arylation was reported by MacMillan and Doyle in 2014 using NiCl2 and Ir complexes as PCs.136 This nickel/photoredox dual catalysis is a general tool for the cross-coupling of carboxylic acids and aromatic electrophiles.137 It has been found that the presence of oxygen is important for catalyst activation when air-stable Ni(II) precatalysts were used to promote rapid reduction of Ni(II) to Ni(0) by Ir(II).138 A wide range of secondary, benzylic, α-amino and α-oxy carboxylic acids reacted with aryl iodides, bromides and chlorides under metallaphotoredox conditions. Notably, an enantioselective arylation of α-AAs by employing bis-oxazoline 164 as a chiral ligand was reported by MacMillan, Fu and co-workers.139 Starting from racemic α-AAs and aryl bromides an enantioconvergent140 arylation provided chiral benzylic amines 165 with good yields and up to 93% ee (Scheme 59). | ||
| Scheme 59 Enantioconvergent decarboxylative arylation of AAs under Ir/Ni photocatalysis. | ||
Luo and Zhang141 employed 4CzIPN (25) instead of Ir complexes as a PC for the cross-coupling of AAs with aryl halides via Ni/photoredox dual catalysis. Under this class of catalysis an enantioconvergent140 cross-coupling of α-heterocyclic carboxylic acids 166 with aryl bromides has been described (Scheme 60).142 In the presence of catalytic amounts of 4CzIPN (25), NiBr2·DME and chiral ligand 164, with Cs2CO3 as a base in acetone under blue LED irradiation the corresponding N-benzyl heterocycles 167 were obtained in modest to good yields. The presence of a directing group at the C2 position in the heterocyclic carboxylic acid increased the enantioselectivity from up to 58% ee to 88% ee. Hypothetical chelation of the alkyl radical to Ni in intermediate A increasing the rigidity of the complex has been postulated.
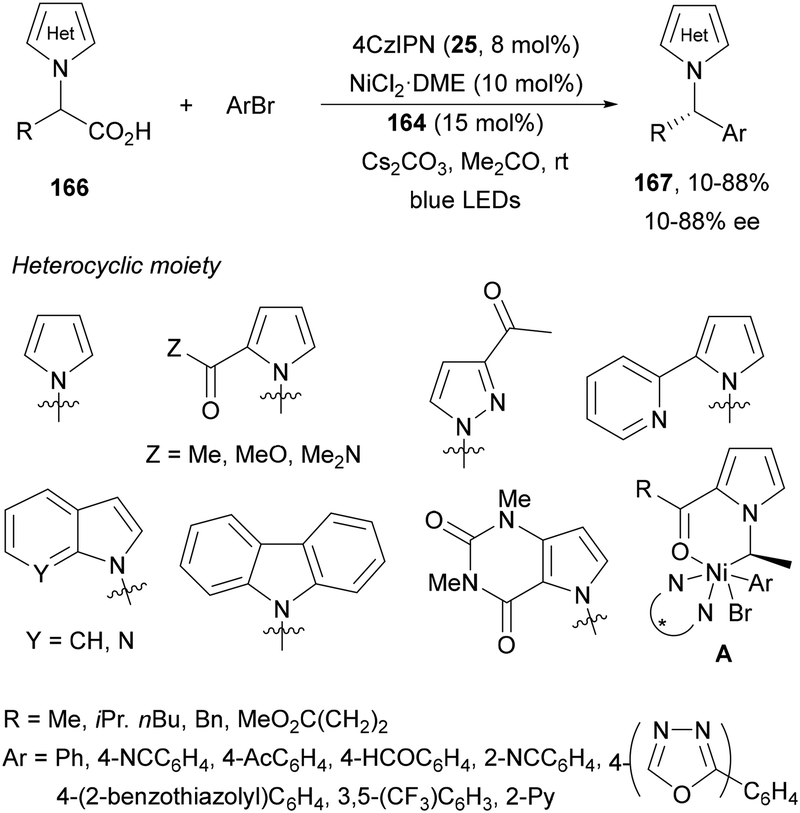 | ||
| Scheme 60 Enantioconvergent decarboxylative arylation of heterocyclic carboxylic acids 166 under 4CzIPN (25)/Ni photocatalysis. | ||
Recently, Itami and co-workers143 reported the cross-coupling of N-protected glycines 168 with aryl bromides to obtain N-protected primary benzylamines 169 under 4CzIPN (25) photoredox conditions (Scheme 61). This decarboxylative process was performed with NiCl2·DME and 2,2′-bipyridine (bpy) as a ligand and Cs2CO3 as a base in DMF under blue LED irradiation to provide N-protected benzylamines 169 with up to 73% yield. Only N-tosyl glycine failed to react.
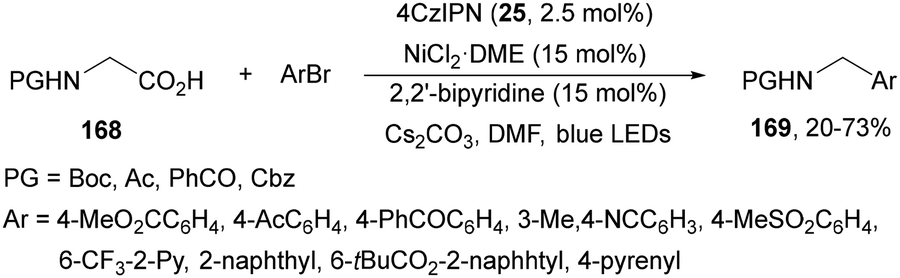 | ||
| Scheme 61 Decarboxylative arylation of N-protected glycines 168 with aryl bromides under 4CzIPN (25)/Ni photocatalysis. | ||
Formylation of aryl halides and triflates under 4CzIPN (25)/Ni photocatalysis was carried out by Mariano, Wang and co-workers144 using diethoxy acetic acid 26 as a source of radical (EtO)2CH˙. Using 20 mol% of 25 and 5 mol% of NiCl2·6H2O as catalysts and 4,4′-di-tert-Bu-2,2′-bipyridine (dtbbpy, 24 mol%) as a ligand and Cs2CO3 as a base under blue LED irradiation, aromatic aldehydes were obtained after acetal deprotection in 45–85% yield. Direct decarboxylative formylation of aryl and hetaryl iodides with glyoxylic acid was carried out by Shang, Fu and co-workers.145 Dual photoredox conditions with 4CzIPN (25) as an organic PC and Pd(Xantphos)Cl2 instead of Ni as a metallaphotocatalyst and CsOAc as a base in DMF at room temperature under blue LED irradiation afforded directly aromatic aldehydes with good yields (Scheme 62). In this case, the aryl iodide is firstly added to a Pd(0) catalyst to produce the Pd(II) intermediate A. Subsequent addition of the formyl radical affords the Pd(III) intermediate B, which after a SET process with 4CzIPN− produces the Pd(II) intermediate C and 4CzIPN. Final reductive elimination of intermediate C generates the aromatic aldehyde. The presence of an excess of CsOAc (150 mol%) is crucial for the in situ generation of the active cesium salt CsO2C–CHO.
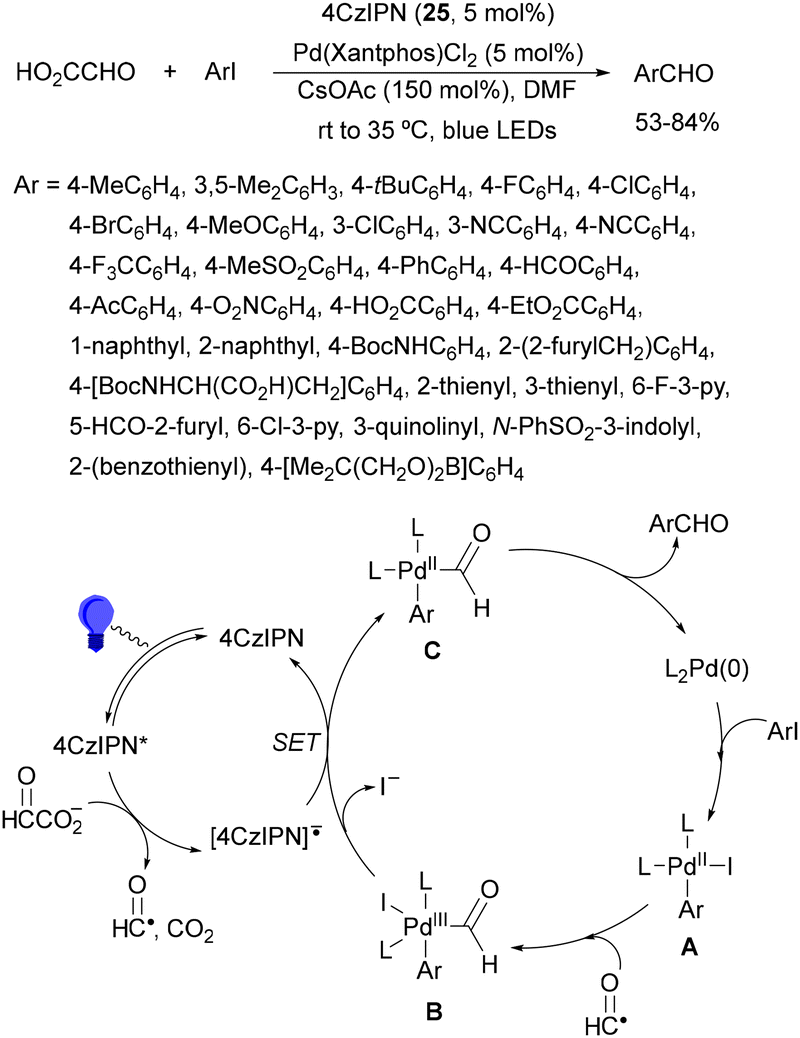 | ||
| Scheme 62 Decarboxylative formylation of aryl halides with glyoxylic acid under 4CzIPN (25)/Pd(0) photocatalysis. | ||
Ribosyl and deoxyribosyl acids 170 underwent Ni/photoredox decarboxylative cross-coupling reaction with aryl/heteroaryl bromides to give aryl/heteroaryl-C-nucleosides 171 (Scheme 63).146 This process was performed using 4CzIPN as an organocatalyst, NiBr2 with bpy as a ligand, K2CO3 as a base in DMF at 30 °C under blue LED irradiation. Products 171 were isolated in moderate to good yields and high stereoselectivity. In the proposed mechanism, the initial excitation of 4CzIPN produces photoexcited (4CzIPN)*, which promotes the photooxidative decarboxylation of 170a to form the anomeric radical A. In the Ni catalytic cycle the active LnNi(0) species is generated via two SET reduction processes of Ni(bpy)Br2 by the PC. This Ni(0) species undergoes oxidative addition to the aryl bromide forming the Ni(II) intermediate B, which reacts with radical A to generate a Ni(III) complex C. Reductive elimination of C produces product 167 and the Ni(I) complex Ni(bpy)Br, which is reduced by (4CzIPN)−˙ to the Ni(0) species.
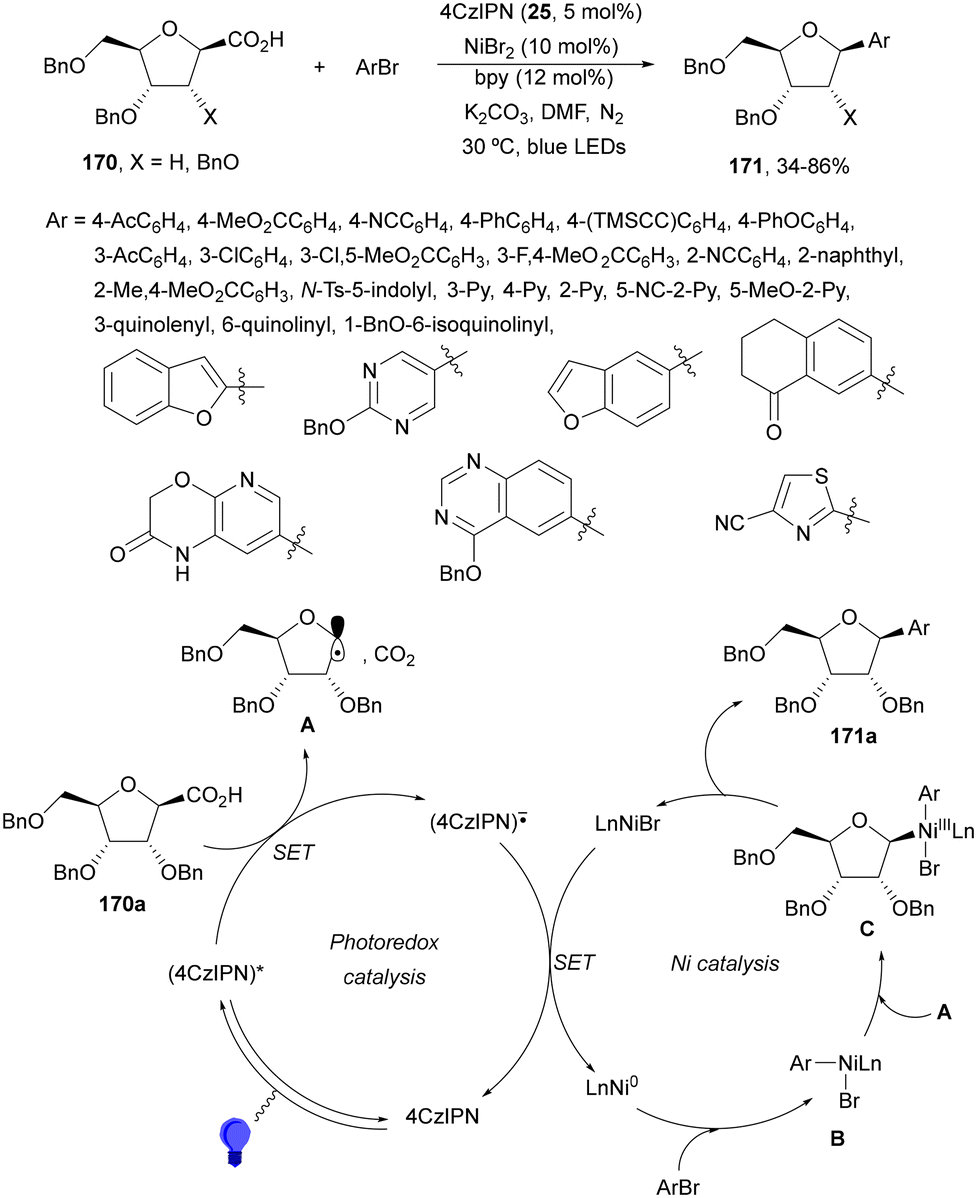 | ||
| Scheme 63 Decarboxylative arylation of ribosyl/deoxyribosyl acids 170 under 4CzIPN (25)/Ni photocatalysis. | ||
Molander and co-workers147,148 reported the cross-coupling of DNA-encoded libraries (DELs), conjugates with aromatic groups 172, with AA derivatives under Ir/Ni photocatalysis. Coupling with aryl bromides and aryl iodides enabled satisfactory incorporation of alkyl groups to provide compounds 173 (Scheme 64). This process was carried out in partially aqueous DMSO as solvent (23:77), with 1,1,3,3-tetramethylguanidine (TMG) as a base during 10 minutes irradiation under blue LED irradiation using Ir complex 4 as a PC and Ni(II) bis(2,2,6,6)tetramethyl-3,5-heptanedioate (TMHD). In this case, Ir complex 4 gave better results than 4CzIPN (25).
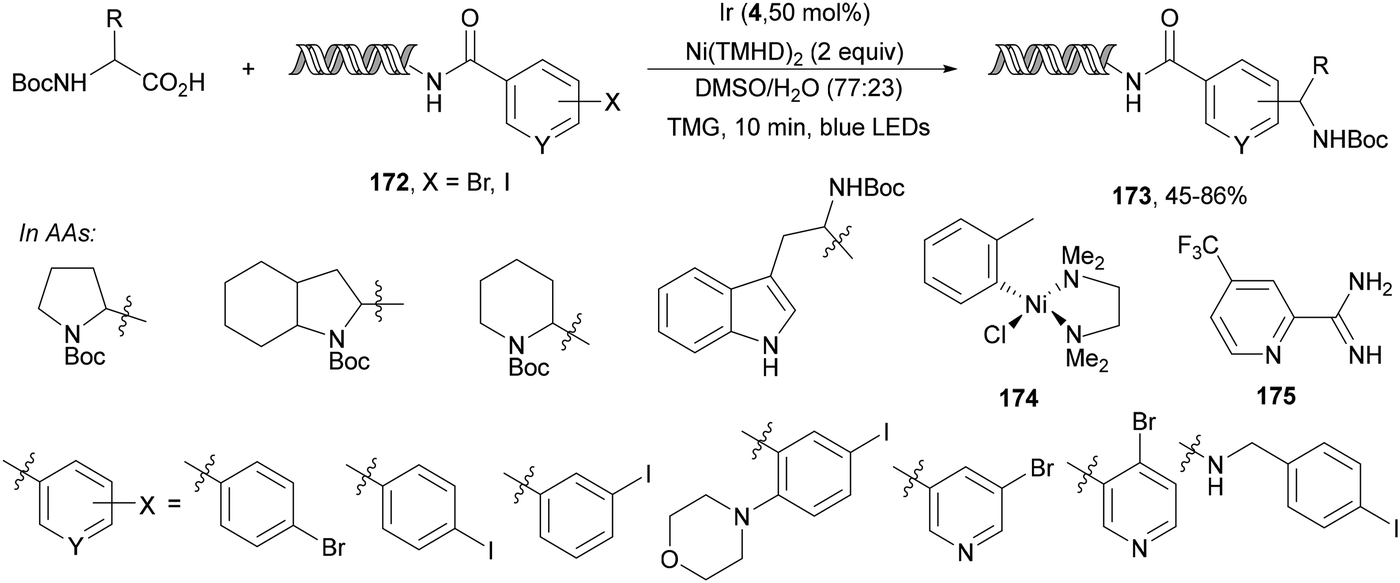 | ||
| Scheme 64 Decarboxylative cross-coupling of DNA-encoded library 172 with AA derivatives 6 under Ir/Ni photocatalysis. | ||
Kölmel, Flanagan and co-workers149 simultaneously described this transformation in DMSO/H2O (40:60) using DNA-tagged and heteroaryl iodides 172 and N-Boc AA derivatives 6 in the presence of Ir complex 4/Ni complex 174 and 4-CF3-2-pyridyl carboxamidine 175 (Scheme 64) as a ligand. This cross-coupling occurred in the presence of K2HPO4 as a base at room temperature during 40 minutes blue LED irradiation to afford products 173 in 9–93% yields. Recently, Chheda and co-workers150 reported the cross-coupling of DELs, 172 bromides, with carboxylic acids using Ir complex 4 and NiCl2·dtbbpy in DMSO in the presence of phthalimide as an additive151 and Barton base [2-tert-butyl-1,1,3,3-tetramethylguanidine, (BTMG)] under a N2 atmosphere and blue LED irradiation. In this process, DNA-cationic surfactant complexation enables dissolution and reaction on-DNA in anhydrous organic solvents. Products of type 173 were obtained with 12–96% yields.
Photoredox Ir/Ni dual catalyzed decarboxylative arylation cross-coupling has been translated from the batch to the continuous flow reactor using a ‘microslug’ screening platform by Jensen, Robinson and co-workers.152 The group of Merck153 has applied cross-coupling reactions under Ir/Ni photoredox catalysis to 18 pharmaceutical relevant aryl halides using an integrated photoreactor. This integrated photoreactor improves the success rate and reduces the reaction time with excellent results in its discovery programs. Using a microscale high-throughput experimentation (HTE), MacMillan and co-workers154 were able to perform a rapid optimization of several photoredox reactions including cross-couplings. This approach has been translated to several commercial flow reactors. Gesmundo and co-workers155 developed chemical-coated glass beads (Chembeads) as a parallel bead dispenser to expedite HTE processes namely C(sp2)–C(sp3) decarboxylative cross-couplings and libraries under Ir/Ni photoredox conditions.
Oxetanes are employed in medicinal chemistry as carbonyl or gem-dimethyl bioisosteres. On the other hand, 1-amino-3-oxetanes can function as amide replacement in druglike molecules and exhibits comparable pKa values.156 In addition, they are also employed as surrogates to gem-dimethyl groups with a similar molecular volume but significant increase of solubility being liponeutral. Terrett, Huestis and co-workers,157 through the combination of an Ir/Ni dual photoredox catalyzed strategy, accomplished the synthesis of aryl aminooxetanes 176 by arylation of oxetanyl amino acids 176. Aryl halides reacted with AAs 178 using Ir complex 4 and [Ni(dtbbpy)(H2O)4]Cl2 (177) as catalysts and Barton's base (BTMG) in anhydrous DMSO at room temperature under blue LED irradiation to provide products 174 in moderate to good yields (Scheme 65). Intermediate tertiary radicals coupled more efficiently than other tertiary acids such as 1-(Boc-amino)cyclobutanecarboxylic acid or cyclopropane derived α-amino acid or 1-(Boc-amino)-3,3-difluorocyclobutanecarboxylic acid. DFT calculations of two possible pathways A and B for the aminooxetanyl radical suggested that the initial oxidative addition of the aryl halide to Ni(0) to give intermediate A1 (ΔG≠ = 11.8 kcal mol−1) becomes the turnover-limiting step. Subsequent radical addition to Ni(II) forms intermediate A2. However, in path B – typical of benzylic radicals – a higher barrier of oxidative addition to the Ni(I)-oxetanyl species to form intermediate B1 was calculated to be 23.9 kcal mol−1, higher than that for intermediate A1.
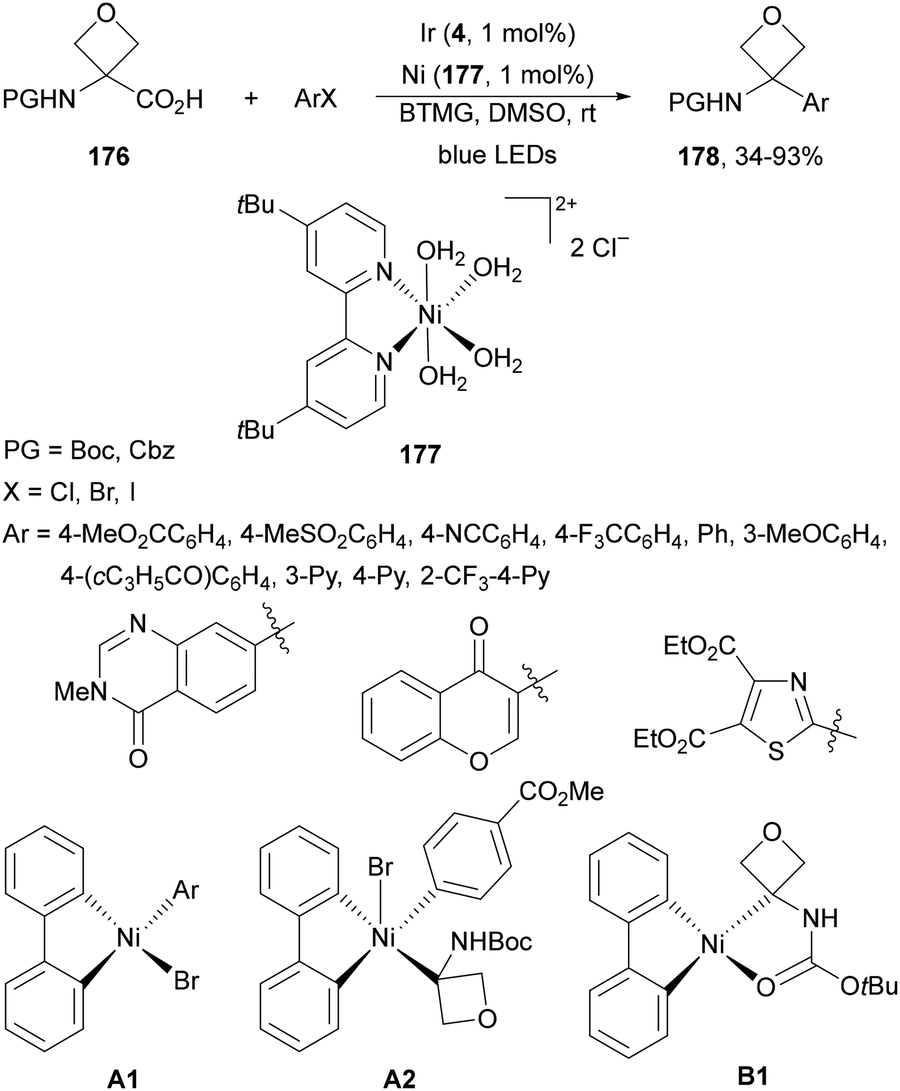 | ||
| Scheme 65 Decarboxylative oxetanylation of aryl halides with oxetanyl amino acids 176 under Ir (4)/Ni (177) dual photocatalysis. | ||
The synthesis of benzylamines by alkylation with benzyl halides and electron-deficient amines suffers from a lack of commercial availability of these electrophiles. However, using easily accessible aryl bromides as electrophiles in decarboxylative cross-coupling metallaphotoredox reactions should allow the synthesis of N-benzylamine derivatives. Hopkins and co-workers158 at Merck have described the cross-coupling of AAs 179 with aryl bromides under Ir/Ni photoredox conditions (Scheme 66). A great diversity of N-benzylic derivatives 180 were obtained in moderate to good yields using DBU as a base in DMA under blue LED irradiation. Working in a continuous flow photoreactor, the representative product 180a was obtained on a gram-scale with 70% yield.
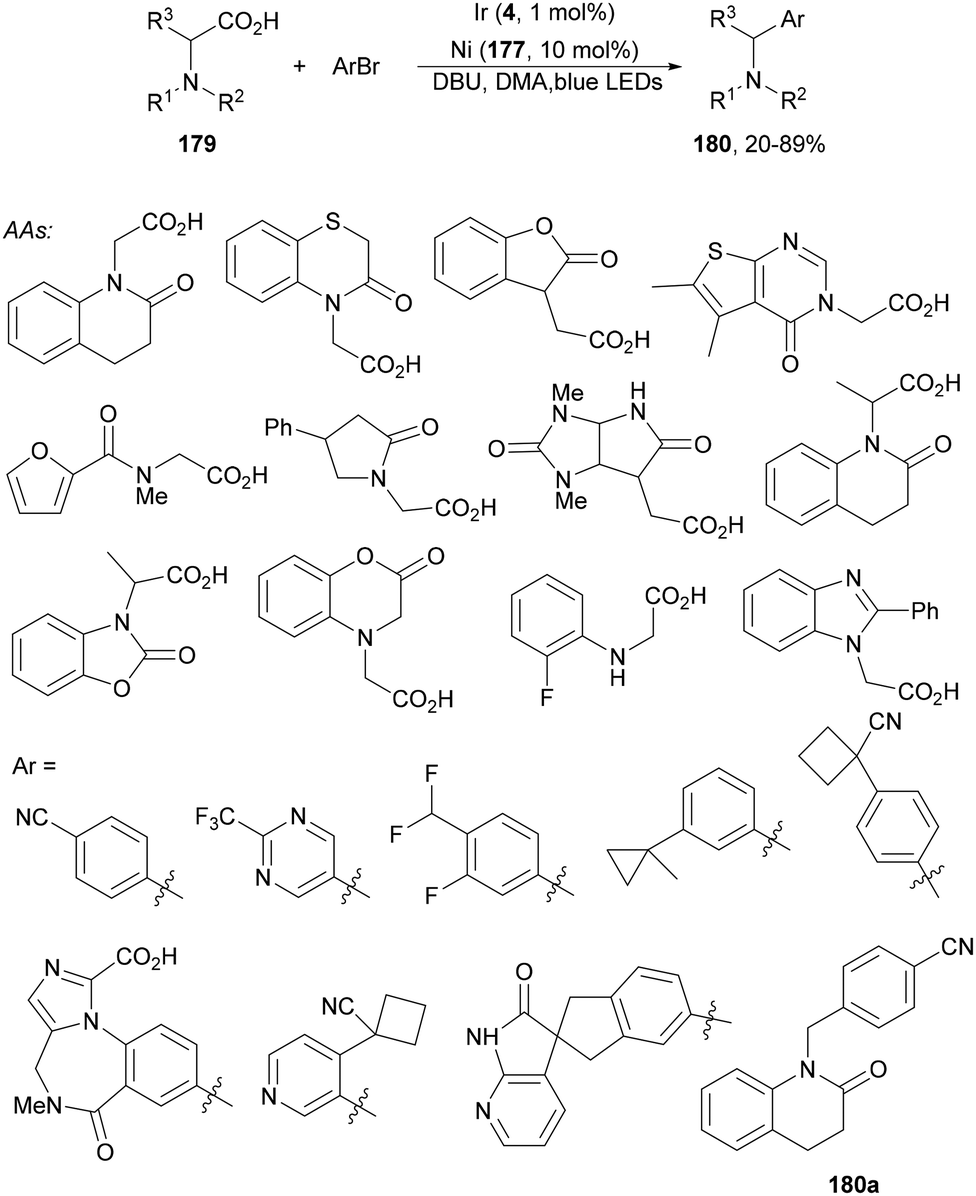 | ||
| Scheme 66 Decarboxylative arylation of α-amino acids 179 with aryl bromides under Ir/Ni (177) dual photocatalysis. | ||
Oderinde and co-workers159 have extensively studied the synthesis of amides 181 by decarboxylative arylation of N-benzoylamino acids of type 6 under Ir/Ni metallaphotoredox conditions (Scheme 67). A wide range of AAs and aryl bromides were applied to the synthesis of N-aryl amides 181 in good yields using Cs2CO3 as a base and DMA as a solvent under violet LED irradiation (400 nm). They observed that only one electron-poor aryl bromide participated in the cross-coupling conditions as it was also observed by MacMillan's group.136,139 The presence of phthalimide151 as an additive with electron-rich aryl bromides gave low yields or no product formation. Mechanistic investigations and DFT calculations revealed that in the Ni catalytic cycle reductive elimination via a Ni(II) species, rather than via a Ni(III) species, is thermodynamically favorable and also the reduction of Ni(II)ArBr to Ni(I)Ar by Ir(II). This alternative mechanism is outlined in Scheme 67.
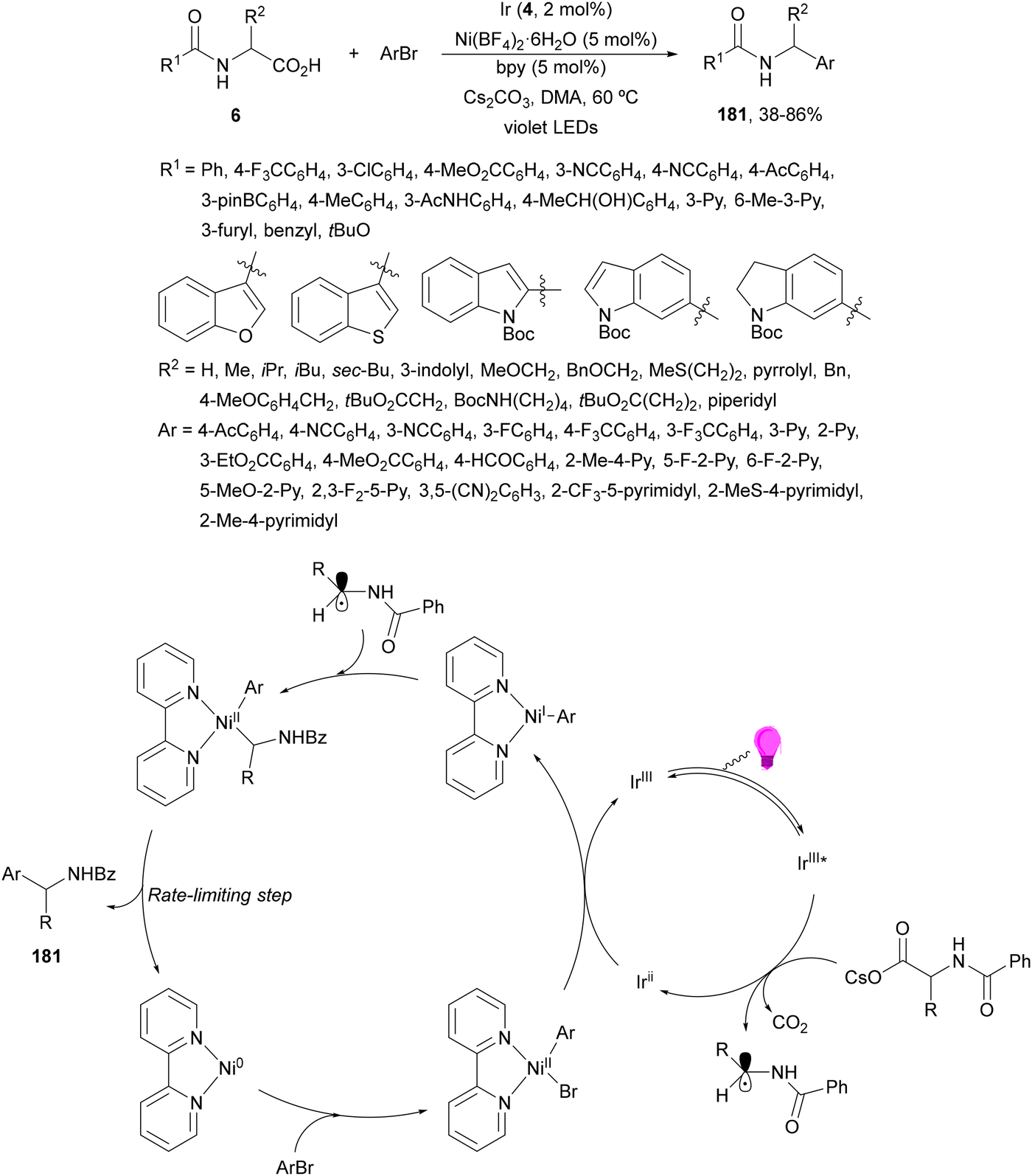 | ||
| Scheme 67 Decarboxylative arylation of N-acylamino acids with aryl bromides under Ir/Ni photocatalysis. | ||
The same group recently reported the stereoselectivity aspects of Ir/Ni metallaphotoredox decarboxylative arylation of substituted cyclic carboxylic acids with aryl bromides and chlorides.160 This process with acids 182 and 183 with aryl chloride 184 took place with Ir (4) as a PC, NiCl2·DME and 4,4′-dimethoxybipyridine (185) as a ligand, BTMG as a base, phthalimide as an additive161 in DMSO at room temperature and under blue LED irradiation to furnish products 186 and 187, respectively, as trans-diastereomers with in general good yields and up to 99:1 dr (Scheme 68a and b). In the case of aryl bromides, hydroxyproline derivative 188 was transformed into trans-pyrrolidines 189 with 23–84% yields and up to >99:1 dr (Scheme 68c). This methodology was applied to the synthesis of iptacopan, LNPO23, a recently approved drug prescribed to adults with C3G, PNH and IgA nephropathy.161 It was prepared enantioselectively on a 200 mmol scale in flow in only 4 steps. In this case, DFT calculations supported that the L2ArXNi(III)R species A is the diastereodetermining intermediate and undergoes reductive elimination to afford trans-products.
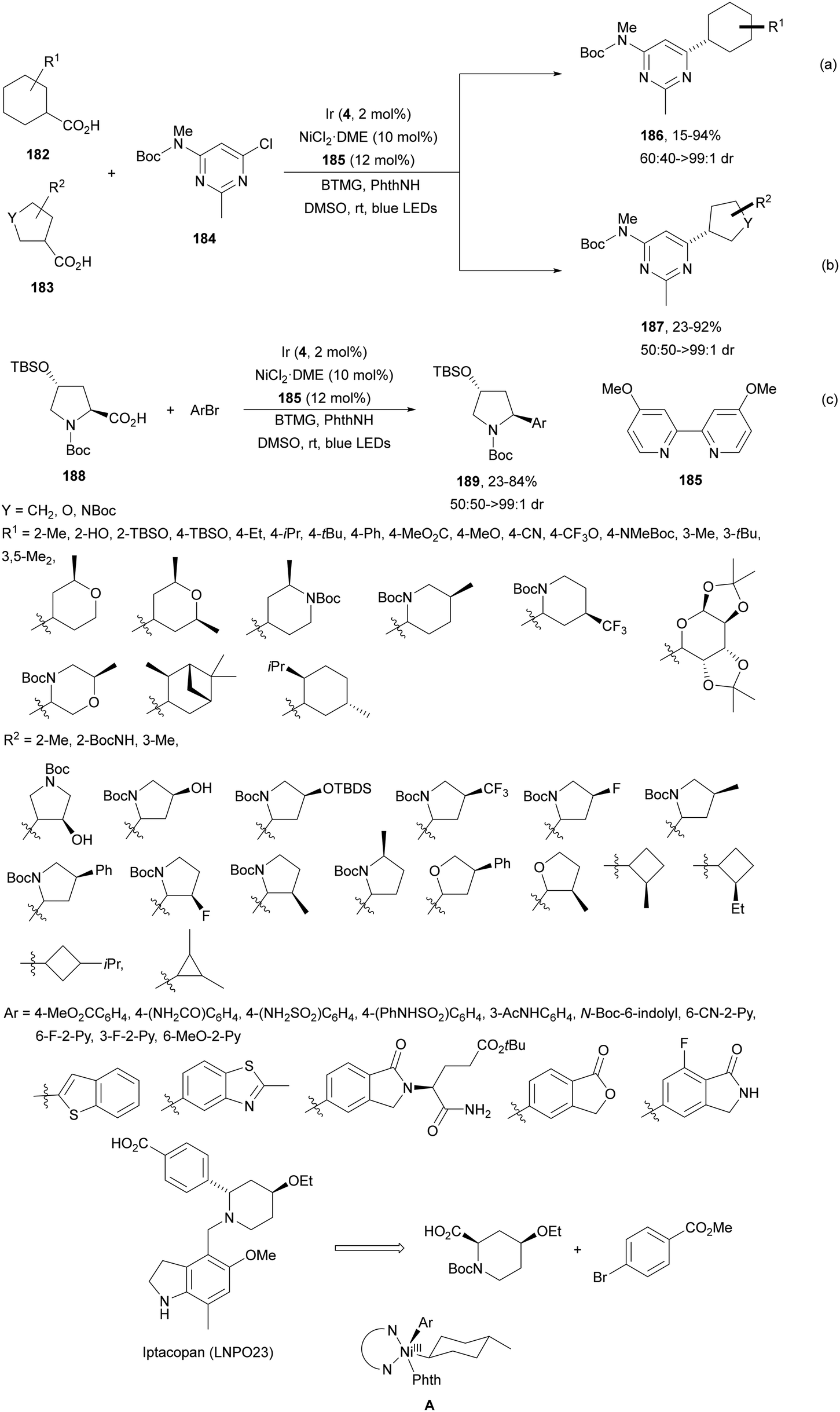 | ||
| Scheme 68 Decarboxylative arylation of cyclic carboxylic acids 182 (a), 183 (b) and 188 (c) under Ir/Ni metallaphotocatalysis. | ||
For the synthesis of secondary and tertiary fluorides 191 an efficient decarboxylative cross-coupling of α-fluoro carboxylic acids 190 with aryl halides was developed by Gutierrez, Koh and co-workers.162 In the presence of catalytic amounts of Ir complex 4, NiBr2·DME and ligand 185 in DMA with TMG and KOTf as bases at room temperature under blue LED irradiation, fluorides 191 were obtained in good yields (Scheme 69). DFT calculations rationalized the less effective decarboxylative cross-coupling for α-fluoro carboxylic acids than that for the nonfluorinated carboxylic acids due to the higher energy barrier for the formation of α-fluoroalkyl radicals.
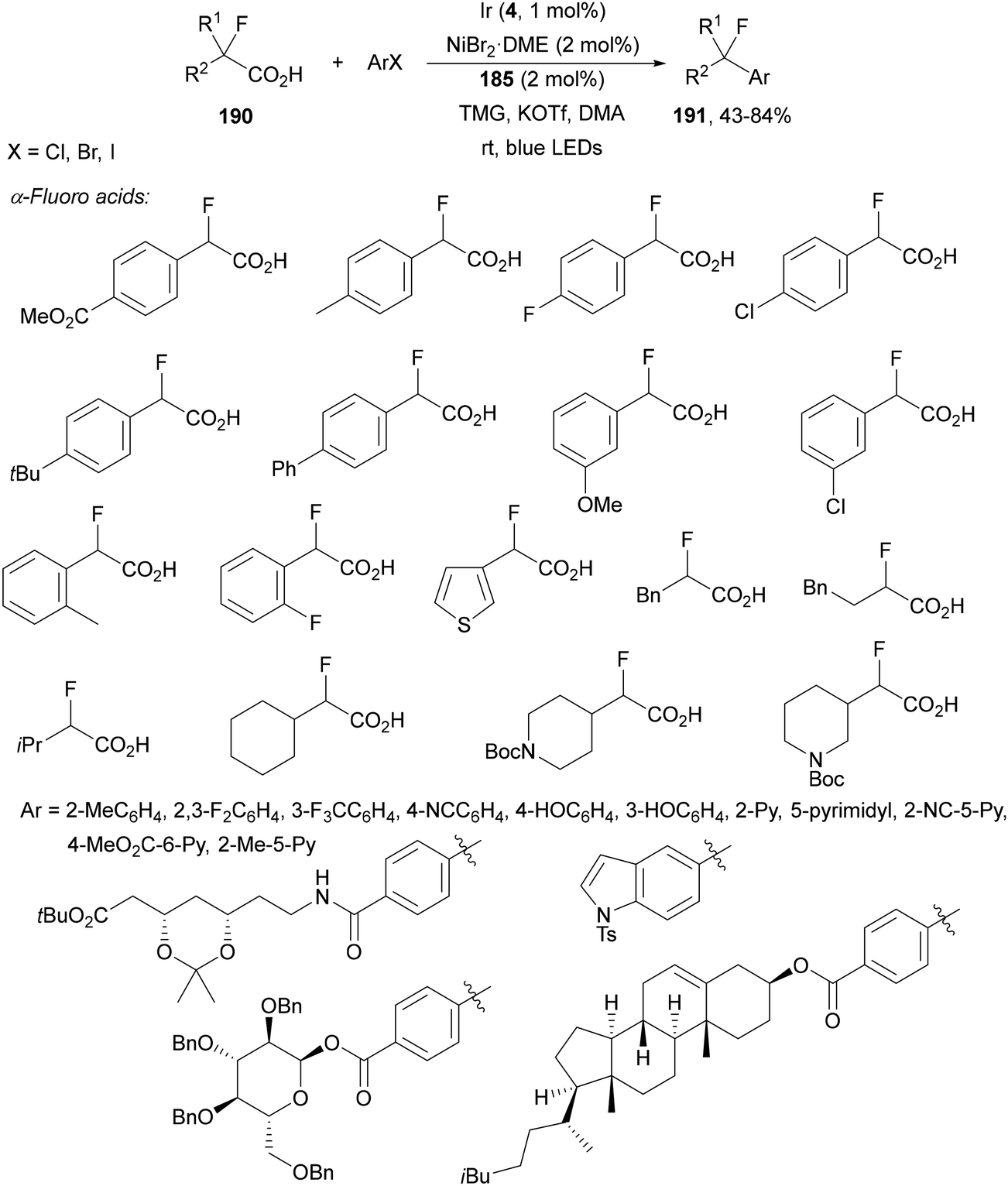 | ||
| Scheme 69 Decarboxylative cross-coupling of aryl halides with α-fluoro carboxylic acids 190 under Ir (4)/NiBr2·185 dual photocatalysis. | ||
Coldham and co-workers163 have prepared orthogonally protected 2-arylpiperazines 193 by decarboxylative arylation of piperazine carboxylic acid 192 with aryl bromides and iodides under Ir (4)/NiCl2·dtbbpy photocatalysis in moderate to good yields (Scheme 70). These products were subjected to kinetic resolution with n-BuLi/(+)-sparteine as a chiral ligand for lithiated intermediates to provide a mixture of (R)-193 and (S)-194 by reaction with methyl chloroformate. This methodology has been applied to the synthesis of glycogen synthase kinase (GSK)-3β inhibitor 195. The direct C–H lithiation of the piperazine ring followed by Negishi coupling to give 2-aryl piperazines was very low-yielding.164
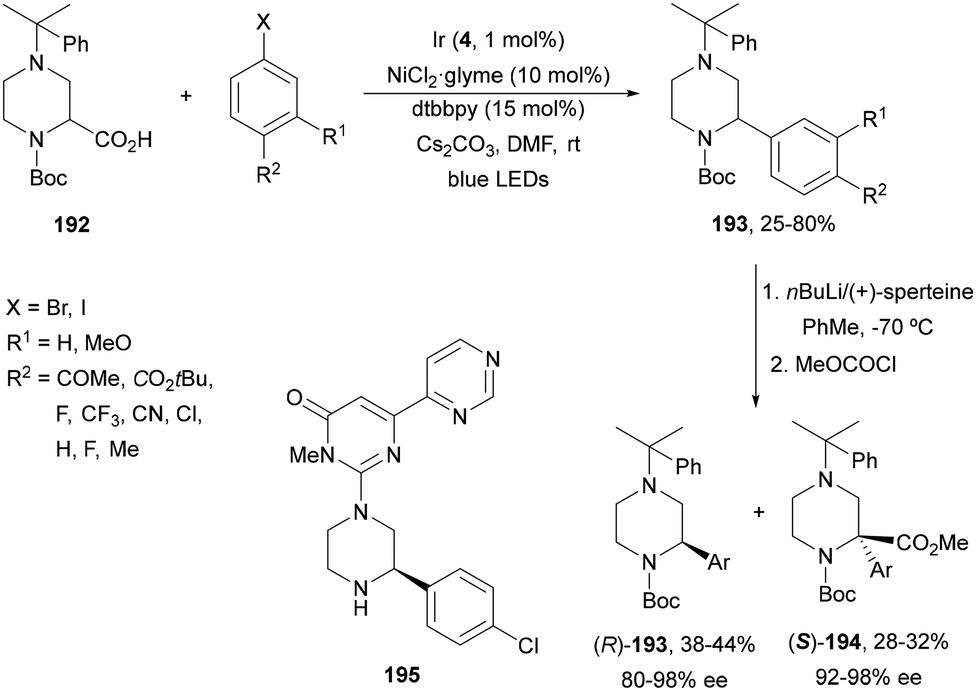 | ||
| Scheme 70 Decarboxylative arylation of 2-piperazine carboxylic acid 192 under Ir/Ni photocatalysis and subsequent kinetic resolution. | ||
Decarboxylative arylation of aryl acetic acids 103via metallaphotoredox catalysis has been employed for the synthesis of heterodiarylmethanes 196 (Scheme 71).165 Different substituted aryl and hetaryl bromides reacted with aryl acetic acids 103 using Ir (4)/NiCl2·dtbbpy as catalysts and K2CO3 as a base in DMF under blue LED irradiation in the presence of air to provide heterodiarylmethanes 196 in moderate yields.
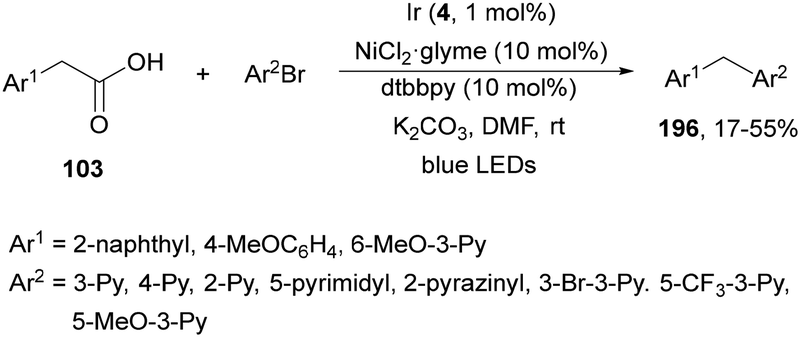 | ||
| Scheme 71 Decarboxylative arylation of (hetero)aryl acetic acids with (hetero)aryl bromides under Ir (4)/Ni photocatalysis. | ||
Noël and co-workers166 employed heterogeneous semiconductor graphite carbon nitride (gCN) 120 as a PC instead of Ir complex 4 for decarboxylative cross-coupling of (hetero)aryl acetic acids 103 with aryl bromides to obtain (hetero)diarylmethanes 196. In this case, 2.5 mg mL−1 of gCN, 5 mol% of NiBr2·glyme and 4,4′-diphenylbipyridyl ligand (7.5 mol%) were used as catalysts. Moreover, phthalimide (1 equivalent)151 was added in order to accelerate the rate of decarboxylative coupling, Cs2CO3 was employed as a base, and MeCN was employed as solvent at 43 °C under 390 nm irradiation. A wide range of products 196 were isolated in 23–96% yield and gCN could be easily recovered and reused multiple times without a loss in reactivity.
Inexpensive FeCl3 as metallaphotoredox, Ni(NO3)2·6H2O and piperidine carboxamidine 197 (4tBuPyCamCN) have been used in decarboxylative cross-coupling of a wide range of aliphatic carboxylic acids 1 with aryl iodides.167 The corresponding products were obtained in the presence of DIPEA as a base and TBAI in dioxane at 390 nm under Ar at room temperature with modest to good yields (Scheme 72). In the proposed catalytic cycles, firstly Fe carboxylate complex A can be formed, which under irradiation undergoes an LMCT process54,55 giving a carboxy radical and Fe(II) species B. Decarboxylation of the carboxy radical gives R˙, which adds to LnNi(I)X (C) to form LnNi(II)RX intermediate D. This species D undergoes SET by B or C to give intermediate E. Reversible oxidative addition of ArI forms F, which by reductive elimination forms the product and regenerates C.
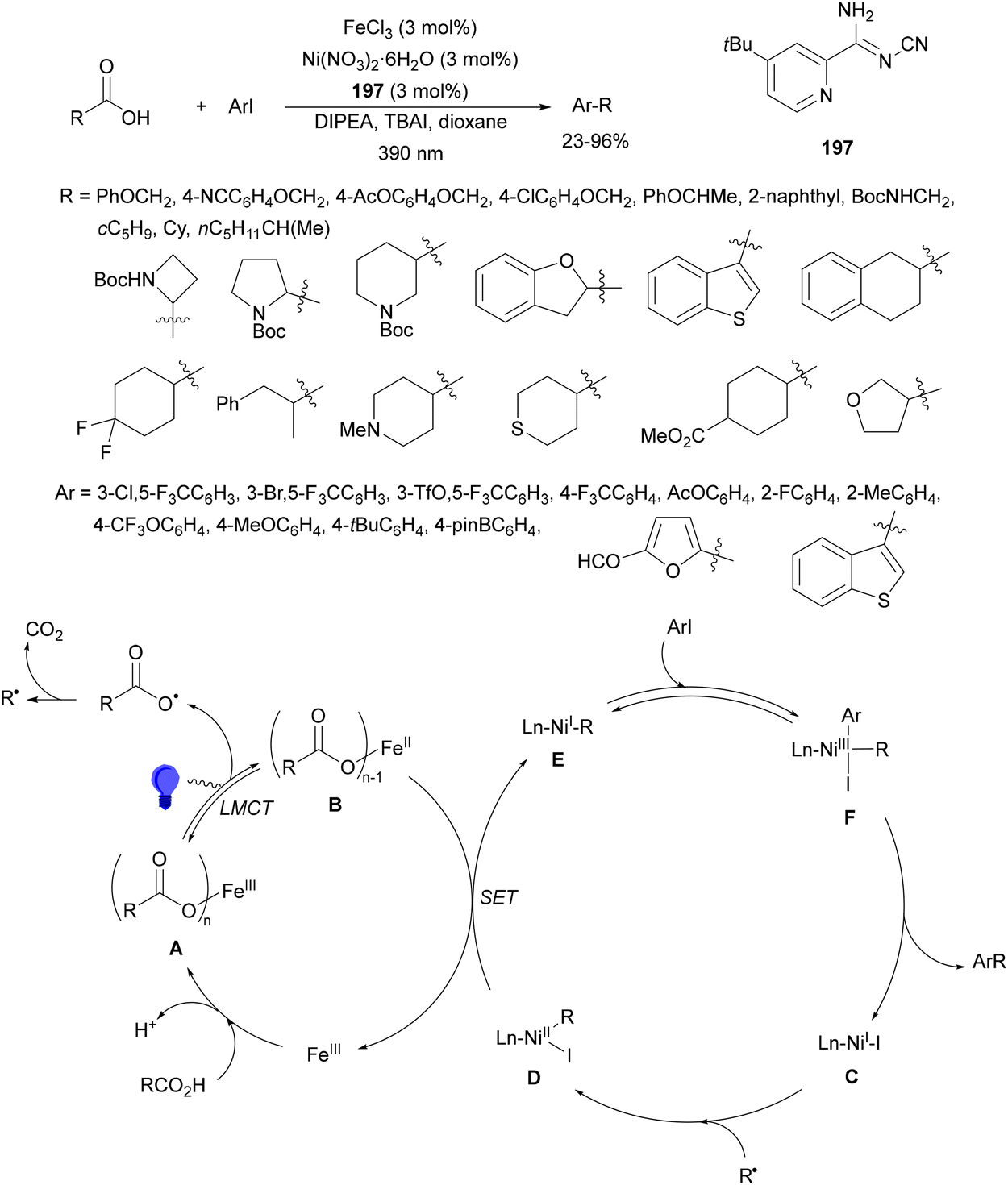 | ||
| Scheme 72 Decarboxylative arylation of aliphatic carboxylic acids under Fe/Ni metallaphotocatalysis. | ||
Redox-active esters19–21 such as NHPI esters have been used as alkylating reagents in cross-coupling decarboxylative arylations under Ir/Ni metallaphotoredox conditions as an alternative to carboxylic acids. Aliphatic NHPI esters 69 reacted with pyrimidine and pyridine heterocyclic chlorides, bromides and iodides using Ir complex 83, NiCl2·glyme and dtbbpy as a ligand, DIPEA as a base in DMSO or dioxane at room temperature under blue LED irradiation (Scheme 73).168 The corresponding alkylated heterocycles 198 were obtained with modest to good yields. In the plausible mechanism, three catalytic cycles were proposed, the radical generation cycle by an Ir(III) PC, the photoexcited Ir(III)* species is reduced by the Hünig's base to generate a N-centered radical cation A and Ir(II) species able to transform the NHPI ester to a C-centered radical, PhthN− and CO2. The initial Ni(0) species undergoes oxidative addition to heteroaryl halides to form the Ni(II) complex B, which reacts with R1 to provide the Ni(III) intermediate C. Subsequent reductive elimination gives the alkylated heterocycle and Ni(I) species which is reduced by Ir(II) species.
 | ||
| Scheme 73 Decarboxylative cross-coupling of NHPI esters 69 with heteroaryl halides under Ir/Ni metallaphotocatalysis. | ||
Decarboxylative coupling of NHPI esters 69 with bromopolyfluoroarenes 199 has been carried out through the synergetic action of Hantzch ester (HE), Ir complex 4 as a PC and NiBr2·DME in DMA at 30 °C under blue LED irradiation to obtain products 200 with good yield (Scheme 74).169 This method was applied not only to primary and secondary alkyl carboxylic NHPI esters but also to unnatural AAs and drug molecules like isoxepac, indomethacin and chlorambucil. In the proposed mechanism, the NHPI ester reacts with HE under blue irradiation to give R˙, which reacts with the Ni(II) species A to give the Ni(III) species B. Subsequently, reductive elimination provides the product and the Ni(I) species C. Final SET from Ir(II), obtained by reductive quenching of Ir(III) with HE, to Ni(I) species provides the Ni(0) species able to react by oxidative addition of ArBr to yield intermediate A.
 | ||
| Scheme 74 Decarboxylative arylation of NHPI esters 69 with bromopolyfluoroarenes 199 under Ir/Ni metallaphotocatalysis. | ||
Molander and co-workers170 reported the arylation of NHPI esters 69 with (hetero)aryl bromides using HE and NiBr2·dtbbby (201) for the decarboxylative cross-coupling reaction. Under light irradiation at 390 nm (purple light) the HE promoted the radical generation through electron donor–acceptor (EDA) complex activation of the NHPI ester. Primary, secondary, benzylic, α-oxy and α-amino acid carboxylic acid derived NHPI esters were transformed into the corresponding arylated products 202 in DMA as solvent with good yields (Scheme 75). Based on experiments, the irradiation of colored EDA complex A triggers an intra-coupling SET process generating a dihydropyridine radical cation and a phthalimide radical anion. Subsequently decarboxylation yields radical B, which by reaction with the Ni(0) species gives the Ni(I) intermediate C. Oxidative addition of aryl halide to C produces the Ni(III) species D, which undergoes reductive elimination to form the product the Ni(I) species E. This intermediate E gives by a SET process from excited HE* the Ni(0) species. A process involving oxidative addition of ArBr to Ni(0) to give intermediate F cannot be ruled out.
 | ||
| Scheme 75 Decarboxylative cross-coupling of NHPI esters 69 with (hetero)aryl bromides under Hantzsch ester and NiBr2(dtbbpy) 201 photocatalysis. | ||
Peptides on a solid phase (Rink amide resin) bearing a NHPI ester derived from aspartic or glutamic acid 203 have been arylated under the same reaction conditions than those from Molander's group.170 By avoiding multistep solution-phase AA synthesis, this greener alternative was applicable to differently supported di-, tri-, tetra- and pentapeptides, which after cross-coupling decarboxylation and hydrolysis provided free C-terminus peptides 204 (Scheme 76).171 These peptides were obtained in good yields considering that the process started from the allyl ester resin-linked peptide.
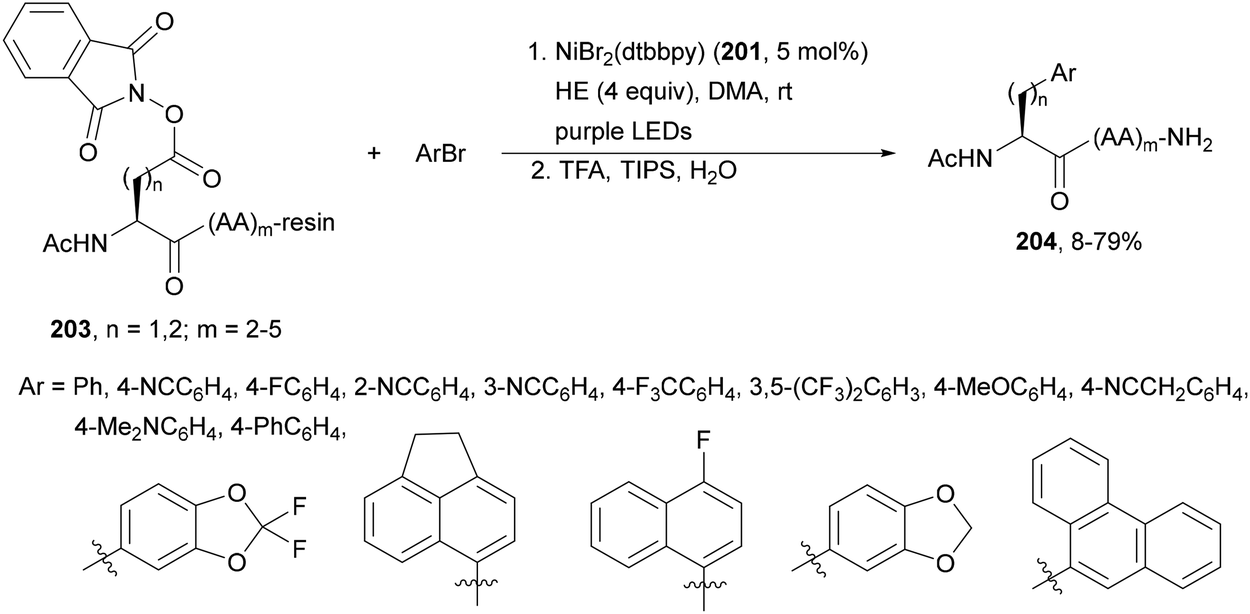 | ||
| Scheme 76 Decarboxylative arylation of supported peptides 203 bearing a NHPI ester in the C-terminus under HE/NiBr2(dtbbpy) (201) photocatalysis. | ||
Rovis and co-workers172 recently reported a low-energy orange-light absorbing Ir(III) photocatalyst 205, which can be used in Ir/Ni metallaphotoredox catalysis in both carboxylic acids and NHPI esters. This PC 205 showed complementary modes of dual oxidative and reductive decarboxylative arylation. Aliphatic α-amino carboxylic acids react with aryl bromides to furnish cross-coupled products 202 with good yields using 205/NiBr2(dtbbpy) 201 as a catalyst and BTMG as a base in DMF at 55 °C (Scheme 77a). In the case of carboxylic acids without the α-heteroatom the reaction failed. However, NHPI esters 69 derived from secondary and tertiary carboxylic acids could be arylated even generating in situ these esters with a coupling reagent. In the presence of DIPEA as a base this cross-coupling gave products 202 in very good yields (Scheme 77b). The proposed catalytic cycles, oxidative and reductive pathways, are depicted in Scheme 77. The oxidative activation of the carboxylic acid to carboxy radical followed by decarboxylation gives the C-centered radical R˙. Subsequent oxidative addition of aryl bromide to Ni(II) and reaction with the radical gives the Ni(III) complex, which by reductive elimination affords the product 202 and a Ni(I) species. In the reductive pathway DIPEA acts as a sacrificial reductant to quench Ir(III)* after irradiation. The resulting Ir(II) species reduces the NHPI ester to liberate the alkyl radical. The Ni cycle proceeds as before for the oxidative pathway.
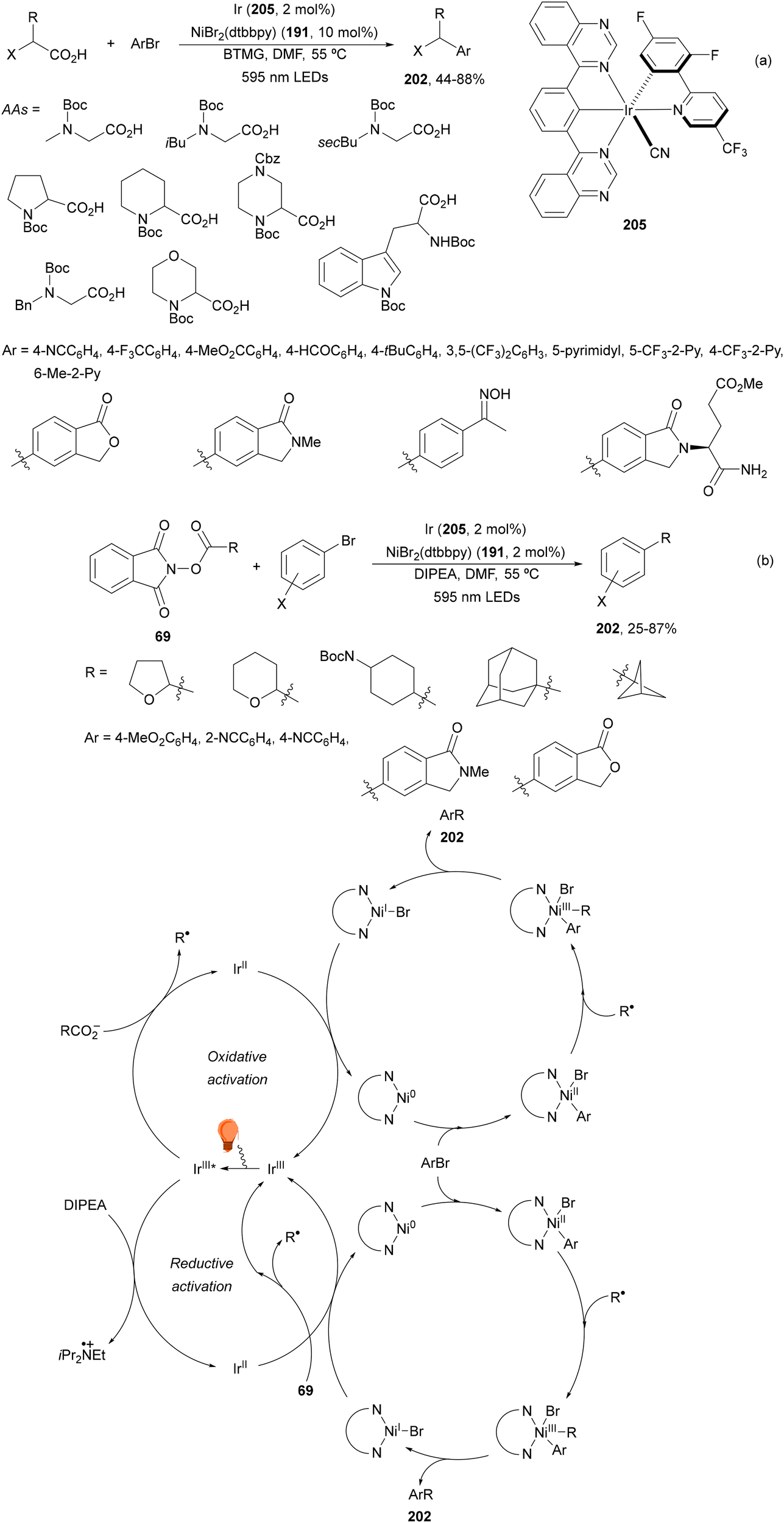 | ||
| Scheme 77 Decarboxylative arylation of carboxylic acids (a) and NHPI esters (69) (b) under Ir (205)/Ni (201) low-energy-light photocatalysis. | ||
1,4-Dialkylbenzene and derivatives 207 can be prepared from 1,4-diiodobenzene analogues 206 by decarboxylative arylation photoredox cross-coupling with NHPI esters 69 under mild reaction conditions (Scheme 78a).173 Zhang and co-workers employed HE as a donor and in situ prepared Ni(II) complex 201, NaHCO3 as a base and NHP as solvent at 30 °C under 390–395 nm purple LED irradiation. Products 207 were obtained in moderate yields and 3,6-dialkylcarbazole analogues 209, useful optoelectronic and medicinal materials, were prepared from 3,6-diiodocarbazoles 208 in modest yields (Scheme 78b).
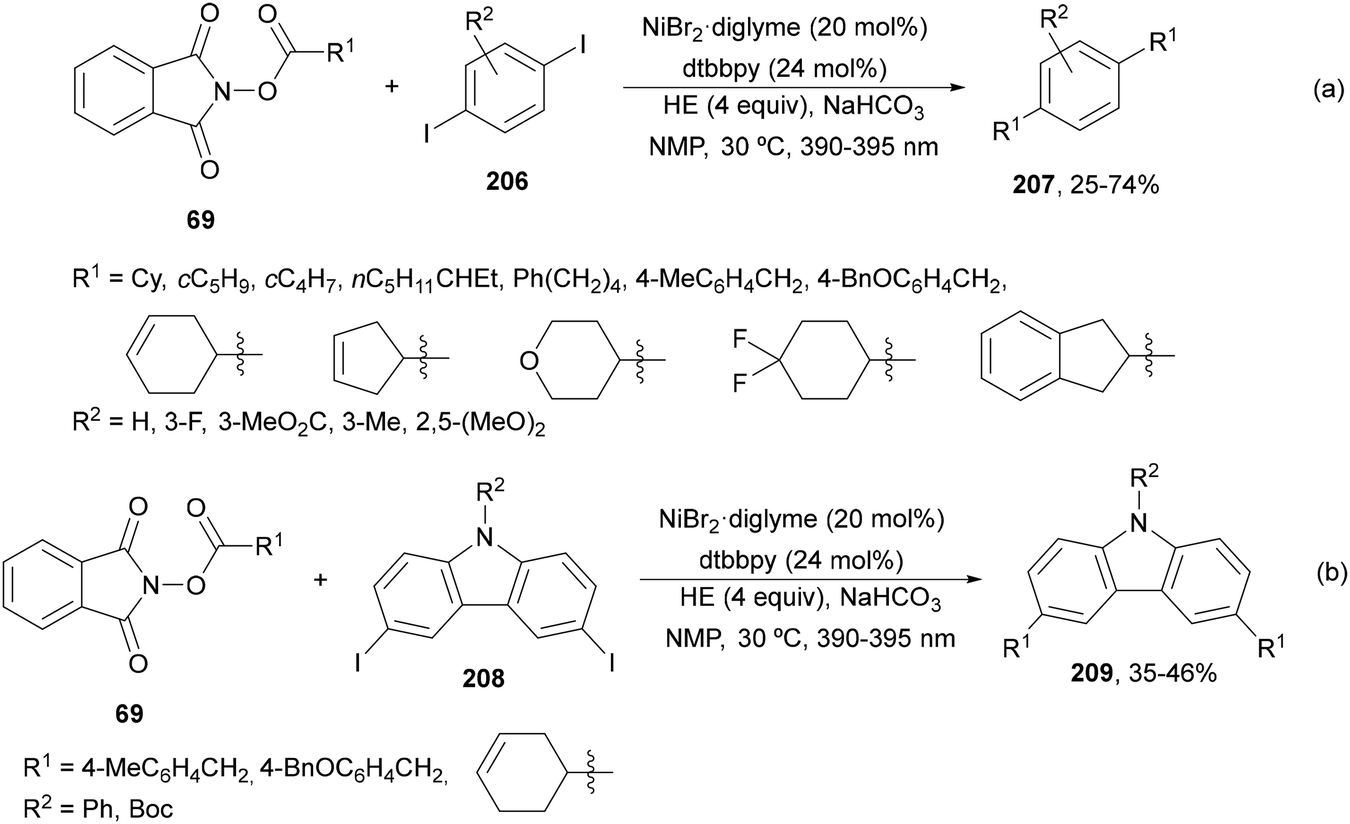 | ||
| Scheme 78 Decarboxylative cross-coupling of diiodoarenes 206 (a) and 208 (b) with NHPI esters (69) under HE/Ni 201 photocatalysis. | ||
Arylation of carbohydrates can be carried out by cross-coupling of DHP-derived esters 37 with aryl bromides under 4CzIPN (25) and Ni(II) dual photocatalysis. Diao and co-workers174,175 prepared aryl C-glycosides 211 using Na2CO3 as a base in dioxane at 84 °C under blue LED irradiation with in general good yields and moderate to high diastereoselectivity (Scheme 79). Furanoses and pyranoses were transformed into dihydropyridine derived esters 37 by reaction with DHP acid 210 using diisopropyl carbodiimide (DIC). In the proposed mechanism oxidation of 37 with 4CzIPN* followed by deprotonation affords intermediate A, which by subsequent fragmentation gives radical B and HE. Upon ejection of CO2, B was transformed into glycoxyl radical C, which enters in the Ni catalytic cycle Ni(0) → Ni(I) → Ni(II) → Ni(III) to give the cross-coupling product with the aryl bromide. D-Mannofuranose derivatives gave mainly the α-anomeric glycosides. D-Ribofuranoses with common protecting groups, such as benzyl, silyl and benzoyl, gave mainly the β-anomers except D-galactofuranose and D-arabinofuranose, which favored the α-anomer due to the dominating effect of the C2 substituent. 2-Deoxy-D-ribofuranoses gave mixtures of α and β anomers. Pyranoses provided mainly α-selectivity due to the kinetic anomeric effect.
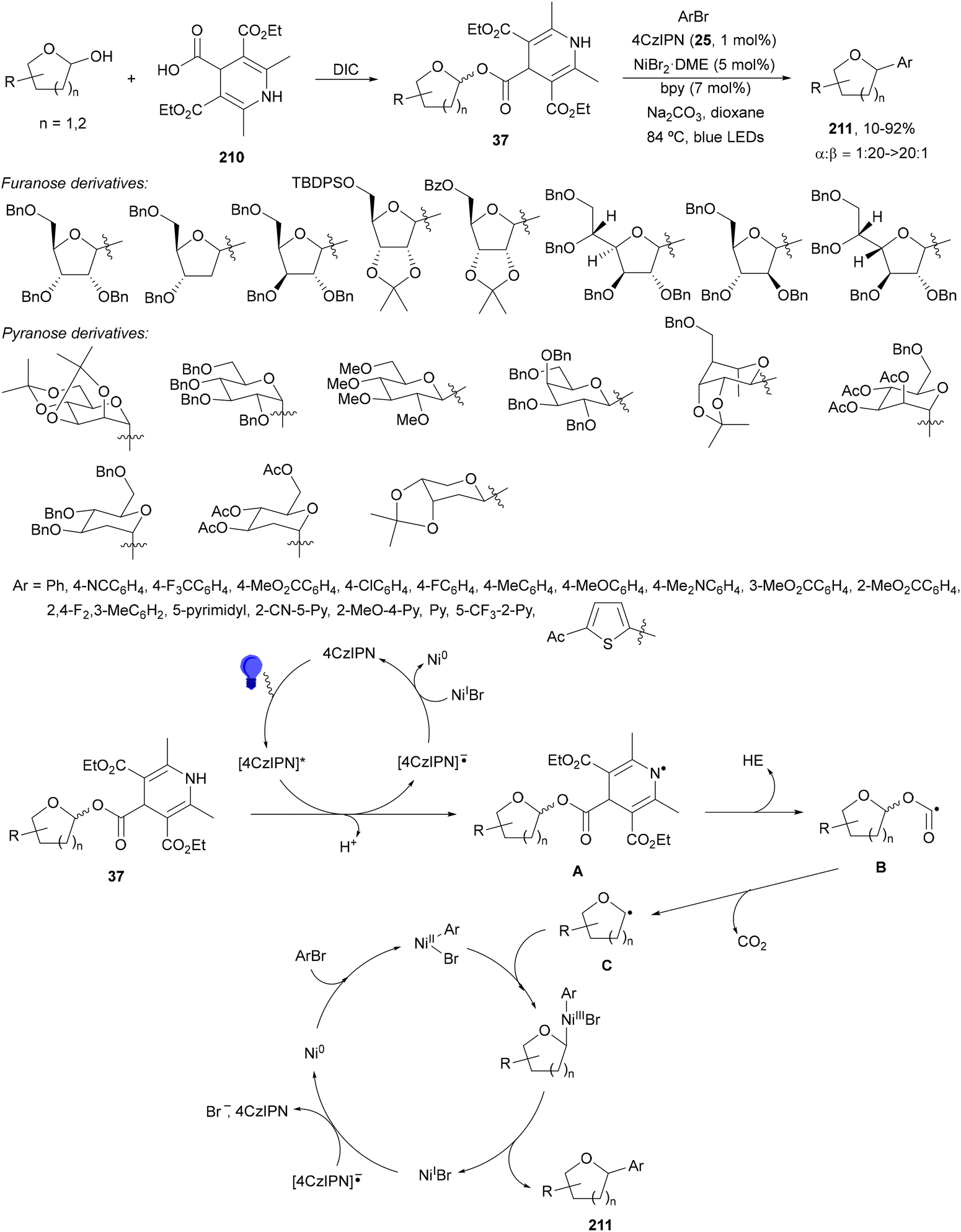 | ||
| Scheme 79 Decarboxylative arylation of DHP-derived glycosyl esters 37 under 4CzIPN (25)/NiBr2 photocatalysis. | ||
Wang and co-workers176 reported the arylation of α-AAs 179 and α-oxy carboxylic acids 65 with aryl nitriles 63 using only 3-aminofluorene-2,4-dicarbonitrile (AFDC, 212) as acceptor–donor PC like 4CzIPN (25). Upon light excitation, the photoexcited state of these compounds showed both strong oxidative and reductive capabilities even higher than Ir complexes and organic dyes. This arylation process was performed in the absence of Ni(II) complexes using CsF in DMSO at room temperature under 390–395 nm LED irradiation to afford arylated products 180 and 213, respectively, with good yields (Scheme 80). In the proposed mechanism, the excited PC* oxidizes the α-AA to form the carboxy radical, which after decarboxylation generates the α-amino radical intermediate A. The reduced PC−˙ reacts with the aryl nitrile to give by a SET process radical anion B followed by regeneration of the PC. Radical–radical coupling of A with B and subsequent aromatization with release of cyanide deliver the benzylic amine 180.
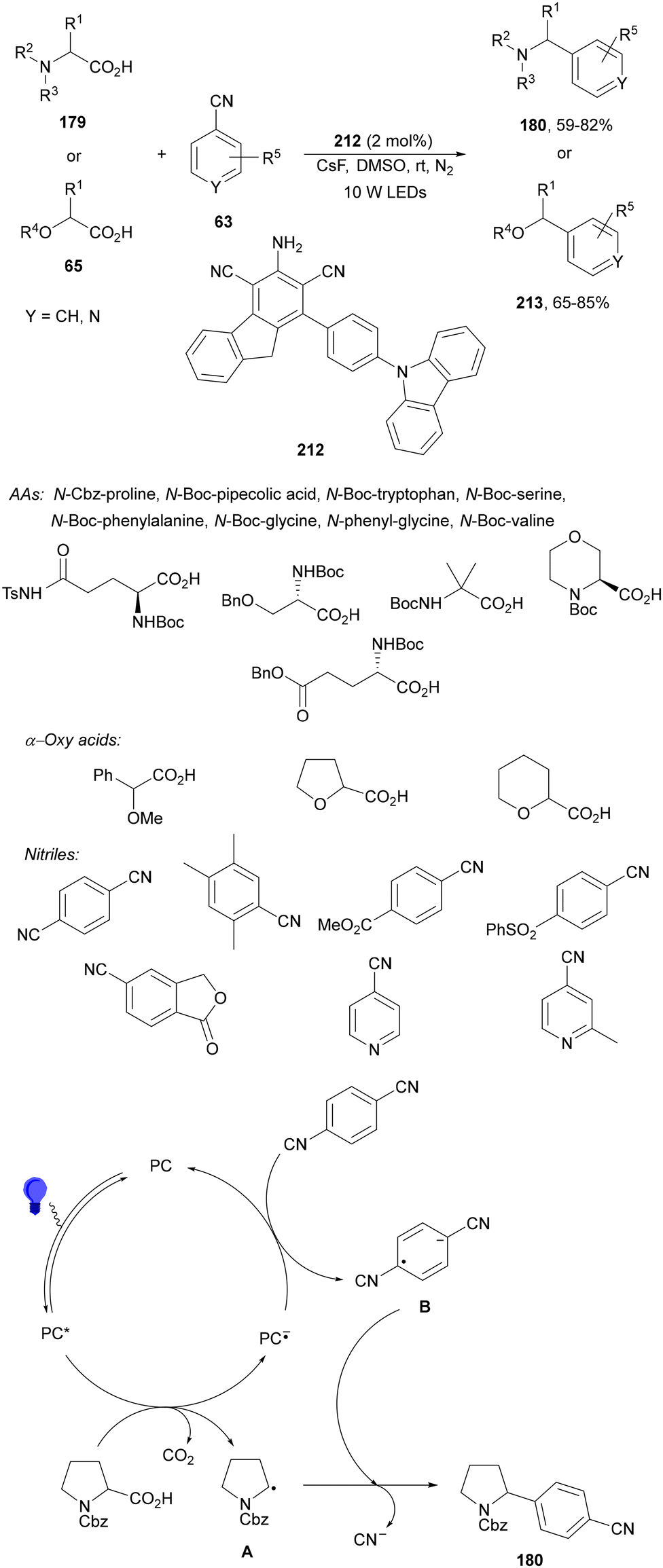 | ||
| Scheme 80 Decarboxylative arylation of α-AAs and α-alkoxy acids with aryl nitriles under 3-amino-fluorene-2,4-dicarbonitrile 212 photocatalysis. | ||
Decarboxylative cross-coupling of aryl halides with alkyl carboxylic acids under metallaphotoredox catalysis has become a general method for the construction of C(sp2)–C(sp3) bonds. Ir(III) complexes as photocatalysts and Ni complexes converted into Ni(0) species by SET events are able to undergo oxidative addition with aryl halides. The resulting Ni-aryl species undergoes oxidative radical capture, from the decarboxylation of the carboxylate ion by a photoexcited Ir(III) complex, resulting in a Ni(III))–aryl–alkyl complex, which undergoes reductive elimination to give the coupled product. This methodology has been extensively applied to the synthesis of benzylamines from α-AAs and DNA-encoded libraries, α-fluorinated benzylic alkanes from α-fluoro carboxylic acids and heterodiarylmethanes from aryl acetic acids. NHPI esters can be used alternatively for the generation of alkyl radicals and have been employed in the presence of stoichiometric amounts of Hantzsch ester or under Ir(III)/Ni(II) metallaphotoredox catalysis under mild reaction conditions. Several processes have been implemented in continuous flow reactors. In addition it has been applied to the synthesis of pharmaceuticals such as GSK-3β inhibitors, iptacopan (LNPO23), arylated peptides and arylated glycosides. These reaction conditions are compatible with enantioselective processes using chiral bisoxazoline ligands for Ni(II) complexes.
2.6. Alkylation reactions
Decarboxylative cross-coupling of carboxylic acids and NHPI esters with alkyl electrophiles under photoredox conditions is a direct method for C(sp3)–C(sp3) carbon bond formation. MacMillan and co-workers reported in 2016177 the decarboxylative alkylation of aliphatic carboxylic acids with alkyl halides by merging photoredox and Ni catalysis. As shown in Section 2.4 for this metallaphotoredox catalysis, the first SET process transformed the carboxylic acid into CO2 and the alkyl radical A by excited Ir(III)* and the other SET process generated a Ni(0) species. This Ni(0) complex reacts with the radical A to produce an alkyl–Ni(I) intermediate B, which by subsequent oxidative addition with alkyl halide forms the Ni(III) species C. Final reductive elimination creates a C(sp3)–C(sp3) bond and the Ni(I) species D, which after a SET process regenerates Ni(0) species (Scheme 81). | ||
| Scheme 81 Proposed mechanism for the metallaphotoredox cross-coupling of carboxylic acids with alkyl bromides. | ||
Gutierrez, Koh and co-workers162 employed α-fluoro carboxylic acids 190 in decarboxylative cross-coupling reactions with aryl halides (Scheme 69) and also alkyl chlorides. Under the same reaction conditions and in the presence of KOTf, the corresponding fluorinated products 214 were obtained in up to 60% yield and moderate diastereoselectivity (Scheme 82).
 | ||
| Scheme 82 Decarboxylative alkylation of α-fluoro carboxylic acids 190 under Ir (4)/NiBr2·185 dual catalysis. | ||
C–Alkyl glycosides are particularly important for the synthesis of glycopeptide analogues and drugs.178 Glycosyl carboxylic acids 170 have been used as sources of glycosyl radicals by Ni/photoredox decarboxylative cross-coupling with aryl bromides using 4CzIPN (25) as a PC (Scheme 63).146 Recently, C-alkyl glycosides 216 have been prepared by Ir/Ni metallaphotocatalytic decarboxylative cross-coupling of acids 170 with alkyl bromides (Scheme 83).179 In this case, Ru(bpy)2Cl2 or 4CzIPN (25) failed as photocatalysts. However, in the presence of Ir (4), NiCl2·DME and ligand dMebpy (215), K2CO3 as a base in aqueous MeCN under blue LED irradiation, C-glycosides 216 were obtained with moderate yields mainly as β-diastereomers. A wide variety of alkyl bromides were employed including those bearing drug molecules or natural product fragments. On the other hand, only uronic acid forms of furanoses were suitable as substrates for this cross-coupling reaction. However, other furanosyl and pyranosyl carboxylic acids failed. Under gram-scale conditions product 216a was obtained in 42% yield instead of 63% yield for the millimole scale. In the proposed catalytic cycle, based on DFT calculations, Ni(0) species reacts firstly with alkyl bromide to give intermediate A, which after reaction with anomeric radical generates Ni(III) species B.
 | ||
| Scheme 83 Decarboxylative alkylation of glycosyl carboxylic acids 170 under Ir/Ni dual photocatalysis. | ||
Asymmetric photoredox cross-coupling has been studied by Jiang and co-workers180 by enantioconvergent140 substitution of α-bromo ketones 217 with N-aryl α-AAs. Single-electron reductive debromination of racemic α-bromo ketones generates achiral alkyl radicals, which participate in asymmetric C(sp3)–C(sp3) bonds forming cross-coupling with α-amino radicals (Scheme 84a). This process took place under metal-free conditions using the dicyanopyrazine-derived chromophore (92, DPZ) as a PC and SPINOL 218 as CPA. Similar reaction conditions were used by the same group104 in the enantioselective decarboxylative reaction of N-aryl glycines with 1,2-dicarbonyl compounds 90 and to isatins 91 (Scheme 33). Products 219 were obtained under blue LEDs irradiation at 0 °C in 1,2-dimethoxyethane with good yields and up to 97% ee. This enantioconvergent cross-coupling was applied to α-bromo-α-fluorophenones 220 to obtain fluorinated β-amino phenones 221 in up to 80% yield and up to 94% ee (Scheme 84b). Tertiary α-bromo ketones 222 were transformed into β-amino phenones 224 using SPINOL CPA 93b, 145 or 223 as chiral catalysts (Scheme 84c). In Scheme 33, the role of the CPA as a bifunctional H-bonding catalyst in the cross-coupling of radicals A and B is depicted.
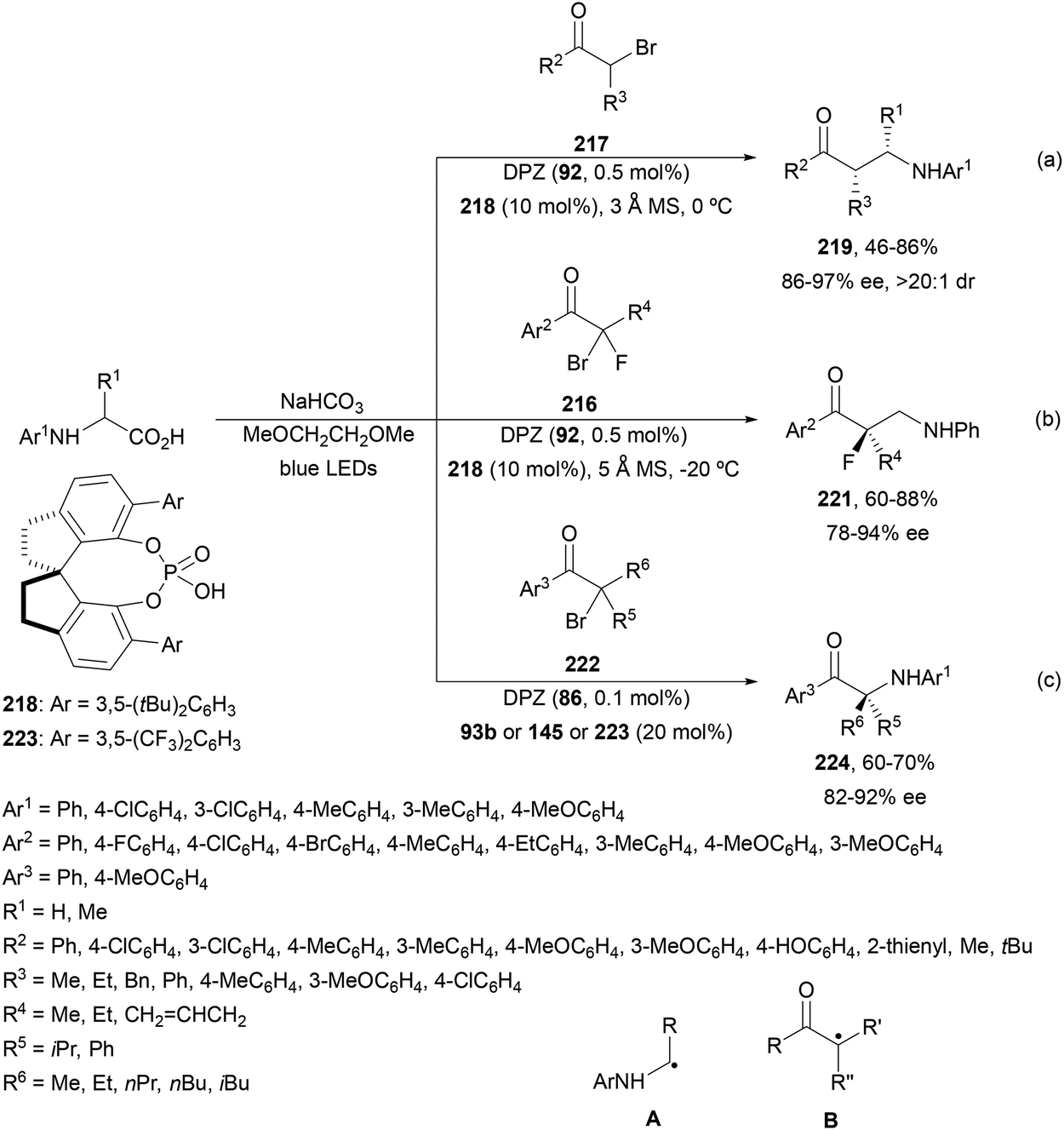 | ||
| Scheme 84 Enantioconvergent decarboxylative alkylation of α-bromo ketones 217 (a), 220 (b) and 222 (c) with N-aryl AAs under DPZ (92)/SPINOL-CPA (93b, 145, 218, and 223) photocatalysis. | ||
Zhao, Jiang and co-workers181 applied the enantioconvergent140 substitution with N-aryl glycines to 3-chloroindoles under visible light irradiation. A cooperative catalysis using DPZ (92) and SPINOL (226) as chiral Brønsted acid was employed for this cross-coupling reaction between 3-chlorooxindoles 225 and N-aryl glycines working in a 5:1 mixture of MTBE/THF at −42 °C under Ar to give chiral 3-aminomethylene-3-alkyl oxindoles 227 in up to 95% yield and up to 93% ee (Scheme 85a). When 3-aryl-substituted oxindoles 228 were allowed to react with N-aryl glycines in the presence of DPZ (92) as a photoredox and SPINOL-CPA 229 as a chiral Brønsted acid in a 2:1 mixture of MTBE/THF at −55 °C under Ar, the corresponding 3-aminomethylene-3-aryl oxindoles 230 were obtained in up to 97% yield and up to 95% ee (Scheme 85b). A ternary TS for the formation of the C–C bond has been proposed.
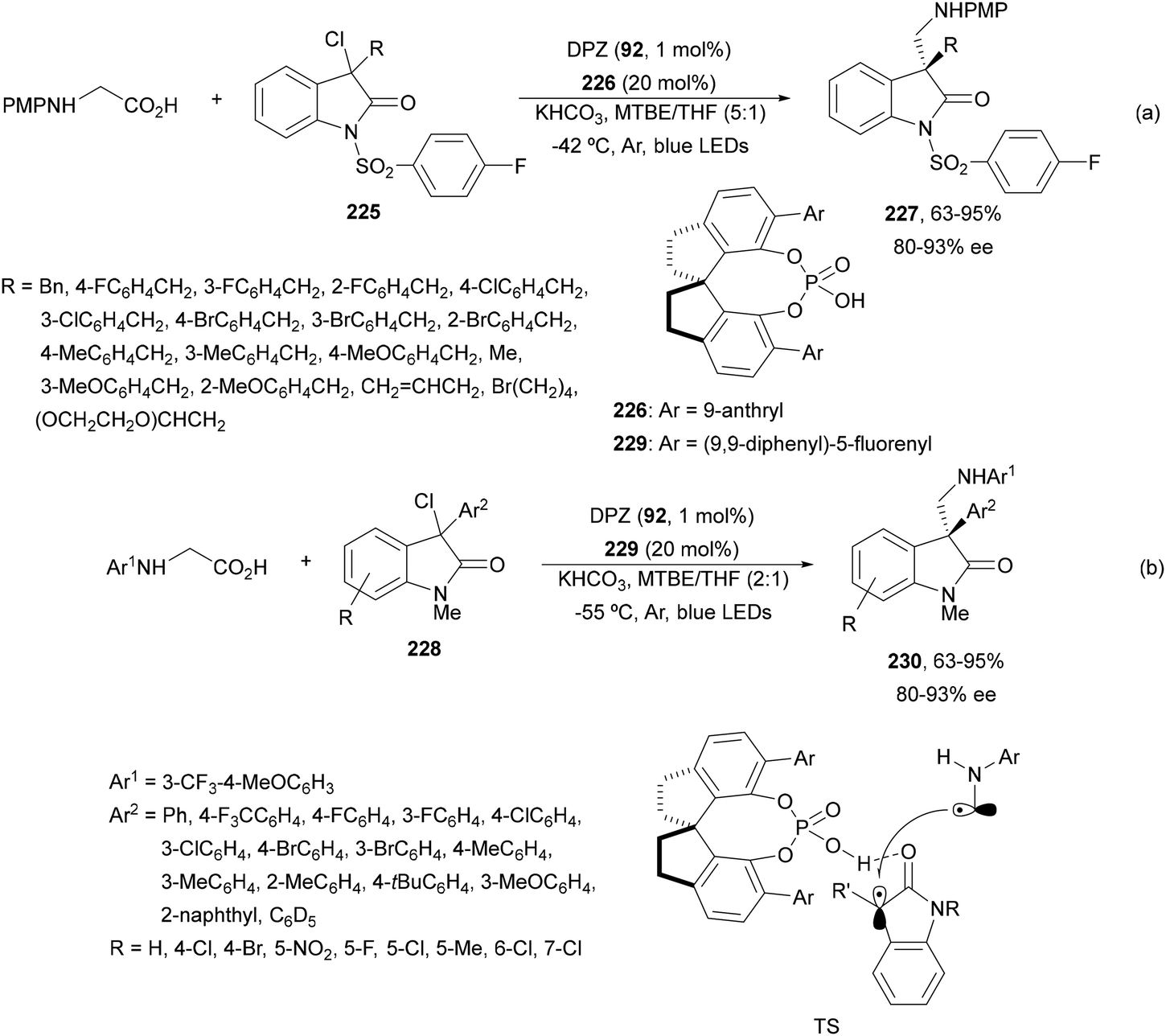 | ||
| Scheme 85 Enantioconvergent decarboxylative alkylation of 3-chloro substituted oxindoles 225 (a) and 228 (b) with N-aryl glycines under DPZ (92)/SPINOL-CPAs (226 and 229) photocatalysis. | ||
Meggers and co-workers182 reported an enantioconvergent140 cross-coupling of α-chloro imidazol-2-yl ketones 231 with N-aryl glycines photocatalyzed by chiral bis-cyclometalated chiral-at-Rh 232 in the presence of 4 Å MS, NaHCO3 as a base in DME at 5–7 °C to provide products 233 in up to 80% yield and up to 98% ee (Scheme 86). In the proposed TS, the complex formed by N,O-bidentate coordination of 2-acyl imidazole substrate to Rh catalyst gives the corresponding radical, which reacted with the aminomethyl radical to give the product.
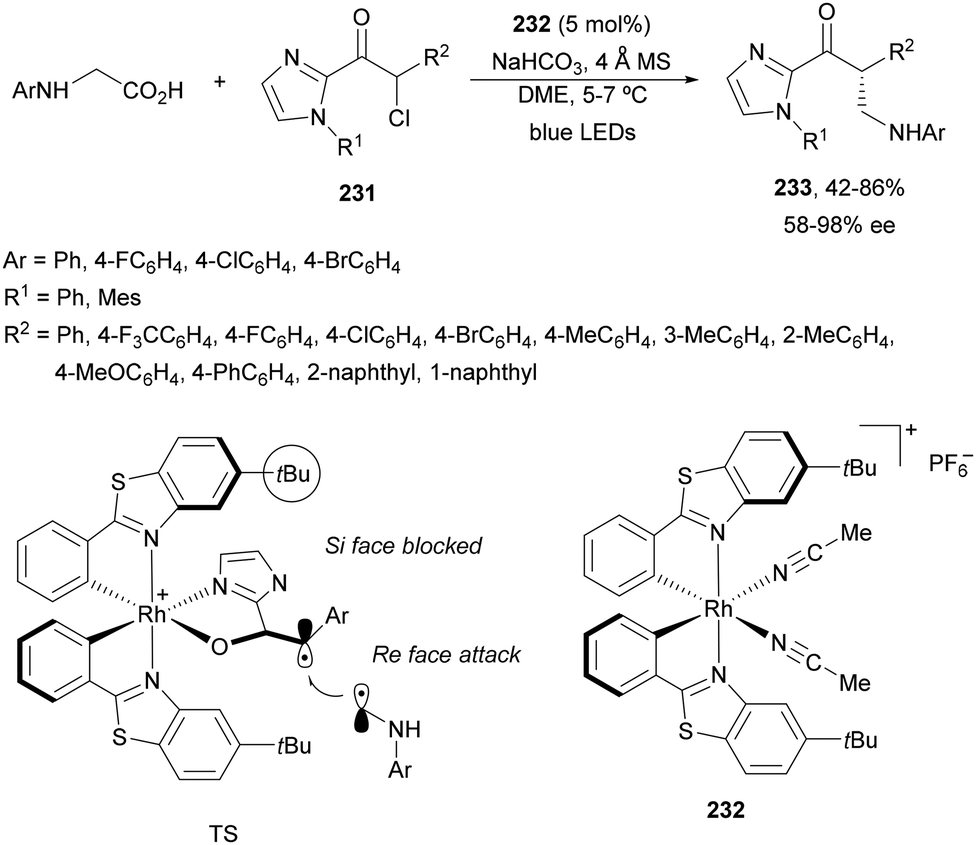 | ||
| Scheme 86 Enantioconvergent decarboxylative alkylation of α-chloro imidazolyl ketones 231 with N-aryl glycines under chiral Rh complex 232 photocatalysis. | ||
Decarboxylative trifluoromethylation of aliphatic carboxylic acids has been achieved by MacMillan and co-workers183 by combination of Ir/Cu dual photocatalysis. Togni's reagent 234 reacted with primary, secondary and tertiary carboxylic acids in the presence of Ir complex 4, CuCN, bathophenanthroline (Bphen) as a ligand, BTMG as a base in aqueous EtOAc at room temperature under blue LED irradiation to furnish alkyl-CF3 products 235 in moderate to good yields (Scheme 87). According to experimental studies, formation of CuCF3 was not detected and a copper-mediated decarboxylation is likely operative. Therefore, the carboxylate ion would ligate the Cu(II) catalyst and by a SET from intermediate A to Ir(III)* would deliver the Cu(III) carboxylate B. The dissociated form of B gives a carboxy radical and Cu(III) complex C. Extrusion of CO2 generates an alkyl radical and Cu(II) D, which recombines to form the Cu(III) species E. A second SET process between E and the reduced Ir(II) affords an alkylcopper(II) species F. Subsequent reaction of F with Togni's reagent delivers an alkyl-CF3 product 229via reductive elimination and complex G, which undergoes ligand exchange with the acid to give intermediate A. Several medicinal agents and natural products were decarboxylative trifluoromethylated by this protocol (Scheme 87).
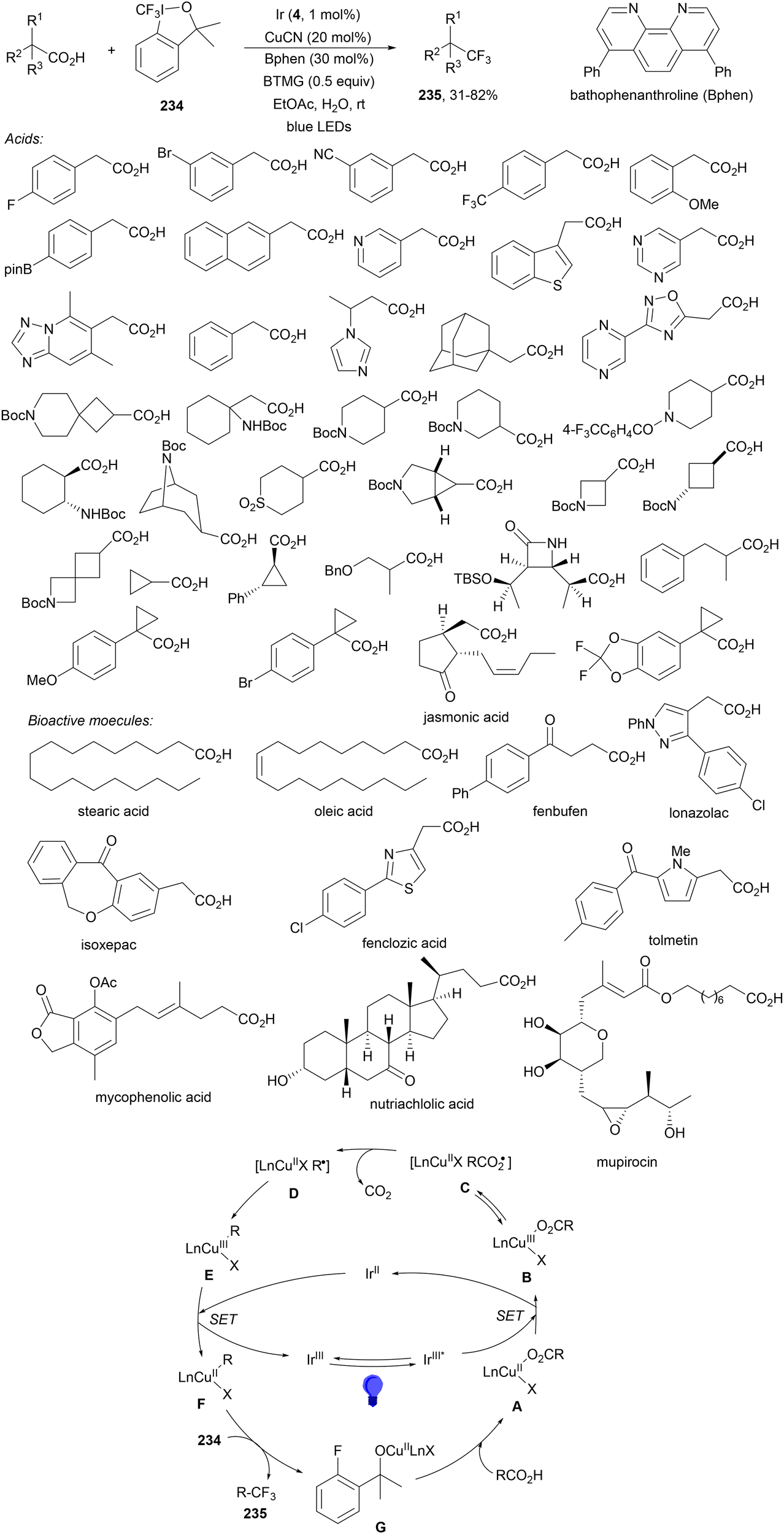 | ||
| Scheme 87 Decarboxylative trifluoromethylation of aliphatic carboxylic acids with Togni's reagent 234 under dual Ir/Cu photocatalysis. | ||
Decarboxylative cross-coupling of alcohols and carboxylic acids through dual combination of N-heterocyclic carbenes (NHC)-mediated deoxygenation and hypervalent iodine-mediated decarboxylation has been achieved by Sakai and MacMillan184 to form C(sp3)–C(sp3) bonds. Firstly, carboxylic acid was premixed with iodomesitylene diacetate to afford the activated iodonium dicarboxylate 236, which was used without additional purification. The alcohol was allowed to react with benzoxazolinium salt 237 to form the activated NHC–alcohol adduct 238 under basic conditions. Visible light irradiation of 230/232, using Ir complex 4 and the Ni complex 239 as catalysts gave coupled products 240 (Scheme 88). These alkyl-alkyl cross-coupled products include highly congested quaternary carbon centers. This methodology has been applied to functionalization of D-glucopyranose, isoandrosterone, cedrol and gemfibrozil. In the proposed catalytic cycle, Ir(III)* oxidized adduct 238a to give intermediate A, which after β-scission liberated the alkyl radical B, which was trapped by Ni catalyst 239 to form the Ni-alkyl intermediate C. Concurrently, reduction of the iodonium dicarboxylate 236a by Ir(II) gives radical D after CO2 extrusion and MesI, and regenerates the Ir(III) catalyst. Finally, Ni-catalyzed bond formation of radicals B and C gives product 240a and regenerates the Ni(II) catalyst.
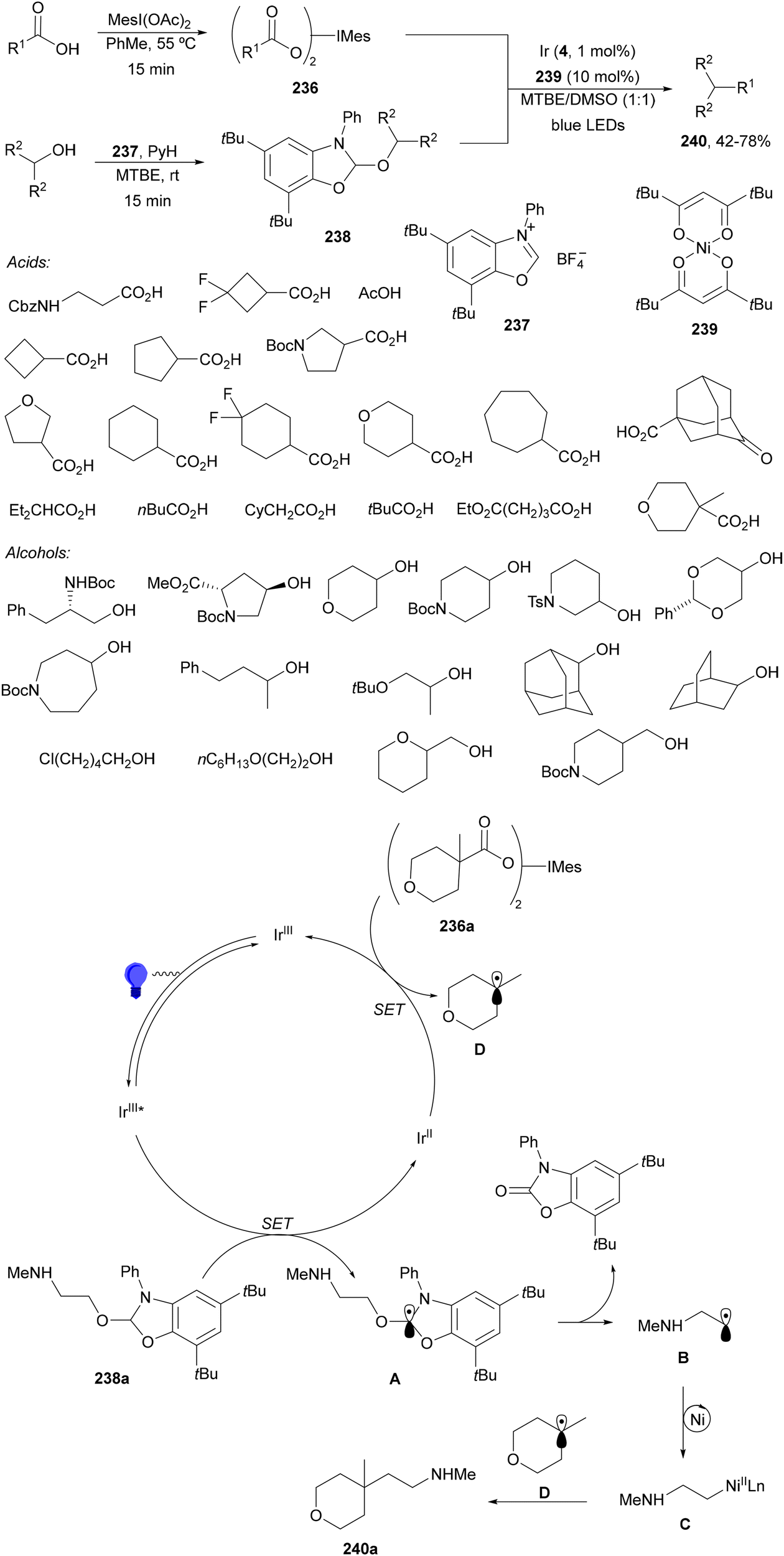 | ||
| Scheme 88 Decarboxylative cross-coupling of alcohols and carboxylic acids under Ir/Ni photocatalysis. | ||
Double decarboxylative cross-coupling of aliphatic carboxylic acids via a complementary sequence involves free radical generation, radical sorting by selective binding to Ni(II) and bimolecular homolytic substitution (SH2) at a Ni(III) alkyl complex. MacMillan and co-workers185 have achieved this elusive transformation by reaction of different primary, secondary and tertiary acids with MesI(OAc)2 to form in situ iodonium dicarboxylate 236, Ni(acac)2, potassium tri-(2,5-dimethyl-1-pyrazolyl) borohydride (K[Tp*]) as the ligand under 365 nm UV irradiation with thioxanthone (TX) as a sensitizer (Scheme 89a). A second protocol was set up for substrates which decomposed upon UV irradiation. For these reaction conditions, 450 nm light with 4CzIPN (25) as a PC was used. Apart from acetic acid other preactivated small alkyl carboxylic acids as iodomesitylene dicarboxylates were also employed to provide cross-coupled products with N-tosyl-4-pipecolic acid in 44–82% yields (Scheme 89b). In the photosensitization catalytic cycle, after irradiation with UV or visible light of TX or 4CzIPN, respectively, a long-lived, high energy triplet state is formed (2.8 or 2.6 eV, respectively) able to transfer energy to the hypervalent iodine(III) species A with a mixture of both carboxylate ions. After I–O homolysis and extrusion of CO2 two alkyl radicals B and C are formed. At this point, Ni(II) complex D would capture the less substituted radical B generating Ni(III) alkyl complex E, which undergoes SH2 homolytic substitution with radical C to yield the cross-coupled product and regenerates the Ni(II) catalyst D.
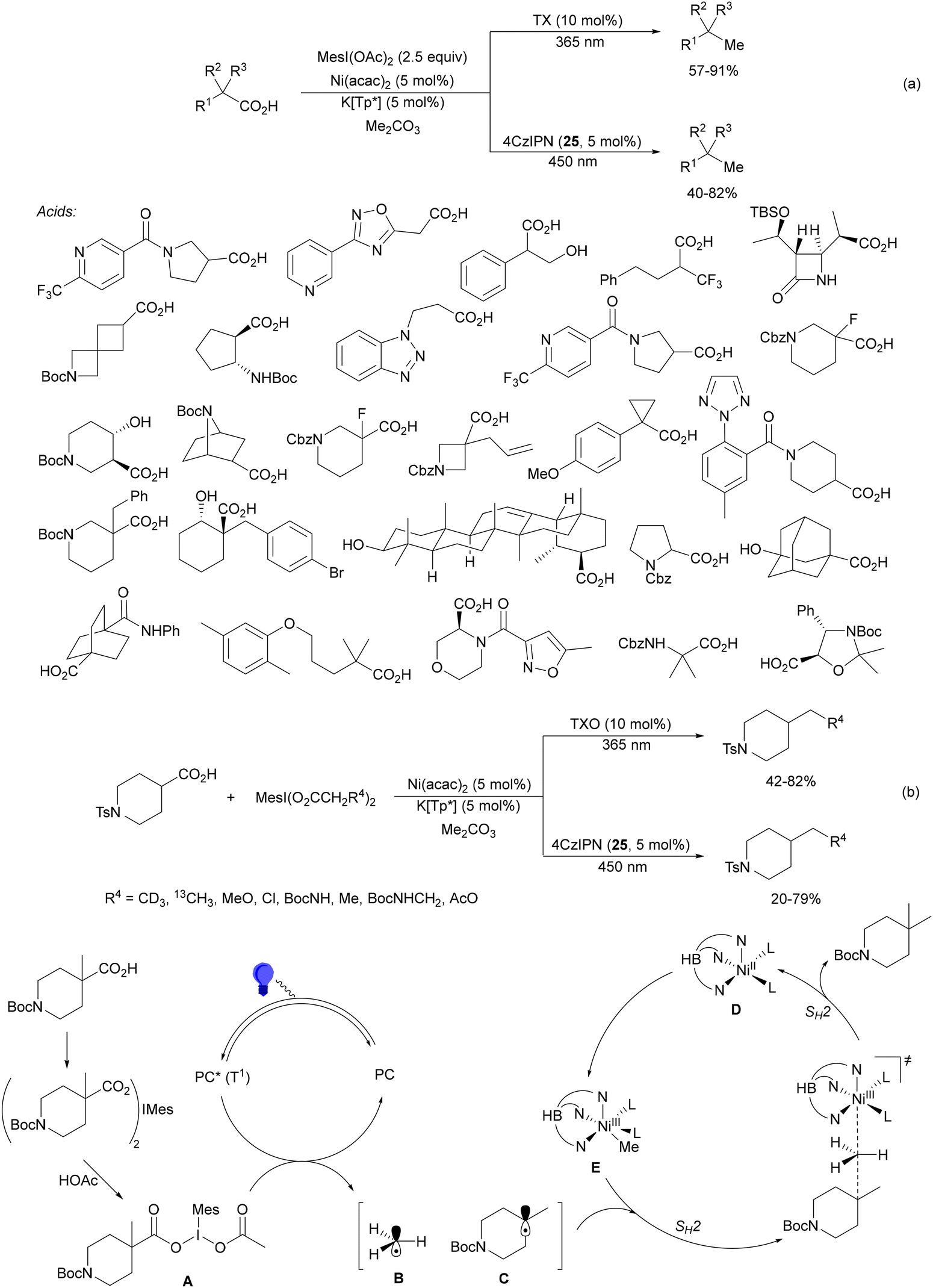 | ||
| Scheme 89 Double decarboxylative cross-coupling of aliphatic acids (a, b) under TX or 4CzIPN (25) and Ni(acac)2 photocatalysis. | ||
Murakami and co-workers186 recently performed a decarboxylative homocoupling of iodonium dicarboxylates 241, prepared from aryl acetic acids and phenyliodine(III) diacetate (PIDA), using 4CzIPN (25) as a PC under blue LED irradiation (Scheme 90). This process works as an energy transfer pathway to form 1,2-diarylethanes 242 with good yields. This radical homocoupling occurs thanks to the high reduction ability of 4CzIPN (25), giving two benzyl radicals A by decarboxylation, which reacted to give products 242. On the other hand, when [Ru(bpy)3]2PF6 was used as a PC, esters 243 were obtained through a SET pathway, which generated firstly a carboxylate ion B and a benzyl radical A. Subsequent single-electron oxidation of A gives cation C, which reacts with carboxylate ion B forming esters 243.
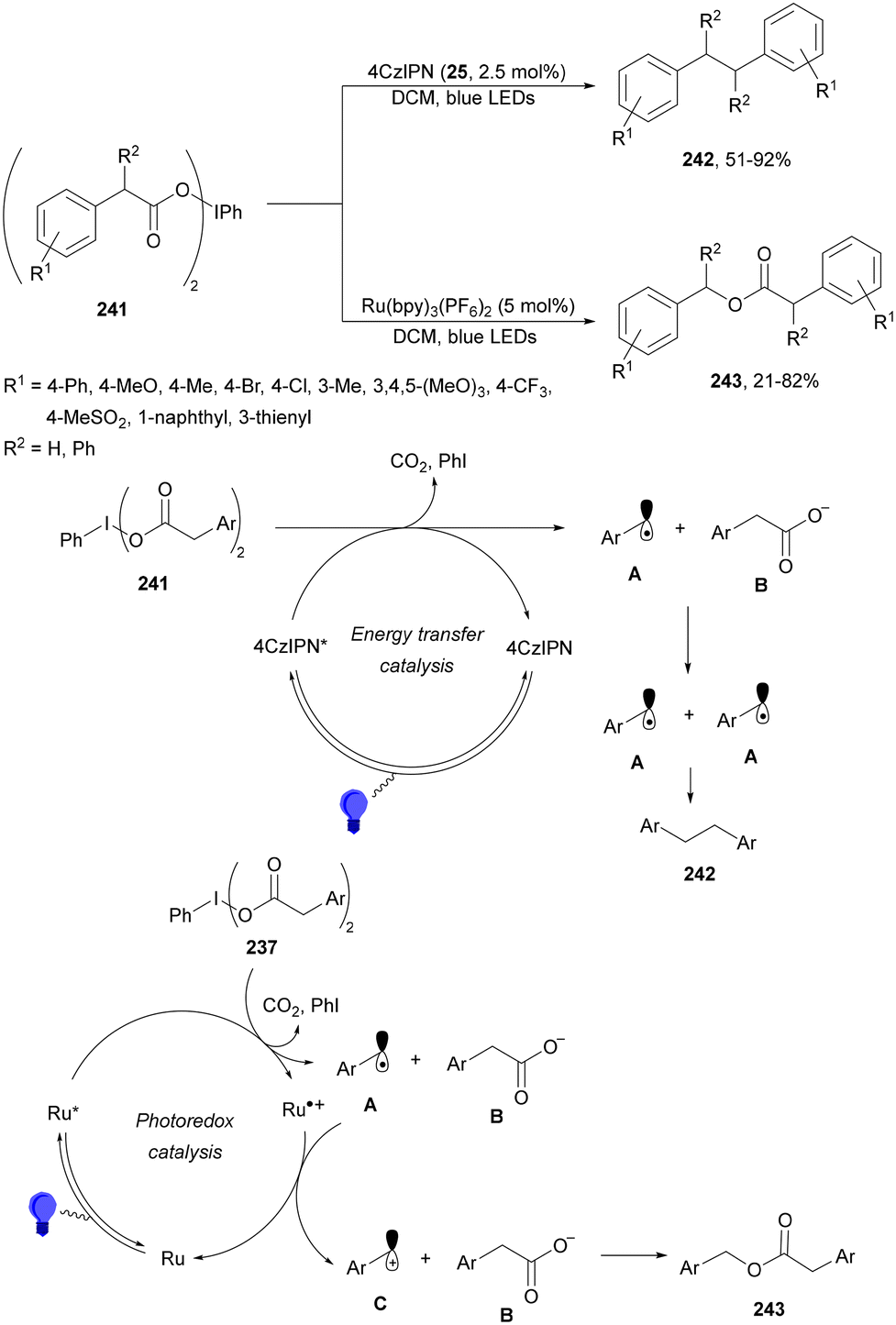 | ||
| Scheme 90 Decarboxylative homocoupling of iodonium dicarboxylates 241 under 4CzIPN (25) photocatalysis and esterification under Ru(bpy)3(PF6)2 photocatalysis. | ||
Redox active tertiary NHPI esters 69 have been coupled with alkyl bromides via iron porphyrin catalysis. This biomimetic C(sp3)–C(sp3) bond formation was reported by MacMillan and co-workers187via dual photoredox and iron catalysis. The reaction of esters 69 derived from AAs and α-oxy acids and primary alkyl bromides was performed with Ir complex 244 as a PC, Fe(OEP)Cl (245), amino silane 71, KOAc as a base in acetone/iPrOH (1:1) under blue LED irradiation to furnish cross-coupled products with good yields (Scheme 91). In the proposed mechanism, after irradiation of 244 this oxidizing Ir complex undergoes SET with the amino silane 71 to give a reduced Ir(II) complex. The oxidized silane reagent generates a silyl radical A, which abstracts a bromine atom from the alkyl bromide to give a primary alkyl radical B. This radical is captured by Fe(II) porphyrin 245 to furnish intermediate C. The Ir(II) complex reduces the NHPI ester via a SET to provide the tertiary radical D upon extrusion of CO2 and phthalimide. Intermediate C and radical D led to an SH2 reaction, giving the cross-coupled product and regenerating the Fe(II) catalyst.
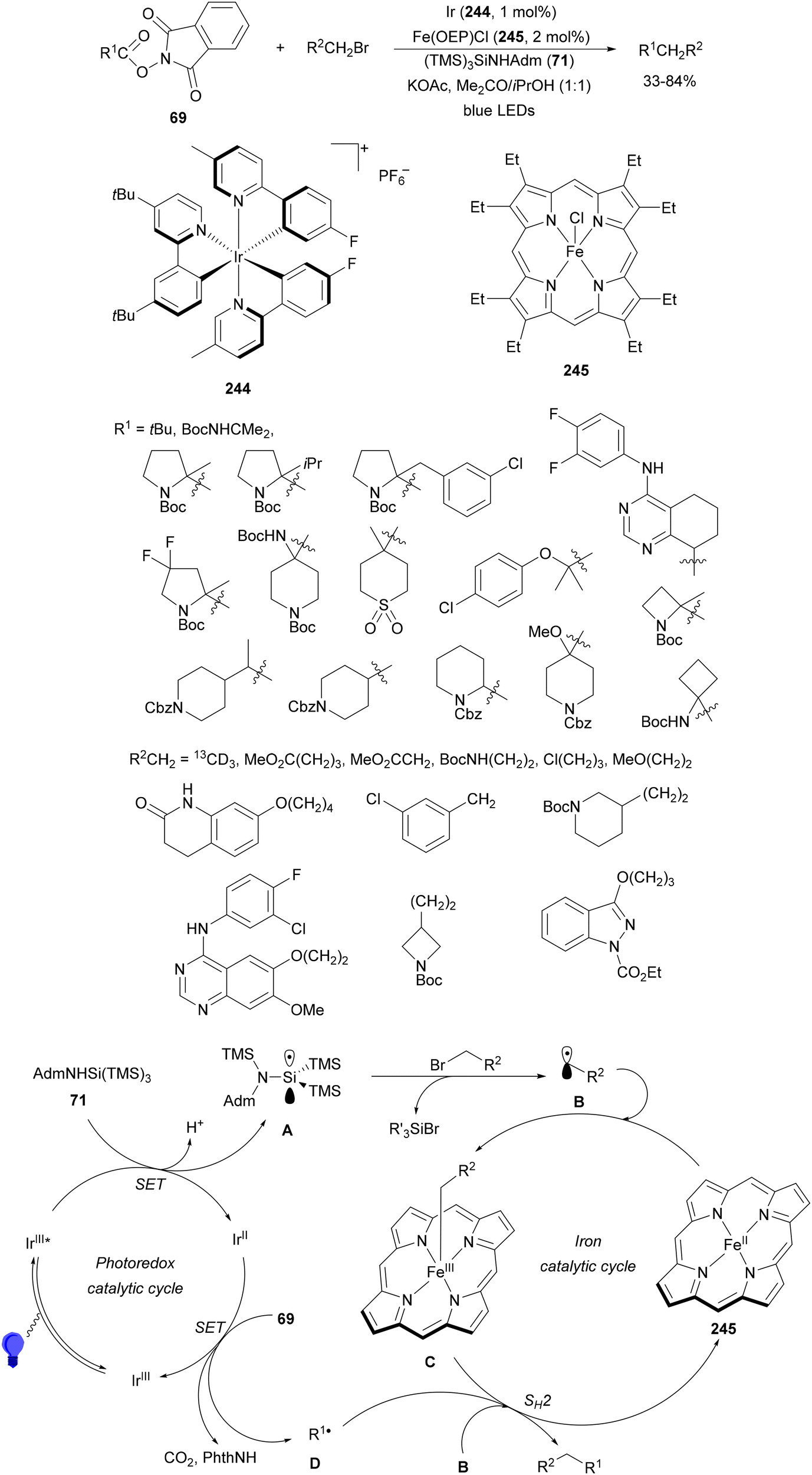 | ||
| Scheme 91 Decarboxylative cross-coupling of NHPI esters 69 with alkyl bromides under Ir (244)/Fe porphyrin (245) photocatalysis. | ||
An umpolung strategy (see Scheme 83) for the synthesis of C-alkyl glycosides 216 has been developed based on a Ni-catalyzed cross-coupling of glycosyl halides 246 and aliphatic NHPI esters 69 in the presence of HE and LiI (Scheme 92).188 In this case, NiBr2·diglyme and ligand 247 were used in DMA/MTBE as solvents at room temperature under blue LED irradiation to give glycosides 216 with moderate to good yields. As glycosyl halides, D-mannofuranose, D-ribofuranose, D-galactofuranose and L-rhamnopyranose derivatives were employed to give after reaction with 3,4-(OCH2O)C6H4CH2CH2NHPI the corresponding glycosides 216 in 39–80% yields and moderate to excellent control of diastereoselectivity. Based on previous disclosures by Molander170 (Scheme 75), a tentative mechanism was proposed for the photoinduced Ni-catalyzed glycosylation reaction. NHPI esters in the presence of HE and LiI generates an EDA complex A. Upon blue LED irradiation B and C species were formed. Intermediate C suffers decarboxylative fragmentation to afford D and radical species E. In the Ni-catalyzed cycle, Ni(II) reacts with glycosyl halide to deliver F and a glycosyl radical, which reassociates with the Ni centre to generate the Ni(II)-species G. Radical E is trapped by G to the Ni(III)-species H, which after reductive elimination provides the Ni(I)-species I and the C-alkyl glycoside product. Ni(I)-species I regenerates the Ni(0)-species by reaction with B to form the pyridine by-product J.
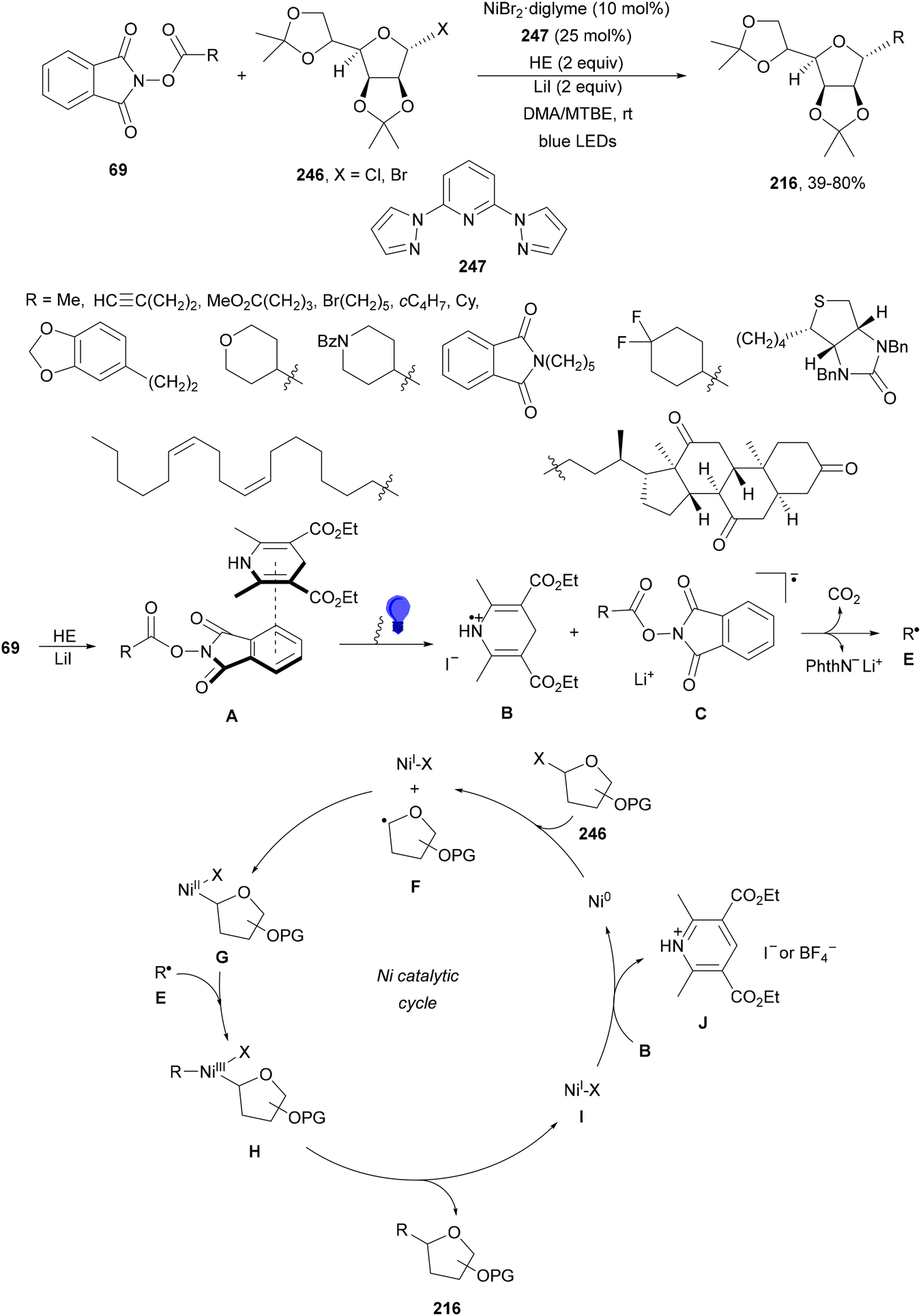 | ||
| Scheme 92 Decarboxylative cross-coupling of NHPI esters 69 with D-mannofuranose derivative 246 under HE and NiBr2·247 photocatalysis. | ||
Photoredox Cu-catalyzed decarboxylative C(sp3)–C(sp3) cross-coupling of aliphatic NHPI esters 69 with diborylalkyl organometallic reagents 248 provided gem-diborylalkanes 249 (Scheme 93).189 This process was carried out using Ir complex 4. CuCl, DBU as a base in DCE at room temperature and (diborylalkyl)lithium 248 were allowed to react in situ with ZnBr2 to form the corresponding (diborylalkyl)zinc species. Products 249 were obtained with moderate to good yields for primary, secondary and tertiary carboxylic acid derivatives 69, including bioactive molecules such as pharmaceuticals and natural compounds, namely, gemfibrozil, enoxolom, oleanolic acid, betulinic acid and lithocholic and dehydrocholic acids, and APIs such as isoxepac, tolmetin, oxaprozin, indomethacin and gabapentin derivatives have been successfully employed. The prepared gem-diborylalkanes are valuable building blocks in organic synthesis, especially in cross-coupling transformations. Based on mechanistic experiments, two catalytic cycles have been proposed to explain this photocatalytic cross-coupling. Diborylmethylzinc halide reagents react with Cu(I) to form diborylmethylcopper(I) complex A by a transmetallation process. Subsequently, via SET with PC* A gives the Cu(II) intermediate B and PC−˙. SET from PC−˙ to NHPI ester 69 gives the alkyl radical C, which can be trapped by B to generate the Cu(III) intermediate D. Final reductive elimination affords the gem-diboryl product.
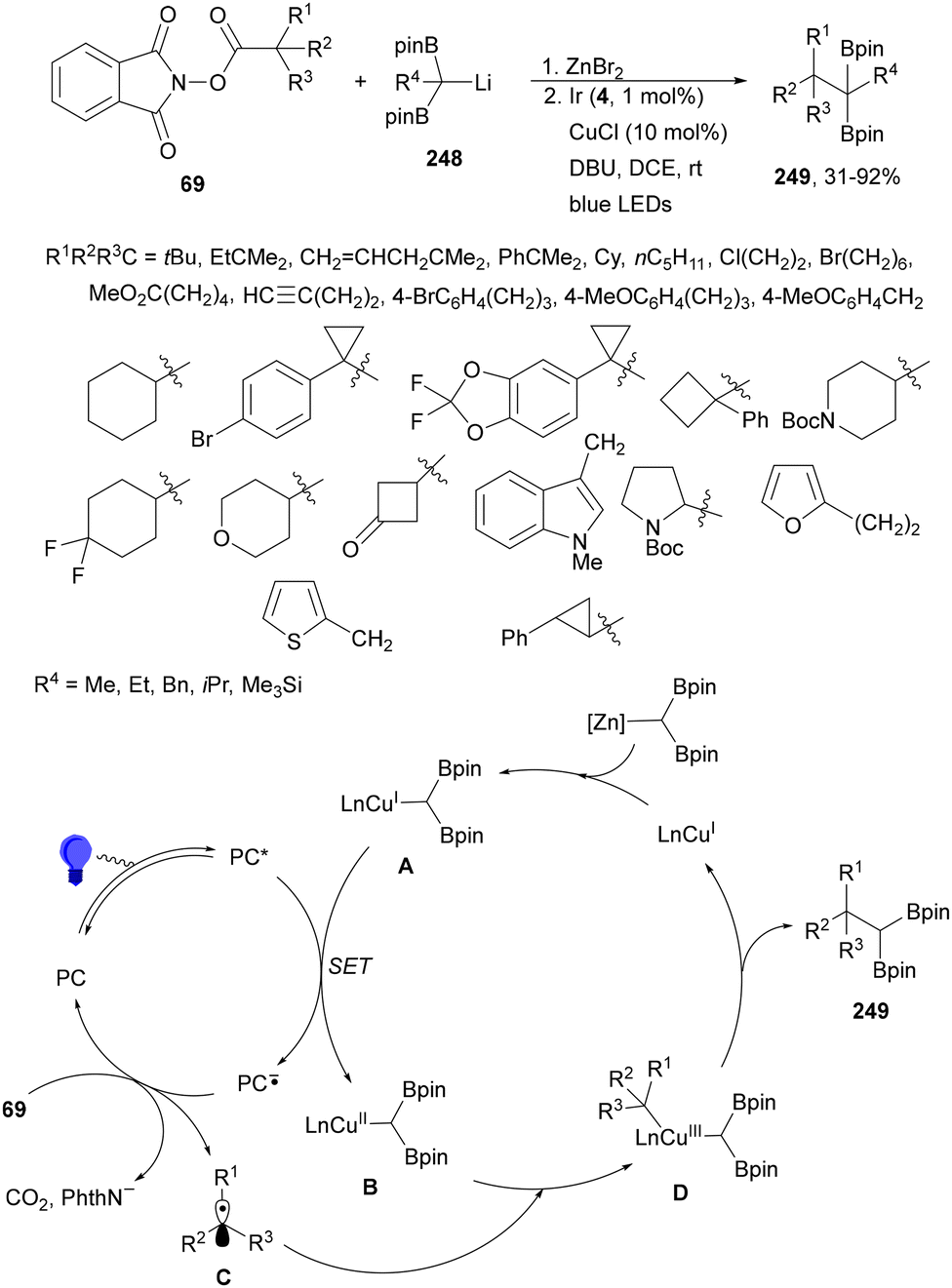 | ||
| Scheme 93 Decarboxylative C(sp3)–C(sp3) cross-coupling of aliphatic esters 69 with diborylalkyl organozinc compounds under Ir/Cu photocatalysis. | ||
Liu and co-workers190 recently reported enantioselective decarboxylative difluoromethylation of aliphatic NHPI esters 69 under Ir/Ni dual photocatalysis. As a nucleophile, (DMPU)2Zn(CF2H)2 (250)191 was used for difluoromethylation of the Ni(II) catalyst. Under blue LED irradiation, Ni(ClO4)2·6H2O, chiral oxazoline ligand 251 and Ir complex 4 as a PC enable enantioconvergent140 preparation of cross-coupled products 252 with up to 89% yield and >99% ee (Scheme 94). This asymmetric difluoromethylation protocol was applied to the synthesis of fluorinated analogues of bioactive molecules. Difluoromethylation of dipeptides, tripeptides and the pentapeptide Leu-enkephalin afforded the corresponding fluorinated derivatives. Other drug analogues of lisdexafematine, dobutamine and ixazomib were difluoromethylated. In the proposed mechanism, the Ir catalytic cycle generates the alkyl radical A from the NHPI ester. In the Ni-catalytic cycle, complex B reacts with 250 to give a Ni(I)–CF2H species C, which by oxidation with Ir(IV) provides a Ni(II)–CF2H complex D. Oxidative substitution of D by the alkyl radical A produces a high-valent alkylNi(III)–CF2H species E. Reductive elimination of the product and regeneration of the Ni(I) catalyst B closes the catalytic cycle. An alternative pathway, involving the quenching of Ir(III)* by Ni(I) species, cannot be ruled out.
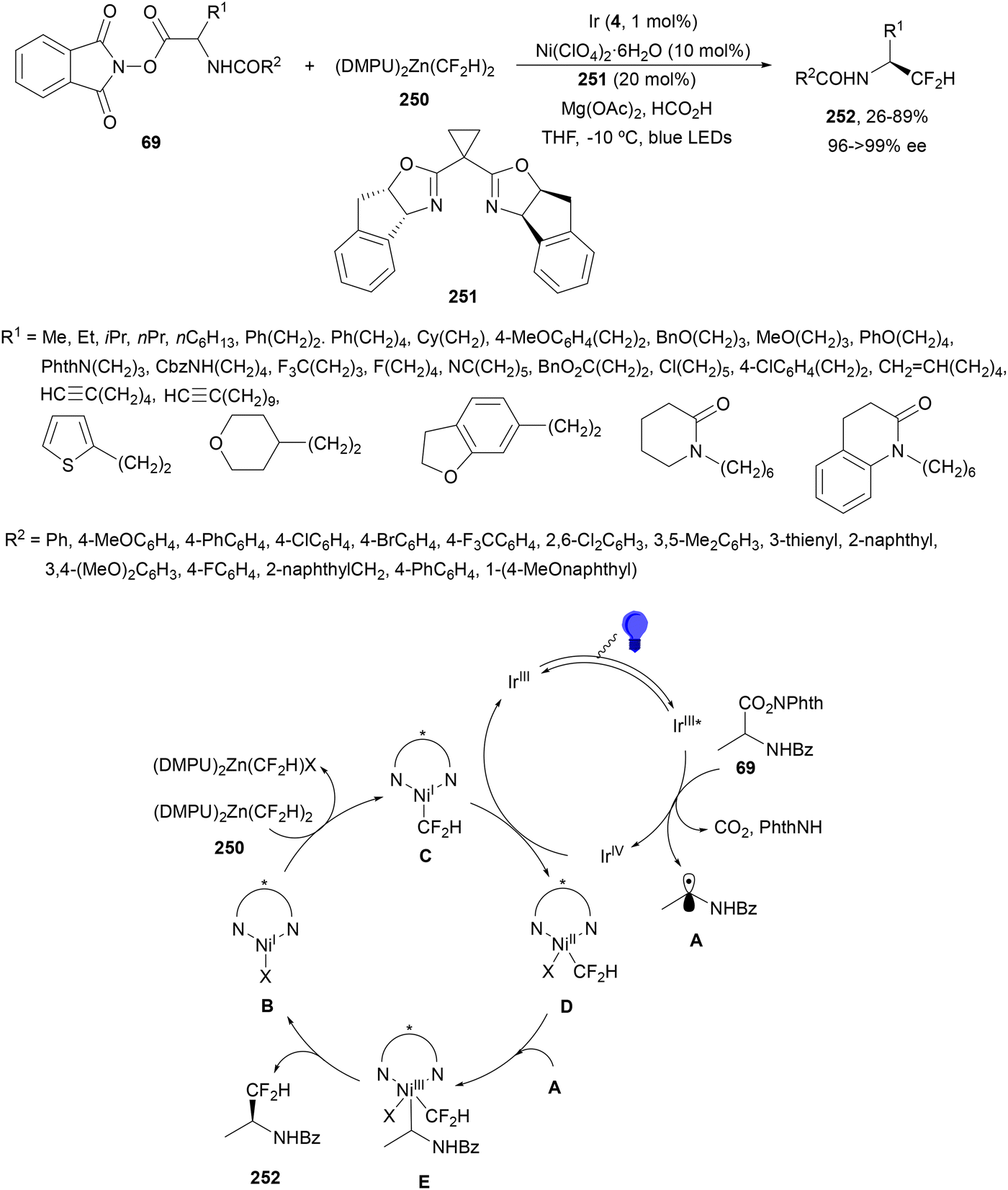 | ||
| Scheme 94 Enantioconvergent decarboxylative difluoromethylation of NHPI esters 69 under Ir/Ni photocatalysis. | ||
Decarboxylative alkylation of 1,3-dicarbonyl compounds with NHPI esters 69 has been carried out under photoredox/Fe/chiral primary amine triple catalysis. This enantioconvergent140 process employed Ir complex 4, iron porphyrin 245 and (S)-253 as a chiral primary amine in a 1:1 mixture of MTBE and PhCF3 at 20 °C under blue LED irradiation to provide products 254, bearing a quaternary stereocenter with good yields and enantioselectivities (Scheme 95).192 Different functionalized NHPI esters 69 were compatible with these reaction conditions. In the case of 1,3-dicarbonyl compounds, 2-oxocycloalkyl esters and 2-oxocyclopentanecarboxamides were appropriate nucleophiles and also acyclic β-keto esters. In the proposed mechanism, the reaction commences by formation of the enamine A between the β-keto ester and the primary amine (S)-253, which is transformed into radical species B by a SET from Ir(III)* and Ir(II). This Ir(II) reduces the NHPI ester to give after decarboxylation the primary alkyl radical C, which is captured by porphyrin Fe(II) D to give alkyl–Fe(OEP). Then, an outer-sphere radical rebound takes place between B and E to form imine F and Fe(II) complex D. Finally, hydrolysis of F yields the corresponding product.
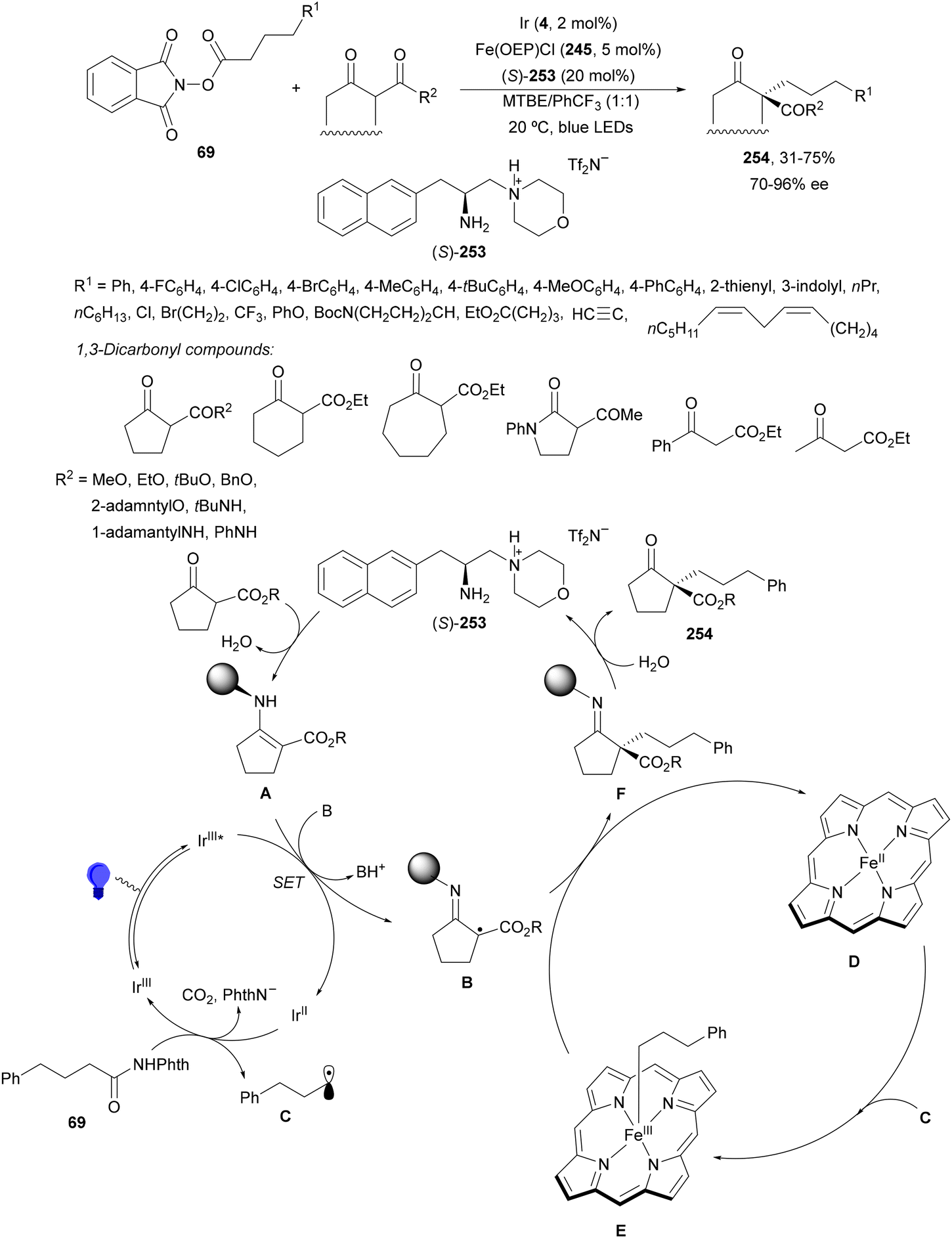 | ||
| Scheme 95 Enantioconvergent decarboxylative alkylation of 1,3-dicarbonyl compounds with NHPI esters 69 under photoredox/Fe/chiral amine 253 triple catalysis. | ||
Silyl enol ethers 255 can also be alkylated with aliphatic NHPI esters 69 under photocatalytic decarboxylation with a combination of triphenylphosphine and sodium iodide.193 α-Alkylated phenones 256 were obtained with good yields using 20 mol% of PPh3 and 150 mol% of NaI in MeCN at room temperature under blue LED irradiation (Scheme 96). This metal-free, low-cost catalytic system is appealing for a large-scale synthesis. DFT calculations and natural bond orbital analysis suggest that complexation to give a charge transfer complex (CTC) A, of NHPI ester, NaI and PPh3 is endergonic (−3.8 kcal mol−1) but after irradiation, the favorable formation of the Ph3P–I˙ (B) radical is 11.9 kcal mol−1 exergonic. In the catalytic cycle, radical C, from the NHPI ester, reacts with silyl enol ether to give radical D, which after SET provides cationic intermediate E, a precursor of product 256.
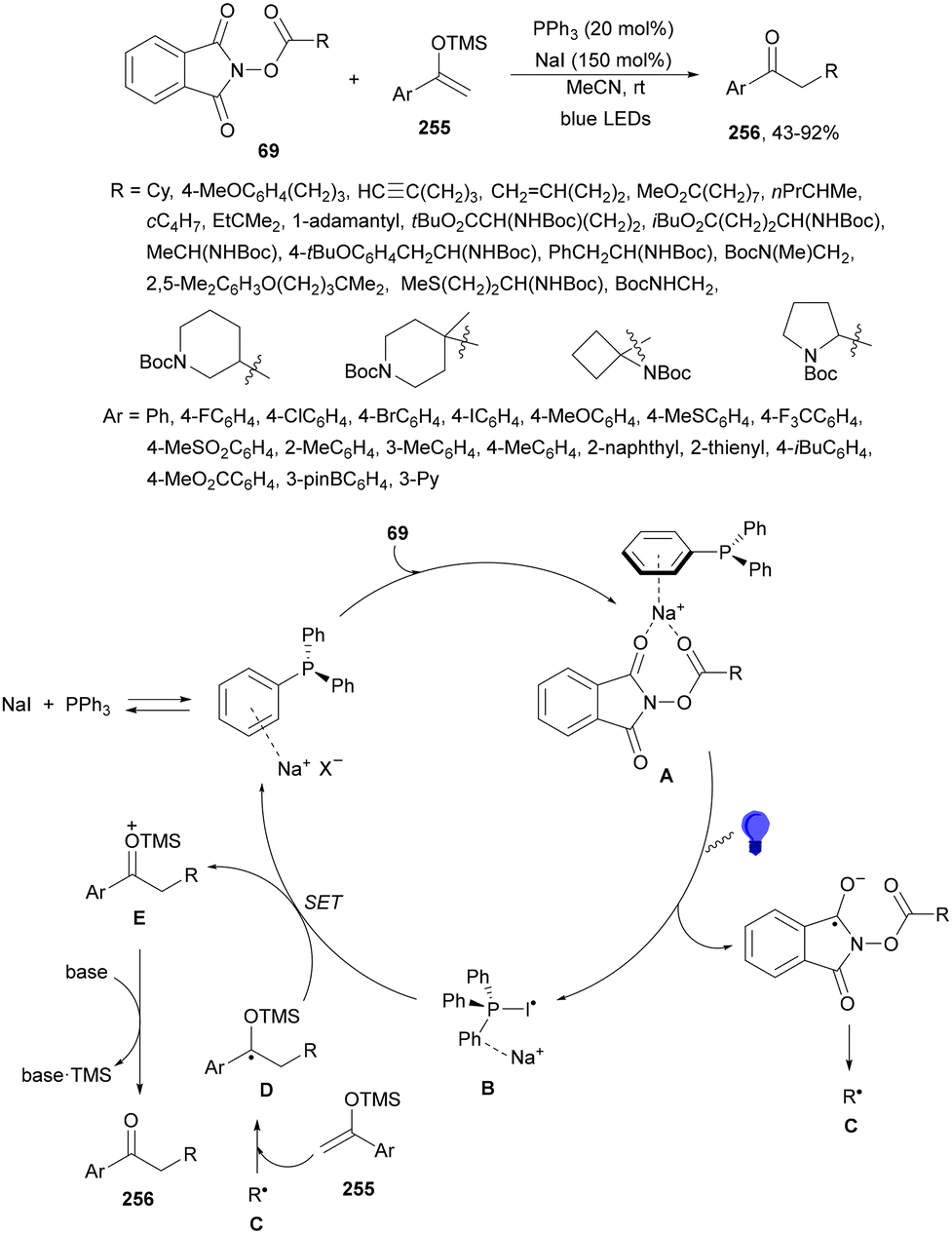 | ||
| Scheme 96 Decarboxylative alkylation of silyl enol ethers 255 with NHPI esters 69 under PPh3/NaI photocatalysis. | ||
Decarboxylative alkylation of difluoroenoxysilanes 257 with NHPI esters using Ir photocatalyst 89 gave α,α-difluoro phenones 258.194 This transformation has been carried out in DMF at 60–65 °C under blue LED irradiation giving products 258 with very good yields even on a gram-scale (Scheme 97). In the case of cyclopropanecarboxylic acid derivatives, both trans- and cis-1-phenyl-2-carboxycyclopropanes gave trans-alkylated products. In the proposed catalytic cycle, radical A formed by photocatalysis of NHPI esters reacts with difluoroenoxysilane 257 to form radical B, which after SET gives cationic species C, the precursor of product 258.
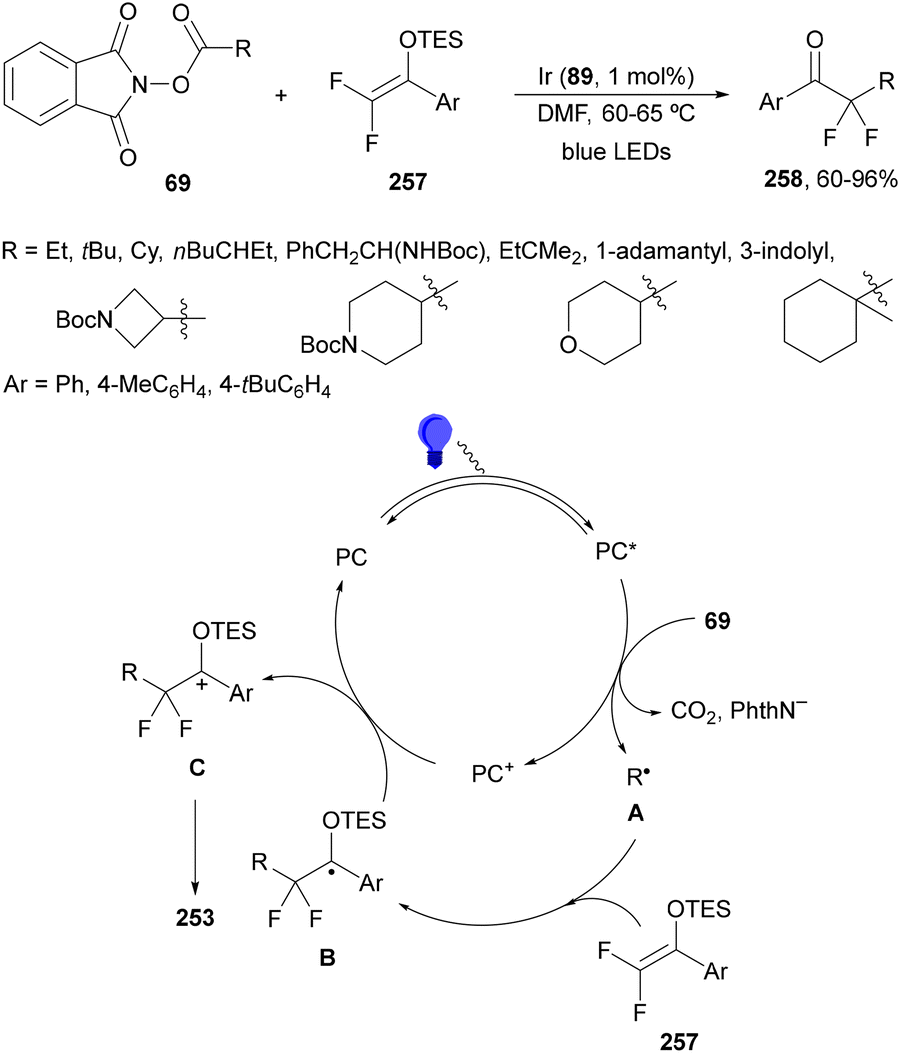 | ||
| Scheme 97 Decarboxylative alkylation of difluoroenoxysilanes 257 with NHPI esters 69 under Ir(ppy)3 (89) photocatalysis. | ||
Decarboxylative cross-coupling of aliphatic carboxylic acids with alkyl halides has been achieved under Ni/Ir dual photoredox catalysis. Enantioconvergent processes have been carried out between amino acids and α-bromo ketones or 3-chloro isoindoles using DPZ and SPINOL as CPA. In the case of α-chloro imidazolyl ketones a chiral Rh complex was employed. Hypervalent iodine reagents were used as intermediates for the trifluoromethylation of carboxylic acids under Ir/Cu photocatalysis. For the alkylation of alcohols and for the double decarboxylation Ni/organocatalyst have been used. In the case of homocoupling of iodonium dicarboxylates 4CzIPN or a Ru complex were employed as photocatalysts. Concerning cross-coupling of aliphatic NHPI esters with alkyl halides, iron porphyrin or NiBr2/Hantzsch ester has been used as a PC. Difluoromethylation of NHPI esters has been performed with organozinc reagents under Ni photocatalysis. Enantioconvergent alkylation of 1,3-dicarbonyls with aliphatic NHPI esters was achieved with Ir/Fe porphyrin as a catalyst in the presence of a chiral primary amine. Silyl enol ethers have been alkylated with NHPI esters with PPh3/NaI or an Ir complex as a PC.
2.7. Allylation and benzylation reactions
Decarboxylative allylation by photoredox catalysis is a direct strategy to build C(sp3)–allyl bonds from aliphatic carboxylic acids. Zhu and co-workers195 reported in 2017 a redox-neutral process under mild reaction conditions based on the allylation of N-aryl AAs with allyl sulfone 259, acting as a radical acceptor, under visible light irradiation. This process was carried out using Ir complex 260 and Cs2CO3 as a base in aqueous MeCN at room temperature to give products 261 with in general good yields (Scheme 98). In the proposed mechanism, Ir(III)* promotes a SET process to N-aryl glycine, which upon loss of H+ and CO2 delivers the α-amino radical A and Ir(II). Radical addition/fragmentation processes of radical A and the allyl sulfone provides the product and sulfinyl radical B, which after oxidation of Ir(II) completes the catalytic cycle. | ||
| Scheme 98 Decarboxylative allylation of N-aryl AAs with allylsulfone 259 under [Ir(ppy)2(bpy)]PF6 (260) photocatalysis. | ||
Dual catalytic decarboxylative allylation and benzylation methods employing carbonates as π-electrophiles have been performed using Pd(0) and 4CzIPN (25) as catalysts by Cartwright and Tunge.196 A variety of carboxylic acids such as acyclic and cyclic AAs, α-alkoxy acids, aryl acetic acids and other aliphatic carboxylic acids reacted with carbonate 262 to give the corresponding allylated products 263 with good yields under blue LED irradiation (Scheme 99a). Branched acyclic allylic carbonates 265 were allowed to react with N-Boc AA 264 to provide stereoselectively products 266 with good yields (Scheme 99b). In the case of styryl carbonates 267, a mixture of Z/E diastereoisomers 268 were obtained (Scheme 99c). Decarboxylative benzylation worked with benzylic carbonates 269 bearing an extended conjugation to provide products 270 in moderate yields (Scheme 99d). The proposed catalytic cycle for allylation proceeds via a reductive quenching pathway in which the excited 4CzIPN* is quenched by a π–allylPd carboxylate species A. SET from the carboxylate ion to Pd gives species B, which by decarboxylation forms a carbon radical. Rebound of this radical with the π–allylPd species results in the formation of an allylated product and Pd(I), which can be reduced by 4CzIPN˙− to complete the cycle.
 | ||
| Scheme 99 Decarboxylative allylation and benzylation of aliphatic carboxylic acids (a–d) with allylic and benzylic carbonates under Pd/4CzIPN (25) dual photocatalysis. | ||
Regio- and enantioconvergent140 allylation of aryl acetic acids has been performed under dual Pd/Ir photoredox catalysis.197 Allylic acetates 271 reacted with aryl acetic acids 103 in the presence of Pd2(dba)3 and chiral ligand 273 with Ir(dFMeppy)2(dtbbpy)PF6 (272) as a PC and K2CO3 as a base in DMA as solvent to furnish products 274 with good yields and up to 94% ee (Scheme 100a). This decarboxylative allylation took place with regioselectivity but other carboxylic acids did not undergo this AAA. Vinyl epoxides 275 were benzylated under the same photocatalytic conditions using MeO-biphep (276) as a chiral ligand and Cs2CO3 as a base in MeCN, affording homoallylic alcohols 277 bearing all-carbon quaternary stereocenters in moderate to good yields with good regio- and enantioselectivities (Scheme 100b). In the proposed plausible pathway, the aryl acetic acid is reduced by the excited PC to give the benzyl radical A and CO2. In the Pd catalytic cycle, oxidative addition between Pd(0) and allylic acetate 271 leads to π–allylPd intermediate B. Trapping of radicals A and B produces the Pd(III) species C, which after reductive elimination gives product 274 and Pd(I) species D. Finally, the Ir(II) complex reduced D to Pd(0) and also regenerated the PC.
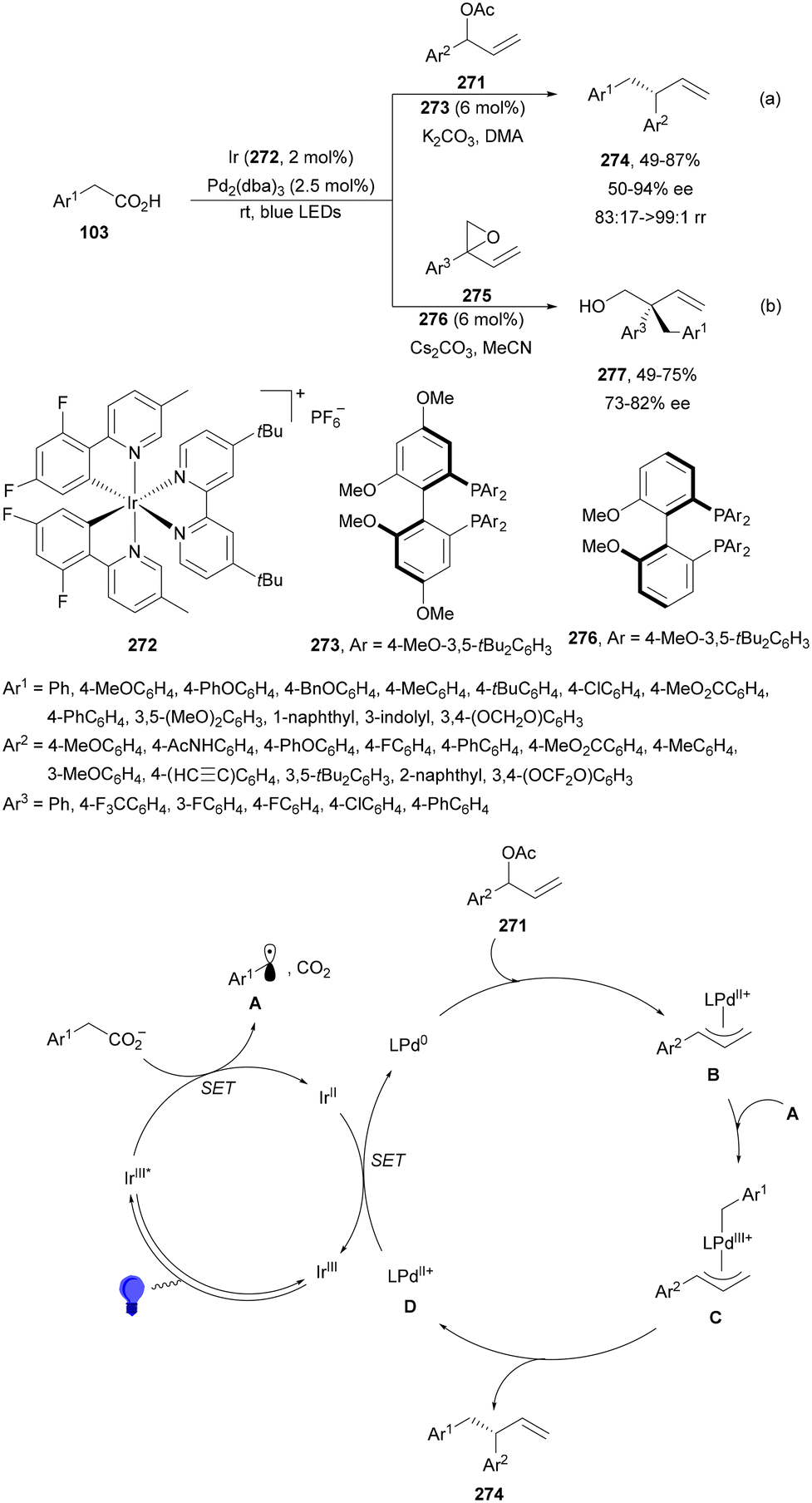 | ||
| Scheme 100 Decarboxylative enantioconvergent allylation of aryl acetic acids with allylic acetates 271 (a) and vinyl epoxides 275 (b) under dual Pd/Ir (272) photocatalysis. | ||
Breit and co-workers198 studied decarboxylative allylation of N-aryl AAs with allylic carbonates using dual Ni/Ir photoredox catalysis. Homoallylic amines 279 were obtained by reaction of N-aryl AAs with allylic carbonates 278 in the presence of NiCl2·glyme and 2,2′-bipyridine as a ligand, Ir(ppy)2(dtbbpy)PF6 (52) as a PC, Cs2CO3 as a base in MeCN at room temperature under blue LED irradiation (Scheme 101a). Compounds 279 were isolated with, in general, very good yields and up to >95:5 dr. When carbonates 280 derived from cinnamyl alcohol and derivatives were allowed to react with N-aryl AAs, branched products 281 were prepared in moderate yields and excellent diastereoselectivities (Scheme 101b). These homoallylic amines were further subjected to cyclization reactions to obtain five- and six-membered rings. Control experiments and DFT calculations supported the proposed mechanism depicted in Scheme 101. After generation of radical A from N-aryl AA, the Ni(0) complex B reacts with allylic carbonate 278 to give by oxidative addition the σ-allyl species C in equilibrium with the π–allyl intermediate D. Radical capture of C forms the Ni(II) intermediate E, which by an oxidative proton coupled electron transfer (PCET) process, the α-amino radical is oxidized to an imine, whereas Ni(II) is reduced to Ni(I) species F. Subsequently, a nucleophilic attack from the Ni-bound allylic moiety at the imine occurs to lead to intermediate G through an eight-membered ring TS. Afterward, the product is liberated and the Ni(I) species H is reduced to Ni(0) by the PC.
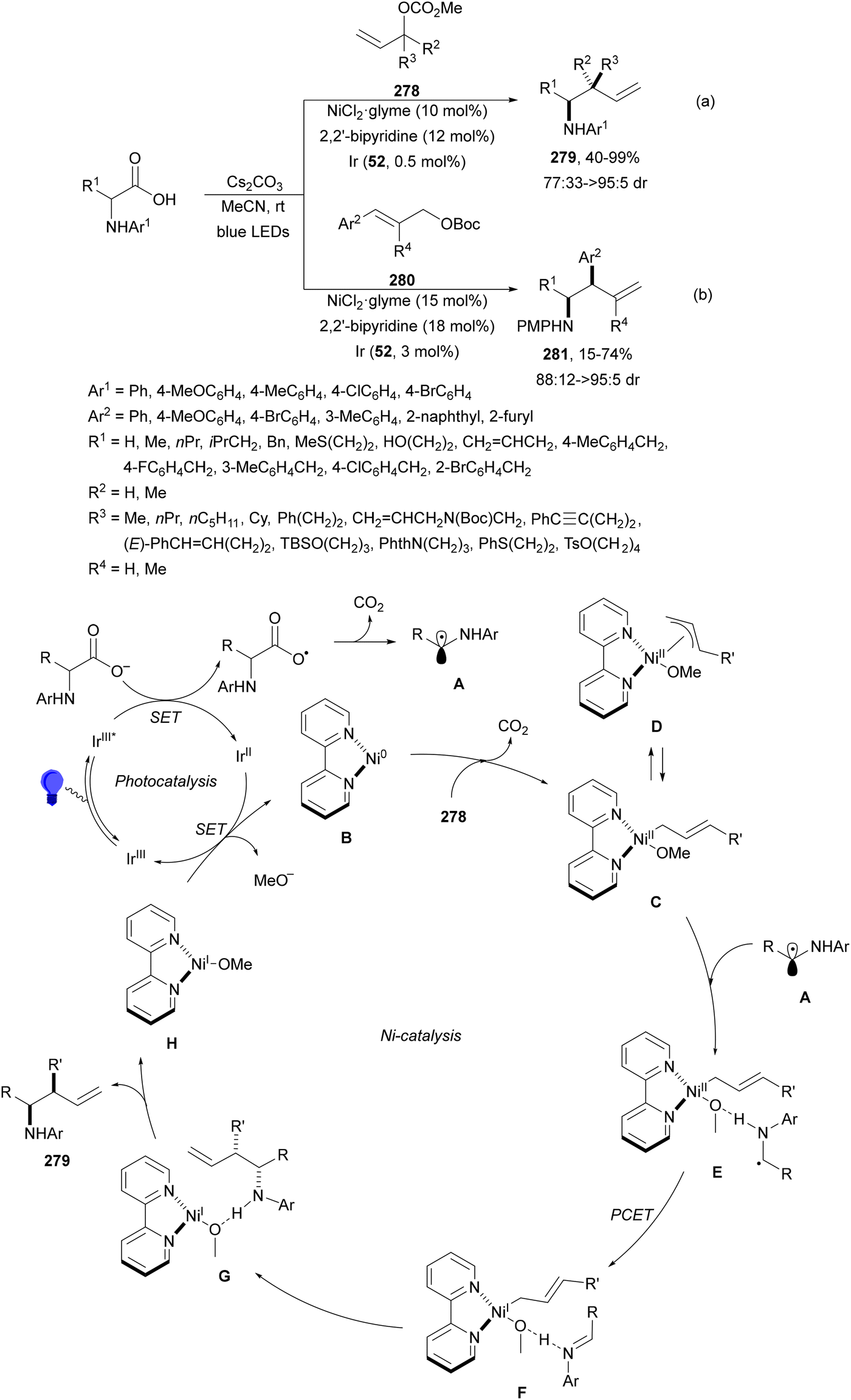 | ||
| Scheme 101 Decarboxylative allylation of N-aryl AAs with allylic carbonates 278 (a) and 280 (b) under Ni/Ir (52) photocatalysis. | ||
Decarboxylative allylation of carboxylic acids with vinyl cyclopropanes 282 has been reported by Sureshkumar and co-workers199 under Ir complex 4 photocatalysis. Different aliphatic carboxylic acids including α-AAs reacted with vinyl cyclopropanes 282 using Cs2CO3 as a base in DCE under blue LED irradiation to give products 283 with 48–94% yields (Scheme 102). The resulting γ,δ-unsaturated diesters and homoallyl amino acid derivatives were obtained mainly as E-diastereomers. A plausible mechanism is proposed, which starts with the formation of radical A after oxidation by photoexcited Ir(III)*. This radical A reacts with 282 to form the radical species B, which after ring-opening affords radical C, which undergoes a SET process to form Ir(II) and anionic intermediate D. Finally, protonation of D gives product 283.
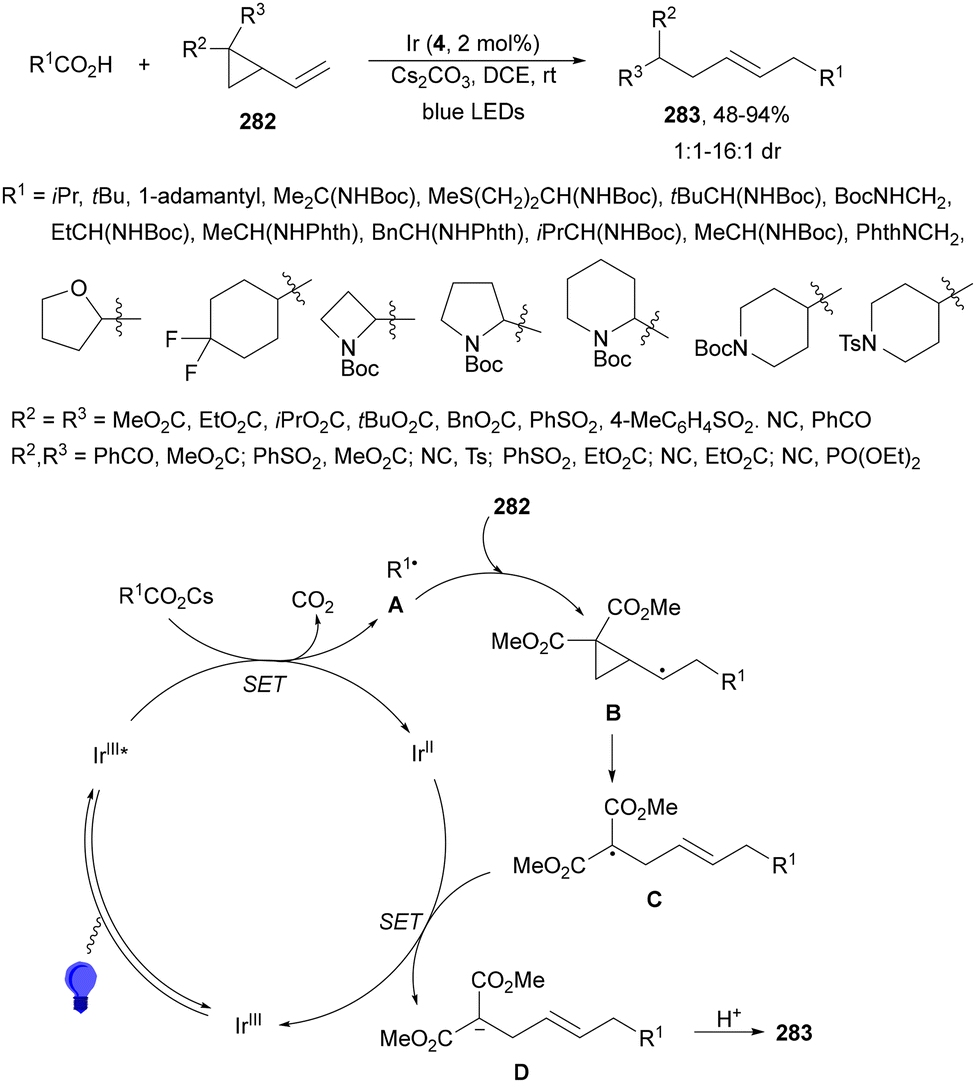 | ||
| Scheme 102 Decarboxylative allylation of aliphatic carboxylic acids with vinyl cyclopropanes 282 under Ir (4) photocatalysis. | ||
Recently, Yang, Xia and co-workers200 reported photoinduced ligand-to-copper charge transfer for decarboxylative allylation of aromatic carboxylic acids. Different allyl sulfones 259 reacted with aromatic carboxylic acids to give allyl arenes 284 in moderate to good yields using Cu(OTf)2 under 390 nm LED irradiation (Scheme 103). In the plausible mechanism, the carboxylic acid coordinates Cu(II) to give the complex A by LMCT54,55 providing after irradiation the carboxy radical B and reduced Cu(I). Radical B undergoes decarboxylation to release an aryl radical C, which is captured by the allyl sulfone to give radical D, Finally, radical D loses the sulfonyl radical to yield the allylated product 284.
 | ||
| Scheme 103 Decarboxylative allylation of aromatic carboxylic acids with allylic sulfones 259 under Cu photoinduced LMCT. | ||
Morita–Baylis–Hillman (MBH) acetates 286 have been recently employed in allylation reactions of aliphatic carboxylic acids by decarboxylative photoredox processes. Xie, Loh and co-workers201 reported an organophotoredox/DABCO catalytic system for the radical–radical coupling of MBHAs 286 with sodium α-fluor and α,α-difluoro carboxylate ions 285 (Scheme 104a). In the presence of Mes-Acr+Ph (BF4)−287 and DABCO under blue LED irradiation the corresponding mono- and difluoro homoallylic compounds 288 were obtained with good yields and in general high E/Z-diastereoselectivities. Several bioactive molecules, FDA-approved drugs and AA derivatives were transformed into the corresponding fluorine-containing molecules. In the proposed mechanism, after oxidation of 286 by PC* under a SET process, radical A is formed after decarboxylation to give PC−˙. MBHA reacts with DABCO producing the allylic ammonium salt B, which is reduced by PC˙− to provide radical C regenerating PC and DABCO. Radical–radical coupling of A and C gives product 288. However, because the reaction can be performed in the absence of DABCO, an alternative mechanism has been proposed. Radical A can be added to MBHA delivering intermediate D, which is reduced by PC˙− to give anion E. Finally, the elimination of the acetate group yields product 288. The same group202 performed the same allylation using N-Boc AAs 6 and MBHAs 286 in the presence of Mes-Acr+Ph (BF4)−287 as an organophotocatalyst and 2.5 equivalents of Na3PO4 as a base in DCE at room temperature under Ar (Scheme 104b). The corresponding homoallylic amines 289 were isolated in good yields with mainly the E-configuration. This method was applied to biologically active peptides such as anserine, triglyceride and acetyltetrapeptide-11. In the proposed mechanism, the α-amino alkyl radical A′ from the N-Boc AA reacts with MBHA 286 to give radical D′, which after reduction by PC˙− and acetate elimination of anion E′ affords product 289.
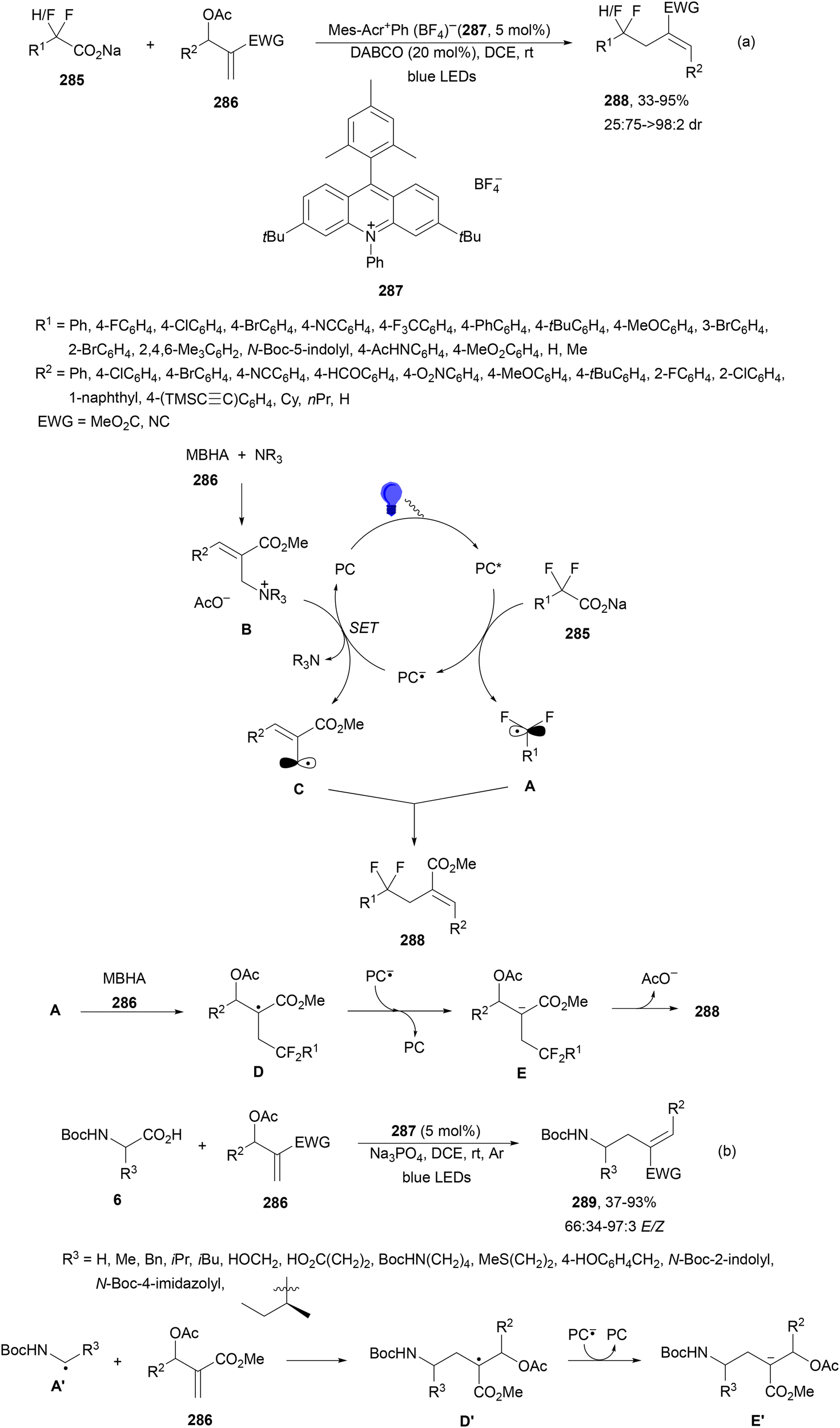 | ||
| Scheme 104 Decarboxylative allylation of sodium mono- and difluoro carboxylate (a) and N-Boc AAs (b) with MBHAs 286 under MesAcr+Ph (BF4−) 287 photocatalysis. | ||
Allylic alkylation of carboxylic acids with MBHAs 286 under visible light has been recently achieved by Akondi and co-workers203 under photoinduced LMCT54,55 using Fe(OTf)2/2,4,6-collidine as a catalyst. Trisubstituted alkenes 290 having mainly the E-configuration have been obtained with moderate to very good yields (Scheme 105). The proposed mechanism involves a similar pathway to that shown in Scheme 104 for N-Boc AAs except the initial oxidation of Fe(II) to Fe(III) and the formation of Fe(III)-carboxylate complex, which upon irradiation with blue LEDs gives the carboxy radical and Fe(III) by a LMCT process. A similar transformation was further described by Loh and co-workers204 using FeBr3 as a photocatalyst, K3PO4 as a base in MeCN at room temperature under 390 nm LED irradiation to give alkenes 290 in 31–96% yields and up to 19:1 E/Z-diastereoselectivity.
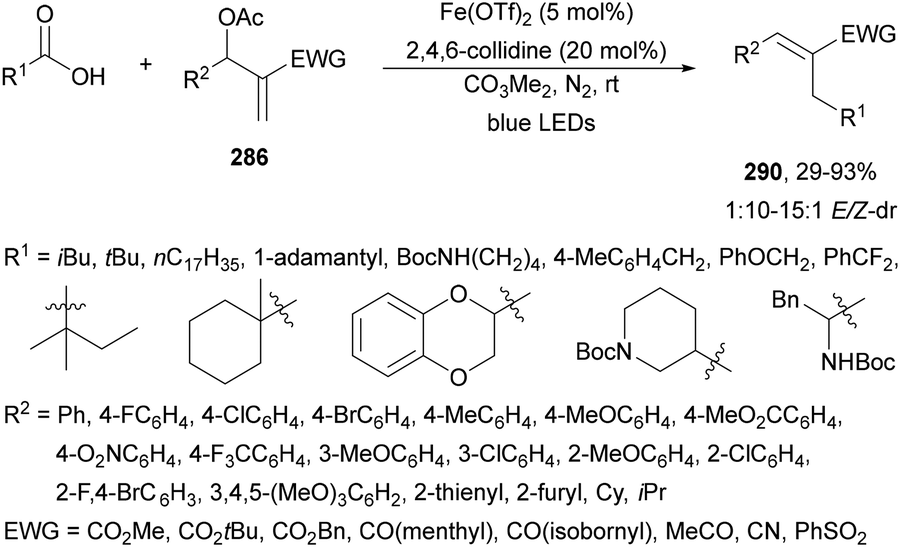 | ||
| Scheme 105 Decarboxylative allylation of aliphatic carboxylic acids with MBHAs (286) under Fe(OTf)2/2,4,6-collidine photocatalysis. | ||
When NHPI esters 69 were used instead of carboxylic acids their allylation with MBHAs 286 took place under very mild reaction conditions in the presence of rose Bengal (RB) as a PC, DIPEA (2 equivalents) as a sacrificial reagent in aqueous DCE at room temperature under blue LED irradiation.205 The corresponding substituted alkyl acrylates 290 were isolated with, in general, good yields and up to >99:1 E/Z-diastereoselectivity (Scheme 106). In the proposed mechanism photoexcited RB* is reductively quenched by DIPEA to give DIPEA+˙ and RB−˙, which reduced NHPI ester to form radical anion A and RB. After N–O bond splitting of intermediate A, radical B is formed as well as CO2 and PhthN−. Radical B undergoes radical addition to MBHA 286 to form radical C, which after β-acetoxy radical elimination gives product 290.
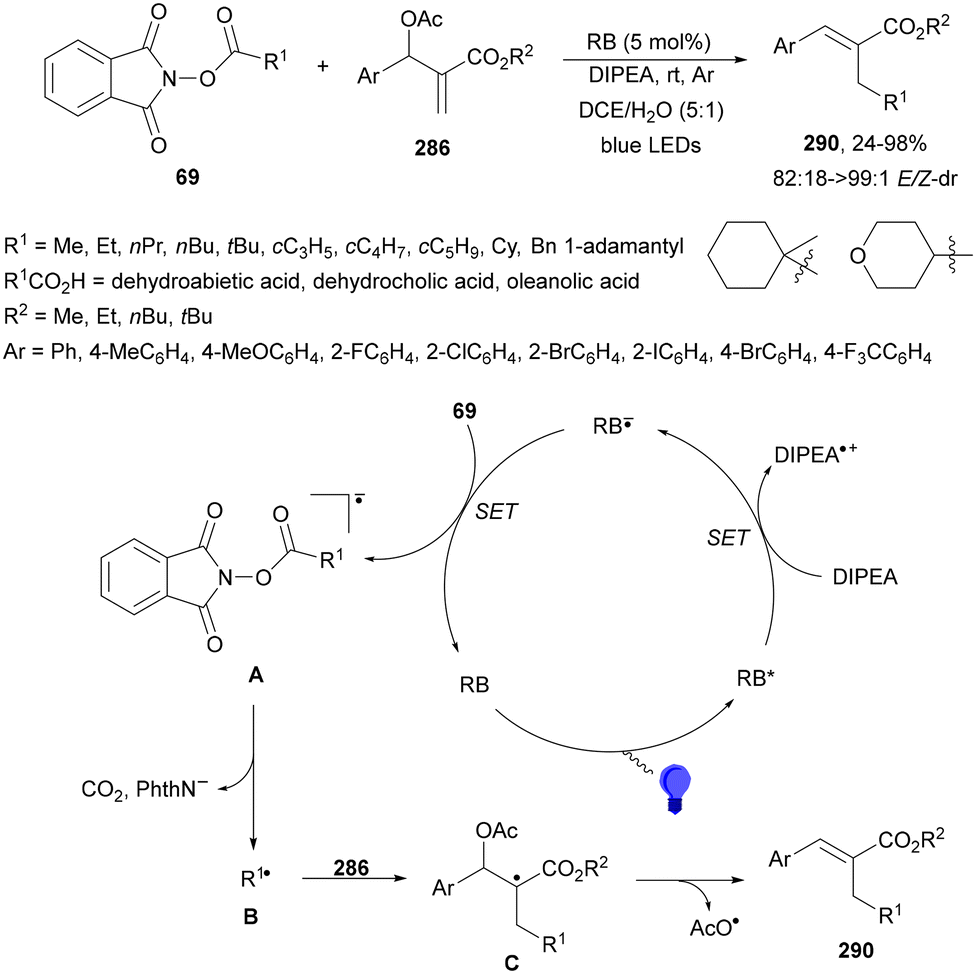 | ||
| Scheme 106 Decarboxylative allylation of aliphatic NHPI esters 69 with MBHAs 286 under RB photocatalysis. | ||
Decarboxylative allylation and benzylation of aliphatic carboxylic acids can be carried out under Pd/organophotocatalysis with carbonates. Allylic carbonates have also been employed using Ni/Ir as a dual photocatalyst and only Ir in the case of vinyl cyclopropanes. Allylic sulfones are appropriate allylic reagents of aromatic carboxylic acids under Cu photoinduced LMCT. Recently, Morita–Baylis–Hillman acetates (MBHAs) have been used as allylation reagents using Mes-Acr+Ph (BF4)− as an organophotocatalyst or Fe(OTf)2 in a LMCT process. In the case of NHPI esters and MBHAs, rose Bengal under aqueous conditions was used to perform the corresponding allylation with high E-diastereoselectivity.
2.8. Alkenylation reactions
Initial studies about decarboxylative vinylation of carboxylic acids were described by MacMillan and co-workers by reaction with vinyl sulfones206 under Ir photocatalysis and also by cross-coupling with vinyl halides207 under Ir/Ni dual photocatalysis. Fu and co-workers208 employed gem-difluoroalkenes 291 for decarboxylative monofluoroalkylation of α-AAs using Ir complex 4 as a PC and a compact fluorescent light (CFL) bulb to obtain products 292 (Scheme 107). This alkenylation reaction took place with Li2CO3 as a base, in DMSO at room temperature under an Ar atmosphere giving products 292 with, in general, good yields and moderate diastereoselectivity. In the proposed mechanism, once the α-aminoalkyl radical A is generated by a SET process followed by decarboxylation, gem-difluoroalkene is reduced by Ir(II) via a SET to a radical anion, which decomposes to radical B and the fluoride anion regenerating Ir(III). Finally, radical–radical cross-coupling between A and B affords product 292.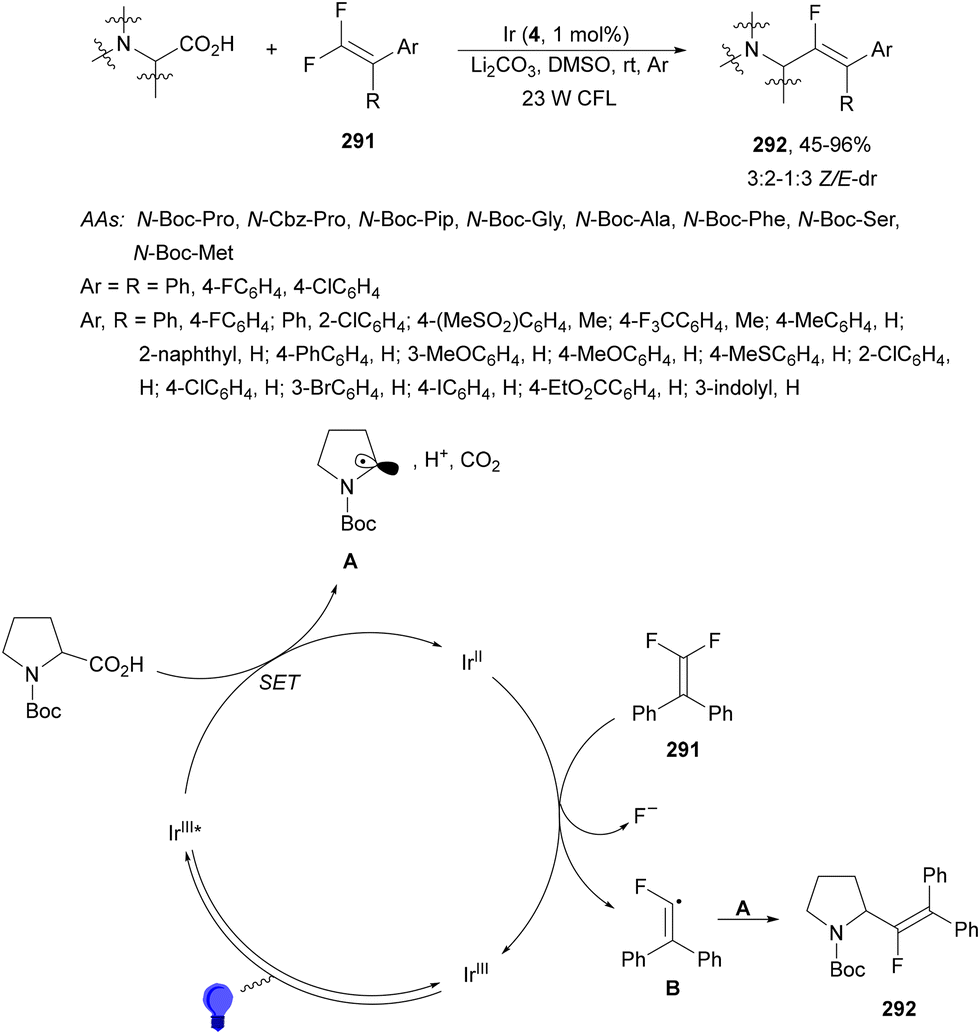 | ||
| Scheme 107 Decarboxylative vinylation of α-AAs with gem-difluoroalkenes 291 under Ir (4) photocatalysis. | ||
Formylation of vinyl bromides 293 was carried out by Mariano, Wang and co-workers144 using diethoxyacetic acid 26 by Ni/4CzIPN (25)-mediated photoredox reaction. This cross-coupling vinylation took place with NiCl2/4,4′-(MeO)2bpy (185) and the organocatalyst 4CzIPN (25) under blue LED irradiation in DMF at room temperature to furnish, after HCl deprotection, the corresponding α,β-unsaturated aldehydes 294 (Scheme 108). A plausible mechanism has been proposed in which the oxidation of diethoxyacetic acid by 4CzIPN* gives CO2 and radical A. Reduction of the Ni(I) intermediate by SET forms Ni(0) species, which reacts with radical A to provide intermediate B. Oxidative addition of vinyl bromide 293 to B generates the Ni(III) complex C. Final reductive elimination of intermediate C produces acetal D and Ni(I)-species. Aqueous acidic work-up converts acetal D into aldehyde 294.
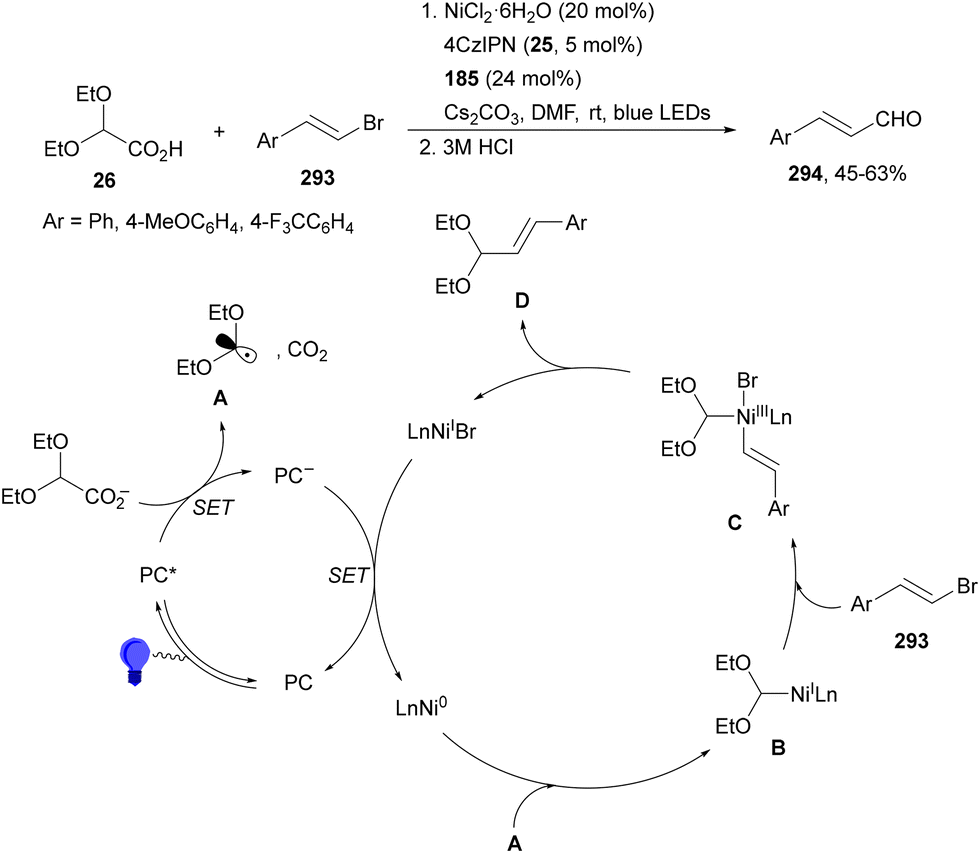 | ||
| Scheme 108 Decarboxylative alkenylation of diethoxyacetic acid 26 with vinyl bromides 293 under Ni/4CzIPN (25) photocatalysis. | ||
Decarboxylative alkenylation of aliphatic carboxylic acids with vinyl sulfones was also carried out in the presence of an organic photocatalyst instead of Ir as it was previously described by MacMillan.206 In this case, riboflavin tetraacetate (RFTA) 296 was used as a PC for the reaction of carboxylic acids with styryl sulfones 295 under blue LED irradiation (Scheme 109).209 The resulting (E)-alkenes 297 were regio- and diastereoselectively obtained with moderate to good yields. This process can be explained by addition of radical A, formed by decarboxylation of the carboxy radical, to styryl sulfone to form radical B followed by β-elimination of the sulfonyl radical under thermodynamic control.
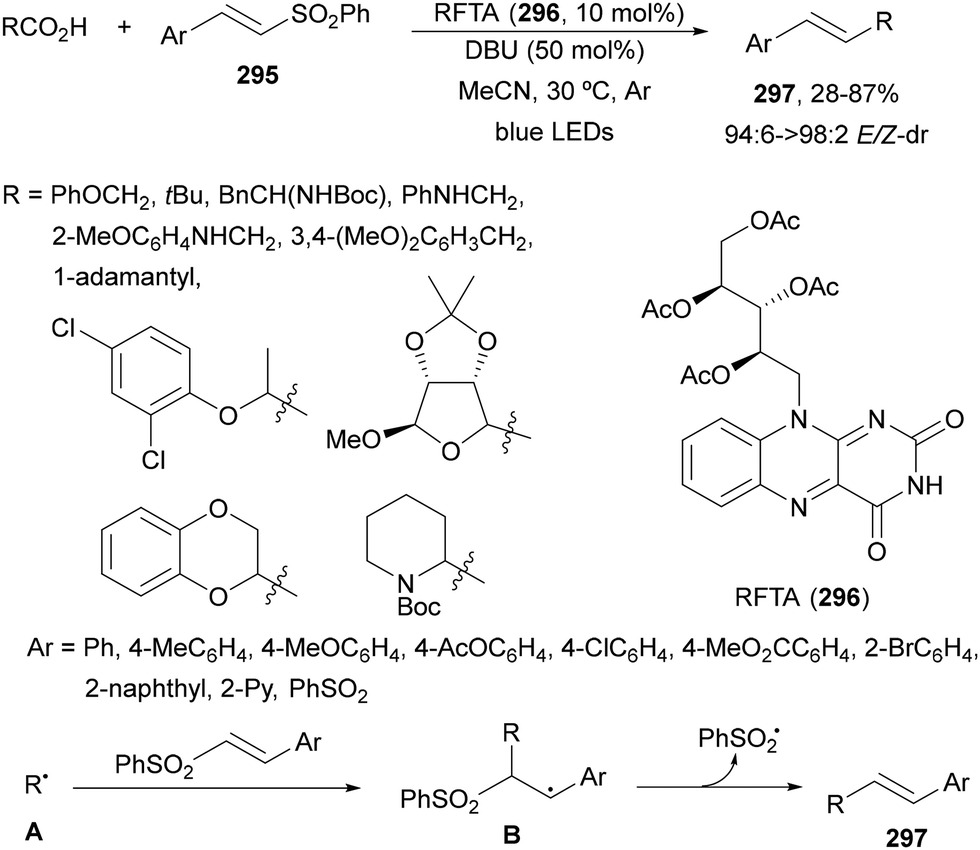 | ||
| Scheme 109 Decarboxylative alkenylation of aliphatic carboxylic acids with styryl sulfones 295 under RFTA 296 photocatalysis. | ||
Recently, a decarboxylative photocatalytic alkenylation of carboxylic acids with styryl sulfones 295via iron LMCT54,55 activation has been reported.210 Aliphatic carboxylic acids reacted with styryl sulfone 295a (Ar = Ph) using Fe(NO3)3, ligand 298, Na2CO3 as a base in DCM at room temperature under 390 nm LED irradiation to provide alkenes 297 with moderate yields and total E-diastereoselectivity (Scheme 110). In this case, the iron carboxylate converts after irradiation to the corresponding photoexcited state A, which by a fragmentation process generates the carboxy radical B and Fe(II) species. Subsequent decarboxylation to C and addition to styryl sulfone followed by β-elimination of the sulfonyl radical gives the product.
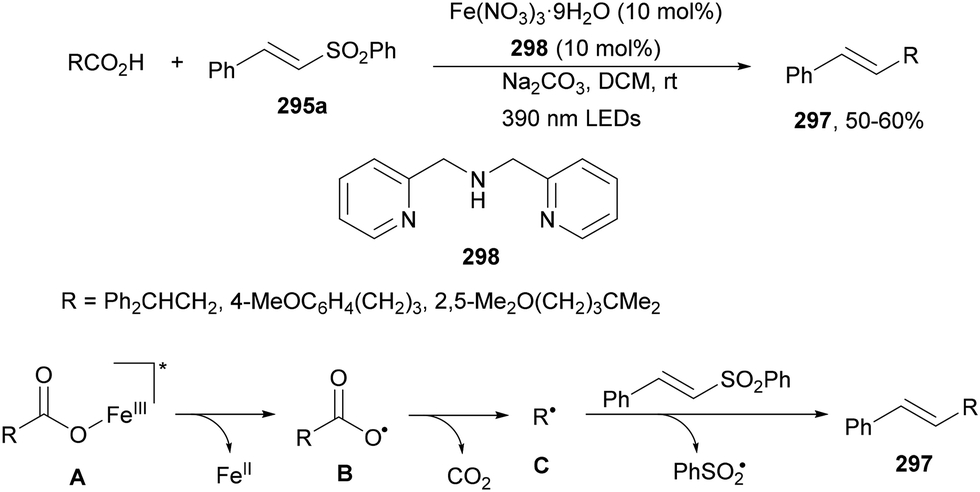 | ||
| Scheme 110 Decarboxylative alkenylation of aliphatic carboxylic acids with styryl sulfone 295a under Fe(III) LMCT photocatalysis. | ||
Vinyl sulfonium salts 299 have been used as radical acceptors for decarboxylative alkenylation of NHPI esters 69 in the presence of Eosil Y 139 as an organophotocatalyst.211 A broad range of primary, secondary and tertiary carboxylic acid derivatives 69 reacted with vinyl sulfonium salts 299 under mild conditions to give alkenes 297 with moderate to good yields and E-configuration (Scheme 111). In this procedure, radical A obtained by decarboxylation of the NHPI ester adds at the α-position of the vinyl sulfonium salts to provide intermediate B. Subsequently, DIPEA+˙ transforms radical B into anion C, which undergoes β-elimination to afford 297.
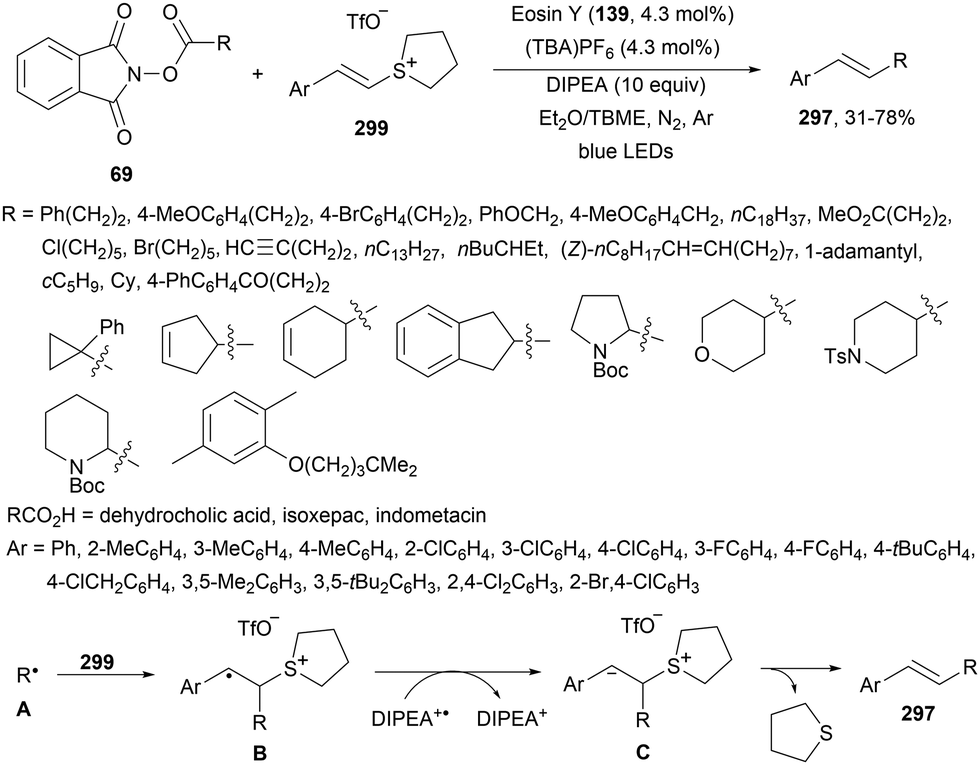 | ||
| Scheme 111 Decarboxylative alkenylation of NHPI esters 69 with vinyl sulfonium salts 299 under Eosin Y 139 photocatalysis. | ||
Aliphatic carboxylic acids have been utilized in a Heck process by a photoredox/Pd catalysis with vinyl arenes.212 A cis-selective decarboxylative alkenylation of tertiary and secondary aliphatic carboxylic acids with styrenes occurred in the presence of Ir complex 4 as a PC, Pd(OAc)2, 3,4,7,8-tetramethyl-1,10-phenanthroline (TMP, 300) as a ligand, K2HPO4 as a base in chlorobenzene as solvent under Ar and blue LED irradiation to deliver β-alkylated styrenes 297 with Z-selectivity (Scheme 112). The observed stereoselectivity is attributed to the Ir PC, which promotes a SET and energy-transfer to sensitize E-olefin to its triplet state isomerizing E-olefin to its thermodynamically less favored Z-isomer. Products Z-297 were isolated in up to 84% yield and up to 99:1 dr. In the proposed mechanism, according to experimental studies, the phenanthroline-supported Pd(II) catalyst reacts with the alkyl radical A and styrene to give a benzylic Pd(III) species B. Oxidation of Ir(II) by B regenerates Ir(III) and gives Pd(II) species C, which by β-H elimination delivers product E-297 and Pd(II)–H species D. This intermediate D inserts into an excess of styrene, as a hydrogen acceptor, to give complex E, which after protonation forms ethylbenzene and regenerates LPd(II)X. Simultaneously, Ir(III)* in its triplet state sensitizes E-297 to Z-297.
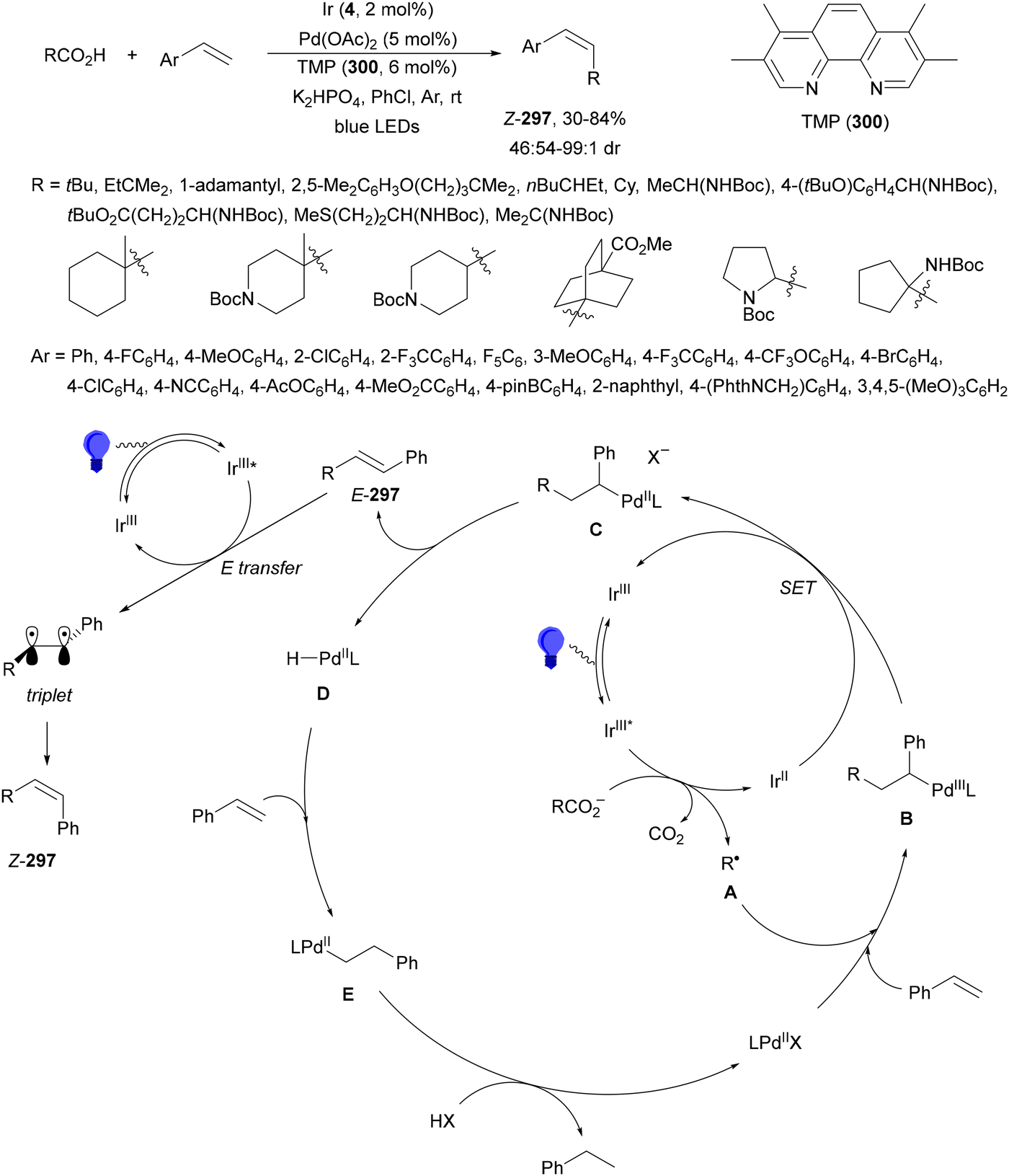 | ||
| Scheme 112 Decarboxylative Heck-type reaction of aliphatic carboxylic acids with styrenes under Ir/Pd photocatalysis. | ||
Decarboxylative Heck-type reaction of aliphatic carboxylic acids with styrenes, vinyl silanes and vinyl boronates in the absence of external oxidants was carried out in the presence of Mes-Acr+ClO4−301 as an organophotocatalyst and cobaloxime catalyst 302.213 The corresponding alkenes, 1,3-dienes and conjugated enynes were obtained with good yields (Scheme 113a). However, β-substituted styrenes and aliphatic alkenes gave very low yields (<10%). In the case of vinyl silanes and boronates, they were efficiently transformed into substituted vinyl silanes and boronates 303 (Scheme 113b). A tentative mechanism has been proposed: in the photoredox catalytic cycle the carboxylate ion was oxidized by [Mes-Acr+]* to deliver radical A, which reacts with styrene to form benzylic radical B. The reduced PC (Mes-Acr˙) can be oxidized by Co(III) catalyst 302 to complete this catalytic cycle. The generated Co(II) can capture radical B to form a Co(III) intermediate C, which delivers the product and LnCo(III)–H. Finally, this LnCo(III)–H may react with a H+ or another molecule of Co(III)–H to release H2 and regenerate the catalyst. A multicomponent coupling of aliphatic carboxylic acids, acrylates and styrenes was achieved using the same reaction conditions (Scheme 113c). In this case, the alkyl radicals derived from carboxylic acids reacted firstly with the acrylate to give an α-acyl radical, which then added to the vinyl arene to provide products 304 with moderate yields and high E-diastereoselectivity.
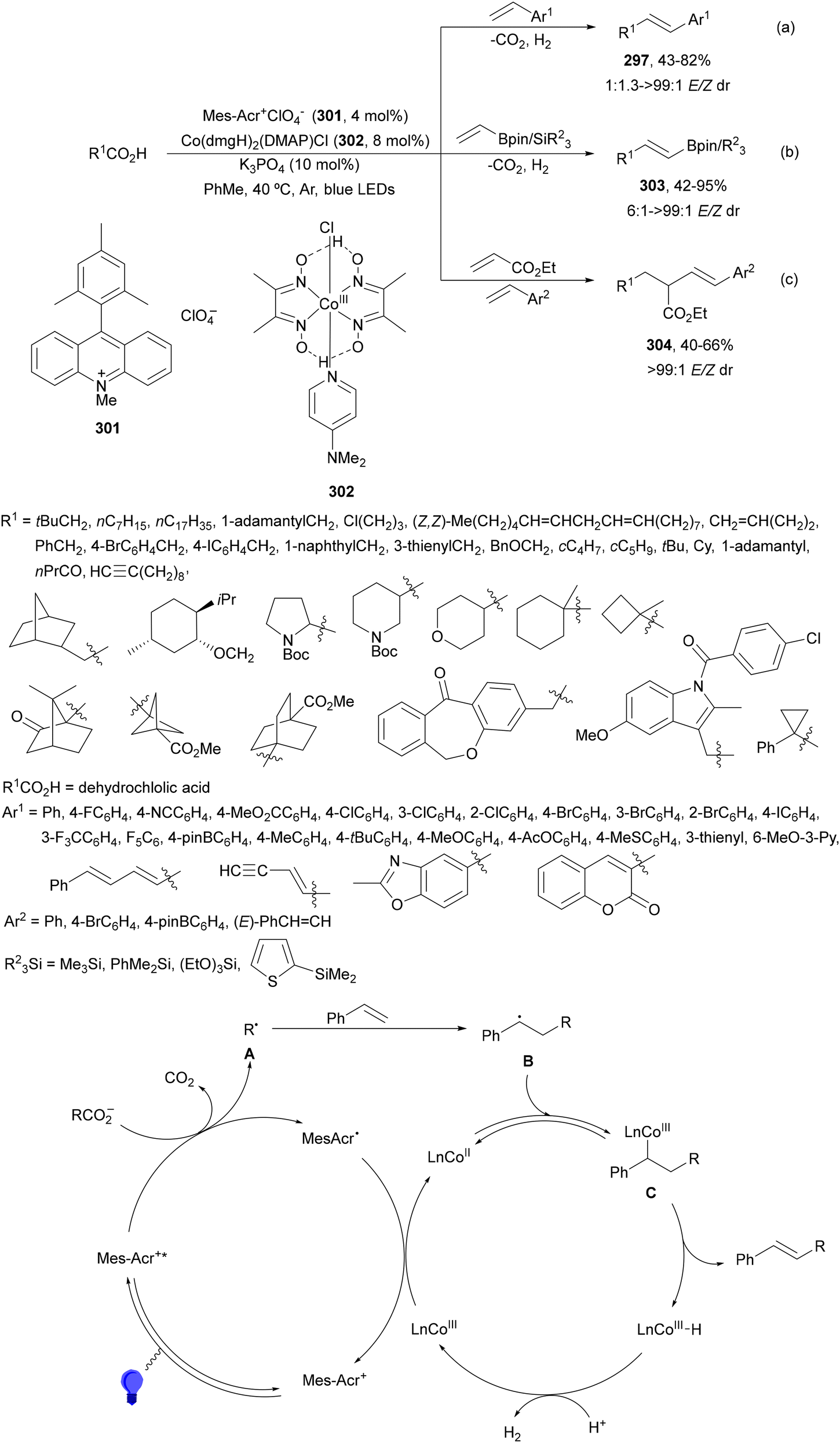 | ||
| Scheme 113 Decarboxylative Heck-type reaction of aliphatic carboxylic acids and terminal alkenes (a–c) under Mes-Acr+ClO4 (301) and cobaloxime 302 photocatalysis. | ||
Decarboxylative Heck-type reaction of aliphatic NHPI esters 69 and styrenes was initially studied by Fu214 and Glorius215 groups independently using Pd as a catalyst under photoredox conditions. The Chinese group214 employed Pd(PPh3)Cl2 (5 mol%) and Xantphos (6 mol%) as a ligand and K2CO3 as a base in aqueous DMA at room temperature under blue LED irradiation to obtain compounds 297 with good yields and diastereoselectivities (Scheme 114a). In the case of Glorius and co-workers,215 Pd(PPh3)4 (5 mol%) in THF at room temperature under blue LED irradiation afforded products E-297 with good yields (Scheme 114b). They proposed a catalytic cycle215 initiated by photoexcitation of Pd(0) followed by SET to NHPI ester, which forms the corresponding alkyl radical A upon release of CO2 and PhthN−. Addition of this radical to styrene gives the benzylic radical B, which after β-hydride elimination releases the product and Pd(II)–H species. Reductive elimination of phthalimide regenerates the catalyst.
 | ||
| Scheme 114 Decarboxylative Heck-type reaction of aliphatic NHPI esters 69 with styrenes under Pd photocatalysis (a, b). | ||
Iridium complexes were used as PCs in decarboxylative Heck-type reactions with alkyl NHPI esters 69 with styrenes. Gao, Ye and co-workers216 employed Ir(ppy)3 (89) as a photocatalyst in the presence of TfOH and DMSO as solvent at room temperature under blue LED irradiation to give the corresponding alkenes 297 with moderate to high yields (Scheme 115). The plausible mechanism involves the formation of the alkyl radical A by means of photoexcited Ir(III)* and subsequent addition to styrene gives radical B, which is oxidized to a carbocation C by Ir(IV). In the presence of TfOH, β-elimination of a proton affords alkene 297.
 | ||
| Scheme 115 Decarboxylative Heck-type reaction of alkyl NHPI esters 69 with styrenes under Ir(ppy)3 photocatalysis. | ||
Zhao, Loh and co-workers217 performed decarboxylative Heck-type reaction of alkyl NHPI esters 69 with enamides 305 using Ir(ppy)3 (89) as a PC (Scheme 116). This process was carried out in DMF at room temperature under blue LED irradiation to provide β-alkylated enamides 306 with good yields and excellent E-diastereoselectivities. In the proposed mechanism, the alkyl radical A from the NHPI ester adds to the enamide to give the α-aminoalkyl radical B, which is oxidized by Ir(III)+ species to form cationic intermediate C. Finally, deprotonation of C gives the product. The resonance form of C gives the iminium ion D with two possible conformers D and D′. Conformer D explains the observed stereochemistry.
 | ||
| Scheme 116 Decarboxylative Heck-type reaction of aliphatic NHP esters 69 with enamides 305 under Ir(ppy)3 photocatalysis. | ||
Sodium iodide and triphenylphosphine have been used as PCs for decarboxylative Heck-type reaction of NHPI esters 69 with 1,1-diarylethylenes217 and with enamides 305.218 Sang, Fu and co-workers217 employed NHPI esters derived from α-AAs, α-alkoxy acids and thioglycolic acid with 1,1-diarylethylenes in acetone at room temperature under blue LED irradiation to obtain allylic products 307 (Scheme 117a). In the case of Fu and co-workers,218 alkyl NHPI esters including α-AA derivatives reacted with enamides 305 to give enamides 306 in good yields and E-diastereoselectivity (Scheme 117b). The proposed mechanism193 for the PPh3/NaI photoredox process is depicted in Scheme 96.
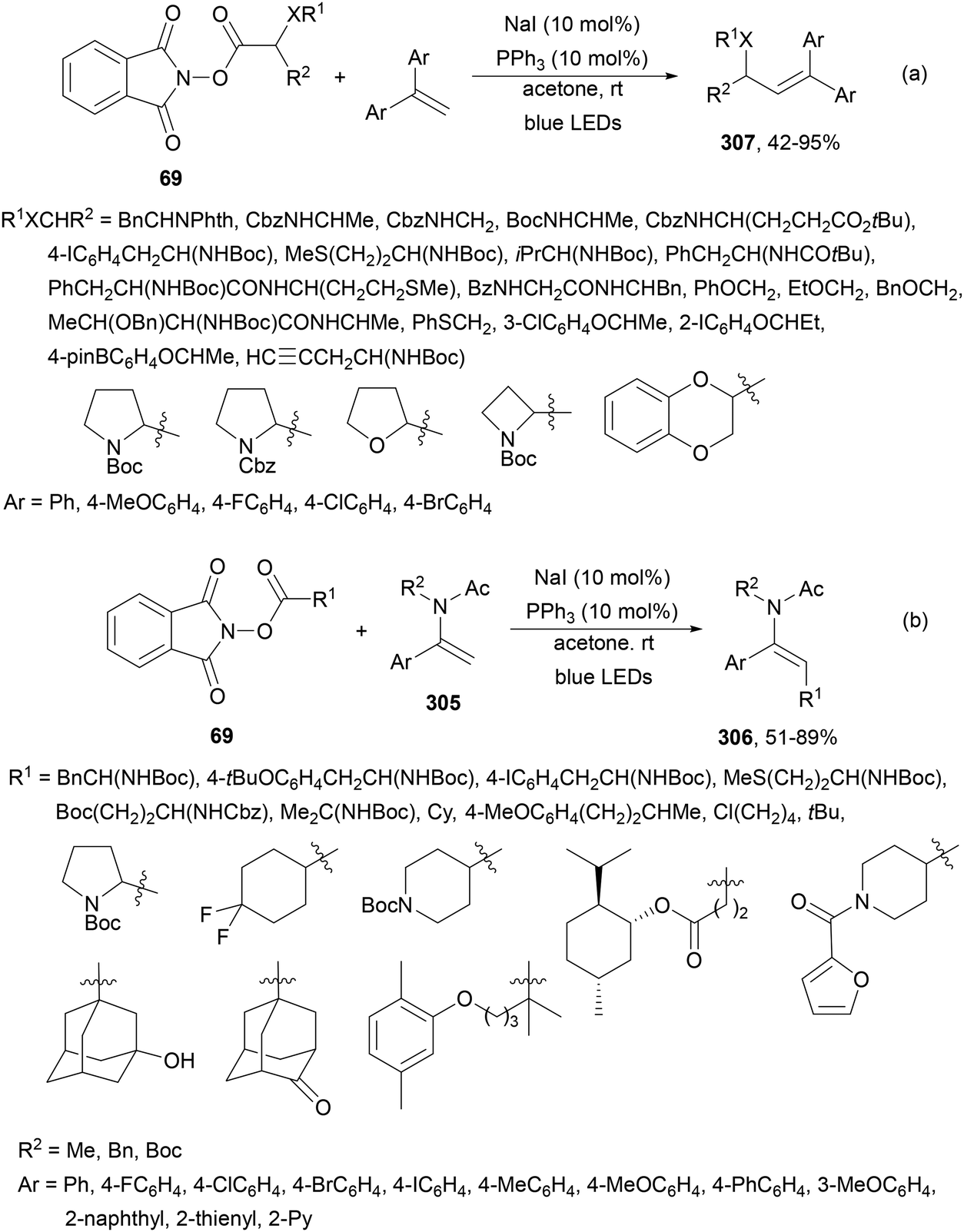 | ||
| Scheme 117 Decarboxylative Heck-type reaction of NHPI esters with 1,1-diarylethylenes (a) or enamides 305 (b) under PPh3/NaI photocatalysis. | ||
A third strategy for C(sp2)–C(sp3) bond formation through decarboxylative vinylation reactions under photoredox conditions used α,β-unsaturated carboxylic acids.219 Duan and co-workers220 reported a dual decarboxylative coupling of α,β-unsaturated carboxylic acids with alkyl NHPI esters 69 using Ir(ppy)3 (89) as a PC and Mg(ClO4)2 as an additive in NMP at room temperature under 23 W fluorescent light (CFL) bulb irradiation. A broad range of substituted alkenes 297 were obtained with, in general, good yields and E-diastereoselectivity (Scheme 118). A plausible mechanism was proposed involving the formation of the radical A from the NHPI ester by Ir(III)* and subsequent attack on the CC bond of the α,β-unsaturated carboxylic acids to give radical B. This is followed by deprotonation of B by the phthalimide anion from radical-carboxylate C, which might be oxidized by PC* to radical D. This radical D delivers CO2 and the final product.
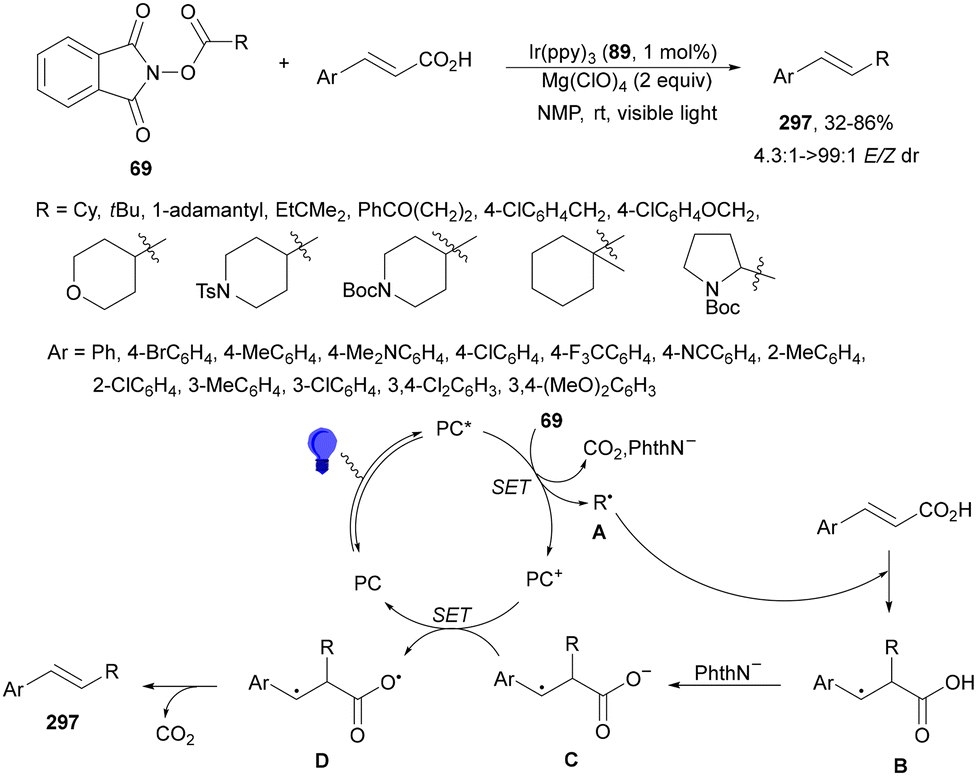 | ||
| Scheme 118 Decarboxylative alkenylation of alkyl NHPI esters 69 with cinnamic acids under Ir(ppy)3 (89) photocatalysis. | ||
Under similar reaction conditions described for the Heck-type reaction of 1,1-diarylethylenes with NHPI esters using NaI/PPh3 as a PC217 (Scheme 117a), a vinylation of NHPI esters with cinnamic acids (Scheme 119) was carried out. Working with N-Boc-protected phenylalanine and α,β-unsaturated carboxylic acids, the corresponding allylic amines 67 were obtained with good yields and E-diastereoselectivity.217
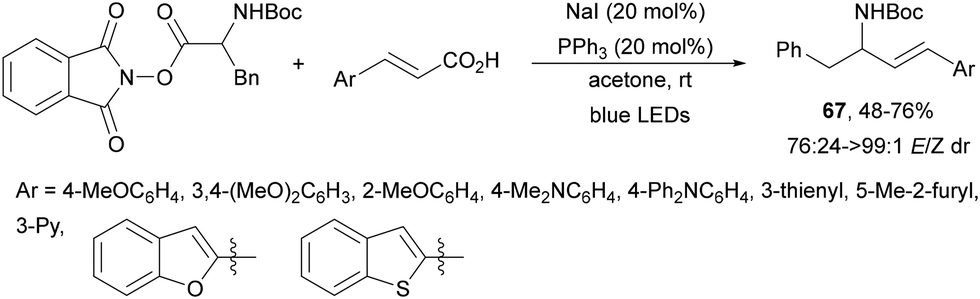 | ||
| Scheme 119 Decarboxylative alkenylation of a NHPI ester with α,β-unsaturated carboxylic acids under NaI/PPh3 photocatalysis. | ||
Lu and co-workers221 reported the synthesis of monofluoroalkenes 309via dual decarboxylative cross-coupling of α-fluoro acrylic acids 308 with alkyl NHPI esters 69 using Ir(ppy)3 (89) as a PC and DABCO as a base in DMAc at room temperature under blue LED irradiation. Starting from Z/E mixtures of α-fluoro acrylic acids 308, the corresponding products 309 were obtained with good yields and high Z-diastereoselectivity (Scheme 120). Primary, secondary and tertiary NHPI esters as well as biologically active molecules containing carboxylic acids or α-fluoro acrylic acids 308 were successfully employed. In the proposed mechanism, radical B resulting from the addition of radical A to 308 is transformed by SET into cation C, which undergoes decarboxylation to form product 309.
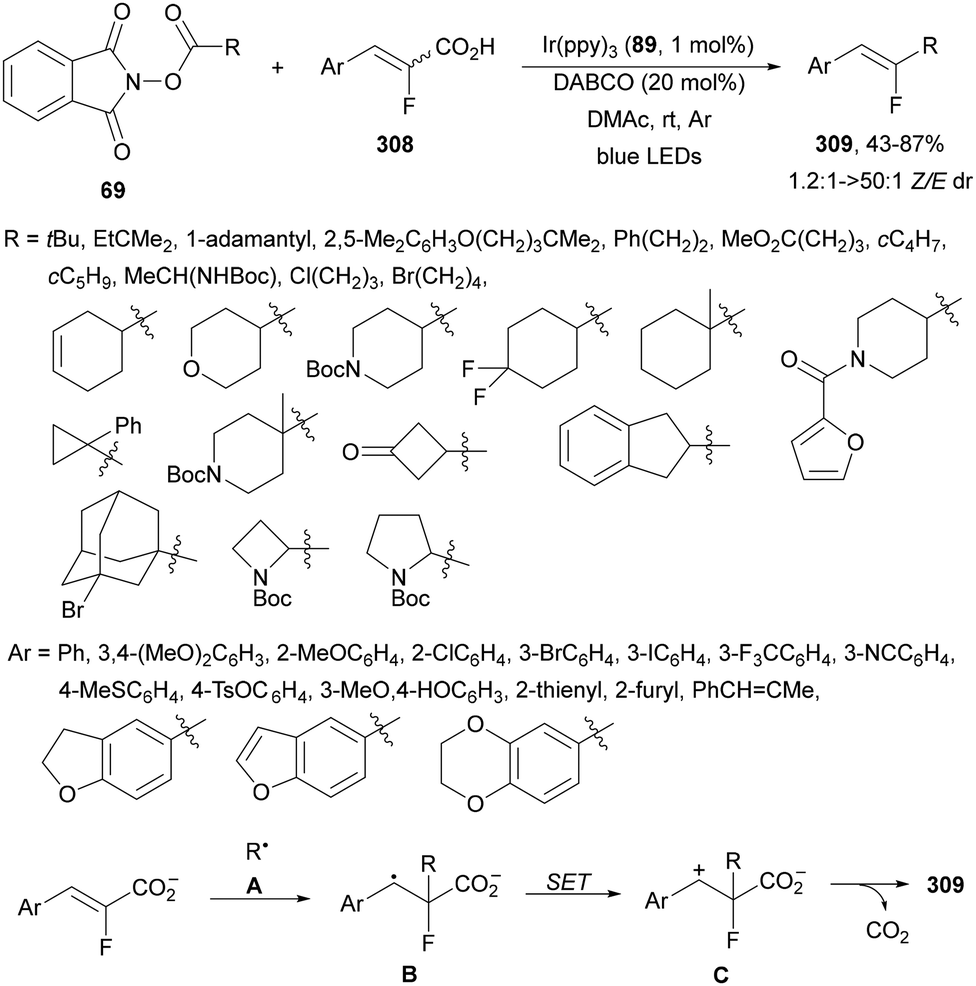 | ||
| Scheme 120 Decarboxylative alkenylation of alkyl esters 69 with α-fluoro acrylic acids 308 under Ir(ppy)3 (89) photocatalysis. | ||
The same group222 recently reported a photoinduced decarboxylative difluoroalkylation and perfluoroalkylation of α-fluoro acrylic acids 308. Using Ru(bpy)3Cl2·6H2O as a PC and DABCO as a base in DMAc at room temperature under blue LED irradiation, tert-butyl 2-bromo-2,2-difluoroacetate 310 gave difluoromethylene-substituted monofluoroalkenes 311 with moderate yields (37–68%) and good Z-diastereoselectivity (Scheme 121a). When bromodifluoro acetamides 312 were employed as alkylating agents, the corresponding products 313 were isolated in 53–63% yields and >20:1 dr (Scheme 121b). Moreover, the reaction was expanded to polyfluoroalkyl bromides 314 to obtain polyfluoroalkylated monofluoroalkenes 315 (Scheme 121c). In the proposed catalytic cycle, photoexcited Ru(bpy)32+* species is reduced by DABCO yielding Ru(bpy)3+, able to reduce bromodifluoroacetate to radical A through a SET. This radical A adds to the α-fluoroacetic acid anion to form radical B, which after oxidation by DABCO+˙ leads to the formation of cation C. Final decarboxylation of C generates product 311.
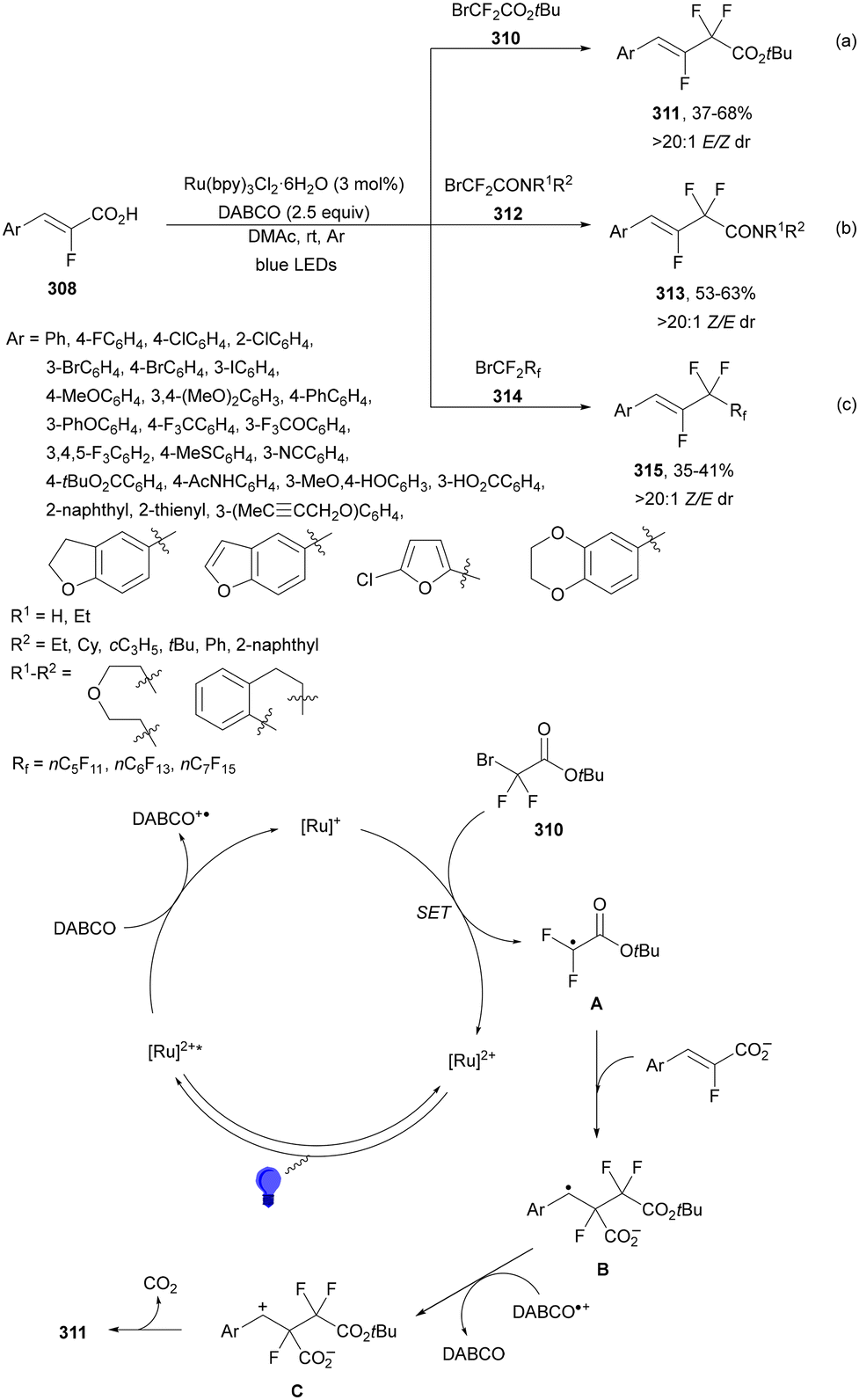 | ||
| Scheme 121 Decarboxylative alkenylation of 2-bromo-2,2-difluoroalkane derivatives 310 (a), 312 (b) and 314 (c) with α-fluoroacrylic acids 308 under Ru(bpy)3Cl2·6H2O photocatalysis. | ||
Alcohols have been used as alkylating agents of α-fluoroacrylic acids 308 under photoinduced decarboxylative cross-coupling by Lu and co-workers.223 In this case, Ir(ppy)3 (89) was used as a PC, DABCO as a base and an electron mediator, and tert-butyl peroxybenzoate (TBPB) as an oxidant in MeCN at room temperature under blue LED irradiation to furnish allylic alcohols 316 in 40 to 75% yields and up to 30:1 Z/E diastereoselectivity (Scheme 122). In the proposed catalytic cycle, reduced Ir(II)(ppy)3 was oxidized by TBPB through a SET to generate Ir(III)(ppy)3 and the tert-butyloxy radical, which abstracts the α-hydrogen of the alcohol to form radical A. Addition of radical A to the α-fluoroacrylate anion generates radical B, which gives an electron to DABCO+˙ to afford cationic intermediate C and DABCO. Finally, decarboxylation of C gives the thermodynamically more stable Z-product 316.
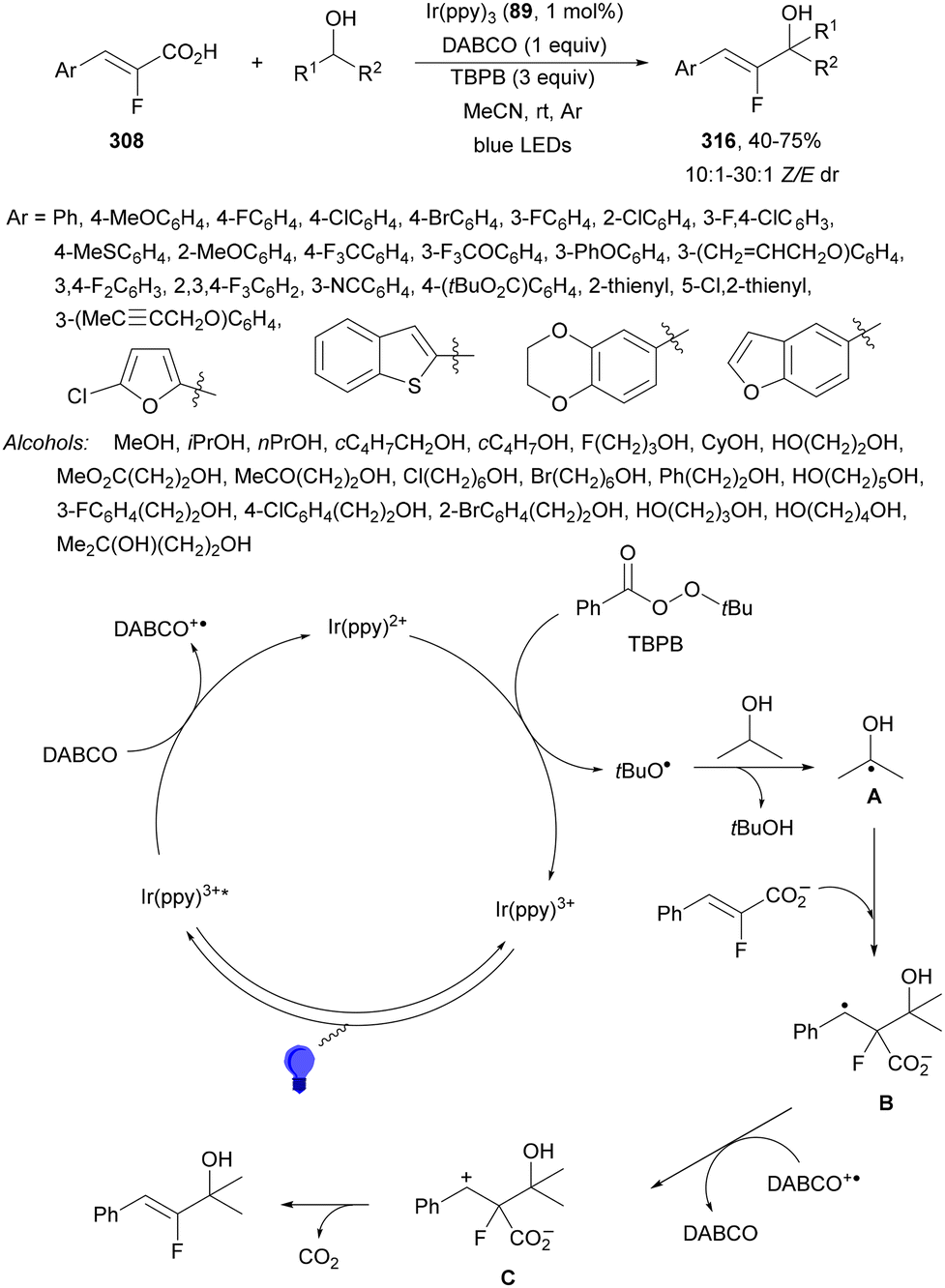 | ||
| Scheme 122 Decarboxylative alkenylation of alcohols with α-fluoroacrylic acids 308 under Ir(ppy)3 (89) photocatalysis. | ||
Three main strategies can be used for decarboxylative alkenylation reactions of alkyl carboxylic acids and derivatives under photoredox conditions: (a) vinyl halides or vinyl sulfones as alkenylation agents, (b) Heck-type alkylation of styrenes and enamides, and (c) α,β-unsaturated carboxylic acids as alkenylating reagents of NHPI esters or alkyl halides or alcohols, In the first case, Ir complexes, NiCl2/4CzIPN, Fe(NO3)2 and organophotocatalysts such as riboflavin tetraacetate and Eosin Y have been used. Heck-type processes with carboxylic acids react in the presence of Ir/Pd or Mes-Acr+/cobaloxime as a dual catalyst, whereas with NHPI esters they have been alkenylated under Pd or Ir or NaI/PPh3 photocatalysis. Cinnamic acids and derivatives react with NHPI esters under Ir or NaI/PPh3 photocatalysis, whereas for the reaction with 2-bromo-2,2-difluoro carboxylate derivatives a Ru complex or an Ir complex for alcohols in the presence of DABCO has been employed as a photocatalyst.
2.9. Alkynylation reactions
Photoredox-mediated decarboxylative alkynylation of aliphatic carboxylic acids has been carried out using hypervalent iodine reagents,224,226 alkynyl sulfones or bromides and terminal alkynes. In 2015, Jiao227 and Waser228 groups reported independently alkynylation of alkyl carboxylic acids with hypervalent iodine reagents such as ethynylbenziodoxolone (EBX) using Ir complex 4 as a PC. In 2016, Li, Chen and co-workers229 employed 9,10-dicyanoantreacene (DCA) as an organophotocatalyst also under blue LED irradiation.Alkyl nitrile radicals generated from carboxylic acid derived oxime ethers 317 have been alkynylated with R–EBX 318 by Waser and co-workers230 using 4ClCzIPN 319 as a PC to provide nitriles 320 under blue LED irradiation (Scheme 123). Azetidinone, cyclobutanone and cyclopentanone oxime ethers 317 gave the corresponding alkynyl nitriles 320 in moderate to good yields. A one-pot protocol started from cyclobutanone and the hydroxylamine derivative; after condensation, two equivalents of Ph–EBX and 5 mol% of 319 were added followed by 1 hour irradiation delivering 6-phenyl-5-hexynenitrile in 71% yield instead of 79% by isolation of the oxime ether. It was proposed that the reaction starts by oxidation of potassium carboxylate by the excited state of the organophotocatalyst 319 to give carboxy radical A and PC−˙. Decarboxylation of A releases the α-oxy radical B, which can either lead to iminyl radical C after acetone extrusion or can be trapped by the EBX reagent forming D. The iminyl radical C fragments into alkyl nitrile radical E, which reacts with EBX to give, through TS in a concert mechanism, the product and radical F. Final reduction of F by PC˙− allows the generation of PC and potassium carboxylate G.
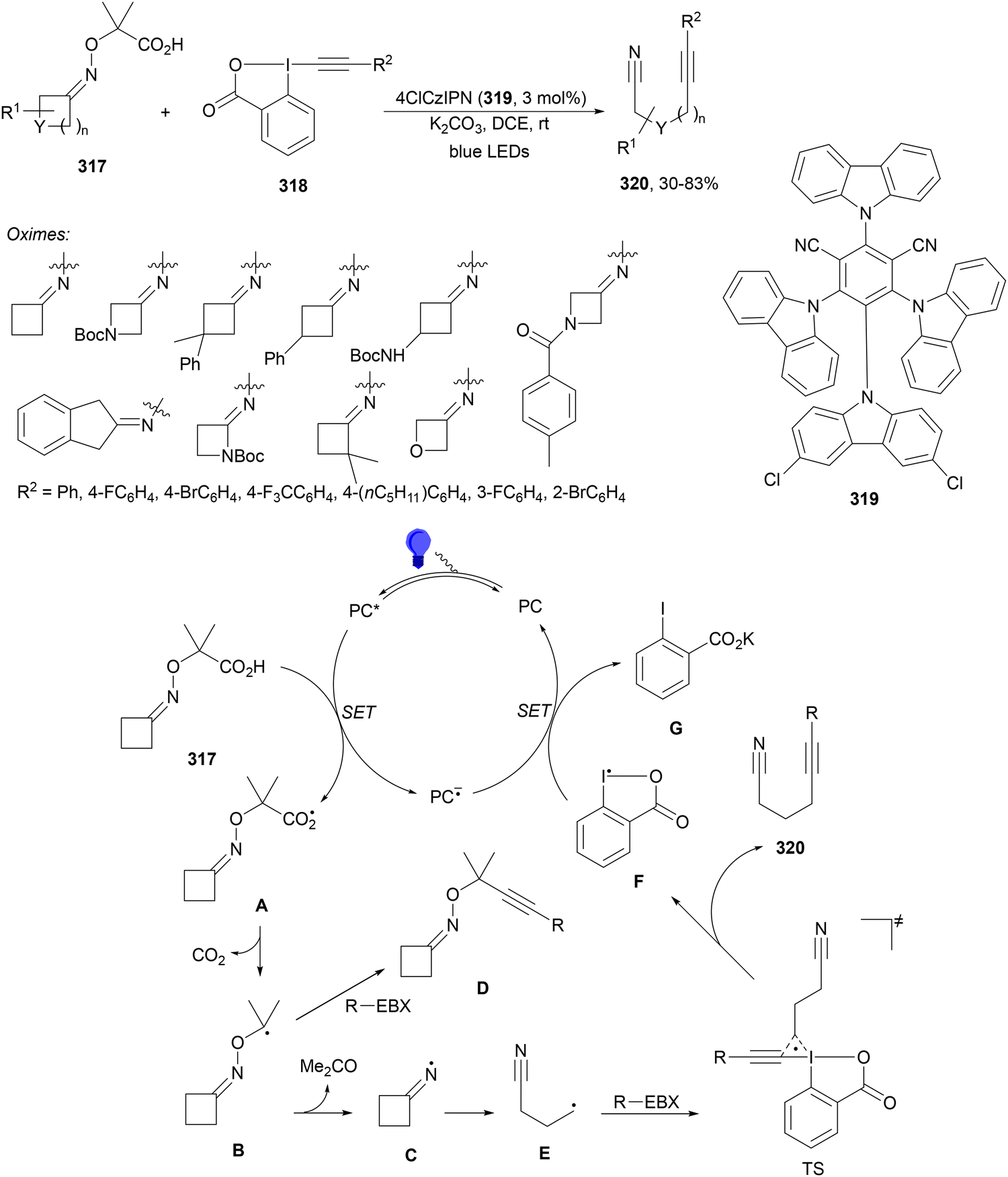 | ||
| Scheme 123 Decarboxylative alkynylation of oxime ethers 317 with R–EBX (318) under 4ClCzIPN (319) photocatalysis. | ||
Waser and co-workers231 performed alkynylation of peptides 321 with R–EBX 318 using 4CzIPN (25) as a PC and K2HPO4 as a base in degassed aqueous DMF at room temperature under blue LED irradiation (Scheme 124). Arylated R–EBX 318 gave the best results providing alkynylated peptides at the C-terminus 322 in moderate to high yields. Di-, tetra- and hexapeptides even unprotected peptides were efficiently alkynylated as well as in the presence of carboxylic acid side chains except tryptophan, which needed to be protected. According to the previously described proposed mechanism,228 after decarboxylation of the peptide by photoexcited 25, the resulting radical A adds to R–EBX at the α-position to the iodine radical leading to adduct B. Subsequently, β-elimination of iodine radical C gives also the alkynylated product. This radical C will be reduced by 4CzIPN˙− to give 2-iodobenzoate D. This methodology was applied to vinylation reactions using vinyl-benziodoxolane as the reagent.
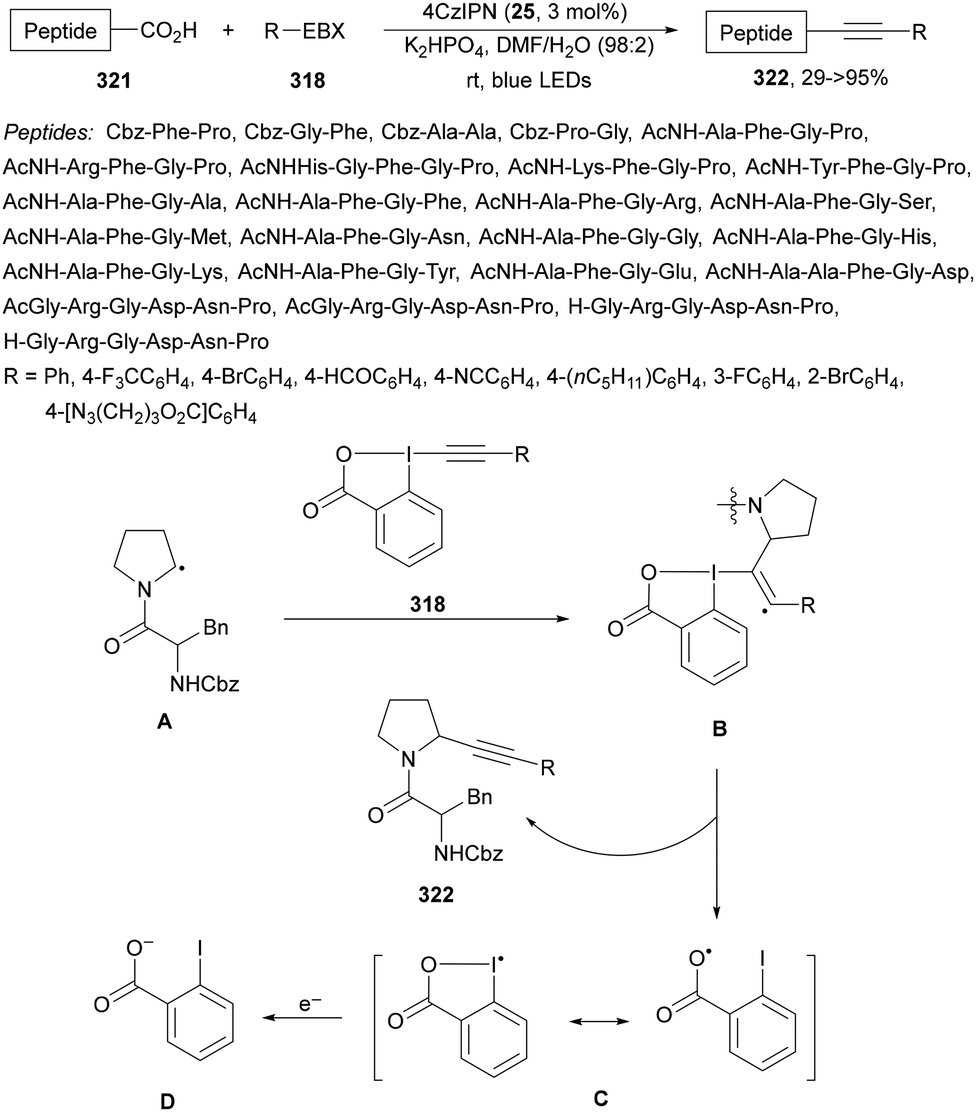 | ||
| Scheme 124 Decarboxylative alkynylation of peptides 321 with alkynylbenziodoxolones (318) under 4CzIPN (25) photocatalysis. | ||
Alkynylation of different glycosylic acids 323 by decarboxylative photoredox reaction with EBXs 318 took place in the presence of Ir complex 324 to provide alkynyl C-glycosides 325 in up to 95% yield (Scheme 125).232 Under visible-light, acids 323 reacted with EBXs in the presence of K2CO3 at 40 °C with excellent diastereoselectivity, except for deoxyribosylic acid, which gave a mixture of β:α = 1/2 anomers. In the proposed catalytic cycle, the glycosyl radical adds to 318 followed by radical elimination to give products 325.
 | ||
| Scheme 125 Decarboxylative alkynylation of glycosylic acids 323 with Ar–EBX 318 under Ir complex 324 photocatalysis. | ||
Decarboxylative alkynylation of aliphatic carboxylic acids with R–EBX 318 under 4CzIPN (25) photocatalysis has been implemented in batch and continuous flow.233 Under both reaction conditions α-AAs and dipeptides were alkynylated using DBU as a base in DMSO at room temperature to give products 326 with similar moderate to high yields (Scheme 126a). Scale-up under flow conditions for the reaction of N-Boc-Pro with Ph–EBX provided the corresponding product in 88% yield. Aliphatic and α-oxy carboxylic acids were decarboxylative alkylated via batch conditions in the presence of Cs2CO3 as a base to give products 327 in moderate to good yields (Scheme 126b).
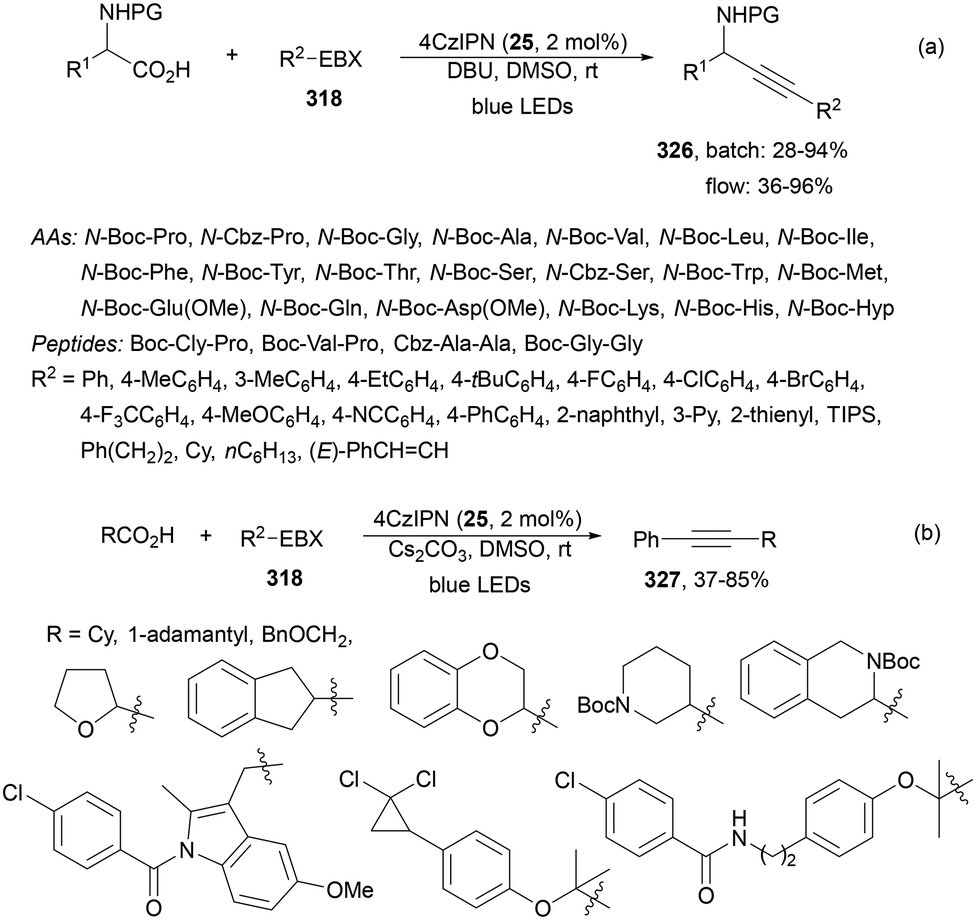 | ||
| Scheme 126 Decarboxylative alkynylation of α-AAs, peptides (a), α-oxy acids and aliphatic carboxylic acids (b) with R–EBX (318) under 4CzIPN (25) photocatalysis under batch and flow conditions. | ||
Along this manuscript it has been shown that unsaturated sulfones are excellent SOMO-philes to trap alkyl radicals.209 Recent applications of alkynyl sulfones in decarboxylative photoredox processes have been described by Wei, Hu and co-workers210 using Fe(NO3)3via a LMCT54,55 mechanism (Scheme 110). They reported a couple of examples using alkynyl sulfone 328 and alkyl carboxylic acids under similar reaction conditions to that for styryl sulfones 295a to provide products 327 in moderate yields (Scheme 127).
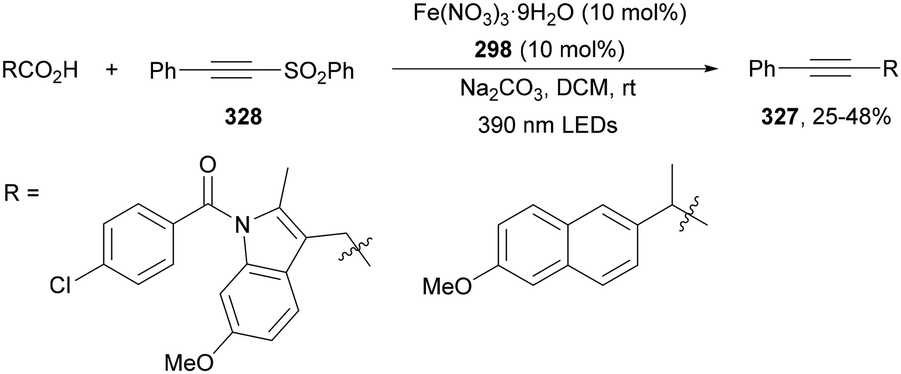 | ||
| Scheme 127 Decarboxylative alkynylation of aliphatic carboxylic acids with alkynyl sulfone 328 under Fe(NO3)3 photocatalysis. | ||
Zhao, Xia and co-workers234 recently reported an iron-catalyzed LMCT photoredox via fragmentation–alkynylation of carboxylic acid derived oxime ethers 317 to give alkynes 320 using alkynyl sulfones 329 as reagents (Scheme 128). The resulting products 320 were obtained in the presence of Fe(acac)3 as a PC and KOH as a base in toluene under N2 at 30 °C and 390 nm LED irradiation. This procedure was performed under batch conditions and a gram-scale reaction was carried out using cyclobutanone derived oxime 317a and methyl phenylethynyl sulfone to give 6-phenyl-5-hexynenitrile in 75% yield. A plausible mechanism involves the photoexcitation of a carboxylate-iron(III) complex B, which by the LMCT event produces the reduced Fe(II) complex and an aroyloxy radical C. Subsequent decarboxylation of C releases a radical intermediate D, which generates after fragmentation the iminyl radical E followed by a radical transposition to radical F. Addition of radical F to alkynyl sulfone gives radical G, which after methylsulfonyl radical elimination leads to product 320. This methodology has been applied to styryl sulfones and allyl sulfones to obtain the corresponding unsaturated nitriles.
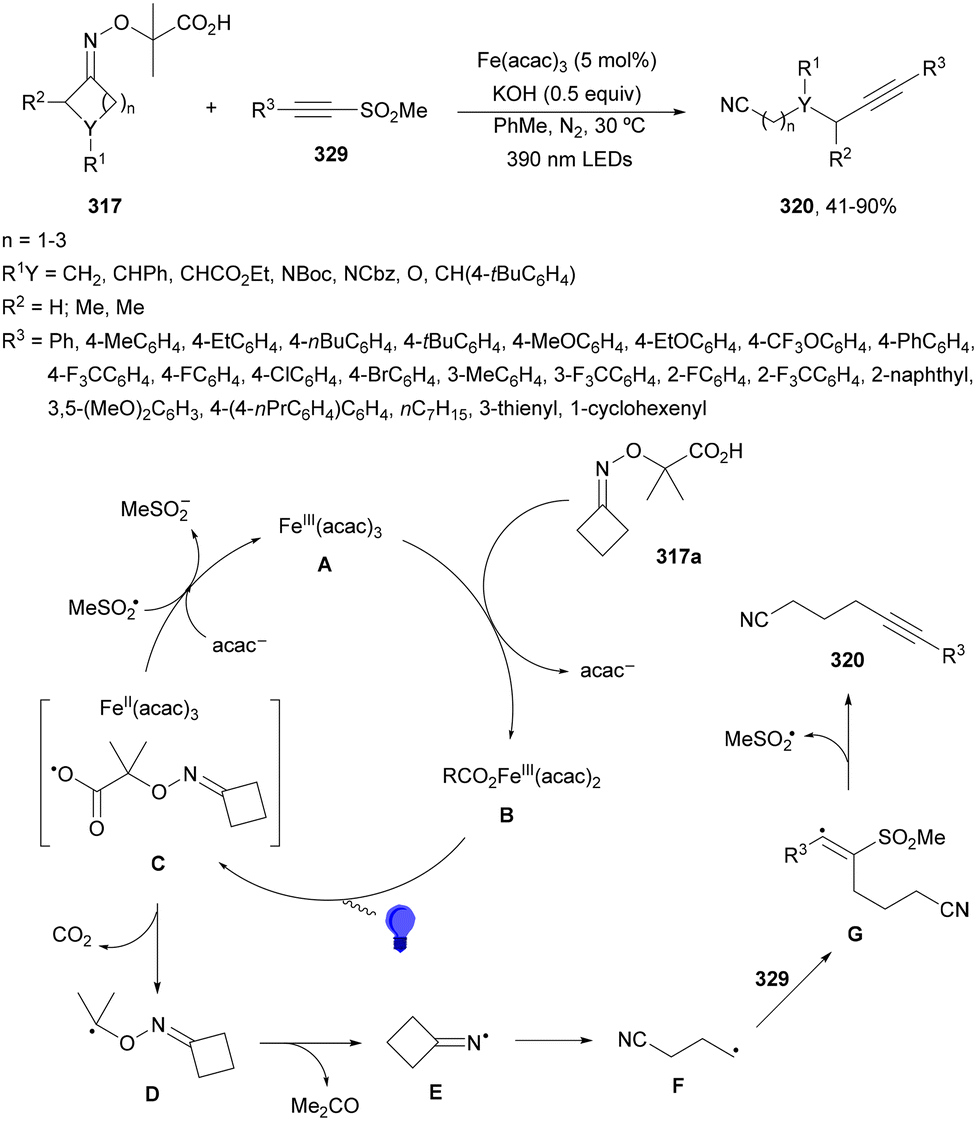 | ||
| Scheme 128 Decarboxylative alkynylation of carboxylic acid derived oxime-ethers 317 with alkynyl sulfones 329 under Fe(acac)3 photoredox conditions. | ||
Alkynyl bromides 330 reacted with glyoxylic acid acetal 26 in the presence of 4CzIPN (25) as a PC and Cs2CO3 as a base in a mixture of DMF/MeCN at room temperature under blue LED irradiation to give alkynyl acetal products 331 with moderate yields (Scheme 129).235 Radical intermediate A can lose CO2 to form acetal radical B, which adds to alkynyl bromide 330 to give radical C which forms the product and a bromine radical. This protocol can be applied to the synthesis of acetylenic aldehydes by acid hydrolysis.
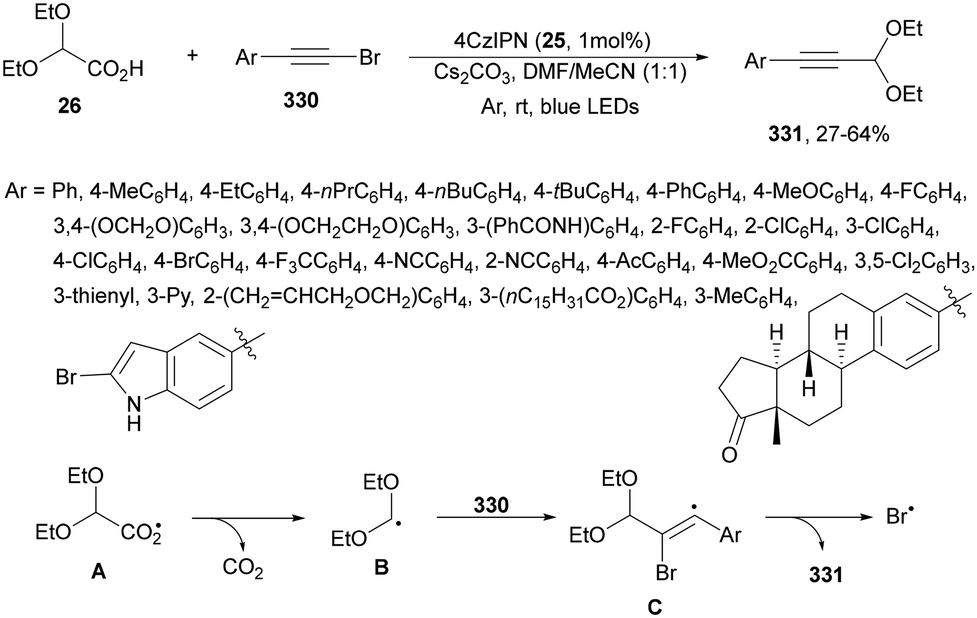 | ||
| Scheme 129 Decarboxylative alkynylation of glyoxylic acid acetal 26 with alkynyl bromides 330 under 4CzIPN (25) photocatalysis. | ||
Alkynyl C-nucleosides 325 have been directly prepared by decarboxylative alkynylation of ribosyl carboxylic acids 323 with terminal alkynes under Ir/Cu dual photocatalysis by Zhu and Messaoudi.236 This coupling was carried out using CuOAc and 4,4′-di-tBu-bpy 322 as a ligand, Ir complex 4 as a PC, and CsOAc as a base in DMA at room temperature under air and blue LED irradiation to furnish products 325 with, in general, good yields and high diastereoselectivity (Scheme 130). Aryl-substituted alkynes with electron-withdrawing and electron-donating groups successfully reacted under these reaction conditions as well as heteroaromatic alkynes. Challenging aliphatic alkynes afforded the corresponding products in yields ranging from 52 to 93%. With respect to carboxylic acid protecting groups, benzoyl and acetonide produced products 325 in an α,β ratio of 4:1, whereas benzyl protected 2-deoxy-D-ribose gave 325 in a 6:1 α,β ratio. On the other hand, OBn-protected glucopyranosyl acid failed. The presence of oxygen inhibits the competitive decarboxylative hydroalkylation of the alkyne under dual copper-photoredox catalysis.98 In the proposed mechanism, the photocatalytic cycle generates the anomeric radical A and the reduced Ir(II) complex. In the copper-catalyzed cycle the monomeric Cu(I)-acetylide complex B is formed, which by a SET gives the Cu(II)-complex C. At this stage, two possible pathways may be involved: (a) oxidation of the Cu(I) complex by oxygen, or (b) photoexcitation of LCu(I) to LCu(I)* followed by SET. Subsequently, the anomeric radical A can be captured by Cu(II) complex C to form the Cu(III)-species D, which after reductive elimination delivers product 325 and regenerates the Cu(I) catalyst. Reoxidation of the Ir(II) complex by a molecule of dioxygen regenerates the Ir PC and closes the catalytic cycle.
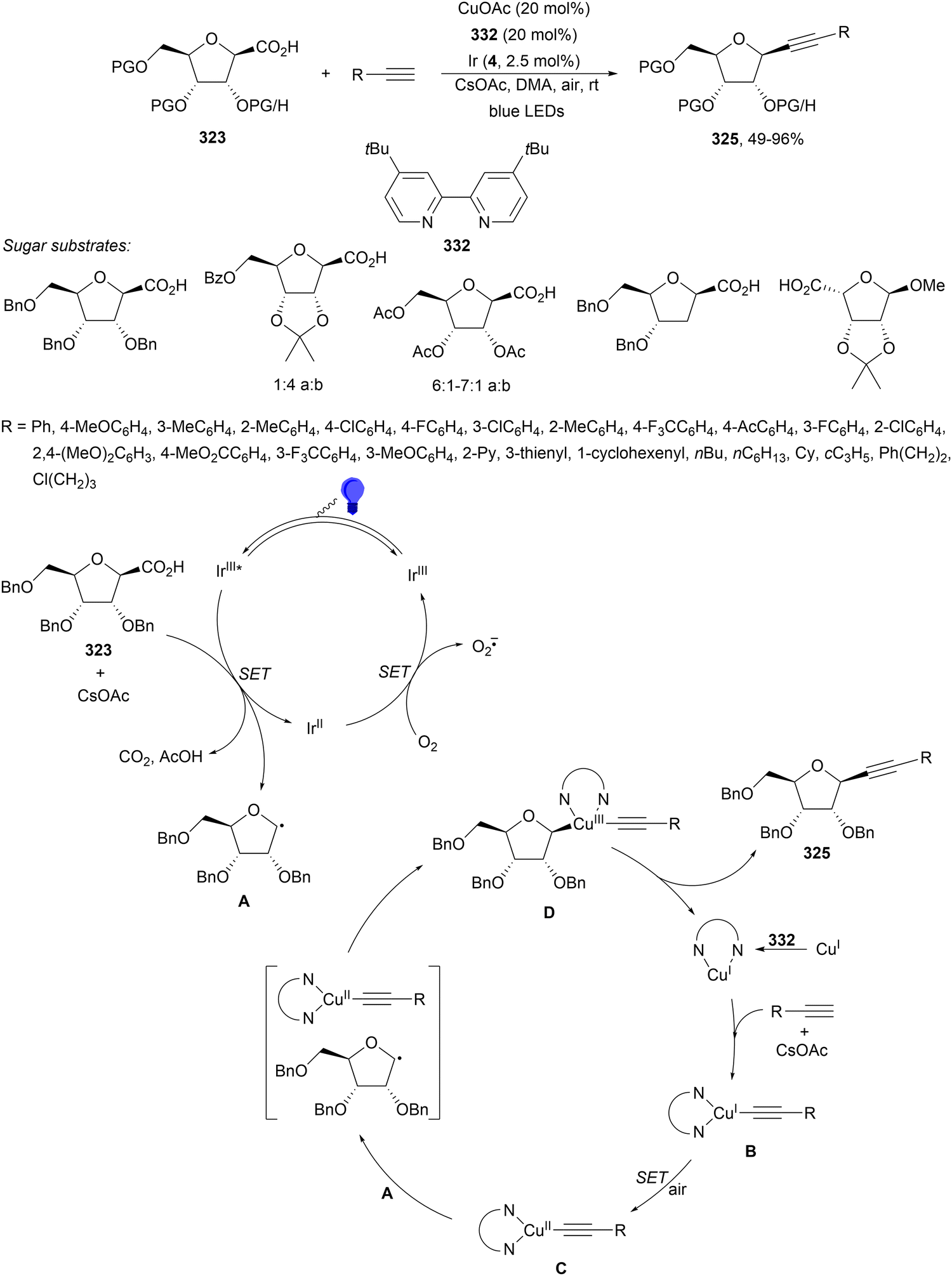 | ||
| Scheme 130 Decarboxylative alkynylation of furanosyl carboxylic acids 323 with terminal alkynes under Ir/Cu dual photocatalysis. | ||
Double decarboxylative coupling of alkynoic acids 333 and alkyl carboxylic acids has been reported by Lee and co-workers237 to obtain the corresponding internal alkynes 327 (Scheme 131). This process was carried out with Fe(NO3)3·9H2O or FeCl2/tris(2-pyridylmethyl)amine (TPA) as a catalyst and PhI(OAc)2 as an oxidizing agent in MeCN at room temperature under blue LED irradiation. Initial studies were performed with different alkynoic acids 333 and 2,3-dihydro-1,4-benzodioxine-2-carboxylic acid 334 to afford products 327 with good yields (Scheme 131a). However, when diverse alkyl carboxylic acids were allowed to react with phenyl propiolic acid the corresponding alkynes 327 were isolated with good yields (Scheme 131b). According to control experiments a plausible mechanism was proposed. Initially, the alkyl carboxylic acid reacts with PhI(OAc)2 to give intermediate A, which reacts with the photoactivated Fe(III)* or Fe(II)* catalyst by SET to give after decarboxylation the radical anion species B. This intermediate evolves to radical C, which adds to the alkynoic acid to form radical D. Sequential SET and deprotonation of D provides intermediate E, which after decarboxylation affords the acetylenic product 327. The formation of radical C by a LMCT54,55 pathway has been also postulated.
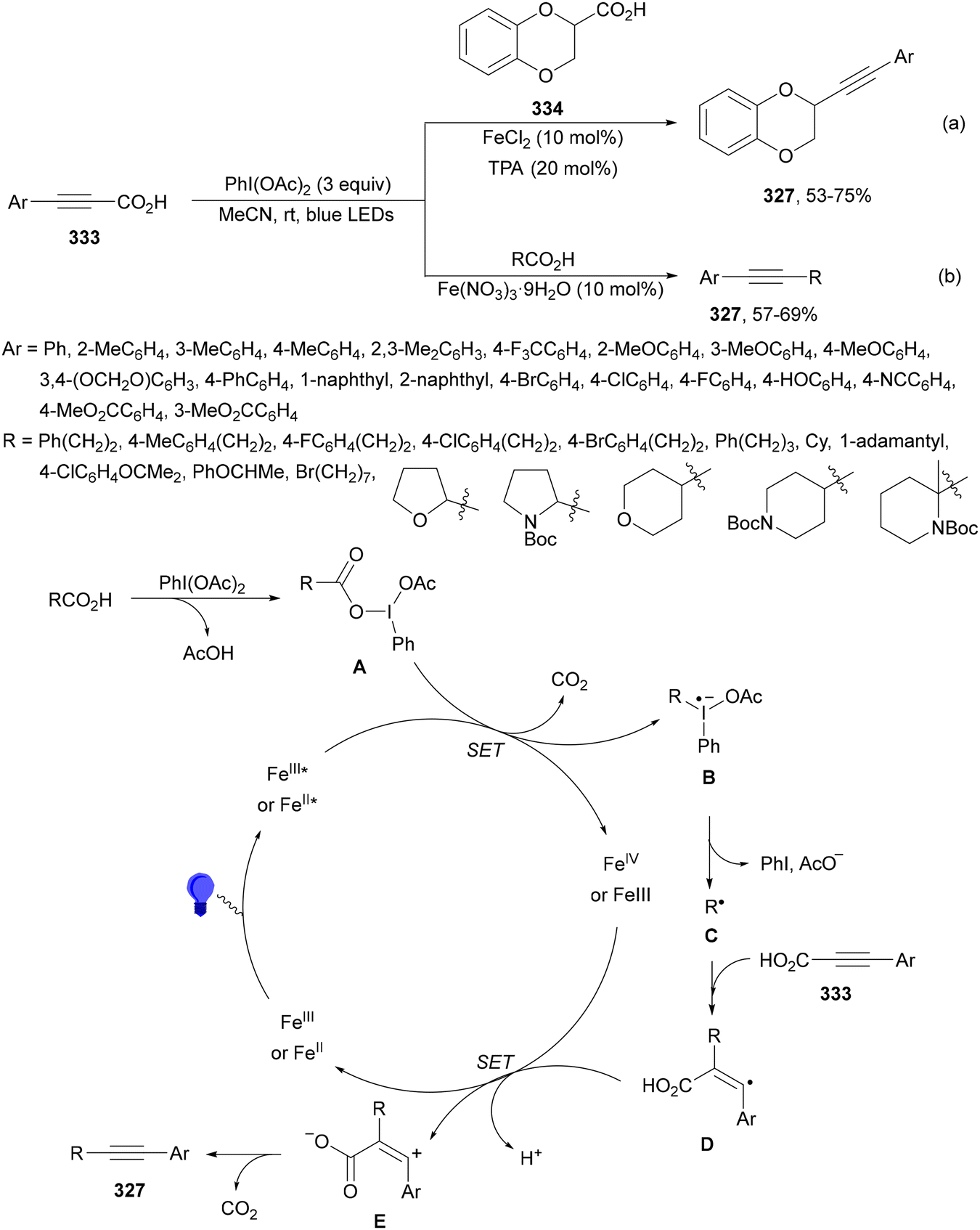 | ||
| Scheme 131 Double decarboxylative alkynylation of alkyl carboxylic acids with alkynoic acids 333 under iron photocatalysis (a, b). | ||
Decarboxylative alkynylation of NHPI esters 69 has been performed using alkynyl sulfones or directly terminal alkynes under mild reaction conditions. Under visible light irradiation alkynyl sulfones 329 reacted with NHPI esters using different photocatalysts such as Ru(bpy)3(PF6)2.226,238–241 Recently, Xu, Song and co-workers242 described the alkynylation of gem-borylsilyl NHPI ester 334 with alkynyl sulfones 328 using Ir(ppy)3 as a PC, Hantzsch ester (HE) as a reductant, and DIPEA as a base in DCM at room temperature under Ar and blue LED irradiation to obtain alkynes 335 with good yields (Scheme 132). In the proposed mechanism, the Ir(II) complex, formed by reduction of Ir(III)* in the presence of HE via a SET process, reduced NHPI ester 334 to give the β-borylsilyl radical A releasing CO2 and PhthN−. This intermediate A adds to alkynyl sulfone 328 affording radical B. Final β-elimination of the sulfonyl radical provides the product. The same process was carried out with allyl sulfones to furnish the corresponding allylated products.
 | ||
| Scheme 132 Decarboxylative alkynylation of gem-borylsilyl NHP esters 334 with alkynyl sulfones 328 under Ir(ppy)3 photocatalysis. | ||
Decarboxylative alkynylation of NHPI esters 69 with terminal alkynes, as in the case of carboxylic acids, has been carried out under copper catalysis. Fu and co-workers243 employed Ru(bpy)2Cl2/CuI and visible light at room temperature with α-AAs to give the corresponding propargyl amines in good yields. Later, Zhang and Zhang244 reported the use of CuI (10 mol%) and a tridentate ligand 336 for the alkynylation of NHPI and N-hydroxytetrachlorophthalimide (TCNHPI) esters derived from primary, secondary and tertiary carboxylic acids with terminal alkynes to give internal alkynes 327 with good yields (Scheme 133). Experimental studies reveal that the Cu–acetylide A gives after irradiation excited complex B, which by a SET to the NHPI ester generates complex C and radical D. Both intermediates C and D form the Cu(III) species E, which undergoes reductive elimination to furnish the product.
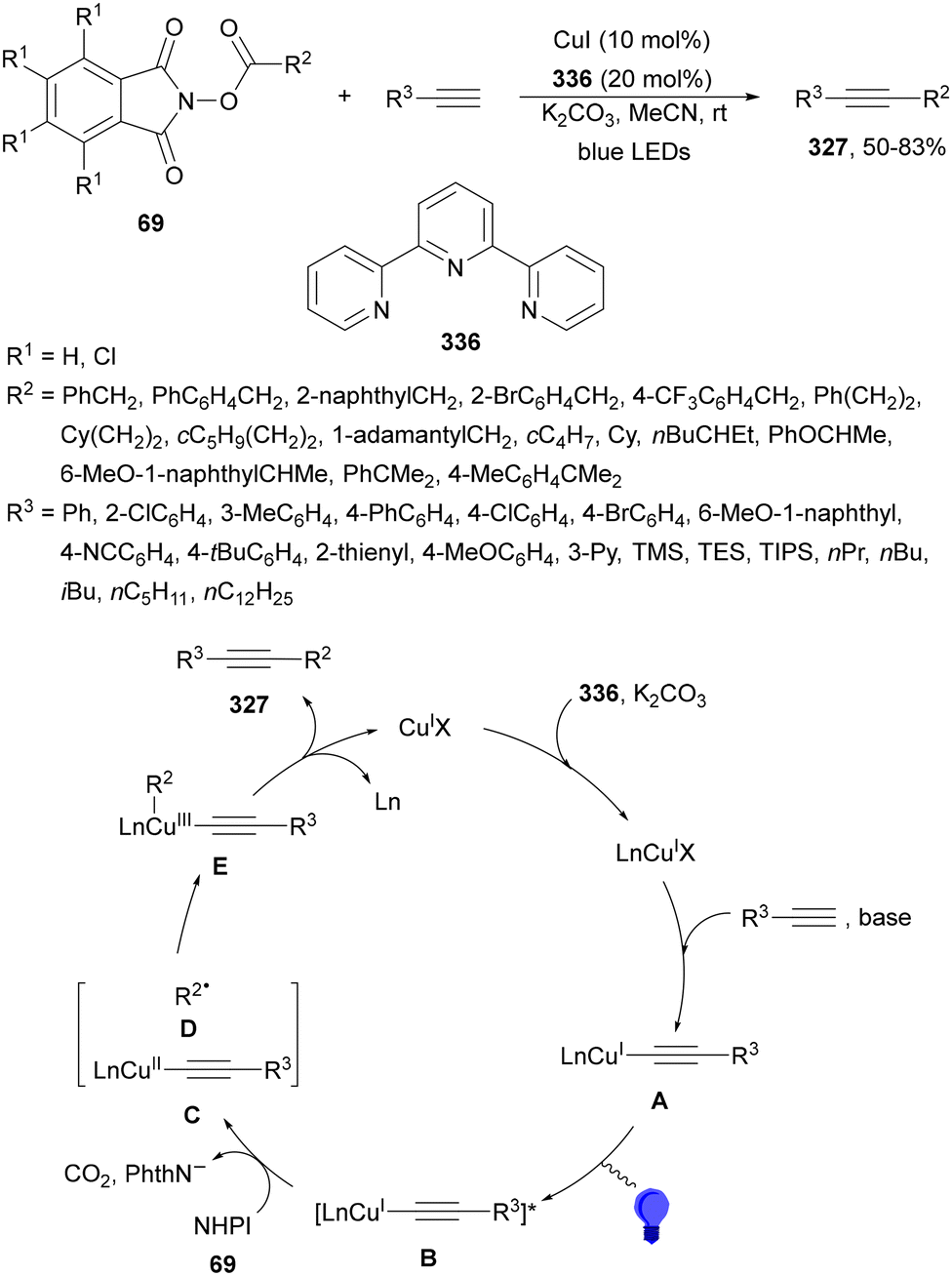 | ||
| Scheme 133 Decarboxylative alkynylation of NHPI esters 69 with terminal alkynes under Cu(I) photocatalysis. | ||
Wang, Liang, Ni and co-workers245 reported the same alkynylation process using CuCl and Cu(acac)2 as catalysts in the presence of Et3N as a ligand in THF at room temperature under Ar and blue LED irradiation. The corresponding internal alkynes 327 were obtained in 26–76% yields with a wide range of carboxylic acid derivatives and terminal alkynes (Scheme 134a). Cyclohexanecarboxylic acid was allowed to react with N-hydroxyphthalimide using DCC in DCM and the formed NHPI ester was alkynylated in situ with 4-MeC6H4C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH under the above reaction conditions providing the corresponding alkyne in 70% yield. For the coupling of non-aryl alkynes, 10% of PhCC–Cu should be added giving products 327 in 45–98% yield (Scheme 134b). Experimental and theoretical DFT calculations supported a reaction mechanism which starts by formation of complex A by reaction of CuCl with Et3N. Subsequently, complex A reacts with the terminal acetylene to form the corresponding acetylide B, which is photoexcited to intermediate C. This photosensitive Cu(I) acetylide C undergoes a SET process with the NHPI ester to afford, after decarboxylation, alkyl radical D and Cu(II)-acetylide E. Ligand exchange of E with Cu(acac)2(NEt3) gives the copper complex F. Final addition of radical D to F provides the Cu(III) intermediate G, which after reductive elimination furnishes acetylene 327 and H. For photoexcitation of copper acetylide the use of a conjugate aromatic terminal alkyne is required to work in the visible light energy range. Secondly, the ligand NEt3 promotes the electron transfer from the copper acetylide to NHPI ester. Finally, the acac ligand intervenes in the generation of bi-copper complexes, which inhibits the homocoupling of the terminal alkyne.
CH under the above reaction conditions providing the corresponding alkyne in 70% yield. For the coupling of non-aryl alkynes, 10% of PhCC–Cu should be added giving products 327 in 45–98% yield (Scheme 134b). Experimental and theoretical DFT calculations supported a reaction mechanism which starts by formation of complex A by reaction of CuCl with Et3N. Subsequently, complex A reacts with the terminal acetylene to form the corresponding acetylide B, which is photoexcited to intermediate C. This photosensitive Cu(I) acetylide C undergoes a SET process with the NHPI ester to afford, after decarboxylation, alkyl radical D and Cu(II)-acetylide E. Ligand exchange of E with Cu(acac)2(NEt3) gives the copper complex F. Final addition of radical D to F provides the Cu(III) intermediate G, which after reductive elimination furnishes acetylene 327 and H. For photoexcitation of copper acetylide the use of a conjugate aromatic terminal alkyne is required to work in the visible light energy range. Secondly, the ligand NEt3 promotes the electron transfer from the copper acetylide to NHPI ester. Finally, the acac ligand intervenes in the generation of bi-copper complexes, which inhibits the homocoupling of the terminal alkyne.
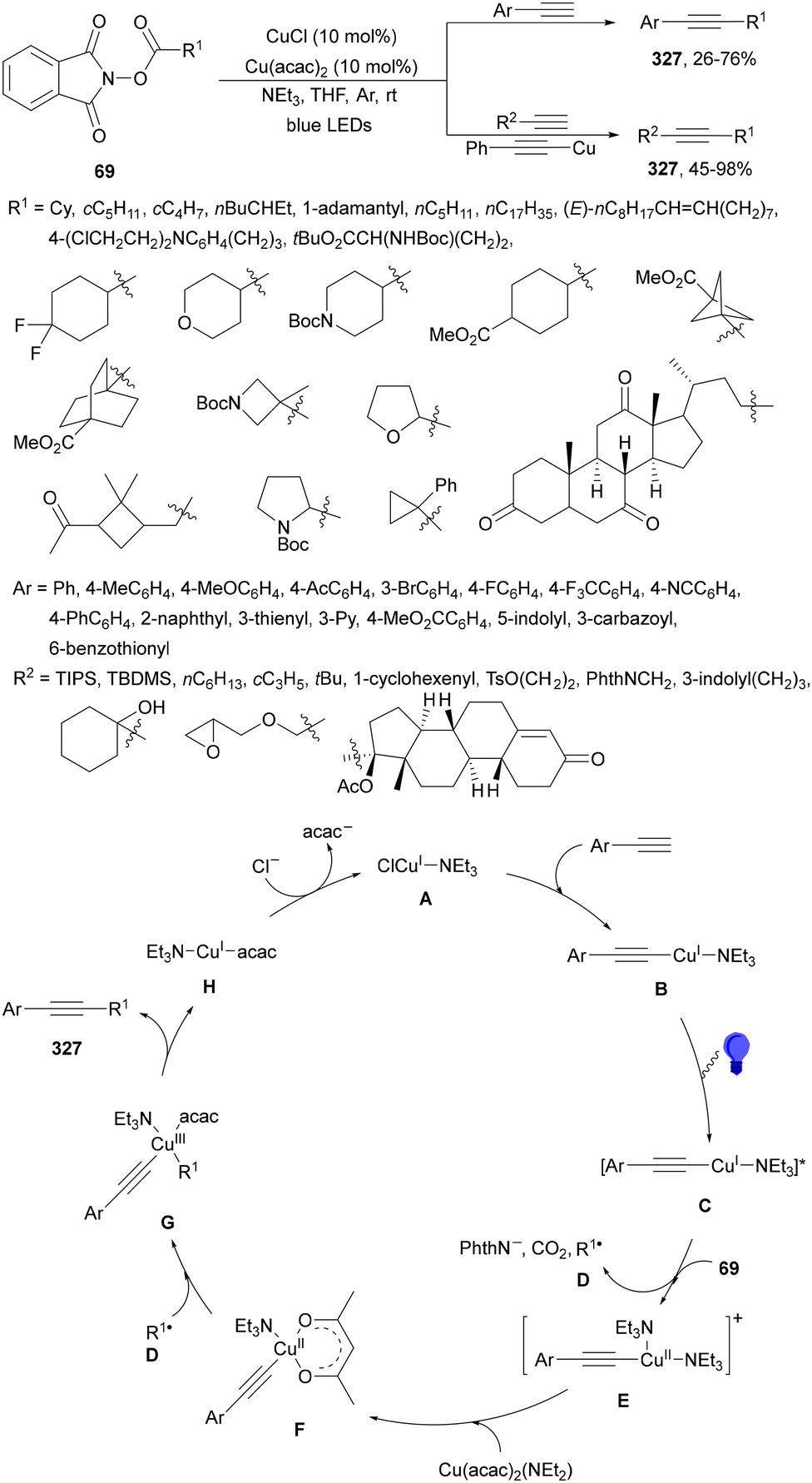 | ||
| Scheme 134 Decarboxylative alkynylation of NHPI esters 69 with terminal alkynes under Cu(I) photocatalysis. | ||
Enantioconvergent140 decarboxylative alkynylation of NHPI esters with terminal alkynes has been described by Liu and co-workers.246N-Hydroxy-2,3-naphthalimide 337 derived from carboxylic acids bearing at the α-position a stereocenter reacted with aliphatic and aromatic terminal alkynes using Cu(I) as a PC, chiral phosphine 338 as a ligand, and Cs2CO3 as a base in trifluoromethylbenzene at room temperature under blue LED irradiation. The corresponding enantioenriched alkynes 339 were obtained in up to 81% yield and up to 99% ee (Scheme 135). A wide range of NHPI esters 337 and terminal alkynes were employed, and a one-pot synthesis and gram-scale preparation were successfully performed. These NHPI esters 337 quenched the excited copper acetylide A very efficiently enabling Glaser coupling.
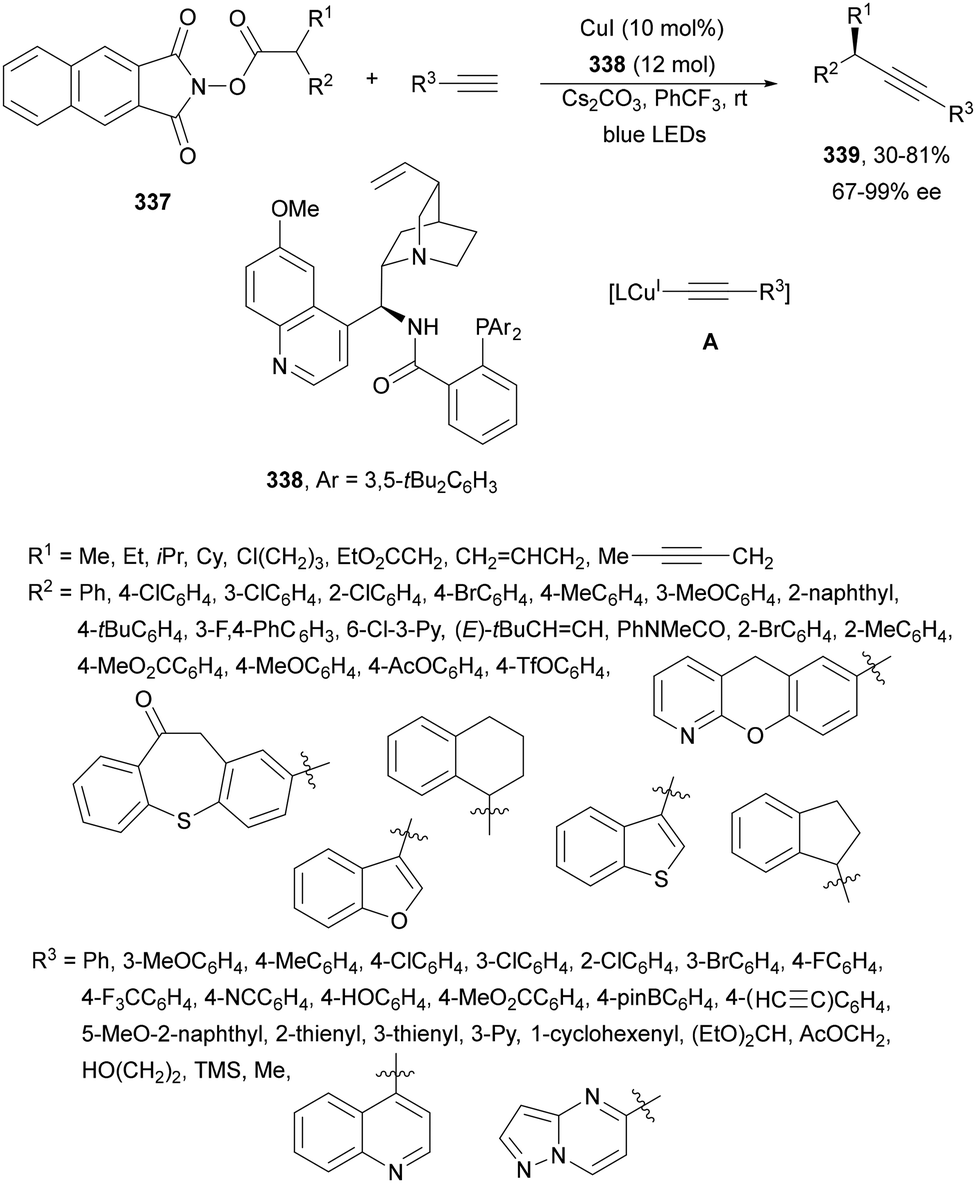 | ||
| Scheme 135 Enantioconvergent decarboxylative alkynylation of NHPI esters 337 under CuI/338 photocatalysis. | ||
Decarboxylative alkynylation of aliphatic carboxylic acids or their NHPI esters under visible-light photoredox conditions is an excellent protocol for the formation of C(sp3)–C(sp) bonds using mild reaction conditions. Hypervalent iodine reagents, alkynyl sulfones and terminal alkynes have been employed for the synthesis of internal alkynes. Peptides can be alkynylated at the C-terminus and glycosylic acids at the anomeric carbon atom with high diastereoselectivity. All these alkynylation reactions can be carried out using mainly 4CzIPN as an organocatalyst. However, copper-catalyzed processes are more effective with terminal alkynes and have been applied to the enantioconvergent decarboxylative alkynylation of NHPI esters using an amino phosphine as a chiral catalyst.
2.10. Acylation reactions
C(sp3)–C(sp2) bond formation using acyl radicals allowed the synthesis of ketones and carbamoyl radicals giving amides by decarboxylation of α-keto acids and oxamic acids, respectively. Acyl radicals reacted with alkenes and imines by addition reactions and can be arylated, allylated and alkynylated. Zhu and co-workers247 developed a visible-light-mediated decarboxylative acylation of styrenes 2 with α-keto acids 15 using Ir complex 4 as a PC (Scheme 136). The corresponding α,β-unsaturated phenones 18 were obtained with good yields in the presence of Selectfluor and NaOAc as a base in a 1:1 mixture of MeCN/H2O at room temperature. In the proposed mechanism, photoexcited Ir(III) oxidized the α-keto acid by a SET process to an acyl radical A after decarboxylation. This radical A adds to styrene to deliver the benzylic radical B. Subsequently, Selectfluor reacts with B to provide a β-fluoro ketone and the radical C, which oxidizes Ir(II) by a SET procedure to give Ir(III). The α-fluoro ketone eliminates HF in the presence of base to provide the final α,β-unsaturated ketone 18. Tunge and co-workers63 employed cobaloxime 16 and [Mes-Acr-Ph]BF417 as dual catalysis (see Scheme 6).
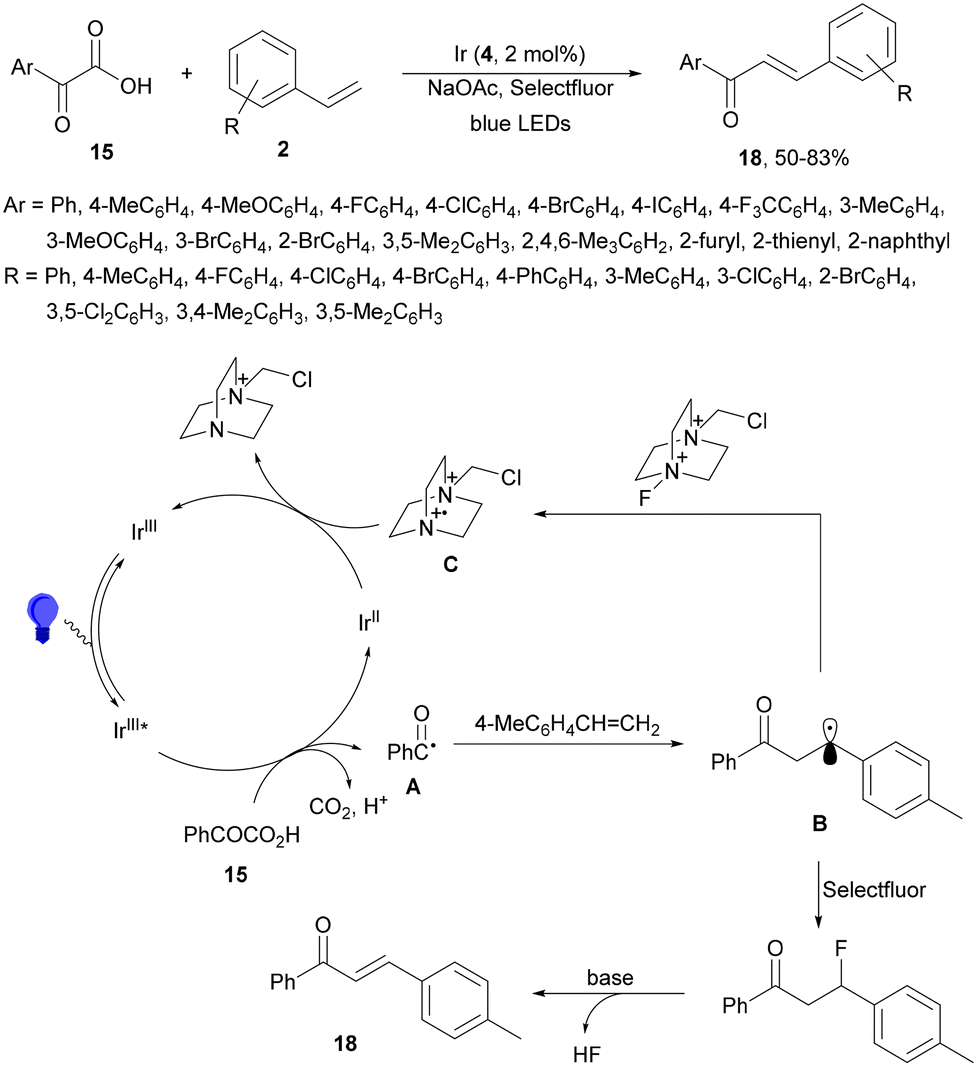 | ||
| Scheme 136 Decarboxylative acylation of styrenes 2 with α-keta acids 15 under Ir photocatalysis. | ||
Alkylation of N-acyl saccharins 340 by photoredox decarboxylation of NHPI esters 69 under visible-light has been reported by Opatz and co-workers.248 In the presence of Hantzsch ester (HE) as a photoreductant to promote radical generation and to participate in the Ni-catalyzed cycle to restore the reaction species, the corresponding ketones were formed in moderate to good yields (Scheme 137). Aroyl and alkanoyl moieties were alkylated with primary, secondary, benzylic, α-oxy and α-amino radicals. According to computational (COSNAR method) and spectroscopic studies, a catalytic cycle was proposed. The Ni(II) complex is reduced by HE to generate Ni(0) species, which undergoes oxidative addition to the N-acyl saccharin 340 to lead to intermediate A. This intermediate trapps radical B from the NHPI ester 69 to give the Ni(II) intermediate C, which after reductive elimination provides the ketone and the Ni(II) species D. Final reduction of D by photoexcited HE closes the catalytic cycle.
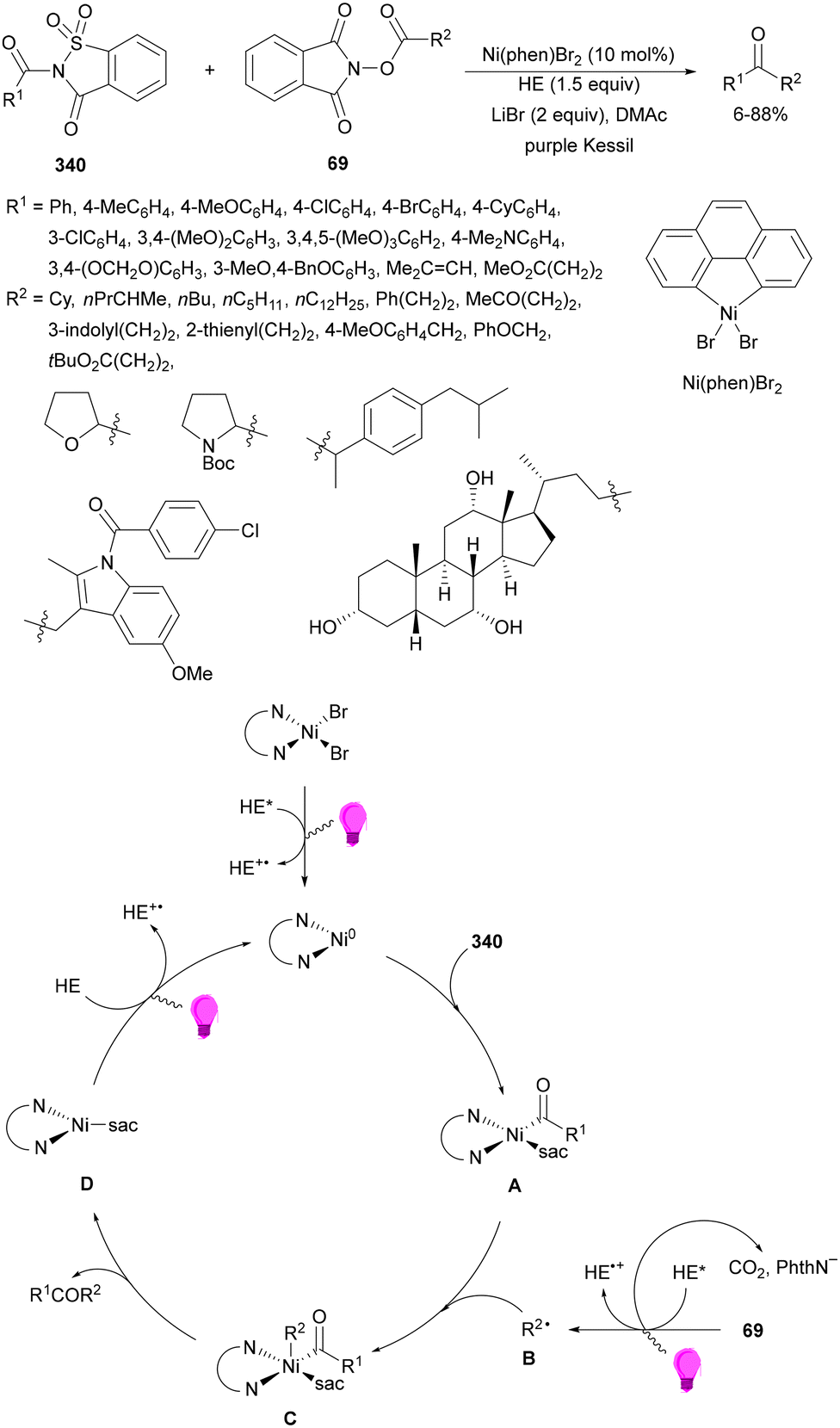 | ||
| Scheme 137 Decarboxylative acylation of NHPI esters 69 with N-acyl saccharins 340 under HE and Ni(phen)Br2 photocatalysis. | ||
Glycosyl esters 37 have been acylated with carboxylic acids to the corresponding C-acyl furanosides using 4CzIPN (25) and NiCl2·DME with 2,2′-bis-2-oxazoline 341 as the ligand in the presence of diethyl dicarbonate (DEDC) in dioxane under blue LED irradiation. Diao and co-workers249 employed a large number of furanoses, pyranoses and carboxylic acids and obtained acylated furanosides 342 with, in general, good yields including a thymidine analogue, and diplobifuranylone B and the modification of (+)-sclareolide (Scheme 138). In the proposed mechanism, the dihydropyridine (DHP) esters undergo anomeric homolysis under photoexcited PC 25 through CO2 and HE-derived pyridine evolution to form a glycosyl radical A, which reacts with the Ni(II) intermediate B to form the Ni(III) complex C. Subsequent reductive elimination gives the product and Ni(I)Br, which is reduced by PC˙− to Ni(0). Reaction of diethyl dicarbonate with the acid provides the mixed anhydride D, which reacts with Ni(0) to regenerate B and CO2.
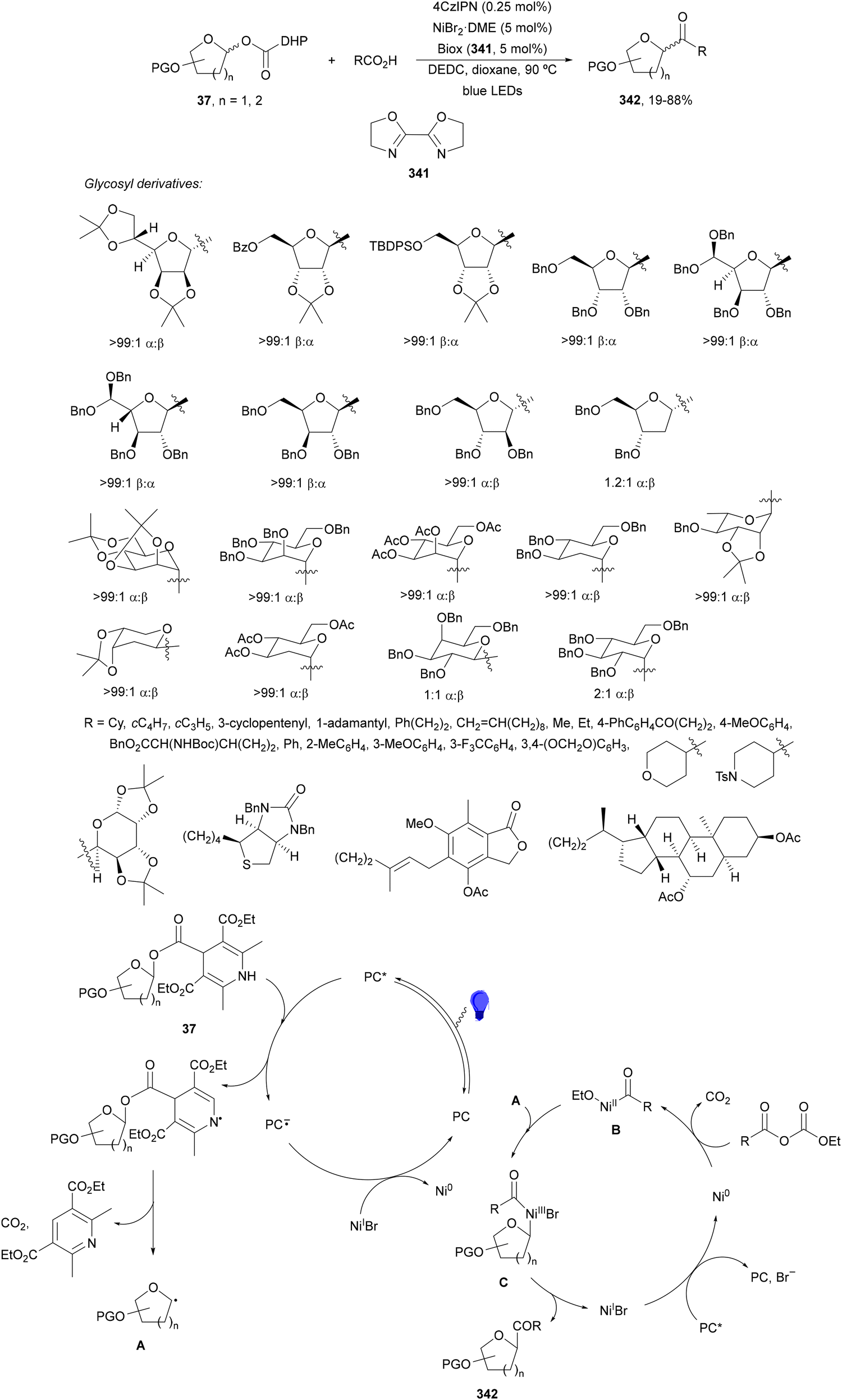 | ||
| Scheme 138 Decarboxylative acylation of glycosyl esters 37 with carboxylic acids under 4CzIPN (25) and NiBr2 photocatalysis. | ||
Molander and co-workers250 performed the synthesis of C-acyl glycosides 342 using furanosyl and pyranosyl acids as acylating agents and NHPI esters 69 as sources of alkyl radicals. This Ni-mediated reaction took place through an electron donor–acceptor (EDA) complex between HE and NHPI ester170 (see Scheme 75). In the presence of HE and dimethyl dicarbonate (DMDC) using complex Ni(dtbpy)Br2 (210) as a catalyst, this photoredox acylation provided products 342 with moderate to good yields (Scheme 139). In the proposed mechanism, HE and NHPI ester forms a molecular aggregate A, which after irradiation at 390 nm triggers SET to give HE+˙ and radical R˙. DMDC activates the saccharide acid affording in situ a carbonic anhydride B. The alkyl radical can follow two pathways based on computational studies, initial oxidative addition of a Ni(0) species to B, forming the Ni(II) intermediate C, which undergoes radical addition to afford the Ni(III) complex D. Subsequent reductive elimination from D gives the product and the Ni(I) complex F, and the Ni(0) catalyst is regenerated from photoexcited HE. The other possible mechanistic pathway starts with the reaction of the alkyl radical and Ni(0) affording the Ni(I) intermediate E, which undergoes oxidative addition of anhydride B to give the Ni(III) species D.
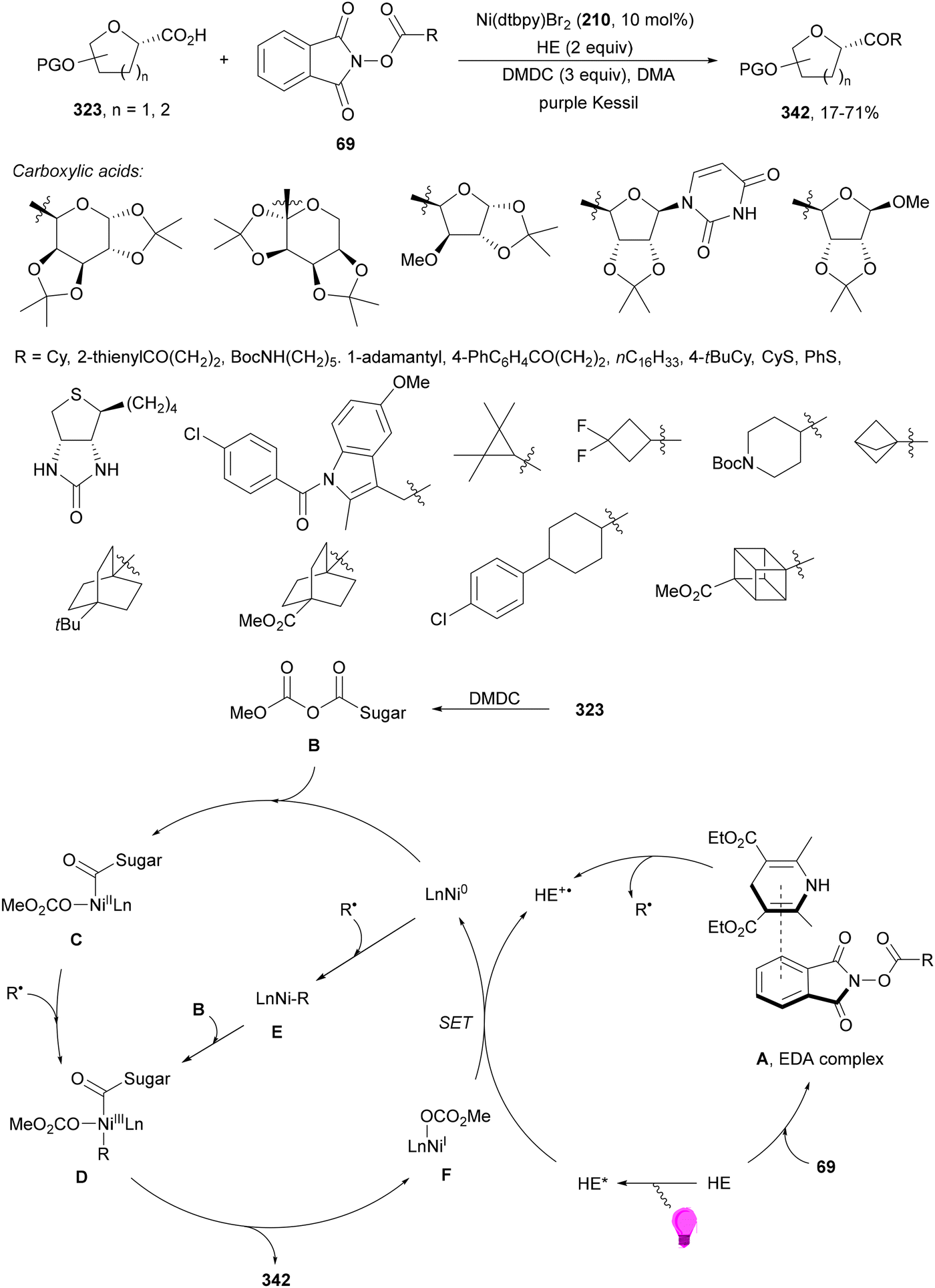 | ||
| Scheme 139 Decarboxylative acylation of NHPI esters 69 with glycosyl acids 323 under HE and Ni complex 210 photocatalysis. | ||
The reductive decarboxylative acyl cross-coupling between 2-pyridyl esters 343 and NHPI esters 69 has been carried out by Qi, Yuan and co-workers251 using 4CzIPN (25) and NiBr2·glyme with 344 as a ligand under dual photocatalytic conditions to provide ketones (Scheme 140). A broad range of primary, secondary and tertiary alkyl NHPI esters 69 as radical precursors and 2-pyridyl esters 343 as acylating components were employed. The presence of HE was crucial to form the photoactive electron donor–acceptor (EDA) complex between NHPI ester and HE170 (see Schemes 75 and 139), which by irradiation evolves to give the corresponding radical, CO2 and phthalimide anion. This radical enters the Ni-catalytic cycle Ni(0)/Ni(I)/Ni(III) to give after final reductive elimination of the Ni(III) intermediate, the ketone.
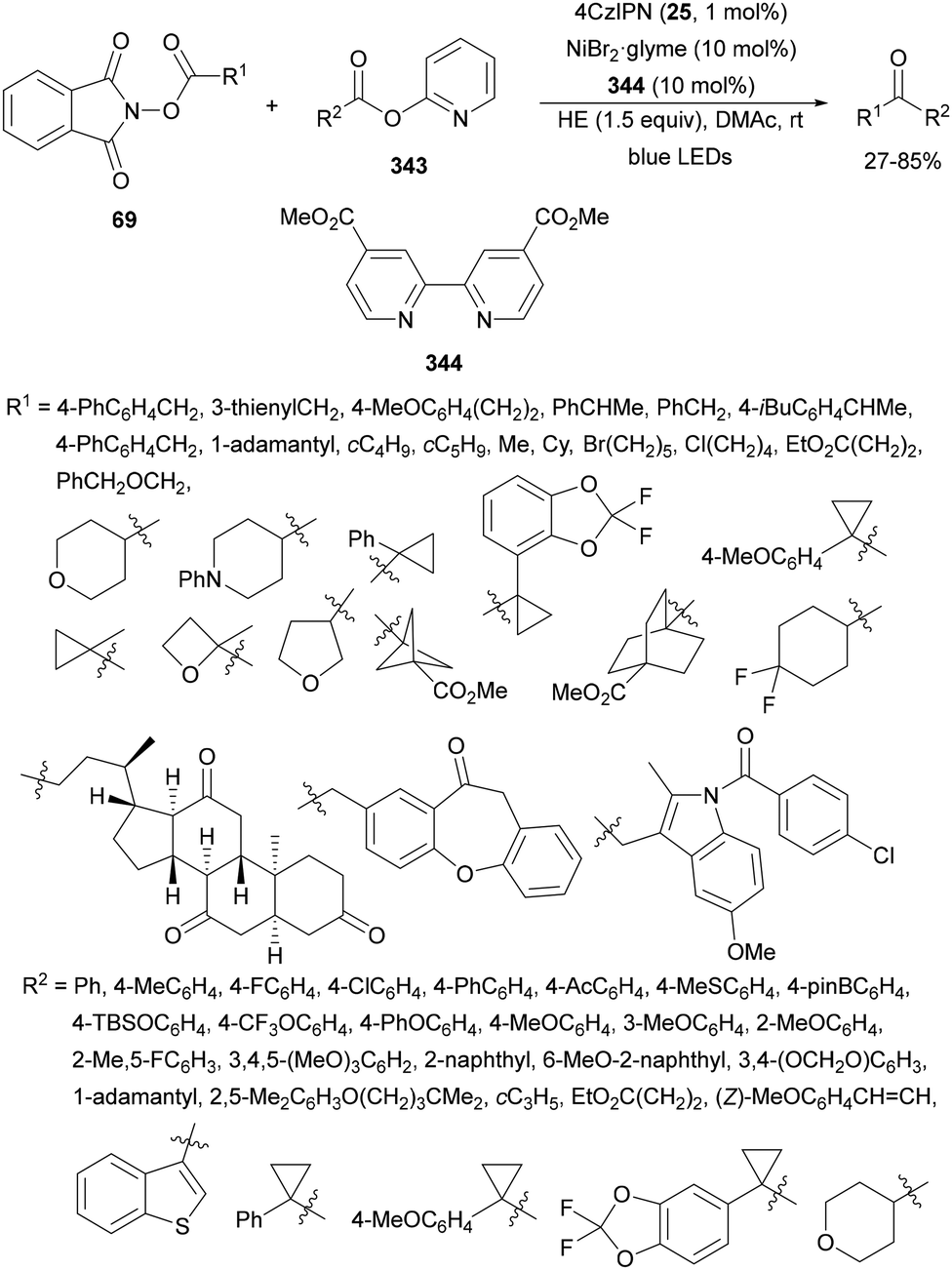 | ||
| Scheme 140 Decarboxylative acylation of NHPI esters 69 with 2-pyridyl esters 343 under 4CzIPN (25)/NiBr2 photocatalysis. | ||
A related procedure has been recently reported using S-2-pyridyl thioesters 345 instead of 2-pyridyl esters 343. Guo, Wu and co-workers252 performed the alkylation of S-2-pyridyl (SPy) aryl thioesters 345 with alkyl NHPI esters 69 in the presence of HE, NiCl2·DME, and ligand 344 as a catalyst to furnish alkyl aryl ketones with good yields (Scheme 141). In the proposed mechanism, HE is excited by a blue LED irradiation and promotes the formation of the alkyl radical A from the NHPI ester. This radical A reacts with Ni(0) to form the Ni(I) intermediate B, which undergoes oxidative addition to SPy to give the Ni(III) intermediate C. Alternatively, Ni(0) gives oxidative addition to SPy followed by radical recombination with A. Reductive elimination of C regenerates Ni(I) intermediate D and the product. The oxidized HE+˙ undergoes deprotonation to form a PyH derivative from HE as a by-product.
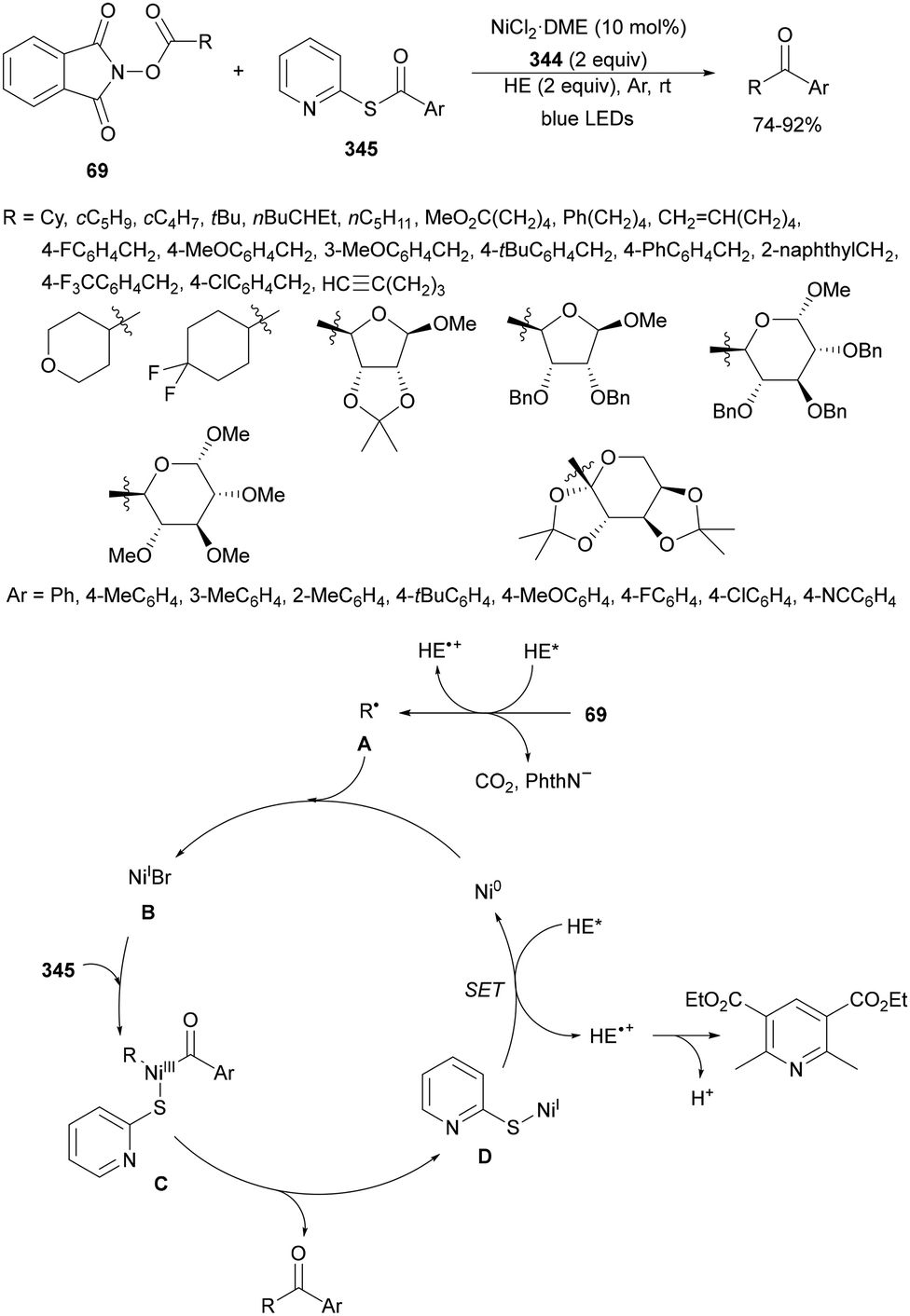 | ||
| Scheme 141 Decarboxylative acylation of alkyl NHPI esters 69 with S-2-pyridyl thioesters 345 under HE and NiCl2 photocatalysis. | ||
Acylation of aryl halides with α-keto acids 15 was initially described by MacMillan253 and Fu254 groups under photocatalytic decarboxylation conditions to provide the corresponding phenones. These C(sp2)–C(sp2) cross-coupling reactions were carried out using Ir/Ni253 and Ir/Pd254 photoredox dual catalysis. The organic dye 2-chlorothioxanthen-9-one (ClTXO, 346) has been used instead of an Ir PC by Li and co-workers.255 This process was effective for the acylation of aryl bromides with aryl and some alkyl α-keto acids using NiCl2·glyme and the ligand dtbbpy as a catalyst in aqueous DMF to give diaryl ketones with good yields under visible light (Scheme 142a). These reaction conditions in the absence of Ni have been employed for the hydroacylation of electron-deficient alkenes, such as acrylates and α,β-unsaturated ketones to furnish the corresponding 1,4-dicarbonyl compounds (Scheme 142b).
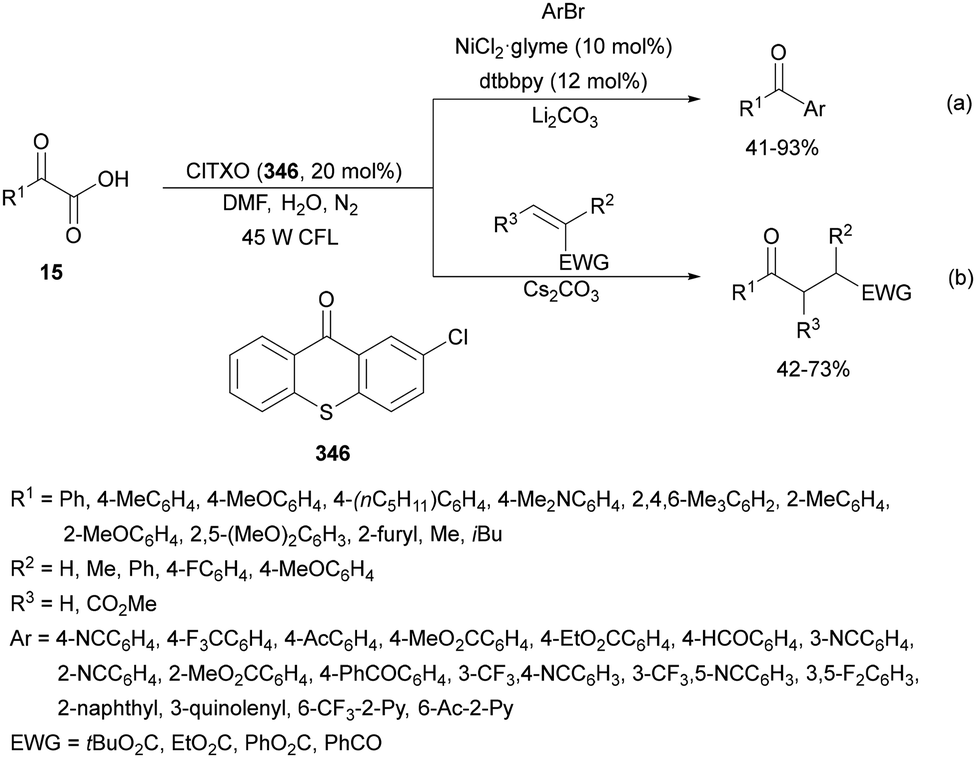 | ||
| Scheme 142 Decarboxylative acylation of aryl bromides (a) and electrophilic olefins (b) with α-keto acids 15 under ClTXO (346) photocatalysis. | ||
Oxalates derived from alcohols were used by Zhang and MacMillan256 for the cross-coupling of aryl halides with alkyl radicals. More recently, Xia and co-workers257 employed potassium oxalates 347 for the alkoxycarbonylation of aryl and alkenyl bromides with Ir complex 4 as a PC and NiBr2(PCy3)2 and dtbbpy as a catalyst under blue LED irradiation. This method gave the corresponding esters with good yields using 1,4-dioxane as solvent at room temperature (Scheme 143). Some synthetic applications allowed the synthesis of probenecid, mefenamic and adapalene, which were prepared with good yields. According to mechanistic studies a plausible mechanism was proposed, starting with the generation of an alkoxycarbonyl radical A by reduction of the hemioxalate via SET in the Ir catalyzed cycle. This radical A (path b) is captured by the LnNi(0) species to give the (alkoxycarbonyl)Ni(I) species B. Subsequent oxidative addition of organic bromide to B generates the Ni(III) intermediate C, which by reductive elimination furnish the ester product and the Ni(I) complex D. Final reduction of D by Ir(II) regenerates Ni(0) species. An alternative possible pathway a proceeds by oxidative addition of the organic halide to Ni(0) species to give Ni(II) species E, which was trapped by the alkoxycarbonyl radical to provide the same Ni(III) species C. However, the stoichiometric coupling experiment may not support this pathway.
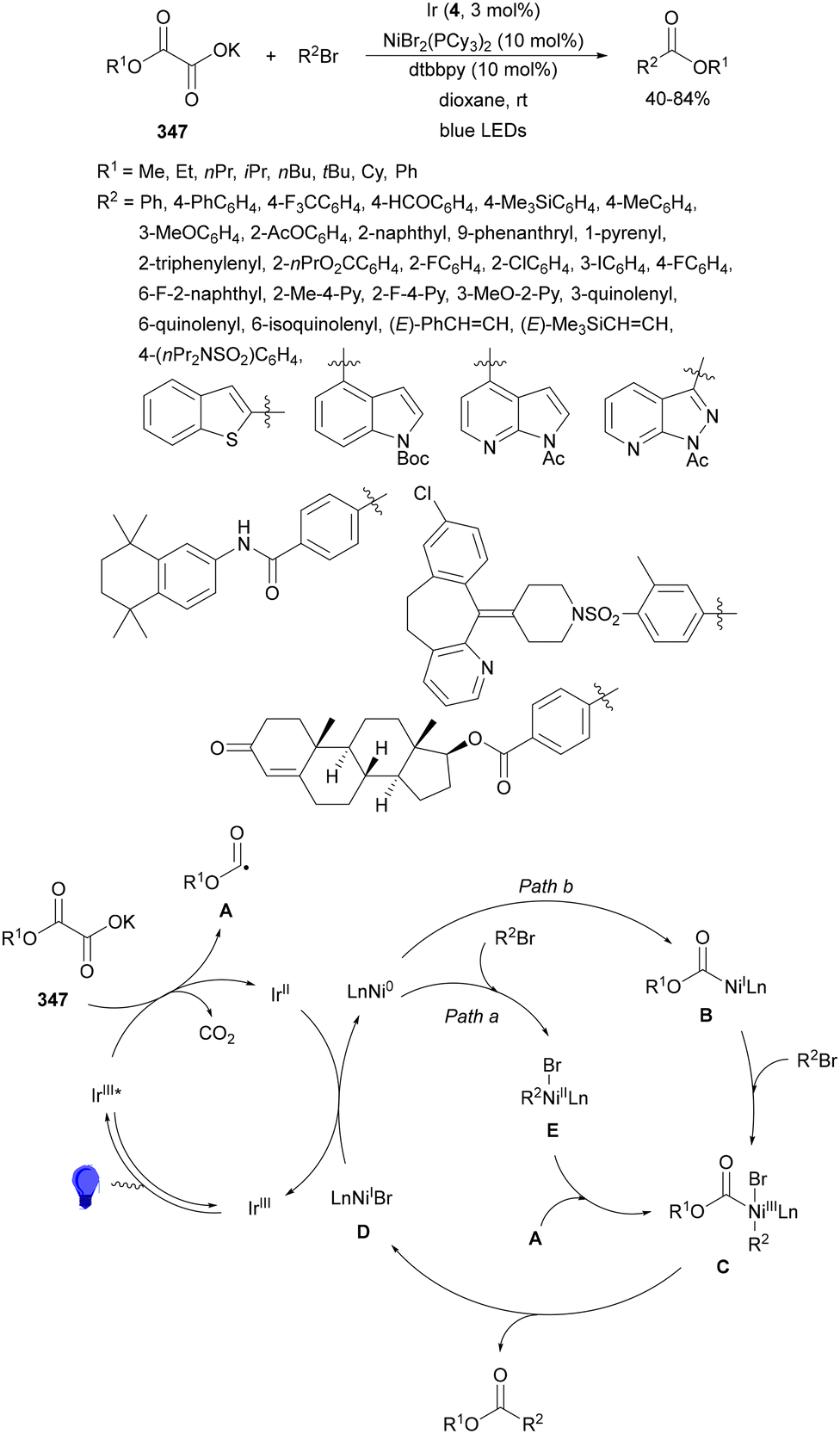 | ||
| Scheme 143 Decarboxylative acylation of organic bromides with potassium oxalates 347 under Ir/Ni photocatalysis. | ||
Photoredox decarboxylation of α-keto acids 15 in the presence of 4CzIPN (25) as a PC and cobaloxime 348 as a catalyst has been employed by Zhong and co-workers258 for the allylation reactions with methacrylates to furnish allylic ketones 349 (Scheme 144). These allylated compounds 349 were formed by dehydrogenation promoted by the cobaloxime Co(dmgBF2)2(H2O)2 (348) as a catalyst. Products 349 were mainly formed using 2,6-lutidine as a base with good yields, whereas in the presence of tBuOK as a base the corresponding saturated 1,4-dicarbonyl compounds 350 were obtained. In the proposed mechanism, the excited 4CzIPN is reductively quenched by the anion of the keto acid leading to a benzoyl radical A with CO2 extrusion and the reductive species 4CzIPN−˙. Benzoyl addition of A to ethyl methacrylate generates radical B, which is captured by Co(II) to form C, an intermediate Co(III). The cleavage of the Co–C bond and subsequent β-H elimination gives the allylic ketone 349 and Co(III)–H. This hydride reacts with ethyl methacrylate to generate Co(III) and a reductive by-product. Subsequent SET from 4CzIPN˙− to Co(III) regenerates the photoredox catalytic cycle and the cobaloxime catalytic cycle. Co(III)–H can also react with a proton to form H2 but only 23% of H2 was detected.
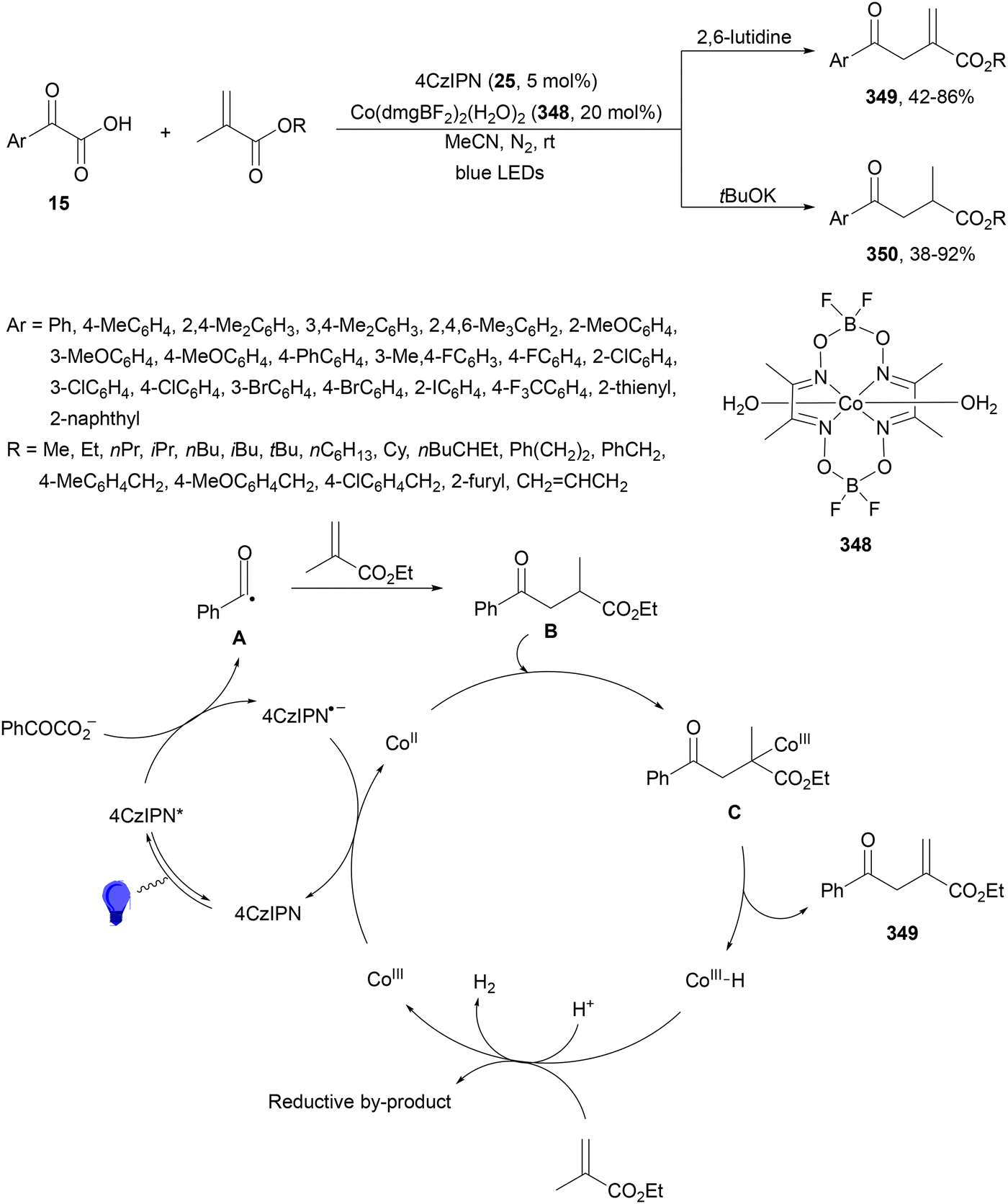 | ||
| Scheme 144 Decarboxylative acylation of methacrylates with α-keto acids 15 under 4CzIPN (25) and cobaloxime 348 photocatalysis. | ||
Decarboxylative acylation of Morita–Baylis–Hillman carbonates 351 with α-keto acids 15 has been carried out with Ru(bpz)3(PF6)2 (352) as a PC under blue LED irradiation.259 Different aromatic α-keto acids were employed as acylating agents, whereas aliphatic ones failed. The corresponding α,β-unsaturated esters 353 were obtained with moderate yields. In some cases, carboxylation products 354 were formed by oxidation of radical A with Ru2+* to give the corresponding cation, which reacted with water to give the carboxylic acid that adds to 351 in a Michael-type process (Scheme 145). The formation of the acyl radical A after oxidation of the α-keto acid by Ru2+* and decarboxylation has been postulated. Subsequent addition to 351 generates radical B, which is reduced by Ru+ to give anion C. Final elimination of BocO− as CO2 and tBuO− provides product 352.
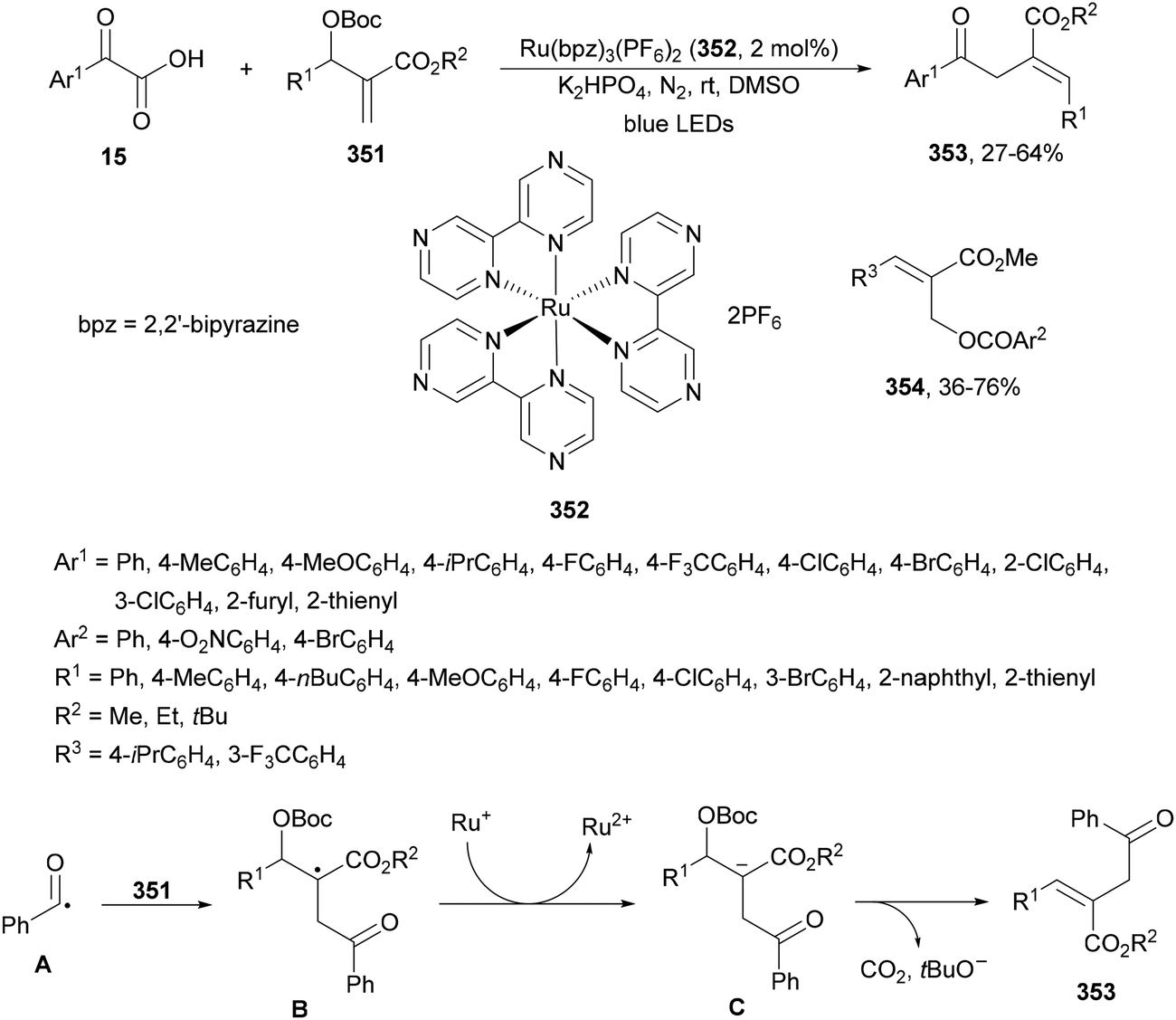 | ||
| Scheme 145 Decarboxylative acylation of MBH carbonates 351 with α-keto acids 15 under Ru2+ photocatalysis. | ||
Recently, Xie, Loh and co-workers260 employed MBHAs 286 as allylating reagents for decarboxylative coupling with α-keto acids 15 using triphenylphosphine and Mes-Acr+Ph(BF4) 17 as catalysts under blue LED irradiation (Scheme 146). When oxamic acids (R1 = NR2′) were used as acylating agents the corresponding amides 353 were formed in 42–96% yield. Products 353 were obtained with moderate to high yields and, in general, with >98:2 E/Z diastereoselectivity. In the plausible mechanism two possible pathways were proposed. Initial oxidation by PC* via SET of α-keto acid 15 gives radical A and CO2. In path a, MBHA reacts with PPh3 to give the phosphonium salt B, which after SET with PC˙− leads to allylic radical C. Subsequently, radical–radical coupling of A and C gives the product. In path b, radical A adds to MBHA to afford radical D, which accepts an electron from PC˙− to form anion E. Final elimination of acetate yields the product. MBHAs bearing α-D-galactopyranose, L-(−)-menthol and cholesterol were successfully employed.
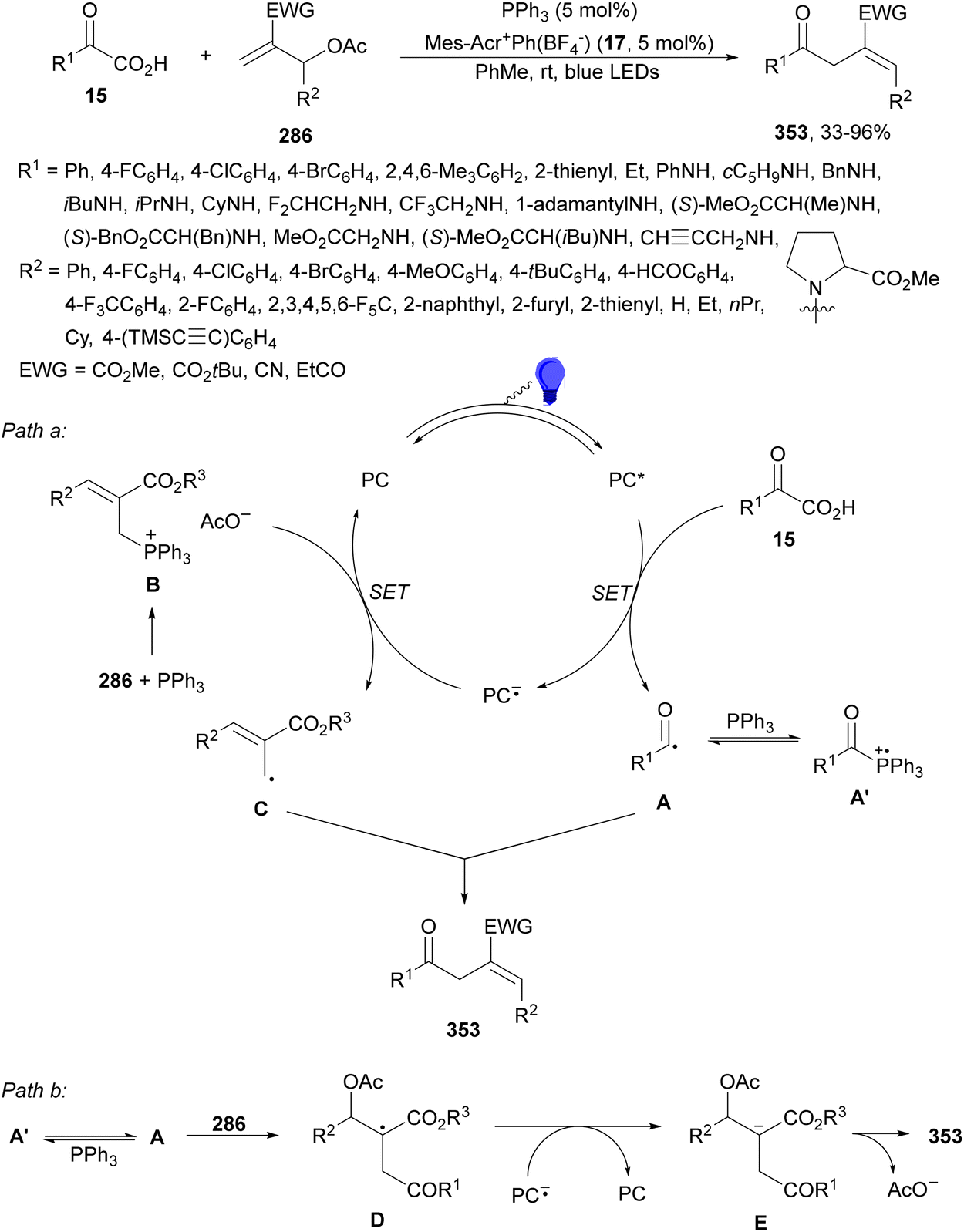 | ||
| Scheme 146 Decarboxylative acylation of MBHAs 286 with α-keto acids 15 under PPh3 and Mes-Acr+Ph(BF4−) 17 photocatalysis. | ||
C(sp2)–C(sp) bond formation by decarboxylative alkynylation of α-keto acids to provide ynones261 was described independently by Chen and co-workers262 and by Li, Wang and co-workers263 in 2015. Hypervalent iodine(III) reagents and Ru(bpy)3(PF6)2 as a PC under blue LED irradiation were used by the first group to give the corresponding ynones with good yields. Oxamic acids were also employed for the synthesis of ynamides. The second Chinese group263 performed the synthesis of enones by alkynylation of α-keto acids with bromoacetylenes 330 photocatalyzed by hypervalent iodine(III) reagent BI–OH.
Oxamic acids 15 (R1 = R2N) generate, by single-electron oxidation, the corresponding carbamoyl radicals after decarboxylation and have been applied to the synthesis of amides.264,265 Decarboxylative arylation allows the synthesis of benzamides under photoredox conditions. Jiang, Yu and co-workers266 employed 4-cyanopyridines 63 for the arylation of oxamic acids 15 (R1 = R2N) with 4CzIPN (25) as a PC and Cs2CO3 as a base in DMSO at room temperature under purple LED irradiation to furnish isonicotinamides 355 with good yields (Scheme 147). In the proposed catalytic cycle, the carbamoyl radical A adds to radical B to form intermediate C, which after decyanation yields product 355.
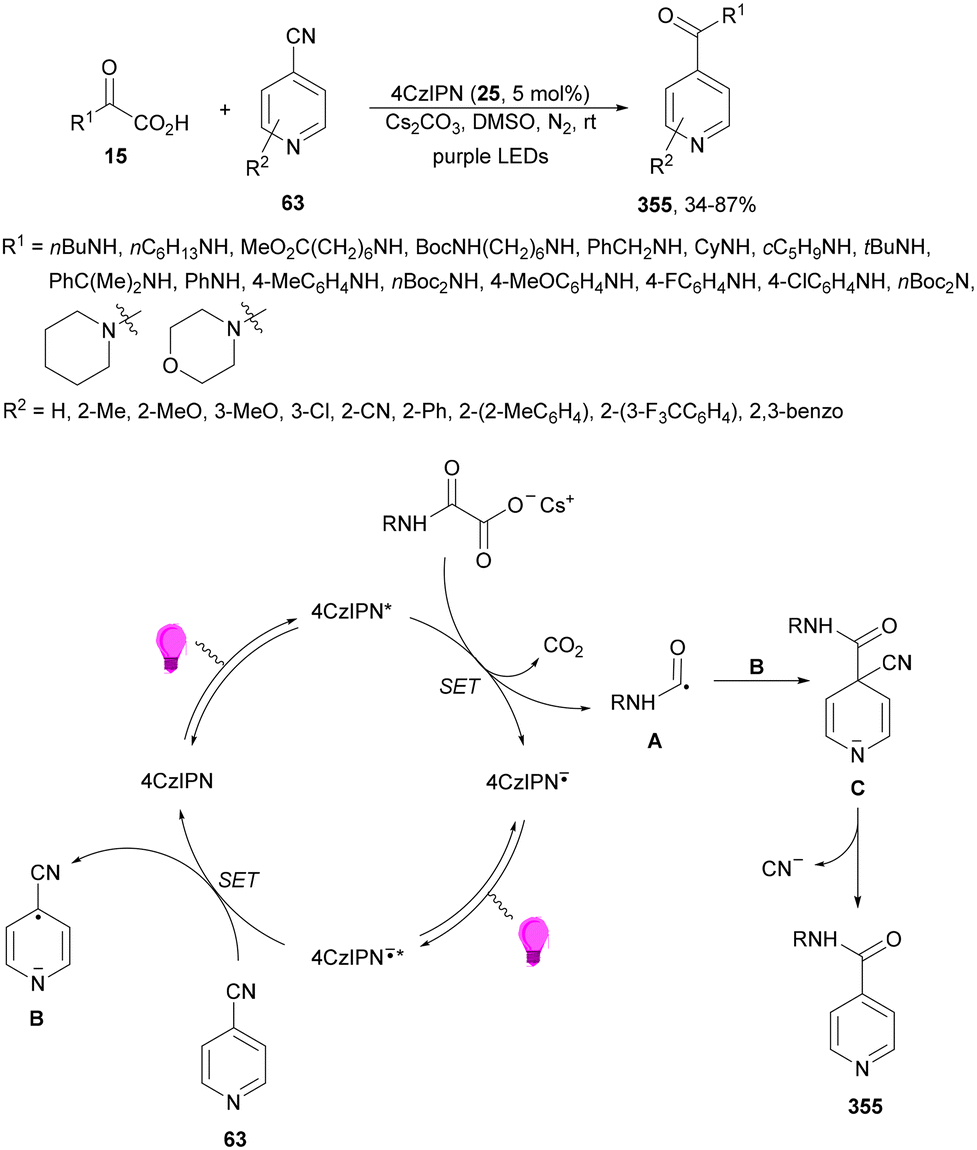 | ||
| Scheme 147 Decarboxylative carbamoylation of 4-cyanopyridines 63 with oxamic acids under 4CzIPN (25) photocatalysis. | ||
Decarboxylative arylation of oxamic acids with aryl bromides has been independently reported by Song267 and Sparr268 groups. In the first case, 4CzIPN (25) and NiBr2·glyme and 4,4′-dimethoxy-2,2′-bipyridine (185) as ligands were used as catalysts to give aromatic and heteroaromatic amides 356 with 35–94% yields (Scheme 148a).267 In the second protocol, they employed 4CzIPN (25) and NiCl2·glyme and dtbbpy as ligands and catalysts providing amides 356 with very modest to good yields (17–87%) (Scheme 148b).268 In both methods, two catalytic cycles have been proposed, the photoredox cycle, which generates a carbamoyl radical, and the Ni catalytic cycle starting from Ni(0) to give a Ni(III) intermediate, which by reductive elimination furnishes the product.
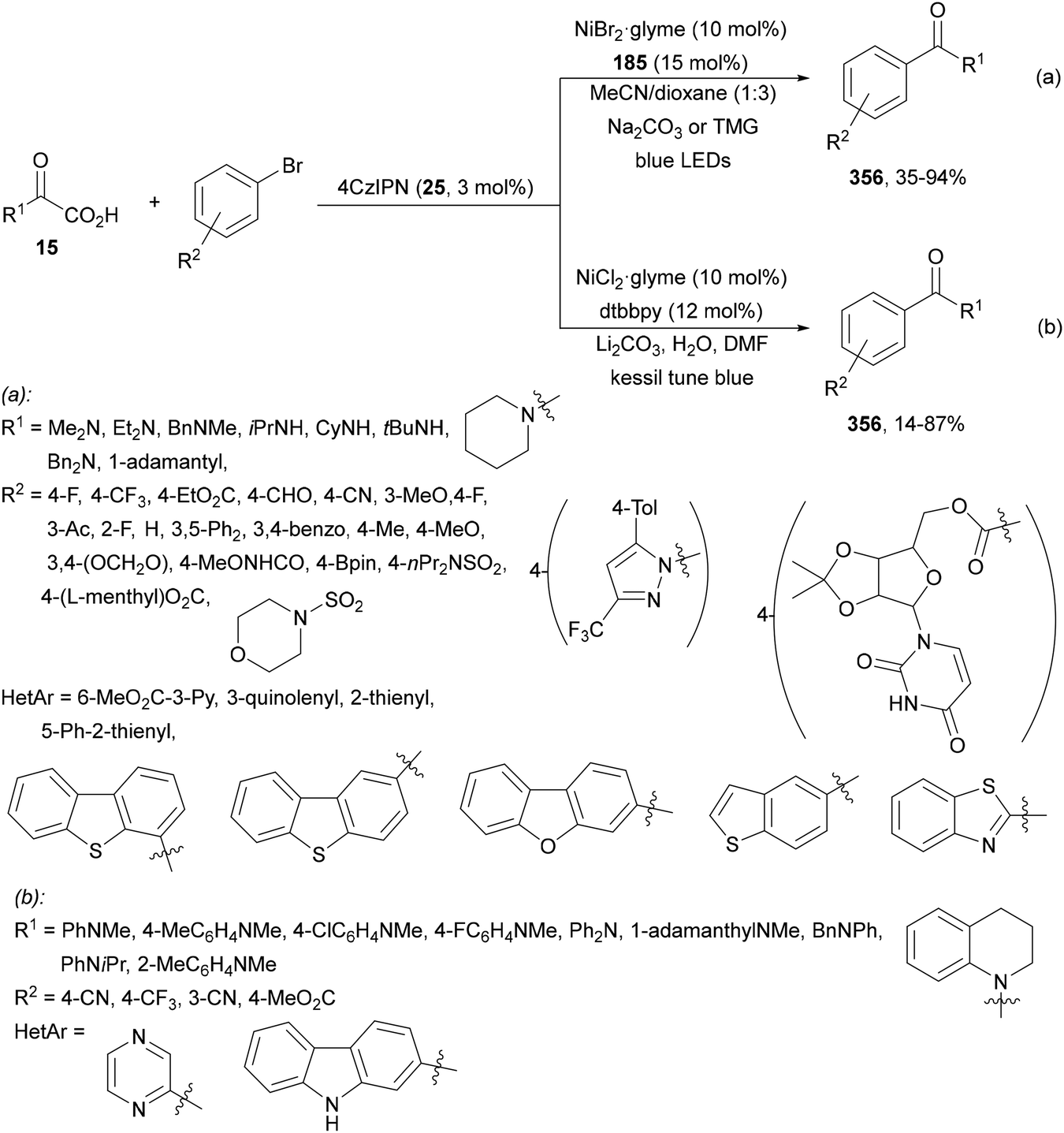 | ||
| Scheme 148 Decarboxylative carbamoylation of aryl bromides with oxamic acids 15 under 4CzIPN (25) (a) and Ni photocatalysis (b). | ||
Recently, Landais and co-workers269 reported decarboxylative addition of oxamic acids to imines 108 in the presence of ferrocene acting both as a PC under visible-light and as a Lewis acid. This process took place in the presence of 2-picolinic acid, which likely provides mixed Cp2Fe-picolinate complexes, and KBrO3 as an oxidant to generate the catalytically active species Fe(III)Ln in DCE at room temperature. A broad range of α-amino acid derived amides 357 were obtained in good yields (Scheme 149a). A 3-component strategy using oxamic acids, aromatic aldehydes and anilines afforded amides 357 in moderate to good yields (28–73%) (Scheme 149b). According to control experiments it was proposed that oxamic acid reacts with catalyst Fe(III)Ln to give the iron carboxylate A. Photoexcitation of A by a LMCT54,55 process gives carboxy radical B and Fe(II)Ln. Decarboxylation of B forms the carbamoyl radical C, which adds to imine D activated by a Brønsted (TFA, picolinic acid, or oxamic acid) or a Lewis acid [BF3 or Fe(III)Ln] to provide the radical cation E. Reduction of E by Fe(II)Ln (path a) and subsequent protonation give product 357 and regenerate the catalyst. The presence of KBrO3 is needed to reoxidize Fe(II)Ln into Fe(III)Ln in order to maintain sufficient Fe(III) in the catalytic cycle (path b).
 | ||
| Scheme 149 Decarboxylative carbamoylation of imines 108 with oxamic acids 15 under ferrocene photocatalysis (a, b). | ||
As acylating agents, α-keto acids, N-acylsaccharins, carboxylic acids and 2-pyridyl esters have been alkylated with styrenes and NHPI esters to provide the corresponding ketones by photoredox decarboxylation. The presence of Hantzsch ester as a photoreductant and for the formation of an electron donor–acceptor (EDA) complex with NHPI esters generates alkyl radicals, whereas Ni complexes form alkyl radicals. In these cases, the corresponding ketones are formed by a C(sp3)–C(sp2) cross-coupling. Arylation of α-keto acids with aryl bromides needs a Ni complex as a catalyst to achieve the C(sp2)–C(sp2) bond formation. Allylation reactions of α-keto acids with methacrylates and NBH esters give the corresponding unsaturated 1,4-dicarbonyl compounds. Oxamic acids generate by a SET process the corresponding carbamoyl radicals after decarboxylation and are arylated with 4-cyanopyridines or aryl bromides giving benzamides. Addition to imines provides α-amino carboxamides using ferrocene as a PC by a LMCT process.
2.11. Cyanation reactions
Direct cyanation of carboxylic acids and derivatives by decarboxylative photoredox catalysis is a very efficient approach. Waser and co-workers270 reported in 2017 a room temperature cyanation of carboxylic acids using cyanobenziodoxolones (CBX)224,225 as cyanation reagents. Asymmetric cyanation of carboxylic acids has been carried out using a photoelectrochemical method by Song, Xu and co-workers271 with cerium/copper relay catalysis and trimethylsilyl cyanide with a chiral bisoxazoline ligand. A similar enantioconvergent140 cyanation was described by Fu and co-workers272 and by Zhang and co-workers.273Enantioconvergent decarboxylative cyanation was initially described with NHPI esters 69 and TMSCN by Lin, Liu and co-workers274 employing Ir(ppy)3 as a PC and CuBr with bisoxazoline 246 as a catalyst under blue LED irradiation. A broad range of enantioenriched alkyl nitriles were obtained both with good yields and enantiomeric excesses (Scheme 150). Theoretical mechanistic studies of this cyanation method was performed by Guan and co-workers275 suggesting a radical mechanism merging oxidative quenching Ir(III)–Ir(III)*–Ir(IV)–Ir(III) and Cu(III)–Cu(I) catalytic cycles. Photoexcited Ir(ppy)* is oxidatively quenched by NHPI ester 69 to give after decarboxylation radical A. Meanwhile, LCuCN B can reduce Ir(IV) to provide Ir(III) and the Cu(II) complex C. Then, complex C and TMSCN undergo cyanide exchange to give dicyanide complex D, which reacts with radical A providing the Cu(III) intermediate E. Finally, the enantio-determining C–CN reductive elimination of E occurs with an energy barrier difference of 2.1 kcal mol−1 to deliver the nitrile and regenerates the copper catalyst B. This energy barrier difference corresponds to a 94% ee in favor of the R-enantiomer in agreement with the observed 84% ee. The most favorable TS is consistent with the observed enantioselectivity and the intramolecular π–π interaction stabilizes this R-type TS.
 | ||
| Scheme 150 Enantioconvergent decarboxylative cyanation of NHPI esters 69 with TMSCN under Ir/Cu photocatalysis. | ||
Photoredox and copper catalysis have been employed for the cyanoalkylation of alkenes,276 1,3-dienes277 and 1,3-enynes278 with NHPI esters 69 and TMSCN. Enantioselective cyanoalkylation of styrenes with alkyl NHPI esters 69 has been carried out by Han and co-workers276 using Ir(ppy)3 as a PC and CuBr/chiral box 358 as a catalyst to furnish nitriles 359 with good yields and enantioselectivities (Scheme 151). This procedure took place in a three-component reaction in NMP/PhCl at room temperature under blue LED irradiation. Radical A, generated from NHPI esters in the photoredox catalytic cycle, adds to styrene to give benzylic radical B, which reacts with the Cu(II) complex to form a Cu(III) intermediate C after reaction with TMSCN. Subsequently, complex C undergoes reductive elimination to give product 359 and regenerates the Cu(I) catalyst.
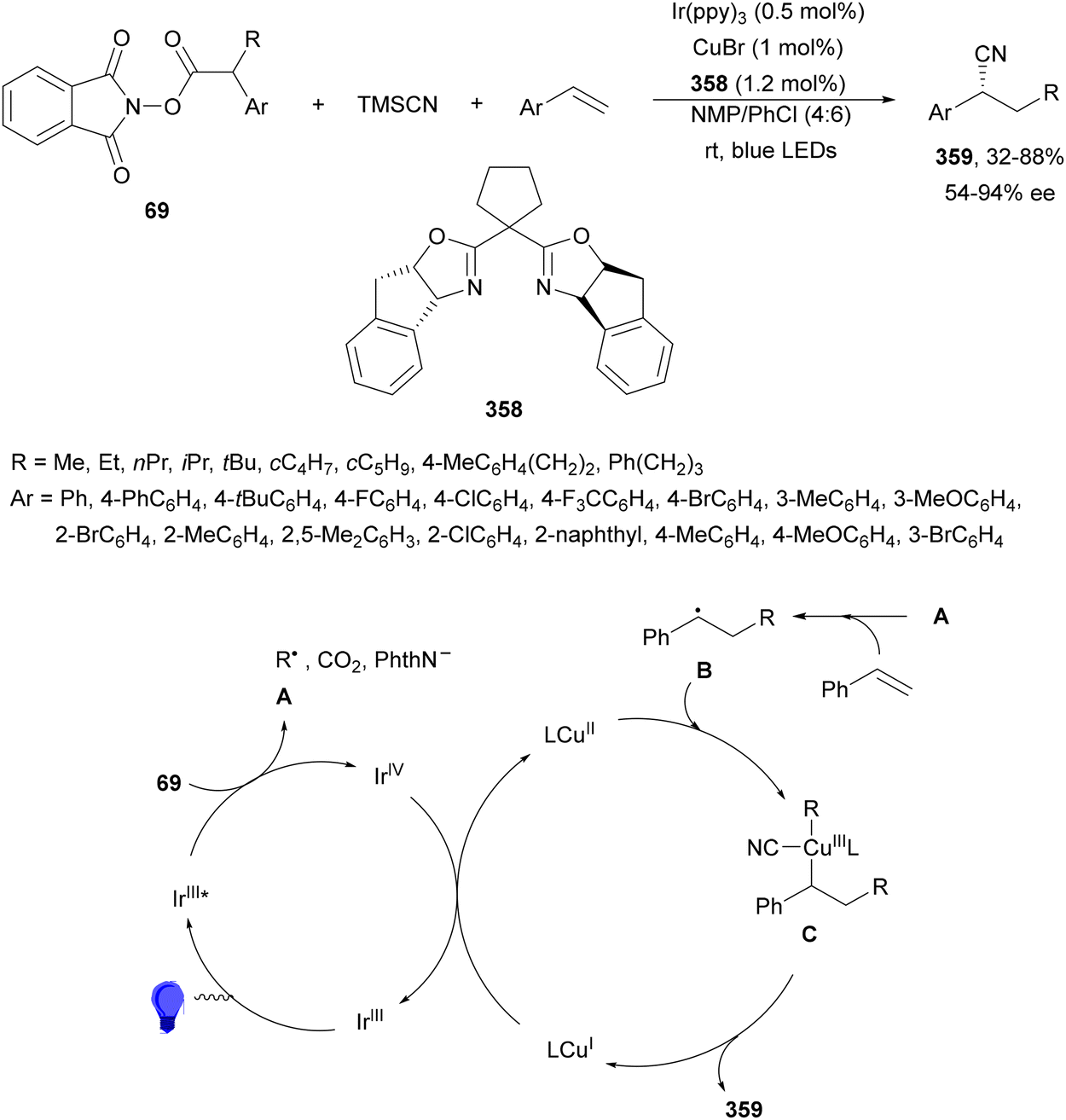 | ||
| Scheme 151 Decarboxylative enantioselective cyanoalkylation of styrenes with NHPI esters 69 and TMSCN under Ir(ppy)3/CuBr/358 photocatalysis. | ||
Xiao and co-workers277 described the enantioselective decarboxylative carbocyanation of 1,3-dienes by using NHPI esters 69 and trimethylsilyl cyanide. In the presence of perylene as an organic PC and Cu(MeCN)4BF4 with 246 as a chiral ligand in anhydrous DCE at 29 °C, the corresponding chiral allyl cyanides 360 were obtained in good yields and high enantioselectivities (Scheme 152a). In the proposed mechanism, radical A generated from NHPI ester adds to (E)-1-phenyl-1,3-butadiene to give allyl radical B, which reacts with LCu(II)CN to provide the chiral allyl Cu(III) species C. Subsequently, intermediate C undergoes reductive elimination to furnish product 360 and the Cu(I) catalyst. Using these reaction conditions, 1,3-enynes reacted with NHPI acetate and TMSCN to provide propargyl cyanides 361 in up to 65% yield and up to 87% ee (Scheme 152b).
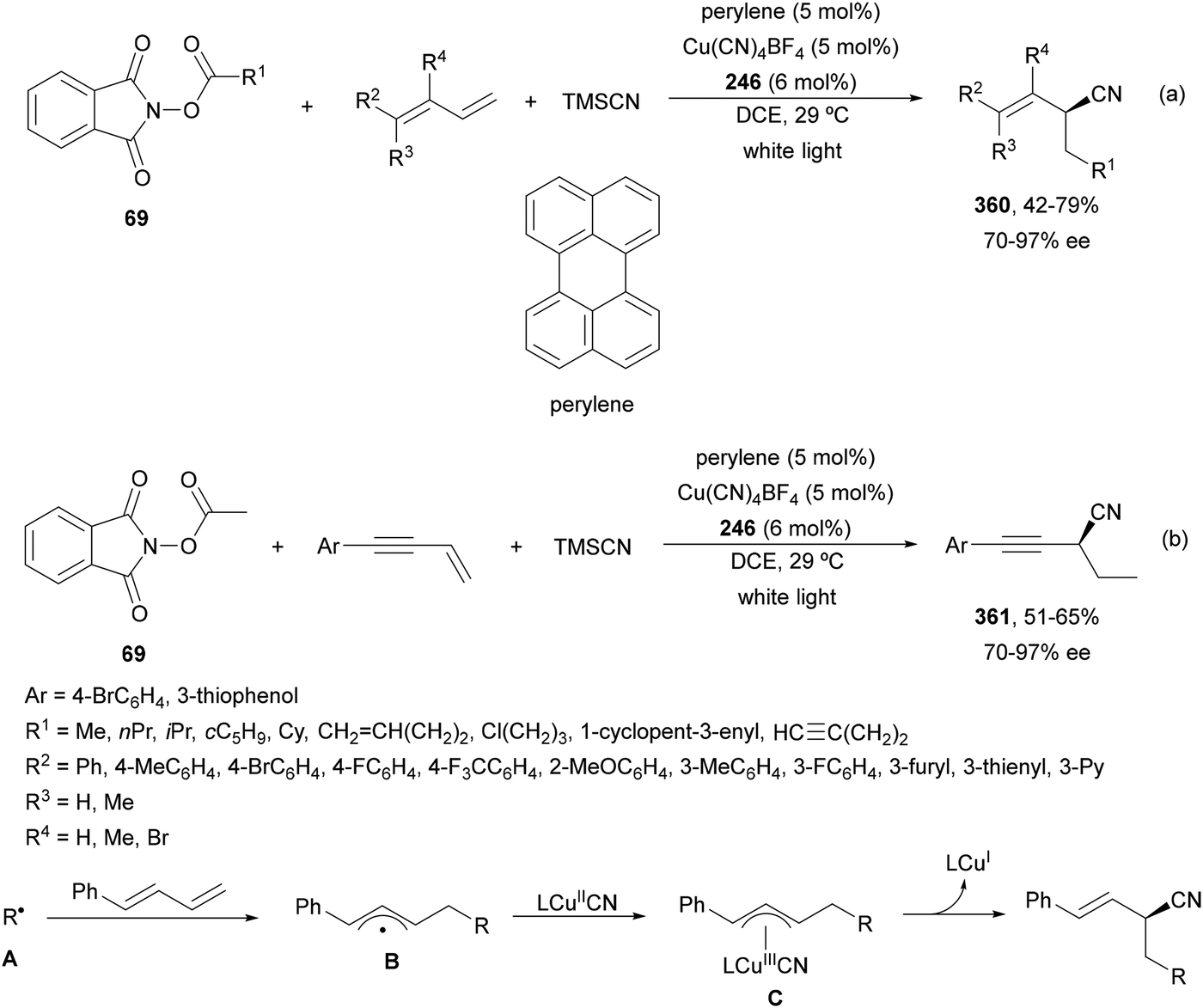 | ||
| Scheme 152 Decarboxylative enantioselective carbocyanation of 1,3-dienes (a) and 1,3-enynes (b) with NHPI esters and TMSCN under perylene/Cu(I) photocatalysis. | ||
In the case of disubstituted 1,3-enynes 362, decarboxylative 1,4-carbocyanation took place with Ir(ppy)3 and Cu(MeCN)4PF6 photoredox dual catalysis using NHPI esters 69 and TMSCN to provide tetrasubstituted allenes 363 (Scheme 153). Lu and co-workers278 performed this protocol in DMA and a N2 atmosphere at room temperature under blue LED irradiation giving the corresponding allenes 363 with, in general, good yields. In the proposed mechanism, radical A from NHPI ester generated in the Ir catalytic cycle, adds to 1,3-enyne to form a propargyl radical B and its resonance form the allene radical B′. Subsequently, this radical reacts with LCu(II) species and TMSCN to provide the Cu(III) species C, which suffers reductive elimination forming the allene 363.
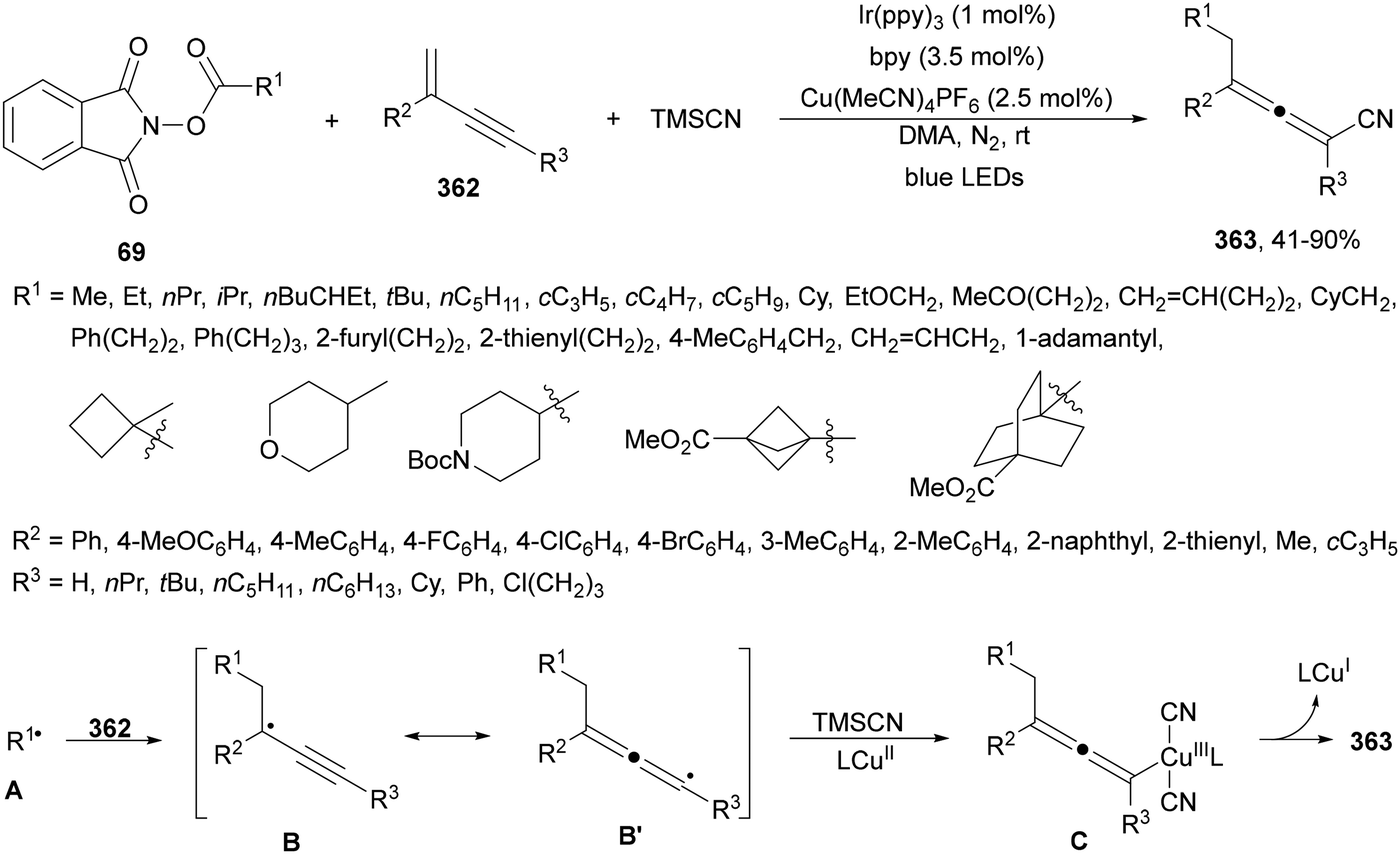 | ||
| Scheme 153 Decarboxylative 1,4-carbocyanation of 1,3-enynes 362 with NHPI esters 69 and TMSCN under Ir(ppy)3/Cu(MeCN)4PF6 dual photocatalysis. | ||
Allenenitriles 365 have been prepared from propargylic oxalates 364 employing a photoredox and copper dual catalysis by Zhang and co-workers.279 In the presence of Ir(ppy)389 as a PC, CuBr and 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) as a ligand, propargylic oxalates 364 reacted with TMSCN in MeCN at room temperature under blue LED irradiation to give exclusively allenenitriles 365 with, in general, good yields (Scheme 154). Experimental and theoretical studies suggested that this process occurred via a visible light-induced redox-neutral reductive quenching radical mechanism instead of the well-established oxidative quenching mechanism. In the photoredox catalytic cycle, photoexcited Ir(III) is quenched by an LCu(I)CN catalyst to generate LCu(II)CN and Ir(III)− species. Oxalate 364 can be reduced by Ir(III)− to form anionic radical intermediate Avia one electron reduction. This intermediate releases the oxalate anion to form propargylic radical B and its resonance structure the allenyl radical B′, which binds to LCu(II)(CN)2 followed by reductive elimination to deliver allenenitrile 365.
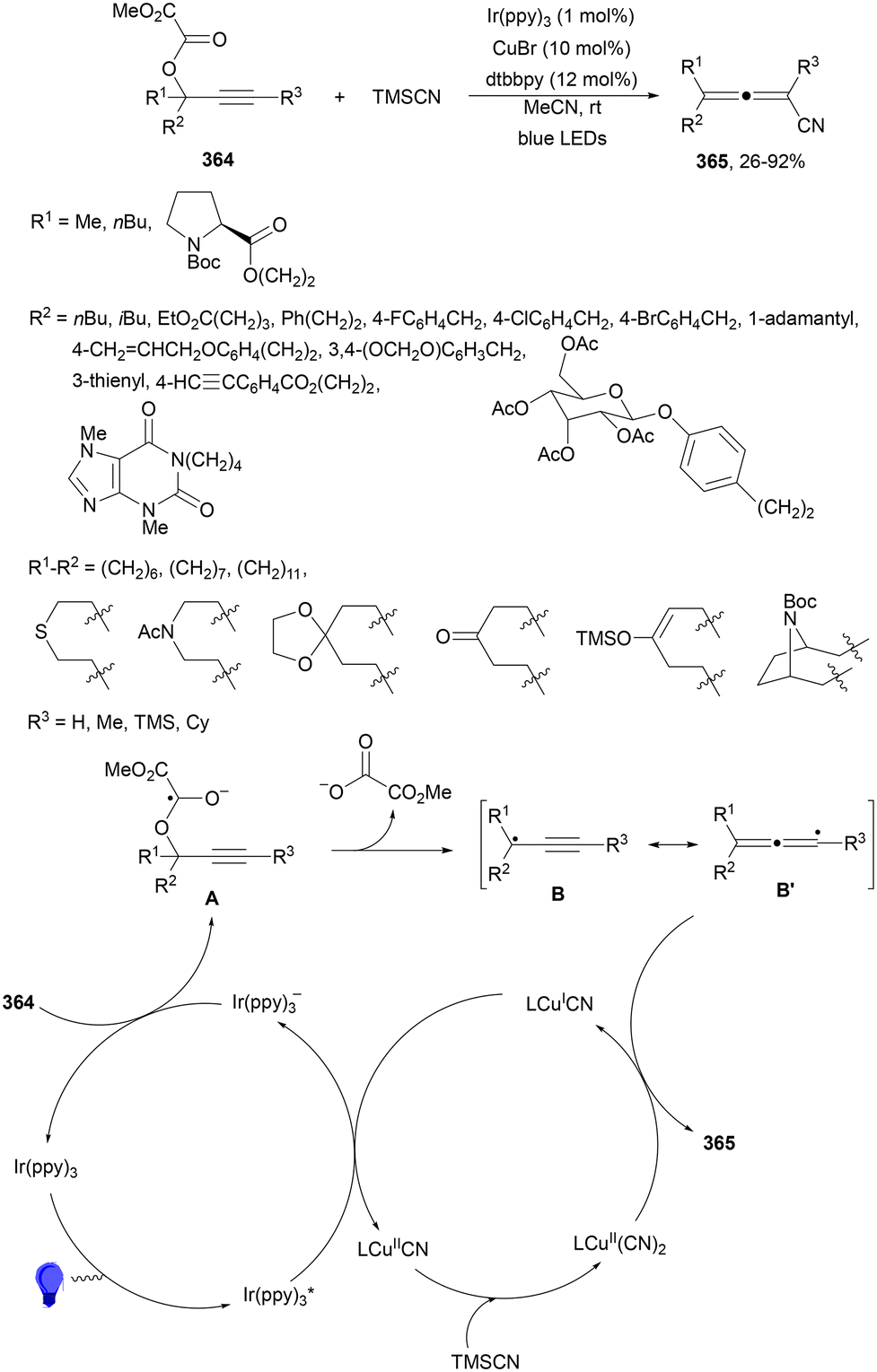 | ||
| Scheme 154 Decarboxylative cyanation of propargylic oxalates 364 with TMSCN under Ir/Cu photocatalysis. | ||
Decarboxylative cyanation reactions performed with carboxylic acids use cyanobenziodoxolones and recently by photoelectrochemical methods. Generally, NHPI esters react with trimethylsilyl cyanide under Ir/Cu dual catalysis and in the presence of chiral ligands such as bixoxazolines to give enantioenriched nitriles. Cyanoalkylation of styrenes, 1,3-dienes and 1,3-enynes can be carried out through three-component reactions with NHPI esters and TMSCN under PC and Cu dual catalysis. In the case of propargylic oxalates, the reaction with TMSCN provides allene nitriles.
2.12. C–H functionalization reactions
Photoredox decarboxylative Minisci reactions have been used mainly for the alkylation, acylation and arylation of aromatic and heterocyclic compounds by C–H functionalization.280,281 These reactions involve addition of carbon-centered radicals to CC and CN bonds followed by hydrogen atom loss under mild reaction conditions using metal and organic-based photoredox catalytic systems or just under visible-light irradiation.
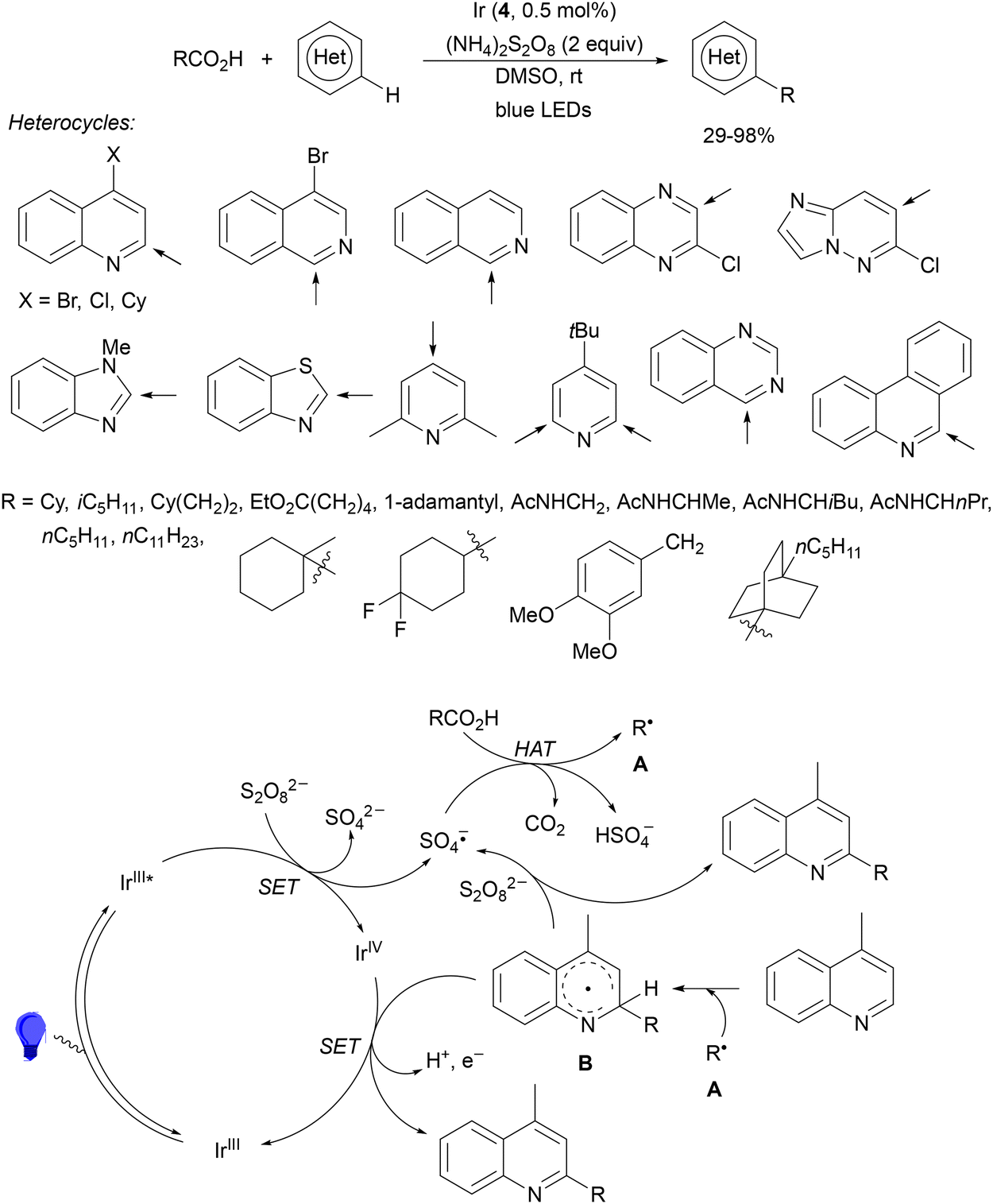 | ||
| Scheme 155 Decarboxylative alkylation of heteroarenes with carboxylic acids under Ir (4)/(NH4)2S2O8 photocatalysis. | ||
Minisci alkylation of N-heteroarenes with aliphatic carboxylic acids has been performed using hypervalent iodine-promoted decarboxylation.284,285 Genovino, Frenette and co-workers284 employed bis(trifluoroacetoxy)iodo benzene (PIFA) and MesAcrMe ClO4 (301) as a PC in MeCN at room temperature under blue LED irradiation to provide alkylated heteroarenes (Scheme 156a). 4-Substituted quinolines, benzothiazole, benzimidazole, 2,6-dichloropurine, 2,6-diphenylpyridine, 4-tert-butylpyridine, phthalazines, 4-phenylpyrimidine, 2,3,5-trimethylpyrazine and drugs such as voriconoazole, varenicline and quinine were alkylated with 17–94% yields. Independently, Li, Chen and co-workers285 described this type of Minisci alkylation using acetoxybenziodoxole (BI-OAc) and Ru(bpy)2Cl2 as a PC in HFIP at 30 °C under CFL irradiation (Scheme 156b). In this case, 4-chlorosubstituted quinolines, isoquinolines, pyrazine, pyridines, 3,6-dichloropyridazine, benzothiazole, benzimidazole, a O-Ac protected purine nucleoside derivative and bioactive molecules such as quinoxylen and fasudil were alkylated at the 2-position of the nitrogen atom with 38–97% yields. In the first case,284 experimental and theoretical calculations (DFT) supported an initial chain reaction, with the carboxylate ion oxidized only in part by MesAcrMe+* to give reduced MesAcrMe*, which transfers one electron to PhI(TFA)2 to initiate the catalytic cycle. The [PhI(O2CR)2]−˙ generates the radical A and CO2. Subsequently, radical A adds to protonated lepidine to give radical cation B, which reacts with the carboxylate ion (RCO2− or TFA−) providing radical C. Finally, C reacts with PIFA to give the alkylated product and regenerates [PhI(O2CR)2]−˙. In the second case,285 a possible mechanism would be that the carboxylic acid reacts with BI-OAc in situ to provide the iodonium ester D. This ester D is reduced by Ru(II)* by a SET to form radical E and 2-iodobenzoate. Decarboxylation of E, addition to protonated pyridine, SET oxidation by Ru(III) and deprotonation affords the product and regenerates the PC Ru(II).
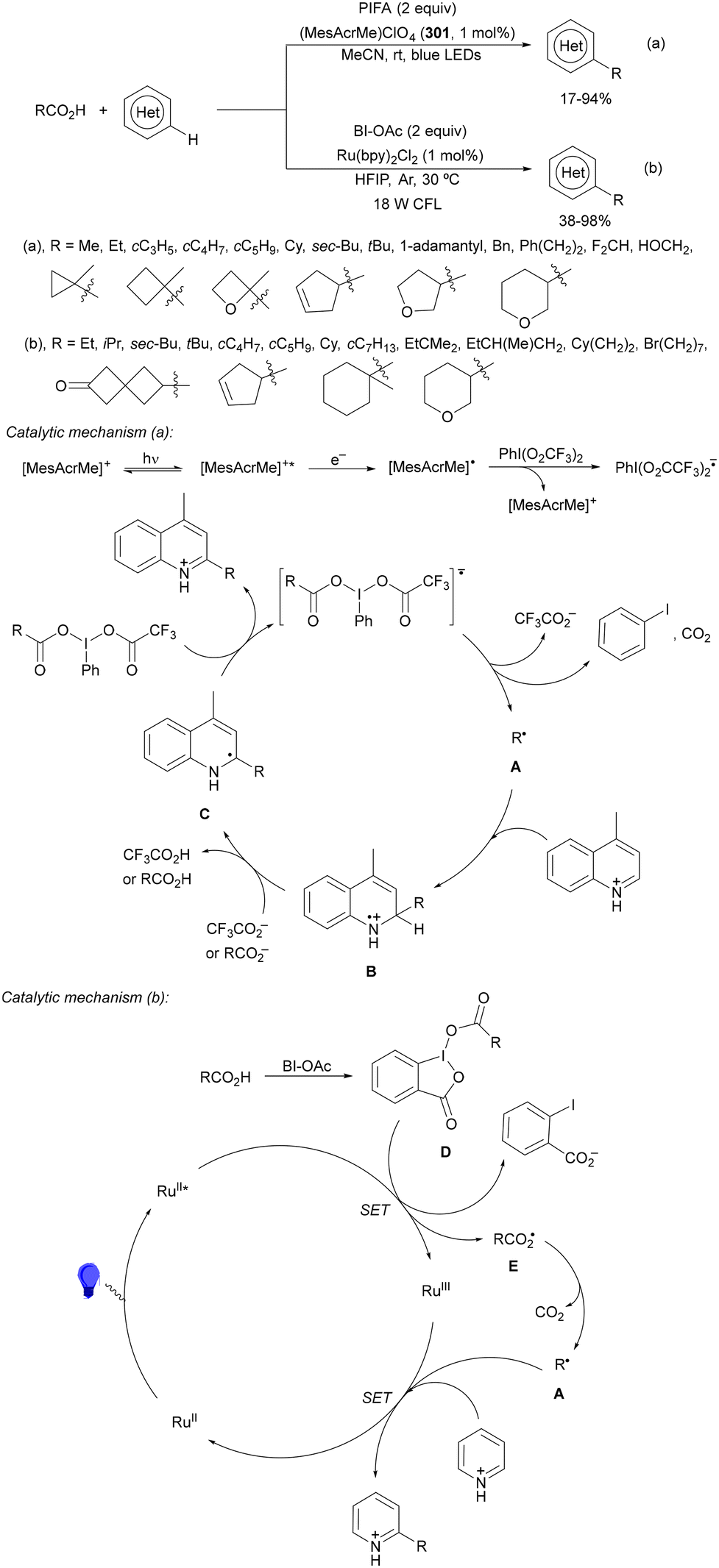 | ||
| Scheme 156 Decarboxylative alkylation of heteroarenes with carboxylic acids using hypervalent iodine reagents and [MesAcrMe]ClO4 (301) (a) or Ru(bpy)2Cl2 (b) photocatalysis. | ||
Yang, Zhang and co-workers286 described a hypervalent iodine-promoted decarboxylation with PIFA in the absence of a PC under blue LED irradiation. The coupling of aliphatic carboxylic acids was carried out with 4-methylquinoline, 4,7-dichloroquinoline, 4-chloro and 4-bromoquinoline, 3-methylquinoline, 3,4-diphenylisoquinoline, 3-chloroisoquinoline, 4-tert-butyl and 4-cyanopyridine, quinazoline, 4,6-dimethylpyrimidine, 5-bromopyrimidine, 2-chloroquinoxaline, phthalazine, benzothiazole and phenanthridine to provide alkylated heterocycles with 30–96% yields.
Another general method for decarboxylative alkylation of heterocycles with carboxylic acids was reported by Jin and co-workers287via iron photocatalysis by an intramolecular charge transfer pathway of iron carboxylate complexes (LMCT).54,55 In the presence of FeSO4·7H2O (5 mol%), picolinic acid (10 mol%) and 2 equivalents of NaBrO3 or NaClO3 as oxidants in aqueous DMSO at room temperature under blue LED irradiation, a broad range of heteroarenes were alkylated with 30–97% yields. Several 4-substituted quinolines 3 and 4-substituted isoquinolines, phenanthridine, quinazolines, phthalazines, quinoxalines, pyridines, pyrimidines, pyridazines, pyrazines, benzothiophenes, benzothiazoles, benzimidazoles and purine derivatives were satisfactorily alkylated at the vicinal carbon atom of the heteroatom. Recently, Li and co-workers288 described this Minisci reaction using palladium-loaded gallium nitride (3 mol%) as a heterogeneous PC working in MeCN, at room temperature under Xe light irradiation. In this case, 4-methylquinoline and 2-substituted quinolines were alkylated at the 2- and 4-position, respectively with 33–95% yields. Other heterocycles such as 1-methylquinoxaline and benzoquinoline were mono- and dialkylated, respectively.
Shen, Zhang and co-workers289 reported a general alkylation of heteroarenes in the presence of TBHP (2 equivalents) and dimethyl carbonate (DMC) as solvent at room temperature under 395 nm LED irradiation. This green and efficient methodology was applied to the alkylation of quinoxalinones, phenanthridine, isoquinoline, quinazoline, quinoxaline, phthalazine, benzoxazinone, and azauracil to provide the corresponding products in 17–84% yields. Other substrates such as quinolines, pyridines, benzothiazoles, theophylline and purine failed. Primary, secondary and tertiary aliphatic carboxylic acids as well as N-protected α-, β-, γ- and δ-AAs gave the corresponding aminoalkylated quinoxalinones with 50–64% yields. In the proposed mechanism, the t-BuO˙ radical is formed by irradiation of TBHP in the presence of light, which starts the radical mechanism of this transformation.
Decarboxylative alkylation of heteroarenes with carboxylic acids without the requirement of stoichiometric amounts of oxidants was reported by Li and co-workers.290 In the presence of Ir complex 4 as a PC and Co(dmgH)2PyCl (16) as a H2 release catalyst, n-Bu4NOAc as a base in EtOAc at room temperature under blue LEDs irradiation, the corresponding alkylated heterocycles were obtained in 37–94% yields (Scheme 157). Benzothiazoles, 4,5-dimethylthiazole, benzoxazoles, oxazoles, benzimidazoles, N-Boc-imidazole, benzofuran, benzothiophenes, 3-substituted thiophenes, quinazolin-4(3H)-one, phenanthridine and quinazoline were alkylated with secondary and tertiary carboxylic acids and with α-AAs. In the proposed reaction pathway, the Ir catalytic cycle generates the corresponding radical by decarboxylation of the carboxylic radical and forms Ir(II). This Ir(II) transfers one electron to Co(III) and produces Co(II) species, which is further reduced to Co(I) by accepting one electron. This Co(I) is transformed into Co(III)–H and reacts with another proton to release H2 and closes the cobalt cycle (path a). In path b, Co(III)–H is reduced to Co(II)–H, which after protonation forms H2. Alternatively, Ir(III)* is oxidatively quenched by Co(III) via SET to form Ir(IV) and Co(II). Subsequently, Ir(IV) abstracts one electron from RCO2− to give radical RCO2˙ and regenerates Ir(III) to close the catalytic cycle. Then, RCO2˙ and Co(II) follow the similar process as described in path a to afford the product with H2 release.
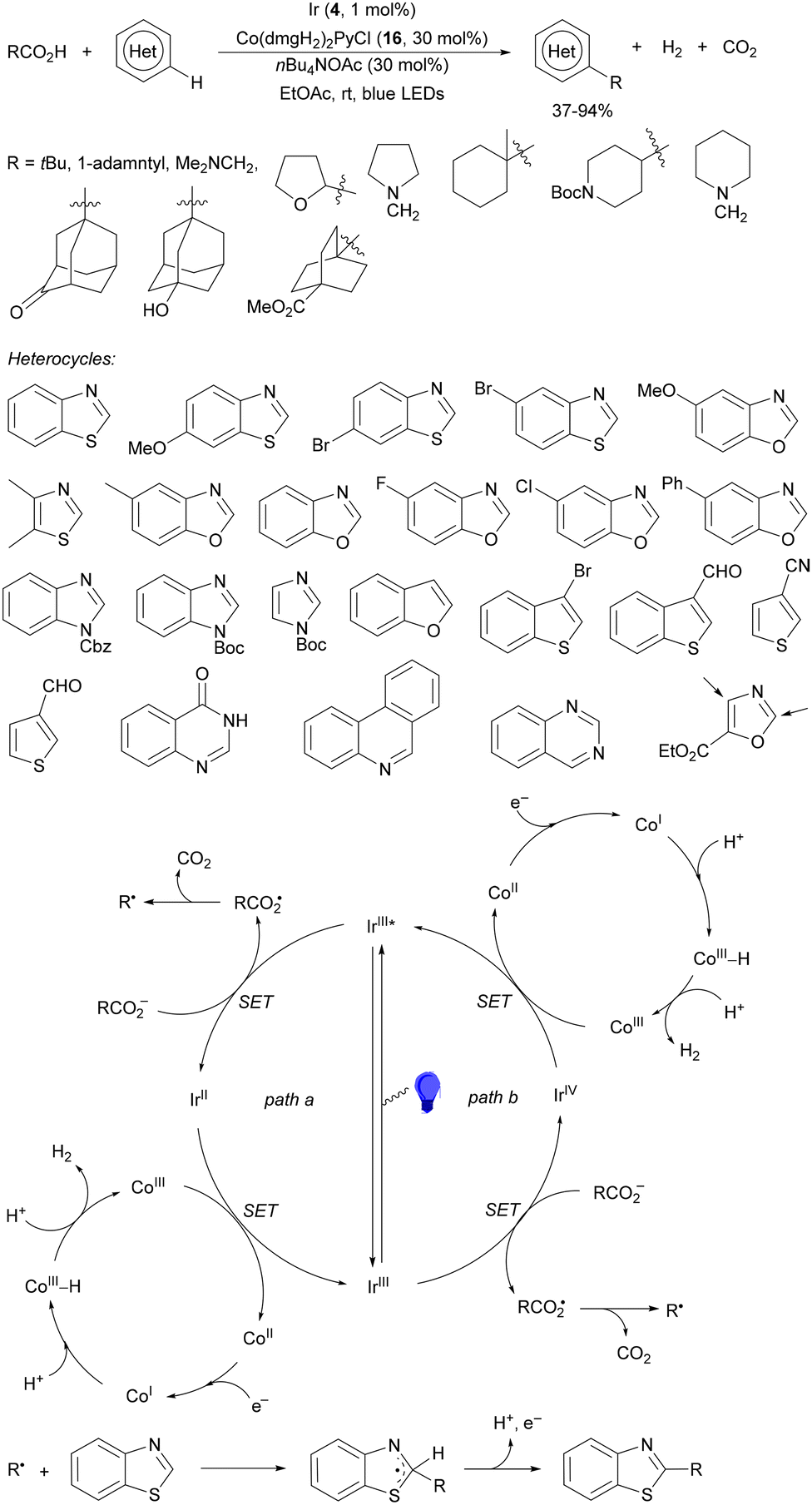 | ||
| Scheme 157 Decarboxylative alkylation of heterocycles with carboxylic acids under Ir/Co photocatalysis. | ||
A general method for the aminoalkylation of a wide range of heterocycles has been described by Liu, Sun and co-workers.291N-Protected α-AAs reacted with these heterocycles in the presence of 4CzIPN as a PC and Cs2CO3 as a base in DMA at room temperature under blue LED irradiation to furnish the corresponding products (Scheme 158). Quinoxalines, 3,6-dichloropiridazine, 2-methyl-3-acetylpyrazine, quinazoline, 2-methoxycarbonylquinoline, benzothiazole and coumarin provided the aminoalkylated derivatives with in general good yields (37–85%). In the proposed mechanism, the deprotonated α-AA was oxidized by 4CzIPN* to deliver the α-aminoalkyl radical A, which adds to 1-methylquinoxalin-2(1H)-one to form radical intermediate B. Subsequent 1,2-H migration in radical B produces radical C. Meanwhile, 1O2 was generated through energy transfer from 4CzIPN* to 3O2 followed by the reduction of 4CzIPN to produce O2−˙. Finally, O2−˙ abstracts a hydrogen atom from radical C to give the alkylated quinoxaline.
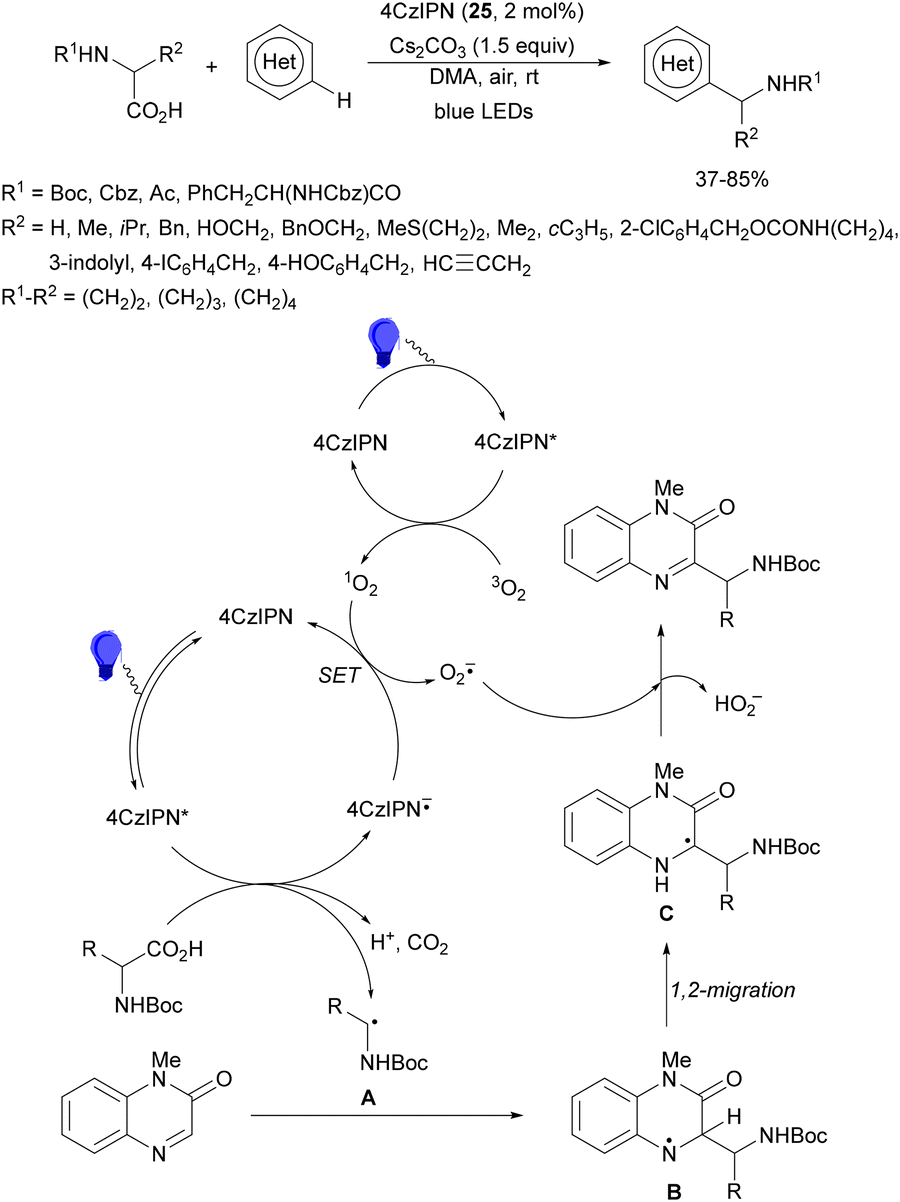 | ||
| Scheme 158 Decarboxylative alkylation of heteroarenes with α-AAs under 4CzIPN (25) photocatalysis. | ||
Decarboxylative trifluoromethylation of aromatic and heteroaromatic compounds with trifluoroacetic acid under photoredox conditions was described in 2017 by Su, Li and co-workers.292 In the presence of Rh-modified anatase TiO2 nanoparticles as a PC and Na2S2O8 (10–40 mol%) as an external oxidant at room temperature in trifluoroacetic acid under 365 nm ultraviolet irradiation, the corresponding trifluoromethylated products were obtained in 34–75% yields. Qing and co-workers293 reported a general method for the trifluoromethylation of arenes and heteroarenes using C6F5I(O2CCF3)2 (FPIFA) (2.5 equivalents), Ru(bpy)3(PF6)2 as a PC in MeCN at 35 °C under blue LED irradiation (Scheme 159a). A wide range of aromatic compounds and heterocycles such as furans, thiophenes, pyridines, pyrimidines, pyrazines and thiazoles were trifluoromethylated with moderate to good yields with excellent regioselectivity at the electron-rich position. Recently, simple and general decarboxylation of trifluoroacetates has been described by Juliá-Hernández and co-workers,294 for the trifluoromethylation of arenes and heterocyclic compounds. Using Fe(OTf)2 as a PC, 4,4′-dimethoxy-2,2′-bipyridine (185) as a ligand, K2S2O8 as an external oxidant in MeCN at room temperature under 405 nm irradiation afforded the corresponding products (Scheme 159b). Electron-rich arenes and substituted pyrroles, thiophenes, pyrimidine, N-methylpyridone, coumarin, indoles and ferrocene were functionalized with modest to good yields. Natural products and drug-like molecules such as caffeine, theophylline, pentoxifylline, uracil, trifluridine, griseofulvin, metaxalone, visnagin, indomethacin and melatonin were also trifluoromethylated with 18–67% yields. This method involves LMCT photocatalysis54,55 by intermediacy of FeIIIOCOCF3, which after irradiation provides a trifluoroacetate radical. Subsequent decarboxylation affords the trifluoromethyl radical.
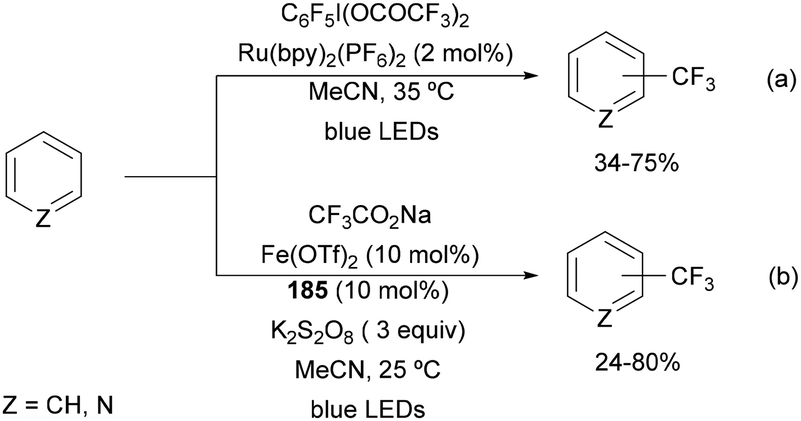 | ||
| Scheme 159 Decarboxylative trifluoromethylation of arenes and heterocyclic compounds under Ru (a) or Fe (b) photocatalysis. | ||
Iron-mediated decarboxylative alkylation of arenes and heteroarenes with carboxylic acids represents a general strategy for C–C bond formation. Yoon and co-workers295 employed FeCl3 and NaOAc as a base in DCM at room temperature under blue LED irradiation. Alkylated arenes and heterocycles such as substituted indole, benzothiophene, furan, benzofuran, pyrrole and pyridine were isolated with moderate to good yields (Scheme 160). It has been postulated that alkyl chlorides are formed as intermediates by reaction of the alkyl radical with FeCl3. In addition, FeCl3 can act as Lewis acid to facilitate the Friedel–Crafts alkylation of the arenes or heteroarenes.
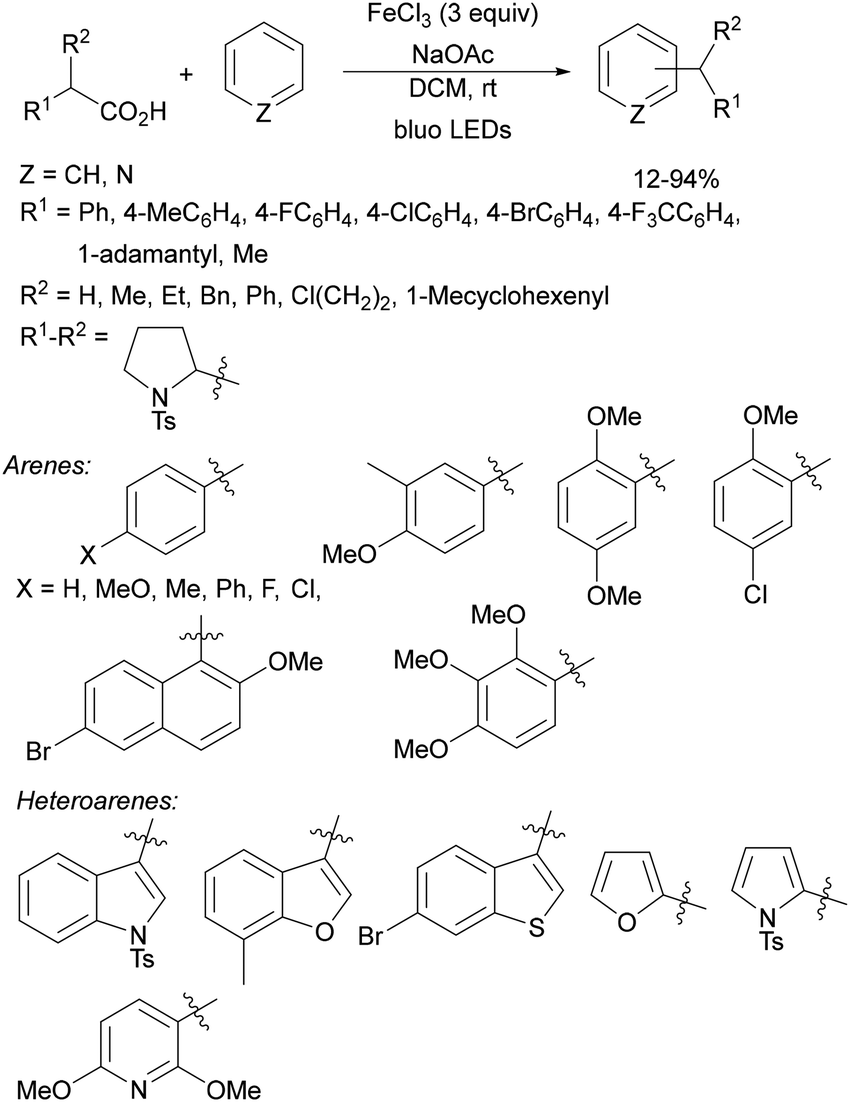 | ||
| Scheme 160 Decarboxylative alkylation of arenes and heteroarenes with carboxylic acids under FeCl3 photocatalysis. | ||
Specific alkylation of quinoxaline-2-(1H)-ones with aliphatic carboxylic acids in aqueous conditions was reported by Qin, Li and co-workers.296 In this case, Ir complex 4, K2S2O8 as an oxidant, Li2CO3 as a base, in DMSO/H2O (1:1) at room temperature under blue LED irradiation were efficient reaction conditions to give the corresponding products in 30–93% yields (Scheme 161). Drug molecule and natural product derived aliphatic acids were also employed such as indomethacin and dehydrochlolic acid. An aldose-reductase inhibitor precursor was prepared by reaction of N-(ethoxycarbonyl)methyl quinoxaline with 4-bromophenylacetic acid. The biological activity of several alkylated quinoxalinones as antifungal agents against Magnaporthe grisea GD08-T19 has been studied.
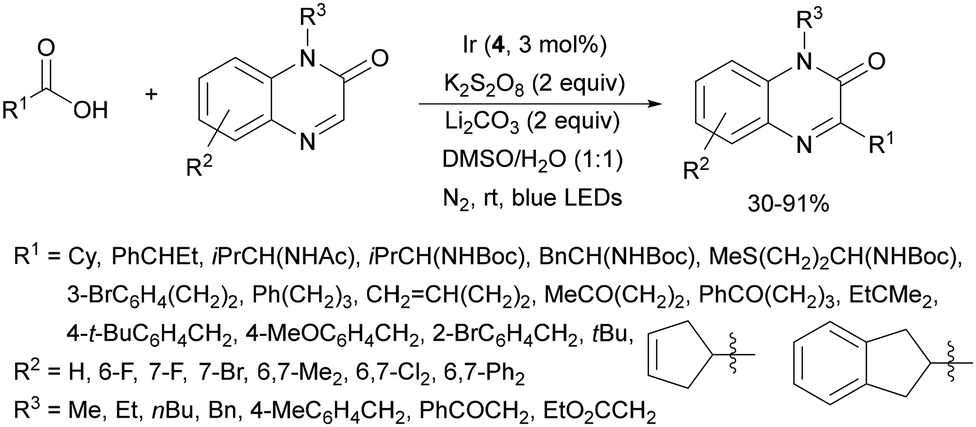 | ||
| Scheme 161 Decarboxylative alkylation of quinoxaline-2(1H)-ones with carboxylic acids under Ir and K2S2O8 photocatalysis. | ||
Roy and co-workers297 performed a decarboxylative alkylation of quinoxaline-2(1H)-ones by a photoinduced ligand to metal charge transfer (LMCT)54,55 using CeCl3 and t-BuOK as a base in MeCN at room temperature under blue LED irradiation. A broad range of primary, secondary and tertiary carboxylic acids were employed to provide alkylated quinoxalinones in 36–92% yields.
Decarboxylative C–H adamantylation of azoles, such as benzothiazoles, benzoxazoles, benzimidazoles and caffeine derivatives, was carried out with [MesAcrMe]ClO4 (301) as an organic PC and Co(dmgH)(dmgH2)Cl2 as a H2 release catalyst, K2HPO4 as a base, in aqueous DCM at 25–30 °C under blue LED irradiation.298 The resulting adamantly azoles were obtained with 40–83% yields.
Imidazo[1,2a]pyridines 366299 have been alkylated with N-arylglycines under photocatalyst-free conditions. Zhu, Le and co-workers300 reported the aminoalkylation of these imidazopyridines in toluene at room temperature under blue LED irradiation to afford the corresponding products 367 with good yields (Scheme 162). The reaction has to be performed in air involving a radical process. Oxidation of N-aryl glycine with 1O2, generated under visible light from molecular oxygen, generates an aminyl radical cation A and a superoxide radical anion (O2−˙). Further protonation of A gives the α-amino radical B after decarboxylation induced by O2−˙. Subsequent oxidation of B by HO2˙ leads to the imine intermediate C, which undergoes electrophilic addition with the imidazopyridine to produce product 367.
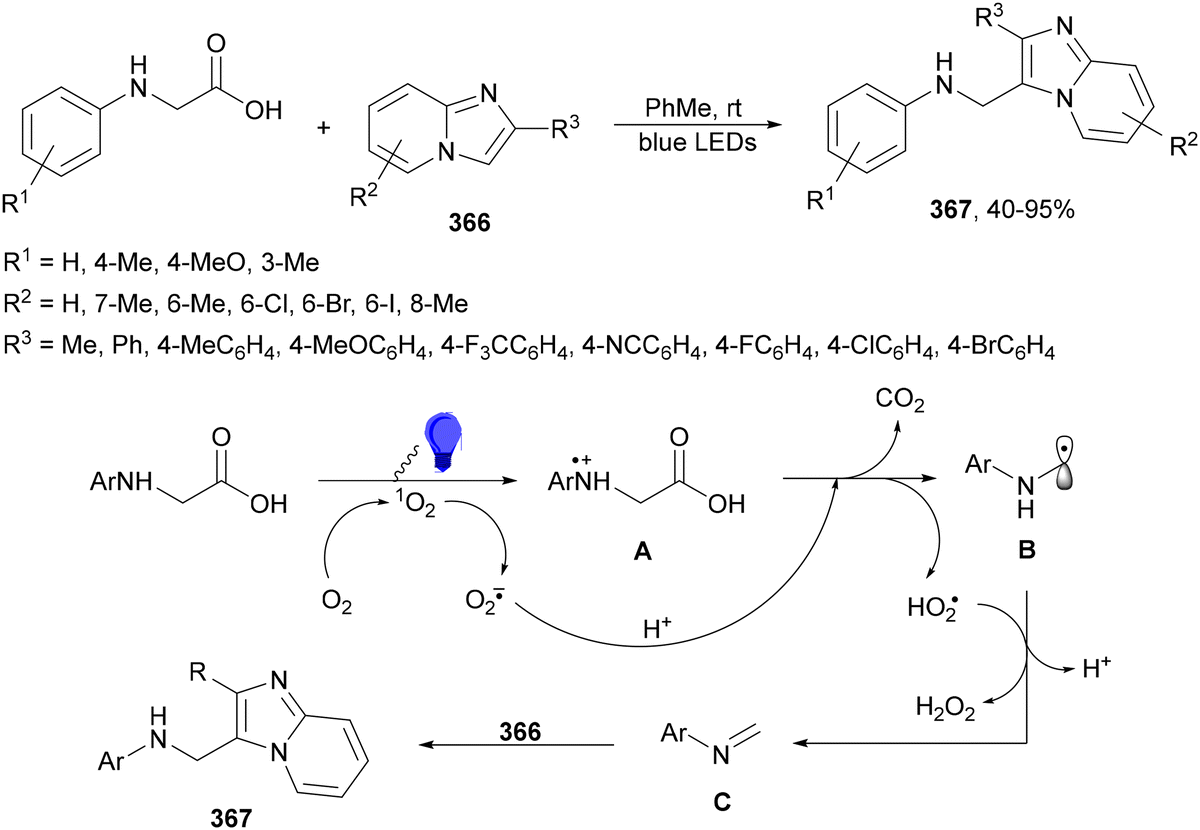 | ||
| Scheme 162 Decarboxylative aminoalkylation of imidazo[1,2a]pyridines (366) with N-arylglycines under visible light irradiation. | ||
The aminoalkylation of imidazopyridines 366 and benzo[d]imidazo[2,1-b]thiazole has been reported by Chen, Yu and co-workers301 using 5 mol% of recyclable perovskite (CsPbBr3) as a heterogeneous PC. This process was carried out in DCE under 25 W white LED irradiation under aerobic conditions with 44–94% yields and was applied for the gram-scale preparation of product 367 (R1 = R2 = H; R3 = Ph).
Photocatalytic decarboxylative alkylation of pyridines at the four position has been achieved using N-amidopyridinium salts 368. Hong and co-workers302 performed the regioselective synthesis of 4-substituted pyridines 369 in the presence of (tBu2MesAcrPh)BF4287 as a PC and K2HPO4 as a base in toluene at room temperature under N2 and blue LED irradiation (Scheme 163). A wide range of pyridines 369 were obtained using primary, secondary and tertiary aliphatic carboxylic acids including α-AAs with good yields. Based on experimental observations, a plausible mechanism was proposed involving the formation of the carboxylic radical MesAcr+*. After decarboxylation radical A is formed, which adds at the 4-position of the N-amidopyridinium salt to give B. Intermediate B undergoes deprotection and homolytic cleavage to furnish the product 369 and amidyl radical C by a SET process and regenerate the PC and give anion C.
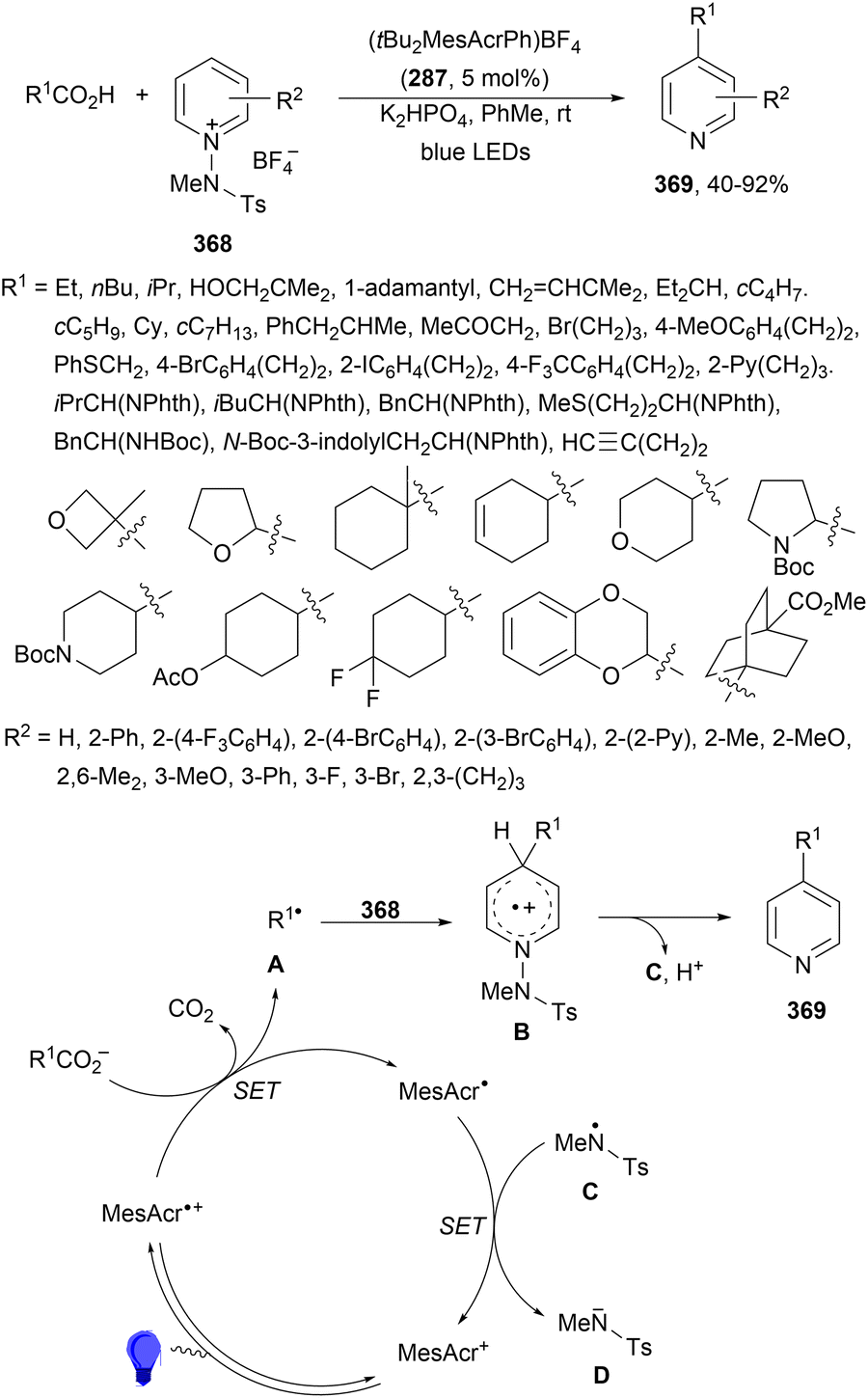 | ||
| Scheme 163 Decarboxylative alkylation of N-amidopiridinium salts 368 with carboxylic acids under MesAcr+287 photocatalysis. | ||
In 2017, as in the case of decarboxylative C–H alkylation of heteroarenes with carboxylic acids reported by Glorius,283 Shang, Fu and co-workers303 reported the same reaction with NHPI esters 69. An Ir-photoredox catalyst 4 in the presence of TFA or In(OTf)3 in DMA at room temperature under blue LED irradiation enabled a general alkylation of a broad range of N-heteroarenes (Scheme 164). Quinolines, quinoxaline, phthalazine, isoquinoline, pyridine, pyrimidine, pyrazine, bipyridine, phenanthroline, and purine reacted with primary, secondary and tertiary alkyl NHPI esters 69 in the presence of an excess of TFA to provide the corresponding alkylated products at the most electrophilic position in 45–92% yields. When 10% of Lewis acid was used for sensitive substrates to Brønsted acids, the resulting products were obtained in 32–94% yields. In the proposed mechanism, the alkyl radical from the NHPI ester adds to protonated heteroarene, e.g. lepidine, to give a radical cation A, which is oxidized by Ir(III)* to regenerate Ir(II) and the alkylated heterocycle.
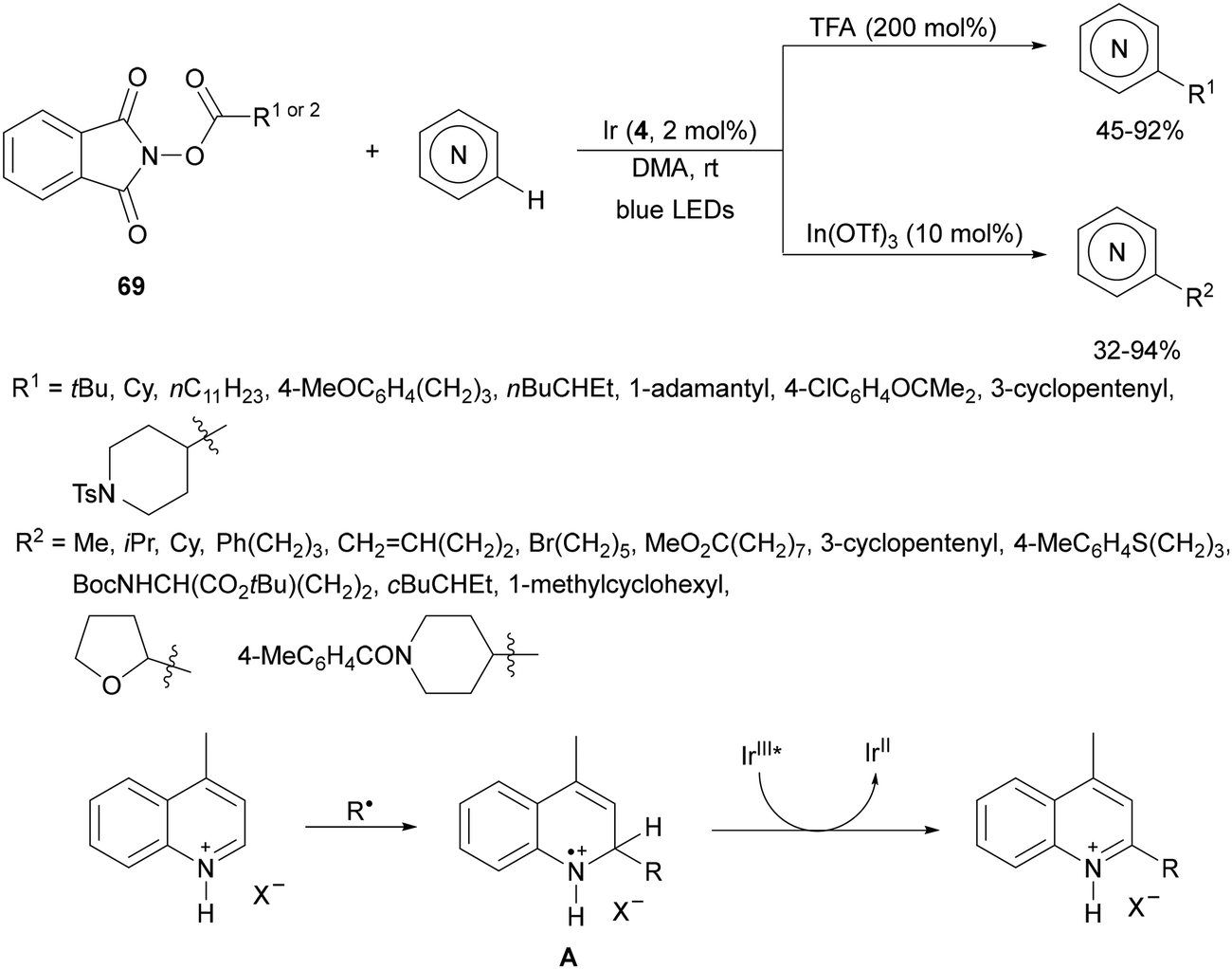 | ||
| Scheme 164 Decarboxylative alkylation of N-heterocycles with NHPI esters 69 under Ir/acid photocatalysis. | ||
Several enantioselective Minisci-type addition reactions of α-aminoalkyl radicals to heteroarenes have been described.304–306,308–311 Phipps and co-workers304 employed pyridines and quinolines as heteroarenes and α-AA derived NHPI esters 143 in the presence of Ir complex 4 as a PC and (R)-TRIP (370) or (R)-TCYP (371) as CPAs under blue LED irradiation to give the corresponding aminoalkylated products 372 in up to 98% yield and up to 97% ee (Scheme 165). In the proposed mechanism, upon formation of the α-aminoalkyl radical A from NHPI ester 143 under photocatalysis, addition to pyridine takes place to give by participation of the CPA, intermediates B and C. Intermediate C forms radical D and liberates the CPA. Subsequent oxidation of D and deprotonation gives product 372 and regenerates Ir(II). Further studies by a predictive mathematical model through evaluation of catalyst/substrate training sets and parameter acquisition platform suggested that other heteroarenes should be amenable for the same enantioselective Minisci reaction.305 Specific predictions through multivariate linear regression (MLR) analysis for pyrimidines and pyrazines were in agreement with experimental results. Under the same reaction conditions several pyrimidines and pyrazines were alkylated with N-aryl AAs derived NHPI esters to furnish aminomethylated products in 23–93% yields.
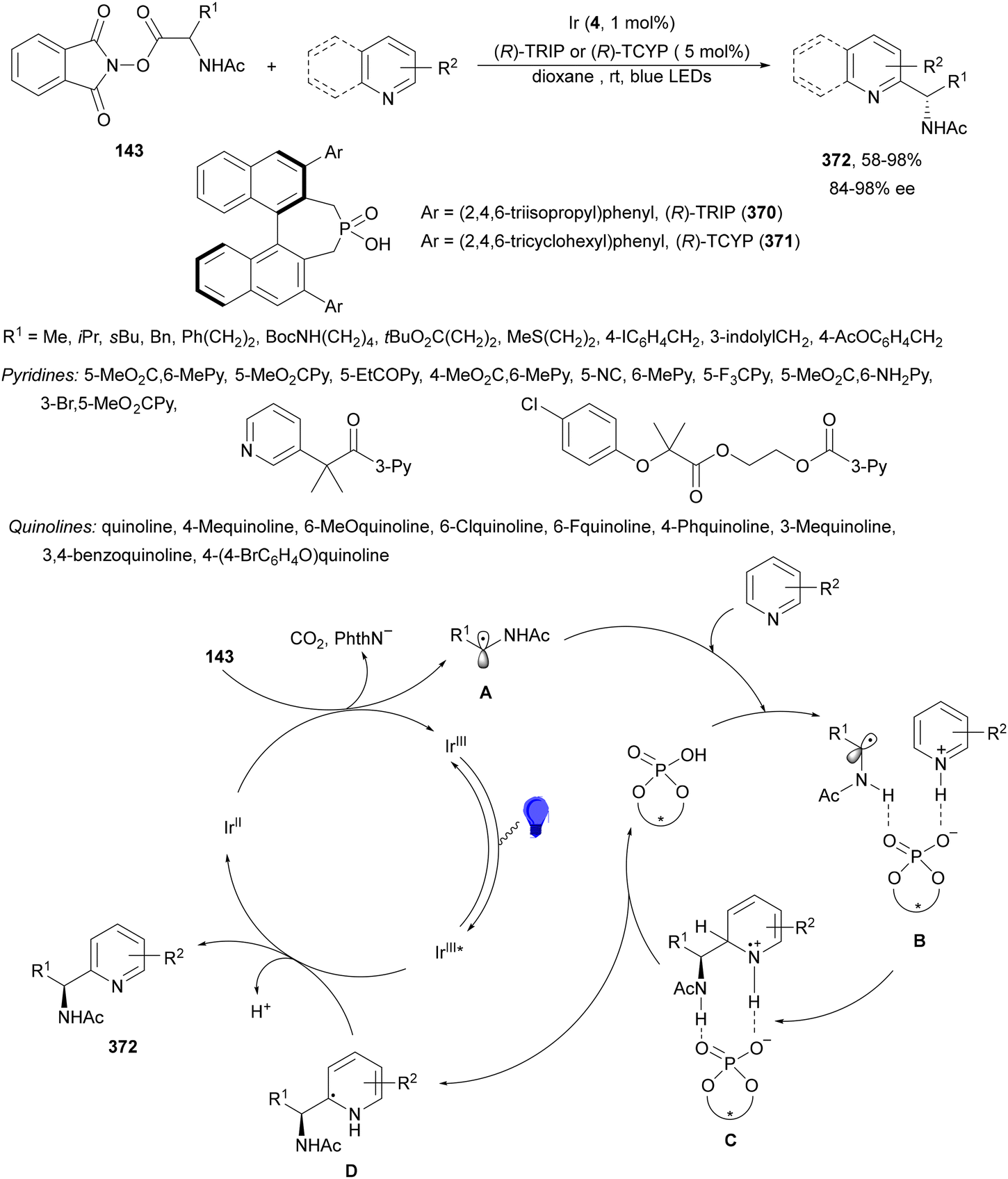 | ||
| Scheme 165 Decarboxylative asymmetric alkylation of quinolines and pyridines with α-AAs derived NHPI esters 143 under Ir/CPAs 370 and 371 photocatalysis. | ||
Computational (DFT calculations) and experimental investigations were performed to elucidate the origin of selectivity in collaboration with Ermanis and Goodman.306 The Curtin–Hammett principle was in operation: a fast and reversible radical addition followed by a slower irreversible enantioselective deprotonation, which determined the enantioselectivity of this Minisci-type reaction. In Scheme 166 is depicted the proposed reaction mechanism: firstly, the α-aminoalkyl radical A generated by reduction of NHPI ester interacts with the quinolinium-TRIP complex B to give complex C. Addition to quinolinium via one of the four possible diastereomeric C-TS gives intermediate D. Deprotonation by the carbonyl oxygen of the N-acetyl group assisted by the phosphate through D-TS provides intermediate E. Deprotonation of E by another quinoline molecule regenerates B and forms radical F, which by a SET process undergoes oxidation giving the product 372 after proton loss. Chain processes303,307 and direct HAT processes308 have been proposed also for the Minisci reactions involving photoredox catalysis.
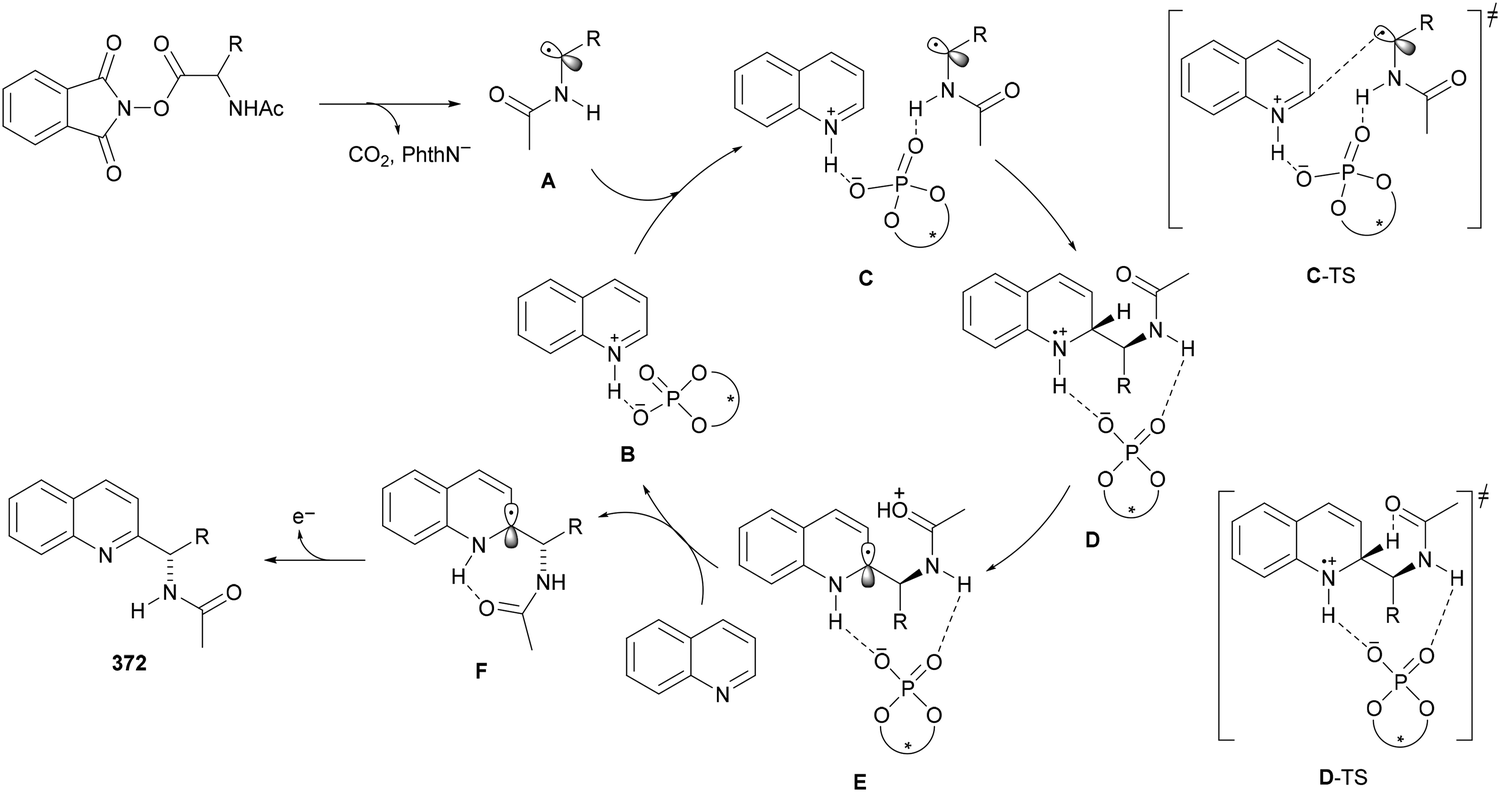 | ||
| Scheme 166 Mechanism for CPA-catalyzed Minisci reaction of NHPI esters with quinoline. | ||
Jiang and co-workers309 reported a similar decarboxylative enantioselective alkylation of isoquinolines with α-AA-derived NHPI esters 143 in the presence of DPZ 92 as an organic PC and SPINOL-CPA 373 to provide 1-isoquinoline-substituted chiral secondary amines 374 in high yields with good to excellent enantioselectivities (Scheme 167). This Minisci-type asymmetric reaction was carried out in the presence of 4 Å MS, in DME at −10 °C under blue LED irradiation.
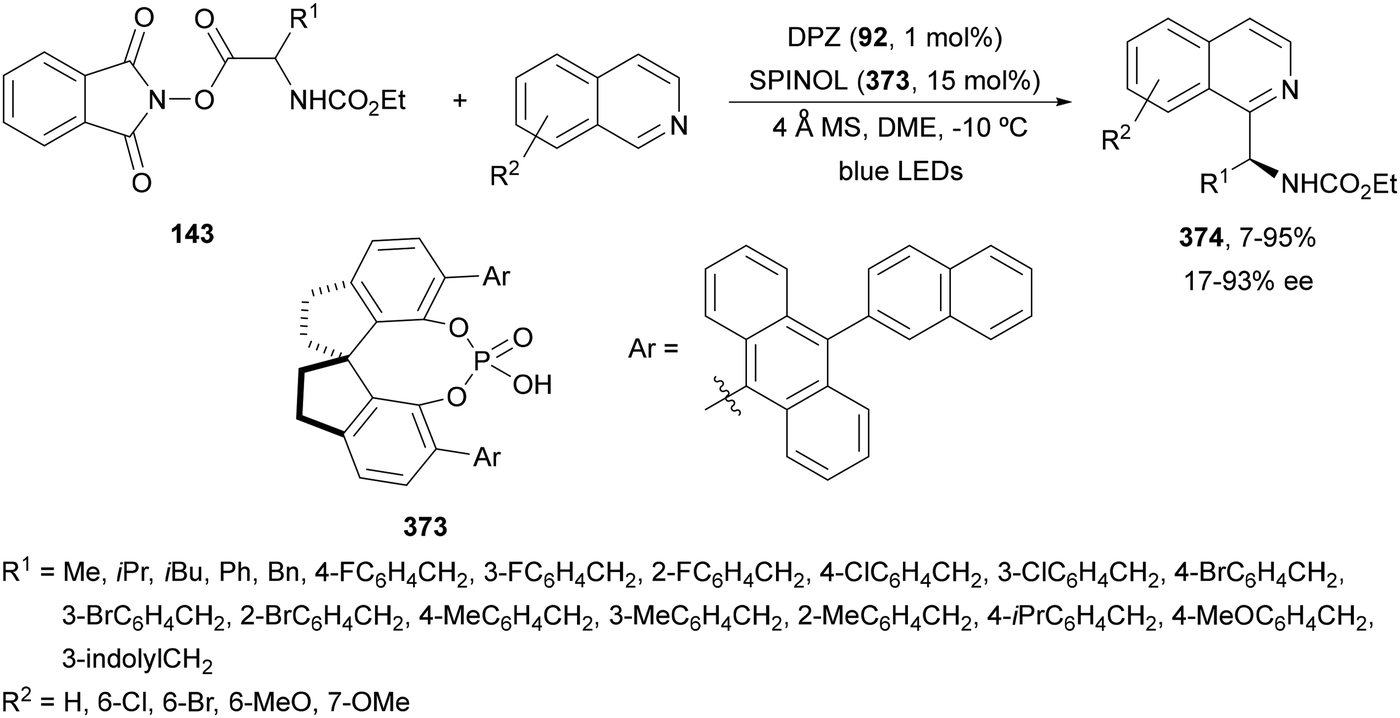 | ||
| Scheme 167 Decarboxylative asymmetric alkylation of isoquinolines with α-AA derived NHPI esters 143 under DPZ (92)/SPINOL-CPA 373 photocatalysis. | ||
Wang and co-workers310 reported a similar enantioselective Minisci reaction by decarboxylative photoredox of α-AA-derived NHPI esters 143 and β-carbolines 375 (Scheme 168). In the presence of Ir complex 4 and SPINOL-CPA 376 in THF at −40 °C under blue LED irradiation, enantioenriched products 377 alkylated at the 1-position were obtained in up to 87% yield and up to 97% ee. This method was applied to the total synthesis of marine alkaloids eudistomin X, (+)-eudistomidin B and (+)-eudistomidin I. In the chiral Brønsted acid cycle, the α-aminoalkyl radical A, generated from 143 through the Ir photoredox cycle, interacts with β-carboline by the bifunctional phosphoric acid catalyst affording intermediate B, which after radical addition gives radical cation species C. Intermediate C undergoes an internal proton abstraction promoted by the carbonyl oxygen of the N-acetyl group providing intermediate D. Deprotonation of D by an external β-carboline forms the radical intermediate E, which is oxidized by Ir(III)* to give product 376. This deprotonation is an irreversible and enantiodetermining step in this transformation.306
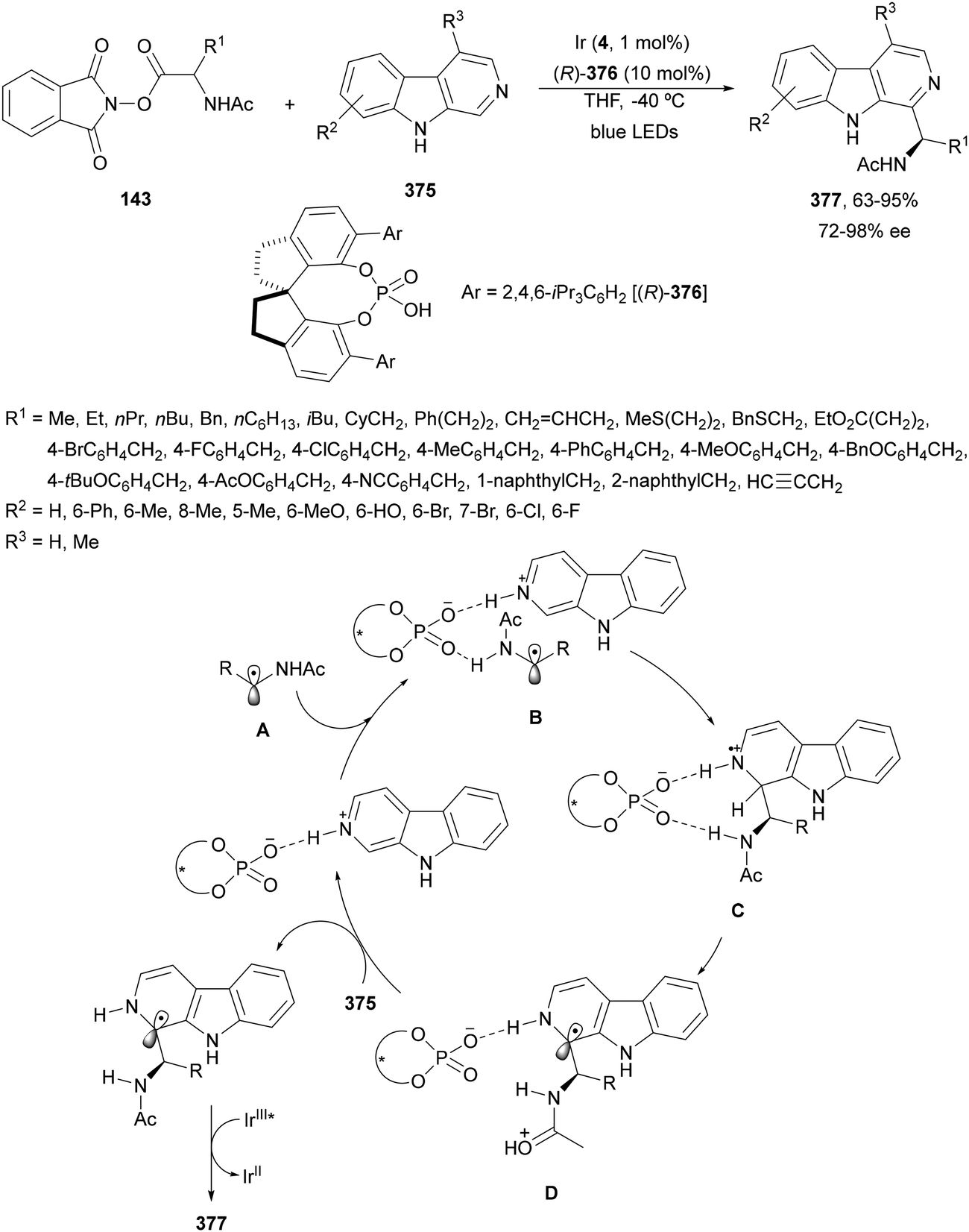 | ||
| Scheme 168 Decarboxylative asymmetric alkylation of β-carbolines 375 with α-AA derived NHPI esters 143 under Ir/SPINOL-CPA 376 photocatalysis. | ||
Synthesis of oxalyl chiral heterocyclic molecules has been achieved by asymmetric Minisci reaction of 5-arylpyrimidines and α-AA-derived NHPI esters 143.311 In the presence of 4CzIPN as a PC and CPAs such as (R)-379 and (R)-380 in dioxane at 15 °C under blue LED irradiation, pyrimidines 378 were transformed into products 381 in up to 82% yield, >19:1 dr and >99% ee (Scheme 169). This desymmetrization process312,313 was also carried out on a gram-scale and also under sunlight irradiation. Radical A undergoes chiral phosphoric acid-catalyzed regio- and enantioselective addition to the pyrimidine unit to provide phosphoric acid-coordinated radical cation B through the depicted TS.
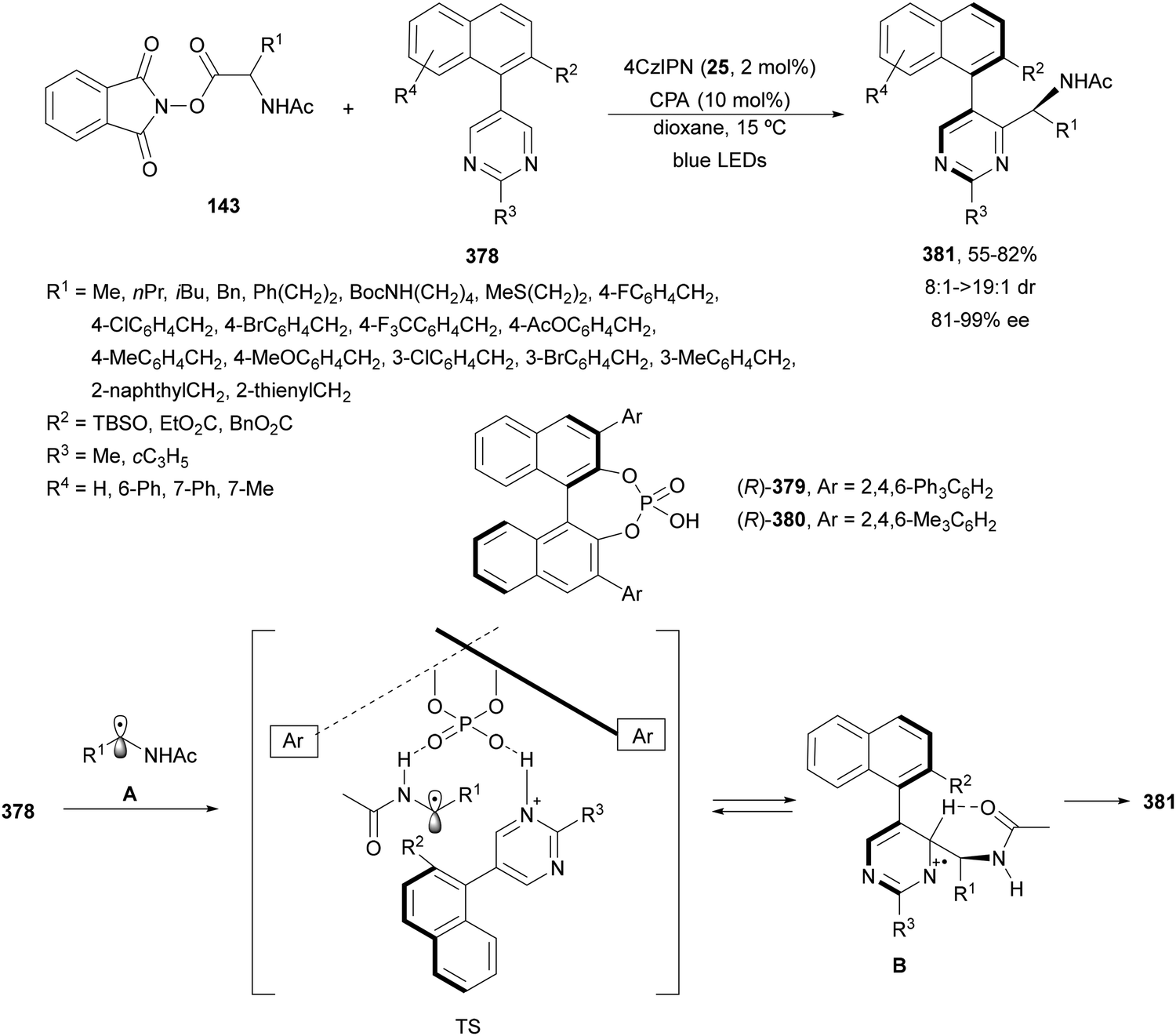 | ||
| Scheme 169 Decarboxylative asymmetric alkylation of 5-arylpyrimidines 378 with α-AA-derived NHPI esters 143 under 4CzIPN/CPA 370, 379 or 380 photocatalysis. | ||
A general method for decarboxylative alkylation of N-heteroarenes with AAs and peptides derived NHPI esters 143 was reported in 2017 by Fu and co-workers.314 As in the case of aliphatic carboxylic acids (Scheme 164),303 Ir complex 4 (2 mol%) in the presence of BINOL derived phosphoric acid (10 mol%) as a Brønsted acid was employed under blue light irradiation with different heterocycles such as quinoline, quinoxaline, phthalazine, isoquinoline, quinazoline, pyrimidine, phenanthridine, phenanthroline and purine. The corresponding aminoalkylated products were obtained with 24–95% yields and this process was also applied to the functionalization of drugs such as fasudil hydrochloride, caffeine and famciclovir. Another general method for the alkylation with aliphatic carboxylic acids derived NHPI esters 69 of heterocyclic compounds was described by Opatz and co-workers.307 In this case, Ru(bpy)3Cl2 (1 mol%) was used as a PC and p-TsOH as a Brønsted acid in DMF at room temperature under blue LED irradiation. Heterocyclic compounds such as isoquinoline, benzothiazole, pyrazine and quinoline gave the corresponding products, which were obtained with 16% to quantitative yields. Mechanistic investigations revealed a radical chain mechanism, which in some cases can proceed even if no PC is added. Under organocatalyzed (4CzIPN, 25) visible-light photoredox conditions, via the intermediacy of the in situ generated NHPI esters, in the presence of TFA or CSA as Brønsted acids, Sherwood and co-workers315 performed the alkylation with aliphatic acids and protected AAs and hydroxy acids of quinoline, pyridine, isoquinoline, quinaldine, phthalazine, quinoxaline, quinazolinone, azaindole, benzimidazole, benzothiazole and caffeine to provide the resulting products in general with modest yields (5–76%). Other bioactive compounds such as nebularine and peracetylated nebularine, adenosine, camptothecin, vemuraferib and imatinib were alkylated providing derivatives to establish SAR. Copper-catalyzed decarboxylative alkylation of isoquinolines, quinolines, pyridine, pyrimidine, quinazoline, phthalazine, phenanthridine and pyridazine derivatives has been accomplished by Wang and co-workers.316 They employed Cu(MeCN)4PF6 (10 mol%), 2,9-dimethyl-1,10-phenanthroline (dmp, 15 mol%) and Xantphos (15 mol%) as ligands, Zn(OTf)2 (10 mol%) as a Lewis acid in DMA at room temperature under blue LED irradiation providing products with in general good yields (18–99%). [Cu(dmp)(Xantphos)]BF4, which was formed in situ, was the PC.
A general method for the decarboxylative alkylation of electron-rich heteroarenes with NHPI esters 69 was reported by Deng, Tang and co-workers.317 In the presence of Ir(ppy)3 (0.5 mol%) as a PC in DMSO at room temperature under white LED irradiation, furans, benzofurans and thiophenes were alkylated at the 2-position with in general moderate yields (21–84%).
Decarboxylative alkylation of quinoxalin-2(1H)-ones with NHPI esters has been carried out by Jin and co-workers318 with Eosin Y-Na2 (139, 1 mol%) as a PC and TFA in DMSO under white LED irradiation. These milder reaction conditions, compared to those described with carboxylic acids (Scheme 161),296,297 gave the corresponding 3-alkylated products in higher yields (63–99%). Independently, Li and coworkers319 performed the same alkylation using Ir(ppy)3 (2 mol%) and TFA in DMSO under white LED irradiation providing 3-alkylated quinoxaline-2(1H)-ones also with in general very good yields (30–95%). More recently, Yatham and co-workers320 described the same photoinduced decarboxylative alkylation just with K2CO3 as a base in DMF at room temperature under 395–400 nm irradiation to furnish the corresponding 3-alkylated quinoxaline-2(1H)-ones with 40–91% yields. These simple reaction conditions were implemented using medicinally important carboxylic acid derived NHPI esters, for instance, fenofibric acid, gemfibrozil, loxoprofen and levulinic acid.
Papaioannou, Fray and co-workers321 carried out regioselective photoredox amido methylation of 4-chloro-3-fluoropyridine (382) with α-AA derived NHPI esters 143. In this case, 4CzIPN (25) was employed as a PC and DMA as solvent at 36 °C by irradiation using a Kessil H150 blue grow light (400–520 nm). The resulting trisubstituted pyridines 383 were obtained with 39–74% yield and 87:13–97:3 isomeric ratios (Scheme 170). The best results were obtained with N-Boc protected esters 143, which were scaled-up to 77 mmol.
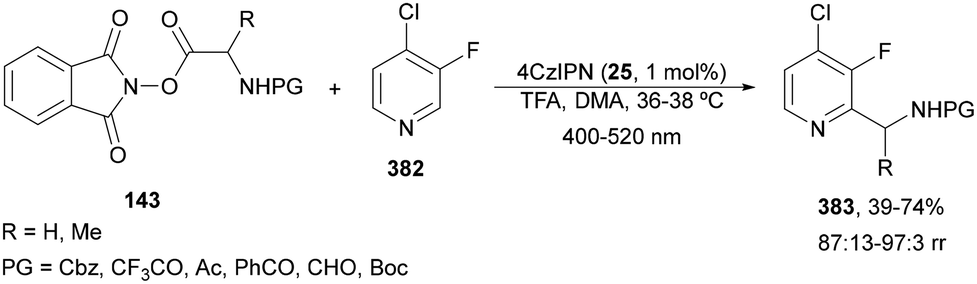 | ||
| Scheme 170 Decarboxylative alkylation of 4-chloro-3-fluoropyridine (382) with α-AA derived NHPI esters (143) under 4CzIPN photocatalysis. | ||
8-Acylaminoquinoline (384) has been alkylated regioselectively at the 2-position with aliphatic primary and tertiary carboxylic acid derived NHPI esters 69 in the presence of Ir(ppy)3 as a PC and TFA in DMSO under white LED irradiation.322 The resulting 2-alkylated quinolines 385 were isolated in good yields using N-acylated derivatives, whereas 9-aminoquinoline failed (Scheme 171). When secondary alkyl NHPI esters were employed, dialkylated quinolines 386 were obtained with good yields.
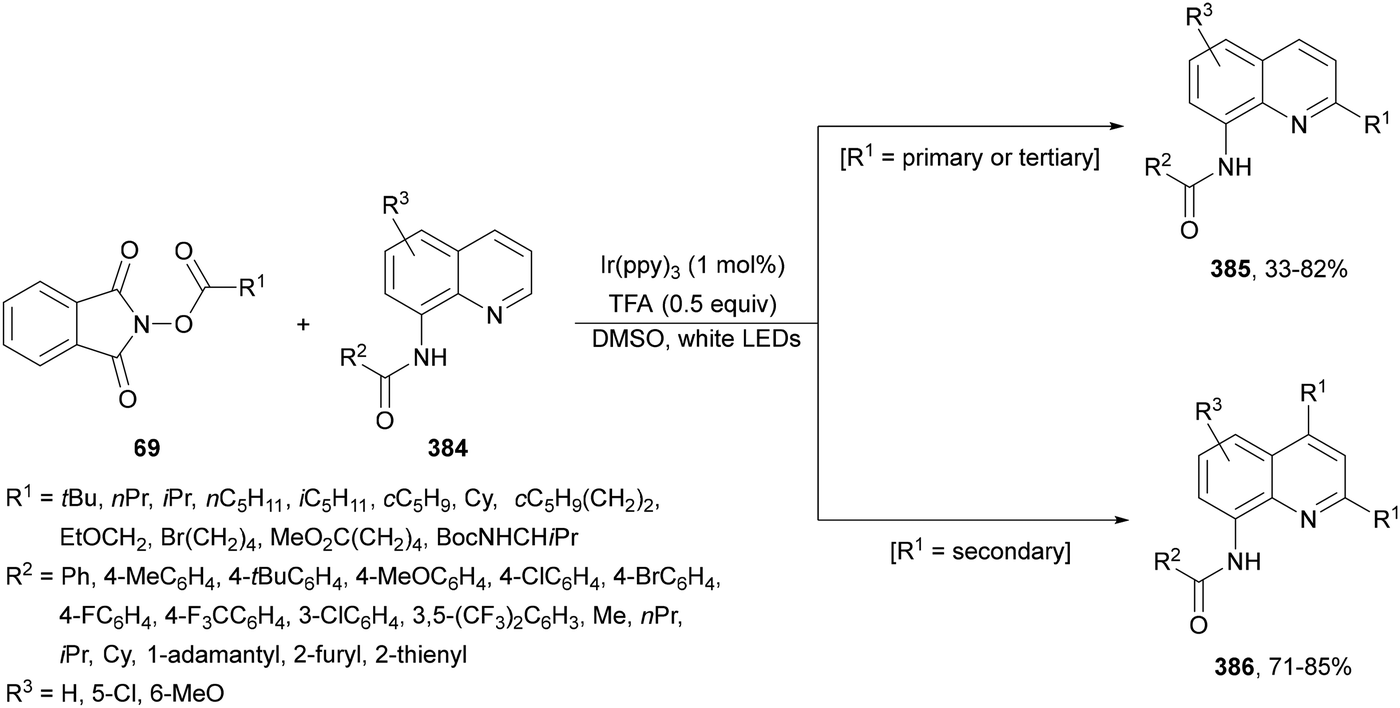 | ||
| Scheme 171 Decarboxylative mono- and di-alkylation of 8-acylaminoquinoline (384) with aliphatic NHPI esters 69 under Ir(ppy)3 photocatalysis. | ||
The 2H-indazole unit is present in many drugs and bioactive molecules and also in materials science. Jiang, Yu and co-workers323 developed a photoredox decarboxylative alkylation of 2-aryl-2H-indazoles 387 with alkyl NHPI esters 69 in the presence of 4CzIPN (25) as a PC and DABCO as a base in dimethyl carbonate (DMC) at 35 °C under N2 and blue LED irradiation (Scheme 172). The resulting 3-alkylated indazoles 388 were obtained under these mild and green reaction conditions with good yields. This protocol was applied to the late-stage modification of drug molecules such as elaidic acid, levulinic acid and gemfibrozil.
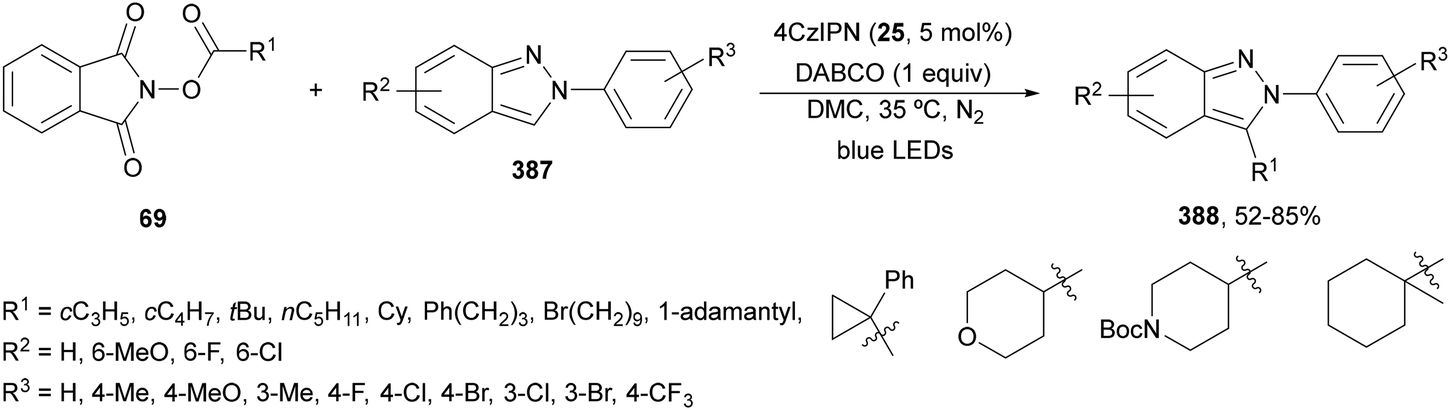 | ||
| Scheme 172 Decarboxylative alkylation of 2-aryl-2H-indazoles 387 with alkyl NHPI esters 69 under 4CzIPN (25) photocatalysis. | ||
The same group324 recently reported the regioselective alkylation of 2,1,3-benzothiazoles 389 under the above described reaction conditions. In this case, the alkylation took place in N,N-dimethylacetamide (DMAc) at the four position of the heterocycles affording products 390 with moderate to good yields (Scheme 173). The reaction of compound 389a (R2 = H) with the cyclohexyl derived NHPI ester was carried out on a gram scale to give product 390a (R1 = Cy; R2 = H) in 60% yield. NHPI esters derived from biologically active molecules such as gemfibrozil, elaidic acid and dehydrochlolic acid were also employed.
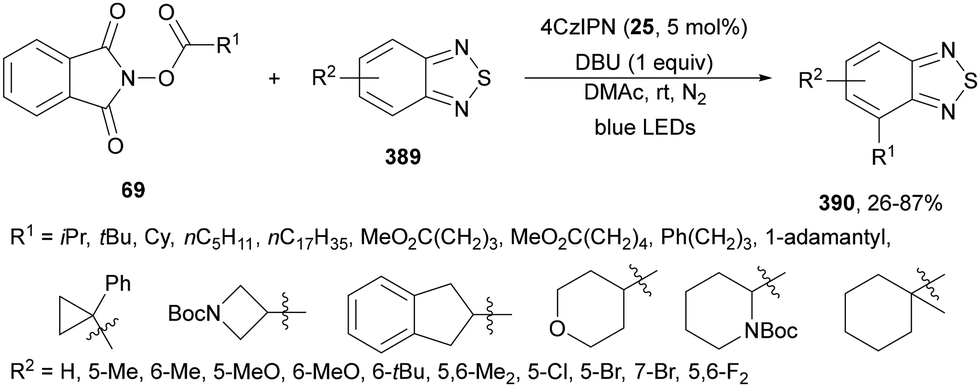 | ||
| Scheme 173 Decarboxylative C-4 alkylation of 2,1,3-benzothiadiazoles 389 with alkyl NHPI esters 69 under 4CzIPN photocatalysis. | ||
Decarboxylative alkylation of imidazo[1,2a]pyridines 366 was also carried out with alkyl NHPI esters 69325 instead of carboxylic acids as it was previously described300 (Scheme 162). In this case, Eosin Y was used as an organic PC in the presence of TfOH at room temperature in DMSO under blue LED irradiation to afford products 367 alkylated at the five position with 38–86% yields.
Sun, Zhou and co-workers326 reported a C-2 alkylation of quinoline 391 and pyridine N-oxides 392 with alkyl NHPI esters 69 in the presence of Eosin Y as a PC and Cs2CO3 as a base in DMF at room temperature under blue LED irradiation. Products 393 and 394 were obtained in moderate to good yields (Scheme 174). Quinoxaline and quinazoline N-oxides were also alkylated at the 2-position with 51 and 26% yields, respectively. Glycosyl-based NHPI ester as well as dehydroabietic acid and quinine derived esters were employed for the alkylation of lepidine N-oxide at the two position.
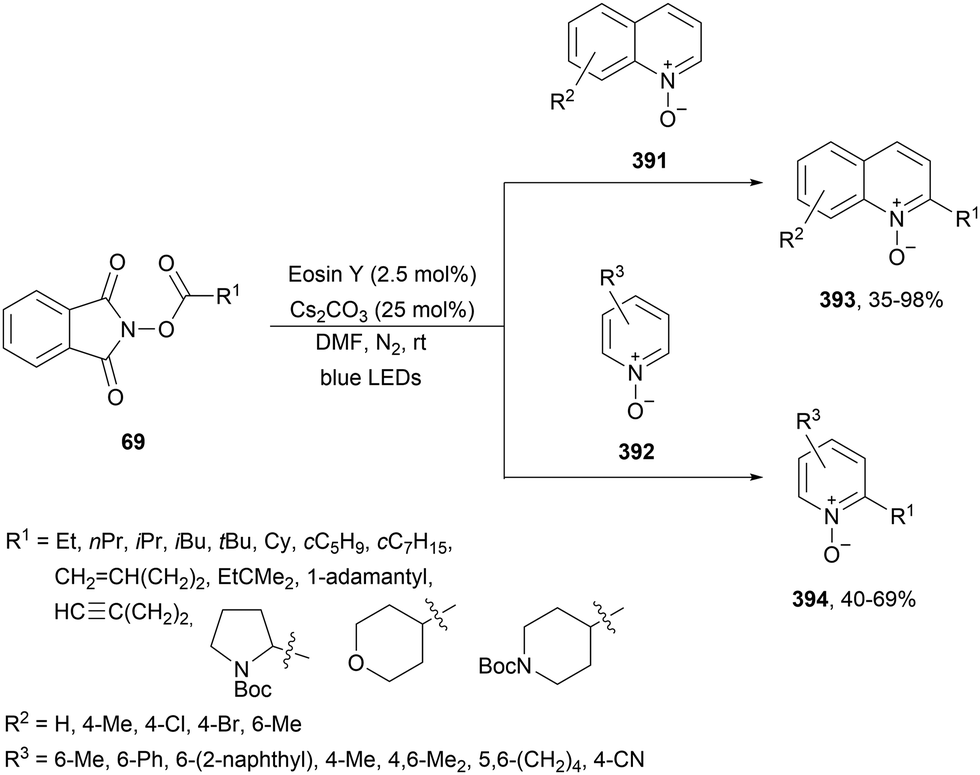 | ||
| Scheme 174 Decarboxylative C-2 alkylation of heterocyclic N-oxides with alkyl NHPI esters 69 under Eosin Y photocatalysis. | ||
C–H alkylation of azauracils 395 has been carried out by Murarka and co-workers327 using alkyl NHPI esters 69 in the presence of NaI/PPh3 and TMEDA as a base in MeCN at room temperature under blue LED irradiation (Scheme 175). This alkylation took place to give products 396 with very good yields and was also carried out with other heterocycles such as cinnolinone, quinoxalinone and pyrazinone. In the proposed mechanism, the reaction begins by the formation of a charge-transfer complex (CTC) assembly A based on cation–π and electronic interactions. Upon irradiation, a SET process from an iodine anion to NHPI ester leads to the generation of alkyl radical B and iodide radical C, after CO2 elimination. Subsequent addition of B to the 6-position of azauracil gives radical D, which after oxidation by C forms intermediate E. Finally, TMEDA or PhthN− deprotonates E to provide the product.
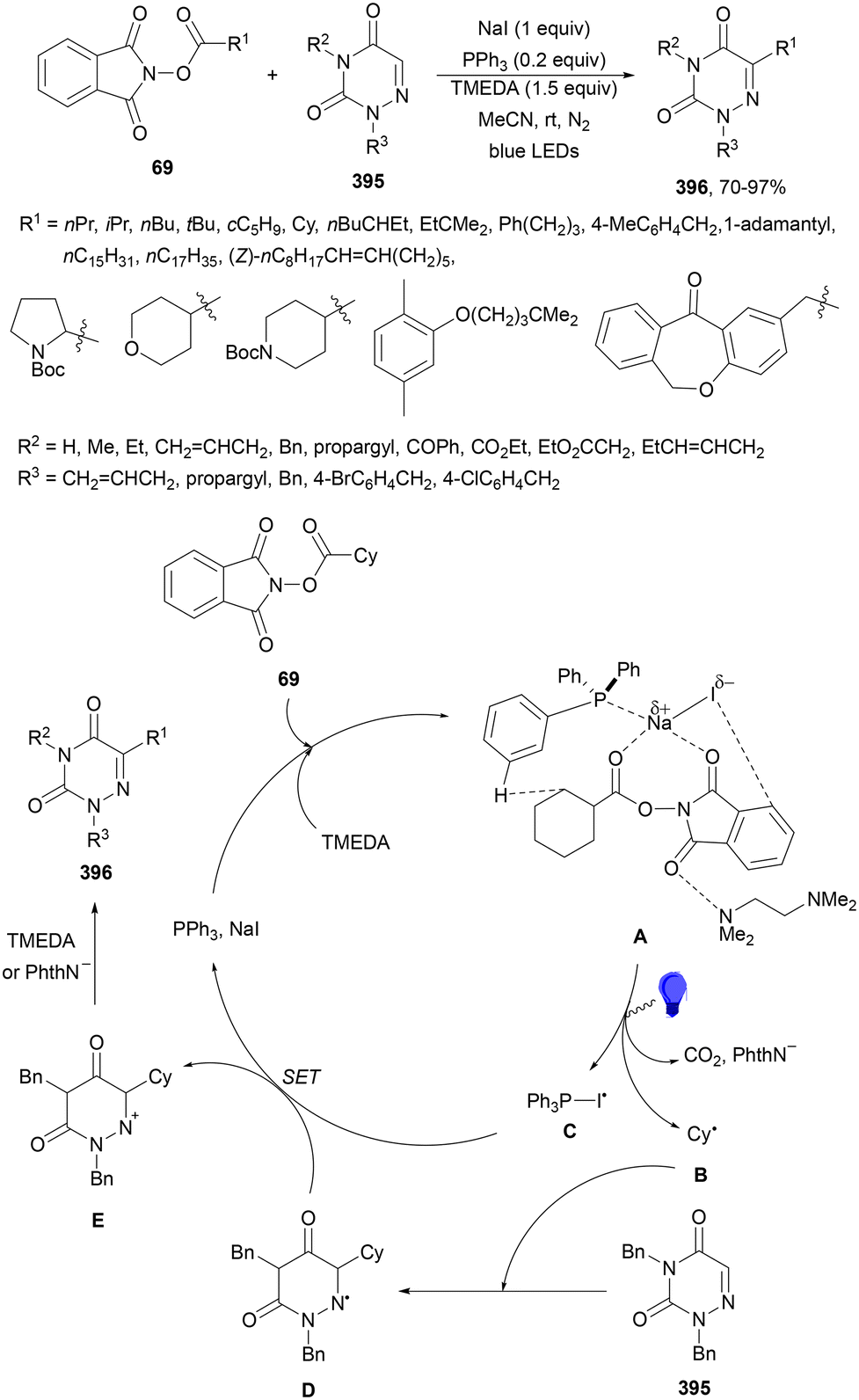 | ||
| Scheme 175 Decarboxylative C-6 alkylation of azaurazils 395 with alkyl NHPI esters under PPh3/NaI photocatalysis. | ||
Non-aromatic heterocycles such as coumarins 397 have been alkylated by carboxylic acid decarboxylation291 and also by means of alkyl NHPI esters.328 This alkylation took place regioselectively at the 3-position using Ir(ppy)3 as a PC and TFA as a Brønsted acid in DMSO at room temperature under N2 and blue LED irradiation (Scheme 176). Products 398 were obtained with 30–92% yields and it was proposed that the alkyl radical A, generated from NHPI ester 69, adds to coumarin to give radical B, which was further oxidized by Ir(IV) to carbocation C. Final deprotonation by trifluoroacetate gives product 398. Independently, Dong, Zhou and co-workers329 reported this alkylation using Ru(bpy)2Cl2·6H2O (0.2 mol%) as a PC and DABCO as a base in DMAc at room temperature under sunlight or blue LED irradiation to give products 398 in 40–81% yields. Other heterocyclic compounds such as quinolinones or quinoxalinones were alkylated in 60–73% yields.
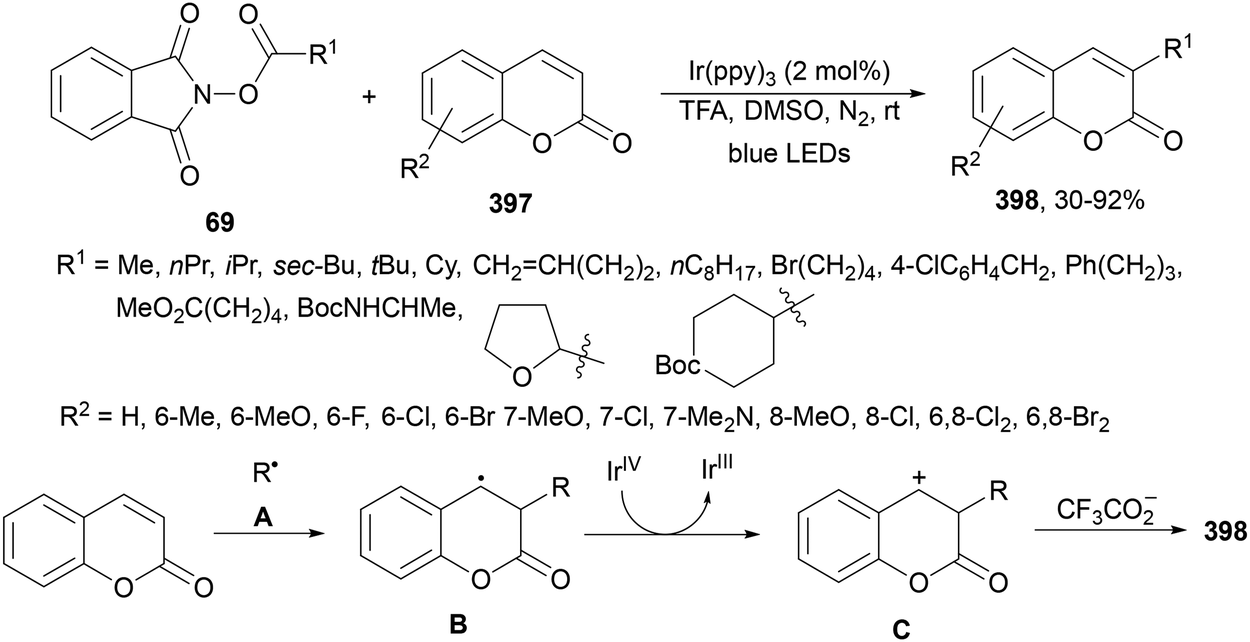 | ||
| Scheme 176 Decarboxylative C-3 alkylation of coumarins 397 with alkyl NHPI esters (69) under Ir(ppy)3 photocatalysis. | ||
Albrecht and co-workers330 employed catalytic amounts of PPh3/KI for the decarboxylative alkylation of 3-cyanochromones 399 with alkyl NHPI esters 69 in acetone as solvent and under blue LED irradiation to give 2-alkylated derivatives 400 (Scheme 177). This process took place by a CTC assembly as depicted in Scheme 175.
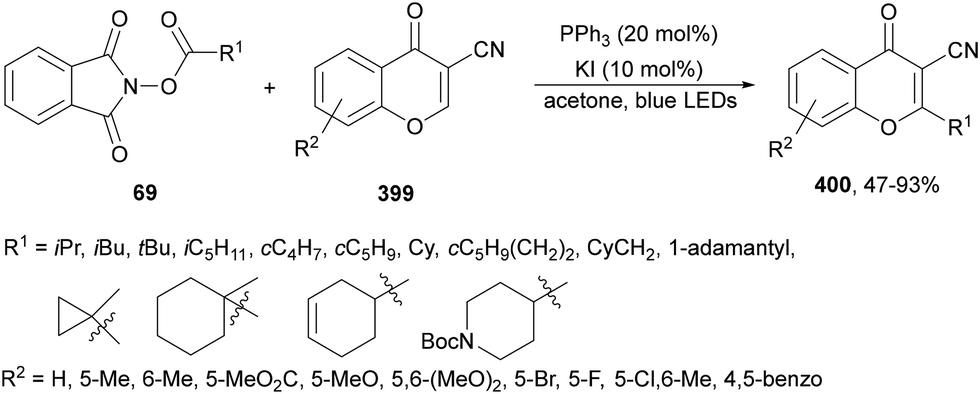 | ||
| Scheme 177 Decarboxylative C-2 alkylation of 3-cyanochromones 399 with alkyl NHPI esters 69 under PPh3/KI photocatalysis. | ||
Other esters have been used as sources of alkyl radicals instead of NHPI esters. In 2015, Overman and MacMillan groups60 reported that tert-alkyl oxalate salts, such as 347, can generate tertiary radicals for 1,4-addition to electrophilic alkenes. In 2019, Overman and co-workers331 incorporated tertiary alkyl substituents into a variety of heterocyclic substrates by means of tert-alkyl oxalate salts 347. They employed Ir complex 4 as a PC and (NH4)2S2O8 as an external oxidant and one equivalent of HCl for this alkylation (Scheme 178). Heteroarenes such as quinolines, pyridines, quinoxaline, quinazolin-4(3H)-one, 7-azaindole, phenanthroline, benzothiazole and 1-methylbenzimidazole were alkylated with moderate yields. This method was also applied to biologically relevant heteroarenes such as purine riboside, quinine, the rho-kinase inhibitor and vasodilator fasudil.
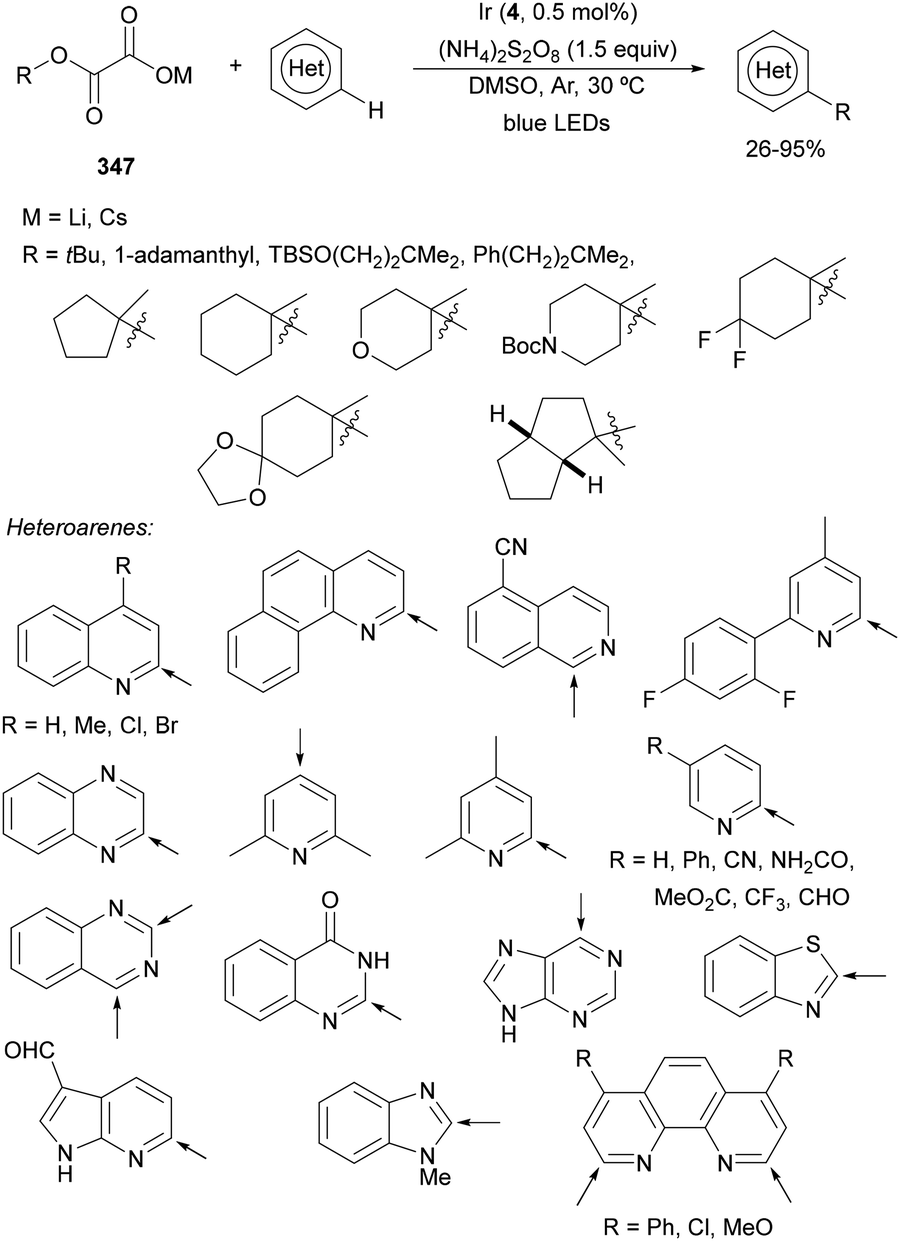 | ||
| Scheme 178 Decarboxylative alkylation of heteroarenes with tert-alkyl oxalate salts 347 under photocatalysis. | ||
Decarboxylative alkylation of heteroarenes has been carried out with N-hydroxybenzimidoyl chloride (NHBC) esters 401 as RAEs for the coupling of α-aminoalkyl radicals.332 This type of RAE was previously employed for the photoinduced hydrofluoroalkylation of unactivated alkenes.333 In the presence of Ir complex 4 and in the absence of external oxidant, in MeCN at room temperature under blue LED irradiation, the corresponding α-aminoalkylated products were obtained with good yields (Scheme 179). Quinolines, isoquinolines, pyridines and phenanthridine were α-aminoalkylated with a broad variety of N-Boc α-AAs. This protocol has been applied to the functionalization of thymopentin, a pentapeptide with potential biomedical applications. NHBC esters 401 generate, by photoexcited Ir(III), the corresponding α-amino radicals, chloride, CO2 and benzonitrile.
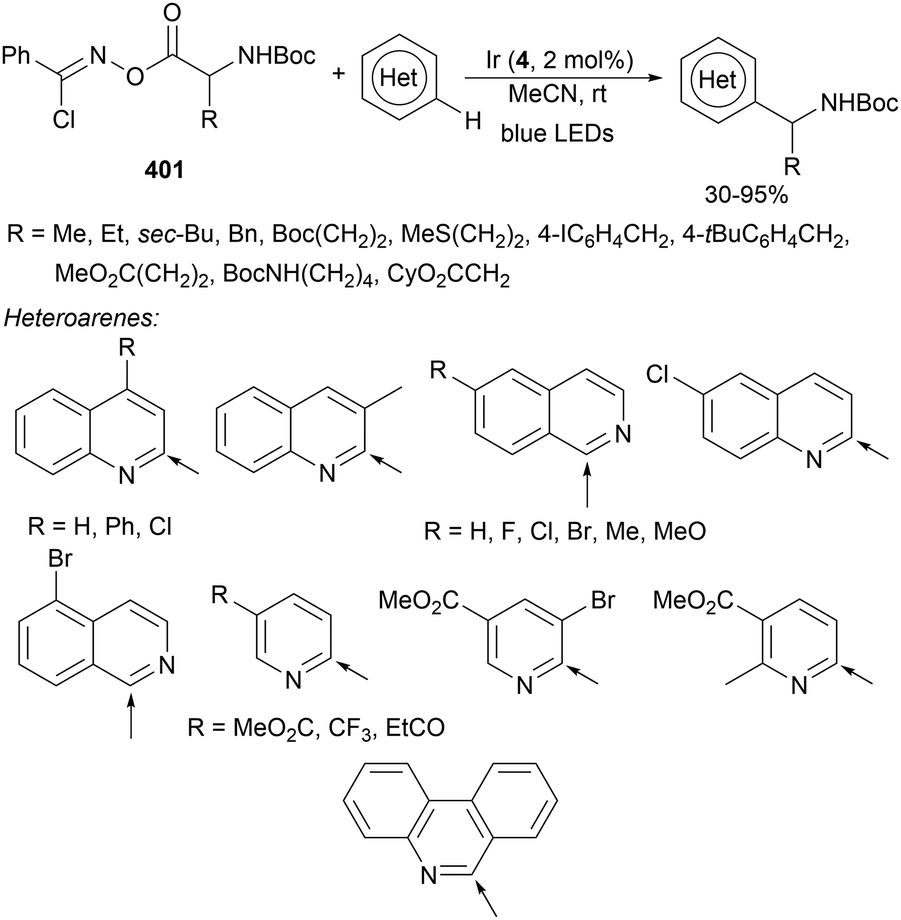 | ||
| Scheme 179 Decarboxylative α-aminoalkylation of N-heteroarenes with N-hydroxy benzimidoyl chloride esters 401 under Ir photocatalysis. | ||
Alkylation of aliphatic C–H bonds with NHPI esters was described by Ren and Cong334 using N-aryl tetrahydroisoquinolines 402. In this decarboxylative alkylation, erythrosine B (403) and P25-type TiO2 were used as visible light PC in 2,2,2-trifluoroethanol (TFE) at 40 °C (Scheme 180). The corresponding 1-substituted tetrahydroisoquinolines 404 were isolated with in general good yields employing primary, secondary and tertiary alkyl NHPI esters 69.
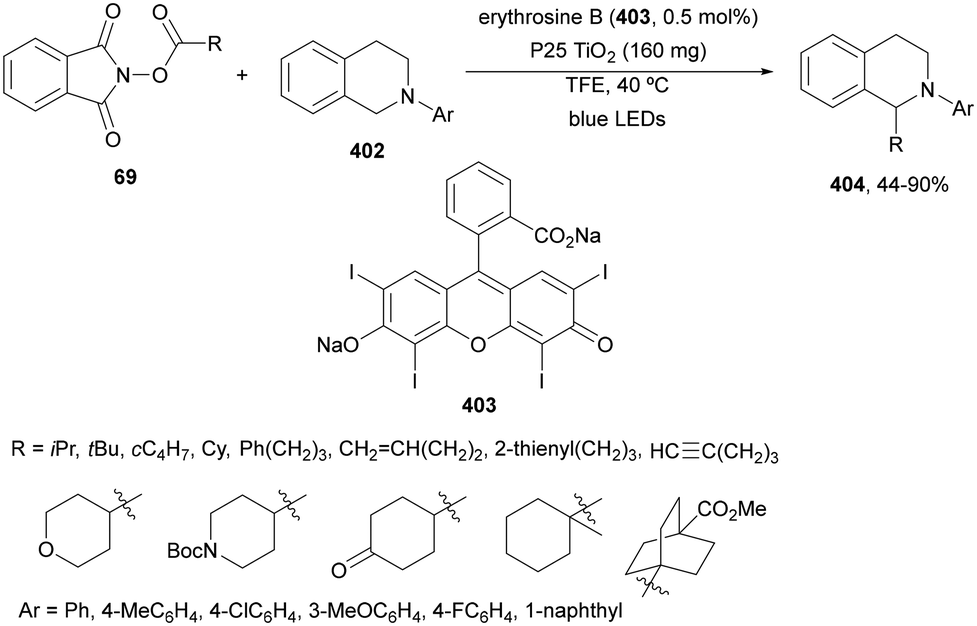 | ||
| Scheme 180 Decarboxylative alkylation of N-aryl tetrahydroisoquinolines 402 with alkyl NHPI esters 69 under erythrosine B (403) sensitized TiO2 photocatalysis. | ||
C(sp3)–H alkylation of N-aryl glycines with NHPI esters 69 for preparing α-alkylated unnatural α-AAs has been carried out under copper-catalyzed decarboxylative conditions.335 Working with 10 mol% of Cu(MeCN)4PF6, 15% of dmp or Xantphos as ligands, DABCO as a base in DMF under blue LEDs irradiation, different glycine derivatives were alkylated with very good yields (Scheme 181a). This process was also applied for the modification of peptides which were regioselectively alkylated at the α-position of the glycine unit with good yields and low diastereoselectivity (Scheme 181b). In the proposed mechanism, the LCu(I) complex is excited to LCu(I)*, which undergoes a SET with alkyl NHPI ester 69 followed by generation of the alkyl radical A and LCu(II). The glycine derivative is oxidized by LCu(II) generating a radical cation B and regenerating the catalyst. Intermediate B is deprotonated by the base or PhthN− and after a 1,2-H shift process the stable α-carbon radical C is formed. Finally, radical–radical coupling of C and A gives the product.
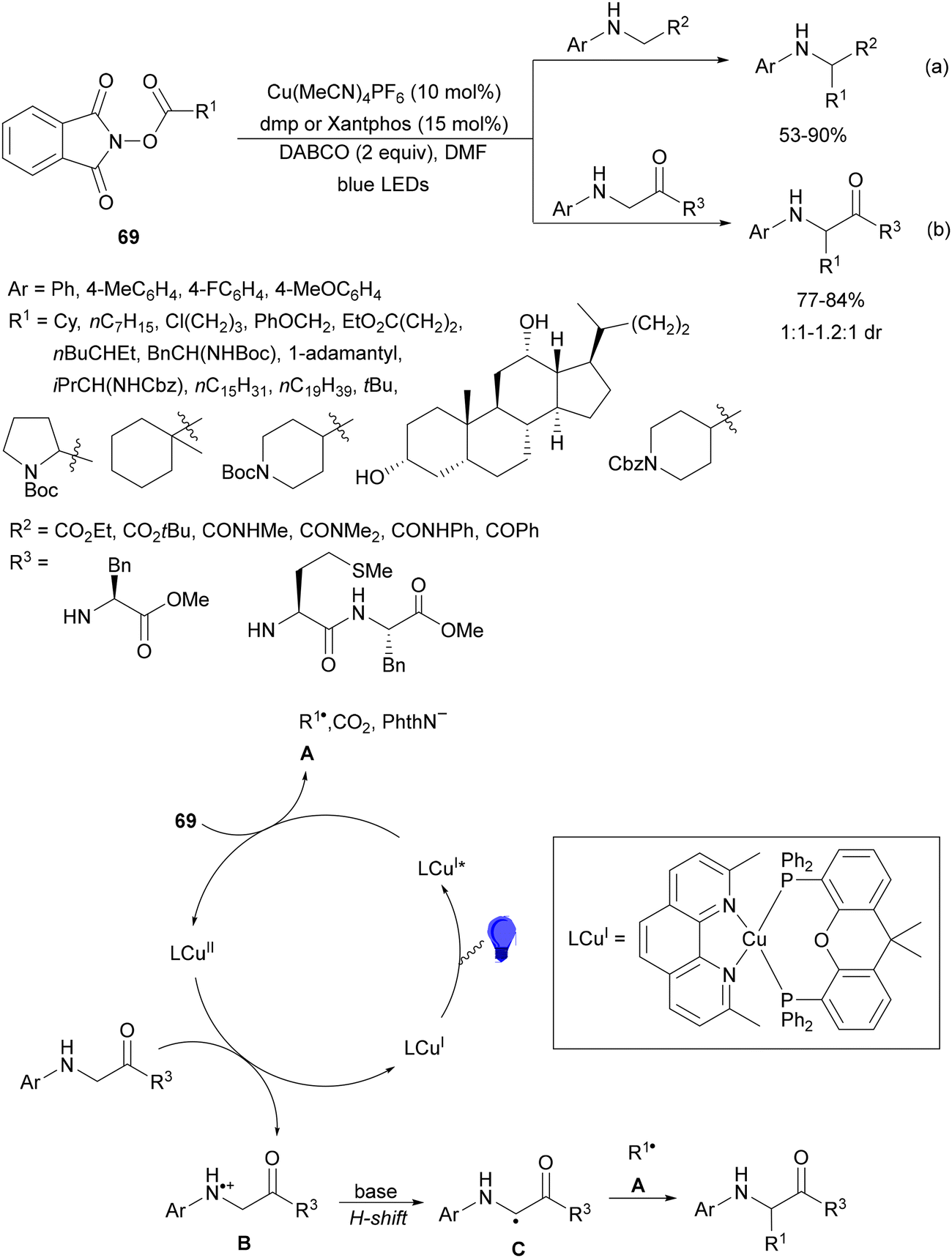 | ||
| Scheme 181 Decarboxylative C(sp3)–H alkylation of glycines (a) and peptides (b) with NHPI esters 69 under Cu(I) photocatalysis. | ||
Glycosylation with α-AAs to construct C-glycopeptides has been achieved by visible-light copper-catalyzed cross-coupling by Liang and co-workers.336 This asymmetric C(sp3)–C(sp3)–H reaction was carried out between glycosyl NHPI esters 405 and N-(8-quinolyl)glycine esters 406 using Cu(OTf)2 and (S)-XylBINAP (407) as a chiral ligand and DABCO as a base in DMF under blue LED irradiation (Scheme 182). Resulting C-glycosides 408 were obtained with moderate to good yields and excellent diastereoselectivity. Different α-AAs as well as dipeptides formed by condensation of glycine with Val, Phe, Ala and Met, and tripeptides such as Gly–Phe–Phe–OMe, Gly–Met–Phe–OMe and Gly–Phe–Val–OMe were also employed. Protected ribose and pyranoses including galactose, mannose and glucose derived NHPI esters proceeded in excellent dr and good yields. The proposed mechanism starts with the Cu(I)-AA complex A, which after irradiation via a SET process reacts with 405 to give the glycosyl-Cu(III) species Bvia glycosyl radical formation. Intramolecular LMCT54,55 of B forms a radical intermediate C, which by a 1,2-H shift produces species D. Subsequent intramolecular recombination of alkyl radical promotes the formation of the chiral Cu(III) E, which evolves to give product 408 through a reductive elimination step and regenerates the catalyst.
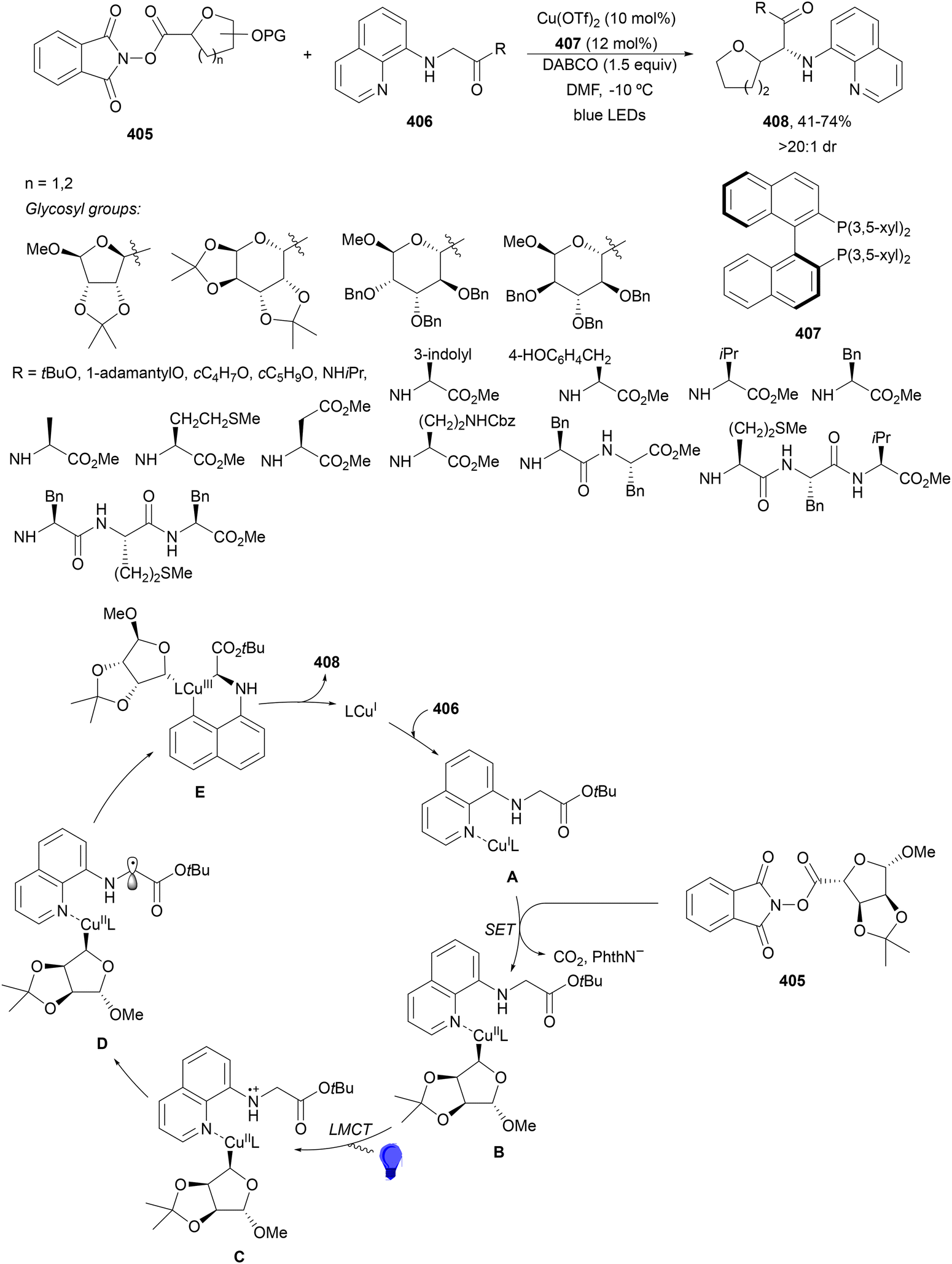 | ||
| Scheme 182 Decarboxylative asymmetric C(sp3)–H alkylation of N-quinolineglycine 406 with glycosyl NHPI esters 405 under Cu photocatalysis. | ||
MacMillan and co-workers337 performed the C(sp3)–C(sp3)–H cross-coupling of bicyclo[1.1.1]pentane (BCP, 157) with N-heterocycles and other nucleophiles and alkyl carboxylic acids via iodonium dicarboxylates 236. The three-component reaction was carried out under Cu(acac)2 and bathophenanthroline (BPhen) catalysis using Ir(ppy)3 as a PC and BTMG as a base in dioxane under blue LED irradiation to provide diverse functionalized bicyclopentanes 409 with good yields (Scheme 183). In the proposed mechanism, photoexcited Ir(III)* reduces iodonium dicarboxylate 236 to generate radical A upon CO2 extrusion. Subsequent radical addition to BCP (157) generates radical B, which reacts with complex C to give the Cu(III) intermediate D. After subsequent reductive elimination, product 409 is formed and the Cu(I) complex E regenerated. Finally, reaction of E with the nucleophile provides the Cu(I) species F, which after oxidation by Ir(IV) forms the PC. These products are valuable pharmaceutical bioisosteres of benzene.
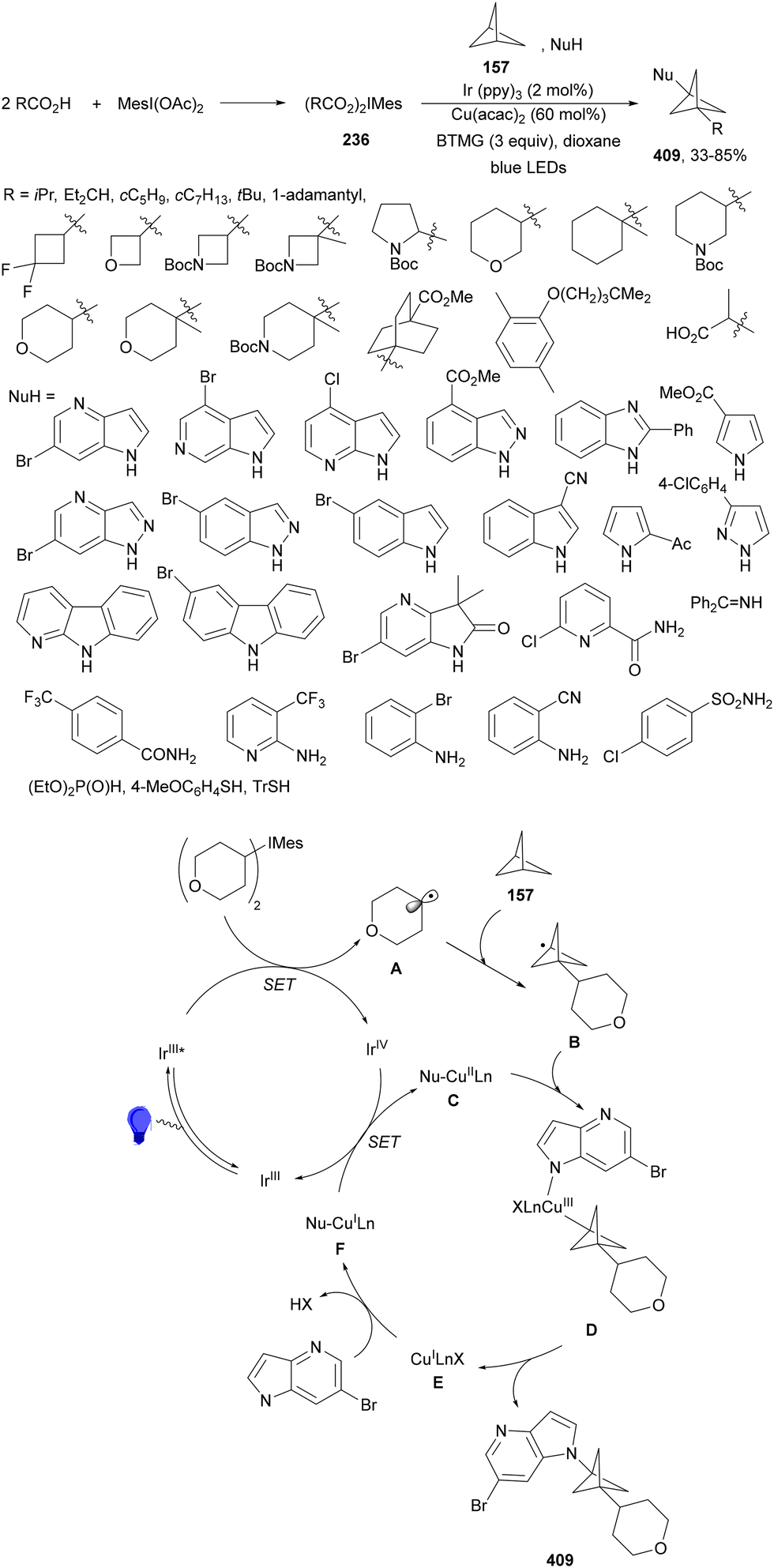 | ||
| Scheme 183 Decarboxylative alkylation of bicyclo[1.1.1]pentane 157 and heteroatom nucleophiles under Ir/Cu photocatalysis. | ||
Decarboxylative alkylation of arenes and heteroarenes can be carried out directly with carboxylic acids or with activated esters such as NHPI. In the first case, different photoredox catalysts such as Ir complexes used S2O82− as an external oxidant, whereas organocatalysts were used in the presence of acids or bases. Metal salts as Fe and Ce worked via photoinduced LMCT. In the case of NHPI esters derived from α-AAs, asymmetric α-aminoalkylation of heterocycles can be performed in the presence of chiral phosphoric acids under Ir or organophotocatalysis. Reaction conditions for NHPI esters are, in general, milder than with carboxylic acids either with Ir, Ru or organic PCs or with Cu complexes. Alternatively, PPh3 and metal iodides have been employed by formation of a charge-transfer complex for azauracils and 3-cyanochromones. Alkylation of C(sp3)–H bonds especially of glycine derivatives has been carried out with copper salts by LMCT processes.
Wencel-Delord and co-workers338 performed a general photoinduced acylation of N-heterocycles in the absence of a PC. In the presence of K2S2O8 as external oxidant in a 1:2 mixture of MeCN/H2O at room temperature, different N-heterocycles including 2- and 4-methylquinolines, isoquinolines, pyridines, pyrimidines, acridine, quinoxaline, quinazoline, phthalazine, 1,10-phenanthroline, benzothiazole and caffeine were acylated with 23–86% yields (Scheme 184). According to mechanistic studies, three different pathways were proposed: (a) direct generation of the acyl radical; (b) generation of the EDA complex A absorbing the visible light and thus enhancing photodecarboxylation; and (c) generation of the EDA complex B promoting homolytic cleavage of S2O82− to produce SO4−˙ triggering decarboxylation of the α-keto acid and generation of the acyl radical.
 | ||
| Scheme 184 Decarboxylative acylation of N-heteroarenes with α-keto acids in the presence of K2S2O8 under photocatalysis. | ||
In the case of electron-deficient N-heterocyclic compounds, the presence of Ir complex as a PC is crucial for the acylation with α-keto acids. Manna and Prabhu339 employed 2 mol% of Ir complex 52 and 2 equivalents of Na2S2O8 as an external oxidant in a 1:1 mixture of MeCN/H2O at room temperature under CFL irradiation for the acylation of isoquinolines, quinolines, pyridines and quinoxaline (Scheme 185). In the proposed mechanism, the acyl radical A is generated through hydrogen atom transfer (HAT) between the α-keto acid and the sulfate radical anion followed by decarboxylation. Addition of A to isoquinoline gives the radical cation B, which after deprotonation affords the α-amino radical C. Subsequent reduction by Ir(IV) forms the product and regenerates the PC.
 | ||
| Scheme 185 Decarboxylative acylation of N-heterocycles with α-keto acids under Ir 52 photocatalysis. | ||
Decarboxylative formylation of heterocycles has been achieved with 2,2-diethoxyacetic acid 26 without a PC, just with (NH4)2S2O8 as an external oxidant. Yang, Xia and co-workers340 performed the formylation of isoquinolines, quinolines, quinoxaline, phenanthridine, benzimidazole, benzothiazole and caffeine using 2 equivalents of (NH4)2S2O8 and Cs2CO3 as a base in N2 under blue LED irradiation followed by hydrolysis with 3 M HCl (Scheme 186a). However, when α-keto acids were used, the presence of 0.2 mol% of Ir complex 4 as a PC was needed, as well as 0.2 mol% of (NH4)2S2O8 (Scheme 186b).
 | ||
| Scheme 186 Decarboxylative formylation (a) and acylation (b) of N-heteroarenes with 2,2-diethoxyacetic acid and α-keto acids, respectively, under photocatalysis. | ||
In the case of acylation of quinoxalin-2(1H)-ones with α-keto acids, Yue, Wei and co-workers341 used Acridine Red (410) as a PC in DCE and air as an oxidant under blue LED irradiation (Scheme 187). These mild reaction conditions with favorable functional group tolerance afforded 3-acylquinoxalin-2(1H)-ones with good yields. In the proposed mechanism, acridine is photoexcited in the presence of visible-light irradiation and interacts with the α-keto acid to produce the acyl radical A, after decarboxylation, and the hydroperoxyl radical. Addition of A to quinoxaline-2(1H)-one produces radical B, which undergoes a 1,2-H shift to radical C. Further oxidation of C by HO2˙ provides cation intermediate D, which after deprotonation gives the product.
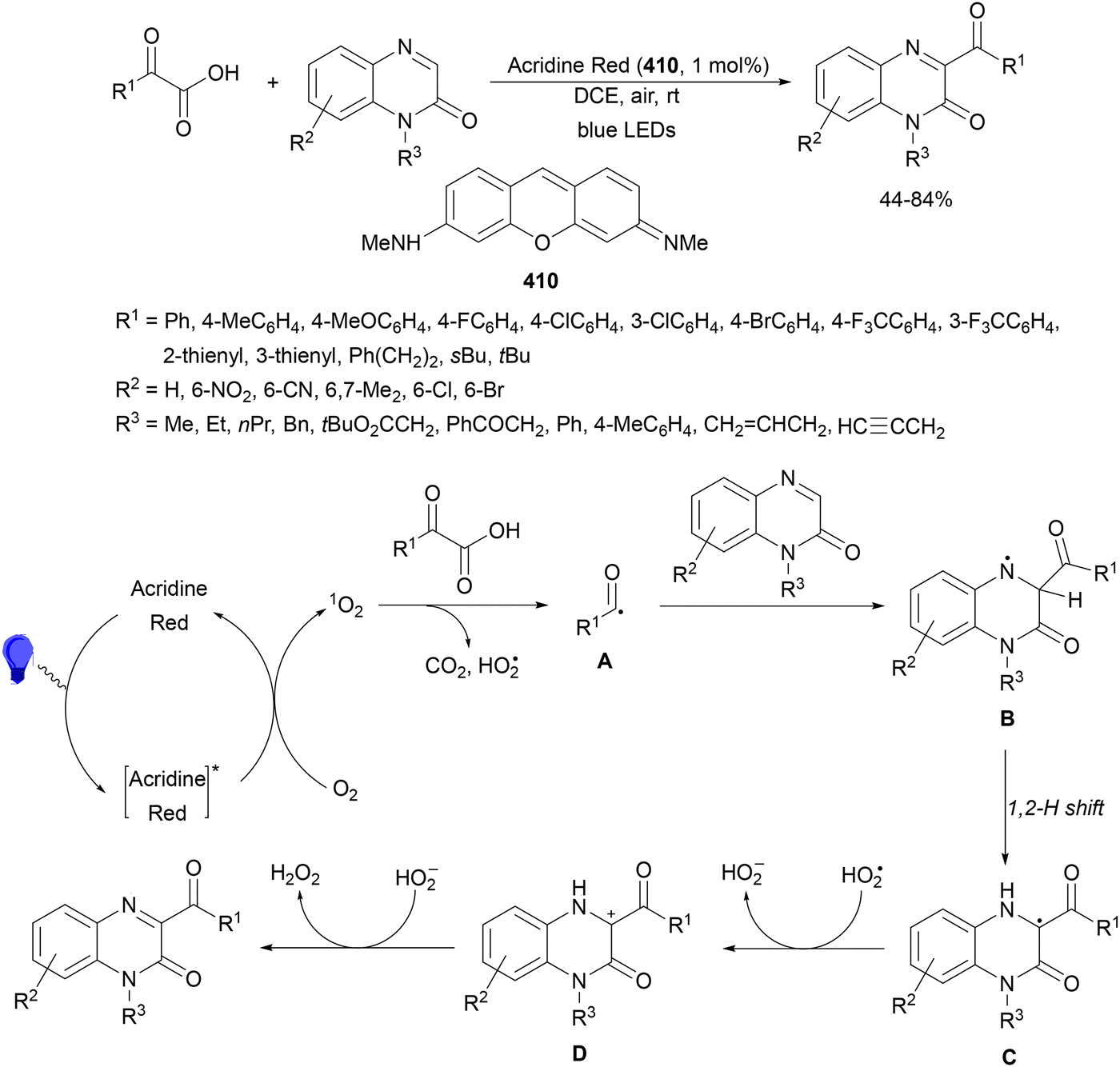 | ||
| Scheme 187 Decarboxylative acylation of quinoxaline-2(1H)-ones with α-keto acids under Acridine Red (410) photocatalysis. | ||
Acylation of quinoxaline-2(1H)-ones has been carried out with 2,2-diethoxyalkylcarboxylic acids 411 in the presence of 4CzIPN (25) as a PC and Cs2CO3 as a base in DMF at 35 °C under air and green LED irradiation.342 The corresponding 3-substituted quinoxaline-2(1H)-ones were isolated as diethyl acetals 412 in good yields (Scheme 188). The quinoxaline-2(1H)-ones carrying drug molecules including dehydrocholic acid (choleretics), indomethacin (a nonsteroidal anti-inflammatory drug) and a levulinic acid derivative were acetylated with moderate yields. In the proposed mechanism, 4CzIPN* was reductively quenched by quinoxaline-2(1H)-one to give 4CzIPN−˙ and the radical cation A. On the other hand, the glyoxylic acid acetal Cs salt is oxidized by A to generate the acetal radical B. Meanwhile, 4CzIPN−˙ is oxidized by O2 in the air to generate 4CzIPN and the photoredox cycle. Radical B attacks the 3-position of quinoxaline-2(1H)-one to afford radical C, which after 1,2-H shift forms radical D. Subsequently radical D is oxidized by a SET process by 4CzIPN* generating cation E. Final deprotonation of E gives the product. In addition, radical C could be deprotonated by Cs2CO3 to form radical anion F, which by a SET process with 4CzIPN* affords product 412.
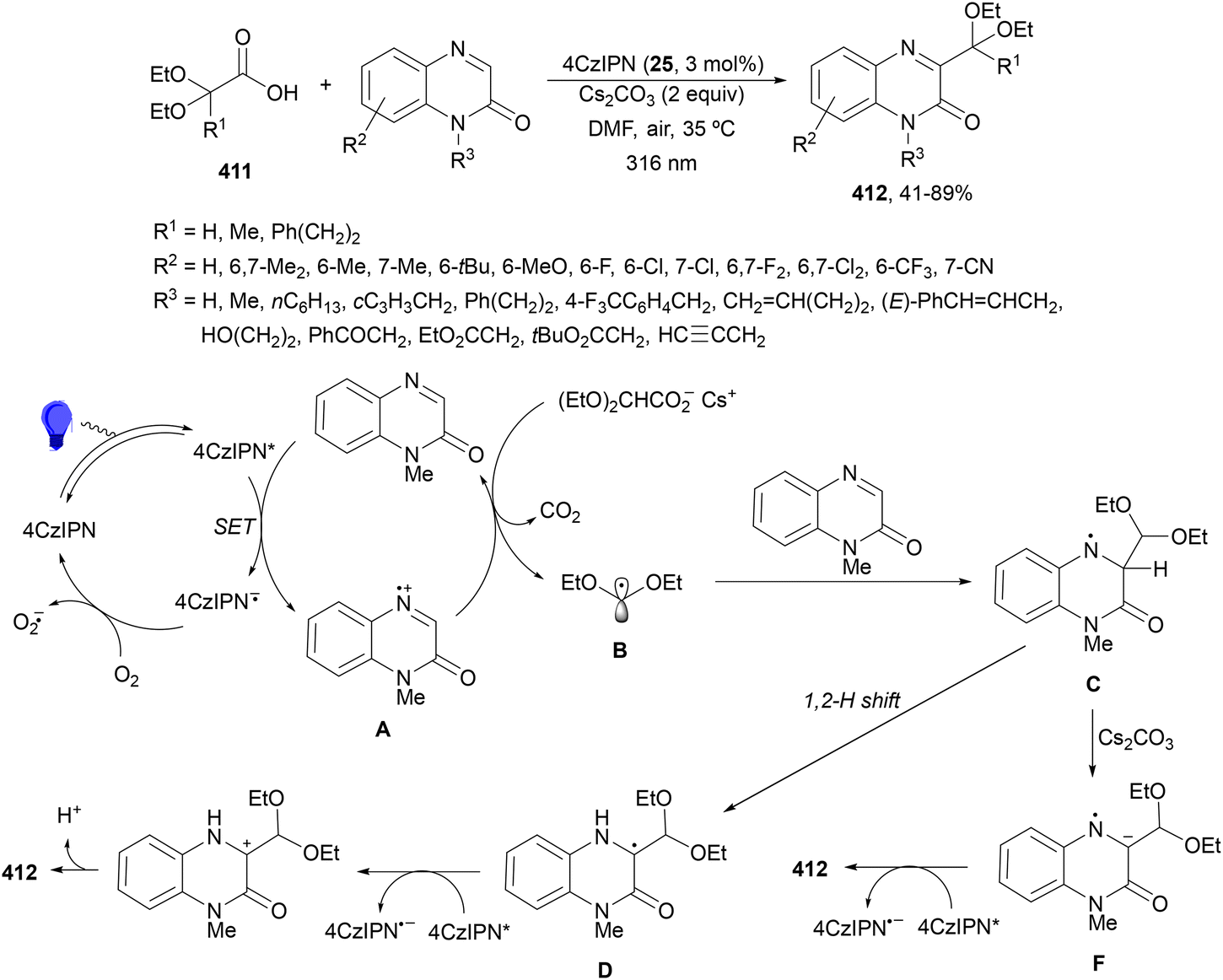 | ||
| Scheme 188 Decarboxylative acetalation of quinoxaline-2(1H)-ones with 2,2-diethoxyalkyl carboxylic acids 411 under 4CzIPN photocatalysis. | ||
Recently, Sing and co-workers343 reported the regioselective decarboxylative acylation of N-methyl-3-arylquinoxalin-2(1H)-ones 413 with α-keto acids at the aryl substituent via dual palladium-photoredox catalysis. In the presence of Pd(OAc)2, tert-butyl peroxybenzoate (TBPB) as an oxidant in air, fluorescein dye as a PC, in EtOH at room temperature under blue LED irradiation, the corresponding acylated products 414 were obtained with good yields (Scheme 189). On the basis of control experiments a plausible mechanism starts with the photoexcitation of fluorescein dye (FI) to give FI*. Meanwhile, cyclopalladation of 413 gives palladacycle B, which reacts with acyl radical A to give the Pd(III) intermediate C. Then, intermediate C can undergo a SET process to form the Pd(IV) intermediate D with simultaneous reduction of FI* to FI−˙. This radical anion FI−˙ closes the photocatalytic cycle by generating tBuO˙ and benzoate. The radical tBuO˙ reacts with α-keto acid to create the acyl radical A after decarboxylation. Intermediate D gives after reductive elimination the product and Pd(II).
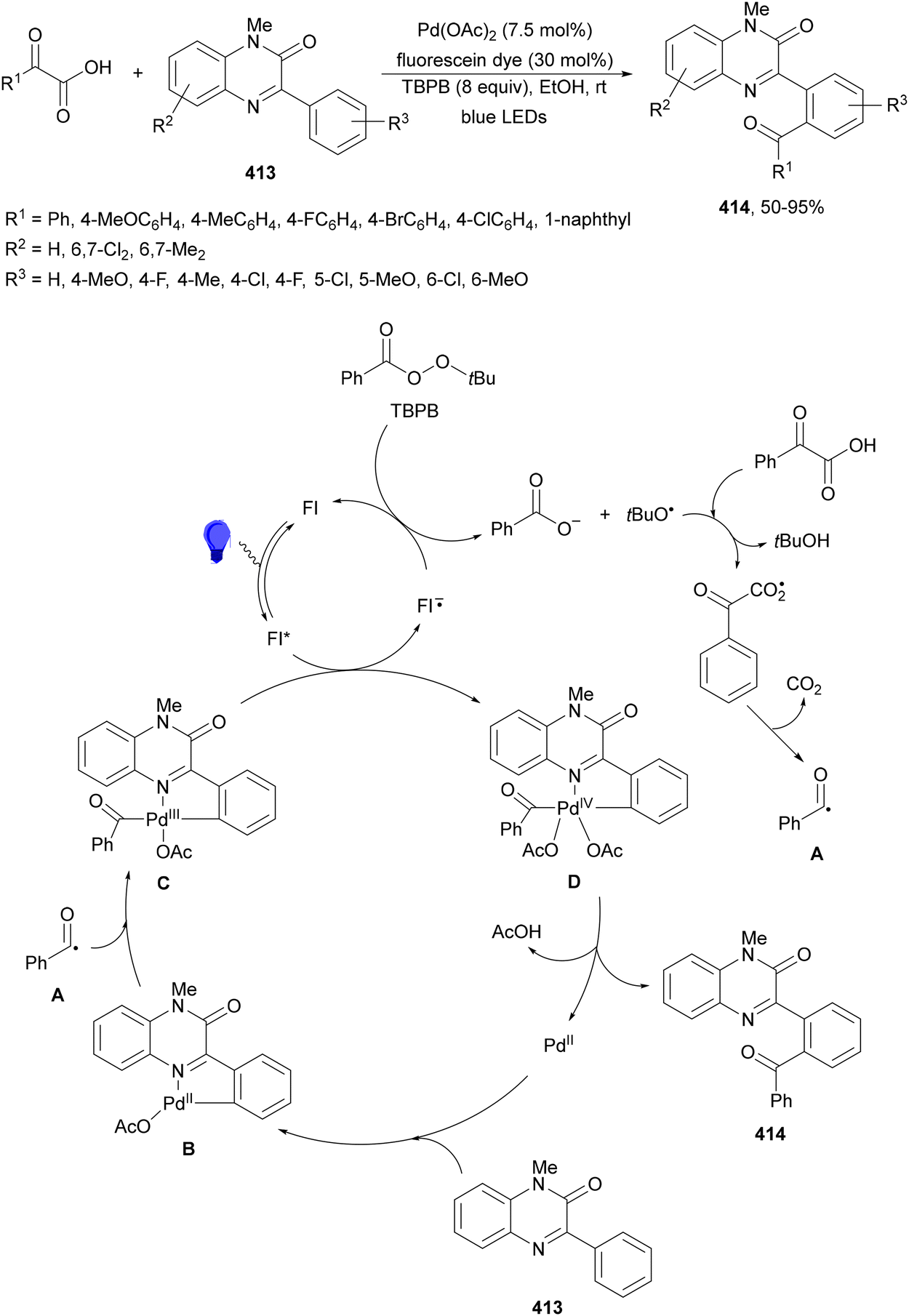 | ||
| Scheme 189 Decarboxylative acylation of N-methyl-3-arylquinoxalin-2(1H)-ones 413 with α-keto acids under Pd(OAc)2/fluorescein dye photocatalysis. | ||
Decarboxylative acylation of 2H-indazoles 387 with α-keto acids has been carried out with visible-light in the absence of PC and oxidants.344 The reaction took place in a 3:1 mixture of MeCN and HFIP as solvents under 420–425 nm irradiation at room temperature to furnish 3-acylated products 415 in moderate to good yields (Scheme 190). It has been proposed that the 2H-indazole absorbs visible-light transitioning to an excited state, which by energy transfer to the α-keto acid facilitates the homolysis to form an acyl radical after decarboxylation.
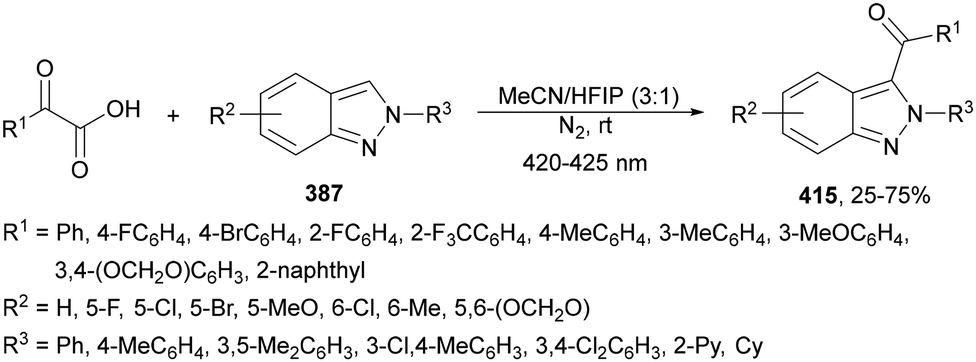 | ||
| Scheme 190 Decarboxylative acylation of 2H-indazoles 387 with α-keto acids under visible light photocatalysis. | ||
Decarboxylative hydroxyalkylation of quinolines with α-keto acids has been described by Ji and co-workers.345 In the presence of Ir complex 4 as a PC, Zn(OTf)2 and TfOH as Lewis and Brønsted acids, respectively, in aqueous MeOH under N2 and blue LED irradiation, hydroxyalkylated products 416 are obtained with, in general, good yields (Scheme 191). 2-Substituted quinolines gave 4-hydroxyalkylated derivatives 416a, whereas 4-substituted quinolines provided the corresponding 2-hydroxyalkylated ones 416b. 6-Bromoisoquinoline reacted at the 1-position with benzoylformic acid and 2-methylpyridine reacted at the 6-position to give the corresponding product in 60 and 25% yield, respectively. In the plausible reaction mechanism, the acyl radical A from the α-keto acid is formed by the oxidized PC Ir(IV). Addition of A to protonated quinoline gives radical cation B, which after protonation leads to the α-amino radical C. This radical C undergoes a spin center shift (SCS) process to afford the α-oxy radical D. The subsequent SET process of D with Ir(III)* followed by protonation provides the product.
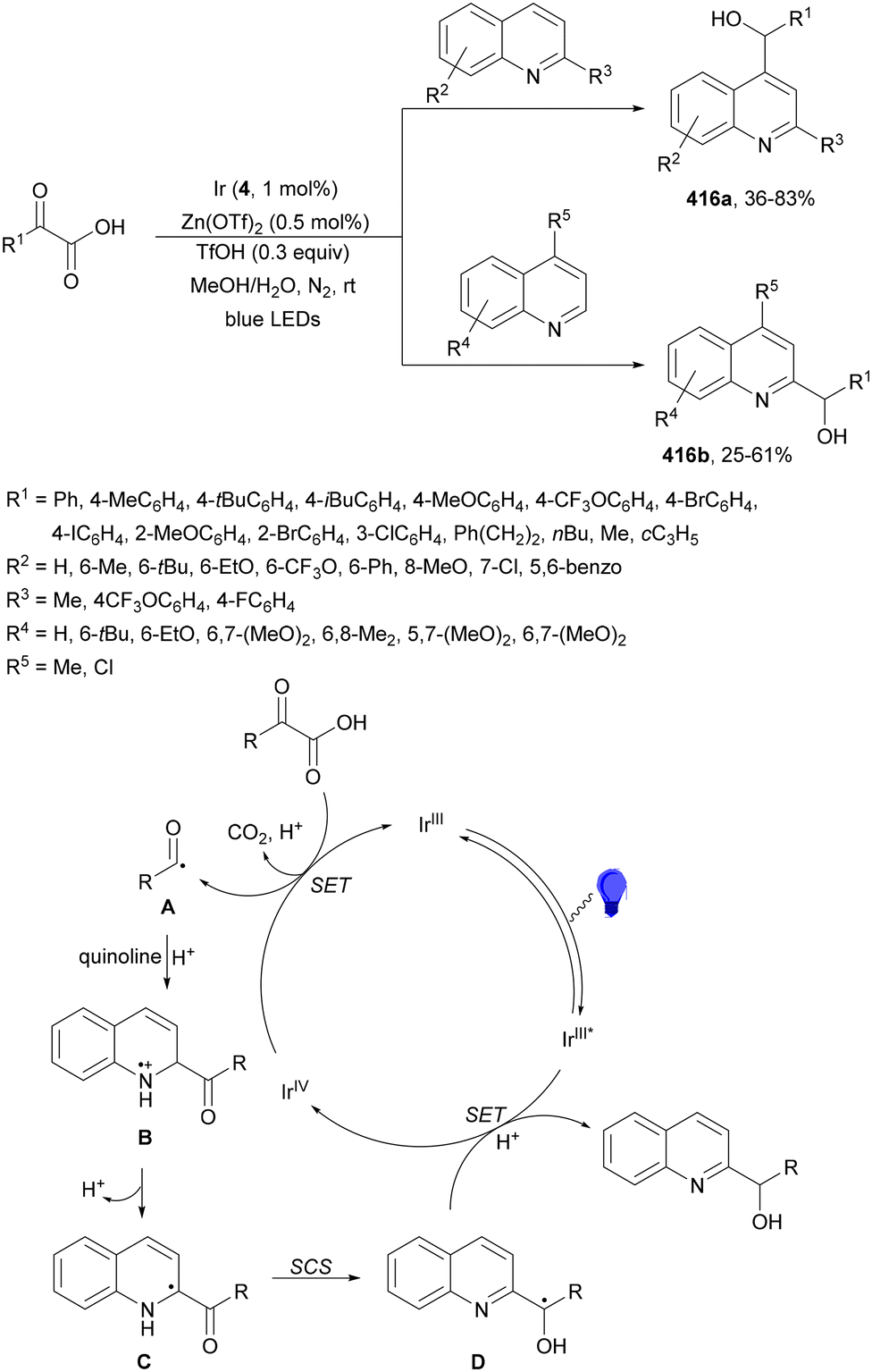 | ||
| Scheme 191 Decarboxylative hydroxyalkylation of heteroarenes with α-keto acids under Ir photocatalysis. | ||
Carbamoylation of heteroarenes with oxamic acids 15 was carried out by Landais and co-workers346 in the presence of 4CzIPN (25) as a PC and acetoxybenziodoxole (BI-OAc) as an external oxidant in DCM at room temperature under blue LED irradiation (Scheme 192). Quinolines, isoquinolines, pyridines, phenanthridine, quinoxaline, benzothiazole and benzimidazole gave the corresponding carboxamides with moderate to good yields. In the tentative mechanism, oxamic acid reacts with BI-OAc to form the hypoiodite species A, which after reaction with 4CzIPN* gives a radical anion B. Cleavage of the O–I bond in B leads to the amidocarbonyl radical C, CO2 and o-iodobenzoic acid. Radical addition of C to quinoline forms intermediate D, which after oxidation with 4CzIPN+˙ generates cationic species E and after deprotonation the product.
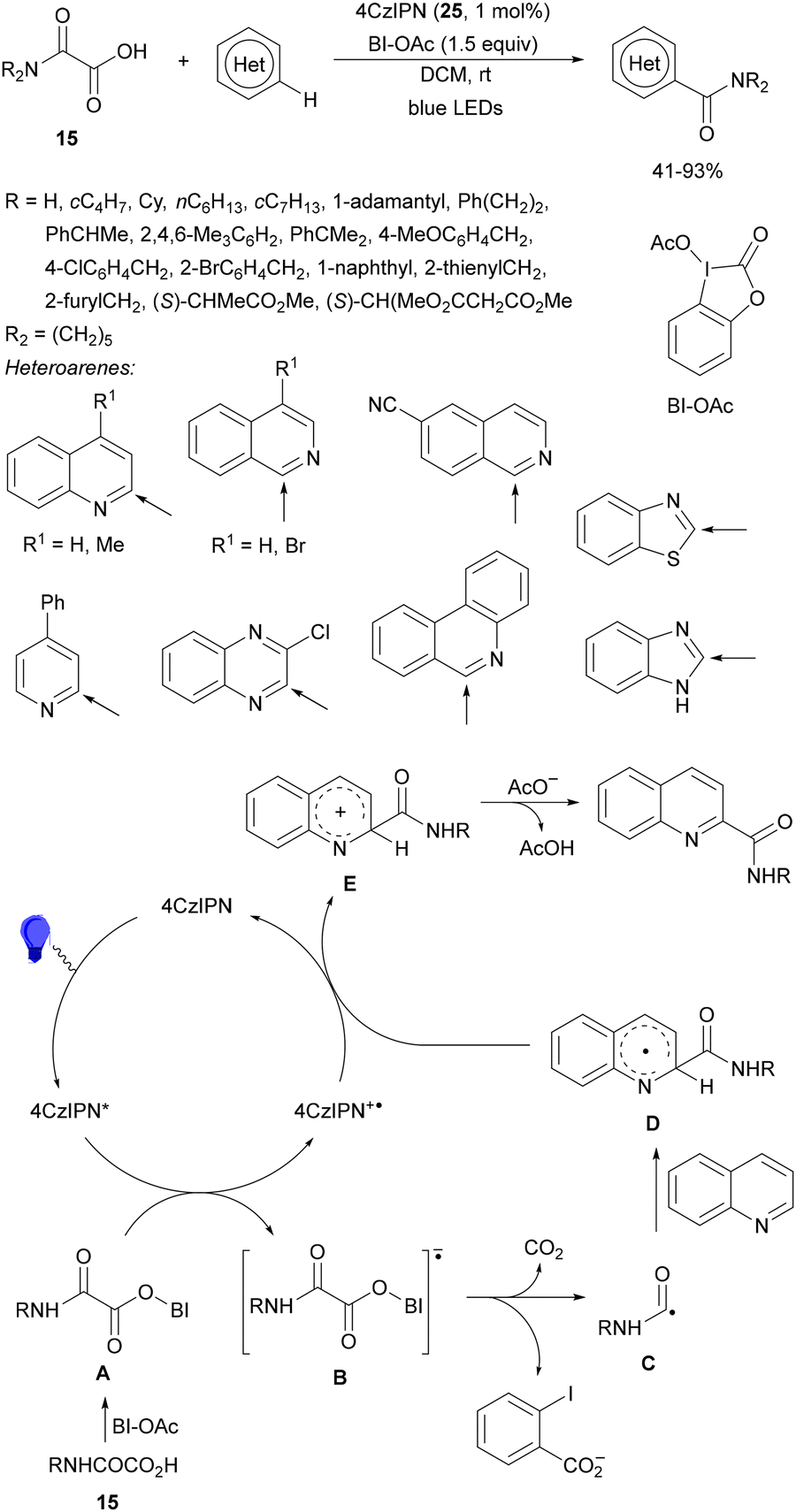 | ||
| Scheme 192 Decarboxylative carbamoylation of heteroarenes with oxamic acids 15 under 4CzIPN (25) and BI-OAc photocatalysis. | ||
Jouffroy and Kong at Merck347 employed oxamic acids or potassium oxamates for the C–H carbamoylation of heterocycles. In the presence of acridinium tetrafluoroborate 287 as a PC, K2S2O8 as an external oxidant, and TFA in aqueous DMSO at 30 °C under blue LED irradiation, heterocyclic carboxamides were obtained with good yields (Scheme 193). Quinolines, isoquinolines, pyridines, pyrimidines, quinoxaline, phthalazine and caffeine performed well in the reaction. In this case, it was proposed that the PC* oxidizes the oxamate salt to the carbamoyl radical A after decarboxylation. Radical addition of A to protonated quinoline provides radical cation B. Simultaneously PC˙− could be oxidized by persulfate to the sulfate dianion and the sulfate radical anion regenerating the PC. This radical anion SO4−˙ oxidizes B through HAT to yield the product after deprotonation.
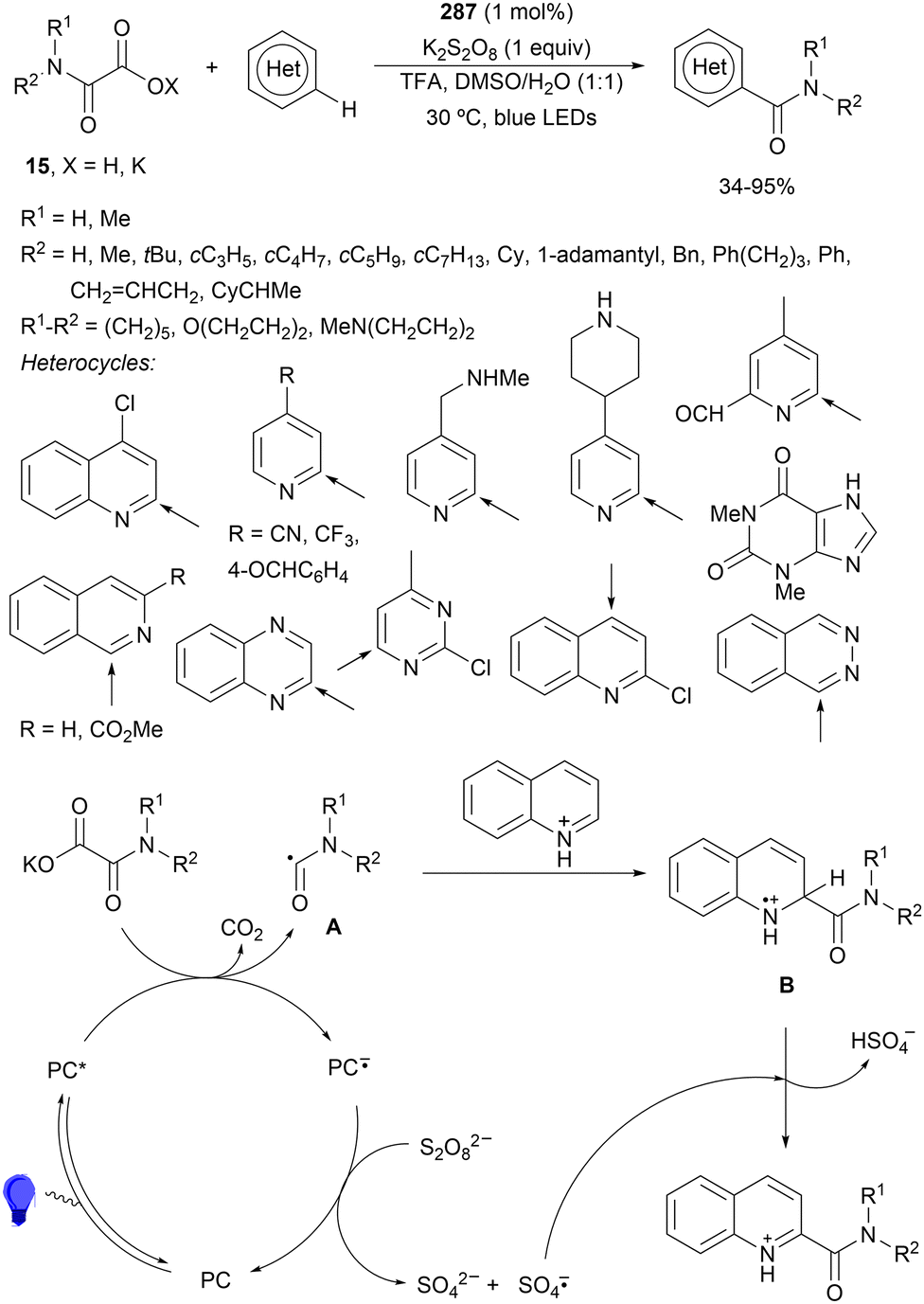 | ||
| Scheme 193 Decarboxylative carbamoylation of heteroarenes with oxamic acids 15 or oxamates under acridinium tetrafluoroborate 287 photocatalysis. | ||
Decarboxylative acylation reactions of heterocyclic compounds with α-keto acids have been carried out in the presence of oxidants such as PIFA, persulfates or air and Ir or organic photocatalysts. The corresponding acylated heterocycles were obtained with good yields and only in the case of quinolines hydroxyalkylation products were formed. General methods for carbamoylation reactions employed organic photocatalysts and external oxidants such as BI-OAc or K2S2O8 to give the corresponding heteroaromatic carboxamides.
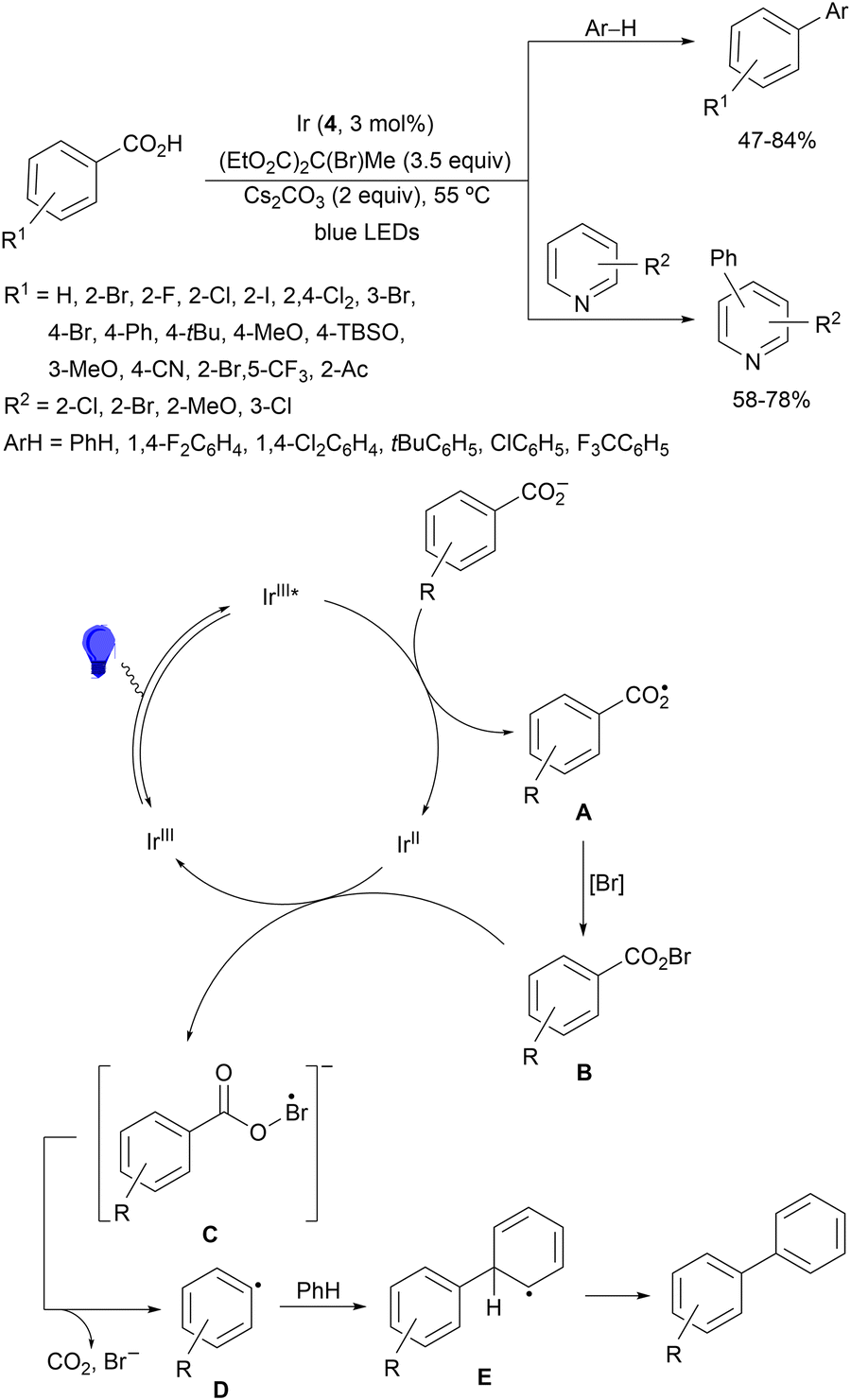 | ||
| Scheme 194 Decarboxylative arylation of arenes and heteroarenes with benzoic acids under Ir photocatalysis. | ||
Direct arylation of quinoxalin-2(1H)-ones has been achieved with acyl peroxides under visible light.349 In the absence of PCs and additives these heterocycles reacted with different dibenzoyl peroxides (BPO) in acetone, under air at room temperature to give 3-arylquinoxalin-2(1H)-ones with good yields (Scheme 195). Arylation of 2H-benzo[b]oxazin-2-one took place with 53% yield. In the proposed mechanism, BPO generates, after decarboxylation, the phenyl radical A, which adds to quinoxalinone to give a N-centered radical B. Final abstraction of the hydrogen atom by a PhCO2˙ generates the 3-phenylquinoxalin-2(1H)-one.
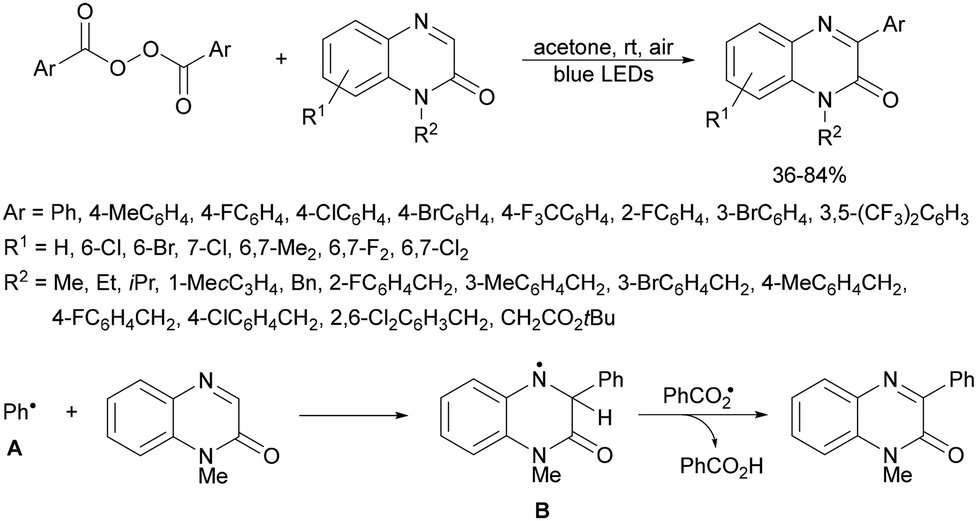 | ||
| Scheme 195 Decarboxylative arylation of quinoxalin-2(1H)-ones with aryl peroxides under visible-light irradiation. | ||
Independently, He and co-workers350 reported the same arylation of quinoxaline-2(1H)-ones with aryl peroxides under visible-light irradiation working in AcOEt instead of acetone. The corresponding 3-arylated quinoxalin-2(1H)-ones were obtained with 63–82% yields.
A general method for decarboxylative arylation of arenes and pyridines involves diethyl bromo-α-methylmalonate as a brominating agent forming the corresponding hypobromite in the presence of Ir as a PC. Acyl and aryl peroxides have been employed for the arylation of quinoxaline-2H-ones only under visible-light irradiation.
2.13. Cyclization reactions
This subject has been recently reviewed351–353 and therefore it will be not considered in this survey.3. Carbon-heteroatom bond forming reactions
In this section photodecarboxylative heterofunctionalization of carboxylic acids and derivatives will be considered including C–Hal, C–O, C–S, C–N, C–P, C–B and C–Si bond formation. Recent reviews until 2021354–356 cover partially this topic, and thus new achievements made in the last four years will be considered.3.1. Carbon–halogen bond forming reactions
Decarboxylative halogenations have been achieved by photochemical methods. Organofluorine compounds are very important in medicinal chemistry and 18F-labeled molecules act as contrast agents for positron emission tomography (PET). Ir complex,357 [Mes-AcrMe]ClO4 (301)358 and carbonitride359 have been employed as PCs in the presence of Selectfluor for decarboxylative fluorination of aliphatic carboxylic acids. In the case of NHPI esters 69, triethylamine trihydrofluoride (Et3N·3HF) or K18F under Ir photocatalysis has been employed.360In recent studies, carboxylic acids have been transformed into organofluorides via ligand-to-metal charge transfer (LMCT).54,55 Ritter and co-workers361 performed decarboxylative fluorination of benzoic acids in the presence of TBAF·(tBuOH)4 complex as a fluoride source, Cu(OTf)2 and Cu(MeCN)4BF4 as PCs in MeCN at 35 °C under purple LED irradiation (Scheme 196). Electron-neutral and rich benzoic acids and some heterocyclic compounds reacted smoothly to give the corresponding aryl fluorides with good yields. In the proposed mechanism, photoactive copper(II) benzoate A generates by photoinduced LMCT carboxy radical B and Cu(I), which by homolysis releases CO2 and forms the aryl radical C. This radical C is captured by Cu(II) salts to afford the arylCu(II) fluoride D, which is oxidized to ArCu(III)F (E). Final reductive elimination gives the aryl fluoride.
 | ||
| Scheme 196 Decarboxylative fluorination of aromatic and heteroaromatic carboxylic acids with TBAF·(tBuOH)4 under Cu photocatalysis. | ||
MacMillan and co-workers362,363 performed a general approach to obtain aryl halides by decarboxylative halogenation of (heteroaryl)carboxylic acids via a LMCT54,55 mechanism. In this case, Cu(MeCN)4BF4 (20 mol%) was used as a PC and 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT, 417) or dicumyl peroxide as an external oxidant in MeCN under 365 nm LED irradiation. When 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 418) or CCl3Br was used, the corresponding brominated products were obtained in 41–85% yields (Scheme 197). In the presence of ZnCl2 heteroaryl chlorides were obtained with 41–>99% yield and with N-iodosuccinimide (NIS) heteroaryl iodides were formed with 56–89% yields. Fluorodecarboxylation reaction was carried out with 2 equivalents of NFTPT (417) as a source of fluoride and in some cases CsF was added with 3 equivalents of Cu(MeCN)4BF4 to give the corresponding fluorides in 41–81% yields.
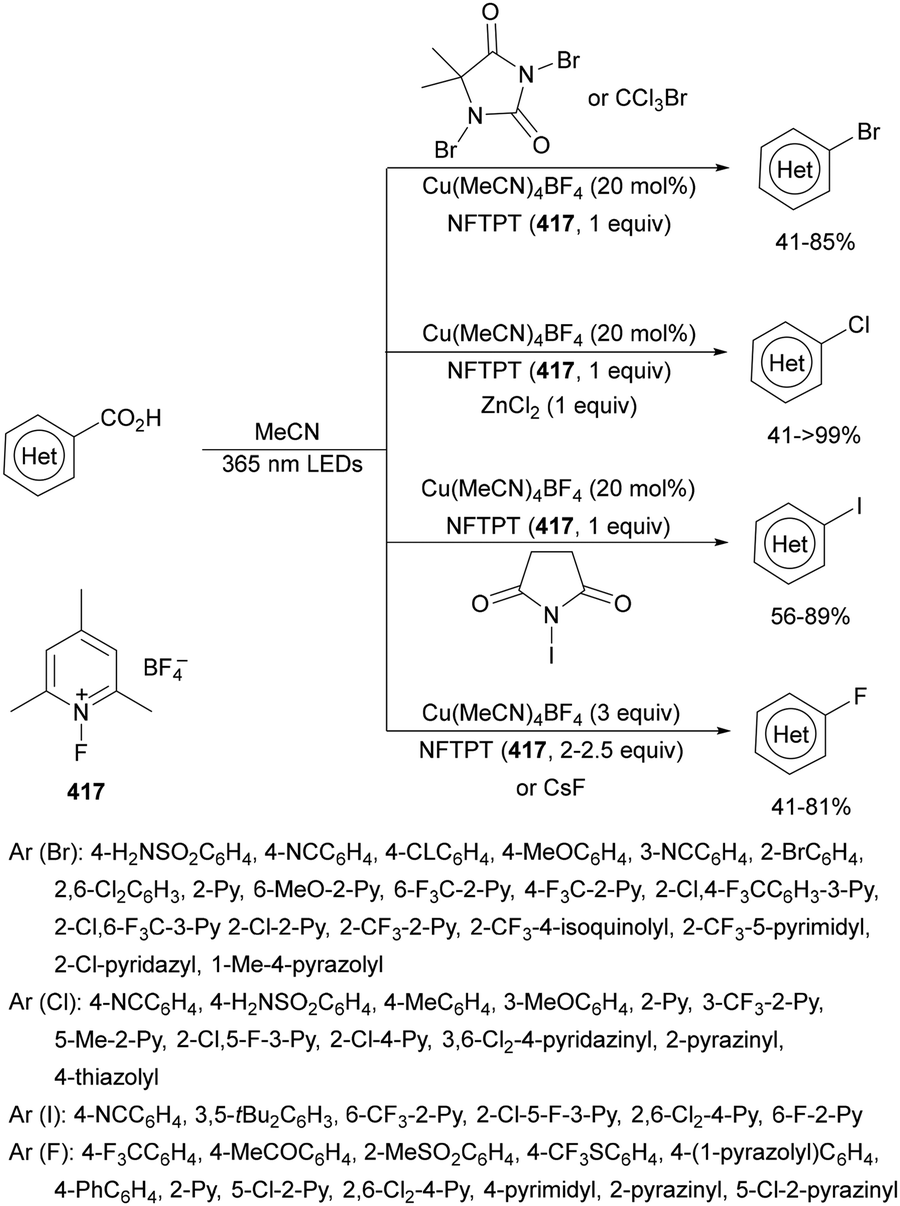 | ||
| Scheme 197 Decarboxylative halogenation of aromatic and heteroaromatic carboxylic acids in the presence of NFTPT (417) under Cu photocatalysis. | ||
Decarboxylative fluorination of aliphatic carboxylic acids has been achieved with Fe(OAc)2 as a PC by a LMCT54,55 mechanism.364 In the presence of bipyridine ligand 185, 2,6-lutidine as a base and Selectfluor in aqueous MeCN at room temperature under N2 and blue LED irradiation, the corresponding alkyl fluorides were isolated in up to 92% yield (Scheme 198). In the proposed mechanism, firstly the iron(III) carboxylate A is formed by oxidation with Selectfluor. This complex A undergoes photoexcitation via LMCT to produce the iron(II) species B and the carboxy radical C. Upon decarboxylation of C an alkyl radical D is formed, which is fluorinated by Selectfluor to afford the product. The iron(II) species B was oxidized by Selectfluor or its radical cation to give the Fe(III) species E. Coordination of E with another alkyl carboxylic acid in the presence of base generates A.
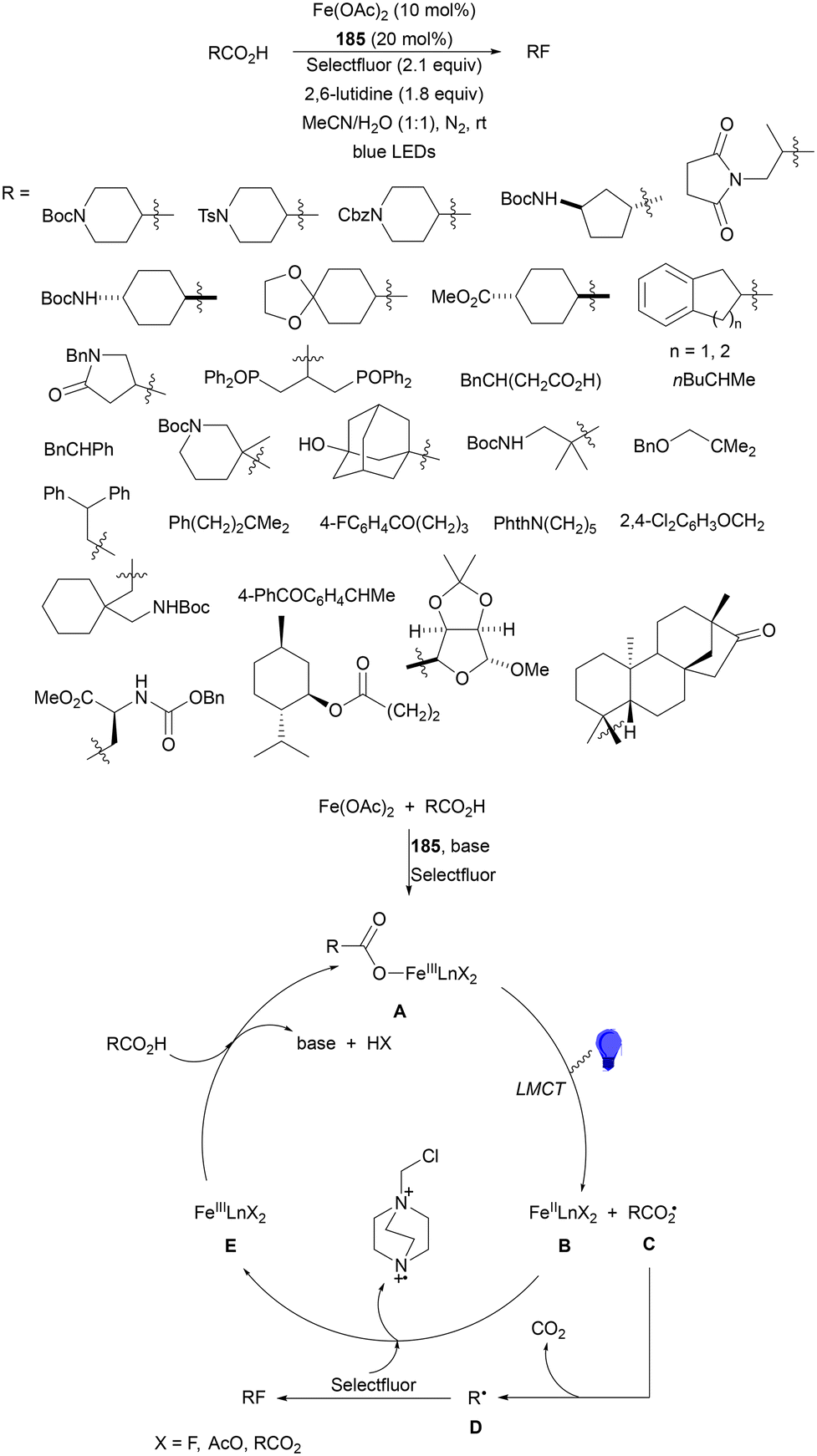 | ||
| Scheme 198 Decarboxylative fluorination of aliphatic carboxylic acids with Selectfluor under Fe photocatalysis. | ||
Iron salts have been recently used also by Hu and co-workers365 as PCs for decarboxylative chlorination, bromination and iodination of aliphatic carboxylic acids. Under similar reaction conditions to that for fluorination using 2,4,6-collidine as a base and MeCN under N2 and NCS, N-bromosaccharine and NIS as source of halogens (Scheme 199), primary, secondary and tertiary carboxylic acids were transformed into alkyl chlorides, bromides and iodides with 27–94% yields.
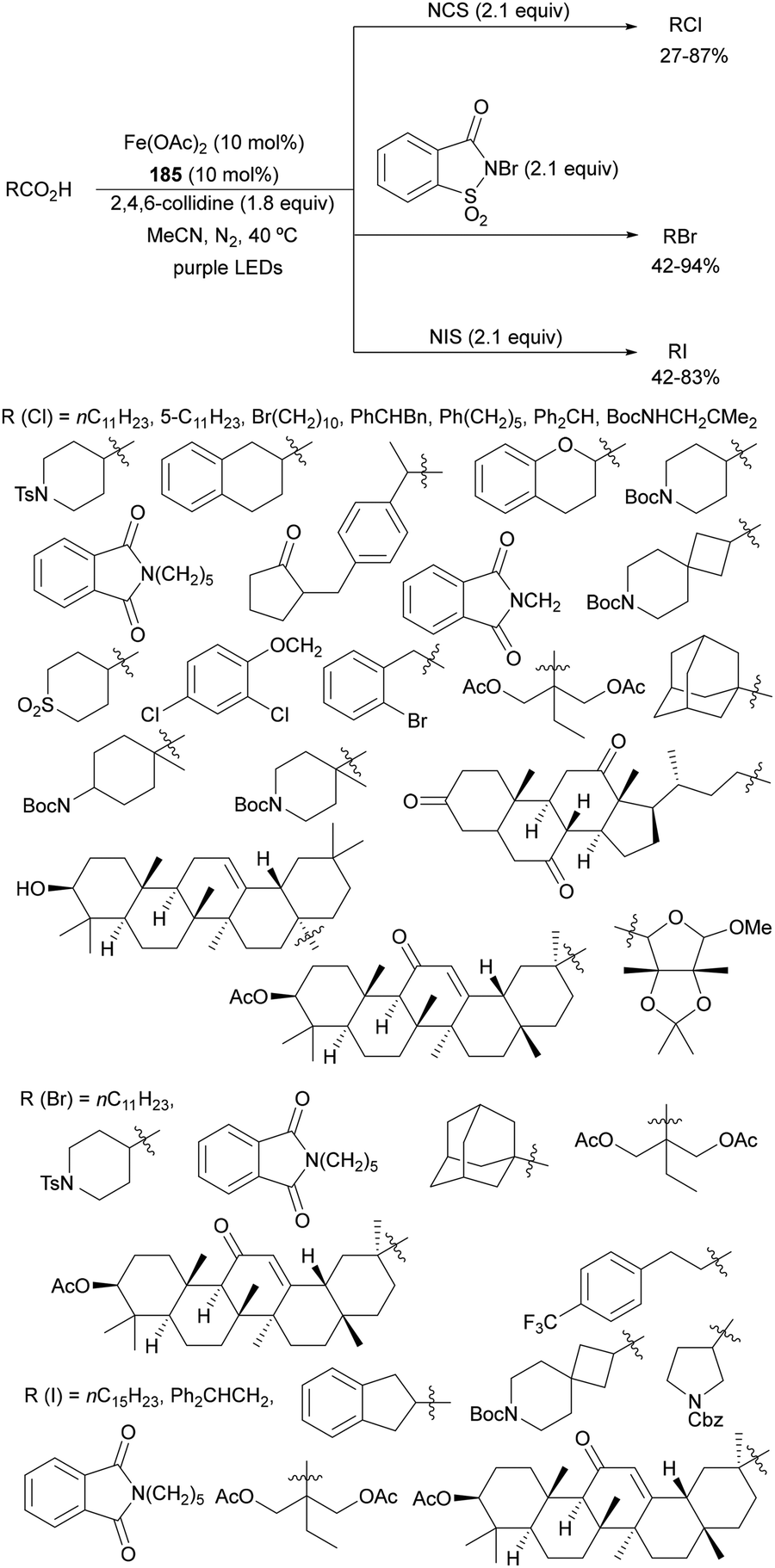 | ||
| Scheme 199 Decarboxylative chlorination, bromination and iodination of carboxylic acids with N-haloamides under Fe photocatalysis. | ||
A general protocol for the photohalodecarboxylation of aliphatic carboxylic acids by a LMCT54,55 mechanism with CeCl3 has been described by Sun, Jin and co-workers.366 Primary, secondary and tertiary carboxylic acids were transformed into the corresponding chloro-, bromo- and iodoalkanes using tBuONa as a base, H2O as solvent at room temperature under air and blue LED irradiation (Scheme 200). As halogenating reagents, trichloroisocyanuric acid (TCCA), NBS and NIS, respectively, were employed. Based on experimental studies, a proposed mechanism is depicted in Scheme 200. Initially, tBuONa (O2−˙ or HO2−) abstracts a proton from the acid and the resulting carboxylate ion reacts with Ce(III) to produce complex A followed by oxidation with O2 (or HO2˙) to generate the Ce(IV) species B. This species B undergoes a phototriggered LMCT process releasing the alkyl radical C, after decarboxylation, and Ce(III). Subsequent halogen atom transfer from the N-haloamide produces the product.
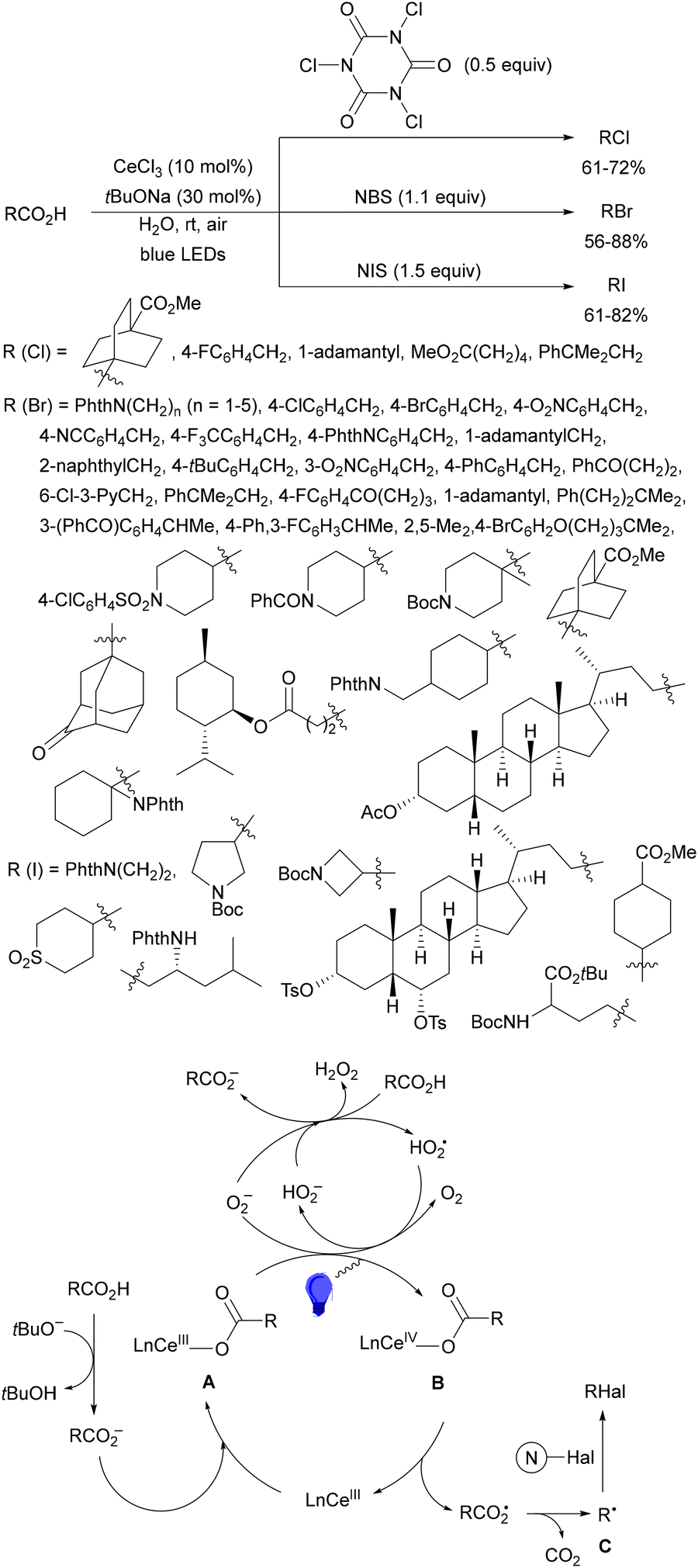 | ||
| Scheme 200 Decarboxylative halogenation of aliphatic carboxylic acids under CeCl3 photocatalysis. | ||
Photoinduced LMCT decarboxylative bromination of aromatic carboxylic acids has been carried out by Xia and co-workers,200 using 2 equivalents of Cu(OTf)2. This process took place in the presence of Li2CO3 as a base, in MeCN at room temperature under N2 and 390 nm LED irradiation to give the corresponding aryl bromides in 63–75% yields.
A covalent organic framework (COF)-based PC has been employed for the decarboxylative fluorination of aliphatic carboxylic acids by Banerjee, Maj and co-workers.367 This anthraquinone-based COF PC TpACl exhibits high durability and can be reused multiple times without significant activity loss (>80% after eight cycles). This fluorination was carried out with Selectfluor in the presence of 2,6-lutidine as a base in aqueous MeCN (1:1) at 40 °C under N2 and purple LED irradiation to give alkyl fluorides with 39–96% yields. Gram-scale fluorination of ketoprofen was performed under batch and flow reaction conditions.
Enantiopure fluoropiperidine 420 has been prepared by decarboxylative fluorination of carboxylic acid 419 using photoredox conditions.368 Starting acid 419 was prepared on a >400 g scale by biocatalytic desymmetrization369 of N-Cbz diethyl 3,5-piperidinedicarboxylate using the Candida antartica lipase A Novocar ADC with 94.3% ee. Subsequent decarboxylative fluorination of 419 with Ir complex 4 as a PC in the presence of Selectfluor under blue LED irradiation using flow conditions gave diastereoselectively product 420 in 52% yield and 99.3% ee, after chiral SFC purification (Scheme 201).
 | ||
| Scheme 201 Decarboxylative fluorination of carboxylic acid 419 with Selectfluor under Ir photocatalysis. | ||
Molander and co-workers370 reported a metal-free decarboxylative chlorination of aliphatic carboxylic acids by their activation with (diacetoxyiodo)benzene (PIDA). This process used 1,2-dichloroethane (DCE) as a halogen source, with the halogen-atom transfer (XAT) being the key step. In this case, 4CzIPN (25) was used as an organophotocatalyst and DCE also as solvent at room temperature under Ar and blue LED irradiation (Scheme 202). Primary, secondary and tertiary carboxylic acids were transformed into alkyl chlorides in modest to good yields. In the proposed mechanism photoexcited 4CzIPN* reacts with (diacetoxyiodo) complex A, generated in situ, to give by a SET process the carboxy radical B. Decarboxylation of B affords the alkyl radical C. This radical reacts with DCE via a XAT to form the alkyl chloride and the chlorinated radical D. This radical evolves to give ethyl chloride or 1,4-dichlorobutane. Radical D can give ethylene and, by a SET process, the anion chloride, which abstracts a hydrogen atom to give HCl and the carboxylate ion.
 | ||
| Scheme 202 Decarboxylative chlorination of aliphatic carboxylic acids mediated by XAT under 4CzIPN (25) photocatalysis. | ||
Cheng371 and Shang372 groups described independently decarboxylative iodination under visible-light of aliphatic NHPI esters 69 with NHC/NaI for secondary alkyl iodides and PPh3/LiI for primary alkyl iodides, respectively. Noble and Aggarwal373 reported chlorination and bromination of primary, secondary and tertiary aliphatic NHPI esters 69 using LiCl and LiBr as halogen sources (Scheme 203). Chlorination reactions were carried out in the presence of Ir complex 4, CuCl2 and 2,9-dimethyl-1,10-phenanthroline (dmp) as a ligand in MeCN at 30 °C under N2 and blue LED irradiation to provide alkyl chlorides with 33–92% yields, whereas for bromination reactions 4CzIPN was used as a PC giving alkyl bromides with modest to good yields (26–94%). In the proposed mechanism for chlorodecarboxylation reactions the NHPI ester is reduced by Ir(II) to form the alkyl radical A, CO2 and PhthN−. This radical A reacts with CuCl2 and LiCl to give the alkyl chloride and CuCl via reductive elimination of an alkylCu(III)X2 species or through an outer-sphere pathway involving atom transfer. In the case of bromodecarboxylation reactions, 4CzIPN* oxidizes Br− to Br˙. Subsequently, the alkyl radical is formed by reduction of NHPI ester with 4CzIPN−.·Trapping of radical A could occur by radical–radical coupling of A with Br˙. Alternatively, Br˙ can be stabilized by Br− generating dibromide radical Br2−˙, which generates the alkyl bromide. A third possible pathway involves dimerization of two Br˙ to form Br2, which is unlikely because substrates with CC bonds are not affected.
 | ||
| Scheme 203 Decarboxylative chlorination and bromination of alkyl NHPI esters 69 with LiCl and LIBr respectively under photocatalysis. | ||
A general method for decarboxylative halogenation of aliphatic, aromatic and heteroaromatic carboxylic acids is based on a metal photocatalyzed LMCT mechanism.
3.2. Carbon–oxygen bond-forming reactions
Decarboxylative oxygenation, etherification and acyloxylation of carboxylic acids or photoactive esters under photoredox conditions will be considered. The corresponding products can be further converted into hydroxylated compounds. If oxamic acids were used, the corresponding urethanes were obtained.Huang, Xiao and co-workers380 employed more recently a non-heme manganese 421 as a PC for decarboxylative oxygenation of carboxylic acids with oxygen in MeCN at 45 °C under blue LED irradiation (Scheme 204). Primary and secondary aliphatic carboxylic acids including pharmaceutical compounds were transformed into aldehydes and ketones, respectively, with modest to good yields. A range of amino acids and a dipeptide were oxidized selectively to amide products in 48–67% yields. Based on experimental studies, a plausible mechanism was proposed starting from the formation of intermediate A by reaction of the catalyst and phenylacetic acid. This complex A is oxidized by O2 under blue LED irradiation to give species B. The superoxide radical attacks intramolecularly the benzylic carbon releasing CO2 and forming the Mn(II)–peroxide C. Subsequent decomposition of C produces benzaldehyde and a Mn(II)–OH species D, which reacts with phenylacetic acid regenerating A.
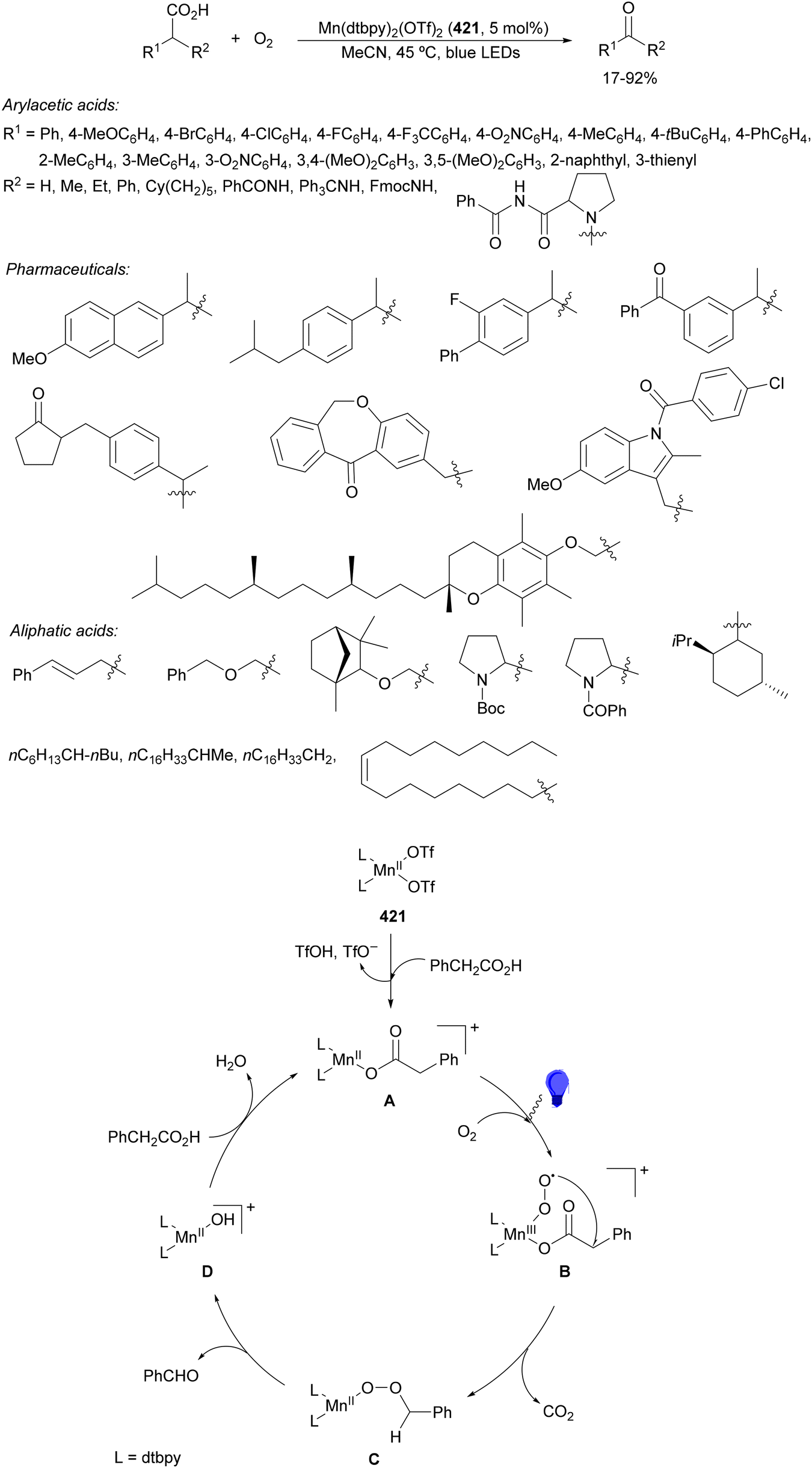 | ||
| Scheme 204 Decarboxylative oxygenation of carboxylic acids with oxygen under Mn 421 photocatalysis. | ||
Chemodivergent381 decarboxylative oxygenation of carboxylic acids using CeCl3 as a PC and O2 as the oxidant was further reported by Huang, Xiao and co-workers.382 By changing the base employed, this process gave hydroperoxides 422 with NaOAc or carbonyl compounds with 2,6-lutidine (Scheme 205). The proposed simplified mechanism suggests that Ce(III) reacts with a carboxylic acid forming complex A, which is oxidized by O2 to afford a Ce(IV) superoxide species B, under blue LED irradiation. This species B undergoes decarboxylation to form the Ce(III) peroxide species C, which reacts with another molecule of acid to afford the alkyl hydroperoxide 422, stable in the presence of NaOAc. However, this hydroperoxide is transformed into the corresponding carbonyl compound in the presence of 2,6-lutidine as a base under blue LED irradiation.
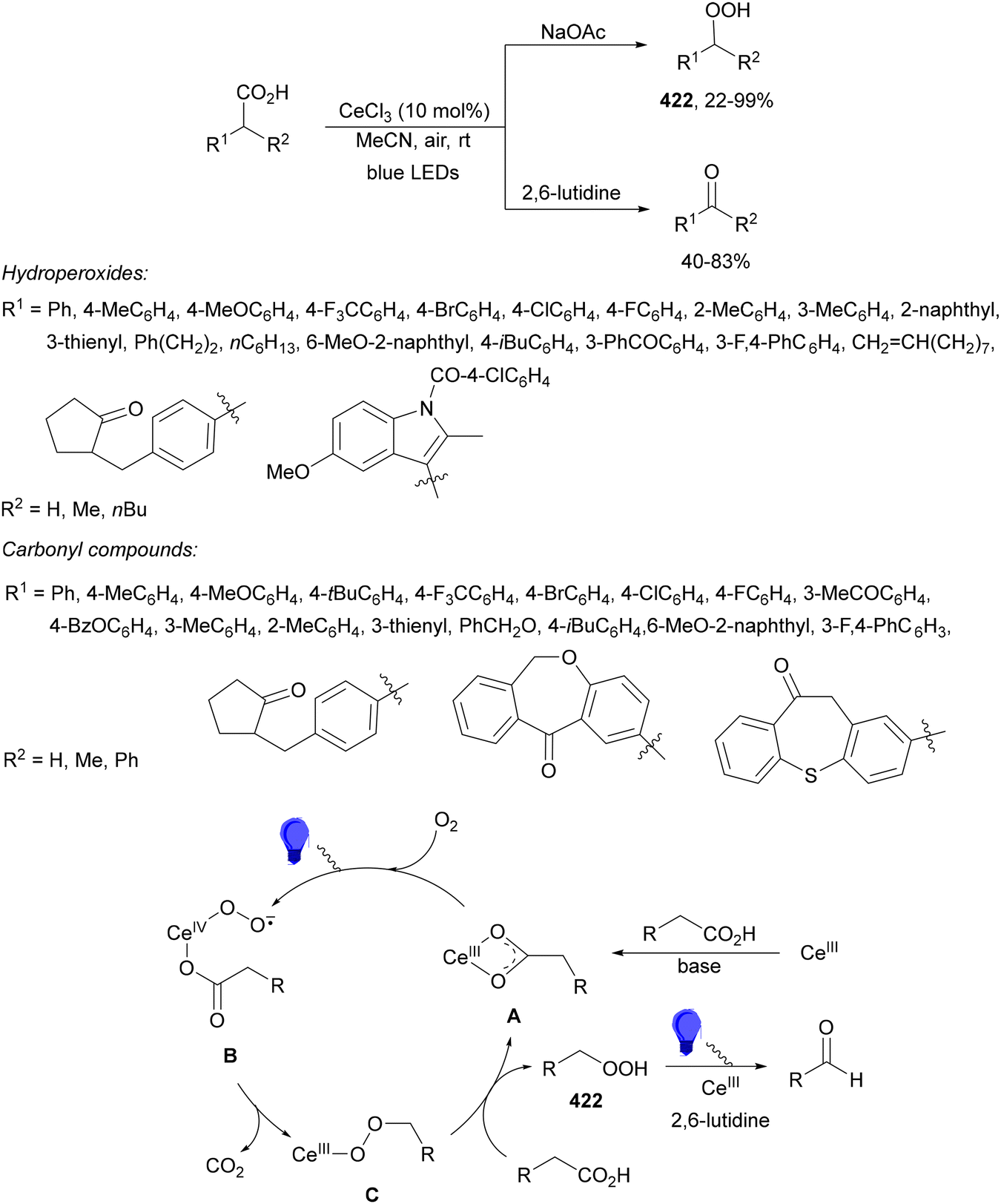 | ||
| Scheme 205 Decarboxylative oxygenation of carboxylic acids under CeCl3 photocatalysis. | ||
Iron-catalyzed photoinduced oxygenative decarboxylation has been independently reported by Guo and Xia383 and Guérinot384 groups via LMCT54,55 processes. In the first case, a decarboxylative ring-opening of cyclic tertiary carboxylic acids 423 gave 1,n-dicarbonyl compounds 424 through homolytic C–C bond cleavage (Scheme 206). By employing Fe(acac)3, DABCO as a base in MeCN at 35 °C under air and 390 nm LED irradiation this protocol was also applied to acyclic primary, secondary and tertiary carboxylic acids to obtain the corresponding aldehydes and ketones with 49–99% yields. In the proposed reaction mechanism for cyclic carboxylic acids, the coordination to iron gives the carboxylate-iron complex A. Photoexcitation of A gives the reduced Fe(II) complex and the aryloxy radical B. Subsequent decarboxylation of B gives the tertiary radical C, whereas via a SET process between dioxygen and Fe(acac)2 the Fe(III) catalyst is regenerated. Intermediate C would be trapped by oxygen to give the peroxyl radical D. This species D undergoes an intramolecular HAT to give the β-carbon radical E, which releases the dioxetane F with the aid of the superoxide radical anion. Dioxetane F undergoes thermal cleavage yielding the dicarbonyl product.
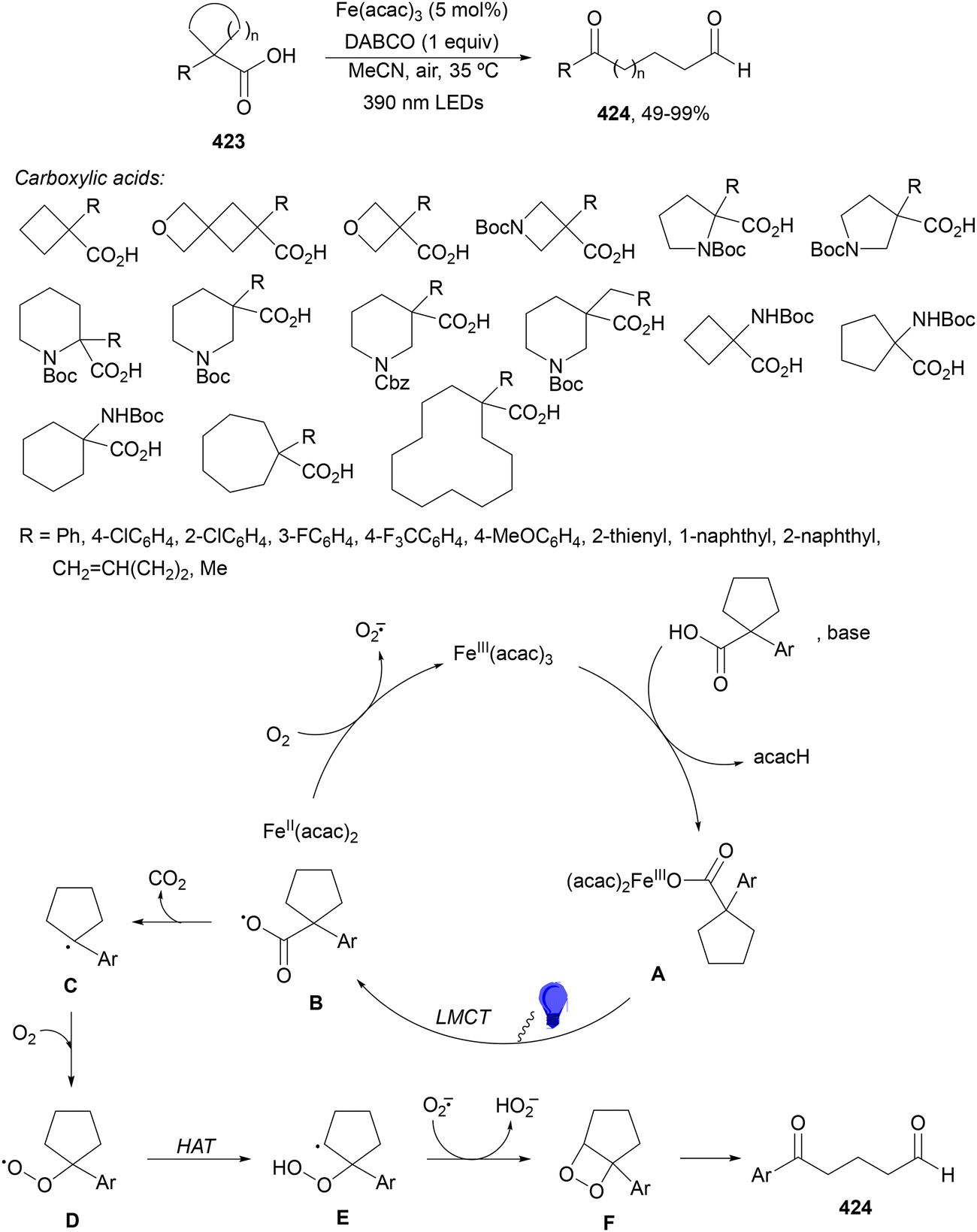 | ||
| Scheme 206 Decarboxylative ring-opening oxygenation of cyclic carboxylic acids 423 under Fe(acac)3 photocatalysis. | ||
In the case of Guérinot and co-workers,384 Fe(NH4)2(SO4)2·6H2O (5 mol%) as a PC and di-(2-picolyl)amine (10 mol%) as a ligand in MeCN at room temperature under air and blue LED irradiation were used for decarboxylative oxygenation. Arylacetic acid was transformed into aromatic aldehydes in 80–94% yields and α-AAs into amides in 77–80% yields. Recently, an iron-catalyzed decarboxylative oxygenation of a broad range of aliphatic carboxylic acids has been performed using TEMPO as an oxidant.385 In the presence of Fe(OTf)3 as a PC and N,N-bis(pyridyn-2-ylmethyl)ethane-1,2-diamine (425) as a ligand, in MeCN at room temperature under air and blue LED irradiation, the corresponding TEMPO adducts 426 were obtained in 53–99% yields (Scheme 207). This protocol was employed for the late-stage functionalization of ibuprofen derivatives in 78–99% yields as well as the sweetener Neotame, cholic acid and the plant hormone indole-3-butyric acid in 24–56% yields. These TEMPO derived products 426 were further transformed into ketones, by treatment with MCPBA, and into ether derivatives. Different kinetic, electrochemical, EPR, UV/vis, HRMS and DFT studies revealed the possible mechanism depicted in Scheme 207. Complex A undergoes ligand substitution with the carboxylate ion to form the Fe(III) intermediate B. Photoexcitation of B induces LMCT54,55 to give C, which after decarboxylation generates the alkyl radical D. Reaction of this radical D with TEMPO forms the product 426 and the complex E, which is oxidized by a SET process regenerating the catalyst. The same group386 recently reported the same process using FeBr3 (2.5 mol%) as a PC to give the corresponding arylacetic acid TEMPO adducts 426 with high yields (70–99%). The reaction between phenylacetic acid and TEMPO was scaled up to 5 mmol with 76% yield. Other primary, secondary and tertiary aliphatic carboxylic acids were transformed into TEMPO adducts 426 in modest to high yields (16–92%).
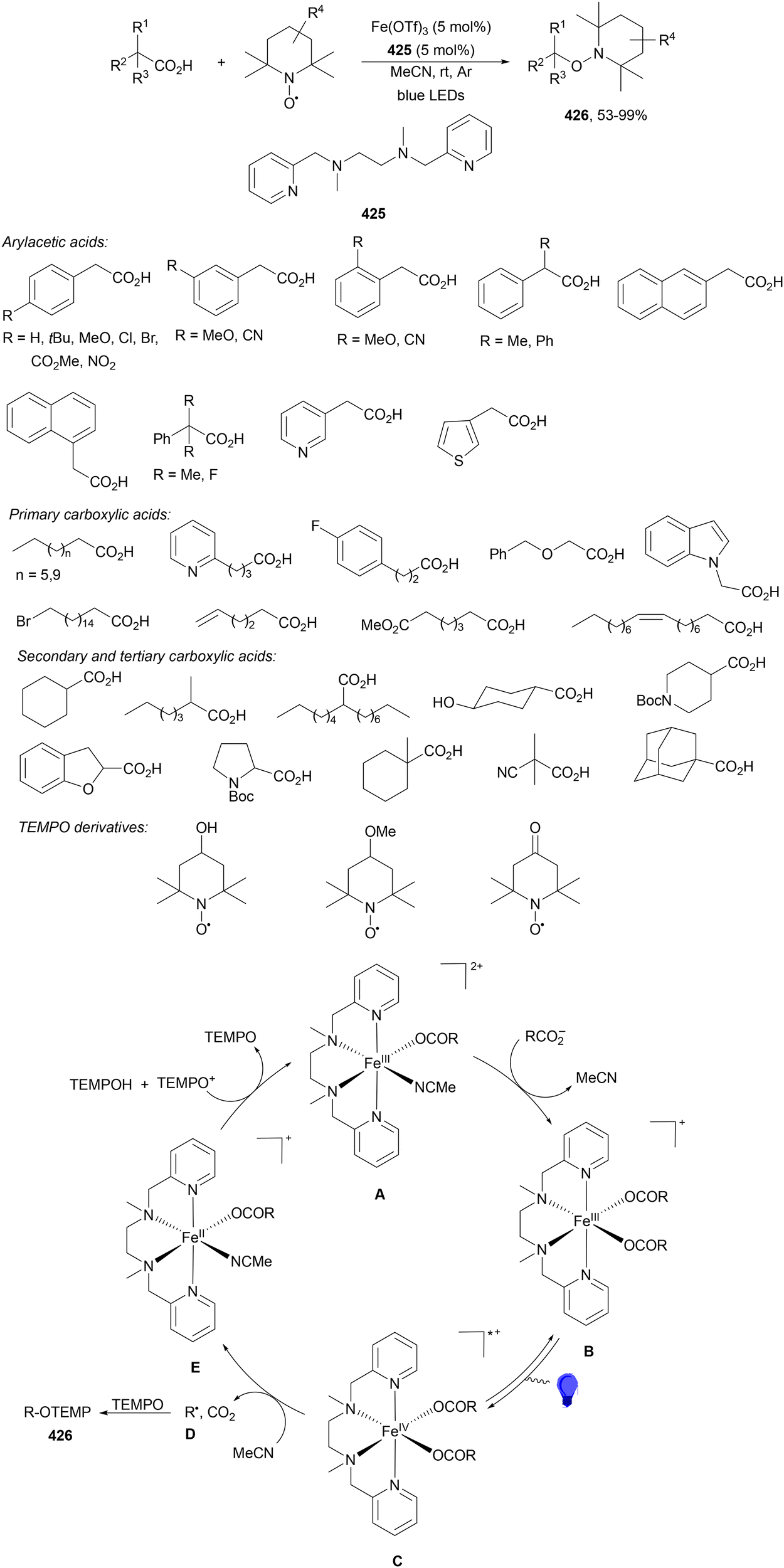 | ||
| Scheme 207 Decarboxylative oxygenation of aliphatic carboxylic acids with TEMPO under Fe(OTf)3 photocatalysis. | ||
Wang and co-workers387 have performed a high yield decarboxylative oxygenation of 2-phenylpropionic acid to acetophenone using (Mes-Acr-Me)ClO4301 as a PC in oxygen-liquid flow. This process was carried out in a photomicroreactor in continuous gas-liquid flow and represents an efficient, green and mild industrial production of ketones from carboxylic acids.
 | ||
| Scheme 208 Decarboxylative etherification of peptides with BI-OAc as an oxidant under Ru(bpy)3Cl2 photocatalysis. | ||
Terret and coworkers389 reported a similar etherification of carboxylic acids in the presence of hypervalent iodine reagent 428 and Ru(dtbbpy)3(PF6)2429 as a PC in a 2:1 mixture of DCE/HFIP at room temperature under N2 and blue LED irradiation (Scheme 209). The corresponding dialkyl ethers 430 were isolated with good yields by intermediacy of transient carbocations from carboxylic acid substrates. In the proposed mechanism, the carboxylic acid is activated by the iodine(III) oxidant 428 to form the iodo-carboxylate species A with loss of AcOH. Photoexcited Ru(II)* species reduces Avia a SET process to generate the radical B after formation of CO2 and the aryl iodide C. The highly oxidizing Ru(III) species would oxidize radical B to carbocation D and regenerate Ru(II). Finally, carbocation D reacts with an alcohol to form the C–O bond.
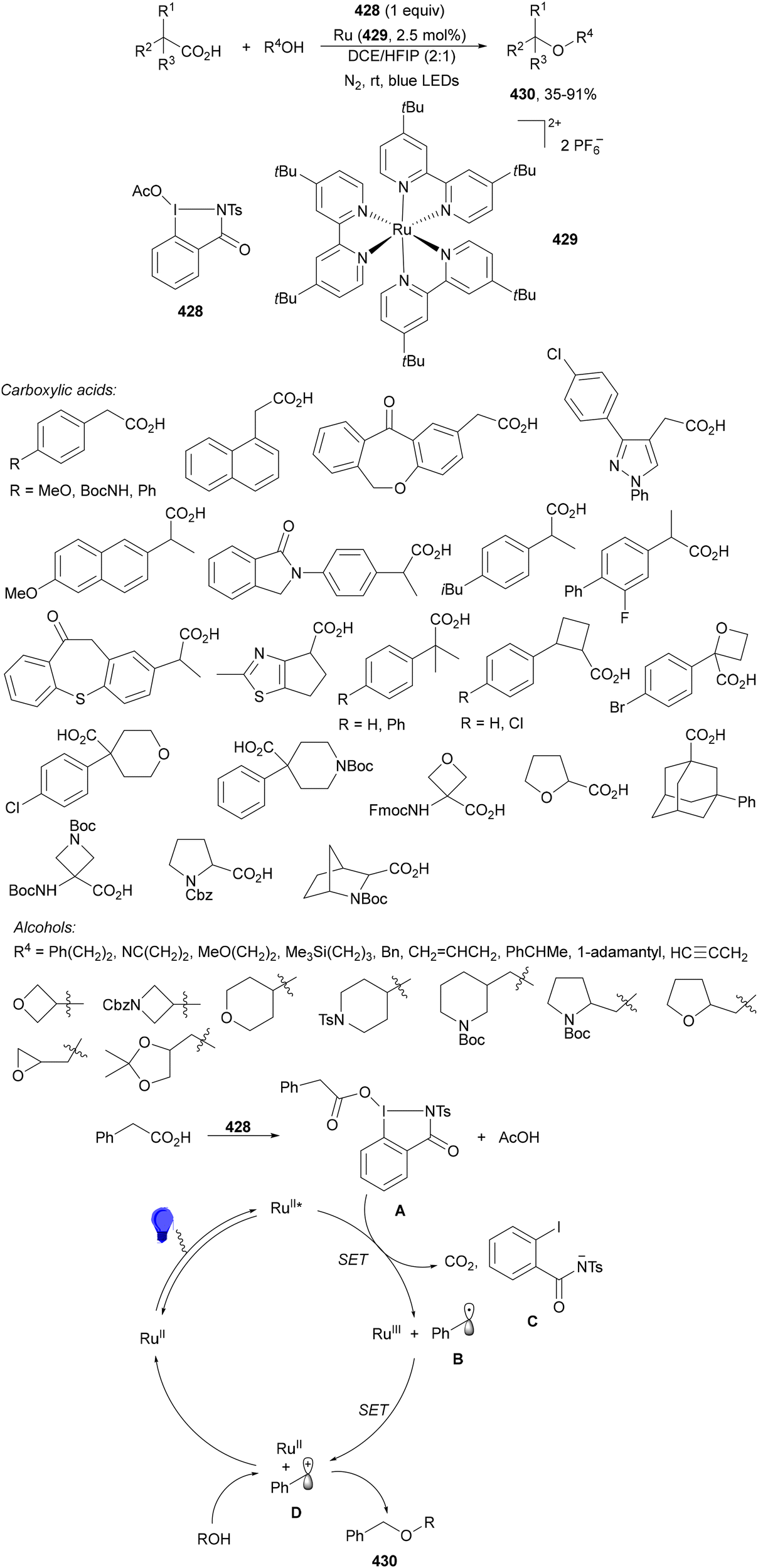 | ||
| Scheme 209 Decarboxylative etherification of carboxylic acids with alcohols and 428 as an oxidant under Ru 429 photocatalysis. | ||
Under similar reaction conditions previously described for the iron-mediated decarboxylative alkylation of arenes with carboxylic acids (Scheme 160),295 FeCl3 or Fe(OTf)3 (3 equiv.) and K2HPO4 as a base in DCM at room temperature under air and blue LED irradiation were employed for the etherification of arylacetic acids with alcohols, which took place with 42–97% yields.
Etherification of NHPI esters under decarboxylation was described in 2018 by Hu and co-workers390 in the presence of a dual Ir complex 52 (1 mol%) and Cu(OTf)2·C6H6 (10 mol%). Aliphatic carboxylic acid derived NHPI esters 69 and phenols gave the corresponding ethers in 49–94% yields. Tertiary benzylic and α-heteroatom aliphatic carboxylic acid derived NHPI esters have been etherified with aliphatic alcohols in 25–96% yields using phenothiazine (10 mol%) as a PC and LiBF4 (10 mol%) in MeCN at room temperature under blue LED irradiation.391 Decarboxylative hydroxylation was also carried out in the presence of water. Li, Guan and co-workers392 employed PPh3/NaI as a photoredox catalyst, and [Cu(BPhen)]Br as a catalyst and BTMG as a base in dioxane at room temperature under blue LED irradiation for the reaction of alkyl NHPI esters with phenols (Scheme 210). The corresponding aryl alkyl ethers 430 were obtained with high yields (≥90%). Theoretical and experimental studies support the formation of the ester–NaI–PPh3 acceptor–donor complex, which is converted to a triplet excited species and after irradiation a subsequent decarboxylation generates the alkyl radical. In the Cu(I)–Cu(II)–Cu(I) catalytic cycle, the radical is captured, which by SET, base-mediated proton transfer and reductive elimination affords the C–O cross-coupling product.
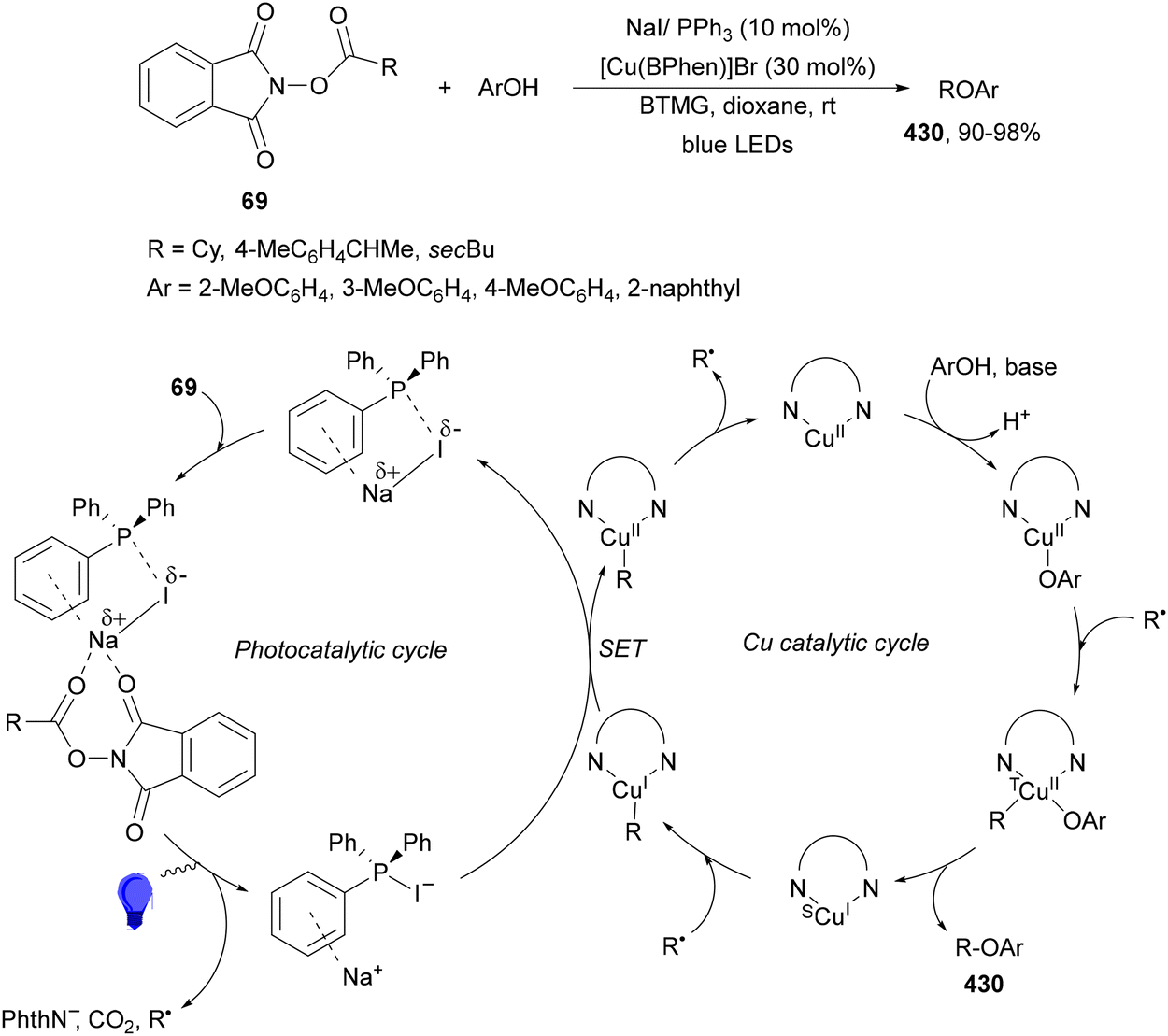 | ||
| Scheme 210 Decarboxylative etherification of alkyl NHPI esters 69 with phenols under Cu(I) and NaI/PPh3 photocatalysis. | ||
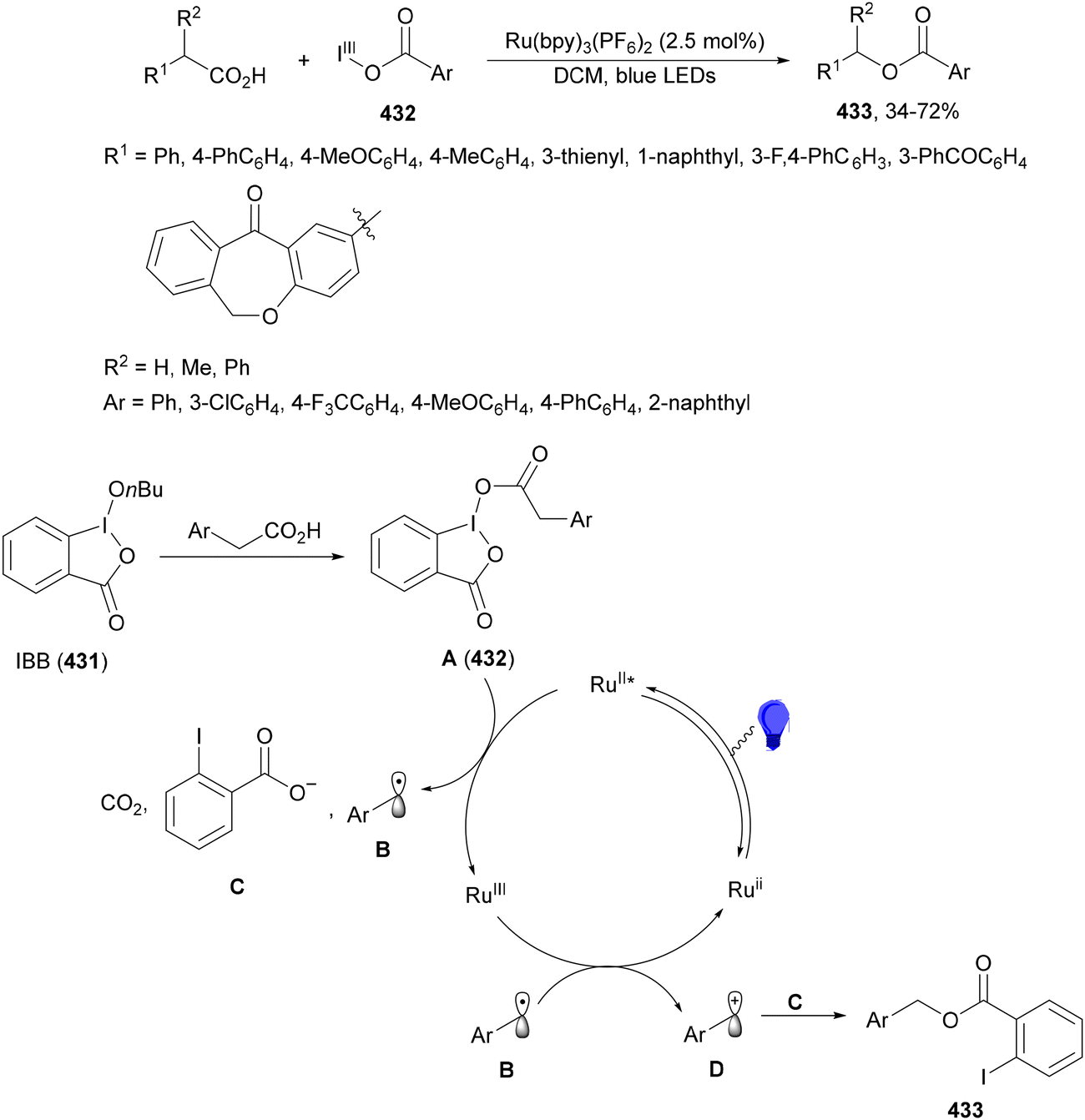 | ||
| Scheme 211 Decarboxylative acyloxylation of carboxylic acids with hypervalent iodine reagents 432 under Ru(II) photocatalysis. | ||
Acyloxylation of benzoic acids has been reported by Ritter and co-workers396via photoinduced LMCT54,55 radical decarboxylation. In the presence of 1.5 equivalents of CuTC and 2.5 equivalents of Cu(OTf)2 at 35 °C under purple LED irradiation, a broad range of benzoates were converted into esters 434, which by hydrolysis with LiOH provided the corresponding phenols (Scheme 212). This two-step procedure is a practical decarboxylative hydroxylation of benzoic acids under mild reaction conditions. The proposed mechanism starts with the formation of Cu(II) carboxylate A, which after irradiation gives the carboxy radical B. Subsequent decarboxylation provides the aryl radical C captured by copper. While the LMCT from Cu(II)TC gives TC˙, which by BET or HAT forms TC−. Intermediate D results by reaction of aryl radical C with Cu(II)TC with subsequent oxidation by Cu(II) to give the arylcopper(III)TC D. Final reductive elimination of D yields an ester and Cu(I).
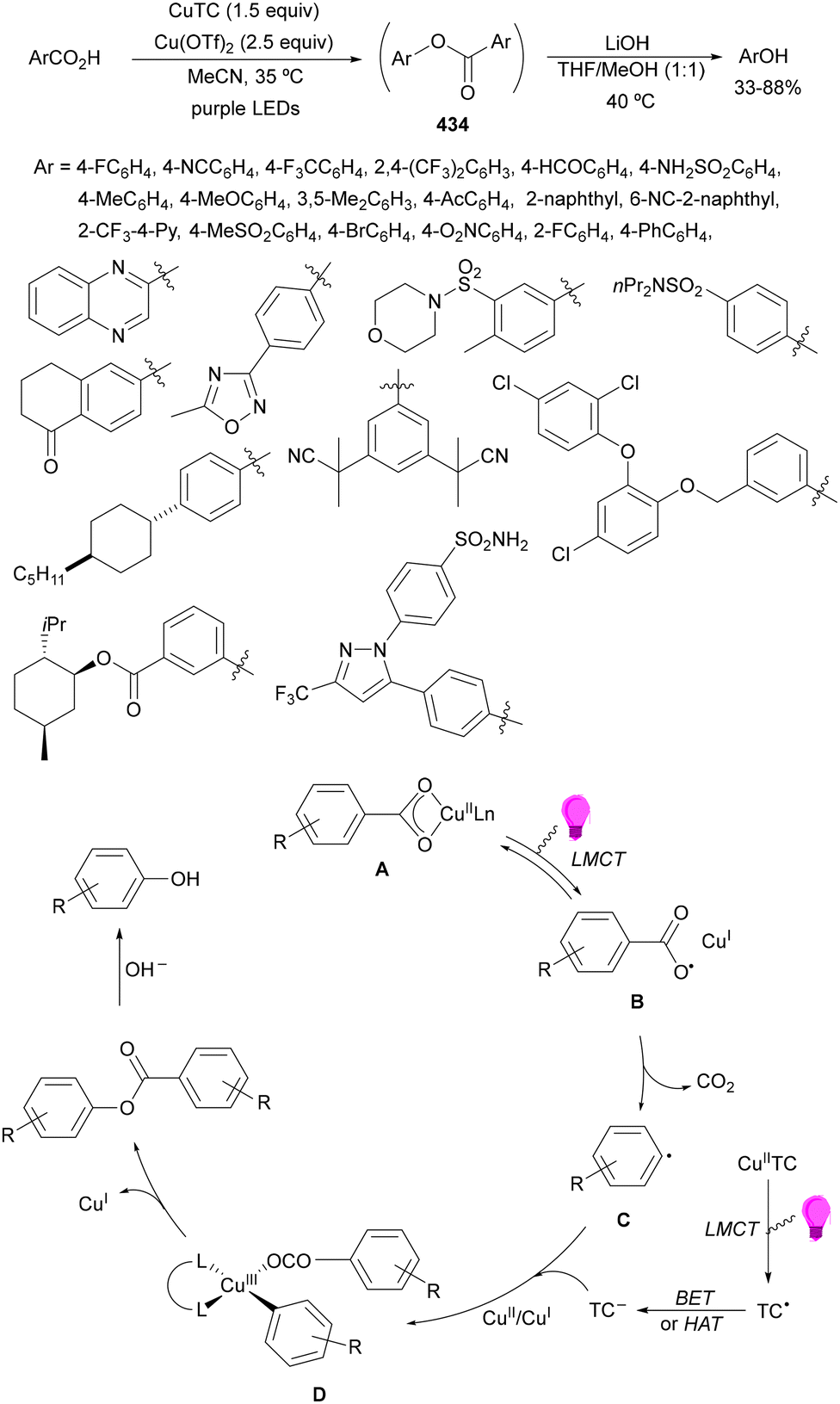 | ||
| Scheme 212 Decarboxylative acyloxylation/hydroxylation of benzoic acids under CuTC/Cu(OTf)2 photocatalysis. | ||
Mandal, Das and co-workers397 recently reported a photoinduced decarboxylative C–O bond functionalization of isatoic anhydride 345 with phenols to give 2-aminobenzoates 436 (Scheme 213). This process took place in the presence of 0.2 equivalents of KI and TBHP as an oxidant in MeCN at room temperature under blue LED irradiation to furnish products 436 with good yields.
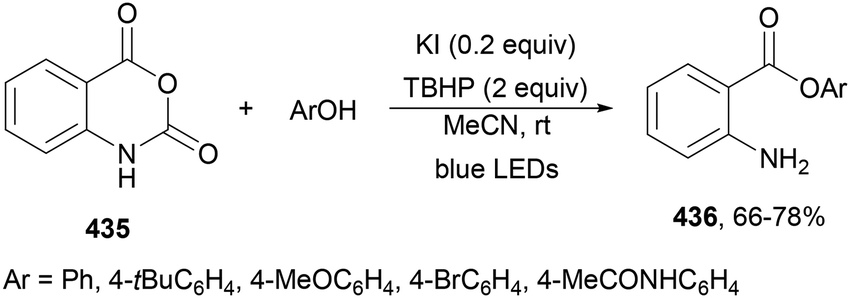 | ||
| Scheme 213 Decarboxylative C–O bond formation of isatoic anhydride (435) and phenols under KI photocatalysis. | ||
Oxidative decarboxylation of oxamic acids 15 has been described by Landais' group.398–400 In the presence of 4CzIPN (25) as a PC and BI-OAc, a hypervalent iodine reagent, as an oxidant in DCE at room temperature under blue LED irradiation, oxamic acids reacted with alcohols to give the corresponding urethanes 437 with 30–90% yields (Scheme 214a).398 A plausible mechanism has been proposed involving the formation of isocyanates 162 as intermediates. Initially, intermediate A, resulting by reaction of oxamic acid with BI-OAc, is reduced by PC* to give the radical anion B. Subsequent decarboxylation of B gives the amidocarbonyl radical C and 2-iodobenzoic acid. Radical C would be oxidized by the PC+˙ to form the isocyanate 162, which by addition of alcohol generates the urethane. This transformation was further carried out using Os(bptpy)2(PF6)2438 as a PC under near-infrared (660 nm) irradiation to give the resulting urethanes in 40–87% yields.399 In addition, in a third alternative for the preparation of urethanes 437, ferrocene was used as a PC, 2-picolinic acid as a ligand and KBrO3 as an oxidant in DCE at room temperature under blue LED irradiation (Scheme 214b).400 In this case, Fe(pinacolate) or FeCp-pinacolate complexes give upon oxidation with potassium bromate the catalytically active species Fe(III)Ln. This complex reacts with oxamic acid to provide a Fe(III) carboxylate complex D, which after irradiation a LMCT54,55 process gives the carboxy radical E. Subsequent decarboxylation forms the amidocarbonyl radical C, which undergoes further oxidation by Fe(III)Ln to give isocyanate 162 and by reaction with the alcohol urethane 437. These processes avoid the isolation of carcinogenic isocyanates.
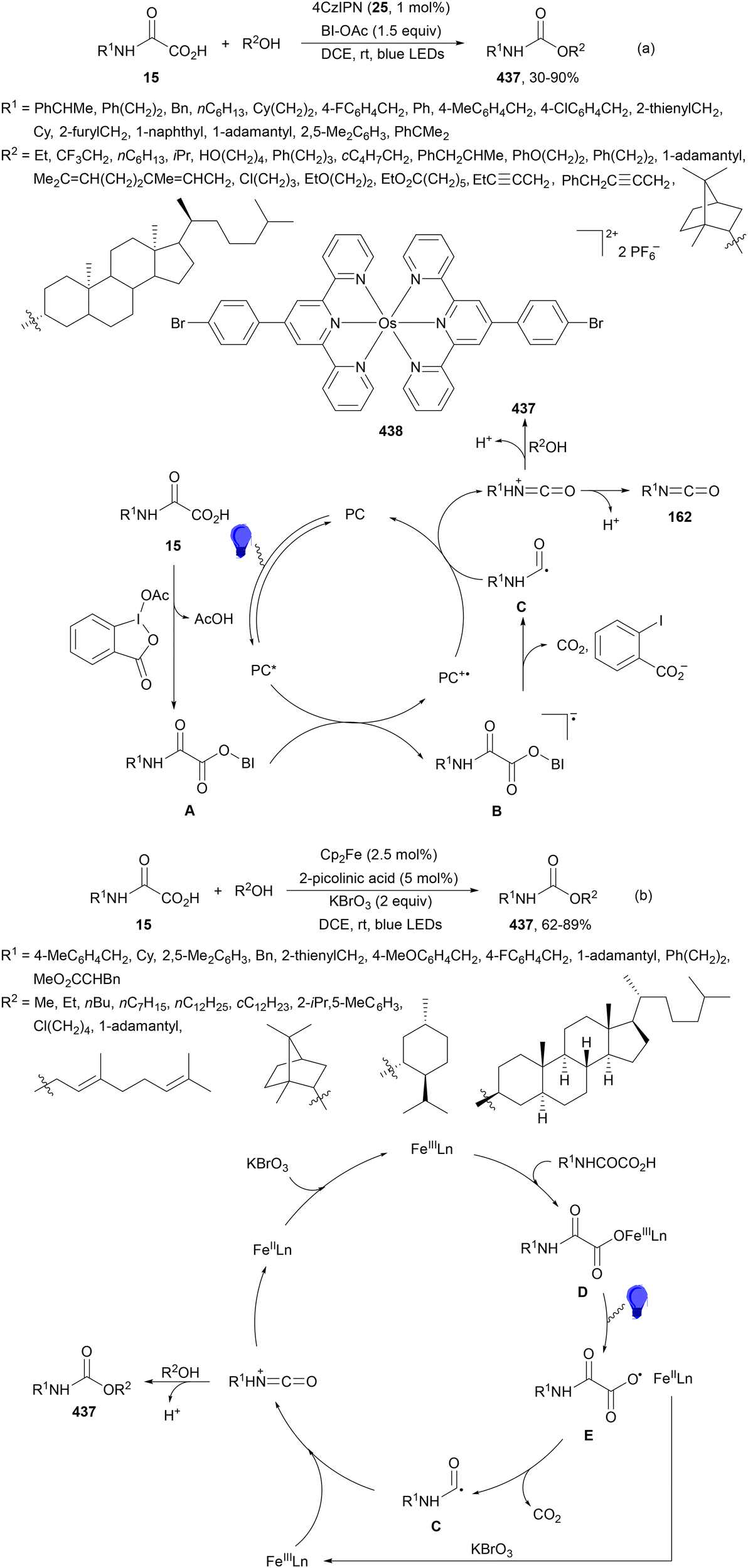 | ||
| Scheme 214 Decarboxylative oxidation of oxamic acids under 4CzIPN (25) (a) or Cp2Fe (b) photocatalysis. | ||
In summary, decarboxylative oxygenation of carboxylic acids to carbonyl compounds can be carried out using Mn, Ce or Fe salts as PCs in the presence of oxygen. In the case of α-AAs, the corresponding amides are prepared. Etherification of carboxylic acids with alcohols required Ru or Fe complexes as PCs and hypervalent iodine reagents as oxidants. Acyloxylation of carboxylic acids has been carried out with Ru complexes as PCs and hypervalent iodine reagents. Benzoic acids have been transformed into phenols by reaction with Cu complexes. In the case of oxamic acids, 4CzIPN and BI-OAc or Cp2Fe and KBrO3 were converted into urethanes by intermediacy of in situ generated isocyanates.
3.3. Carbon–sulfur bond-forming reactions
Decarboxylation of carboxylic acids or their active esters has been employed under photoredox conditions for thiolation, thioetherification, thioesterification, sulfilimination, sulfinylation and sulfonylation reactions.S bond of 439 with subsequent N–O bond cleavage to form the phthalimide radical, which regenerates the acridine PC through consecutive electron and proton transfer steps.
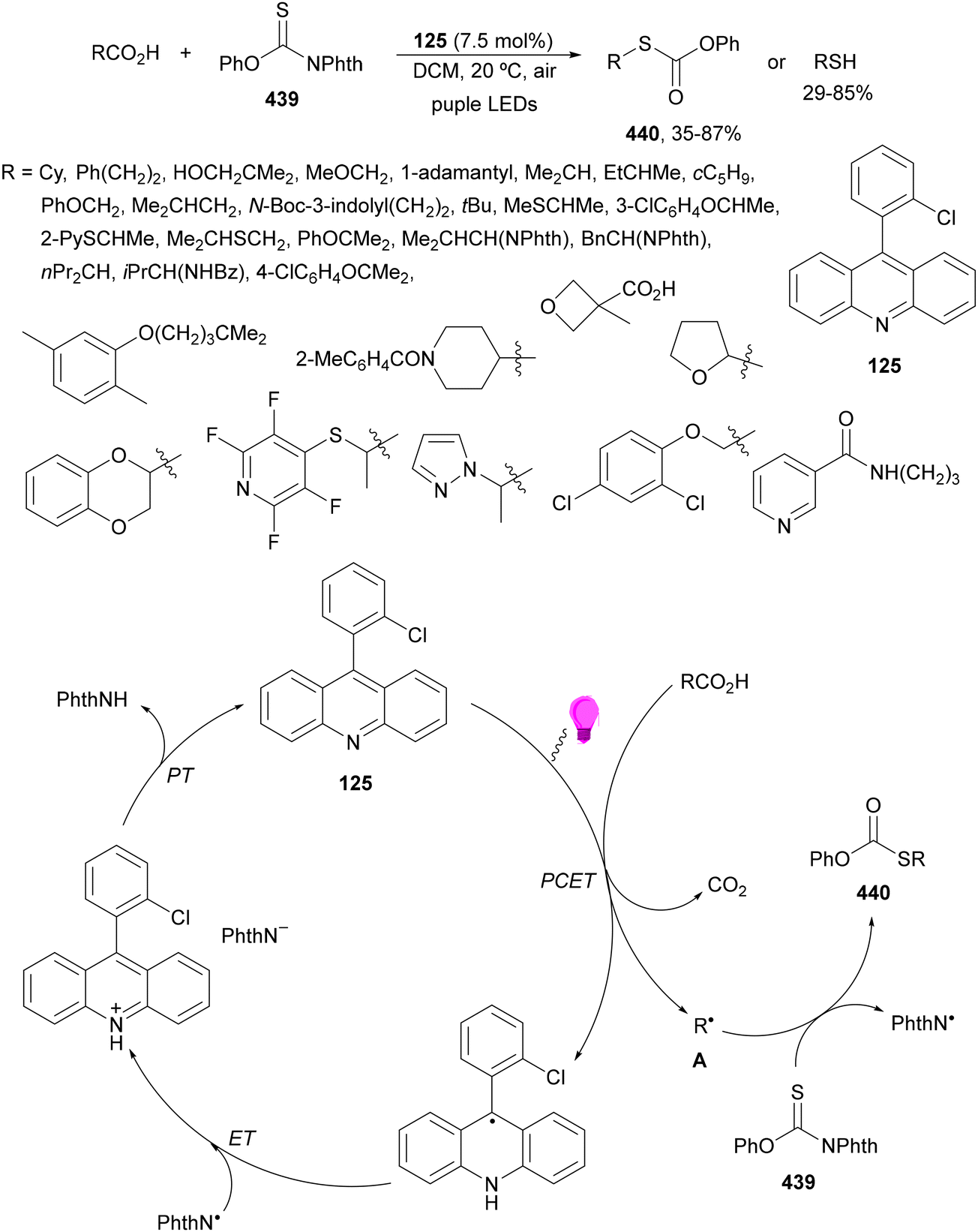 | ||
| Scheme 215 Decarboxylative thiolation of carboxylic acids with thiocarbonate 439 under acridine 125 photocatalysis. | ||
Larionov and co-workers402 performed acridine 441 photocatalysis for the direct access to thiols from carboxylic acids and elemental sulfur. This economical thiolation took place in MeCN at 100 °C under 400 nm LED irradiation followed by transformation of the oligosulfide intermediates into thiols by means of phenylsilane in 45–71% yields (Scheme 216a). Alternatively, disulfide derivatives 442 can be obtained by in situ treatment with diphenyl disulfide in 42–75% yields (Scheme 216b). A wide range of carboxylic acids and medicinally relevant acids as well as natural products have been employed.
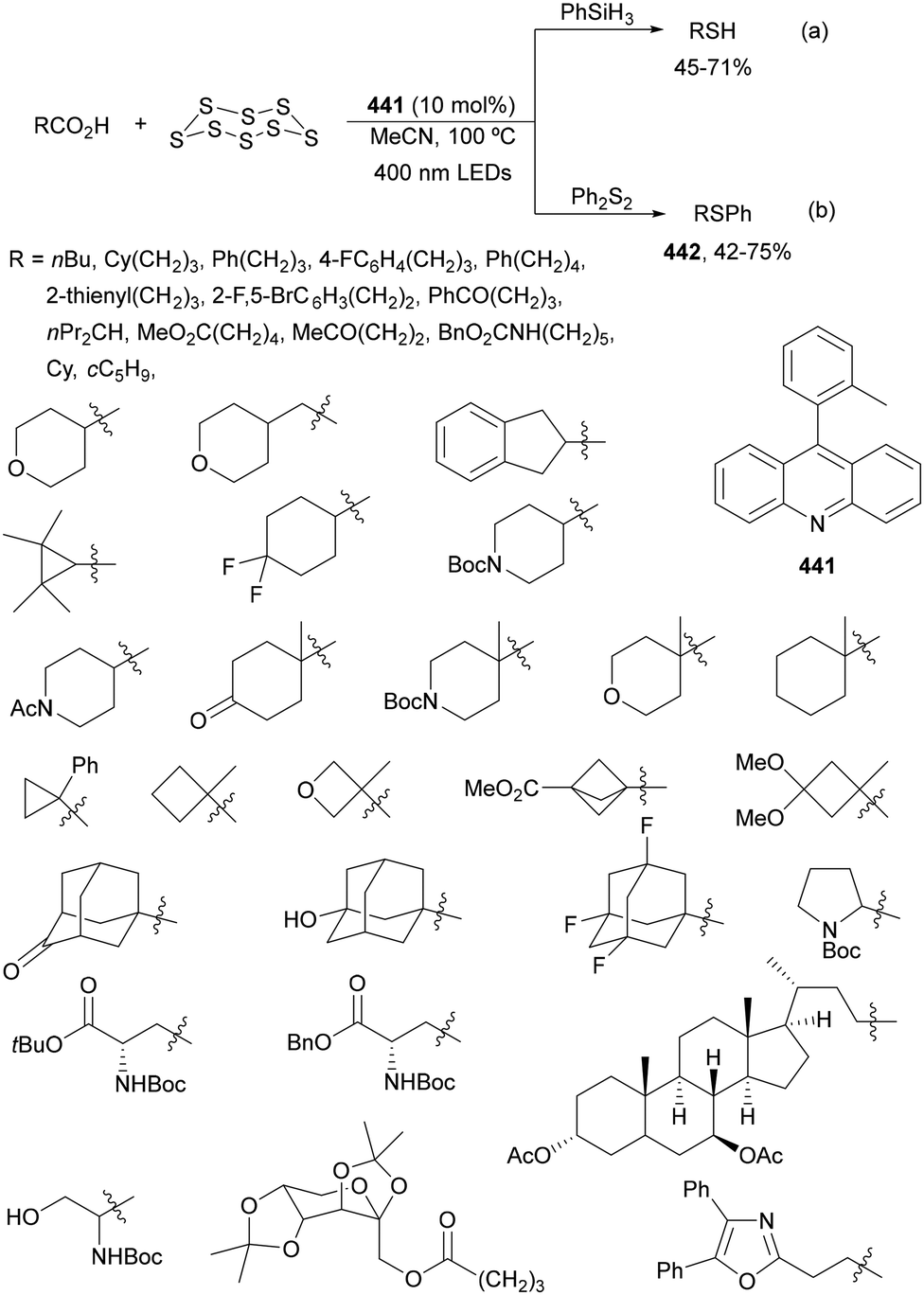 | ||
| Scheme 216 Decarboxylative thiolation of carboxylic acids with elemental sulfur under acridine 441 photocatalysis (a, b). | ||
Thiolation of alkyl NHPI esters 69 has been previously described by Liao and co-workers403 using 4-methoxybenzothioamide (443) as a sulfur donor, Eosin Y-Na2 (139) as an organophotocatalyst, and DIPEA as a base in MeCN under Ar and blue LED irradiation (Scheme 217). A wide range of primary, secondary and tertiary alkyl NHPI esters gave the corresponding thiols in 10–81% yields. Volatile thiols were isolated as disulfides 442 by in situ trapping with diphenyl disulfide in 24–80% yields. As a plausible mechanism, excited PC* is quenched by DIPEA or by thioamide 443 affording PC−˙. Subsequent SET from PC−˙ to NHPI esters forms the radical anion A and regenerates the PC. This intermediate A undergoes fragmentation to give CO2, PhthN− and radical B. Addition of B to 443 generates intermediate C, which can be oxidized to imine D by a SET from PC*. Imine D gives after ArCN elimination the corresponding thiol.
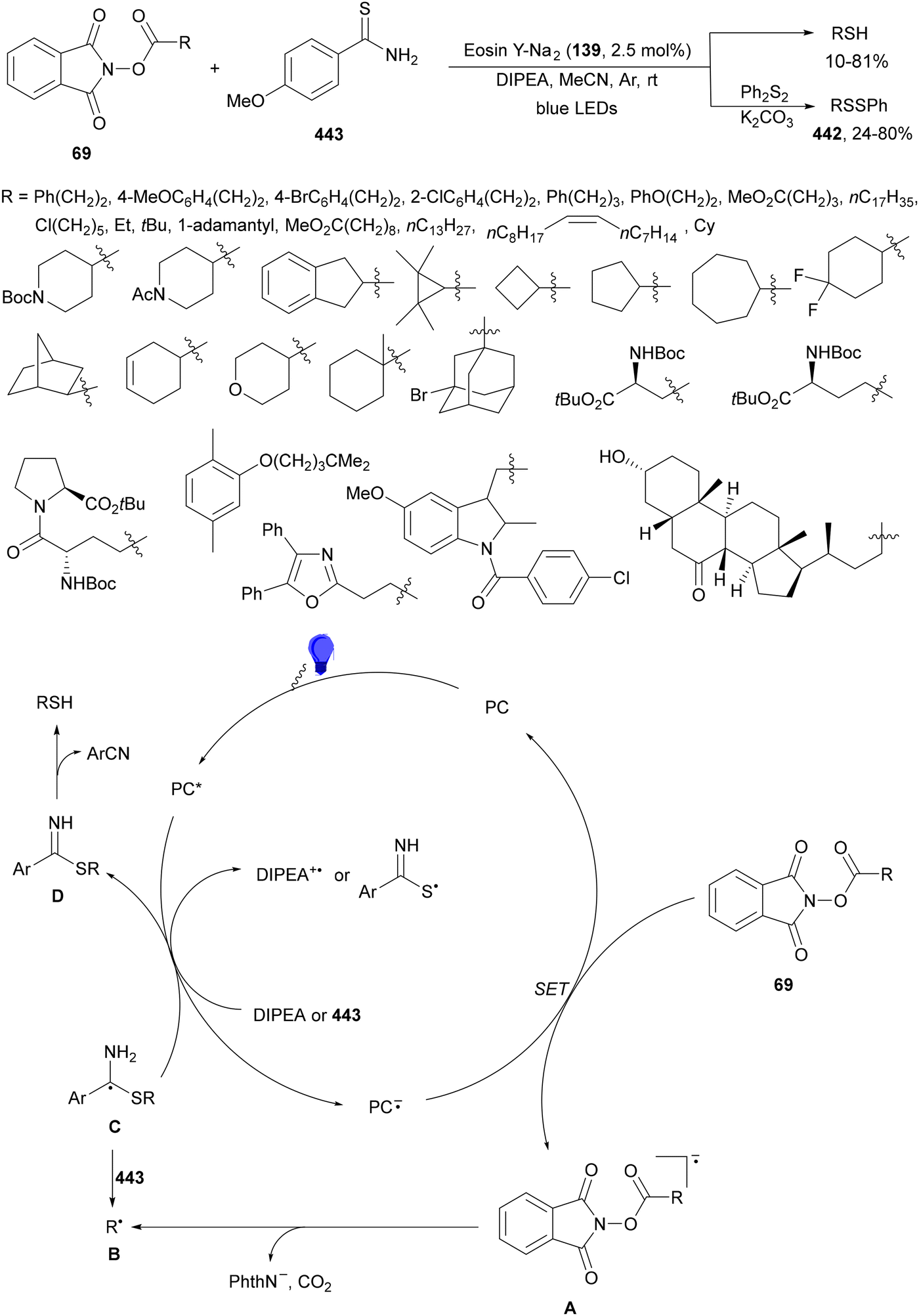 | ||
| Scheme 217 Decarboxylative thiolation of alkyl NHPI esters with 4-methoxybenzothioamide (433) under Eosin Y-Na2 (139) photocatalysis. | ||
A more recent thioetherification procedure involved trisulfide dioxides 444 as disulfuration agents of carboxylic acids. Wu and Pratt406 performed this process with [Mes-Acr-Ph]BF4287 as a PC and CsF as a base in EtOAc at room temperature under N2 and blue LED irradiation to give disulfides 442 with good yields (Scheme 218). A range of primary, secondary and tertiary carboxylic acids including α-AAs were efficiently converted into the corresponding disulfides 442. These trisulfide-1-dioxides 444 undergo homolytic substitution with the alkyl radical generated from the carboxylic acid at the PhSO2–S bond to give the phenylsulfonyl radical and the product.
 | ||
| Scheme 218 Decarboxylative thioetherification of aliphatic carboxylic acids with trisulfide dioxides 444 under [Mes-Acr-Ph]BF4287 photocatalysis. | ||
Guo, Xia and co-workers407 employed thiosulfonates 445 as thioetherification reagents for the iron-catalyzed LMCT54,55 chemodivergent381 process of aliphatic and heteroaromatic carboxylic acids into thioethers 446 or sulfoxides 447. In the presence of 10 mol% of Fe(NO3)3·9H2O and K2CO3 as a base in MeCN at 35 °C under N2 and LED (390 nm) irradiation, the corresponding thioethers 446 were obtained with 32–94% yields (Scheme 219a). Furthermore, it was found that in the presence of air a decarboxylative sulfinylation of heteroaromatic carboxylic acids occurred giving sulfoxides 447 with modest yields (Scheme 219b). Diverse drug active molecules, natural products and α-AAs were modified at the late-stage to obtain the corresponding thioethers with 39–63% yields. In the plausible pathway proposal for decarboxylative thiolation reaction, firstly the base deprotonates the carboxylic acid and forms a carboxylate-iron(III) intermediate A. Subsequent photoinduced LMCT gives Fe(NO3)2 and the carboxy radical B, which after decarboxylation generates radical R˙ (C). This radical C attacks the thiosulfonate 445 to form the thioether 446 and a sulfonyl radical D. Radical D converts Fe(II) into Fe(III) regenerating the catalyst. Wei, Hu and co-workers210 employed also Fe(NO3)3 and bis[(2-pyridyl)methyl]amine (298) as a ligand for the thiolation reaction of carboxylic acids with thiosulfonates 445 to give thioethers 446 in 50–89% yields.
 | ||
| Scheme 219 Decarboxylative thioetherification of aliphatic and heteroaromatic carboxylic acids with thiosulfonates 445 under Fe(NO3)3 photocatalysis (a, b). | ||
A similar decarboxylative thiolation of aromatic carboxylic acids with thiosulfonates 445 has been recently performed by Xia and co-workers.200 In this case, Cu(OTf)2 (2 equivalents) was used for the LMCT process and Li2CO3 as a base in MeCN at 35 °C under N2 and 390 nm LED irradiation to obtain thioethers 446 with 43–73% yields.
Dilman and co-workers408 reported that a perfluorinated disulfide 448 can be employed for the thiolation of carboxylic acids. In the presence of acridine 449 as a PC for PCET catalysis, sodium perborate as a reoxidant of the forming thiol in aqueous DCM at room temperature under 400 nm LED irradiation resulted in sulfides 450 in moderate to good yields (Scheme 220). A wide range of primary, secondary and tertiary aliphatic carboxylic acids were employed as well as α-AAs and drugs such as ketoprofen, naproxen, indomethacin, ibuprofen and gemfibrozil. As natural products, pinonic and ursodeoxycholic acids also delivered the corresponding sulfides in good yields.
 | ||
| Scheme 220 Decarboxylative thioetherification of aliphatic carboxylic acids with perfluorinated disulfide 448 under acridine 449 photocatalysis. | ||
Yoon and co-workers295 employed FeCl3-mediated decarboxylative photoredox conditions for C–H activation of arenes and heteroarenes (Scheme 160). By photochemical decarboxylation followed by radical-polar crossover, diverse C–O, C–N and C–S bonds can be formed. In the case of thioetherification reactions, phenylacetic acid, as the only example, was allowed to react with cyclohexanethiol to give the corresponding sulfide in 53% yield.
Recently, He and Dydio409 reported thianthrenation of aryl and heteroaryl carboxylic acids by photoinduced LMCT54,55 mediated by Cu(II) salts. The reaction of carboxylic acids with thianthrenes 451 in the presence of Cu(OTf)2 and Cu(MeCN)4OTf with NaF as a base in MeCN under purple LED irradiation provided thianthrenium salts 452 in modest to good yields (Scheme 221). These salts 452 were further transformed into 13C-labeled carboxylic acids by reaction with 13CO2 and ZnCl2 in boronates, sulfonamides, phosphate esters and by Negishi-type coupling to alkyl derivatives.
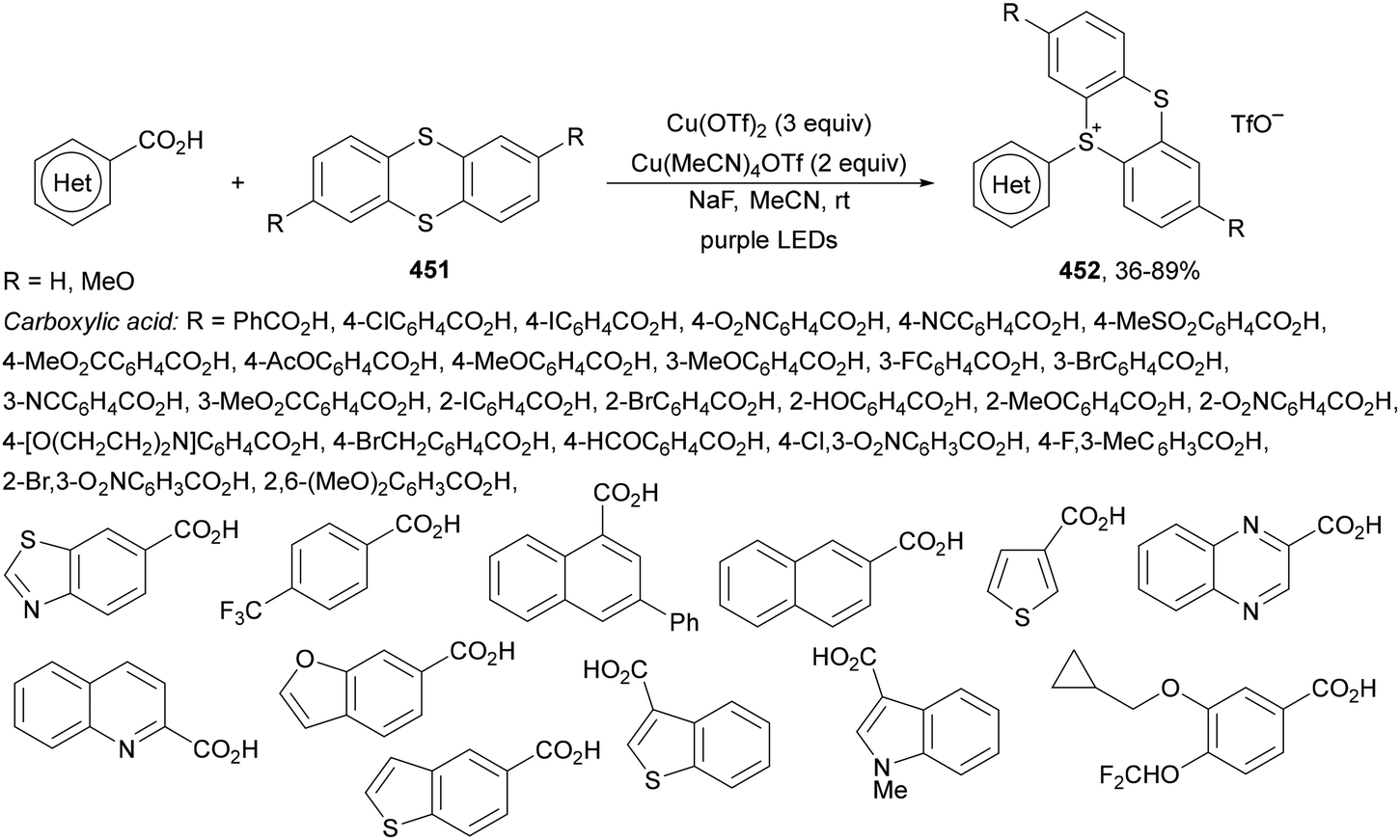 | ||
| Scheme 221 Decarboxylative thianthrenation of aromatic and heteroaromatic carboxylic acids with thianthrenes 451 under Cu(II) photocatalysis. | ||
The use of NHPI esters in thioetherification reactions was described in 2016 by Fu and co-workers410 using thiophenols and Cs2CO3 as a base under 40 W CFL irradiation to give alkyl aryl sulfides in 44–94% yields. Zheng and co-workers411 described a Ru-photoredox catalysis for the thioetherification of NHPI esters with alkyl and aryl disulfides under blue LED irradiation to give the corresponding sulfides in 24–98% yields. Wang and co-workers412 used thiosulfonates and 4-TolSO2SR as a thiolating reagent, giving sulfides in the presence of 4CzIPN as a PC under LED irradiation with 43–91% yields.412
Oxime esters derived from benzophenone RCO2NCPh2 were employed by Glorius and co-workers413 for trifluoromethylthiolation with Ir complex 4 as a PC under blue LED irradiation. Later, Wu and Pratt414 prepared unsymmetrical disulfides by reaction of benzophenone oxime esters with tetrasulfides with Ir complex 4 as a PC under blue LED irradiation.
Recently, Noble, Aggarwal and co-workers373 reported a decarboxylative thiocyanation of NHPI esters 69 with potassium thiocyanate under blue LED irradiation. In the presence of Ir complex 4 as a PC, CuSCN, 2,9-dimethyl-1,10-phenanthroline (dmp) as a ligand in acetone at 30 °C, the corresponding alkyl thiocyanates 453 were obtained in 44–79% yields accompanied by small amounts of isothiocyanates (Scheme 222). The proposed mechanism was similar to the one depicted in Scheme 203 for the chlorination of NHPI esters 69.
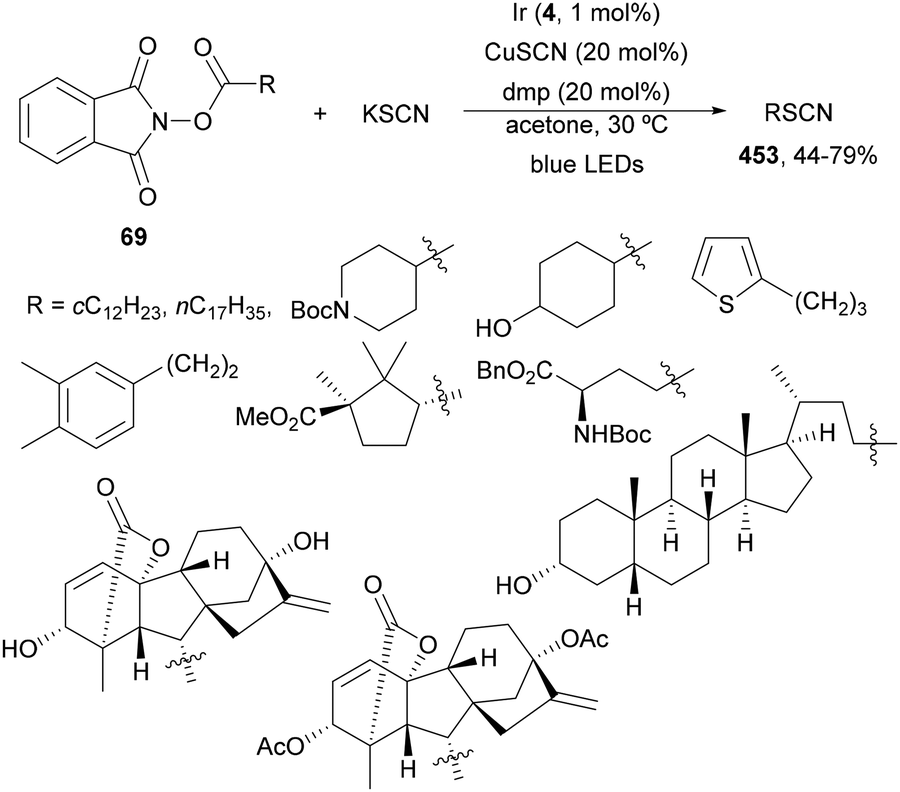 | ||
| Scheme 222 Decarboxylative thiocyanation of alkyl NHPI esters 69 with KSCN under Ir complex 4 photocatalysis. | ||
Prieto and co-workers417 employed benzophenone oxime esters 454 and xanthate dimer 455 for the synthesis of xanthates with moderate to good yields (Scheme 223). This xanthylation process was carried out in the presence of thioxanthone (TX) and EtOH as a solvent at room temperature under purple LED (390 nm) irradiation. Primary, secondary and tertiary esters 454 including gibberellic acid, gemfibrozil and enoxolone were converted into the corresponding thioxanthates. Mechanistic investigations revealed that only xanthate dimer 455 is involved giving the xanthyl radical A, which reacts with iminyl radical B to give intermediate C. Subsequent β-scission of C by reacting with R˙ gives product 456.
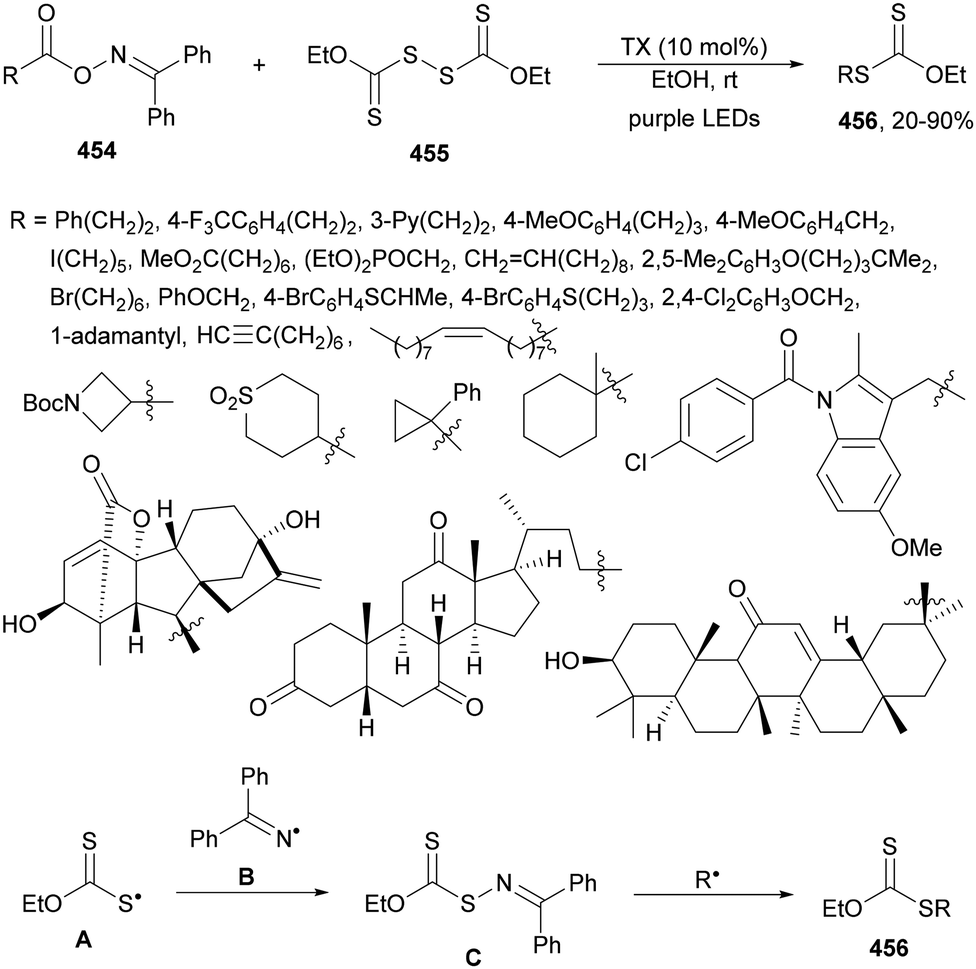 | ||
| Scheme 223 Decarboxylative xanthylation of benzophenone oxime esters 454 with xanthate dimer 455 under thioxanthone photocatalysis. | ||
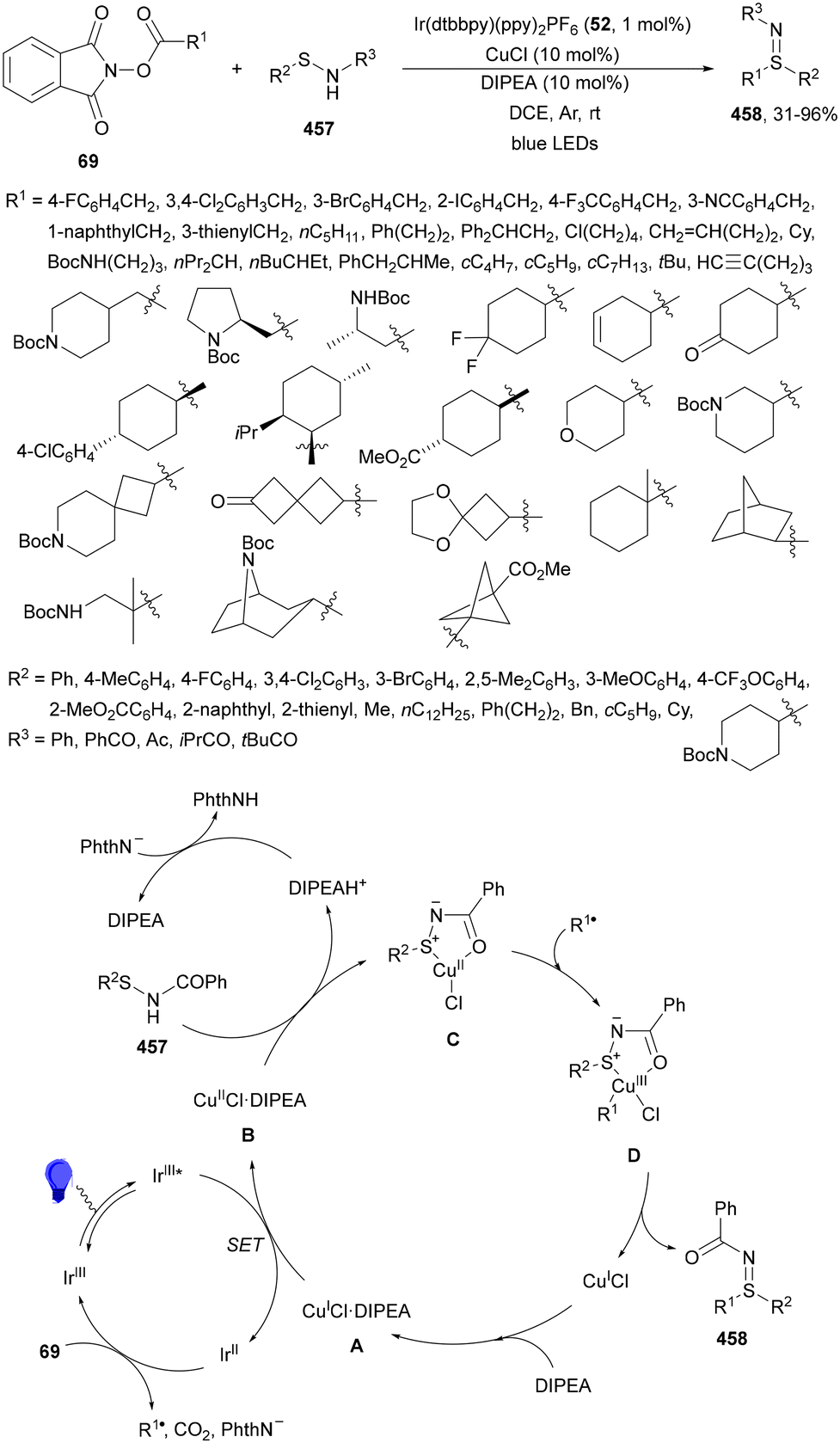 | ||
| Scheme 224 Decarboxylative sulfilimination of alkyl NHPI esters 69 with sulfenamides 457 under Ir complex 52 and CuCl photocatalysis. | ||
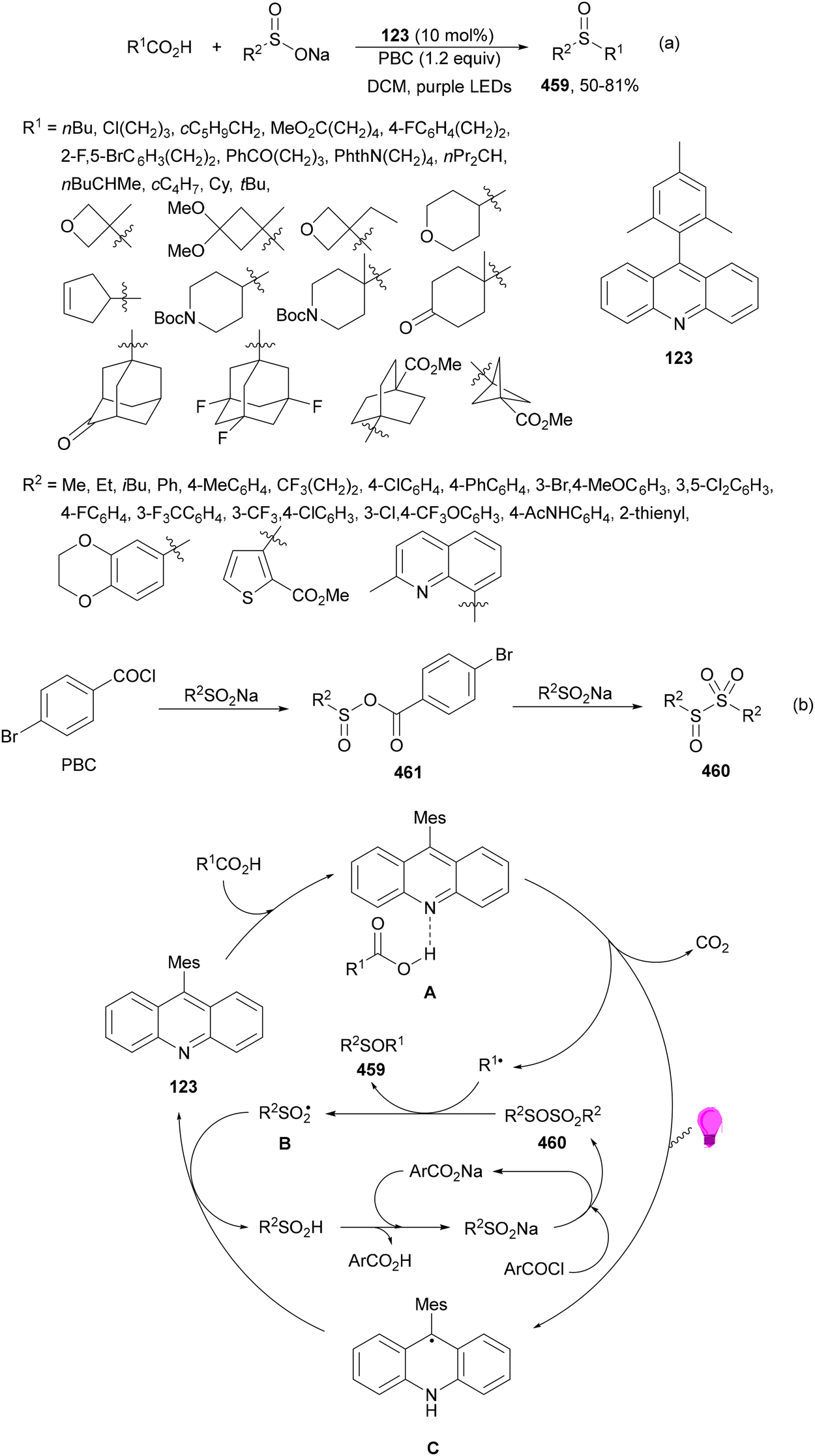 | ||
| Scheme 225 Decarboxylative sulfinylation of aliphatic carboxylic acids with sodium sulfinates and PBC under acridine 123 photocatalysis. | ||
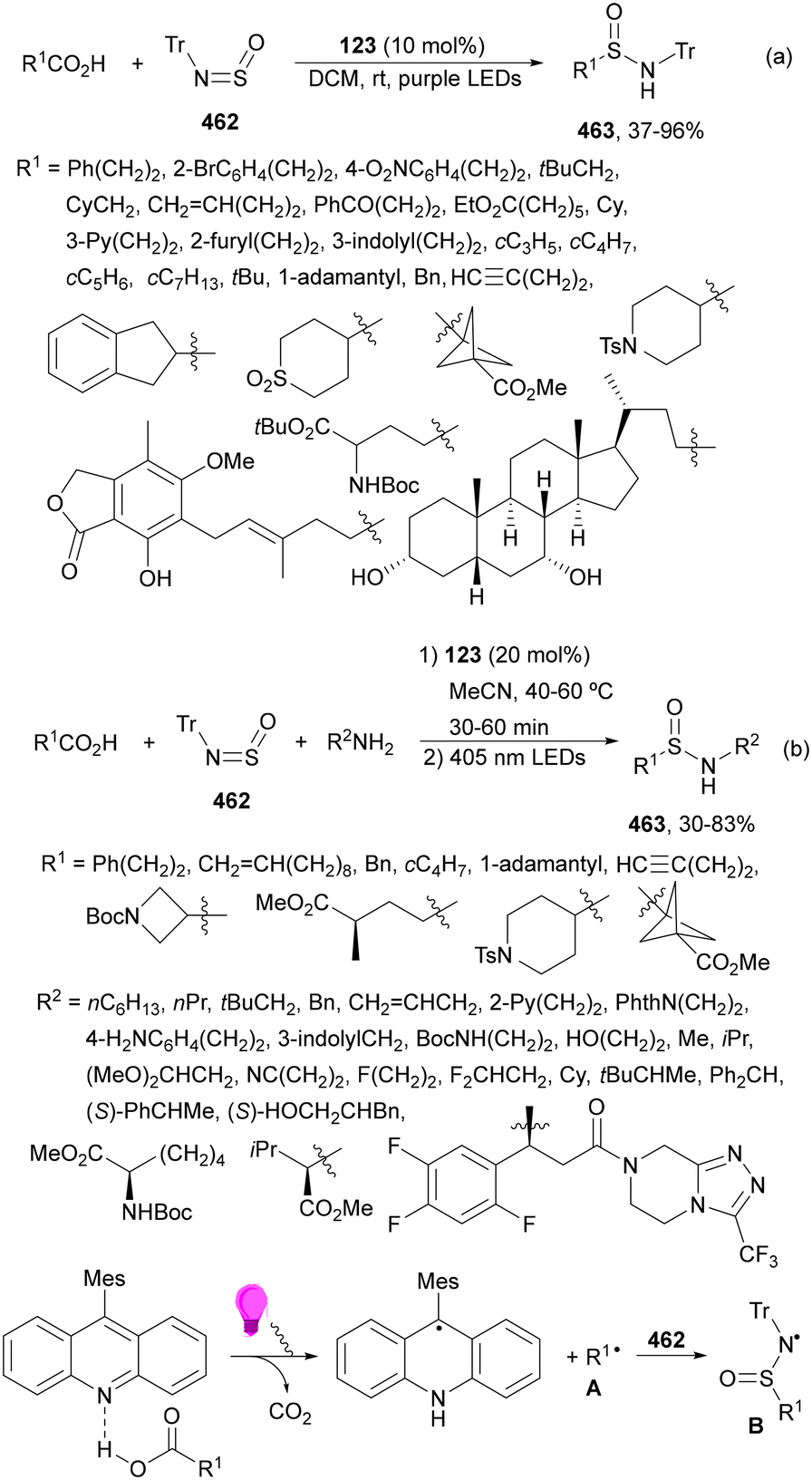 | ||
| Scheme 226 Decarboxylative sulfinamidation of alkyl carboxylic acids with TrNSO (462) under acridine 123 photocatalysis. | ||
In the case of Larionov and co-workers,423 acridine 125 was used as a PC and Cu(MeCN)4BF4 as a catalyst with dtbpy as a ligand in a 2:1 mixture of DCM and MeCN under 400 nm LED irradiation (Scheme 227a). Primary, secondary and tertiary aliphatic carboxylic acids and N-aryl or heteroaryl sulfinylamines 462 gave sulfinamides 463 with very good yields. Primary sulfinamides 464 were prepared using silyl sulfinylamines followed by treatment with TBAF (Scheme 227b). Active pharmaceutical ingredients (APIs) and natural products such as gemfibrozil, mycophenolic acid, N-Boc-proline and aspartic acid derivative, oxaprozin, isoxepac, erucic acid, linoleic acid, 10,12-pentacosadinoico acid, cis-pinonic acid, oleanolic acid, glycyrrhetinic acid and unprotected chenodeoxycholic acid were transformed into the corresponding sulfinamides in 51–99% yields. In the case of gemfibroxil, the reaction with N-phenyl sulfinylamine was carried out on a gram-scale with 96% yield. Based on experimental and computational data the proposed catalytic cycle starts with the formation of intermediate A between acridine 125 and the acid. Photoinduced proton-coupled electron transfer (PCET) of A forms, after decarboxylation, the alkyl radical B, which reacts with the sulfinylamine 462 to give the amino sulfinyl radical C. This radical C is stabilized by the Cu(I) catalyst to form complex D. Subsequent PCET with acridinyl radical E releases the product 463 and the Cu(I) catalyst.
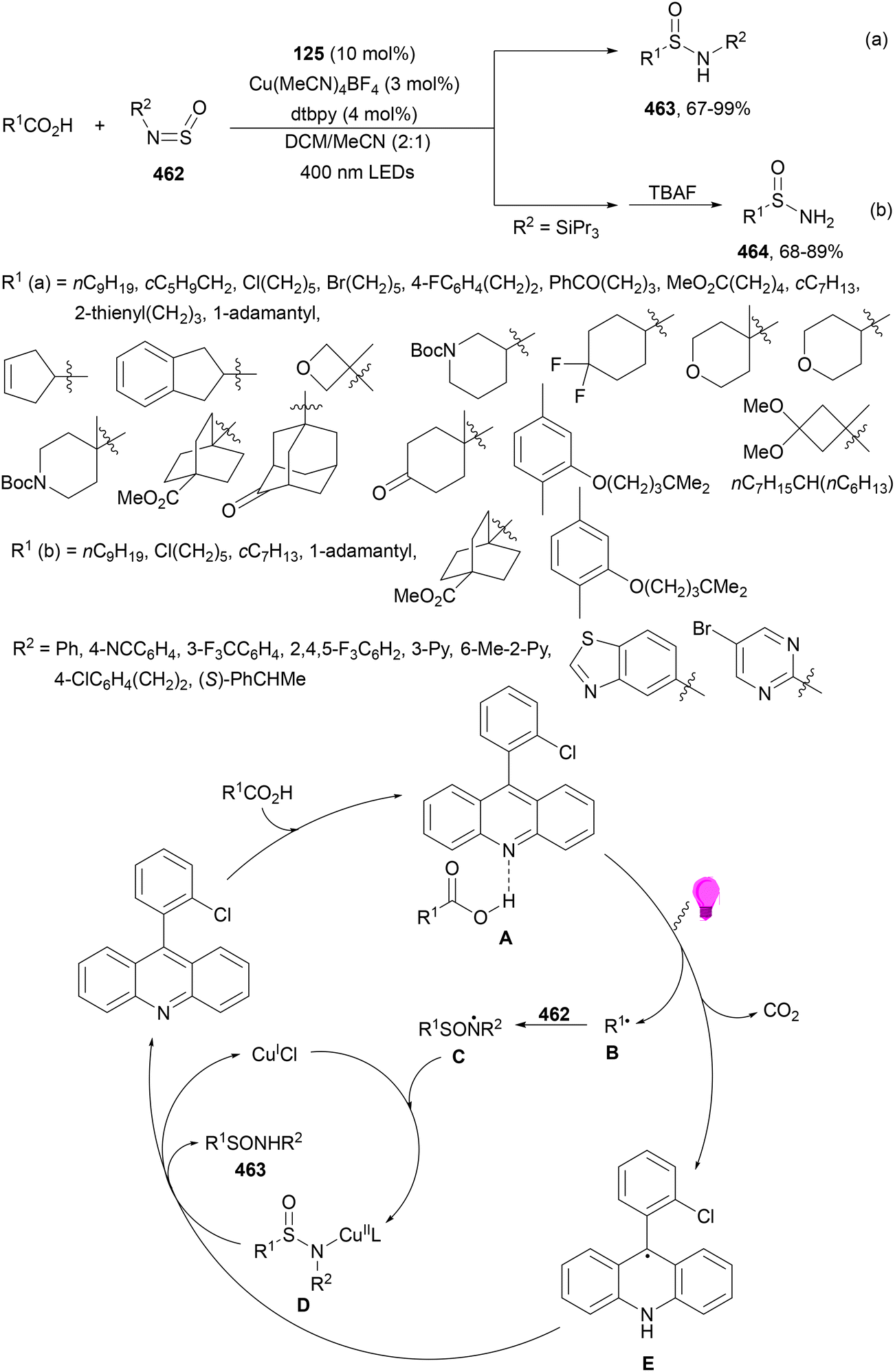 | ||
| Scheme 227 Decarboxylative sulfinamidation of alkyl carboxylic acids with RNSO (462) under acridine 125 and Cu(MeCN)4PF4 photocatalysis. | ||
More recently, Larionov and co-workers425 reported a three-component reaction for the synthesis of sulfones 466 using carboxylic acids, 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO, 465) or sodium metabisulfite and alkyl or heteroaryl halides or electrophilic alkenes with acridine 125 as a PC under LED irradiation (400 nm) (Scheme 228a). A wide range of sulfones 466–468 have been prepared including those derived from natural products and drugs in 52–95% yields. This decarboxylative sulfonylation was applied to the preparation of sodium sulfinates 469 after a simple basic work-up in 54–96% yields (Scheme 228b). Moreover, sulfonyl chlorides 470 and fluorides 471 can be prepared under the same reaction conditions by addition of NCS followed by an aqueous solution of KHF2 and NFSI, respectively (Scheme 228c and d). Mechanistic studies showed that the photocatalytic decarboxylation takes place in the excited state of the acridine-carboxylic acid complex via a PCET process (see Scheme 207).
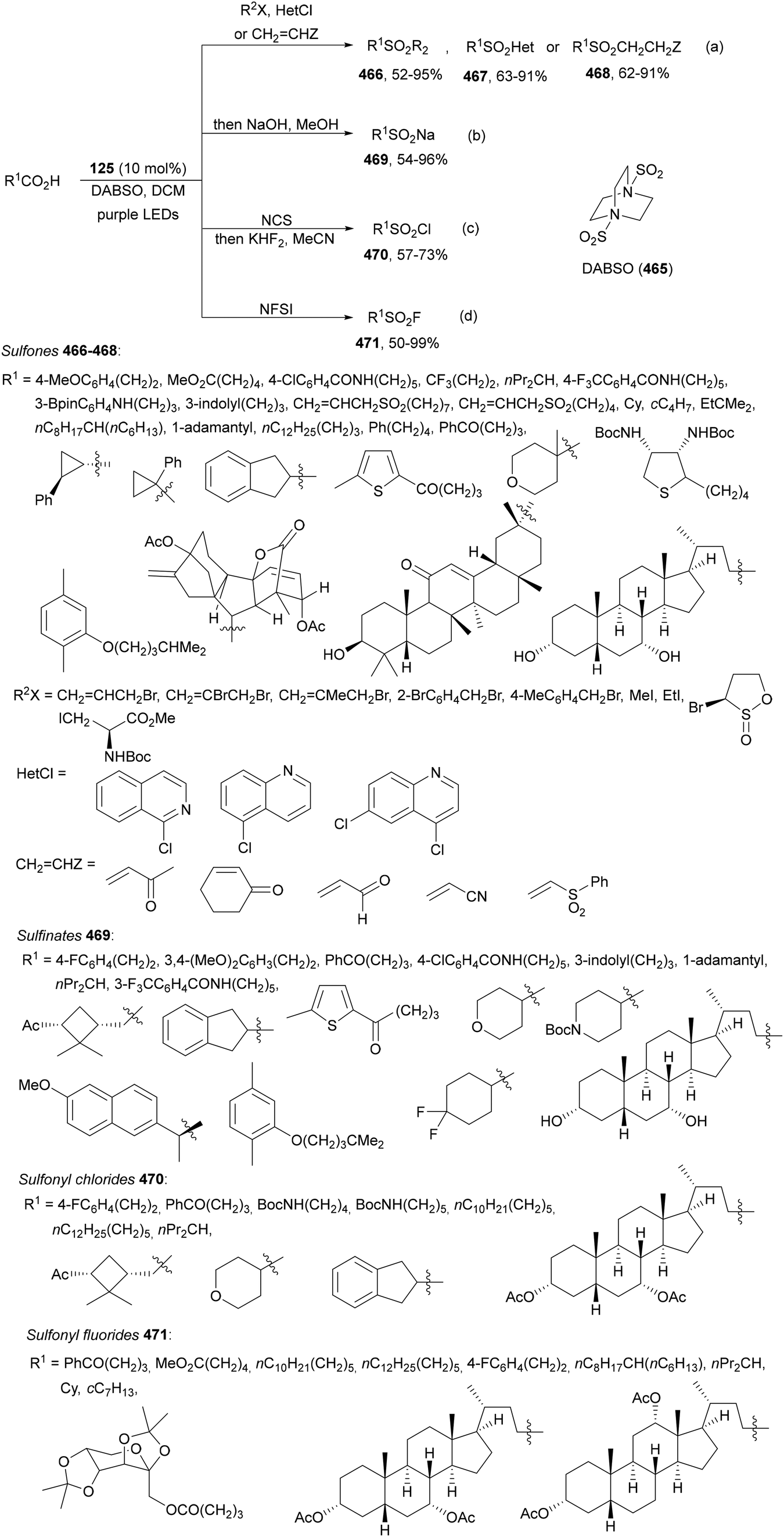 | ||
| Scheme 228 Decarboxylative sulfonylation of aliphatic carboxylic acids with DABSO (465) under acridine 125 photocatalysis. | ||
The same group426 developed a dual acridine/copper photocatalytic system for the tricomponent decarboxylative sulfonylation of carboxylic acids to sulfones. Acridine 472 and CuOTf/diamine 473 were used as catalysts for the synthesis of alkyl aryl or heteroaryl sulfones 467 with good yields in the presence of K2S2O5 or DABSO as a stable sulfur dioxide donor and Cs2CO3 or DABCO as a base, in MeCN at 100 °C under purple LED (400 nm) irradiation (Scheme 229). This method was applied to primary, secondary and tertiary carboxylic acids (see Scheme 228) as well as natural products and drugs such as gemfibrozil, oxaprozin, mycophenolic acid, Boc–Glu–OMe, a biotin derivative, D-fructose, ursodeoxycholic acid, cholic acid, deoxycholic acid, chenodeoxycholic acid, picamilon, bedaquiline, empagliflozin, canagliflozin and lapatinib in 43–92% yields.
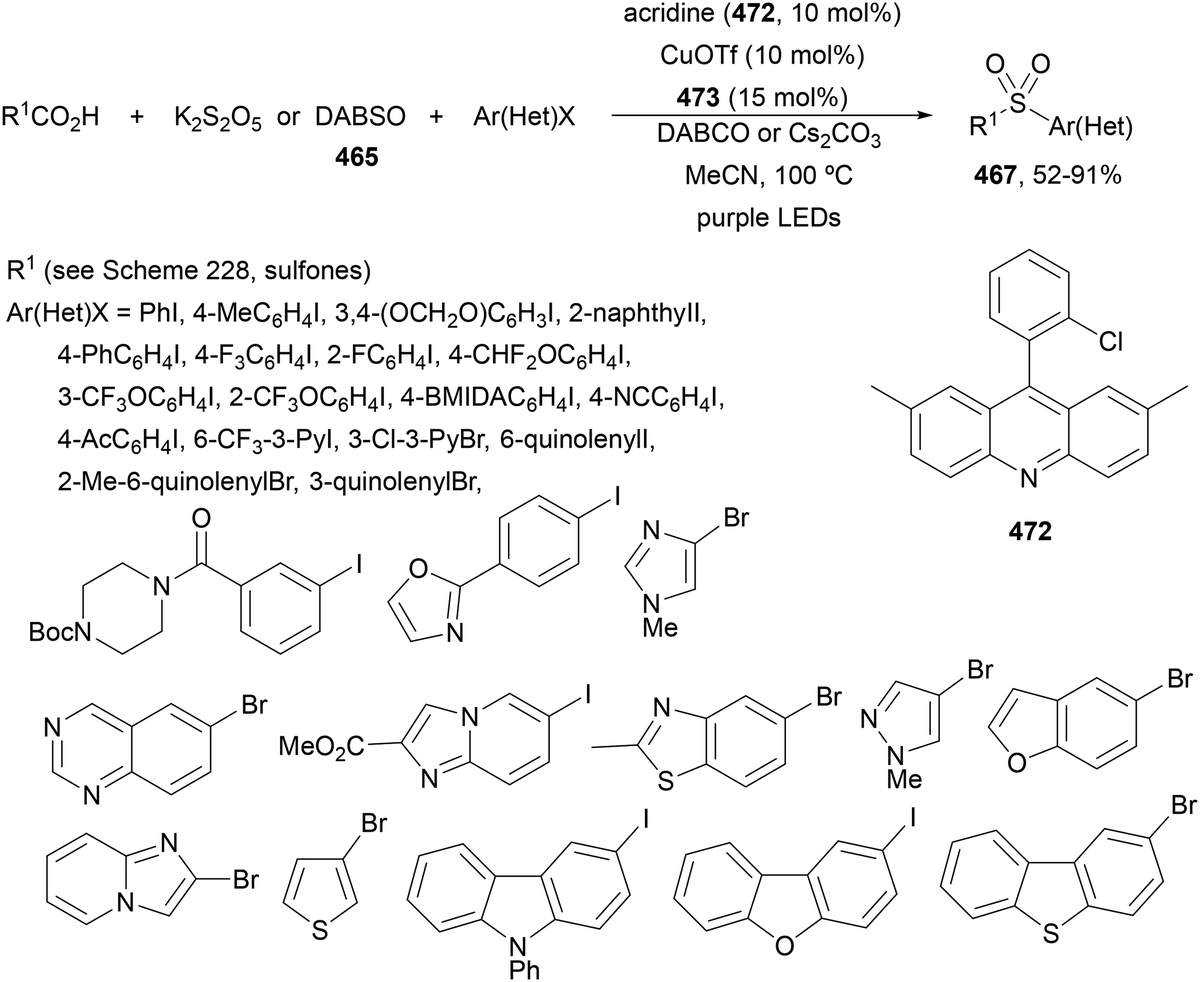 | ||
| Scheme 229 Decarboxylative sulfonylation of aliphatic carboxylic acids with DABSO (465) under acridine 472 and CuOTf dual photocatalysis. | ||
Zeng and co-workers427 reported recently a similar three-component reaction between aliphatic carboxylic acids, DABSO (465) and alkyl halides using TBAFeCl4 as a PC, Et3N as a base in DCM at room temperature under Ar and 390 LED irradiation (Scheme 230). The corresponding dialkyl sulfones 466 were obtained in modest to good yields. In the proposed mechanism, a SET between the carboxylate ion and Fe(III) generates Fe(II) and RCO2˙. A subsequent decarboxylation of A forms the alkyl radical B, which reacts with DABSO (465) to give RSO2˙ species C. This radical C regenerates Fe(III) and furnishes the sulfinate anion D. The final reaction of D with allyl bromide produces the sulfone.
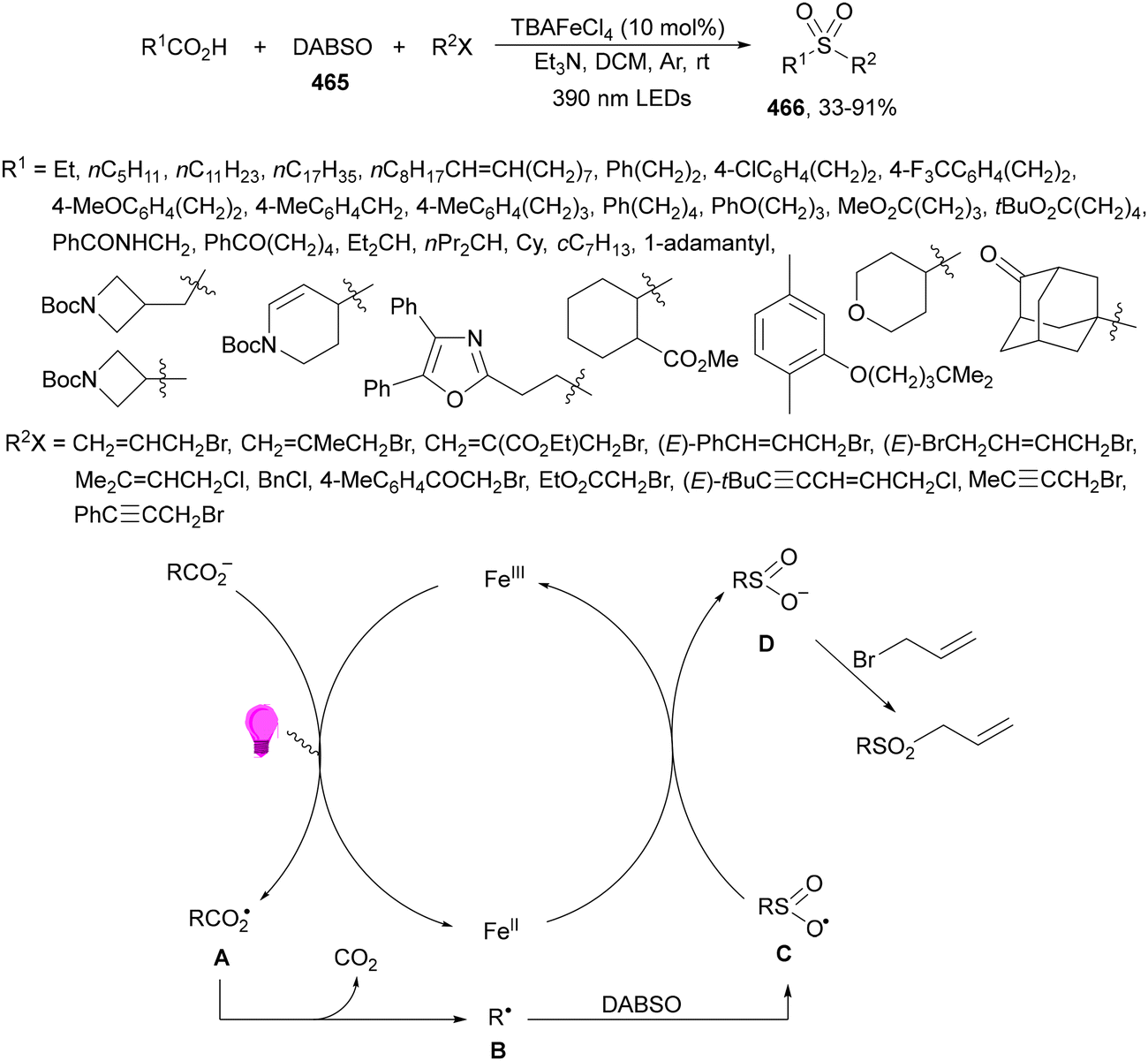 | ||
| Scheme 230 Decarboxylative sulfonylation of aliphatic carboxylic acids with DABSO under TBAFeCl4 photocatalysis. | ||
Vinyl sulfones have been prepared by decarboxylative sulfonylation of cinnamic acids. In 2016, Cai and co-workers428 described this sulfonylation using sulfonyl hydrazides in the presence of oxygen as an oxidant and Eosin Y (139) as a PC in aqueous DMF under visible light to afford styryl sulfones in 35–92% yields. Later, arylsulfonate phenol esters 473 have been used for the decarboxylative sulfonylation of cinnamic acids only under visible-light in the absence of PC and oxidant (Scheme 231).429 The corresponding styryl aryl sulfones 295 were obtained stereoselectively with good yields in the presence of Cs2CO3 as a base at room temperature in DMA under blue LED irradiation. As a plausible mechanism, the initial formation of an electron donor–acceptor complex A between aryl sulfonate phenol ester 473 and DMA with the assistance of Cs2CO3 is postulated. After irradiation, the excited complex A* undergoes a SET process to deliver radical anion B and the carbon-centered radical C. Fragmentation of B forms the sulfonyl radical D, which adds to cinnamic acid to give the benzyl radical E. SET of E by A* affords F and regenerates radical anion B. Finally, decarboxylation of F produces the sulfone 295.
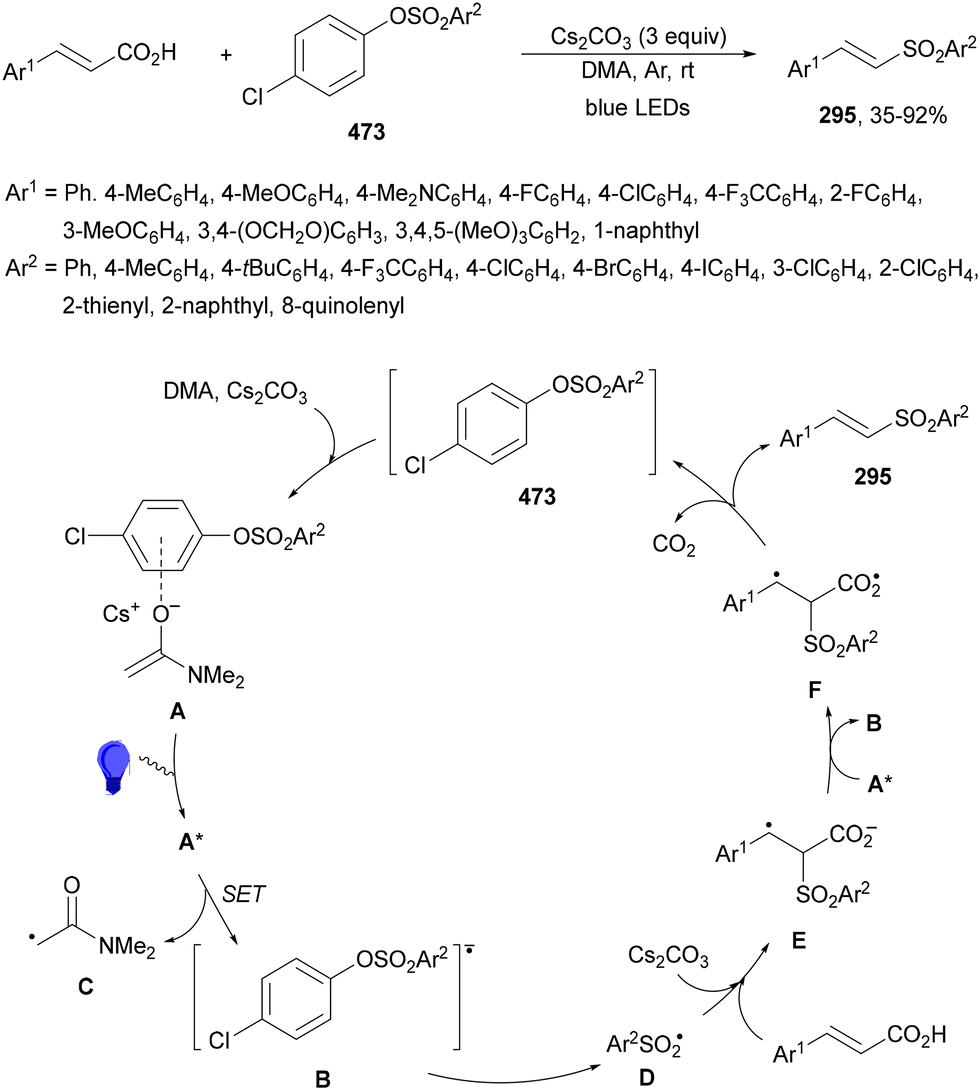 | ||
| Scheme 231 Decarboxylative sulfonylation of cinnamic acids with aryl sulfonate phenol esters 473 under photocatalysis. | ||
Recently, Sing and co-workers430 employed sulfonylazides 474, tosylmethyl isocyamide 475 (TosMIC) and β-keto sulfones 476 as sulfonating agents of cinnamic acids in the presence of rhodamine B (477) or Eosin Y (139) as a PC, TBHP as an oxidant and Cs2CO3 as a base. In the case of sulfonylazides 474, rhodamine B (477) was used as a PC and DMSO as solvent to give styryl aryl sulfones 295 in 42–87% yields (Scheme 232a). Propane-2-sulfonylazide gave styryl isopropyl sulfone in 72% yield. When tosylmethyl isocyanide 475 was employed in DMF as solvent and Eosin Y (139) as a PC, the corresponding styryl 4-tolyl sulfones 295 were obtained with 67–97% yields (Scheme 232b). In addition, under the same reaction conditions, β-keto sulfones 476 afforded styryl sulfones 295 in 69–89 yields (Scheme 232c). In the proposed mechanism, an aryl sulfonyl radical is formed in the three cases, which undergoes conjugate addition to the cinnamic acid as shown in Scheme 231.
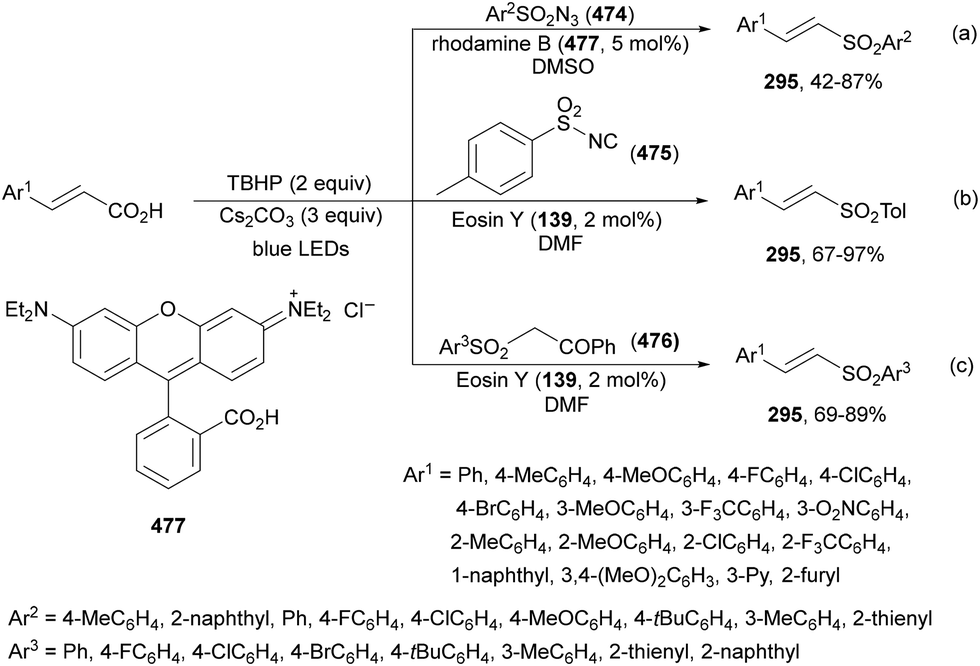 | ||
| Scheme 232 Decarboxylative sulfonylation of cinnamic acids with sulfonylazides 474, tosyl metyl isocyanide 475 and β-keto sulfones 476 under rhodamine B (477) or Eosin Y (139) photocatalysis. | ||
Fluorosulfonylation of primary and secondary aliphatic carboxylic acids by decarboxylative photoredox reaction was described by Larionov and co-workers425 using acridine 125 as a PC and DABSO (465) (see Scheme 228). Lu, Weng and co-workers431 performed a copper-catalyzed decarboxylative fluorosulfonylation of primary, secondary and tertiary aliphatic carboxylic acids by two methods with different N-centered HAT reagents. Firstly, Cu powder, sodium metabisulfite and N-fluorobenzene sulfonimide (NFSI) at room temperature gave sulfonyl fluorides (471) derived from 3-arylpropionic acid in 5–65% yields. In the second method, 9-mesitylacridine (123) as the HAT reagent and PC, DABSO (465) and Selectfluor, Cu powder, 5,5′-dimethyl-2,2′-bipyridine (478) as a ligand in DCM at room temperature under 400 nm LED irradiation were employed giving sulfonyl fluorides 471 in 35–66% yields (Scheme 233). In the proposed mechanism, the acridine catalyst activates the carboxylic acid after irradiation to give a carboxy radical A, which after decarboxylation forms the alkyl radical B. After trapping of radical B by SO2, the alkylsulfonyl radical C is formed. The Cu(II) complex undergoes SET with Selectfluor to generate a Cu(II) complex and radical D. The last step is a fluorine atom transfer (FAT) process to yield the product and regenerate the Cu(I) complex.
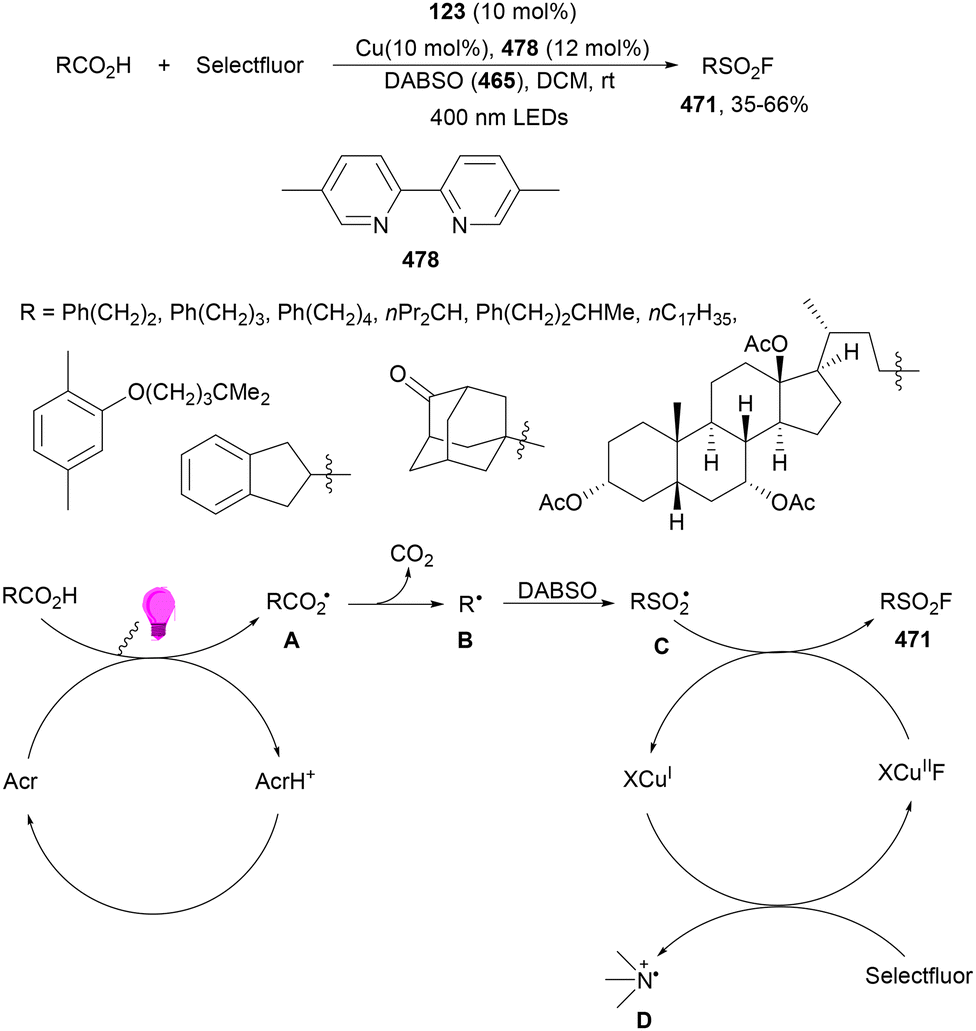 | ||
| Scheme 233 Decarboxylative fluorosulfonylation of aliphatic carboxylic acids with selectfluor and copper under photocatalysis. | ||
The same group432 reported the decarboxylative fluorosulfonylation of aldoxime esters 479 with DABSO (465) and NFSI in the presence of Ir complex 4 as a PC, K3PO4 as a base in a 2:1 MeCN/DCM mixture at room temperature under blue LED irradiation (Scheme 234). Primary and secondary alkyl esters were transformed into sulfonyl fluorides 471 with good yields. In the proposed mechanism, photoexcited PC* sensitizes the oxime esters 479 to the excited state A through a triple-triplet energy-transfer process. Subsequent homolysis of the N–O bond affords the iminyl radical B and the alkyl radical C by releasing CO2. Radical C is trapped by SO2 to deliver the alkylsulfonyl radical D, which is captured by NFSI through the FAT process to give the sulfonyl fluoride 471.
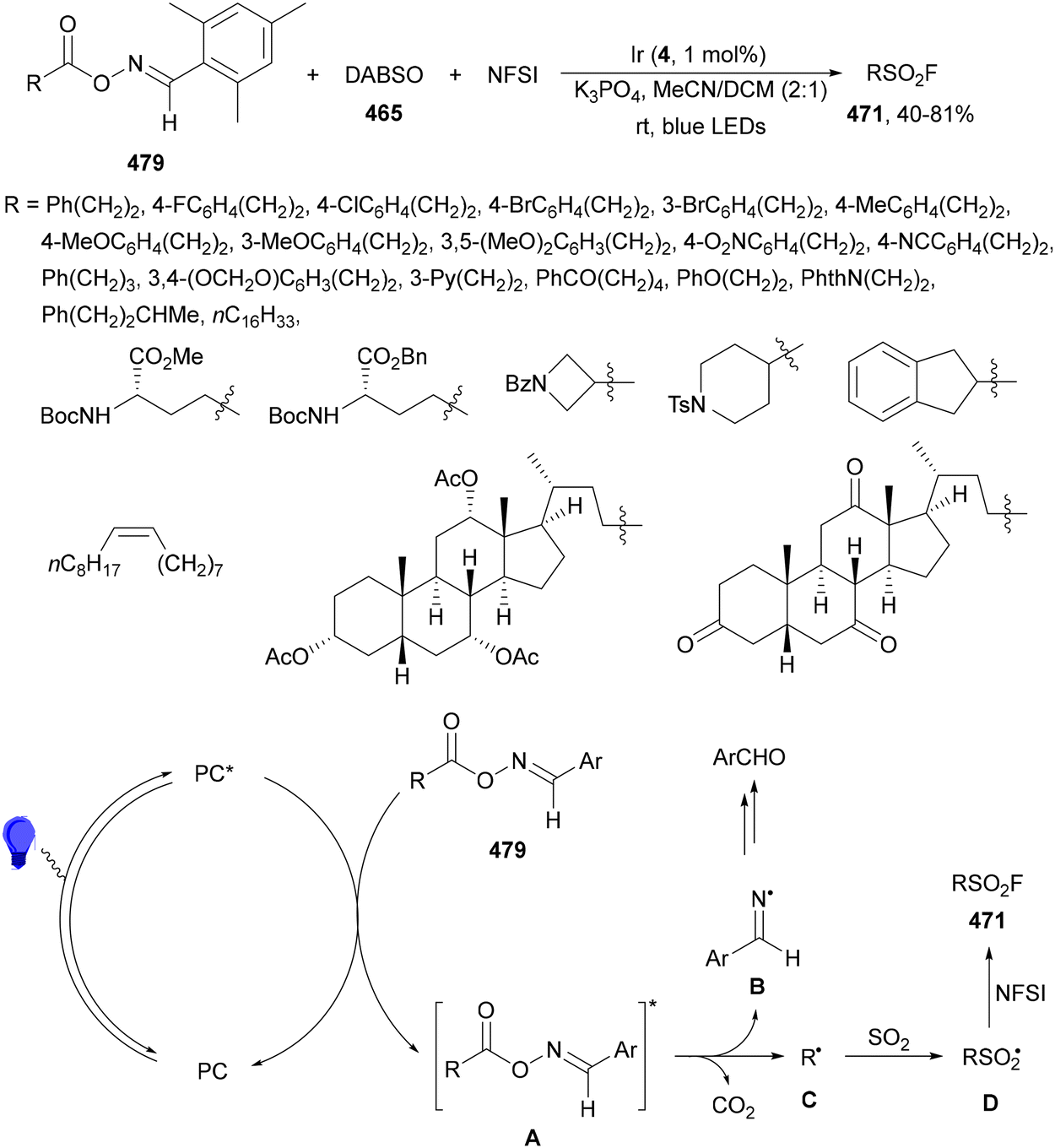 | ||
| Scheme 234 Decarboxylative fluorosulfonylation of aldoxime esters 479 with DABSO (465) and NFSI under Ir 4 photocatalysis. | ||
Copper LMCT54,55 has been employed by MacMillan and co-workers433 for the synthesis of sulfonyl fluorides 471 and chlorides 470 from aromatic and heteroaromatic carboxylic acids. Decarboxylative fluorosulfonylation was carried out using Cu(MeCN)4BF4 (50 mol%), Selectfluor, a solution of SO2 in MeCN under high intensity source of 365 nm LED irradiation to provide the corresponding sulfonyl fluorides 471 in 48–82% yields (Scheme 235a). This method was applied to late stage modification of drugs such as ataluren, lumacaftor and celebrex in 41–62% yields. In the case of sulfonyl chlorides 470, apart from Cu(MeCN)4BF4 (20 mol%) and SO2, 1-fluor-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) as an oxidant and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) as a chlorine atom source were used (Scheme 235b). The resulting sulfonyl chlorides 470 were transformed into sulfonamides by adding morpholine and DIPEA to the crude reaction mixture after irradiation and removal of unreacted SO2. The addition of LiBF4 was beneficial to avoid the formation of the sulfonyl fluoride byproduct. The corresponding amine was then added and MeCN or THF was used as solvent providing sulfonamides 479 with 45–64% overall yields.
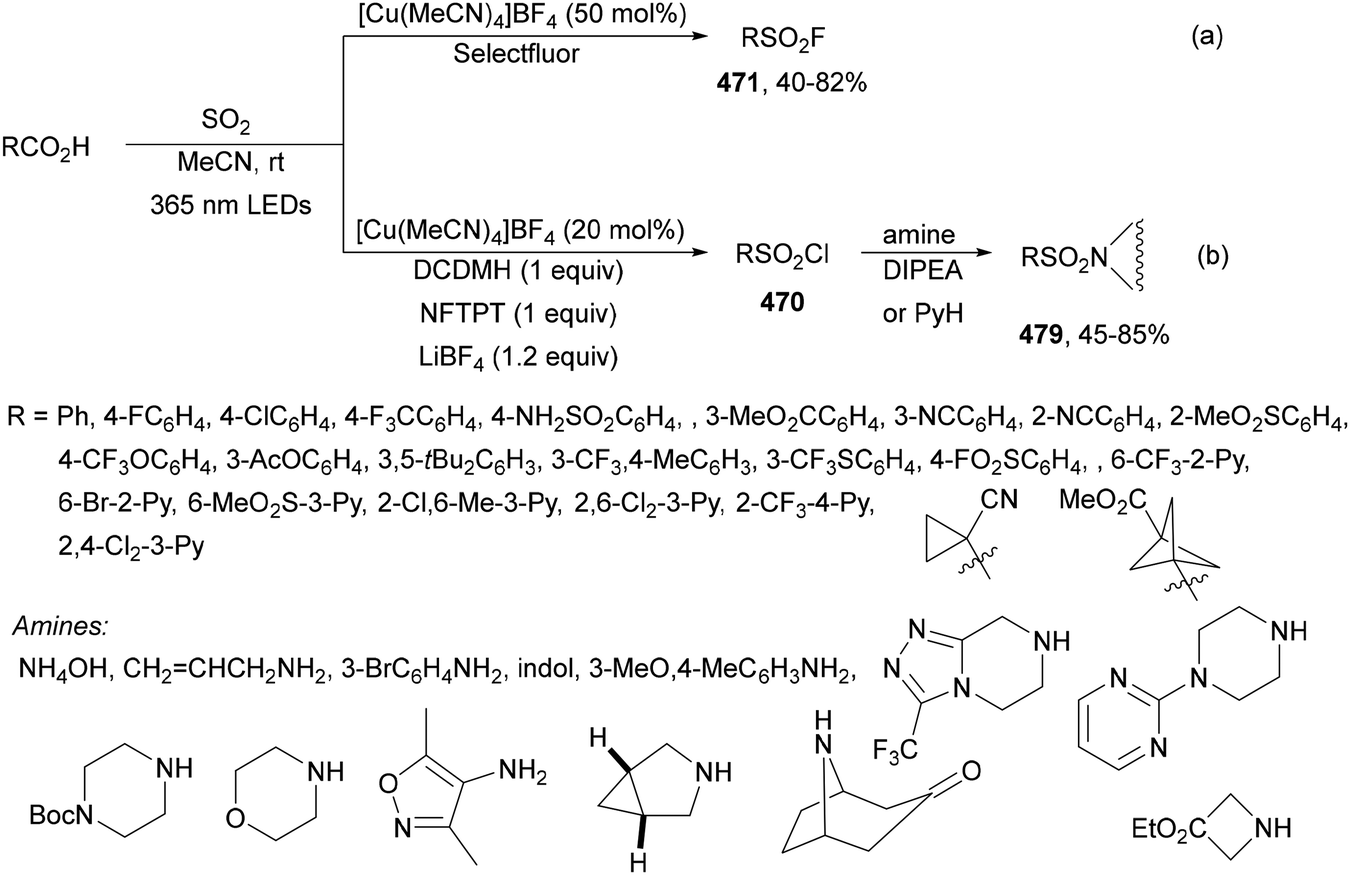 | ||
| Scheme 235 Decarboxylative fluoro- and chlorosulfonylation of aromatic and heteroaromatic carboxylic acids under Cu LCTM photocatalysis. | ||
Larionov and co-workers434 reported a direct amidosulfonylation of aliphatic carboxylic acids by in situ generation of sulfinic acids using acridine and Cu dual photocatalysis. In the presence of acridine 123, DABSO (465), CuF2 and O-benzylhydroxylamine as a ligand for aliphatic amines in DCM under 400 nm LED irradiation, the corresponding sulfonamides 479 were isolated in 50–92% yields (Scheme 236a). When aromatic or heteroaromatic amines were used, CuOTf, acridine 125, DABSO (465) and tert-butyl perbenzoate as oxidants gave sulfonamides 479 in 51–91% yields (Scheme 236b). Sulfonyl azides 474 were prepared using copper 2-thiophene carboxylate (CuTC) and acridine 123 as catalysts in the presence of DABSO as a SO2 source, tBuO2Bz as an oxidant and NaN3 in a 3:1 mixture of PhCF3/MeCN under 400 nm LED irradiation (Scheme 236c). Sulfonamides derived from chenodeoxycholic acid, aleuritic acid, gibberellic acid and carbohydrate derivatives as well as pinonic acid have been produced in 52–89% yields.
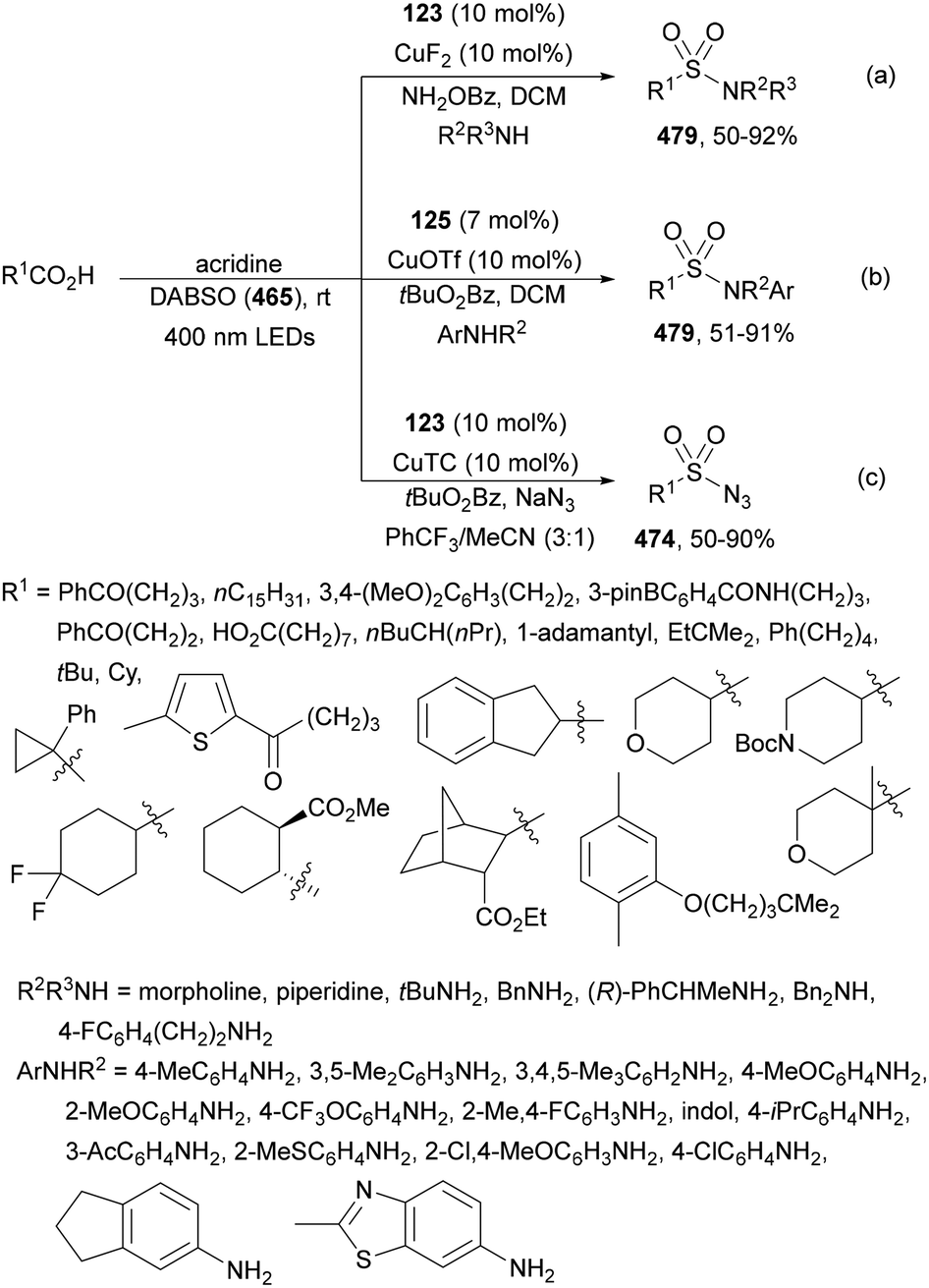 | ||
| Scheme 236 Decarboxylative amido- and azidosulfonylation of aliphatic carboxylic acids using DABSO (465) under dual acridine/Cu photocatalysis. | ||
Willis and co-workers421 employed sulfinylamine TrNSO (462) for the synthesis of sulfinamides 463 (Scheme 226). When N-t-butoxysulfinamide (tBuONSO, 480) was used as a sulfinamidation reagent, intermediate sulfinamides 481 were formed. Subsequent in situ transformation by hydrolysis with NaOH in IPA or an amine in toluene under MW irradiation at 90 °C, sulfonamides 479 or sulfonimidamides 482, respectively, were isolated (Scheme 237a or b).
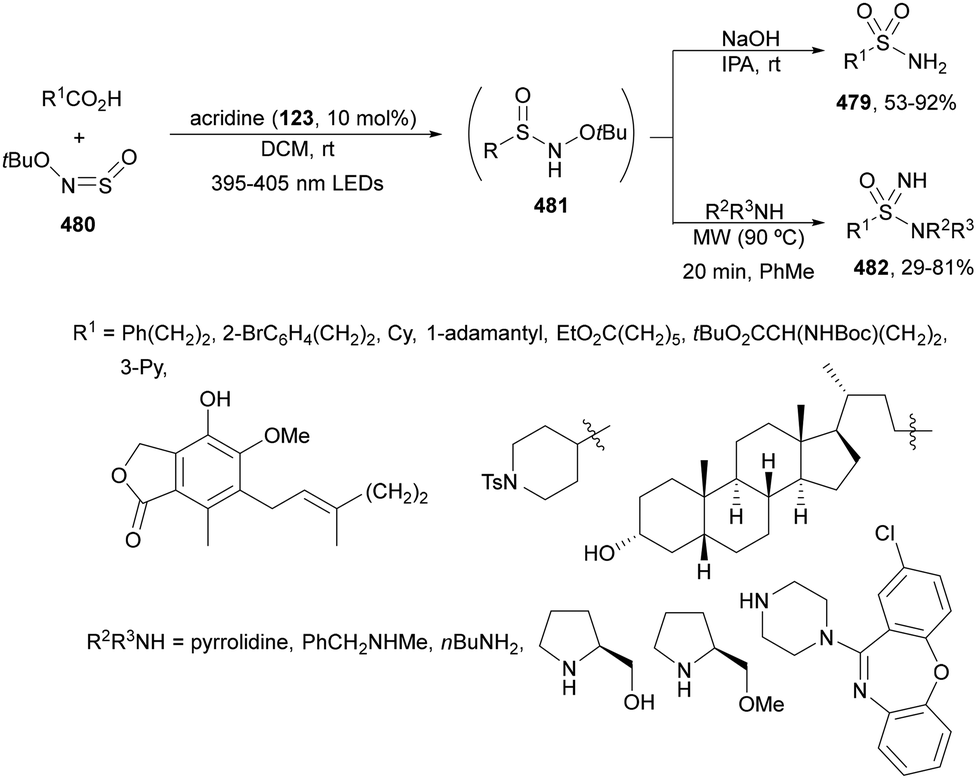 | ||
| Scheme 237 Decarboxylative sulfinamidation of aliphatic carboxylic acids with tBuONSO (480) under acridine photocatalysis followed by hydrolysis or aminolysis. | ||
Aminosulfonylation of aromatic and heteroaromatic carboxyl oxime esters 454 under visible-light induced decarboxylation has been recently reported by Zhang, Yang and co-workers.435 Working with Ir complex 4 as a PC, in the presence of DABSO (465) and NH4Cl in MeCN under Ar and blue LED irradiation, the corresponding N-sulfonyl ketimines 483 were formed. These products were transformed into sulfonamides 479 by deprotection with 4 M HCl in dioxane at room temperature (Scheme 238). This procedure was applied to the large-stage modification of drugs and natural products such as acipimox, dehydrochlolic acid, fusaric acid, a picolinafen intermediate and a PHA-543613 (selective α7-nAChR) intermediate, sorafenib and flazasulfuron.
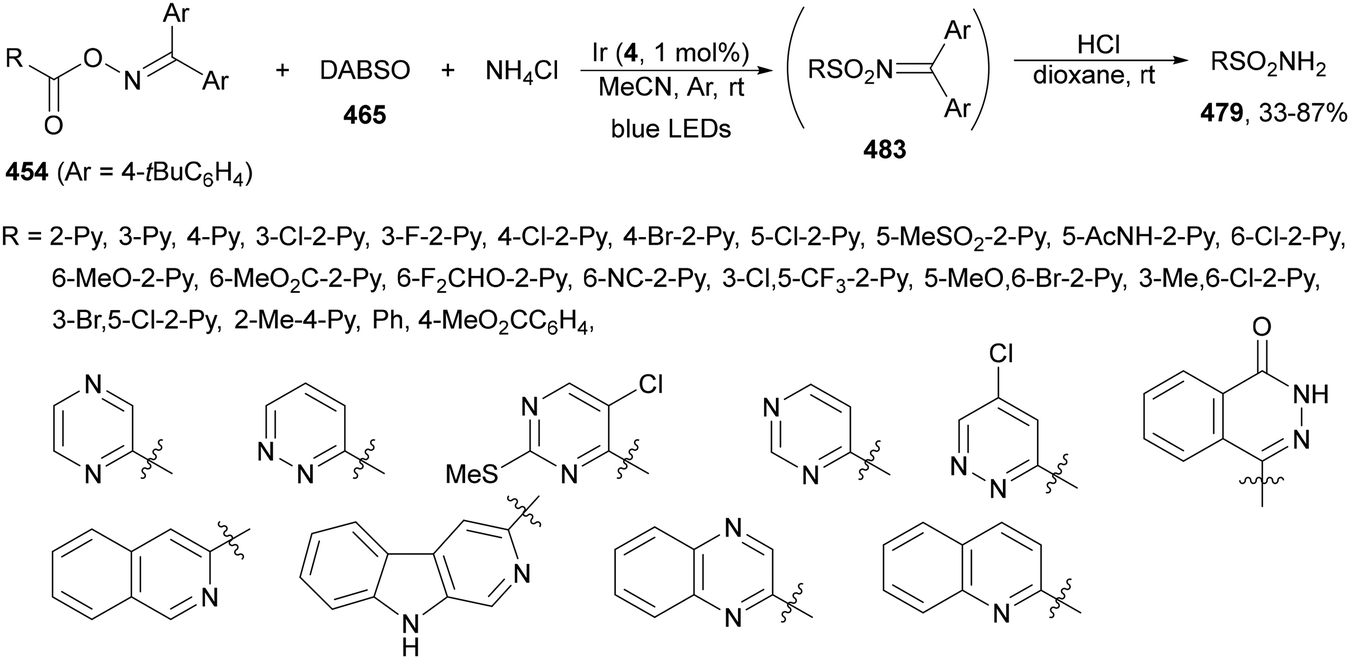 | ||
| Scheme 238 Decarboxylative aminosulfonation of aromatic and heteroaromatic oxime esters 454 with DABSO (465) and NH4Cl under Ir 4 photocatalysis. | ||
Chen, Wu and co-workers436 performed a similar aminosulfonylation of aliphatic and aromatic carboxylic oxime esters 454 derived from benzophenone using 4,4-dimethoxybenzophenone as a PC. When the reaction was carried out in MeCN at 40–50 °C under N2 and 390 nm Kessil lamp irradiation, the corresponding N-sulfonyl ketimines 483 were isolated in 28–77% yields (Scheme 239a), whereas in the presence of NH4Cl resulted in sulfonamides 479 in 46–95% yields (Scheme 239b). Both procedures were applied to natural products and drugs such as stearic acid, α-tocopheryl succinate and dehydrochlolic acid.
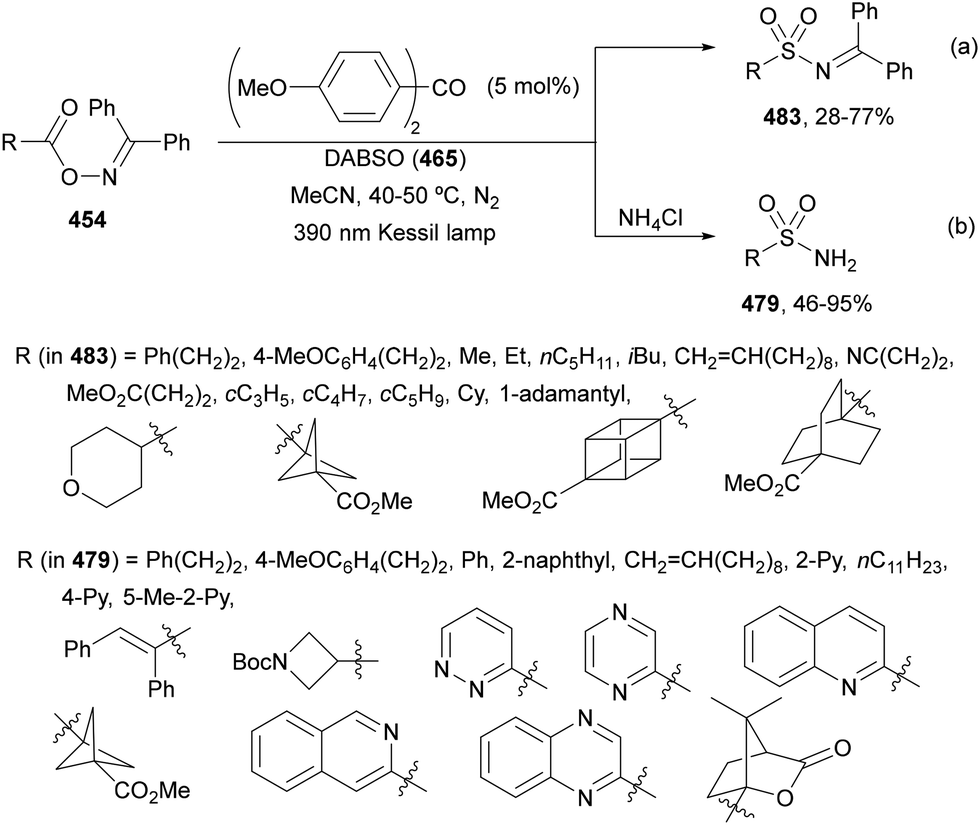 | ||
| Scheme 239 Decarboxylative aminosulfonylation of aliphatic and aromatic carboxylic oxime esters 454 with DABSO (465) under (4-MeOC6H4)2CO photocatalysis. | ||
As a summary of Section 3.3, acridine-photocatalyzed thiolations have been employed for alkyl carboxylic acids using thionocarbonates or a more economical elemental sulfur. In the case of alkyl NHPI esters, Eosin Y-Na2was used as a PC and 4-methoxybenxylamide as a sulfur source. Recent thioetherification of aliphatic carboxylic acids has been performed with trisulfide dioxides, thionosulfonates and a perfluorinated disulfide. The most sustainable method used Fe(NO3)2 as a LMCT catalyst not only for aliphatic but also for heteroaromatic carboxylic acids for the thioetherification with thiosulfonates. Aliphatic NHPI esters have been recently transformed into thiocyanates under Ir photocatalysis. Xanthylation photodecarboxylative processes have been used for thioesterification of carboxylic acids and benzophenone oxime esters. Sulfilimination of alkyl NHPI esters have been recently described under Ir and Cu dual photocatalysis using sulfenamides. Direct sulfinylation of aliphatic carboxylic acids has been achieved with sodium sulfinates and p-bromobenzoyl chloride, which generates in situ the sulfinyl sulfone. Acridine-photocatalyzed reaction provides the corresponding sulfoxides. Sulfinamidation of aliphatic carboxylic acids can be performed using sulfinylamine reagents RNSO and acridine as photocatalysts. Sulfonation of aliphatic carboxylic acids to give sulfones employed DABSO as a source of SO2 and acridines or TBAFeCl4 as PCs. In the case of vinyl sulfones decarboxylative sulfonylation of cinnamic acids used aryl sulfonate phenol ethers without a PC. When arylsulfonyl azides, TosMIC or β-keto sulfones were used as sulfonylating reagents, vinyl aryl sulfones can also be prepared with rhodamine or Eosin Y as PCs. Fluorosulfonylation of aliphatic carboxylic acids has been carried out with DABSO and N-fluorobenzene sulfonamide using acridine as a PC or with Selectfluor with acridine and copper. Amidosulfonylation of carboxylic acids employed DABSO, Cu salts and acridines as PCs. N-t-Butoxy sulfinamide can also be used in the presence of amines.
3.4. Carbon–nitrogen bond-forming reactions
Decarboxylative C–N bond formation has been achieved by amination of aliphatic carboxylic acids and NHPI esters with aliphatic, aromatic and heteroaromatic amines. Amidation has been reported with sulfonamides, sulfoximines ureas and isocyanates, and by amination of α-keto acids. Other reactions used azides, nitro compounds, nitrites, diaziridines and azodicarboxylates delivering C–N and CN bonds.
The direct decarboxylative N-alkylation of aliphatic carboxylic acids under Ru(dtbbpy)3(PF6)2 (429) photocatalysis in the presence of hypervalent iodine reagent 428 was described by Terrett and co-workers.439 Similar reaction conditions were previously applied to etherification of carboxylic acids (Scheme 209).389 A wide range of azoles were alkylated with primary, secondary and tertiary aliphatic carboxylic acids in DCE/HFIP (2:1) at room temperature under visible-light irradiation to give products 484 with good yields (Scheme 240).
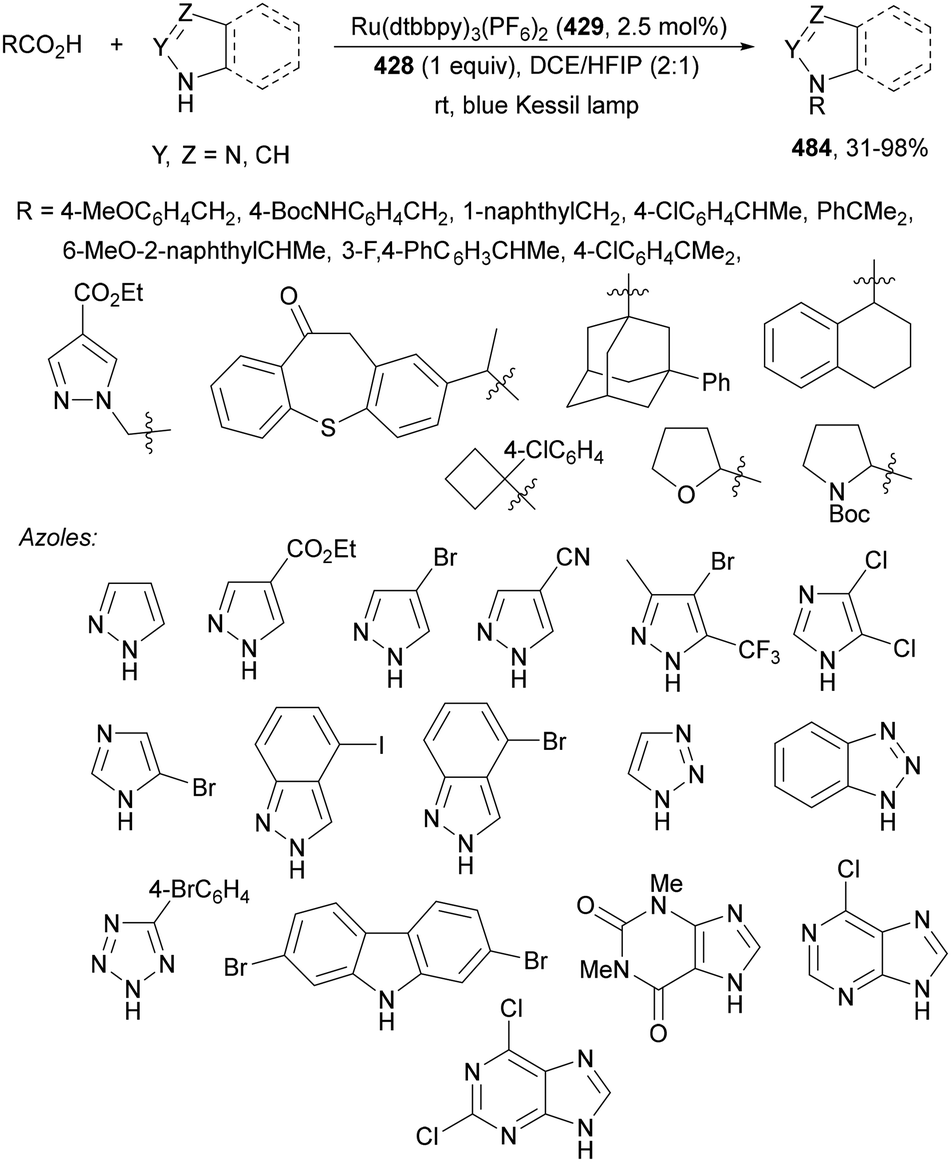 | ||
| Scheme 240 Decarboxylative amination of aliphatic carboxylic acids with azoles under Ru 429 photocatalysis. | ||
Photoinduced iron/copper dual catalysis was applied to C–C bond formation by decarboxylative Giese reaction of carboxylic acids and electron-deficient alkenes (Scheme 2).53 These reaction conditions were applied to form C–N bonds by amination of carboxylic acids. In the presence of Fe(OAc)2 and Cu(acac)2 with DTBP as an oxidant, DBU as a base in EtOAc at room temperature under N2 and purple LED irradiation, the corresponding amines were obtained in moderate to very good yields (Scheme 241). Aromatic amines were alkylated by decarboxylation of primary, secondary and tertiary aliphatic carboxylic acids. In the proposed mechanism, in the iron catalytic cycle, Fe(II) is oxidized to Fe(III) in the presence of DTBP and DBU. Then, the LMCT process under 390 nm irradiation generates R1CO2˙ (A) and subsequently R1˙ after decarboxylation. In the Cu catalytic cycle, the amine and Cu(II) forms intermediate B after deprotonation with DBU. This species B is trapped by radical R1˙ to form an alkyl Cu(III) species C, which after reductive elimination formed the product and Cu(I). In the last step, Cu(I) is oxidized by DTBP to Cu(II).
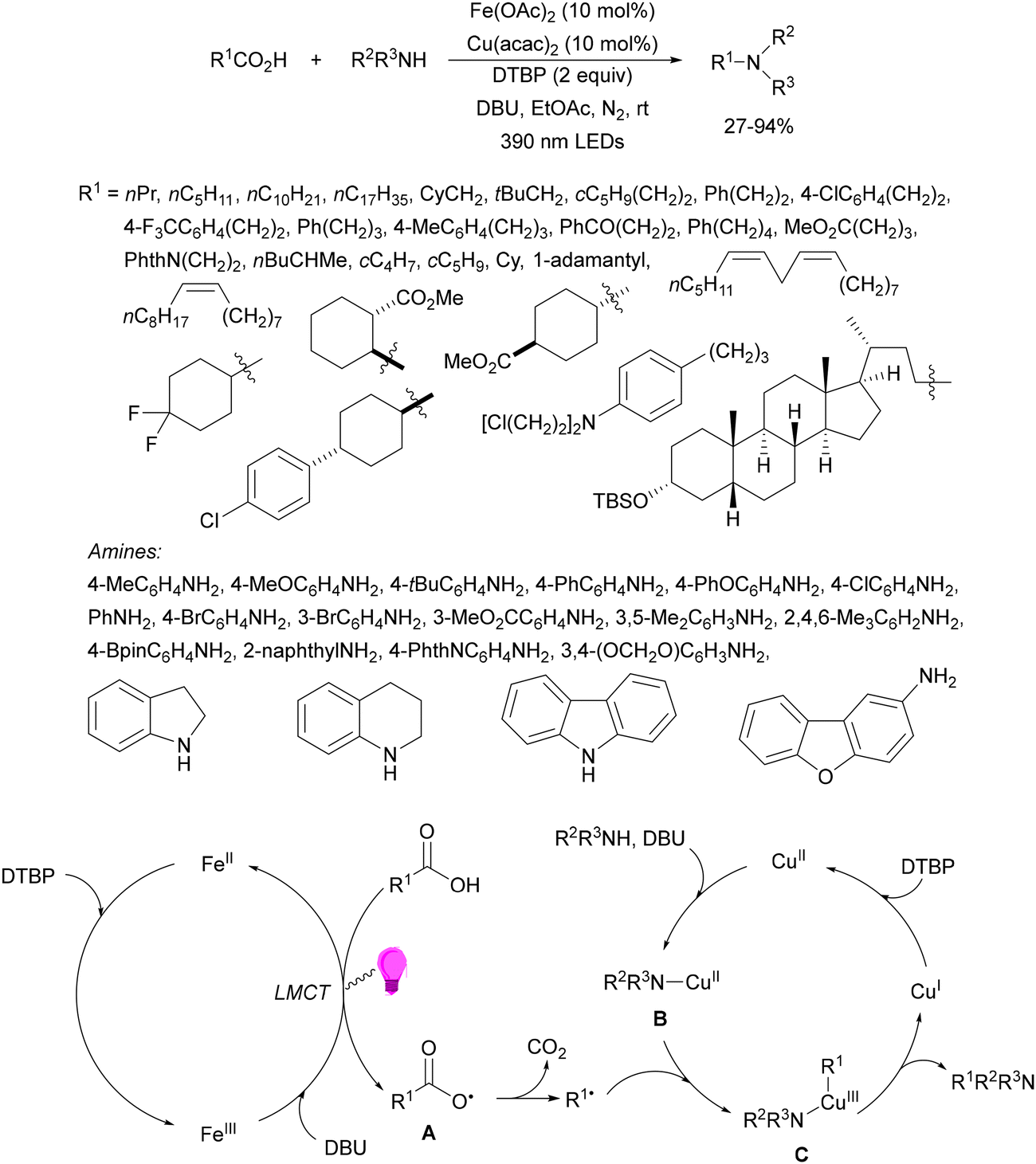 | ||
| Scheme 241 Decarboxylative amination of aliphatic carboxylic acids with amines and azoles under Fe and Cu photocatalysis. | ||
Yoon and co-workers295 employed 3 equivalents of FeCl3 for the amination of flurbiprofen with aliphatic and aromatic amines. In this case, the amine should be added once to the reaction mixture and irradiated with 427 nm blue LEDs in order to avoid binding of the amine with FeCl3. This salt acted as a PC through the LMCT process and also as a terminal oxidant. Working in the presence of Na3PO4 as a base in MeCN at room temperature, the corresponding amines, including imidazole, were obtained in 59–94% yields (Scheme 242).
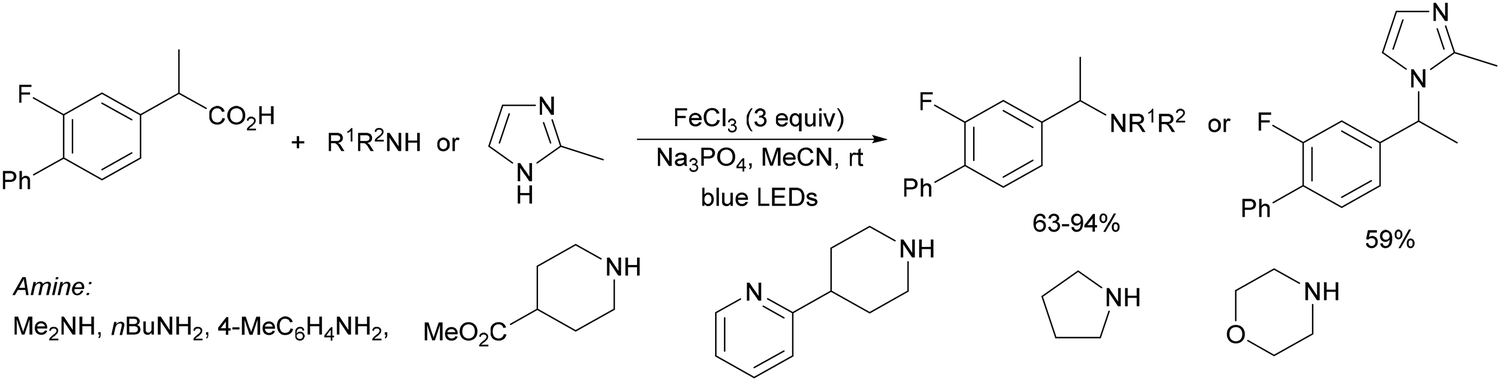 | ||
| Scheme 242 Decarboxylative amination of flurbiprofen with amines in the presence of FeCl3 photocatalysis. | ||
Decarboxylative amination of alkyl NHPI esters 69 was described in 2018 by Hu and co-workers440 using Ru(bpy)3(PF6)2 (10 mol%) and CuBr (20 mol%) as catalysts under blue LED irradiation. Alkyl NHPI esters and primary anilines gave the corresponding products in 28–96% yields. When benzophenone-derived imines were employed as nucleophiles and Ir complex 4 and Cu(MeCN)4PF6 as catalysts even hindered alkyl NHPI esters were iminated with 36–99% yields.441 This process can also be carried out under metal-free conditions using 4CzIPN (25) as a PC.442
Li, Guan and co-workers392 described not only etherification reactions with NHPI esters 69 (see Scheme 210), but also amination with indoles, indazoles and azaindoles using NaI-PPh3 and CuBr as a dual metallaphotoredox catalytic system (Scheme 243). The resulting alkylated heterocyclic amines were obtained with excellent yields working with BTMG as a base, in dioxane at room temperature under blue LED irradiation. Mechanistic proposal is depicted in Scheme 210 for the etherification process.
 | ||
| Scheme 243 Decarboxylative amination of alkyl NHPI esters 69 with heterocyclic amines under NaI-PPh3/CuBr photocatalysis. | ||
Decarboxylative N-alkylation of azoles with NHPI esters 69 has been carried out with N-phenylbenzo[b]phenothiazine (PTH, 484)443 as an organophotoredox catalyst. Ohmiya and co-workers444 performed this amination in the presence of the pyridinium salt of 2,4,6-collidine (485) in DCE under blue LED irradiation to give the corresponding N-alkylated azoles in moderate to good yields (Scheme 244). The proposed mechanism for this radical-polar crossover starts with the formation of a charge-transfer complex A between the phenothiazine (PTH) and the NHPI ester. Irradiation induces SET from PTH to ester affording radical cation species from PTH B and the radical anion of NHPI ester C. In the presence of collidine/HBF4, C generates an alkyl radical D after CO2, PhthN− and collidine release. Combination of B and D through SET affords alkylsulfonium intermediate E, which reacts with azole to give the product and regenerate 485 and PTH 484.
 | ||
| Scheme 244 Decarboxylative amination of NHPI esters 69 with azoles under PTH 484 and 485 photocatalysis. | ||
Glycosyl NHPI esters 405 have been used by Yang, Li and co-workers445 for the synthesis of nucleoside analogues under Ir/Cu photoredox conditions. This reaction was performed with Ir(ppy)3 complex 89 and Cu(MeCN)4PF6 as a catalyst and Et3N as a base at room temperature in MeCN under blue LED irradiation. Different nucleobase derivatives were used as nucleophiles to provide the corresponding nucleoside analogues in 40–74% yields (Scheme 245). This method was applied to the total synthesis of the acid-labile oxetanocin A, a natural product isolated from Bacillus megaterium, which exhibits inhibitory activity against a wide range of viruses including HIV and HBV.446 Bristol-Myers-Squibb developed the broad-spectrum antiviral agent lobucavir based on oxetanocin A structure.
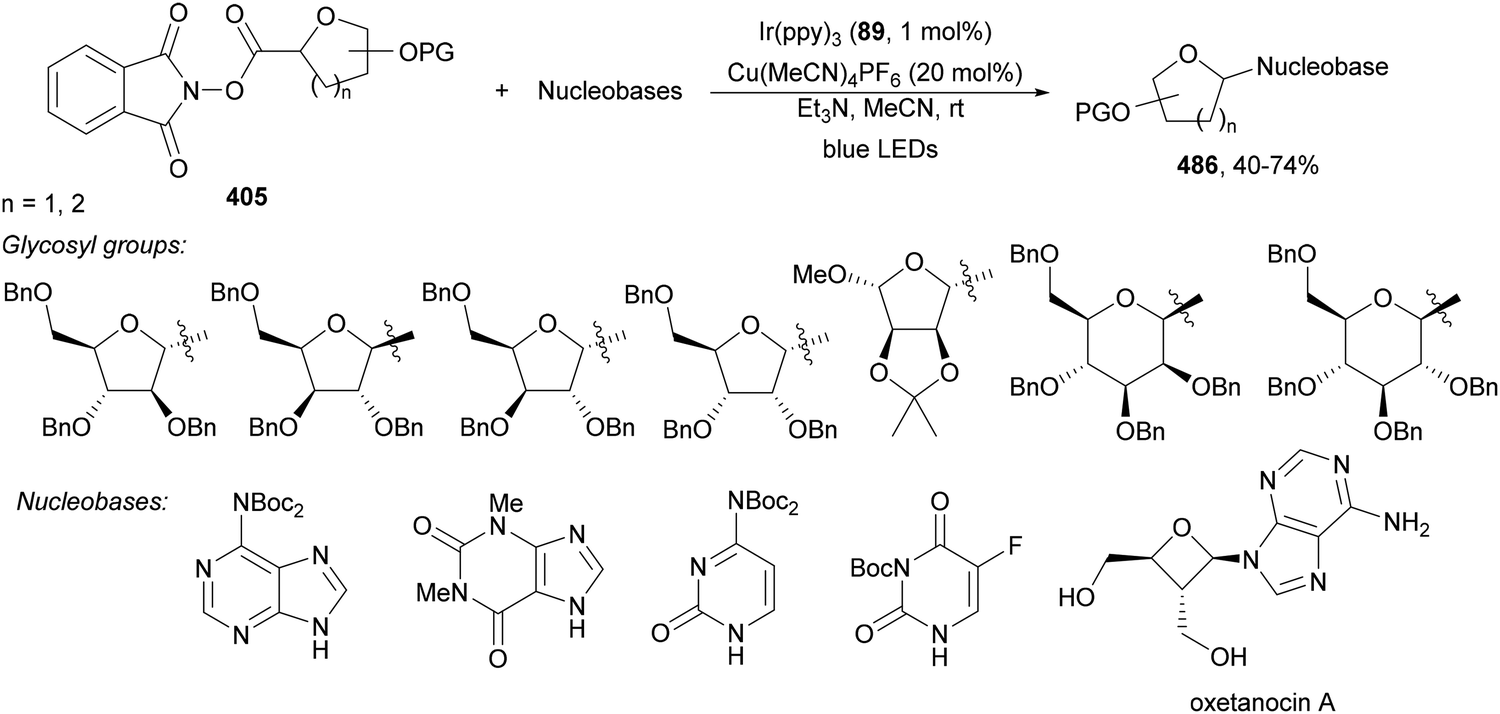 | ||
| Scheme 245 Decarboxylative amination of glycosyl NHPI esters 405 with nucleobases under Ir/Cu photocatalysis. | ||
In the case of carboxylic acids, Terrett and co-workers439 applied similar reaction conditions already described for azoles (Scheme 240), for ureas, carbamates and sulfonamides. 6-MeO-2-naphthyl-1-ethanecarboxylic acid reacted with these nucleophiles in the presence of iodine(III) reagent 428 as an oxidant of the benzylic radical intermediate to a carbocation by Ru 429 as a PC (Scheme 209). The resulting products were obtained with modest to good yields and in the case of Cbz-carbamate the subsequent hydrogenation step gives the primary amine in 45% yield after two steps (Scheme 246).
 | ||
| Scheme 246 Decarboxylative amidation of carboxylic acids with sulfonamides, ureas and carbamates in the presence of iodine reagent 428 under Ru 429 photocatalysis. | ||
Yoon and co-workers447 reported decarboxylative cross-coupling of carboxylic acids with sulfonamides, carboxamides and carbamates under copper-mediated visible-light irradiation (Scheme 247). Aliphatic secondary and tertiary carboxylic acids were amidated in the presence of 2.5 equivalents of Cu(OTf)2, Na3PO4 as a base in MeCN at room temperature under blue LED irradiation to afford the corresponding products with, in general, very good yields. In this reaction a LMCT process54,55 is operating, with Cu(II) carboxylate species being the chromophore.
 | ||
| Scheme 247 Decarboxylative amidation of aliphatic carboxylic acids with sulfonamides, carboxamides and carbamates under Cu(OTf)2 photocatalysis. | ||
Decarboxylative sulfonamidation of carboxylic acids was described by the same group295 using 3 equivalents of FeCl3 as in the case of amination reactions (see Scheme 242). In this case, different aliphatic carboxylic acids and sulfonamides were allowed to react under blue LED irradiation to give the corresponding N-alkylated sulfonamides with modest to high yields (Scheme 248).
 | ||
| Scheme 248 Decarboxylative sulfonamidation of aliphatic carboxylic acids under FeCl3 photocatalysis. | ||
Sulfoximination of benzoic acids has been achieved by a photoinduced LMCT54,55 process by Ritter and co-workers.448N-Arylated sulfoximines 488 were prepared by decarboxylative arylation of sulfoximines 487 with lithium benzoates in the presence of 2.5 equivalents of Cu(OTf)2, LiOMe as a base, DTBP as an oxidant in MeCN at 35 °C under purple LED irradiation (Scheme 249). Enantiopure sulfoximines gave the corresponding N-arylated products 488a and 488b without racemization. In the proposed mechanism, photoinduced LMCT of copper carboxylates A gives aryl carboxy radical intermediates B, which after decarboxylation affords aryl radicals C. Subsequent capture of C by copper generates the arylcopper(III) intermediate D. Final reductive elimination of D provides products 488 and Cu(I), which can be oxidized by DTBP regenerating Cu(II).
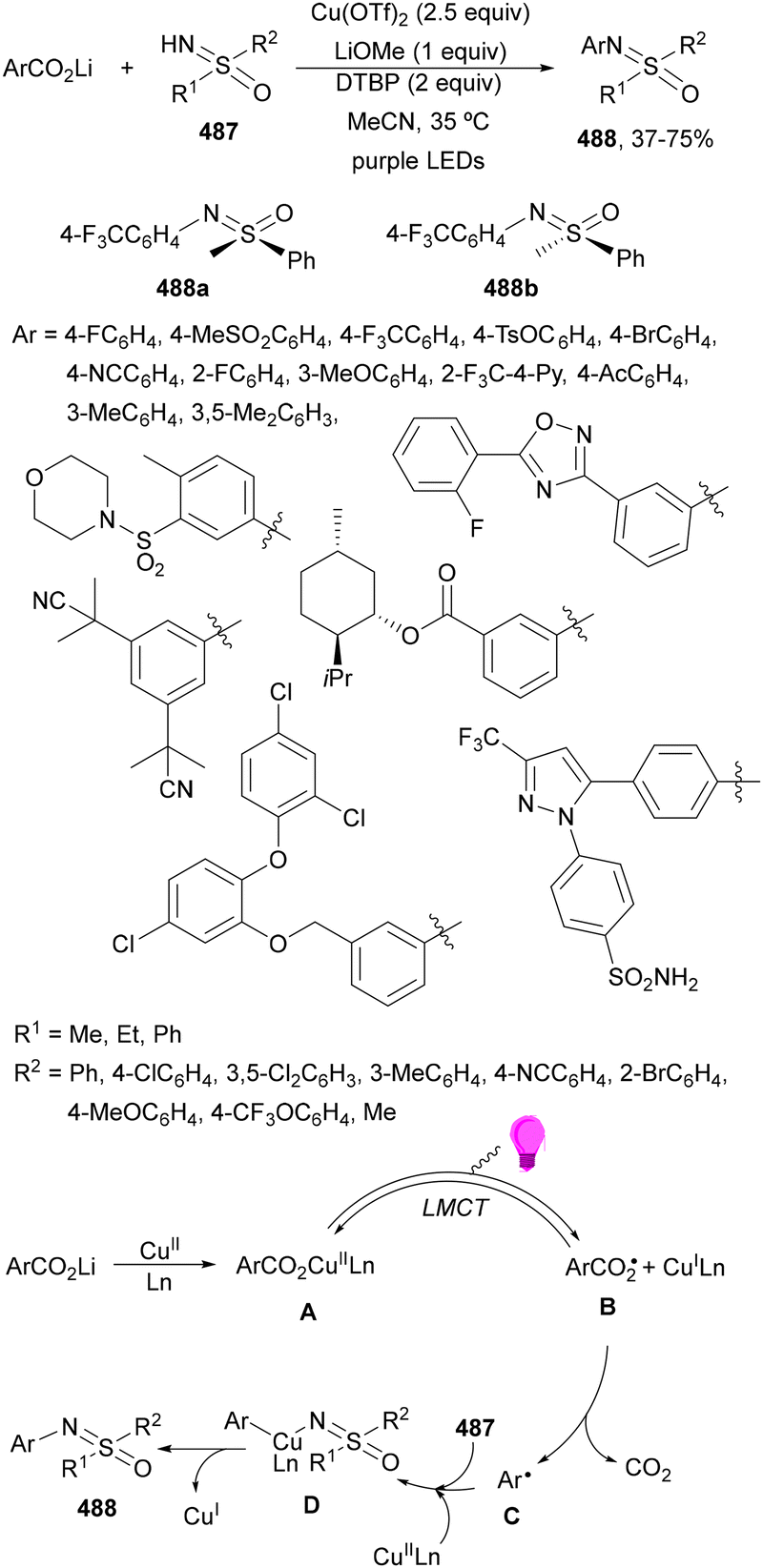 | ||
| Scheme 249 Decarboxylative sulfoximination of lithium benzoates with sulfoximines 487 under Cu(OTf)2 photocatalysis. | ||
Decarboxylative amidation of α-keto acids with primary aliphatic and aromatic amines was performed by Lan, Lei and co-workers449 in 2014 under Ru(phen)3Cl2 visible-light photocatalysis. The same amidation was further carried out in the absence of PC by Xu and co-workers450 under an O2 atmosphere.
Landais and co-workers398 reported the synthesis of ureas by decarboxylation of oxamic acids 15 in the presence of aliphatic primary amines. This amidation procedure was carried out with 4CzIPN (25) as a PC and BI-OAc as an oxidant in DCE at room temperature under blue LED irradiation followed by adding Et3N as a base and the amine in the same reaction media (Scheme 250). In the first step, decarboxylation takes place to give the carbamoyl radical, which by oxidation generates intermediate isocyanate 162 (see Scheme 214). In the second step, the amine was added and reacted with isocyanate to give the corresponding urea.
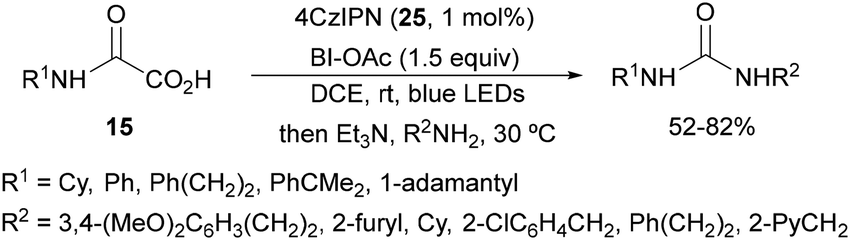 | ||
| Scheme 250 Decarboxylative amidation of oxamic acids 15 with primary aliphatic amines under 4CzIPN (25) and BI-OAc photocatalysis. | ||
Decarboxylation of carboxylic acids under iron photocatalysis via a photoinduced LMCT54,55 mechanism allowed the formation of alkyl radicals210 (Schemes 110 and 127). When phenyl isocyanate 162 was employed as a radical acceptor, the corresponding amides were obtained in 43–97% yields (Scheme 251). In this case, the reaction was performed in the absence of ligand 298.
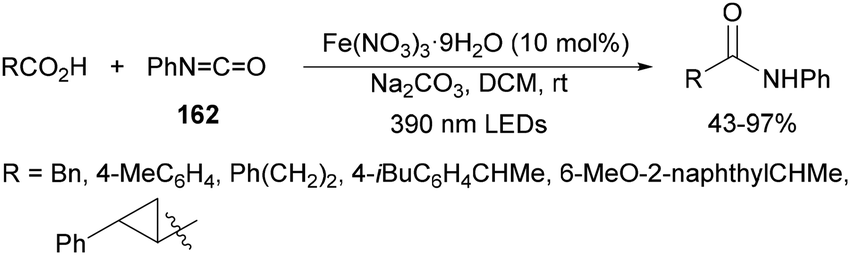 | ||
| Scheme 251 Decarboxylative amidation of aliphatic carboxylic acids with phenyl isocyanate 162 under Fe(NO3)3 photocatalysis. | ||
Midya, Ghosh and co-workers451 recently reported an oxidative decarboxylative cross-coupling of α,β-unsaturated acids with aromatic amines under Ir/Pd dual photocatalysis. This α-ketoamidation took place with Ir complex 4 as a PC, Pd(TFA)2 as a catalyst, K2HPO4 as a base in aqueous DMSO in open air at room temperature under 465 nm LED irradiation affording α-keto amides 489 with good yields (Scheme 252). In the photocatalytic cycle, Ir(III)* is reduced by a SET with cinnamic acid to generate a radical cationic species A and Ir(II). SET from Ir(II) to molecular oxygen promoted superoxide radical anion O2−˙ generation and regenerated an Ir(III) PC. Addition of water to species A in the presence of K2HPO4 forms the benzyl alcohol intermediate B. Oxidation of B by O2−˙ gives the β-keto radical species C, which by oxidative addition to Pd catalyst D forms the Pd(III) intermediate E. Subsequent SET of D by O2−˙ provides Pd(IV) intermediate F and peroxide O22−. Reductive elimination of species F gives intermediate G with autogeneration of precatalyst Pd(TFA)2. Intermediate G undergoes decarboxylative protonation giving H, which by oxidation with O2 forms imine I and O2−˙ addition gives J. Kornblum de la Mare rearrangement of J forms the keto amide 489. This mechanism was supported through intermediate trapping and isotope labeling experiments.
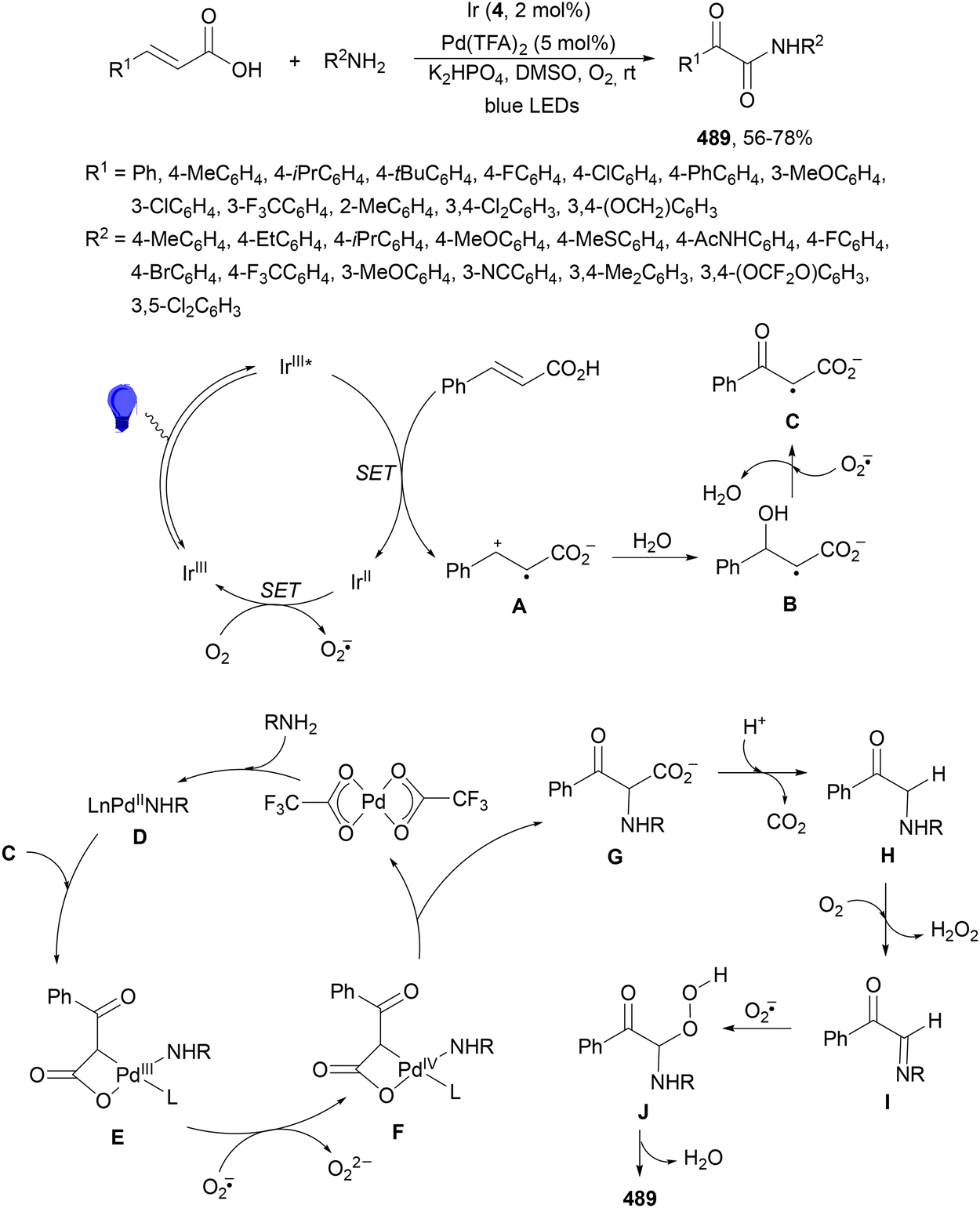 | ||
| Scheme 252 Decarboxylative α-keto amidation of α,β-unsaturated acids with aromatic amines under Ir/Pd photocatalysis. | ||
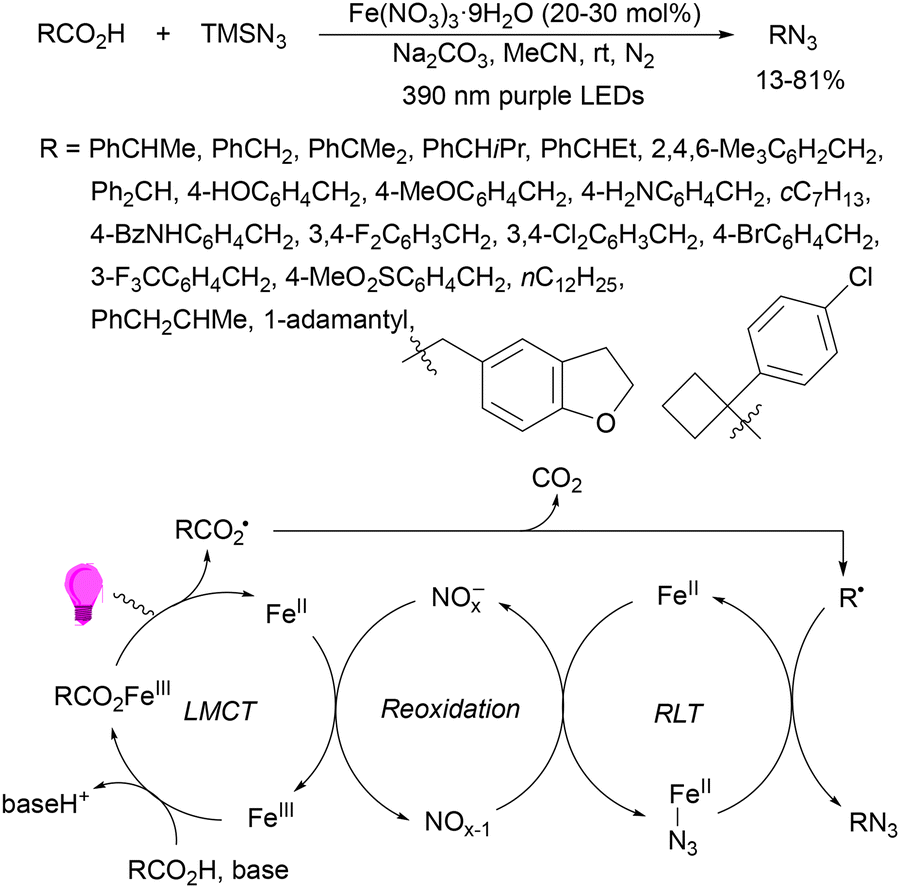 | ||
| Scheme 253 Decarboxylative azidation of aliphatic carboxylic acids with TMSN3 under Fe(NO3)3 photocatalysis. | ||
Decarboxylative amination with nitroarenes has been developed by Xie and co-workers.454,455 Working with arylacetic acid and nitroarenes in the presence of 4CzIPN (25) as a PC and FeI2/tetraphenylporphirin (TPP) as a catalyst, (EtO)3SiH as a reductant, and 2,6-lutidine as a base in MeCN at 65 °C under blue LED irradiation, the corresponding aromatic tertiary amines 490 were obtained with good yields (Scheme 254a).454 In this case, the carboxylic acid and the nitroarene were used in a 3:1 molar ratio. Moreover, when two different arylacetic acids and a nitroarene were employed in a 3:3:2 molar ratio a three-component reaction provided non-symmetrical aromatic tertiary amines 491 with moderate yields (Scheme 254b).454 However, when α-alkyl arylacetic acids were used, under similar reaction conditions, secondary aromatic amines 492 (Scheme 254c) were obtained. In the proposed mechanism, the alkyl radical A can be produced via either SET between the carboxylate ion and (4CzIPN)* or direct LMCT pathway. The reduction of nitroarenes with Fe(II) and (EtO)3SiH leads to nitrosoarene intermediate B, which reacts with radical A to form the Fe(III) complex C. Subsequently, intermediate C undergoes ligand exchange with the carboxylic acid to give D and E. Intermediate E is further reduced by Fe(II) and (EtO)3SiH to give the secondary aromatic amine 492.454
 | ||
| Scheme 254 Decarboxylative amination of carboxylic acids with nitroarenes under 4CzIPN/FeI2 photocatalysis. | ||
Xu and co-workers456 reported an iron-catalyzed decarboxylative C–N coupling of alkyl carboxylic acids with sodium nitrite. In the presence of Fe(NO3)3·9H2O, AcOH (2 equivalents) in aqueous THF at room temperature under N2 and 400 nm LED irradiation the corresponding oximes were obtained with very good yields (Scheme 255). This simple and efficient procedure seems to take place by initial ligand exchange of Fe(NO3)2 with NaNO2 to give complex A, which forms complex B by reaction with the carboxylic acid. Photoexcitation of B involves a LMCT process to afford complex C and the carboxy radical D. Fe(II)NO2 (C) leads to after irradiation Fe(II)NO+ (F) in equilibrium with Fe(III)–NO (G). Subsequent NO releases from G and regenerates Fe(III)NO3 (A). The alkyl radical E reacts with NO producing the nitroso compound H, which tautomerizes to the oxime product.
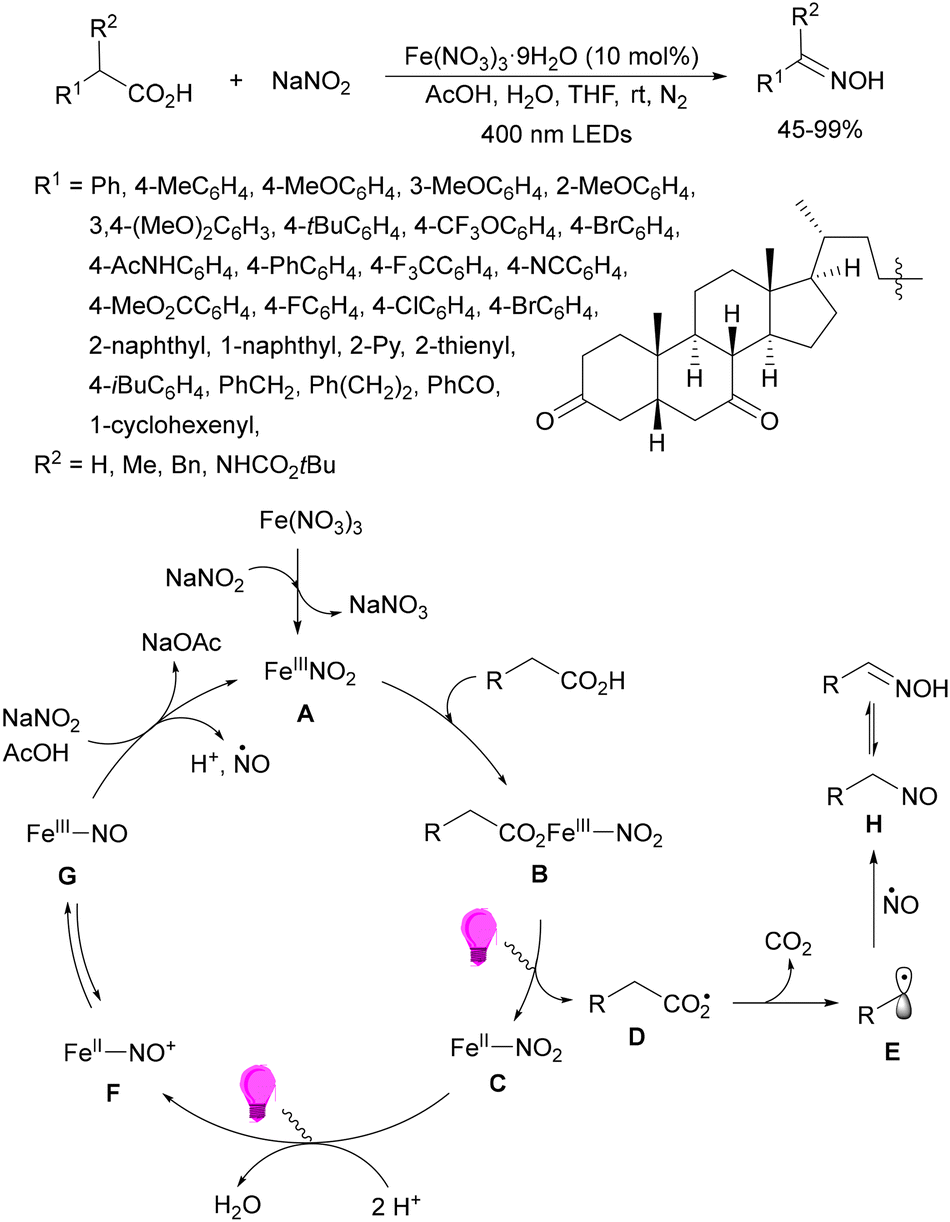 | ||
| Scheme 255 Decarboxylative nitrosation of aliphatic carboxylic acids with NaNO2 under Fe(NO3)3 LMCT photocatalysis. | ||
Diazirine 493 has been employed for the decarboxylative amination of alkyl NHPI esters 69 by Liao and co-workers.457 A chemodivergent381 transformation was achieved depending on the reaction conditions. Thus, working with Eosin Y-Na2 (139), DIPEA as a base in tBuOH under Ar at room temperature and green LED irradiation, imines 494 were isolated in 31–86% yields (Scheme 256a). When DIPEA was replaced by Hantzsch ester (HE) and DME was used as solvent under blue LED irradiation, diaziridines 495 were formed in 36–92% yields (Scheme 256b). These experimental results were explained by the plausible mechanism depicted in Scheme 256. Eosin Y-Na2 (139) is photoexcited to PC* and then is reductively quenched by DIPEA or HE through SET processes affording PC−˙. This PC˙− reduces NHPI ester 69 to A by SET followed by N–O scission and CO2 release to give the alkyl radical B. Addition of B to 493 gives intermediate C, which dimerizes to form tetraazo intermediate D. Subsequently, D undergoes N2 extrusion to produce two molecules of imine 494. On the other hand, intermediate C abstracts a hydrogen from HE+˙ to give diaziridine 495.
 | ||
| Scheme 256 Decarboxylative divergent amination of NHPI esters with diazirine 493 under Eosin Y-Na2 (139) photocatalysis. | ||
Photoredox decarboxylative hydrazination has been carried out by addition of alkyl radicals of aliphatic carboxylic acids to azodicarboxylates.458–462 Wang and co-workers463 have reported the addition of N-aryl glycines to symmetrical azobenzenes using methylene blue (MB, 496) as a PC in MeCN at room temperature under air and blue LED irradiation providing 1,2,4-triaryl-1,2,4-triazolidines 497 with modest to very good yields (Scheme 257). In the case of unsymmetrical azobenzenes, the corresponding thiazolidines 498 were obtained in good yields under the same reaction conditions (Scheme 257b). The proposed mechanism for this decarboxylative addition/cyclization process starts by photoexcitation of MB+ to MB+*, which promotes the oxidative decarboxylation of N-phenyl glycine to produce the aminomethyl radical A and MB˙. Then, addition of A to azobenzene gives radical B, which couples with another radical A to form intermediate C. Subsequent protonation of C gives intermediate D, which after releasing an aniline molecule forms iminium cation E. Intramolecular cyclization of E and deprotonation provides product 497. In the case of MB˙, after oxidation by O2 is back to MB+ generating superoxide radical O2−˙. This O2˙− combines with a proton to give the hydroxyperoxyl radical (HOO˙), which abstracts a proton from N-phenyl glycine to form H2O2. Alternatively, oxidation of N-phenyl glycine by photoexcited azobenzene, generated by energy transfer between azobenzene and MB+*, cannot be ruled out.
 | ||
| Scheme 257 Decarboxylative hydrazination of N-aryl glycines with azobenzenes under methylene blue (496) photocatalysis. | ||
Recent progress in decarboxylative amination reactions of aliphatic carboxylic acids used Fe(OAc)2/Cu(acac)2 and DTBP as an oxidant with aromatic and heteroaromatic amines. Aliphatic amines are used by FeCl3-mediated decarboxylation of arylacetic acids. In the case of NHPI esters, amination with heteroaromatic amines can be carried out under NaI-PPh3 and CuBr photocatalysis. A benzophenothiazine (PHT) can also be used as an organophotoredox catalyst with a broad range of azoles. Glycosyl NHPI esters react with nucleobases under dual Ir/Cu photocatalysis. Decarboxylative amidation of aliphatic carboxylic acids has been performed by LMCT processes mediated by Cu(OTf)2 or FeCl3 and sulfonamides, ureas, carbamates and sulfoximines. Recent amination of oxamic acids with amines to the corresponding ureas is carried out under 4CzIPN and BI-OAc photocatalysis. Aliphatic carboxylic acids can be transformed into carboxamides using phenyl isocyanate under FeCl3 photocatalysis. Amidation of cinnamic acids with aromatic amines under Ir/Pd dual photocatalysis provides α-keto amides. Other nucleophiles such as trimethylsilyl azide and nitroarenes have transformed aliphatic acids into azides and arylamines, respectively, under Fe photocatalysis. In the case of sodium nitrite, the corresponding oximes are prepared also under Fe(III) nitrate LMCT photocatalysis. N-Aryl glycines react with azodicarboxylates under methylene blue photocatalysis forming triazolidines. Diaziridine derived from trifluoroacetophenone has been employed as a nucleophile with aliphatic NHPI esters under an Eosin Y-Na2 organophotoredox catalyst to give either imines in the presence of DIPEA or N-alkyl diaziridines in the presence of Hantzsch ester.
3.5. Carbon-phosphorous bond-forming reactions
Decarboxylative C–P bond formation has been carried out with different phosphorous compounds such as chlorophosphines, phosphites, phosphine oxides, phosphonites and white phosphorus mainly with NHPI esters 69.Larionov and co-workers464 reported in 2019 the coupling of NHPI esters 69 with chlorophosphines using Ir complex 260 as PC under 400 nm LED irradiation to give phosphines. Aggarwal and co-workers465 reported the decarboxylative phosphonylation of N-protected α-amino acid derived NHPI esters 143 with trimethyl phosphite to provide α-amino phosphonates 499 with modest to high yields (Scheme 258). This procedure took place with 4CzIPN (25) as a PC, trifluoroacetic acid in MeCN at room temperature under blue LEDs irradiation and was also applied to dipeptides Gly-Pro, acetyl captopril and fosinopril. In the proposed mechanism, the protonated NHPI ester by TFA undergoes SET to provide after decarboxylation the α-aminoalkyl radical A. Subsequent oxidation of A by PC+˙ by a polar crossover process gives rise to N-acyliminium ion B and regenerates the PC. Cation B reacts with trimethyl phosphite leading to phosphonium ion C, which undergoes Arbuzov-type demethylation promoted by trifluoroacetate to provide the α-amino phosphonate 499.
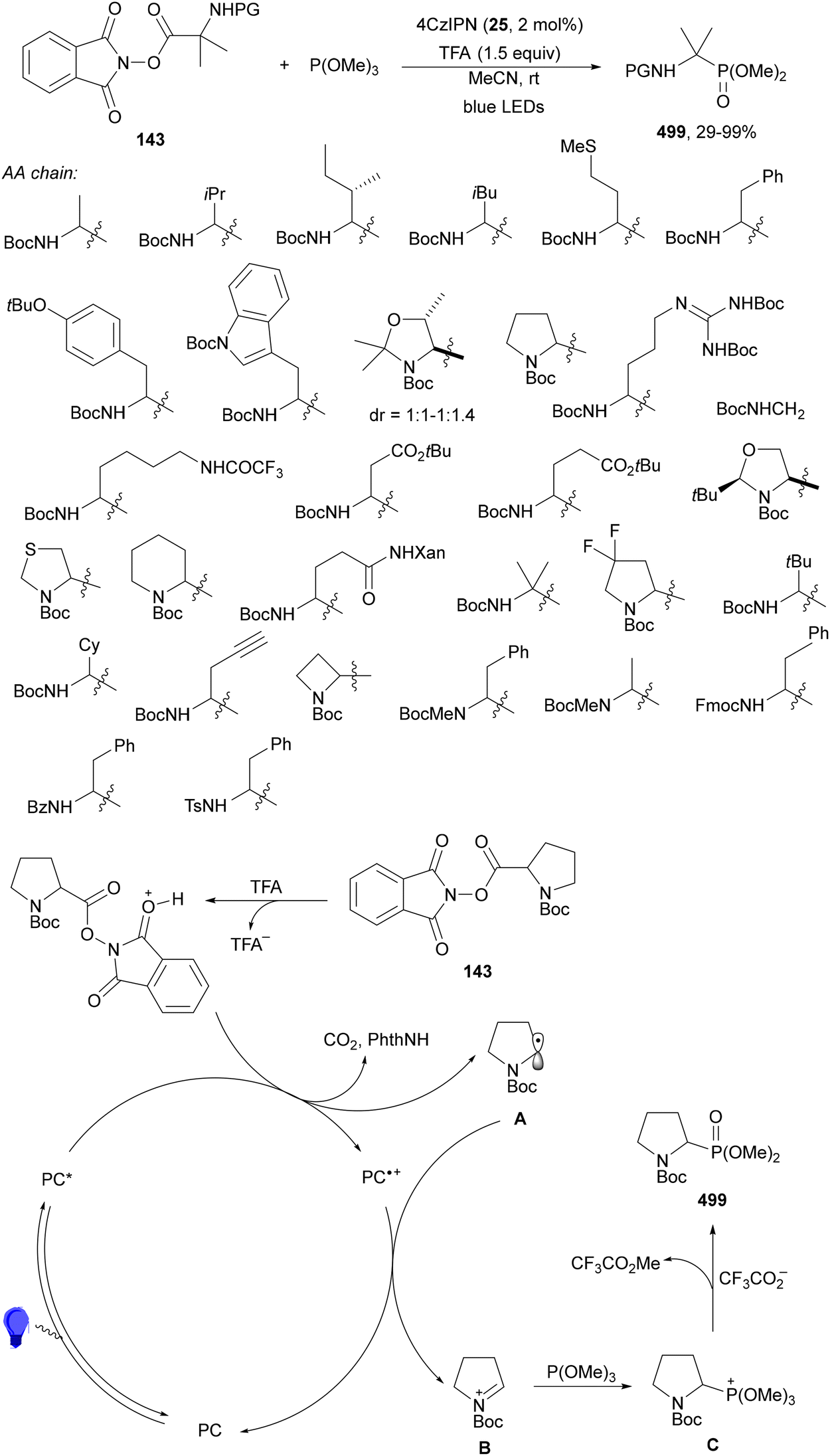 | ||
| Scheme 258 Decarboxylative phosphonylation of α-AA-derived NHPI esters 143 with trimethyl phosphite under 4CzIPN (25) photocatalysis. | ||
Decarboxylative phosphonylation of alkyl NHPI esters 69 with benzhydryl-catechol-phosphite (Beca P, 500) was performed by Aggarwal and co-workers.466 This phosphite reagent 500 enables a more efficient phosphonylation than common trialkyl phosphites. In this case, Ir complex 4 was used as a PC in the presence of 4 equivalents of MeOH in MeCN at 30 °C under blue LED irradiation to give phosphonates 501 with good yields for primary and secondary alkyl NHPI esters (Scheme 259). This procedure was applied to the large-stage functionalization of several natural products and drugs. In addition, MeOH can be replaced by other alcohols to provide phosphonates 501 with ethyl, isopropyl, and benzyl groups. The proposed mechanism starts with SET reduction of NHPI ester by Ir(III)* to form after decarboxylation the alkyl radical A. Beca P 500 reacts with radical A to form phosphonate radical B, which after β-scission gives intermediate C and benzhydryl radical D. Intermediate C reacts with MeOH to give the product. Radical D can be oxidized to cation E by the oxidized state of the catalyst, and after trapping by MeOH provides by-products PhthNH and Ph2CHOMe.
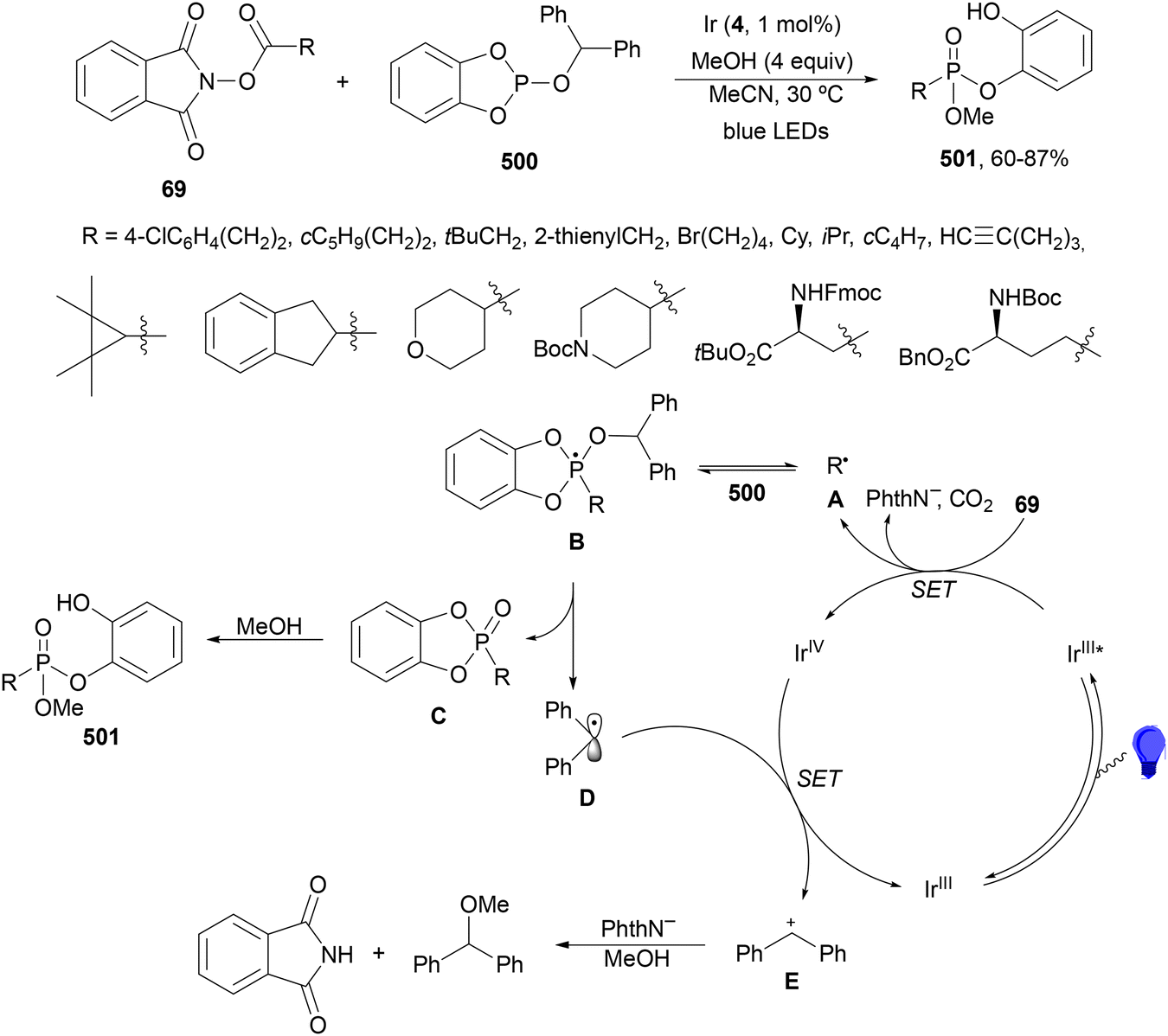 | ||
| Scheme 259 Decarboxylative phosphonylation of NHPI esters 69 with Beca P (500) under Ir complex 4 photocatalysis. | ||
Phosphonylation of alkyl NHPI esters 69 with trialkyl phosphites was also achieved using 1 mol% of 1,2,3,5-tetrakis(diphenylamino)-4,6-dicyanobenzene (4DPAIPN, 502) as a PC, Cu(OAc)2, lithium benzoate as a base in chlorobenzene at room temperature under blue LEDs irradiation (Scheme 260).467 The corresponding primary and secondary alkylphosphonates 503 were isolated with good yields. In the proposed mechanism, the photoexcited 502* is oxidatively quenched by NHPI ester to give 502+˙ and NHPI ester radical anion, which after decarboxylation gives PhthN− and radical A. Complex B is formed by reaction of Cu(I) with P(OEt)3, which by SET from radical cation 502+˙ forms the Cu(II) complex C and regenerates the PC 502. Complex C reacts with radical A providing the phosphonium intermediate D, which is de-ethylated by nucleophilic attack of benzoate (Arbuzov type) to furnish the product 503 regenerating the Cu(I) catalyst.
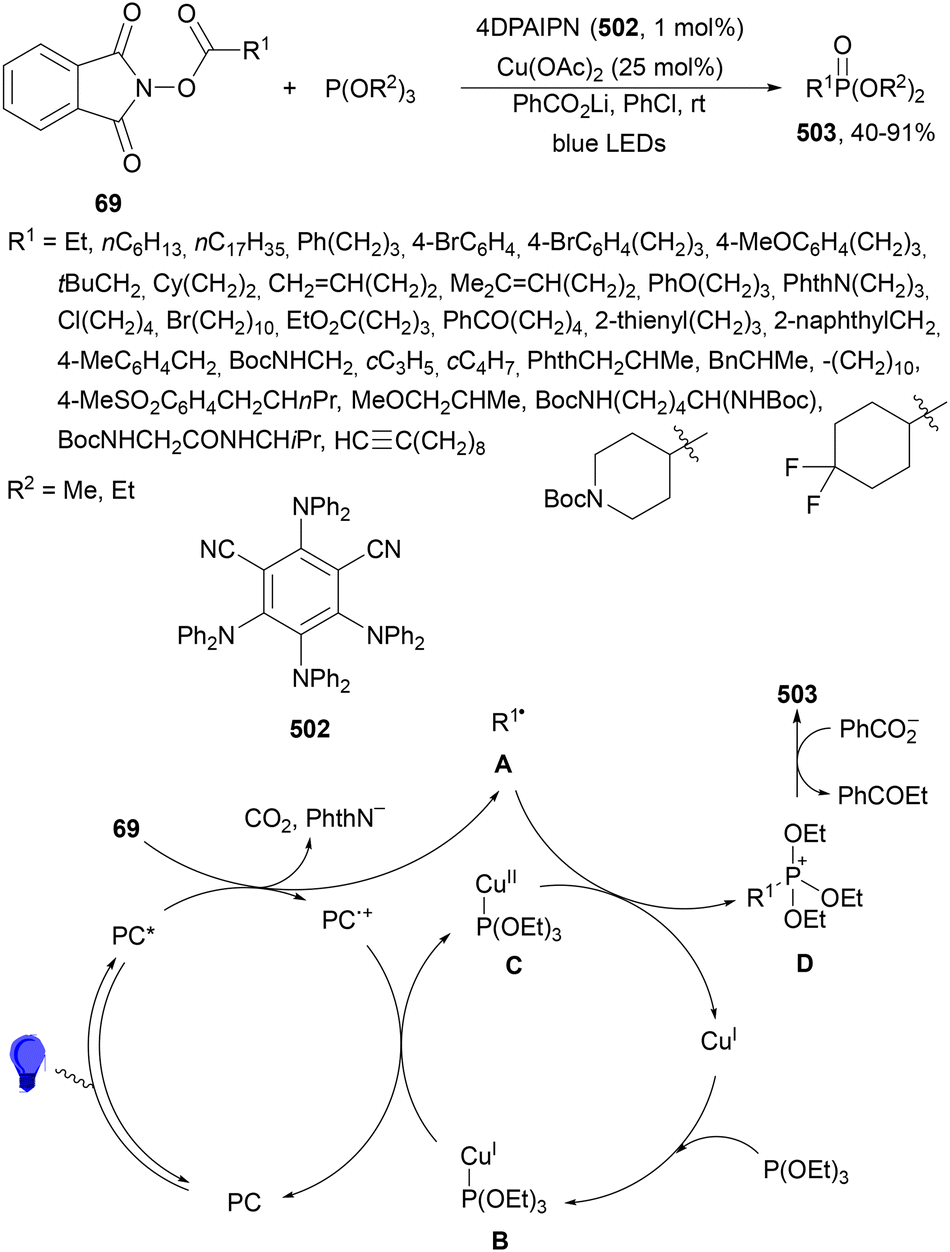 | ||
| Scheme 260 Decarboxylative phosphonylation of alkyl NHPI esters 69 with trialkyl phosphites under 4DPAIPN (502) and Cu(OAc)2 photocatalysis. | ||
Photoinduced decarboxylative phophinylation of aliphatic NHPI esters 69 with dialkylaryl or alkyl phosphonites 504 has been reported by the same Chinese group.468 Working with 4CzIPN (25) as a PC in DMA at room temperature under blue LED irradiation resulted in phosphinates 505 with, in general, very good yields (Scheme 261). This method was applied to the synthesis of bioactive phosphinic acids such as kynureninase inhibitor and glutamine synthetase inhibitor phosphinothricin. A plausible mechanism was proposed involving the generation of radical A, which reacts with dimethyl phenyl phosphonite to give the phosphoranyl radical B. A subsequent SET process of radical B with (4CzIPN)+˙ forms the phosphonium cation C and the ground state of 4CzIPN. Arbuzov demethylation of C with PhthN− gives the product PhthNMe.
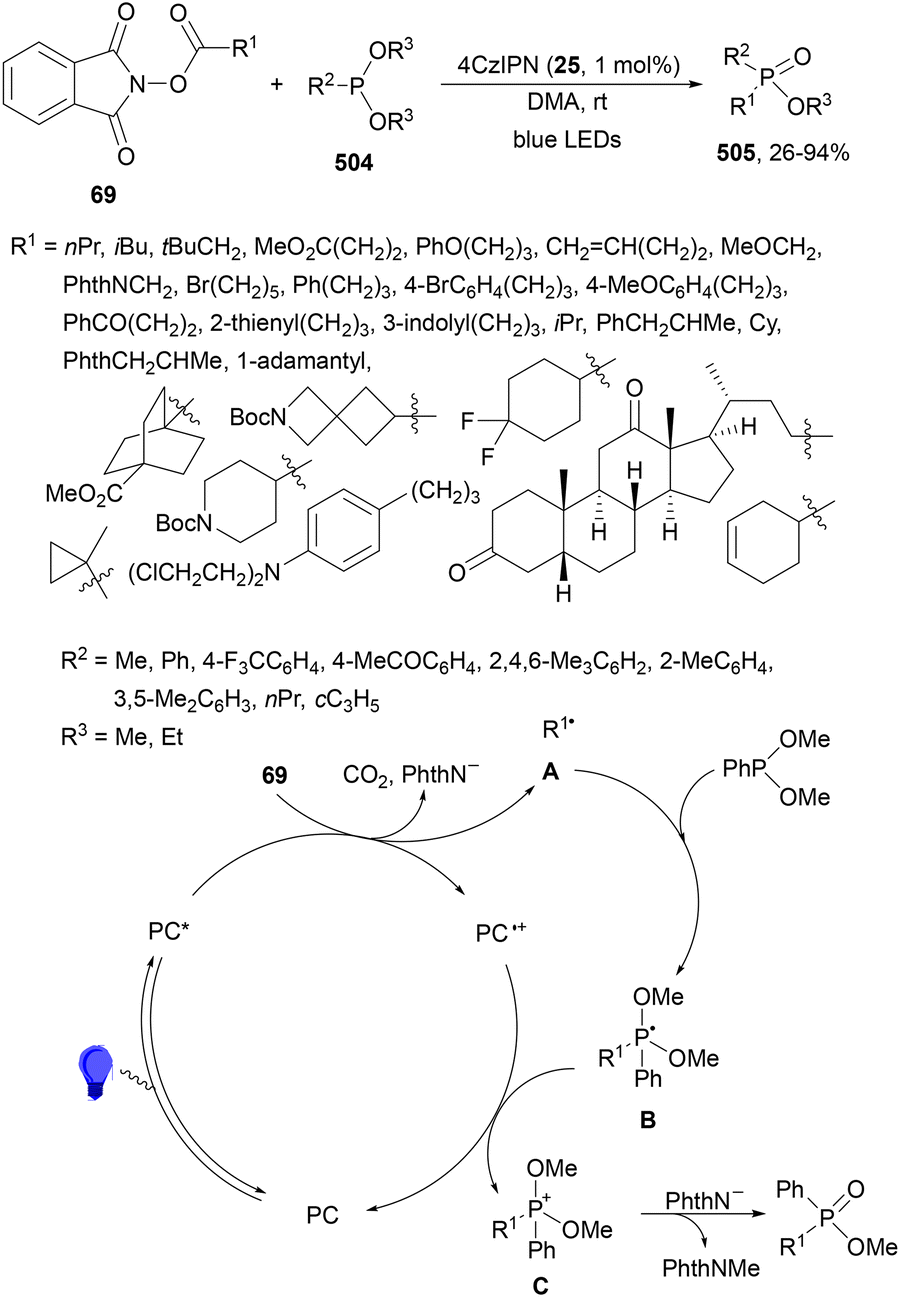 | ||
| Scheme 261 Decarboxylative phosphinylation of aliphatic NHPI esters 69 with phosphonates 504 under 4CzIPN (25) photocatalysis. | ||
White phosphorus has been alkylated with alkyl NHPI esters 69 in the presence of HE and NaI in DMF/toluene at room temperature under Ar and visible-light irradiation without PC.469 Dialkyl phosphines were isolated as dialkyl phosphine oxides (DAPOs) in air with high yields (52–82%) for primary alkyl groups (Scheme 262a). However, secondary alkyl NHPI esters gave lower yields (12–84%) and tertiary ones failed. Several scale-up experiments afforded the corresponding products in good yields. When the amount of NHPI ester was increased from 3.5 to 8.0 equivalents, in the presence of air, the corresponding trialkyl phosphines (TAPOs) were obtained with yields ranging from 47 to 87% (Scheme 262b). In the proposed mechanism, an EDA complex between NHPI ester, an iodide anion and HE is formed (see Scheme 92), which after photoexcitation resulted in an alkyl radical. This radical reacts with P4 yielding an unstable phosphorus-centered radical A, which absorbs a H atom from HE+˙ to give species B. Subsequent alkylation provides the primary phosphine (RPH2) C. Finally, the alkyl radical abstracts a H atom to form the hydro phosphonyl radical D. A radical coupling between D and R˙ yields the secondary phosphine E, which after oxidation forms product DAPO.
 | ||
| Scheme 262 Decarboxylative phosphorylation of alkyl NHPI esters 69 with P4 under NaI/HE photocatalysis. | ||
Direct decarboxylative/dehydrogenative coupling of α-fluoro-α,β-unsaturated carboxylic acids 506 with diaryl phosphine oxides 507 was described by Lu, Zhou and co-workers.470 Working with Ru(bpy)3Cl2 as a PC, tert-butyl peroxybenzoate (TBPB) as an oxidant, DABCO as a base in MeCN at room temperature and blue LED irradiation, monofluoroalkenyl phosphine oxides 508 were obtained in 46–80% yields and 30:1 E/Z diastereoselectivity (Scheme 263a). This procedure was also applied to the late-stage modification of an estrone derivative and a vitamin E derivative. In the case of alkoxy phosphine oxides 509 the reaction was sluggish giving the corresponding phosphonates 510 with modest yields and lower E/Z diastereoselectivity owing to the low stability of the alkoxy substituted phosphinoyl radical compared to the diaryl substituted phosphinoyl radical (Scheme 263b). In the proposed mechanism, photoexcited [Ru3+]* oxidizes DABCO to form DABCO+˙ and [Ru]2+via a SET. Then, [Ru]2+ was oxidized by TBPB regenerating [Ru]3+ and tBuO˙, which is captured by 507 to give the diphenyl phosphinoyl radical A. Subsequent addition of A to α-fluoro cinnamic acid forms the β-radical carboxylate intermediate B. This radical B releases an electron to DABCO+˙ to form intermediate C. Final decarboxylation of C gives product 508.
 | ||
| Scheme 263 Decarboxylative coupling of α-fluoro-α,β-unsaturated carboxylic acids 506 with diaryl phosphine oxides 507 and dialkoxy phosphine oxides 509 under Ru photocatalysis. | ||
Phosphorus derivatives such as phosphonates and phosphinates are accessible by photoredox decarboxylative C–P bond forming reactions working with NHPI esters under LED irradiation. Phosphites react with NHPI esters using an Ir complex as a PC or 4CzIPN or 4DPSIPN as organic photoredox catalysts to provide phosphonates. In the case of using phosphonites as nucleophiles with 4CzIPN as a PC and Cu(OAc)2 as a catalyst resulted phosphinates. Dialkyl and trialkyl phosphine oxides are accessible by reaction of NHPI esters with phosphorus under blue LED irradiation. Diaryl phosphine oxides react with α-fluoro-α,β-unsaturated carboxylic acids using Ru(III) and DTPB as an oxidant to give monofluoroalkenyl phosphine oxides.
3.6. Carbon–boron bond-forming reactions
Decarboxylative borylation reactions were reported in 2017 by Aggarwal471 and Li472 groups using alkyl NHPI esters 69 and B2cat2 under blue LED irradiation,471 whereas in the second case B2pin2 or B2(OH)6/KHF2 with an Ir complex as PC under 45 W CFL irradiation, were employed. Aryl NHPI esters were borylated with B2pin2 under blue LED irradiation in the absence of a PC by Glorius and co-workers.473 In 2020, Wang and co-workers474 employed N-hydroxybenzimidoyl chloride (NHBC) esters 401 for the borylation with B2cat2 or bis(dimethylpentanediol)diboron (B2dmpd2) for aliphatic or aromatic carboxylic acid derivatives, respectively, using an Ir complex as a PC under blue LED irradiation.Recent developments in decarboxylative borylation of aliphatic NHPI esters under visible-light conditions (developed by the Aggarwal group471) were carried out by Masson and co-workers.475 Borylation of NHPI esters 69 containing α- or β-heteroatoms, including α- and β-amino acids, was carried out with B2cat2 in DMA followed by in situ transamination with 1,8-diaminonaphthalene (DANH2) to provide stable α- and β-substituted boronamides 510 in moderate to excellent yields (Scheme 264). Carboxy group-containing drugs such as baclofen and isoxepac were transformed into the corresponding DAN-boronates 510. In the mechanism proposed by Aggarwal and co-workers,471 under photochemical conditions, NHPI ester, B2cat2 and DMA form complex A, which after irradiation cleaves the B–B bond to deliver a DMA-stabilized boryl radical C and O-boryl-NPhth ester radical B. This radical B gives, after decarboxylation and release of O-borylphthalimide, an alkyl radical D. Reaction of D with DMA-coordinated B2cat2E yields an alkylboronate ester and DMA-stabilized boryl radical C. Subsequently, NHPI ester reacts with C to give radical F, which regenerates radical D.
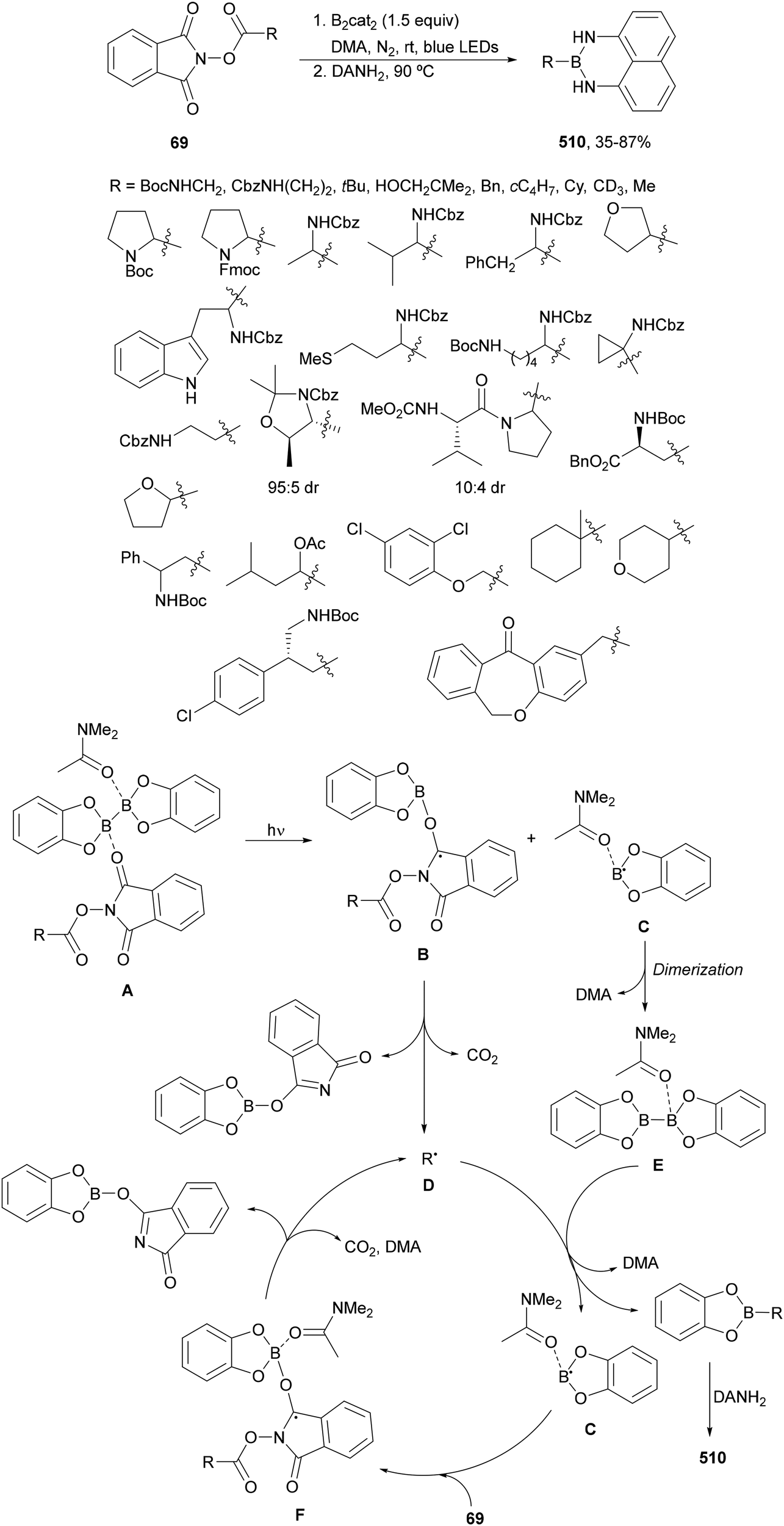 | ||
| Scheme 264 Decarboxylative borylation of NHPI esters 69 with B2cat2 under visible light photocatalysis followed by amination with DANH2. | ||
Recently, Tolnai, Novák and co-workers476 employed hypoboric acid for the borylation of alkyl NHPI esters 69 under Ar and visible-light irradiation in DMF at room temperature. After in situ addition of 1.5 equivalents of pinacol the corresponding boronic esters 511 were isolated with good yields (Scheme 265). This borylation did not proceed under dark conditions even at 110 °C. In the proposed mechanism, complex A between hypoboric acid and DMF can be formed, which by irradiation generates the boron radical B. This radical B reacts with NHPI ester to form radical C. Subsequent decarboxylation of C generates an alkyl radical D, which reacts with complex A to give the alkylboronic acid and radical B.
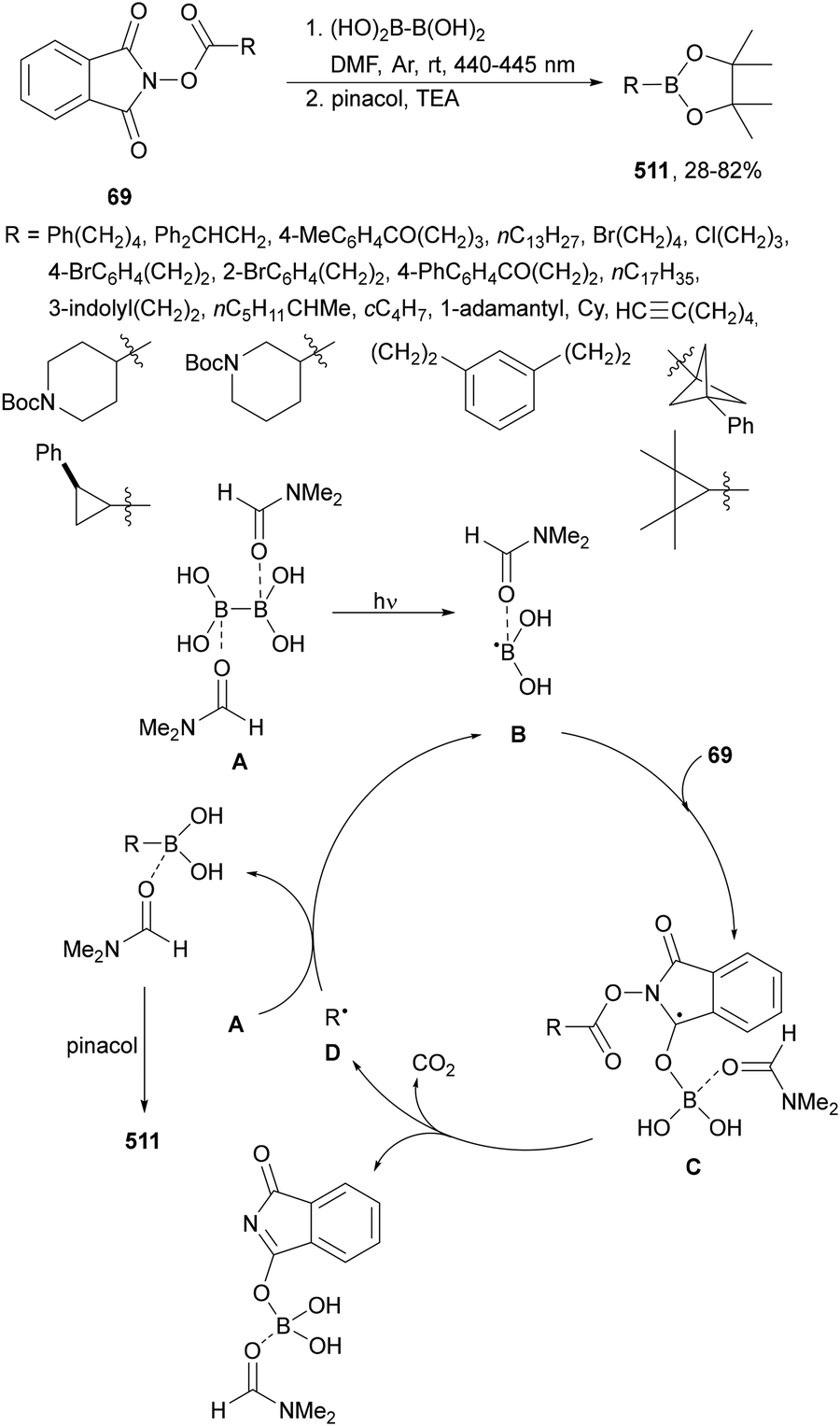 | ||
| Scheme 265 Decarboxylative borylation of alkyl NHPI esters 69 with hypoboric acid under 440–445 nm light followed by esterification with pinacol. | ||
Direct decarboxylative borylation of (hetero)aryl acids was reported in 2022 by MacMillan and coworkers477 by copper LMCT photocatalysis. This borylation was performed using B2pin2 in the presence of Cu(MeCN)4BF4, NFSI as an oxidant, NaF and LiClO4 as MeCN-soluble ion sources to generate the activated lithium fluoroborate in an integrated photoreactor with a 365 nm LED module. The corresponding borylated arene products 512 were isolated with good yields under these mild reaction conditions (Scheme 266). In addition, this borylation was combined with a Suzuki–Miyaura reaction and also with a double decarboxylative coupling of two (hetero)aryl acids under parallel Cu-LMCT bromination and borylation procedures. A plausible mechanism for the borylation process involves the formation of the Cu(I) carboxylate, which after oxidation gives complex A. Under near-UV irradiation this complex A undergoes LMCT from the carboxylate ion to the Cu(II) center to produce Cu(I) and an aryloxy radical B. Subsequent decarboxylation of B gives aryl radical C, which reacts with an activated metal boronate, generated in situ from B2pin2 and metal salt additives, to deliver the arylboronic ester 512.
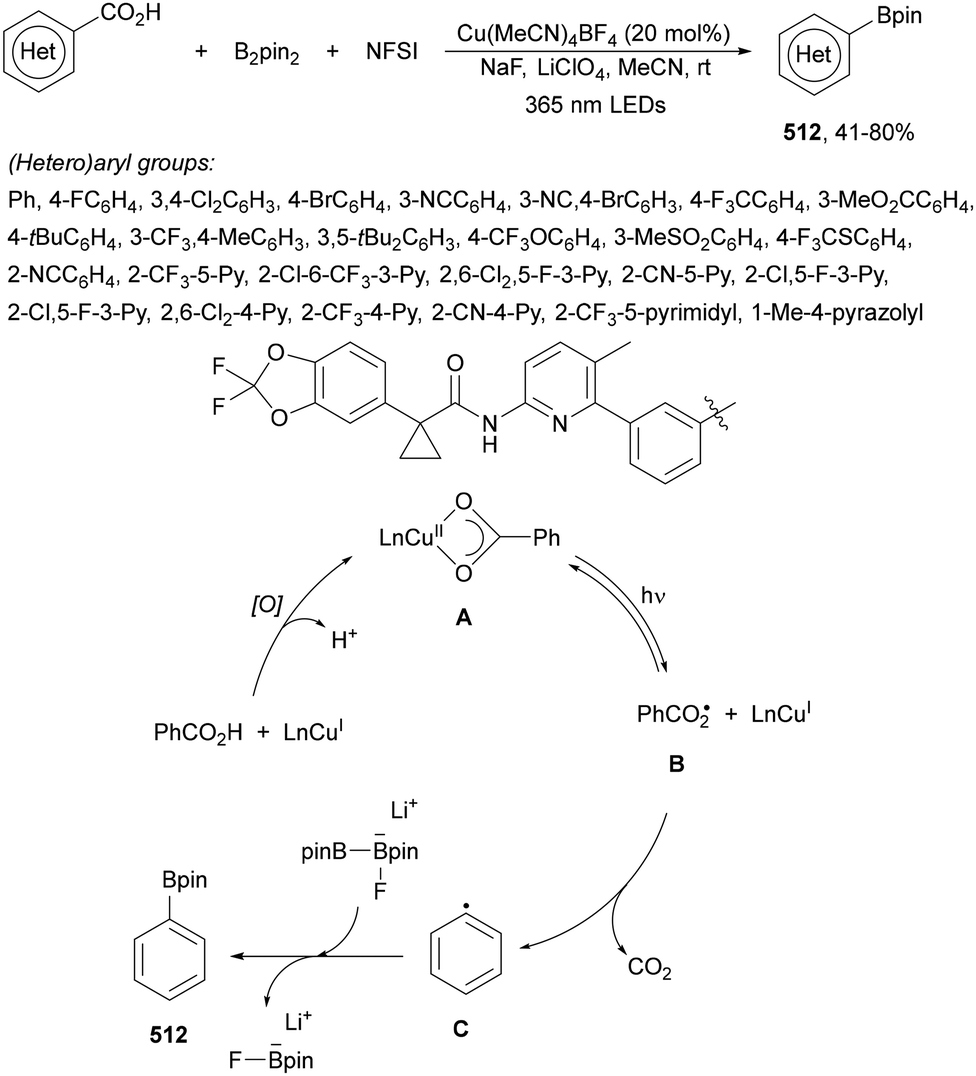 | ||
| Scheme 266 Decarboxylative borylation of (hetero)aryl acids with B2pin2 under Cu-LMCT photocatalysis. | ||
Liu and co-workers478 reported the borylation of aromatic acids with B2pin2 using Ir complex (324) as a PC, Co(dmgH)2PyCl (16) as an oxidant, and TMG as a base, to activate the carboxylic acid in tBuOAc under air and blue LED irradiation (Scheme 267). The resulting aryl boronates 512 were isolated in moderate to good yields and this procedure was also applied to bioactive molecules. In the proposed mechanism, TMG forms a complex A with the acid and Ir(III) was photoexcited to Ir(III)*, which after oxidative quenching with Co(III) gives Ir(IV). Then, complex A is oxidized to complex B and Ir(III) is regenerated. Decarboxylation of B gives the aryl radical C and TMGH+. Borylation of radical C takes place by complex D, formed by reaction of B2pin2 and TMG.
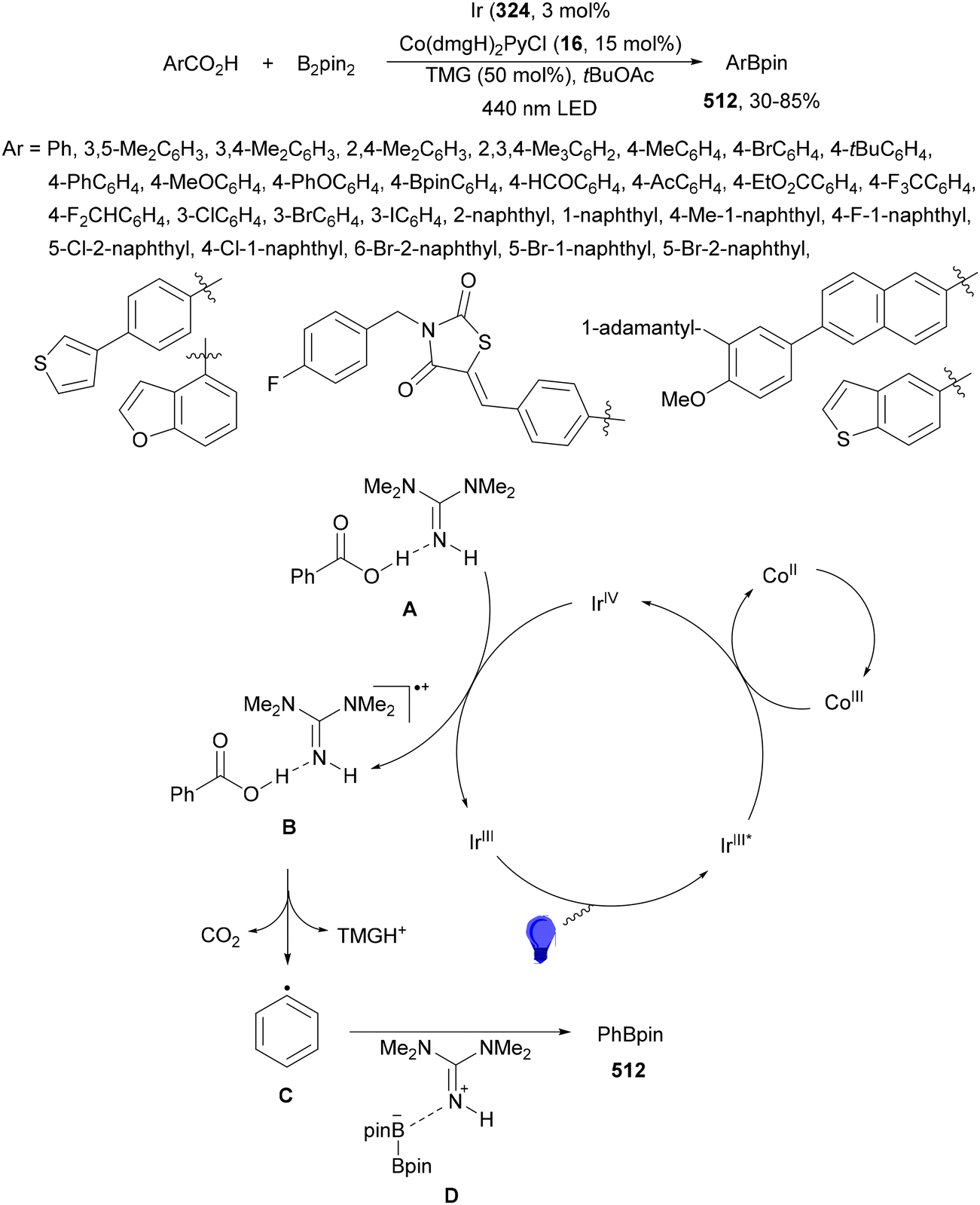 | ||
| Scheme 267 Decarboxylative borylation of aromatic carboxylic acids with B2pin2 under Ir/Co photocatalysis. | ||
Decarboxylative borylation has been performed with alkyl NHPI esters in the absence of photocatalyst either with B2cat2 or hypoboric acid followed by esterification with pinacol. (Hetero)aryl acids have been transformed into (hetero)arylboronates with Cu LMCT photocatalysis and NFSI as an oxidant or with Ir as a PC and a cobalamin as an oxidant.
3.7. Carbon–silicon bond-forming reactions
The first decarboxylative silylation of silacarboxylic acids 513 was described in 2020 by Uchiyama and co-workers479 under 4CzIPN (25) photocatalysis. Silicon-based radicals were trapped by alkenes to give the hydrosilycated products in good yields. More recently Jin, Ren and co-workers480 performed this silylation reaction in the presence of α-trifluoromethylaryl alkenes 514, which underwent a photoredox defluorinative silylation to furnish silylated gem-difluoroalkenes 515 in moderate to good yields (Scheme 268). This defluorinative silylation was carried out on a 2 molar scale to give 515a (R1 = R2 = Me; R3 = Ph) in 74% yield. A plausible mechanism was proposed starting by oxidative decarboxylation of silacarboxylate by PC* to give radical A. Subsequent radical addition of A to trifluoroalkene 514 provides a benzyl radical B, which undergoes a SET forming benzyl anion C. After a defluorination process of C product 515 is obtained.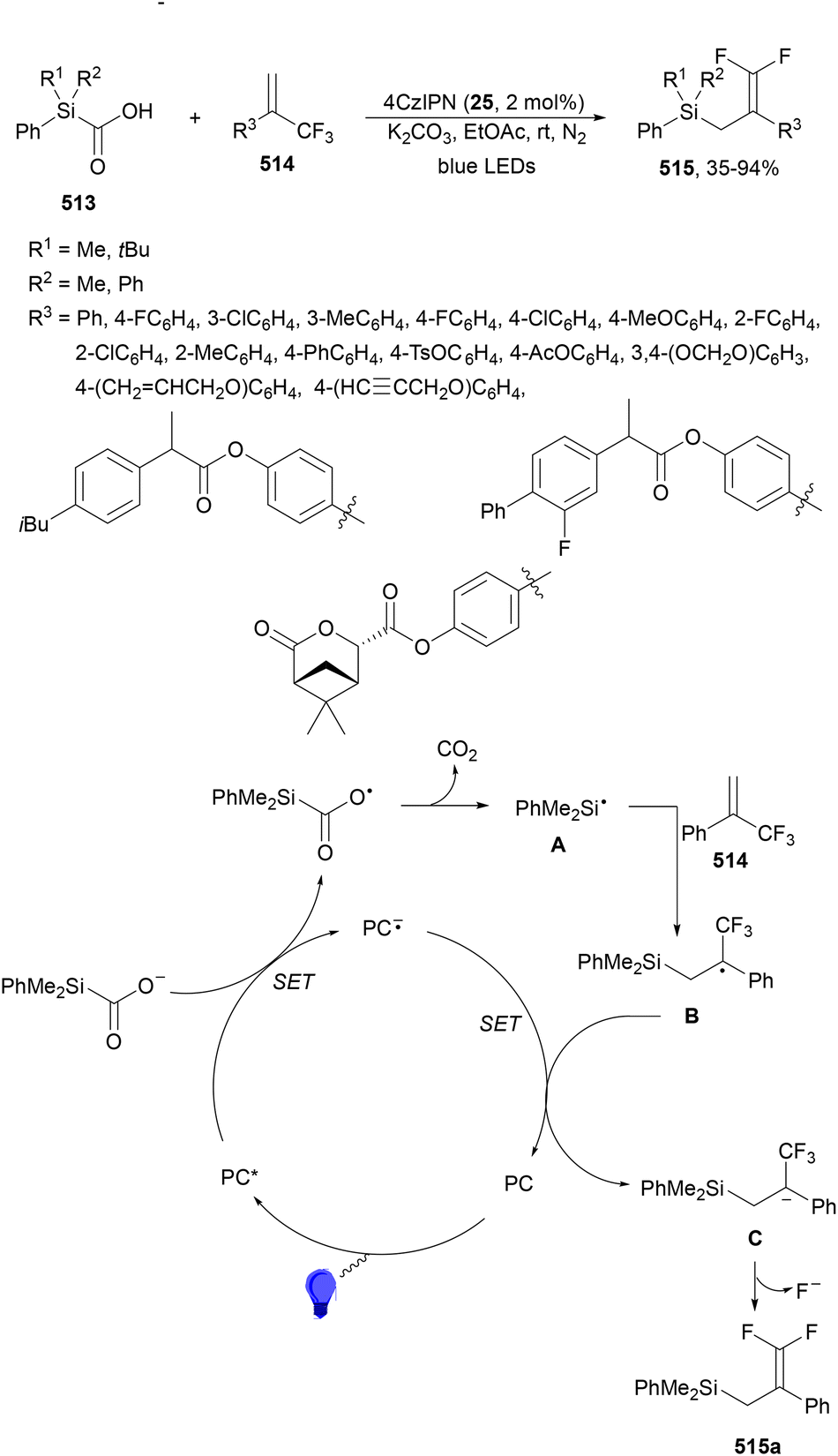 | ||
| Scheme 268 Decarboxylative defluorinative silylation of α-trifluoromethyl alkenes 514 with silacarboxylic acids 513 under 4CzIPN (25) photocatalysis. | ||
Lu, Zhou and co-workers470 reported a decarboxylative coupling of α-fluoro-α,β-unsaturated carboxylic acids 506 with diaryl and dialkyl phosphine oxides 507 and 509 (Scheme 263). When this photoinduced decarboxylation was carried out with triethylsilane instead of 507, in the presence of Ru(bpy)3Cl2 and TBPB as an oxidant in DMSO under Ar and blue LED irradiation monofluoroalkenyl silanes 516 were obtained in good yields and excellent diastereoselectivity (Scheme 269).
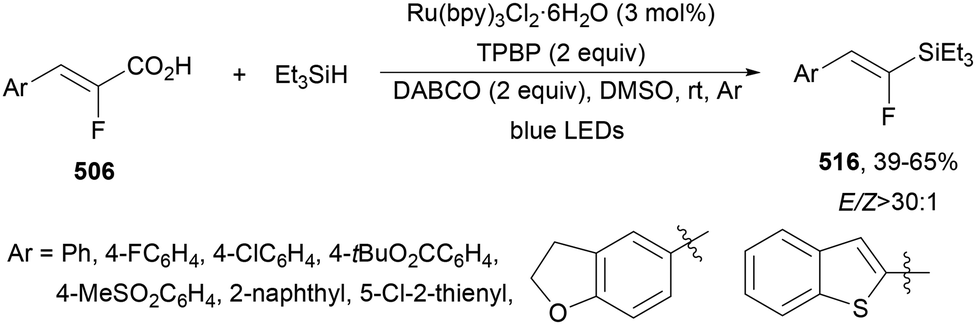 | ||
| Scheme 269 Decarboxylative coupling of α-fluoro-α,β-unsaturated acids 506 with Et3SiH under Ru photocatalysis. | ||
4. Hydro- and deuterodecarboxylation
Photomediated decarboxylative protonation or deuteration of carboxylic acids and derivatives is a mild procedure for the C–H481 and C–D482 bond formation. In 2014, Wallentin and co-workers483 employed an acridinium (Mes-Acr-Me)BF4 (1 mol%) as a strong oxidizing PC and phenyl disulfide (10 mol%) as a source of a hydrogen donor for the protonation of α-AAs and α-hydroxy acids. Nicewicz and co-workers484 reported a similar hydrodecarboxylation of aliphatic carboxylic acids using (Mes-Acr-Ph)BF4 (17) as a PC.More recent applications of decarboxylative protonation or deuteration have been described using mainly acridinium salts for aliphatic carboxylic acids and Cu or Fe LMCT processes also for aromatic acids. Li, Zhu and co-workers485 reported a deuterodecarboxylation procedure from aliphatic carboxylic acids including erdosteine, ambrisentan, gemfibrozil and oleanic acid. They employed (Mes-Acr-Me)ClO4 (301) as a PC and 2,4,6-triisopropylbenzenethiol as a HAT catalyst, 2,4,6-collidine as a base in a 4:1 mixture of DCM/D2O for benzylic and α-heteroatom substituted carboxylic acids under LED irradiation in moderate to good yields and good D-incorporation (up to 99%). Alternatively, CsOH and Ir complex 4 (1 mol%) as a PC have also been employed (Scheme 270). A scalable process was carried out using a recirculation reaction with a peristaltic pump up to a 50 mmol scale. The proposed mechanism starts with the generation of an alkyl radical A after oxidation of the carboxylate ion by PC*. Then, deuterated thiol acts as a HAT catalyst and transfer D to radical A generating a thiyl radical B, which can accept an electron from the PC−˙ and regenerate the PC.
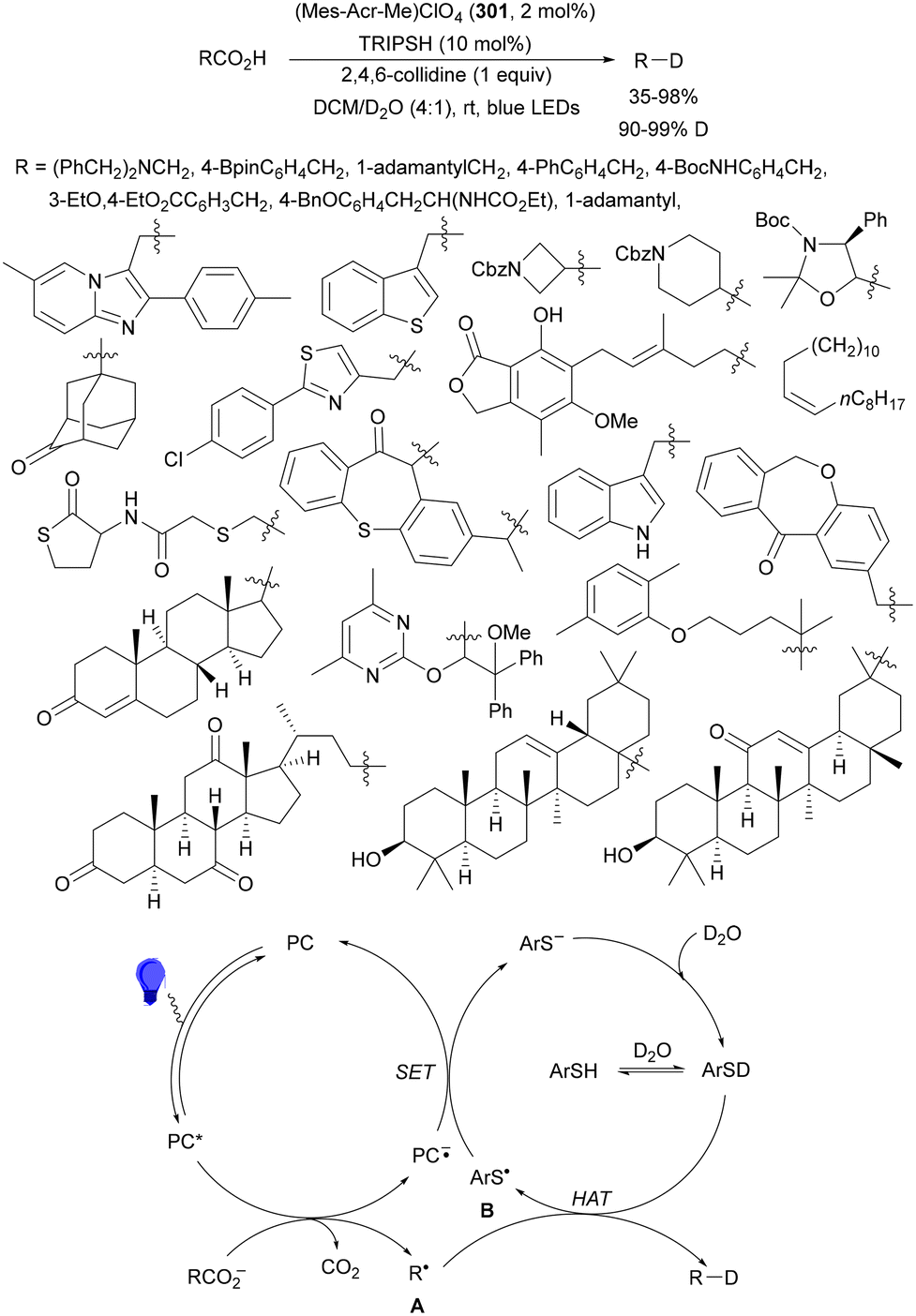 | ||
| Scheme 270 Deuterodecarboxylation of aliphatic carboxylic acids under (Mes-Acr-Me)ClO4 (301) photocatalysis. | ||
Fatty acids underwent decarboxylative protonation and deuteration using (Mes-1,3,6,8-tetramethoxy-Acr-3′′,5′′-dimethoxyPh)BF4 (517) as a PC, (4-methylphenyl) disulfide as a HAT catalyst in EtOAc and in EtOAc/D2O (4:1), respectively.486 In this case, n-Bu4NOAc (TBAA) was employed as a base at 35–40 °C under Ar and blue LED irradiation to give the corresponding alkanes and deuterated alkanes with good yields. This procedure was scaled-up to the gram-scale (Scheme 271).
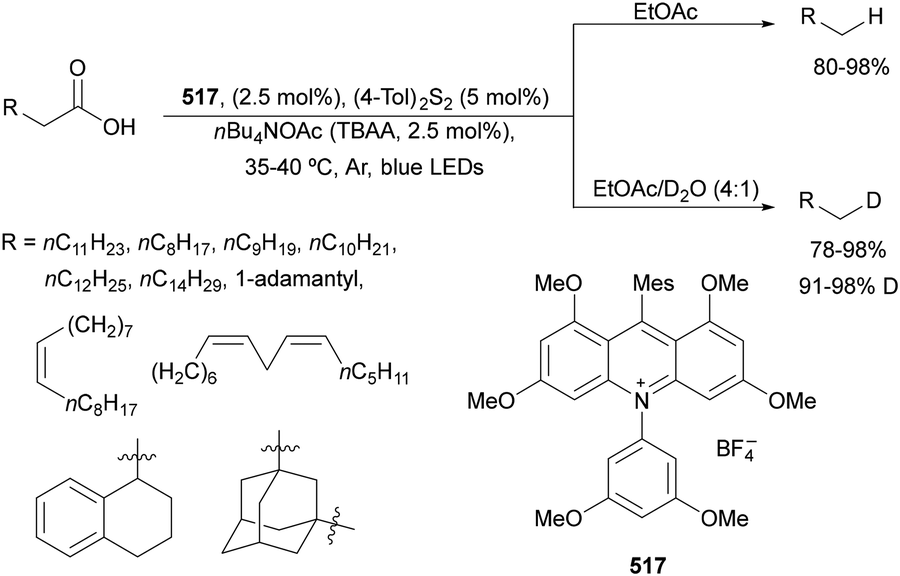 | ||
| Scheme 271 Proto- and deuterodecarboxylation of fatty acids under acridinium salt 517 photocatalysis. | ||
Cavalcanti and co-workers487 described the hydrodecarboxylation of fatty acids using 10 mol% of acridine 125 as a PC, benzenethiol (20 mol%) as a HAT catalyst at room temperature in aqueous DCM under N2 and LED irradiation. The fatty acids were converted into hydrocarbons in excellent yields (95–99%). This protocol was applied to a mixture of fatty acids obtained from the hydrolysis of Licuri oil affording a mixture of C9–C17 hydrocarbons in quantitative yield and has potential application to produce drop-in biofuels.
Perfluorinated disulfide 448, previously described by Dilman and co-workers408 for decarboxylative thiolation of aliphatic carboxylic acids (Scheme 220) providing effective hydrodecarboxylation of gemfibroxil and ursodeoxycholic acid. In this case, 5 mol% of mesylacridine 123 as a PC, sodium perborate as oxidant in aqueous DCM at room temperature under blue LEDs irradiation furnished the corresponding products in 72 and 92%, respectively.
Okamoto and co-workers488 have recently described the stereoselective synthesis of baloxavir marboxil (BXM), an inhibitor of cap-dependent endonuclease used for treating influenza infections. The synthesis of the triazinone core 519 was carried out under mild reaction conditions by photoredox hydrodecarboxylation of compound 518. In this step, (Mes-Acr-Me)ClO4 (301) was used as a PC, (4-ClC6H4)2S2 as a HAT catalyst, DBU as a base in MeOH at room temperature under blue LED irradiation to give compound 519 in 92% yield (Scheme 272).
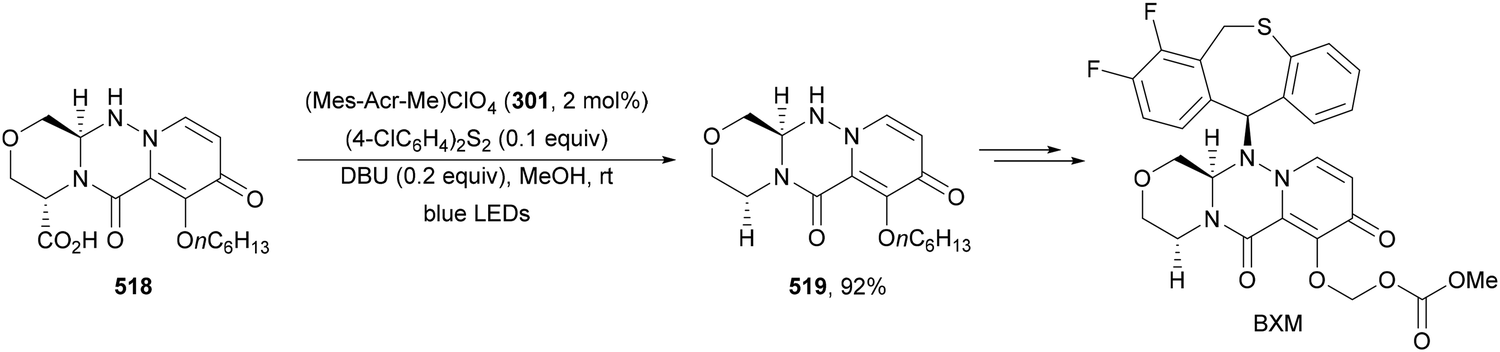 | ||
| Scheme 272 Hydrodecarboxylation of compound 518 under acridinium salt 301 photocatalysis. | ||
Pospech and co-workers489 performed hydro- and deuterodecarboxylation of carboxylic acids using an organic pyrimidopteridine 520 as a photoredox catalyst. Primary, secondary and tertiary aliphatic biologically active carboxylic acids were treated with 520, K3PO4 as a base in MeCN/H2O or MeCN/D2O at 50 °C under 396 nm irradiation to provide the corresponding products in modest to high yields (Scheme 273).
 | ||
| Scheme 273 Hydro- and deuterodecarboxylation of aliphatic carboxylic acids under pyrimidopteridine 520 photocatalysis. | ||
A cooperative iron/thiol catalyst has been employed for decarboxylative protonation of aliphatic carboxylic acids. Lu and West490 employed Fe(NO3)3·9H2O, di(2-pinacol)amine as a ligand and TRIP disulfide as a catalyst, and Na2CO3 as a base in DCE/H2O (1:1) under 390 LED irradiation. A broad range of carboxylic acids including natural products and drugs were efficiently transformed into decarboxylated products in excellent yields (Scheme 274). A couple of examples were treated with DCE/D2O to give deuterated products with 95 and 98% deuterium incorporation. These processes took place via a photoinduced LMCT mechanism by initial formation of Fe(III) carboxylate, which after irradiation gives a carboxy radical A and a Fe(III) species. After decarboxylation of A an alkyl radical B is formed, which can be reduced by HAT from the thiol co-catalyst to produce the product.
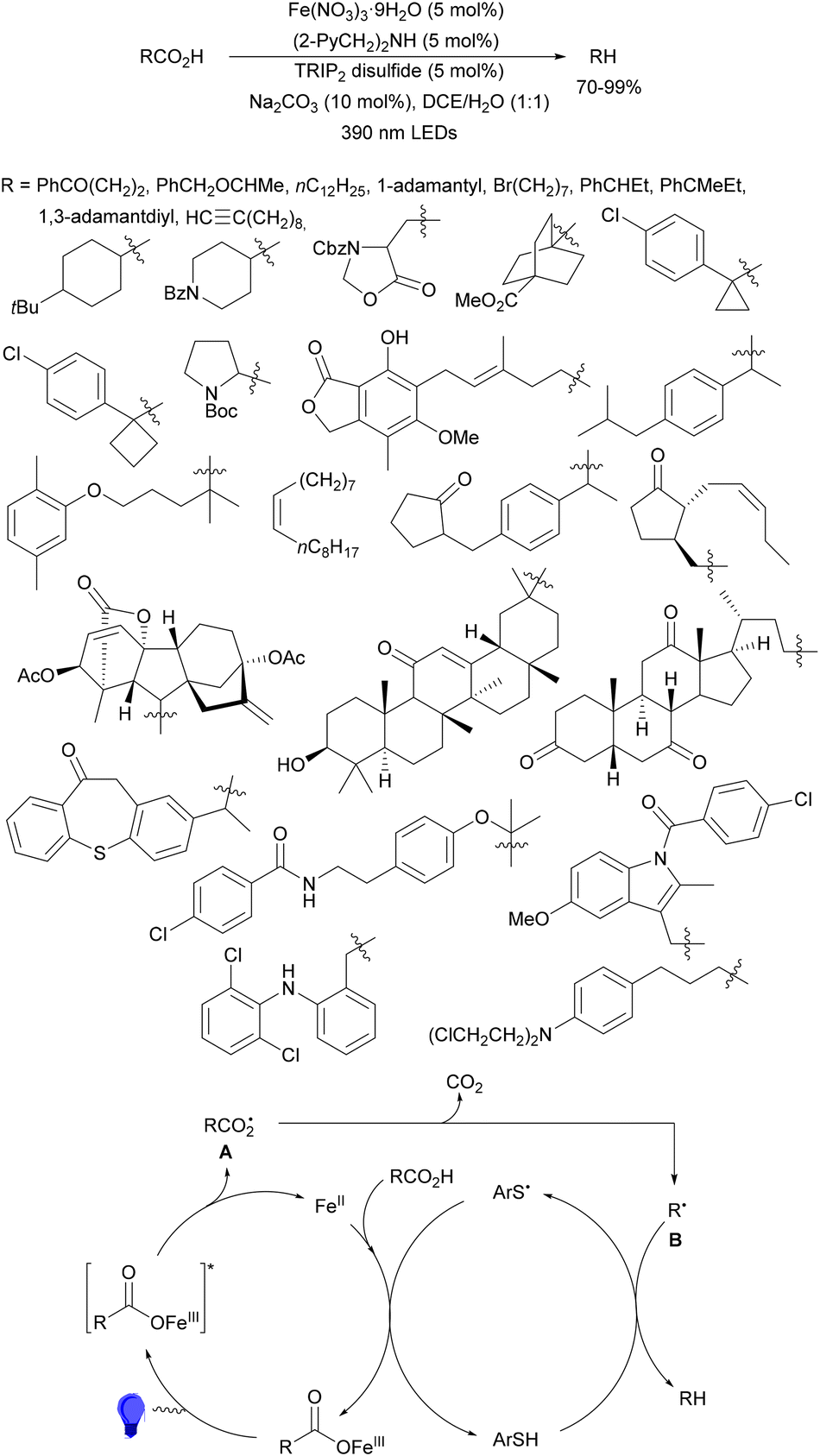 | ||
| Scheme 274 Protodecarboxylation of aliphatic carboxylic acids under Fe/thiol photocatalysis. | ||
In the case of α-keto acids 15, Hu and Li491 described their hydro- and deuterodecarboxylation using an acridinium salt 521 as a PC and TRIPSH as co-catalyst, TBAA as a base in DCE/H2O and DCE/D2O (10:3), respectively, under blue LED irradiation. These processes led to the formation of aldehydes and deuterated aldehydes, respectively (Scheme 275). In this case, carbonyl radicals are formed after decarboxylation of the α-keto acid, which reacts by HAT with TRIPSH or TRIPSD to give the products.
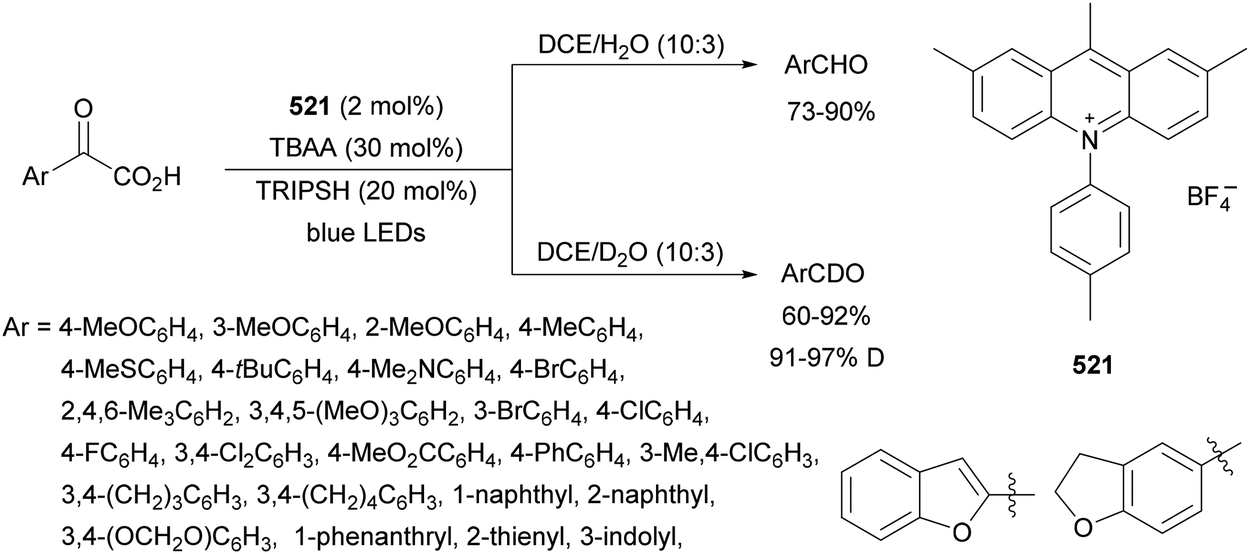 | ||
| Scheme 275 Hydro- and deuterodecarboxylation of α-keto acids 15 under acridinium salt 521 photocatalysis. | ||
Photoredox hydrodecarboxylation of carboxylic acids has been carried out also under heterogeneous conditions, Bayzar and Hosseini-Sarvari492 employed Au@ZnO core-shell nanoparticles (NPs) as semiconductor photocatalyst for aliphatic and aromatic carboxylic acids protodecarboxylation. Working with 4 mg (1.89 wt% Au) per 1 mmol of carboxylic acid and K2CO3 as a base in CHCl3 at room temperature under Ar and blue LED irradiation, the corresponding products were isolated in 51–96% yields. These NPs exhibited excellent reusability without appreciable decrease of activity after 5 runs. Wang and co-workers493 reported a photocatalytic decarboxylation of fatty acids to long-chain alkanes in high yields (>90%) using Pt/TiO2 as a PC under a H2 atmosphere and 365 nm LED irradiation. Hu and García groups494 performed a photocatalytic hydrodecarboxylation of octanoic acid by Ni NPs deposited on TiO2 previously treated with NaBH4 using UV/vis light irradiation with a 300 W lamp. The photocatalytic performance was maintained for six consecutive runs. A metal-free heterogeneous semiconductor was developed by Wang and co-workers495 for visible light photocatalytic hydro- and deuterodecarboxylation of aliphatic and aromatic carboxylic acids. Ceramic boron carbon nitrides (BCN, 30 mg per 0.2 mmol of acid) in MeOH or CD3OD at 40 °C under Ar and 420 nm LED irradiation furnished the corresponding products in 25–93% yields and 86–99% deuterium incorporation. Recycling tests show a slight decrease on activity after 5 recycles. Recently, Gao and co-workers496 described protodecarboxylation of fatty acids over α-Fe2O3 under visible light-induced self-heating.
Enzymatic decarboxylation of carboxylic acids to alkanes has been reported by Hollmann and co-workers.497,498 By using a photodecarboxylase from Chlorella variabilis NC64A (CvFAP),499 fatty acids497 and short-chain aliphatic carboxylic acids498 were transformed into the corresponding alkanes working in DMSO at 30 °C under blue LED irradiation with modest turnover numbers. Wu and co-workers500 developed a divergent protein engineering of WT-CvFAP as more efficient photodecarboxylase for decarboxylative deuteration of fatty acids, working in 20% DMSO or MeCN and D2O under blue LED irradiation with high yields and excellent D incorporation. Recently, a triple mutant CvFAP (Y466T/P460A/G4621) has shown excellent performance in biobased ethylbenzene production from β-phenylpropionic acid derived from phenylalanine in 82% conversion.501
Redox active esters (RAEs) derived from 4-fluorobenzophenone oximes 454 were employed by Glorius and co-workers502 as substrates for decarboxylative deuteration in the presence of Ir complex 4 as a PC in CDCl3 under blue LED irradiation. Aliphatic and aromatic carboxylic acid derivatives 454 were deuterated in good yields and >97% deuterium incorporation (Scheme 276).
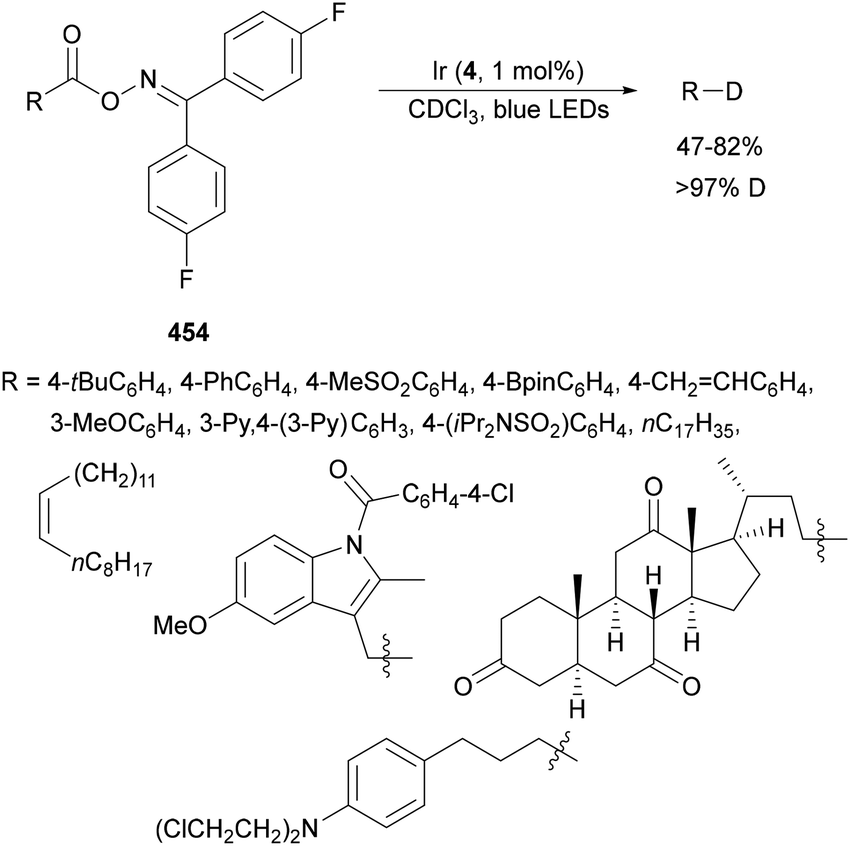 | ||
| Scheme 276 Decarboxylative deuteration of 4-fluorobenzophenone oxime aromatic and aliphatic esters (454) under Ir complex 4 photocatalysis. | ||
Constantini and Mendoza503 synthesized cis-cyclopropanes 525 by decarboxylation of NHPI esters 523 in the presence of benzothiazoline 524 in DMSO at room temperature under blue LED irradiation (Scheme 277). These NHPI esters 523 were prepared by enantioselective cyclopropanation504,505 of styrenes with diazo compounds 522 under Rh2[(S)-TPCP)]4 catalysis and then subjected to decarboxylation. This stereoselective decarboxylation was explained by a stereoretentive HAT. NHPI esters 523 and benzothiazoline 524 associate in solution to form the EDA complex A, which undergoes a photoinduced electron transfer (PET) in the excited state to form the radical ion par between B and C. Subsequent fragmentation and decarboxylation gives the cyclopropyl radical D, which abstracts a hydrogen atom from the benzyl C–H bond of the benzothiazoline radical cation C. cis-Cyclopropane product 525 is kinetically preferred.
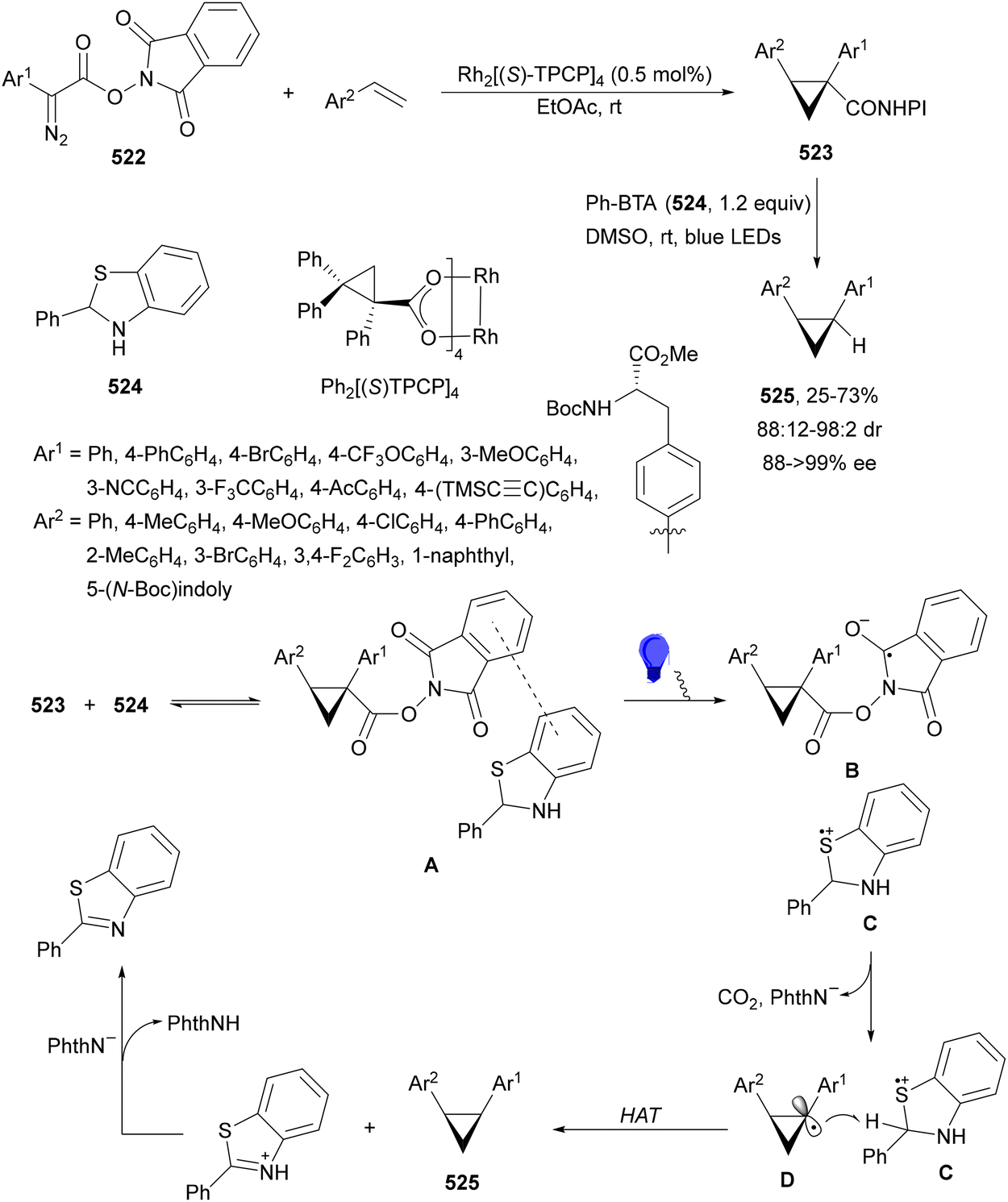 | ||
| Scheme 277 Stereoselective hydrodecarboxylation of cis-cyclopropanes 523 under benzothiazine 524 photocatalysis. | ||
Carboxylic acids can be subjected to photoinduced decarboxylative protonation under homogeneous, heterogeneous and enzymatic conditions. For aliphatic carboxylic acids acridinium salts are the most appropriate PC in combination with thiols as HAT. In the case of α-keto acids aldehydes and deuteroaldehydes are obtained by hydro- and deuterodecarboxylation, respectively. For heterogeneous conditions Au@ZnO NPs and ceramic boron carbon nitrides are the best catalysts for aliphatic and aromatic carboxylic acids. Enzymatic processes are based on CvFAP photodecarboxylase for aliphatic acids. Redox active esters derived from benzophenone oximes and aliphatic or aromatic acids have been deuterated under Ir complex as a PC. I the case of NHPI esters derived from cyclopropane carboxylic acids, the decarboxylation takes place under blue LED irradiation and a benzothiazoline as a HAT reagent to give diastereoselectively cis-1,2-diarylcyclopropanes.
5. Decarboxylative eliminations
In this section photoredox retro-hydrocarboxylation of aliphatic carboxylic acids and their NHPI esters to afford alkenes pioneered by Kochi506 will be considered. Ring-opening of cyclic tertiary carboxylic acids by photoredox C–C bond cleavage will be included as well.5.1. Retro-hydrocarboxylation
Ritter and co-workers507 reported a decarboxyolefination of primary, secondary and tertiary aliphatic carboxylic acids by a cooperative interplay between a cobaloxime complex Co(dmgH)2(4-MeO-Py)Cl (526), as a proton reduction catalyst, and a Ir complex 4 as a photoredox catalyst. The resulting alkenes were obtained in good yields using Cs2CO3 as a base in DME/H2O (18:1) at 35 °C under blue LED irradiation (Scheme 278). A broad range of carboxylic acids including jasmonic acid, picamilon, aleuritic acid, quinic acid, ciprofibrate, benzafibrate, tianeptine, haloxyfop, ramipril, Cbz–Phe–Leu, 18β-glycyrrhetinic acid and cholic acid were transformed into the corresponding alkenes. In the proposed mechanism, the carboxylate ion is oxidized by Ir(IV) to give radical A, which after decarboxylation generates an alkyl centered radical B. In the dehydrogenative catalytic cycle, Co(III)-hydride can release H2 by protonation forming Co(III). This is reduced to Co(II) by Ir(III)*. This Co(II) complex accepts radical B providing complex C. Upon photolysis, homolytic cleavage of the Co–C bond followed by β-hydrogen abstraction by Co(II) produces the olefin and the Co(III)-hydride complex, which after protonation by the carboxylic acid regenerates the Co(III) catalyst.
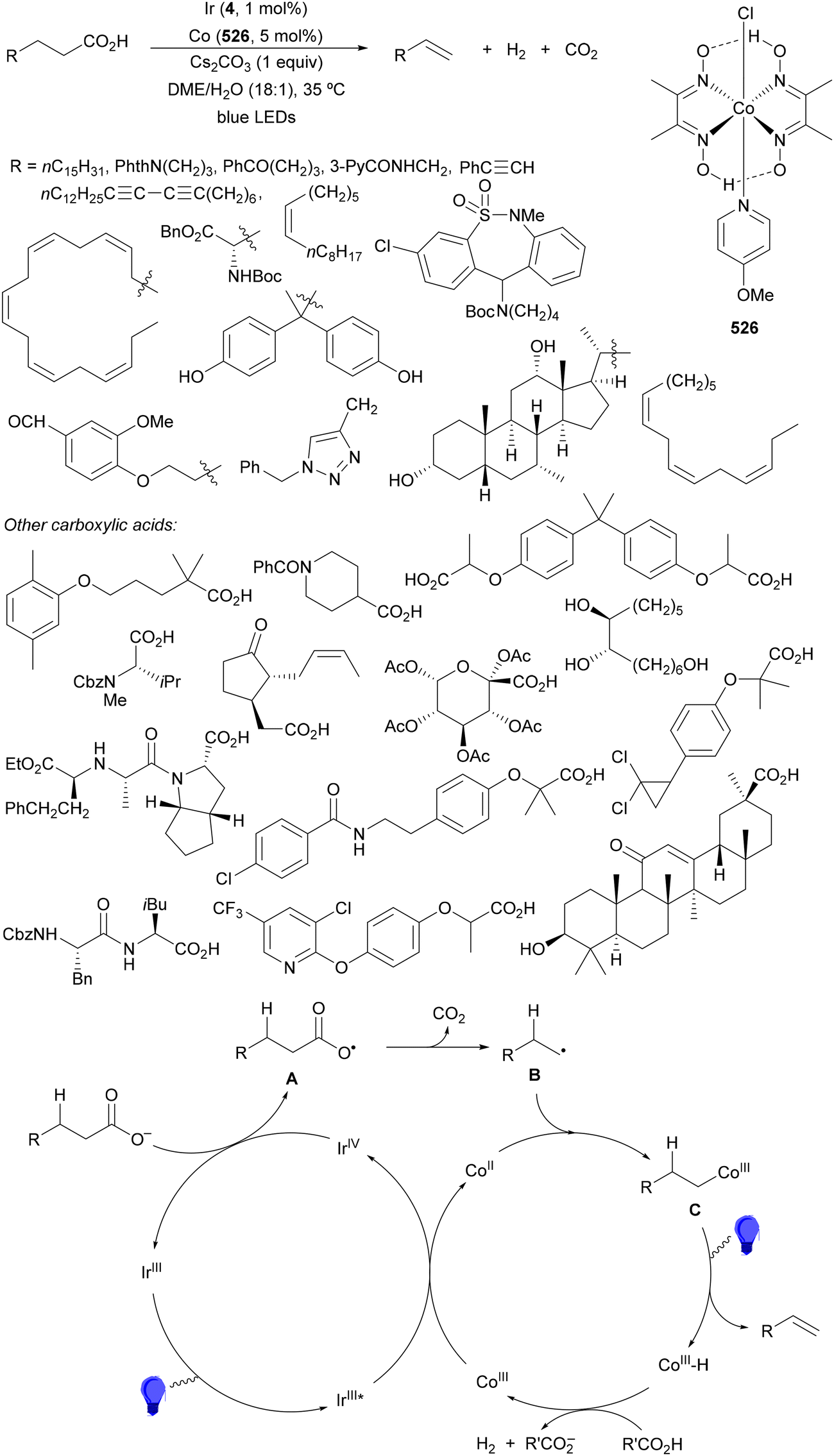 | ||
| Scheme 278 Decarboxylative olefination of aliphatic carboxylic acids under dual Ir/Co photocatalysis. | ||
The former catalytic strategy was simultaneously reported by Cartwright and Tunge508,509 for the decarboxylative elimination of N-acyl AAs to enamides 306. They used (Mes-2,7-Me2-Acr-Ph)BF4 (527) as a PC and cobaloxime Co(dmgH)2ClPy (16) as a catalyst in aqueous MeOH under Ar and blue LED irradiation (Scheme 279). In this case, Co(III) was previously reduced to Co(I) by sodium triacetoxyborohydride (STAB) and Na2CO3 in refluxing methanol. Enamides 306 were obtained with good yields and moderate to good diastereoselectivities. This decarboxylative elimination would take place by protonation of the Co(I) complex by an α-AAs to give the Co(III)–H species and the α-AA carboxy radical A. Photooxidation of A followed by decarboxylation gives radical B. The reduced PC− can reduce Co(III) to Co(II), which is a HAT acceptor of the β-C–H bond of the radical B to produce the product and Co(III)H2, which after H2 evolution regenerates the Co(I) catalyst. These reaction conditions were also applied to a variety of aliphatic carboxylic acids.510
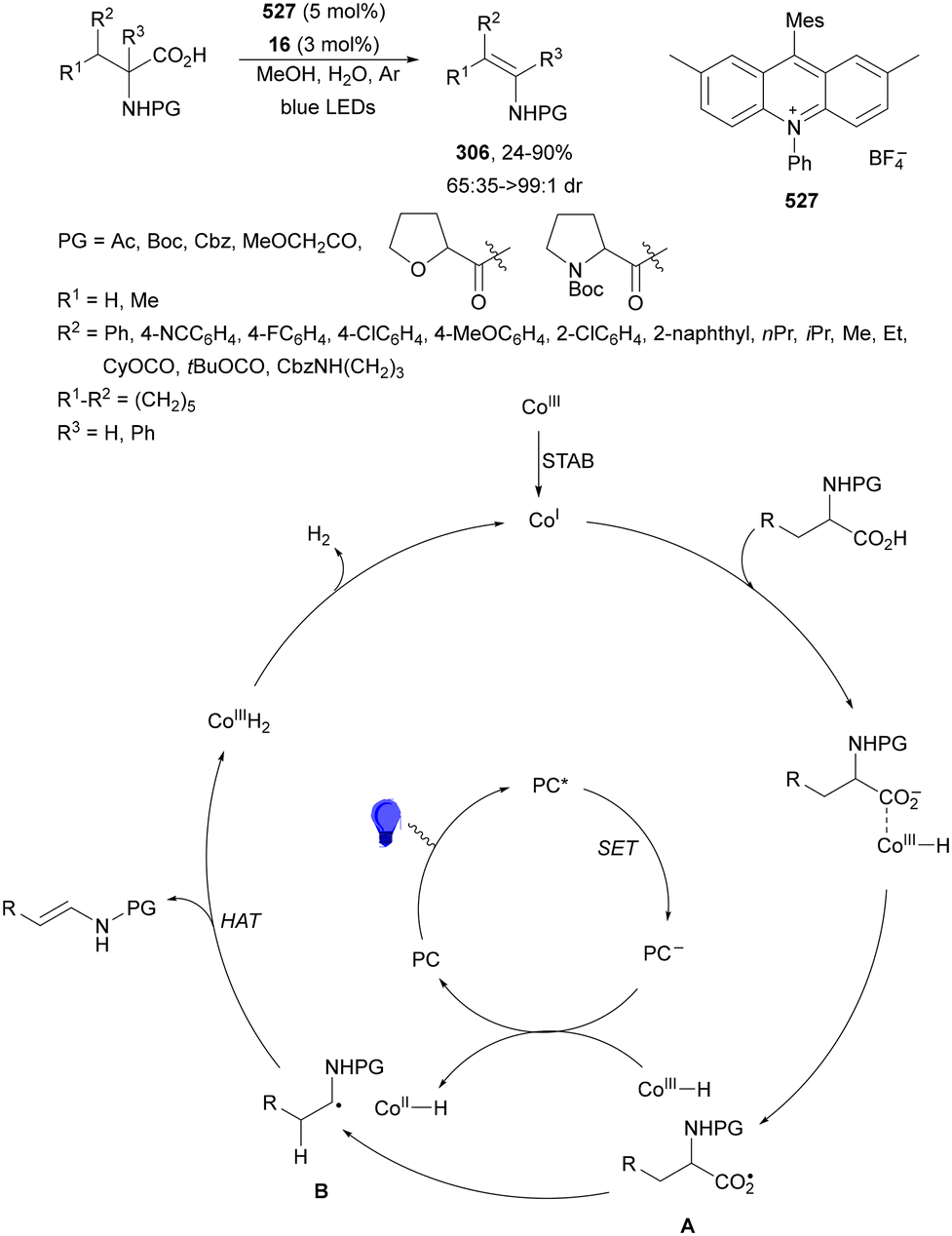 | ||
| Scheme 279 Decarboxylative olefination of α-AAs under dual acridinium salt 16 and Co 527 photocatalysis. | ||
Larionov and co-workers511 reported a cooperative dehydrodecarboxylation of numerous aliphatic carboxylic acids including biomass and bioderived chemicals using acridine 123 and cobaloxime 16 as catalysts. The corresponding alkenes were obtained with, in general, very good yields working in DCM/MeCN (2:1) at room temperature under 400 nm LEDs irradiation (Scheme 280a). These reaction conditions are compatible with an enzymatic process LACo (lipase–acridine–cobaloxime) for conversion of triglycerides to long-chain alkenes. Amano lipase PS from Burkholderia cepacia was used in the cooperative chemoenzymatic LACo process in DCM/MeCN at pH 7 buffer solution of stearin 528 (glyceryl tristearate) to give alkene 529 in 74% yield (Scheme 280b). This LACo process was extended to other triglycerides, for instance glycerol tripalmitate (palmitin, 83% yield) and also to sunflower, canola, corn and soybean oils.
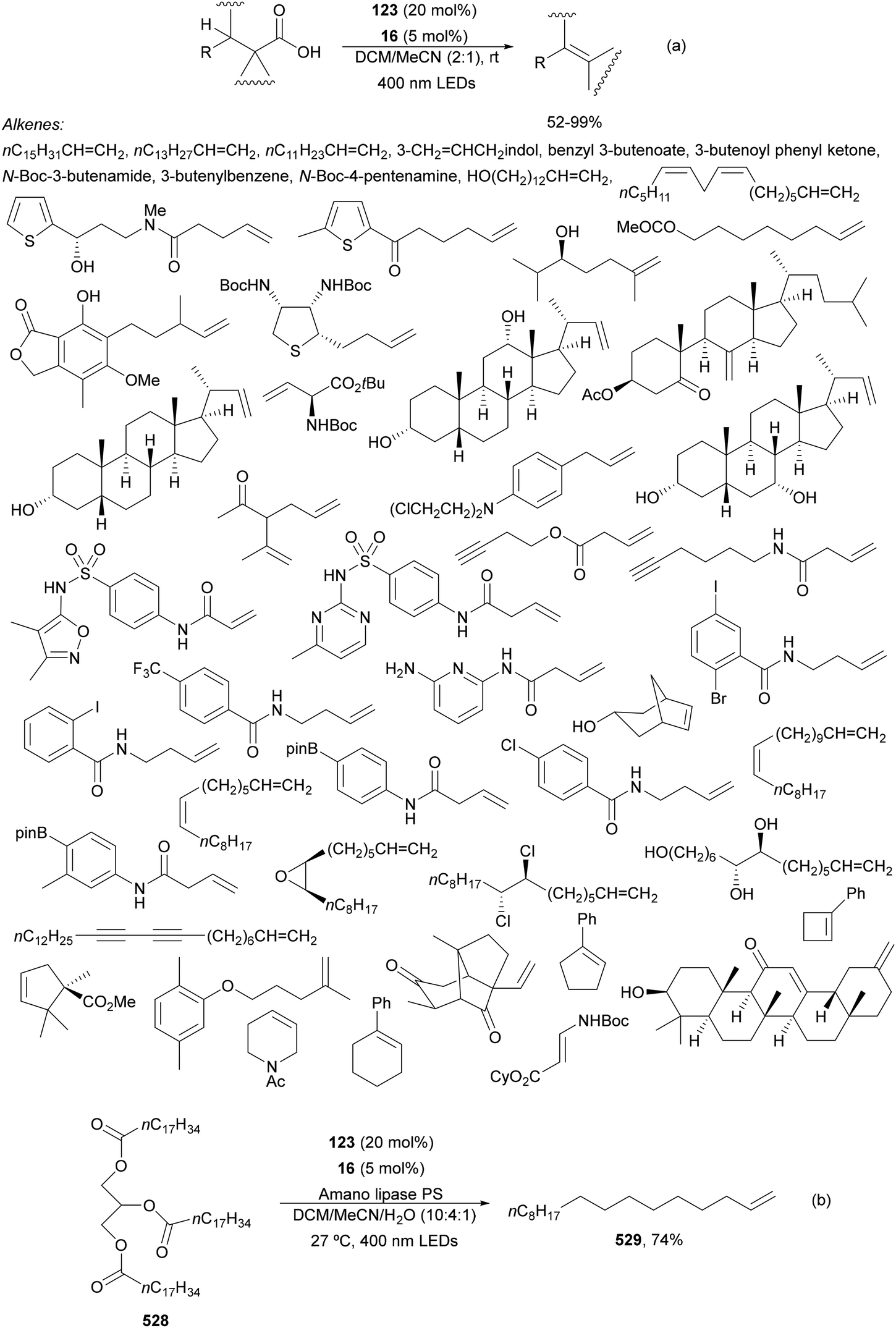 | ||
| Scheme 280 Decarboxylative olefination of aliphatic carboxylic acids under acridine 123 and cobaloxime 16 photocatalysis. | ||
Hydrophobic two-dimensional lead and tin halide perovskites, prepared by intercalating 1-hexadecylammonium (HDA) cation between the inorganic layers, have been employed as PCs in the decarboxylative dehydrogenation of indoline acids to indoles.512 As a conceptual demonstration (HDA)2PbI4 or (HDA)2SnI4 were employed as photoredox catalysts (1 mol%) in DCM under white LEDs irradiation in the presence of oxygen. The lead catalyst gave the best results, with 20–84% yields for indole and 5-bromo- and 5-chloroindole.
Photoinduced decarboxylative elimination of AAs or acyclic tertiary carboxylic acids has been achieved using Fe(OAc)2 and Cu(acac)2 as PCs, DTBP as an oxidant, DBU as a base in EtOAc at room temperature under 390 nm LED irradiation (Scheme 281).53 This LMCT process has been applied to Giese reaction (Scheme 2) and amination reactions (Scheme 241). In the case of AAs, the efficient N–atom might stabilize the copper intermediate and promote the β-elimination instead of C–N bond reductive elimination to give enamines 306. When the nitrogen is protected by an acetyl group a Z/E mixture of products was obtained. However, when a Boc group was used as a protecting group to form styryl derivatives only Z-products were obtained, what is an alternative to Tunge's work.508,509
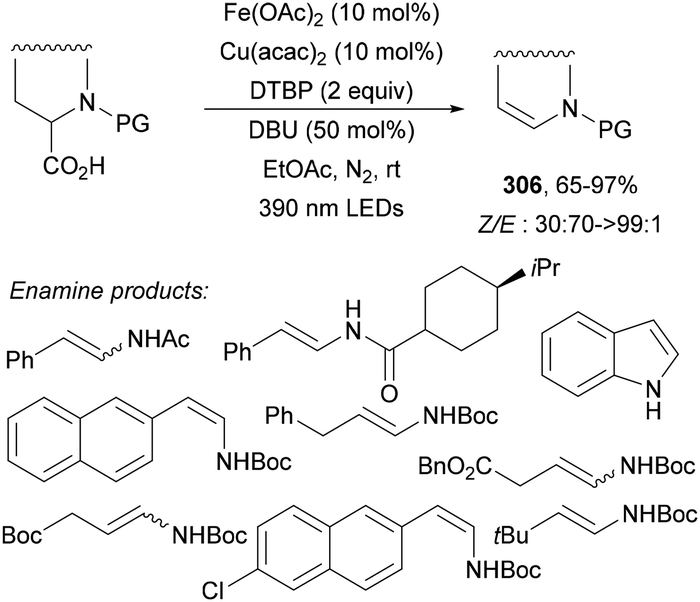 | ||
| Scheme 281 Decarboxylative elimination of α-AAs under Fe/Cu LCMT photocatalysis. | ||
Seidel, Sumerlin and co-workers513 performed the degradation of polyacrylates by photoredox dehydrodecarboxylation followed by one-pot ozonolysis. Under Tunge's reaction conditions508 (Scheme 279) carboxylic acids were converted into internal alkenes and oxidatively cleaved.
Decarboxylative olefination of NHPI esters 69 was reported by Glorius and co-workers514 in 2018. Using a dihydrophenazine 530 as an organophotocatalyst and Cu(II) 2-ethylhexanoate as a catalyst in toluene at room temperature under Ar and 400 LED irradiation, the corresponding alkenes were obtained in general with good yields (Scheme 282). The mechanistic proposal started with a SET process by a photoexcited PC of the NHPI esters to give an alkyl radical A after CO2 and PhthN− extrusion. This radical A is trapped by the Cu(II) catalyst leading to an alkylCu(/III) species B, which gives rise to an olefin and the Cu(I) intermediate Cvia an oxidative elimination process. This dual catalytic cycle is closed by SET between PC+˙ and the Cu(I) species C.
 | ||
| Scheme 282 Decarboxylative elimination of alkyl NHPI esters 69 under dihydrophenazine 530 and Cu(II) 2-ethylhexanoate photocatalysis. | ||
Palladium-catalyzed decarboxylative alkenylation of NHPI esters 69 has been achieved by a dual ligand system under blue LED irradiation.515 Working with PdCl2 as a metal source, Xantphos and Cy-Johnphos as ligands, 2,4,6-collidine as a base in DMA at room temperature under Ar, aliphatic alkenes, styrenes, enol ethers, enamides and peptide enamides were obtained in good yields and moderate to good diastereoselectivity (Scheme 283). This process was applied to a three-step synthesis of cytotoxic chondriamide A and C in 68% overall yield. Based on mechanistic studies, it was proposed that Pd(0) complex A coordinated with both Xantphos and Cy-Johnphos transfers one electron to NHPI ester inducing decarboxylation to form an alkyl radical and a Pd(I) intermediate (B). This intermediate B can dissociate one weakly coordinate phosphine ligand under irradiation to allow alkyl binding to undergo β-H elimination. The dissociate monodentate phosphine rebinds the Pd(0) catalyst after releasing the olefin.
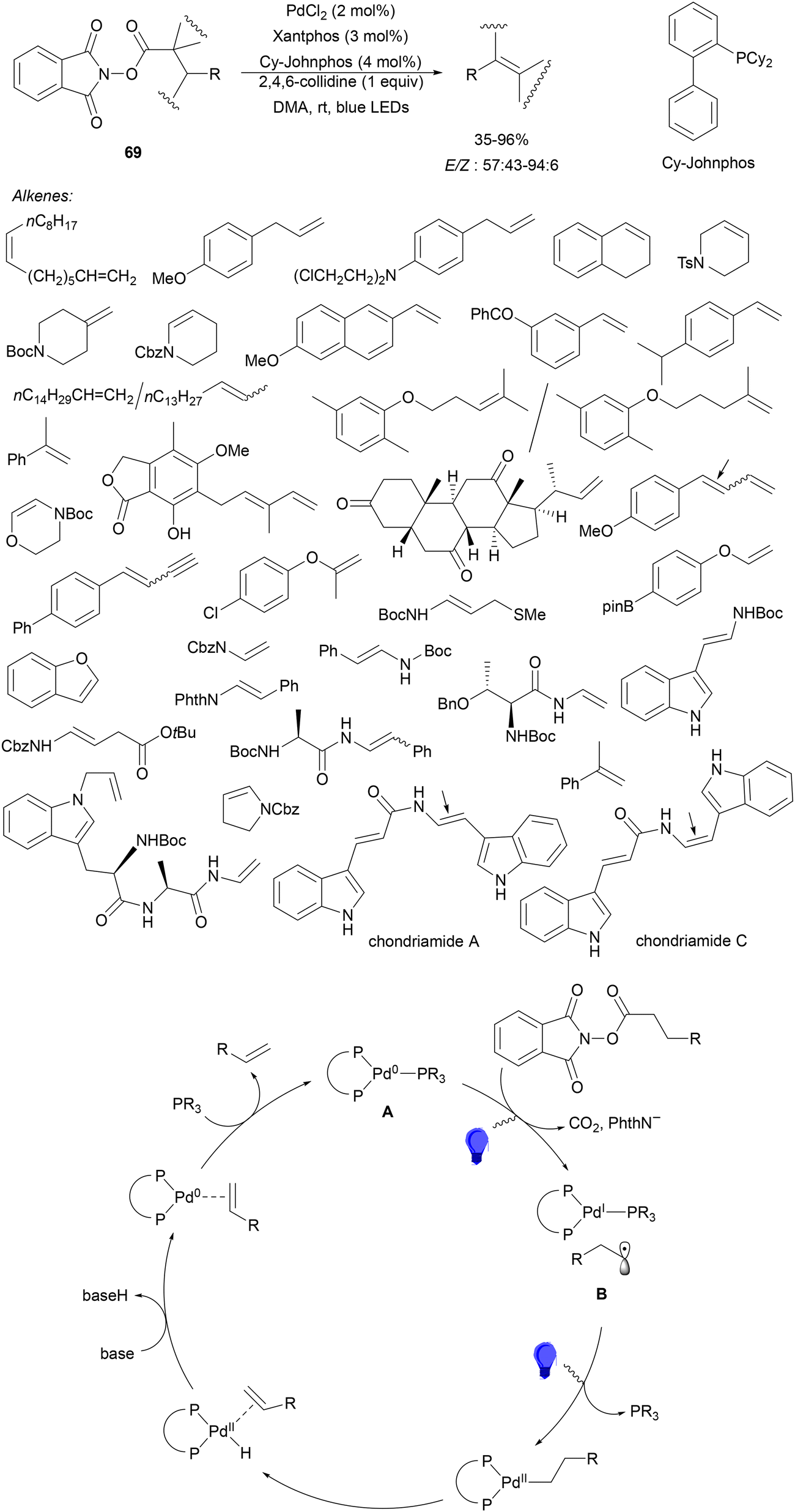 | ||
| Scheme 283 Decarboxylative olefination of alkyl NHPI esters 69 under Pd and synergistical ligands photocatalysis. | ||
An alternative method for the synthesis of olefins from NHPI esters 69 is based on an electron donor-acceptor (EDA) complex.516 Using NaI (2 equivalents) in acetone under blue LED irradiation, alkyl NHPI esters were transformed into the corresponding alkenes with very good yields (Scheme 284). This process probably takes place by formation of the EDA complex A between the NHPI ester and NaI, which after irradiation provides carbon centered radical B and iodine radical C. Subsequently, the iodine radical abstracts a hydrogen atom from radical B to form the olefin.
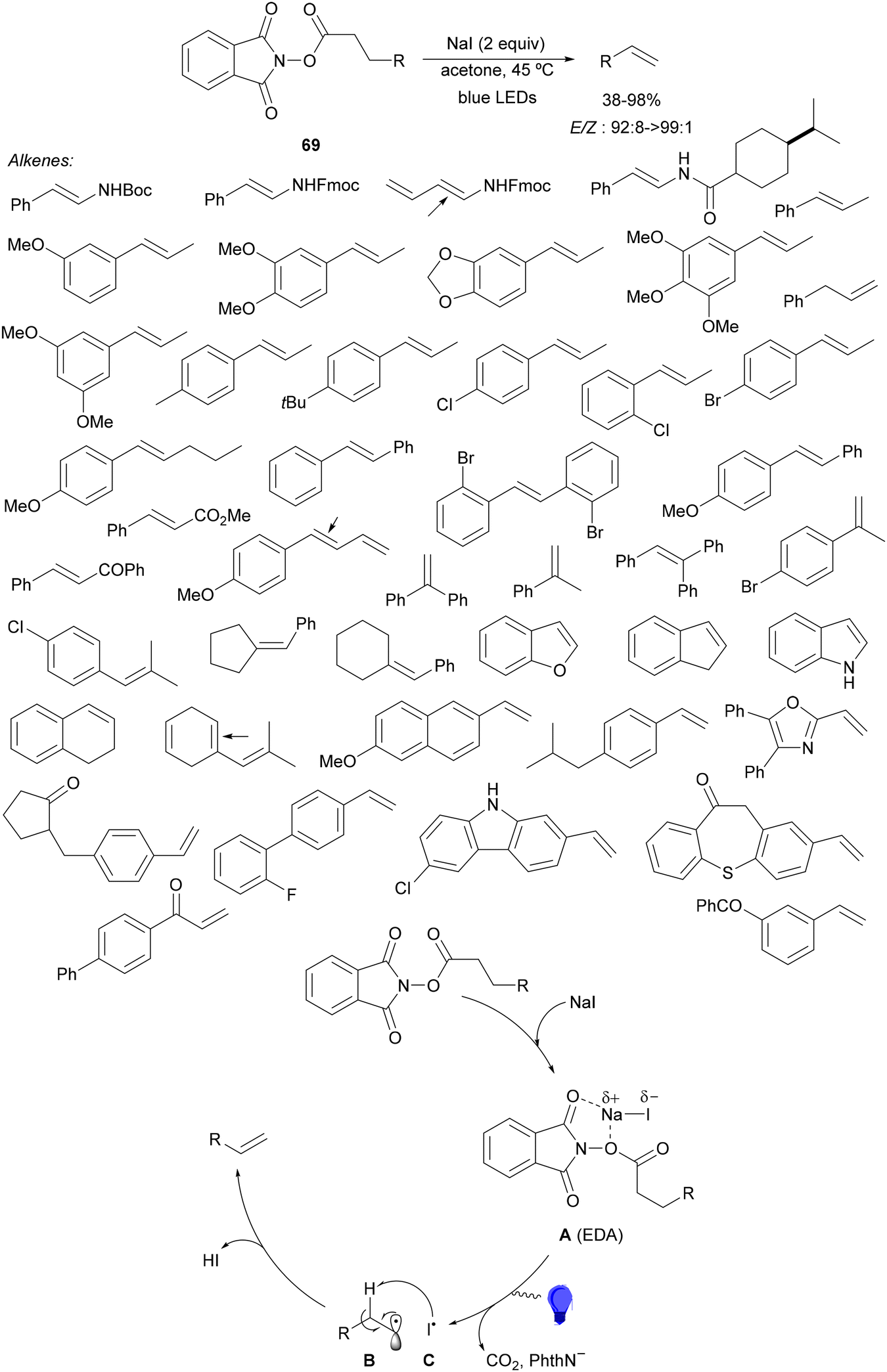 | ||
| Scheme 284 Decarboxylative olefination of NHPI esters under NaI photocatalysis. | ||
5.2. Decarboxylative C–C bond cleavage
Homolytic C–C bond cleavage of carbo- and heterocyclic carboxylic acids has been achieved by photoinduced ligand-to-metal charge transfer (LMCT, see Scheme 200).383 This process was carried out with Fe(acac)3 as a catalyst and DABCO as a base in MeCN at 35 °C under air and 390 nm LED irradiation to give 1,n-dicarbonyl compounds 531 with very good yields (Scheme 285a). In the proposed mechanism, the carboxylate–iron(III) complex A is formed, which after photoexcitation forms by LMCT the reduced Fe(II) complex and the carboxy radical B. Subsequent decarboxylation of B releases the carbon centered radical C. A SET process between dioxygen and Fe(II) regenerates the Fe(III) catalyst. Intermediate C can be trapped by oxygen to provide the peroxyl radical D, which by an intramolecular HAT gives intermediate E. Intermediate E reacts with the superoxide radical anion to form dioxetane F, which undergoes cleavage to yield the dicarbonyl product. When this Fe-catalyzed process was carried out in the presence of cobaloxime 532 and Cs2CO3 as a base a decarboxylative elimination process took place giving the corresponding alkenes with good yields (Scheme 285b). As a plausible mechanism, it was proposed that radical C reacts with Co(II) species to form the complex G, which after a β-H elimination step furnishes the alkene and the Co(III)–H species. This hydride reacts with a proton to form hydrogen and the Co(III) species serves to convert Fe(II) to Fe(III) to close the iron catalytic cycle.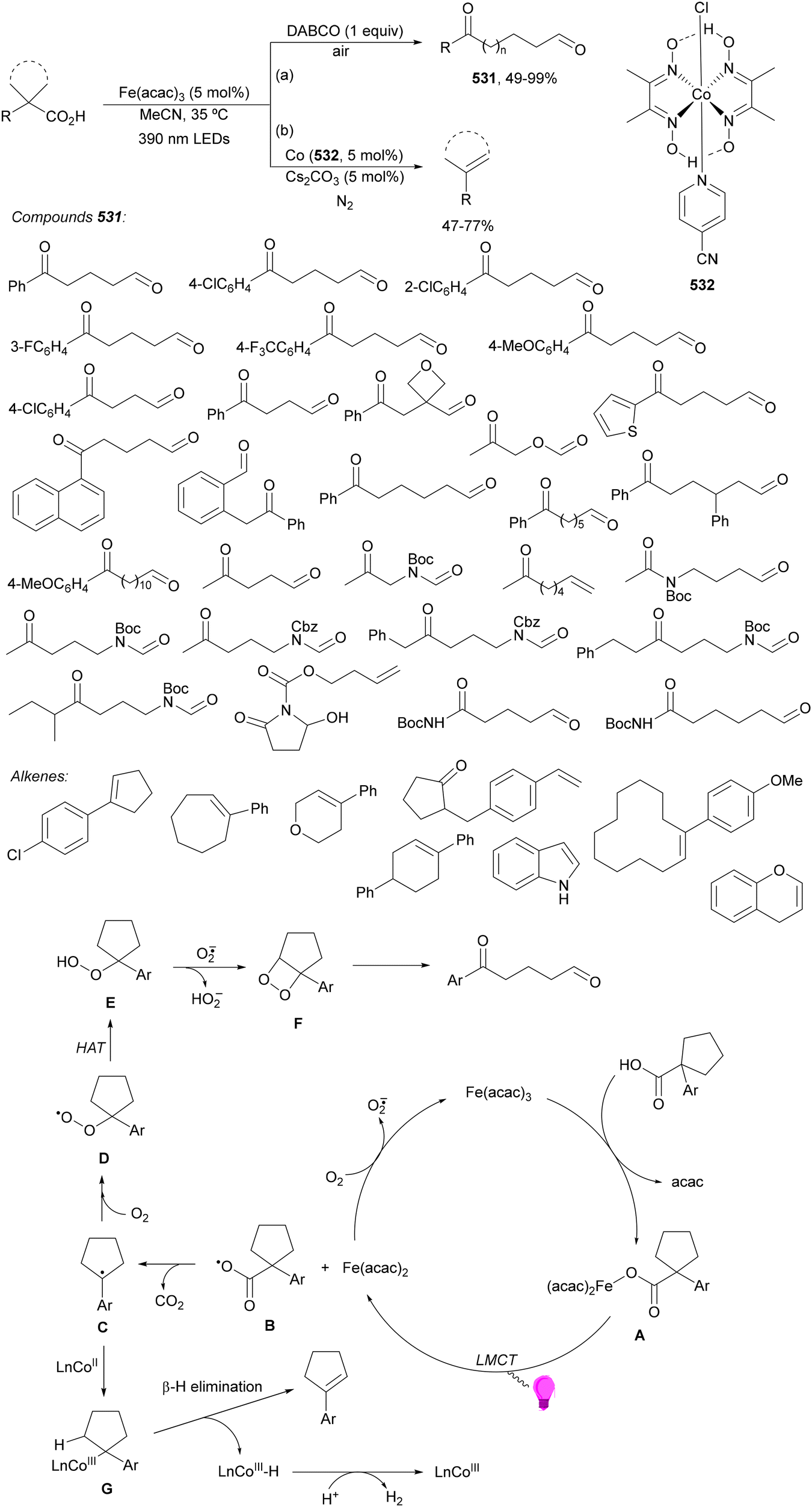 | ||
| Scheme 285 Decarboxylative ring-opening of cyclic carboxylic acids under Fe(acac)3 LMCT photocatalysis and decarboxylative elimination under Fe/Co photocatalysis. | ||
Sun and co-workers517 reported simultaneously the C–C bond cleavage not only of cyclic carboxylic acids but also of α-trisubstituted carboxylic acids. In this case, [dF(CF3)(ppy)2-Ir-μCl]2 complex 533 was used as a PC, Cu(OAc)2 and 2,2′-bipyridine as a ligand, and Cs2CO3 as a base at 30 °C under air and blue LED irradiation provided 1,n-dicarbonyl compounds 531 and ketones for cyclic acids (Scheme 286a and b). Under these reaction conditions, cyclic β-hydroxy carboxylic acids were also transformed into 1,n-dicarbonyl compounds 531 (Scheme 286c). In addition, cyclic carboxylic acids were transformed into n-hydroxy ketones 534 when Selectfluor was added to the reaction mixture (Scheme 286d). All these transformations took place with, in general, good yields and were applied to the synthesis of drugs such as primaperone, melperone and haloperidol and the natural product sertraline. The proposed mechanism is similar to the one proposed by Xia's group.383 In this case, Cu-catalysis promotes a last additional oxidative transformation by a second C–C bond cleavage.
 | ||
| Scheme 286 Decarboxylative C–C bond cleavage of carboxylic acids under Ir 533 and Cu(OAc)2 photocatalysis. | ||
Photoredox decarboxylative olefination of carboxylic acids can be carried out under Ir, acridinium salts and acridine as PCs and cobaloximes as proton reduction catalysis to give the corresponding alkenes. In the case of α-AAs, Fe(OAc)2 and Cu(acac)2 as dual catalysts and DTBP as an oxidant the corresponding enamides were obtained with moderate to high diastereoselectivity. NHPI esters have been transformed into alkenes using a dihydrophenazine as a PC and a Cu carboxylate under a LMCT process. Palladium-catalyzed processes need two type of ligands, a bidentate and a monodentate ones, allowing a broad scope of decarboxylative alkenylation. A simpler method is based on the formation of an EDA complex of the NHPI ester with NaI giving E-alkenes with very good diastereoselectivity. Decarboxylative C–C cleavage of cyclic carboxylic acids under Fe(acac)3 photocatalysis and oxygen can be used for the synthesis of 1,n-dicarbonyl compounds under LMCT conditions. This process, in the presence of cobaloxime, allows the decarboxylative elimination. In the presence of Ir and Cu(OAc)2, cyclic carboxylic acids and β-hydroxy acids were transformed into 1,n-dicarbonyl compounds.
6. Conclusions
Carbon–carbon bond-forming reactions by decarboxylative photocatalysis of carboxylic acids or active esters can be carried out by addition reactions to electron-deficient alkenes, hydroalkylation of alkenes and of alkynes and by addition to carbon heteroatom multiple bonds. C(sp2)–C(Csp3) cross-coupling reactions are performed by arylation of carboxylic acid or active ester derived radicals with aryl halides. Alkylation reactions are appropriate for C(sp3)–C(sp3) carbon–carbon bond-formation between alkyl, allyl and benzyl electrophiles and aliphatic carboxylic acids. For vinylation reactions, alkenyl halides and sulfones give the corresponding alkenes. A second strategy is a decarboxylative Heck-type reaction of aliphatic acids or their active esters with alkenes, and the third one employs cinnamic acids and active esters for this type of C(sp2)–C(sp3) bond-forming reaction. Photoredox-mediated decarboxylative alkynylation of aliphatic carboxylic acids can be carried out using hypervalent iodine reagents. Acylation reactions of aryl halides are mainly performed with α-keto acids, oxalates and oxamic acids. Decarboxylative cyanations are carried out between TMSCN and active esters. For C–H functionalization reactions, decarboxylative alkylation, acylation and arylation procedures, mainly for heterocyclic compounds, can be performed.The third section involves carbon–heteroatom bond-forming reactions by photoredox-mediated decarboxylation of carboxylic acids or their active esters. Aromatic, heteroaromatic and aliphatic acids have been halogenated mainly under metallocatalysis under different halogen sources. In the case of C–O bond-forming reactions, carboxylic acid oxygenation affords carboxy compounds. Decarboxylative etherification of carboxylic acids with alcohols resulted in the presence of hypervalent iodine reagents as well as acyloxylation. For C–S bond-forming reactions, thiolation, thioetherification, thioesterification, sulfilimination, sulfinylation and sulfonylation processes are possible for aliphatic carboxylic acids and their active esters. The corresponding thiols, thioethers, thioesters, sulfilimines, sulfinimides, sulfones, sulfinates, sulfonyl halides, sulfonamides and sulfonyl ketimines can be prepared. Decarboxylative C–N bond forming reactions involve mainly amination and amidation reactions and also azidation and hydrazination processes. In the case of C–P bond-forming reactions, chlorophosphines, phosphites, phosphine oxides and phosphonites are appropriate reagents mainly for active esters. White phosphorus can be employed for aliphatic carboxylic acids. Aliphatic and aromatic carboxylic acids and their active esters are transformed into boronates. For C–Si bond-formation, silacarboxylic acids transfer silyl radicals as well as trialkyl silanes.
Hydro- and deuterodecarboxylation reactions are considered in Section 4 for aliphatic carboxylic acids in the presence of thiols as HAT agents and water of deuterium oxide. In the case of α-keto acids, aldehydes or deuterated ones can be prepared. Heterogeneous conditions can be used for aliphatic and aromatic carboxylic acids. Enzymatic processes based on photodecarboxylase allow protodecarboxylation of aliphatic carboxylic acids.
In Section 5, decarboxylative elimination reactions such as retro-hydrodecarboxylation and decarboxylative C–C bond cleavage are described. In the first case, terminal alkenes and enamides can be prepared from aliphatic carboxylic acids and their active esters. This methodology has been applied to biomass and a broad range of bioactive molecules. Homolytic C–C bond cleavage of cyclic carboxylic acids and cyclic γ-hydroxy acids gives 1,n-dicarbonyl compounds.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| AA | α-Amino acid |
| AAA | Asymmetric allylic alkylation |
| Ac | Acetyl |
| acac | Acetylacetonate |
| Acr | Acridine |
| Adm | Adamantyl |
| Admnsilane | Adamantylaminosupersilane |
| AFDC | 3-Aminofluorene-2,4-dicarbonitrile |
| alk | Alkyl |
| amyl | Pentyl |
| API | Active pharmaceutical ingredient |
| AQ | Anthraquinone |
| Ar | Argon, aryl |
| BBI | n-Butoxybenziodoxole |
| BCN | Boron carbon nitrides |
| Beca P | Benzydryl-catechol-phosphite |
| BCP | Bicyclo[1.1.1]pentane |
| BET | Back electron transfer |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl |
| BI-OAc | Acetoxybenziodoxole |
| BI-OH | Hydroxybenziodoxole |
| Biox | 2,2′-Bis-2-oxazoline |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| BP | Biphenyl |
| Bphen | Bathophenantroline, 4.7-diphenyl-1,10-phenantroline |
| Bpin | Pinacolate boron |
| BPO | Dibenzoyl peroxide |
| bptpy | 2,6-Di(2-pyridyl)-3-(4-bromophenyl)pyridine |
| bpy | 2,2′-Bipyridine |
| bpz | 2,2′-Bipyrazine |
| BrettPhos | Dicyclohexyl-[2′,4′,6′-triisopropyl-3,6-dimethoxy-(1,1′-biphenyl)-2-yl]phosphine |
| BTMG | Barton base, 2-tert-butyl-1,1,3,3-tetramethylguanidine |
| BXM | Baloxavir marboxil |
| Bz | Benzoyl |
| C3G | Cyanidin-3-O-glucoside |
| CBX | Cyanobenziodoxolone |
| Cbz | Benzyloxycarbonyl |
| CDCB | Carbazolyldicyanobenzene |
| CFL | Compact fluorescent lamp |
| Chembead | Chemical-coated glass bead |
| 10Ci-C3H4 | Heterocatalyst from melamine and glyoxal |
| ClTXO | 2-Chlorothioxanthem-9-one |
| CMA | 9-Cyano-10-methoxycarbonylanthracene |
| CN | Cyano, carbon nitride |
| COF | Covalent organic framework |
| collidine | Trimethylpyridine |
| COSNAR | Carbonyl substitution nitrogen atom replacement |
| Cp | Cyclopentadienyl |
| CPA | Chiral phosphonium acid |
| CPME | Cyclopentyl methyl ether |
| CSA | Camphor sulfonic acid |
| CTC | Charge transfer complex |
| Cy | Cyclohexyl |
| 4CzIPN | 1,2,3,5-Tetrakis(carbazole-9-yl)-4,6-dicyanobenzene |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DABSO | 1,4-Dithiabicyclo[2.2.2]octane bis(sulfur dioxide) |
| DANNH2 | 1,8-Diaminonaphthalene |
| DAPO | Dialkyl phosphine oxide |
| dba | Dibenzylideneacetone |
| DBC | Dibenzo[g,p]chrysene |
| DBDMH | 1,3-Dibromo-5,6-dimethylhydantoin |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCA | 9,10-Dicyanoanthracene, dicyanoarene |
| DCB | Dicyanobenzene |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DCE | 1,2-Dichloroethylene |
| DCM | Dichloromethane |
| DCN | 1,4-Dicyanonaphthalene |
| DEDC | N,N-Diethylthiocarbamate |
| DEL | DNA-encoded library |
| dF | Difluor |
| dFMeppy | 2-(2,4-Difluorophenyl)-5-methylpyridine |
| DFT | Density functional theory |
| DGR | Decarboxylative Giese reactions |
| DHP | 1,4-Dihydropyridine, 3,4-dihydropyran |
| DIC | Diisopropyl carbodiimide |
| DIPEA | Diisopropyl ethyl amine |
| DMA | N,N-Dimethylacetamide |
| DMAc | N,N-Dimethylacetamide |
| DMAP | 4-Dimethylaminopyridine |
| DMC | Dimethyl carbonate |
| DMDC | Dimethyl dicarbonate |
| DME | Dimethoxyethane |
| dMebpy | 4.4′-Dimethyl-2,2′-bipyridine |
| DMF | Dimethyl formamide |
| dmgBF2 | Difluoroboryl dimethylglyoxymate |
| dmgH2 | Dimethylglyoxime |
| dmp | 2,9-Dimethyl-1,10-phenanthroline |
| DMP | Dimethoxyphenyl |
| DMPU | 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidone |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| 4DPAIPN | 1,2,3,5-Tetrakis(diphenylamino)-4,6-dicyanobenzene |
| DPZ | 2,3-Dicyano-5,6-di[2′-(5′-methoxythienyl)]pyrazine |
| dr | Diastereomers ratio |
| dtbbpy | 4,4′-Di-tert-butyl-2,2’dipyridyl |
| DTBP | Di-tert-butyl peroxide |
| EA | Electron acceptor |
| EBX | Ethylbenziodoxolone |
| ED | Electron donor |
| EDA | Electron donor-acceptor |
| ee | Enantiomeric excess |
| EPR | Electron paramagnetic resonance |
| equiv. | Equivalent(s) |
| ET | Energy transfer |
| EWG | Electron withdrawing group |
| EYNa2 | Disodium EosinY |
| FAP | Fatty acid photodecarboxylase |
| FAT | Fluorine atom transfer |
| FI | Fluorescein dye |
| FL | Flavin |
| Fmoc | 9-Fluorenylmethyloxycarbonyl |
| FPIFA | Bis(trifluoroacetoxy)iodo pentafluorobenzene |
| GABA | γ-Amino butyric acid |
| gCN | Graphite carbon nitride |
| GSK | Glycogen synthase kinase |
| HAT | Hydrogen atom transfer |
| HBV | Hepatitis B viruses |
| HDA | Hexadecylammonium cation |
| HE | Hantzsch ester |
| Het | Heterocycle, heterocyclic |
| HFIP | Hexafluoroisopropanol |
| HIV | Human immunodeficiency viruses |
| HOMO | Highest occupied molecular orbital |
| HP | 2,6-Dimethyl-3,5-di-(ethoxycarbonyl)pyridine |
| HRMS | High resolution mass spectroscopy |
| THE | High-throughput experimentation |
| IgA | Inmunoglobulin A |
| IPA | Isopropanol |
| Johnphos | (2-Biphenyl)di-tert-butylphosphine |
| kDa | Kilodalton |
| KHMDS | Potassium hexamethyldisilazide |
| L (Ln) | Ligand(s) |
| LACo | Lipase-acridine-cobaloxime |
| LED | Light-emitting diode |
| Leu | Leucine |
| LG | Leaving group |
| LMCT | Ligand to metal change transfer |
| LNPO23 | Iptacopan, 4-{(2S,4S)-4-ethoxy-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl}benzoic acid |
| LUMO | Lowest unoccupied molecular orbital |
| lutidine | Dimethylpyridine |
| MB | Methylene blue |
| MBH | Morita–Baylis–Hilman |
| MBHA | Morita–Baylis–Hilman acetate |
| MCPBA | meta-Chloroperbenzoic acid |
| Mes | Mesityl, 2,4,6-trimethylphenyl |
| MIC | Methylisocyanide |
| MLR | Multivariate linear regression |
| M n | Number-average molecular weight |
| Ms | Methanesulfonyl |
| MS | Molecular sieves |
| MW | Microwave irradiation |
| M x /Mn | Polydispersity |
| NBS | N-Bromosuccinimide |
| NFSI | N-Fluorobenzene sulfonimide |
| NFTPT | 1-Fluoro-1,3,5-trimethylpyridinum tetrafluoroborate |
| NHBC | N-Hydroxybenzimidoyl chloride |
| NHC | N-Heterocyclic carbenes |
| NHPI | N-(Acyloxy)phthalimide |
| NIS | N-Iodosuccinimide |
| NMP | N-Methylpyrrolidone |
| NMR | Nuclear magnetic resonance |
| NP | Nanoparticle(s) |
| Nu, NuH | Nucleophile |
| OEP | Octaethylporphirin |
| P25 | Commercial photocatalyst |
| PARSE | Prospective analysis of the reaction space |
| PBC | para-Bromobenzoyl chloride |
| PC | Photocatalyst |
| PCET | Proton coupled electron transfer |
| Pent | Pentyl |
| PET | Photoinduced electron transfer, positron emission tomography |
| PG | Protecting group |
| PHA | Polyhydroxy acid |
| Phe | Phenylalanine |
| Phen | Phenanthrene |
| PHT | Benzophenothiazine |
| PhthN | Phthalimido |
| PIDA | Phenyliodine(III) diacetate |
| PIFA | Bis(trifluoroacetoxy)iodo benzene |
| pinB | Pinacolateboryl |
| PMB | para-Methoxybenzyl |
| PMP | para-Methoxyphenyl |
| PNH | Paroxysmal nocturnal hemoglobinuria |
| PP | Polypeptide |
| PPTNO | Pyrimidopteridine N-oxide |
| ppy | 2-Phenylpiridine |
| Pro | Proline |
| PTH | N-Phenylbenzo[b]phenothiazine |
| PVAc | Polyvinyl acetate |
| Py | Pyridyl |
| PyCam | Pyridine carboxamide |
| rac | Racemic |
| RAE | Redox active ester |
| RAFT | Reversible addition-fragmentation chair-transfer |
| RFTA | Riboflavin tetraacetate |
| RLT | Radical-ligand transfer |
| rr | Regiosomers ratio |
| RS | Rose Bengal |
| rt | Room temperature |
| Sac | Saccharine |
| SAR | Structure–activity relationship |
| Selectfluor | Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) |
| SET | Single-electron transfer |
| SPINOL | 3,3-Diaryl-1,1′-spiroblindane-7,7′-diol |
| Spy | S-2-Pyridyl |
| STAB | Sodium triacetoxyborohydride |
| TAPO | Trialkyl phosphine oxide |
| TBA | Tetra-n-butylammonium |
| TBAA | Tetra-n-butylammonium acetate |
| TBAC | Tetra-n-butylammonium chloride |
| TBADT | Tetra-n-butylammonium decatungstate |
| TBAF | Tetra-n-butylammonium fluoride |
| TBAI | Tetra-n-butylammonium iodide |
| TBDP | Tetra-n-butylphosphonium bromide |
| TBDPS | tert-Butyldiphenylsilyl |
| TBHP | tert-Butyl hydroperoxide |
| TBPB | tert-Butyl peroxybenzoate |
| TBS | tert-Butyldimethylsilyl |
| TCCA9 | Trichloroisocynamic acid |
| TCNHPI | N-Hydroxytetrachlorophthalimide |
| TCYP | 3,3′-Bis(2,4,6-tricyclohexylphenyl)-1,1′-bi-2-naphthol cyclic monophosphate |
| TEA | Triethylamine |
| TEBACl | Benzyltriethylammonium chloride |
| TEMPO | 2,2,6,6-Tetramethylpiperidinyloxy |
| TES | Triethylsilyl |
| Tf | Triflic, trifluoromethylsulfonyl |
| TFA | Trifluoroacetic acid, trifluoroacetate |
| TFE | 2,2,2-Trifluoroethanol |
| TIPS | Triisopropylsilyl |
| TMEDA | Tetramethylethylenediamime |
| TMG | 1,1,3,3-Tetramethylguanidine |
| TMHD | Bis-(2,2,6,6)-tetramethyl-3,5-heptanedioate |
| TMP | 3,4,7,8-Tetramethyl-1,10-phenanthroline |
| TMS | Trimethylsilyl |
| Tol | 4-Methylphenyl |
| Tp | 1,3,5-Trifromylphloroglucinol |
| Tp* | Tri-(2,5-dimethyl-1-pyrazolyl)borohydride |
| TPA | Tris-(2-pyridylmethyl)amine |
| TPP | Tetraphenylporphirine |
| Tr | Trityl, triphenylmethyl |
| TRIP | 3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate |
| Ts | Tosyl, 4-methylphenylsulfonyl |
| TS | Transition state |
| TX | Thioxanthone |
| UV | Ultraviolet |
| Vis | Visible light |
| W | Watt, decatungstate |
| Xantphos | (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine) |
| XAT | Halogen atom transfer |
| XylBINAP | 2,2′-Bis[di(3,5-xylyl)phosphino]-1,1′-binaphthyl |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We thank the Ministerio de Ciencia, Innovación y Universidades (RED2018-102387-T, RED2022-134287-T, and PID2019-107268GB-100) funded by MCIN/AEI/10.13039/501100011033 and Conselleria de Educación, Cultura, Universidades y Empleo (IDIFEDER/2021/013, GVA-COVID19/2021/079 CIAPOT/2022/11, and APOTIP/2021/020), the University of Alicante (VIGROB-050; UADIF17-42 UAUSTI16-02, VIGROB-068 and UAUSTI21-05) and Medalchemy S. L. (Medalchemy 22T).References
- J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 Search PubMed.
- H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983–4988 Search PubMed.
- Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 Search PubMed.
- T. Patra and D. Maiti, Chem. – Eur. J., 2017, 23, 7328–7401 Search PubMed.
- Y. Li, L. Ge, M. T. Muhammad and H. Bao, Synthesis, 2017, 5263–5284 Search PubMed.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 Search PubMed.
- S. Mondal and S. Chowdhury, Adv. Synth. Catal., 2018, 360, 1884–1909 Search PubMed.
- J.-Q. Liu, A. Shatskly, B. S. Matsuura and M. D. Kärkäs, Synthesis, 2019, 2759–2791 Search PubMed.
- M. Rahman, A. Mukherjee, I. S. Kovalev, D. S. Kopchuk, G. V. Zyranov, M. V. Tsurkan, A. Majee, B. C. Ranu, V. N. Charushin, O. N. Chupakhin and S. Santra, Adv. Synth. Catal., 2019, 361, 2161–2214 Search PubMed.
- L. McMurray, T. M. McGuire and R. I. Howells, Synthesis, 2020, 1719–1737 Search PubMed.
- X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, iScience, 2020, 23, 101266 Search PubMed.
- L. Li, Y. Yao and N. Fu, Eur. J. Org. Chem., 2023, e202300166 Search PubMed.
- S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Adv. Synth. Catal., 2021, 363, 3693–3736 Search PubMed.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 Search PubMed.
- A. M. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, P. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 Search PubMed.
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 Search PubMed.
- T. Shao, X. Ban and Z. Jiang, Chem. Rec., 2023, 23, e202300122 Search PubMed.
- Y. Sakakibara, K. Itami and K. Murakami, J. Synth. Org. Chem. Jpn., 2023, 81, 1050–1061 Search PubMed.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1751 Search PubMed.
- P. Niu, J. Li, Y. Zhang and C. Huo, Eur. J. Org. Chem., 2020, 5801–5812 Search PubMed.
- S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 Search PubMed.
- S. He, H. Li, X. Chen, I. B. Krylov, A. O. Terent’ev, L. Qu and B. Yu, Chin. J. Org. Chem., 2021, 41, 4661–4689 Search PubMed.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 15598–15627 Search PubMed.
- M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576–9580 Search PubMed.
- D. S. Müller, N. L. Untiedt, A. P. Dieskau, G. L. Lackner and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 660–663 Search PubMed.
- J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 Search PubMed.
- Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 Search PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 Search PubMed.
- A. Gualandi, E. Matteucci, F. Monti, A. Baschieri, N. Armaroli, L. Sambri and P. G. Cozzi, Chem. Sci., 2017, 8, 1613–1620 Search PubMed.
- G. H. Lovett and B. A. Sparling, Org. Lett., 2016, 18, 3494–3497 Search PubMed.
- A. Millet, Q. Lefebvre and M. Rueping, Chem. – Eur. J., 2016, 22, 13464–13468 Search PubMed.
- S. Inuki, K. Sato, T. Fukuyama, I. Ryu and Y. Fujimoto, J. Org. Chem., 2017, 82, 1248–1253 Search PubMed.
- S. Zhang, Z. Tan, H. Zhang, J. Liu, W. Xu and K. Xu, Chem. Commun., 2017, 53, 11642–11645 Search PubMed.
- T. Guo, L. Zhang, Y. Fang, X. Jin, Y. Li, R. Li, X. Li, W. Chen, X. Liu and Z. Tian, Adv. Synth. Catal., 2018, 360, 1352–1357 Search PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 Search PubMed.
- D. K. Kölmel, R. P. Loachi, T. Knauber and M. E. Flanagan, ChemMedChem, 2018, 13, 2159–2164 Search PubMed.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2158 Search PubMed.
- J. C. DeForest, S. A. Samame, G. Suryn, A. Burtea and S. D. Rychnovsky, J. Org. Chem., 2018, 83, 8914–8925 Search PubMed.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6088 Search PubMed.
- I. C. S. Wan, M. D. Witte and A. J. Minnaard, Org. Lett., 2019, 21, 7669–7673 Search PubMed.
- R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 Search PubMed.
- L. Gingipalli, J. Boerth, D. Emmons, T. Grebe, H. Hatoum-Mokdad, B. Peng, L. Sha, S. Tentarelli, H. Wang, Y. Wu, X. Zheng, S. Edmondson and A. Gopalsamy, Org. Lett., 2020, 22, 3418–3422 Search PubMed.
- G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J.-C. Cintrat, Angew. Chem., Int. Ed., 2020, 59, 2618–2622 Search PubMed.
- N. P. Ramírez and J. C. González-Gómez, Eur. J. Org. Chem., 2017, 2154–2163 Search PubMed.
- G. Z. Wang, R. Shang, W. M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4883 Search PubMed.
- T. Yamamoto, T. Iwasaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2018, 83, 3702–3709 Search PubMed.
- A. A. Shah, M. J. Kelly III and J. J. Perkins, Org. Lett., 2020, 22, 2196–2200 Search PubMed.
- F. El-Hage, C. Schöll and J. Pospech, J. Org. Chem., 2020, 85, 13853–13867 Search PubMed.
- H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 Search PubMed.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 Search PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 Search PubMed.
- L. L. Lao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D. G. Yu, J. Am. Chem. Soc., 2021, 143, 2812–2821 Search PubMed.
- N. Xiong, Y. Li and R. Zeng, ACS Catal., 2023, 13, 1678–1685 Search PubMed.
- V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 Search PubMed.
- S. Gavelle, M. Innocent, T. Aubineau and A. Guérinot, Adv. Synth. Catal., 2022, 364, 4189–4230 Search PubMed.
- S. O. Klein, A. A. Baniahmad and M. Jung, Chem. Commun., 2023, 59, 1971–1974 Search PubMed.
- M. A. J. Dubois, J. J. Rojas, A. J. Sterling, H. C. Broderick, M. A. Smith, A. J. P. White, P. W. Miller, C. Choi, J. J. Mousseau, F. Duarte and J. A. Bull, J. Org. Chem., 2023, 88, 6476–6488 Search PubMed.
- K. Anwar, F. J. Aguilar Troyano, A. H. Abazid, O. El Yarroudi, I. Funes-Ardoiz and A. Gómez-Suárez, Org. Lett., 2023, 25, 3216–3221 Search PubMed.
- K. Anwar, L. Capaldo, T. Wan, T. Noël and A. Gómez-Suárez, Chem. Commun., 2024, 60, 1456–1459 Search PubMed.
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 Search PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 Search PubMed.
- J.-Q. Chen, R. Chang, Y.-L. Wei, J. N. Mo, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2018, 83, 253–259 Search PubMed.
- A. M. Davies, R. D. Hernandez and J. A. Tunge, Chem. – Eur. J., 2022, 28, e202202781 Search PubMed.
- A. Vega-Peñalaza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell’Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 Search PubMed.
- Y. Yoshimi, J. Photochem. Photobiol., A, 2017, 42, 116–130 Search PubMed.
- Y. Yoshimi, Chem. Rec., 2024, 24, e202300326 Search PubMed.
- S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2020, 85, 5362–5369 Search PubMed.
- Y. Tajimi, Y. Nachi, R. Inada, R. Hashimoto, M. Yamawaki, K. Ohkubo, T. Morita and Y. Yoshimi, J. Org. Chem., 2022, 87, 7405–7413 Search PubMed.
- M. Yamawaki, R. Hashimoto, Y. Kawabata, M. Ichihashi, Y. Nachi, R. Inari, C. Sakamoto, T. Morita and Y. Yoshimi, Eur. J. Org. Chem., 2022, e202201225 Search PubMed.
- R. Hashimoto, T. Furutani, H. Suzuki and Y. Yoshimi, Synlett, 2024, 357–361 Search PubMed.
- Y. Shinkawa, T. Furutani, T. Ikeda, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2022, 87, 11816–11825 Search PubMed.
- M. Yamawaki, K. Matsumoto, T. Furutani, S. Sugihara and Y. Yoshimi, Polymer, 2024, 308, 127336 Search PubMed.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Fu, Chem. Commun., 2019, 55, 5408–5412 Search PubMed.
- O. Zhang and J. W. Schubert, J. Org. Chem., 2020, 85, 6225–6232 Search PubMed.
- K. Merkens, F. J. Aguilar Troyano, J. Djossou and A. Gómez-Suárez, Adv. Synth. Catal., 2020, 362, 2354–2359 Search PubMed.
- H. Yang, Y. Yao, Q. Yang, Y. Yao, J. Sun and S. Sun, Org. Lett., 2024, 26, 4194–4199 Search PubMed.
- P. Ji, J. Chen, X. Meng, F. Gao, Y. Dong, H. Xu and W. Wang, J. Org. Chem., 2022, 87, 14706–14714 Search PubMed.
- D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 Search PubMed.
- A. Abas, J. Mass-Roselli, M. M. Amer, D. J. Durand, R. R. Groleau, N. Fey and J. Clayden, Angew. Chem., Int. Ed., 2019, 58, 2418–2422 Search PubMed.
- A. M. Davies, S. S. Londhe, E. R. Smith and J. A. Tunge, Org. Lett., 2023, 25, 8634–8639 Search PubMed.
- M. Ayurini, D. Haridas, D. J. Mendoza, G. Garnier and J. F. Hooper, Angew. Chem., Int. Ed., 2024, 63, e202317071 Search PubMed.
- D. M. Kitcatt, E. Pogacar, L. Mi, S. Nicolle and A.-L. Lee, J. Org. Chem., 2024, 89, 16055–16059 Search PubMed.
- A. Chen, S. Zhao, Y. Han, Z. Zhou, B. Yang, L.-G. Xie, M. A. Walczak and F. Zhu, Chem. Sci., 2023, 14, 7569–7580 Search PubMed.
- R. Talukdar, D. Chong and A. J. Fairbanks, Org. Lett., 2024, 26, 10536–10541 Search PubMed.
- Y.-Z. Cheng, X.-L. Huang, W.-H. Zhuang, Q.-R. Zhao, X. Zhang, T.-S. Mei and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 18062–18067 Search PubMed.
- C. Zhou, M. Li, J. Sun, J. Cheng and S. Sun, Org. Lett., 2021, 23, 2895–2899 Search PubMed.
- J. Zheng, A. Nikbakht and B. Breit, ACS Catal., 2021, 11, 3343–3350 Search PubMed.
- J. Zheng, N. Tang, H. Xie and B. Breit, Angew. Chem., Int. Ed., 2022, 61, e202200105 Search PubMed.
- C.-H. Ma, Y. Ji, J. Zhao, X. He, S.-T. Zhang, Y.-Q. Jiang and B. Yu, Chin. J. Catal., 2022, 43, 571–583 Search PubMed.
- A. Wang, Y. Ma, J.-H. Jin, L.-Q. Wang, D.-D. Li, Z.-W. Xi, C. Chen and Y.-M. Shen, J. Org. Chem., 2024, 89, 17382–17388 Search PubMed.
- J.-Z. Wang, E. Mao, J. A. Nguyen, W. L. Lyon and D. W. C. MacMillan, J. Am. Chem. Soc., 2024, 146, 15693–15700 Search PubMed.
- K.-J. Bian, Y.-C. Lu, D. Nemoto Jr, S.-C. Kao, X. Chen and J. G. West, Nat. Chem., 2023, 15, 1683–1692 Search PubMed.
- X.-K. Qi, L.-J. Yao, M.-J. Zheng, L. Zhao, C. Yang, L. Guo and W. Xia, ACS Catal., 2024, 14, 1300–1310 Search PubMed.
- X. Jiang, Y. Lan, Y. Hao, K. Jiang, J. He, J. Zhu, S. Jia, J. Song, S.-J. Li and L. Niu, Nat. Commun., 2024, 15, 6115 Search PubMed.
- W. Liao and X. Ni, Photochem. Photobiol. Sci., 2020, 16, 1211–1219 Search PubMed.
- N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 Search PubMed.
- H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 5738–5745 Search PubMed.
- M. M. Mastandrea, S. Cañellas, X. Caldentey and M. A. Pericàs, ACS Catal., 2020, 10, 6402–6408 Search PubMed.
- I. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 Search PubMed.
- G.-L. Dai, S.-Z. Lai, Z. Luo and Z.-Y. Tang, Org. Lett., 2019, 21, 2269–2272 Search PubMed.
- C. Yang, X. Sheng, L. Zhang, J. Yu and D. Huang, Asian J. Org. Chem., 2020, 9, 23–41 Search PubMed.
- S. Sharma, S. Sultan, S. Devari and B. A. Shah, Org. Biomol. Chem., 2016, 14, 9645–9649 Search PubMed.
- A. G. Griesbeck, J.-M. Neudörfl, B. Goldfuss and S. Molitor, ChemPhotoChem, 2017, 1, 355–362 Search PubMed.
- Y. Liu, X. Liu, J. Li, X. Zhao, B. Qiao and Z. Jiang, Chem. Sci., 2018, 9, 8094–8098 Search PubMed.
- S. Pan, M. Jiang, J. Hu, R. Xu, X. Zeng and G. Zhong, Green Chem., 2020, 22, 336–341 Search PubMed.
- H. Wen, R. Ge, Y. Qu, J. Sun, X. Shi, W. Cui, H. Yan, Q. Zhang, Y. An, W. Su, H. Yang, L. Kuai, A. L. Satz and X. Peng, Org. Lett., 2020, 22, 9484–9489 Search PubMed.
- X. Huang, Y.-Q. Hu, C. Zhou, Y. Zheng and X. Zhang, Green Chem., 2022, 24, 5764–5769 Search PubMed.
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 Search PubMed.
- K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242–252 Search PubMed.
- G. Liu, Y. Gao and W. Su, J. Org. Chem., 2023, 88, 6322–6332 Search PubMed.
- C.-L. Dong, H.-C. Liu, Z. Guan and Y.-H. He, J. Org. Chem., 2024, 89, 10929–10938 Search PubMed.
- A. F. Garrido-Castro, H. Choubane, M. Daaou, M. C. Maestro and J. Alemán, Chem. Commun., 2017, 53, 7764–7767 Search PubMed.
- J. Guo, Q.-L. Wu, Y. Xie, J. Weng and G. Lu, J. Org. Chem., 2018, 83, 12559–12567 Search PubMed.
- S. Pan, M. Jiang, G. Zhong, L. Dai, Y. Zhou, K. Wei and X. Zeng, Org. Chem. Front., 2020, 7, 4043–4048 Search PubMed.
- L. Dai, Q. Zhu, J. Zeng, Y. Liu, G. Zhong, X. Han and X. Zeng, Org. Chem. Front., 2022, 9, 2994–2999 Search PubMed.
- F. Foubelo, C. Nájera, M. G. Retamosa, J. M. Sansano and M. Yus, Chem. Soc. Rev., 2024, 53, 7983–8085 Search PubMed.
- C. Nájera, J. M. Sansano and M. Yus, Org. Biomol. Chem., 2015, 13, 8596–8636 Search PubMed.
- A. Fall, M. Magdei, M. Savchuk, S. Oudeyer, H. Beucher and J.-F. Brière, Chem. Commun., 2024, 60, 6316–6319 Search PubMed.
- J. Tian, L. Zhao, C. Yang, C. Yang, L. Gao and W. Xia, ACS Catal., 2023, 13, 866–876 Search PubMed.
- S. Kim, B. Park, G. S. Lee and S. H. Hong, J. Org. Chem., 2023, 88, 6532–6537 Search PubMed.
- Z. M. Rubanov, V. V. Levin and A. D. Dilman, Org. Lett., 2024, 26, 3174–3178 Search PubMed.
- X. Sui, H. T. Dang, A. Porey, R. Trevino, A. Das, S. O. Fremin, W. B. Hughes, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2024, 15, 9582–9590 Search PubMed.
- J. Jia, Q. Lefebvre and M. Rueping, Org. Chem. Front., 2020, 7, 602–608 Search PubMed.
- Z. M. Rubanov, V. V. Levin and A. Dilman, Org. Lett., 2023, 25, 8751–8755 Search PubMed.
- G. Wu, J. Wang, C. Liu, M. Sun, L. Zhang, Y. Ma, R. Cheng and J. Ye, Org. Chem. Front., 2019, 6, 2245–2248 Search PubMed.
- P. Ji, Y. Zhang, Y. Wu, H. Huang, W. Hu, P. A. Marino and W. Wang, Org. Lett., 2019, 21, 3086–3090 Search PubMed.
- J. Wang, Z. Shao, K. Tan, R. Tang, Q. Zhou, M. Xu, Y.-M. Li and Y. Shen, J. Org. Chem., 2020, 85, 9944–9954 Search PubMed.
- A. Shatskiy, A. Axelsson, E. V. Stepanova, J.-C. Liu, A. Z. Temerdashev, B. P. Kore, B. Blomkvist, J. M. Gardner, P. Dinér and M. D. Kärkäs, Chem. Sci., 2021, 12, 5430–5437 Search PubMed.
- S. Bonciolini, A. Pulcinella, M. Leone, D. Shiroli, A. Luguera Ruiz, A. Sorato, M. A. J. Dubois, R. Gopalakrishnan, G. Massonv, N. Della Ca’, S. Protti, M. Fagnoni, E. Zysman-Colman, M. Johansson and T. Noël, Nat. Commun., 2024, 15, 1509 Search PubMed.
- M.-L. Shen, Y. Shen and F.-S. Wang, Org. Lett., 2019, 21, 2993–2997 Search PubMed.
- P. R. Sultane, T. B. Mete and R. G. Bhat, Org. Biomol. Chem., 2014, 12, 261–264 Search PubMed.
- H. Qiu, A. Matsumoto and K. Maruoka, J. Am. Chem. Soc., 2024, 146, 35478–35485 Search PubMed.
- K. Blatt, A. Adili, A. H. Tran, K. M. Elmallah, I. Ghiviriga and D. Seidel, J. Am. Chem. Soc., 2024, 146, 26331–26339 Search PubMed.
- H.-H. Li, J.-Q. Li, X. Zheng and P.-Q. Huang, Org. Lett., 2021, 23, 876–880 Search PubMed.
- H.-C. Li, G.-N. Li, K. Sun, X.-L. Chen, M.-X. Jiang, L.-B. Qu and B. Yu, Org. Lett., 2022, 24, 2431–2435 Search PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terret, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 Search PubMed.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 Search PubMed.
- M. S. Oderinde, A. Varela-Alvarez, B. Aquila, D. W. Robbins and J. W. Johannes, J. Org. Chem., 2015, 80, 7642–7651 Search PubMed.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 Search PubMed.
- M. Yus, C. Nájera, F. Foubelo and J. M. Sansano, Chem. Rev., 2023, 123, 11817–11892 Search PubMed.
- J. Lao and J. Zhang, ACS Catal., 2016, 6, 873–877 Search PubMed.
- C. Pezzetta, D. Bonifazi and R. W. M. Davidson, Org. Lett., 2019, 21, 8957–8961 Search PubMed.
- J. Jung, T. Kinoshita, Y. Makihara, Y. Sakakibara, K. Amaike, K. Murakami and K. Itami, Synlett, 2024, 337–341 Search PubMed.
- H. Huang, X. Li, C. Yu, Y. Zang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 Search PubMed.
- B. Zhao, R. Shang, W.-M. Cheng and Y. Fu, Org. Chem. Front., 2018, 5, 1782–1786 Search PubMed.
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Chem. Commun., 2019, 55, 14657–14660 Search PubMed.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 Search PubMed.
- B. Matsuo, A. Granados, G. Levitre and G. A. Molander, Acc. Chem. Res., 2023, 56, 385–401 Search PubMed.
- D. K. Kölmel, J. Meng, M.-H. Tsai, J. Que, R. P. Loach, T. Knauber, J. Wan and M. E. Flanagan, ACS Comb. Sci., 2019, 21, 588–597 Search PubMed.
- P. R. Chheda, N. Simons and Z. Shi, Org. Lett., 2024, 26, 4365–4370 Search PubMed.
- C. N. Prieto Kullner, J. A. Kautzky, S. W. KrsKa, T. Nowak, S. D. Dreher and D. W. C. MacMillan, Science, 2022, 376, 532–539 Search PubMed.
- H.-W. Hsieh, C. W. Coley, L. M. Baumgartner, K. F. Jensen and R. I. Robinson, Org. Process Res. Dev., 2018, 22, 542–550 Search PubMed.
- R. Zhang, G. Li, M. Wismer, P. Vachal, S. L. Colletti and Z.-C. Shi, ACS Med., Chem. Lett., 2018, 9, 773–777 Search PubMed.
- M. González-Esguevillas, D. F. Fernández, J. A. Rincón, M. Barberis, O. de Frutos, C. Mateos, S. García-Cerrada, J. Agejas and D. W. C. MacMillan, ACS Cent. Sci., 2021, 7, 1126–1134 Search PubMed.
- N. J. Gesmundo, N. P. Tu, K. A. Sarris and Y. Wang, ACS Med. Chem. Lett., 2023, 14, 521–529 Search PubMed.
- G. Wuitschik, M. Roger-Evans, A. Buckl, M. Bernasconi, M. Märkl, T. Gödel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. M. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512–4515 Search PubMed.
- K. Kolahdouzan, R. Khalaf, J. M. Grandner, Y. Chen, J. A. Terrett and M. P. Huestis, ACS Catal., 2020, 10, 405–411 Search PubMed.
- M. D. Shea, U. F. Mansoor and B. A. Hopkins, Org. Lett., 2020, 22, 1052–1055 Search PubMed.
- M. Pitchai, A. Ramirez, D. M. Mayder, S. Ulaganathan, H. Kumar, D. Aulakh, A. Gupta, A. Marthur, J. Kempson, N. Meanwell, Z. M. Hudson and M. S. Oderinde, ACS Catal., 2023, 13, 647–658 Search PubMed.
- R. Nallagonda, R. Quan, L. Grant, C. Jorge, S. Yip, D.-R. Wu, T. G. Dhar, J. Kempson, A. Mathur and M. S. Oderinde, ACS Catal., 2024, 14, 13439–13450 Search PubMed.
- N. Mainolfi, T. Ehara, R. G. Karki, K. Anderson, A. MacSweeney, S.-M. Liao, U. A. Argikar, K. Jendza, C. Zhang, J. Powers, D. W. Klosowski, M. Crowley, T. Kawanami, J. Ding, M. April, C. Forster, M. Serrano-Wu, M. Capparelli, R. Ramqaj, C. Solovay, F. Cumin, T. M. Smith, L. Ferrara, W. Lee, D. Long, M. Prentiss, A. De Erkenez, L. Yang, F. Liu, H. Sellner, F. Sirockin, E. Valeur, P. Erbel, D. Ostermeier, P. Ramage, B. Gerhartz, A. Schubart, S. Flohr, N. Gradoux, R. Feifel, B. Vogg, C. Wiesmann, J. Maibaum, J. Eder, R. Sedrani, R. A. Harrison, M. Mogi, B. D. Jaffee and C. M. Adams, J. Med. Chem., 2020, 63, 5697–5722 Search PubMed.
- H. Wang, C.-F. Liu, Z. Song, M. Yuan, Y. A. Ho, O. Gutierrez and M. J. Koh, ACS Catal., 2020, 10, 4451–4459 Search PubMed.
- A. Das, A. Choi and I. Coldham, Org. Lett., 2023, 25, 987–991 Search PubMed.
- J. D. Firth, P. O’Brian and L. Ferris, J. Org. Chem., 2017, 82, 7023–7031 Search PubMed.
- A. Yanez and D. M. Jeanneret, Synlett, 2024, 1728–1732 Search PubMed.
- F. Lukas, M. T. Findlay, M. Fillols, J. Templ, E. Savino, B. Martin, S. Allmendinger, M. Furegati and T. Noël, Angew. Chem., Int. Ed., 2024, 63, e202405902 Search PubMed.
- R. Nsouli, S. Nayak, V. Balakrishnan, J.-Y. Lin, B. K. Chi, H. G. Ford, A. V. Tran, I. A. Guzei, J. Bacsa, N. R. Armada, F. Zenov, D. J. Weix and L. K. G. Ackerman-Biegasiewicz, J. Am. Chem. Soc., 2024, 146, 29551–29559 Search PubMed.
- N. E. Behnke, Z. S. Sales, N. Li and A. T. Herrmann, J. Org. Chem., 2021, 86, 12945–12955 Search PubMed.
- M. Tao, L.-Y. Zeng, W. Li, G. Pu, J. Jia, Q. Yao, X. Li and C.-Y. He, Adv. Synth. Catal., 2023, 365, 854–859 Search PubMed.
- L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 Search PubMed.
- S. Pal, J. Openy, A. Krzyzanowski, A. Noisier and P. ’t Hart, Org. Lett., 2024, 26, 2795–2799 Search PubMed.
- K. A. Xie, E. Bednarova, C. L. Joe, T. C. Sherwood, E. R. Welin and T. Rovis, J. Am. Chem. Soc., 2024, 146, 25780–25787 Search PubMed.
- Y.-Y. Tang, L. Yang, S.-Y. Gao, Y. Guo and P.-L. Zhang, J. Org. Chem., 2024, 89, 18472–18476 Search PubMed.
- Y. Wei, B. Ben-zvi and T. Diao, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 Search PubMed.
- G. A. Dawson, E. H. Spielvogel and T. Diao, Acc. Chem. Res., 2023, 56, 3640–3653 Search PubMed.
- Y. Chen, P. Lu and Y. Wang, Org. Lett., 2019, 21, 2130–2133 Search PubMed.
- C. P. Johnston, R. T. Schmith, S. Allmendiger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 Search PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 Search PubMed.
- W.-Y. Shi, J.-J. Ma, H.-Y. Li, D. Chen, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2024, 89, 11136–11147 Search PubMed.
- J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Nat. Commun., 2018, 9, 2445 Search PubMed.
- G. Zeng, Y. Li, B. Qiao, X. Zhao and Z. Jiang, Chem. Commun., 2019, 55, 11362–11365 Search PubMed.
- Z. Zhou, X. Nie, K. Harms, R. Riedel, L. Zhang and E. Meggers, Sci. China Chem., 2019, 69, 1512 Search PubMed.
- J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522–6526 Search PubMed.
- H. A. Sakai and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6185–6192 Search PubMed.
- A. V. Tsymbal, L. Delarme Bizzini and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 21278–21286 Search PubMed.
- Y. Sakakibara, K. Itami and K. Murakami, J. Am. Chem. Soc., 2024, 146, 1554–1562 Search PubMed.
- W. Liu, M. N. Lavagnino, C. A. Gould, J. Alcázar and D. W. C. MacMillan, Science, 2021, 374, 1258–1263 Search PubMed.
- Y. Wei, Q. Wang and M. J. Koh, Angew. Chem., Int. Ed., 2023, 62, e202214247 Search PubMed.
- M. Huang, H. Sun, F. Seufert, A. Friedich, T. M. Marder and J. Hu, Angew. Chem., In. Ed., 2024, 63, e202401782 Search PubMed.
- X. Zhao, C. Wang, L. Yin and W. Liu, J. Am. Chem. Soc., 2024, 146, 29297–29304 Search PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536–2539 Search PubMed.
- L.-J. Li, J.-C. Zhang, W.-P. Li, D. Zhang, K. Duanmu, Q. Ping and Z.-P. Yang, J. Am. Chem. Soc., 2024, 146, 9404–9412 Search PubMed.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 Search PubMed.
- H. Song, R. Cheng, Q.-Q. Min and X. Zhang, Org. Lett., 2020, 22, 7747–7751 Search PubMed.
- Y. Duan, M. Zhang, R. Ruzi, Z. Wu and C. Zhu, Org. Chem. Front., 2017, 4, 525–528 Search PubMed.
- K. C. Cartwright and J. A. Tunge, Chem. Sci., 2020, 11, 8167–8170 Search PubMed.
- C. Song, H.-H. Zhang and S. Yu, ACS Catal., 2022, 12, 1426–1432 Search PubMed.
- J. Zheng, C. Nopper, R. Bibi, A. Nikbakht, F. Bauer and B. Breit, ACS Catal., 2022, 12, 5949–5960 Search PubMed.
- P. Chandu, D. Das, K. G. Gosh and D. Sureshkumar, Adv. Synth. Catal., 2022, 364, 2340–2345 Search PubMed.
- A.-H. Hu, J.-L. Tu, K. Wang, J. Yin, L. Guo, C. Chang and W. Xia, Org. Lett., 2024, 26, 8572–8576 Search PubMed.
- X. Zhang, Y. Zhang, X. Li, B. Li, S. Xiao, Y. Tang, P. Xie and T.-P. Loh, Org. Lett., 2023, 25, 6863–6868 Search PubMed.
- Y. Zang, Y. Lu, C. Ju, Z. Zang, D. Wang, S. Wang, P. Xie and T.-P. Loh, Org. Lett., 2024, 26, 8121–8127 Search PubMed.
- D. Golagani, K. K. Prakash, P. S. Thapa, M. B. Soi’Naik and S. M. Akondi, Org. Lett., 2024, 26, 8583–8588 Search PubMed.
- Y. Yin, F. Chen, D. Chen, P. Xie, D. Wang and T.-P. Loh, Org. Lett., 2025, 27, 269–274 Search PubMed.
- D.-R. Zhang, L.-P. Hu, F.-L. Liu, X.-H. Huang, X. Li, B. Liu and G.-L. Huang, Green. Chem., 2022, 24, 6840–6844 Search PubMed.
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 Search PubMed.
- A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 Search PubMed.
- J. Li, Q. Lefebvre, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2017, 53, 10299–10302 Search PubMed.
- S.-J. Burlingham, D. Guijarro, I. Bosque, R. Chinchilla and J. C. Gonzalez-Gomez, Org. Biomol. Chem., 2022, 20, 7923–7928 Search PubMed.
- L.-J. Li, Y. Wei, Y.-L. Zhao, Y. Gao and X.-Q. Hu, Org. Lett., 2024, 26, 1110–1115 Search PubMed.
- Y.-L. Zhang, L. Yang, J. Wu, C. Zhu and P. Wang, Org. Lett., 2020, 22, 7768–7772 Search PubMed.
- C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 Search PubMed.
- H. Cao, H. Jiang, H. Feng, J.-M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360–16366 Search PubMed.
- G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888–891 Search PubMed.
- M. Koy, F. Sandfort, A. Tlahuext-Aca, L. Quach, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2018, 24, 4552–4555 Search PubMed.
- Z.-H. Xia, C.-L. Zhang, Z.-H. Gao and S. Ye, Org. Lett., 2018, 20, 3496–3499 Search PubMed.
- Y.-T. Wang, M.-C. Fu, B. Zhao, R. Shang and Y. Fu, Chem. Commun., 2020, 56, 2495–2498 Search PubMed.
- J.-X. Wang, Y.-T. Wang, H. Zhang and M.-C. Yu, Org. Chem. Front., 2021, 8, 4466–4472 Search PubMed.
- A. J. Borah and G. Yang, Org. Biomol. Chem., 2015, 13, 8094–8114 Search PubMed.
- J.-J. Zhang, J.-C. Yang, L.-N. Guo and X.-H. Duan, Chem. – Eur. J., 2017, 23, 10259–10263 Search PubMed.
- X.-Y. Lu, A. Gao, M.-Y. Ge, Z.-J. Xia, Q.-L. Liu, T.-H. Tao and X.-M. Sun, J. Org. Chem., 2022, 87, 4654–4669 Search PubMed.
- X.-Y. Lu, R. Huang, Z.-Z. Wang, X. Zhang, F. Jiang, G.-X. Yang, F.-Y. Shui, M.-X. Su, Y.-X. Sun and H.-L. Sun, J. Org. Chem., 2024, 89, 6494–6505 Search PubMed.
- X.-Y. Lu, M.-T. Gao, L.-J. Yu, H.-Y. Pan, X. Zhang, R. Huang, K. Yang, F.-Y. Shui, Y.-W. Song and G.-X. Yang, Org. Chem. Front., 2023, 10, 1788–1795 Search PubMed.
- F. de Vaillant and J. Waser, Chimia, 2017, 71, 226–230 Search PubMed.
- C. Chen, X. Wang and T. Yang, Front. Chem., 2020, 8, 551159 Search PubMed.
- J. Schwarz and B. König, ChemPhotoChem, 2017, 1, 237–242 Search PubMed.
- Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 Search PubMed.
- F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., In. Ed., 2015, 54, 11200–11204 Search PubMed.
- C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 Search PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Grin’ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 Search PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8185 Search PubMed.
- K. Lu, Y. Ma, S. Liu, S. Guo and Y. Zhang, Chin. J. Chem., 2022, 40, 681–686 Search PubMed.
- L. Qin, X. Zhang, H. Sun, X. Duan, J. Liu, M. Wu, X. Yuan, J. Qiu and K. Guo, Green Synth. Catal., 2024, 5, 20–24 Search PubMed.
- Y. Zhu, H. Gao, J.-L. Tu, C. Yang, L. Guo, Y. Zhao and W. Xia, Org. Chem. Front., 2024, 11, 1729–1735 Search PubMed.
- X.-L. Lyu, S.-S. Huang, H.-J. Song, Y. Liu and Q.-M. Wang, RSC Adv., 2019, 9, 36213–36216 Search PubMed.
- M. Zhu and S. Messaoudi, ACS Catal., 2021, 11, 6334–6342 Search PubMed.
- H. Kang, S. An and S. Lee, Org. Chem. Front., 2023, 10, 5151–5157 Search PubMed.
- J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 Search PubMed.
- J. Li, H. Tian, M. Jiang, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2016, 52, 7292–7294 Search PubMed.
- C. Gao, J. Li, J. Yu, H. Yang and H. Fu, Chem. Commun., 2016, 52, 8862–8864 Search PubMed.
- M. Jiang, Y. Jin, H. Yang and H. Fu, Sci. Rep., 2016, 6, 26161 Search PubMed.
- G. Lin, Y. Guo, M. Zhang, Q. Xuan, J. Xu and Q. Song, Org. Lett., 2024, 26, 9097–9102 Search PubMed.
- H. Zhang, P. Zhang, M. Jiang, H. Yang and H. Fu, Org. Lett., 2017, 19, 1016–1019 Search PubMed.
- Y. Zhang and D. Zhang, Org. Biomol. Chem., 2020, 18, 4479–4482 Search PubMed.
- Y. Mao, W. Zhao, S. Lu, L. Yu, Y. Wang, Y. Liang, S. Ni and Y. Pan, Chem. Sci., 2020, 11, 4939–4947 Search PubMed.
- H.-D. Xia, Z.-L. Li, Q.-S. Gu, X.-Y. Dong, J.-H. Fang, X.-Y. Du, L. L. Wang and X.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 16926–16931 Search PubMed.
- H. Zhang, J. Xi, R. Ruzi, N. Li, Z. Wu, W. Li and C. Zhu, J. Org. Chem., 2017, 82, 9305–9311 Search PubMed.
- J. Brauer, E. Quraishi, L. M. Kammer and T. Opatz, Chem. – Eur. J., 2021, 27, 18168–18174 Search PubMed.
- Y. Wei, J. Lam and T. Diao, Chem. Sci., 2021, 12, 11414–11419 Search PubMed.
- M. Escolano, M. J. Cabrera-Alonso, M. Ribagorda, S. O. Bader and G. A. Molander, J. Org. Chem., 2022, 87, 4981–4990 Search PubMed.
- X. Xi, Y. Luo, W. Li, M. Xu, H. Zhao, Y. Chen, S. Zheng, X. Qi and W. Yuan, Angew. Chem., Int. Ed., 2022, 61, e202114731 Search PubMed.
- J.-H. Liu, Z.-Y. Tian, Z.-Y. Wu, T.-L. Huang, Z. Lin, L. Zhang, J. Chen, L. Hai, L. Guo and Y. Wu, J. Org. Chem., 2024, 89, 17059–17068 Search PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7930 Search PubMed.
- W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Chem. – Eur. J., 2015, 21, 13191–13195 Search PubMed.
- D.-L. Zhu, Q. Wu, D. J. Young, H. Wang, Z.-G. Ren and H.-X. Li, Org. Lett., 2020, 22, 6832–6837 Search PubMed.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 13862–13865 Search PubMed.
- W.-D. Li, Y. Q. Jiang, Y.-L. Li and J.-B. Xia, JCS Chem., 2022, 4, 1326–1336 Search PubMed.
- H. Zhang, Q. Xiao, X.-K. Qi, X.-W. Gao, Q.-X. Tong and J.-J. Zhong, Chem. Commun., 2020, 56, 12530–12533 Search PubMed.
- H. Zhao, N. Ni, X. Li, D. Cheng and X. Xu, Polyhedron, 2021, 206, 115337 Search PubMed.
- X. Zhang, F. Hou, Y. Zhang, B. Li, P. Xie and T.-P. Loh, Org. Lett., 2024, 26, 10696–10701 Search PubMed.
- C. Nájera, L. K. Sydnes and M. Yus, Chem. Rev., 2019, 119, 11110–11244 Search PubMed.
- H. Huang, G. Zhang and Y. Chen, Angew. Chem., In. Ed., 2015, 54, 7872–7876 Search PubMed.
- H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374–8377 Search PubMed.
- I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2022, 59, 7593–7607 Search PubMed.
- B. T. Matsuo, P. H. R. Oliveira, E. F. Pissinati, K. B. Vega, I. S. de Jesus, J. T. M. Correia and M. Paixao, Chem. Commun., 2022, 58, 8322–8339 Search PubMed.
- C. Ma, Y. Tian, J. Wang, X. He, Y. Jiang and B. Yu, Org. Lett., 2022, 24, 8265–8270 Search PubMed.
- D. Duan and L. Song, Org. Chem. Front., 2024, 11, 47–52 Search PubMed.
- V. Hutskalova, F. B. Hamdan and C. Sparr, Org. Lett., 2024, 26, 2768–2772 Search PubMed.
- M. Badufle, F. Robert and Y. Landais, RSC Adv., 2024, 14, 12528–12532 Search PubMed.
- F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790–1800 Search PubMed.
- X.-L. Lai, M. Chen, Y. Wang, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 20201–20206 Search PubMed.
- K. Yang, Y. Wang, S. Luo and N. Fu, Chem. – Eur. J., 2023, 29, e20223962 Search PubMed.
- Y. Yuan, J. Yang and Z. Zhang, Chem. Sci., 2023, 14, 705–710 Search PubMed.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 Search PubMed.
- Y. Liang, G. Sun, Z. Su and W. Guan, Dalton Trans., 2020, 49, 15276–15284 Search PubMed.
- W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489–7494 Search PubMed.
- F.-D. Lu, L.-Q. Lu, G.-F. He, J.-C. Bai and W.-J. Xiao, J. Am. Chem. Soc., 2021, 143, 4168–4173 Search PubMed.
- Y. Chen, J. Wang and Y. Lu, Chem. Sci., 2021, 12, 11316–11321 Search PubMed.
- Q. Liu, J. Zheng, X. Zhang and S. Ma, Nat. Commun., 2022, 13, 3302 Search PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 Search PubMed.
- H. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 Search PubMed.
- F. Minisci, R. Bernardi, R. Galli and M. Perchinumm, Tetrahedron, 1971, 27, 3575–3579 Search PubMed.
- R. A. Garza-Sanchez, A. Tlahvext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 Search PubMed.
- J. Genovino, Y. Lian, Y. Zhang, T. O. Hope, A. Juneau, Y. Gagné, G. Ingle and M. Frenette, Org. Lett., 2018, 20, 3229–3232 Search PubMed.
- J. Wang, G.-X. Li, G. He and G. Chen, Asian J. Org. Chem., 2018, 7, 1307–1310 Search PubMed.
- X.-Y. Zhang, W.-Z. Weng, H. Liang, H. Yang and B. Zhang, Org. Lett., 2018, 20, 4686–4690 Search PubMed.
- Z. Li, X. Wang, S. Xia and J. Jin, Org. Lett., 2019, 21, 6930–6936 Search PubMed.
- L. Tan, H. Kang, M. Liu, H. Su, J.-T. Han and C.-J. Li, Precis. Chem., 2023, 1, 437–442 Search PubMed.
- J. Xu, C. Liang, J. Shen, Q. Chen, W. Li and P. Zhang, Green Chem., 2023, 25, 1975–1981 Search PubMed.
- W.-F. Tian, C.-H. Hu, K.-H. He, X.-Y. He and Y. Li, Org. Lett., 2019, 21, 5930–5935 Search PubMed.
- Y. Li, C. Dai, S. Xie, P. Liu and P. Sun, Org. Lett., Org. Lett., 2021, 23, 5906–5910 Search PubMed.
- J. Lin, Z. Li, S. Huang, W. Su and Y. Li, Nat. Commun., 2017, 8, 14353 Search PubMed.
- B. Yang, D. Yu, X.-H. Xu and F.-L. Qing, ACS Catal., 2018, 8, 2839–2843 Search PubMed.
- S. Fernández-García, V. O. Chantzakou and F. Juliá-Hernández, Angew. Chem., Int. Ed., 2024, 63, e202311984 Search PubMed.
- G. A. Lutovsky, S. N. Gockel, M. W. Bundesmann, S. W. Bagley and T. P. Yoon, Chem., 2023, 9, 1610–1621 Search PubMed.
- M. Shao, H. Liang, Y.-L. Liu, W. Qin and Z. Li, Asian J. Org. Chem., 2020, 9, 782–787 Search PubMed.
- S. Sing, N. Dagar and S. R. Roy, Chem. Commun., 2022, 58, 3831–3834 Search PubMed.
- J. Koeller, P. Gandeepan and L. Ackermann, Synthesis, 2020, 1719–1727 Search PubMed.
- A. K. Bagdi and A. Hajra, Org. Biomol. Chem., 2020, 18, 2611–2631 Search PubMed.
- J.-J. Li, Z.-Q. Zhu, L.-J. Xiao, D. Guo, X. Zhu, J. Tang, J. Wu, Z.-B. Xie and Z.-G. Le, Org. Chem. Front., 2019, 6, 3693–3697 Search PubMed.
- T. Shi, K. Sun, X.-L. Chen, Z.-X. Zhang, X.-Q. Huang, Y.-Y. Peng, L.-B. Qu and B. Yu, Adv. Synth. Catal., 2020, 362, 2143–2148 Search PubMed.
- C. Kim, J. Jeong, M. Vellakkaran and S. Hong, ACS Catal., 2022, 12, 13225–13233 Search PubMed.
- W.-M. Cheng, R. Sang, M.-C. Fu and Y. Fu, Chem. – Eur. J., 2017, 23, 2537–2541 Search PubMed.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 Search PubMed.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 19178–19185 Search PubMed.
- K. Ermanis, A. C. Colgan, R. S. J. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 21091–21101 Search PubMed.
- L. M. Kammer, A. Rahman and T. Opatz, Molecules, 2018, 23, 764 Search PubMed.
- A. Feng, Y. Yang, Y. Liu, C. Geng, R. Zhu and D. Zhang, J. Org. Chem., 2020, 85, 7207–7217 Search PubMed.
- X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Org. Lett., 2018, 20, 6298–6301 Search PubMed.
- M. P. Luo, Y.-J. Gu and S.-G. Wang, Chem. Sci., 2020, 14, 251–256 Search PubMed.
- D. Liang, J.-R. Chen, L.-P. Tan, Z.-W. He and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 6040–6049 Search PubMed.
- J. A. Carmona, C. Rodriguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 Search PubMed.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 Search PubMed.
- W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 Search PubMed.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. G. M. Dhar, J. Org. Chem., 2018, 83, 3000–3013 Search PubMed.
- X.-L. Lyu, S.-S. Huang, H.-J. Song, Y.-X. Liu and Q.-M. Wang, Org. Lett., 2019, 21, 5728–5732 Search PubMed.
- H.-W. Hu, C. Zhang, Y.-M. Yang, H.-Q. Deng and Z.-Y. Tang, Tetrahedron Lett., 2022, 103, 153966 Search PubMed.
- Z. Yan, B. Sun, X. Zhang, X. Zhuang, J. Yang, W. Su and C. Jin, Chem. – Asian J., 2019, 14, 3344–3348 Search PubMed.
- H. Zhang, J. Xu, M. Zhou, J. Zhao, P. Zhang and W. Li, Org. Biomol. Chem., 2019, 17, 10201–10208 Search PubMed.
- A. Bisoyi, A. R. Tripathy, G. S. Yedase, S. Sim P, U. Choudhury and V. R. Yatham, J. Org. Chem., 2023, 88, 2631–2641 Search PubMed.
- N. Papaioannou, M. J. Fray, A. Rennhack, T. J. Anderson and J. E. Stokes, J. Org. Chem., 2020, 85, 12067–12079 Search PubMed.
- B. Sun, D. Li, X. Zhuang, R. Zhu, A. Aisha and C. Jiu, Synlett, 2020, 677–682 Search PubMed.
- C. Ma, Z. Feng, J. Li, D. Zhang, W. Li, Y. Jiang and B. Yu, Org. Chem. Front., 2012, 8, 3286–3291 Search PubMed.
- G. Xu, J. Lv, Q. Ding, C. Ma, Y. Jiang and B. Yu, J. Org. Chem., 2024, 89, 2777–2781 Search PubMed.
- B. Sun, T. Xu, L. Zhang, R. Zhu, J. Yang, H. Xu and C. Jin, Synlett, 2020, 363–368 Search PubMed.
- P.-T. Qin, J. Sun, F. Wang, J.-Y. Wang, H. Wang and M.-D. Zhou, Adv. Synth. Catal., 2020, 362, 4707–4712 Search PubMed.
- S. P. Panda, S. K. Hota, R. Dash, L. Roy and S. Murarka, Org. Lett., 2023, 25, 3739–3744 Search PubMed.
- C. Jin, Z. Yan, B. Sun and J. Yang, Org. Lett., 2019, 21, 2064–2068 Search PubMed.
- L. Liu, N. Pan, W. Sheng, L. Su, L. Liu, J. Dong, Y. Zhou and S.-F. Yin, Adv. Synth. Catal., 2019, 361, 4126–4132 Search PubMed.
- M. Moczulski, A. Artelska, L. Albrecht and A. Albrecht, Eur. J. Org. Chem., 2022, e202200630 Search PubMed.
- S. P. Pitre, M. Muuronen, D. A. Fishman and L. E. Overman, ACS Catal., 2019, 9, 3413–3418 Search PubMed.
- X. Li, Q. Zhang, W. Zhang, Y. Wang, Y. Wang and Y. Pan, J. Org. Chem., 2019, 84, 14360–14368 Search PubMed.
- W. Wang, Z. Zhou, Y. Wang, Y. Wang, Y. Liang, Z. Zhu, Y. Zheng and Y. Pan, Angew. Chem., Int. Ed., 2019, 58, 624–627 Search PubMed.
- L. Ren and H. Cong, Org. Lett., 2018, 20, 3225–3228 Search PubMed.
- C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 15841–15846 Search PubMed.
- Y.-N. Ding, N. Li, Y.-C. Huang, Y. An and Y.-M. Liang, Org. Lett., 2022, 24, 4519–4523 Search PubMed.
- X. Zhang, R. T. Smith, C. Le, S. J. McCarves, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220 Search PubMed.
- L. Guillemard, F. Colobert and J. Wencel-Delord, Adv. Synth. Catal., 2018, 360, 4184–4189 Search PubMed.
- S. Hanna and K. R. Prabhu, J. Org. Chem., 2019, 84, 5067–5077 Search PubMed.
- W. Jia, Y. Jian, B. Huang, C. Yang and W. Xia, Synlett, 2018, 18881–18886 Search PubMed.
- P. Bao, F. Liu, Y. Lv, H. Yue, J.-S. Li and W. Wei, Org. Chem. Front., 2020, 7, 492–498 Search PubMed.
- C. Ma, M. Heng, J. Li, X. Yang, Y. Jiang and B. Yu, Chin. J. Chem., 2022, 40, 2655–2662 Search PubMed.
- M. Prince, P. Kumar and B. K. Singh, ACS Omega, 2024, 9, 651–657 Search PubMed.
- M. Niu, C. Yang, M. Leng, Q. Cao, M. Li and Z. Shen, J. Org. Chem., 2024, 89, 6159–6168 Search PubMed.
- X. Ji, Z. Yang, X. Wu, G.-J. Deng and H. Huang, J. Org. Chem., 2022, 87, 4168–4182 Search PubMed.
- A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466–469 Search PubMed.
- M. Jouffroy and J. Kong, Chem. – Eur. J., 2019, 25, 2217–2220 Search PubMed.
- L. Candish, M. Freitag, T. Gensch and F. Glorius, Chem. Sci., 2017, 8, 3618–3622 Search PubMed.
- J. Xu, H. Zhang, J. Zhao, Z. Ni, P. Zhang, B.-F. Shi and W. Li, Org. Chem. Front., 2020, 7, 4031–4042 Search PubMed.
- L.-Y. Xie, S. Peng, L.-H. Yang, C. Peng, Y.-W. Lin, X. Yu, Z. Cao, Y.-Y. Peng and W.-M. He, Green Chem., 2021, 23, 374–378 Search PubMed.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 Search PubMed.
- L. McMurray, T. M. McGuire and R. L. Howells, Synthesis, 2020, 1719–1737 Search PubMed.
- S. Das, Org. Biomol. Chem., 2025, 23, 1016–1066 Search PubMed.
- D. Petzold, M. Giedyk, A. Chatterjee and B. König, Eur. J. Org. Chem., 2019, 1193–1244 Search PubMed.
- Z. Zeng, A. Feceu, N. Sivendran and L. J. Gooβen, Adv. Synth. Catal., 2021, 363, 2678–2722 Search PubMed.
- S. Barata-Vallejo, D. E. Yerien and A. Postigo, ACS Sustainable Chem. Eng., 2021, 9, 10016–10047 Search PubMed.
- S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 Search PubMed.
- X. Wu, C. Meng, X. Yuan, X. Jia, W. Qian and J. Ye, Chem. Commun., 2015, 51, 11864–11867 Search PubMed.
- G. Tarantino and C. Hammond, ACS Catal., 2018, 8, 10321–10330 Search PubMed.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493–9500 Search PubMed.
- P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 Search PubMed.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, Chem. Rxiv., preprint, chemrxiv:14451117.v1 DOI:10.26434/chemrxiv.14451117.v1.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296–8305 Search PubMed.
- Y. Zhang, J. Qian, M. Wang, Y. Huang and P. Hu, Org. Lett., 2022, 24, 5972–5976 Search PubMed.
- J. Qian, Y. Zhang, W. Zhao and P. Hu, Chem. Commun., 2024, 60, 2764–2767 Search PubMed.
- X. Xu, P. Huang, Y. Jiang, C. Lv, P. Li, J. Wang, B. Sun and C. Jin, Green. Chem., 2023, 25, 8741–8747 Search PubMed.
- A. Jati, K. Mahato, D. Chanda, P. Kumar, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2024, 146, 23923–23932 Search PubMed.
- C. A. Blakemore, J. M. Humphrey, E. Yang, J. T. Kohrt, P. D. Morse, R. M. Howard, H. G. Yayla, T. Knauber, L. Xie, T. Makowski, J. W. Raggon, R. B. Watson, C. W. am Ende, T. Ryder, O. White, M. R. M. Koos, R. Kumar, F. Shi, J. Li, H. Wang, L. Chen and J. Wang, Org. Process Res. Dev., 2024, 28, 3801–3807 Search PubMed.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Tetrahedron, 2022, 106–107, 132629 Search PubMed.
- G. Levitre, A. Granados and G. A. Molander, Green Chem., 2023, 25, 560–565 Search PubMed.
- K.-Q. Chen, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2020, 22, 8059–8064 Search PubMed.
- M.-C. Fu, J.-X. Wang and R. Sang, Org. Lett., 2020, 22, 8572–8577 Search PubMed.
- J. Wu, C. Shu, Z. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2023, 62, e202309684 Search PubMed.
- H. T. Song, W. Ding, Q.-Q. Zhou, J. Liu, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2016, 81, 7250–7255 Search PubMed.
- Y. Sakakibara, P. Cooper, K. Murakami and K. Itami, Chem. – Asian J., 2018, 13, 2410–2413 Search PubMed.
- T. M. Faraggi, W. Li and D. W. C. MacMillan, Isr. J. Chem., 2020, 60, 410–415 Search PubMed.
- S. N. Khan, M. K. Zaman, R. Li and Z. Sun, J. Org. Chem., 2020, 85, 5019–5026 Search PubMed.
- S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 Search PubMed.
- S. He, X. Chen, F. Zeng, P. Lu, Y. Peng, L. Qu and B. Yu, Chin. Chem. Lett., 2020, 31, 1863–1867 Search PubMed.
- R. Guan, E. L. Bennett, Z. Huang and J. Xiao, Green Chem., 2022, 24, 2946–2952 Search PubMed.
- I. P. Beletskaya, C. Nájera and M. Yus, Chem. Soc. Rev., 2020, 49, 7101–7166 Search PubMed.
- R. Guan, G. Chen, E. L. Bennett, Z. Huang and J. Xiao, Org. Lett., 2023, 25, 2482–2486 Search PubMed.
- J.-L. Tu, H. Gao, M. Luo, L. Zhao, C. Yang, L. Guo and W. Xia, Green Chem., 2022, 24, 5553–5558 Search PubMed.
- M. Innocent, G. Labande, F. Cam, T. Aubineau and A. Guérinot, Eur. J. Org. Chem., 2023, e202300892 Search PubMed.
- L. M. Denkler, M. A. Shekar, T. S. Ngan, L. Wylie, D. Abdullin, M. Engeser, G. Schnakenburg, T. Hett, F. H. Pilz, B. Kirschner, O. Schiemann, P. Kielb and A. Bunescu, Angew. Chem., Int. Ed., 2024, 63, e202403292 Search PubMed.
- M. Innocent, C. Tanguy, S. Gavelle, T. Aubineau and A. Guérinot, Chem. – Eur. J., 2024, 30, e202401252 Search PubMed.
- Q. Chen, Y. Wang and G. Guo, Chem. Eng. J., 2023, 461, 141767 Search PubMed.
- E. Le Du, M. Garreau and J. Waser, Chem. Sci., 2021, 12, 2467–2473 Search PubMed.
- P. Li, J. R. Zbieg and J. A. Terret, ACS Catal., 2021, 11, 10997–11004 Search PubMed.
- R. Mao, J. Balon and X. Hu, Angew. Chem., In. Ed., 2018, 57, 13624–13628 Search PubMed.
- S. Shibutani, T. Kodo, M. Takeda, K. Nagao, N. Tokunaga, Y. Sasaki and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 1211–1216 Search PubMed.
- R.-H. Li, Y.-L. Zhao, Q.-K. Shang, Y. Geng, X.-L. Wang, Z.-M. Su, G.-F. Li and W. Guan, ACS Catal., 2021, 11, 6633–6642 Search PubMed.
- S. Senaweera, K. C. Cartwright and J. A. Tunge, J. Org. Chem., 2019, 84, 12553–12561 Search PubMed.
- B. Maeda, Y. Sakakibara, K. Murakami and K. Itami, Org. Lett., 2021, 23, 5113–5117 Search PubMed.
- Y. Sakakibara, K. Itami and K. Murakami, J. Synth. Org. Chem. Jpn., 2023, 81, 1050–1061 Search PubMed.
- W. Su, P. Xu and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 24012–24017 Search PubMed.
- R. D. Mandal, D. Das, A. Sarkar, M. Saha, A. R. Das, A. Mahato and A. Pramanik, J. Org. Chem., 2024, 89, 18069–18080 Search PubMed.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337–9340 Search PubMed.
- I. M. Ogbu, D. M. Bassani, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 8802–8805 Search PubMed.
- G. Kurtai, J. Lusseau, F. Robert and Y. Landais, Synlett, 2023, 342–346 Search PubMed.
- D. L. Lipilin, M. O. Zubkov, M. D. Kosobokov and A. D. Dilman, Chem. Sci., 2024, 15, 644–650 Search PubMed.
- A. Porey, S. O. Fremin, S. Nand, R. Trevino, W. B. Hughes, S. K. Dhakal, V. D. Nguyen, S. G. Greco, H. D. Arman and O. V. Larionov, ACS Catal., 2024, 14, 6973–6980 Search PubMed.
- T. Cao, T. Xu, X. Shu and S. Liao, Nat. Commun., 2020, 11, 5340 Search PubMed.
- L. Candish, L. Pitzer, A. Gómez-Suárez and F. Glorius, Chem. – Eur. J., 2016, 22, 4753–4756 Search PubMed.
- L. Wei, C. Wu, C.-H. Tung, W. Wang and Z. Xu, Org. Chem. Front., 2023, 6, 3224–3227 Search PubMed.
- Z. Wu and D. A. Pratt, Angew. Chem., Int. Ed., 2021, 60, 15596–15605 Search PubMed.
- A.-M. Hu, J.-L. Tu, M. Luo, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2023, 10, 4764–4773 Search PubMed.
- M. O. Zubkov, M. D. Kosobokov, V. V. Levin and A. D. Dilman, Org. Lett., 2022, 24, 2354–2358 Search PubMed.
- Z. He and P. Dydio, Angew. Chem., Int. Ed., 2024, 63, e202410616 Search PubMed.
- Y. Jin, H. Yang and H. Fu, Chem. Commun., 2020, 52, 12909–12912 Search PubMed.
- Z. Xiao, L. Wang, J. Wei, C. Ran, S. H. Liang, J. Shang, G.-Y. Chen and C. Zheng, Chem. Commun., 2020, 56, 4164–4167 Search PubMed.
- Y. Dong, P. Ji, Y. Zhang, C. Wang, X. Meng and W. Wang, Org. Lett., 2020, 22, 9562–9567 Search PubMed.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 Search PubMed.
- Z. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 Search PubMed.
- C. G. Na, D. Ravelli and E. J. Alexanian, J. Am. Chem. Soc., 2020, 142, 44–49 Search PubMed.
- T. Xu, T. Cao, M. Yang, R. Xu, X. Nie and S. Liao, Org. Lett., 2020, 22, 3692–3696 Search PubMed.
- L. Geniller, C. Souche, M. Taillefer, F. Jaroschik and A. Prieto, Org. Lett., 2024, 26, 9574–9579 Search PubMed.
- M. Zhang, L. Liu, Y. Tan, Y. Jing, Y. Liu, Z. Wang and Q. Wang, Angew. Chem., Int. Ed., 2024, 63, e202318344 Search PubMed.
- V. D. Nguyen, G. C. Haug, S. G. Greco, R. Trevino, G. B. Karki, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2022, 61, e202210525 Search PubMed.
- S.-H. He, G.-L. Chen, X.-Y. Gong, G.-Z. Ao and F. Liu, J. Org. Chem., 2023, 88, 6671–6681 Search PubMed.
- J. A. Andrews, J. Kalepu, C. F. Palmer, D. L. Poole, K. E. Christensen and M. C. Willis, J. Am. Chem. Soc., 2023, 145, 21623–21629 Search PubMed.
- J. A. Andrews, R. G. Woodger, C. F. Palmer, D. L. Poole and M. C. Willis, Angew. Chem., Int. Ed., 2024, 63, e202407970 Search PubMed.
- H. T. Dang, A. Porey, S. Nand, R. Trevino, P. Manning-Lorino, W. B. Hughes, S. O. Fremin, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2023, 14, 13384–13391 Search PubMed.
- J. He, G. Chen, B. Zhang, Y. Li, J.-R. Chen, W.-J. Xiao, F. Liu and C. Liu, Chem., 2020, 6, 1149–1159 Search PubMed.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Wuong, C. B. Karki, H. D. Arman and O. V. Larionov, Chem. Sci., 2022, 13, 4170–4179 Search PubMed.
- V. T. Nguyen, R. Trevino, S. G. Greco, H. D. Arman and O. V. Larionov, ACS Catal., 2022, 12, 8729–8739 Search PubMed.
- Y. Dong, N. Xiong, Z. Rong and R. Zeng, Org. Lett., 2024, 26, 2381–2386 Search PubMed.
- S. Cai, Y. Yu, D. Chen, L. Li, Q. Chen, M. Huang and W. Weng, Org. Lett., 2016, 18, 2990–2993 Search PubMed.
- Q.-Q. Ge, J.-S. Qian and J. Xuan, J. Org. Chem., 2019, 84, 8691–8701 Search PubMed.
- K. Ishiu, M. Kumar and K. N. Sing, J. Org. Chem., 2024, 89, 10919–10928 Search PubMed.
- J.-T. Yi, X. Zhou, Q.-L. Chen, Z.-D. Chen, G. Lu and J. Weng, Chem. Commun., 2022, 58, 9409–9412 Search PubMed.
- Z.-D. Chen, X. Zhou, J.-T. Yi, H.-J. Diao, Q.-L. Chen, G. Lu and J. Weng, Org. Lett., 2022, 24, 2474–2478 Search PubMed.
- P. S. Pedersen, D. J. Blakemore, G. M. Chinigo, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 21189–21196 Search PubMed.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, H. D. Arman and O. V. Larionov, Chem. Sci., 2021, 12, 6429–6436 Search PubMed.
- Z. Zhuang, Y. Sun, Y. Zhong, Q. He, X. Zhang and C. Yang, Org. Lett., 2024, 26, 713–718 Search PubMed.
- Z. Zong, J. Yang, L. Yuan, X. Wang, J.-Q. Chen and J. Wu, Org. Lett., 2024, 26, 8625–8631 Search PubMed.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 Search PubMed.
- V. T. Nguyen, G. C. Haug, N. T. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2020, 59, 7921–7927 Search PubMed.
- P. Li, J. R. Zbieg and J. A. Terrett, Org. Lett., 2021, 23, 9563–9568 Search PubMed.
- R. Mao, A. Frey, J. Balon and X. Hu, Nat. Can., 2018, 1, 120–126 Search PubMed.
- R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 9501–9504 Search PubMed.
- G. Barzano, R. Mao, M. Carreau, J. Waser and X. Hu, Org. Lett., 2020, 22, 5412–5416 Search PubMed.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Chem. – Eur. J., 2017, 23, 5972–5977 Search PubMed.
- R. Kobayashi, S. Shibutani, K. Nagao, Z. Ikeda, J. Wang, I. Ibáñez, M. Reynolds, Y. Sasaki and H. Ohmiya, Org. Lett., 2021, 23, 5415–5419 Search PubMed.
- R. Wang, H. Hu, A. Banerjee, Z. Cui, Y. Ma, W. G. Wittingham, P. Yang and A. Li, Org. Lett., 2024, 26, 2691–2696 Search PubMed.
- Y.-C. Yan, H. Zhang, K. Hu, S.-M. Zhou, Q. Chen, R.-Y. Qu and G.-F. Yang, Bioorg. Med. Chem., 2022, 72, 116968 Search PubMed.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 Search PubMed.
- P. Xu, W. Su and T. Ritter, Chem. Sci., 2022, 13, 13611–13616 Search PubMed.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 Search PubMed.
- W.-T. Xu, B. Huang, J.-J. Dai, J. Xu and H.-J. Xu, Org. Lett., 2016, 18, 3114–3117 Search PubMed.
- S. Mondal, S. Das, S. Mondal, S. P. Midya and P. Ghosh, J. Org. Chem., 2024, 89, 16750–16758 Search PubMed.
- D. C. Marcote, R. Street-Jeakings, E. Dauncey, J. J. Douglas, A. Ruffoni and D. Leonori, Org. Biomol. Chem., 2019, 17, 1839–1842 Search PubMed.
- S.-C. Kao, K.-J. Bian, X.-W. Chen, Y. Chen, A. A. Martí and J. G. West, Chem. Catal., 2023, 3, 100603 Search PubMed.
- S. Wang, T. Li, C. Gu, J. Han, C.-G. Zhao, C. Zhu, H. Tan and J. Xie, Nat. Commun., 2022, 13, 2432 Search PubMed.
- M. Ding, S. Zhou, S. Yao, C. Zhu, W. Li and J. Xie, Chin. J. Chem., 2024, 42, 351–355 Search PubMed.
- S. Yang, Y. Wang, W. Xu, X. Tian, M. Nao and X. Yu, Org. Lett., 2023, 25, 8834–8838 Search PubMed.
- X. Shu, Y. Wang, W. Xu, X. Tian, M. Bao and X. Yu, Org. Lett., 2023, 25, 8834–8838 Search PubMed.
- S. B. Lang, K. C. Cartwright, R. S. Welter, T. M. Locascio and J. A. Tunge, Eur. J. Org. Chem., 2016, 3331–3334 Search PubMed.
- M.-J. Zhang, G. M. Schroeder., Y.-H. He and Z. Guan, RSC Adv., 2016, 6, 96693–96699 Search PubMed.
- G. Feng, X. Wang and J. Jin, Eur. J. Org. Chem., 2019, 6728–6732 Search PubMed.
- V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 Search PubMed.
- R. R. Merchant, S. B. Lang, T. Yu, S. Zhao, Z. Qi, T. Suzuki and J. Bao, Org. Lett., 2020, 22, 4180–4184 Search PubMed.
- J. Yang, M. Song, H. Zhou, Y. Qi, B. Ma and X.-C. Wang, Green Chem., 2021, 23, 5806–5811 Search PubMed.
- S. Jin, G. C. Haug, V. T. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9764–9774 Search PubMed.
- D. Reich, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202207063 Search PubMed.
- S. K. Pagire, C. Shu, D. Reich, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2023, 145, 18649–18657 Search PubMed.
- J. Jin, X. Lin, L. Chai, C.-Y. Wang, L. Zhu and C. Li, Chem., 2023, 9, 1945–1954 Search PubMed.
- Y. Cheng, J. Zheng, L. Chai, J. Wang, J. Yin, L. Zhu and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202316764 Search PubMed.
- F. Chen, M. Bai, Y. Zhang, W. Liu, X. Huangfu, Y. Liu, G. Tang and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202210334 Search PubMed.
- X.-Y. Lu, H.-Y. Pan, R. Huang, K. Yang, X. Zhang, Z.-Z. Wang, Q.-Q. Tao, G.-X. Yang, X.-J. Wang and H.-P. Zhou, Org. Lett., 2023, 25, 2476–2481 Search PubMed.
- A. Faweett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 Search PubMed.
- D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 Search PubMed.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 Search PubMed.
- Q. Zhang, X. Li, W. Zhang, S. Ni, Y. Wang and Y. Pan, Angew. Chem., Int. Ed., 2020, 59, 21875–21879 Search PubMed.
- A. Serafino, H. Pierre, F. Le Vaillant, J. Boutet, G. Guillamot, L. Neuville and G. Masson, Org. Lett., 2023, 25, 9249–9254 Search PubMed.
- B. Nagy, Z. Gonda, T. Földesi, P. P. Fehér, A. Stirling, G. L. Tolnai and Z. Novák, Org. Lett., 2024, 26, 2292–2296 Search PubMed.
- N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6163–6172 Search PubMed.
- Q. Wei, Y. Lee, W. Liang, X. Chen, B.-S. Mu, X.-Y. Cui, W. Wu, S. Bai and Z. Liu, Nat. Commun., 2022, 13, 7112 Search PubMed.
- N.-X. Xu, B.-X. Li, C. Wang and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 10639–10644 Search PubMed.
- Y. Zhang, G. Nie, Z. Jin and S. Ren, Synlett, 2024, 347–351 Search PubMed.
- Y. Yoshimi, T. Itou and M. Hatanaka, Chem. Commun., 2007, 5244–5246 Search PubMed.
- I. Itou, Y. Yoshimi, K. Nishikawa, T. Morita, Y. Okada, N. Ichinose and N. Hatanaka, Chem. Commun., 2010, 46, 6177–6179 Search PubMed.
- C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228–4231 Search PubMed.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 Search PubMed.
- N. Li, Y. Ning, X. Wu, J. Xie, W. Li and C. Zhu, Chem. Sci., 2021, 12, 5505–5510 Search PubMed.
- Y.-L. Sun, F.-F. Tan, R.-C. Hu, C.-H. Hu and Y. Li, Chin. J. Chem., 2022, 40, 1903–1908 Search PubMed.
- J. G. L. de Araujo, N. S. B. da Silva, J. C. C. V. Bento, A. M. de Azevêdo, A. M. M. Araújo, A. S. D. dos Anjos, C. A. Martínez-Huitte, E. V. dos Santos, A. D. Gondim and L. A. Cavalcanti, Chem. – Eur. J., 2023, 29, e202302330 Search PubMed.
- K. Okamoto, T. Ueno, Y. Hato, Y. Kawaguchi, T. Hakogi, S. Majima, T. Ohara, M. Hagihara, N. Tanimoto and T. Tsuritani, J. Org. Chem., 2024, 89, 9927–9948 Search PubMed.
- T. S. Mayer, T. Taeufer, S. Brandt, J. Rabeah and J. Pospech, J. Org. Chem., 2023, 88, 6347–6353 Search PubMed.
- Y.-C. Lu and J. G. West, Angew. Chem., Int. Ed., 2023, 62, e202213055 Search PubMed.
- C.-H. Hu and Y. Li, J. Org. Chem., 2023, 88, 6401–6406 Search PubMed.
- Z. Bazyar and M. Hosseini-Sarvari, J. Org. Chem., 2019, 84, 13503–13515 Search PubMed.
- Z. Huang, Z. Zhao, C. Zhang, J. Lu, H. Liu, N. Luo, J. Zhang and F. Wang, Nat. Catal., 2020, 3, 170–178 Search PubMed.
- X. Du, Y. Peng, J. Albero, D. Li, C. Hu and H. García, ChemSusChem, 2022, 15, e202102107 Search PubMed.
- J. Shi, T. Yuan, M. Zheng and X. Wang, ACS Catal., 2021, 11, 3040–3047 Search PubMed.
- C. Hao, J. Wen, H. Song, B. Huang, G. Gao and S. An, Appl. Catal., B, 2024, 354, 124122 Search PubMed.
- M. M. E. Huijbers, W. Zhang, F. Tonin and F. Hollmann, Angew. Chem., Int. Ed., 2018, 57, 13648–13651 Search PubMed.
- W. Zhang, M. Ma, M. M. E. Huijbers, G. A. Filonenko, E. A. Pidko, M. van Schie, S. de Boer, B. O. Burek, J. Z. Bloh, W. J. H. van Berkel, W. A. Smith and F. Hollmann, J. Am. Chem. Soc., 2019, 141, 3116–3120 Search PubMed.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 Search PubMed.
- J. Xu, J. Fan, Y. Lou, W. Xu, Z. Wang, D. Li, H. Zhou, X. Lin and Q. Wu, Nat. Commun., 2021, 12, 3983 Search PubMed.
- Z. Qin, Y. Zhou, Z. Li, M. Höhner, U. T. Bornschener and S. Wu, Angew. Chem., Int. Ed., 2024, 63, e202314566 Search PubMed.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10519 Search PubMed.
- M. Constantini and A. Mendoza, ACS Catal., 2021, 11, 13312–13319 Search PubMed.
- M. Montesinos-Magraner, M. Constantini, R. Ramirez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930–5935 Search PubMed.
- Z. Yu and A. Mendoza, ACS Catal., 2019, 9, 7870–7875 Search PubMed.
- J. D. Bacha and J. K. Kochi, Tetrahedron, 1968, 24, 2215–2226 Search PubMed.
- X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 Search PubMed.
- K. C. Cartwright and J. A. Tunge, ACS Catal., 2018, 8, 11801–11806 Search PubMed.
- K. C. Cartwright, S. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84, 2933–2940 Search PubMed.
- K. C. Cartwright, E. Joseph, C. G. Comadoll and J. A. Tunge, Chem. – Eur. J., 2020, 26, 12454–12470 Search PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. Flores-Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9485–9496 Search PubMed.
- Z. Hong, W. K. Chong, A. Y. R. Ng, M. Li, R. Ganguly, T. C. Sum and H. S. Soo, Angew. Chem., In. Ed., 2019, 58, 3456–3460 Search PubMed.
- A. G. Korspusik, A. Adili, K. Bhatt, J. E. Anatot, D. Seidel and B. S. Sumerlin, J. Am. Chem. Soc., 2023, 145, 10480–10485 Search PubMed.
- A. Tlahuext-Aca, L. Candish, R. A. Garza-Sanchez and F. Glorius, ACS Catal., 2018, 8, 1715–1719 Search PubMed.
- W.-M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 Search PubMed.
- K.-Q. Chen, J. Shen, Z.-X. Wang and X.-Y. Chen, Chem. Sci., 2021, 12, 6684–6690 Search PubMed.
- R. Li, Y. Dong, S. N. Khan, M. K. Zaman, Z. Zhou, P. Miao, L. Hu and Z. Sun, Nat. Commun., 2022, 13, 7061 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |