
Intracellular metal ion-based chemistry for programmed cell death
Yawen
You
 abc,
Zhaochen
Guo
d,
Tyler
Wolter
abce and
Quanyin
Hu
*abc
abc,
Zhaochen
Guo
d,
Tyler
Wolter
abce and
Quanyin
Hu
*abc
aPharmaceutical Sciences Division, School of Pharmacy, University of Wisconsin Madison, Madison, WI 53705, USA. E-mail: qhu66@wisc.edu
bCarbone Cancer Center, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI 53705, USA
cWisconsin Center for NanoBioSystems, School of Pharmacy, University of Wisconsin-Madison, Madison, WI 53705, USA
dDepartment of Biochemistry, College of Agriculture and Life Science, University of Wisconsin-Madison, Madison, WI 53706, USA
eInstitute for Clinical and Translational Research, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI 53705, USA
First published on 2nd January 2025
Abstract
Intracellular metal ions play essential roles in multiple physiological processes, including catalytic action, diverse cellular processes, intracellular signaling, and electron transfer. It is crucial to maintain intracellular metal ion homeostasis which is achieved by the subtle balance of storage and release of metal ions intracellularly along with the influx and efflux of metal ions at the interface of the cell membrane. Dysregulation of intracellular metal ions has been identified as a key mechanism in triggering programmed cell death (PCD). Despite the importance of metal ions in initiating PCD, the molecular mechanisms of intracellular metal ions within these processes are infrequently discussed. An in-depth understanding and review of the role of metal ions in triggering PCD may better uncover novel tools for cancer diagnosis and therapy. Specifically, the essential roles of calcium (Ca2+), iron (Fe2+/3+), copper (Cu+/2+), and zinc (Zn2+) ions in triggering PCD are primarily explored in this review, and other ions like manganese (Mn2+/3+/4+), cobalt (Co2+/3+) and magnesium ions (Mg2+) are briefly discussed. Further, this review elaborates on the underlying chemical mechanisms and summarizes these metal ions triggering PCD in cancer therapy. This review bridges chemistry, immunology, and biology to foster the rational regulation of metal ions to induce PCD for cancer therapy.
 Yawen You | Yawen You is a postdoctoral fellow in Professor Hu's laboratory at the School of Pharmacy, University of Wisconsin-Madison. She received her PhD degree at the University of Science and Technology of China. Her research focuses on inorganic catalysis, drug delivery, and cell therapy. |
 Quanyin Hu | Quanyin Hu is an Assistant Professor at School of Pharmacy, University of Wisconsin-Madison (UW-Madison). He received his PhD degree in Biomedical Engineering at University of North Carolina at Chapel Hill (UNC-CH) and North Carolina State University. Before he joined UW-Madison, he was a postdoc associate at Koch Institute for Integrative Cancer Research at Massachusetts Institute of Technology (MIT). His research group focuses on drug delivery, cell therapy, immunotherapy and personalized therapy. |
1. Introduction
As one of the most common heterogeneous diseases, cancer has been characterized as a cell death disorder. Cell death plays a significant role in the homeostasis of multicellular organisms. According to the type of triggered stimulus, cellular context, and morphological criteria, cell death is categorized into accidental cell death (ACD) and programmed cell death (PCD).1 ACD is an out-of-control biological process. However, PCD relies on elaborate regulations that can be modulated by genetic, pharmacological, and biochemical interventions.2,3 Current PCD has been critically summarized into molecularly oriented definitions by the Nomenclature Committee on Cell Death (NCCD), including necroptosis, pyroptosis, ferroptosis, cuproptosis,4 entotic cell death, netotic cell death (NETosis), parthanatos, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis, and oxeiptosis (Fig. 1). The molecular mechanisms responsible for triggering and transmitting distinct PCD are intricately interconnected. Therefore, different lethal subroutines within PCD can significantly impact cancer progression and treatment outcomes. In the early stages of oncogenesis, cancer cells may resist cancer treatments due to mutations disrupting the PCD pathway, thereby underscoring the evasion of PCD as a hallmark of cancer. The strategic initiation and management of PCD signals, either individually or synergistically, presents a means to surmount such resistance in specific cancer subtypes.5,6 Collectively, there is an urgent need to clarify the deep pathogenesis of cancer diseases combining various mechanisms of PCD for combating cancers.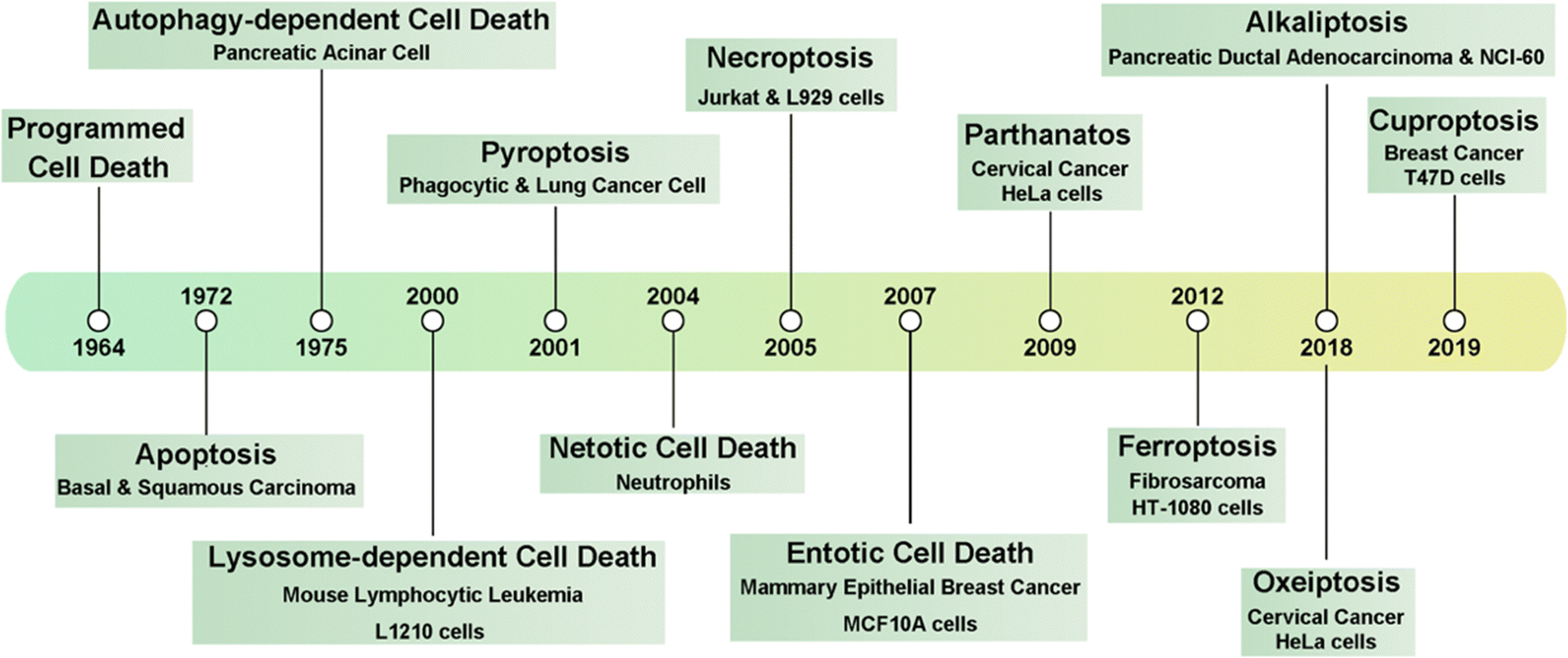 | ||
| Fig. 1 Timeline of the discovery of multiple forms of PCD. | ||
Recently, knowledge of the mechanisms underlying PCD has increased substantially, with multiple PCD-inducing targets being identified and exploited as new therapeutic modalities. Dyshomeostasis of intracellular metal ions, i.e., a deficiency or excess, can alter intracellular signaling thereby contributing to diverse PCDs. For example, Fe2+/3+, Cu+/2+, Mn2+/3+/4+, and Co2+/3+ can disrupt redox homeostasis to produce excessive reactive oxygen species (ROS) and further induce cancer cell death by apoptosis, ferroptosis, or other PCDs.7–10 Furthermore, excess Cu+ can destabilize iron–sulfur (Fe–S) proteins, resulting in cuproptosis.11 Ca2+, Zn2+, and Mg2+ play vital roles in enzyme catalysis and cellular metabolism.12,13 Particularly, Zn2+ and Mg2+ can induce apoptosis through the starvation effect by inhibiting the activity of glycolysis-related enzymes.14 In addition, Ca2+ can trigger the release of different caspases to induce apoptosis and pyroptosis.15 Therefore, it is promising to design metal ion-based chemistry to either disrupt or maintain intracellular metal ions homeostasis to alter cell fate with the goal of boosting or suppressing PCD.
To leverage the vital role of metal ions in PCD for cancer treatment, in this review, we will elaborate on the signaling pathways of each type of PCD with a particular focus on the contribution of intracellular metal ions. This review will first describe the importance of intracellular metal ions in living systems. Next, the impacts of dyshomeostasis of intracellular metal ions will be outlined. Then, we will introduce the latest research progress in different types of PCD (Fig. 2). Further, we will introduce metal ion-induced chemical reactions and illustrate how they can help regulate cancer cell death mechanisms. To that end, we will discuss the rational development and prospects of precision anti-tumor therapeutic strategies through regulating intracellular metal ions for PCD.
 | ||
| Fig. 2 A schematic illustration of metal ion-triggered programmed cell death (PCD) for cancer cells. | ||
2. Intracellular metal ion-related chemistry reaction
Metal ions, through catalyzing or initiating various chemical reactions, contribute to the production of diverse chemicals, species, and products crucial for sustaining normal biological functions.16,17 Specifically, numerous metal ions assume pivotal roles, influencing enzyme catalytic reactions, inflammatory responses, oxidative stress and cellular physiological behaviors.18 Notably, calcium (Ca2+), iron (Fe2+/3+), copper (Cu+/2+), and zinc (Zn2+) ions, among others, are indispensable for the complexities of tumorigenesis, impacting both developmental processes and the sensitivity or resistance of tumors to treatment.19,20 This underscores the increasing significance of metal ions in cancer biology and therapy.2.1 Calcium
 | ||
| Fig. 3 Schematic illustration of intracellular calcium ion-related chemistry reaction. | ||
Intracellular Ca2+ release channels,28 exemplified by the ryanodine receptor (RyR) and inositol 1,4,5-triphosphate receptor (IP3R) channels, play pivotal roles in cellular signaling.26 Activation of RyR channels is triggered by heightened [Ca2+]i or through protein signaling events. In a canonical signal transduction cascade, a G protein-coupled receptor (GPCR) stimulates the generation of IP3, subsequently activating IP3R channels through IP3 binding, thereby facilitating Ca2+ release from the ER and SR.29 The ER lumen serves as the principal reservoir for intracellular calcium; its depletion instigates calcium accumulation in the mitochondrial matrix, NADPH oxidase complex assembly, and ROS production, culminating in the initiation of NETosis.30 Calcium-dependent interactions with key molecular chaperones and enzymes within the ER, including binding immunoglobulin protein (BiP), calreticulin (CRT), and transmembrane and coiled-coil domains 1 (TMCO1), are crucial for maintaining calcium homeostasis and enzymatic activities.31–34 Moreover, when cytosolic calcium overload, oxidative stress, ER stress, or other cellular stimuli occur, they can activate pro-apoptotic and ferroptotic proteins that induce apoptosis and ferroptosis.35,36
2.1.3.1 Oxidative phosphorylation. [Ca2+]i is meticulously regulated both temporally and spatially, playing a crucial role in signal transduction pathways. Mitochondria, integral to this regulation, actively buffer [Ca2+]i levels through the dynamic uptake and release of Ca2+, concurrently facilitating ATP production by the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS).37 Precise modulation of OXPHOS activity by appropriate Ca2+ levels in the mitochondrial matrix is indispensable for maintaining the optimal rate of ATP production. In physiological states, the accumulation of Ca2+ within mitochondria enhances oxidative metabolism by modulating Ca2+-sensitive dehydrogenases and metabolite carriers.38 However, mitochondrial Ca2+ overload leads to excessive ROS generation, diminished ATP production, and mitochondrial outer membrane permeabilization (MOMP). This phenomenon may culminate in cell death facilitated by the release of pro-apoptotic factors including cytochrome C (cytoC) and activated caspases.39
The Ca2+-permeable channel transient receptor potential melastatin 2 (TRPM2) establishes a unique linkage between the intracellular redox status and the adenine nucleotide metabolic network.40 Thus, TRPM2 emerges as a critical regulator by influencing ROS production and the antioxidant response. This modulation is achieved by facilitating Ca2+ entry via channel activation, ultimately influencing cell survival and the progression of tumors in various cancers.
2.1.3.2 Inflammatory reactions. In the pathogenesis of cardiovascular and related diseases, inflammation assumes a contributory role. Specifically, Ca2+-calmodulin-dependent protein kinase II (CaMKII) mediates inflammation within the myocardial microenvironment, precipitating cellular dysfunction, heightened inflammatory responses, and cellular demise. The overactivation of CaMKII disrupts normal myocardial functioning by engaging various inflammatory pathways. Furthermore, CaMKII exerts influence over mitochondrial Ca2+ homeostasis, with its hyperactivation fostering the opening of the mitochondrial permeability transition pore (mPTP).41 Activation of the NOD-like receptor (NLR) protein 3 (NLRP3) inflammasome constitutes a crucial mechanism that connects the functionality and integrity of mitochondria to innate immunity.42 Following cardiomyocyte damage, an inflammatory cascade is triggered by the generation of cytokines, damage-associated molecular patterns (DAMPs), and extracellular vesicles.43 This inflammatory process is underpinned by the release of DAMPs, crucial for orchestrating immunogenic cell death (ICD), with constituents such as the ER chaperone calreticulin and ATP.44 The release of DAMPs relies on a danger signaling module induced by select anticancer treatments that are characterized by oxidative-ER stress.
2.2 Iron
 | ||
| Fig. 4 Schematic illustration of intracellular iron ion-related chemistry reaction. | ||
Lipid peroxidation denotes the dioxygenation process leading to the formation of lipid (hydro)peroxides.54 Mitochondria, serving as key regulators of OXPHOS, are pivotal in iron metabolism and homeostasis.55 Mitochondrial iron is primarily involved in two critical processes: Fe–S cluster proteins and heme synthesis.56 The accumulation of lipoylated enzymes and depletion of Fe–S cluster proteins in mitochondria leads to cuproptosis. Additionally, a pool of free Fe2+ exists within mitochondria, actively contributing to the accumulation of mitochondrial ROS (mitoROS). Hence, the accrual of iron within mitochondria is linked to the induction of mitochondrial dysfunction. The resulting lipid peroxidation within the mitochondria yields lipid hydroperoxides and reactive aldehydes, including malondialdehyde and 4-hydroxynonenal.54,57
Iron is a crucial trace element vital for enzymatic activity, certain iron-containing proteins exhibit the capacity to generate substantial levels of ROS. Specifically, Fe2+ acts as a cofactor for lipoxygenase (LOX), an enzyme implicated in the peroxidation of polyunsaturated fatty acids (PUFAs).58 LOXs are non-heme iron-dependent dioxygenases with a substrate preference for PUFAs direct oxygenation, and they extend this activity to PUFA-containing lipids within biological membranes. This observation raises the possibility that LOXs might serve as mediators in ferroptosis. Enzymes housing [4Fe–4S] clusters become susceptible to attack by ROS, resulting in the transformation of these clusters into [3Fe–4S] configurations. This conversion process is accountable for the liberation of labile iron and subsequent enzyme inactivation.59 The cytotoxic potential of labile iron underscores its deleterious impact on cellular components, with both labile iron and ROS-generating iron-containing enzymes posing threats to DNA, lipids, and proteins. This toxicity, exacerbated by ROS from iron-containing enzymes, leads to oxidative stress that predominantly triggers apoptosis and ferroptosis via ROS signaling.
2.3 Copper
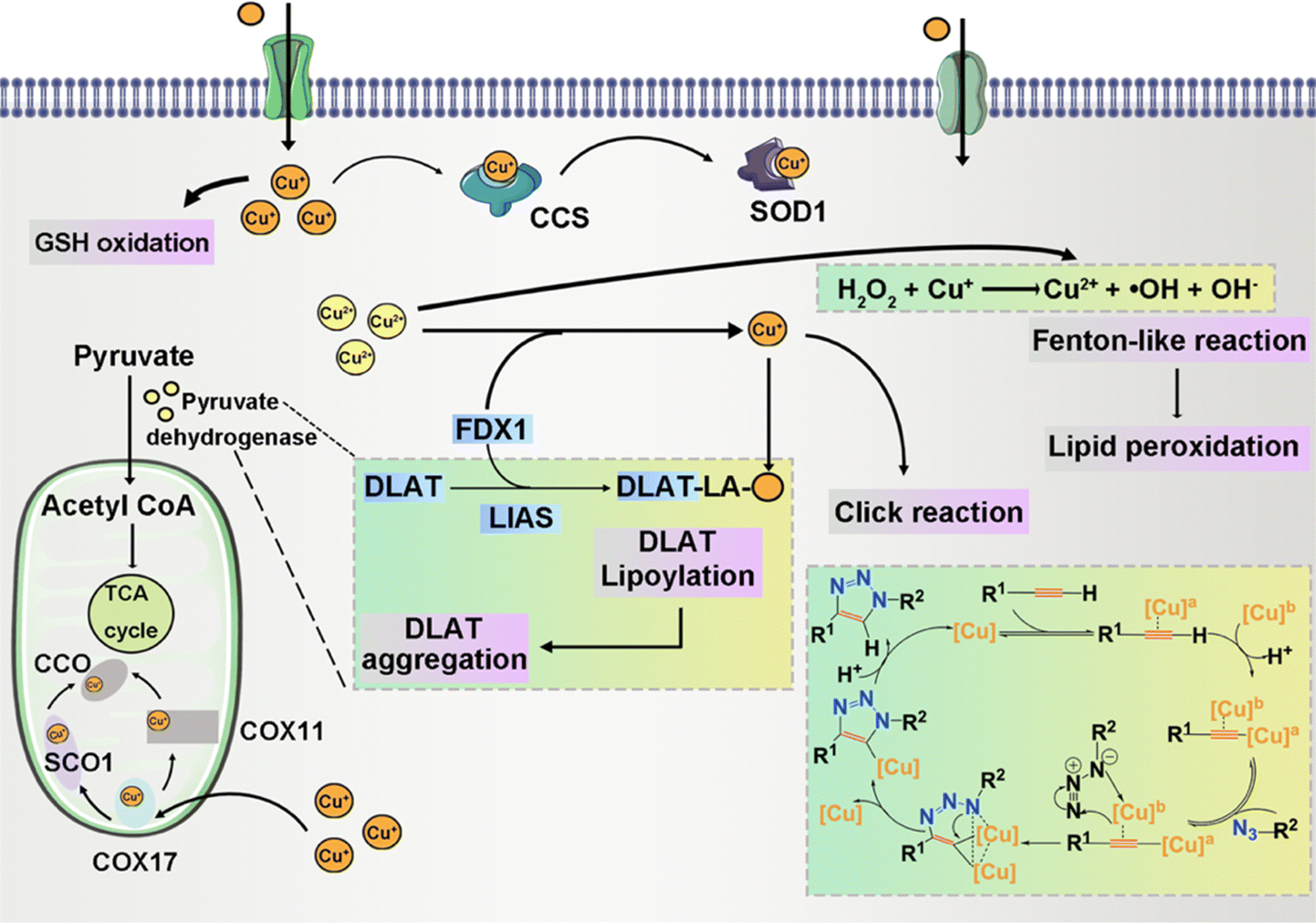 | ||
| Fig. 5 Schematic illustration of intracellular copper ion-related chemistry reaction. | ||
Cu chaperone for superoxide dismutase (CCS) functions as a copper transport protein, facilitating the involvement of Cu+ in diverse physiological processes encompassing oxidation, protein synthesis, and protein secretion.81 Specifically, CCS acts as the mediator for copper delivery to superoxide dismutase 1 (SOD1).82 SOD1, also known as Cu, Zn-SOD, is an enzyme instrumental in the dismutation of superoxide radical anions, affecting their conversion into H2O2. In this catalytic role, SOD1 is integral to the regulation of intracellular ROS homeostasis.83 Its precise function is paramount in preserving the integrity of essential biomolecules such as DNA, lipids, and proteins, against oxidative damage. Notably, the cytosolic presence of Cu, Zn-SOD holds great importance in safeguarding against ROS damage, especially concerning cardioprotection in ischemia-reperfusion injury.84,85 During the mild formation of ROS in ischemic preconditioning, Cu, Zn-SOD assumes a significant regulatory role in modulating apoptosis.
2.4 Zinc
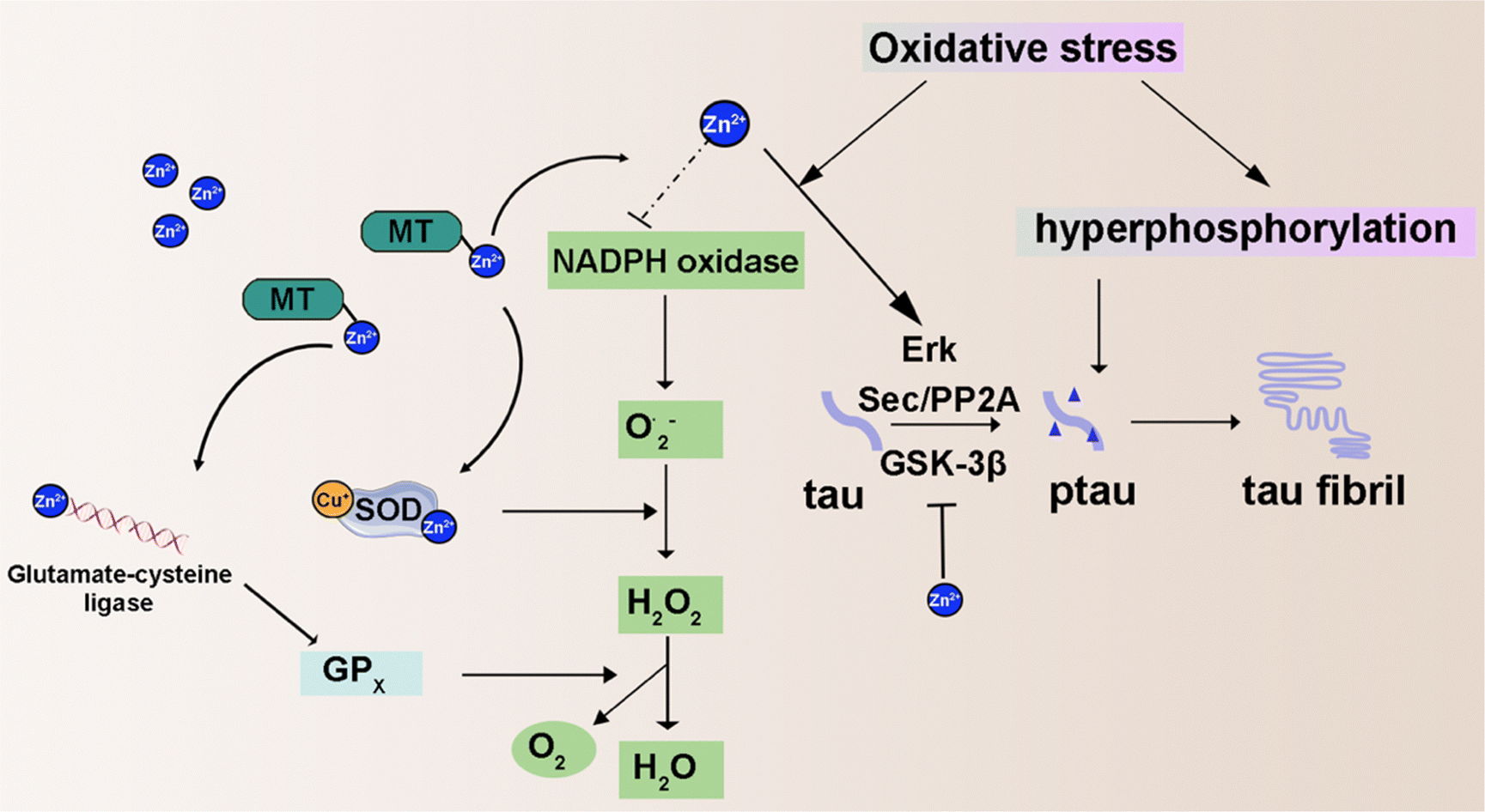 | ||
| Fig. 6 Schematic illustration of intracellular zinc ion-related chemistry reaction. | ||
2.5 Other intracellular metal ions
Several other intracellular metal ions are proposed as essential for physiological functions (Fig. 7). Mg, the second most abundant intracellular metal, functions as a calcium channel blocker, modulating vasodilation, bronchodilation, acetylcholinesterase release, and anti-inflammatory responses.115 Mn and Co are redox-active transition metals, participate in the Fenton reaction, and are crucial components of numerous enzyme-active centers enabling chemical reactions at physiologically compatible rates.116,117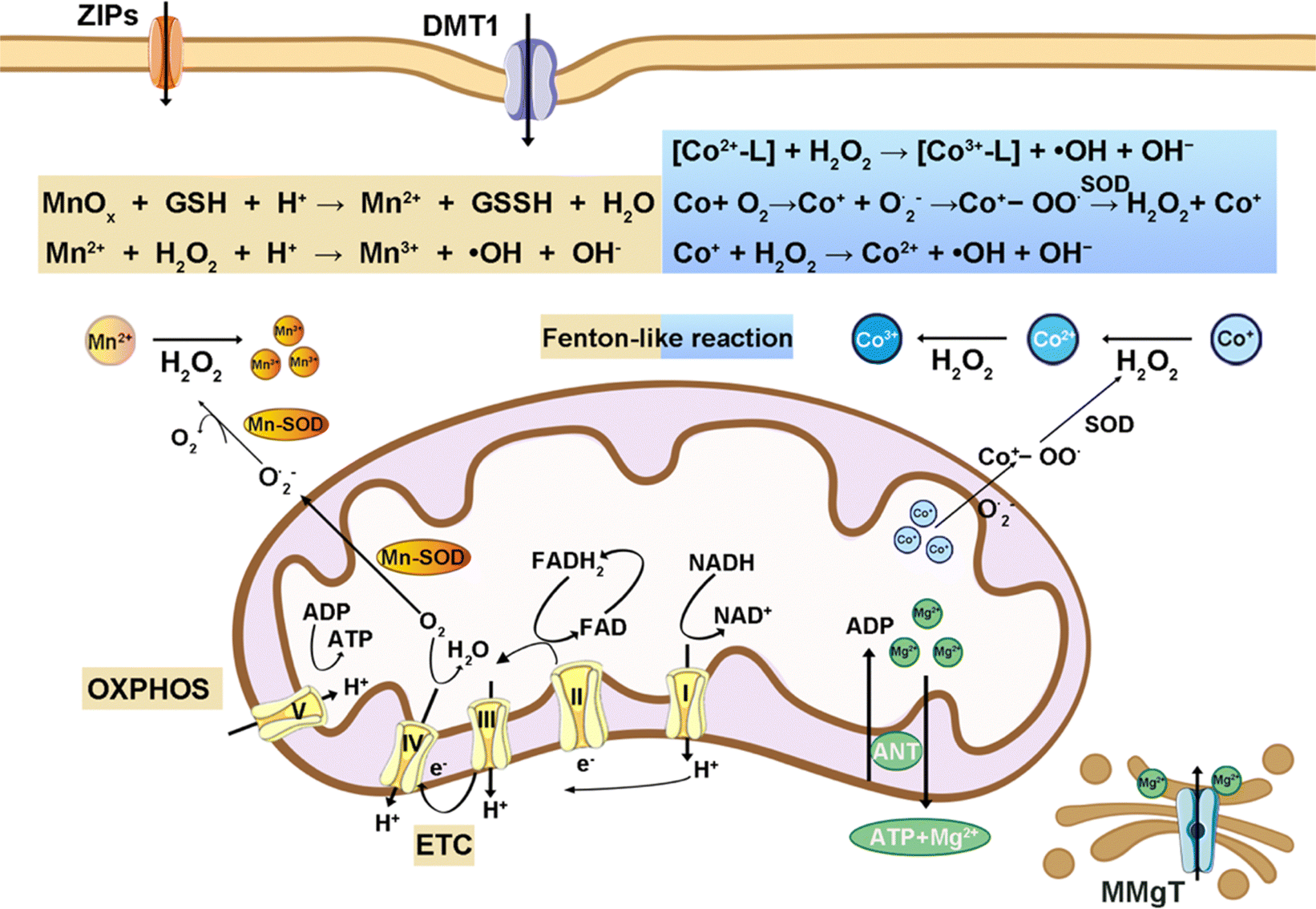 | ||
| Fig. 7 Schematic illustration of intracellular manganese, cobalt, and magnesium ions-related chemistry reaction. | ||
Mg ions exhibit an oxidation state +2 and coordinate with six oxygen donor atoms.118 Mg2+ is indispensable for enzymes that catalyze ATP-dependent reactions. As a physiological antagonist to Ca, the Mg/Ca ratio significantly influences the activity of Ca-ATPases and other calcium transport proteins. Mild fluctuations in cellular magnesium levels can alter calcium signaling or lead to toxicity.119 Therefore, elucidating the pathways and mechanisms of Mg2+ transport is vital for advancing therapeutic strategies against cancer and related pathologies.
Mn exists in biological systems in oxidation states ranging from +2 to +4, adopting various coordination geometries and contributing to oxidation reactions.120,121 Mn can be transported from the bloodstream into cells via voltage-regulated or glutamate-activated calcium channels. Mn-SOD (SOD2) in mitochondria is pivotal for increasing gene expression linked to radiation-induced adaptive responses and is influenced by tumor protein p53 (TP53 or p53), underscoring its significance in cancer mechanisms and treatment strategies. Pyruvate carboxylase is a mitochondrial enzyme that facilitates the conversion of pyruvate to oxaloacetate and involves a biotin group and Mn2+ or Mg2+. Furthermore, arginase is an Mn-dependent enzyme in the urea cycle that processes L-arginine into urea and L-ornithine.122 Given the multifunctional role of Mn, its homeostasis is critically maintained to prevent toxicity while ensuring its biological availability.
Co is crucial for forming the Co–C bond in the coenzyme vitamin B12; however, excessive Co is carcinogenic.123,124 Its standard oxidation states are +2 and +3. It disrupts DNA repair and promotes damage through cobalt-catalyzed free radical generation via the Fenton reaction. In the presence of SOD, Co contributes to oxidative stress by reacting with dioxygen, forming ˙OH through cobalt peroxy radicals (Co+-OO˙). Co and Mn can occupy the same nonheme site, influencing heme-copper oxidase activity in heme-nonheme biosynthetic myoglobin models. Furthermore, cobalt can substitute zinc in Zn-dependent enzymes and increase the activity of certain liver enzymes like heme oxidase.
Perturbations in intracellular metal ion homeostasis can trigger chemical reactions and influence diverse physiological functions. Table 1 briefly summarizes the representative intracellular metal ions and their corresponding chemical reactions, key pathways, and major localization. This understanding inspires a promising cancer treatment strategy by intentionally disrupting metal ion homeostasis to destabilize tumor environments. In addition, intracellular metal ions are mechanistically linked to diverse PCDs. Therefore, it is promising to investigate how to leverage intracellular metal ions to treat cancer by PCD.
| Metal ion | Reaction category | Key pathways | Major localization | Ref. | |
|---|---|---|---|---|---|
| Calcium | Enzyme activation, signal transduction | RyR, IP3R; TRPM2, CaMKII | Endoplasmic reticulum, mitochondria, nuclear | 21–26 | |
| Iron | Fenton and lipid peroxidation reaction, TCA cycle | Fe with H2O2, GSH oxidation, GPX4, Fe–S, LOX; glutamine, ETC | Mitochondria, lysosomes, cytosol, nuclei | 45–47,49–51,54,61 | |
| Copper | Fenton-like reaction, copper-containing enzyme catalysis, click reaction | Cu with H2O2, GSH oxidation; COX, CCO, SCO1, CCS | Mitochondria, nuclei, cytosol | 66–71,86,87 | |
| Zinc | Zinc-containing enzyme catalysis, hyperphosphorylation, oxidative stress | MT, ZnT, ZIP; p38, ErK, Sec/PP2A; NADPH oxidation | Mitochondria, nuclei, cytosol, golgi apparatus | 20,95–98 | |
| Others | Cobalt | Fenton-like reaction | Co with H2O2, SOD, B12 | Cytosol, nuclei, | 116,117 |
| Magnesium | ATP-dependent catalysis | ATP, pyruvate carboxylase | Mitochondria, nuclei, endoplasmic, sarcoplasmic reticulum | 118,119 | |
| Manganese | Fenton-like reaction | Mn with H2O2, GPX4, SOD2, pyruvate carboxylase, arginase | Mitochondria, lysosome, cytosol, golgi apparatus | 120–122 | |
3. Programmed cell death in cancers
3.1 Apoptosis
Apoptosis represents the principal cellular death pathway actively involved in developmental processes and the maintenance of tissue homeostasis.125,126 Apoptosis is the biological mechanism responsible for the removal of damaged or superfluous cells from the body.127,128 This process is characterized by a well-organized sequence of events, including cellular condensation, nuclear fragmentation, and nucleolysis, leading to the demise of apoptotic cells. The induction of apoptosis is governed by the precise regulation of key signaling pathways and targets,129 which encompass tumor necrosis factor (TNF)-related ligands and receptors, B cell lymphoma 2 (BCL-2) family members, apoptotic peptidase activating factor 1 (APAF1), cytoC, the nuclear factor-kappa B (NF-κB) pathway, and p53, among others.130 Notably, the transfer of Ca2+ from the ER to mitochondria emerges as a prerequisite for specific apoptotic stimuli. The redox-active transition metals (Fe, Cu, Mn, and Co) and Zn2+ emerge as potent inducers of ROS generation to trigger apoptosis by the Fenton reaction. Failure to regulate or resist this process often results in tumorigenesis. To this end, a Cu+-catalyzed click reaction was able to activate apoptosis by the generation of an anti-tumor drug (Fig. 8(a)).93 Hence, the meticulous modulation of the apoptotic signaling pathway emerges as a pivotal approach to enhancing cancer treatment.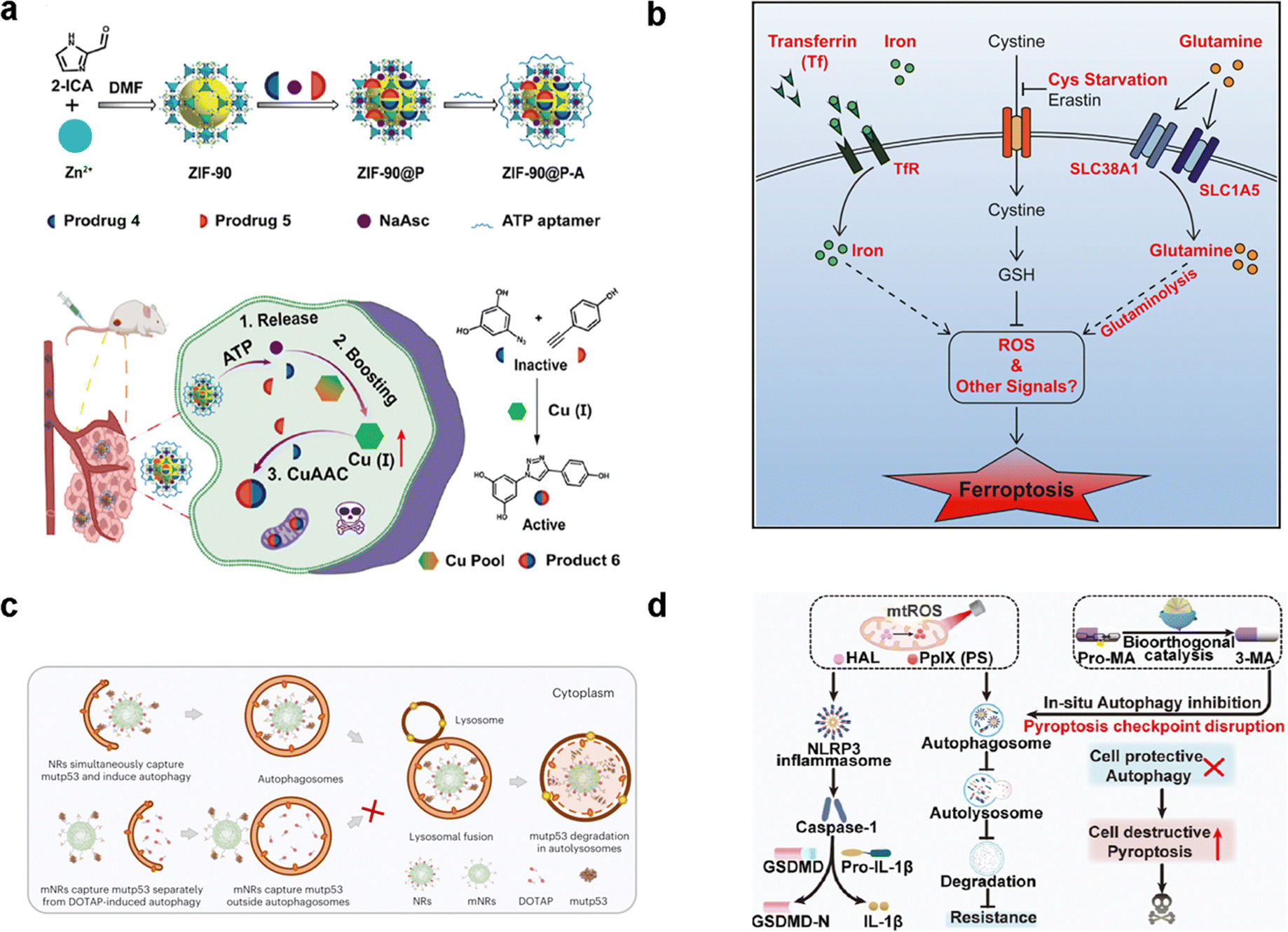 | ||
| Fig. 8 The application of PCD in cancer cells. (a) Schematic of an endogenous copper(I)-based bioorthogonal catalysis system for cancer therapy by apoptosis. Reproduced with permission.93 Copyright 2023, American Chemical Society. (b) Diagram the transferrin and the glutaminolysis is required for ferroptosis.60 Reproduced with permission. Copyright 2015, Elsevier. (c) Schematic diagram of two nanoreceptors within the cytoplasm, binding mutant p53 protein and inducing autophagy. Reproduced with permission.131 Reproduced with permission. Copyright 2024, Springer Nature. (d) Illustration of preparation of a ZPHM nanoregulator and the boosted pyroptosis for cancer therapy. Reproduced with permission.132 Copyright 2023, American Chemical Society. | ||
In apoptosis, two main pathways—intrinsic and extrinsic—are governed by caspases, a family of cysteine proteases.133 Intrinsic apoptosis is instigated by disturbances within the cellular microenvironment, such as DNA damage, ER or oxidative stress, and cytosolic calcium overload. MOMP is governed by a balance in the BCL-2 protein family.134 Certain anti-apoptotic members of the BCL2 family are believed to promote cellular survival through various mechanisms. These mechanisms include (1) regulating Ca2+ homeostasis within the ER, (2) boosting bioenergetic metabolism via F1FO ATP synthase interaction, and (3) maintaining redox homeostasis. However, extrinsic apoptosis is commonly initiated by death receptors (DRs) upon engagement with their respective ligands.135 DRs, following ligand binding, facilitate the assembly of DISC (death-inducing signaling complex) and complex II, culminating in caspase-8 (CASP8) activation and subsequent extrinsic apoptosis.136
Intrinsic apoptosis is pivotal in preventing oncogenesis, and dysregulation of this process has been implicated in numerous cancers, such as chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML).137 Particularly, the upregulation of BCL2, BCL-XL, or MCL-1 accelerates leukemia and lymphoma development, often in conjunction with overexpression of the MYC proto-oncogene. The pharmacological targeting of BCL2 proteins effectively combats MYC-driven tumors, even when p53 function is impaired. Notably, p53 serves as a direct and indirect regulator of apoptotic gene expression and links apoptosis to cell cycle arrest.138 Extensive research has unveiled the tumor-suppressive role of the intrinsic apoptotic pathway across a spectrum of cancer types.139,140 Additionally, distinct roles within the BCL2 protein family have been ascribed to specific members in cancer. These results encourage researchers to investigate and better understand why the inhibition of apoptotic cell death promotes tumorigenesis in certain scenarios while inhibiting it in others.
Recent research has unveiled tumor-suppressive role of CASP8 in the liver and select tissues, impacting the DNA damage response and chromosomal instability in early-stage tumorigenesis.141,142 However, TNF-R1 and Fas exhibit contrasting roles in hepatic and ovarian oncogenesis compared to other tumor types. TNF blockade attenuates hepatic cancer onset in experimental cholestatic hepatitis. Iron plays a pivotal role in regulating Fas-mediated extrinsic apoptosis by modulating alternative splicing in an iron-dependent manner.143,144 The apparent variability in outcomes across different cancer models may be attributed to cancer-specific predilections for either apoptotic, necroptotic, inflammatory, or pro-invasive signaling pathways elicited by FasL.
3.2 Necroptosis
Necroptosis is governed by RIPK1 and can be induced by various triggers.145–147 Significantly, it is intriguing to observe that the same ligands are responsible for initiating the extrinsic apoptosis pathway, either by caspase inhibitors like Z-VAD-FMK or by depleting Fas-associated death domain protein (FADD).148 Notably, necroptosis is distinct from necrosis, as it strictly adheres to intracellular signaling regulation within cancer cells and involves active energy consumption.149 RIPK3 phosphorylates mixed lineage kinase domain-like protein (MLKL), culminating in necroptosis characterized by disruption of the plasma membrane and cell lysis.150 Necroptotic signaling serves a diverse function in tumorigenesis, necrosis, metastatic progression, and immune interactions.Necroptosis is a common occurrence within the central regions of solid tumors, primarily attributed to inadequate vascularization leading to oxygen and nutrient deprivation. Recent research findings indicate deletion of MLKL leads to a substantial reduction in tumor necroptosis, with the remaining areas exhibiting a shift in cell death mode to apoptosis.151 The RIPK3 expression has a distinct pattern in various solid cancer cell lines, especially in human solid tumor models.152,153 Notably, necroptosis mediated by RIPK3 can yield both anti-tumorigenic and pro-tumorigenic outcomes that are dependent on tumor type, stage, and progression.154,155
3.3 Ferroptosis
Ferroptosis describes an iron-dependent, non-apoptotic form of PCD initiated by erastin.51 A defining feature of ferroptosis is cell mortality driven by iron-dependent lipid peroxide accumulation in mitochondria, alongside the deactivation of GPX4.156 The functionality of GPX4 is intricately tied to glutathione (GSH), a molecule derived from cysteine and glutamate, which are regulated by the amino acid antiporter, system xc-. Three pivotal hallmarks categorize ferroptosis: (1) oxidative damage to membrane phospholipids containing polyunsaturated fatty acids (PUFA); (2) the availability of redox-active iron; (3) the diminished capacity to repair lipid hydroperoxides (LOOH).54 Cells undergoing erastin-induced ferroptosis exhibit mitochondria with aberrant morphology, specifically reduced cristae and compromised outer membranes, evidenced by condensation and rupture.157 The onset of ferroptosis is intimately tied to intracellular iron accumulation, free radical generation, fatty acid availability, and lipid peroxidation.158,159 The overarching objective of elucidating the mechanistic underpinnings of ferroptosis is to enhance the spectrum of therapeutic avenues for cancer treatment.Ferroptosis, functioning as an adaptive mechanism for the eradication of malignant cells, represents an emerging tumor-suppressing pathway.160,161 For example, Interferon-γ, traditionally recognized for its inhibitory effect on system xc- activity, is secreted from CD8+ T-cells.162 This secretion leads to the suppression of both subunits of system xc- within tumor cells which further enhances the efficacy of cancer immunotherapy. Additionally, iron paves the way to produce ROS by the Fenton reaction (Fig. 8(b)).60 Both traits make cancer cells particularly receptive to ferroptosis induction. Furthermore, the augmentation of cellular iron availability through the autophagic degradation of ferritin has been observed to promote ferroptosis. Conversely, mechanisms that facilitate the export of cellular iron have demonstrated the capacity to enhance cell resistance to ferroptosis.
Diverse cancer cell lines show an inherent sensitivity to ferroptosis, and a multitude of molecular mechanisms contributing to these susceptibilities. For instance, in p53-dependent cancers, ALOX12 can instigate ferroptosis independently of GPX4 function.163 In addition, ACSL4-overexpressed cells display susceptibility to ferroptosis inducers erastin and RSL3, whereas ACSL4-deficient cancer cells exhibit high resistance.164 The sensitivity or resistance of a specific cancer to ferroptosis induction is fundamentally governed by its distinct genetic landscape.
3.4 Autophagy-dependent cell death
Autophagy,165 a type II PCD, comprises four fundamental stages: induction, involving the initiation of autophagy and formation of autophagic vesicles; autophagosome formation; fusion between autophagosomes and lysosomes; and lastly, the degradation occurring within the autolysosome.166 Autophagy exhibits a dual role in tumor initiation and progression. During the early stages of tumor formation, autophagy serves as a safeguard by regulating and eliminating cancer cells. However, once tumors have developed, autophagy shifts its function to sustain the survival of tumors and facilitate their growth. The orchestration of the autophagic process involves a suite of key genes, including Unc-51-like kinase 1 (ULK1), p62, Beclin-1, forkhead box O (FoxO), light chain 3 (LC3), and other autophagy-related (ATG) genes.167 Additionally, the phosphatidylinositol 3-kinase/protein (PI3K) kinase-B(Akt)/mammalian target of the rapamycin (mTOR) pathway is a crucial signaling cascade. Simultaneously, AMP-activated protein kinase (AMPK) serves as a pivotal energy sensor, regulating cellular metabolism and preserving energy equilibrium.168 A significant contributor to autophagy regulation is the tumor suppressor p53, whose location determines its function.169 Nuclear p53 enhances autophagy by activating upstream of mTOR, whereas cytoplasmic p53 inhibits autophagy. The precise function of autophagy in cancer remains a subject of ongoing debate. Moreover, elevated intracellular metal ions will simulate the autophagic regulators and induce the autophagic process.Autophagy-dependent cell death represents a PCD type reliant on the autophagic machinery. The molecular machinery shows some differences with adaptative autophagy. Proficient autophagic responses primarily function at the core of stress adaptation, thereby conferring cytoprotective effects. Consequently, interference with autophagy, through pharmacological or genetic means, tends to expedite cellular death in response to stress. Furthermore, inherent or temporary defects in autophagy have been linked to conditions such as embryonic lethality, developmental anomalies, and a spectrum of pathological disorders, especially cancer. As numerous elements within the autophagy machinery serve functions distinct from autophagy, it is advisable to confirm the participation of a minimum of two distinct ATG proteins. Furthermore, in instances of autophagy-dependent cell death, when the inhibition of autophagy averts cell death, it is essential to provide substantiating evidence that rules out the involvement of other PCD modalities, including FAS-driven extrinsic apoptosis, necroptosis, and ferroptosis.
At a fundamental level, the cell-independent impact of autophagy is associated with tumor-promoting functions (Fig. 8(c)).131,170 ULK1 is a multifunctional target that triggers the autophagy process. Both ULK1 inhibition and activation have substantial effects on tumor treatment outcomes.171 The inhibition of ULK1 to impede early autophagy has shown promise in suppressing cell growth and combatting drug resistance in tumor therapy.172 Conversely, ULK1 activation is associated with adverse tumor prognosis. This observation implies that activating ULK1 to induce autophagy may represent a promising therapeutic strategy for inhibiting tumor growth in select cancer types.
3.5 Pyroptosis
Pyroptosis,173 a PCD mechanism with an associated inflammatory response, encompasses two pathways: the canonical pathway, mediated by caspase-1 (CASP 1), and the noncanonical pathway, involving caspase-4, 5, and 11.174,175 Nod-like receptors (NLRs) or absent in melanoma 2 (AIM2)-like receptors (ALRs) instigate the assembly of inflammatory bodies in response to specific stimuli, culminating in the formation of activated CASP 1.176 Conversely, lipopolysaccharide (LPS) induces the activation of caspase-4, -5, and -11.177 These caspases cleave gasdermin-D (GSDMD) thereby instigating pyroptosis.178 In both pyroptotic pathways, caspase-1/4/5 cleaves GSDMD protein, facilitating the conversion of pro-IL-1β and pro-IL-18 to their respective mature forms.179 Conversely, GSDME is cleaved by caspase-3, resulting in pyroptotic death. These proteins create nano-scale pores (10 to 20 nm) in the cell membrane, facilitating the gradual release of cellular contents through these membrane apertures. Consequently, pyroptosis manifests through nuclear condensation, concurrent cell swelling, and substantial plasma membrane blebbing, ultimately leading to rupture.Pyroptosis exhibits suppressive effects on the initiation and progression of cancers.180,181 Robust evidence from diverse studies underscores the capacity of chemotherapeutic drugs, natural compounds, and specific reagents to instigate pyroptosis across various cancer types.182 Noteworthy examples include actinomycin-D, doxorubicin (DOXO), and bleomycin, which have demonstrated the ability to induce pyroptosis in lung cancer (LC) and melanoma cell lines.183,184 Lobaplatin treatment demonstrates a notable capacity to induce pyroptosis in colorectal cancer (CRC) cell lines, evidenced by the cleavage of GSDMD and GSDME.185,186 These findings lay the groundwork for understanding the diverse abilities of distinct chemotherapeutic agents in inducing pyroptosis across cancer cell lines expressing GSDME. Emerging nanomaterials designed to target pyroptosis show promise in tumor therapy. Recently, a bioorthogonal pyroptosis nanoregulator was shown to not only induce pyroptosis, but also disrupt the construction of checkpoints, presenting a significant advancement in the pursuit of safe and efficacious pyroptosis-based cancer therapy (Fig. 8(d)).132 These investigations provide innovative perspectives endorsing pyroptosis as a promising mechanism for eradicating oncogene-addicted tumor cells.
3.6 Cuproptosis
Cuproptosis is set in motion by an excess of Cu2+.187,188 This Cu-induced PCD pathway stands apart from other cell death mechanisms. Intracellular copper ions exhibit a distinct affinity for fatty-acylated proteins within the tricarboxylic acid (TCA) cycle associated with mitochondrial respiration, leading to consequential fatty-acylation modifications.11 The accumulation of lipoylated mitochondrial proteins bound to copper, in conjunction with the subsequent depletion of Fe–S clusters, imposes proteotoxic stress, culminating in cell demise. The elucidation of the cuproptosis mechanism paves the way for future drug research, with a specific focus on copper ionophores and compounds associated with cuproptosis.71,189 These findings introduce novel perspectives on potential applications in cancer treatment. Disulfiram (DSF) and elesclomol (ES) have emerged as focal points of research and have been the subject of clinical trials. DSF, when combined with Cu2+, demonstrates noteworthy anti-tumor activity across diverse cancer types.190 The gene most significantly linked to ES sensitivity, Ferredoxin 1 (FDX1), was identified as a direct target of ES–Cu.191 ES–Cu binding directly inhibits FDX1 function in Fe–S cluster formation. Ammonium tetrathiomolybdate (TTM) effectively counteracts these changes in mitochondrial membrane potential.192 ES triggers cell death through the translocation of copper ions into both cells and mitochondria, thereby increasing intracellular copper ion levels. This, in turn, leads to dihydrolipoamide S-acetyltransferase (DLAT) oligomerization, diminished Fe–S stability, and interaction with Npl4.193,194 These discoveries suggest that the intracellular delivery of copper via ionophores holds promise as a therapeutic approach for specific tumor subtypes.3.7 Entotic cell death
Entotic cell death (Entosis), a manifestation of cell cannibalism, is marked by the engulfment and demise of one cell by another.195 Initially identified in human mammary epithelia, then this phenomenon is prevalent in epithelial and other human tumors.196 Central to the regulation of entosis induction are the pathways governing cell adhesion and cytoskeletal rearrangements. Entosis is distinguished by the digestion of engulfed cells by host cells, a process independent of BCL2 proteins and caspases. Instead, it predominantly relies on LC3-associated phagocytosis (LAP) and lysosomal degradation pathway mediated by cathepsin B (CTSB).197,198 LAP serves as an intermediary linking phagocytosis and autophagy.199,200 The recruitment of the essential LC3 lipidation machinery, comprising ATG5, ATG7, PIK3C3, and PIK3 regulatory subunit 4 (PIK3R4), to the cytosolic aspect of vesicles containing entotic cells is instrumental in facilitating their fusion with lysosomes.Entosis is facilitated by cell-in-cell structures and has been observed across various cancers, such as breast, colon, and pancreatic carcinoma (PACA).201,202 Entosis can impede tumor advancement by inducing death in internalized cells, while concurrently, may foster tumor progression by promoting competition between tumor cells. Thus, entosis presents a dualistic role in tumorigenesis, characterized by both tumor-inhibitory and tumor-promoting effects. It functions as an inhibitor by eliminating matrix-detached tumor cells, impeding the advancement of tumors. Conversely, it may contribute to tumor progression through its ability to induce changes in cell ploidy.203 This disruption leads to the formation of binucleate-engulfing cells. Consequently, entosis is identified as a cell death process with notable involvement in critical biological events associated with cancer.
3.8 Netotic cell death
Netotic cell death (NETosis), instigated by neutrophil extracellular traps (NETs) release, stands as a distinct form of PCD.204 NETs are released by activated neutrophils, but other epithelial cells can also generate NETs in response to diverse stresses, such as leukocyte populations, and cancer cells. NETosis is a dynamic process involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)-driven ROS generation, the induction of autophagy, and the release/translocation of granular enzymes and peptides, mainly belonging to the cathelicidin family.205 NETosis occurs via NOX-dependent and NOX-independent mechanisms, and the specific mechanism is dependent on the inducer. NOX-dependent NETosis is triggered by agents like phorbol 12-myristate 13-acetate and LPS, which promote NOX assembly and ROS production, thereby initiating NETosis.206 Conversely, NOX-independent NETosis is instigated by ionomycin, which induces NETosis through Ca overload without NOX activation.207 This overload activates PAD4, leading to histone citrullination, while Gasdermin D contributes to chromatin decondensation, nuclear disintegration, and cell rupture.208,209The formation of neutrophil NETs occurs within the tumor microenvironment.210 Malignant tumors can secrete granulocyte colony-stimulating factor (G-CSF) to promote NETosis.211 Upregulation of G-CSF correlates with an increased neutrophil count in the bloodstream, leading to ROS production and subsequent NET formation. NETosis has emerged as a potential contributor to the reawakening of dormant tumor cells, playing a pivotal role in tumor recurrence and metastasis. Thus, NETosis exhibits dualistic characteristics, manifesting both tumor-promoting and antitumor effects. The outcome is contingent upon the immune system's status and the dynamic interplay within the tumor microenvironment.212 Consequently, there is an imperative need to channel further research into finely balancing the regulation and occurrence of NETosis.
3.9 Parthanatos
Poly(ADP-ribose)polymerase-1 (PARP-1)-dependent cancer cell death is a novel form of PCD termed “parthanatos.”213 Parthanatos is instigated in response to oxidative stress, inducing DNA damage and chromatin degradation. While it shares certain characteristics with necroptosis, autophagy, and apoptosis, the underlying molecular mechanisms of NETosis are distinct. Noteworthy features of parthanatos include plasma membrane rupture, the prompt activation of PARP-1, early poly ADP-ribose (PAR) accumulation,214,215 depletion of cellular nicotinamide adenine dinucleotide (NAD+) and ATP, mitochondrial depolarization, apoptosis-inducing factor (AIF) release from the mitochondria, recruitment of migration inhibitory factor (MIF), nuclear translocation of the AIF/MIF complex, and DNA fragmentation following DNA damage.216 Consequently, the pivotal events in parthanatos entail the transduction of PAR polymer signaling to the mitochondria and the translocation of AIF from the mitochondria to the nucleus.PARP-1 has a multifaceted role in tumorigenesis. The clinical implementation of PARP-1 inhibitors has exhibited favorable outcomes in specific cancer types and among individuals with distinct genetic susceptibilities.217 PARP-1 serves as a DNA damage receptor during tumorigenesis, rapidly activating its enzyme activity following DNA damage, culminating in pronounced PARP-1 activity and eventual parthanatos.218 Elevated levels of PARP-1 have been observed in BC,219 ovarian cancer (OC),220 and head and neck cancer (HNC)221 tissues in comparison to their normal counterparts, underscoring the intimate association between parthanatos and these malignancies. Conversely, studies have revealed that PARP-1 knockout mice exhibit a substantially reduced risk of epithelial cancer.222 Presently, clinical cancer treatment has benefited from the approval of four PARP inhibitors: olaparib,223 rucaparib,224 niraparib,225 and talazoparib.226 Among them talazoparib exhibits superior efficacy, particularly in advanced BC.227 Given the integral role of PARP-1 in multiple DNA repair pathways and genomic stability maintenance, the regulation of PARP-1 activity emerges as a vital clinical strategy for interventions in related cancers.228
3.10 Lysosome-dependent cell death
Lysosome-dependent cell death (LDCD) is characterized by the destabilization of lysosomes and necessitates lysosomal membrane permeabilization (LMP).229 Lysosomes, acidic cellular organelles, are proficient in degrading diverse cargos derived from heterophagy and autophagy processes.230 Lysosomes undergo LMP when exposed to lysosomotropic detergents, ROS, dipeptide methyl esters, and lipid metabolites.231,232 ROS, notably via H2O2-driven luminal hydroxyl radical production in Fenton reactions and the resultant lipid peroxidation, assume a pivotal role in destabilizing the lysosomal membrane during LMP.233 Additionally, ROS facilitates the activation of lysosomal Ca2+ channels, namely TRPM2.234 As cell death executioners, cathepsins play varied roles in different contexts.235 The specific cathepsins involved in initiating and executing LCD depend on the context of LMP. Inhibiting cathepsin expression or activity effectively impedes LCD. Nevertheless, cathepsins do not singularly govern LCD, as this cellular demise pathway can manifest diverse features, such as apoptotic, necrotic, autophagic, or ferroptosis-like characteristics, contributing to the intricate landscape of LCD.236The lysosome, acting as a central regulator, plays a crucial role in meeting catabolic demands for growth and supporting neoplastic anabolic processes.237,238 Numerous agents designed to disrupt lysosomal function are currently subjects of investigation or in various stages of clinical development for cancer therapy. One notable example is chloroquine, which induces LMP to modulate lysosomal function.239 This, in turn, has the potential to reinstate sensitivity to cisplatin in non-small cell lung cancer cells that had exhibited resistance. These findings point to an increased fragility of lysosomes in tumor cells relative to normal cells, predisposing them to LMP and subsequent LDCD.240 Hence, it is imperative to gain a comprehensive understanding of the intricate regulatory processes governing LMP in cancer cells in order to leverage LDCD for cancer therapy.
3.11 Alkaliptosis
Alkaliptosis, identified as a newly recognized pH-dependent form of PCD,241 demonstrates robust anti-tumor efficacy, notably against pancreatic ductal adenocarcinoma.242 Song et al. screened 254 G-protein-coupled receptors (GPCRs)-interacted compounds to identify a GPCR-targeted small molecule JTC801 with cytotoxic activity against a human pancreatic cancer (PC) cell line. The anticancer mechanism attributed to JTC801 is specifically associated with the induction of alkaliptosis, distinguishing it from apoptosis, necroptosis, ferroptosis, and autophagy-dependent cell death. Notably, the alkaliptosis-inducing properties of JTC801 exhibit promise as a selectively lethal agent for tumor cells, devoid of significant side effects.242 Alkaliptosis, marked by a fatal elevation in intracellular pH, is orchestrated by the coordinated activity of ion channels as well as transporters within both intracellular and extracellular pathways.243,244Intracellular alkalization-dependent alkaliptosis emerges as a promising cell death mechanism for cancer therapy, especially in drug-resistant cancers.245 In human PC cells, the heightened expression of ATP6V0D1 has been identified as a facilitator of JTC801-induced alkaliptosis, showcasing a distinct interaction with STAT3. This underscores the pro-death role of ATP6V0D1 in alkaliptosis, offering insights for the development of PCD strategies in tumor treatment.
3.12 Oxeiptosis
Oxeiptosis, characterized as a ROS-induced noninflammatory PCD, operates independently of caspases.246 The key mediators in the oxeiptotic process include kelch-like ECH-associated protein 1 (KEAP1), PGAM family member 5 (PGAM5), and AIFM1.247 The KEAP1-NFE2L2 pathway is recognized for orchestrating cytoprotective responses in the face of oxidative injury when ROS concentrations are low. In contrast, heightened ROS concentrations prompt KEAP1 to disengage from PGAM5 and engage in an interaction with AIFM1, instigating death signaling.Recent investigations have unveiled that macrophages can instigate oxeiptosis in mesothelioma cells by activating PGAM5 through a ROS-dependent mechanism. Notably, sanguinarine has emerged as an efficacious agent in curtailing tumor growth in human CRC cells.248 This suppression occurs through the induction of oxeiptosis, driven by ROS, with a specific focus on H2O2-dependent activation of the KEAP1-PGAM5-AIFM1 pathway.249 Moreover, alloimperatorin has been identified as a modulator of the Keap1/PGAM5/AIFM1 pathway, facilitating oxeiptosis and effectively hindering the survival of BC cells.250,251
As shown in Table 2, twelve representative PCDs capable of inducing different types of cancer cell demise through specific targets have been briefly summarized, including their key regulator molecules and the corresponding intracellular metal ions. However, some PCDs that are triggered by intracellular metal ions have yet to be further explored. Collectively, the relationship between intracellular metal ions and PCD remains to be further investigated.
| Cell death mode | Morphological features | Key regulators | Cancer cell line | Metal ion | Ref. |
|---|---|---|---|---|---|
| Apoptosis | Cell and nuclear condensation, apoptotic body formation | BCL-2 family (BAX/BAK, BOX, BCL2, BID), p53, Caspase-2/8/9/10, Caspase-3/6/7, APAF1, NF-κB | Lung cancer, gastric cancer, prostate cancer | Ca, Fe, Cu, Zn, Co, Mg | 127–130,133,134 |
| Necroptosis | Cell swelling, nuclear chromatin loss, plasma membrane rupture | RIPK-1, RIPK-3, MLKL | Colon cancer | Fe | 145–153 |
| Ferroptosis | Cell swelling, mitochondrial condensation and rupture, plasma membrane collapse | GPX4, SLC7A11, ALOX12/15, ACSL4, NCOA4 | Triple-negative breast cancer, colorectal cancer | Ca, Fe, Cu, Co, Mn | 51,156–164 |
| Autophagy-dependent cell death | Autophagosomes and lysosomes formation, autophagosome–lysosome fusion | ULK1, p62, Beclin-1, FoxO, LC3, ATG, p53, PI3K/Akt | Breast cancer | Ca, Cu, Zn | 131,165–172 |
| Pyroptosis | Nuclear condensation, cell swelling, plasma membrane rupture, bubbling, chromatin fragmentation | Caspase-1, Caspase-4, Caspase-5, Caspase-11, GSDMD, GPX4, ESCRT-III | Human melanoma, colon cancer, lung cancer, breast cancer, gastric cancer, hepatocellular carcinoma | Ca, Fe, Mn | 132,173–186 |
| Cuproptosis | Cell swelling, chromatin fragmentation, mitochondrial condensation | P53, FDX1, DLAT, glutamine | Glioblastoma, non-small cell lung cancer | Cu | 187–194 |
| Entosis | Cell-in-cell structure | ATG5, ATG7, PIK3C3, PIK3 | Breast cancer, colon cancer, pancreatic cancer | 195–202 | |
| Netosis | Nuclear swelling, plasma membrane rupture, chromatin fiber release | PAD4, MPO, NAPDH/ROS | Pancreatic cancer | 204–211 | |
| Parthanatos | Nuclear condensation, membrane rupture, large DNA fragmentation | PARP-1, AIF, MIF | Breast cancer, ovarian cancer, head and neck cancer, epithelial cancer | 213–222 | |
| Lysosome-dependent cell death | Lysosome and plasma membrane rupture | Cathepsins, p53 | Non-small cell lung cancer | Ca, Zn | 229–240 |
| Alkaliptosis | Necroptosis-like morphology | NF-κB, STAT3 | Pancreatic ductal adenocarcinoma, prostate cancer | 241–245 | |
| Oxeiptosis | Apoptosis-like morphology | KEAP1, PGAM5, AIFM1 | Mesothelioma, colorectal cancer, breast cancer | 246–251 |
4. Intracellular metal ion-triggered programmed cell death
PCD represents a promising strategy for the eradication of cancer by orchestrating the process of PCD that is integral to maintaining cellular homeostasis by disposing of aged, impaired, or undesirable cells.252 The pervasive role of intracellular metal ions as universal cell signaling entities underscores their regulation of numerous physiological activities across all biological domains. Consequently, exploring intracellular metal ion dyshomeostasis is poised to provide novel insights into PCD.253 This discussion primarily focuses on Ca2+, Fe2+/3+, Cu+/2+, Zn2+, elucidating their interplay in different signaling pathways in PCD.2544.1 Calcium-triggered programmed cell death
The [Ca2+]i, as a ubiquitous second messenger, plays a pivotal role in PCD, especially in the pathogenesis and progression of tumors.255 Notably, dysregulation in [Ca2+]i homeostasis emerges as a critical determinant influencing proliferation, invasion, and metastasis within cancerous tissues.256 The depletion of ER Ca2+ stores plays a crucial role in the regulation of diverse cellular processes, encompassing cell growth, proliferation, exocytosis, enzymatic activity and motility modulation, and immune responses. A prevalent observation notes the swift escalation of cytoplasmic Ca2+ concentration in response to oxidative stress across diverse cell types. This necessitates an examination of the roles of Ca2+ transport systems within the ER and mitochondria in cellular fate. Like other intracellular second messengers, calcium possesses a dose-dependent capacity to induce hormesis or cell death processes, such as apoptosis, necroptosis, pyroptosis, ferroptosis, autophagy-dependent cell death, and so on (Fig. 9).257–259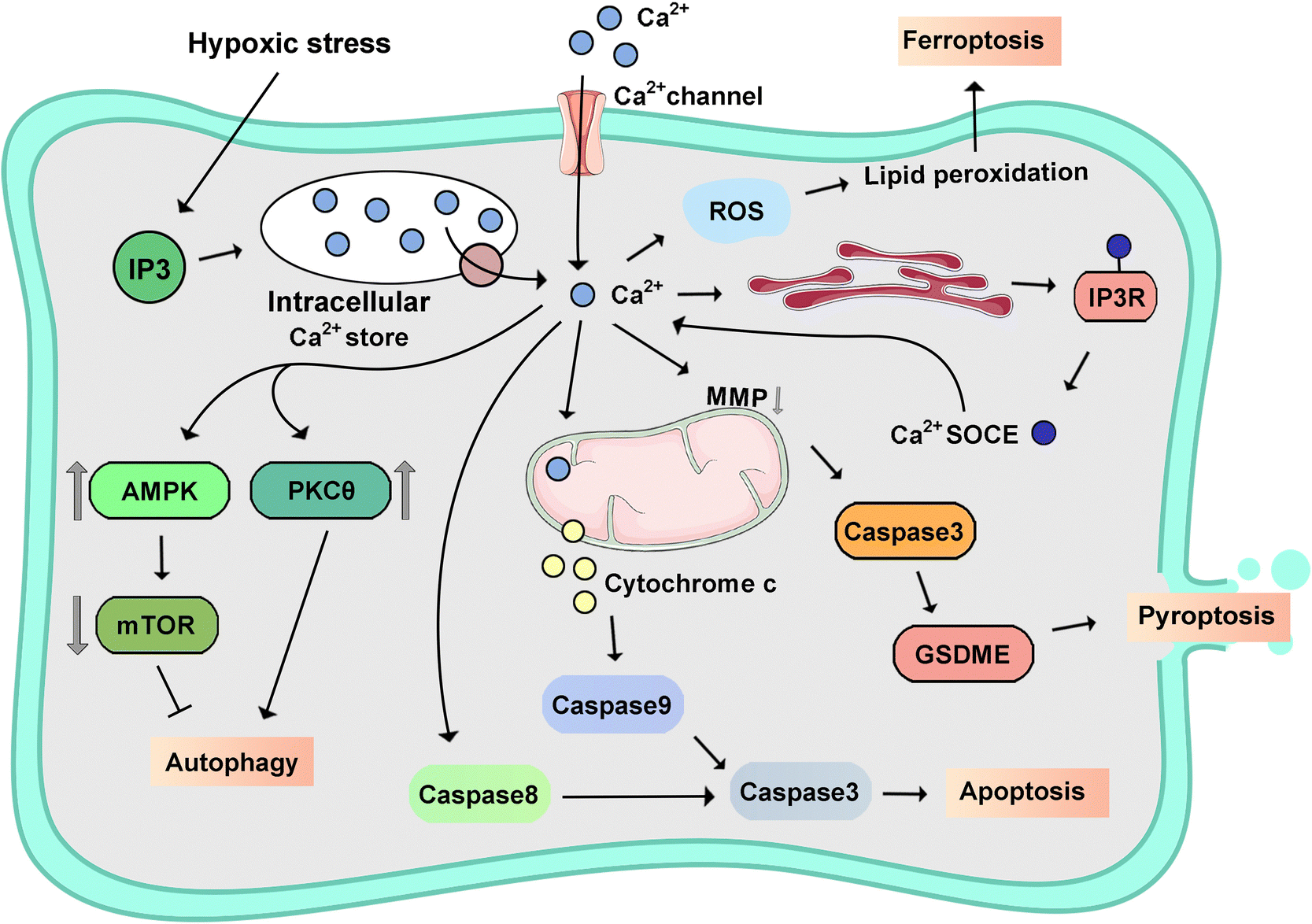 | ||
| Fig. 9 Schematic signaling pathway of representative calcium-triggered programmed cell death. | ||
Significantly, [Ca2+]i signaling pathways exhibit dual roles, with IP3R-mediated signaling promoting proliferation and oncogenic processes, whereas ryanodine receptor (RyR)-mediated signaling has been identified as a pro-apoptotic mechanism, particularly in lung cancer cells.262 Ca2+-mediated apoptosis is induced by Bcl-2 IP3 receptor disrupter-2 (BIRD-2), a phenomenon previously documented in lymphoid malignancies, and subsequently in diffuse large B-cell lymphoma marked by heightened Bcl-2 expression.263,264
Store-operated Ca2+ (SOC) channels are the primary contributors to the influx of Ca2+ that leads to calcium overload in pancreatic acinar cells.277 Activation of SOC channels induces the initiation of a cascade involving the activation of calcineurin (CaN), which, in turn, stimulates activation of the transcription factor EB (TFEB).278 This activation of TFEB has enduring effects on autophagy and vacuolization, notably influencing the development of acute pancreatitis (AP). The mitochondrial calcium uniporter regulator 1 (MCUR1), a 40 kDa protein situated in the IMM, interacts with the MCU.279 Silencing MCUR1 significantly inhibits agonist-induced mitochondrial Ca2+ uptake. Notably, MCUR1 depletion precipitates a cellular bioenergetic crisis, triggering the autophagic process. The regulatory role of promyelocytic leukemia protein (PML) extends to the modulation of IP3R3 activity, governing the autophagic response.280 This modulation is integral to the requirement for efficient Ca2+ transfer from the ER to mitochondria, which is essential for meeting cellular energy demands and facilitating mitochondrial metabolism. Diminished IP3 receptor activity leads to reduced Ca2+ transfer which compromises mitochondrial function and ATP generation, ultimately instigating autophagy.
4.2 Iron-triggered programmed cell death
Iron ions are crucial transition metal ions ubiquitous in living organisms. However, excessive accumulation of iron is regarded as a risk factor for cancer, contributing to both carcinogenesis and the metastasis of cancer cells.284 The principal detrimental effects arising from the induction of free radicals, facilitated by iron ions, are evidenced through the manifestation of diverse DNA oxidation products. The process of lipid peroxidation is intricately associated with the catalytic influence of Fe2+ and Fe3+, giving rise to alkoxyl radicals (RO·) and peroxyl radicals (ROO·).285 The cyclization of RO· originating from arachidonic acid results in the formation of 4-hydroxynonenal. This molecule has been implicated as a putative causative factor in various diseases, including multiple cancer types. Consequently, the scrutiny of iron-triggered PCD has become a focal point, particularly within the realms of apoptosis, necroptosis, ferroptosis, and pyroptosis (Fig. 10).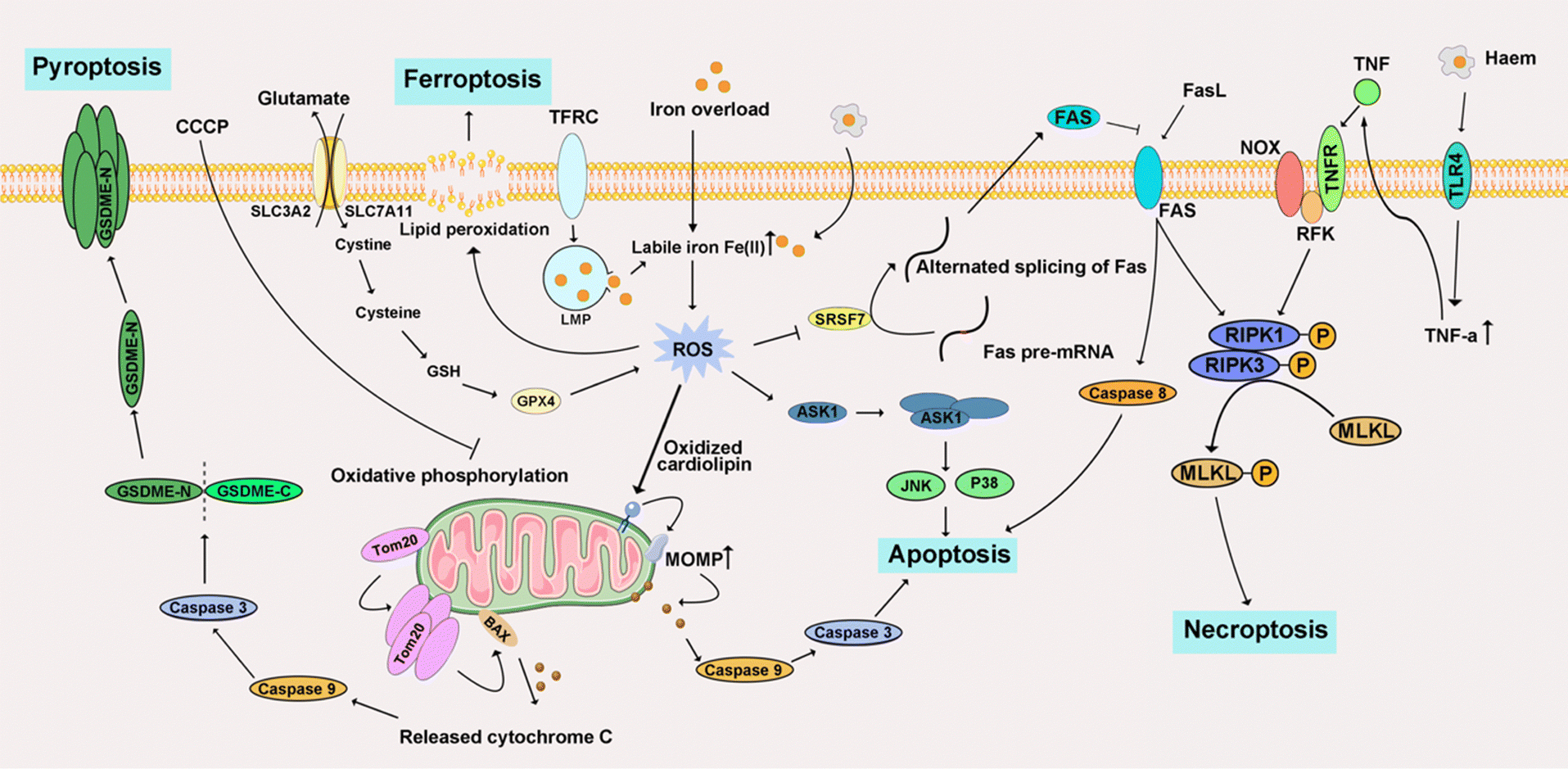 | ||
| Fig. 10 Schematic signaling pathway of representative iron-triggered programmed cell death. | ||
4.3 Copper-triggered programmed cell death
Copper is implicated in the modulation of multiple signaling pathways within tumor cells.308 Emerging evidence underscores copper's role in activating enzymes and signaling cascades associated with metastasis, thereby propagating malignancy.309 Its direct involvement is manifested through the binding and activation of key molecules. Noteworthy is the direct activation of PI3K by copper, instigating downstream AKT activation. Simultaneously, copper interacts with mitogen-activated protein kinase kinase 1 (MEK1), facilitating the phosphorylation of ERK1/2. This, in turn, triggers c-JNK, thereby orchestrating regulatory mechanisms in tumor growth. Consequently, this discussion summarizes recent research advancements elucidating Cu-triggered PCD (Fig. 11).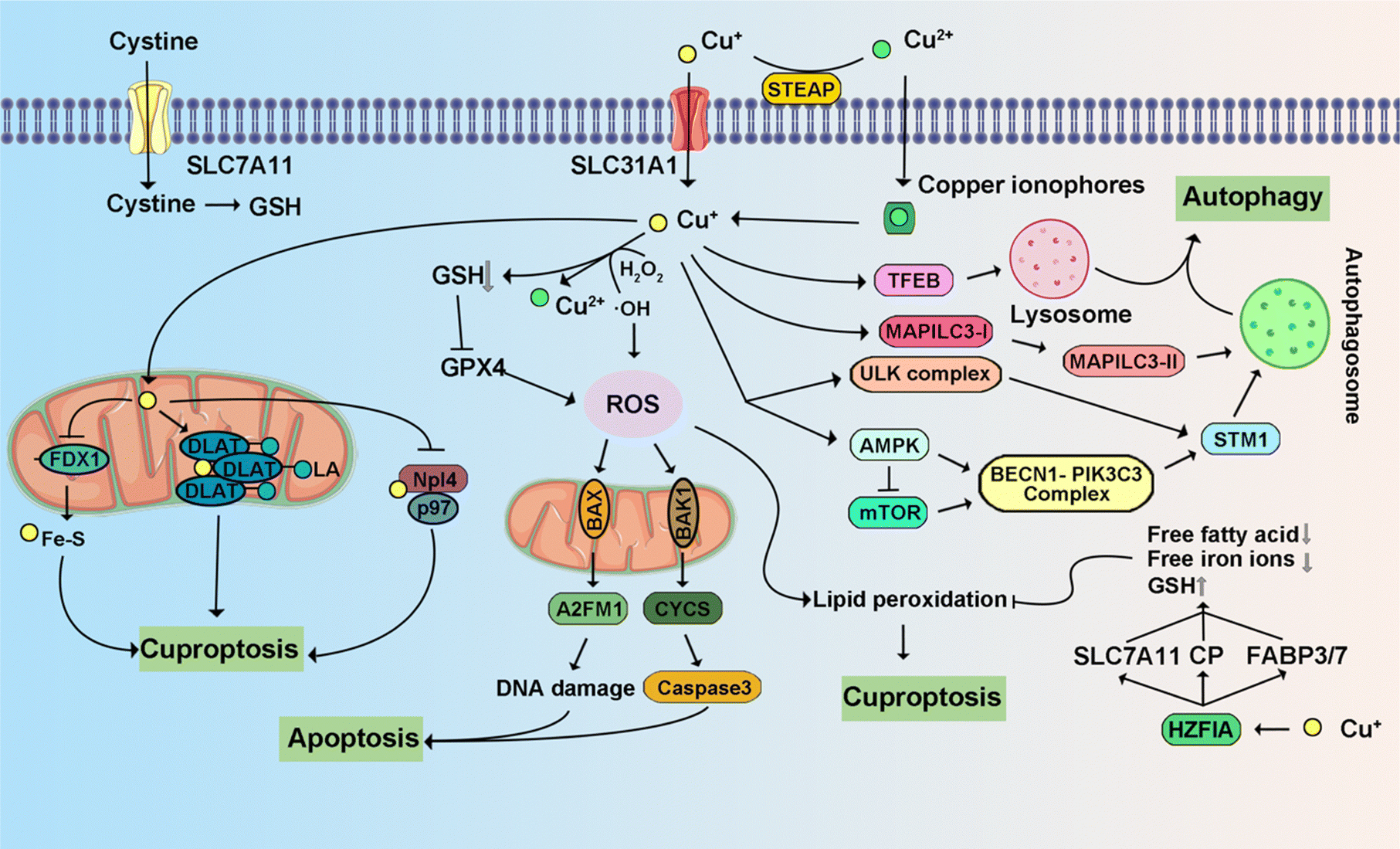 | ||
| Fig. 11 Schematic signaling pathway of representative copper-triggered programmed cell death. | ||
4.4 Zinc-triggered programmed cell death
Zinc, a robust inhibitor of caspases 3, -7, and -8, intricately regulates caspase-mediated inflammatory cascades.330 The intricate impact of zinc on tumor growth manifests in both promotion and inhibition with diverse mechanisms operative across various cancer types.331 Specifically, activation of the extracellular ERK and c-JNK pathways by zinc, both integral to tumorigenesis, underscores its significant contributory role. The dysregulation of ZnT has been extensively validated in numerous studies, showcasing its impact on cell proliferation and apoptosis. Concurrently, this dysregulation precipitates changes in multiple signaling pathways, ultimately contributing to the advancement of cancer.332 The exploration of zinc's pharmacological mechanisms across various PCDs and its prospective application in PCD is an avenue for further research (Fig. 12). | ||
| Fig. 12 Schematic signaling pathway of representative zinc-triggered programmed cell death. | ||
4.5 Other metal-triggered programmed cell death
Besides these four primary intracellular metals that can trigger different types of PCD in cancers, other intracellular metals (Fig. 13), such as Mn2+, Mg2+, and Co2+, play an important role in inducing cancer cell death by PCD. Briefly, Table 3 highlights metal ions and their related cell death modes in cancer treatments. Mg2+ is preferentially permeable to transmembrane cation channels formed by MLKL protein, a key factor in TNF-induced necroptosis.350 Increasing intracellular Mg2+ has been demonstrated to induce apoptosis in breast cancers.351 Mn2+ not only induced neurological apoptosis by enhancing endoplasmic reticulum stress but triggered pyroptosis through ROS-activated caspase-3/GSDME.116,352 Increasing intracellular Mn2+ could also induce apoptosis in cervical cancer.353 Co2+/3+ could trigger apoptosis in various cancers by mimicking the hypoxic factor and Co3+-ligand to induce ROS, respectively.354,355 Notably, some of these intracellular metal ions are integral to various types of PCD because they are implicated in different signaling pathways to regulate cell death, a process further complicated by its diverse subroutines and external influences.356 Zn2+, Ca2+, and Mg2+ help stabilize cell membrane integrity by crosslinking with organic “building blocks,” counterbalanced by organic anions. Ca2+ plays a unique role in information transmission, influenced by its variable ionic characteristics. Furthermore, metalloenzymes utilize Mg2+ and Zn2+ for catalytic activity, whereas metal centers such as Fe2+/3+, Cu+/2+, Mn2+/3+/4+, and Co2+/3+, driven by specific ligand interactions, facilitate electron transfers.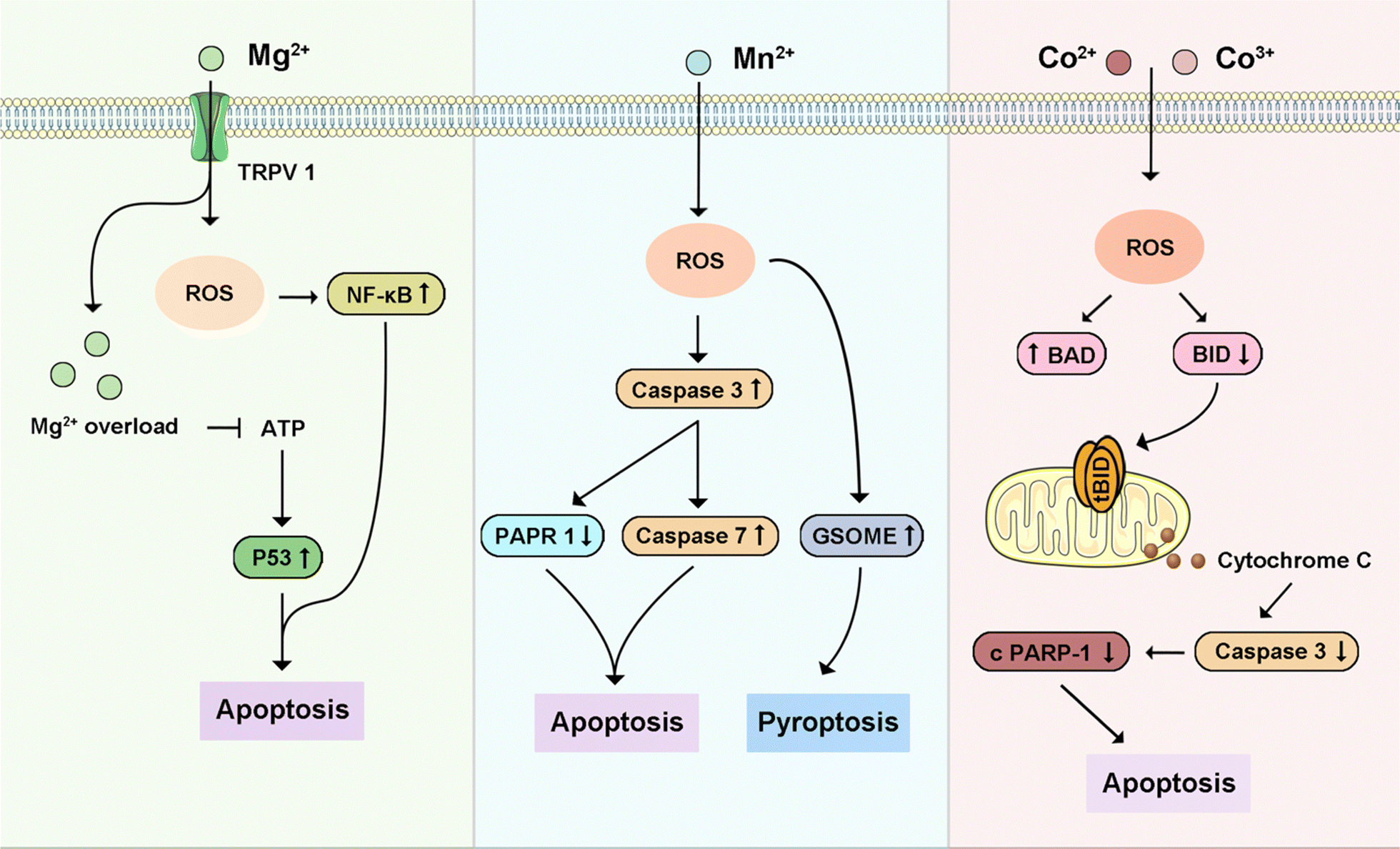 | ||
| Fig. 13 Schematic signaling pathway of representative other intracellular metal ions-triggered programmed cell death. | ||
| Metal ion | Cell death mode | Key pathways | Tumor type | Ref. | |
|---|---|---|---|---|---|
| Calcium | Apoptosis | SERCA, RyR, IP3R, BIRD-2, MFN2, CaMKII | Prostate cancer, lung cancer, osteosarcoma | 260–264 | |
| Pyroptosis | NLRP3, Caspase 3, CaSR | 265–269 | |||
| Ferroptosis | ESCRT-III | 270–275 | |||
| Autophagy-dependent cell death | CaMKKβ/AMPK, mTORC1, SOC, MCUR1 | 276–280 | |||
| Necroptosis | mPTP, RIP1 and MLKL | 281 | |||
| LDCD | Ca-calpain axis | 282 | |||
| Entosis | MLCK-actomyosin axis | 283 | |||
| Iron | Apoptosis, | ASK1, Fas; | Renal cell carcinoma, colorectal cancer, lung cancer, melanoma | 286–289 | |
| Necroptosis, | STAT3, GRIM-19, NOX; | 291–294 | |||
| Ferroptosis, | PLOOH, LOX, POR, NCOA4, NRF2, SLC39A14; | 295–302 | |||
| Pyroptosis, | Tom20-Bax-caspase-GSDM; | 303,304 | |||
| LDCD | NAADP-regulated 2-pore, RAB7A | 305,306 | |||
| Copper | Apoptosis, | p53, JNK, p38MAPK, Cyt C, Apaf-1, Caspase 9; | Pancreatic cancer, breast cancer, lung cancer | 310–315 | |
| Ferroptosis, | GPX4, TAX1BP1, CP; | 316–320 | |||
| Cuproptosis, | Elesclomol, FDX1; | 192,321 | |||
| Autophagy-dependent cell death, | ULK1/2, ATG5, BECN1, CRIP2, UBE2D2; | 322–326 | |||
| Necroptosis, | ROS, | 327 | |||
| Pyroptosis | CASP 1 | 328,329 | |||
| Zinc | Apoptosis, | ZIP10, Caspase 3, Caspase 6, Caspase 8, Zfp260; | Breast cancer, colorectal cancer, gastric cancer | 333–335 | |
| LDCD, | MT-3 | 336–338 | |||
| Autophagy-dependent cell death, | ERK, Beclin 1 | 339–343 | |||
| Necroptosis, | ROS | 344 | |||
| Ferroptosis, | ABCC9 | 345,346 | |||
| Pyroptosis | CASP 1, GSDMD | 347–349 | |||
| Others | Magnesium | Apoptosis | TRPV1, TRPM7 | Breast cancer | 350,351 |
| Manganese | Apoptosis, | cGAS, STING | Cervical cancer | 116 | |
| Pyroptosis | Caspase 3, GSDME | 352,353 | |||
| Cobalt | Apoptosis | phosphatidylserine, BAX, BID, BAD, BAK, BCL-2, BCL-xL, MCL-1 | Breast cancer, glioblastoma, lung cancer, cervical cancer, colon cancer | 354,355 | |
4.6 Multiple metal ions-triggered programmed cell death
As described previously, the ability of single metal ions to induce multiple forms of PCD, coupled with the observation that various metal ions can activate the same PCD pathways, highlights their complex roles in cancer therapy. Intracellularly, metal ions have been shown to interact synergistically or competitively, thereby affecting each other's capacity to induce PCD and playing a critical role in cancer treatment strategies. Ca2+ is a ubiquitous mediator of cell functions, which has been reported to be modulated by several intracellular metal ions; therefore, it is selected as a specific target to trigger PCD strategically. Fe2+ disrupts homeostasis of Ca2+ by lipid peroxidation, overlapping effects on HIF-1, ROS-mediated activation of IP3R/RyR channels, and excitotoxicity.357 Cu increases the [Ca2+]i to activate the exchange of Na+/Ca2+ and results non-specific membrane leaks, which further enhances Cu-toxicity to trigger cell death.358 Zn2+, as a second messenger, can modulate L-type Ca2+ and K+ channels.359 ZIP7 releases Zn2+ from the ER to the cytosol, leading to mitochondrial Ca2+ uptake for dually triggering ferroptosis.346 Jiang et al. also demonstrated that overload Zn2+ disrupted [Ca2+]i homeostasis, inhibited ETC, and promoted ROS to trigger apoptosis.360 In addition, Mg2+ regulates the mitochondrial calcium uniporter (MCU) and mediates mitochondrial Ca2+ uptake. Moreover, Ca2+ overloading occurs with a decrease of mitochondrial matrix Mg2+. However, with the opening of the mPTP, MCU activity has been increased.361 Recently, Zn2+/Fe2+/Mg2+-ion-chelated L-phenylalanine nanostructures induce Ca2+ influx to trigger the inflammasome pathway by K+ efflux, activating the NF-κB pathway and further augmenting immunotherapy.362 Fe and Cu can also interact with each other. For example, Cu always positively influences the transport of Fe. Fe may antagonize Cu metabolism.363 Additionally, Cu+ replaces other metal sites in protein structures, such as disruption of Zn2+ binding ligand in zinc-finger DNA and metalloprotein X-linked inhibitor of apoptosis protein (XIAP).330,364 Intracellular metal ions thus serve multifaceted roles in biological systems, and their dysregulation can lead to different types of PCD.Modulating intracellular metal ions presents a novel avenue for regulating PCD, as the dyshomeostasis of various metals can positively impact therapeutic outcomes. The successful application of metal-focused strategies in PCD heavily relies on leveraging the overexpression of intracellular metal ions. Intracellular redox-active metals, such as Fe2+/3+, Cu+/2+, Mn2+/3+/4+, and Co2+/3+, are cytotoxic due to their role in ROS generation. Metal ions such as Ca2+, Zn2+, and Mg2+ are essential cofactors in enzymatic catalysis and cellular metabolism. These distinct properties provide opportunities for tailoring PCD mechanisms, offering new directions for cancer treatment. A deeper understanding of the mechanisms underlying the interaction between intracellular metal ions and PCD is crucial for advancing direct antitumor strategies. Moreover, the induction of PCD by multiple intracellular metal ions highlights the complex interplay among these ions. The observed ability of multiple metal ions to trigger PCD points to intricate inter-ion interactions within cells. This insight advocates a broader focus on modulating intracellular metal ion networks, positioning their regulation as a strategic approach to inducing PCD for cancer treatment.
5 Conclusion: challenges and future directions
The signaling of intracellular metal ions is crucial in maintaining cellular homeostasis and plays a significant role in regulating physiological activity, especially programmed cell death (PCD).365 Understanding the mechanisms of these ion-based chemical reactions is essential for regulating PCD in disease and the advancement of novel treatments.366 As cancer is one of the most severe diseases and can be very challenging to treat, this review is directed towards PCD via metal-based chemistry reactions that ultimately induce the demise of cancer cells.Intracellular metal ions present significant prospects in biological and biochemical research, yet their application is fraught with challenges. A primary concern is their double-edged sword in cellular processes, which has yielded conflicting clinical outcomes regarding disease development. This underscores the need for more robust clinical trials. Despite significant advances in understanding the mode of action of metal-based chemistry reactions that induce PCD, the relationship between anti-tumor concentration and reactivity remains elusive. In addition, key questions remain unanswered, including the specifics of the death phenotype, the underlying mechanisms of cell destruction, and the potential roles of these ions in subcellular organelles. Moreover, designing metal-based chemistry reactions to target specific cell death pathways is particularly challenging due to the intricate signaling networks involved in cell death modes. The ideal intracellular metal ion concentration varies between organs, which is critical for refining metal ion-based drug therapies. Future developments in intracellular ion regulators should prioritize organ and cell specificity, as well as controlled release mechanisms.
Advancements understanding of diverse PCD types and their complex molecular interrelations have added complexity to the field. This interconnectedness often results in concurrent activation of multiple cell death pathways, rendering molecular markers less distinct in identifying specific types.367 Four key metals—Ca, Fe, Cu, and Zn are frequently reported to induce cancer cell death by triggering distinct PCDs. Other metals, such as Mg, Mn, and Co, also activate PCD in tumors. Inspired by identified ferroptosis, cuproptosis, and the emerging concept of calcicoptosis,13 we envision that other metal-based new cell death modes could emerge as targets of cell death pathways for future cancer treatment. What's more, the effectiveness and specificity of intracellular metal ions in modulating these distinct cell death pathways continue to be a significant concern in the field.
Addressing the challenges posed by intracellular metal ion dyshomeostasis and leveraging these ions for PCD necessitates a comprehensive understanding of their role in cancer-related cell death via PCD pathways.368 This understanding is fundamental for evolving new cancer treatment methodologies. Although a series of metal-based chemistry reactions have been reported to induce common PCD, such as apoptosis, ferroptosis, and pyroptosis, some molecular mechanisms of other PCDs that were triggered by intracellular metal ions remain to be explored. For example, it remains unclear whether necroptosis exclusively underlies tumor necrosis in all solid tumors. However, emerging evidence suggests that the impact of necroptosis on tumorigenesis can be either promotive or inhibitory, contingent upon the specific tumor type. Therefore, the factors behind the ability of intracellular metal ions to drive necroptosis demand further in-depth investigations. Integrating metal ion-triggered biochemical reactions with diverse PCD mechanisms can potentially overcome limitations of traditional apoptosis-based cancer treatments, including adaptive drug resistance, high recurrence, and poor targeting in hard-to-treat cancers. Considering the complex interactions among different PCDs, note that a single ion can activate multiple PCDs and that multiple ions can initiate the same PCD. Collectively, a thorough understanding of cell death processes and types is essential to comprehend cellular homeostasis. Future research should adopt an integrated approach to explore various cell death types with different intracellular metal ions, focusing on discovering new patterns and their interrelations.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Q. H. acknowledges the start-up package support from the University of Wisconsin-Madison. T. W. was supported by TL1TR002375 within the Institute for Clinical and Translational Research at the University of Wisconsin-Madison, supported by U54TR002373.References
- J. C. Ameisen, Science, 1996, 272, 1278–1279 CrossRef CAS.
- S. Nagata and M. Tanaka, Nat. Rev. Immunol., 2017, 17, 333–340 CrossRef CAS PubMed.
- F. Peng, M. Liao, R. Qin, S. Zhu, C. Peng, L. Fu, Y. Chen and B. Han, Signal Transduction Targeted Ther., 2022, 7, 286 CrossRef CAS PubMed.
- P. Tsvetkov, A. Detappe and K. Cai, et al. , Nat. Chem. Biol., 2019, 15, 681–689 CrossRef CAS.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- I. Vitale, F. Pietrocola and E. Guilbaud, et al. , Cell Death Differ., 2023, 30, 1097–1154 CrossRef.
- T. Song, G. Yang and H. Zhang, et al. , Nano Today, 2023, 51, 101896 CrossRef CAS.
- X. Dai, Y. Xie, W. Feng and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309160 CrossRef CAS.
- W. Xu, J. Qian, G. Hou, T. Wang, J. Wang, Y. Wang, L. Yang, X. Cui and A. Suo, Adv. Funct. Mater., 2022, 32, 2205013 CrossRef CAS.
- K. Zhang, C. Qi and K. Cai, Adv. Mater., 2023, 35, e2205409 CrossRef PubMed.
- P. Tsvetkov, S. Coy and B. Petrova, et al. , Science, 2022, 375, 1254–1261 CrossRef CAS.
- Y. Huang, G. Qin, T. Cui, C. Zhao, J. Ren and X. Qu, Nat. Commun., 2023, 14, 4647 CrossRef CAS PubMed.
- M. Zhang, R. Song and Y. Liu, et al. , Chem, 2019, 5, 2171–2182 CAS.
- C. C. Daw, K. Ramachandran and B. T. Enslow, et al. , Cell, 2020, 183, 474–489 CrossRef CAS.
- Y. Deng, F. Jia, P. Jiang, L. Chen, L. Xing, X. Shen, L. Li and Y. Huang, Biomaterials, 2023, 301, 122293 CrossRef CAS.
- J. F. Seeler, A. Sharma and N. J. Zaluzec, et al. , Nat. Chem., 2021, 13, 683–691 CrossRef CAS PubMed.
- L. A. Finney and T. V. O’Halloran, Science, 2003, 300, 931–936 CrossRef CAS PubMed.
- K. Jomova, M. Makova, S. Y. Alomar, S. H. Alwasel, E. Nepovimova, K. Kuca, C. J. Rhodes and M. Valko, Chem. – Biol. Interact., 2022, 367, 110173 CrossRef CAS.
- H. Lin, Y. Chen and J. Shi, Chem. Soc. Rev., 2018, 47, 1938–1958 RSC.
- M. Valko, K. Jomova, C. J. Rhodes, K. Kuca and K. Musilek, Arch. Toxicol., 2016, 90, 1–37 CrossRef CAS PubMed.
- D. E. Clapham, Cell, 1995, 80, 259–268 CrossRef CAS PubMed.
- M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11–21 CrossRef CAS PubMed.
- R. Bagur and G. Hajnoczky, Mol. Cell, 2017, 66, 780–788 CrossRef CAS.
- N. Xu, M. Francis, D. L. Cioffi and T. Stevens, Am. J. Physiol.: Cell Physiol., 2014, 306, C636–C638 CrossRef CAS PubMed.
- A. Bononi, C. Giorgi and S. Patergnani, et al. , Nature, 2017, 546, 549–553 CrossRef CAS PubMed.
- S. Fujii, R. Ushioda and K. Nagata, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216857120 CrossRef CAS PubMed.
- S. Linse and S. Forsén, Adv. Second Messenger Phosphoprotein Res., 1995, 30, 89–151 CAS.
- M. D. Bootman, M. J. Berridge and H. L. Roderick, Curr. Biol., 2002, 12, R563–R565 CrossRef CAS.
- M. J. Berridge, Physiol. Rev., 2016, 96, 1261–1296 CrossRef CAS PubMed.
- S. Gupta and M. J. Kaplan, Nat. Rev. Nephrol., 2016, 12, 402–413 CrossRef CAS.
- N. Schauble, S. Lang and M. Jung, et al. , EMBO J., 2012, 31, 3282–3296 CrossRef PubMed.
- S. Luo, P. Baumeister, S. Yang, S. F. Abcouwer and A. S. Lee, J. Biol. Chem., 2003, 278, 37375–37385 CrossRef CAS PubMed.
- M. Obeid, A. Tesniere and F. Ghiringhelli, et al. , Nat. Med., 2007, 13, 54–61 CrossRef CAS PubMed.
- S. Zheng, D. Zhao and G. Hou, et al. , Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2111380119 CrossRef CAS PubMed.
- T. H. Murphy, A. T. Malouf, A. Sastre, R. L. Schnaar and J. T. Coyle, Brain Res., 1988, 444, 325–332 CrossRef CAS.
- Y. P. Rong, G. Bultynck and A. S. Aromolaran, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14397–14402 CrossRef CAS.
- M. Calvo Rodriguez and B. J. Bacskai, Trends Neurosci., 2021, 44, 136–151 CrossRef CAS PubMed.
- X. He, C. Hawkins, L. Lawley, M. Wunderlich, B. Mizukawa, X.-M. Zha, S. Halene and J. Fang, Blood, 2019, 134, 2661 CrossRef.
- J. C. Goldstein, N. J. Waterhouse, P. Juin, G. I. Evan and D. R. Green, Nat. Cell Biol., 2000, 2, 156–162 CrossRef CAS.
- C. Zhong, J. Yang and Y. Zhang, et al. , Research, 2023, 6, 0159 CrossRef CAS PubMed.
- M. L. Joiner, O. M. Koval and J. Li, et al. , Nature, 2012, 491, 269–273 CrossRef CAS PubMed.
- H. Zhang, T. Zhu and W. Liu, et al. , J. Mol. Med., 2015, 93, 1033–1043 CrossRef CAS PubMed.
- A. Murao, M. Aziz, H. Wang, M. Brenner and P. Wang, Apoptosis, 2021, 26, 152–162 CrossRef CAS.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- J. Wang and K. Pantopoulos, Biochem. J., 2011, 434, 365–381 CrossRef CAS PubMed.
- U. Rauen, A. Springer, D. Weisheit, F. Petrat, H. G. Korth, H. de Groot and R. Sustmann, ChemBioChem, 2007, 8, 341–352 CrossRef CAS PubMed.
- P. J. Aggett, Clin. Endocrinol. Metab., 1985, 14, 513–543 CrossRef CAS.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS.
- S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS PubMed.
- B. D'Autreaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef PubMed.
- S. J. Dixon, K. M. Lemberg and M. R. Lamprecht, et al. , Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- D. Shin, J. Lee, J. H. You, D. Kim and J. L. Roh, Redox Biol., 2020, 30, 101418 CrossRef CAS PubMed.
- I. Poursaitidis, X. Wang and T. Crighton, et al. , Cell Rep., 2017, 18, 2547–2556 CrossRef CAS.
- M. Conrad and D. A. Pratt, Nat. Chem. Biol., 2019, 15, 1137–1147 CrossRef CAS PubMed.
- B. T. Paul, D. H. Manz, F. M. Torti and S. V. Torti, Expert Rev. Hematol., 2016, 10, 65–79 CrossRef.
- O. Stehling and R. Lill, Cold Spring Harb Perspect. Biol., 2013, 5, a011312 Search PubMed.
- S. Dalleau, M. Baradat, F. Gueraud and L. Huc, Cell Death Differ., 2013, 20, 1615–1630 CrossRef CAS.
- H. Kuhn, S. Banthiya and K. van Leyen, Biochim. Biophys. Acta, 2015, 1851, 308–330 CrossRef CAS.
- L. Vernis, N. El Banna, D. Baille, E. Hatem, A. Heneman and M. E. Huang, Oxid. Med. Cell. Longevity, 2017, 2017, 3647657 CrossRef PubMed.
- M. Gao, P. Monian, N. Quadri, R. Ramasamy and X. Jiang, Mol. Cell, 2015, 59, 298–308 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli and H. Bayir, et al. , Cell, 2017, 171, 273–285 CrossRef CAS.
- M. Gao and X. Jiang, Curr. Opin. Chem. Biol., 2018, 51, 58–64 CrossRef CAS.
- M. Conrad, V. E. Kagan, H. Bayir, G. C. Pagnussat, B. Head, M. G. Traber and B. R. Stockwell, Genes Dev., 2018, 32, 602–619 CrossRef CAS.
- A. M. Battaglia, R. Chirillo, I. Aversa, A. Sacco, F. Costanzo and F. Biamonte, Cells, 2020, 9, 1505 CrossRef CAS.
- M. Gao, J. Yi, J. Zhu, A. M. Minikes, P. Monian, C. B. Thompson and X. Jiang, Mol. Cell, 2019, 73, 354–363 CrossRef CAS.
- A. Grubman and A. R. White, Expert Rev. Mol. Med., 2014, 16, e11 CrossRef PubMed.
- B. Halliwell, K. Zhao and M. Whiteman, Free Radical Res., 2000, 33, 819–830 CrossRef CAS.
- N. M. Garza, A. B. Swaminathan, K. P. Maremanda, M. Zulkifli and V. M. Gohil, Trends Endocrinol. Metab., 2023, 34, 21–33 CrossRef CAS PubMed.
- M. Ralle, D. Huster and S. Vogt, et al. , J. Biol. Chem., 2010, 285, 30875–30883 CrossRef CAS PubMed.
- I. F. Scheiber, J. F. B. Mercer and R. Dringen, Prog. Neurobiol., 2014, 116, 33–57 CrossRef CAS PubMed.
- L. Chen, J. Min and F. Wang, Signal Transduction Targeted Ther., 2022, 7, 378 CrossRef CAS.
- N. Husain and R. Mahmood, Environ. Sci. Pollut. Res., 2019, 26, 20654–20668 CrossRef CAS.
- B. Blades, S. Ayton, Y. H. Hung, A. I. Bush and S. La Fontaine, Biochim. Biophys. Acta, Gen. Subj., 2021, 1865, 129979 CrossRef CAS.
- J. Chen, C. Lan and H. An, et al. , Sci. Total Environ, 2021, 760, 143375 CrossRef CAS PubMed.
- M. H. Alqarni, M. M. Muharram, S. M. Alshahrani and N. E. Labrou, Int. J. Biol. Macromol., 2019, 128, 493–498 CrossRef CAS.
- L. M. Ruiz, A. Libedinsky and A. A. Elorza, Front. Mol. Biosci., 2021, 8, 711227 CrossRef CAS.
- N. Girerd, J. Intern. Med., 2022, 291, 710–712 CrossRef.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS.
- S. Puig and D. J. Thiele, Curr. Opin. Chem. Biol., 2002, 6, 171–180 CrossRef CAS.
- E. Nyvltova, J. V. Dietz, J. Seravalli, O. Khalimonchuk and A. Barrientos, Nat. Commun., 2022, 13, 3615 CrossRef CAS PubMed.
- J. Bertinato and M. R. L'Abbe, J. Biol. Chem., 2003, 278, 35071–35078 CrossRef CAS.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O’Halloran, Science, 1999, 284, 805–808 CrossRef CAS PubMed.
- L. Miao and D. K. St. Clair, Free Radical Biol. Med., 2009, 47, 344–356 CrossRef CAS.
- S. R. Powell, D. Hall and A. Shih, Circ. Res., 1991, 69, 881–885 CrossRef CAS.
- X. Chen, Q. Cai and R. Liang, et al. , Cell Death Dis., 2023, 14, 105 CrossRef.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS.
- M. Yang, Y. Yang and P. R. Chen, Top. Curr. Chem., 2016, 374, 2 CrossRef PubMed.
- Y. You, Q. Deng, Y. Wang, Y. Sang, G. Li, F. Pu, J. Ren and X. Qu, Nat. Commun., 2022, 13, 1459 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- Y. You, H. Liu, J. Zhu, Y. Wang, F. Pu, J. Ren and X. Qu, Chem. Sci., 2022, 13, 7829–7836 RSC.
- X. Xue, C. Qian and Q. Tao, et al. , Natl. Sci. Rev., 2020, 8, nwaa286 CrossRef PubMed.
- J. Zhu, Y. You, W. Zhang, F. Pu, J. Ren and X. Qu, J. Am. Chem. Soc., 2023, 145, 1955–1963 CrossRef CAS PubMed.
- C. E. Outten and T. V. O’Halloran, Science, 2001, 292, 2488–2492 CrossRef CAS PubMed.
- T. Hara, T. A. Takeda, T. Takagishi, K. Fukue, T. Kambe and T. Fukada, J. Physiol. Sci., 2017, 67, 283–301 CrossRef CAS PubMed.
- L. Huang and J. Gitschier, Nat. Genet., 1997, 17, 292–297 CrossRef CAS PubMed.
- L. Guo, L. A. Lichten, M. S. Ryu, J. P. Liuzzi, F. Wang and R. J. Cousins, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2818–2823 CrossRef CAS PubMed.
- S. Choi, X. Liu and Z. Pan, Acta Pharmacol. Sin., 2018, 39, 1120–1132 CrossRef CAS PubMed.
- Y. Y. Chen and T. V. O’Halloran, Cell, 2022, 185, 2013–2015 CrossRef CAS.
- A. Krezel and W. Maret, Chem. Rev., 2021, 121, 14594–14648 CrossRef CAS PubMed.
- H. Ma, L. Su, H. Yue, X. Yin, J. Zhao, S. Zhang, H. Kung, Z. Xu and J. Miao, Sci. Rep., 2015, 5, 15121 CrossRef CAS.
- W. Ohashi, S. Kimura and T. Iwanaga, et al. , PLoS Genet., 2016, 12, e1006349 CrossRef.
- T. Miyai, S. Hojyo and T. Ikawa, et al. , Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11780–11785 CrossRef CAS.
- D. Liang, L. Xiang, M. Yang, X. Zhang, B. Guo, Y. Chen, L. Yang and J. Cao, Cell. Signalling, 2013, 25, 1126–1135 CrossRef CAS PubMed.
- P. Thomas, Y. Pang, J. Dong and A. H. Berg, Endocrinology, 2014, 155, 4250–4265 CrossRef.
- J. Sterling, S. Guttha, Y. Song, D. Song, M. Hadziahmetovic and J. L. Dunaief, Exp. Eye Res., 2017, 155, 15–23 CrossRef CAS PubMed.
- O. Florey, S. E. Kim, C. P. Sandoval, C. M. Haynes and M. Overholtzer, Nat. Cell Biol., 2011, 13, 1335–1343 CrossRef CAS.
- S. Krishna, W. Palm, Y. Lee, W. Yang, U. Bandyopadhyay, H. Xu, O. Florey, C. B. Thompson and M. Overholtzer, Dev. Cell, 2016, 38, 536–547 CrossRef CAS PubMed.
- Y. Zhang, E. Aizenman, D. B. DeFranco and P. A. Rosenberg, Mol. Med., 2007, 13, 350–355 CAS.
- I. Kim, E. J. Park, J. Seo, S. J. Ko, J. Lee and C. H. Kim, NeuroReport, 2011, 22, 839–844 CrossRef CAS.
- X. Y. Sun, Y. P. Wei and Y. Xiong, et al. , J. Biol. Chem., 2012, 287, 11174–11182 CrossRef CAS.
- Q. Sun, W. Zhong, W. Zhang, Q. Li, X. Sun, X. Tan, X. Sun, D. Dong and Z. Zhou, Am. J. Physiol.: Gastrointest. Liver Physiol., 2015, 308, G757–G766 CrossRef CAS.
- K. Homma, T. Fujisawa and N. Tsuburaya, et al. , Mol. Cell, 2013, 52, 75–86 CrossRef CAS.
- R. A. Yokel, J. Alzheimer's Dis., 2006, 10, 223–253 Search PubMed.
- R. H. Kretsinger., V. N. Uversky and E. A. Permyakov, Encyclopedia of Metalloproteins, 2013 Search PubMed.
- C. Wang, Y. Guan and M. Lv, et al. , Immunity, 2018, 48, 675–687 CrossRef CAS PubMed.
- D. Osman, A. W. Foster, J. Chen, K. Svedaite, J. W. Steed, E. Lurie Luke, T. G. Huggins and N. J. Robinson, Nat. Commun., 2017, 8, 1884 CrossRef PubMed.
- W. Yang, J. Y. Lee and M. Nowotny, Mol. Cell, 2006, 22, 5–13 CrossRef CAS PubMed.
- F. Y. Li, B. Chaigne-Delalande and C. Kanellopoulou, et al. , Nature, 2011, 475, 471–476 CrossRef CAS.
- T. E. Gunter, C. E. Gavin, M. Aschner and K. K. Gunter, Neurotoxicology, 2006, 27, 765–776 CrossRef CAS.
- J. Lee, D. A. Kitchaev and D. H. Kwon, et al. , Nature, 2018, 556, 185–190 CrossRef CAS.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS.
- M. Giedyk and D. Gryko, Chem. Catal., 2022, 2, 1534–1548 CrossRef CAS.
- R. Ortega, C. Bresson and A. Fraysse, et al. , Toxicol. Lett., 2009, 188, 26–32 CrossRef CAS PubMed.
- A. B. Moshnikova, S. A. Moshnikov, V. N. Afanasyev, K. E. Krotova, V. B. Sadovnikov and I. P. Beletsky, Int. J. Biochem. Cell Biol., 2001, 33, 1160–1171 CrossRef CAS PubMed.
- B. A. Carneiro and W. S. El-Deiry, Nat. Rev. Clin. Oncol., 2020, 17, 395–417 CrossRef.
- R. Valentin, S. Grabow and M. S. Davids, Blood, 2018, 132, 1248–1264 CrossRef CAS PubMed.
- A. Jain, J. Gosling and S. Liu, et al. , Nat. Nanotechnol., 2024, 19, 106–114 CrossRef CAS.
- E. C. Josefsson, D. L. Burnett and M. Lebois, et al. , Nat. Commun., 2014, 5, 3455 CrossRef PubMed.
- T. Xu, Q. Ma and Y. Li, et al. , Signal Transduction Targeted Ther., 2022, 7, 354 CrossRef CAS.
- X. Huang, Z. Cao and J. Qian, et al. , Nat. Nanotechnol., 2024, 19, 545–553 CrossRef CAS.
- W. Zhang, Z. Liu, J. Zhu, Z. Liu, Y. Zhang, G. Qin, J. Ren and X. Qu, J. Am. Chem. Soc., 2023, 145, 16658–16668 CrossRef CAS.
- B. Z. Carter, W. Tao and L. B. Ostermann, et al. , Blood, 2021, 138, 3332 CrossRef.
- S. T. Diepstraten, M. A. Anderson, P. E. Czabotar, G. Lessene, A. Strasser and G. L. Kelly, Nat. Rev. Cancer, 2021, 22, 45–64 CrossRef PubMed.
- E. A. Bowling, J. H. Wang and F. Gong, et al. , Cell, 2021, 184, 384–403 CrossRef CAS.
- J. Hou, R. Zhao and W. Xia, et al. , Nat. Cell Biol., 2020, 22, 1264–1275 CrossRef CAS PubMed.
- K. A. Sarosiek and K. C. Wood, Trends Cancer, 2023, 9, 96–110 CrossRef CAS PubMed.
- Y. Ge, S. Wu, Z. Zhang, X. Li, F. Li, S. Yan, H. Liu, J. Huang and Y. Zhao, Protein Cell, 2019, 10, 808–824 CrossRef CAS PubMed.
- J. A. Malla, R. M. Umesh, S. Yousf, S. Mane, S. Sharma, M. Lahiri and P. Talukdar, Angew. Chem., Int. Ed., 2020, 59, 7944–7952 CrossRef CAS.
- J. J. McGuire, J. S. Frieling, C. H. Lo, T. Li, A. Muhammad, H. R. Lawrence, N. J. Lawrence, L. M. Cook and C. C. Lynch, Nat. Commun., 2021, 12, 723 CrossRef CAS PubMed.
- Y. Boege, M. Malehmir and M. E. Healy, et al. , Cancer Cell, 2017, 32, 342–359 CrossRef CAS.
- G. Liccardi, L. Ramos Garcia and T. Tenev, et al. , Mol. Cell, 2019, 73, 413–428 CrossRef CAS PubMed.
- G. Ichim and S. W. Tait, Nat. Rev. Cancer, 2016, 16, 539–548 CrossRef CAS.
- J. R. Tejedor, P. Papasaikas and J. Valcarcel, Mol. Cell, 2015, 57, 23–38 CrossRef CAS.
- J. Yuan, P. Amin and D. Ofengeim, Nat. Rev. Neurosci., 2019, 20, 19–33 CrossRef CAS PubMed.
- D. Bertheloot, E. Latz and B. S. Franklin, Cell. Mol. Immunol., 2021, 18, 1106–1121 CrossRef CAS PubMed.
- Y. S. Cho, S. Challa, D. Moquin, R. Genga, T. D. Ray, M. Guildford and F. K. Chan, Cell, 2009, 137, 1112–1123 CrossRef CAS PubMed.
- E. W. Lee, J. H. Kim and Y. H. Ahn, et al. , Nat. Commun., 2012, 3, 978 CrossRef PubMed.
- A. Degterev, Z. Huang and M. Boyce, et al. , Nat. Chem. Biol., 2005, 1, 112–119 CrossRef CAS.
- J. Yan, P. Wan, S. Choksi and Z. G. Liu, Trends Cancer, 2022, 8, 21–27 CrossRef CAS PubMed.
- D. Jiao, Z. Cai and S. Choksi, et al. , Cell Res., 2018, 28, 868–870 CrossRef CAS.
- R. J. Thapa, S. Nogusa, P. Chen, J. L. Maki, A. Lerro, M. Andrake, G. F. Rall, A. Degterev and S. Balachandran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3109–E3118 CrossRef CAS PubMed.
- N. Robinson, S. McComb, R. Mulligan, R. Dudani, L. Krishnan and S. Sad, Nat. Immunol., 2012, 13, 954–962 CrossRef CAS PubMed.
- R. Thijssen, S. Alvarez-Diaz, C. Grace, M.-Y. Gao, D. H. Segal, Z. Xu, A. Strasser and D. C. S. Huang, Cell Death Differ., 2020, 27, 2531–2533 CrossRef.
- U. Höckendorf, M. Yabal and T. Herold, et al. , Cancer Cell, 2016, 30, 75–91 CrossRef.
- W. S. Yang, R. SriRamaratnam and M. E. Welsch, et al. , Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- J. P. Friedmann Angeli, M. Schneider and B. Proneth, et al. , Nat. Cell Biol., 2014, 16, 1180–1191 CrossRef CAS.
- M. J. Hangauer, V. S. Viswanathan and M. J. Ryan, et al. , Nature, 2017, 551, 247–250 CrossRef CAS PubMed.
- X. Jiang, B. R. Stockwell and M. Conrad, Nat. Rev. Mol. Cell Biol., 2021, 22, 266–282 CrossRef.
- N. Kang, S. Son, S. Min, H. Hong, C. Kim, J. An, J. S. Kim and H. Kang, Chem. Soc. Rev., 2023, 52, 3955–3972 RSC.
- W. S. Yang, K. J. Kim, M. M. Gaschler, M. Patel, M. S. Shchepinov and B. R. Stockwell, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4966–E4975 CAS.
- W. Wang, M. Green and J. E. Choi, et al. , Nature, 2019, 569, 270–274 CrossRef CAS PubMed.
- B. Chu, N. Kon, D. Chen, T. Li, T. Liu, L. Jiang, S. Song, O. Tavana and W. Gu, Nat. Cell Biol., 2019, 21, 579–591 CrossRef CAS.
- S. Doll, B. Proneth and Y. Y. Tyurina, et al. , Nat. Chem. Biol., 2017, 13, 91–98 CrossRef CAS.
- T. J. Nevalainen, Virchows Arch. B Cell Pathol., 1975, 18, 119–127 CrossRef CAS PubMed.
- D. Denton and S. Kumar, Cell Death Differ., 2019, 26, 605–616 CrossRef CAS.
- Z. Zhang and M. Costa, Semin. Cancer Biol., 2021, 76, 267–278 CrossRef CAS.
- J. Kim, M. Kundu, B. Viollet and K. L. Guan, Nat. Cell Biol., 2011, 13, 132–141 CrossRef CAS.
- M. C. Maiuri, L. Galluzzi, E. Morselli, O. Kepp, S. A. Malik and G. Kroemer, Curr. Opin. Chem. Biol., 2010, 22, 181–185 CrossRef CAS.
- H. Zuo, C. Chen and Y. Sa, Pharmacol. Rep., 2023, 75, 499–510 CrossRef PubMed.
- M. Zachari and I. G. Ganley, Essays Biochem., 2017, 61, 585–596 CrossRef.
- D. F. Egan, M. G. H. Chun and M. Vamos, et al. , Mol. Cell, 2015, 59, 285–297 CrossRef CAS.
- M. A. Brennan and B. T. Cookson, Trends Microbiol., 2001, 9, 113–114 Search PubMed.
- S. K. Hsu, C. Y. Li and I. L. Lin, et al. , Theranostics, 2021, 11, 8813–8835 CrossRef CAS.
- N. Kayagaki, I. B. Stowe and B. L. Lee, et al. , Nature, 2015, 526, 666–671 CrossRef CAS.
- T. D. Kanneganti, M. Lamkanfi and G. Nunez, Immunity, 2007, 27, 549–559 CrossRef CAS.
- G. P. Amarante-Mendes, S. Adjemian, L. M. Branco, L. C. Zanetti, R. Weinlich and K. R. Bortoluci, Front. Immunol., 2018, 9, 2379 CrossRef.
- J. Shi, Y. Zhao and K. Wang, et al. , Nature, 2015, 526, 660–665 CrossRef CAS PubMed.
- N. M. de Vasconcelos, N. Van Opdenbosch, H. Van Gorp, E. Parthoens and M. Lamkanfi, Cell Death Differ., 2019, 26, 146–161 CrossRef CAS.
- A. Al Mamun, A. A. Mimi, M. A. Aziz, M. Zaeem, T. Ahmed, F. Munir and J. Xiao, Eur. J. Pharmacol., 2021, 910, 174444 CrossRef CAS.
- X. Zhu and S. Li, Adv. Sci., 2023, 10, 2300824 CrossRef CAS.
- Z. Zheng and G. Li, Int. J. Mol. Sci., 2020, 21, 1456 CrossRef CAS.
- Y. Wang, W. Gao, X. Shi, J. Ding, W. Liu, H. He, K. Wang and F. Shao, Nature, 2017, 547, 99–103 CrossRef CAS.
- P. Yu, H. Y. Wang, M. Tian, A. X. Li, X. S. Chen, X. L. Wang, Y. Zhang and Y. Cheng, Acta Pharmacol. Sin., 2019, 40, 1237–1244 CrossRef CAS PubMed.
- J. Yu, S. Li, J. Qi, Z. Chen, Y. Wu, J. Guo, K. Wang, X. Sun and J. Zheng, Cell Death Dis., 2019, 10, 193 CrossRef.
- C. C. Zhang, C. G. Li and Y. F. Wang, et al. , Apoptosis, 2019, 24, 312–325 CrossRef CAS.
- J. Xie, Y. Yang, Y. Gao and J. He, Mol. Cancer, 2023, 22, 46 CrossRef CAS PubMed.
- D. Tang, G. Kroemer and R. Kang, Nat. Rev. Clin. Oncol., 2024, 21, 370–388 CrossRef CAS PubMed.
- P. Pei, Y. Wang and W. Shen, et al. , Aggregate, 2024, e484 CrossRef CAS.
- X. Lun, J. C. Wells and N. Grinshtein, et al. , Clin. Cancer Res., 2016, 22, 3860–3875 CrossRef CAS PubMed.
- M. Zulkifli, A. N. Spelbring and Y. Zhang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216722120 CrossRef CAS.
- M. Buccarelli, Q. G. D’Alessandris and P. Matarrese, et al. , J. Exp. Clin. Cancer Res., 2021, 40, 228 CrossRef CAS PubMed.
- F. Paredes, K. Sheldon and B. Lassegue, et al. , Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1789–1794 CrossRef CAS.
- E. A. Rowland, C. K. Snowden and I. M. Cristea, Curr. Opin. Chem. Biol., 2018, 42, 76–85 CrossRef CAS PubMed.
- M. Overholtzer, A. A. Mailleux, G. Mouneimne, G. Normand, S. J. Schnitt, R. W. King, E. S. Cibas and J. S. Brugge, Cell, 2007, 131, 966–979 CrossRef CAS.
- Q. Sun, E. S. Cibas, H. Huang, L. Hodgson and M. Overholtzer, Cell Res., 2014, 24, 1288–1298 CrossRef CAS PubMed.
- P. A. Kreuzaler, A. D. Staniszewska and W. Li, et al. , Nat. Cell Biol., 2011, 13, 303–309 CrossRef CAS.
- T. J. Sargeant, B. Lloyd-Lewis, H. K. Resemann, A. Ramos-Montoya, J. Skepper and C. J. Watson, Nat. Cell Biol., 2014, 16, 1057–1068 CrossRef CAS PubMed.
- M. A. Sanjuan, C. P. Dillon and S. W. Tait, et al. , Nature, 2007, 450, 1253–1257 CrossRef CAS.
- S. E. Kim and M. Overholtzer, Semin. Cancer Biol., 2013, 23, 329–336 CrossRef CAS.
- X. Wang, Y. Li and J. Li, et al. , Front. Cell Dev. Biol., 2019, 7, 311 CrossRef.
- J. Song, R. Xu, H. Zhang, X. Xue, R. Ruze, Y. Chen, X. Yin, C. Wang and Y. Zhao, Gastroenterology, 2023, 165, 1505–1521 CrossRef CAS.
- M. Krajcovic and M. Overholtzer, Cancer Res., 2012, 72, 1596–1601 CrossRef CAS.
- V. Brinkmann, U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D. S. Weiss, Y. Weinrauch and A. Zychlinsky, Science, 2004, 303, 1532–1535 CrossRef CAS.
- H. R. Thiam, S. L. Wong, D. D. Wagner and C. M. Waterman, Annu. Rev. Cell Dev. Biol., 2020, 36, 191–218 CrossRef CAS PubMed.
- D. Azzouz, M. A. Khan and N. Palaniyar, Cell Death Discovery, 2021, 7, 113 CrossRef CAS.
- P. Li, M. Li, M. R. Lindberg, M. J. Kennett, N. Xiong and Y. Wang, J. Exp. Med., 2010, 207, 1853–1862 CrossRef CAS.
- M. Su, C. Chen and S. Li, et al. , Nat. Cardiovasc. Res., 2022, 1, 732–747 CrossRef CAS.
- K. W. Chen, M. Monteleone and D. Boucher, et al. , Sci. Immunol., 2018, 3, eaar6676 CrossRef PubMed.
- V. Poli and I. Zanoni, Trends Microbiol., 2023, 31, 280–293 CrossRef CAS.
- M. Demers, D. S. Krause, D. Schatzberg, K. Martinod, J. R. Voorhees, T. A. Fuchs, D. T. Scadden and D. D. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13076–13081 CrossRef CAS PubMed.
- S. Sangaletti, C. Tripodo and C. Vitali, et al. , Cancer Discovery, 2014, 4, 110–129 CrossRef CAS.
- K. K. David, S. A. Andrabi, T. M. Dawson and V. L. Dawson, Front. Biosci., 2009, 14, 1116–1128 CrossRef CAS.
- S. A. Andrabi, H. C. Kang and J. F. Haince, et al. , Nat. Med., 2011, 17, 692–699 CrossRef CAS PubMed.
- M. Mashimo, J. Kato and J. Moss, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18964–18969 CrossRef CAS.
- Y. Wang, R. An and G. K. Umanah, et al. , Science, 2016, 354, aad6872 CrossRef.
- Y. Zhou, L. Liu, S. Tao, Y. Yao, Y. Wang, Q. Wei, A. Shao and Y. Deng, Pharmacol. Res., 2021, 163, 105299 CrossRef CAS.
- S. A. Andrabi, T. M. Dawson and V. L. Dawson, Ann. N. Y. Acad. Sci., 2008, 1147, 233–241 CrossRef CAS PubMed.
- P. Donizy, A. Halon, P. Surowiak, G. Pietrzyk, C. Kozyra and R. Matkowski, Eur. J. Histochem., 2013, 57, e35 CrossRef CAS PubMed.
- N. Pillay, A. Tighe and L. Nelson, et al. , Cancer Cell, 2019, 35, 519–533 CrossRef CAS PubMed.
- D. Li, Y. Kou, Y. Gao, S. Liu, P. Yang, T. Hasegawa, R. Su, J. Guo and M. Li, Aging, 2021, 3, 4242–4257 CrossRef.
- J. Q. Wang, Y. Tang, Q. S. Li, M. Xiao, M. Li, Y. T. Sheng, Y. Yang and Y. L. Wang, Oncol. Rep., 2019, 41, 2657–2666 CAS.
- U. A. Matulonis, P. Harter and C. Gourley, et al. , Cancer, 2016, 122, 1844–1852 CrossRef CAS.
- P. Li, Y. Du, J. Qiu, D. Li, G. Li and G. Shan, Adv. Healthcare Mater., 2023, 12, e2301517 CrossRef PubMed.
- M. R. Mirza, B. J. Monk and J. Herrstedt, et al. , N. Engl. J. Med., 2016, 375, 2154–2164 CrossRef CAS PubMed.
- J. K. Litton, H. S. Rugo and J. Ettl, et al. , N. Engl. J. Med., 2018, 379, 753–763 CrossRef CAS PubMed.
- H. Rugo, E. Shtivelman and A. Perez, et al. , Breast Cancer Res. Treat., 2007, 105, 17–28 CrossRef CAS.
- A. A. Fatokun, V. L. Dawson and T. M. Dawson, Br. J. Pharmacol., 2014, 171, 2000–2016 CrossRef CAS PubMed.
- H. Nakamura, T. Tanaka, C. Zheng, S. A. Afione, B. M. Warner, M. Noguchi, T. Atsumi and J. A. Chiorini, Arthritis Rheumatol., 2023, 75, 1586–1598 CrossRef CAS PubMed.
- G. Kroemer and M. Jaattela, Nat. Rev. Cancer, 2005, 5, 886–897 CrossRef CAS PubMed.
- Z. Zuo, H. Yin, Y. Zhang, C. Xie and Q. Wang, Nat. Commun., 2023, 14, 5456 CrossRef CAS PubMed.
- P. Phadatare and J. Debnath, J. Clin. Invest., 2023, 133, e169240 CrossRef CAS.
- T. Kurz, A. Terman, B. Gustafsson and U. T. Brunk, Histochem. Cell Biol., 2008, 129, 389–406 CrossRef CAS.
- A. Sumoza-Toledo and R. Penner, J. Physiol., 2011, 589, 1515–1525 CrossRef CAS.
- J. Brojatsch, H. Lima, Jr., D. Palliser, L. S. Jacobson, S. M. Muehlbauer, R. Furtado, D. L. Goldman, M. P. Lisanti and K. Chandran, Cell Cycle, 2015, 14, 964–972 CrossRef CAS PubMed.
- K. F. Hsu, C. L. Wu, S. C. Huang, C. M. Wu, J. R. Hsiao, Y. T. Yo, Y. H. Chen, A. L. Shiau and C. Y. Chou, Autophagy, 2009, 5, 451–460 CrossRef CAS.
- M. Tang, B. Chen and H. Xia, et al. , Nat. Commun., 2023, 14, 5888 CrossRef CAS.
- M. E. Guicciardi, M. Leist and G. J. Gores, Oncogene, 2004, 23, 2881–2890 CrossRef CAS PubMed.
- M. Mauthe, I. Orhon and C. Rocchi, et al. , Autophagy, 2018, 14, 1435–1455 CrossRef CAS.
- H. Du Rietz, H. Hedlund, S. Wilhelmson, P. Nordenfelt and A. Wittrup, Nat. Commun., 2020, 11, 1809 CrossRef CAS PubMed.
- F. Chen, R. Kang, J. Liu and D. Tang, Front. Cell Dev. Biol., 2023, 11, 1213995 CrossRef PubMed.
- X. Song, S. Zhu and Y. Xie, et al. , Gastroenterology, 2018, 154, 1480–1493 CrossRef CAS.
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, Cell Res., 2019, 29, 347–364 CrossRef CAS PubMed.
- J. Liu, F. Kuang, R. Kang and D. Tang, Cancer Gene Ther., 2020, 27, 267–269 CrossRef CAS.
- X. Chen, H. J. Zeh, R. Kang, G. Kroemer and D. Tang, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 804–823 CrossRef PubMed.
- C. Holze, C. Michaudel and C. Mackowiak, et al. , Nat. Immunol., 2018, 19, 130–140 CrossRef CAS PubMed.
- P. Kang, J. Chen and W. Zhang, et al. , Cell Death Discovery, 2022, 8, 70 CrossRef CAS PubMed.
- S. Pallichankandy, F. Thayyullathil and A. R. Cheratta, et al. , Cell Death Discovery, 2023, 9, 94 CrossRef CAS PubMed.
- B. Pan, B. Zheng, C. Xing and J. Liu, Cancers, 2022, 14, 3309 CrossRef CAS PubMed.
- Y. Zou, J. Xie and S. Zheng, et al. , Int. J. Surg., 2022, 107, 106936 CrossRef PubMed.
- J. Zhang, R. F. Gao, J. Li, K. D. Yu and K. X. Bi, Biochem. Cell Biol., 2022, 100, 213–222 CrossRef CAS.
- H. Hu, Q. Xu, Z. Mo, X. Hu, Q. He, Z. Zhang and Z. Xu, J. Nanobiotechnol., 2022, 20, 457 CrossRef.
- Q. Li, Y. Feng, R. Wang, R. Liu, Y. Ba and H. Huang, Toxicol. Res., 2023, 39, 355–372 CrossRef CAS.
- Y. Luo, Y. Fu, Z. Huang and M. Li, J. Cell. Physiol., 2021, 236, 7144–7158 CrossRef CAS PubMed.
- Y. Guo, Q. Bao, P. Hu and J. Shi, Nat. Commun., 2023, 14, 7306 CrossRef CAS PubMed.
- R. Zimmermann, J. Clin. Immunol., 2016, 36, 5–11 CrossRef CAS.
- P. Yu, X. Cai, Y. Liang, M. Wang and W. Yang, Molecules, 2020, 25, 4826 CrossRef CAS.
- Y. D. Zheng, Z. He, Z. C. Su, H. Wang, X. H. Jiang, X. Fang, S. L. Lu and Y. Li, Cell Biol. Int., 2023, 47, 1344–1353 CrossRef CAS PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS.
- S. Kuchay, C. Giorgi and D. Simoneschi, et al. , Nature, 2017, 546, 554–558 CrossRef CAS PubMed.
- O. M. de Brito and L. Scorrano, Nature, 2008, 456, 605–610 CrossRef PubMed.
- T. Vervliet, I. Pintelon and K. Welkenhuyzen, et al. , Biochem. Pharmacol., 2017, 132, 133–142 CrossRef CAS.
- H. Akl, G. Monaco and R. La Rovere, et al. , Cell Death Dis., 2013, 4, e632 CrossRef CAS.
- M. Kerkhofs, R. La Rovere, K. Welkenhuysen, A. Janssens, P. Vandenberghe, M. Madesh, J. B. Parys and G. Bultynck, Cell Calcium, 2021, 94, 102333 CrossRef CAS PubMed.
- B. Zhou, Y. Lin, S. Chen, J. Cai, Z. Luo, S. Yu and J. Lu, Bull. Exp. Biol. Med., 2021, 171, 297–304 CrossRef CAS PubMed.
- E. M. Brown, G. Gamba and D. Riccardi, et al. , Nature, 1993, 366, 575–580 CrossRef CAS.
- J. Wang, R. Zhang and Y. Huo, et al. , Ecotoxicol. Environ. Saf., 2019, 175, 251–262 Search PubMed.
- Y. Zhu, G. Xu, P. Chen, K. Liu, Y. Xu, Y. Liu and J. Liu, Ecotoxicol. Environ. Saf., 2019, 179, 257–264 CrossRef CAS PubMed.
- Q. Zhong, T. I. Roumeliotis, Z. Kozik, M. Cepeda-Molero, L. A. Fernandez, A. R. Shenoy, C. Bakal, G. Frankel and J. S. Choudhary, PLoS Biol., 2020, 18, e3000986 CrossRef CAS PubMed.
- P. Maher, K. van Leyen, P. N. Dey, B. Honrath, A. Dolga and A. Methner, Cell Calcium, 2018, 70, 47–55 CrossRef CAS PubMed.
- L. Pedrera, U. Ros and A. J. Garcia-Saez, Cell Calcium, 2023, 114, 102778 CrossRef CAS.
- Q. Xue, D. Yan and X. Chen, et al. , Autophagy, 2023, 19, 1982–1996 CrossRef CAS PubMed.
- E. Dai, L. Meng, R. Kang, X. Wang and D. Tang, Biochem. Biophys. Res. Commun., 2020, 522, 415–421 CrossRef CAS.
- L. Pedrera, R. A. Espiritu, U. Ros, J. Weber, A. Schmitt, J. Stroh, S. Hailfinger, S. von Karstedt and A. J. Garcia-Saez, Cell Death Differ., 2021, 28, 1644–1657 CrossRef CAS.
- J. Zhao, Y. Wu and K. Zhou, et al. , Biochim. Biophys. Acta, Mol. Cell Res., 2023, 1870, 119452 CrossRef CAS PubMed.
- A. Woods, K. Dickerson, R. Heath, S. P. Hong, M. Momcilovic, S. R. Johnstone, M. Carlson and D. Carling, Cell Metab., 2005, 2, 21–33 CrossRef CAS.
- Z. D. Zhu, T. Yu, H. J. Liu, J. Jin and J. He, Cell Death Dis., 2018, 9, 50 CrossRef PubMed.
- Y. Tong and F. Song, Autophagy, 2015, 11, 1192–1195 CrossRef CAS.
- K. Mallilankaraman, C. Cardenas and P. J. Doonan, et al. , Nat. Cell Biol., 2012, 14, 1336–1343 CrossRef CAS PubMed.
- S. Missiroli, M. Bonora and S. Patergnani, et al. , Cell Rep., 2016, 16, 2415–2427 CrossRef CAS.
- T. Zhang, Y. Zhang and M. Cui, et al. , Nat. Med., 2016, 22, 175–182 CrossRef CAS PubMed.
- M. Milani, P. Pihan and C. Hetz, Cell Calcium, 2023, 113, 102751 CrossRef CAS.
- A. R. Lee and C. Y. Park, Adv. Sci., 2023, 10, e2205913 CrossRef.
- L. Lin, S. Wang and H. Deng, et al. , J. Am. Chem. Soc., 2020, 142, 15320–15330 CrossRef CAS.
- G. Spiteller, Free Radical Biol. Med., 2006, 41, 362–387 CrossRef CAS PubMed.
- V. E. Kagan, V. A. Tyurin and J. Jiang, et al. , Nat. Chem. Biol., 2005, 1, 223–232 CrossRef CAS.
- J. E. Ricci, C. Munoz-Pinedo, P. Fitzgerald, B. Bailly-Maitre, G. A. Perkins, N. Yadava, I. E. Scheffler, M. H. Ellisman and D. R. Green, Cell, 2004, 117, 773–786 CrossRef CAS PubMed.
- H. Ichijo, E. Nishida and K. Irie, et al. , Science, 1997, 275, 90–94 CrossRef CAS PubMed.
- M. Ahamed, M. J. Akhtar, M. A. Siddiqui, J. Ahmad, J. Musarrat, A. A. Al-Khedhairy, M. S. AlSalhi and S. A. Alrokayan, Toxicology, 2011, 283, 101–108 CrossRef CAS PubMed.
- J. Cheng, T. Zhou, C. Liu, J. P. Shapiro, M. J. Brauer, M. C. Kiefer, P. J. Barr and J. D. Mountz, Science, 1994, 263, 1759–1762 CrossRef CAS.
- T. Vanden Berghe, A. Linkermann, S. Jouan-Lanhouet, H. Walczak and P. Vandenabeele, Nat. Rev. Mol. Cell Biol., 2014, 15, 135–147 CrossRef CAS PubMed.
- C. Xie, N. Zhang, H. Zhou, J. Li, Q. Li, T. Zarubin, S. C. Lin and J. Han, Mol. Cell. Biol., 2005, 25, 6673–6681 CrossRef CAS PubMed.
- Y. S. Kim, M. J. Morgan, S. Choksi and Z. G. Liu, Mol. Cell, 2007, 26, 675–687 CrossRef CAS PubMed.
- G. B. Fortes, L. S. Alves and R. de Oliveira, et al. , Blood, 2012, 119, 2368–2375 CrossRef CAS PubMed.
- J. Zhu, Y. Xiong, Y. Zhang, J. Wen, N. Cai, K. Cheng, H. Liang and W. Zhang, Oxid. Med. Cell. Longevity, 2020, 2020, 8810785 Search PubMed.
- F. Ursini, M. Maiorino, M. Valente, L. Ferri and C. Gregolin, Biochim. Biophys. Acta, Lipids Lipid Metab., 1982, 710, 197–211 CrossRef CAS.
- Y. Zou, H. Li and E. T. Graham, et al. , Nat. Chem. Biol., 2020, 16, 302–309 CrossRef CAS PubMed.
- A. Anandhan, M. Dodson, A. Shakya, J. Chen, P. Liu, Y. Wei, H. Tan, Q. Wang, Z. Jiang, K. Yang, J. G. Garcia, S. K. Chambers, E. Chapman, A. Ooi, Y. Yang-Hartwich, B. R. Stockwell and D. D. Zhang, Sci. Adv., 2023, 9, eade9585 CrossRef CAS PubMed.
- W. Hou, Y. Xie, X. Song, X. Sun, M. T. Lotze, H. J. Zeh Iii, R. Kang and D. Tang, Autophagy, 2016, 12, 1425–1428 CrossRef CAS.
- M. Gao, P. Monian, Q. Pan, W. Zhang, J. Xiang and X. Jiang, Cell Res., 2016, 26, 1021–1032 CrossRef CAS.
- K. Hattori, H. Ishikawa, C. Sakauchi, S. Takayanagi, I. Naguro and H. Ichijo, EMBO Rep., 2017, 18, 2067–2078 CrossRef CAS PubMed.
- Y. Yu, L. Jiang and H. Wang, et al. , Blood, 2020, 136, 726–739 CrossRef CAS PubMed.
- B. Zhou, J. Y. Zhang and X. S. Liu, et al. , Cell Res., 2018, 28, 1171–1185 CrossRef CAS PubMed.
- Y. Chen, W. Li, S. Kwon, Y. Wang, Z. Li and Q. Hu, J. Am. Chem. Soc., 2023, 145, 9815–9824 CrossRef CAS PubMed.
- J. Feng, Z. X. Wang, J. L. Bin, Y. X. Chen, J. Ma, J. H. Deng, X. W. Huang, J. Zhou and G. D. Lu, Cancer Lett., 2024, 587, 216728 CrossRef CAS.
- B. Fernández, E. Fdez, P. Gómez-Suaga, F. Gil, I. Molina-Villalba, I. Ferrer, S. Patel, G. C. Churchill and S. Hilfiker, Autophagy, 2016, 12, 1487–1506 CrossRef.
- Y. Wang, M. Wang, Y. Liu, H. Tao, S. Banerjee, S. Srinivasan, E. Nemeth, M. J. Czaja and P. He, Redox Biol., 2022, 55, 102407 CrossRef CAS PubMed.
- E. J. Ge, A. I. Bush and A. Casini, et al. , Nat. Rev. Cancer, 2022, 22, 102–113 CrossRef CAS PubMed.
- H. M. Alvarez, Y. Xue, C. D. Robinson, M. A. Canalizo-Hernández, R. G. Marvin, R. A. Kelly, A. Mondragón, J. E. Penner-Hahn and T. V. O’Halloran, Science, 2010, 327, 331–334 CrossRef CAS PubMed.
- D. Cen, D. Brayton, B. Shahandeh, F. L. Meyskens and P. J. Farmer, J. Med. Chem., 2004, 47, 6914–6920 CrossRef CAS.
- N. C. Yip, I. S. Fombon and P. Liu, et al. , Br. J. Cancer, 2011, 104, 1564–1574 CrossRef CAS PubMed.
- R. Gupta, V. Luxami and K. Paul, Bioorg. Chem., 2021, 108, 104633 CrossRef CAS.
- L. I. Victoriano, Coord. Chem. Rev., 2000, 196, 383–398 CrossRef CAS.
- K. G. Daniel, D. Chen, S. Orlu, Q. C. Cui, F. R. Miller and Q. P. Dou, Breast Cancer Res., 2005, 7, R897 CrossRef CAS PubMed.
- Z. Zhang, H. Wang, M. Yan, H. Wang and C. Zhang, Mol. Med. Rep., 2017, 15, 3–11 CrossRef CAS PubMed.
- T. Hirschhorn and B. R. Stockwell, Free Radical Biol. Med., 2019, 133, 130–143 CrossRef CAS PubMed.
- X. Ren, Y. Li and Y. Zhou, et al. , Redox Biol., 2021, 46, 102122 CrossRef CAS PubMed.
- Y. Shang, M. Luo, F. Yao, S. Wang, Z. Yuan and Y. Yang, Cell. Signalling, 2020, 72, 109633 CrossRef CAS.
- G. Lei, Y. Zhang and P. Koppula, et al. , Cell Res., 2020, 30, 146–162 CrossRef CAS.
- M. Yang, X. Wu and J. Hu, et al. , J. Hepatol., 2022, 76, 1138–1150 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Biochem. J., 1984, 219, 1–14 CrossRef CAS PubMed.
- T. Tsang, J. M. Posimo, A. A. Gudiel, M. Cicchini, D. M. Feldser and D. C. Brady, Nat. Cell Biol., 2020, 22, 412–424 CrossRef CAS.
- F. Wan, G. Zhong and Z. Ning, et al. , Ecotoxicol. Environ. Saf., 2020, 190, 110158 CrossRef CAS.
- H. Guo, Y. Ouyang and H. Yin, et al. , Redox Biol., 2022, 49, 102227 CrossRef CAS PubMed.
- K. A. Peña and K. Kiselyov, Biochem. J., 2015, 470, 65–76 CrossRef.
- L. Chen, N. Li and M. Zhang, et al. , Angew. Chem., Int. Ed., 2021, 60, 25346–25355 CrossRef CAS.
- W. Chen, X. Wang and B. Zhao, et al. , Nanoscale, 2019, 11, 12983–12989 RSC.
- Y. Zhang, Q. Jia and J. Li, et al. , Adv. Mater., 2023, 35, e2305073 CrossRef.
- G. Zhu, Y. Xie, J. Wang, M. Wang, Y. Qian, Q. Sun, Y. Dai and C. Li, Adv. Mater., 2024, 36, 2409066 CrossRef CAS.
- G. Briassoulis, P. Briassoulis, S. Ilia, M. Miliaraki and E. Briassouli, Antioxidants, 2023, 12, 1942 CrossRef CAS PubMed.
- E. Bafaro, Y. Liu, Y. Xu and R. E. Dempski, Signal Transduction Targeted Ther., 2017, 2, 17029 CrossRef.
- B. Chen, P. Yu and W. N. Chan, et al. , Signal Transduction Targeted Ther., 2024, 9, 6 CrossRef CAS PubMed.
- P. Chandler, B. S. Kochupurakkal, S. Alam, A. L. Richardson, D. I. Soybel and S. L. Kelleher, Mol. Cancer, 2016, 15, 2 CrossRef PubMed.
- W. Alker and H. Haase, Nutrients, 2018, 10, 976 CrossRef PubMed.
- K. Thambiayya, A. M. Kaynar, C. M. S. Croix and B. R. Pitt, Pulm. Circ., 2012, 2, 443–451 CrossRef CAS.
- S. J. Lee, M. H. Park, H. J. Kim and J. Y. Koh, Glia, 2010, 58, 1186–1196 CrossRef PubMed.
- L. Tan, C. He, X. Chu, Y. Chu and Y. Ding, Chem. Eng. J., 2020, 395, 125177 CrossRef CAS.
- F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo and G. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS.
- J. P. Liuzzi and R. Pazos, J. Trace Elem. Med. Biol., 2020, 62, 126636 CrossRef CAS PubMed.
- H. Fang, S. Geng and M. Hao, et al. , Nat. Commun., 2021, 12, 109 CrossRef CAS PubMed.
- A. Lahiri and C. Abraham, Gastroenterology, 2014, 147, 835–846 CrossRef CAS.
- K. M. Taylor, P. Vichova, N. Jordan, S. Hiscox, R. Hendley and R. I. Nicholson, Endocrinology, 2008, 149, 4912–4920 CrossRef CAS PubMed.
- K. W. Kim, C. K. Speirs, D. K. Jung and B. Lu, J. Thorac. Oncol., 2011, 6, 1542–1552 CrossRef.
- M. Farasat, F. Niazvand and L. Khorsandi, Biologia, 2020, 75, 161–174 CrossRef CAS.
- Y. Li, C. Jiang, X. Zhang, Z. Liao, L. Chen, S. Li, S. Tang, Z. Fan and Q. Zhang, Cancer Nanotechnol., 2022, 13, 3 CrossRef CAS.
- P. H. Chen, J. Wu, Y. Xu, C. K. C. Ding, A. A. Mestre, C. C. Lin, W. H. Yang and J.-T. Chi, Cell Death Dis., 2021, 12, 198 CrossRef CAS.
- P. Zheng, B. Ding, G. Zhu, M. Wang, C. Li and J. Lin, Chem. Eng. J., 2022, 450, 137967 CrossRef CAS.
- B. Ding, H. Chen, J. Tan, Q. Meng, P. Zheng, P. A. Ma and J. Lin, Angew. Chem., Int. Ed., 2023, 62, e202215307 CrossRef CAS PubMed.
- X. Su, B. Liu, W. J. Wang, K. Peng, B. B. Liang, Y. Zheng, Q. Cao and Z. W. Mao, Angew. Chem., Int. Ed., 2023, 62, e202216917 CrossRef CAS PubMed.
- B. Xia, S. Fang, X. Chen, H. Hu, P. Chen, H. Wang and Z. Gao, Cell Res., 2016, 26, 517–528 CrossRef CAS.
- X. Shao, C. Qu, G. Song, B. Wang, Q. Tao, R. Jia, J. Li, J. Wang and H. An, Adv. Funct. Mater., 2023, 33, 2306585 CrossRef CAS.
- J. He, X. Ma and J. Zhang, et al. , Food Chem. Toxicol., 2024, 184, 114322 CrossRef CAS.
- P. Wang, C. Liang, J. Zhu, N. Yang, A. Jiao, W. Wang, X. Song and X. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 41140–41147 CrossRef CAS PubMed.
- N. de Dios, R. Riedel and M. Schanton, et al. , Biol. Reprod., 2024, 111, 708–722 CrossRef.
- B. Deka, T. Sarkar, A. Bhattacharyya, R. J. Butcher, S. Banerjee, S. Deka, K. K. Saikia and A. Hussain, Dalton Trans., 2024, 53, 4952–4961 RSC.
- X. Zhao, Y. Wang, T. Zhu, H. Wu, D. Leng, Z. Qin, Y. Li and D. Wu, Int. J. Nanomed., 2022, 17, 5187–5205 CrossRef CAS.
- M. T. Nunez and C. Hidalgo, Front. Neurosci., 2019, 13, 48 CrossRef PubMed.
- C. Manzl, J. Enrich, H. Ebner, R. Dallinger and G. Krumschnabel, Toxicology, 2004, 196, 57–64 CrossRef CAS.
- Z. Pan, S. Choi, H. Ouadid-Ahidouch, J.-M. Yang, J. H. Beattie and I. Korichneva, Front. Biosci.-Landmark, 2017, 22, 623–643 CrossRef CAS.
- Y. Jiang, K. Shao, F. Zhang, T. Wang, L. Han, X. Kong and J. Shi, Aggregate, 2023, 4, e321 CrossRef CAS.
- C. A. Blomeyer, J. N. Bazil, D. F. Stowe, R. K. Dash and A. K. S. Camara, J. Bioenerg. Biomembr., 2016, 48, 175–188 CrossRef CAS.
- M. Tan, G. Cao and R. Wang, et al. , Nat. Nanotechnol., 2024, 19, 1903–1913 CrossRef CAS.
- Y. Li, Y. Du, Y. Zhou, Q. Chen, Z. Luo, Y. Ren, X. Chen and G. Chen, Cell Commun. Signaling, 2023, 21, 327 CrossRef CAS.
- K. E. Splan, S. R. Choi, R. E. Claycomb, I. K. Eckart-Frank, S. Nagdev and M. E. Rodemeier, JBIC, J. Biol. Inorg. Chem., 2023, 28, 485–494 CrossRef CAS PubMed.
- C. Liu, L. Guo, Y. Wang, J. Zhang and C. Fu, Coord. Chem. Rev., 2023, 494, 215332 CrossRef CAS.
- W. Park, S. Wei, B.-S. Kim, B. Kim, S.-J. Bae, Y. C. Chae, D. Ryu and K.-T. Ha, Exp. Mol. Med., 2023, 55, 1573–1594 CrossRef CAS.
- L. Y. Wang, X. J. Liu and Q. Q. Li, et al. , Mol. Cell. Biochem., 2023, 479, 2255–2272 CrossRef PubMed.
- M. N. Messmer, A. G. Snyder and A. Oberst, Cell Death Differ., 2019, 26, 115–129 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |