
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-enhanced Raman spectroscopy: a half-century historical perspective†
Jun
Yi‡
 a,
En-Ming
You‡
b,
Ren
Hu‡
a,
De-Yin
Wu
a,
Guo-Kun
Liu
a,
Zhi-Lin
Yang
a,
Hua
Zhang
a,
Yu
Gu
a,
Yao-Hui
Wang
a,
Xiang
Wang
a,
Hao
Ma
a,
Yang
Yang
a,
Jun-Yang
Liu
a,
Feng Ru
Fan
a,
Chao
Zhan
a,
Jing-Hua
Tian
a,
Yu
Qiao
a,
Hailong
Wang
a,
Si-Heng
Luo
a,
Zhao-Dong
Meng
a,
Bing-Wei
Mao
a,
Jian-Feng
Li
a,
Bin
Ren
a,
Javier
Aizpurua
c,
Vartkess Ara
Apkarian
d,
Philip N.
Bartlett
e,
Jeremy
Baumberg
f,
Steven E. J.
Bell
g,
Alexandre G.
Brolo
hi,
Louis E.
Brus
j,
Jaebum
Choo
k,
Li
Cui
l,
Volker
Deckert
mn,
Katrin F.
Domke
o,
Zhen-Chao
Dong
pq,
Sai
Duan
r,
Karen
Faulds
s,
Renee
Frontiera
t,
Naomi
Halas
u,
Christy
Haynes
t,
Tamitake
Itoh
v,
Janina
Kneipp
w,
Katrin
Kneipp
w,
Eric C.
Le Ru
x,
Zhi-Peng
Li
y,
Xing Yi
Ling
zaa,
Jacek
Lipkowski
ab,
Luis M.
Liz-Marzán
acadae,
Jwa-Min
Nam
af,
Shuming
Nie
ag,
Peter
Nordlander
u,
Yukihiro
Ozaki
ah,
Rajapandiyan
Panneerselvam
ai,
Jürgen
Popp
mn,
Andrea E.
Russell
e,
Sebastian
Schlücker
aj,
Yang
Tian
ak,
Lianming
Tong
al,
Hongxing
Xu
amanao,
Yikai
Xu
ap,
Liangbao
Yang
aq,
Jianlin
Yao
ar,
Jin
Zhang
ar,
Yang
Zhang
pq,
Yao
Zhang
pq,
Bing
Zhao
as,
Renato
Zenobi
at,
George C.
Schatz
au,
Duncan
Graham
s and
Zhong-Qun
Tian
*a
a,
En-Ming
You‡
b,
Ren
Hu‡
a,
De-Yin
Wu
a,
Guo-Kun
Liu
a,
Zhi-Lin
Yang
a,
Hua
Zhang
a,
Yu
Gu
a,
Yao-Hui
Wang
a,
Xiang
Wang
a,
Hao
Ma
a,
Yang
Yang
a,
Jun-Yang
Liu
a,
Feng Ru
Fan
a,
Chao
Zhan
a,
Jing-Hua
Tian
a,
Yu
Qiao
a,
Hailong
Wang
a,
Si-Heng
Luo
a,
Zhao-Dong
Meng
a,
Bing-Wei
Mao
a,
Jian-Feng
Li
a,
Bin
Ren
a,
Javier
Aizpurua
c,
Vartkess Ara
Apkarian
d,
Philip N.
Bartlett
e,
Jeremy
Baumberg
f,
Steven E. J.
Bell
g,
Alexandre G.
Brolo
hi,
Louis E.
Brus
j,
Jaebum
Choo
k,
Li
Cui
l,
Volker
Deckert
mn,
Katrin F.
Domke
o,
Zhen-Chao
Dong
pq,
Sai
Duan
r,
Karen
Faulds
s,
Renee
Frontiera
t,
Naomi
Halas
u,
Christy
Haynes
t,
Tamitake
Itoh
v,
Janina
Kneipp
w,
Katrin
Kneipp
w,
Eric C.
Le Ru
x,
Zhi-Peng
Li
y,
Xing Yi
Ling
zaa,
Jacek
Lipkowski
ab,
Luis M.
Liz-Marzán
acadae,
Jwa-Min
Nam
af,
Shuming
Nie
ag,
Peter
Nordlander
u,
Yukihiro
Ozaki
ah,
Rajapandiyan
Panneerselvam
ai,
Jürgen
Popp
mn,
Andrea E.
Russell
e,
Sebastian
Schlücker
aj,
Yang
Tian
ak,
Lianming
Tong
al,
Hongxing
Xu
amanao,
Yikai
Xu
ap,
Liangbao
Yang
aq,
Jianlin
Yao
ar,
Jin
Zhang
ar,
Yang
Zhang
pq,
Yao
Zhang
pq,
Bing
Zhao
as,
Renato
Zenobi
at,
George C.
Schatz
au,
Duncan
Graham
s and
Zhong-Qun
Tian
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, School of Electronic Science and Engineering, College of Environment and Ecology, State Key Laboratory of Marine Environmental Science, Department of Physics, iChEM, IKKEM, Xiamen University, Xiamen 361005, China. E-mail: zqtian@xmu.edu.cn
bSchool of Ocean Information Engineering, Fujian Provincial Key Laboratory of Oceanic Information Perception and Intelligent Processing, Jimei University, Xiamen 361021, China
cDonostia International Physics Center, DIPC, and Ikerbasque, Basque Agency for Research, and University of the Basque Country (UPV/EHU), San Sebastian, Spain
dDepartment of Chemistry, University of California Irvine, Irvine, California 92697, USA
eSchool of Chemistry and Chemical Engineering, University of Southampton, Highfield, Southampton SO17 1BJ, UK
fNanoPhotonics Centre, Cavendish Laboratory, Department of Physics, University of Cambridge, JJ Thompson Avenue, Cambridge, UK
gSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, BT9 5AG Belfast, UK
hDepartment of Chemistry, University of Victoria, Victoria, BC V8N 4Y3, Canada
iCentre for Advanced Materials and Related Technologies (CAMTEC), University of Victoria, Victoria, BC V8P 5C2, Canada
jDepartment of Chemistry, Columbia University, New York, 10027, USA
kDepartment of Chemistry, Chung-Ang University, Seoul 06974, South Korea
lXiamen Key Laboratory of Indoor Air and Health, Key Lab of Urban Environment and Health, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China
mLeibniz Institute of Photonic Technology, Albert-Einstein-Str. 9, 07745 Jena, Germany
nInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
oFaculty of Chemistry, University of Duisburg-Essen, Universitätsstr. 5, 45141 Essen, Germany
pHefei National Research Center for Physical Sciences at the Microscale and Synergetic Innovation Center of Quantum Information and Quantum Physics, School of Physics and Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, China
qHefei National Laboratory, University of Science and Technology of China, Hefei 230088, China
rCollaborative Innovation Center of Chemistry for Energy Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, MOE Key Laboratory of Computational Physical Sciences, Department of Chemistry, Fudan University, Shanghai 200433, China
sCentre for Nanometrology, Department of Pure and Applied Chemistry, Technology and Innovation Centre, University of Strathclyde, Glasgow G1 1RD, UK
tDepartment of Chemistry, University of Minnesota, 207 Pleasant St SE, Minneapolis, Minnesota 55455, USA
uDepartment of Chemistry, Department of Electrical and Computer Engineering, Department of Physics & Astronomy, Department of Materials Science and Nanoengineering, Laboratory for Nanophotonics Rice University, Houston, Texas 77005, USA
vHealth and Medical Research Institute (HRI), National Institute of Advanced Industrial Science and Technology (AIST), Takamatsu, Kagawa 761-0395, Japan
wDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
xThe MacDiarmid Institute for Advanced Materials and Nanotechnology, School of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington 6140, New Zealand
yBeijing Key Laboratory for Nano-Photonics and Nano-Structure (NPNS), Department of Physics, Capital Normal University, Beijing 100048, China
zSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, Singapore, 637371, Singapore
aaSchool of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China
abElectrochemical Technology Center, Department of Chemistry, University of Guelph, Guelph, Ontario N1G 2W1, Canada
acCIC biomaGUNE, Basque Research and Technology Alliance (BRTA), 20014 Donostia-San Sebastián, Spain
adIkerbasque, Basque Foundation for Science, 48009 Bilbao, Spain
aeCinbio, University of Vigo, 36310 Vigo, Spain
afDepartment of Chemistry, Seoul National University, Seoul 08826, South Korea
agDepartment of Bioengineering, Department of Electrical and Computer Engineering, Department of Materials Science and Engineering and Department of Chemistry, University of Illinois at Urbana – Champaign, Champaign, Illinois 61801, USA
ahSchool of Biological and Environmental Sciences, Kwansei Gakuin University, 1 Gakuen-Uegahara, Sanda, Hyogo 669-1330, Japan
aiDepartment of Chemistry, SRM University AP, Amaravati, Andhra Pradesh 522502, India
ajPhysical Chemistry I, Department of Chemistry, and Center of Nanointegration Duisburg-Essen (CENIDE) & Center of Medical Biotechnology (ZMB), University of Duisburg-Essen (UDE), 45141 Essen, Germany
akShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Dongchuan Road 500, Shanghai 200241, P. R. China
alCenter for Nanochemistry, Beijing Science and Engineering Center for Nanocarbons, Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, 100871 Beijing, China
amSchool of Physics and Technology and Key Laboratory of Artificial Micro- and Nano-structures of Ministry of Education and School of Microelectronics, Wuhan University, Wuhan 430072, China
anWuhan Institute of Quantum Technology, Wuhan 430206, China
aoHenan Academy of Sciences, Zhengzhou 450046, China
apKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China
aqAnhui Province Key Laboratory of Medical Physics and Technology, Institute of Health and Medical Technology, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, China
arCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, China
asState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
atDepartment of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland
auDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208-3113, USA
First published on 23rd December 2024
Abstract
Surface-enhanced Raman spectroscopy (SERS) has evolved significantly over fifty years into a powerful analytical technique. This review aims to achieve five main goals. (1) Providing a comprehensive history of SERS's discovery, its experimental and theoretical foundations, its connections to advances in nanoscience and plasmonics, and highlighting collective contributions of key pioneers. (2) Classifying four pivotal phases from the view of innovative methodologies in the fifty-year progression: initial development (mid-1970s to mid-1980s), downturn (mid-1980s to mid-1990s), nano-driven transformation (mid-1990s to mid-2010s), and recent boom (mid-2010s onwards). (3) Illuminating the entire journey and framework of SERS and its family members such as tip-enhanced Raman spectroscopy (TERS) and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) and highlighting the trajectory. (4) Emphasizing the importance of innovative methods to overcome developmental bottlenecks, thereby expanding the material, morphology, and molecule generalities to leverage SERS as a versatile technique for broad applications. (5) Extracting the invaluable spirit of groundbreaking discovery and perseverant innovations from the pioneers and trailblazers. These key inspirations include proactively embracing and leveraging emerging scientific technologies, fostering interdisciplinary cooperation to transform the impossible into reality, and persistently searching to break bottlenecks even during low-tide periods, as luck is what happens when preparation meets opportunity.
 Jun Yi | Jun Yi is an associate professor of Electronic Science and Engineering at Xiamen University. He obtained his BSc in Physics from University of Science and Technology of China in 2012 and completed his PhD in Physical Chemistry from Xiamen University in 2019 under the supervision of Prof. Zhong-Qun Tian. He conducted postdoctoral research at the University of California, Berkeley, with Prof. Xiang Zhang from 2019 to 2021, and joined Xiamen University as a faculty member in 2021. His research focuses on SERS, plasmonics and nano-spectroscopy. |
 En-Ming You | En-Ming You is an associate professor at the School of Ocean Information Engineering at Jimei University. He earned his BSc in Physics from Nanjing University and obtained his PhD in Physical Chemistry at Xiamen University in 2020, under the supervision of Prof. Zhong-Qun Tian. Following this, he conducted postdoctoral research in Prof. Tian's group. He joined Jimei University in 2023, and his research interests include plasmon-enhanced Raman spectroscopy, plasmon-enhanced infrared spectroscopy, and infrared nanospectroscopy and imaging. |
 Ren Hu | Ren Hu is a senior engineer of State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University. She obtained her PhD in Physical Chemistry at Xiamen University in 2005. She conducted postdoctoral research in LIA CNRS XiamENS (with Professor Christian Amatore's group at École Normale Supérieure, Paris and Professor Zhong-Qun Tian in Xiamen University). She joined Xiamen University as a senior engineer in 2011. Her current research interest focuses on micro-/nano-electrochemistry and spectroelectrochemistry. |
 De-Yin Wu | De-Yin Wu received his BSc at Shaanxi's Normal University in 1992 and his PhD in Physical Chemistry at Sichuan University in 1998. Then he worked as a postdoc fellow with Professor Zhong-Qun Tian on theories of SERS. Now he is a full professor of Department of Chemistry, College of Chemistry and Chemical Engineering (2006-), Xiamen University. His research area focuses on theories of surface-enhanced Raman spectroscopy, spectroelectrochemistry and plasmon-mediated chemical reactions. He has published about 250 papers. |
 Guo-Kun Liu | Guo-Kun Liu is a professor at the College of Environment and Ecology at Xiamen University. He earned his BSc in Chemical Education from Soochow University and obtained his PhD in Physical Chemistry at Xiamen University in 2006, under the supervision of Prof. Zhong-Qun Tian. He conducted postdoctoral research with Prof. Ru-Qin Yu at Hunan University, then Prof. Peng Chen at Cornell University and finally Dr Thomas Moffat at NIST USA. He joined Xiamen University in 2015, and his research interest focuses on various applications of SERS. |
 Zhong-Qun Tian | Zhong-Qun Tian received his BSc from Xiamen University in 1982 and completed a PhD in Electrochemical SERS under Prof. Martin Fleischmann (FRS) at the University of Southampton in 1987. He became a full professor at Xiamen University in 1991, and Member of the Chinese Academy of Sciences and Fellow of the Royal Society of Chemistry in 2005. He served as President of the International Society of Electrochemistry (2019–2020), associate editor of Chem. Soc. Rev. (2012–2024), and is on the advisory boards of 16 international journals. He received several scientific awards, including the Raman lifetime award. His research focuses on SERS and SHINERS, spectroelectrochemistry, nanochemistry, plasmonics, and catalytic assembly. |
1. Introduction
Half a century ago, in 1974, M. Fleischmann, P. J. Hendra, and A. J. McQuillan of the University of Southampton reported unexpected and high-quality potential-dependent surface Raman spectra from pyridine adsorbed on a silver (Ag) electrode roughened by an electrochemical method.1 Stunned by the results, R. P. Van Duyne and D. L. Jeanmaire of Northwestern University meticulously reproduced the experiments, and determined that the effective Raman scattering cross-section from adsorbed pyridine was enhanced by 105–106 times. Their paper was published at the end of 1977 after a multi-year and multiple journal review process, as the so-called surface enhancement effect was initially met with strong skepticism.2 In the same year, J. A. Creighton and M. G. Albrecht of the University of Kent independently reported a similar result.3 Following these discoveries, several mechanisms were proposed to explain the abnormally intense Raman scattering on the surface. The widely accepted surface plasmon mechanism was put forward by M. Moskovits of the University of Toronto in 1978.4 Subsequently, the effect was officially named surface-enhanced Raman scattering (SERS) in 1979 by Van Duyne.5The process of making a groundbreaking discovery by these key pioneers that open a new scientific field was intricate and challenging. It often involved pushing the boundaries of existing beliefs and theories, requiring individuals to think outside the box and challenge conventional understanding. Four critical elements in the process including experiments, new concepts, theoretical explanations, and naming of the observed effect are vital for significant scientific breakthroughs such as SERS, as illustrated in Fig. 1.
 | ||
| Fig. 1 Four key elements in the process of discovering the surface-enhanced Raman scattering effect. | ||
1. A new phenomenon is observed in experiments and documented faithfully: by conducting experiments and carefully observing unexpected phenomena, scientists can uncover new information that challenges existing knowledge or reveals previously unknown patterns or behaviors. The experimental observation was the starting point of the discovery journey to SERS.
2. Novel concepts are introduced: once unexpected phenomena are observed, scientists must develop new concepts or ideas to explain these observations. This often involves thinking creatively and outside the box to propose novel theories that can account for observed data.
3. Theoretical models/explanations are developed to support these concepts: after introducing new concepts, scientists must then develop theoretical explanations to support these ideas. This involves creating models, equations, or frameworks that can explain the underlying mechanisms behind the observed phenomena and predict future outcomes.
4. An appropriate name is provided for the observed effect: naming the observed effects is an important step in solidifying scientific discovery and communicating it to the wider scientific community. The name should accurately reflect the nature of the effect and make it easier for other researchers to reference and build upon the discovery.
In the scientific community, every individual who contributes to these key steps plays a vital role. Without the collective efforts of these pioneers, the emergence of new research areas and the expansion of scientific knowledge would not have been possible.
Similar to that of SERS, the discovery of the Raman effect about a century ago was not accomplished in one stroke. Smekal predicted the inelastic scattering of light after interactions with molecules utilizing classical quantum theory in 1923.6 However, the experimentalists at that time did not take note of the theory, probably because the concepts of quantum mechanics were not completely understood. In 1928, Raman and Krishnan observed the scattered secondary radiation with degraded frequency in 60 liquids and vapors and later named the inelastic scattering the Raman effect.7,8 In the same year, Landsberg and Mandelstam accidentally observed a similar effect in quartz and Iceland spar while studying the change of wavelength in scattering based on the Debye's theory of specific heat in solids.9,10 Generally, the publications by Raman7 and Landsberg9 are considered landmarks in the discovery of the Raman effect.11 The discovery not only promoted the development of quantum theory, but also fostered an important method for qualitative and quantitative chemical analysis. Two years later, Raman was awarded the Nobel Prize in physics for his work on the scattering of light and the discovery of the Raman effect. However, Raman scattering is a weak effect, with a typical cross-section of about 106 and 1014 times lower than that of the infrared absorption and fluorescence processes, respectively.12 Since the birth of Raman spectroscopy, detection sensitivity has always been a major challenge hindering the smooth development and broad application of Raman spectroscopy.
Breaking developmental bottlenecks in a scientific field often requires the introduction of new science and technology. In the 1960s, the invention and introduction of lasers had greatly improved sensitivity and expanded applications of Raman spectroscopy. However, it was still extremely difficult to apply Raman spectroscopy in the study of surface science until the discovery of SERS (Fig. 2). The emergence of SERS revolutionized surface spectroscopy and analytical science by eliminating the inherent limitation of low detection sensitivity in surface Raman spectroscopy. Nevertheless, the upsurge following the discovery of SERS was regrettably short-lived. From the mid-1970s to the mid-1980s, SERS could only be realized on roughened substrates and colloids composed of “free-electron-like” metals, mainly Ag, Au and Cu. The lack of substrate and surface generalities severely limited the acceptance of SERS by the community of surface science, electrochemistry, corrosion, catalysis and other industries. Many research groups were gradually exiting the field, causing a low-tide from the mid-1980s to the mid-1990s.
 | ||
| Fig. 2 A timeline of historical advances in Raman effect, SERS, and related revolutionizing science and technologies. The invention and introduction of the lasers in the 1960s expanded the applications of Raman spectroscopy. Advancements in nanoscience in the 1990s boosted the second upsurge of the SERS field. EF: enhancement factor; LSP: localized surface plasmon; NP: nanoparticle; SM: single-molecule; TERS: tip-enhanced Raman spectroscopy; SHINERS, shell-isolated nanoparticle-enhanced Raman spectroscopy; SE-FSRS: surface-enhanced femtosecond stimulated Raman spectroscopy. | ||
Similar to the reactivation of Raman spectroscopy by SERS, the rapid advancements in nanoscience since the 1990s provided a boost for SERS, which regained widespread interest in the mid-1990s and thereafter experienced a resurgence until today. In this period, innovative nanomaterial synthesis, nanofabrication and characterization techniques enabled the evolution of SERS substrates from micrometer-scale ill-defined rough electrodes to tens of nanometer-scale well-defined nanoparticles.13 High-quality SERS spectra from a single molecule adsorbed on Ag and Au nanoparticles had been obtained, marking SERS as one of the most promising tools for single-molecule science and trace analysis.14,15
In parallel, the urgent need to develop high-spatial resolution techniques and characterize atomically flat single-crystals in surface science gave rise to two important variants of SERS: tip-enhanced Raman spectroscopy (TERS)16–19 and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS).20 Both methods produced intense Raman signals from undisturbed surfaces of general materials, breaking the bottlenecks of surface and material generalities of SERS.21 Thereafter, the application fields of SERS have been expanded from surface chemistry to material characterization, bioanalysis, drug analysis, food safety, and environmental monitoring, etc.,21–34 which have enormously increased the impact and promise of SERS.
In the past 50 years, SERS has gone through a tortuous path to develop into a powerful diagnostic method. Due to the length of the review, this retrospect describes four important stages of the SERS field from the view of experimental and theoretical methods in the fifty-year progression. First, we recall the history of the birth of SERS and discuss the milestone works in the initial 10 years, i.e., from 1974 to the ∼mid-1980s, during which the main experimental and theoretical foundations of SERS were established. Then we discuss the low-tide period (mid-1980s to mid-1990s) and the second upsurge of SERS research profoundly boosted by nanoscience (nano-driven SERS), which laid the key foundation and triggered an exponential increase in interest in the SERS field for the subsequent 2 decades. We conclude by presenting the notable methods of recent decades, which have significantly pushed the boundaries in terms of detection sensitivity and spatial resolution of SERS, TERS, and SHINERS.
We believe that a deep analysis of the discovery and breakthroughs of SERS highlights the invaluable spirit of groundbreaking innovation and perseverance demonstrated by the pioneers and trailblazers over the past 50 years. The lessons contemporary researchers can draw from this history are rich and profound: they involve not blindly believing in authority but being willing to challenge established knowledge to break developmental bottlenecks; proactively embracing and leveraging emerging scientific technologies to lead to revolutionary advancements; encouraging interdisciplinary cooperation to transform the seemingly impossible into reality; and bravely recognizing that great discoveries and observations are often achieved even with incomplete or incorrect explanations.
There have been over 800 reviews and books on SERS from different perspectives, so it is impossible for us to comprehensively present the whole story of the birth and advancement of SERS in all its details. Instead, we focus on innovative experimental and theoretical methods, as they form the core of this new field and continuously break developmental bottlenecks. We hope this review serves as a treasured reminder of the importance of innovation, perseverance, and collaboration in any scientific research, offering invaluable insights and inspiration for future generations of researchers.
2. The foundation of surface-enhanced Raman spectroscopy (mid-1970s–mid-1980s)
SERS has become one of the most important branches of Raman spectroscopy and vibrational spectroscopy in the past two decades, providing “molecular fingerprint” information with exceptionally high sensitivity.35 It greatly transformed the capabilities and applications of Raman spectroscopy. Thus, we briefly recall the early days of the Raman effect before reviewing the discovery of SERS, drawing inspiration by comparing the discovery history of these great scientific fields.2.1 Theoretical prediction and experimental discovery of the Raman effect
About a century ago, the Austrian physicist A. G. Smekal, whose scientific interests were focused on applying new quantum theories to different areas of physics, theoretically predicted the inelastic scattering of light from gases, liquids, or solids at wavelengths different from those of the usual monochromatic incident radiation. Smekal utilized the classical quantum theory, by which he assumed that light had a quantum structure and showed that scattered monochromatic light would consist of its original wavelength as well as higher and lower wavelengths. The related paper was subsequently published in a German-language journal in 1923.6 The key equations are shown as followed,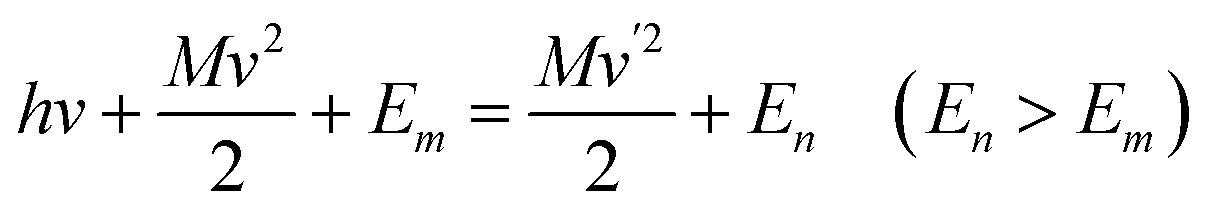 | (1) |
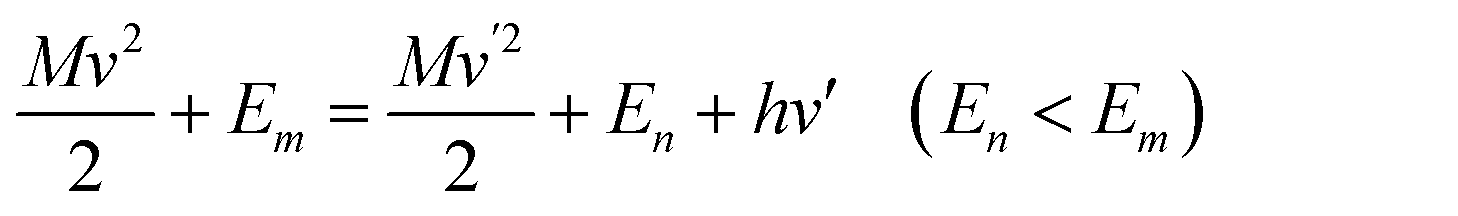 | (2) |
Five years later, the phenomenon was first discovered experimentally by the Indian scientist C. V. Raman. In the paper published in the Indian J. Phys., Raman described the experimental phenomenon of scattered secondary radiation and gave spectroscopic evidences.8 The paper was delivered on March 16, 1928, and published on March 31, 1928. Meanwhile, Raman and Krishnan's article “A New type of secondary radiation”, published in Nature,7 reported the observation of frequency-varying secondary scattering radiation in 60 solutions and vapors. The work was sent out on February 16, 1928, and published on March 31. They pointed out that the scattered light was polarized and of great intensity to rule out the possibility of fluorescence. The effect was then named after Raman as the Raman effect, which was also known as the Smekal-Raman effect in some German-speaking districts.36
Almost at the same time, the former Soviet Union (now Russian) scientists G. S. Landsberg and L. I. Mandelstam, accidentally observed the similar effect in quartz and Iceland spars while studying the Brillouin scattering in solids.9 The work was first published in the French-language journal Comptes Rendus on July 9, 1928,10 and then in the German-language journal Die Naturwissenschaften four days later,9 which were the earliest articles reporting Raman effects in solids. In some reviews,11 these two pieces of work by Raman and Landsberg are considered as the landmark of the discovery of the Raman effect. The discovery of the Raman effect had far-reaching consequences for scientific research in physics and chemistry. Two years after the experimental discovery, C. V. Raman was awarded the 1930 Nobel Prize in Physics “for his work on the scattering of light and for the discovery of the effect named after him.”
Anything that is strong and excellent often also has its weaknesses and less favorable aspects. Raman scattering, classified as a two-photon process, exhibits a scattering cross-section for molecules that is approximately 106 to 1014 times smaller than that associated with infrared and fluorescence processes,35 respectively. As a result, Raman spectroscopy is fundamentally limited by its low detection sensitivity.
In the early experiments, scientists used filtered sunlight or mercury lamps as the light source and the eye or photographic film as the detector, with very long exposure times, often up to several days. Because of the very low intensity of the Raman scattering signals and the lack of monochromatic and high-brightness light sources, the efficiency of the technique remained quite low until the 1960s. The invention of laser in 1960 and its rapid introduction to Raman spectroscopy therefore really revolutionized the field. The first laser-based Raman scattering experiment was realized in 1962.37 Thereafter, Raman spectroscopy became a more versatile technique that utilizes lasers ranging from ultraviolet (UV) to near-infrared (IR) to analyze samples at the molecular level. This flexibility allows for the examination of various materials, including lunar rocks, ocean ore, silicon chips, plastic, proteins, and drugs.38–43
2.2 Why was surface Raman spectroscopic research initiated in the 1970s?
In the 1970s, two of the main challenges for Raman spectroscopy were its potential application in surface science and trace analysis. They involved studying a much smaller number of probe molecules compared to the bulk materials or solutions typically used in Raman measurements. For instance, even if a flat surface was fully covered with sample molecules like pyridine, there would only be between 1011 and 1013 molecules within the 1 mm diameter of the laser spot. This was the typical illuminated area before the widespread use of the confocal microscope in the mid-1990s. Given that usually one Raman photon is produced by approximately 106–109 incident photons for the surface adsorbates of with monolayer coverage,12 it remained difficult to detect it using even the most advanced Raman instruments available at that time. Moreover, increasing the power of the laser cannot be of great help because it will ultimately damage the probed sample. Increasing the sample concentration is a common way to enhance the intensity, which proved effective for studying bulk signals in liquid phases and transparent samples. However, this approach had limitations when it came to surface adsorption, where only one monolayer of molecules is typically present. At this concentration level, the signals for most adsorbates without a resonance Raman effect fell below the detection limit. Consequently, due to the inherent weakness of the scattering mechanism, Raman spectroscopy was rarely used for characterizing surface species, with only a few papers addressing highly powdered oxide catalysts (which have surface areas many orders of magnitude larger than those of flat surface samples) before the mid-1970s.44–46Nevertheless, Raman spectroscopy held great appeal for surface scientists as it had the potential to provide valuable insights into various chemical, physical, and biological surfaces and interfaces at the molecular level.35 It could determine surface bonding, molecular conformation, and adsorption orientation. Additionally, Raman spectroscopy could be employed for in situ investigations of various interfaces, including but not limited to solid–liquid, solid–gas, and solid–solid interfaces. It is particularly useful in scenarios where many other surface techniques are not applicable. These significant advantages strongly motivated some researchers to challenge established knowledge and explore ways to enhance the applicability of Raman spectroscopy in surface science and related analytical fields, ultimately leading to the birth of surface-enhanced Raman spectroscopy (SERS).21
2.3 Why was the birth of SERS rooted in an electrochemical system?
Between the mid-1960s and 1970s, electrochemistry was undergoing a significant transition from a macroscopic to a microscopic level. During this period, spectro-electrochemistry emerged to provide mechanistic and dynamic insights into electrochemical interfaces at a molecular level.47–52 Several leading electrochemists seized the opportunity provided by the rapid development of spectroscopy at that time, introducing several optical spectroscopy techniques to electrochemical systems in sequence, including ellipsometry, UV-vis absorption spectroscopy, and IR spectroscopy.52 The university of Southampton was one of the leading institutes of spectro-electrochemistry. Nevertheless, Raman spectroscopy appeared to be the most promising optical techniques for analyzing the chemical structure of species on the most commonly used metal electrode surfaces,48 given that the widely used aqueous solutions in electrochemistry do not significantly interfere with the surface signal compared to IR spectroscopy.51However, initiating interdisciplinary collaboration between Raman spectroscopy and electrochemistry was challenging. The primary obstacle was designing an electrochemical cell that could effectively measure both Raman scattering signals and electrochemical signals simultaneously and efficiently. A significant challenge was the Raman process's inherent low detection sensitivity. In an electrochemical cell, the deflection of the optical path by the liquid phase further reduced Raman spectroscopy's detection efficiency. These issues made electrochemical Raman experiments impractical and unfeasible. It took the bravery of a small group with diverse expertise in electrochemistry and Raman spectroscopy. Their expert cross-disciplinary cooperation, courageous efforts, and methodological advancements ultimately transformed what was once considered impossible into reality.
Fleischmann, a very creative scientist in electrochemistry in the 1970s, played a key role in establishing electrochemical Raman spectroscopy. According to the formula for Raman intensity in textbooks, it was not possible to detect a surface adsorbate with monolayer coverage even with state-of-the-art Raman instruments. However, he was highly eager to break through this limitation by developing a new method that utilizes Raman spectroscopy to measure the signals of molecules that are adsorbed on electrode surfaces. However, bravery alone is not sufficient to achieve breakthroughs in science; rational design, a detailed scientific plan, and finding the best collaborators are also essential. Encouraged by Graham Hills, the Chair of Physical Chemistry at the University of Southampton, Fleischmann initiated a successful collaboration with Hendra, an expert in Raman spectroscopy who had pioneered the study of adsorption on oxide powder catalysts with much larger surface areas than electrodes, and with McQuillan, a young and energetic postdoc.
McQuillan documented the collaboration between the two scientists in his note,53 “Both were innovative scientists, enjoyed competing with each other in scientific brainstorming sessions, and were excited by the prospect of audacious experiments.” Recalling the history, Hendra wrote that54 “Back in 1972 we discussed and argued over the possibility of recording Raman spectra from an electrode surface. Problem, of course, is that the amount of material present is minimal. So what system to use?” “My ears duly flapped because Calomel (Hg2Cl2) is nearly the strongest Raman scatterer known to man (C60is better!), Martin came up with the idea of generating a high area mercury surface – so we were hopefully ‘in business’.” Fleischmann and McQuillan generated a surface coated with micro globular mercury, which increased the surface area to ten times that of a smooth surface. Then they successfully recorded Raman spectra from multiple layers of Hg2Cl2, Hg2Br2 and HgO on small mercury droplets formed by nucleation onto a platinum disc electrode. This work was reported in early 1973 in a brief paper without figures in Chemical Communications.55 Hg2Cl2 has a very strong Hg–Hg stretch Raman signal, but detection limits for Hg2Cl2 multilayers were still significantly above the more intriguing monolayer level. Nevertheless, this approach was a logical first step in scientific exploration, particularly in venturing into the unknown. The authors successfully demonstrated that Raman signals could be obtained from their designed electrochemical cell.
Encouraged by these results, they continued to advance their methods to achieve detection at the monolayer level. As noted by McQuillan,53 “Thus, the focus of work turned to methods of enhancing the Raman signal. The possibilities of using phase-sensitive detection, resonance Raman, and large-surface-area electrodes were considered. As Pat had pioneered the use of Raman to study species adsorbed on large-area oxide catalysts, so large surface area became the favoured direction.” The collaboration between the electrochemist and the Raman spectroscopist facilitated the combination of Ag electrodes and pyridine molecules. McQuillan wrote down the words about the choice of pyridine,53 “Pyridine had long been used as a probe molecule for infrared spectroscopic studies of acidic sites on metal oxide catalysts and had also found use in corresponding Raman studies that Pat's group had conducted.” Fleischmann and McQuillan prepared various roughed electrodes by an electrochemical oxidation reduction cycle (ORC) method, as written by Hendra,56 “Martin's Group came up with many roughened surfaces – platinum, palladium, iron, lead, you name it – and studied them in electrodes containing good Raman scattering prospects. Among numerous ideas, it was decided to try silver using an electrolyte containing pyridine.”
2.4 First observation of SERS spectra of molecules adsorbed on electrode
On 15 May 1974, the first potential-dependent surface Raman spectra of pyridine adsorbed on an electrochemically roughened Ag electrode were reported by Fleischmann, Hendra and McQuillan (Fig. 3a–c) in Chem. Phys. Lett., 1974, 26(2), 163–166.1 The abstract of the article states that,1 “Raman spectroscopy has been employed for the first time to study the role of adsorption at electrodes. It has been possible to distinguish two types of pyridine adsorption at a silver electrode. The variation in intensity and frequency of some of the bands with potential in the region of the point of zero charge has given further evidence as to the structure of the electrical double layer; it is shown that the interaction of adsorbed pyridine and water must be taken into account.” Ahead of this publication, McQuillan gave a very brief report and presented a single Raman spectrum of pyridine adsorbed at Ag electrode in the Faraday Discussions of the Chemical Society on Intermediates in Electrochemical Reactions at Oxford on 19 September, 1973.57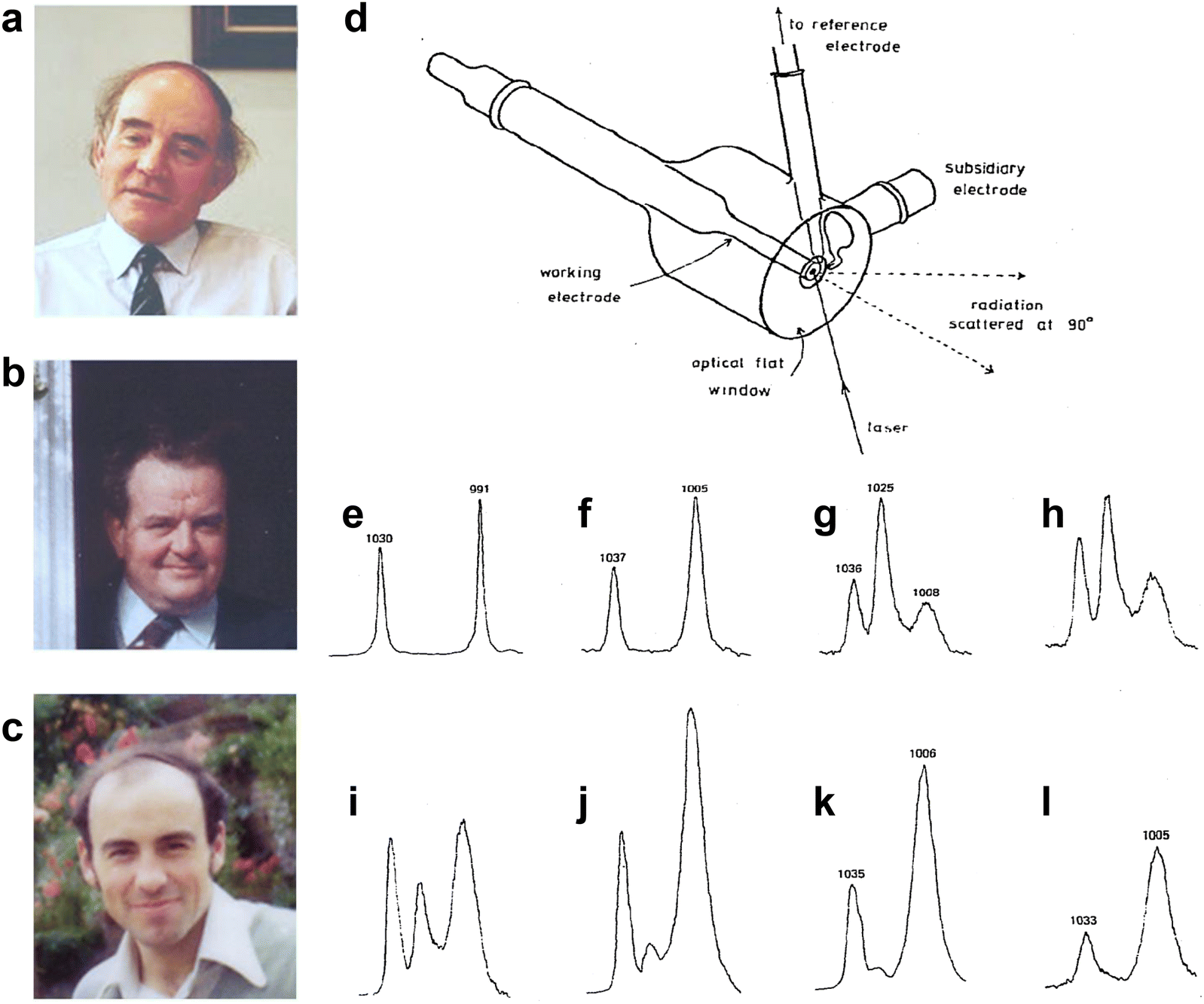 | ||
| Fig. 3 (a)–(c) Photographs of Martin Fleischmann, Patrick Hendra and Jim McQuillan, respectively. (d) Illustration of the electrochemical cell used for the Raman detection of pyridine adsorbed on Ag electrode. And Raman spectra of pyridine in solution and on the Ag electrode at different potentials: (e) liquid pyridine; (f) 0.05 M aqueous pyridine; (g) Ag electrode 0 V vs. saturated calomel electrode (S.C.E.); (h) −0.2 V; (i) −0.4 V; (j) −0.6 V; (k) −0.8 V; (l) −1.0 V. (a)–(c) are reproduced from ref. 56 with permission. Copyright 2016 Royal Society of Chemistry. (d)–(l) are reproduced from ref. 1 with permission. Copyright 1974 Elsevier B.V. | ||
The experiments were conducted in a specially designed electrochemical cell (Fig. 3d), well-matched with the spectrometer.53 The collecting window for both the incident and scattered light was optically flat, ensuring optimal efficiency. The surface consisted of a Ag rod electrode sheathed in Teflon, which could be easily adjusted in position relative to the window. Additionally, the focused argon ion laser beam could be incident at variable angles. The obtained Raman spectra were of very high quality and clearly indicated the presence of a surface species, as evidenced by the distinctive dependence of spectral features, particularly the relative intensity, on the electrode potential, as shown in Fig. 3g–l.
According to McQuillan's note,53 “the first really significant evidence which could well be due to adsorbed pyridine” … “a shoulder about 1008 cm−1under the 1003 cm−1(solution) band.” … “On 4 July I spent a day on the Cary 82 using the 5145 Å laser line looking at Ag deposited onto Pt and included some cyclic voltammetry of system containing pyridine. Once again evidence for adsorbed (?) pyridine as on 22 June … quite a distinct variation in the size of the adsorbed species peak with potential.” … “Short discussion with Martin about results. Quite enthusiastic.”
Fleischmann and McQuillan attended the Faraday Discussions on Intermediates in Electrochemical Reactions at Oxford on 19 September 1973. During the discussion session, McQuillan gave a very brief report with a single spectrum about their observations.57 Then the paper was prepared for publication at the end of 1973, as noted by McQuillan.53 “…, and began putting a paper together for Chemical Physics Letters. This was finally sent to David Buckingham on 21 February 1974 after considerable discussion over unresolved aspects, such as the unusually high observed Raman intensities. In the end no comment was included on the intensities. The response from David Buckingham less than a week later was to say that the paper had been accepted and that ‘you appear to have found an interesting phenomenon’.”
In retrospect, this was indeed the first SERS measurement, and the roughened electrode can be considered the first SERS-active nanostructure (albeit mixed with microstructures), although it was not recognized as such at the time. They initially believed that the electrochemical roughening procedure had significantly increased the electrode's surface area and, consequently, the number of adsorbed molecules. This increase was thought to be responsible for the intense surface Raman signal of pyridine, a molecule with a very large Raman cross-section.
Through persistent and broad searching, logically followed by a focus on the pyridine/Ag system, the high-level collaboration between electrochemists and Raman spectroscopists led to the first Raman measurements of adsorbed molecules on electrode surfaces. This initial experimental observation marked the right entrance door, i.e., the beginning of the discovery journey of SERS.
2.5 First determination of the surface enhanced Raman effect
The deep understanding of the essence of phenomena is crucial for further transforming a preliminary phenomenon observation into a scientific progress and even a new field. The abnormally intense Raman spectra of pyridine adsorbed on a roughened Ag electrode had garnered significant attention from Van Duyne. This was not only due to its capability to measure surface-adsorbed monolayers of molecules, but primarily because their intensities were about a million times stronger than the same species in solution, an astonishing claim first made by Van Duyne when he was a young assistant professor (Fig. 4a). | ||
| Fig. 4 (a) Photograph of Richard P. Van Duyne. Reproduced from ref. 58 with permission. Copyright 2020 Wiley. (b)–(e) Raman spectra of pyridine adsorbed on a Ag electrode. The solution contained 50 mM pyridine and 0.1 M KCI. Electrode potentials are −0.2 V, −0.4 V, −0.6 V and −0.8 V for (b)–(e). The Raman spectra were enhanced by image processing methods as the original spectra were blurry. Reproduced from ref. 2 with permission. Copyright 1977 Elsevier B.V. | ||
He described how he was surprised when he first read the paper by Fleischmann et al.,59 “Imagine my surprise in early May 1974 when I opened the May 4th issue of Chemical Physics Letters and found the report by Fleishmann et al. describing their observation of normal Raman signals from a monolayer of pyridine adsorbed on a roughened silver electrode immersed in a simple aqueous electrolyte solution in an electrochemical cell. I was stunned! They observed count rates of ∼500–1000 counts per second using 100 mW of 514.5 nm argon ion laser light.”
Van Duyne's big surprise stemmed from his own interest in using Raman spectroscopy to study electrochemical systems since 1973.60 He had estimated that the Raman signals of monolayer molecules adsorbed on surfaces could not be measured using the state-of-art instrument of the time.59 “I estimated that for 1 monolayer of a non-resonant adsorbate such as pyridine adsorbed in a vertical orientation on a platinum (111) single crystal and excited with 1 watt of argon ion laser light at 488.0 nm, the total Raman signal for the 991 cm−1fundamental integrated over the vibrational band would be something like 25 counts per second on a (then) state-of-the-art scanning double monochromator with a cooled photomultiplier tube as a detector. I concluded that this experiment was just barely possible and therefore was of limited practicality.” Subsequently, he just focused on improving the Raman sensitivity by several methods, especially by taking advantages of the resonance Raman effect of dye molecules.59
To resolve the confusion in his mind, Van Duyne then visited Fleischmann's laboratory at the University of Southampton after the International Society of Electrochemistry meeting in Brighton, England in September 1974. As written in ref. 59, “I was very keen to understand how it was possible to observe surface Raman signals from pyridine because I had just calculated how unlikely this experiment was to succeed. I learned from Jim that surface roughness was likely to be the key factor.”
Although Fleischmann's article attributed the enhancement of the pyridine Raman signal on the surface of rough Ag electrodes to the increased surface area of the rough electrodes, Van Duyne still raised several questions about the renowned electrochemist's interpretation upon his return to Northwestern:59 “1. Why was the Raman intensity of 500–1000 counts s−1so high? 2. If the surface area was made ∼1000 times greater, should not the double-layer capacitance of the electrode/solution interface have increased in proportion to the area, yielding a huge, easily measurable value of ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 μF cm−2?”
000 μF cm−2?”
He persuaded his graduate student David Jeanmaire to check the questions via scrutinizing the Fleischmann experiments, and they successfully repeated, and largely extended the experiments over the next two years. As surface roughness was speculated to be a key factor, they started with the surface roughness dependence of Raman intensity and found unexpected results. As written in ref. 59, “To test the surface area hypothesis, we repeated the Fleischmann electrochemical roughening procedure and carefully measured pyridine surface Raman intensity vs. the number of coulombs of charge passed. Surprisingly, we found that, starting from roughness conditions used by Fleischmann, the surface Raman signals increased as the surface roughness decreased! Eventually, we found that the ‘optimum’ level of roughness corresponded to the oxidation and re-reduction of approximately 100 monolayers of silver.”
The surface roughening procedure for Ag electrodes was modified by using a slightly treated electrode that underwent a few cycles of electrochemical oxidation–reduction. This electrode displayed a yellowish surface and produced Raman signals that were either equivalent to or even stronger than those obtained from deeply roughened Ag electrodes used in the Fleischmann experiments. The latter electrodes, subjected to over a hundred cycles of treatment, exhibited a very dark color. This observation suggests that the unexpectedly intense Raman signals were likely attributed to specific surface properties of the roughened electrode, rather than an increase in surface area.
Chance favors the prepared mind. Utilizing his previous research experience on electrochemical Raman spectroscopy based on the resonance Raman effect, Van Duyne and Jeanmaire calculated the enhancement factor.59 “Subsequently, David and I devised a procedure to measure the surface enhancement factor which compared the signal intensity per molecule on the surface to the signal intensity per molecule for the same molecule in free solution and discovered the 105–106enhancement factor associated with SERS.” This result led them to propose the surface enhancement effect, as stated in their article published in J. Electroanal. Chem., 1977, 84(1), 1–20:2 “Given that the experimentally observed intensities of NR scattering from adsorbed pyridine in our laboratory are 5–6 orders of magnitude greater than expected, we felt that some property of the electrode surface or the electrode/solution interface is acting to enhance the effective Raman scattering cross section for these adsorbed amines.”
They compiled the results and sent it out for publication in 1976. However, their paper underwent a multi-year, multi-journal review process, as the so-called surface enhancement was initially met with strong skepticism. How could the Raman signal from molecules adsorbed on rough metal surfaces be dramatically and substantially increased compared to their solution state? This notion was indeed too surprising and difficult for the scientific community to accept. Their paper was submitted to J. Electroanal. Chem. (received 7 October 1976) and accepted on 12 May 1977. Subsequently, the paper was published on November 1977.2
Independently, Creighton and Albrecht reported a similar result in J. Am. Chem. Soc., 1977, 99(15), 5215–5217,3 which was submitted 6 months later than Van Duyne's. Both papers presented compelling evidence indicating that the enormously intense surface Raman signal is attributable to a true enhancement in the efficiency of Raman scattering itself.35
The effect was then designated as surface-enhanced Raman scattering (SERS) in 1979 by Van Duyne.5 “Such enormous enhancements of the normal Raman (NR) scattering process completely overcome the traditional limitation of SRS – its low sensitivity. The experimental context in which surface enhanced Raman scattering (SERS) was first recognized was that of an electrochemical cell.”
After five decades, it may be essential to draw inspiration by delving deep into the starting point and asking three correlated questions. Why was the discovery of the SERS effect first made in pyridine that was adsorbed on an electrochemically roughened Ag electrode? Why did Van Duyne and other colleagues continue their investigations in the same Ag–pyridine system? More surprisingly, why does the combination of Ag and pyridine, proposed half a century ago, remain one of the best electrochemical SERS systems with the strongest signal among all non-resonance Raman molecules? Nowadays, every newcomer to this electrochemical SERS may be asked to first repeat this model system (but do not blindly follow, be careful about the 1025 cm−1 peak, see Section 4.8 for detailed discussions) to ensure the correct experimental procedure.
Upon closer examination, it becomes clear that this was not just a coincidence or a stroke of luck, but a result of a very successful and high-quality collaboration, including mutual brainstorming by experts in different fields: an electrochemist well-versed in electrochemical cells and roughening electrode procedures and a Raman spectroscopist knowledgeable about the instrument and the best probe molecules with large Raman cross sections.
The birth of SERS sparked enthusiastic interests from scientists and engineers, and attracted researchers from various fields such as physics, analytical chemistry, electrochemistry, heterogeneous catalysis, materials science, etc. Compared with the exploration on the various materials and feasible systems meeting recurrent obstacles, the advancement of mechanisms was more fruitful in the early days of SERS.
2.6 Foundation of enhancement mechanisms of SERS
From the late 1970s to the mid-1980s, research into theoretical mechanisms saw remarkable growth. Many active groups in physics and physical chemistry were fully captivated by the serendipitous discovery, eager to unravel the mysteries of this new phenomenon. Such was the excitement that some viewed SERS as a divine gift to surface spectroscopy. The field thrived with an abundance of innovative ideas, diverse models, and lively, constructive debates.Meanwhile, as shown in Fig. 5, a number of mechanisms were proposed and developed in the late 1970s and the mid-1980s to account for SERS.4,61–69 It should be noted that, according to the current understanding, surface plasmons can be categorized into two types: propagating surface plasmon polaritons (SPPs) and localized surface plasmons (LSPs). The “surface plasmons” mentioned in earlier works largely refer to what is now recognized as SPPs.
 | ||
| Fig. 5 The timeline of the mechanistic studies on SERS in the first decade. SPP: surface plasmon polariton; ATR: attenuated total reflectance; LSP: localized surface plasmon; EM: electromagnetic; CT: charge transfer. | ||
As we can see, Fleishmann and colleagues observed the first SERS spectrum on a rough Ag electrode, while they misattributed it to an increased surface adsorption area. Van Duyne and Creighton discovered the significant enhancement effect of SERS, nevertheless, their interpretations concerning the origin of this colossal enhancement remained erroneous. In fact, recalling the whole history of modern natural sciences, a common phenomenon is that of “Great discoveries/observations with incorrect/incomplete explanations”, especially in the early stages of establishing disciplines across many fields. This is because great discoveries are often about making the impossible possible and extending the boundaries of the knowledge, and thus, explanations based on past established knowledge are usually wrong or incomplete. In fact, this phenomenon has been recurring throughout the history of SERS, particularly when significant breakthroughs were achieved. Over the subsequent decades, almost every new milestone in SERS research was followed by a period of confusion and debate as the scientific community grapples with understanding the true implications and underlying mechanisms of the discovery.
 | ||
| Fig. 6 (a) Photograph of Martin Moskovits. (b) The dependence of the quantity Fa/v2 on exciting frequency v0 for various values of λR, which is the resonance wavelength. The Raman intensity is proportional to Fa/v2. q is the volume fraction of the metal within a dielectric of dielectric constant ε0. The quantity plotted is actually 6.69 × 107Fa/v2. (c) The solid line is the one of λR = 929 nm in (b). Points are taken from ref. 72 and refer to the 1008 cm−1 ring vibration of pyridine. Reproduced from ref. 4 with permission. Copyright 1978 AIP publishing. | ||
 | ||
| Fig. 7 (a) Photograph of J. Alan Creighton, taken in his laboratory in the early 1980s. (b)–(e) Extinction spectra and Raman spectra of Ag and Au colloids with added pyridine. (b) Extinction spectrum of Ag sol (1.5 cm3) (i) freshly prepared; (ii) 5 min after adding 0.25 cm3 0.01 mol dm−3 pyridine; (iii) 1 min and (iv) 5 min after adding 0.1 cm3 10−1 mol dm−3 pyridine. (c) Raman spectra of Ag sol with added pyridine. (d) Extinction spectrum of Au sol (1.5 cm3) (i) freshly prepared; (ii) 3 h after adding 0.05 cm3 0.1 mol dm−3 pyridine. (e) Raman spectra of Au sol with added pyridine. Figures are reproduced from ref. 70 with permission. Copyright 1979 Royal Society of Chemistry. | ||
Recalling that history, Moskovits wrote that:73 “In the spring of 1978 I wrote a paper in which I jotted down my early ideas regarding the relationship between SERS and the excitation of localized surface plasmons (LSPs) in microrough silver. Focusing on the original reports on SERS from electrochemically roughened silver, I suggested that the submicroscopic silver roughness features (nano-features in today's idiom) were subwavelength and sustained plasmons, and it was the plasmon resonance that was responsible for the enhancement.”
Based on the mechanism, Moskovits also proposed several forecasts including that similar enhancements would be observed in Ag and Cu colloids covered with adsorbates, as described in his Note published in 2012:73 “I also made several predictions: (i) that intense SERS would be observed primarily with nanostructured systems composed of the coinage metals (and the alkalis) because only nanoparticles of those metals would possess the high-Q LSP resonances required for large enhancement; (ii) that subwavelength nanostructure was an essential ingredient for observing SERS; and (iii) that true colloids of those metals should also produce SERS.”
At about the same time, in March 1978, and unaware of Moskovits’ predictions, Creighton et al. for the first time demonstrated SERS from Ag and Au colloids experimentally (Fig. 7a). This pioneering experimental work was submitted to J. Chem. Soc. on 19th July 1978 and published in 1979. (J. Chem. Soc., Faraday Trans., 2, 1979, 75, 790–798).70 Their work on this began with the synthesis of Ag and Au colloids. In his Note published on 2009,74 he recalled that: “Working together one afternoon, having done no previous literature work on metal colloids, we tried adding a solution of sodium borohydride, the only reducing agent that happened to be on our reagent shelves, to a silver nitrate solution. We found that provided that the silver nitrate solution was sufficiently dilute (less than about 10−3mol dm−3) this gave a beautifully clear yellow silver colloid. When we then added pyridine to this yellow colloid (to a concentration of about 10−2mol dm−3, the same concentration as in the pyridine silver electrode experiments) the colour slowly changed through various greenish shades, finally becoming blue-grey before silver was deposited as a black precipitate. A similar procedure using Na[AuCl4] in place of AgNO3gave a wine-red gold colloid that also changed colour gradually when pyridine was added.”
And then they tried Raman measurements in the colloid system and obtained enhanced Raman signals from pyridine, as shown in Fig. 7b–e.74 Creighton wrote that:74 “We straight away put these pyridine-containing colloids into the Raman spectrometer and observed the same 1008 and 1036 cm−1Raman bands of adsorbed pyridine that we had previously seen from the roughened silver electrodes. Although the bands were not as intense as in the spectra from roughened silver electrodes, it was clear that they were nevertheless considerably enhanced, because with the same spectrometer settings we could scarcely see the ca. 1000 cm−1bands from a pyridine solution of the same concentration in the absence of the colloidal metal particles. The Raman bands were also substantially depolarized, a characteristic of SERS bands, whereas for pyridine in solution the strong bands around 1000 cm−1are very strongly polarized.”
The first experimental correlation between the optimized excitation wavelength for SERS and the wavelength of the surface plasmon resonance (SPR) was also reported in the same paper,70 and it was from this correlation that it was independently proposed that plasmon resonances in metal nanoparticles or roughened electrodes are responsible for SERS. “During that most productive weekend, the moment when we realized that the SERS excitation profiles were sharply peaked and that the SERS excitation profile maximum was tracking the absorption maximum as the colloid slowly changed colour was the most exciting research moment I can recall.”
Moskovits and Creighton's pioneering works extended the concept of SERS active substrates from roughened electrodes or island films to metal colloid systems. Colloidal substrates offered several advantages over electrodes: they were easy to prepare, and their particle size, shape, and aggregation could be adjusted during preparation. Although it was very challenging to control and characterize them during that time, this variability allowed for the exploration of the relationship between surface morphology and SERS enhancement. Also, at that time there was already a good theoretical understanding of elastic light scattering by colloidal dispersions, and the introduction of colloidal SERS substrates therefore quickly stimulated theoretical work on the SERS of metallic nanoparticles. It's worthy to note that their pioneering works in colloidal SERS4,70 also inferred the close relationship between SERS and nanoscience, heralding its second upsurge in the era of nanoscience.
Kerker and coworkers employed a molecular dipole and spherical nanoparticle mutual polarization model (Fig. 8a and b),80,90 and comprehensively calculated the influence of particle size, molecular dipole location, and incident light wavelength on the SERS EF. It is noteworthy that Kerker simplified the geometry to a sphere, thus the results were analytically solved with high accuracy by deriving the Lorenz–Mie theory.
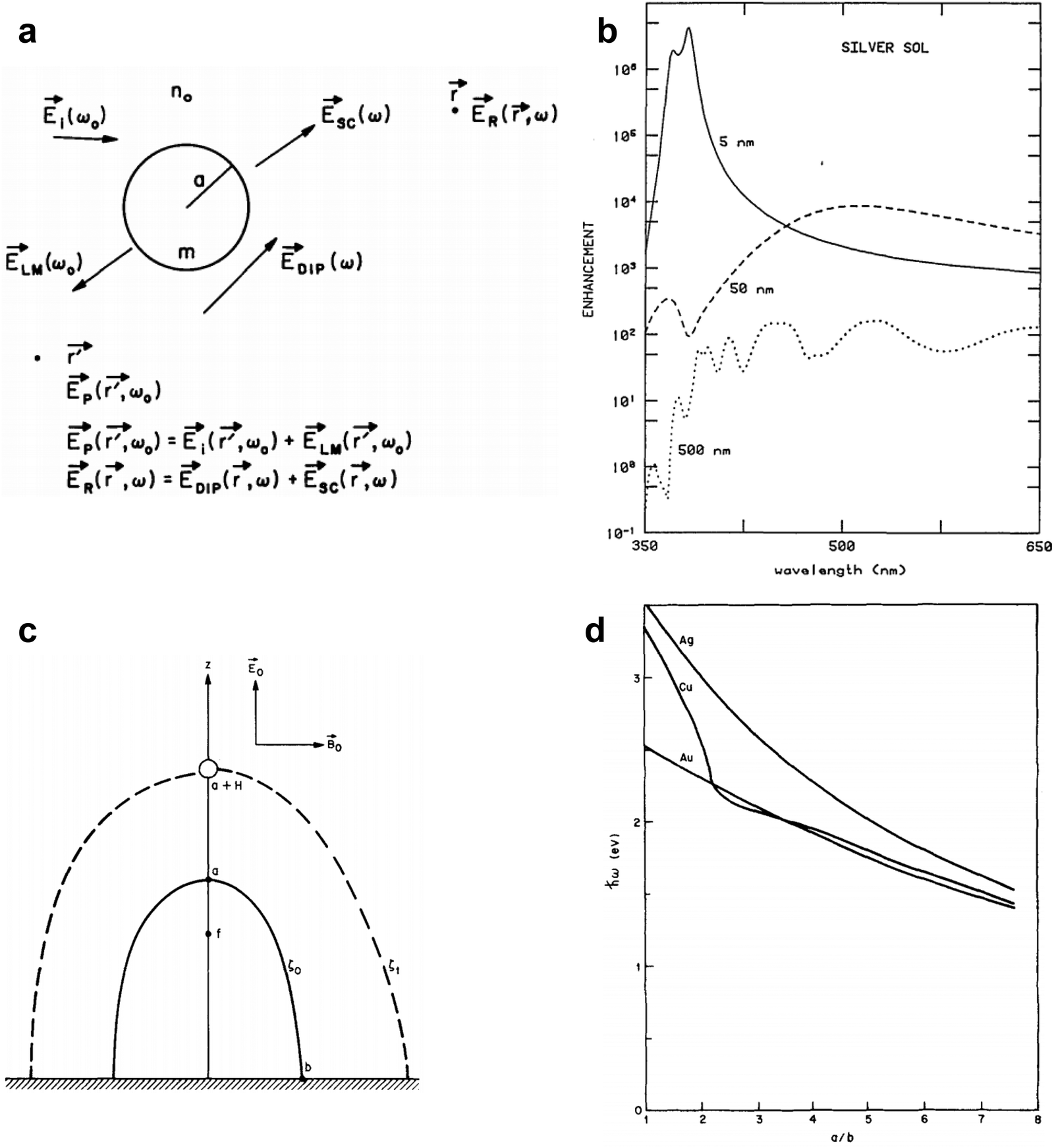 | ||
| Fig. 8 (a) Schematic view of electric fields near sphere of radius a and refractive index m. At the incident frequency ω0 there are the incident, Lorenz–Mie, and primary fields, Ei(ω0), ELM(ω0), and Ep(r, ω0). At the shifted frequency ω there are the dipole, scattered, and Raman fields, EDIP(ω), Esc(ω), and ER(r, ω), where r′ and r are the positions of the dipole and observer, respectively. (b) Calculated enhancement for a 1010 cm−1 Raman band of molecules that are adsorbed on Ag spheres of radii 5, 50, and 500 nm. Reproduced from ref. 80 with permission. Copyright 1980 Optica publishing. (c) Geometry of surface protrusion. The semimajor axis is a and the semi minor axis is b. The spheroid surface is ξ = ξ0 and that surface passing through the molecule is ξ = ξ1. (d) Energy of surface plasmons for Ag, Cu, and Au as a function of the aspect ratio of the spheroid. Reproduced from ref. 81 with permission. Copyright 1980 AIP publishing. | ||
Gersten and Nitzan simplified the metallic rough surface as hemispheroid protruding from a conducting plane, and employed a molecular induced dipole model to investigate the mirror field effect of molecular dipoles on the semi-ellipsoid (Fig. 8c and d).81 They revealed that in addition to the excitation of surface plasmons, the image dipole enhancement effect as well as the “lightning rod” effect at the tip of the ellipsoid also contributes to the Raman enhancement.77,81 McCall et al. developed a spheroidal model of the rough metallic surface and revealed the EM interactions between spheroidal metals and molecular dipoles.84 It is worth noting that Gersten and Nitzan, along with Kerker independently, were the first show via calculations that the SERS enhancement is proportional to the fourth power of the local electromagnetic field.
Subsequently, Metiu and coworkers were the first to study the electromagnetic (EM) coupling of a nanosphere dimer77 (Fig. 9a). The electromagnetic interaction between the two spheres significantly amplifies the local field in the space between them (Fig. 9b). Later on, the calculation based on electrostatic field theory for nanosphere-on-plane coupled structure78 demonstrated the production of very large local enhancements of the electric fields exceeding those obtained from the isolated sphere or isolated flat surface (Fig. 9c–e). The importance of colloidal dimers was not fully recognized until the discovery of single-molecule SERS.14,15 And the nanoparticles on mirror configuration set the early theoretical foundation of TERS and SHINERS. Laor and Schatz examined clusters of metal hemispheroids on a perfectly conducting flat surface and found significant SERS enhancement arising from the electromagnetic coupling between the random distributed metal hemispheroids, which was termed as the multiple plasmon resonance effect.75,76
 | ||
| Fig. 9 (a) Schematic of the 2-sphere system. Shown here are the relative positions of the two spheres, the orientation of the external field E0 and the observation point at which the field enhancement is calculated. (b) The upper curve is for the 2-sphere system λ = 0.4545, is a plot of I (on a log scale) versus distance d/D as one proceeds from the surface of one sphere to the other along their common axis. The lower curve is for the isolated sphere λ = 0 and the abscissa for this case should be read 5d/R0 rather than d/D as shown. The external frequency ω = 3.48 eV is the resonance of the isolated sphere. Reproduced from ref. 77 with permission. Copyright 1981 Elsevier. (c) Schematic of the particle on substrate system. (d) Resonance spectrum of the Ag–Ag system (i.e., Ag substrate with Ag sphere on top) with parameters R = 10 nm, D = 0.5 nm. The intensity enhancement ratio I is plotted as a function of the incident laser frequency in eV. The laser beam is incident at an angle of 45°. The solid curve is for p-polarized incident light and the dotted curve for s-polarized light. The value of FA indicated for each curve is the factor by which the vertical axis must be multiplied to obtain the value of I for that curve. (e) The variation of the resonance frequencies with system geometry (λ) for the Ag–Ag system. Dashed curve: λ = 0.03, FA = 2000; solid curve: λ = 0.05, FA = 1000; dotted curve: λ = 0.1, FA = 100. In each case the light beam is incident at 45° and is p-polarized. Reproduced from ref. 78 with permission. Copyright 1983 Elsevier. | ||
 | ||
| Fig. 10 (a) and (b) Configuration of the ATR method. (a) The metal contacts the prism and couples the SPPs with the evanescent field of the totally reflected light wave, Kretschmann–Raether configuration. (b) The dielectric (air) lies between the prism and the metal surface, Otto configuration. Reproduced from ref. 93 with permission. Copyright 1988 Springer Nature. (c) Time-averaged and normalized square of the electric field strength as a function of distance d from the electrode–electrolyte interface under surface plasmon resonance conditions. λ0 = 514.5 nm; φ1 = 50°; dAg = 50 nm, nhemi = l.906; nH2O = 1.336; the components in x- and z-direction of the enhanced field are also shown. The insert shows the geometry for the observation of Raman scattered light. (d) Angle dependence of the Raman intensity at 1010 cm−1 from pyridine on Ag electrode with ATR excitation by a Kretschmann configuration. λ0 = 514.5 nm. Reproduced from ref. 94 with permission. Copyright 1979 Elsevier. | ||
Burstein and coworkers theoretically predicted a substantial hundred-fold enhancement for ATR-Raman and ATR-coherent anti-Stokes Raman spectroscopy (CARS) in 1976.95 Building on this theoretical framework, Pettinger et al. experimentally demonstrated the excitation of surface plasmons at Ag electrodes through ATR94 in 1979 (Fig. 10c), resulting in an enhanced Raman signal from adsorbed pyridine (Fig. 10d). Subsequent research underscored the significant influence of excitation frequency and incident light angle on surface Raman intensity for roughened Au, Ag, and Cu electrodes.96 Subsequently, Dornhaus et al. performed a comparative investigation of attenuated total reflection Raman spectroscopy (ATR-Raman) using hemicylindrical prisms coated with silver films, alongside surface Raman spectra obtained from silver islands. Their findings highlighted the pivotal role of surface plasmon excitation in SERS in 1980.97 Furthermore, in 1982, Shen and coworkers investigated SERS of pyridine adsorbed onto weakly roughened Ag films using a well-characterized SPP excitation.98 They demonstrated that the SPP was the primary contributing factor contributing to the additional enhancement in the Raman signal.
Smooth metal films can also be excited by metallic gratings or molecular dipoles to generate surface plasmons. In 1975, Philpott investigated the interaction of excited molecules with the surface plasmon of metallic film and developed a theoretical framework for explaining the effect of surface plasmons on molecular electronic transition.99 Tsang et al. and Girlando et al. reported the Raman spectra enhanced by the SPP on a Ag grating in 1979100 and 1980.101,102 The EF provided by surface plasmons is quite limited compared to the LSP mechanism, primarily due to the minor electric field enhancement of SPP.
 | ||
| Fig. 11 (a) The optical probe particle (i) intercepts an incident laser beam, of frequency ωin, and concentrates the field in a region adjacent to the sample surface (ii). The Raman signal from the sample surface is reradiated into the scattered field at frequency ωout. The surface is scanned by moving the optically transparent probe-tip holder (iii) by piezoelectric translators (iv). (b) Transverse dependence of coherent Raman enhancement for several values of D/a, where D is the distance from the sample to the center of curvature of the probe tip and a is the radius of curvature at the probe tip. Figures are reproduced from ref. 103 with permission. Copyright 1985 Optica publishing. | ||
The electromagnetic mechanism, represented by the LSPR, contributes dominantly to the anomalous enhancement in SERS, which constitutes a distinctive feature of SERS distinguishing it unambiguously from the previous surface Raman spectroscopy and the surface resonance Raman spectroscopy. The surface plasmon enhancement mechanism, however, fails to account for modifications such as shifts in peak positions or changes in relative intensities in the SERS spectrum.
These feature was first observed in Fleischmann's experiment.1 Subsequently, in 1977, Van Duyne and Jeanmaire2 documented in detail that the Raman intensities of the adsorbed pyridine are a function of electrode potential with a laser excitation wavelength of 514.5 nm, which presented a resonance shaped profile as shown in Fig. 12a. Since no known molecular transition existed in that spectral region, it was speculated that when considering the molecule and metal system as a whole, the reason for the spectral features was probably due to the surface structure, applied potential as well as charge transfer (CT). Similar potential/photon dependence phenomena of SERS were also observed by Otto,105–107 Ueba,108,109 Furtak,110,111 Lombardi,112 Weaver113 and coauthors in pyridine and other SERS molecular probes on Ag electrode systems under different excitation wavelengths of laser, which elicited a surge of studies on chemical enhancement (CE) mechanisms in early years of SERS.106–109,113–120
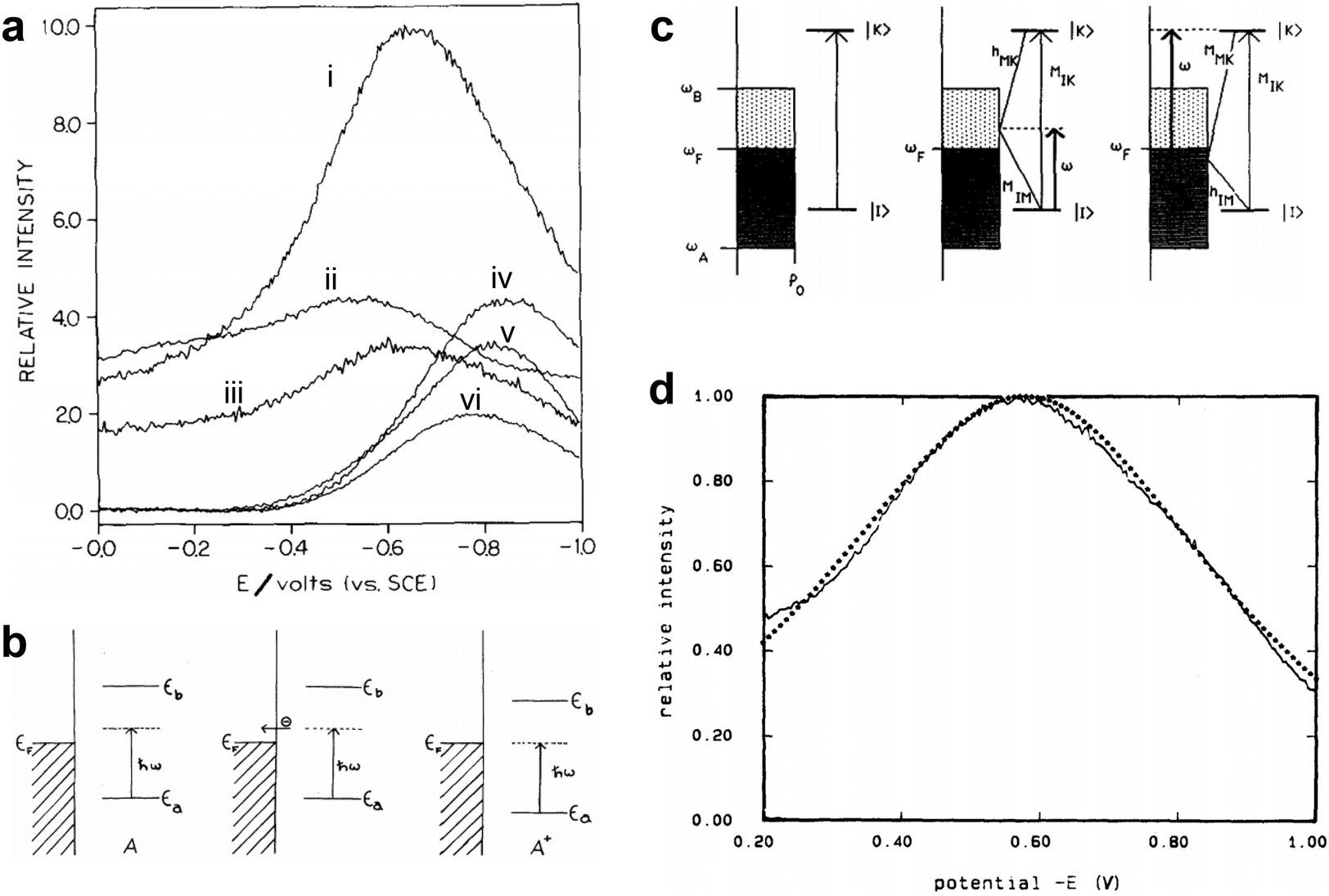 | ||
| Fig. 12 (a) The SERS intensity of the adsorbed pyridine on Ag as a function of the electrode potential, (i) 1006 cm−1, (ii) 1035 cm−1, (iii) 3056 cm−1, (iv) 1215 cm−1, (v) 1594 cm−1, (vi) 623 cm−1. (b) Conceptual model of the energy levels changing with the electrode potential in the CT process from molecule to metal in the metal–molecule system: metal–neutral molecule A on illumination immediately before tunneling (left), electron tunneling to the metal (middle), and adjustment of energy levels on formation of A+ for the resonant tunneling case (right). (c) The relevant energy states and the vibronic coupling involved in the Franck–Condon term and the Herzberg–Teller terms in the CT process. (d) The potential-intensity profile fitting from the CT enhancement mechanism for pyridine adsorbed on a Ag electrode. Figures are reproduced from ref. 2, 115 and 118 with permissions. Copyright 1977 Elsevier. Copyright 1979 American Physical Society. Copyright 1986 AIP publishing. | ||
The CE mechanism mainly focused on the increase or change of spectral intensity from chemical active site properties on the surface such as the bonding and molecular adsorption orientation between probe molecules and surfaces. The CE mechanism developed in the early years of SERS centered essentially in the CT resonance Raman enhancement. The CT processes necessitate the association of the CT state between the metal and the molecule. When the CT processes are resonant with the exciting radiation, it resembles an electronic resonance of the normal resonance Raman effect, which could partially explain the observed Raman scattering cross sections on the order of resonance Raman spectra.
In 1979, Gersten, Birke and Lombardi adopted a model involving CT process between adsorbed molecule and Fermi level of metal (Fig. 12b) and proposed a theory predicting that the CT between surface molecule and metal would induce a resonance in the vicinity of Fermi surface and lead to an enhancement of Raman scattering.115 Lombardi and coauthors in 1986 re-investigated the model (Fig. 12c) and presented a comprehensive development of the CT theory of SERS on the basis of direct CT mechanism.118 Based on the theory of Tang and Albrecht121 and the work of Adrian,120 they incorporated the Herzberg–Teller mixing of zero-order Born–Oppenheimer electronic states by using of vibronic interaction terms in the Hamiltonian. Considering both Franck–Condon and Herzberg–Teller contributions, they predicted the potential dependent Raman intensity profiles that fit very well to the observed intensity vs. voltage profile for pyridine adsorbed on a Ag electrode as shown in Fig. 12d, although the surface coverage was not strictly considered.
Aside from the previously studied enhancement by collective (surface plasmon) excitations, Burstein et al. in 1979 emphasized the role of electron–hole excitations which, due to their localized nature, coupled strongly with the adsorbed molecules.114 They further proposed the four types of electron–hole excitations related enhancement mechanisms, including the three-step Frohlich scattering (Fig. 13a-i) and the four-step CT mechanism (Fig. 13a-ii).114 The former energy transfer Frohlich mechanism was developed later by Pettinger.122 While the latter four-step CT mechanism was integrated to the adatom model proposed by Otto et al.107 Burstein et al. in their work114 depicted a relatively comprehensive physical picture of CE mechanism, despite that the quantitative estimation was not given, since at the time the theory of the radiative excitation of electron–hole pairs was not complete as well as precise information of interface energy level alignment was missing.
 | ||
| Fig. 13 Chemical enhancement mechanisms of SERS. (a) Schemes of (i) the three-step Frohlich Raman scattering mechanism and (ii) the CT mechanism. (i) Step 1, and step 3 and 3′ correspond to the radiation excitation and recombination, of the electron–hole pairs, respectively. Steps 2 and 2′ correspond to the scattering of excited electrons and excited holes by the vibrations of adsorbed molecule, respectively. (ii) Steps 1 and 4 correspond to the radiative excitation and recombination of an electron–hole pair, respectively. Steps 2 and 3 correspond to the hopping of the excited electron to and back from the virtual bound state of adsorbed molecule, respectively. An energy versus configuration curve for the virtual bound state, in which Q0 and Q′ are the equilibrium and perturbated configuration of the molecule, is also shown. (b) the four-step CT processes for pyridine adsorbed on a Ag electrode (i) and cyanide anion adsorbed on a Ag electrode (ii). Figures are reproduced from ref. 107 and 114 with permissions. Copyright 1979 & 1984 Elsevier. | ||
In 1981, Persson proposed a model for Raman scattering from molecules chemisorbed on surfaces and found that CT excitations between the Ag and the adsorbed pyridine molecules could produce an EF of up to 102.117 In the same year, CT states were experimentally observed for pyridine chemisorbed on Ag(111) by Demuth and Sanda utilizing high-resolution electron energy loss spectroscopy, which bolstered the CT mechanism.116 Later, Adrian120 used the semiempirical Wolfsberg–Helmholz method to treat the molecule–surface interactions and showed that CT Raman enhancements were of the order of 10 to 1000 for the ethylene adsorbed on silver model system. In this work, both Franck–Condon and Herzberg–Teller terms were derived, despite that the Herzberg–Teller term was considerably smaller than the Franck–Condon term for this model system.
Interfacial energy level alignment plays an important role in the CT enhancement mechanism. Ueba et al.108,109 conducted a detailed re-examination on the pyridine–silver system and constructed an energy diagram of pyridine adsorbed on Ag. They explained SERS as adsorbate-induced resonance Raman scattering through CT excitations and used the Newns–Anderson model of chemisorption to obtain a large EF about 103 when an extremely narrower CT state were assumed.
Following the CT mechanism proposed by Burstein et al.,114 Otto and coauthors further proposed the four-step CT mechanism based on the adatom model for pyridine and cyanide anion adsorbed on Ag surfaces in electrochemical interfaces,107 which predicted resonant-like Raman scattering by adsorbate vibrations facilitated by photon-excited charge-transfer transitions from localized electronic states at atomic-scale roughness sites (e.g., “adatoms”) on Ag surfaces to the affinity levels of the adsorbates (Fig. 13b). For pyridine adsorbed on a Ag electrode (Fig. 13b-i), they considered that the CT process proceeds in four steps, such as the generation of the surface plasmon excitation, the formation of the negative ionic molecule, electron transfer back to metal equal to the energy and final emission as radiating photons. The CT took place from the Ag to the unoccupied molecular orbital of pyridine. While for cyanide anions adsorbed on Ag electrode (Fig. 13b-ii), the CT took place from adsorbed cyanide anion to the unoccupied state above the Fermi level in Ag.
Although the CE, represented by the CT, processes typically provide EFs about one to three orders of magnitude for non-resonance Raman molecules, this phenomenon involves intricate interactions between surfaces, photons and molecules. Thus, it plays a crucial role in investigating molecular adsorption and chemical reactions on surfaces and cannot be overlooked. Furthermore, to accurately quantify the SERS intensity, a profound understanding of the CE effects arising from surface interactions at the molecular level is essential.
In 1982, Chang and Furtak edited and published the first book on SERS, which for the first time collected representative theoretical and experimental contributions in the development of the SERS field.62 There were some experimental exploration of various SERS active substrates by chemical and physical means.123–128 In 1985, after nearly a decade of exploration and competition, the electromagnetic theory represented by surface plasmon mechanism became the dominant SERS mechanism in the academic community, marked by the famous review article by Moskovits.69
Overall, in the initial decade of its foundation, the theoretical framework of SERS gradually took shape, laying robust groundwork for the field. Though it fell short of precisely quantifying the SERS effect or fully unveiling the nature of SERS-active structures, this foundational science served as the bedrock upon which the next four decades of progress would stand, guiding its evolution and inspiring further exploration.
3. The decade of persistent exploration (mid-1980s–mid-1990s)
The journey of SERS had been fraught with significant challenges and limitations during its second decade. The generality and versatility of SERS have been frequently questioned by the broader scientific community beyond the communities of Raman spectroscopy, materials and surface science.According to the electromagnetic mechanism, the SERS effect mainly arises from the excitation of surface plasmons in free-electron-like metals with subwavelength structures.21,69 Nevertheless, only a few metals like Ag, Au, and Cu, along with some alkali metals can provide the significant enhancement required for strong Raman signal amplification.4,69 The limitation, that SERS rely on specific metals for giant enhancement, severely restricts the breadth of practical applications involving other materials, thus curtailing the versatility of SERS in various scientific and industrial contexts.
Even these “free electron metals” require specific surface morphologies to achieve the significant enhancement characteristic of SERS. Surfaces must possess roughness, island-like structure, or contain metal colloids in the sub-micrometer size range.62,69 In contrast, the atomically flat and structurally well-defined surfaces commonly used in fundamental research in surface science and interface engineering could not support the necessary localized surface plasmon resonances.69 As a result, SERS could not be effectively applied to these structurally well-defined surfaces, further limiting its adoption and utility. This was especially awkward for SERS, given its name emphasizes “surface-enhanced,” yet it struggled to gain acceptance and make itself a useful tool across the scientific and industrial communities.
Consequently, many research groups gradually abandoned the field from the late 1980s to the mid-1990s, primarily due to difficulties in securing research funding. Some young researchers even faced the risk of being dismissed. However, a few scientists persisted in their efforts to break through the seemingly impossible barriers. The SERS effect, though incompletely understood, remained an incredibly attractive phenomenon due to its extremely high surface sensitivity. Those who persevered in exploring new approaches believed that SERS shouldn't be so easily sentenced to death or exile. The breakthrough might eventually occur, even though no one could predict when.
3.1 The “borrowing SERS activity” strategy
During the mid-1980s and early 1990s, several research groups tried to overcome the key limitations of generality. They aimed to create SERS activity from metallic substrates beyond the traditional materials of Ag, Au, and Cu, and to obtain Raman signals from atomically flat single-crystal surfaces. Some researchers have reported achieving both unenhanced129–131 and enhanced132–134 Raman signals from adsorbates on either roughened or mechanically polished Rh and Pt electrodes and, thereafter, on porous Pt, Pd, Ni, Co and Ti films.135 Nonetheless, the SERS spectra documented in these studies have either proven challenging to replicate or exhibited minimal detectability.As a result, these efforts were largely unacknowledged by the Raman spectroscopy and surface science communities, casting a bleak outlook on the potential of SERS. The mainstream consensus was that SERS was not as important as initially expected when it was discovered, and it seemed unlikely to evolve into a generalized and powerful analytical tool.
Fortunately, a few determined groups continued their efforts during this low period. They believed that the discovery of SERS, which made highly surface-sensitive analysis possible, suggested that finding ways to transform SERS into a versatile tool was achievable,21,35,136 because when one door closes, another window of opportunity often opens, revealing new paths and possibilities. While the electromagnetic mechanism of the SERS effect defined the limitations of substrates and surfaces, it also left room for potential solutions to circumvent these obstacles.
One of the highlights in that dark period is an innovative strategy with long-term significance, i.e., exploring SERS signals of adsorbates on semiconductor or transition metal surfaces, see Fig. 14. This strategy utilized the long-range effect of the electromagnetic field and deposited small Au or Ag islands on the SERS-inactive substrate.137–142 The probed molecules on the non-SERS-active surface can still experience the electromagnetic field from the SERS-active-islands thus their Raman signals were enhanced accordingly. This strategy allowed researchers to exploit the SERS effect on the traditionally non-SERS-active surfaces, which was later referred to as “borrowing SERS activity” strategy.143
 | ||
| Fig. 14 Schematic illustrations of the two strategies for borrowing SERS activity: (a) deposit discontinuous SERS-active metal islands on the substrate to study the molecules adsorbed on the non-SERS-active substrate; (b) coat an ultrathin non- or weak-SERS-active layer on the SERS-active substrate to study the molecules adsorbed on the layers. (c) Surface-enhanced resonance Raman spectra of Ru(bpy)32+-treated n-GaAs: without (upper) and with (lower) discontinuous Ag islands overlayer by strategy illustrated in (a). (d) Raman spectra in the C–N stretching region before and after zinc deposition onto a Ag electrode at the potentials (i) −1.2 V, (ii) −1.25 V, (iii) −1.25 V, after 3 min, (iv) −1.30 V, (v) −1.4 V, (vi) −1.4 V after 1 min, (vii) − 1.4 V after 3 min, (viii) – 1.4 V after 16 min. (e) Potential-dependent SER spectra in C–O stretching region for CO adsorbed on a platinum-coated Au electrode. (d) and (e) were obtained by the strategy illustrated in (b). Panel a and b are reproduced from ref. 35. Copyright 2007 Royal Society of Chemistry. Panel c is reproduced from ref. 137, copyright 1983 American Chemical Society. Panel d is reproduced from ref. 138, copyright 1987 Elsevier. Panel e is reproduced from ref. 139, copyright 1987 American Chemical Society. | ||
The first investigation that introduced the concept of “borrowing SERS activity” strategy was conducted and reported by Van Duyne group in 1983.137 They deposited discontinuous Ag overlayers or islands on non-SERS-active semiconductor surfaces pre-adsorbed with the molecules of interest, as illustrated in Fig. 14a. Despite their poor nanostructure aggregate, the SERS-active Ag islands created the long-range effect of strong electromagnetic fields, enhancing the resonance Raman scattering of adsorbates on the nearby semiconductor surface. Consequently, surface-enhanced resonance Raman signals of the adsorbed transition metal complexes molecules on n-GaAs electrodes (Fig. 14c) were observed.137,140
In this preliminary trial of the “borrowing SERS activity” strategy, it was challenging to ensure that the species of interest were selectively adsorbed only on the non-SERS-active substrate other than on the high-SERS-active Ag islands. In most cases, molecules tend to undergo surface diffusion and eventually adsorb on the high-SERS-active materials, which generate stronger signals. The signals overshadowed the smaller signals from the non-SERS-active surfaces of interest, thus complicating the analysis and reducing the effectiveness of the “borrowing SERS activity” strategy.
A more effective and reliable approach, which involved electrochemical deposition of an ultrathin layer of weak or non-SERS-active metal materials on Ag and Au roughened substrates (Fig. 14b), was independently developed in 1987 by Fleischmann group138,141 and Weaver group.139,142,144 By borrowing the strong electromagnetic field generated by inner Ag or Au nanostructures, Raman signals of target molecules in vicinity of their surfaces are enhanced even if the molecules are not contact with the SERS active substrate directly. For example, as shown in Fig. 14d(i)–(iv), the peak ascribed to CN− adsorbed on Ag was gradually replaced by a new peak ascribed to CN− adsorbed on zinc during the process of underpotential deposition of zinc. Subsequently the peak intensity of CN− adsorbed on zinc gradually decreased upon the further progress of overpotential deposition of the thick film as shown in Fig. 14d(v)–(viii).138Fig. 14e illustrates that using an ultrathin Pt film over roughened Au surfaces effectively reveals SER spectra for carbon monoxide adsorbed on Pt thin film (ca. one to three monolayers) with potential-dependence. Since the enhancement decreases exponentially with increased distance from the SERS-active substrate, the transition metal layer must be ultrathin, typically only a few atomic layers, to effectively borrow/experience the electromagnetic field. Meanwhile, the ultrathin metal film must be pinhole-free to prevent direct molecule adsorption on the underlying SERS-active Ag or Au substate, which could lead to misleading spectral interpretations.
It is worth noting that preparing ultrathin and pinhole-free metal layers was quite challenging due to the irregular and undulating surface of electrochemically roughened electrodes. Despite their clear limitations at the time, these early efforts in exploring the “borrowing SERS activity” strategy set a foundation for solving generality problems and challenging seemingly impossible boundaries.
3.2 Explorations on bio-SERS and electrochemical-SERS
During these challenging times, it is widely recognized that the standard benchmarks and reproducibility are lacking in SERS studies without extending the research to weak- or non-SERS-active material, even with the use of roughened Ag and Au electrodes or colloids. Furthermore, the messy and disorderly SERS-active sites, which include various ad-atoms, ad-clusters, and surface complexes, undermines the rigorous scientific standards required for systematic and reproducible studies. Therefore, the coherent recognition and full support from spectroscopy, analytical chemistry, biology, electrochemistry and other fields were lacked in the SERS studies.Fortunately, some researchers persisted in exploring various research methods and application areas to enhance the advantages and mitigate the disadvantages of SERS-active metals like Ag and Au. Cu, being highly reactive and prone to forming copper oxide, complicates the establishment of a reliable benchmark. One approach is to focus on the detection sensitivity for probing surfaces species because normally only monolayer or even sub-monolayer species (1013–1015 per cm2) are presented, especially for some weak Raman scatterers.145–147 They developed several ways to improve the detection sensitivity: (1) employing a high numerical aperture lens to optimize collection efficiency; (2) increasing laser power to the maximum permissible level without exceeding the damage threshold; (3) utilizing the potential difference method to isolate the faint interfacial signal from the stronger bulk signal; (4) selecting an appropriate excitation wavelength to leverage the resonance Raman effect; and (5) maximizing the SERS effect through appropriate modifications of the electrode surfaces.148–150
Another crucial approach is to develop various methods for creating new substrates that produce strong and stable SERS signals. These methods include: roughening of electrode surfaces through multiple oxidation–reduction cycles (ORCs),141,151–154 chemical etching of surfaces in acidic solutions,155–157 deposition of island films on substrates at elevated temperatures,158 as well as the preparation of films via evaporation or sputtering in a vacuum onto cryogenic substrates (100 K),159 and electrochemical deposition.141,160 Additionally, colloids—particularly aggregated ones, can be generated through chemical methods161 or laser ablation.162 Sub-microstructure can also be prepared using lithographic, self-assembly, or template techniques.163
A highlight was the development of a highly stable SERS-active Au substrate using an asymmetric ORC process, which was proposed by Weaver and his colleagues.164 The Au surface showed a distinct brown color after repeating the process for about 15 minutes, contrasting with the yellowish hue of highly SERS-active Ag electrodes. Although the nanostructured surfaces were poorly defined at the time, their effectiveness was evident, and the differences could be distinguished by the naked eye, which often provides quicker and more sensitive detection than a simple visible spectrometer in that time.
From methodologic view, SERS research could be broadly divided into two trends: species identification from an analytical perspective and structure/process characterization from a physicochemical perspective.165 The analytically oriented studies primarily focused on whether SERS could detect and identify the target species in complex systems such as the biologic systems. In contrast, physicochemical-oriented studies require a more comprehensive understanding of the way and the reason that target species interact with surfaces and their surroundings by analyzing spectral features. To achieve this, more systematic characterizations are needed to estimate all the physical and chemical mechanisms involved, while considering surface selection rules. Additionally, it is necessary to develop preparation methods for simpler SERS-active surface, as the spectral features of the same probed molecules, whether tightly trapped in a gap or freely adsorbed on a surface, can differ significantly. These efforts are essential for providing meaningful insights into adsorption configurations or reaction mechanisms, such as in electrochemistry.49,50,130,166,167
During the 1980s, there was an increasing interest in the utilization of the “SERS reporter” technique, which significantly enhances the weak signals from targeted species by employing reporters exhibiting strong (resonant) Raman signals. This approach has been demonstrated to be effective for the trace detection of molecules in solutions, as well as for the identification of specific molecules within live cells or animal models. With the effort of Cotton et al.,168,169 Koglin and Sequaris,170 Nabiev et al.171 and other groups,172–175 SERS was also extended to biological systems, including DNA (from base to mononucleotide, double-stranded polynucleotides and modified DNA, as shown in Fig. 15), chromosomes, amino acid and protein (e.g., lysozyme and BSA) and various porphyrin chromophores. Although there are some criticisms of SERS or surface enhanced resonance Raman spectroscopy (SERRS) as a technique for biological systems regarding the possibility of denaturation by the interaction with the SERS substrate surface, the in vivo applicability of SERS was promisingly anticipated with the further improvements in SERS substrates and instrumentation.168,170
 | ||
| Fig. 15 (a) SERS spectra of native DNA (top) and of poly(dA-dT)·poly(dA-dT) (bottom). DNA concentration 50 μg ml−1 and 80 μg ml−1, respectively; 0.15 M KCl, 10−3 M Tris pH 8; adsorption potential −0.6 V vs. Ag/AgCl. (b) SERS spectra of native and γ-irradiated DNA. 0.1 M KCl + 2 × 10−3 M Na2HPO4; DNA concentration: 300 μg ml−1; pH 8.0; irradiated with γ-rays from a 60Co-source. Figures are reproduced from ref. 170 with permissions. Copyright 1986 Springer Nature. | ||
This perseverance and creative problem-solving kept the field of SERS research alive, eventually leading to its resurgence and broader acceptance as a powerful analytical tool for various branches of SERS.
3.3 Interfacial water as an unusual target by SERS
Regarding molecular generality and electrochemical application, SERS also underwent a slow and intricate journey. In the early exploration of molecules chemically or physically adsorbed at surfaces, which can be probed by SERS, the surface water molecules on electrodes stood out as one of the most surprising and unique systems. Since water is one of the most important molecules and the most essential solvent, playing significant roles in interfacial processes such as electrocatalysis, researchers were eager to obtain the SERS of water after discovering the SERS effect. Although water is naturally a very weak Raman scatterer, the presence of multiple layers of water molecules at the interface often results in a significant Raman signal. It can be reasonably assumed that the SERS signal from the total amount of water molecules should not be too weak. However, surface/interfacial water gave no detectable SERS signals despite its universal presence at most interfaces, which remained a puzzle.In 1981 Fleischmann and coworkers were the first to obtain SERS of water with a paper entitled “Enhanced Raman spectra from species formed by the coadsorption of halide ions and water molecules on silver electrodes”.176 For this they adopted a special experimental condition with a high concentration halide salt aqueous solution, such as 1 mol dm−3 KCl, to obtain the SERS signal from water, as shown in Fig. 16. In the experiment, the Ag electrode underwent electrochemical oxidation–reduction treatment in a KCl aqueous solution, and in situ observation was immediately conducted in the same solution under controlled electrochemical potentials. This procedure is effective in avoiding the influence of surface contaminants. It was later identified as a typical SERS-detectable coadsorption system containing halide anions and water molecules.
 | ||
| Fig. 16 (a) The Raman spectra in the OH stretching frequency region of molar KCl/H2O with the laser focused on a polished Ag electrode. All spectra were measured at a potential of −0.2 V. (i) Polished electrode; (ii) roughened electrode; (iii) roughened electrode pushed up against the cell window; (b) a comparison of the Raman spectra in the OH stretching region and using the same intensity scale of: (i) pure H2O; (ii) 4 M KCl; (iii) a Ag electrode roughened in 1 M KCl, with the electrode pushed up against the cell window. Figures are reproduced from ref. 176 with permission. Copyright 1981 Elsevier B.V. | ||
In the same year, Pettinger et al. attributed SERS of water and halide ions in a similar solution to the formation of surface complexes involving Ag adatoms, halide ions, and water molecules.177 The further experimental results provided a strong evidence that the SERS signal of water was irreversibly diminished when the applied potential was moved to highly negative potentials. The desorption of halide ions from the negatively charged surface led to decomposition of the SERS-active site.178 Therefore, the SERS of water in surface complex forms is most likely related to chemical enhancement. These water molecules are atypical in the double layer of ordinary electrolytes and this aspect of the SERS of water undermined the utility of SERS for characterizing the water molecules typically present in the double layer, dealing a heavy blow to researchers' confidence in using SERS as a powerful tool to study electrochemical interfaces.
This great difficulty, however, only fueled the determination of a small group of researchers to persistently unravel this mystery. They thought that if we cannot understand SERS of water, no one can claim SERS is truly useful and the SERS mechanism is fully understood. In 1987, Funtikov et al. reported an intriguing and atypical finding regarding the surface-enhanced Raman scattering (SERS) of water at silver electrodes immersed in sodium sulfate solutions devoid of (pseudo-)halide ions.179 They asserted that detection was limited to the SERS signal associated with the bending vibration of water, while the characteristic stretching mode remained undetectable. Furthermore, the study was confined to a narrow potential range of −1.1 V to −1.45 V (with reference to a saturated silver chloride electrode) owing to the interference caused by hydrogen evolution at more negative potentials.
After seven years, progress was made through the cooperation of Tian group and Funtikov group in 1994. They employed a special ORC procedure to achieve a highly SERS-active surface and obtain high quality SERS spectra of both the stretching and bending vibration modes of water at Ag electrodes.180 Additionally, by positioning the electrode as close as possible to the optical window of the Raman spectroscopic cell to form a thin layer cell, the interference of hydrogen bubbles and Raman signal from bulk water was efficiently minimized. This allowed for the investigation of the potential dependence of the SERS intensity and frequency over a much wider potential range from −0.5 V to −2.0 V (referred to saturated calomel electrode, SCE), including potentials with severe hydrogen evolution. However, to obtain high-quality SERS signals, a highly concentrated electrolyte, such as 8 mol dm−3 of NaClO4, was needed, leaving some aspects of the puzzle unsolved.
Because of the efforts of these dedicated researchers through the mid-1990s, the interfacial water systems probing by SERS were systematically expanded to various transition metal surfaces in different dilute electrolytes by Tian group, via the several approaches including special ORC roughening procedure, the use of a thin layer cell, as well as the difference spectrum method when confocal Raman microscopy with much higher detection sensitivity at surfaces was invented and commercialized.181 These works provided basic methods for the later and wider exploration of interfacial water structures on single-crystal surfaces of different metals in the past decade.
Looking backward to the first two decades of SERS, the journey of SERS was fraught with significant challenges and limitations. A broader historical and fundamental view is needed to understand the reason why SERS had such a challenging growth and took a difficult and intricate journey. It is now much clearer that SERS is truly a branch of nanoscience, as its sensitivity and spectral features critically depend on the interaction between nanostructures and incident light. However, SERS was discovered about two decades before the advent of nanoscience, thus lacking suitable experimental and theoretical methods that were finally developed in the 1990s and beyond.
The first and second chapters of the SERS story are thus unique and encouraging compared to many other scientific fields because it would have been much easier to get started in the mid-1990s when nanoscience was booming. However, the early hard exploration was worthy as it marks SERS as one of the oldest branches of plasmon-enhanced spectroscopy, plasmonics, and even the broader field of nanoscience. The pioneers who laid the foundation of SERS and their persevering spirits in those early decades deserve great respect.
4. SERS breakthroughs enabled by advancements in nanoscience (mid-1990s–mid-2010s)
Undoubtedly, the significant advances and breakthroughs in the SERS field were driven by nanoscience. To understand this further, we need to examine why nanoscience gained momentum in the 1990s rather than in the 1960s. In 1959, Richard Feynman had laid the conceptual foundation of nanoscience with his famous lecture, “There's Plenty of Room at the Bottom”.182 He proposed the potential for manipulating and controlling matter at atomic and molecular scales, laying the intellectual groundwork for what would later become nanotechnology. However, despite his groundbreaking vision, Feynman was unable to develop nanotechnology tools or devices directly, leaving the field open for further exploration and advancement in the 1990s.The three decades’ delay in opening the field of nanoscience is mainly due to lack of adequate characterization methods. From the perspective of the history of scientific development and methodology, to precisely control and manipulate matter at any scale, it is essential first to develop methods and tools capable of observing that scale. The starting point could be the 1986 Nobel Prize in Physics awarded jointly to Ernst Ruska, Gerd Binnig, and Heinrich Rohrer for their groundbreaking contributions to microscopy techniques at the nano- and atomic scales. These revolutionary methods provided unprecedented resolution, enabling scientists to image and manipulate matter, effectively opening the door to nanotechnology. The widespread recognition of any new field and its integration into various scientific branches typically took 5 to 10 years, driven by the development of in-house solutions or the commercialization of new instruments.
For the younger generation today, it may be hard to imagine that in the 1980s, obtaining a nanometer-level scanning electron microscope (SEM) image was far more difficult for most research groups worldwide than achieving a sub-angström resolution with a transmission electron microscope (TEM) is today. Scanning probe microscopes (SPM), such as the scanning tunneling microscope (STM) and atomic force microscope (AFM), were still in their infancy, with no commercial products available. At that time, experimental techniques and theoretical capabilities were inadequate to overcome the obstacles hindering SERS development. Highly active and stable SERS substrates could not be rationally prepared or accurately characterized at the nanoscale, leading to challenges in quantitatively analyzing and establishing mechanical models of SERS. These challenges illustrate just how early researchers were in the field, highlighting the crucial role of developing new methods and instruments in emerging areas like nanoscience and its one of the oldest branches, SERS.
4.1 The significant rise in SERS
The rapid development of nanoscience served as a booster for SERS,35 which rose like a phoenix from the ashes in the mid-1990s to 2000s, as evidenced by the significant increase in annual publications (Fig. 17). A series of new synthesis methods for SERS-active nanoparticles (NPs), nanofabrication techniques for structured surfaces, and advanced analysis methods for characterizing the prepared nanostructures in the 1990s contributed significantly to the progress of SERS substrates, which evolved from roughened Au and Ag electrodes, island films deposited on cold surfaces in the ultrahigh vacuum, and colloidal aggregates,1–3,5,62,70,183,184 to Au, Ag and Cu185,186 nanoparticles featuring nanometer-sized gaps, shells, tips, edges, and structured surfaces with nanometer-sized holes, voids, bumps, grooves, or ridges.14,15,20,22,26,35,187–198 The characteristic dimensions of the above structures are progressively decrease from the ill-defined and disordered micrometer and sub-micrometer scale to the defined and ordered tens of nanometers scale structure, as shown in Fig. 18. The landmark work marking the entry of SERS into the nano era was the demonstration of SERS using monodisperse Au and Ag nanospheres by Natan and coworkers in 1995,187 heralding the phase when SERS substrates transitioned into a well-defined structural stage. | ||
| Fig. 17 The number of publications in the 1974–2024 period searched through the ISI Web of Sciences (top panel) and the ratio of the number of publications (bottom panel). The keywords used for SERS include “Surface-enhanced Raman”, “SERS”, “Surface-enhanced Resonance Raman” and “SERRS”. The keyword used for nano is “Nano”. The 50-years history of SERS was divided into four periods: 1974–1985: the foundation period; 1985–1995: the perseverance period; 1995–2015: the explosive increase period; 2015–2024: the recent progresses. | ||
 | ||
| Fig. 18 The evolution and milestones related to (a) representative SERS-active substrates and (b) development of Raman instrument. Figures of panel a are reproduced from ref. 14, 20, 35, 187 and 199–201 with permissions. Copyright 1981 & 1982 Elsevier B.V. Copyright 1995 & 1997 AAAS. Copyright 1999 APS. Copyright 2007 Royal Society of Chemistry. Copyright 2010 Springer Nature. Figures of panel b are reproduced from ref. 18, 165 and 202 with permissions. Copyright 1962 Optica publishing. Copyright 2002 American Chemical Society. Copyright 2000 Elsevier. | ||
4.2 Demonstration of single-molecule sensitivity in SERS
A significant milestone in the advancements of SERS in nano era was the achievement of single-molecule SERS (SM-SERS) detection. In 1997, Nie and Emory14 and Kneipp and colleagues15 independently reported experimental evidence observing SERS from single dye molecules on Ag nanoparticle colloids (Fig. 19). This breakthrough not only pioneered a new single-molecule spectroscopic method beyond single-molecule fluorescence but also provided high energy-resolved molecular vibrational fingerprint information while avoiding the common photobleaching issues in single-molecule fluorescence.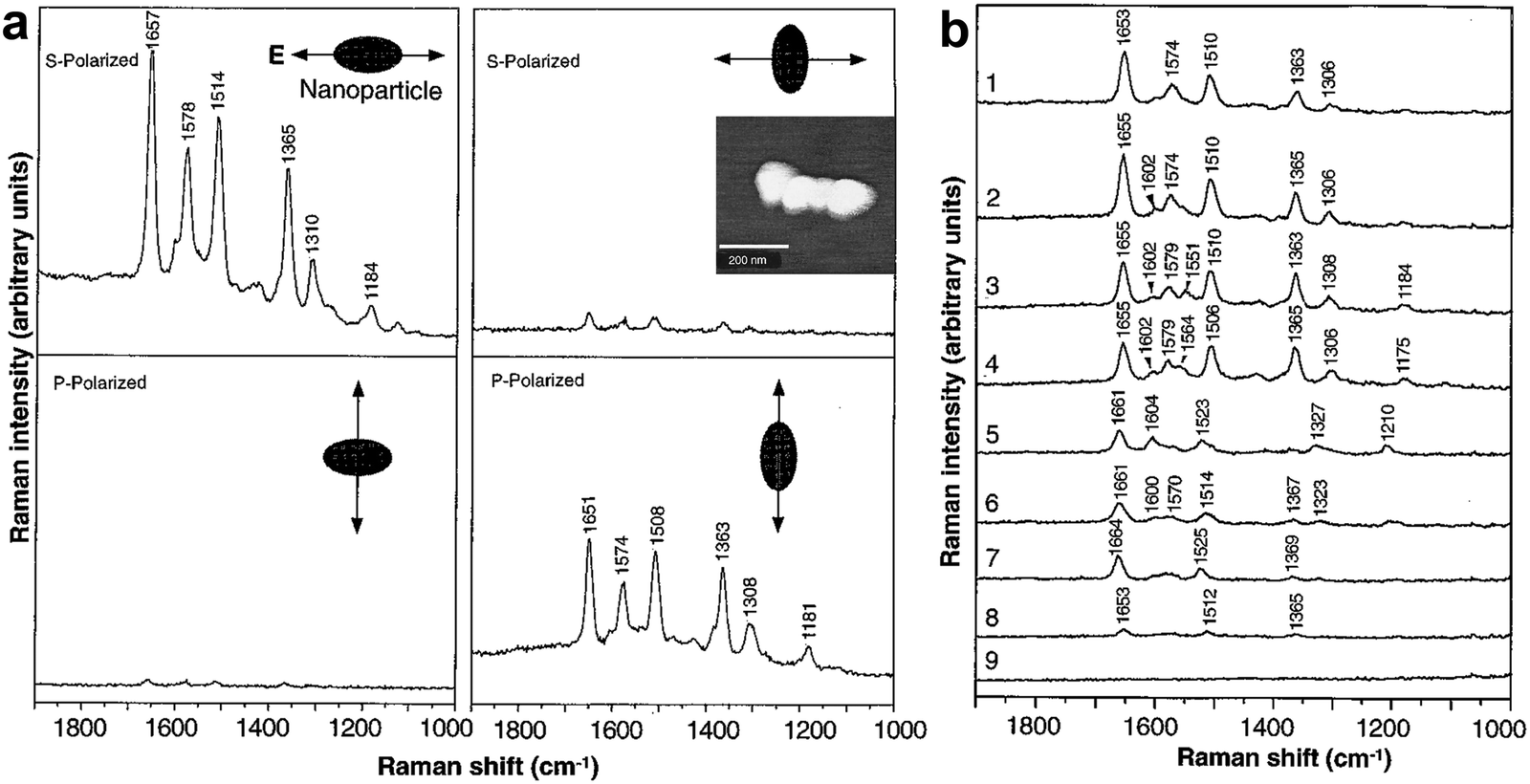 | ||
| Fig. 19 Experimental demonstration of single-molecules SERS. (a) SERS of rhodamine 6G (R6G) obtained with a linearly polarized confocal laser beam from Ag nanoparticles. (b) Time-resolved surface-enhanced Raman spectra of a single R6G molecule recorded at 1-s intervals. The Raman signals abruptly changed in both frequency and intensity three times, as shown in spectra 2, 5, and 8. Figures are adapted from ref. 14 with permission. Copyright 1997 AAAS. | ||
At that moment, Nie had already noted that achieving SM-SERS required suitable nanostructures and excitation conditions, and he estimated a SERS EF of 1014–1015 in their experiments. (Note that these were wrongly referenced against non-resonance Raman cross-section and were likely closer to 1010 when using the corrected single-molecule EF definition.203 see Section 4.7) Nie wrote,14 “An unexpected finding during this work was that a very small number of nanoparticles exhibited unusually high enhancement efficiencies. These particles emitted bright, Stokes-shifted (toward longer wavelengths) light and are called hot particles.” While Nie performed resonant SERS experiments, Kneipp applied near infrared (NIR) excitation non-resonant to the target molecules and explained their single molecule experiments by very strong electromagnetic enhancement, related to Ag clusters (aggregates of Ag nanoparticles), which is particularly effective at NIR excitation.15
The specific structure of these hot particles seems to be a key to unveil the unexpected signal enhancement observed in SM-SERS experiments. Nie investigated the morphology of these hot particles using High-resolution AFM and found that “High-resolution AFM images of selected hot Ag nanoparticles show that the majority of them are well-separated, single particles with a narrow size range of 110 to 120 nm in diameter, indicating a strong correlation between enhancement efficiency and particle size.”14 Experiments by Kneipp and colleagues showed that huge SERS enhancement is related to colloidal clusters and NIR excitation.15,204,205 This observation was in agreement with computations by Stockman et al. which showed that electromagnetic enhancement increases with longer excitation wavelengths in the fractal clusters.206 By using electron microscopy, Kneipp found that “Electron micrographs of the sol… show that the colloidal solution is slightly aggregated and consists of small 100–150 nm sized clusters”.15 However, it was not clear how the clusters/single particles cause the efficient Raman enhancement, and such SM level enhancement were not widely accepted by the community at that time.
Aiming to understanding the SM-SERS mechanism, in 1999, detailed electromagnetic calculations and experiments by Käll and coworkers201,207 and Brus and coworkers208,209 proved that it's the giant electromagnetic field enhancement in the nanogaps between coupled Ag nanoparticles that are essential for achieving SM-SERS. As shown in Fig. 20, the Raman spectra of single hemoglobin in Ag nanoparticle dimer show significant temporal fluctuation, which is consider as the direct evidence of SM-SERS event. Fig. 20b and c confirmed the giant electromagnetic field enhancement (E4) in the nanogap of nanoparticle dimer, which is highly sensitive to the gap distance as well as the incident polarizations (Fig. 20d and e). These results clearly elucidated the observations from the 1997 SM-SERS experiments by Nie and Emory, and Kneipp and colleagues, revealing the electromagnetic mechanism underlying the SM-SERS, which also initiated the field of nanogap plasmonics.
 | ||
| Fig. 20 (a) SEM image of a hot dimers and corresponding single Hb molecule spectra in the inset. The double arrow indicates the polarization of the incident laser field. (b) EM enhancement for nanoparticle dimers with separations from top to bottom 5.5 nm and 1 nm, respectively. (c) Calculated enhancement of the two-particle system (configuration shown in the inset) compared to the single-particle case, as a function of particle coupling. (z/R) 0 is at one particle center, and (z/R) + 1 is on the particle surface at the particle–particle interface side. (d) Calculated electromagnetic EF for the midpoint between two Ag spheres separated by d = 5.5 nm and for a point d/2 outside a single sphere. Diameters of spheres D = 60 nm (dashed curves), 90 nm (solid curves) and 120 nm (dotted curves). Double arrows indicate the incident polarizations. Inset shows the enhancement versus D for λ = 514.5 nm. (e) Schematic representation of SERS active aggregate geometry. A single R6G molecule adsorbed on the Ag particle covered with a citrate layer. The xanthene plane is aligned along the local field direction. Panel a and d are reproduced from ref. 201. Copyright 1999 American Physical Society. Panel b is reproduced from ref. 207. Copyright 2000 American Physical Society. Panel c and e are adapted from ref. 209. Copyright 2003 American Chemical Society. | ||
It is important to mention that actually Metiu and coworkers77 had pointed out in 1981 that the gap between nanostructures could lead to significant electromagnetic interactions and produce enormous SERS enhancement inside the gap. These “rare sites” were further conceptualized by Shalaev et al. as the hotspots.210 However, due to the limitations in precise fabrication and characterization of such nanostructures at that time, the significance of gap mode in SERS was not widely recognized until after the seminal single-molecule experiments in 1997. Via rigorous theoretical analysis, Käll and Xu further proposed that the gap between nanoparticle dimers can generate significant optical trapping forces.211 The optical forces create an optical trap for molecules in the hotspots, thereby make the SM-SERS detection possible.
It is noteworthy that the initial SM-SERS studies were based on ultra-low concentration of analytes, whereby the concentration was diluted to the extent that only a single molecule was present in the detection area. However, for a molecule to contribute a signal, it must adsorb onto the “rare hot particles/sites,” making it challenging to accurately estimate the number of adsorbed molecules. The convolution of “rare sites” (or hotspots) with ultra-low concentrations results in poor statistical reliability and soundness, thus aroused numerous doubts and led to debates regarding the interpretation and even the reality of the observations of SM-SERS.14,15 These debates were only fully settled with the development of the bi-analyte SERS technique for an unambiguous demonstration of single-molecule SERS detection.212–214
While the electromagnetic theories developed in the first phase of SERS history by numerous pioneers provided a solid foundation for the resurgence of SERS,4,62,64,65,67,69,75–90 it is essential to recognize that the substantial advancements in nanostructure preparation, characterization, and theoretical and computational methods during the mid-1990s to 2000s contributed to the beginning of the resurgence of SERS.14,15,187,201,207–210,212–215 Subsequently a substantial amount of research further propelled the revival of SERS in the nanotechnology era.21,32
An international symposium on Progress in Surface Raman Spectroscopy was held in Xiamen University, Xiamen, China during Aug. 14–17, 2000. The symposium brought together approximately 80% of the most active and leading researchers in SERS worldwide, as depicted in the group photo in Fig. 21 and the symposium program, to present cutting-edge methods and advancements. This symposium provided a dynamic platform for discussing and even debating SERS mechanisms, SM-SERS, the newly invented TERS, and their applicability to broader systems. The detailed topics can be found in the program as shown in Table 1.
 | ||
| Fig. 21 Group photo of International Symposium on Progress in Surface Raman Spectroscopy, held in Xiamen University, Xiamen, China during Aug. 14–17, 2000. Attendees include (from the front row to the back row, counting from right to left): 1st row: Shu-Ming Nie (1st), Nai-Teng Yu (3rd), James Durig (4th), Richard Van Duyne (5th), Martin Moskovits (8th), Andreas Otto (9th), Mildred Dresselhaus (10th), Aleksey Polubotko (12th), Ricardo Aroca (13th), Vladimir Shalaev (14th), John Lombardi (15th), Zhong-Qun Tian (16th); 2nd row: Linda Prinsloo (1st), Alexandre Brolo (3rd), Joel Rubim (8th), Kwan Kim (11th); 3rd row: Alan Creighton (2nd), Bernhard Schrader (5th), Wieland Hill (6th), Andrzej Kudelski (10th), Christy Haynes (11th); 4th row: Katrin Kneipp (1st), Yan-Xia Chen (3rd), Marek Prochazska (5th), Jürgen Popp (6th), Ralf Gessner (7th), Kiki Gessner (8th), Volker Deckert (11th), Wei-Qing Xu (15st); 5th row: Andreas Emge (6th), Jörg Kottmann (8th), Jian-Lin Yao (9th), Louis Brus (11th), Bin Ren (13th), Juan Otero (17th), Bing Zhao (18st). | ||
|
In 1997, skepticism regarding the 1014 EF claimed for SM-SERS from the mainstream community was quite similar to the strong doubts about the 6th power EF claimed for SERS in 1977. Therefore, validations and mechanisms of SM-SERS became a major focus of the symposium. To delve deeply into this theme, round table discussion 1 was arranged immediately following the presentations by Kneipp and Nie, to examine and discuss the challenges and opportunities within SM-SERS. Round table discussion 2 was held after presentations by Campion, Shalaev, Otero, and Kottman to highlight the latest progress in physical and chemical enhancement mechanisms. These heated discussions and debates were instrumental in exchanging various views and advancing theoretical methods.
Notably, the symposium coincided with newly reported TERS results, and Deckert, one of the inventors, delivered an acclaimed plenary presentation despite being an early-career researcher at that time. Coincidently, the dual advances of SM-SERS and TERS methods have collectively captured significant attention from experimenters and theorists, focusing on the same ‘hotspot’—the singular behavior of molecules, nanoparticles, and nano-tips with profoundly high local electromagnetic fields. This focus has ultimately positioned SERS as a new scientific Frontier, extending the boundaries of knowledge beyond traditional surface spectroscopy.
Moreover, efforts to broaden the materials generality of SERS, such as transition metal SERS using confocal microscopy reported by Ren and semiconductor SERS by Quagliano, also invoked some interests. Many topics have continued to evolve over the subsequent decades of SERS research. The collective contribution by all participants reinvigorated research momentum in the field, marking a shift from a low-tide phase to a dynamic, nano-driven SERS era.
4.3 Key methods of SERS since the mid-1990s
SM-SERS demonstrated the astonishing sensitivity of SERS, fueling the belief that SERS, when combined with well-defined nanostructures and characterization techniques, can usher in a new era of development opportunities. This realization sparked widely renewed interest among researchers in the field of SERS. Leveraging rapid advancements in nanotechnology, numerous methods in SERS research have flourished, creating a vibrant and dynamic landscape. We meticulously and comprehensively organize these methods from the past decades in a matrix format, covering aspects such as detection sensitivity, spatial and temporal resolution, materials and morphology generalities for SERS, and quantitative analysis of SERS (see Fig. 22). These achievements have greatly propelled the three decades of continuous explosive growth of SERS. | ||
| Fig. 22 The timeline of key methods since 1990s (a) toward well-ordered SERS-active micro-/nano-structures fabrication for high-sensitivity of SERS; (b) key methods for high spatial and temporal resolution; (c) key methods for expanding the generalities of SERS substrate materials and morphology. (d) key methods for quantification of SERS EF and quantitative analysis. The abbreviations: TM: transition metals; SHINs: shell-isolated nanoparticles; NPoM: nanoparticle on mirror; MCBJ: mechanically controllable break junction; STM: scanning tunneling microscope; SESORS: surface-enhanced spatially offset Raman spectroscopy; LT-UHV: low temperature-ultrahigh vacuum; tr: time-resolved; TE-CARS: tip enhanced coherent anti-Stokes Raman spectroscopy; SE-FSRS: surface enhanced femtosecond stimulated Raman spectroscopy; tr-SE-CARS: time-resolved surface enhanced CARS; tr-TERS: time-resolved TERS; SM tr-SE-CARS: single molecule tr-CARS; fs-tr-TERS: femtosecond time-resolved TERS; UPD: underpotential deposition; ATR-SERS: attenuated total reflection-SERS; EC-TERS: electrochemical TERS; NC: nanocrystal; EF: enhancement factor; CT: charge transfer. | ||
Nano-driven SERS has also opened new avenues for pushing the limits of capability, reliability, and applicability. With the ability to precisely engineer and control nanostructures at the atomic level, researchers can now tailor the plasmonic properties of SERS substrates to enhance the signal, improve reproducibility, and expand the range of analytes that can be detected.21,30 Moreover, the integration of SERS with spatial-resolved and time-resolved spectroscopic techniques, such as TERS and ultrafast spectroscopy, has enabled the investigation of chemical and biological processes with unprecedented spatial and temporal resolution.104,216,217 These developments have not only deepened our understanding of the fundamental mechanisms underlying SERS but have also paved the way for its widespread adoption in various fields.21,32
It is important to highlight that SERS is the eldest member of the broader family of nanostructure-based plasmon-enhanced spectroscopy techniques.21 By harnessing the plasmon enhancement effect of nanostructures, other spectroscopic methods such as surface-enhanced infrared absorption,218 surface-enhanced fluorescence spectroscopy,219 and surface-enhanced nonlinear spectroscopy217 (including surface-enhanced second harmonic generation,220 sum frequency generation,221 third harmonic generation,222 four wave mixing,223 and coherent anti-Stokes Raman scattering224) were also realized. These advancements clearly demonstrate the tremendous progress and boundless possibilities that the field of surface-enhanced spectroscopy has experienced and driven by the development of nanoscience and technology.
In the development of SERS, the researchers have enjoyed exchanging ideas and establishing deep cooperation via international meetings including two Faraday discussions on SERS (held in 2005,225 2017226), the biennial International Conference on Raman Spectroscopy (ICORS) (of which, SERS is the largest branch), and other SERS or TERS symposia. Readers are referred to these proceeding papers especially the records of two Faraday discussions to witness the full collision of ideas.225–227
4.4 Key methods for high sensitivity
| GSERS = G1(ω0)G2(ωR) = (|Eloc(ω0)|2|Eloc(ωR)|2)/(|E0(ω0)|2|E0(ωR)|2) ≈ |Eloc(ω0)|4/|E0(ω0)|4 | (3) |
 | ||
| Fig. 23 (a) Representative nanostructures for gap-mode SERS/TERS, including nanoparticle dimer, nanoparticle on substrate and tip on substrate (b) The two-step process of enhancement of the electromagnetic field in SERS for nanoparticle dimer. (c) The spectra of the maximum Raman EFs for nanoparticle dimers with different gap widths. | ||
The electromagnetic field surrounding such nanostructures is highly localized in specific areas referred to as SERS hotspots (Fig. 23). These hotspots can include configurations such as nanotips, interparticle nanogaps, or gaps between particles and substrates.21,77,78,209,210,228–237 The local electromagnetic field in the nanogaps between Au or Ag nanoparticle dimers with nanogaps is extremely intense and highly sensitive to the interparticle gap distance. For instance, when the gap size of a dimer composed of gold nanospheres is decreased from 10 nm to 1 nm, the SERS EF improves dramatically from 104 to 109 (Fig. 23c). Consequently, the SERS signals are significantly amplified from these hotspots, even enabling single-molecule detection.14,15,201,215,238–240
In 2006, Xia and coworkers developed a method for synthesizing monodisperse single-crystalline Ag nanocubes (Fig. 24a).244,245 García de Abajo, Liz-Marzán, and coworkers introduced a simple synthetic method for zeptomol SERS detection of non-resonant molecule by sandwiching the analyte between the tips of Au nanostar nanoparticles and a flat Au surface (Fig. 24b).246 This was accomplished by exploiting the lightning rod effect of individual nanostars and the plasmonic coupling between the nanostars and the Au film.
 | ||
| Fig. 24 SERS-active nanoparticles synthesized using bottom-up method. (a) SEM image of a Ag nanocube. Reproduced from ref. 245 with permission. Copyright 2007 American Chemical Society. (b) High-resolution STEM dark-field image of a single Au nanostar. Reproduced from ref. 246 with permission. Copyright 2009 American Chemical Society. (c) A TEM image of SiO2@Au nanoshells. Reproduced from ref. 198 with permission. Copyright 2007 American Chemical Society. (d) TEM image of a SERS-active Au nanoparticle coated with a 40 nm thick silica shell. Reproduced from ref. 247 with permission. Copyright 2003 American Chemical Society. (e) TEM image of the Au core-molecular tag-Ag shell nanoparticles. Reproduced from ref. 248 with permission. Copyright 2015 Wiley. (f) TEM image of a SiO2 shell-isolated nanoparticle. Reproduced from ref. 20 with permission. Copyright 2010 Springer Nature. (g) SEM image of gapped nanowire structures with dimers, trimers and tetramers. Reproduced from ref. 197 with permission. Copyright 2006 by The National Academy of Sciences of the USA. (h) SEM image of a dimer of Ag nanospheres. Reproduced from ref. 191 with permission. Copyright 2009 American Chemical Society. (i) High-resolution TEM image of a single-DNA-tethered Au–Ag core–shell dimer. Reproduced from ref. 249 with permission. Copyright 2013 American Chemical Society. (j) SEM image of a Langmuir–Blodgett Ag nanowire monolayer deposited on a silicon wafer. Reproduced from ref. 250 with permission. Copyright 2003 American Chemical Society. (k) SEM image of Au nanorods 3D-supercrystals. Reproduced from ref. 193 with permission. Copyright 2011 by The National Academy of Sciences of the USA. (l) TEM image showing the size distribution of the 20 nm Ag nanoparticles. Reproduced from ref. 251 with permission. Copyright 2005 American Chemical Society. | ||
In addition to nanostructures with varying morphologies, core–shell nanoparticles represent a significant category of SERS substrates due to the wide range of possible combinations of core and shell materials, different shell thicknesses, and distinct optical properties (see Fig. 25), which offers a new degree of freedom to modify the optical properties of nanostructures. These variations, particularly in dielectric indices, can lead to diverse SERS activities and novel applications, which play the a key role in overcoming the limitations of the material and morphology generality of traditional SERS.
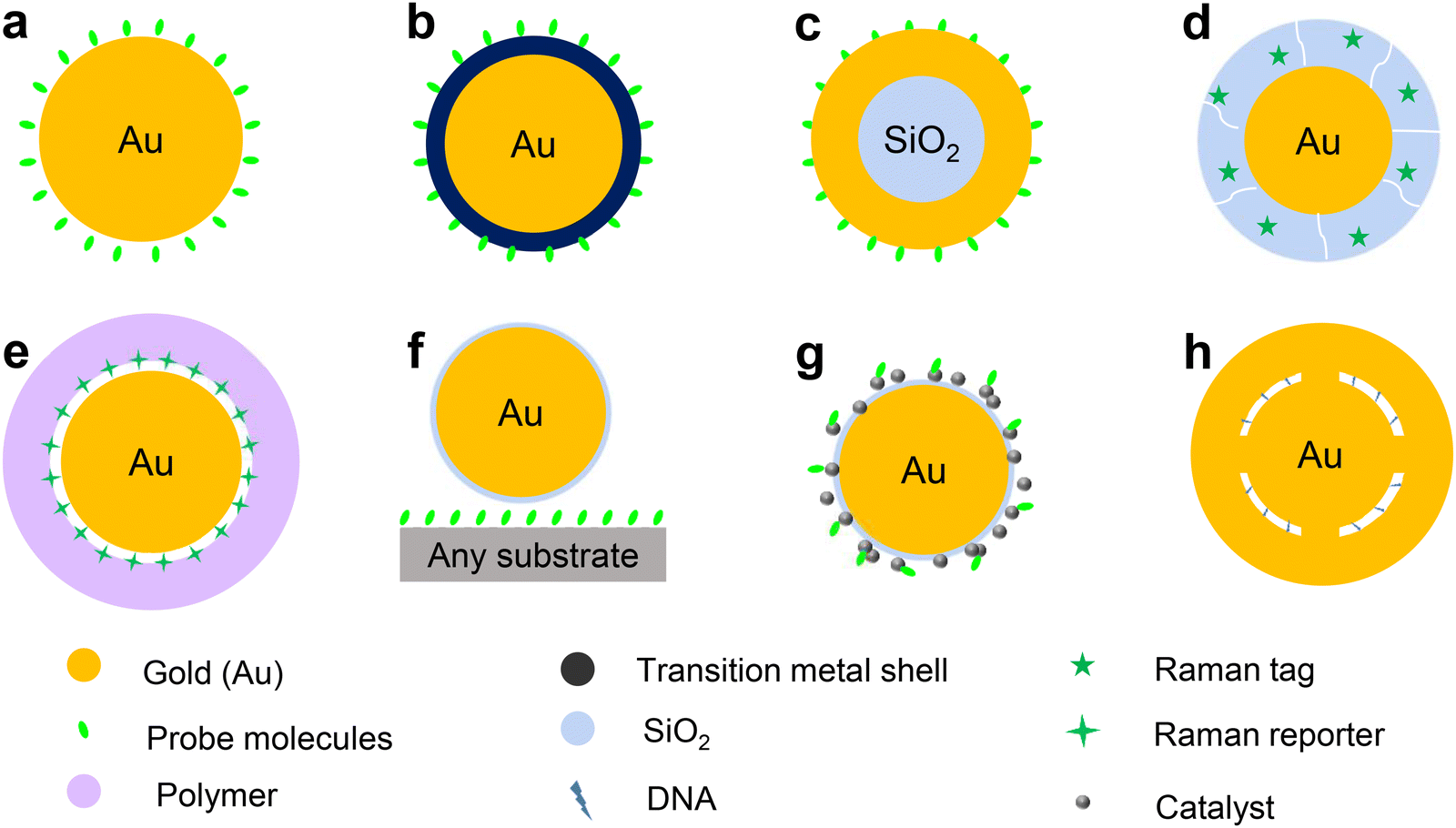 | ||
| Fig. 25 Schematic of eight different strategies used for SERS measurements of probe molecules: (a) pure Au nanoparticles developed by Natan group.187 (b) Au nanoparticle coated with an ultrathin transition metal shell by Weaver group.252 (c) Au nanoshells by Halas group. (d) Au core coated with a porous and thick silica shell and Raman tags by Nie and Natan groups.247,253 (e) Au core bonded with Raman reporters and coated with thiol-modified PEG and heterofunctional PEG (SH-PEG-COOH).254 (f) Au core coated with a dense and ultra-thin silica shell by Tian group.20 (g) Au core coated with a dense and ultra-thin silica shell and modified nano-catalysts by Li group.255 (h) DNA-anchored nanobridged nanogap particles by Suh and Nam groups.237 | ||
In 1997, Halas and colleagues pioneered the development of nanoshells, consisting of a dielectric core and a metallic shell.256,257 By manipulating the dimensions of the core and shell, the resonance wavelength of the structure can be tailored across the visible to near-infrared spectrum (Fig. 24c and 25c). Subsequently, Nordlander, Halas, and coworkers introduced the plasmon hybridization theory,258 drew from molecular orbital theory, which effectively explained and predicted the optical properties of nanoshell and other plasmonic structures.198 Nanoshells offer two key advantages as SERS substrates: (1) the plasmon resonance can be easily tuned to match the wavelength of the pump laser, and (2) nanoshells exhibit significantly larger field enhancements compared to solid metal cores due to hybridized plasmon resonances, which we will discuss in detail in Section 4.4.10.
Another breakthrough was made in 1999 by Porter and coworkers,259 which employed dielectric shell coated Au nanoparticles with embedded molecules (Raman tag) that possess a high Raman scattering cross-section in the shell as Raman labels for bioanalysis. They259 and Mirkin and coworkers260 developed a Raman tag system, known as extrinsic Raman labels (ERLs) by functionalizing Au nanoparticles with a Raman reporter molecule. The ERLs are applied in sandwich immunoassay of biomolecules, where the SERS signals do not come directly from the biological analyte, but from the tag. Therefore, the sensitivity can be greatly improved and a library of ERLs can be implemented for analytical multiplexing.261 In some cases, this Raman tagging system suffers from weak stability and easy aggregation. Although the aggregation issue can be addressed by proper surface modification,262 the competitive adsorption between the tag molecules and the antibody or DNA on the Au surfaces may pose limitations in certain applications.
To address this limitation, Natan and coworkers further developed Au@tag@SiO2 nanoparticles, incorporating Raman tags within a silica shell,253 adapting Liz-Marzán's method.263 These particles featured a 35 nm Au core and a 40 nm thick silica shell.253 The silica shell protected the Raman tags, significantly enhancing the system's stability. Additionally, antibodies or DNA used in sandwich immunoassays could be assembled on the silica shell, preventing competitive adsorption with the Raman tags. This approach enabled the preparation of a large library of unique Raman tags, facilitating the probing of multiple biological components in both in vitro and in vivo environment (Fig. 25e).264
Nie and coworkers further enhanced the detection sensitivity of the Au@tag@SiO2 system by replacing the tag with dye molecules (Fig. 24d and 25d).247 In their system, SERS enhancement was combined with the SERRS effect of the dyes, resulting in total EFs up to 1013–1014, thereby opening new possibilities for ultrahigh-sensitive bioanalysis. They also explored alternative shell materials, such as multiple ligands265 and polymers,266 to accommodate different detection scenarios. These tagged core–shell nanoparticles have been widely used in SERS bioanalysis, including in vitro diagnostics, in vivo spectroscopic detection, and image-guided cancer surgery.28 For instance, they achieved the specific Raman detection of tumor cells in vitro and in vivo by conjugating these core–shell tag nanoparticles to tumor-targeting ligands.254
Building on the core–shell tag strategy, Ren and colleagues developed a quantitative SERS method in 2015, utilizing the tag as an internal standard (IS) (Fig. 24e).248 Quantitative analysis in SERS is often challenging due to the low uniformity and reproducibility of SERS signals, which arise from the sensitivity of signal enhancement to the local structure of hot spots. In their systems, the IS molecules were embedded within the shell while the target molecules were adsorbed onto the outer surface. This configuration prevents competition for surface binding sites, ensuring that the IS signal remains unaffected by the surrounding detection environment. The IS signal effectively corrects for signal fluctuations caused by variations in the SERS substrate and detection conditions, enabling accurate quantitative SERS analysis across diverse environments (see Section 4.8 for details).
However, it should be noted that strong Raman enhancements have predominantly been obtained on Au, Ag, Cu nanoparticles or nanostructures, a limitation commonly referred to as the “material limitation of SERS”. This limitation has significantly hindered the applications of SERS in the study of important surface and interface processes of a great diversity of materials. To address this long-standing issue, the “borrowing SERS activity” strategy was developed.137–139,141,142,144 Early research primarily focused on depositing transition metal shells on rough Au or Ag electrode surfaces (see Section 3.1).
In 2002, Weaver group took the advantage of the advancement of nanoscience, introducing a synthesis nanoparticle overcoated electrode technique to replace the electrochemically roughened electrode using underpotential deposition (UPD) and redox replacement to coat Au nanoparticles with a transition metal layer, thereby creating more versatile nanoparticle-based SERS substrates.252 This work, for the first time, transformed the SERS substrate from roughened electrodes to self-assembled nanoparticle structures, moving from ill-defined structures to well-defined ones. Initially, gold nanoparticles were assembled on an indium tin oxide (ITO) surface using a coupling agent that interacted with amine groups. A monolayer of Cu was then electrochemically deposited via the UPD method, then was chemically replaced by Pt or Pd. This process resulted in a pinhole-free, single-atomic-layer thick transition metal coating on the gold nanoparticles. As a result, the strong electromagnetic fields generated by the ordered nanostructure enhance the Raman signals adsorbed on the transition metal shells. Compared with traditional rough electrodes, such a nanoparticle-based substrate is more ordered, offer better control, and exhibit higher Raman enhancement capability. However, this process was highly complex, involving surface functionalization, electrochemical UPD, and redox replacement for the coating of each monolayer.
To address the issue of the cumbersome nature of the above method, Zhang and colleagues developed a more versatile and simplified wet-chemical synthesis technique to create Au@Pt core–shell nanoparticles and collaborated with Tian group to test the SERS performance.267 By combining two approaches from Weaver group and Zhang group, Tian and colleagues have systematically developed the “borrowing SERS activity” strategy based on the Au core-transition metal shell (Au@TM, TM = Pt, Pd, Rh, Ru, Co, Ni, etc.) nanoparticles, extending SERS applications to transition metal electrode surfaces. By precisely controlling the ratio of gold nanoparticles to transition metal precursors in the solution, accurate modulation of the transition metal shell can be achieved, thereby avoiding the cumbersome procedures associated with the above UPD method.21,35 The transition metal shell comprised only a few atomic layers, with the inner Au nanoparticle serving as a plasmonic core to enhance Raman signals from molecules adsorbed on the shell.
This approach provided a strong enhancement (around 4–5 orders of magnitude), sufficient for molecular-level analysis.35 A key advantage of this method was the ability to easily adjust the shell thickness by varying the ratio of Au nanoparticles to transition metal precursors, making the process both simple and flexible.143,268 Additionally, the synthesized nanoparticles could be directly assembled on electrode surfaces without interference from coupling agents, thus facilitating their use in electrochemical studies.
It must be emphasized that although the “borrowing SERS activity” strategy has somewhat expanded material versatility, many metals, alloys, semiconductors, and polymers cannot be effectively utilized with such ultrathin coatings. Consequently, a fundamentally different approach is needed. Tian and colleagues identified this bottleneck:21,35,269 all previous SERS-active substrates inherently functioned as both signal amplifiers and sites for molecular adsorption, creating a conflict between these two roles.269 For example, it's nearly impossible to use one hand to cook while simultaneously using the other to write a paper! In other words, the traditional working mechanism of SERS is based on a contact mode, where the analyte must be directly adsorbed onto the surface of the Raman-enhancing material. However, for many studies, it is necessary to develop a non-contact mode, in which the analyte adsorbs onto the surface of the material of interest rather than directly contacting the SERS-active material, thereby avoiding signal interference.
The optimal solution is to separate these two functions: the SERS-active core should be coated by an inert layer that does not adsorb any probed molecules, allowing it to act solely as an electromagnetic field enhancer. This separation could overcome the limitations related to material and morphology, thereby improving the versatility and applicability of SERS studies. However, finding an effective method to coat a chemically and electrically inert ultrathin layer remains a significant challenge.
To address this issue, Tian group developed a new technique named shell-isolated nanoparticle-enhanced Raman spectroscopy or SHINERS by using Au nanoparticles coated with ultrathin, pinhole-free silica shells as Raman amplifiers (see Section 4.6.5).20 The key technical challenge was synthesizing an ultrathin, pinhole-free dielectric shell. They devoted over two years optimizing the synthesis of ultra-thin and dense SiO2 coated Au nanoparticles (Fig. 24f and 25f).20 Characterization of such ultra-thin silica shells on the Au nanoparticles was also very challenging at that time. Collaborated with Wang on high-resolution TEM, they demonstrated the pinhole-free shells with a thickness of ∼2 nm have been homogeneously coated on the Au nanoparticles.20
The principle of SHINERS is fundamentally different from the Au@SiO2 particles with thick and porous shells developed by Natan and Nie, as ultrathin but pinhole-free silica shells coated Au nanoparticles are employed as the Raman amplifiers. Meanwhile, the signals for the core–shell tag strategy are from the Raman markers, while the core–shell tag strategy produces signals from Raman markers, SHINERS detects signals directly from the target species, making it particularly promising for in situ monitoring of structural evolution and chemical reactions of target species (see Section 5.2.2).
However, widely used nanocatalysts are typically on a support (oxide, carbon, etc.) with a high specific surface area, and the concentration of active sites is relatively low. For oxide bulk materials, there is no strong plasmonic coupling effect between shell-isolated nanoparticles (SHINs) and supported nanocatalysts, leading to weaker enhancement (generally only 103–104). To address this issue, Li, Chen, Fu and colleagues developed a SHINERS-satellites strategy to achieve an in situ Raman study on practical catalysis,255 by assembling nanocatalysts on the surface of SHINs, forming a nanostructure with Au as the core, silica as the shell, and nanocatalysts as satellites (Fig. 25g). Such a structure ensures that the nanocatalysts are within the enhancement range of Au, and the plasmonic coupling effect generated by hotspots of the SHINs particles leads to a Raman EF 2–3 orders of magnitude higher than that of dispersed SHINs. This configuration enables in situ study of the reaction processes on the surface of the nanocatalysts. Using this SHINERS-satellite strategy, the effects of catalyst composition, size, and support on the surface reaction processes and catalyst performance were studied at the molecular level.270
Another interesting class of core–shell nanoparticles comprises the coating of SERS-active by thermosensitive polymer shells (e.g. poly-N-isopropylacrylamide, pNIPAM).271 Through changes in the hydrophilic/hydrophobic character of the polymer shell, Liz-Marzán and co-workers demonstrated the possibility of reversibly trapping and detecting molecules without affinity for the metal surface, such as 1-naphthol.271
As the SERS signals from coupled nanoparticles are highly sensitive to the interparticle distance and orientation, to achieve uniform and reproducible SERS signals essential for the quantitative analysis, precise control of particle configuration is crucial. Controlling the coupling between nanoparticles is an effective approach to generate SERS hotspots. Mirkin and colleagues fabricated controllable Au disk dimers, trimers, tetramers, and pentamers using an on-wire-lithography method (Fig. 24g).197 Xia and co-workers controllably synthesized Ag nanosphere dimers with a diameter of around 30 nm and an interparticle gap size of approximately 1.8 nm (Fig. 24h).191 Controlled assembly of nanoparticles for SERS have also been demonstrated using DNA origami,272 polymeric self-driven arrangements273 and supramolecular chemistry274 among other approaches. Nam and coworkers synthesized DNA-tethered Au nanosphere dimers overgrown with a conformal Ag nanoshell (Fig. 24i).236,249,275 These Au@Ag nanodumbbells with a ∼1 nm nanogap exhibited high EFs capable of even single-molecule detection due to the ultra-narrow gap coupled with the Ag shell.
By uniformly synthesizing core–gap–shell nanoparticles with ∼1 nm nanogaps at high yield, Nam group achieved significant SERS enhancement with a narrow EF distribution.237 For the synthesis of the Au nanobridged-nanogap particles (AuNNPs), an Au core was grafted with dye-modified DNA followed by the budding and growth of an Au shell while leaving 1-nm gap inside the particle (Fig. 25h). The DNA strands acted as surface blocking ligands to generate the nanogap, with thickness equal to the width (∼1 nm) of DNA.276 The dual role of DNA – as both a morphology-determining agent and a carrier for incorporating Raman tags into the gap – became the basis for the design and synthesis of a series of nanoparticles with controlled interior and exterior nanogaps.
Solid SERS substrates require consistent SERS signals across a sufficiently large area. Therefore, achieving moderate reproducibility requires the nanoparticles on a solid support to be arranged in a close-packed manner. Yang and coworkers prepared a monolayer of Ag nanowire thin film using the Langmuir–Blodgett method (Fig. 24j),250 while Liz-Marzán and coworkers synthesized a 3D superlattice of Au nanorods (Fig. 24k),193 achieving uniform electric field enhancement as well as high reproducibility and sensitivity for SERS detection. Lee and coworkers prepared a high-density nanoparticle thin film (Fig. 24l) by self-assembling through the Langmuir–Blodgett (LB) technique on a water surface, serving as an excellent substrate for SERS.251
 | ||
| Fig. 26 Array substrate fabricated using top-down methods for SERS. (a) SEM image of a Ag particle array produced by evaporating Ag onto sides of SiO2 posts. (b) AFM image of single-layer periodic particle arrays fabricated by NSL. (c) SEM image of closed Au nanofingers after molecule trapping. (d) SEM image of a periodic array of Ag particles, fabricated by electron beam lithography and reactive ion etching. (e) SEM image of a periodic arrays of sub-wavelength nanoholes in Au films, fabricated by focused ion beam milling. (f) SEM images of a sculpted substrate prepared using 600 nm diameter spheres. Reproduced from ref. 184, 192, 241 and 278–280 with permission. Copyright 1981 Elsevier. Copyright 2001&2010 American Chemical Society. Copyright 1998 Elsevier. Copyright 2004 American Chemical Society. Copyright 2005 Royal Society of Chemistry. | ||
In 1995, Van Duyne and coworker developed nanosphere lithography (NSL), a cost-effective, easy-to-implement, inherently parallel, high-throughput, and versatile nanofabrication technique capable of producing a wide variety of nanoparticle structures and well-ordered 2D nanoparticle arrays (Fig. 26b).278 Importantly, NSL allowed for systematic tuning of the localized surface plasmon resonance, enabling detailed studies on the relationship between nanoparticle optical properties and SERS EFs.190
Nanolithography techniques, such as electron beam lithography (EBL) and focused ion-beam (FIB) milling, provide precise control over the size, morphology, and interparticle distance of nanostructures,279–282 though achieving feature spacings below 10 nm remains challenging. Substrates produced by these methods are typically highly uniform and reproducible but offer moderate SERS enhancement. Li and colleagues employed imprint lithography to fabricate nanofinger arrays, allowing for the inexpensive duplication of identical structures over a large area (Fig. 26c).192 Kahl and colleagues fabricated an array of Ag nanoparticles using EBL technique (Fig. 26d),279 achieving a Raman EF one order of magnitude higher than that of island films. Brolo and colleagues utilized FIB technique to prepare periodic nanoholes in a Au film (Fig. 26e),280 enabling SERS detection through extraordinary optical transmission.282 Russell, Baumberg, Bartlett and coworkers fabricated sculpted SERS-active substrates by assembling a close packed monolayer of uniform polystyrene colloidal particles (ranging from 350 to 800 nm in diameter) on a Au-coated surface.241 Subsequently, they electrodeposited Au through this template to create films with controlled thickness. Selecting suitable void diameters and film thicknesses allows for customization of the optical characteristics of SERS substrates, so as to achieve maximal enhancement (Fig. 26f).241 Lipkowski and colleagues developed a Au nanowire array in which only transverse plasmons were exited, allowing for the measurement of temporal changes in the passive layer during Au dissolution in Na2S2O3 solutions unaffected by the length of the nanowire.283,284
One major drawback of nanolithographic methods based on FIB and EBL is that they are not suitable for mass fabrication. However, they generate high quality nanostructures in silicon that can be used for a nanofabrication method based on template stripping.285 In template stripping, a plasmonic metal (Au or Ag, for instance) is deposited in nanopatterned silicon substrates (prepared by methods such as EBL), and then an adhesive layer is applied to the deposited metal. Since the metal–silicon interaction is weak, the metal film can be simply separated from the silicon surface by pulling them apart. The resulting nanopatterned metal surface is ultra-smooth and mass fabrication with high reproducibility can be achieved using this method.286
A common technique for generating high-density hotspots involves the formation of solid films from colloidal nanostructures. In 2001, Nie group pioneered the creation of nanostructured thin films by depositing monodisperse Ag nanoparticles onto polycarbonate membranes.287 In 2005, Halas group fabricated highly ordered Au nanoparticle arrays with sub-10 nm spacing between adjacent nanoparticles, this is the beginning of the assembly of ordered nanostructures which exhibits high, stable, and reproducible SERS activity.288 Fabrizio group demonstrated the super-hydrophobic surface to realize a concentration factor of at least 104 in comparison with conventional flat plasmonic substrate and break the diffusion limit with super-hydrophobic delivery of molecules to plasmonic SERS structures.289 Notably, particle anisotropy and substrate coverage significantly impact the SERS enhancement of assembled solid films. Large-scale simulations by García de Abajo, Obelleiro and coworkers290 showed that, for monolayer films composed of nanorods or nanospheres, the average SERS EF increases significantly with higher particle coverage due to stronger interparticle coupling (Fig. 27a and b). In contrast, for nanostars, the EF remains nearly constant.
 | ||
| Fig. 27 (a) Simulation model of molecular coverages on a randomly arranged monolayer of Au nanospheres (GNPs) deposited on glass. (b) Density dependence of SERS performance in planar monolayers of nanoparticles with varying morphologies. Particle coverage is defined as the fraction of the area occupied by the projection of the metal along the plane normal. Figures a and b are reproduced from ref. 290 with permission. Copyright 2016 American Chemical Society. (c) 3D hotspot matrix of Ag sols generated during the water evaporation process. The sketches representing (I) a droplet of Ag sols on a hydrophobic surface, (II) the adhesive-force-constructed closely packed particles in 3D space formed in the water-evaporation process, and (III) the aggregation and deposition of particles on the dried substrate, which quenches the 3D hotspot matrix. Figure c is adapted from ref. 291 with permission. Copyright 2014 American Chemical Society. (d) Spontaneous and collective vertical alignment of GNRs (GNRs) at the oleic acid–water interface. (e) Interfacial liquid-state label-free detection method consists of three steps including mixing GNRs and targets, molecules, adding oleic acid, and conducting Raman measurements at the clearly visible interface. Figures d and e are reproduced from ref. 292 with permission. Copyright 2013 Springer Nature. | ||
These dry solid films can generate high-density hotspots, but they are often easily prone to damage from prolonged laser illumination, leading to challenges in spectral reproducibility and stability. To improve stability, Yang, Liu and coworkers proposed a dynamic SERS (D-SERS) method in 2014.291 This technique relies on the transition of nanoparticles from a wet to a dry state, with measurements taken during the colloidal phase, including the solvent and evaporation processes. They discovered that during evaporation, a 3D hotspot matrix is formed with minimal particle size polydispersity and maximum uniformity in particle spacing-features distinct from the dry state (Fig. 27c). In addition to assembling films on solid substrates, methods have been developed to control the assembly of nanoparticles at liquid interfaces, which provides a self-healing and reproducible approach for the detection of multi-analytes in the aqueous. In 2012, Edel, Kornyshev and coworkers293 successfully demonstrated the formation of densely packed nanoparticle arrays through a process of self-assembly at liquid–liquid or liquid–air interfaces. They were able to modulate the density of the nanoparticle arrays by altering the functional groups present on the nanoparticles, the pH level of the surrounding solution, and the concentration of salts within the medium. Subsequently, Kang and coworkers292 demonstrated a versatile liquid–liquid interface capable of functioning as a substrate for the orientation of colloidal Au nanorods (GNRs), as shown in Fig. 27d and e.
An alternative approach to enhancing SERS detection is to guide molecules into hotspots. Conventional methods often fail to position analytes in these regions due to contamination by impurities or ligands during the preparation process. Additionally, spatial constraints can prevent molecules from entering the tiny gaps between nanoparticles. Fang, Ren and coworkers introduced the buoyant plasmonic-particulate-based few-to-single particle-nano-sensors method, leveraging force analysis at three-phase interfaces and the Young–Laplace equation.294 This approach not only mitigates the coffee-ring effect but also enhances analyte enrichment in plasmonic sensitive sites. Inspired by transpiration in plants, where capillary action and osmotic pressure facilitate water transport, Yang and coworkers proposed the nanocapillary pumping model, which exploits capillary action and solvent evaporation to promote rapid and sensitive SERS detection.295 This method ensures the ubiquity of hotspots and the inevitability of molecular entry, resulting in comprehensive hotspot activation. The nanocapillary pumping effect demonstrates versatility in detecting various analytes with high sensitivity and maintains stable signals for 1–2 minutes during dynamic detection processes.
The rapid advancement of nanotechnology has significantly propelled the development of SERS. However, SERS has also inherited some inherent limitations of nanotechnology, particularly the instability of nanostructures, leading to unstable SERS spectra. To address these challenges, a Faraday Discussions meeting was convened in London in 2005, focusing on several critical scientific issues: (1) what is the magnitude of the chemical enhancement mechanism, if it indeed exists? (2) Are SERS hotspots present, and if so, what are their origins and compositions? (3) How should SERS EFs be defined and quantitatively assessed? (4) Can nanotechnology facilitate the fabrication of substrates? The detailed topics are listed in Table 2.

|
In the concluding remarks of the conference, Natan posited a SERS version of the “uncertainty principle”:296 the greater the enhancement, the less we can know about the atomic-level details of the substructures involved. It's primarily due to the mobility of atoms on noble metal surfaces and the mobility of analyte molecules on such surfaces, which contributes to the phenomenon of SERS ‘‘blinking’. This principle underscores the complex relationship between enhancement and structural stability in SERS.
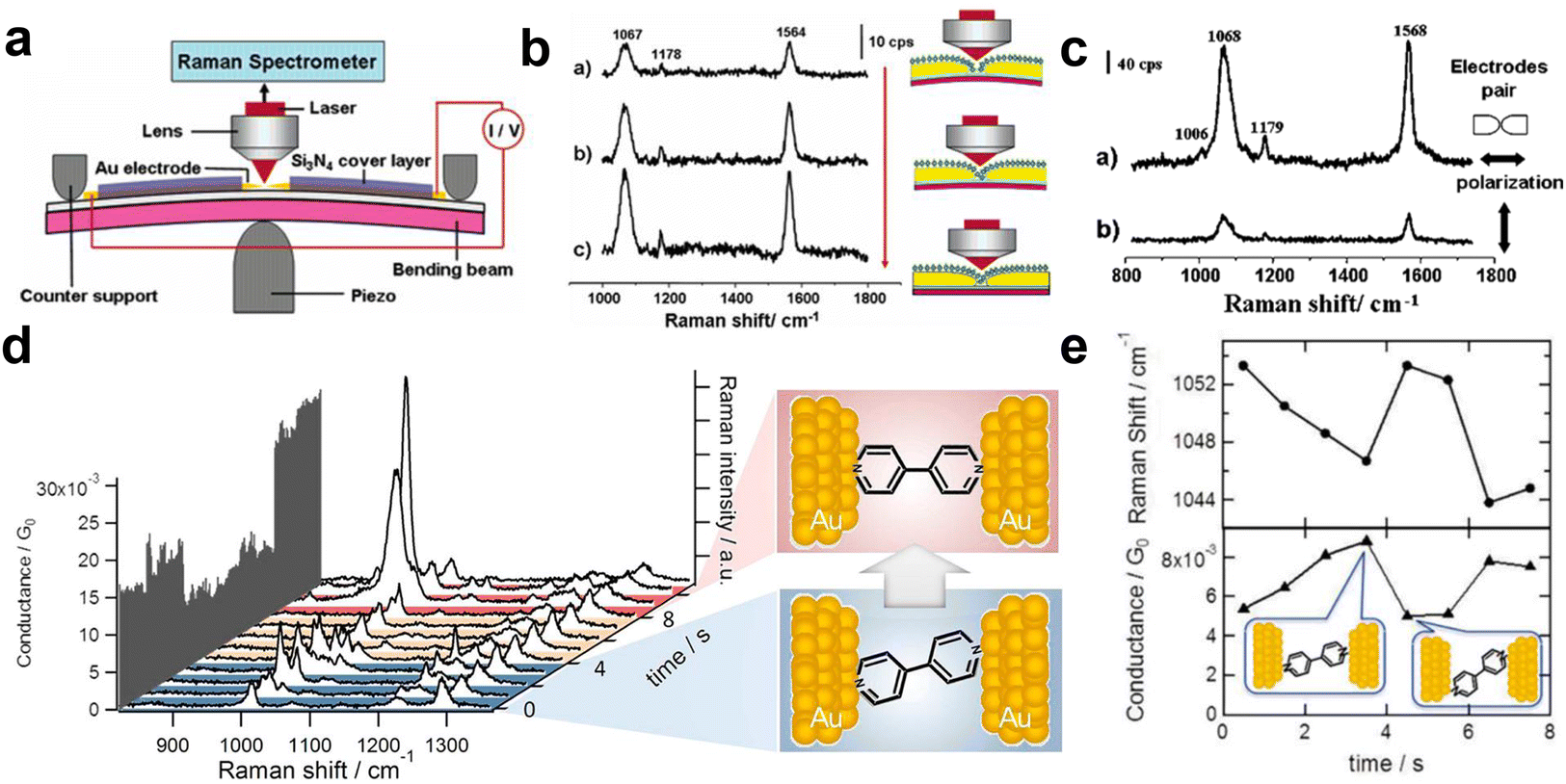 | ||
| Fig. 28 Combined mechanically controllable break junction (MCBJ) and SERS. (a) Schematic of the setup. The gap distance between two Au electrodes can be precisely controlled by the bottom piezoelectric transducer. With laser illumination, the gap also hosts as the hotspots for SERS measurement. (b) SERS of 1,4-benzenedithiol in the nanogap with the process of bending the metallic electrodes pair. The gap width 8 Å, 6 Å, 4 Å, respectively (from top to bottom panel). (c) The distinct different SERS signals when the Au electrodes pair has its axis parallel and perpendicular to the incident polarization. (d) Three types of SERS spectra of a single 4,4′-bipyridine molecule junction and possible geometrical structures. (e) The SERS signal fluctuation along with the conductance changes resulting from the junction configuration changes. Figures are reproduced from ref. 297 and 298 with permission. Copyright 2006 & 2012 American Chemical Society. | ||
In 2013, Murakoshi, Kiguchi and colleagues used the MCBJ technique to simultaneously measure the conductivity and the SERS of a single 4,4′-bipyridine molecular junction in solution at room temperature (Fig. 28d and e).298 The significant changes in SERS intensity and Raman vibrational band selectivity are related to the current fluctuations during the fracture and formation of single-molecule junctions. Through the mode switching between b1 and b2 in SERS, the authors observed the movement of 4,4′-bipyridine between vertical and tilted configurations in the nanogap inside the Au electrode pair. This study indicates that individual molecules at metal gaps generate Raman spectra, which contain detailed information on molecular electronic and geometric structure.298
The MCBJ technique allows for precise and stable adjustment of the separation between the electrodes on a chip by using a piezoelectric transducer, ranging from a few angströms to several nanometers with a resolution of one angström. Therefore, this method also makes it possible to monitor chemical reactions at the single-molecule level inside the nanogap. In 2018, Hong, Yang and colleagues investigated the charge transport through a BDT molecular junction with a combined MCBJ and SERS approach.299 They observed Raman characteristic peaks of S–S bonds in different conductance states of the BDT molecule, revealing that the BDT molecules had undergone dimerization. This finding demonstrated that the low conductance state originated from dimerized-BDT, thereby resolving a long-standing controversy regarding the relationship between BDT molecular junction configurations and their multiple conductance states.299
In 2016, the first work on selective adsorption sites on the single-molecule scale was carried out using this technique by Kiguchi group.300 The SERS signals distinguished three substates of 1,4-benzenedithiol generated by different adsorption sites, corresponding to the bridge, hollow, and atop sites, respectively. This study reveals that the SERS intensity depends on the strength of the molecule–metal interaction, showing the interdependence between the optical and electronic properties of single-molecule junctions.300 On this basis, they reported in 2019 that the modulation and monitoring of aminobenzenethiol and benzenedithiol single-molecule adsorption sites was achieved by means of regulating the applied bias voltage, which provides new perspectives for studying and controlling single-molecule circuits.301
The radiative de-excitation rate of surface-enhanced fluorescence (SEF) has been assumed to be smaller than the vibrational decay rate of the electronic excited state as shown in Fig. 29a and b. However, the radiative de-excitation rate for a molecule inside the hotspots is largely enhanced by plasmon resonance and can become greater than the vibrational decay rate.303 Thus, in this situation SEF light can be emitted from a vibrational excited state as shown in Fig. 29c, resulting in broadband light emitted in SEF which appears as a background in SERS and TERS spectra. This type of SEF process means the breakdown of Kasha's rule, in which an excited molecule is supposed to emit fluorescence from the vibrational ground state.304 This type of SEF is useful for investigating ultrafast electronic dynamics in the electronic excited state of molecules inside the hotspots.
 | ||
| Fig. 29 Simplified Jablonski diagrams depicting the fluorescence processes in the three possible situations: (a) Standard fluorescence; (b) SEF in which the vibrational decay rate is faster than the SEF rate; and (c) SEF in which the vibrational decay rate is slower than the SEF rate. Reprinted with permission from ref. 303 Copyright 2007 American Chemical Society. | ||
In free space, the length of oscillation of the electromagnetic fields in an optical cycle is much larger than that of a molecule (and thus the extension of the electronic transitions). This assumption is referred to as the long wavelength approximation, in which the excitation selection rule does not depend on the spatial extension and structure of light. However, the volume of the electromagnetic field mode can be compressed to nanometer-size within hotspots. Within the hotspots, the gradient of the electric field escalates to dimensions commensurate with the electronic transition scale of a molecule. This escalation precipitates the failure of the long-wavelength approximation.305 Hence, the excitation of multi-poles, such as a quadrupole, becomes non-negligible as shown in Fig. 30.306 In such a situation, the excitation selection rule needs to be modified. This modification was investigated through the observation of forbidden transition modes, such as the activation of certain IR modes in SERS and TERS spectra. For example, Murakoshi and colleagues reported the observation of the dipole forbidden electronic transition through the SERRS of a carbon nanotube located in a plasmonic nanogap. They presented evidence suggesting the field-gradient effect would affect SERS.305
 | ||
| Fig. 30 Selection-rule breakdown in plasmon-induced electronic excitation of an isolated single-walled carbon nanotube (SWNT). (a) Illustration of an SWNT lying in the nanogap of a dimer and the enhanced field polarization. (b) SEM images of a well-defined Au nanodimers. (c) SERS spectra showing the radial-breathing (RBM) mode of an isolated SWNT in the nanogap. The applied-field polarization is parallel to the long axis of the nanodimer. (d) Typical resonance Raman scattering spectrum of bundled SWNTs dispersed on flat glass. (e) The forbidden electronic transitions for resonance SERS spectroscopy. The Δn = ±2 transition has a nonzero quadrupole transition moment Θ, which couples to the field gradient to give resonance SERS. Figures are reproduced from ref. 305 and 306 with permission. Copyright 2013 Springer Nature. | ||
In Section 4.5.3, we will address the achievement of sub-nanometric resolution in TERS thanks to the use of atomic-scale localization of electromagnetic fields. Such localization also affects Raman selection rules, by activating forbidden vibrational modes for homogeneous illumination. An example of such activation of Raman lines by atomic-scale hot spots is shown in Fig. 38d.
 | (4) |
The GSERS and (αρσ)mn terms represent the contributions from electromagnetic fields and from adsorbed molecules, respectively, while QTmT0 represent instrumental contributions. Accordingly, SERS sensitivity can be improved from different aspects including increasing the SERS activity of the substrates (electromagnetic fields, the GSERS term), utilizing molecules with large Raman cross-sections including those with resonance Raman effects (the (αρσ)mn term), as well as continuously improving the collection and detection efficiency of instruments, etc. Therefore, integrating experiments, theory, and instrumentation into a cohesive trinity is essential and even decisive for making breakthroughs. A comprehensive and systematic consideration of the entire SERS process is necessary. This holistic approach extends beyond merely selecting appropriate nanostructures, molecules, and excitation wavelengths. It necessitates the rational optimization of efficiency at each stage of the instrumental system, encompassing excitation, collection, grating dispersion and detector detection.
For example, in the STM-Raman setup as we explain in the following section, optical fibers were arranged in a circular shape to better collect scattered light and improve sensitivity. In early confocal Raman systems as well as the first EC-TERS systems, instrument improvements were crucial for achieving high sensitivities.
It is important to clarify that physical enhancement based on surface plasmons can increase the Raman scattering cross-section by a factor of 106 to 108 (the GSERS) term in eqn (4) playing a dominant role.35,307,308 In contrast, chemical enhancement typically results in an increase of 1–3 orders of magnitude (the (αρσ)mn) term in eqn (4), even higher SERS EF were predicted.309–311 Physical enhancement is primarily responsible for the enhancement of the SERS effect; without the plasmonic enhancement mechanism, the establishment and advancement of the SERS field would not have occurred.
However, chemical enhancement should not be overlooked. A comprehensive understanding and effective utilization of the SERS effect necessitates consideration of both physical and chemical enhancement mechanisms. Moreover, the study of chemical enhancement mechanisms has practical applications. From a surface chemistry perspective, comprehending the interaction between molecules and surfaces via SERS requires knowledge of chemical enhancement mechanisms. Crucially, for applying SERS in diverse analytical fields, particularly in quantitative or semi-quantitative analysis systems aimed at commercialization, a clear grasp of chemical enhancement mechanisms is indispensable. Hence, researchers continue to make sustained efforts and contributions in exploring these chemical mechanisms, including unified mechanisms that incorporate both physical and chemical enhancement.312–314
The utilization of the chemical enhancement is intrinsically linked to a profound understanding of the CE mechanism in SERS. During this period, significant theoretical advancements were also made on the basis of the first-principle ab initio quantum chemical calculations. It is summarized that the CE mechanism mainly involves three scenarios contributing to the total polarizability part of  in eqn (4):166,310,315 (1) enhancement arising from chemical bonding interactions in the ground state between the molecule and the nanoparticle, which occur independently of any excitations within the nanoparticle-molecule system. (2) Resonance Raman enhancement, where the excitation wavelength aligns with an internal transition of the molecule. (3) CT resonance Raman enhancement, characterized by the excitation wavelength being resonant with charge-transfer transitions between the metal and the molecule.
in eqn (4):166,310,315 (1) enhancement arising from chemical bonding interactions in the ground state between the molecule and the nanoparticle, which occur independently of any excitations within the nanoparticle-molecule system. (2) Resonance Raman enhancement, where the excitation wavelength aligns with an internal transition of the molecule. (3) CT resonance Raman enhancement, characterized by the excitation wavelength being resonant with charge-transfer transitions between the metal and the molecule.
The non-resonant chemical mechanism is challenging to detect experimentally due to its contribution to the overall SERS EF being strongly dependent on physical or chemical adsorption of probe molecules, despite ongoing experimental endeavors to quantify it. Theoretical investigations are expected to play a crucial role in clarifying this mechanism, as it is likely shaped by the local molecular environment.310 This environment can be effectively modeled using small metal clusters, which allow for the representation of the localization of chemical bonds as determined through density functional theory calculations. Consequently, numerous electronic structure investigations have been conducted on the impact of small metal clusters on the Raman characteristics of small molecules.315–317 Experimentally, a significant chemical EF was observed in the specific wagging vibrational mode of benzyl radical and aniline adsorbed on Ag electrodes and Ag colloids.311,318
The resonance Raman effect is observed when the wavelength of the incident laser light corresponds to the electronic absorption energy of the sample being analyzed, significantly boosting the likelihood of Raman scattering by several orders of magnitude. When a molecule is adsorbed onto the surface of a plasmonic-active nanostructure, SERRS can be utilized for detection.14,24,319,320 The added signal amplification stemming from this molecular electronic resonance renders SERRS an exceptionally sensitive vibrational spectroscopic method. Although SERRS can achieve higher sensitivity, resonance Raman imposes limitations on the universality of molecules as well as laser excitation wavelengths and may introduce significant fluorescence background.
It should be noticed that two distinct types of electric fields, the optical electromagnetic field and the electrochemical electrostatic field, co-exist in electrochemical interfaces systems with various nanostructures (Fig. 31).166 Both chemical and physical enhancements can be influenced to some extent by applying an electrode potential, making EC-SERS one of the most complex systems in SERS. Significant efforts have been made to thoroughly understand SERS and analyze EC-SERS spectra based on chemical and physical enhancement mechanisms, providing valuable insights into the mechanisms of electrochemical adsorption and reactions.
 | ||
| Fig. 31 (a) Schematic diagrams of the photon-driven CT from a metal electrode to an adsorbed molecule in the EC-SERS system. Left: The conceptual model of the energy levels changed with the electrode potential in the CT process. Right: The relevant energy states involved in the electronic levels and the vibrational levels in the CT process. Right bottom: The corresponding SERS intensity-potential profile and Vi denotes the applied potential. Reproduced from ref. 166 with permission. Copyright 2008 Royal Society of Chemistry. (b) Displaced harmonic potential energy curves of both electronic ground state and CT state. (c) The relative Raman intensities of the ring breathing mode and triangle deformation mode of pyridine binding to Ag dimer (Ag2). Figure c is reproduced from ref. 322 with permission. Copyright 2004 Elsevier. (d) The potential-intensity profile of the ring breath mode for pyridine adsorbed on Ag electrodes reproduced from DFT calculations and Raman scattering theory. Reproduced from ref. 323 with permission. Copyright 2005 AIP publishing. (e) Pyridine–Ag20 complex model. (f) Simulated Raman spectrum and (g) CT resonant-like Raman spectrum from pyridine–Ag20 complex. Figures (e)–(g) are reproduced from ref. 324 with permission. Copyright 2006 American Chemical Society. | ||
The intensity of SERS observed in various EC-SERS systems is notably influenced by both the applied potential and the nature of the metal substrate. This observation provides compelling evidence for a synergistic relationship between CT and EM enhancement. As depicted in Fig. 31a, HOMO and LUMO refer to the highest occupied molecular orbital and the lowest unoccupied molecular orbital of the adsorbed species, respectively.166 Additionally, Ψg(Vi) represents the molecular adsorption ground state, while ΨCT(Vi) denotes the photon-induced CT excited state, which is generated by the transition from a filled metallic level to the LUMO of the adsorbed molecule at an applied potential, Vi. The right part of Fig. 31a presents a conceptual picture relevant to not only the energy level but also the vibronic energy states along the vibrational coordinates.166 It displays much more clearly the change of the surface electronic ground state and corresponding vibrational levels of the adsorbate with electrode potential. Please note that each energy level represents the total energy of the combined system of the adsorbed molecule and interacting metal electrode (molecule/metal) in the excited or ground electronic state that can be influenced by applied potentials.
It is assumed that the energy position of the photon-driven CT state remains constant regardless of the applied potential. The observed SERS signal initially increases and then decreases as the electrode potential is adjusted to more negative values. This variation in SERS intensity can be attributed to the photon-driven CT from the metal to the molecule, as explained by the energy levels or states associated with interfacial energy level alignment. As illustrated on the left side of the figure (energy level concept), at the applied potential V1, the conditions are inadequate to generate photon-driven CT states on the surface with an excitation energy of hν. Consequently, the observed changes in relative SERS intensity are primarily governed by bonding effects. When the potential is adjusted to V2, the increase in the Fermi level of the metal electrode aligns the excitation photon energy with the required energy for photon-driven CT, resulting in resonance-like Raman scattering and a pronounced enhancement in the SERS intensity of the associated vibrational modes. However, if the potential is further shifted to V3 in the negative direction, the excitation energy no longer falls within the optimal resonance conditions, leading to a reduction in the contribution of the CT states to the overall SERS signal.
These early studies were based mainly on phenomenological theoretical models to consider the mechanism of CT enhancement. In the early 2000s, the theoretical models were investigated on the basis of DFT calculations combining with the molecule–metallic cluster model or the first-principles periodic slab model.315 By combining the DFT calculations and Raman scattering theory, the CT enhancement mechanism was found to be closely associated with the displacement of the minima of the excited state with respect to the ground states along a given normal coordinate with the totally symmetric representation (Fig. 31b).310 When the effect of applied potentials was considered to be equivalent to tuning the Fermi level of metal electrodes, the resonance-like CT enhancement mechanism was realized to match the energy levels from excitation laser wavelength through applied potentials at electrochemical interfaces (Fig. 31c).322 In this case, the ring breathing mode of pyridine interacting with a silver cluster displayed a stronger Raman signal than that of the triangle deformation mode when the photon-driven CT mechanism occurs.
For pyridine adsorbed on Ag electrodes, the CT mechanism combining with the chemical adsorption interaction describes the potential-dependent Raman intensity profile of the ring breathing mode (Fig. 31d) well even using a small metallic cluster.323,325 Such a simple model is reasonable to describe the interfacial chemisorption bond from the basic bonding principles, i.e., energy similarity, symmetry matching, and maximum overlap of the interacting orbitals. Furthermore, the potential energy surfaces of the photon-driven CT excited state should be predicted well based on the TD-DFT approach for pyridine adsorbed on silver surfaces.
The resonant-like Raman effect needs to accurately consider the interfacial energy state alignment. Schatz and coworkers made significant contribution to understand the chemical enhancement mechanism.326,327 Their TD-DFT approach can predict the significant enhancement of several CT excited states contributing to the SERS intensities of pyridine–Ag20 to mimicking the pyridine adsorbed on Ag colloids. The use of large metallic clusters can improve the utility of the calculations in describing the characters of interfacial chemical bonds at different surface sites. Jensen and colleagues found that the simulated SERS spectra still strongly depend on the specific chemisorption bond for pyridine adsorbed on surface active sites (Fig. 31e and f).324 The interfacial energy level alignment also directly influences the relative Raman intensities of pyridine–Ag20 (Fig. 31g).324
The idea further was extended by Jensen and colleagues to investigate the molecule–surface chemical coupling SERS effect from electronic structures of meta- and para-substituted pyridines interacting with a Ag20 cluster.316 They found that the magnitude of chemical enhancement is governed to a large extent by the energy difference between the highest occupied energy level (HOMO) of the metal and the lowest unoccupied energy level (LUMO) of the molecule. The trend was verified by considering substituted benzenethiols, small molecules, and silver clusters of varying sizes. The findings provide the framework for designing new molecules which exhibit high chemical enhancements.
The experimental and theoretical research by Aranda, Otero, Santoro and colleagues328,329 have led to the development of a computational model for electrochemical surface-enhanced Raman scattering (EC-SERS). In this model, the surface excess charge induced by the electrode potential (Vel) was introduced by applying an external electric field to a series of clusters [Agn]q, specifically with (n = 20, q) values of (19, ±1) or (n = 20, 0), onto which a pyridine molecule is adsorbed (Fig. 32a). They estimated the EFs involved in SERS mechanisms, including chemical or non-resonant, and resonance Raman with bright states of the adsorbate, charge-transfer (CT) states, and plasmon-like excitations on the metal cluster (Fig. 32b). Employing DFT and TD-DFT calculations, the metal–molecule complexes were categorized based on the partial charge of the adsorbate. The primary properties influenced by Vel were analyzed concurrently, facilitated by vibronic resonance Raman calculations that identified shifts in vibrational wavenumbers and relative intensities.
 | ||
| Fig. 32 Electrochemical SERS enhancement mechanism of pyridine on silver electrodes. (a) Molecular model for SERS of pyridine that is adsorbed on Ag electrodes in surface S-complexes. (b) The charges on the adsorption site are included explicitly in the metal cluster when considered, while an external electric field applied along the Z-direction reproduces the effect of the nearby surface charges. (c) Experimental SERS and (d) calculated resonance Raman spectra for the bright states for matched values of Vel and qPy. Notice that each panel has a different Y-axis scale. Stick lines were convoluted with a Lorentzian of HWHM = 5 cm−1. All E in 10−4 a.u. CAM-B3LYP-D3/6-31+G(2d,2p)/LANL2DZ level of theory. Figures are adapted from ref. 328 with permission. Copyright 2022 American Chemical Society. | ||
By inspecting detailed experimental EC-SERS data (Fig. 32c), they tested the mentioned above model for pyridine adsorbed on Ag electrode. They noted that the electrode potential Vel has a remarkable effect on the simulated Raman spectra. The findings successfully replicated the majority of experimental observations, while also offering a critical evaluation of the approach's limitations (Fig. 32d). The explicit consideration of surface charges is essential for EC-SERS modeling, and the highest calculated EFs, ranging from 107 to 108, were attributed to the coupling between bright local excitations of the metal cluster and CT states. These results indicated that the importance of nonadiabatic effects in SERS and the capabilities of EC-SERS as the theoretical approach to study the coupling between the CT excited-state and plasmon-like states by tuning applied potentials.
To investigate the CT mechanism has opened the question of whether the possibility to induce chemical transformations by plasmonic hot carriers (hot electrons and hot holes). This can be rationalized as the reactive analogue of the well-established concept of optical hot spots to the chemical hot spots, which have drawn a great deal of attention to plasmonic nanostructures for their ability to tune the photochemical reaction efficiency and selectivity.330–335 Thus it should be careful sometimes the observed SERS signals were products of photochemical reactions rather than the initially added probe molecules, such as p-aminothiophenol,310,336 adenine,337 and 8-bromoadenine.338
The photon-driven CT process was discovered that it can lead to profound changes in Raman spectra, in addition to changes of vibrational wavenumbers and relative intensities. In the initial stage of SERS study, these changes were often considered as negative examples of the photodecomposition or thermal decomposition caused by the focused lasers. Recently, it was found that these changes in spectra resulted from the selective chemical reactions mediated by the photon-driven CT process, namely plasmon-mediated chemical reactions (PMCRs).332,339 In such process, reactions happen on the charged state to form targeted chemicals under mild conditions, but during which usually a longer lifetime of the charged state is needed. More detailed discussions are shown in relevant reviews.332,340,341
Now the chemical reactivity of plasmonic systems is deeply influenced by the dynamics of photons, plasmon-polaritons, carriers, phonons, and molecular states. These degrees of freedom can affect the reaction rates, the product selectivity, or the spatial localization of a chemical reaction in nanostructures.330 It is important to tailor the generation of non-thermal carriers and phonons in metallic nanostructures and their dissipation relaxation is shown as a promise to understand and exploit thermal photocatalysis at the nanoscale. This is useful to control these processes for the rational design of solar nanometric photocatalysts.
For nanoshells, the theory provided an intuitive understanding of both the tunability and their large field enhancements. In Fig. 33a–c, it showed that the hybridized plasmon modes arise due to a coupling between the plasmon modes on the inner and outer surfaces of the shell.198 For a thin shell, the interaction between the plasmons is very large, leading to a strong tunable redshifted “bonding” plasmon. The wave function of this plasmon has the charge density associated with the cavity and the inner and outer surfaces aligned, resulting in a larger charge density than for a solid sphere, corresponding to large field enhancements. More efficient SERS substrates can be constructed using nanoshells as building blocks for composite structures. The prime example is the nanoshell dimer,343 providing larger field enhancements and SERS sensitivities in orders of magnitude. The large field enhancements explicitly showed in Fig. 33d and e. The nanoshell dimer exhibits large field enhancements in the junction, and also illustrates that the field enhancements can be made even larger by texturing the surface of the nanoshells, resulting in a much larger hot spot volume.
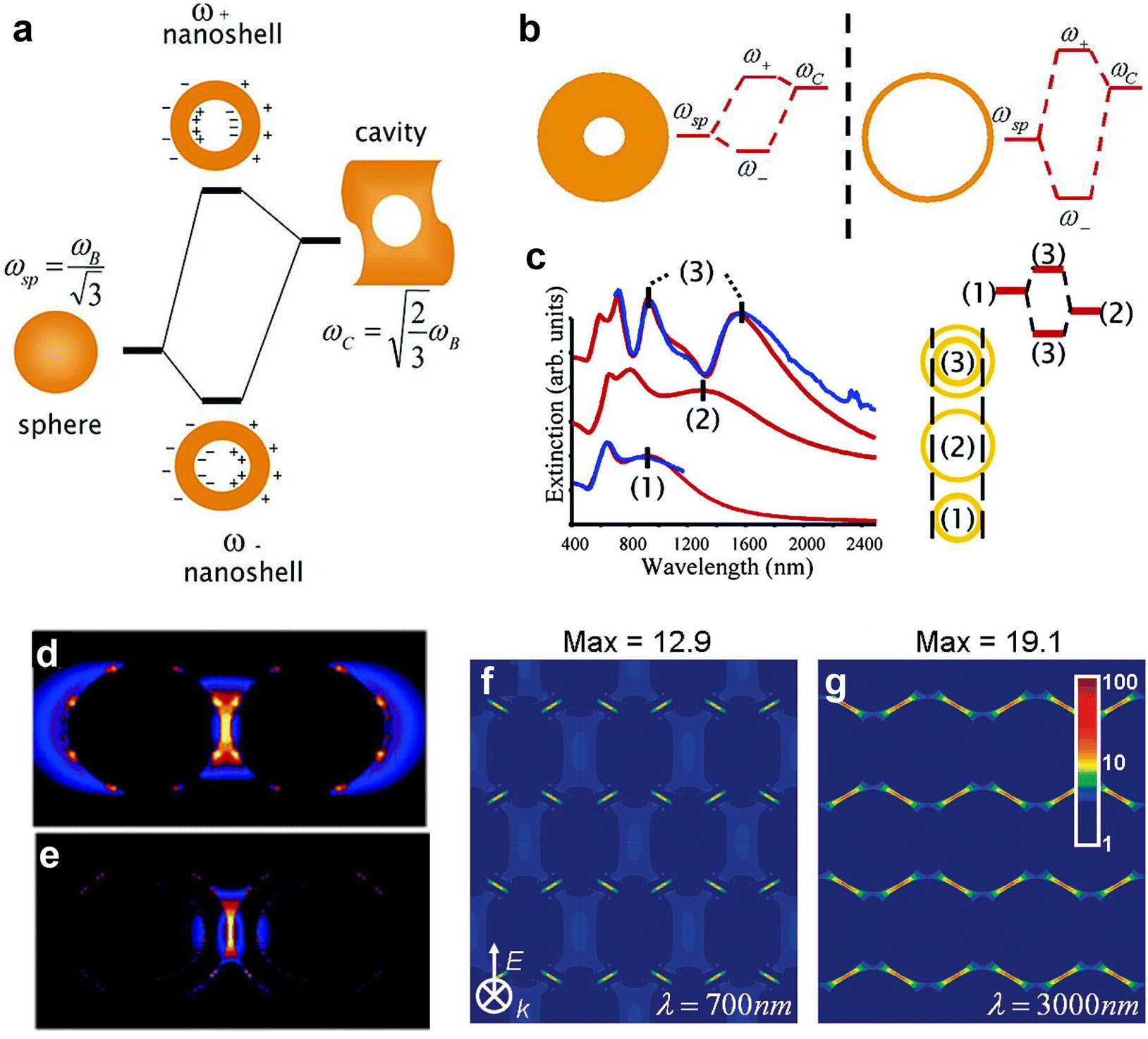 | ||
| Fig. 33 (a) Energy level diagrams depicting plasmon hybridization in metal nanoshells resulting from interacting sphere and cavity plasmons with the two hybridized plasmon modes being an anti-symmetric or “antibonding” plasmon resonance (ω+) and a symmetric or “bonding” plasmon resonance (ω−). (b) Illustrating the dependence of nanoshell plasmon energies on the strength of the interaction between the sphere and cavity plasmons, determined by the thickness of the metallic shell. (c) Experimental (blue) and theoretical (red) extinction spectra for concentric nanoshells (3) compared to the inner (1) and outer (2) nanoshell plasmon resonances. (d) Electromagnetic near field enhancement at the excitation laser's wavelength (633 nm) for an adjacent nanosphere pair with interparticle axis parallel to the incident polarization, and (e) a nanoshell dimer with interparticle axis parallel to the incident polarization. (f) FDTD simulation results of the local electric field enhancement for a hexagonally close-packed (HCP) Au nanoshell array, at wavelength of 700 nm and (g) 3000 nm, respectively. Figures are reproduced from ref. 198, 258, 343 and 344 with permissions. Copyright 2003 AAAS. Copyright 2005 & 2007 & 2008 American Chemical Society. | ||
Based on the hybridization theory, it's possible to design nanostructures with continuously tunable plasmon resonance from the visible to the mid-infrared. Another highly useful example is the hexagonally close-packed (HCP) nanoshell array.344 As illustrated in Fig. 33f and g, which shows the optical properties of this structure, the difference in hybridization between the dipolar and the quadrupolar nanoshell resonances is shown. Here the quadrupolar plasmon modes in the visible/near IR interact only weakly, resulting in a nondispersive plasmon mode similar to the d band in a solid. In contrast, the dipolar plasmons interact very strongly, resulting in a broad, strongly redshifted mode in the mid infrared. Both of those modes provide large field enhancements: the quadrupolar modes, at 700 nm, and the dipolar modes, at 3.0 microns. What is remarkable about these field enhancements is that they appear at the same spatial region of the arrays, enabling the possibility of doing both surface-enhanced Raman and IR absorption spectroscopy (SEIRA) on the same molecules on the substrate. Combined SERS and SEIRA provides a much more comprehensive chemical fingerprint of an analyte molecule for accurate chemical identification and this dual functionality has been used extensively for chemical fingerprinting applications.345
4.5 Key methods for high spatial and temporal resolution
 | ||
| Fig. 34 The combined STM and SERS experiments. (a) Schematics of the combined STM with Raman instrument based on the remote operation of optical fiber, which enables the simultaneous measurements of STM and SERS of an electrochemical cell. (b) STM images of a HOPG surface after the stripping of Ag. (c) A SERS spectrum of thiourea adsorbed on the surface of (b). Figures are adapted from ref. 347 and 348 Copyright 2007 Wiley. Copyright 1996 Elsevier. | ||
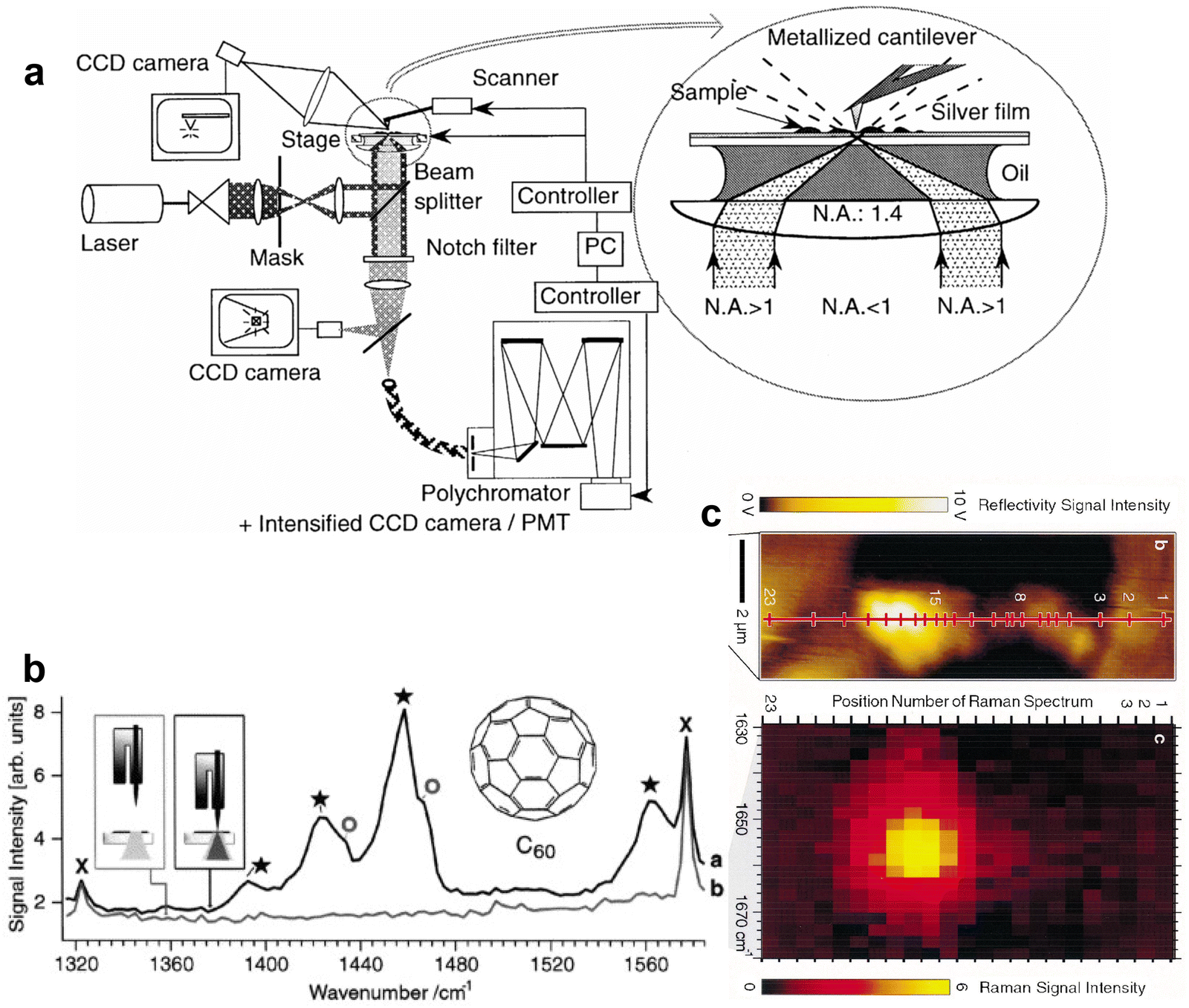 | ||
| Fig. 35 The first demonstration of TERS. (a) The configuration of the TERS setup developed based on a near-field Raman microscope. (b) The demonstration of TERS based on a combined Raman and AFM instrument using a metallic AFM tip in shear-force mode. The Raman spectra of C60 is shown with an Ag tip in contact with the sample (trace a), and tip retracted (trace b). Circles mark C60 normal modes, stars mark C60 modes due to CT with an Au tip. The crosses mark Raman bands of the immersion oil. (c) Demonstration of the spatially resolved Raman measurement by TERS, with a standard silver island coated AFM cantilever tip. The Raman spectra of an ultra-flat homogeneous brilliant cresyl blue (BCB) sample were measured along the line in top panel at the marked positions, and the position dependent Raman spectra are displayed in the bottom panel. No additional cavity/gap-mode effect was required. Note that the silver film was not present in the examples in (b) and (c). And a tuning-force setup was used in (b). Figures are adapted from ref. 16 and 18 with permissions. Copyright 2000 Elsevier. | ||
Later on, by optimizing the tip structure, excitation and collection conditions, and controlling the tip–substrate coupling distance, researchers could confirm the extremely high spatial resolution of TERS,349–351 reaching a level of approximately 10 nm. At that time, whether the spatial resolution limit of TERS could break the 1 nm barrier to achieve single-molecule resolution was an intriguing but unresolved question. Theoretical predictions suggested that due to quantum tunneling and non-local effects of electrons,352,353 the confinement of light cannot be reduced to 1 nm at least not in the preferred gap-mode configuration. Additionally, the mobility of molecules on the surface and the thermal drift of the instrument also hindered further improvements in TERS spatial resolution.
 | ||
Fig. 36 (a) Schematic of single-molecule TERS at LT-UHV condition. (b) The typical Raman spectrum taken from different positions of molecules. (c) Height profile of a line trace in the inset STM topograph. (d) TERS intensity profile of the same line trace for the inset Raman map associated with the 817![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−1 Raman peak. Figures are reproduced from ref. 354 with permission. Copyright 2013 Springer Nature. cm−1 Raman peak. Figures are reproduced from ref. 354 with permission. Copyright 2013 Springer Nature. | ||
This experiment also triggered the reconsideration of plasmonic optical processes at atomic scales. Subsequently, Aizpurua, Sánchez-Portal and coworkers reported detailed time-dependent density functional theory calculations of the optical response of metal nanoparticles with atomic-scale features, and they showed that several atomistic protrusions could confine the optical field to sub-nanometer or even atomic scales356,357 as shown in Fig. 37a–c. This was spurred by their collaboration with Baumberg and coworkers, who demonstrated experimentally that a picocavity can be formed by a few adatoms in a nanoparticle-on-mirror (NPoM) configuration,358 boosting new Raman lines, as shown in Fig. 38.
 | ||
| Fig. 37 (a) Schematics of atomistic near field generated by the atomistic protrusions at metallic interfaces in clusters. (b) Calculated electromagnetic field enhancement of a Na dimer with a gap distance of 0.6 nm, with tip-to-facet configuration and (c) tip-to-tip configuration. Maximum enhancement value of ∼40 is colored in red. (d) Schematics of the molecular optomechanical description of SERS process. Figures are adapted from ref. 356 and 359 with permissions. Copyright 2015 & 2016 American Chemical Society. | ||
 | ||
| Fig. 38 (a) The schematics of picocavity formed by adatoms on Au nanoparticles. (b) Near field enhancement in the cavity. (c) Time-series anti-Stokes and Stokes SERS in the picocavity. (d) Comparison of two experimental spectra (bottom) with DFT simulated spectra (top) under the assumption of atomic-scale field confinement (inset shows geometries, red sphere shows field ocalization for simulations). Figures are reproduced from ref. 358 with permission. Copyright 2016 AAAS. | ||
Since then, the importance of atomic-scale structures on nanostructure surfaces has gained increasing attention in the SERS and plasmonic community. At the same time, a different approach was proposed based on the assumption that even without any external fields the energy levels and consequently the vibrations must change with respect to the positioning of the tip. Theoretical modelling of Raman spectra of a silver atom scanning over an adenine molecule at different heights clearly showed this chemical effect.360 Deckert and colleagues showed in classical electromagnetic models that atomic scale protrusions on TERS tips exist and can act as the previously mentioned enhancing sites, which are further responsible for the chemically induced band shifts and leading to a higher resolution.361
This atomistic-scale lightning rod effect leads to highly intense and localized hot spots, which differs from the general understanding of localization of plasmonic fields. Although the calculation model was simplified as a Na380 dimer with a size of 2 nm, which is much smaller than commonly used nanostructures, those results solidly indicate that the presence of atomic-scale structures at plasmonic interfaces,356 additionally enhancing the underlying plasmonic field, appears as a new arena of research in extreme filed localization and attracts tremendous attention to investigations of plasmon–matter interactions at the atomic scale. More recently, another transient phenomenon observed in all SERS measurements was also understood by the Aizpurua and Baumberg teams, from an entire patch of the surface atomic monolayer lifting, which generates intense broadband light modulated by plasmonic resonances, and known as flares.362,363
Shortly after, Galland, Kippenberg and colleagues364 and Aizpurua, Schmidt and colleagues359 independently developed an optomechanical model of SERS and provided a quantum electrodynamics (QED) description of the coherent interaction of plasmons and molecular vibrations (Fig. 37d). They showed that the optomechanical interaction between the molecules and extremely trapped optical fields can explain the high spatial resolution in TERS as well as the nonlinear behavior of the Raman signal with laser intensity. Other important effects from these optomechanical interactions are also seen such as SERS peak shifts.365 The dynamics of the formation and destruction of these adatoms under laser illumination leads to fluctuations in SERS intensities that can be observed when using high-speed acquisition rates.366,367 Based on these achievements, recently the spatial resolution of TERS have been successfully pushed to single-bond368,369 and single-atom limits.370 We will discuss the details of these recent advances in atomistic TERS in Section 5.1.
It's noted that in the first SM-SERS experiments14 in 1997 and the first sub-nanometer TERS experiments in 2013, the resonance Raman effect was employed to achieve the best sensitivity. This is because the intensity of the Raman signal is collectively determined by the structural EF, the intrinsic cross-section of the molecule, and the optimization of the instrumentation, as discussed in eqn (4). Therefore, for establishing new methods, or exploring/optimizing new nanostructures/systems, it is beneficial to test with resonance Raman molecules for high detection sensitivity. When pursuing further improvement on the landmark work, however, testing non-resonance Raman molecules and expanding molecular generality in more applications and systems are inevitable. From a methodological point of view, resonance Raman and non-resonance Raman are two different aspects in the same development line approaching the high sensitivity of SERS.
In recognition of this relationship, attention should be paid to the related consideration of EFs (see Section 4.7). There is no need to emphasize how high the EF is when resonance Raman molecules are used, as it is not the practical goal of the research method. It cannot be compared with the EF of non-resonance Raman systems, because the purpose of the two is different, moreover in the end non-resonant molecules are more relevant to general SERS applications.
In 2015, Ren and coworkers developed an EC-TERS approach based on STM by directing light horizontally into an EC-TERS cell to minimize optical distortion (Fig. 39a).371 They reported potential-dependent behaviors of an aromatic molecule adsorbed on a well-defined Au(111) surface (Fig. 39b), highlighting significant alterations in the molecular configuration due to protonation and deprotonation phenomena. Simultaneously, Van Duyne and coworkers developed the AFM-based EC-TERS with a bottom-illumination configuration and investigated the nanoscale redox behavior of Nile Blue (NB) on a transparent ITO substrate.372 The disappearance of the 591 cm−1 peak of NB upon reduction and its reversible reappearance during oxidation was monitored and used to form the TERS voltammetry (TERS-CV) (Fig. 39c and d). They observed a shift in the redox potential between the TERS-CV and the conventional CV, which was attributed to the perturbation of the electric double layer by the TERS tip. Thereafter, EC-TERS images over a Au plate on ITO electrode was performed. Following the fitting of EC-TERS spectra obtained at various potentials for each position, the site-dependent formal potentials corresponding to the Au and ITO electrodes were derived. Notably, a more negative formal potential was observed at the Au electrode, along with a heterogeneous distribution of formal potentials across the ITO electrode.373
 | ||
| Fig. 39 The electrochemical TERS (EC-TERS). (a) The schematics of EC-TERS setup. (b) The potential dependent EC-TERS spectra of 4-PBT adsorbed on Au(111) surface. Tip potential was −200 mV, and the tunneling current was 500 pA. (c) EC-TER spectra of NB acquired at different potentials during cyclic voltammetry showing reversible reduction and oxidation of NB. P = 110 μW, t = 1 s, λ = 633 nm, scan rate = 10 mV s−1. The asterisk denotes the Si Raman band from the AFM tip. (d) Potential dependence of the integrated TERS intensity of the 591 cm−1 band of NBOX. Figures are reproduced from ref. 371 and 372 with permissions. Copyright 2015 American Chemical Society. | ||
In recent years, the introduction of water-immersion objectives by Ren and coworkers,374,375 and Maisonhaute, Lucas and coworkers376 fully avoided the optical distortion, significantly improving the sensitivity of EC-TERS. As a result, EC-TERS can be used to study more electrochemical processes, such as electrocatalysis. In 2019, Domke and colleagues investigated the site-dependent variations over the Au(111) surface during the oxygen evolution reaction by EC-TERS.377 They observed the formation of AuOx over the defect sites before the oxidation of the terrace sites, indicating varying activities over the Au(111) surface. In 2024, Ren, Tan, Wang and colleagues utilized EC-TERS to study the active sites of MoS2 during the hydrogen evolution reaction (HER).378 They observed that the edge induced electron transition region and lattice reconstruction region kept changing during the HER process until the highest activity was obtained, revealing the generation and dynamic variation of active sites during electrocatalytical reactions.
SORS capitalizes on the principle that Raman photons emitted from deeper regions of a sample experience increased lateral diffusion owing to multiple scattering events, which differentiate them from those originating in superficial layers. In practice, SORS collects Raman spectra at a spatial offset relative to the laser incidence point. This method, augmented by sophisticated multivariate data analysis and processing techniques, enables the differentiation and deconvolution of signals arising from various depths. The deconvoluted Raman spectra unveils layer-specific information, thereby facilitating the analysis of subsurface layers within complex samples.
Different from the backscattering collection mode commonly utilized in Raman measurements, TRS features a configuration where the laser is incident on one side of the sample, and the Raman scattered light is collected from the opposing side, allowing TRS to probe deeper regions within the sample, effectively extending the penetration depth beyond conventional capabilities.
Stone and colleagues further combined SORS and TRS with SERS and developed surface-enhanced spatially offset Raman spectroscopy (SESORS)381 and surface-enhanced transmission Raman spectroscopy (SETRS).382 The two techniques have significantly extended the penetration depth of SERS up to the millimeter and even centimeter level. This breakthrough has paved the way for in vivo diagnostic applications, allowing for the non-invasive examination of biological tissues at greater depths than ever before.
Temporal resolution is also a crucial aspect of SERS.217 Ultrafast Raman spectroscopy often employs modulation techniques, such as Kerr gating, to provide background-free Raman spectra with high signal-to-noise ratios and rapid acquisition times. In 1999, Matousek et al. demonstrated the efficacy of picosecond Kerr gating in rejecting fluorescence background interference by obtaining time-resolved spectra of p-quaterphenyl from solutions that were contaminated with a fluorescent dye.390 Subsequently, researchers developed other ultrafast SERS techniques, including time-resolved coherent anti-Stokes Raman spectroscopy (tr-CARS)391,392 and time-resolved stimulated Raman spectroscopy (tr-SRS).393,394 A significant milestone was achieved in 2011 by Van Duyne group, who pioneered the integration of SERS with femtosecond stimulated Raman spectroscopy (FSRS).394 They presented the first demonstration of surface-enhanced femtosecond stimulated Raman spectroscopy (SE-FSRS), using an experimental setup featuring picosecond pulses for the Raman pump, femtosecond pulses for the broadband Raman probe, and Au nanoantennae for signal enhancement (Fig. 40). This configuration achieved an EF estimated to be between 104 and 106. They further found that the signal-to-noise ratio can be significantly improved by choosing approximate repetition rates of the fs-laser.395 Latter work used picosecond time-resolved SERS to probe processes like plasmon-driven electron transfer and plasmonic heating of adsorbates.396,397 These results open new avenues for exploring the ultrafast dynamics of plasmonic materials and small homogeneous molecular subsets.
 | ||
| Fig. 40 The demonstration of surface-enhanced femtosecond stimulated Raman spectroscopy (SE-FSRS). (a) Schematic depiction of the nanoantennas used in the SE-FSRS experiments. Bottom panel show the extinction spectrum of the trans-1,2-bis(4-pyridyl) ethylene (BPE)-functionalized nanoantennas. (b) SERS spectrum (red curves) and SE-FSRS spectrum (black curves) of BPE-functionalized nanoantennas. The SE-FSRS spectrum shows dispersive peaks due to resonance effects from the plasmon. (c) SE-FSRS spectra obtained from BPE-nanoparticle assemblies with 100 kHz (red) and 1 MHz (blue) repetition rates of pulse. The pump pulse energy and average pump power are equal in both experiments. The spectra obtained at 100 kHz exhibit noise in the spectral region of 1000–1200 cm−1 that is approximately 10× greater than that obtained at 1 MHz. Figures are adapted from ref. 394 and 395 with permission. Copyright 2011 & 2016 American Chemical Society. | ||
The integration of ultrafast spectroscopy with TERS offers a powerful approach for investigating the nanoscale molecular dynamics with both spatial and temporal resolution. One of the earliest and most advanced applications in this field is tip-enhanced coherent anti-Stokes Raman spectroscopy (TECARS), which was firstly demonstrated by Inouye and colleagues in 2004.398 They successfully coupled 5 ps pulses with a plasmonic tip and operated at an 800 kHz repetition rate. This technique allowed them to acquire high-resolution TECAR images of adenine molecules within DNA clusters, capitalizing on the localized electric field enhancement beneath the metallic nanoprobe (Fig. 41a–d).
 | ||
| Fig. 41 Nonlinear and ultrafast TERS. (a) Schematics of the first tip-enhanced CARS (TE-CARS) of molecules near the metallic tips. (b) TE-CARS imaging of the DNA clusters at resonant frequency. (c) The topographic image of DNA clusters. (d) Far-field CARS image of the corresponding area obtained without the Ag tip. (e) Schematics of a picosecond pulse excited TERS (in a UHV environment). (f) Comparison of the TERS spectra of R6G molecules, under continuous wave (cw) and picosecond pulse (ps) excitation, respectively. Figures are adapted from ref. 398 and 399 with permissions. Copyright 2004 American Physical Society. Copyright 2014 American Chemical Society. | ||
In 2014, Van Duyne group reported the integration of picosecond-pulsed irradiation from an optical parametric oscillator (OPO) with a UHV-TERS system (Fig. 41e and f).399 They demonstrated that picosecond UHV-TERS of resonant adsorbates can be observed without permanent signal loss, which typically affects picosecond TERS in ambient conditions. This suggests that a UHV environment is a valuable asset for temporally resolved TERS experiments. Recently, Garg group demonstrated ultrafast LT-UHV TERS excited by ultrashort laser pulses (500 fs),400 laying the foundation for future experiments involving time-resolved femtosecond TERS.
4.6 Key methods for expanding the generalities of SERS substrate materials and morphologies
 | ||
| Fig. 42 (a) Schematic diagram of a confocal microprobe Raman system. Insertion: two possible configurations of the experimental setup between the collection microscope objective and the electrode surface. (b) Illustration of the roughening procedure for Pt and Rh surfaces by using square-wave potential (by controlling the E1/E2) or current (by controlling the I1/I2) oxidation–reduction method. The solution used is 0.5 M H2SO4 (c) a comparison of the cyclic voltammograms of roughened (solid line) and smooth (dotted line) Pt electrodes in 0.5 M H2SO4 solution. Scan rate: 50 mV s−1. (d) The STM image of a Pt electrode after electrochemical ORC roughening treatment. (e) Potential dependent surface Raman spectra of (i) hydrogen adsorption in 0.5 M H2SO4 + H2O and (ii) deuterium adsorption in 0.5 M H2SO4 + D2O at roughened Pt surfaces. (f) The AFM image of an Rh electrode after electrochemical ORC roughening treatment. (g) Surface-enhanced Raman spectra of a roughened Rh surface in 0.1 M CH3OH + 0.1 M Na2SO4. 0.2 M methanol was added into a Raman cell containing 0.2 M Na2SO4 solution with the electrode controlled at −0.6 V to form the final measuring solution. (h) Raman spectra of pure pyridine, pyridine solution, and pyridine that is adsorbed on noble metal and transition metal surfaces at open circuit potentials. Figures are adapted from ref. 165, 181 and 403 with permission. Copyright 1996 Elsevier. Copyright 2002 & 2003 American Chemical Society. | ||
While there was experimental progress in transition metal SERS, how to understand the enhancement mechanism behind it remained an important issue in the SERS field. SERS is significantly influenced by the electromagnetic enhancement mechanism, which is predominantly attributed to the LSPR exhibited by nanoparticles composed of coinage metals. When exposed to visible light, the conduction electrons within these nanoparticles can be induced to oscillate collectively, thereby producing a robust electromagnetic field in proximity to the nanoparticle's surface. A high-quality SPR effect should meet two conditions: first, the real part of the dielectric constant of the nanoparticle must be negative; second, the imaginary part of the dielectric constant should be as close to 0 as possible.404 In contrast to noble and alkali metals, transition metals exhibit distinct electronic structures characterized by the positioning of the Fermi level within the d band. This configuration results in a high probability of interband excitations occurring within the visible light spectrum. The interaction between conduction electrons and interband electronic transitions significantly diminishes the quality of the SPR in transition metals.269 This considerably reduces the SERS effect, as observed in many experiments.
Based on the two-dimensional particle array model, Tian group carefully studied the correlation between the SERS EF and the morphology of the ellipsoidal particles, especially the aspect ratio of the particles and the frequency of the excitation light.405 The maximum electromagnetic EF for SERS on Ni is observed to range between two to four orders of magnitude, with a marked increase corresponding to higher aspect ratios. This phenomenon can be elucidated through the lightning rod effect, which typically leads to significant electric field enhancement in regions with pronounced curvature on the surface. Furthermore, the data indicates a pronounced increase in the SERS EF for Ag when the frequency of the incident light approaches the frequency of surface plasmon resonance. In contrast, the EF for the Ni electrode remains relatively across the same orders of magnitude, provided the aspect ratio of the particles is maintained. This observation suggests that the SERS response of Ni does not exhibit the distinctive characteristics associated with SPR, with the EF demonstrating only minor fluctuations in response to variations in incident photon energy. Experimental findings corroborate this, revealing that the SERS enhancement for the Ni substrate remains nearly unchanged when the excitation laser wavelength is adjusted from 632.8 nm (1.96 eV) to 514.5 nm (2.41 eV), assuming all other experimental conditions are held constant. The critical role of morphology was also emphasized by Moskovits and coworkers in 2017, they showed that any metal can produce intense SERS when properly structured.406
Everything that has been learned regarding single molecule SERS since 1997 has led scientists to believe that the SERS-active system is also strongly dependent on the particle–particle, particle–substrate or tip–substrate near-field coupling effect. Therefore, the quantitative evaluation of SERS EFs in actual transition metal systems inevitably involves non-spherically symmetric particles and complex near field coupling between particles. Thus, it was important that the research model for the EM enhancement mechanism of SERS for transition metals changed its focus from highly symmetric particles to particle aggregates of arbitrary size and shape.
In order to conduct electrodynamic calculations of light scattering from complex SERS-active systems, the application of numerical methods is essential. Various techniques, such as the finite difference time domain (FDTD), finite element method (FEM), boundary element method (BEM), and discrete dipole approximation (DDA), have been developed to analyze the electromagnetic fields surrounding individual non-spherical metallic nanoparticles as well as nanoparticle arrays in the context of surface-enhanced spectroscopy. Among these approaches, the FDTD method stands out as a comprehensive, systematic, and versatile tool for evaluating the optical response of nanostructures with arbitrary geometries and symmetries to incident light waves. It has been effectively employed in SERS research involving transition metals. A typical simulated SERS enhancement for transition metal systems with a cauliflower-like morphology is shown in Fig. 43a and b.407 Two distinct models were employed to simulate the cauliflower-like nanoparticles: one model consisted of a Rh sphere with a diameter of 120 nm, which was coated with 20-nm semispherical particles, while the other model featured a flat Rh surface adorned with 20-nm nanohemispheres, as illustrated in Fig. 43a. The computed electric field distribution across the surface is depicted using color visualization. The greatest enhancement of the electric field is typically observed at the apex of the smaller hemispheres within the “cauliflower” structure, which contrasts with the maximum field enhancement typically found at the junctions of two nanoparticles. However, when the cauliflower structure is separated and arranged into a planar grating-like configuration, the location of the field maximum shifts the crevices, as demonstrated in Fig. 43b. These findings suggest that the electromagnetic enhancement is highly sensitive not only to the polarization of the incident light, the electronic characteristics of the metal, and the surface morphology, but also to the symmetrical properties of the SERS nanostructures.
 | ||
| Fig. 43 (a) FDTD simulated electric-field distribution for a cauliflower-like Rh nanoparticle and (b) a flat Rh surface decorated with small nanohemispheres. The laser beam illuminates along the y-direction with x polarization. The scale bar is given at the bottom of (b). Figures are adapted from ref. 405 and 407 with permission. Copyright 2006 Springer Nature. Copyright 2006 Royal Society of Chemistry. | ||
Enhanced Raman scattering based on semiconductors stretched back to 1980s. In 1982 Yamada et al. reported enhanced Raman signal of pyridine adsorbed on the surface of single-crystal NiO(110).411 They also made similar observation for the surface of Ti and single-crystal TiO2(001).135 Although the mechanism of enhancement was attributed to the CT mechanism at that time, it was actually difficult to distinguish between CT and resonance Raman effects in the experiments.69 In 1988, Hayashi et al. found the enhanced Raman scattering derived from electromagnetic mechanism for copper phthalocyanine molecules with strong resonance Raman effect on GaP NPs.412 The development of semiconductor enhanced Raman scattering stagnated in the 1990s probably due to very low EF. Semiconductor-enhanced Raman scattering was looked at again after 2000. For example, Zhao, Lombardi and colleagues reported semiconductor-enhanced Raman scattering for metal oxides, metal tellurides, metal sulfides, and so on.413–415 Based on these studies they proposed a clear CT model to explain the enhanced Raman on semiconductor materials.
There are several kinds of semiconductor materials for enhanced Raman scattering. Metal oxides such as TiO2 and ZnO are representative of these. Metal chalcogenides such as CdS, CdSe, PbS, and ZnSe are also popular materials for semiconductor-enhanced Raman scattering. Other semiconductor materials for enhanced Raman scattering are doped semiconductors like doped TiO2, graphene, Si, and Si and Ge nanowires. The representative enhanced Raman active semiconductor materials are summarized in ref. 34, 243, 416 and 417. The EF of semiconductor-enhanced Raman scattering is, in general, 103–108.
The advantages of semiconductor enhanced Raman scattering are summarized as follows:34,243,416,417
(1) Semiconductor materials have excellent high chemical and thermal stability. Thus, they can avoid the problem of signal reproducibility caused by substrate oxidation. Moreover, they have broadened the applicable media of enhanced Raman technique, such as strong oxidation medium, strong acid medium, high-pressure medium and so on.
(2) Semiconductor materials have superb surface modifiability. In contrast to metal materials, most of common functional groups can be directly adsorbed on semiconductor materials surfaces. In other words, the surface of semiconductor materials is modified more flexibly than that of metal materials.
(3) Semiconductor materials have good biocompatibility. They have several hydroxyl groups on their surface, allowing them to combine with proteins through amino groups and carboxyl groups. Such binding ability solves the problem of biocompatibility of metal-based SERS technique.
(4) Semiconductor materials encompass a diverse range of species that are characterized by their low cost and established preparation methods. The energy levels and structural properties of these semiconductors can be finely tuned through various techniques, including adjustments to size, shape, material composition, and doping. This versatility facilitates the selection of appropriate semiconductor substrates tailored to meet specific detection requirements.
(5) Semiconductor materials have been used extensively as active components in a variety of practical devices. The wide applicability of semiconductor materials provides more possibilities for the application of enhanced Raman scattering.
The SERS enhancement mechanisms for dielectrics, particularly semiconductors, also encompass both electromagnetic and chemical enhancement.21 Unlike metals, which exhibit LSPR due to high-density of free electrons, the dielectric materials primarily achieve electromagnetic enhancement through light-trapping and subwavelength-focusing capabilities, as well as morphology-dependent resonances such as Mie resonance. These enhancement effects are closely related to the morphology of the dielectric material. For highly doped semiconductors, plasmonic resonances can be induced in the low-frequency range (generally in the near-infrared to mid-infrared regime).
Additionally, CT process between semiconductors and molecules constitutes an important chemical enhancement mechanism in semiconductor SERS substrates. In fact, most of the current CT mechanism models in semiconductor SERS originate from Lombardi's pioneering work in 1979.115 The CT process is influenced by the vibronic coupling between the conduction band (CB) and the valence band (VB), as well as between the excited and ground states of the molecules involved.415 CT models applicable to semiconductor-enhanced Raman scattering can be categorized into three distinct types: ground-state CT, electronic resonant transitions, and excited-state CT. The first two categories align with the CT models established for metal-based surface-enhanced Raman scattering (SERS), whereas the excited-state CT model for semiconductor SERS exhibits greater complexity. For example, a CT complex (similar as a new molecule) formed by the strong chemical bonding between the molecule and semiconductor generally possesses new energy levels, providing additional CT pathways.418 This is even more important when the excitation light does not have enough energy to excite CT transitions between two original energy levels in a semiconductor–molecule system. Fig. 44 shows various mechanisms of a photo-induced CT process (blue line) and coupling scheme (purple line) in semiconductor–molecule systems.
 | ||
| Fig. 44 Various mechanisms of a photo-induced CT process (blue line) and coupling scheme (purple line) in semiconductor–molecule systems. (a) Resonant excitation of the CT complex, (b) molecular HOMO-to-CB, (c) CT complex-to-CB, (d) VB-to-molecular LUMO, (e) surface state to molecular LUMO, and (f) molecular excited state-to-CB transitions. Figures are adapted from ref. 417 with permission. Copyright 2018 World Scientific Publishing Europe Ltd. | ||
From a methodological perspective, it is important to note that highly efficient CT transitions typically occur in semiconductor-molecule systems at the nanometer scale due to the size-dependent behavior of electronic properties in semiconductor nanomaterials. Therefore, theoretical modeling should account for the size effect. Although various CT pathways have been proposed based on DFT calculations, experimental investigations into semiconductor CT mechanisms remain relatively limited.243 To elucidate the specific enhancement mechanisms, more systematic and correlated experiments, such as ultrafast dynamics studies, are essential.
Another challenge lies in the size mismatch issue. While CT transitions typically occur in nanoscale semiconductors, effective regulation of the visible and near-infrared electromagnetic fields requires semiconductor materials with feature sizes ranging from sub-micrometers to micrometers. To address this size mismatch issue, a rational approach is to design semiconductor superstructures, which allows for the integration and further optimization of the synergistic effect between electromagnetic and chemical enhancement.419,420 In such a superstructure, the engineering of size and morphology can induce light-trapping effects, subwavelength focusing capabilities, and morphology-dependent resonances. These modifications contribute to a substantial electromagnetic enhancement of SERS. Additionally, a highly efficient CT contribution arising from the structure of the superstructure further amplifies the SERS signals.
Note that SPR in semiconducting materials could be contributable to SERS on semiconductors.421 In such case, plasmons may lower the material stability. The carrier density of semiconductors can span several orders of magnitude based on doping techniques, increasing opportunities for SERS-based applications.422 In addition, noble metal SPR is found to be capable of promoting CT in semiconductor–molecule systems, and their synergistic contribution enables giant Raman signal enhancement on metal–semiconductor heterostructures.423,424
Semiconductor-enhanced Raman scattering has opened a variety of new research fields in photoelectrochemistry, catalysis research, bioscience, and sensing technology. The high chemical stability of semiconductor materials enhances the sensing performance including spectral reproducibility increasing the reliability of enhanced Raman scattering analytical experiments. The superior biocompatibility has opened a variety of new applications in life sciences.
For example, Hildebrandt, Weidinger and colleagues reported the redox process of heme proteins on nanostructured TiO2 electrodes.425 It has also become possible to explore the photoelectrical properties of adsorbents on the semiconductor surfaces such as dye-sensitized solar cells, lithium–ion batteries, and semiconductor-supported photocatalytic process. A number of new applications have emerged; for example, submicron-sized ZnO superstructures have demonstrated excellent SERS performances with the synergistic effect of Mie resonance-induced electric field with CT.419 Furthermore, ZIF-8 coated ZnO superstructures could further concentrate the electric field around particle surface, permitting signal enhancement for the detection of non-adsorbing volatile organic compounds.426 Besides, Ji, Ozaki, Xie and colleagues, demonstrated the feasibility of large-area fabricating SERS-active semiconductor substrates for the assessment of heavy metal ion toxicity based on screen-printing of micron-sized TiO2 inks synthesized through a simple flame thermal-assisted method.427 The resultant TiO2 microspherical arrays (MSAs) exhibit extraordinary SERS sensitivity with an EF of 3.28 × 107, which represents one of the highest sensitivity and are the easiest strategy among the reported SERS-active semiconductor substrates.
Note that caution must be exercised when quantifying EFs in these systems, particularly when using widely employed fluorescent molecules, as resonance Raman effects and fluorescence quenching may lead to a significant overestimation of the SERS EF (see Section 4.7.1 for detailed discussions). For a comprehensive discussion on this topic, we recommend the reviews by Lombardi and Alessandri,243 and Ozaki, Itoh, Procházka, Dong and colleagues.34
M via resonance design.432 Nevertheless, Raman enhancement on two-dimensional materials provides a platform to study material properties, and more efforts are necessary to move to practical SERS detection.
The position of the Fermi level in graphene plays a crucial role in determining the EF of GERS.433 Consequently, modulating the Fermi level in graphene provides a valuable strategy for controlling the EF of GERS. In recent years, dynamic modulation approaches have emerged as an effective means to address the limitations posed by the fixed optical properties of conventional SERS substrates post-preparation. Notably, photo modulation has been employed to dynamically control SERS signals by manipulating the Fermi level of graphene or other materials through UV-induced adsorption and desorption of gas molecules, as evidenced in studies utilizing reduced graphene oxide (rGO) and polystyrene (PS) nanoarrays.434 Additionally, electrical modulation techniques, including piezoelectric,435 thermoelectric,436 and pyroelectric methods,437 have garnered significant attention. For example, piezoelectric substrates convert mechanical pressure into electrical energy, thereby enhancing SERS signals through charge injection into plasmonic nanostructures.435 Likewise, thermoelectric substrates, such as those incorporating GaN with Ag nanoparticles, capitalize on temperature gradients to regulate CT, significantly improving SERS signal intensity.436 Furthermore, ferroelectric materials have been utilized to modulate the energy band structure of graphene oxide, facilitating flexible tuning of CT processes and enhancing SERS responses across various analytes.438 Collectively, these advancements underscore the potential of active modulation techniques to enable real-time adjustments in SERS performance, thereby expanding the applicability of this sensitive analytical tool in diverse fields, including environmental monitoring and biomedical diagnostics.
Near-infrared Fourier transform surface-enhanced Raman spectroscopy (NIR-FT-SERS) microprobe spectroscopy offers distinct advantages over visible SERS due to the reduced likelihood of surface photochemical changes induced by near-infrared radiation, potential avoidance of sample fluorescence, and an EF that is approximately an order of magnitude greater than that achieved with visible excitation. In NIR-FT-SERS, the excitation of Raman scattering is facilitated by a Nd:YAG laser operating at wavelength of 1064 nm, with spectrum acquisition conducted using a Fourier Transform Infrared (IR) spectrometer that operates within the near-infrared spectral range. Owing to these advantages, significant interest has emerged in utilizing NIR-SERS spectroscopy as a novel analytical technique for acquiring structural insights regarding adsorption and reactivity within the initial adsorption layer. Fig. 45a and b depicts the NIR-SERS microprobe spectra of pyridine adsorbed on a roughened Au or electrode in a 0.5 M LiCl aqueous solution under 1064 nm excitation at various characteristic potentials in 1988.439
 | ||
| Fig. 45 (a) and (b) NIR-SERS spectrum of (a) a pyridine/Au electrode and (b) a pyridine/Ag electrode at −0.6 V versus Ag/AgCl 0.05 M pyridine, 0.5 M LiCl after roughening. UV-SERS spectra of SCN− on Rh (c) and Ru (d) electrodes as a function of the applied potential. (e) Deep-UV SERS spectrum of a 1 × 10−4 M crystal violet solution on a 50 nm thick aluminum surface, the wavelength of the incident laser was 244 nm. Reproduced from ref. 439–441 with permissions. Copyright 1988 Society for Applied Spectroscopy, Copyright 2003 American Chemical Society. Copyright 2007 Wiley. | ||
However, this strategy cannot be applied to the UV region. This is because the SPR of SERS-active materials such as Ag and Au is difficult to extend to the UV range, where the interband transition from d band to sp band is dominated. The development of new substrate materials is necessary for such an extension. Tian and colleagues were the first to observe SERS excited by UV light on transition-metal electrodes (Fig. 45c and d).441 The adsorption of pyridine and SCN− on roughened Rh and Ru electrodes, respectively, was investigated with 325 nm laser excitation. The experimental UV-SERS findings in 2003 align with initial theoretical calculations grounded in the electromagnetic enhancement mechanism, indicating an EF of approximately two orders of magnitude for Rh and Ru electrodes under 325 nm excitation.
Popp group found later that Al acts as a SERS-active substrate for deep-UV excitation wavelengths in 2008. They reported a UV-SERS spectrum of a 1 × 10−4 M crystal violet solution on a 50 nm thick Al surface, excited with a 244 nm laser (Fig. 45e).440 Al with different nanostructures, such as nanovoids and Al film-over-nanosphere substrates, was then gradually developed into a main deep-UV SERS-active substrate. In 2014, Halas group reported the fabrication of Al nanodisks using electron beam lithography, demonstrating tunable plasmon resonance from UV to visible region, as shown in Fig. 46a.442 However, individual nanodisks struggled to generate sufficiently strong hot spots for SERS detection. In 2016, Van Duyne group reported the first fabrication of Al film-over nanosphere (AlFON) substrates for UV SERRS at the deepest UV wavelength (λext = 229 nm).443
 | ||
| Fig. 46 (a) Aluminum plasmon generated by aluminum nano disk. (b) Experimental and theoretical extinction spectra of monodisperse Al nanocrystals in isopropanol, the inset shows the SEM image of an Al dimer (scale bar = 200 nm). (c) Size control of aluminum nanocrystals. Increasing the fraction of tetrahydrafuran (THF) in a 1,4-dioxane/THF solution yields larger nanocrystals. The top panel shows the reaction scheme for synthesis of aluminum nanocrystals. The bottom panel shows the representative TEM images from synthesis with 0, 0.5, 0.6, and 0.8 THF volume fractions in a THF/1,4-dioxane solution (from left to right) (scale bar = 100 nm). (d) SERS spectra of APhS and (e) PABA on Al (red) and Au (blue) substrates with normal Raman spectra (black) as reference. Figures are adapted from ref. 442, 444 and 445 with permissions. Copyright 2014, 2015 & 2017 American Chemical Society. | ||
Subsequently, in 2017, Halas group achieved SERS measurements using Al nanocrystals (NCs), which involved a chemical synthesis with uniform shape and controllable size, as shown in Fig. 46b.445 Interest in Al nanocrystal plasmonics for SERS is due to the sustainable and earth-abundant nature of Al (20000 times less expensive than Au). Most importantly, the development of bottom-up chemical syntheses resulted in highly pure Al with far fewer impurities than top-down fabrication methods. For SERS, this results in substrates with significantly larger field enhancements than obtainable with Al SERS substrates fabricated using film deposition methods. Other advantage of Al is the remarkable size-tunability, ranging from the UV into the mid-infrared and the thin oxide surface of Al, which facilitates far more binding chemistries than Au-based SERS substrates.446 In contrast to Ag nanoparticles, the self-terminating surface oxide passivates the Al nanocrystal surface, making the metal nanoparticle more stable. Fig. 46c shows how Al nanocrystals of different sizes can be synthesized by a straightforward change in their growth conditions (solvent used during synthesis).444Fig. 46d and e compares the SERS spectrum obtained with both Al and Au substrates and with the normal Raman spectrum of the molecules under study.445 It is worth pointing out that in the case of Al SERS substrates, the analyte spectrum is more similar to that of the unenhanced Raman spectrum than for Au substrates.
One potential approach involves the excitation of SPPs on the flat metallic surface through the implementation of a specialized optical configuration. Although the ATR-SERS had been developed since the 1980s to study the smooth single-crystal surfaces, the EF is limited to around one to two orders of magnitude. The invention of TERS in 2000 provided a solution to the limited surface generality of SERS. In TERS, the LSPR effect is harnessed to amplify the electromagnetic field at the apex of a plasmonic probe. When the probe is irradiated with a laser source of appropriate wavelength, the resultant enhanced electromagnetic field can interact with a sample positioned in close proximity to the probe's apex. In principle, TERS is promising for solving the issue of substrate and surface generalities as the Raman signals from any substrate regardless of material and surface smoothness can be increased by borrowing from the tip apex enhancement, as shown in Table 3. However, TERS is strongly dependent on sophisticated instrumentation, and its total Raman signals are very low as only a singular nanotip works as the Raman amplifier. These issues make TERS impractical for many applications.
| Localized surface plasmon, LSP | Raman spectroscopy | SERS (LSP + Raman) | TERS (LSP + Raman + tip) | SHINERS (LSP + Raman + shell) | |
|---|---|---|---|---|---|
| Sensitivity | Excellent | Poor | Excellent | Excellent | Excellent |
| Single molecules | Many molecules | Single molecules | Single bond | Single molecules | |
| Energetic resolution | Poor | Excellent | Excellent | Excellent | Excellent |
| ∼10−2 eV | ∼10−4 eV (1 cm−1) | ∼10−4 eV (1 cm−1) | ∼10−4 eV (1 cm−1) | ∼10−4 eV (1 cm−1) | |
| Spatial resolution | Good-excellent | Good | Good | Excellent | Good |
| ∼200 nm (far field) | ∼200 nm | ∼200 nm | ∼3 nm (Ambient) | ∼200 nm | |
| ∼3 nm (near field) | Sub-Å (UHV, LT) | ||||
| Materials generality | Poor | Excellent | Poor | Poor | Poor |
| Au, Ag, Cu, Li, … | Au, Ag, Cu, Li, … | Au, Ag, Cu, Li, … | Au, Ag, Cu, Li, … | ||
| Morphology generality | Nanostructure | Any morphologies | Nanostructure | Flat surface | Any morphologies |
| Molecule generality | Adsorbed molecules | Any molecules | Adsorbed molecules | Adsorbed molecules | Adsorbed molecules |
| Nanostructure | Poor | — | Poor | Poor | Good |
| Stability | Atomic migration | Atomic migration | Atomic migration | Shell protected |
In 2010, SHINERS20 was invented to tackle the above-mentioned limitations of SERS in study of single-crystal surfaces and SERS-inactive materials (Fig. 47). SHINERS is the latest version of the “borrowing SERS activity” strategy and utilizes shell-isolated nanoparticles that feature plasmonic cores made of Au or Ag and are encased in ultrathin (1–5 nm) shells that are chemically and electrically inert, such as SiO2 or Al2O3. In SHINERS, the Au cores can generate strong electromagnetic fields to enhance the Raman signals of molecules nearby, while the chemically and electrically inert silica shells can prevent direct contact between the Au/Ag core and probe materials, thus avoiding interference from the metal cores and improving the universality of probed materials and surface morphology (Fig. 24f and 25f). In principle, SHINERS can achieve trace analysis on any surfaces by simply spreading many Au@SiO2 core–shell nanoparticles – typically around a thousand within the light spot – on the surface to be probed, thus solving, at least to some extent, the long-standing materials and morphology limitations of traditional SERS.
 | ||
| Fig. 47 Schematic of the mode of SERS, TERS, and SHINERS. (a) Bare Au nanoparticles: contact mode. (b) Au core–transition metal shell nanoparticles adsorbed by probed molecules: contact mode. (c) TERS: noncontact mode. (d) SHINERS: shell-isolated mode. Figures are reproduced from ref. 20 with permission. Copyright 2010 Springer Nature. | ||
The benefits of SHINERS are multifaceted. Firstly, the ultrathin, pinhole-free shells effectively isolate the core particles from the surrounding material surface and environment, thereby reducing interference from the Au and Ag cores. Secondly, the chemically inert nature of the shell inhibits fusion of both interparticle and particles with the metal substrate, thus significantly improving the stability of the nanoparticles and probe configurations. Thirdly, the thickness of the shell can be manipulated to regulate the nanogap between the Au or Ag core particles and the substrate, which in turn influences the electromagnetic coupling between the particles and the substrate. Finally, the Au and Ag cores enhance the local electromagnetic field, thereby amplifying Raman signals from the probe substrate while maintaining its structural integrity.
SHINERS is distinguished from contact-mode SERS and TERS by the presence of an inert, ultrathin shell, which acts as an insulating barrier, preventing direct interaction between the probe materials and the noble-metal cores composed of Au and Ag (shell-isolated mode). SERS implicitly involves direct contact between the SPR-active nanostructure surface and the probe target (molecules or materials) (contact mode), as shown in Fig. 47a and b. Therefore, SHINERS cannot be classified as a SERS technique. This distinction also applies to TERS. However, since SERS, TERS, and SHINERS all involve local field enhancement primarily from SPR enhancement, each can be considered as a unique PERS (plasmon-enhanced raman spectroscopy) technique. The detailed comparison of the LSP, Raman and those PERS techniques are shown in Table 3. Based on SHINERS, Tian group successfully acquired Raman signals of molecules adsorbed on Au and Pt single-crystals. The average EFs for Au(110) and Pt(110) reach 106 and 105, respectively, which are found to be equivalent to, or in some instances surpass, the EFs achieved with bare Au nanoparticles.447 Moreover, this approach enables in situ Raman measurements of weakly adsorbed species like water molecules on single-crystal transition metal surfaces, a feat previously unattainable (see Section 5.2.1).
It should be noted that, although SHINERS addresses several limitations associated with contact-mode SERS for surface analysis, it is accompanied by a reduction in Raman enhancement.21,269 Specifically, the interparticle plasmonic coupling and the coupling between the nanoparticles and the substrate in shell-isolated nanoparticle-on-substrate configurations are relatively moderate when compared with bare nanoparticle-on-substrate systems. Consequently, dependent on the thickness and dielectric constant of the shells, the average enhancement factor achieved through SHINERS may be lower than that obtained from a bare gold nanoparticle dimer situated on silicon and platinum substrates, respectively. However, theoretical studies have shown that by optimizing the thickness and refractive index of the SHIN shell, the sensitivity of SHINERS can even surpass that of traditional bare Au nanoparticles. Nevertheless, achieving this goal remains experimentally challenging due to the need to fabricate ultrathin yet dense shells.
Another important development of SHINERS is that the ultrathin inert shell-isolated strategy is also applied to TERS by Li and Zenobi group and named as shell-isolated tip-enhanced Raman spectroscopy (SITERS).448 In traditional TERS, although its working principle is based on the non-contact mode (someone called as the gap mode), the exposed tip may still lead to direct adsorption of the analyte molecules onto the tip surface, causing signal interference, especially in studying different facets of single crystal surfaces. This issue is particularly evident in solution systems. Therefore, coating the tip with an inert shell to shift from the traditional non-contact mode to a shell-isolated mode can prevent the adsorption of analyte molecules on the tip, thereby expanding the application range of TERS. In addition, the shell of SHINs can be functionalized by modifying its composition to meet specific detection requirements, such as acid and alkali resistance or anti-sintering properties.
4.7 Key methods for evaluation of enhancement factors
To ensure the precision and reliability of SERS, it is essential to develop quantitative analysis techniques for SERS. The emergence of the SERS field can be traced back to Van Duyne's quantitative analysis of spectral EFs.2 In 1983, Moskovits and colleagues observed an enhancement exceeding 4 × 106 for colloidal Ag nanoparticles (Ag NPs) by comparing the Raman intensity of the SERS signal with that of normal Raman spectroscopy under the same experimental conditions.449 From the current definition, this article was the first to calculate the average Raman EF.Notably, the EF encompasses not only electromagnetic enhancement from nanostructures but also chemical enhancement mechanisms, including resonant Raman effects and charge transfer processes (see eqn (4) and Fig. 49). Consequently, EF can be defined and quantified differently depending on the specific aspects under consideration, leading to varying values across different analytical frameworks. In 1984, Stockburger and colleagues introduced the concept of EF to assess the SERS performance of colloidal substrates.450 They defined EF as
 | (5) |
In 1997, two groups14,15 reported the observation of SERS spectra from single dye molecules on Ag nanoparticle colloids independently. The single-molecule EF, which is the SERS enhancement of a given molecule at a specific point, is influenced by various factors, such as the Raman tensor of the molecule, its orientation on the SERS substrate, and the local electric field at the specific point. Additionally, the orientation of the SERS substrate in relation to the incident laser polarization and direction also plays a significant role in determining EF. Therefore, a precise definition of the SERS substrate geometry and the accurate positioning and orientation of the molecule on the substrate are essential in calculating EF. The single molecule EF is defined as
| EF = ISMSERS/ISMRaman | (6) |
It should be noted that in the original SM-SERS report, EFs of the order of 1014ISMSERS were taken for a resonant molecule.14 Even in the absence of plasmonic nanostructures, the Raman EFs of resonant molecules induced by resonance Raman effects can achieve 104–106. Therefore, more realistic EFs of SM-SERS in terms of EM enhancement are of the order of 108–1010, which is sufficient for SM-SERS detection in the best conditions with dye molecules such as R6G with resonance Raman enhancement of 106. It is crucial to distinguish whether the study concerns SERS (non-resonant molecules) or SERRS (resonant molecules) when quantifying the Raman EF. If the process under investigation is SERRS, it is essential to exclude the Raman enhancement resulting from the resonance Raman effect in the resonant molecules to obtain an accurate Raman EF from nanostructures.
In 1998, Tian and coworkers402 proposed a similar EF for evaluating solid SERS substrates as
 | (7) |
The confocal microscope possesses excellent depth discrimination, ensuring that only a minuscule effective volume of the solution is probed. This capability significantly reduces the interference from robust Raman signals that may arise from the bulk phase of the solution. Furthermore, the integration of the confocal microscope system facilitates a considerable enhancement in the numerical aperture (N.A.) of the objective lens, which in turn greatly increases the solid angle available for the collection of scattered photons. The direct determination of the term NRaman should be derived by considering the confocal properties of the microscope. Fig. 48 illustrates the spatial intensity distribution, specifically the waist contour, of a laser beam that has been focused within an aqueous medium. Conventionally, it is assumed that all molecules within the irradiated volume of the solution contribute to the quantity denoted as NRaman. However, the efficiency of photon collection from molecules dispersed throughout the solution exhibits variation in accordance with the confocal depth, which influences the efficiency of scattered photon detection across different planes within the illuminated volume. Molecules located at the ideally focused plane (where z equals zero) contribute the most to the overall intensity. Thus the NRaman should be written as NRaman = AhcNA, where A is the area of the focal spot of laser. h is the height of the effective layer, depending on the pinhole size and the objective lens. c is the concentration of the molecules and NA is the Avogadro constant.
 | ||
| Fig. 48 (a) Illustration of the contribution to IRaman from a certain plane of illuminated solution. (b) and (c) Illustration of vertical movement of a flat Si waver for obtaining the confocal depth dependence of the Raman signal. Reproduced from ref. 402 with permission. Copyright 1998 Elsevier. | ||
In 2007, Etchegoin, Le Ru and coworkers introduced the analytical EF (AEF) as
 | (8) |
TERS significantly enhances signals due to the intensified electromagnetic field at the tip apex, a result of the LSP and the lightning rod effect. The technique detects a rapidly diminishing near-field signal from a minuscule scattering volume, which is scattered by the tip in the presence of a far-field background emanating from the entire illuminated region. The expression of the EF in TERS is defined as452
 | (9) |
From a fundamental research perspective, there remains a considerable discrepancy between theoretical predictions and experimental measurements of SERS/TERS EFs. For instance, experimentally calculated EFs are often significantly lower than those predicted theoretically. Accurate quantification necessitates accounting for not only the electromagnetic mechanism in nanostructures, which can be strongly orientation-dependent, but also the chemical enhancement mechanism. This includes considering both resonant and non-resonant effects, as well as the average effect of single and multiple hot spots, which remains a substantial challenge in the field.
This challenge also partly arises from an inherent contradiction between the ultra-sensitivity of SERS substrates and their capability for quantitative detection. The ultra-sensitive SERS “hotspots” required for high sensitivity detection can dramatically amplify molecular signals, making it exceedingly difficult to accurately estimate the number of molecules. Furthermore, metal colloids and solid substrates are prone to aggregation, contamination, surface reconstruction, and degradation when exposed to ambient environments. These factors, along with variations of laser and optical alignment, lead to instability in SERS signals, thereby affecting the accuracy and reproducibility of quantitative analysis.
Overcoming the primary challenge in achieving quantitative analysis with SERS involves addressing the uniformity and reliability of the SERS substrates, which requires the standardized production of uniform SERS substrates, such as nanoarrays with specific arrangements.454 Bell, Xu, and coworkers showcased emulsion-templated self-assembly as a method capable of producing consistently spherical colloidal aggregates that are free of surface modifications and maintain stability for extended periods.455 The robust stability and potent plasmonic enhancement of these aggregates enable precise and quantitative detection of weakly adsorbing analytes in protein-rich biomedia through SERS.
Therefore, when analyzing the total SERS EF (Fig. 49), it is crucial to consider the contributions of SERS enhancement from different aspects quantitively and distinguish between average EF and maximal EF. The former is typically associated with large-area SERS substrates, while the latter is commonly estimated in single-molecule or single-particle experiments. For instance, the widely accepted SERS EF for colloidal Au and Ag nanoparticles is often cited as 106,456 which generally refers to the average EF. In contrast, single-molecule SERS experiments frequently report factors of up to 1015,14,457 which are typically defined as the maximal EF. It is important to note that such maximal EF usually encompass the combined contributions of multiple enhancement mechanisms, including electromagnetic field enhancement, resonance effects, and chemical enhancement, as shown in Fig. 49.
 | ||
| Fig. 49 Typical ranges of EM and CE contributions to the total SERS EF. The total SERS EF of Au, Ag, Cu and nontraditional materials have several contributions, including EM mechanism, resonance Raman effect and CT mechanism. The error bars represent the typical range of each contribution. For Au, Ag and Cu, the typical contribution of EM mechanism is 6 to 12 orders of magnitude, while that for nontraditional SERS materials is 1 to 4 orders of magnitude. The typical SERS EF resulted from the resonance Raman effect is approximately 2 to 6 orders of magnitude, while the that contributed by CT mechanism is about 1 to 3 orders of magnitude. | ||
In the context of EM mechanism, the quality factor (or the loss of the plasmonic mode) and the mode volume of the plasmonic mode are the key factors to determine the EM enhancement. For individual Ag, Au nanoparticles, average SERS EF typically range from 10 to 103, with maximal EF from 102 to 104.21,456 Notably, plasmonic coupling between nanostructures can significantly reduce the mode volume, thereby generating intense hotspots. In such coupled systems, maximal SERS EF can reach 108–1010, with average EFs exceeding 106.21,456 In extreme cases involving atomic-scale protrusions within the hotspots, such as the picocavity, maximal EF can surpass 1012.34,358,457
Conversely, for non-traditional metals other than Ag and Au, increased plasmonic losses lead to decreased plasmonic resonance quality, resulting in reduced SERS EFs. For instance, typical average EFs for these materials generally fall within the range of 10 to 104.165 In addition to metallic materials, the SERS EF of dielectric substrates, utilizing Mie scattering and the lightning rod effect, can reach up to 103.458 Further, materials such as graphene achieve an SERS EF by a factor of approximately 10 via CE mechanism while the SERS EF on semiconductors can reach up to 107 or even 108.408
Another significant yet often overlooked effect in SERS is the resonance effect, which is associated with molecular electronic state transitions and excitation wavelength. This effect can provide additional signal enhancement beyond the electromagnetic (EM) mechanism. For instance, in the case of common R6G dye molecules, resonant excitation can contribute a resonance EF of up to 106.456,457 This resonance effect plays a crucial role in compensating for signal enhancement in SM-SERS experiments. However, as previously discussed, the resonance effect is not general in SERS analytes. Besides, it should be noted that the resonance EFs for most fluorescent molecules beyond R6G are typically limited from 102 to 104. Moreover, upon surface adsorption, the anticipated resonance enhancement is usually attenuated due to chemisorption induced quenching, leading to less pronounced enhancement than expected. Consequently, an excessive focus on the substantial EFs or absolute signal intensities resulting from resonance Raman effects often has limited significance in broader SERS applications.
The CT mechanism in SERS is influenced by various factors, including the adsorption state, molecular orientation, and CT processes. While the contribution of chemical enhancement to the overall SERS enhancement is typically modest, ranging from 10 to 1000,19,456,457 its significance should not be underestimated. From a surface chemistry perspective, understanding the CE mechanism is crucial for elucidating molecule–surface interactions via SERS. Moreover, for the application of SERS across diverse analytical fields, particularly in the development of quantitative or semi-quantitative analysis systems aimed at commercialization, a comprehensive grasp of chemical enhancement mechanisms is indispensable.
Moreover, for specific materials and systems under investigation, it is crucial to comprehensively evaluate the interplay between EM and CE mechanisms on enhancement factors. Notably, this interaction cannot be simplified as a mere product of EM and CE contributions; rather, it demands comprehensive analysis of their complex interdependence. A prime example is the phenomenon of molecular adsorption at interfaces, which can simultaneously modify the interfacial refractive index,459 thereby affecting both the optical response of nanostructures and the EM EF at hotspots. Consequently, the development of a self-consistent theoretical framework that integrates both physical and chemical enhancement effects355,460,461 remains crucial for accurate SERS enhancement factor evaluation, although significant challenges persist in this endeavor.
4.8 Key methods for quantitative analysis
Quantitative analysis of molecular concentration and number is crucial for both fundamental research and practical applications of SERS. For example, in the development of SM-SERS, following its initial observation, SM-SERS faced considerable controversy within the scientific community. It wasn’t until a decade after its first reported detection that SM-SERS was unequivocally validated. This confirmation came through rigorous quantitative analysis based on bi-analyte SERS experiments utilizing isotopically edited molecules.212–214,462,463Quantitative analysis can be traced back to 1981, as shown in Fig. 22d, in which the first attempt was tried by Pemberton and Buck.464 As they stated that “To this time, little emphasis has been placed on development of analytical implications of this technique…. However, no systematic investigation of SERS + RRS behavior for adsorption from varying starting solution concentrations of a colored adsorbate at an enhancing metal electrode has been made. The work presented here was undertaken in an attempt to demonstrate the analytical utility of SERS + RRS in detecting surface species adsorbed at an enhancing metal electrode from solutions initially of low concentration in adsorbate.”. Normally, according to the surface adsorption isothermal, a linear relation is possible between SERS intensity of a certain Raman peak and the concentration of the analyte, and a much wider linearity range could be realized via a log/log plot. Note that the univariate evaluation relying solely on the peak intensity of a single Raman peak may introduce potential qualitative deviations.
The surface coverage plays a crucial role in the quantitative analysis of SERS. In 1981, Shen and coworkers investigated the adsorption of pyridine adsorbed on a Ag electrode–electrolytic solution by using SERS465 and surface second harmonic generation (SHG).466 The observed adsorption isotherms can be approximated by the Langmuir isotherm, while the transient adsorption following a simple Langmuir kinetic model was difficult to descript surface adsorption of pyridine on Ag electrode.
Similar to fluorescence, SERS exhibits a linear relationship with surface concentration at low to moderate coverage levels. However, deviations from this linearity occur at higher surface coverages. It is important to note that it only applies to surface coverages not to bulk concentrations. When the adsorption is less than 2/3 of a monolayer for various analytes, a linear correlation between SERS intensity and analyte concentration in the bulk solution may be observed, suggesting that solutions should be diluted, akin to conditions in molecular fluorescence.
This conclusion also helps in understanding the early observations of potential-dependent SERS, as discussed in Fig. 12. At that time, this phenomenon was considered direct evidence of the CT mechanism. This phenomenon was considered direct evidence of the CT mechanism. However, in 1991, Stolberg et al. correlated SERS intensity with the independently measured surface concentration for various bulk pyridine concentrations and variable electrode potentials.119
Fig. 50a presents the 1010 cm−1 band intensity as a function of electrode potential for four different bulk pyridine concentrations. These plots display characteristic maxima similar to those observed in Fig. 12a. However, when the same data are plotted against the surface coverage of pyridine, as seen in Fig. 50b, the dependence on bulk concentration is eliminated. This data aligns with a single relationship, featuring a maximum at a surface concentration of 4 × 10−10 mol cm−2, which corresponds to 2/3 of a monolayer coverage. It indicates that observed SERS intensity depends on coverage.
 | ||
| Fig. 50 (a) Plots of the intensity of the 1010 cm−1 band on the electrode potential (E vs. SCE) for the varied bulk pyridine concentrations. (b) Data from panel a plotted versus surface concentration of pyridine Γ. Figures are adapted from ref. 119 with permission. Copyright 1991 Elsevier. | ||
Later in 1999 and 2001, a more solid quantitative analysis was achieved by considering more information from the peak or the whole Raman spectrum with the introduction of multivariate calibration with partial least squares (PLS), one of the mostly commonly used machine learning techniques. The experimental variations (such as reproducibility and uniformity of SERS substrate, the modified interaction between analyte and SERS substrate, and the instrument conditions) make the accurate quantitative analysis challenging, however by careful consideration of the surface adsorption chemistry and control of the reproducibility of the surface used for enhancement, accurate and reliable quantitation can be achieved using SERS. Initially the focus of quantitative SERS analysis using Ag nanoparticles resulted in a series of studies to improve the synthesis of the nanoparticles467 and to understand the surface chemistry of adsorption,468 before extending to the measurement of therapeutic drugs,469 illicit substances470 and biomolecules.471 These select studies showed this approach with many more studies reported covering multiple targets with the focus being on control of the surface adsorption and aggregation of nanoparticles. Many comprehensive reviews are dedicated to this topic and have more information on this key area of performance for utilization of SERS.472–474
An alternative approach is to rely on a calibration strategy via an internal standard. A trusted internal standard should be able compensate for the above variations with a SERS signal free of interference with that of the analyte. The isotopic internal standard method was proposed independently by Bell et al.475 and Zhang et al.476
In 2004, Bell and colleagues used isotopic pyridine as an internal standard to realize quantitative analysis of nicotine over a wide concentration range (0.1–10 ppm) with a R2 of 0.998.475 Independently, in 2005, Zhang et al. used the isotopic rhodamine 6G as internal standard for rhodamine 6G, by which a better than 3% batch-to-batch reproducibility over a concentration range of 200 pM–2 μM was accomplished.476 Instead of using the expensive isotopic reagents, Bell et al. introduced CNS− as the internal standard for the SERS detection of dipicolinic acid, a marker for bacterial spores, and achieved a linear range of 0–50 ppm with a R2 of 0.986.477
An alternative quantitative analysis is to use frequency shift, which originated from the change in relative intensity of probe molecules before and after interaction with analyte. In 2012, Olivo group demonstrated the frequency shift-based quantification of antigen triggered by biomarker-ligand recognition.478
Despite being well demonstrated, it is difficult to avoid the dynamic exchange and competitive adsorption between analyte and the internal standard molecule when their concentrations are significantly different. Thanks to the development of SERS-active core–molecule–shell nanoparticles (CMS NPs),237,247 in which a molecular layer is interposed between the core and shell, the SERS signal of the embedding molecular layer is not influenced by the external environment, and can be used as a novel internal standard for quantitative analysis. The CMS based internal standard strategy was independently developed by Zhang group479 and Ren group in 2015.248 They convincingly demonstrated the capability of these nanoparticles to quantitatively analyze target molecules across a wide concentration range, exhibiting a linear relationship between the relative SERS intensity and the surface coverage. Additionally, enhancing the specificity of substrates towards analytes through surface modification and functionalization also promises to improve the accuracy and save time in quantitative analysis.
It seems the CMS based internal standard strategy for quantitative analysis is the most promising one among the above ones. Nevertheless, realizing a reliable quantitative analysis in real samples remains an unresolved challenge. This endeavor encompasses several key areas, such as the controllable large-scale production of CMS NPs, which serve as the foundation for SERS assays. Additionally, efficient sample pretreatment is a critical preliminary step for SERS analysis of most real samples, as it can significantly influence the qualitative and quantitative analysis. Moreover, the working curve for quantitative analysis may be subject to potential deviations due to the competitive adsorption of residual impurities on the SERS substrate. Addressing these concerns is crucial for advancing the field of SERS-based quantitative analysis.
To apply comparable and reproducible measuring conditions in SERS, microfluidic devices were developed and combined with a Raman microscope for readout, which could help to average SERS signal over either time or space to reduce the signal fluctuation and improve the reliability of quantitative analysis. The first study on quantitative analysis utilizing SERS in microfluidic environments date back to 2002,480 where a model derivative of TNT was developed within the lab-on-a-chip paradigm, employing solely the analyte and readily accessible reagents. In 2006, the quantitative detection of nicotine within the range of 0.1 ppm and 10 ppm481 as well as the quantification of the pesticide methyl parathion between 0.1 ppm and 1 ppm482 was achieved. In 2007, by applying a liquid/liquid segmented flow within a microfluidic device, individual droplets of 180 nL were investigated illustrating the high potential of SERS to quantify substances in sub-μL volumes without the agglomeration of colloidal nanoparticles at the channel walls avoiding the memory effect.483 Later, droplet-based microfluidic SERS approaches prove their potential in drug monitoring by quantification of levofloxacin484 or nitroxoline485 in spiked human urine samples.
It should be noted that the complexity of the multiple interactions among the laser, molecules, and nanostructures that generate the SERS signal may exceed our imagination, often leading to significant differences in the SERS spectra obtained by different experimental groups due to variations in experimental details. For example, since Fleischmann and coworkers reported the first SERS spectra of pyridine on a Ag electrode 50 years ago, over 2000 studies on the SERS of pyridine have been published, some of which reported different spectral features. In Fleischmann et al.'s spectra, a new and strong peak at 1025 cm−1 appeared (Fig. 3g–h), as we mentioned in Section 2.5, which differed from those reported by others later. This discrepancy arose because they electrochemically roughened the Ag electrode under high-power laser irradiation in a solution containing pyridine and KCl, performing nearly 200 oxidation–reduction cycles. Upon further investigation, it was found that the 1025 cm−1 peak most likely originates from a surface complex of pyridine with Ag clusters and chloride ions formed through a laser-induced surface chemical process, rather than from free pyridine molecules adsorbed on the electrode surface.
Kudelski and coworkers also found that for Ag nanoparticle structures, this peak is either very weak or difficult to observe when spherical Ag nanoparticles are used as the SERS substrate, but it is more easily detected on irregular flattened Ag nanoparticles.486 Muniz-Miranda attributed this peak to a complex formed between pyridine and the positively charged Ag32+ cluster, which is related to the chloride ions present in the electrolyte solution.487 Therefore, to accurately analyze the Raman peaks in SERS spectra for nanoparticles or substrates prepared by different experimental methods, it is necessary to precisely correlate all the relationships of interface species formed and all experimental parameters. This is crucial for methodology and establishing detailed benchmark experiments to study the SERS mechanism and facilitate commercialization successfully.
5. Advances in recent years (mid-2010s–mid-2020s)
The rapid advancement of nanoscience and the urgent needs of analytical science, surface science, electrochemistry, biology and other disciplines initiated the second upsurge of SERS. As summarized in Fig. 17, it shows that the number of SERS related publications increased rapidly from 1995–2015. It should be noted that the exponential growth in the number of publications since 2000 was largely due to lower technological threshold for nano synthesis and the increased accessibility of SERS research. However, the key foundations of the outbreak were laid in 1974–2000.14–19,187There have been many excellent reviews about the theories,21,32,34,242,243,269,404,452,488,489 experimental methods,21,25,30–34,166,242,243,269,404,452,489–500 and practical applications21,25,28,30–32,34,242,269,452,490–493,497,501 of SERS, which can be referred to for more detailed progress in the field for this period. We emphasize that the methodological foundations underpinning these advancements were largely established within the first two decades of SERS research. These early contributions laid the groundwork for subsequent developments and applications. Comprehensive discussions of these methodologies and their applications can be found in several seminal books in the field.12,62,502–510
Although the annual output of SERS-related articles remains high, with around four thousand papers published each year, a closer examination reveals a diminishing growth rate since 2015, leading to a recent plateau (see Fig. 17). This trend is explained by the fact that most published papers employ SERS merely as a readily accessible characterization technique or analytical method, rather than generating innovative contributions or breakthroughs in SERS methodology and commercialization. The stagnation in the growth of SERS-related publications may signal the need to reinvigorate scientific momentum and advance SERS research. To achieve this, it is crucial to proactively embrace emerging science, develop novel methodologies, and push the boundaries of current practices.
To achieve a new vibrant state in SERS, it is essential to delineate the three distinct paths that have emerged since its discovery. Each path presents unique development challenges and leads to significant milestones: deep fundamental research, broad application techniques, and extensive instrument commercialization, as illustrated in Fig. 51.
 | ||
| Fig. 51 The different development directions and focus of SERS since its discovery. | ||
In the realm of fundamental research, the focus is on advancing new theories, instruments, and experimental methods to overcome detection sensitivity and resolution limits, thereby expanding the boundaries of what was previously deemed impossible. Researchers in this area often utilize highly idealized model systems and environments, such as LT-UHV settings and resonant molecules, to approach detection limits as closely as possible. For instance, in Section 5.1, we will explore recent breakthroughs in spatial resolution within fundamental research. The implementation of TERS at liquid helium temperatures has enabled unprecedented advancements in spatial resolution, achieving angström-scale and even atomic-level resolution. This remarkable progress marks a significant leap forward in our capacity to probe and characterize materials at the most fundamental scales.
In the context of general applications, the emphasis shifts to the expansion of SERS versatility, including substrate materials, morphologies, and molecular generality beyond the scope of traditional Raman spectroscopy. This approach facilitates the integration of SERS into diverse research fields such as physics, chemistry, biology, and materials science, thereby supporting fundamental research in these areas. Consequently, the priority in this direction is not solely on achieving the highest signal strength with resonant molecules, as their practical and scientific value may be limited except when used as labels. Instead, the focus should be on more realistic scenarios, such as detecting non-resonant molecules in complex environments, to enhance the practical utility of SERS. In Section 5.2, we will discuss recent efforts to extend SERS applications to weakly adsorbed molecules, diverse surface-interface systems, and complex environments like biological research. These advancements have significantly broadened the general applicability of SERS. Notably, in general applications, the quantitative analytical capabilities of SERS may be more critical than its sensitivity, as elaborated in Section 5.3.
The third path poses the greatest challenges, as SERS must be recognized as a significant analytical technique for its relevant instruments to be realized and widely adopted. In the context of commercial and market applications, consistency and stability of spectral signals, as well as cost efficiency, become paramount. Developing reliable, reproducible, and affordable SERS techniques is crucial for their successful integration into the market and everyday use.
During this period, the themes and directions of exploration in SERS Frontiers and applications were highlighted at the Faraday discussions meeting held in Glasgow, UK, in 2017, chaired by Duncan Graham (see Fig. 52 and Table 4). On the first day of the conference, there was an in-depth discussion on the theory of SERS, with a particular focus on the quantum electrodynamics description theory of SERS, especially following the recent demonstration of picocavity SERS.358 The second day commenced with discussions on ultra-sensitive SERS, examining how the technique's sensitivity could be maximized. This was followed by examples of biological SERS. Throughout the day, a clear theme emerged: the SERS community was perhaps underselling itself regarding the reproducibility and quantitative nature of the technique when used in appropriate applications—a strong view that was emphasized in the discussions. On the final day, the focus was on the analytical uses of SERS, discussing real-life examples and the impact of SERS in various fields.
 | ||
| Fig. 52 Group photo of the Faraday discussions meeting on SERS, 30 August–1 September 2017, Glasgow, UK. | ||

|
Notably, Richard Van Duyne, one of the two major pioneers in the SERS field and a leading scientist with over four decades of significant contributions, was honored with the Spiers Medal, presented by Eleanor Campbell, President of the Faraday Division, just before his outstanding opening lecture.
5.1 TERS in the atomistic near field (ATERS)
Currently, both SERS and TERS have achieved remarkable milestones in fundamental studies. In terms of spatial resolution, in the ambient condition, TERS has achieved spatial resolution of 3 nm.511 Furthermore, in LT-UHV condition, individual chemical bonds can be resolved by TERS368,369,511 and even sub-angström (Å) level spatial resolution is achieved.370 This fascinating advance originates from a deep understanding of light–matter interactions in optical cavities.The Å-scale in optical microscopy was reported in 2017 by Apkarian, Jensen and coworkers through TERS carried out in the atomistic near-field (ANF), ATERS for short.512,513 They used a CO terminated tip to map out the electrostatic potential surfaces inside individual metalloporphyrin molecules, along with providing fully resolved images of vicinal CH bonds separated by 2.5 Å.514 The full power of ATERS was illustrated by them in 2019 by visualizing the vibrational normal modes within a single molecule (Fig. 53a and b)368 and by recording atom resolved images of a 2D insulating Cu2N film.515 ATERS image of vibrational normal modes within a single Mg–porphine was also reported in the same year by Dong, Hou, Luo and coworkers.369 In a notable subsequent illustration, they combined STM and noncontact AFM with TERS to image the three pentacene-derivatives obtained by sequentially breaking its individual carbon–hydrogen bonds (Fig. 53c).370 Highlighted in Fig. 53c is the resolved 0.89 Å displacement in peak location of the imaged CH-stretch, which is in excellent agreement with their theoretical simulation. The ATERS image resolution, as defined by the full width at half maximum of observable features, is limited by the Wigner Seitz radius of the terminal atom at the tip apex, demonstrably 1.6 Å for atomically terminated Ag tips.368–370,515
 | ||
| Fig. 53 Representative research work on TERS to achieve ultimate sensitivity and spatial resolution. (a) Schematic of TERS of the Co(II)–tetraphenyl porphyrin (CoTPP) molecule immobilized on Cu(100) in the LT-UHV TERS (6 K) system. (b) Spectra recorded on cobalt (1), pyrrole (2) and a phenyl group (3) are shown. The inset is the corresponding topography (left panel). The TERS mapping of Raman modes at 1156 cm−1 and 3006 cm−1 are shown (right panel). Scale bars are 2 Å. Reproduced from ref. 368 with permission. Copyright 2019 Springer Nature. (c) Measured and simulated maps for the C–H stretching mode of the pentacene species α and β on the Ag(110) surface. Vertical and horizontal lines indicate the long and short molecular axes, respectively (left panel). Line profiles obtained along the short molecular axis. The orange and green curves are the Gaussian fitting of the peaks. Reproduced from ref. 370 with permission. Copyright 2021 AAAS. | ||
The attained resolution verifies atomic confinement of the apex mode on atomically terminated plasmonic tips, as anticipated by two seminal works: Takahara had pointed out that there is no cut-off for TM0 plasmons on nanowires of negative dielectric,516 and Nerkararyan and coworkers517,518 pointed that lateral confinement of plasmons, d, on a conical tip of negative dielectric scales as 1/ρ2, where ρ is the height-dependent cone radius. Therefore, classically, there is no limit to confinement of the photon since d → 0 as ρ → 0; however, quantization of matter sets the limit to the minimum attainable value of ρ to the atomic radius, as realized experimentally.368–370,515 Beyond the confinement of optical field, Luo and colleagues also showed that in non-resonance Raman conditions, Raman images can visualize vibrational modes owing to the accessibility of the Herzberg–Teller contributions.519
Operationally, optical microscopy in the ANF is reached using atomically terminated plasmonic tips, scanned with sub-Å precision afforded by ultrahigh vacuum cryogenic STMs. The paradigm shift in ATERS arises in that the measurements are carried out at junction gaps where tip and substrate wavefunctions overlap, as attested by the dc tunneling current used for tip placement. This is most directly manifested under resonance Raman conditions, where the local optical density of states images molecular orbitals,520,521 also as shown in Fig. 36. Notably, the TERS signal intensity peaks at contact,512,515 which by virtue of its lateral confinement forms a quantum pointed contact (QPC),522 as recognized by Kumagai and coworkers,523–525 and realized in implementations where approach curves are extended to contact,526 in space-time resolved THz microscopy,527 in CARS on graphene nanoribons.528 This is in stark contrast with standard TERS, or more generally, near-field scanning optical microscopy (NSOM), which is formulated and carried out in the electrostatic near-field.529 In the ANF, the external radiation field is excluded from the junction, the scattering is driven by the optical current of tunneling or conductive plasmons, reducing ATERS to the alternating current analog of scanning tunneling electron microscopy.530
It should be emphasized that the novel observable in ATERS is the intramolecular polarization current, which, at contact, extends into the tip. In visualizing vibrational normal modes involving unresolvable atomic displacements of order 0.1–0.01 Å, what is imaged is the nonlocal time-harmonic vibronic polarization current associated with a given mode, which is nicely formulated in the recently introduced Raman bond model (RBM) by Jensen and coworkers.531–533 Indeed, a variety of theoretical adaptations have been implemented to model and interpret the advances cited in this section and to revise the fundamental understanding of the science underlying SERS and TERS on that basis. We should stress that with the extreme confinement, other components in the plasmonic field, including magnetic field534 as well as momentum distribution,535 which are negligible in conventional optical fields, would come into play, opening an entirely new territory for not only SERS and TERS but also nano-optics. We refer the reader to a recent comprehensive review for theoretical predictions in such highly inhomogeneous optical fields.536
5.2 SERS on weakly adsorbed molecules and interfacial structures
To ensure reliable analysis of weakly adsorbed molecules by SERS, it's essential to exclude the effect of surfactants and contaminants, which is distinctly different from the strongly adsorbed systems. Three critical aspects warrant careful consideration during measurement protocols. First, preliminary surface cleaning through either chemical537–539 or physical540,541 methods is essential to eliminate competitive adsorption from surfactants (such as citrate, ascorbic, CTAB and PVP), thereby ensuring measurement accuracy. Chemical cleaning procedures may involve various protocols including acid treatment or plasma cleaning, while physical methods typically encompass thermal or mechanical approaches. Second, comprehensive concentration titration across a broad range is imperative to identify potential interference from ultra-trace strongly adsorbed contaminants, which may originate from reagents used542 or plastic lab containers.543 Third, SERS performance must demonstrate independence from laser power to exclude the possibility of laser-induced photochemical reactions during measurement, which could otherwise compromise data integrity. These methodological considerations are fundamental to achieving reproducible and reliable SERS measurements for weakly adsorbed molecular species.544,545
Lipkowski and colleagues developed an ordered array of ∼50 nm Au nanoparticles by dissolving the shell from an assembly of silica coated SHINs and applied it to measure SERS of sugars (weak adsorbers and weak Raman scatterers) at Au. In this way, they avoided coalescence of Au nanoparticles produced by the citrate method into random clusters.546 Up to now, various strategies have been applied to improve the sensitivity by strengthening interfacial interactions,547 such as electrostatic interactions,539 molecular steric effect,548–551 host–guest recognition,552,553 and introducing probe molecules via biological recognition, chemical derivatization, hydrogen bonding, ion–dipole interactions, and so on.554–558
Water molecules, as special weakly adsorbed species with hydrogen bond network interacting with electrolyte ions extending to several water layers. It exhibits complicated interfacial structures that are intrinsically linked to electrochemical reactions, electrical double layer formation, and water dissociation processes. SHINERS serves as an ideal tool for investigating interfacial water adsorption on single-crystal surfaces. By depositing SHINs onto metal single-crystals, NPoM configurations with significant Raman enhancement can be constructed. The NPoM structure can also be excited through the Kretschmann or Otto configuration, leveraging the coupling between SPP and LSP.489 At the critical angle where SPP excitation at the film/air interface occurs, the SERS intensity at the film surface reaches its peak either with or without the coupled nanoparticles.559–561 This configuration has been termed as the SPR-SERS configuration.
To further enhance the sensitivity of SHINERS, precise control over the coupling of SHINs particles is crucial. Recently, Li and coworkers demonstrated that assembling multiple Ag@SiO2 SHINs particles on an Ag film surface can effectively amplify the Raman signal intensity, enabling simultaneous measurements of single-molecule fluorescence and single-molecule Raman spectra of individual rhodamine B isothiocyanate (RITC) molecules.562
Therefore, leveraging the high sensitivity of the gap mode, SHINERS can be utilized to investigate interfacial dynamic processes across various single-crystal surfaces and reveal the mechanism of important interfacial processes, especially in situ probes of interfacial water by Raman spectroscopy. For instance, the advent of SHINERS has revitalized Raman spectroscopy research on interfacial water, an area that had seen little progress for over a decade. Cheng, Li and coworkers utilized SHINERS to study the interfacial water on Au(111) and Au(100) single-crystals under hydrogen evolution reaction (HER) conditions.563 They observed three types of interfacial water molecules undergoing configurational changes with varying potentials: with the negative shift of the potential, the interfacial water changed from a “parallel” configuration to “one-H-down”, and then to “two-H-down”.
Compared to Au, platinum-group metal surfaces have garnered more attention due to their extensive applications in catalysis and energy. However, the plasmonic coupling effect between SHINs particles and platinum group metal surfaces is weak, leading to a reduction in the EF of SHINERS by 2–3 orders of magnitude.564 This limits the detection of trace species on their surfaces. For example, Pd is one of the most active HER catalytic materials, but the weak plasmonic coupling between Pd and SHINs makes it extremely challenging to study the structure of interfacial water using SHINERS. To address this issue, Li, Pan, and coworkers deposited different atomic layers of Pd on a Au single-crystal surface by alternately using Cu UPD and spontaneous displacement methods. Thus, based on the “borrowing SERS activity” strategy, strong plasmonic coupling effect between the underlying Au single-crystal and SHINs could achieve a Raman EF as high as 107–108. With such a strategy, they further employed SHINERS to in situ investigate the structure and dissociation of interfacial water on a Pd single-crystal surface(Fig. 54),565 and obtained spectral evolutions of hydrogen-bonded and Na+ hydrated water during the hydrogen evolution reactions (HER). At HER potentials, interfacial water undergoes dynamic structural changes, transitioning from random to ordered arrangements facilitated by the bias potential and Na+ ion cooperation. This ordered water structure enhances electron transfer and HER rates. Electrolytes and electrode surface properties influence interfacial water structure, suggesting potential for improved electrocatalytic reaction rates through local cation tuning strategies.565
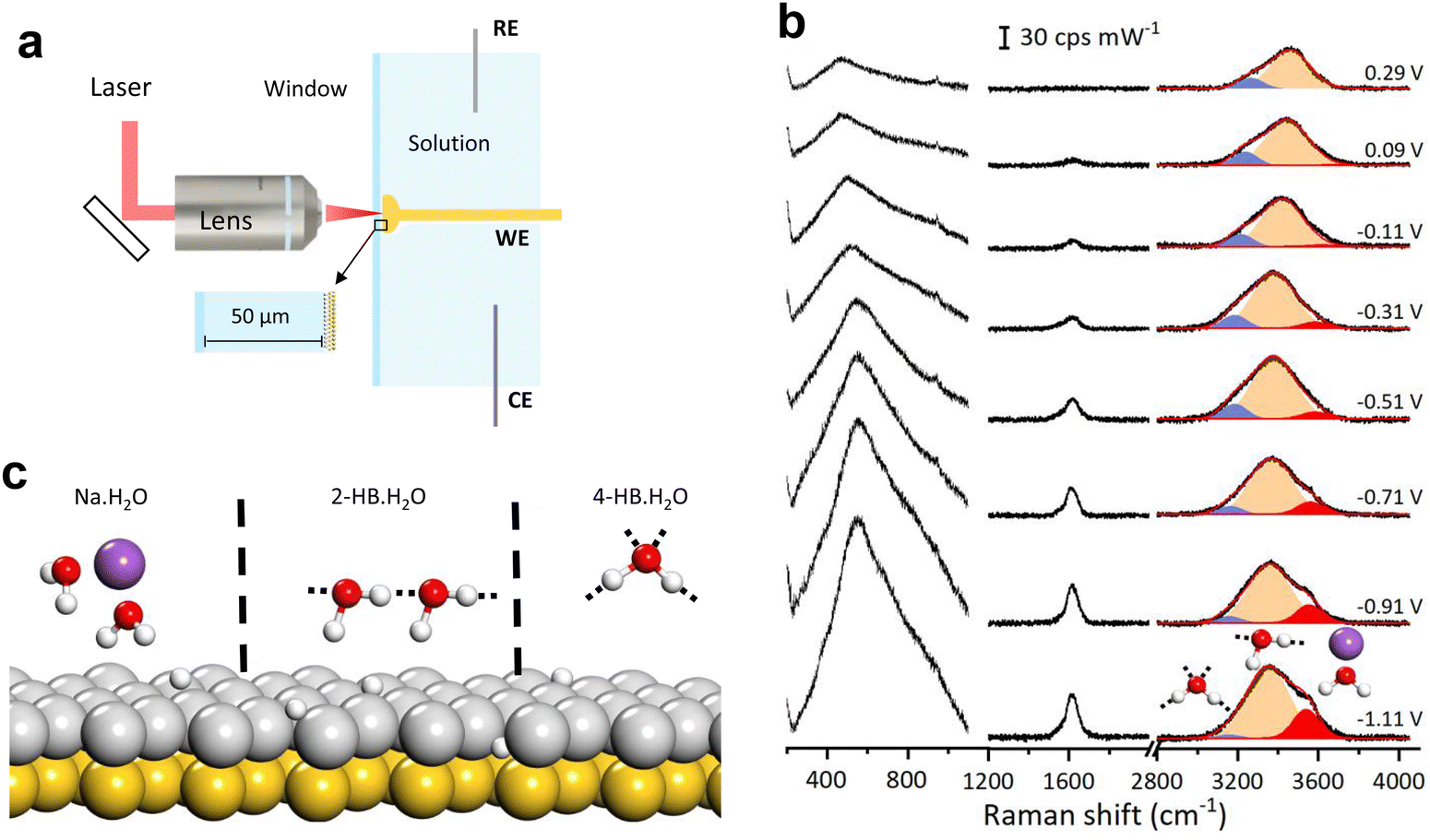 | ||
| Fig. 54 (a) In situ SHINERS of interfacial water on Pd(hkl) single-crystal surface and reveal that interfacial water consists of hydrogen-bonded and Na+ hydrated water. CE, counter electrode; RE, reference electrode; WE, working electrode. (b) The in situ Raman spectra of interfacial water on a Pd(111) electrode in a 0.1 M NaClO4 solution (pH 11) were shown. Gaussian fits of three O–H stretching modes are shown in blue, orange, and red, respectively. (c) Schematic of the three types of interfacial water on the Pd/Au surface. Reproduced from ref. 565 with permission. Copyright 2021 Springer Nature. | ||
Using this strategy, Li, Dong, Wang and coworkers have in situ studied the evolution of the intermediates and the structure of catalysts during hydrogen oxidation reaction (HOR) on PtRu surfaces, which is the main composition of commercial HOR catalysts (Fig. 55a).566 As shown in Fig. 55b–e, Pt shells were first coated on Au cores, and Ru was then deposited on the outer surface of the Pt shell. The resulting Au@PtRu display superior HOR performance compared to Au@Pt and commercial Pt/C (Fig. 55f). In situ SERS reveals Ru(+4)Ox (583 cm−1) surface oxides, and OHad (712 cm−1) species appeared during the HOR process, based on the isotope experimental results (Fig. 55g and h). Meanwhile, Had species were also observed at lower potentials on the catalysts. The observation of OHad species indicates HOR on PtRu follows the bifunctional mechanism. By comparing the in situ spectroscopic results for PtRu with those for pure Pt surfaces, they found that the introduction of RuOx on the Pt surface can optimize the adsorption energy of OHad, thus improving HOR performance. Using the similar “borrowing SERS activity” strategy based on Au core transition shell nanoparticles, Li and coworkers have also investigated the reaction mechanisms of various important catalytic reactions including HOR, oxygen reduction reaction, CO oxidation, etc.420,567–569
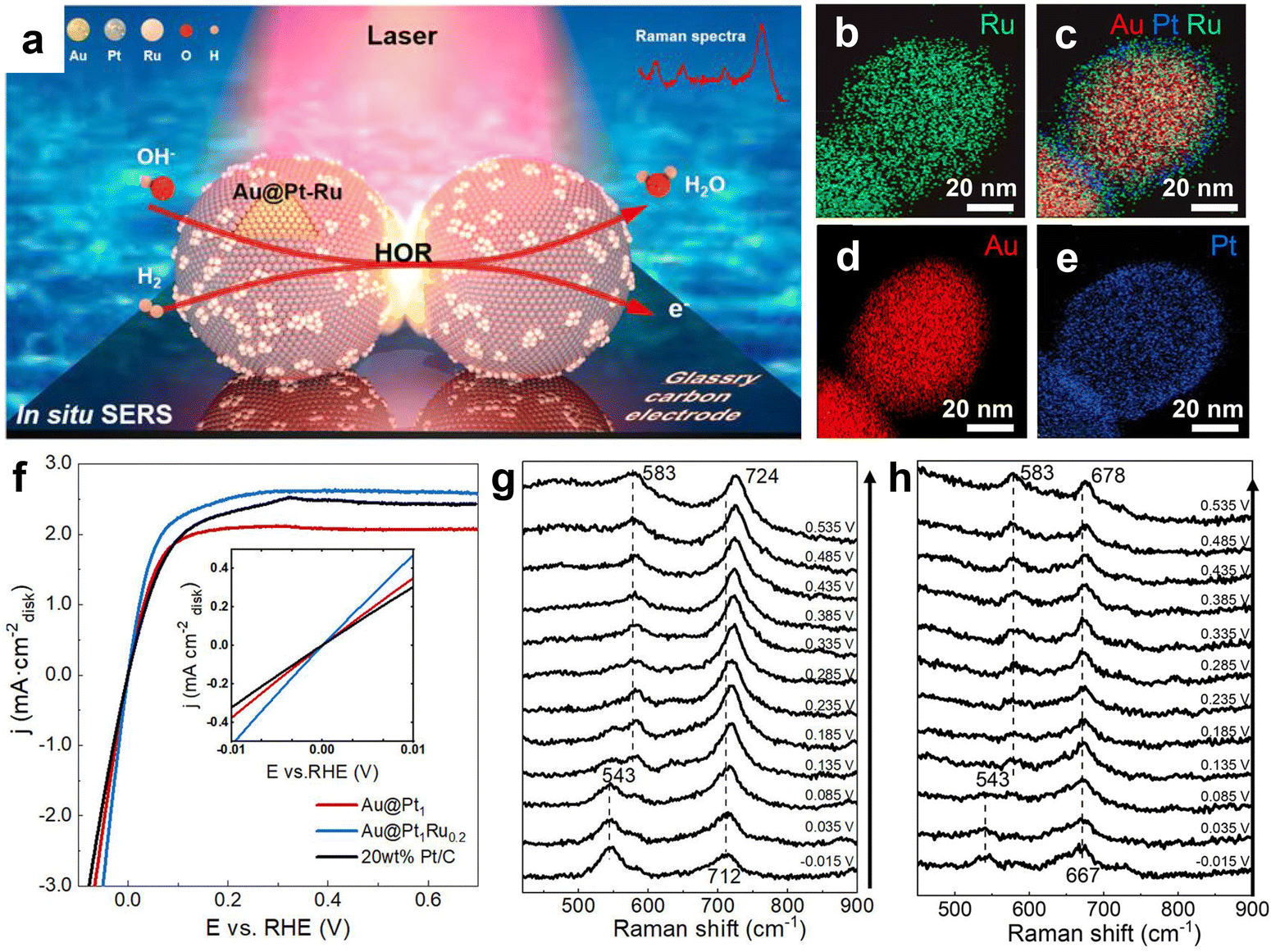 | ||
| Fig. 55 (a) Schematic illustration of in situ SERS study of HOR on Au@PtRu. (b)–(e) Elemental mapping images of the Au@PtRu nanoparticles. (f) HOR polarization curves of commercial 20 wt% Pt/C, Au@Pt, and Au@PtRu. Sweep rate: 10 mV s−1. Rotation rate: 1600 rpm. (g) In situ SERS spectra of HOR on Au@PtRu and (h) the deuterium isotope experimental results. Reproduced from ref. 566 with permission. Copyright 2022 American Chemical Society. | ||
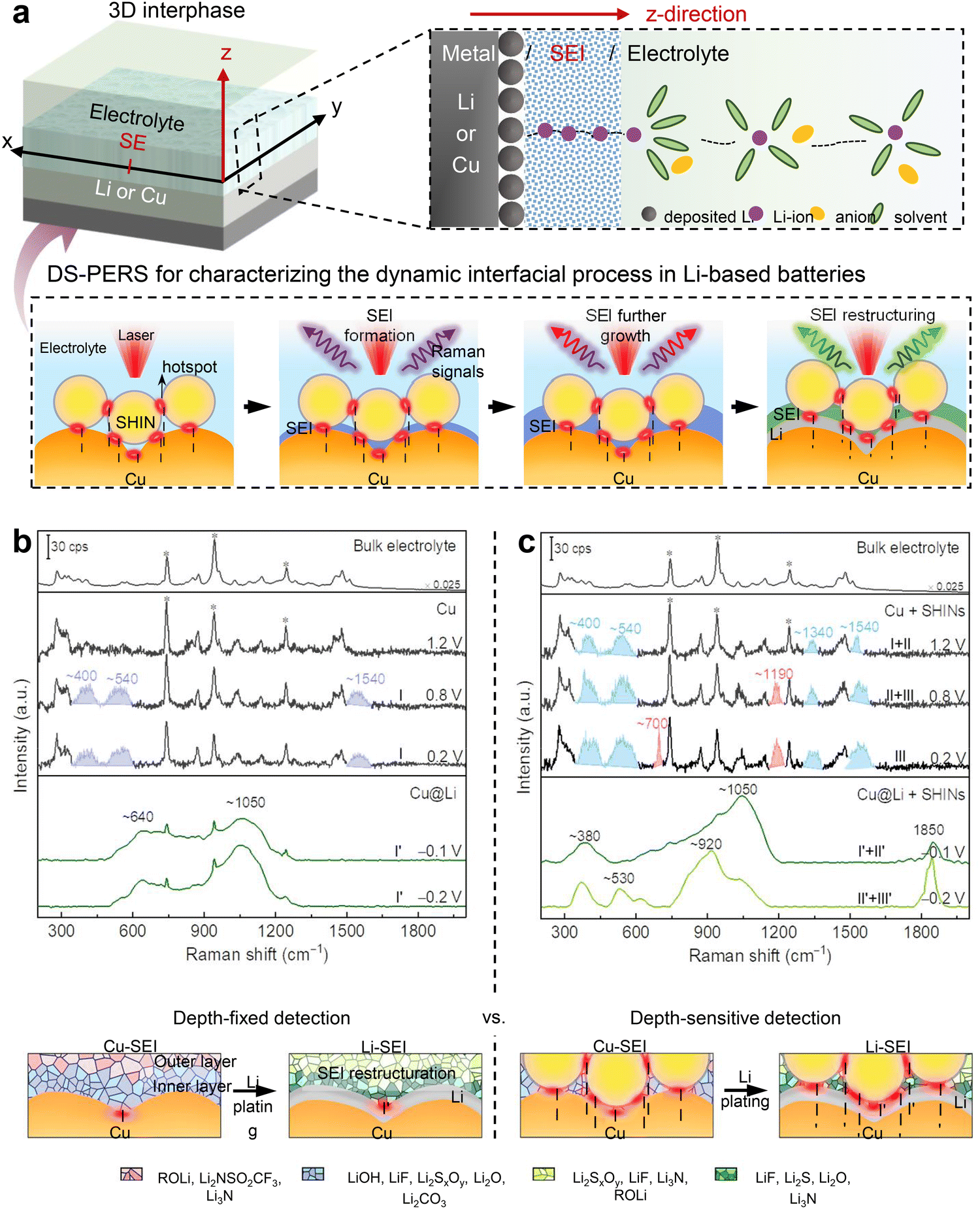 | ||
| Fig. 56 DS-PERS vertical direction sensing of the complex 3D interfacial process. (a) Schematics of DS-PERS for depth-resolved detection of SEI in Li-based batteries. (b) and (c) Comparison of the depth-fixed strategy based on LSPs of nanostructured Cu alone and the depth-sensitive strategy based on synergetic LSPs of integrated Cu-SHINs for in situ characterizing the formation and reconstruction of SEI. In situ Raman spectra showing the formation and evolution of SEIs on the nanostructured Cu substrate (b) and the Cu-SHINs substrates (c) in normal ether-based electrolyte. Reproduced from ref. 568 with permission. Copyright 2023 Springer Nature. | ||
One illustrative example is the spectroscopic analysis of SEI formation on lithium-based battery anodes, which is a prime representation of a complex three-dimensional interphase. The SEI, formed through (electro) chemical reactions between anodes and electrolytes, functions as both an electronic insulator and Li-ionic conductor,571 significantly impacting battery performance.572 However, characterizing SEIs presents significant challenges due to their thin, nonuniform nature, complex composition, and dynamic evolution during cycling. Additionally, analyzing these reactive surfaces within organic electrolytes requires precise spectroscopic measurement techniques.
To tackle this complexity, direct in situ and real-time methods with structure-specific and depth-sensitive features are essential. These methods are crucial for tracking the dynamic interfacial processes of the anode, guiding the rational enhancement of SEIs in lithium-based batteries. Recently, Mao, Cui, Tian and coworkers devised and developed a unique in situ depth-sensitive plasmon-enhanced Raman spectroscopy (DS-PERS) method, integrating SERS and SHINERS for real-time characterization of SEIs of Li metal anodes.568,573 This method integrated two techniques based on the “borrowing SERS activity” strategy and utilized the combined LSP effects of nanostructured Cu, Li deposits and SHINs, as shown in Fig. 56.
The synergistic plasmonic enhancement enables depth-sensitive detection of signals from an SEI of tens of nanometers and from the Cu/SEI and Li/SEI interfaces during SEI formation at different stages. The application of the DS-PERS method revealed that for Li metal anodes initiating from bare Cu as the current collector, SEIs are formed sequentially on Cu prior to Li deposition and then on freshly deposited Li during normal galvanostatic polarization. Notably, the primary SEI formed on Cu contains less stable higher-oxidation-state components and undergoes restructuring to finally form the SEI on Li. They also proposed that reducing the duration time in bare Cu state facilitates the rapid and direct formation of thinner SEI on Li enriched with more desirable lower-oxidation-state components.
This advancement in understanding the formation mechanisms of SEIs and the significant impact of metallic Li offers insights for optimizing operational procedures for Li anodes. Furthermore, the DS-PERS offers a valuable approach for the nondestructive characterization of intricate nanoscale interphases and interfaces, delivering high spatial information. This capability addresses significant challenges encountered across various disciplines, including materials science and energy science and technology. It is crucial to recognize that complex three-dimensional interfacial phases like the SEI are constantly evolving with different battery systems. The spectral information obtained can be highly intricate, and the development of AI-assisted SERS/SHINERS techniques have the potential to illuminate the complexities of interfaces/interphases further.
5.3 Quantitative analysis
A key parameter that is often overlooked is the analyte's surface affinity. In colloidal SERS, the analyte molecules are frequently required to displace a surfactant or traverse a stabilizing/capping layer that is present on the surface of the nanoparticles. As a result, the SERS signal is highly dependent upon the interaction affinity of the analyte with the nanoparticle's surface. Weakly adsorbing molecules will give lower SERS intensities compared to stronger adsorbing ones. This may be interpreted wrongly by a lower SERS EF of the plasmonic substrate. Selecting a ‘neutral’ analyte that exhibits a potentially high binding affinity, along with the capacity to displace existing surface species or to co-adsorb, may present a viable solution. The Table 1 in ref. 473 contains a list of potential SERS reporter/probe molecules that are considered to be useful, together with a short description of their respective strengths and limitations. Briefly, 4-mercaptobenzoic acid (4-MBA), which is deprotonated at pH 10, along with adenine, are recommended as standard Raman reporters due to their strong adsorption characteristics and non-resonant behavior. Dyes such as R6G and CV are not recommended. In the future, the SERS community will have to decide on less-strongly adsorbing compounds that complement this initial suggestion.
The SERS EF is a key figure of merit in factor (ii) above. Variations in the EF can be alleviated through the incorporation of a standard whose response is affected in a manner consistent with that of the target analyte in response to experimental fluctuations. By using such a standard, the target SERS signal can then be normalized to that of the internal standard. Isotopologues are the most obvious internal standards. Alternatively, chemically similar compounds or standard addition may be employed. Importantly, by such an appropriate internal intensity standard, quantitative SERS analysis can be achieved even without very highly reproducible substrates. With respect to good analytical practice, separate calibration and validation steps are essential. SERS-based detection scheme without a validation step should be questioned regarding its relevance to an analytical application. A significant issue with quantitative SERS measurements is reproducibility. Generally, repeated measurements are necessary for confirming reproducibility. Suggestions for both colloidal and solid SERS substrates are available.473
Overall, three aspects may be recommended:473 (a) to characterize the solid or colloidal SERS substrate through the integration of correlative electron microscopy, optical microscopy, and spectroscopy techniques; (b) to evaluate the SERS EF, incorporating appropriate Raman reporter or probe molecules; and (c) to implement stringent analytical practices throughout the research process. Nevertheless, we are just at the beginning and a substantial collaborative effort towards SERS metrology is needed until, in the hopefully not-too-distant future, we may call SERS a truly robust and quantitative tool for molecular sciences.
In 2023, Dong and coworkers utilized the LT-UHV-TERS system to investigate such chemical effects through TERS of a single planar ZnPc molecule with varying but controlled contact environments.575 The TERS signals obtained from zinc phthalocyanine (ZnPc) that was chemisorbed in a flat orientation onto the metal substrates exhibited significant quenching. This phenomenon can be attributed to the screening effect of Raman polarizability, which arises from dynamic CT at the interface, as illustrated in Fig. 57. However, the quenched Raman signals can be “rescued” and enhanced in the tip-molecule point contact configuration. These results suggest that the so-called chemically enhanced Raman signals, typically attributed to CT effects, in fact, is the result of a combination of chemical (CT) and physical (electromagnetic field) effects, rather than solely chemical in origin. Additionally, the commonly considered resonance CT enhancement mechanism may be challenging to achieve in many systems, and the enhancement effect may still arise from the influence of ground-state chemical interactions on the Raman polarizability and its orientation. Thus, a comprehensive understanding and consideration of both electromagnetic and chemical enhancement mechanisms are crucial for realizing precise quantitative analysis in SERS.
 | ||
| Fig. 57 Quenching of TERS signals induced by the face contact with a metal substrate. (a) Quenched TERS signals acquired from ZnPc adsorbed on Ag(100) without tip–molecule contact (black curve), in sharp contrast to the dramatically enhanced TERS spectrum measured on a NaCl-decoupled ZnPc with tip–molecule contact (red curve). (b) Waterfall plot for TERS spectra from ZnPc/Ag(100) as a function of tip displacements showing dramatic spectral changes upon the tip–molecule contact. (c) Upper panel: Schematic illustrating the electromagnetic (left) and chemical (right) quenching mechanisms for ZnPc flatly lying on Ag(100). Lower panel: Schematic illustrating the mechanism of TERS enhancement caused by the tip–molecule point contact. Figures are reproduced from ref. 575 with permission. Copyright 2023 Wiley. | ||
In 2018, Brolo and coworkers demonstrated the concept of digital analysis in SERS measurements by combing SM-SERS and a bi-analyte strategy, as shown in Fig. 58, realizing a LOQ at pM level for enrofloxacin and ciprofloxacin.578 Later, Trau, Wuethrich, Lobb and coworkers extended digital SERS to biomedical research and achieved the attomolar level sensitivity by utilizing a pillar array SERS substrate to hold the single cytokine and single-particle active SERS nanotags for cytokine identification and counting.579
 | ||
| Fig. 58 The digital analysis in SERS measurement. (a) Schematics of the experimental procedure. (b) Histogram showing the frequency of normalized scores from “factor 1”, which corresponds to the spectral signature of the analyte. The data is obtained from high concentration regime and (c) ultralow concentration regime. (d) Digital calibration curve at ultralow concentration. Figures are adapted from ref. 578 with permission. Copyright 2018 American Chemical Society. | ||
Recently, Ye and coworkers further advanced digital SERS to femtomolar-level sensitivity with improved reproducibility for various molecules by using colloidal SERS substrate and leveraging advanced data analysis.577 Shortly, Li, Xu and their colleagues reported a statistical route to SM-SERS quantitation by gauging SERS probability.580 The rise of statistical quantitative spectroscopy by digital counting, that is, gauging SERS probability rather than traditional intensity, would bring a new quantitation tool for ultra-sensitive SERS detection and analysis.
The digital SERS approach can also be applied in immunoassays based on SERS probes.581 Moreover, instead of using only the peak intensity from a single peak in the spectrum to count “0” and “1”, Schultz and coworkers considered the overall contribution of the whole spectral information by introducing multivariate curve resolution with an established score threshold.582 Note that in digital-SERS, the deviation of quantitative analysis is quite large when expanding the lower limit of the SERS linear range to its lowest physical limit, although this error can be minimized by increasing the number of counts.582
5.4 Development of SERS methods in bio research
The high sensitivity and unique fingerprint information provided by SERS justify its lively applications in bioanalysis.31,492,501,583–586 SERS in bio research can be tracked back as early as 1980, when Cotton et al. reported the SERRS from cytochrome c and myoglobin adsorbed on a silver electrode.169 Following the nano-driven SERS starting in mid-1990s, an upsurge of bio-related SERS researches started in about mid-2000s, and the number of related publications is ca. a quarter of total SERS publications in recent five years. The bio-related applications of SERS primarily focus on hierarchical levels of samples, including biomolecules, biofluids, living cells, and living organisms.501,587It is noteworthy that in the adaptation process to bio-related targets/environments, SERS has been evolving in both SERS substrates and combining various contemporary techniques such as microdevices and microfluidics,586 3D printing,501 machine learning587 to strengthen its power in bio-related detections. In particular, the increasing integration of SERS into point-of-care testing (POCT) allows for real-time analysis of on-site samples by SERS.588–590 This progress has been facilitated by advancement in portable Raman spectrometers and microdevices such as lateral flow assay (LFA) strips and microfluidic channels.586 All these advancements have significantly contributed to the steady progress of SERS toward precise clinical diagnostics and high-throughput omics research.
Unlike carbohydrates, proteins bare relatively large cross section; however, they suffer from complex composition and adsorption configuration, leading to broad and irreproducible spectra. Moreover, except proteins with cofactors, most proteins still require higher concentration to achieve detectable signal, necessitating purification from biological fluids to avoid dominance by concentrated and strongly adsorbing species. To address these issues, Ozaki, Zhao and colleagues conducted systematic tests,599,600 and proposed a technique called “Western SERS,” which replaces the standard silver staining step with silver colloid staining.601 This approach allows colloids to adsorb to the protein blots and enables subsequent SERS analysis of the separated proteins. To improve reproducibility, Ren group reported a method by utilizing Ag NPs with iodide monolayers to eliminate protein denaturation for more reproducible label-free SERS detection.602 It is found that iodide's strong adsorption interactions with metal surfaces improved SERS signal reproducibility by preventing structural changes in proteins upon adsorption. Based on this substrate, Yang and colleagues further proposed dynamic SERS to form controllable hotspot taking advantage of solvent evaporation and nanocapillary pumping,295 which has shown promise for dynamic detection of native proteins.603
Regarding nucleic acids detection, Bell group obtained the SERS spectra for all the DNA/RNA mononucleotides with Ag NPs.592 However, DNA detection faces similar problems as that of proteins, which leads to bad spectral reproducibility. In this context, Halas group developed a series of methods to achieve stable conformations and improve reproducibility.591,604 Additionally, surface modification plays a crucial role; for instance, Abell and colleagues utilized mercaptohexanol molecules as spacers to protect the substrate and enhance hybridization,605 while Graham and colleagues introduced spermine to facilitate DNA adhesion and trigger aggregation in colloidal solutions.606 Thereafter, the strategies have been substantially expanded as summarized by a comprehensive review by Guerrini, Alvarez-Puebla and colleagues.607
The constitution of living organisms, particularly living cells, can be directly analyzed as well. By detecting SERS spectra of pathogens or intracellular species, identifying, classifying cells, and monitoring molecular evolution can be achieved. For instance, Popp and colleagues applied SERS-based microfluidic techniques into bacterial strain identification, enabling high spectral acquisition throughput and high reproducibility.608 Moreover, Ziegler group introduced statistical methods and optimized the procedures for monitoring evolution of different cells.609–611 Additionally, SERS-active solid substrates such as nanopipette, have been recently developed for precisely detecting and monitoring of cellular responses in real time.612
To date, these methods have expanded the application of SERS in bioanalysis from the quantitative and qualitative detection of biomolecules to the analysis of cells, paving the way for new applications in structural biology and metabolomics of living cells and organisms.501,613–616 Moreover, recent advancements have demonstrated its application to the discovery of novel communication mechanisms between cancer cells617 and the immune system.618 Nevertheless, the application of direct detection is limited, owing to both the intrinsically low sensitivity of some biomolecules and the complex spectral information involving the entire biological system. This challenge is further compounded when attempting to measure local pH and the temperature inside a cell. Recent developments in this direction have shown that 3D cell models can be constructed by means of 3D printing, allowing not only to replicate the cellular environment but also to incorporate plasmonic nanoparticles as SERS substrates.501 In a representative example, Liz-Marzán and colleagues have developed a microfluidic chip that allows evaluation of drug efficacy by monitoring drug diffusion by SERS in correlation with cell death (see Fig. 59).619
 | ||
| Fig. 59 (a) SEM image of a 3D printed hydrogel-AuNR nanocomposite scaffold. (b) A set-up integrating a nanocomposite scaffold with a tumor cell environment (Matrigel) and a reservoir for drug delivery (right). (c) Methylene blue diffusion patterns along the scaffold containing Matrigel and cancer cells. (d) Confocal fluorescence microscopy evaluation of cell death before (right) and 2 hours after (left) methylene blue injection. Adapted with permission from ref. 619. Copyright 2021 American Chemical Society. | ||
To acquire better analyte SERS signals requires well-designed substrates, in this regard, Nam and coworkers synthesized an Au-Ag bimetallic nanosnowman structure with a nanocrevice at the junction between Au and Ag.620,621 Anisotropic growth of Ag bodies on an Au core was achieved by tuning salt concentration to hinder Ag nucleation. Another method for selective nucleation was based on the intrinsic morphological characteristics of core particles. Because ligand packing density is lower at regions of higher curvature, deposition is facilitated at, for example, nanorod tips and nanocube corners. On nanorods, this principle was used to synthesize Au dual-gap nanodumbbells (AuDGNs), with dumbbell-like head structures grown on both tips (Fig. 60a and b).622 Open nanogaps were present between the core and dumbbell shell, as well as between the dumbbells. Similarly, corner-specific growth on Au nanocubes generated open cross-gap (OXNCs) with eight nanocubes grown onto the central cube (Fig. 60d and e).623 The AuDGN was capable of label-free discrimination of DNA bases (Fig. 60c), while the OXNC could detect the molecular fingerprint Raman spectra of various proteins (Fig. 60f).
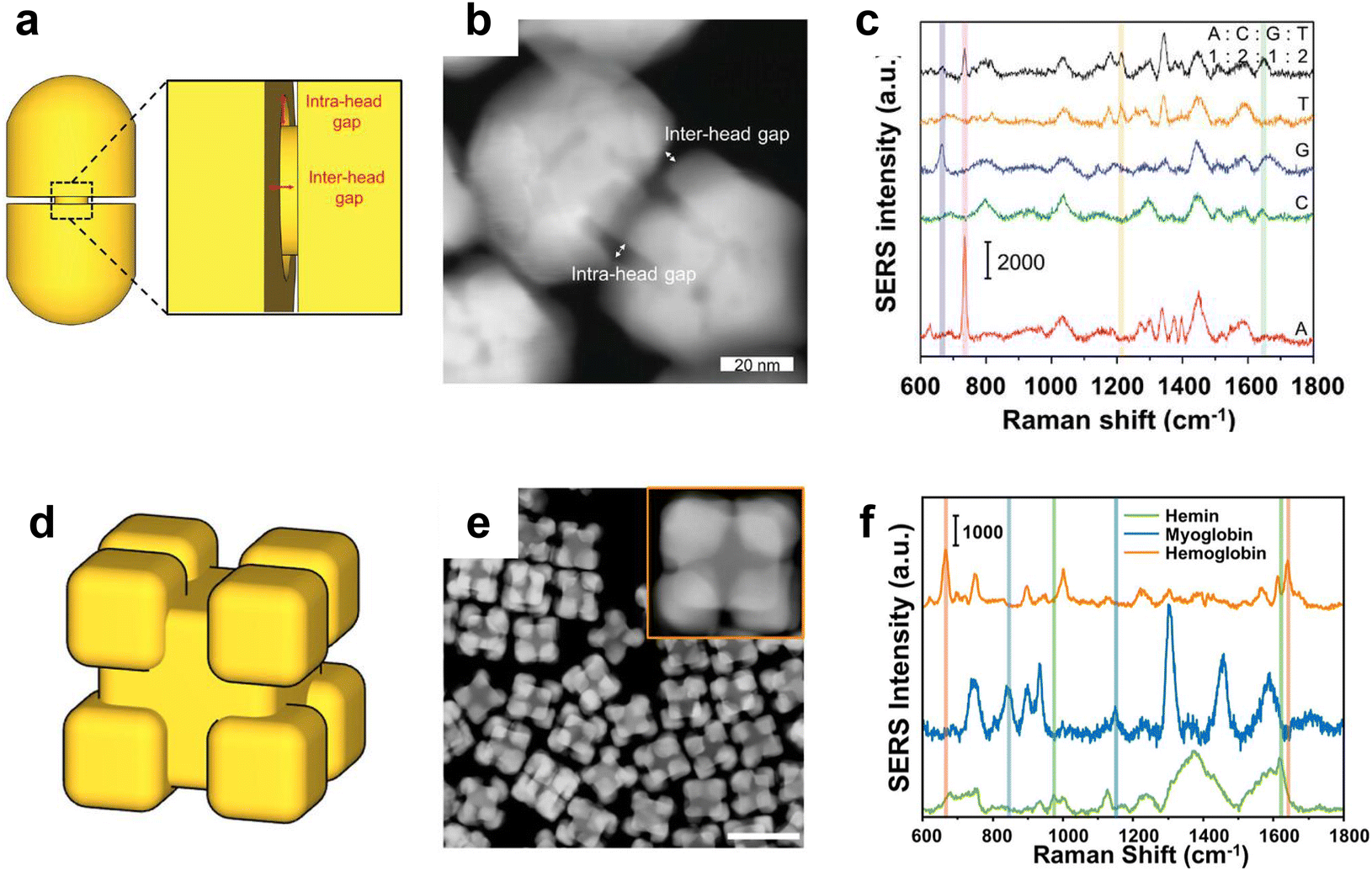 | ||
| Fig. 60 (a) Schematic of AuDGNs with controllable gap sizes. (b) HAADF-STEM image of AuDGN showing both inter and intra-head gaps. (c) SERS spectra of the four nucleobases. Pink, green, blue, and orange lines indicate fingerprint SERS peaks of adenine (A), cytosine (C), guanine (G), and thymine (T), respectively. The concentrations of A, C, G, and T are 0.01, 0.01, 0.01, and 0.1 mM, respectively. The black line indicates the SERS spectrum of the mixture containing four nucleobases with a 1:2:1:2 (A:C:G:T) ratio. Reproduced from ref. 622 with permission. Copyright 2023 Wiley. (d) Schematic of OXNCs. (e) HAADF-STEM image of OXNCs with a gap size of 5.6 nm approximately. The inset shows a magnified image of the structure. The scale bars indicate 100 nm. (f) SERS spectra of hemin mixed with the OXNC with 2.6 nm gaps, myoglobin with the OXNC with 5.6 nm gaps, and hemoglobin mixed with the OXNC with 5.6 nm gaps. Panel (d)–(f) are reproduced from ref. 623 with permission, copyright 2024 American Chemical Society. | ||
Immunoassay is of special interest in biomedical diagnostics due to its superior specificity. Great efforts have been devoted to combine immunoassay with indirect SERS detection. In 1989, the binding between biomolecules with mutual affinity, including antigen–antibody interactions, was measured by SERRS on a roughened silver film.625 With advances in nanoscience, SERS-based immunoassays on Au NPs were reported by Porter and colleagues in 1999.259 They immobilized antibodies onto Au substrates to capture target antigens, forming sandwich immunocomplexes, and then introduced SERS nanotags labeled with detection antibodies and SERS reporter. The further introduction of immunogold labeling demonstrated ultrasensitive SERS detection for prostate-specific antigen, achieving detection limits down to the femtomolar level.492,626 Subsequently, SERS-based immunoassays have rapidly developed, driven by diverse newly prepared substrate materials and SERS reporters, thus enabling multiplex detection. Its platform encompasses a range of options, including microfluidic chips, paper devices, and optical fibers with various combinations of solid substrates and liquid phases, enabling the analysis of various biological species as well as cells.627
On the other hand, the growing demand for high-throughput, multiplex SERS-based bioassays underscores the importance of integrating various microdevices. In 2005, Choo and colleagues developed a SERS-based microfluidic channel capable of detecting two types of dye-labeled DNA nucleotides.628 They effectively mixed two types of dye-labeled sex-determining Y genes with Ag nanoparticles within a microfluidic channel shape similar to an alligator tooth, adsorbed the target genes onto particle surfaces, and then measured the SERS signals. The result, showed that these genes could be detected with high sensitivity at concentrations as low as 10−11 M. In 2007, Freeman and colleagues introduced a concept of SERS based LFA strip.629 They used three glass-coated SERS nanotags on an LFA strip to form sandwich immunocomplexes and measured their Raman signal intensities, demonstrating the ability to distinguish three different viruses simultaneously. Since the publication of this paper, various SERS-based LFA strips capable of diagnosing different diseases on-site have been developed. In 2008, Sha and colleagues introduced the idea of labeling circulating tumor cells in whole blood with SERS nanotags on their surface biomarker proteins, then fixing and separating these cells using magnetic beads to quantitatively analyze their SERS signal.630 After years of development, numerous strategies have been proposed based on the properties of the analytes, enabling high-throughput separation and SERS detection.586
Despite these advancements, challenges remain in ensuring the reproducibility of SERS enhancement levels for accurate quantitative analysis of target biomarkers. Variations primarily stem from difficulties in controlling particle aggregation and achieving uniform distribution of analytes on the substrate surface.631 To address these reproducibility issues, methods such as Raman mapping for LFA strips and average ensemble measurements for microfluidic sensors have been introduced in SERS-based POCT microdevices.586 Choo and colleagues proposed a method to enhance reproducibility by using Raman mapping for accurate quantitative analysis of target antigens at the test and control lines of the LFA strip.632Fig. 61 presents a schematic illustration of the LFA strip's structure. As the sample containing target antigens and SERS nanotags flowed toward the absorption pad and reached the test line, the antigens were captured by antibodies immobilized on the test line, forming sandwich immunocomplexes. To minimize errors caused by spot-to-spot fluctuations brought by the non-uniform distribution of SERS nanotags on the test line, SERS mapping was applied to different target concentrations. The average Raman spectra of the 1600-pixel points enabled precise quantitative analysis of the target antigen in the SERS-LFA system to improve the reproducibility of SERS-LFA strips.
 | ||
| Fig. 61 SERS-based immunoassays using SERS-LFA: (a) schematic illustration of the structure of SERS-LFA strip. SERS mapping images were acquired using a peak intensity at 1615 cm−1 for nine different target concentrations ranging from 0.1 pg mL−1 to 1000 ng mL−1. For each concentration, images were taken with a resolution of 80 × 20 pixels (1 pixel = 10 μm × 10 μm). The color bar at the bottom indicates changes in Raman peak intensity. (b) Average Raman spectra of 1600-pixel points from the SERS mapping zones. Figures are adapted from ref. 632 with permission. Copyright 2016 Royal Society of Chemistry. | ||
Recently, the integration of machine learning (ML) with SERS has become a game-changer in solving complex real life sample problems. ML algorithms overcome the limitations of manual spectra analysis, enabling quick and accurate interpretation of the data. Critically, ML tools can elucidate subtle signal variances and establish prediction models to rapidly classify and quantify target analytes accurately, especially for multiplexed analytes in complex matrices. For example, Ling and coworkers used PLS-DA to differentiate breath samples of COVID-19 patients from healthy individuals in a case-control study.633 An array of SERS platforms, each functionalized with different molecular receptors, was used to capture and detect different volatile organic compounds in breath samples (Fig. 62). The PLS-DA model achieved a classification sensitivity of 96.2% and specificity of 99.9% for both asymptomatic and symptomatic COVID-19 patients.
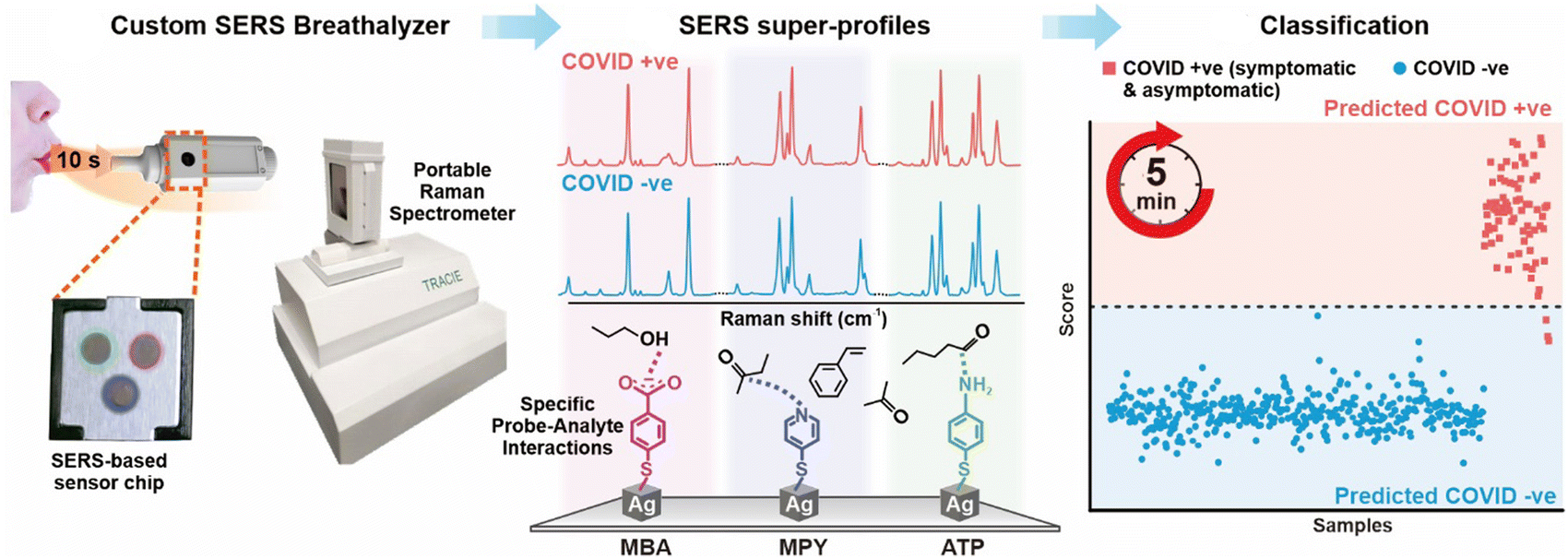 | ||
| Fig. 62 Using multiple molecular receptor-functionalized SERS substrate to capture and detect breath volatile organic compounds related to COVID-19, then using partial least squares discriminant analysis (PLS-DA) to classify SERS spectra based on their COVID-19 infection status. Figures are adapted from ref. 633 with permission. Copyright 2022 American Chemical Society. | ||
To date, practical and reliable bioanalytical SERS have been achievable. However, caution is still warranted regarding SERS bio-detection, owing to the complicated nano-bio-interactions, i.e. chemical and physical interactions between nanomaterials and biological systems, especially in the presence of laser illuminations. These factors also pose significant challenges for spectral analysis and interpretation. As advancements in nanotechnology and machine learning continue to evolve, the future of SERS holds immense potential for even more sophisticated and accessible biosensing solutions, paving the way for next-generation diagnostic tools and comprehensive omics analysis.
Due to space constraints, we are unable to provide an exhaustive review of SERS applications across various fields. Instead, we have compiled a selection of recently published review articles in Table 5 to offer a comprehensive overview of diverse applications of SERS, including surface and interfacial detection, life sciences and medicine, trace analysis and beyond. Readers seeking in-depth discussions on specific SERS applications are encouraged to consult these references for detailed insights into the current state of research and emerging trends in the respective areas.
| Field | Content | Method & strategy | Materials | Reviews | |
|---|---|---|---|---|---|
| Surface & interface | Electrochemistry | Adsorption, reaction process and interfacial structure | Electrochemical roughening, chemical & physical bottom-up or top-down synthesis & assembly | Noble (Au, Ag, Cu) & transition metals (Pt, Pd, Ru, Rh etc.), semiconductors (quantum dots, graphene and MOF etc.) | Ref. 26,32,216,217,269,496,634,635 |
| Catalysis | |||||
| Energy | |||||
| Materials | |||||
| Life science & medicine | Life science & clinic | Biomarker, DNA, protein, cell, microorganism, drug delivery, therapeutic drug monitoring, enzyme catalysis… | Chemical & physical bottom-up or top-down synthesis & assembly, flexible support (paper, membrane, tap, cotton); label-free & labeled method (electrostatic modification, host–guest recognition molecular steric effect, biological recognition, chemical derivatization, stable isotope probing, etc.); lab-on-chip (mill-/micro-/nano-fluidic control) | Metals (Au, Ag, Cu, Pt, Rh, Pd, Ru, Fe, Zn, Al, Li, etc.), semiconductors (0D) (quantum dots), 2D (graphene, MXene, hexagonal boron nitride, graphitic carbon nitride, transition metal dichalcogenide, black phosphorus etc.) and 3D (MOF, COF, etc.), multicomponent (mixture, alloy, core–shell, heterojunction, etc.) | Ref. 31,501,586,589,590,607,627,636–638 |
| Trace analysis | Environmental monitoring | Pollutants in ecosystem (air, water and soil) | Ref. 639–643 | ||
| Food safety | Illegal additive, pesticide, heavy metal ions, bacteria, micro-/nano-plastics | ||||
| Forensics | Natural and synthesized poisons, bacteria | ||||
| Public security | Abused/illicit drugs, explosive, bacterial | ||||
| Others | Art and archaeology | Natural and artificial dyes | Ref. 644,645 | ||
| Aerospace | Biosignatures (e.g., carotenoids, chlorophyll, oxalates), astronaut health monitoring, hazardous contaminants in airborne |
6. Summary and inspirations from the SERS history
The corresponding author of this review was extremely fortunate to enter this field as a PhD student of Fleischmann and to have the opportunity to get to know all the pioneers well starting from 1983. This privileged perspective allowed a deep understanding of how the ‘genetic makeup’ of SERS was shaped by Fleischmann and Van Duyne. These major pioneers together formed the foundation of SERS, turning what seemed impossible into reality and establishing the methodological roots and richness of the field.We meticulously present the history of the discovery of SERS and its foundational era from a methodological standpoint, weaving together the memorial notes of the pioneers themselves, like stringing sparkling pearls into a necklace infused with their scientific spirit and creative methods. We aim to provide a more precise and comprehensive picture, particularly highlighting those who made significant contributions in the first two decades but might be less known to younger generations.
Although there have been over 800 review articles on SERS, our main goal of recounting this long and intricate scientific journey from different perspectives is to raise a fundamental question: What can we learn from the discovery and advancement of SERS for future scientific explorations? We aim to extract the invaluable spirit behind the serendipitous discovery and arduous journey of SERS and distill deep insights and creativity from the pioneers into ten key inspirations, which could serve as treasures for younger generations.
6.1 Collective contribution in opening a new scientific field
Opening a new scientific field, such as the full discovery of the SERS effect, is contributed collectively by several pioneering groups during the intricate early stages of SERS. It constitutes four elementary pieces: The first experimental observation of abnormally intense Raman signals from pyridine adsorbed on a roughened Ag electrode. The first recognition of the huge 105–106 surface enhancement factor. The initial proposal of the surface plasmon mechanism. The naming of SERS. These contributions laid the groundwork for the field and exemplify how collaborative efforts and multiple discoveries can synergistically establish a new area of scientific research.6.2 Challenge to conventional wisdom and transforming the impossible into reality
The story of discovering SERS as the first phase of its 50-year history has been incredibly inspiring and encouraging for younger generations. In the mid-1970s, it was widely believed, based on established textbooks, that Raman spectroscopy could not be a tool for surface analysis. The pioneers, filled with strong curiosity, courageous challenges to impossibilities or authority, expert cross-border cooperation, and perseverance in seeking truth despite negative feedback, proved otherwise. This spirit extended to making other milestones, such as single-molecule SERS, TERS beyond the diffraction limitation, SHINERS for non-SERS active substrates. It demonstrates the power of challenging conventional wisdom and transforming what was once considered impossible into reality.6.3 Learning from the premature and discovery of SERS
It is especially interesting to reflect on the entire journey of SERS, which prematurely emerged about two decades ahead of its time. Without the bold challenges to conventional wisdom posed by its pioneers, the serendipitous discovery of this phenomenon might not have occurred in the mid-1970s. This research would have been much easier to initiate in the mid-1990s, during the boom of nanoscience, as SERS is fundamentally a branch of nanoscience, with its sensitivity and spectral characteristics critically dependent on nanostructures.Accordingly, it becomes much clearer why SERS faced significant challenges and underwent a difficult and complex journey before the mid-1990s. However, this also underscores SERS as one of the oldest branches of plasmon-enhanced spectroscopy, plasmonics, and even the broader field of nanoscience. The first chapter of the SERS story is thus unique and inspiring compared to many other scientific fields. Therefore, all the pioneers and trailblazers who collectively established the foundation of SERS in those early decades deserve our utmost respect and should serve as a source of learning.
6.4 Entering a new field as early as possible
Most of the fundamental concepts and strategies for developing SERS and its relevant subbranches were established between 1974 and 1985. These include the first SERS in roughened electrodes and colloid sols, the first “borrowing SERS activity” strategy, the first dimer as the 'hot spot' concept, and the initial concept of TERS. These pioneering works and far-reaching ideas were recognized and validated only after several decades. In fact, based on the keywords 'SERS' and 'surface-enhanced Raman' in the Web of Science database, only about 860 papers were published during those ten years, while approximately 80000 papers have been published up to now (September 2024). This clearly indicates the significant role played by the pioneers and trailblazers who laid the foundation of SERS. This scenario is common in different fields of science and demonstrates the essence of creativity and pioneering work. It is therefore crucial for anyone to be sensitive to opportunities and enter newly emerging fields as early as possible.
6.5 Significant discoveries are often accompanied by initial incorrect explanations
The phenomenon of great discoveries accompanied by incorrect theories occurs quite often in the history of science because groundbreaking discoveries push the boundaries of what is considered possible, often relying on new mechanisms that extend beyond existing knowledge. As a result, initial explanations based on prior understanding frequently fall short, leading to controversies, and some experimental results may not be reproducible by other groups. However, the scientific community should be more tolerant and open to unconventional views, focusing on scientific discussion and arguments rather than negative or harsh criticism. This openness is essential for healthfully opening a new field, laying groundwork, and making advancements smoothly.6.6 Perseverant searching in times of low tide may lead to triumph
In the second phase (mid-1980s to mid-1990s) of SERS, the wave of excitement was tempered by the realization that the enhancement heavily relied on fine structures (the term “nano” was not yet in use at that time) consisting of specific free-electron metals like Au, Ag, Cu, and Li with poorly characterized and uncontrollable morphologies. The mainstream considered the Achilles' heel of SERS to be its lack of versatility and applicability, which significantly impeded enthusiasm for SERS. Many groups had to leave this field mainly due to a shortage of funding support, while some groups continued to make efforts to break through the seemingly impossible barriers. It is therefore important for anyone not to easily give up in a new and promising field (such as one with extremely high detection sensitivity) even when there are many negative views and uncertainties. Perseverant searching can eventually lead to breakthroughs and significant advancements, although nobody can predict when they will occur. The story of SERS is a testament to the importance of persistence and dedication in scientific research because luck is what happens when preparation meets opportunity.6.7 Proactively embracing and adopting new science and technology
The huge upsurge of SERS from the mid-1990s was accompanied by and driven by the newly established and rapidly developing fields of nanoscience and nanotechnology. This provided an unprecedented opportunity and marked a new era of nano-driven SERS because the SERS effect is one of the phenomena that can truly be described as nanoscience. The SERS activity depends critically on the size, shape, inter-particle space, and optical properties of material nanostructures with tunable extinction, local field enhancement, and excited carriers. With the broad advancement of synthesis, fabrication, and characterization methods for nanomaterials, structural features of SERS-active substrates evolved from discrete size distributions (electrochemically roughened surfaces) to monodisperse nanostructures (nanoparticle-based SERS) and singular nanotips (TERS), from irregular agglomeration (colloid-based SERS) to controlled dimers or multimers (SM-SERS), and from naked surfaces to core–shell nanoparticles (SHINERS). Recent progress in TERS introduced the highest spatial resolution to the angström level, while SHINERS enabled the characterization on various single-crystal surfaces of materials. These advancements allowed for structurally well-defined and rationally designed nanostructures, fostering a deeper understanding of the SERS mechanism.In fact, throughout the entire trajectory of SERS, proactively embracing newly emerging science and technology has been the key to opening new fields and setting milestones. For instance, Raman spectroscopy embraced laser technology in the 1960s, and SERS leveraged nanoscience for rapid and significant enhancements in various aspects in the 1990s. It now appears to be the right time for nano-driven SERS to fully embrace AI science and technology in the 2020s.
6.8 High level and high-quality interdisciplinary collaboration
Given that transformative scientific revolutions may occur about once every half-century, a small number of people can encounter them during the height of their research careers. However, throughout several decades of research, there are still opportunities for breakthroughs in their fields. These require pushing beyond current knowledge boundaries and engaging in interdisciplinary collaboration. Only high-level expert cross-disciplinary collaborations with brainstorming and rational designing can make these breakthroughs successful. Examples include combining electrochemistry with Raman spectroscopy, plasmonics with surface spectroscopy, SERS with scanning probe microscopy, and AI with analytical techniques.It may be necessary to note that nowadays the entry threshold to the SERS field is quite low compared to the very high thresholds for TERS and SHINERS. The synthesis of metal nanoparticles has become so easy and routine that many researchers from different fields widely use SERS-active particles for measurements and publications. Consequently, the high quantity of publications shown in Fig. 17 may not always signify high-quality collaboration or the prosperity of the field. Accordingly, for anyone looking to break developmental bottlenecks in their chosen direction, carefully building their own capabilities and seeking expert collaborators across different disciplines is crucial.
6.9 Comprehensive methodology is the key driver for major advancements
The methodology is always essential for discovery and advancing milestones, a theme prevalent throughout this review. The integration of experimental, theoretical, and instrumental methods is vital for addressing complex challenges and achieving breakthroughs. Nearly a century ago, the discovery of the Raman effect stemmed from what appeared to be a simple, yet meticulously and systematically designed experimental apparatus. Similarly, at the heart of the SERS field is the continuous push to enhance detection sensitivity, evolving from its origins 50 years ago to the latest cutting-edge advancements. This evolution includes the development and design of specialized or entirely new instruments tailored to meet the needs of innovative experiments and to validate emerging theoretical models. These innovations span the full spectrum of instrumental optimization—from macroscopic devices for optical excitation and collection to the microscopic design of plasmon-enhanced nanostructures, as well as instrument-driven data processing software. A prime example is the invention of TERS, where the design and custom construction of scientific instruments played a pivotal role. Thus, the comprehensive and seamless integration of experiment, theory, and instrumentation into a unified methodological approach is not only essential but often decisive for achieving breakthroughs.6.10 Boldly venture out of the comfort zone of your familiar research to tackle more complex challenges
From the broadest perspective, spanning from basic upstream research to market downstream for everyday use by the public, it is regrettable to observe that the transition of SERS into a widely commercially viable technique has been surprisingly sluggish in comparison with the fundamental breakthroughs over the past decades. The reasons for this unexpectedly slow progress may encompass both surface-level issues and deeper underlying factors. Because SERS offers numerous advantages over other optical spectroscopies, researchers often achieve results easily, publish and cite them, and thus remain within their comfort zones without tackling more complex and challenging issues. The deeper-level challenge is the intrinsic instability of nanostructured hot-spots at the 1 to 3 nanometer scale because SERS is scientifically characterized by a tripartite interaction involving nanostructures, photons, and molecules, making the hot-spots even more unstable. Therefore, the high and mixed demands of long-term durability, measurement reproducibility, product batch consistency, and wide viability of commercial SERS technique remain largely unresolved.In fact, this is a grand challenge not only for SERS but also for other nanotechnologies in expediting their transition to commercially viable technologies. However, imagine what pioneers in the SERS field could achieve. They would seize the upcoming AI era as a grand opportunity to spark another revolution. AI-assisted nanostructure guidance and pre-trained AI models can quantitatively establish complex high-dimensional correlations between SERS substrate manufacturing parameters, morphology, and spectral responses. This approach offers the prospect of designing novel SERS nanostructures and devices that have not been explored, addressing the trade-off between sensitivity and stability. Moreover, the consistency of SERS nanostructures can also be dramatically improved by utilizing AI-guided automated manufacturing and measurement systematically. Due to the length limitations of this review, the future AI-Nano-Driven SERS will be presented in detail elsewhere.646
Overall, we hope that all these inspirations encapsulate the essence of the SERS journey and its pioneers, serving as guiding principles for future scientific endeavors as each generation faces grand challenges and emerging opportunities. Looking forward, we foresee that the ongoing emergence of new technologies, such as advancements in nanotechnology over the past 30 years and recent breakthroughs in AI technology, will drive serendipitous discoveries and new applications. These innovations are likely to provide fresh and powerful momentum for the pivotal new phase of SERS development, ensuring its continued prosperity in the coming half-century.
Author contributions
This historical perspective serves as the inaugural paper in the themed collection for Chemical Society Reviews, celebrating the 50th anniversary of SERS, co-edited by Duncan Graham and Zhong-Qun Tian. The writing process occurred in two phases: the structure and main content were first compiled by members of Tian group and collaborators at Xiamen University, while significant enrichment, particularly in Sec. 4 and 5, was collectively contributed by authors from various active groups. The detailed contributions by each author are listed in the ESI.†Data availability
The data of the number of publications in Fig. 17 was searched through publicly available data from the core database of Web of Science. The keywords used for SERS include “Surface-enhanced Raman”, “SERS”, “Surface-enhanced Resonant Raman” and “SERRS”. The keyword used for nano is “Nano”.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to dedicate this commemorative review article specifically to M. Fleischmann and R. P. Van Duyne, the key pioneers and figures of the SERS field. Their groundbreaking work laid the foundation for countless advancements in this area. We also extend our deepest gratitude to other pioneers, P. J. Hendra, J. A. Creighton, M. Moskovits and A. J. McQuillan for their invaluable comments and suggestions regarding the discovery of SERS, and for generously providing their photographs for this review. We also express our heartfelt gratitude to some trailblazers, A. Otto, E. Burstein, J. Lombardi, B. Pettinger, K. Kerker, H. Metiu, A. Nitzan, R. K. Chang, T. M. Cotton, J. E. Pemberton, W. E. Smith, M. J. Natan, M. J. Weaver, S. Kawata, D. Irish, P. G. Etchegoin, V. Shalaev, M. Käll and F. Javier García de Abajo et al. for their important contributions to the SERS field, which have inspired and helped the authors in the past four decades. Whenever work from the authors’ groups is mentioned, we express our deep gratitude to the self-motivated and hard-working students, as well as all other group members, for their invaluable contributions over the past four decades. We sincerely thank L. Tay and R. N. Zare for their valuable contributions and helpful suggestions to the article. The work mentioned has been made possible by continuous financial support on (NSFC 91427304, 21533006, 21321062, 21522508, 20021002, 21473140, 22272140, 22202162, 22272139, 22032004, and 22372072), (MOST 2015CB932300), (CAS 2023-ZW03-A-014). J. Aizpurua acknowledge support from project PID2022-139579NB-I00 funded by Spanish Ministry of Science and Innovation, IT 1526-22 from the Basque Government, and Elkartek project u4Smart of the Department of Economy of the Basque Country. J. Choo acknowledges support from National Research Foundation of Korea (grant numbers 2020R1A5A1018052 and RS-2024-00352256). Z. C. Dong, Yang Zhang, Yao Zhang acknowledge support from Innovation Program for Quantum Science and Technology (2021ZD0303301). N. Halas and P. Nordlander acknowledge support from the Robert A. Welch Foundation under grants C-1220 and C-1222. J.-M. Nam acknowledges the support by the National Research Foundation of Korea (NRF) grants funded by the Korea Government (MSIT) (NRF-2021R1A2C3010083 and No. RS-2024-00397807). V. Deckert acknowledges support from the German Science Foundation via the CRC 1375 NOA and the CRC 234 CataLight.Notes and references
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- M. Moskovits, J. Chem. Phys., 1978, 69, 4159–4161 CrossRef CAS.
- R. P. Van Duyne, in Chemical and Biochemical Applications of Lasers, ed. C. B. Moore, Academic Press, 1979, pp. 101–185 DOI:10.1016/B978-0-12-505404-1.50009-X.
- A. Smekal, Naturwissenschaften, 1923, 11, 873–875 CrossRef CAS.
- C. V. Raman and K. S. Krishnan, Nature, 1928, 121, 501–502 CrossRef CAS.
- C. V. Raman, Indian J. Phys., 1928, 2, 387–398 CAS.
- G. S. Landsberg and L. I. Mandelstam, Naturwissenschaften, 1928, 16, 557–558 CrossRef CAS.
- G. Landesberg and L. Mandelstam, Compted Rendus, 1928, 187, 109–110 Search PubMed.
- R. Singh and F. Riess, Notes Records R. Soc. London, 2001, 55, 267–283 CrossRef.
- E. C. Le Ru and P. G. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy and Related Plasmonic Effects, Elsevier, Amsterdam, 2009 Search PubMed.
- L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085–7107 RSC.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- R. M. Stöckle, Y. D. Suh, V. Deckert and R. Zenobi, Chem. Phys. Lett., 2000, 318, 131–136 CrossRef.
- M. S. Anderson, Appl. Phys. Lett., 2000, 76, 3130–3132 CrossRef CAS.
- N. Hayazawa, Y. Inouye, Z. Sekkat and S. Kawata, Opt. Commun., 2000, 183, 333–336 CrossRef CAS.
- B. Pettinger, G. Picardi, R. Schuster and G. Ertl, Electrochemistry, 2000, 68, 942–949 CrossRef CAS.
- J.-F. Li, Y. F. Huang, Y. Ding, Z.-L. Yang, S. B. Li, X.-S. Zhou, F.-R. Fan, W. Zhang, Z.-Y. Zhou, D.-Y. Wu, B. Ren, Z. L. Wang and Z.-Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- D. Graham and K. Faulds, Chem. Soc. Rev., 2008, 37, 1042–1051 RSC.
- I. A. Larmour and D. Graham, Analyst, 2011, 136, 3831–3853 RSC.
- G. McNay, D. Eustace, W. E. Smith, K. Faulds and D. Graham, Appl. Spectrosc., 2011, 65, 825–837 CrossRef CAS PubMed.
- B. Sharma, R. R. Frontiera, A.-I. Henry, E. Ringe and R. P. Van Duyne, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- S. McAughtrie, K. Faulds and D. Graham, J. Photochem. Photobiol., C, 2014, 21, 40–53 CrossRef CAS.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- S. Laing, L. E. Jamieson, K. Faulds and D. Graham, Nat. Rev. Chem., 2017, 1, 0060 CrossRef CAS.
- R. Panneerselvam, G.-K. Liu, Y.-H. Wang, J.-Y. Liu, S.-Y. Ding, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Chem. Commun., 2018, 54, 10–25 RSC.
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- X. X. Han, R. S. Rodriguez, C. L. Haynes, Y. Ozaki and B. Zhao, Nat. Rev. Methods Primers, 2022, 1, 87 CrossRef.
- T. Itoh, M. Procházka, Z.-C. Dong, W. Ji, Y. S. Yamamoto, Y. Zhang and Y. Ozaki, Chem. Rev., 2023, 123, 1552–1634 CrossRef CAS PubMed.
- Z.-Q. Tian, B. Ren, J.-F. Li and Z.-L. Yang, Chem. Commun., 2007, 3514–3534, 10.1039/B616986D.
- R. Singh and F. Riess, Curr. Sci., 1998, 74, 1112–1115 Search PubMed.
- M. H. Harper, Appl. Opt., 1962, 1, 139–146 CrossRef.
- G. J. Thomas, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 1–27 CrossRef CAS PubMed.
- R. L. McCreery, Raman Spectroscopy for Chemical Analysis, 2000, pp. 373–413 DOI:10.1002/0471721646.ch13.
- L. P. Choo-Smith, H. G. M. Edwards, H. P. Endtz, J. M. Kros, F. Heule, H. Barr, J. S. Robinson Jr, H. A. Bruining and G. J. Puppels, Biopolymers, 2002, 67, 1–9 CrossRef CAS PubMed.
- A. E. Grow, L. L. Wood, J. L. Claycomb and P. A. Thompson, J. Microbiol. Methods, 2003, 53, 221–233 CrossRef CAS PubMed.
- R. Hochleitner, N. Tarcea, G. Simon, W. Kiefer and J. Popp, J. Raman Spectrosc., 2004, 35, 515–518 CrossRef CAS.
- H. G. M. Edwards, P. Vandenabeele and P. Colomban, Raman Spectroscopy in Cultural Heritage Preservation, 2023 Search PubMed.
- P. J. Hendra, J. R. Horder and E. J. Loader, J. Chem. Soc. A, 1971, 1766–1770, 10.1039/J19710001766.
- P. J. Hendra, I. D. M. Turner, E. J. Loader and M. Stacey, J. Phys. Chem., 1974, 78, 300–304 CrossRef CAS.
- T. A. Egerton and A. H. Hardin, Catal. Rev., 1975, 11, 71–116 CrossRef CAS.
- H. Gerischer, R. F. Broman, W. R. Heineman, T. Kuwana, W. J. Albery, M. D. Archer, N. J. Field, A. D. Turner, G. C. Barker, D. McKeown, M. J. Williams, G. Bottura, V. Concialini, Yu. V. Pleskov, Z. A. Rotenberg, V. V. Eletsky, V. I. Lakomov, R. Parsons, A. A. Vlćek, G. Porter, A. Bewick, B. Kastening, B. R. Eggins, R. M. Reeves, Frank R. Smith, J. W. Schultze, I. E. Epelboin, D. J. Schiffrin, A. W. B. Aylmer-Kelly, P. R. Cantrill, A. M. Tuxford, O. R. Brown, B. E. Conway, J. D. E. McIntyre, W. F. Peck, D. M. Kolb, I. Bergman, C. L. Gardner, E. J. Casey, M. A. Barrett, H. Angerstein-Kozlowska, M. Keddam, A. J. McQuillan, J. S. Clarke, A. T. Kuhn, W. J. Orville-Thomas, W. Davison, J. A. Harrison, J. Thompson, P. Bindra, M. Fleischmann, J. W. Oldfield, D. Singleton, A. R. Despić, M. Vuković, V. Pravdić, H. R. Thirsk, F. C. Ho, J. Klinger, B. MacDougall, S. Gottesfeld, E. Gileadi, A. Capon, K. J. Vetter, H. P. Dhar, R. D. Armstrong, R. E. Firman, J. C. Lestrade, S. Bruckenstein, J. Comeau, L. Pospíšil, J. Volke, O. Manousěk, A. Rusina, J. Heitbaum, D. J. Brown, L. N. Nekrasov, A. H. Davis, A. J. Mason, J. K. Dohrmann, F. Gallusser, H. Wittchen, B. Gostiša-Mihelćić, J. Divišek, A. J. Bard, V. J. Puglisi, J. V. Kenkel, A. Lomax, R. E. W. Jansson, R. D. Rieke and J. E. B. Randles, Faraday Discuss. Chem. Soc., 1973, 56, 1–383 RSC.
- R. E. White, J. O. M. Bockris, B. E. Conway and E. Yeager, Comprehensive Treatise of Electrochemistry, Springer US, Boston, MA, 1984 Search PubMed.
- H. D. Abruña, Electrochemical Interfaces: Modern Techniques for In-Situ Interface Characterization, VCH Publishers Inc., New York, Weinheim, Cambridge, 1991 Search PubMed.
- J. Lipkowski and P. N. Ross, Adsorption of Molecules at Metal Electrodes, VCH, Weinheim, New York, 1992 Search PubMed.
- D. Pletcher, Z.-Q. Tian and D. E. Williams, Developments in Electrochemistry, Wiley, online, 2014 Search PubMed.
- C.-Y. Li and Z.-Q. Tian, Chem. Soc. Rev., 2024, 53, 3579–3605 RSC.
- A. J. McQuillan, Notes Records R. Soc., 2009, 63, 105–109 CrossRef.
- P. Hendra, Internet J. Vib. Spectrosc., 1999, 4(2), 1 Search PubMed , https://www.irdg.org/ijvs/volume-4-edition-2/editorial.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, J. Chem. Soc., Chem. Commun., 1973, 80–81, 10.1039/C39730000080.
- P. Hendra, Analyst, 2016, 141, 4996–4999 RSC.
- I. Bergman, B. E. Conway, A. Bewick, T. Kuwana, J. D. E. McIntyre, C. L. Gardner, E. J. Casey, M. A. Barrett, H. Angerstein-Kozlowska, D. M. Kolb, M. Keddam, A. J. McQuillan, J. S. Clarke, A. T. Kuhn and W. J. Orville-Thomas, Faraday Discuss. Chem. Soc., 1973, 56, 152–170 RSC.
- G. C. Schatz, J. Raman Spectrosc., 2021, 52, 268–278 CrossRef CAS.
- C. L. Haynes, C. R. Yonzon, X. Zhang and R. P. Van Duyne, J. Raman Spectrosc., 2005, 36, 471–484 CrossRef CAS.
- D. L. Jeanmaire, M. R. Suchanski and R. P. Van Duyne, J. Am. Chem. Soc., 1975, 97, 1699–1707 CrossRef CAS.
- T. E. Furtak and J. Reyes, Surf. Sci., 1980, 93, 351–382 CrossRef CAS.
- in, Surface Enhanced Raman Scattering, ed. R. K. Chang and T. E. Furtak, Plenum Press, New York and London, 1982 Search PubMed.
- T. E. Furtak, J. Electroanal. Chem., 1983, 150, 375–388 CrossRef CAS.
- M. Kerker, Acc. Chem. Res., 1984, 17, 271–277 CrossRef CAS.
- H. Metiu and P. Das, Annu. Rev. Phys. Chem., 1984, 35, 507–536 CrossRef CAS.
- A. Otto, in Light Scattering in Solids IV: Electronics Scattering, Spin Effects, SERS, and Morphic Effects, ed. M. Cardona and G. Güntherodt, Springer Berlin Heidelberg, Berlin, Heidelberg, 1984, pp. 289–418 DOI:10.1007/3-540-11942-6_24.
- G. C. Schatz, Acc. Chem. Res., 1984, 17, 370–376 CrossRef CAS.
- A. Campion, Annu. Rev. Phys. Chem., 1985, 36, 549–572 CrossRef CAS.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783 CrossRef CAS.
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC.
- M. J. Dignam and M. Moskovits, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 65–78 RSC.
- J. A. Creighton, M. G. Albrecht, R. E. Hester and J. A. D. Matthew, Chem. Phys. Lett., 1978, 55, 55–58 CrossRef CAS.
- M. Moskovits, Notes Records R. Soc., 2012, 66, 195–203 CrossRef.
- J. A. Creighton, Notes Records R. Soc., 2009, 64, 175–183 CrossRef.
- U. Laor and G. C. Schatz, Chem. Phys. Lett., 1981, 82, 566–570 CrossRef CAS.
- U. Laor and G. C. Schatz, J. Chem. Phys., 1982, 76, 2888–2899 CrossRef CAS.
- P. K. Aravind, A. Nitzan and H. Metiu, Surf. Sci., 1981, 110, 189–204 CrossRef CAS.
- P. K. Aravind and H. Metiu, Surf. Sci., 1983, 124, 506–528 CrossRef CAS.
- M. Kerker, Appl. Opt., 1979, 18, 1180–1189 CrossRef CAS PubMed.
- M. Kerker, D.-S. Wang and H. Chew, Appl. Opt., 1980, 19, 4159–4174 CrossRef CAS PubMed.
- J. Gersten and A. Nitzan, J. Chem. Phys., 1980, 73, 3023–3037 CrossRef CAS.
- W. H. Weber and G. W. Ford, Phys. Rev. Lett., 1980, 44, 1774–1777 CrossRef CAS.
- S. L. McCall and P. M. Platzman, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 1660–1662 CrossRef CAS.
- S. L. McCall, P. M. Platzman and P. A. Wolff, Phys. Lett. A, 1980, 77, 381–383 CrossRef.
- F. W. King, R. P. Van Duyne and G. C. Schatz, J. Chem. Phys., 1978, 69, 4472–4481 CrossRef CAS.
- S. Efrima and H. Metiu, J. Chem. Phys., 1979, 70, 1602–1613 CrossRef CAS.
- S. Efrima and H. Metiu, J. Chem. Phys., 1979, 70, 1939–1947 CrossRef CAS.
- S. Efrima and H. Metiu, J. Chem. Phys., 1979, 70, 2297–2309 CrossRef CAS.
- M. Moskovits, J. Chem. Phys., 1982, 77, 4408–4416 CrossRef CAS.
- M. Kerker, D. S. Wang and H. Chew, Appl. Opt., 1980, 19, 3373–3388 CrossRef CAS PubMed.
- E. Kretschmann and H. Raether, Z. Naturf. A, 1968, 23, 2135–2136 CrossRef CAS.
- A. Otto, Z. Phys. A Hadrons Nuclei, 1968, 216, 398–410 CrossRef CAS.
- H. Raether, in Surface Plasmons on Smooth and Rough Surfaces and on Gratings, ed. H. Raether, Springer Berlin Heidelberg, Berlin, Heidelberg, 1988, pp. 4–39 DOI:10.1007/BFb0048319.
- B. Pettinger, A. Tadjeddine and D. M. Kolb, Chem. Phys. Lett., 1979, 66, 544–548 CrossRef CAS.
- Y. J. Chen, W. P. Chen and E. Burstein, Phys. Rev. Lett., 1976, 36, 1207–1210 CrossRef CAS.
- B. Pettinger, U. Wenning and H. Wetzel, Surf. Sci., 1980, 101, 409–416 CrossRef CAS.
- R. Dornhaus, R. E. Benner, R. K. Chang and I. Chabay, Surf. Sci., 1980, 101, 367–373 CrossRef CAS.
- H. W. K. Tom, C. K. Chen, A. R. B. de Castro and Y. R. Shen, Solid State Commun., 1982, 41, 259–262 CrossRef CAS.
- M. R. Philpott, J. Chem. Phys., 1975, 62, 1812–1817 CrossRef CAS.
- J. C. Tsang, J. R. Kirtley and J. A. Bradley, Phys. Rev. Lett., 1979, 43, 772–775 CrossRef CAS.
- A. Girlando, M. R. Philpott, D. Heitmann, J. D. Swalen and R. Santo, J. Chem. Phys., 1980, 72, 5187–5191 CrossRef CAS.
- J. R. Kirtley, S. S. Jha and J. C. Tsang, Solid State Commun., 1980, 35, 509–512 CrossRef CAS.
- J. Wessel, J. Opt. Soc. Am. B, 1985, 2, 1538–1541 CrossRef CAS.
- X. Wang, S.-C. Huang, T.-X. Huang, H.-S. Su, J.-H. Zhong, Z.-C. Zeng, M.-H. Li and B. Ren, Chem. Soc. Rev., 2017, 46, 4020–4041 RSC.
- J. Billmann and A. Otto, Solid State Commun., 1982, 44, 105–107 CrossRef CAS.
- I. Pockrand, J. Billmann and A. Otto, J. Chem. Phys., 1983, 78, 6384–6390 CrossRef CAS.
- A. Otto, J. Billmann, J. Eickmans, U. Ertürk and C. Pettenkofer, Surf. Sci., 1984, 138, 319–338 CrossRef CAS.
- H. Ueba and S. Ichimura, J. Chem. Phys., 1981, 74, 3070–3075 CrossRef CAS.
- H. Ueba, S. Ichimura and H. Yamada, Surf. Sci., 1982, 119, 433–448 CrossRef CAS.
- T. E. Furtak and S. H. Macomber, Chem. Phys. Lett., 1983, 95, 328–332 CrossRef CAS.
- T. E. Furtak and D. Roy, Phys. Rev. Lett., 1983, 50, 1301–1304 CrossRef CAS.
- J. R. Lombardi, R. L. Birke, L. A. Sanchez, I. Bernard and S. C. Sun, Chem. Phys. Lett., 1984, 104, 240–247 CrossRef CAS.
- M. J. Weaver, S. Farquharson and M. A. Tadayyoni, J. Chem. Phys., 1985, 82, 4867–4874 CrossRef CAS.
- E. Burstein, Y. J. Chen, C. Y. Chen, S. Lundquist and E. Tosatti, Solid State Commun., 1979, 29, 567–570 CrossRef CAS.
- J. I. Gersten, R. L. Birke and J. R. Lombardi, Phys. Rev. Lett., 1979, 43, 147–150 CrossRef CAS.
- J. E. Demuth and P. N. Sanda, Phys. Rev. Lett., 1981, 47, 57–60 CrossRef CAS.
- B. N. J. Persson, Chem. Phys. Lett., 1981, 82, 561–565 CrossRef CAS.
- J. R. Lombardi, R. L. Birke, T. Lu and J. Xu, J. Chem. Phys., 1986, 84, 4174–4180 CrossRef CAS.
- L. Stolberg, J. Lipkowski and D. E. Irish, J. Electroanal. Chem. Interfacial Electrochem., 1991, 300, 563–584 CrossRef CAS.
- F. J. Adrian, J. Chem. Phys., 1982, 77, 5302–5314 CrossRef CAS.
- J. Tang and A. C. Albrecht, in Raman Spectroscopy: Theory and Practice, ed. H. A. Szymanski, Springer US, Boston, MA, 1970, pp. 33–68 DOI:10.1007/978-1-4684-3027-1_2.
- B. Pettinger, J. Chem. Phys., 1986, 85, 7442–7451 CrossRef CAS.
- I. Pockrand, Chem. Phys. Lett., 1982, 92, 514–518 CrossRef CAS.
- P. Gao, M. L. Patterson, M. A. Tadayyoni and M. J. Weaver, Langmuir, 1985, 1, 173–176 CrossRef CAS.
- B. Pettinger, M. R. Philpott and J. G. Gordon, II, J. Phys. Chem., 1981, 85, 2746–2751 CrossRef CAS.
- H. Wetzel, H. Gerischer and B. Pettinger, Chem. Phys. Lett., 1981, 78, 392–397 CrossRef CAS.
- M. Fleischmann, I. R. Hill and M. E. Pemble, J. Electroanal. Chem. Interfacial Electrochem., 1982, 136, 361–370 CrossRef CAS.
- L. Moerl and B. Pettinger, Solid State Commun., 1982, 43, 315–320 CrossRef CAS.
- R. P. Cooney, M. Fleischmann and P. J. Hendra, J. Chem. Soc., Chem. Commun., 1977, 235–237, 10.1039/C39770000235.
- B. Pettinger and U. Tiedemann, J. Electroanal. Chem. Interfacial Electrochem., 1987, 228, 219–228 CrossRef CAS.
- M. A. Bryant, S. L. Joa and J. E. Pemberton, Langmuir, 1992, 8, 753–756 CrossRef CAS.
- T. Maeda, Y. Sasaki, C. Horie and M. Osawa, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 381–389 CrossRef CAS.
- S. A. Bilmes, J. C. Rubim, A. Otto and A. J. Arvia, Chem. Phys. Lett., 1989, 159, 89–96 CrossRef CAS.
- C. Shannon and A. Campion, J. Phys. Chem., 1988, 92, 1385–1387 CrossRef CAS.
- H. Yamada and Y. Yamamoto, Surf. Sci., 1983, 134, 71–90 CrossRef CAS.
- R. K. Chang and B. L. Laube, Crit. Rev. Solid State Mater. Sci., 1984, 12, 1–73 CrossRef CAS.
- R. P. Van Duyne and J. P. Haushalter, J. Phys. Chem., 1983, 87, 2999–3003 CrossRef CAS.
- M. Fleischmann, Z. Q. Tian and L. J. Li, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 397–410 CrossRef CAS.
- L. W. H. Leung and M. J. Weaver, J. Am. Chem. Soc., 1987, 109, 5113–5119 CrossRef CAS.
- R. P. Van Duyne, J. P. Haushalter, M. Janik-Czachor and N. Levinger, J. Phys. Chem., 1985, 89, 4055–4061 CrossRef CAS.
- G. Mengoli, M. M. Musiani, M. Fleischmann, B. Mao and Z. Q. Tian, Electrochim. Acta, 1987, 32, 1239–1245 CrossRef CAS.
- L. W. H. Leung and M. J. Weaver, J. Electroanal. Chem., 1987, 217, 367–384 CrossRef CAS.
- J. F. Li, Z. L. Yang, B. Ren, G. K. Liu, P. P. Fang, Y. X. Jiang, D. Y. Wu and Z. Q. Tian, Langmuir, 2006, 22, 10372–10379 CrossRef CAS PubMed.
- L. W. H. Leung and M. J. Weaver, Langmuir, 1988, 4, 1076–1083 CrossRef CAS.
- R. E. Holt and T. M. Cotton, J. Am. Chem. Soc., 1987, 109, 1841–1845 CrossRef CAS.
- H. Feilchenfeld and M. J. Weaver, J. Phys. Chem., 1991, 95, 7771–7777 CrossRef CAS.
- J. Hu, R. S. Sheng, Z. S. Xu and Y. E. Zeng, Spectrochim. Acta, Part A, 1995, 51, 1087–1096 CrossRef.
- A. Helmenstine, M. Uziel and T. Vo-Dinh, J. Toxicol. Environ. Health, 1993, 40, 195–202 CrossRef CAS PubMed.
- M. Osawa, N. Matsuda, K. Yoshii and I. Uchida, J. Phys. Chem., 1994, 98, 12702–12707 CrossRef CAS.
- Y. Yamamoto, H. Nishihara and K. Aramaki, J. Electrochem. Soc., 1993, 140, 436 CrossRef CAS.
- M. Fleischmann and Z. Q. Tian, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 385–395 CrossRef CAS.
- B. Pettinger, Surf. Sci., 1985, 158, 409–410 CrossRef.
- N. A. Cross and J. E. Pemberton, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 93–100 CrossRef CAS.
- J. C. Rubim, J.-H. Kim, E. Henderson and T. M. Cotton, Appl. Spectrosc., 1993, 47, 80–84 CrossRef CAS.
- M. E. Kordesch, W. Stenzel and H. Conrad, Surf. Sci., 1987, 186, 601–623 CrossRef CAS.
- K. T. Carron, G. Xue and M. L. Lewis, Langmuir, 1991, 7, 2–4 CrossRef CAS.
- G. Xue, J. Ding, P. Lu and J. Dong, J. Phys. Chem., 1991, 95, 7380–7384 CrossRef CAS.
- A. Hamelin, L. Stoicoviciu, S.-C. Chang and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1991, 307, 183–194 CrossRef CAS.
- I. Mrozek and A. Otto, J. Electron Spectrosc. Relat. Phenom., 1990, 54–55, 895–911 CrossRef CAS.
- M. Fleischmann, G. Sundholm and Z. Q. Tian, Electrochim. Acta, 1986, 31, 907–916 CrossRef CAS.
- A. Henglein, P. Mulvaney and T. Linnert, Electrochim. Acta, 1991, 36, 1743–1745 CrossRef CAS.
- J. Neddersen, G. Chumanov and T. M. Cotton, Appl. Spectrosc., 1993, 47, 1959–1964 CrossRef CAS.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2011, 693, 7–25 CrossRef CAS PubMed.
- P. Gao, D. Gosztola, L.-W. H. Leung and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1987, 233, 211–222 CrossRef CAS.
- Z.-Q. Tian, B. Ren and D.-Y. Wu, J. Phys. Chem. B, 2002, 106, 9463–9483 CrossRef CAS.
- D.-Y. Wu, J.-F. Li, B. Ren and Z.-Q. Tian, Chem. Soc. Rev., 2008, 37, 1025–1041 RSC.
- M. Fleischmann and I. R. Hill, in Comprehensive Treatise of Electrochemistry: Experimental Methods in Electrochemistry, ed. R. E. White, J. O. M. Bockris, B. E. Conway and E. Yeager, Springer US, Boston, MA, 1984, vol. 8, pp. 373–432 DOI:10.1007/978-1-4613-2679-3_6.
- T. M. Cotton, J.-H. Kim and G. D. Chumanov, J. Raman Spectrosc., 1991, 22, 729–742 CrossRef CAS.
- T. M. Cotton, S. G. Schultz and R. P. Van Duyne, J. Am. Chem. Soc., 1980, 102, 7960–7962 CrossRef CAS.
- E. Koglin and J.-M. Séquaris, Surface enhanced raman scattering of biomolecules, in Analytical Problems. Topics in Current Chemistry, Springer, Berlin, Heidelberg, 1986, vol. 134 Search PubMed.
- I. R. Nabiev, R. G. Efremov and G. D. Chumanov, Phys.-Usp., 1988, 31, 241 CrossRef.
- T. Watanabe, O. Kawanami, H. Katoh, K. Honda, Y. Nishimura and M. Tsuboi, Surf. Sci., 1985, 158, 341–351 CrossRef CAS.
- K. Kneipp and J. Flemming, J. Mol. Struct., 1986, 145, 173–179 CrossRef CAS.
- T. Vo-Dinh, K. Houck and D. L. Stokes, Anal. Chem., 1994, 66, 3379–3383 CrossRef CAS PubMed.
- T. Vo-Dinh, M. Uziel and A. L. Morrison, Appl. Spectrosc., 1987, 41, 605–610 CrossRef.
- M. Fleischmann, P. J. Hendra, I. R. Hill and M. E. Pemble, J. Electroanal. Chem. Interfacial Electrochem., 1981, 117, 243–255 CrossRef CAS.
- B. Pettinger, M. R. Philpott and J. G. Gordon, II, J. Chem. Phys., 1981, 74, 934–940 CrossRef CAS.
- T. E. Furtak and D. Roy, Surf. Sci., 1985, 158, 126–146 CrossRef CAS.
- A. M. Funtikov, S. K. Sigalaev and V. E. Kazarinov, J. Electroanal. Chem. Interfacial Electrochem., 1987, 228, 197–218 CrossRef CAS.
- Z. Q. Tian, S. K. Sigalaev, S. Z. Zou, B. W. Mao, A. M. Funtikov and V. E. Kazarinov, Electrochim. Acta, 1994, 39, 2195–2196 CrossRef CAS.
- B. Ren, Q. J. Huang, W. B. Cai, B. W. Mao, F. M. Liu and Z. Q. Tian, J. Electroanal. Chem., 1996, 415, 175–178 CrossRef.
- R. Feynmann, presented in part at the American Physical Society annual meeting, CalTech, 1959.
- M. Moskovits and D. P. DiLella, J. Chem. Phys., 1980, 73, 6068–6075 CrossRef CAS.
- P. F. Liao, J. G. Bergman, D. S. Chemla, A. Wokaun, J. Melngailis, A. M. Hawryluk and N. P. Economou, Chem. Phys. Lett., 1981, 82, 355–359 CrossRef CAS.
- H. Wang, F. Tam, N. K. Grady and N. J. Halas, J. Phys. Chem. B, 2005, 109, 18218–18222 CrossRef CAS PubMed.
- G. H. Chan, J. Zhao, E. M. Hicks, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2007, 7, 1947–1952 CrossRef CAS.
- R. G. Freeman, K. C. Grabar, K. J. Allison, R. M. Bright, J. A. Davis, A. P. Guthrie, M. B. Hommer, M. A. Jackson, P. C. Smith, D. G. Walter and M. J. Natan, Science, 1995, 267, 1629–1632 CrossRef CAS PubMed.
- S. J. Oldenburg, S. L. Westcott, R. D. Averitt and N. J. Halas, J. Chem. Phys., 1999, 111, 4729–4735 CrossRef CAS.
- L. A. Dick, A. D. McFarland, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2002, 106, 853–860 CrossRef CAS.
- C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2003, 107, 7426–7433 CrossRef CAS.
- W. Li, P. H. C. Camargo, X. Lu and Y. Xia, Nano Lett., 2009, 9, 485–490 CrossRef CAS PubMed.
- M. Hu, F. S. Ou, W. Wu, I. Naumov, X. Li, A. M. Bratkovsky, R. S. Williams and Z. Li, J. Am. Chem. Soc., 2010, 132, 12820–12822 CrossRef CAS PubMed.
- R. A. Alvarez-Puebla, A. Agarwal, P. Manna, B. P. Khanal, P. Aldeanueva-Potel, E. Carbo-Argibay, N. Pazos-Perez, L. Vigderman, E. R. Zubarev, N. A. Kotov and L. M. Liz-Marzan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8157–8161 CrossRef CAS PubMed.
- N. G. Greeneltch, M. G. Blaber, A.-I. Henry, G. C. Schatz and R. P. Van Duyne, Anal. Chem., 2013, 85, 2297–2303 CrossRef CAS PubMed.
- X. Zhang, Y. Zheng, X. Liu, W. Lu, J. Dai, D. Y. Lei and D. R. MacFarlane, Adv. Mater., 2015, 27, 1090–1096 CrossRef CAS PubMed.
- M. J. Mulvihill, X. Y. Ling, J. Henzie and P. Yang, J. Am. Chem. Soc., 2010, 132, 268–274 CrossRef CAS PubMed.
- L. Qin, S. Zou, C. Xue, A. Atkinson, G. C. Schatz and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13300–13303 CrossRef CAS PubMed.
- H. Wang, D. W. Brandl, P. Nordlander and N. J. Halas, Acc. Chem. Res., 2007, 40, 53–62 CrossRef CAS PubMed.
- C. G. Blatchford, J. R. Campbell and J. A. Creighton, Surf. Sci., 1982, 120, 435–455 CrossRef CAS.
- S. G. Schultz, M. Janik-Czachor and R. P. Van Duyne, Surf. Sci., 1981, 104, 419–434 CrossRef CAS.
- H. Xu, E. J. Bjerneld, M. Käll and L. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- S. P. S. Porto and D. L. Wood, J. Opt. Soc. Am., 1962, 52, 251–252 CrossRef CAS.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- K. Kneipp, H. Kneipp, V. B. Kartha, R. Manoharan, G. Deinum, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. E:Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, R6281–R6284 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1996, 76, 2444–2447 CrossRef CAS PubMed.
- M. I. Stockman, V. M. Shalaev, M. Moskovits, R. Botet and T. F. George, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 2821–2830 CrossRef PubMed.
- H. Xu, J. Aizpurua, M. Käll and P. Apell, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2000, 62, 4318–4324 CrossRef CAS PubMed.
- A. M. Michaels, M. Nirmal and L. E. Brus, J. Am. Chem. Soc., 1999, 121, 9932–9939 CrossRef CAS.
- J. Jiang, K. Bosnick, M. Maillard and L. Brus, J. Phys. Chem. B, 2003, 107, 9964–9972 CrossRef CAS.
- V. M. Shalaev and A. K. Sarychev, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 13265–13288 CrossRef CAS.
- H. Xu and M. Käll, Phys. Rev. Lett., 2002, 89, 246802 CrossRef PubMed.
- E. C. Le Ru, M. Meyer and P. G. Etchegoin, J. Phys. Chem. B, 2006, 110, 1944–1948 CrossRef CAS PubMed.
- J. A. Dieringer, R. B. Lettan, K. A. Scheidt and R. P. Van Duyne, J. Am. Chem. Soc., 2007, 129, 16249–16256 CrossRef CAS PubMed.
- P. G. Etchegoin, M. Meyer, E. Blackie and E. C. Le Ru, Anal. Chem., 2007, 79, 8411–8415 CrossRef CAS PubMed.
- E. C. Le Ru and P. G. Etchegoin, Annu. Rev. Phys. Chem., 2012, 63, 65–87 CrossRef CAS PubMed.
- A. B. Zrimsek, N. Chiang, M. Mattei, S. Zaleski, M. O. McAnally, C. T. Chapman, A.-I. Henry, G. C. Schatz and R. P. Van Duyne, Chem. Rev., 2017, 117, 7583–7613 CrossRef CAS PubMed.
- N. L. Gruenke, M. F. Cardinal, M. O. McAnally, R. R. Frontiera, G. C. Schatz and R. P. Van Duyne, Chem. Soc. Rev., 2016, 45, 2263–2290 RSC.
- F. Neubrech, C. Huck, K. Weber, A. Pucci and H. Giessen, Chem. Rev., 2017, 117, 5110–5145 CrossRef CAS PubMed.
- J.-F. Li, C.-Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962–3979 RSC.
- S. Shen, L. Meng, Y. Zhang, J. Han, Z. Ma, S. Hu, Y. He, J. Li, B. Ren, T.-M. Shih, Z. Wang, Z. Yang and Z. Tian, Nano Lett., 2015, 15, 6716–6721 CrossRef CAS PubMed.
- W.-T. Liu and Y. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1293–1297 CrossRef CAS PubMed.
- G. Hajisalem, M. S. Nezami and R. Gordon, Nano Lett., 2014, 14, 6651–6654 CrossRef CAS PubMed.
- M. Danckwerts and L. Novotny, Phys. Rev. Lett., 2007, 98, 026104 CrossRef PubMed.
- X. Hua, D. V. Voronine, C. W. Ballmann, A. M. Sinyukov, A. V. Sokolov and M. O. Scully, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 89, 043841 CrossRef.
- J. A. Dieringer, A. D. McFarland, N. C. Shah, D. A. Stuart, A. V. Whitney, C. R. Yonzon, M. A. Young, X. Zhang, R. Van Duyne, K. Kneipp, H. Kneipp, F. Svedburg, M. Käll, M. Futamata, E. Le Ru, P. G. Etchegoin, R. Maher, L. F. Cohen, B. Vlćková, G. Lucassen, Z.-Q. Tian, B. Pettinger, B. Ren, A. E. Russell, M. J. Natan, J. J. Baumberg, G. McNay, K. Murakoshi, S. Nie, R. Aggarwal, T. A. Kelf, T. Itoh, T. Maeda, A. Kasuya, B. Pergolese, M. Muniz-Miranda, G. Sbrana, A. Bigotto, M. Sládková, P. Mojzeš, M. Šlouf, C. Naudin, G. Le Bourdon, D. G. Cunningham, R. E. Littleford, W. E. Smith, P. J. Lundahl, I. Khan, D. W. McComb, D. Graham, N. Laforest, K. F. Domke, B. D. Alexander, E. Blanch, C. Eliasson, A. Pal, M. J. T. Milton, R. Goodacre, E. M. Kosower, O. Glembocki, Z.-L. Yang, J.-F. Li, Y. Zhang, X.-F. Lin, J.-W. Hu, D.-Y. Wu, S. Lazar, Y. Sawai, B. Takimoto, H. Nabika, K. Ajito, S. H. Cintra, M. E. Abdelsalam, P. N. Bartlett, Y. Sugawara, R. J. C. Brown, J. Wang, R. Tantra, R. E. Yardley, D. Roy, M. E. Welland, W. I. Barnes, J. A. Creighton, M. Green, V. Poponin, N. Stone, D. Richards, R. M. Jarvis, M. Whitby, S. R. Emory, R. A. Jensen, T. Wenda, M. Han, K. J. Faulds, L. Fruk, D. C. Robson, D. G. Thompson, A. Enright, P. Köllensperger, T. Cass, A. Brooker, N. R. Isola, J. P. Alarie, D. L. Stokes, T. Vo-Dinh, A. F. McCabe, R. A. Prasath, A. Hernandez-Santana, L. Stevenson, I. Apple, P. A. G. Cormack, P. Corish, S. J. Lipscomb, E. R. Holland, P. D. Prince, B. Foulger, M. Zoorob, B. Pearson, P. Donaldson, N. E. Cant and T. Pal, Faraday Discuss., 2006, 132, 1–340 RSC.
- A.-I. Henry, T. W. Ueltschi, M. O. McAnally, R. P. Van Duyne, M. K. Schmidt, R. Esteban, F. Benz, J. J. Baumberg, J. Aizpurua, L. Velleman, L. Scarabelli, D. Sikdar, A. A. Kornyshev, L. M. Liz-Marzán, J. B. Edel, N. S. Mueller, S. Heeg, P. Kusch, E. Gaufrès, N. Y.-W. Tang, U. Hübner, R. Martel, A. Vijayaraghavan, S. Reich, J. R. Lombardi, P. Dawson, D. Frey, V. Kalathingal, R. Mehfuz, J. Mitra, R. L. Gieseking, M. A. Ratner, G. C. Schatz, D. Graham, R. Goodacre, H. Arnolds, J.-F. Masson, D.-H. Kim, W. Lum, A. Silvestri, B. de Nijs, Y. Xu, G. D. Martino, M. Natan, S. Schlücker, P. Wuytens, I. Bruzas, C. Kuttner, M. Hardy, R. Chikkaraddy, N. M. Sabanés, I. Delfino, S. Gawinkowski, N. Bontempi, S. Mahajan, B. Hourahine, S. Bell, A. Królikowska, M. Porter, A. Keeler, M. Kamp, A. Fountain, C. Fasolato, F. Giorgis, J. C. Otero, C. Matricardi, V. Deckert, Z. Zhang, M. Richard-Lacroix, C. L. D. Lee, K. C. Hewitt, A. Elizabeth, J. H. K. Pfisterer, K. F. Domke, P. B. Joshi, T. P. Anthony, A. J. Wilson, K. A. Willets, H. Minamimoto, F. Kato, F. Nagasawa, M. Takase, K. Murakoshi, C. Novara, A. Chiadò, N. Paccotti, S. Catuogno, C. L. Esposito, G. Condorelli, V. De Franciscis, F. Geobaldo, P. Rivolo, M. Chisanga, H. Fleming, C. Heck, L. Jamieson, N. Lidgi-Guigui, C. Lightner, H. Muhamadali, J. Popp, Z.-Q. Tian, A. Tripathi, X. Wang, A. Wark, K. (Kallie) Willets, M. Willner, R. Kimber, H. Demol, N. Turk, K. Gevaert, A. G. Skirtach, M. Lamkanfi, R. Baets, J. Langer, I. García, B. Walkenfort, M. König, L. König, S. Kasimir-Bauer, F. Lussier, T. Brulé, M.-J. Bourque, C. Ducrot, L.-É. Trudeau, J. Taylor, J. Milton, M. Willett, J. Wingfield, A. Bonifacio, S. Corsetti, O. Eremina, K. Faulds, T. Keyes, F. Nicolson, E. Nikelshparg, K. Plakas, D. Prezgot, N. Pytlik, N. Stone, S.-Y. Ding, E.-M. You, J. Yi, J.-F. Li, T. A. Galloway, L. Cabo-Fernandez, I. M. Aldous, F. Braga, L. J. Hardwick, H. Wei, A. McCarthy, J. Song, W. Zhou, P. J. Vikesland, I. Szabó, S. J. Barrow, G. Wu, C. Carnegie, W. Wang, W. M. Deacon, E. Rosta, O. A. Scherman, S. Dick, V. A. Turek, C. Tserkezis, A. Lombardi, A. Kuhn, J. A. Guicheteau, E. D. Emmons, S. D. Christesen, H. Aitchison, M. G. Castano, J. Gracie, J. Guicheteau and J. H. Granger, Faraday Discuss., 2017, 205, 1–626 RSC.
- G. Di Martino, H. Fleming, M. Kamp and F. Lussier, Chem. Commun., 2017, 53, 12726–12733 RSC.
- E. C. Le Ru, P. G. Etchegoin and M. Meyer, J. Chem. Phys., 2006, 125, 204701 CrossRef CAS PubMed.
- J. M. McMahon, S. Li, L. K. Ausman and G. C. Schatz, J. Phys. Chem. C, 2012, 116, 1627–1637 CrossRef CAS.
- A. Moreau, C. Ciracì, J. J. Mock, R. T. Hill, Q. Wang, B. J. Wiley, A. Chilkoti and D. R. Smith, Nature, 2012, 492, 86–89 CrossRef CAS PubMed.
- B. Pettinger, P. Schambach, C. J. Villagómez and N. Scott, Annu. Rev. Phys. Chem., 2012, 63, 379–399 CrossRef CAS PubMed.
- S. L. Kleinman, R. R. Frontiera, A.-I. Henry, J. A. Dieringer and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2013, 15, 21–36 RSC.
- H. Wei and H. Xu, Nanoscale, 2013, 5, 10794–10805 RSC.
- A. Shiohara, Y. Wang and L. M. Liz-Marzán, J. Photochem. Photobiol., C, 2014, 21, 2–25 CrossRef CAS.
- J. J. Baumberg, J. Aizpurua, M. H. Mikkelsen and D. R. Smith, Nat. Mater., 2019, 18, 668–678 CrossRef CAS PubMed.
- D.-K. Lim, K.-S. Jeon, H. M. Kim, J.-M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS PubMed.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- P. G. Etchegoin and E. C. Le Ru, Phys. Chem. Chem. Phys., 2008, 10, 6079–6089 RSC.
- J. A. Dieringer, K. L. Wustholz, D. J. Masiello, J. P. Camden, S. L. Kleinman, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2009, 131, 849–854 CrossRef CAS PubMed.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2011, 133, 4115–4122 CrossRef CAS PubMed.
- S. Cintra, M. E. Abdelsalam, P. N. Bartlett, J. J. Baumberg, T. A. Kelf, Y. Sugawara and A. E. Russell, Faraday Discuss., 2006, 132, 191–199 RSC.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- I. Alessandri and J. R. Lombardi, Chem. Rev., 2016, 116, 14921–14981 CrossRef CAS PubMed.
- J. M. McLellan, A. Siekkinen, J. Chen and Y. Xia, Chem. Phys. Lett., 2006, 427, 122–126 CrossRef CAS.
- J. M. McLellan, Z.-Y. Li, A. R. Siekkinen and Y. Xia, Nano Lett., 2007, 7, 1013–1017 CrossRef CAS PubMed.
- L. Rodríguez-Lorenzo, R. A. Álvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stéphan, M. Kociak, L. M. Liz-Marzán and F. J. García de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef PubMed.
- W. E. Doering and S. Nie, Anal. Chem., 2003, 75, 6171–6176 CrossRef CAS PubMed.
- W. Shen, X. Lin, C. Jiang, C. Li, H. Lin, J. Huang, S. Wang, G. Liu, X. Yan, Q. Zhong and B. Ren, Angew. Chem., Int. Ed., 2015, 54, 7308–7312 CrossRef CAS PubMed.
- H. Lee, J.-H. Lee, S. M. Jin, Y. D. Suh and J.-M. Nam, Nano Lett., 2013, 13, 6113–6121 CrossRef CAS PubMed.
- A. Tao, F. Kim, C. Hess, J. Goldberger, R. He, Y. Sun, Y. Xia and P. Yang, Nano Lett., 2003, 3, 1229–1233 CrossRef CAS.
- Y. Lu, G. L. Liu and L. P. Lee, Nano Lett., 2005, 5, 5–9 CrossRef CAS PubMed.
- S. Park, P. Yang, P. Corredor and M. J. Weaver, J. Am. Chem. Soc., 2002, 124, 2428–2429 CrossRef CAS PubMed.
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef CAS.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- H. Zhang, C. Wang, H.-L. Sun, G. Fu, S. Chen, Y.-J. Zhang, B.-H. Chen, J. R. Anema, Z.-L. Yang, J.-F. Li and Z.-Q. Tian, Nat. Commun., 2017, 8, 15447 CrossRef CAS PubMed.
- R. D. Averitt, D. Sarkar and N. J. Halas, Phys. Rev. Lett., 1997, 78, 4217–4220 CrossRef CAS.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- J. Ni, R. J. Lipert, G. B. Dawson and M. D. Porter, Anal. Chem., 1999, 71, 4903–4908 CrossRef CAS PubMed.
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS PubMed.
- G. Wang, H.-Y. Park, R. J. Lipert and M. D. Porter, Anal. Chem., 2009, 81, 9643–9650 CrossRef CAS PubMed.
- F. Burgio, D. Piffaretti, F. Schmidt, U. Pieles, M. Reinert, M.-F. Ritz and S. Saxer, ACS Appl. Nano Mater., 2020, 3, 2447–2454 CrossRef CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef.
- O. E. Eremina, A. T. Czaja, A. Fernando, A. Aron, D. B. Eremin and C. Zavaleta, ACS Nano, 2022, 16, 10341–10353 CrossRef CAS PubMed.
- B. A. Kairdolf and S. Nie, J. Am. Chem. Soc., 2011, 133, 7268–7271 CrossRef CAS PubMed.
- I. Srivastava, R. Xue, J. Jones, H. Rhee, K. Flatt, V. Gruev and S. Nie, ACS Nano, 2022, 16, 8051–8063 CrossRef CAS PubMed.
- L. Lu, G. Sun, H. Zhang, H. Wang, S. Xi, J. Hu, Z. Tian and R. Chen, J. Mater. Chem., 2004, 14, 1005–1009 RSC.
- J.-W. Hu, Y. Zhang, J.-F. Li, Z. Liu, B. Ren, S.-G. Sun, Z.-Q. Tian and T. Lian, Chem. Phys. Lett., 2005, 408, 354–359 CrossRef CAS.
- J.-F. Li, Y.-J. Zhang, S.-Y. Ding, R. Panneerselvam and Z.-Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS PubMed.
- H. Zhang, S. Duan, P. M. Radjenovic, Z.-Q. Tian and J.-F. Li, Acc. Chem. Res., 2020, 53, 729–739 CrossRef CAS PubMed.
- R. A. Álvarez-Puebla, R. Contreras-Cáceres, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2009, 48, 138–143 CrossRef PubMed.
- V. V. Thacker, L. O. Herrmann, D. O. Sigle, T. Zhang, T. Liedl, J. J. Baumberg and U. F. Keyser, Nat. Commun., 2014, 5, 3448 CrossRef PubMed.
- A. Lee, G. F. S. Andrade, A. Ahmed, M. L. Souza, N. Coombs, E. Tumarkin, K. Liu, R. Gordon, A. G. Brolo and E. Kumacheva, J. Am. Chem. Soc., 2011, 133, 7563–7570 CrossRef CAS PubMed.
- J. C. Fraire, V. N. Sueldo Ocello, L. G. Allende, A. V. Veglia and E. A. Coronado, J. Phys. Chem. C, 2015, 119, 8876–8888 CrossRef CAS.
- J.-H. Lee, J.-M. Nam, K.-S. Jeon, D.-K. Lim, H. Kim, S. Kwon, H. Lee and Y. D. Suh, ACS Nano, 2012, 6, 9574–9584 CrossRef CAS PubMed.
- J.-W. Oh, D.-K. Lim, G.-H. Kim, Y. D. Suh and J.-M. Nam, J. Am. Chem. Soc., 2014, 136, 14052–14059 CrossRef CAS PubMed.
- K. Y. Hong and A. G. Brolo, Anal. Chim. Acta, 2017, 972, 73–80 CrossRef CAS PubMed.
- C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2001, 105, 5599–5611 CrossRef CAS.
- M. Kahl, E. Voges, S. Kostrewa, C. Viets and W. Hill, Sens. Actuators, B, 1998, 51, 285–291 CrossRef CAS.
- A. G. Brolo, R. Gordon, B. Leathem and K. L. Kavanagh, Langmuir, 2004, 20, 4813–4815 CrossRef CAS PubMed.
- A. Gopinath, S. V. Boriskina, B. M. Reinhard and L. D. Negro, Opt. Express, 2009, 17, 3741–3753 CrossRef CAS PubMed.
- A. G. Brolo, E. Arctander, R. Gordon, B. Leathem and K. L. Kavanagh, Nano Lett., 2004, 4, 2015–2018 CrossRef CAS.
- S. R. Smith, C. Zhou, J. Y. Baron, Y. Choi and J. Lipkowski, Electrochim. Acta, 2016, 210, 925–934 CrossRef CAS.
- S. R. Smith, J. J. Leitch, C. Zhou, J. Mirza, S.-B. Li, X.-D. Tian, Y.-F. Huang, Z.-Q. Tian, J. Y. Baron, Y. Choi and J. Lipkowski, Anal. Chem., 2015, 87, 3791–3799 CrossRef CAS PubMed.
- P. Nagpal, N. C. Lindquist, S.-H. Oh and D. J. Norris, Science, 2009, 325, 594–597 CrossRef CAS PubMed.
- T. W. Johnson, Z. J. Lapin, R. Beams, N. C. Lindquist, S. G. Rodrigo, L. Novotny and S.-H. Oh, ACS Nano, 2012, 6, 9168–9174 CrossRef CAS PubMed.
- D. J. Maxwell, S. R. Emory and S. Nie, Chem. Mater., 2001, 13, 1082–1088 CrossRef CAS.
- H. Wang, C. S. Levin and N. J. Halas, J. Am. Chem. Soc., 2005, 127, 14992–14993 CrossRef CAS PubMed.
- F. De Angelis, F. Gentile, F. Mecarini, G. Das, M. Moretti, P. Candeloro, M. L. Coluccio, G. Cojoc, A. Accardo, C. Liberale, R. P. Zaccaria, G. Perozziello, L. Tirinato, A. Toma, G. Cuda, R. Cingolani and E. Di Fabrizio, Nat. Photonics, 2011, 5, 682–687 CrossRef CAS.
- D. M. Solís, J. M. Taboada, F. Obelleiro, L. M. Liz-Marzán and F. J. García de Abajo, ACS Photonics, 2017, 4, 329–337 CrossRef PubMed.
- H. Liu, Z. Yang, L. Meng, Y. Sun, J. Wang, L. Yang, J. Liu and Z. Tian, J. Am. Chem. Soc., 2014, 136, 5332–5341 CrossRef CAS PubMed.
- K. Kim, H. S. Han, I. Choi, C. Lee, S. Hong, S.-H. Suh, L. P. Lee and T. Kang, Nat. Commun., 2013, 4, 2182 CrossRef PubMed.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS PubMed.
- D. Zhang, L. Peng, X. Shang, W. Zheng, H. You, T. Xu, B. Ma, B. Ren and J. Fang, Nat. Commun., 2020, 11, 2603 CrossRef CAS PubMed.
- M. Ge, P. Li, G. Zhou, S. Chen, W. Han, F. Qin, Y. Nie, Y. Wang, M. Qin, G. Huang, S. Li, Y. Wang, L. Yang and Z. Tian, J. Am. Chem. Soc., 2021, 143, 7769–7776 CrossRef CAS PubMed.
- M. J. Natan, Faraday Discuss., 2006, 132, 321–328 RSC.
- J.-H. Tian, B. Liu, X. Li, Z.-L. Yang, B. Ren, S.-T. Wu, N. Tao and Z.-Q. Tian, J. Am. Chem. Soc., 2006, 128, 14748–14749 CrossRef CAS PubMed.
- T. Konishi, M. Kiguchi, M. Takase, F. Nagasawa, H. Nabika, K. Ikeda, K. Uosaki, K. Ueno, H. Misawa and K. Murakoshi, J. Am. Chem. Soc., 2013, 135, 1009–1014 CrossRef CAS PubMed.
- J. Zheng, J. Liu, Y. Zhuo, R. Li, X. Jin, Y. Yang, Z.-B. Chen, J. Shi, Z. Xiao, W. Hong and Z.-Q. Tian, Chem. Sci., 2018, 9, 5033–5038 RSC.
- S. Kaneko, D. Murai, S. Marqués-González, H. Nakamura, Y. Komoto, S. Fujii, T. Nishino, K. Ikeda, K. Tsukagoshi and M. Kiguchi, J. Am. Chem. Soc., 2016, 138, 1294–1300 CrossRef CAS PubMed.
- S. Kaneko, E. Montes, S. Suzuki, S. Fujii, T. Nishino, K. Tsukagoshi, K. Ikeda, H. Kano, H. Nakamura, H. Vázquez and M. Kiguchi, Chem. Sci., 2019, 10, 6261–6269 RSC.
- T. Itoh, Y. S. Yamamoto, H. Tamaru, V. Biju, S.-I. Wakida and Y. Ozaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 195436 CrossRef.
- E. C. Le Ru, P. G. Etchegoin, J. Grand, N. Félidj, J. Aubard and G. Lévi, J. Phys. Chem. C, 2007, 111, 16076–16079 CrossRef CAS.
- T. Itoh, Y. S. Yamamoto, H. Tamaru, V. Biju, N. Murase and Y. Ozaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235408 CrossRef.
- M. Takase, H. Ajiki, Y. Mizumoto, K. Komeda, M. Nara, H. Nabika, S. Yasuda, H. Ishihara and K. Murakoshi, Nat. Photonics, 2013, 7, 550–554 CrossRef CAS.
- C. M. Aikens, L. R. Madison and G. C. Schatz, Nat. Photonics, 2013, 7, 508–510 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS PubMed.
- L. L. Zhao, L. Jensen and G. C. Schatz, Nano Lett., 2006, 6, 1229–1234 CrossRef CAS PubMed.
- D.-Y. Wu, X.-M. Liu, S. Duan, X. Xu, B. Ren, S.-H. Lin and Z.-Q. Tian, J. Phys. Chem. C, 2008, 112, 4195–4204 CrossRef CAS.
- S. Tao, L.-J. Yu, R. Pang, Y.-F. Huang, D.-Y. Wu and Z.-Q. Tian, J. Phys. Chem. C, 2013, 117, 18891–18903 CrossRef CAS.
- A. J. Haes and R. P. Van Duyne, Anal. Bioanal. Chem., 2004, 379, 920–930 CrossRef CAS PubMed.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2008, 112, 5605–5617 CrossRef CAS.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
- L. Jensen, C. M. Aikens and G. C. Schatz, Chem. Soc. Rev., 2008, 37, 1061–1073 RSC.
- S. M. Morton and L. Jensen, J. Am. Chem. Soc., 2009, 131, 4090–4098 CrossRef CAS PubMed.
- D. J. Trivedi, B. Barrow and G. C. Schatz, J. Chem. Phys., 2020, 153, 124706 CrossRef CAS PubMed.
- A. Wang, Y.-F. Huang, U. K. Sur, D.-Y. Wu, B. Ren, S. Rondinini, C. Amatore and Z.-Q. Tian, J. Am. Chem. Soc., 2010, 132, 9534–9536 CrossRef CAS PubMed.
- H. M. Dykstra, S. B. Hall and M. R. Waterland, J. Raman Spectrosc., 2017, 48, 405–412 CrossRef CAS.
- D. H. Murgida and P. Hildebrandt, Acc. Chem. Res., 2004, 37, 854–861 CrossRef CAS PubMed.
- C. D. Lindstrom and X. Y. Zhu, Chem. Rev., 2006, 106, 4281–4300 CrossRef CAS PubMed.
- D. Y. Wu, M. Hayashi, S. H. Lin and Z. Q. Tian, Spectrochim. Acta, Part A, 2004, 60, 137–146 CrossRef CAS PubMed.
- M. T. Lee, D. Y. Wu, Z. Q. Tian and S. H. Lin, J. Chem. Phys., 2005, 122, 094719 CrossRef CAS PubMed.
- L. L. Zhao, L. Jensen and G. C. Schatz, J. Am. Chem. Soc., 2006, 128, 2911–2919 CrossRef CAS PubMed.
- J. C. Rubim, P. Corio, M. C. C. Ribeiro and M. Matz, J. Phys. Chem., 1995, 99, 15765–15774 CrossRef CAS.
- L. Jensen and G. C. Schatz, J. Phys. Chem. A, 2006, 110, 5973–5977 CrossRef CAS PubMed.
- N. Valley, N. Greeneltch, R. P. Van Duyne and G. C. Schatz, J. Phys. Chem. Lett., 2013, 4, 2599–2604 CrossRef CAS.
- D. Aranda, F. García-González, F. J. Avila Ferrer, I. López-Tocón, J. Soto and J. C. Otero, J. Chem. Theory Comput., 2022, 18, 6802–6815 CrossRef CAS PubMed.
- F. García-González, J. C. Otero, F. J. Ávila Ferrer, F. Santoro and D. Aranda, J. Chem. Theory Comput., 2024, 20, 3850–3863 CrossRef PubMed.
- J. Gargiulo, R. Berté, Y. Li, S. A. Maier and E. Cortés, Acc. Chem. Res., 2019, 52, 2525–2535 CrossRef CAS PubMed.
- A. Nitzan and L. E. Brus, J. Chem. Phys., 1981, 75, 2205–2214 CrossRef CAS.
- C. Zhan, X.-J. Chen, J. Yi, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Nat. Rev. Chem., 2018, 2, 216–230 CrossRef.
- L. Brus, Acc. Chem. Res., 2008, 41, 1742–1749 CrossRef CAS PubMed.
- E. S. Thrall, A. Preska Steinberg, X. Wu and L. E. Brus, J. Phys. Chem. C, 2013, 117, 26238–26247 CrossRef CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- Y.-F. Huang, H.-P. Zhu, G.-K. Liu, D.-Y. Wu, B. Ren and Z.-Q. Tian, J. Am. Chem. Soc., 2010, 132, 9244–9246 CrossRef CAS PubMed.
- Y.-Q. Su, J. Liu, R. Huang, H.-T. Yang, M.-X. Li, R. Pang, M. Zhang, M.-H. Yang, H.-F. Su, R. Devasenathipathy, Y.-F. Wu, J.-Z. Zhou, D.-Y. Wu, S.-Y. Xie, B.-W. Mao and Z.-Q. Tian, J. Phys. Chem. Lett., 2023, 14, 5163–5171 CrossRef CAS PubMed.
- J. Liu, Z.-Y. Cai, W.-X. Sun, J.-Z. Wang, X.-R. Shen, C. Zhan, R. Devasenathipathy, J.-Z. Zhou, D.-Y. Wu, B.-W. Mao and Z.-Q. Tian, J. Am. Chem. Soc., 2020, 142, 17489–17498 CrossRef CAS PubMed.
- C. Zhan, J. Yi, S. Hu, X.-G. Zhang, D.-Y. Wu and Z.-Q. Tian, Nat. Rev. Methods Primers, 2023, 3, 12 CrossRef CAS.
- S. Linic, S. Chavez and R. Elias, Nat. Mater., 2021, 20, 916–924 CrossRef CAS PubMed.
- A. Stefancu, N. J. Halas, P. Nordlander and E. Cortes, Nat. Phys., 2024, 20, 1065–1077 Search PubMed.
- N. J. Halas, S. Lal, S. Link, W.-S. Chang, D. Natelson, J. H. Hafner and P. Nordlander, Adv. Mater., 2012, 24, 4842–4877 CrossRef CAS PubMed.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS PubMed.
- F. Le, D. W. Brandl, Y. A. Urzhumov, H. Wang, J. Kundu, N. J. Halas, J. Aizpurua and P. Nordlander, ACS Nano, 2008, 2, 707–718 CrossRef CAS PubMed.
- A. B. Sánchez-Alvarado, J. Zhou, P. Jin, O. Neumann, T. P. Senftle, P. Nordlander and N. J. Halas, ACS Nano, 2023, 17, 25697–25706 CrossRef PubMed.
- L. W.-H. Tian Zhong-Qun, M. Ji-Qian, M. Bing-Wei, C. Jie-Guang, Z. Xiang-Dong, Z. Wei, W. Duo and Y. En-Rou, Acta Phys.-Chim. Sin., 1994, 10, 1062–1065 Search PubMed.
- Z. Q. Tian, W. H. Li, B. Ren, B. W. Mao, J. G. Chen, J. Q. Mu, X. D. Zhuo and D. Wang, J. Electroanal. Chem., 1996, 401, 247–251 CrossRef.
- Z.-Q. Tian and B. Ren, Raman Spectroscopy of Electrode Surfaces, in Encyclopedia of Electrochemistry, ed. A. J. Bard, Wiley-VCH, 2007 DOI:10.1002/9783527610426.bard030306.
- J. Stadler, T. Schmid and R. Zenobi, Nano Lett., 2010, 10, 4514–4520 CrossRef CAS PubMed.
- T. Ichimura, S. Fujii, P. Verma, T. Yano, Y. Inouye and S. Kawata, Phys. Rev. Lett., 2009, 102, 186101 CrossRef PubMed.
- T.-a Yano, P. Verma, Y. Saito, T. Ichimura and S. Kawata, Nat. Photonics, 2009, 3, 473–477 CrossRef CAS.
- R. Esteban, A. G. Borisov, P. Nordlander and J. Aizpurua, Nat. Commun., 2012, 3, 825 CrossRef PubMed.
- C. Ciracì, R. T. Hill, J. J. Mock, Y. Urzhumov, A. I. Fernández-Domínguez, S. A. Maier, J. B. Pendry, A. Chilkoti and D. R. Smith, Science, 2012, 337, 1072–1074 CrossRef PubMed.
- R. Zhang, Y. Zhang, Z. C. Dong, S. Jiang, C. Zhang, L. G. Chen, L. Zhang, Y. Liao, J. Aizpurua, Y. Luo, J. L. Yang and J. G. Hou, Nature, 2013, 498, 82–86 CrossRef CAS PubMed.
- S. Duan, G. Tian, Y. Ji, J. Shao, Z. Dong and Y. Luo, J. Am. Chem. Soc., 2015, 137, 9515–9518 CrossRef CAS PubMed.
- M. Barbry, P. Koval, F. Marchesin, R. Esteban, A. G. Borisov, J. Aizpurua and D. Sánchez-Portal, Nano Lett., 2015, 15, 3410–3419 CrossRef CAS PubMed.
- T. Neuman, R. Esteban, D. Casanova, F. J. García-Vidal and J. Aizpurua, Nano Lett., 2018, 18, 2358–2364 CrossRef CAS PubMed.
- F. Benz, M. K. Schmidt, A. Dreismann, R. Chikkaraddy, Y. Zhang, A. Demetriadou, C. Carnegie, H. Ohadi, B. de Nijs, R. Esteban, J. Aizpurua and J. J. Baumberg, Science, 2016, 354, 726–729 CrossRef CAS PubMed.
- M. K. Schmidt, R. Esteban, A. González-Tudela, G. Giedke and J. Aizpurua, ACS Nano, 2016, 10, 6291–6298 CrossRef CAS PubMed.
- F. Latorre, S. Kupfer, T. Bocklitz, D. Kinzel, S. Trautmann, S. Gräfe and V. Deckert, Nanoscale, 2016, 8, 10229–10239 RSC.
- S. Trautmann, J. Aizpurua, I. Götz, A. Undisz, J. Dellith, H. Schneidewind, M. Rettenmayr and V. Deckert, Nanoscale, 2017, 9, 391–401 RSC.
- C. Carnegie, M. Urbieta, R. Chikkaraddy, B. de Nijs, J. Griffiths, W. M. Deacon, M. Kamp, N. Zabala, J. Aizpurua and J. J. Baumberg, Nat. Commun., 2020, 11, 682 CrossRef CAS PubMed.
- J. J. Baumberg, R. Esteban, S. Hu, U. Muniain, I. V. Silkin, J. Aizpurua and V. M. Silkin, Nano Lett., 2023, 23, 10696–10702 CrossRef CAS PubMed.
- P. Roelli, C. Galland, N. Piro and T. J. Kippenberg, Nat. Nanotechnol., 2016, 11, 164–169 CrossRef CAS PubMed.
- L. A. Jakob, W. M. Deacon, Y. Zhang, B. de Nijs, E. Pavlenko, S. Hu, C. Carnegie, T. Neuman, R. Esteban, J. Aizpurua and J. J. Baumberg, Nat. Commun., 2023, 14, 3291 CrossRef CAS PubMed.
- N. C. Lindquist, C. D. L. de Albuquerque, R. G. Sobral-Filho, I. Paci and A. G. Brolo, Nat. Nanotechnol., 2019, 14, 981–987 CrossRef CAS PubMed.
- N. C. Lindquist and A. G. Brolo, J. Phys. Chem. C, 2021, 125, 7523–7532 CrossRef CAS.
- J. Lee, K. T. Crampton, N. Tallarida and V. A. Apkarian, Nature, 2019, 568, 78–82 CrossRef CAS PubMed.
- Y. Zhang, B. Yang, A. Ghafoor, Y. Zhang, Y.-F. Zhang, R.-P. Wang, J.-L. Yang, Y. Luo, Z.-C. Dong and J. G. Hou, Natl. Sci. Rev., 2019, 6, 1169–1175 CrossRef CAS PubMed.
- J. Xu, X. Zhu, S. Tan, Y. Zhang, B. Li, Y. Tian, H. Shan, X. Cui, A. Zhao, Z. Dong, J. Yang, Y. Luo, B. Wang and J. G. Hou, Science, 2021, 371, 818–822 CrossRef CAS PubMed.
- Z.-C. Zeng, S.-C. Huang, D.-Y. Wu, L.-Y. Meng, M.-H. Li, T.-X. Huang, J.-H. Zhong, X. Wang, Z.-L. Yang and B. Ren, J. Am. Chem. Soc., 2015, 137, 11928–11931 CrossRef CAS PubMed.
- D. Kurouski, M. Mattei and R. P. Van Duyne, Nano Lett., 2015, 15, 7956–7962 CrossRef CAS PubMed.
- G. Kang, M. Yang, M. S. Mattei, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2019, 19, 2106–2113 CrossRef CAS PubMed.
- Y.-F. Bao, M.-F. Cao, S.-S. Wu, T.-X. Huang, Z.-C. Zeng, M.-H. Li, X. Wang and B. Ren, Anal. Chem., 2020, 92, 12548–12555 CrossRef CAS PubMed.
- S.-C. Huang, J.-Z. Ye, X.-R. Shen, Q.-Q. Zhao, Z.-C. Zeng, M.-H. Li, D.-Y. Wu, X. Wang and B. Ren, Anal. Chem., 2019, 91, 11092–11097 CrossRef CAS PubMed.
- T. Touzalin, S. Joiret, I. T. Lucas and E. Maisonhaute, Electrochem. Commun., 2019, 108, 106557 CrossRef CAS.
- J. H. K. Pfisterer, M. Baghernejad, G. Giuzio and K. F. Domke, Nat. Commun., 2019, 10, 5702 CrossRef CAS PubMed.
- T.-X. Huang, X. Cong, S.-S. Wu, J.-B. Wu, Y.-F. Bao, M.-F. Cao, L. Wu, M.-L. Lin, X. Wang, P.-H. Tan and B. Ren, Nat. Catal., 2024, 7, 646–654 CrossRef CAS.
- P. Matousek, I. P. Clark, E. R. C. Draper, M. D. Morris, A. E. Goodship, N. Everall, M. Towrie, W. F. Finney and A. W. Parker, Appl. Spectrosc., 2005, 59, 393–400 CrossRef CAS PubMed.
- P. Matousek and N. Stone, J. Biomed. Opt., 2007, 12, 024008 CrossRef PubMed.
- N. Stone, M. Kerssens, G. R. Lloyd, K. Faulds, D. Graham and P. Matousek, Chem. Sci., 2011, 2, 776–780 RSC.
- N. Stone, K. Faulds, D. Graham and P. Matousek, Anal. Chem., 2010, 82, 3969–3973 CrossRef CAS PubMed.
- A. V. Baranov and Y. S. Bobovich, JETP Lett., 1982, 36, 339–343 Search PubMed.
- D. V. Murphy, K. U. Von Raben, R. K. Chang and P. B. Dorain, Chem. Phys. Lett., 1982, 85, 43–47 CrossRef CAS.
- K. Kneipp, H. Kneipp and F. Seifert, Chem. Phys. Lett., 1995, 233, 519–524 CrossRef CAS.
- J. C. Hulteen, M. A. Young and R. P. Van Duyne, Langmuir, 2006, 22, 10354–10364 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp and K. Kneipp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17149–17153 CrossRef CAS PubMed.
- F. Madzharova, Z. Heiner and J. Kneipp, Chem. Soc. Rev., 2017, 46, 3980–3999 RSC.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, ChemPhysChem, 2011, 12, 101–103 CrossRef CAS PubMed.
- P. Matousek, M. Towrie, A. Stanley and A. W. Parker, Appl. Spectrosc., 1999, 53, 1485–1489 CrossRef CAS.
- D. V. Voronine, A. M. Sinyukov, X. Hua, K. Wang, P. K. Jha, E. Munusamy, S. E. Wheeler, G. Welch, A. V. Sokolov and M. O. Scully, Sci. Rep., 2012, 2, 891 CrossRef PubMed.
- S. Yampolsky, D. A. Fishman, S. Dey, E. Hulkko, M. Banik, E. O. Potma and V. A. Apkarian, Nat. Photonics, 2014, 8, 650–656 CrossRef CAS.
- Z. Sun, J. Lu, D. H. Zhang and S.-Y. Lee, J. Chem. Phys., 2008, 128, 144114 CrossRef PubMed.
- R. R. Frontiera, A.-I. Henry, N. L. Gruenke and R. P. Van Duyne, J. Phys. Chem. Lett., 2011, 2, 1199–1203 CrossRef CAS PubMed.
- L. E. Buchanan, N. L. Gruenke, M. O. McAnally, B. Negru, H. E. Mayhew, V. A. Apkarian, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. Lett., 2016, 7, 4629–4634 CrossRef CAS PubMed.
- E. L. Keller and R. R. Frontiera, ACS Nano, 2018, 12, 5848–5855 CrossRef CAS PubMed.
- C. L. Warkentin and R. R. Frontiera, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305932120 CrossRef CAS PubMed.
- T. Ichimura, N. Hayazawa, M. Hashimoto, Y. Inouye and S. Kawata, Phys. Rev. Lett., 2004, 92, 220801 CrossRef PubMed.
- E. A. Pozzi, M. D. Sonntag, N. Jiang, N. Chiang, T. Seideman, M. C. Hersam and R. P. Van Duyne, J. Phys. Chem. Lett., 2014, 5, 2657–2661 CrossRef CAS PubMed.
- Y. Luo, A. Martin-Jimenez, R. Gutzler, M. Garg and K. Kern, Nano Lett., 2022, 22, 5100–5106 CrossRef CAS PubMed.
- J. S. Gao and Z. Q. Tian, Spectrochim. Acta, Part A, 1997, 53, 1595–1600 CrossRef.
- W. B. Cai, B. Ren, X. Q. Li, C. X. She, F. M. Liu, X. W. Cai and Z. Q. Tian, Surf. Sci., 1998, 406, 9–22 CrossRef CAS.
- B. Ren, X.-F. Lin, J.-W. Yan, B.-W. Mao and Z.-Q. Tian, J. Phys. Chem. B, 2003, 107, 899–902 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- Z.-Q. Tian, Z.-L. Yang, B. Ren and D.-Y. Wu, in Surface-Enhanced Raman Scattering: Physics and Applications, ed. K. Kneipp, M. Moskovits and H. Kneipp, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 125–146 DOI:10.1007/3-540-33567-6_7.
- K. N. Kanipe, P. P. F. Chidester, G. D. Stucky, C. D. Meinhart and M. Moskovits, J. Phys. Chem. C, 2017, 121, 14269–14273 CrossRef CAS.
- Z.-Q. Tian, Z.-L. Yang, B. Ren, J.-F. Li, Y. Zhang, X.-F. Lin, J.-W. Hu and D.-Y. Wu, Faraday Discuss., 2006, 132, 159–170 RSC.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2010, 10, 553–561 CrossRef CAS PubMed.
- Z. Q. Tian, B. Ren and B. W. Mao, J. Phys. Chem. B, 1997, 101, 1338–1346 CrossRef CAS.
- J. L. Yao, B. Ren, Z. F. Huang, P. G. Cao, R. A. Gu and Z.-Q. Tian, Electrochim. Acta, 2003, 48, 1263–1271 CrossRef CAS.
- H. Yamada, Y. Yamamoto and N. Tani, Chem. Phys. Lett., 1982, 86, 397–400 CrossRef CAS.
- S. Hayashi, R. Koh, Y. Ichiyama and K. Yamamoto, Phys. Rev. Lett., 1988, 60, 1085–1088 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, H. Jia, M. Li, J. Zeng, B. Yang, B. Zhao, W. Xu and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 996–1000 CrossRef CAS.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2014, 118, 11120–11130 CrossRef CAS.
- X. Wang and L. Guo, Angew. Chem., Int. Ed., 2020, 59, 4231–4239 CrossRef CAS PubMed.
- X. X. Han, W. Ji, B. Zhao and Y. Ozaki, Nanoscale, 2017, 9, 4847–4861 RSC.
- W. Ji, X. X. Han and B. Zhao, Recent Developments in Plasmon-Supported Raman Spectroscopy, 2018, pp. 451–482 DOI:10.1142/9781786344243_0016.
- A. Musumeci, D. Gosztola, T. Schiller, N. M. Dimitrijevic, V. Mujica, D. Martin and T. Rajh, J. Am. Chem. Soc., 2009, 131, 6040–6041 CrossRef CAS PubMed.
- W. Ji, L. Li, W. Song, X. Wang, B. Zhao and Y. Ozaki, Angew. Chem., Int. Ed., 2019, 58, 14452–14456 CrossRef CAS PubMed.
- W. Ji, L. Li, J. Guan, M. Mu, W. Song, L. Sun, B. Zhao and Y. Ozaki, Adv. Opt. Mater., 2021, 9, 2101866 CrossRef CAS.
- P. Li, L. Zhu, C. Ma, L. Zhang, L. Guo, Y. Liu, H. Ma and B. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 19153–19160 CrossRef CAS PubMed.
- E. Sachet, M. D. Losego, J. T. Guske, S. Franzen and J. P. Maria, Appl. Phys. Lett., 2013, 102, 051111 CrossRef.
- Y. Liu, H. Ma, X. X. Han and B. Zhao, Mater. Horiz., 2021, 8, 370–382 RSC.
- X. Wang, P. Li, X. X. Han, Y. Kitahama, B. Zhao and Y. Ozaki, Nanoscale, 2017, 9, 15303–15313 RSC.
- X. X. Han, C. Köhler, J. Kozuch, U. Kuhlmann, L. Paasche, A. Sivanesan, I. M. Weidinger and P. Hildebrandt, Small, 2013, 9(24), 4175–4181 CrossRef CAS PubMed.
- X. Liu, Z. Ye, Q. Xiang, Z. Xu, W. Yue, C. Li, Y. Xu, L. Wang, X. Cao and J. Zhang, Chem, 2023, 9, 1464–1476 CAS.
- J. Li, Q. Xie, J. Li, L. Sun, Y. Xie, Y. Ozaki and W. Ji, Adv. Funct. Mater., 2024, 34, 2400523 CrossRef CAS.
- W. Xu, N. Mao and J. Zhang, Small, 2013, 9, 1206–1224 CrossRef CAS PubMed.
- W. Xu, X. Ling, J. Xiao, M. S. Dresselhaus, J. Kong, H. Xu, Z. Liu and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9281–9286 CrossRef CAS PubMed.
- X. Ling, W. Fang, Y.-H. Lee, P. T. Araujo, X. Zhang, J. F. Rodriguez-Nieva, Y. Lin, J. Zhang, J. Kong and M. S. Dresselhaus, Nano Lett., 2014, 14, 3033–3040 CrossRef CAS PubMed.
- S. Zhang, N. Zhang, Y. Zhao, T. Cheng, X. Li, R. Feng, H. Xu, Z. Liu, J. Zhang and L. Tong, Chem. Soc. Rev., 2018, 47, 3217–3240 RSC.
- H. Kitadai, Q. Tan, L. Ping and X. Ling, npj 2D Mater. Appl., 2024, 8, 11 CrossRef.
- H. Xu, L. Xie, H. Zhang and J. Zhang, ACS Nano, 2011, 5, 5338–5344 CrossRef CAS PubMed.
- L. Zhou, L. Pusey-Nazzaro, G. Ren, L. Chen, L. Liu, W. Zhang, L. Yang, J. Zhou and J. Han, ACS Nano, 2022, 16, 577–587 CrossRef CAS PubMed.
- S. Almohammed, A. Fularz, F. Zhang, D. Alvarez-Ruiz, F. Bello, D. D. O’Regan, B. J. Rodriguez and J. H. Rice, ACS Appl. Mater. Interfaces, 2020, 12, 48874–48881 CrossRef CAS PubMed.
- J. Tan, B. Du, C. Ji, M. Shao, X. Zhao, J. Yu, S. Xu, B. Man, C. Zhang and Z. Li, ACS Photonics, 2023, 10, 2216–2225 CrossRef CAS.
- C. Li, S. Xu, J. Yu, Z. Li, W. Li, J. Wang, A. Liu, B. Man, S. Yang and C. Zhang, Nano Energy, 2021, 81, 105585 CrossRef CAS.
- M. Shao, C. Ji, J. Tan, B. Du, X. Zhao, J. Yu, B. Man, K. Xu, C. Zhang and Z. Li, Opto-Electron. Adv., 2023, 6, 230094 CAS.
- D. B. Chase and B. A. Parkinson, Appl. Spectrosc., 1988, 42, 1186–1187 CrossRef CAS.
- T. Dörfer, M. Schmitt and J. Popp, J. Raman Spectrosc., 2007, 38, 1379–1382 CrossRef.
- B. Ren, X.-F. Lin, Z.-L. Yang, G.-K. Liu, R. F. Aroca, B.-W. Mao and Z.-Q. Tian, J. Am. Chem. Soc., 2003, 125, 9598–9599 CrossRef CAS PubMed.
- M. W. Knight, N. S. King, L. Liu, H. O. Everitt, P. Nordlander and N. J. Halas, ACS Nano, 2014, 8, 834–840 CrossRef CAS PubMed.
- B. Sharma, M. F. Cardinal, M. B. Ross, A. B. Zrimsek, S. V. Bykov, D. Punihaole, S. A. Asher, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2016, 16, 7968–7973 CrossRef CAS PubMed.
- M. J. McClain, A. E. Schlather, E. Ringe, N. S. King, L. Liu, A. Manjavacas, M. W. Knight, I. Kumar, K. H. Whitmire, H. O. Everitt, P. Nordlander and N. J. Halas, Nano Lett., 2015, 15, 2751–2755 CrossRef CAS PubMed.
- S. Tian, O. Neumann, M. J. McClain, X. Yang, L. Zhou, C. Zhang, P. Nordlander and N. J. Halas, Nano Lett., 2017, 17, 5071–5077 CrossRef CAS PubMed.
- A. Bayles, C. J. Fabiano, C. Shi, L. Yuan, Y. Yuan, N. Craft, C. R. Jacobson, P. Dhindsa, A. Ogundare, Y. Mendez Camacho, B. Chen, H. Robatjazi, Y. Han, G. F. Strouse, P. Nordlander, H. O. Everitt and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2321852121 CrossRef CAS PubMed.
- J.-F. Li, S.-Y. Ding, Z.-L. Yang, M.-L. Bai, J. R. Anema, X. Wang, A. Wang, D.-Y. Wu, B. Ren, S.-M. Hou, T. Wandlowski and Z.-Q. Tian, J. Am. Chem. Soc., 2011, 133, 15922–15925 CrossRef CAS PubMed.
- Y.-P. Huang, S.-C. Huang, X.-J. Wang, N. Bodappa, C.-Y. Li, H. Yin, H.-S. Su, M. Meng, H. Zhang, B. Ren, Z.-L. Yang, R. Zenobi, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2018, 57, 7523–7527 CrossRef CAS PubMed.
- J. S. Suh, D. P. DiLella and M. Moskovits, J. Phys. Chem., 1983, 87, 1540–1544 CrossRef CAS.
- P. Hildebrandt and M. Stockburger, J. Phys. Chem., 1984, 88, 5935–5944 CrossRef CAS.
- E. J. Kiefl, R. F. Kiefl, D. P. dos Santos and A. G. Brolo, J. Phys. Chem. C, 2017, 121, 25487–25493 CrossRef CAS.
- X. Wang, S.-C. Huang, S. Hu, S. Yan and B. Ren, Nat. Rev. Phys., 2020, 2, 253–271 CrossRef.
- T. L. Vasconcelos, B. S. Archanjo, B. S. Oliveira, R. Valaski, R. C. Cordeiro, H. G. Medeiros, C. Rabelo, A. Ribeiro, P. Ercius, C. A. Achete, A. Jorio and L. G. Cançado, Adv. Opt. Mater., 2018, 6, 1800528 CrossRef.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- Z. Ye, C. Li, Q. Chen, Y. Xu and S. E. J. Bell, Angew. Chem., Int. Ed., 2019, 58, 19054–19059 CrossRef CAS PubMed.
- M. Moskovits, Phys. Chem. Chem. Phys., 2013, 15, 5301–5311 RSC.
- E. C. Le Ru and B. Auguié, ACS Nano, 2024, 18, 9773–9783 CrossRef CAS PubMed.
- M. Caldarola, P. Albella, E. Cortés, M. Rahmani, T. Roschuk, G. Grinblat, R. F. Oulton, A. V. Bragas and S. A. Maier, Nat. Commun., 2015, 6, 7915 CrossRef CAS PubMed.
- C. Zhan, B.-W. Liu, Z.-Q. Tian and B. Ren, J. Am. Chem. Soc., 2020, 142, 10905–10909 CrossRef CAS PubMed.
- D. J. Masiello and G. C. Schatz, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 78, 042505 CrossRef.
- T. Neuman, J. Aizpurua and R. Esteban, Nanophotonics, 2020, 9, 295–308 CrossRef CAS.
- P. J. G. Goulet and R. F. Aroca, Anal. Chem., 2007, 79, 2728–2734 CrossRef CAS PubMed.
- Y. Sawai, B. Takimoto, H. Nabika, K. Ajito and K. Murakoshi, J. Am. Chem. Soc., 2007, 129, 1658–1662 CrossRef CAS PubMed.
- J. E. Pemberton and R. P. Buck, Anal. Chem., 1981, 53, 2263–2267 CrossRef CAS.
- C. K. Chen, T. F. Heinz, D. Ricard and Y. R. Shen, Chem. Phys. Lett., 1981, 83, 455–458 CrossRef CAS.
- C. K. Chen, T. F. Heinz, D. Ricard and Y. R. Shen, Phys. Rev. Lett., 1981, 46, 1010–1012 CrossRef CAS.
- J. J. Laserna, E. L. Torres and J. D. Winefordner, Anal. Chim. Acta, 1987, 200, 469–480 CrossRef CAS.
- G. McAnally, C. McLaughlin, R. Brown, D. C. Robson, K. Faulds, D. R. Tackley, W. E. Smith and D. Graham, Analyst, 2002, 127, 838–841 RSC.
- C. McLaughlin, D. MacMillan, C. McCardle and W. E. Smith, Anal. Chem., 2002, 74, 3160–3167 CrossRef CAS PubMed.
- K. Faulds, W. E. Smith, D. Graham and R. J. Lacey, Analyst, 2002, 127, 282–286 RSC.
- K. Faulds, W. E. Smith and D. Graham, Anal. Chem., 2004, 76, 412–417 CrossRef CAS PubMed.
- R. Goodacre, D. Graham and K. Faulds, TrAC, Trends Anal. Chem., 2018, 102, 359–368 CrossRef CAS.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. L. de la Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., Int. Ed., 2020, 59, 5454–5462 CrossRef CAS PubMed.
- S. Sloan-Dennison, G. Q. Wallace, W. A. Hassanain, S. Laing, K. Faulds and D. Graham, Nano Convergence, 2024, 11, 33 CrossRef CAS PubMed.
- S. E. J. Bell and N. M. S. Sirimuthu, Analyst, 2004, 129, 1032–1036 RSC.
- D. Zhang, Y. Xie, S. K. Deb, V. J. Davison and D. Ben-Amotz, Anal. Chem., 2005, 77, 3563–3569 CrossRef CAS PubMed.
- S. E. J. Bell, J. N. Mackle and N. M. S. Sirimuthu, Analyst, 2005, 130, 545–549 RSC.
- K. W. Kho, U. S. Dinish, A. Kumar and M. Olivo, ACS Nano, 2012, 6, 4892–4902 CrossRef CAS PubMed.
- Y. Zhou, R. Ding, P. Joshi and P. Zhang, Anal. Chim. Acta, 2015, 874, 49–53 CrossRef CAS PubMed.
- R. Keir, E. Igata, M. Arundell, W. E. Smith, D. Graham, C. McHugh and J. M. Cooper, Anal. Chem., 2002, 74, 1503–1508 CrossRef CAS PubMed.
- J.-H. Jung, J.-B. Choo, D.-J. Kim and S.-H. Lee, Bull. Korean Chem. Soc., 2006, 27, 277–280 CrossRef CAS.
- D. Lee, S. Lee, G. H. Seong, J. Choo, E. K. Lee, D.-G. Gweon and S. Lee, Appl. Spectrosc., 2006, 60, 373–377 CrossRef CAS PubMed.
- K. R. Strehle, D. Cialla, P. Rösch, T. Henkel, M. Köhler and J. Popp, Anal. Chem., 2007, 79, 1542–1547 CrossRef CAS PubMed.
- I. J. Hidi, M. Jahn, M. W. Pletz, K. Weber, D. Cialla-May and J. Popp, J. Phys. Chem. C, 2016, 120, 20613–20623 CrossRef CAS.
- I. J. Hidi, M. Jahn, K. Weber, T. Bocklitz, M. W. Pletz, D. Cialla-May and J. Popp, Anal. Chem., 2016, 88, 9173–9180 CrossRef CAS PubMed.
- J. Krajczewski, K. Kołątaj and A. Kudelski, Chem. Phys. Lett., 2015, 625, 84–90 CrossRef CAS.
- E. Giorgetti, P. Marsili, F. Giammanco, S. Trigari, C. Gellini and M. Muniz-Miranda, J. Raman Spectrosc., 2015, 46, 462–469 CrossRef CAS.
- S.-Y. Ding, E.-M. You, Z.-Q. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- H.-L. Wang, E.-M. You, R. Panneerselvam, S.-Y. Ding and Z.-Q. Tian, Light: Sci. Appl., 2021, 10, 161 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp and K. Kneipp, Chem. Soc. Rev., 2008, 37, 1052–1060 RSC.
- S. Lal, N. K. Grady, J. Kundu, C. S. Levin, J. B. Lassiter and N. J. Halas, Chem. Soc. Rev., 2008, 37, 898–911 RSC.
- M. D. Porter, R. J. Lipert, L. M. Siperko, G. Wang and R. Narayanan, Chem. Soc. Rev., 2008, 37, 1001–1011 RSC.
- N. J. Halas and M. Moskovits, MRS Bull., 2013, 38, 607–611 CrossRef CAS.
- B. Sharma, M. Fernanda Cardinal, S. L. Kleinman, N. G. Greeneltch, R. R. Frontiera, M. G. Blaber, G. C. Schatz and R. P. Van Duyne, MRS Bull., 2013, 38, 615–624 CrossRef CAS.
- E. A. Pozzi, G. Goubert, N. Chiang, N. Jiang, C. T. Chapman, M. O. McAnally, A.-I. Henry, T. Seideman, G. C. Schatz, M. C. Hersam and R. P. V. Duyne, Chem. Rev., 2017, 117, 4961–4982 CrossRef CAS PubMed.
- C. L. Brosseau, A. Colina, J. V. Perales-Rondon, A. J. Wilson, P. B. Joshi, B. Ren and X. Wang, Nat. Rev. Methods Primers, 2023, 3, 79 CrossRef CAS.
- Y.-J. Zhang, H. Ze, P.-P. Fang, Y.-F. Huang, A. Kudelski, J. Fernández-Vidal, L. J. Hardwick, J. Lipkowski, Z.-Q. Tian and J.-F. Li, Nat. Rev. Methods Primers, 2023, 3, 36 CrossRef CAS.
- E. Bailo and V. Deckert, Chem. Soc. Rev., 2008, 37, 921–930 RSC.
- T. Deckert-Gaudig, A. Taguchi, S. Kawata and V. Deckert, Chem. Soc. Rev., 2017, 46, 4077–4110 RSC.
- R. C. Maher, C. M. Galloway, E. C. Le Ru, L. F. Cohen and P. G. Etchegoin, Chem. Soc. Rev., 2008, 37, 965–979 RSC.
- L. Troncoso-Afonso, G. A. Vinnacombe-Willson, C. García-Astrain and L. M. Liz-Márzan, Chem. Soc. Rev., 2024, 53, 5118–5148 RSC.
- in Surface-Enhanced Raman Scattering: Physics and Applications, ed. M. M. Katrin Kneipp and H. Kneipp, Springer Berlin, Heidelberg, 2006 Search PubMed.
- in Tip Enhancement, ed. V. M. S. Satoshi Kawata, Elsevier Science, 2007 Search PubMed.
- in Surface Enhanced Raman Spectroscopy: Analytical, Biophysical and Life Science Applications, ed. S. Schlücker, 2010 Search PubMed.
- in Frontiers of Surface-Enhanced Raman Scattering: Single Nanoparticles and Single Cells, ed. K. K. Yukihiro Ozaki and R. Aroca, 2014 Search PubMed.
- M. Prochazka, Surface-Enhanced Raman Spectroscopy: Biological and Medical Physics, Biomedical Engineering, Springer, Cham, 2016 Search PubMed.
- Y. O. Katrin Kneipp and Z.-Q. Tian, Recent Developments in Plasmon-Supported Raman Spectroscopy, 2018 Search PubMed.
- in Principles and Clinical Diagnostic Applications of Surface-Enhanced Raman Spectroscopy, ed. Y. Wang, Elsevier, 2021 Search PubMed.
- in SERS for Point-of-care and Clinical Applications, ed. A. Fales, Elsevier, 2022 Search PubMed.
- D. A. Long, Raman Eff., 2002, 19–29, DOI:10.1002/0470845767.ch2.
- J.-H. Zhong, X. Jin, L. Meng, X. Wang, H.-S. Su, Z.-L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136 CrossRef CAS PubMed.
- J. Lee, N. Tallarida, X. Chen, P. Liu, L. Jensen and V. A. Apkarian, ACS Nano, 2017, 11, 11466–11474 CrossRef CAS PubMed.
- N. Tallarida, J. Lee and V. A. Apkarian, ACS Nano, 2017, 11, 11393–11401 CrossRef CAS PubMed.
- J. Lee, N. Tallarida, X. Chen, L. Jensen and V. A. Apkarian, Sci. Adv., 2018, 4, eaat5472 CrossRef PubMed.
- K. T. Crampton, J. Lee and V. A. Apkarian, ACS Nano, 2019, 13, 6363–6371 CrossRef CAS PubMed.
- J. Takahara, S. Yamagishi, H. Taki, A. Morimoto and T. Kobayashi, Opt. Lett., 1997, 22, 475–477 CrossRef CAS PubMed.
- K. V. Nerkararyan, Phys. Lett. A, 1997, 237, 103–105 CrossRef CAS.
- A. J. Babadjanyan, N. L. Margaryan and K. V. Nerkararyan, J. Appl. Phys., 2000, 87, 3785–3788 CrossRef CAS.
- S. Duan, G. Tian and Y. Luo, Angew. Chem., Int. Ed., 2016, 55, 1041–1045 CrossRef CAS PubMed.
- S. Duan, Z. Xie, G. Tian and Y. Luo, J. Phys. Chem. Lett., 2020, 11, 407–411 CrossRef CAS PubMed.
- R. B. Jaculbia, H. Imada, K. Miwa, T. Iwasa, M. Takenaka, B. Yang, E. Kazuma, N. Hayazawa, T. Taketsugu and Y. Kim, Nat. Nanotechnol., 2020, 15, 105–110 CrossRef CAS PubMed.
- N. Agraït, A. L. Yeyati and J. M. van Ruitenbeek, Phys. Rep., 2003, 377, 81–279 CrossRef.
- S. Liu, B. Cirera, Y. Sun, I. Hamada, M. Müller, A. Hammud, M. Wolf and T. Kumagai, Nano Lett., 2020, 20, 5879–5884 CrossRef CAS PubMed.
- S. Liu, A. Hammud, M. Wolf and T. Kumagai, Nano Lett., 2021, 21, 4057–4061 CrossRef CAS PubMed.
- B. Cirera, Y. Litman, C. Lin, A. Akkoush, A. Hammud, M. Wolf, M. Rossi and T. Kumagai, Nano Lett., 2022, 22, 2170–2176 CrossRef CAS PubMed.
- R.-P. Wang, C.-R. Hu, Y. Han, B. Yang, G. Chen, Y. Zhang, Y. Zhang and Z.-C. Dong, J. Phys. Chem. C, 2022, 126, 12121–12128 CrossRef CAS.
- T. Siday, J. Hayes, F. Schiegl, F. Sandner, P. Menden, V. Bergbauer, M. Zizlsperger, S. Nerreter, S. Lingl, J. Repp, J. Wilhelm, M. A. Huber, Y. A. Gerasimenko and R. Huber, Nature, 2024, 629, 329–334 CrossRef CAS PubMed.
- Y. Luo, A. Martin-Jimenez, M. Pisarra, F. Martin, M. Garg and K. Kern, Nat. Commun., 2023, 14, 3484 CrossRef CAS PubMed.
- L. Novotny and S. J. Stranick, Annu. Rev. Phys. Chem., 2006, 57, 303–331 CrossRef CAS PubMed.
- J. Lee and V. A. Apkarian, Microsc. Microanal., 2023, 29, 622–623 CrossRef.
- R. Chen and L. Jensen, J. Chem. Phys., 2020, 152, 024126 CrossRef PubMed.
- R. Chen and L. Jensen, J. Chem. Phys., 2020, 153, 224704 CrossRef CAS PubMed.
- R. Chen and L. Jensen, J. Chem. Phys., 2022, 157, 184705 CrossRef CAS PubMed.
- S. Duan, Z. Rinkevicius, G. Tian and Y. Luo, J. Am. Chem. Soc., 2019, 141, 13795–13798 CrossRef CAS PubMed.
- S. Duan and X. Xu, J. Phys. Chem. Lett., 2023, 14, 6726–6735 CrossRef CAS PubMed.
- S. Duan, G. Tian and Y. Luo, Chem. Soc. Rev., 2024, 53, 5083–5117 RSC.
- J. G. Mehtala, D. Y. Zemlyanov, J. P. Max, N. Kadasala, S. Zhao and A. Wei, Langmuir, 2014, 30, 13727–13730 CrossRef CAS PubMed.
- H. He, P. Li, X. Tang, D. Lin, A. Xie, Y. Shen and L. Yang, Spectrochim. Acta, Part A, 2019, 212, 293–299 CrossRef CAS PubMed.
- M. U. Amin, R. Zhang, L. Li, H. You and J. Fang, Anal. Chem., 2021, 93, 7657–7664 CrossRef CAS PubMed.
- M.-D. Li, Y. Cui, M.-X. Gao, J. Luo, B. Ren and Z.-Q. Tian, Anal. Chem., 2008, 80, 5118–5125 CrossRef CAS PubMed.
- J.-Y. Huang, C. Zong, L.-J. Xu, Y. Cui and B. Ren, Chem. Commun., 2011, 47, 5738–5740 RSC.
- S.-Q. Pan, P. Luo, J. Chen, T. Wu, B. Xu, F. Chen, D.-Y. Wu, B. Ren, G.-K. Liu, J. Xie, P. Xu and Z.-Q. Tian, Anal. Chem., 2023, 95, 13346–13352 CrossRef CAS PubMed.
- P.-S. Wang, H. Ma, X.-H. Xi, X. Yue, S.-Q. Pan, M. Cao, G.-K. Liu, X. Wang and B. Ren, J. Raman Spectrosc., 2023, 54, 573–579 CrossRef CAS.
- D. Malyshev, R. Öberg, T. Dahlberg, K. Wiklund, L. Landström, P. O. Andersson and M. Andersson, Spectrochim. Acta, Part A, 2022, 265, 120381 CrossRef CAS PubMed.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- S. R. Smith and J. Lipkowski, J. Phys. Chem. C, 2018, 122, 7303–7311 CrossRef CAS.
- Z.-M. Zhou, H. Zheng, T. Liu, Z.-Z. Xie, S.-H. Luo, G.-Y. Chen, Z.-Q. Tian and G.-K. Liu, Anal. Chem., 2021, 93, 8603–8612 CrossRef CAS PubMed.
- L. He, Y. Liu, J. Liu, Y. Xiong, J. Zheng, Y. Liu and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 3741–3745 CrossRef CAS PubMed.
- X. Qiao, B. Su, C. Liu, Q. Song, D. Luo, G. Mo and T. Wang, Adv. Mater., 2018, 30, 1702275 CrossRef PubMed.
- J. W. M. Osterrieth, D. Wright, H. Noh, C.-W. Kung, D. Vulpe, A. Li, J. E. Park, R. P. Van Duyne, P. Z. Moghadam, J. J. Baumberg, O. K. Farha and D. Fairen-Jimenez, J. Am. Chem. Soc., 2019, 141, 3893–3900 CrossRef CAS PubMed.
- Q.-Q. Chen, R.-N. Hou, Y.-Z. Zhu, X.-T. Wang, H. Zhang, Y.-J. Zhang, L. Zhang, Z.-Q. Tian and J.-F. Li, Anal. Chem., 2021, 93, 7188–7195 CrossRef CAS PubMed.
- J. Mosquera, Y. Zhao, H.-J. Jang, N. Xie, C. Xu, N. A. Kotov and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 1902082 CrossRef CAS.
- S. Kasera, F. Biedermann, J. J. Baumberg, O. A. Scherman and S. Mahajan, Nano Lett., 2012, 12, 5924–5928 CrossRef CAS PubMed.
- D.-W. Li, L.-L. Qu, K. Hu, Y.-T. Long and H. Tian, Angew. Chem., Int. Ed., 2015, 54, 12758–12761 CrossRef CAS PubMed.
- L. Guerrini, E. Pazos, C. Penas, M. E. Vázquez, J. L. Mascareñas and R. A. Alvarez-Puebla, J. Am. Chem. Soc., 2013, 135, 10314–10317 CrossRef CAS PubMed.
- J. Liu, Z. Liu, W. Wang and Y. Tian, Angew. Chem., Int. Ed., 2021, 60, 21351–21359 CrossRef CAS PubMed.
- W. Zhu, E.-L. Cai, H.-Z. Li, P. Wang, A.-G. Shen, J. Popp and J.-M. Hu, Angew. Chem., Int. Ed., 2021, 60, 21846–21852 CrossRef CAS PubMed.
- Y. Zeng, J.-Q. Ren, A.-G. Shen and J.-M. Hu, J. Am. Chem. Soc., 2018, 140, 10649–10652 CrossRef CAS PubMed.
- E.-M. You, H.-L. Wang, J.-R. Zheng, Z.-D. Meng, M.-X. Zhang, S.-Y. Ding and Z.-Q. Tian, J. Raman Spectrosc., 2021, 52, 446–457 CrossRef CAS.
- S. A. Meyer, E. C. Le Ru and P. G. Etchegoin, Anal. Chem., 2011, 83, 2337–2344 CrossRef CAS PubMed.
- S. A. Meyer, B. Auguié, E. C. Le Ru and P. G. Etchegoin, J. Phys. Chem. A, 2012, 116, 1000–1007 CrossRef CAS PubMed.
- C.-Y. Li, S. Duan, J. Yi, C. Wang, P. M. Radjenovic, Z.-Q. Tian and J.-F. Li, Sci. Adv., 2020, 6, eaba6012 CrossRef CAS PubMed.
- C.-Y. Li, J.-B. Le, Y.-H. Wang, S. Chen, Z.-L. Yang, J.-F. Li, J. Cheng and Z.-Q. Tian, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- X.-M. Lin, X.-T. Wang, Y.-L. Deng, X. Chen, H.-N. Chen, P. M. Radjenovic, X.-G. Zhang, Y.-H. Wang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Nano Lett., 2022, 22, 5544–5552 CrossRef CAS PubMed.
- H. Ze, Z.-L. Yang, M.-L. Li, X.-G. Zhang, Y.-L. A, Q.-N. Zheng, Y.-H. Wang, J.-H. Tian, Y.-J. Zhang and J.-F. Li, J. Am. Chem. Soc., 2024, 146, 12538–12546 CrossRef CAS PubMed.
- Y. Gu, E.-M. You, J.-D. Lin, J.-H. Wang, S.-H. Luo, R.-Y. Zhou, C.-J. Zhang, J.-L. Yao, H.-Y. Li, G. Li, W.-W. Wang, Y. Qiao, J.-W. Yan, D.-Y. Wu, G.-K. Liu, L. Zhang, J.-F. Li, R. Xu, Z.-Q. Tian, Y. Cui and B.-W. Mao, Nat. Commun., 2023, 14, 3536 CrossRef CAS PubMed.
- H. Ze, X. Chen, X.-T. Wang, Y.-H. Wang, Q.-Q. Chen, J.-S. Lin, Y.-J. Zhang, X.-G. Zhang, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2021, 143, 1318–1322 CrossRef CAS PubMed.
- K. Xu, J. Power Sources, 2023, 559, 232652 CrossRef CAS.
- Y. Gu, W.-W. Wang, Y.-J. Li, Q.-H. Wu, S. Tang, J.-W. Yan, M.-S. Zheng, D.-Y. Wu, C.-H. Fan, W.-Q. Hu, Z.-B. Chen, Y. Fang, Q.-H. Zhang, Q.-F. Dong and B.-W. Mao, Nat. Commun., 2018, 9, 1339 CrossRef PubMed.
- Y. Gu, S. Tang, J. Yi, S.-H. Luo, C.-Y. Li, G. Liu, J. Yan, J.-F. Li, B.-W. Mao and Z.-Q. Tian, J. Phys. Chem. C, 2023, 127, 13466–13477 CrossRef CAS.
- E.-M. You, Y. Gu, J. Yi, D.-Y. Wu, J.-F. Li and Z.-Q. Tian, Electrochim. Acta, 2024, 498, 144689 CrossRef CAS.
- S. Duan, X. Xu, Y. Luo and Z.-Q. Tian, Chem. Commun., 2011, 47, 11438–11440 RSC.
- B. Yang, G. Chen, A. Ghafoor, Y.-F. Zhang, X.-B. Zhang, H. Li, X.-R. Dong, R.-P. Wang, Y. Zhang, Y. Zhang and Z.-C. Dong, Angew. Chem., Int. Ed., 2023, 62, e202218799 CrossRef CAS PubMed.
- Y. Zhang and H. Noji, Anal. Chem., 2017, 89, 92–101 CrossRef CAS PubMed.
- X. Bi, D. M. Czajkowsky, Z. Shao and J. Ye, Nature, 2024, 628, 771–775 CrossRef CAS PubMed.
- C. D. L. de Albuquerque, R. G. Sobral-Filho, R. J. Poppi and A. G. Brolo, Anal. Chem., 2018, 90, 1248–1254 CrossRef CAS PubMed.
- J. Li, A. A. I. Sina, F. Antaw, D. Fielding, A. Möller, R. Lobb, A. Wuethrich and M. Trau, Adv. Sci., 2023, 10, 2204207 CrossRef CAS PubMed.
- H. Zhang, L. Yang, M. Zhang, H. Wei, L. Tong, H. Xu and Z. Li, Nano Lett., 2024, 24, 11116–11123 CrossRef CAS PubMed.
- A. Tuckmantel Bido and A. G. Brolo, ACS Appl. Nano Mater., 2023, 6, 15426–15436 CrossRef CAS.
- H. C. Schorr and Z. D. Schultz, Analyst, 2024, 149, 3711–3715 RSC.
- N. Choi, Y. Zhang, Y. Wang and S. Schlücker, Chem. Soc. Rev., 2024, 53, 6675–6693 RSC.
- D. Cialla-May, A. Bonifacio, T. Bocklitz, A. Markin, N. Markina, S. Fornasaro, A. Dwivedi, T. Dib, E. Farnesi, C. Liu, A. Ghosh and J. Popp, Chem. Soc. Rev., 2024, 53, 8957–8979 RSC.
- J. Kneipp, S. Seifert and F. Gärber, Chem. Soc. Rev., 2024, 53, 7641–7656 RSC.
- S. Lee, H. Dang, J.-I. Moon, K. Kim, Y. Joung, S. Park, Q. Yu, J. Chen, M. Lu, L. Chen, S.-W. Joo and J. Choo, Chem. Soc. Rev., 2024, 53, 5394–5427 RSC.
- R. Beeram, K. R. Vepa and V. R. Soma, Biosensors, 2023, 13, 328 CrossRef CAS PubMed.
- S. Lee, L. Bi, H. Chen, D. Lin, R. Mei, Y. Wu, L. Chen, S.-W. Joo and J. Choo, Chem. Soc. Rev., 2023, 52, 8500–8530 RSC.
- J. H. Granger, N. E. Schlotter, A. C. Crawford and M. D. Porter, Chem. Soc. Rev., 2016, 45, 3865–3882 RSC.
- S. S. Panikar, D. Cialla-May, E. De la Rosa, P. Salas and J. Popp, TrAC, Trends Anal. Chem., 2021, 134, 116122 CrossRef CAS.
- A. Barhoumi, D. Zhang, F. Tam and N. J. Halas, J. Am. Chem. Soc., 2008, 130, 5523–5529 CrossRef CAS PubMed.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Am. Chem. Soc., 2006, 128, 15580–15581 CrossRef CAS PubMed.
- L.-J. Xu, Z.-C. Lei, J. Li, C. Zong, C. J. Yang and B. Ren, J. Am. Chem. Soc., 2015, 137, 5149–5154 CrossRef CAS PubMed.
- K. E. Shafer-Peltier, C. L. Haynes, M. R. Glucksberg and R. P. Van Duyne, J. Am. Chem. Soc., 2003, 125, 588–593 CrossRef CAS PubMed.
- D. A. Stuart, J. M. Yuen, N. Shah, O. Lyandres, C. R. Yonzon, M. R. Glucksberg, J. T. Walsh and R. P. Van Duyne, Anal. Chem., 2006, 78, 7211–7215 CrossRef CAS PubMed.
- J. M. Yuen, N. C. Shah, J. T. Walsh, Jr., M. R. Glucksberg and R. P. Van Duyne, Anal. Chem., 2010, 82, 8382–8385 CrossRef CAS PubMed.
- C. R. Yonzon, C. L. Haynes, X. Zhang, J. T. Walsh and R. P. Van Duyne, Anal. Chem., 2004, 76, 78–85 CrossRef CAS PubMed.
- K. Ma, J. M. Yuen, N. C. Shah, J. T. Walsh, Jr., M. R. Glucksberg and R. P. Van Duyne, Anal. Chem., 2011, 83, 9146–9152 CrossRef CAS PubMed.
- X. X. Han, G. G. Huang, B. Zhao and Y. Ozaki, Anal. Chem., 2009, 81, 3329–3333 CrossRef CAS PubMed.
- G. G. Huang, X. X. Han, M. K. Hossain and Y. Ozaki, Anal. Chem., 2009, 81, 5881–5888 CrossRef CAS PubMed.
- X. X. Han, H. Y. Jia, Y. F. Wang, Z. C. Lu, C. X. Wang, W. Q. Xu, B. Zhao and Y. Ozaki, Anal. Chem., 2008, 80, 2799–2804 CrossRef CAS PubMed.
- L.-J. Xu, C. Zong, X.-S. Zheng, P. Hu, J.-M. Feng and B. Ren, Anal. Chem., 2014, 86, 2238–2245 CrossRef CAS PubMed.
- P.-S. Wang, H. Ma, S. Yan, X. Lu, H. Tang, X.-H. Xi, X.-H. Peng, Y. Huang, Y.-F. Bao, M.-F. Cao, H. Wang, J. Huang, G. Liu, X. Wang and B. Ren, Chem. Sci., 2022, 13, 13829–13835 RSC.
- A. Barhoumi and N. J. Halas, J. Am. Chem. Soc., 2010, 132, 12792–12793 CrossRef CAS PubMed.
- J. L. Abell, J. M. Garren, J. D. Driskell, R. A. Tripp and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 12889–12892 CrossRef CAS PubMed.
- L. Guerrini, Ž. Krpetić, D. van Lierop, R. A. Alvarez-Puebla and D. Graham, Angew. Chem., Int. Ed., 2015, 54, 1144–1148 CrossRef CAS PubMed.
- E. Garcia-Rico, R. A. Alvarez-Puebla and L. Guerrini, Chem. Soc. Rev., 2018, 47, 4909–4923 RSC.
- A. Walter, A. März, W. Schumacher, P. Rösch and J. Popp, Lab Chip, 2011, 11, 1013–1021 RSC.
- I. S. Patel, W. R. Premasiri, D. T. Moir and L. D. Ziegler, J. Raman Spectrosc., 2008, 39, 1660–1672 CrossRef CAS PubMed.
- W. R. Premasiri, J. C. Lee, A. Sauer-Budge, R. Théberge, C. E. Costello and L. D. Ziegler, Anal. Bioanal. Chem., 2016, 408, 4631–4647 CrossRef CAS PubMed.
- P. Lemler, W. R. Premasiri, A. DelMonaco and L. D. Ziegler, Anal. Bioanal. Chem., 2014, 406, 193–200 CrossRef CAS PubMed.
- F. Lussier, T. Brulé, M. Vishwakarma, T. Das, J. P. Spatz and J.-F. Masson, Nano Lett., 2016, 16, 3866–3871 CrossRef CAS PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- W. R. Premasiri, J. C. Lee and L. D. Ziegler, J. Phys. Chem. B, 2012, 116, 9376–9386 CrossRef CAS PubMed.
- X. Bi, J. Wang, B. Xue, C. He, F. Liu, H. Chen, L. L. Lin, B. Dong, B. Li, C. Jin, J. Pan, W. Xue and J. Ye, Cell Rep. Med., 2024, 5, 101579 CrossRef CAS PubMed.
- H. Ma, S. Yan, X. Lu, Y.-F. Bao, J. Liu, L. Liao, K. Dai, M. Cao, X. Zhao, H. Yan, H.-L. Wang, X. Peng, N. Chen, H. Feng, L. Zhu, G. Yao, C. Fan, D.-Y. Wu, B. Wang, X. Wang and B. Ren, Sci. Adv., 2023, 9, eadh8362 CrossRef CAS PubMed.
- J. Plou, I. García, M. Charconnet, I. Astobiza, C. García-Astrain, C. Matricardi, A. Mihi, A. Carracedo and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 1910335 CrossRef CAS.
- P. S. Valera, J. Plou, I. García, I. Astobiza, C. Viera, A. M. Aransay, J. E. Martin, I. R. Sasselli, A. Carracedo and L. M. Liz-Marzán, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2311674120 CrossRef CAS PubMed.
- J. Plou, B. Molina-Martínez, C. García-Astrain, J. Langer, I. García, A. Ercilla, G. Perumal, A. Carracedo and L. M. Liz-Marzán, Nano Lett., 2021, 21, 8785–8793 CrossRef CAS PubMed.
- J.-H. Lee, G.-H. Kim and J.-M. Nam, J. Am. Chem. Soc., 2012, 134, 5456–5459 CrossRef CAS PubMed.
- J.-H. Lee, M.-H. You, G.-H. Kim and J.-M. Nam, Nano Lett., 2014, 14, 6217–6225 CrossRef CAS PubMed.
- J.-M. Kim, J. Kim, K. Choi and J.-M. Nam, Adv. Mater., 2023, 35, 2208250 CrossRef CAS PubMed.
- J. Kim, J.-M. Kim, K. Choi, J.-E. Park and J.-M. Nam, J. Am. Chem. Soc., 2024, 146, 14012–14021 CrossRef CAS PubMed.
- Y. Zhang, D. Jimenez de Aberasturi, M. Henriksen-Lacey, J. Langer and L. M. Liz-Marzán, ACS Sens., 2020, 5, 3194–3206 CrossRef CAS PubMed.
- T. E. Rohr, T. Cotton, N. Fan and P. J. Tarcha, Anal. Biochem., 1989, 182, 388–398 CrossRef CAS PubMed.
- D. S. Grubisha, R. J. Lipert, H.-Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS PubMed.
- Z. Wang, S. Zong, L. Wu, D. Zhu and Y. Cui, Chem. Rev., 2017, 117, 7910–7963 CrossRef CAS PubMed.
- T. Park, S. Lee, G. H. Seong, J. Choo, E. K. Lee, Y. S. Kim, W. H. Ji, S. Y. Hwang, D.-G. Gweon and S. Lee, Lab Chip, 2005, 5, 437–442 RSC.
- W. E. Doering, M. E. Piotti, M. J. Natan and R. G. Freeman, Adv. Mater., 2007, 19, 3100–3108 CrossRef CAS.
- M. Y. Sha, H. Xu, M. J. Natan and R. Cromer, J. Am. Chem. Soc., 2008, 130, 17214–17215 CrossRef CAS PubMed.
- P. Dey, ACS Measure. Sci. Au, 2023, 3, 434–443 CrossRef CAS PubMed.
- J. Hwang, S. Lee and J. Choo, Nanoscale, 2016, 8, 11418–11425 RSC.
- S. X. Leong, Y. X. Leong, E. X. Tan, H. Y. F. Sim, C. S. L. Koh, Y. H. Lee, C. Chong, L. S. Ng, J. R. T. Chen, D. W. C. Pang, L. B. T. Nguyen, S. K. Boong, X. Han, Y.-C. Kao, Y. H. Chua, G. C. Phan-Quang, I. Y. Phang, H. K. Lee, M. Y. Abdad, N. S. Tan and X. Y. Ling, ACS Nano, 2022, 16, 2629–2639 CrossRef CAS PubMed.
- J. Prakash, S. Sun, H. C. Swart and R. K. Gupta, Appl. Mater. Today, 2018, 11, 82–135 CrossRef.
- C. Zhan, X.-J. Chen, Y.-F. Huang, D.-Y. Wu and Z.-Q. Tian, Acc. Chem. Res., 2019, 52, 2784–2792 CrossRef CAS PubMed.
- L. Cui, D. Zhang, K. Yang, X. Zhang and Y.-G. Zhu, Anal. Chem., 2019, 91, 15345–15354 CrossRef CAS PubMed.
- M. Vezvaie, C. L. Brosseau and J. Lipkowski, Electrochim. Acta, 2013, 110, 120–132 CrossRef CAS.
- N. Choi and S. Schlücker, ACS Nano, 2024, 18, 5998–6007 CrossRef CAS PubMed.
- S. Pang, T. Yang and L. He, TrAC, Trends Anal. Chem., 2016, 85, 73–82 CrossRef CAS.
- M.-L. Xu, Y. Gao, X. X. Han and B. Zhao, J. Agric. Food Chem., 2017, 65, 6719–6726 CrossRef CAS PubMed.
- B. Yu, M. Ge, P. Li, Q. Xie and L. Yang, Talanta, 2019, 191, 1–10 CrossRef CAS PubMed.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2020, 1097, 1–29 CrossRef CAS PubMed.
- A. Hakonen, P. O. Andersson, M. Stenbæk Schmidt, T. Rindzevicius and M. Käll, Anal. Chim. Acta, 2015, 893, 1–13 CrossRef CAS PubMed.
- M. Veneranda, Appl. Spectrosc. Rev., 2023, 59, 883–907 CrossRef.
- F. Pozzi and M. Leona, J. Raman Spectrosc., 2016, 47, 67–77 CrossRef CAS.
- J. Yi, E.-M. You, G.-K. Liu and Z.-Q. Tian, Nat. Nanotechnol., 2024, 19, 1748–1762 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00883a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |