
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGains and losses in zinc-ion batteries by proton- and water-assisted reactions
Yauhen
Aniskevich
and
Seung-Taek
Myung
 *
*
Hybrid Materials Research Center, Department of Nanotechnology and Advanced Materials Engineering & Sejong Battery Institute, Sejong University, Seoul 05006, South Korea. E-mail: smyung@sejong.ac.kr
First published on 31st March 2025
Abstract
Research on aqueous zinc-ion batteries (AZIBs) has expanded significantly over the last decade due to their promising performance, cost, and safety as well as environmentally friendly features. The use of aqueous electrolytes enables promising AZIB properties while simultaneously introducing undesired reactions and processes. This review focuses on fundamental and critical considerations of water-related equilibria and reactions in zinc-ion batteries. First, we examine Zn2+/water ionic equilibria and their consequences for the chemistry of electrodes. Then, we focus on the mechanisms and kinetics of proton and Zn2+ insertion in host frameworks. Next, special attention is given to the water-related dissolution, deposition, and amorphization phenomena of transition-metal-based cathode materials. Finally, we highlight the role of water- and proton-assisted reactions through a systematic comparison of aqueous and nonaqueous zinc-ion batteries.
 Yauhen Aniskevich | Yauhen Aniskevich earned his PhD in physical chemistry from Belarusian State University in Minsk, Belarus, in 2021. He is currently a postdoctoral researcher at the Department of Nano-engineering at Sejong University, South Korea. His research interests mainly focus on the study and development of post-lithium energy storage systems, including Na– and Zn-ion batteries, with an emphasis on electrode kinetics. |
 Seung-Taek Myung | Seung-Taek Myung is a DaeYang Distinguished Professor in the Department of Nanotechnology and Advanced Materials Engineering at Sejong University, South Korea. Before 2011, he was affiliated with VK (principal engineer), 3M (seinor engineer), and Iwate University (assistant professor). He received his PhD degree in Chemical Engineering from Iwate University, Japan, in 2003. His research focuses on electro-active materials and the corrosion of metallic current collectors for rechargeable Li-, Na-, K, and Zn-ion batteries. |
1. Introduction
Rechargeable batteries are an essential part of modern society due to their applications in diverse areas, such as portable electronic devices, grid applications, and electric vehicles, which particularly require safety enhancement.1,2 One of the core technologies is lithium-ion batteries (LIBs) as they offer high volumetric and gravimetric capacity and acceptable cyclability, although unexpected thermal runaway can lead to serious safety concerns. The limited reserves of resources such as lithium, nickel, and cobalt and the refining of these ores have raised several concerns in terms of sustainability and environmental issues. These difficulties have motivated researchers to explore alternative energy storage devices beyond lithium-based systems.3–5Several post-lithium batteries have been introduced by adopting monovalent Na-ion,6–8 K-ion,9,10 or multivalent charge carriers,5,11–13 such as Mg-ion,14 Ca-ion,15 Al-ion,16 and Zn-ion batteries.17–19 Monovalent systems may be paired with high-capacity carbon-based anode materials, typically hard carbon anodes.20,21 Notably, multivalent metal-ion batteries mostly use metal anodes, which theoretically offer high volumetric capacities of the corresponding metals, providing an additional merit over safety (Fig. 1a).
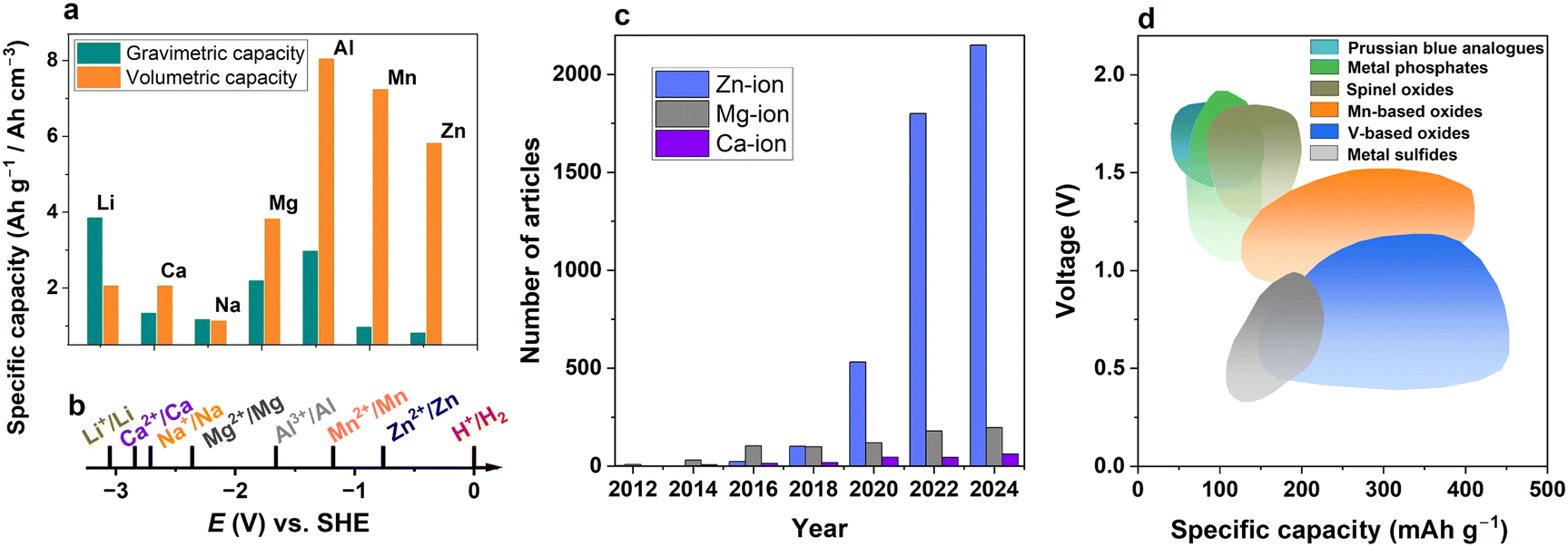 | ||
| Fig. 1 Comparison of several ion storage systems. (a) Volumetric and gravimetric specific capacities of lithium, sodium, and some multivalent metals. (b) Standard reduction potentials of ions to corresponding metals. (c) Number of articles mentioning multivalent-ion battery in title, and (d) operation voltage vs. specific capacity for Zn-ion batteries.18,22,23 | ||
When comparing divalent metals, the standard potentials for Ca2+/Ca and Mg2+/Mg are relatively low (−2.84 V and −2.36 V vs. SHE respectively), indicating the viability of these systems for high-voltage applications (Fig. 1b). The standard potential of Zn2+ reduction is comparatively much higher (−0.76 V); hence, zinc-ion batteries (ZIBs) cannot compete with Ca-ion and Mg-ion batteries for high-voltage applications. In addition, these divalent ion reactions have sluggish kinetics in nonaqueous batteries.
However, the use of water-based electrolytes can mitigate these drawbacks for Zn batteries. Namely, the sluggish reaction becomes faster, and the Zn-metal electrode can be operated near the stability window of the electrolyte, where the pH values of electrolytes range from neutral to slightly acidic. This environment is not available for highly reactive calcium and magnesium metals. Thus, the chemical stability of zinc metals in aqueous solution allows the use of inexpensive water-soluble salts for the investigation of aqueous ZIBs (AZIBs). This explains the recent surge in research on AZIBs compared to the mentioned Ca-ion and Mg-ion systems (Fig. 1c). The achievement of high capacity is thought to be another reason that attracts interest in AZIBs (Fig. 1d). Therefore, AZIBs have a significant advantage over other divalent systems in aqueous solutions due to the participation of water in the electrochemical process.
The history of Zn-ion and Zn-metal batteries dates back to the 19th century with the invention of the Leclanché cell, a primary Zn|MnO2 cell utilizing an ammonium chloride electrolyte (Fig. 2). This work and the subsequent development of alkaline-electrolyte-based cells have met the primary battery demand over the last century. The desire to design a system with similar affordable electrodes in a secondary, rechargeable manner is unsurprising. The first publication on MnO2-based Zn-metal rechargeable non-alkaline batteries was the 1986 work by Yamamoto and Shoji which reported cyclability of up to 30 cycles in an aqueous ZnSO4 solution.24 A year later, in a pioneering work by Gocke and colleagues, the intercalation of Zn2+ was demonstrated in Chevrel phases from a nonaqueous electrolyte.25 Since then, only a few articles on ZIBs or Zn-intercalation were published until the 2010s, when research on ZIBs boomed, with the dominance of aqueous systems (Fig. 2).26–55 Besides pure aqueous and nonaqueous ZIBs, mixed electrolytes have been used in recent years to achieve an appropriate balance between the fast kinetics and stability issues caused by water-related reactions. In addition, research on the redox reactions of organic compounds in Zn2+ electrolytes has been conducted since at least 1972, when the voltammetry of tetrachloro-1,4-benzoquinone (chloranil) was evaluated in aqueous ZnCl2 along with calculations of theoretical energy density of the corresponding secondary battery.48,49 After some time, studies on polyaniline50 and nitroxide-containing polymers51,52 emerged. As interest in zinc batteries grew, the search for new stable organic electrodes gained momentum. Notable examples include calix[4]quinone (C4Q)53 and pyrene-4,5,9,10-tetraone (PTO),54 along with many other oxygen- and nitrogen-based compounds, which offer promising properties for both stationary and miniaturized energy storage solutions.55–57 As the number of ZIB studies has reached the thousands, it is difficult to discuss all achievements in a brief history note; other historical perspectives can be found in ZIB-related reviews elsewhere.18,23
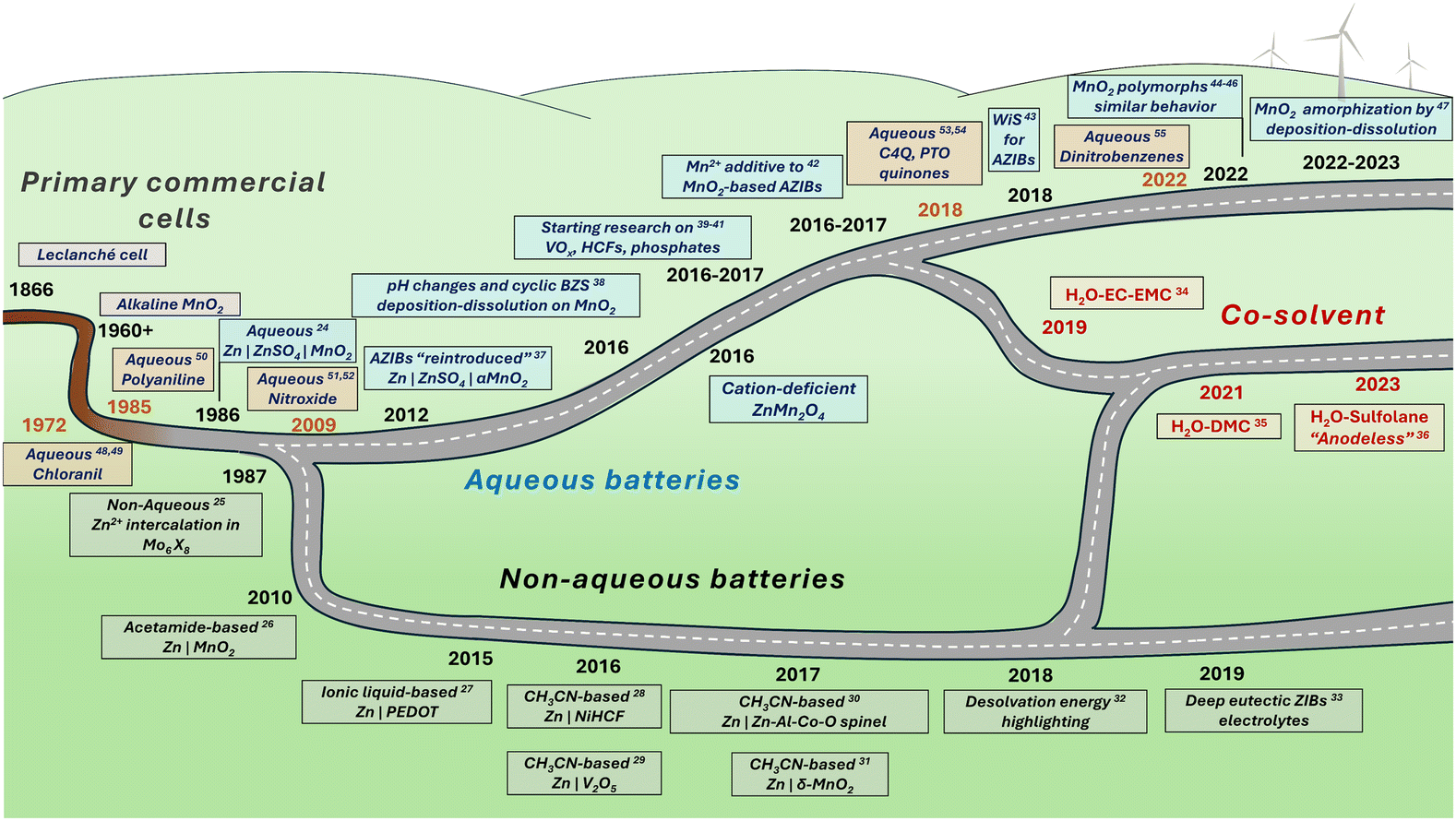 | ||
| Fig. 2 History of research on zinc-ion batteries, including aqueous and non-aqueous systems.24–55 | ||
The associated chemistry of AZIBs has often been considered as the classical rocking chair transport of Zn2+ ions between the anode (negative electrode) and cathode (positive host), often underestimating the role of water and its protolytic reactions, which can completely change the mechanism in comparison with non-aqueous batteries.5,11,17,23,58 The paradigm has changed over the last few years. It is now clear that water-based electrochemical reactions are involved in the main process for charge compensation in various materials in AZIBs.59,60 Recent literature and reviews explain the ambiguous role of H+ in mildly acidic aqueous electrolytes during electrochemical reactions. Under certain conditions, the formation of H+ is not favorable because the active material can be locally damaged by the generation and propagation of H+, which results in a local acidic environment at the interface between the cathode and electrolyte.58 Water reduction at Zn results in H2 gas formation, which can in turn cause an explosion in the hermetically sealed cell, threatening the safety of AZIBs.61,62 Nevertheless, the insertion of H+ provided from Zn2+ aqueous equilibria boosts the capacity and kinetics of aqueous zinc cells.63 This implies that the charge carrier Zn2+ is often not the main species contributing to the charge-compensation process in cathodes.59,60,64 Additionally, Zn2+ participates in reactions leading to the production of layered basic zinc salts (hydroxide salts) on the surface of both electrodes, which eventually increases the cell impedance,65 thereby degrading the electrode performance, and hence considered unfavorable or parasitic.58 In addition, dendritic growth of metallic zinc is unavoidable,61,66 and the process is facilitated by the consumption of H+ ions and Zn2+ on the surface of the Zn anode.67 Alkalization of the surface results in the propagation of Zn dendrites accompanied by passivation through the formation of Zn(OH)2, ZnO, and Zn0 moieties, reducing the cycle life of cells.68
In this review, we highlight the electrochemical reactions in aqueous Zn cells: (I) ionic equilibria that facilitate proton-coupled reactions, including basic salt formation; (II) the effect on kinetics for charge storage; (III) material transformation and “activation” by hydrogen ions (H+(aq)) and water in both cathodic and anodic reactions; and IV) the suggested role of water and protons in AZIBs. Therefore, we first address ionic and electrochemical equilibria in aqueous zinc electrolytes and at the zinc | electrolyte interface; the discussion then expands to consider water-related reactions for cathodes, including intercalation and conversion reactions in aqueous and non-aqueous ZIBs.
2. Zn-H2O chemistry in aqueous Zn2+ solutions
When the study of mildly acidic rechargeable zinc aqueous batteries first began, investigation of the reaction chemistry progressed mainly on the cathode materials using manganese and vanadium oxides to improve their electrode performance and examine the related phase transitions during charge storage.69 In these studies, the mechanism of ion intercalation or compound conversion was studied in cells with a considerable excess of metallic Zn, masking the zinc electrode problems. In addition, the research on the properties of water as a solvent often did not advance beyond its high polarity and solvation ability, leaving behind its protolytic properties. As a result, the mainstream research studies underestimated the role of pH and H+ in defining cell performance and the behavior of electrodes. In this section, we consider aqueous Zn(II) electrolytes and their ionic equilibria, whose consequences affect the use of Zn-metal anodes.2.1. Aqueous Zn2+ ionic equilibria and the formation of basic zinc salts
Aqueous solutions of zinc salts were used for zinc plating before their introduction for AZIBs.70–73 Water-based electrolytes are generally cost-effective and highly ionic conductive, with the resulting conductivity being similar to or better than that of nonaqueous electrolytes (Fig. 3a). In addition, the high conductivity beneficial for mass transport in aqueous solutions is achieved because of the large solubility of zinc salts. Moreover, aqueous electrolytes often exhibit more facile kinetics of zinc deposition/dissolution than their nonaqueous counterparts. Kundu et al.32 showed that the activation energy for charge transfer at the Zn|Zn2+ interface was almost 10 times lower for an aqueous solution than that for an acetonitrile-based one (Fig. 3b). | ||
| Fig. 3 Electrolytes for Zn-ion batteries. (a) Comparison of conductivity for some aqueous and nonaqueous electrolytes.74–76 (b) Charge transfer activation energy at Zn metal interface in nonaqueous vs. aqueous zinc electrolyte. Reproduced from ref. 32 with permission from the Royal Society of Chemistry. Copyright 2018. (c) Distribution of Zn(II) hydroxo-complexes at infinite dilution at 298 K.77 | ||
Water is a highly polar solvent that can dissolve many inorganic salts and provide unique solvation structures. It is also a very attractive solvent due to its low cost and versatility, making aqueous batteries more competitive than nonaqueous systems. Notwithstanding, its intrinsic high polarity and hydration character are not the only distinguishing features of aqueous electrolytes. Water is a protic solvent with a high autoprotolysis constant:
| 2H2O ⇌ H3O+ + OH− (KW = 1 × 10−14) | (1) |
H3O+(aq) – protonated form of water (hydronium ion, hydrated proton, H+(aq)). In general, water molecules can easily donate, accept, or exchange protons.
Some of the most popular electrolytes in AZIB research are zinc sulfate (ZnSO4) and zinc trifluoromethanesulfonate (zinc triflate, Zn(CF3SO3)2, and Zn(OTf)2), whereas zinc perchlorate, zinc nitrate, and zinc acetate are less often studied in the literature.78 Many of these electrolytes are salts of strong acids, which negates anion protonation. However, the hydrated zinc ion, Znaq2+, is a weak Brønsted acid that interacts with water, leading to mild acidity of the Zn2+-related electrolyte:77
| Zn2+ + 2H2O ⇌ ZnOH+ + H3O+ | (2) |
The reported equilibrium constant for this process is ≈ 10−8–10−9,77 resulting in pH values of ≈3.5–5 for commonly used zinc salt concentrations of 0.1 M to 3 M. Here, Zn2+ denotes the hexacoordinated aqua cation Zn(H2O)62+, which corresponds to the most stable ligand configuration in a water environment, whereas ZnOH+ is also a simplified notation for the coordinated aqua cation (Zn(H2O)5OH+).79 Hereafter, Zn2+ and H+ notation will be used to represent the aquacations throughout the work. The acidity of the zinc ion according to reaction (2) is crucial for AZIBs. Despite the equilibrium concentration of H3O+ (H+) being 4–5 orders of magnitude lower than that of Zn2+ and the equilibrium ZnOH+ concentration being low (Fig. 3c), the fast equilibrium can support the generation of protons if they are consumed in chemical or electrochemical reaction, e.g., during intercalation of H+ into cathode materials. Furthermore, the fast H+ diffusion coefficient in water (9.3 × 10−5 cm2 s−1 for H+vs. 0.7 × 10−5 cm2 s−1 for Zn2+)80 makes H+ an important electroactive species in AZIBs and an active cation for charge storage in the cathode host along with or instead of Zn2+.11,17
The hydrolysis of the Zn2+ cation has another effect at higher pH values. Traditionally, a Pourbaix diagram for the Zn–H2O system depicts the formation of Zn(OH)2 around neutral or slightly basic pH depending on the Zn2+ concentration (Fig. 4a). However, in the presence of anions, the formation of basic zinc salts (BZSs) becomes favorable as the pH of the zinc salt solution increases (Fig. 4b);81–85 converting Zn(NO3)2 to Zn5(OH)8(NO3)2·2H2O,83,85 ZnSO4 to Zn4(OH)6SO4·xH2O,84 ZnCl2 to Zn5(OH)8Cl2·H2O,82 and Zn(OAc)2 to Zn5(OH)8(CH3CO2)2·nH2O.81 The other anions, such as triflate, can also form such basic salts.65,86 Traditionally, many reviews and papers on ZIBs provide a Pourbaix diagram similar to that presented in Fig. 4a, whereas the basic salt area diagram more reasonably represents the actual behavior of the system.
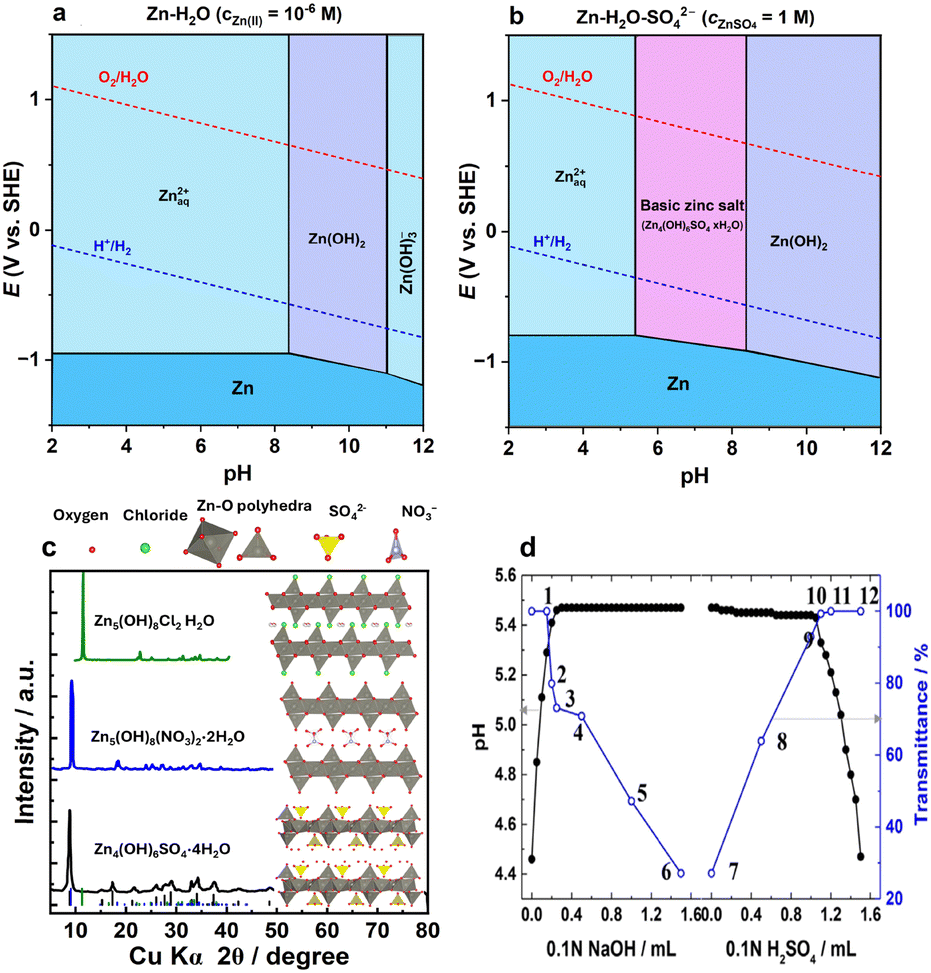 | ||
| Fig. 4 Pourbaix diagrams and formation of basic salts in zinc electrolytes. (a) Pourbaix diagram for the Zn-H2O system without consideration of anions, plotted using data from ref. 87 (b) Pourbaix diagram for the Zn–H2O–SO42− system calculated with respect to basic salt formation, plotted using data from ref. 87 and 88. (c) Typical layered zinc basic salts with their crystal structure and XRD patterns.89–94 (d) Solution pH and transmittance upon NaOH and H2SO4 addition to ZnSO4 solution. Reproduced with permission from ref. 38. Copyright 2016, John Wiley & Sons. | ||
The structure of most basic zinc salts is layered, with anions and water occupying the interlayer space (Fig. 4c). The amount of interlayer water can differ and is usually easily reduced, even upon mild drying. The chemistry of BZSs has been extensively studied aside from the field of batteries, particularly due to their ion exchange abilities.95,96 Some of these basic salts can be used as catalysts97 and for the synthesis of nano-sized oxide particles.81,82,84
The formation of these hydroxy salts is reversible—they can be obtained via direct precipitation by hydroxide addition to Zn2+ solution and, conversely, dissolved by acid treatment (Fig. 4d), as indicated in the equations below:38
| 4Zn2+ + SO42− + 6OH− + nH2O = Zn4(OH)6SO4·nH2O ↓ | (3) |
| Zn4(OH)6SO4·nH2O + 6H+ = 4Zn2+ + SO42− + (n + 6)H2O | (4) |
As the precipitation of the BZS is initiated below pH 7, it can also be rewritten in another form:
| 4Zn2+ + SO42− + (n + 6)H2O = Zn4(OH)6SO4·nH2O + 6H+ | (5) |
The reversible chemistry of BZSs is also observed in AZIBs, where they form and dissolve at the electrode surface during discharge (reduction) and charge (oxidation) of the cathodes, respectively.38,58,65 This phenomenon occurs when cathode materials store a significant amount of protons upon discharge and release them while charging, which is typical for many compounds used in ZIBs.98,99 That is, the electrode serves as a source/sink of protons.
Fig. 5a presents a schematic diagram of the formation of BZSs at the interface between a cathode and an electrolyte. As mentioned above, BZS formation is preferred over Zn(OH)2 deposition; therefore, the Pourbaix diagram should be constructed to account for this. Upon discharge (reduction) of the cathode, protons are consumed. A decrease in the aqueous proton concentration shifts the local pH to a higher value (Fig. 5b and c). When the surface pH of the electrode reaches that of BZS formation, the layered hydroxide salt begins to precipitate on the surface of the cathode depending on the solubility product of the corresponding basic zinc salt in the electrolyte (e.g., reaction (3) for sulfate, Ksp = 2.5 × 10−56). Note that this reaction consumes hydroxide ions; thus, it acts as a buffer, and the pH near the electrode is stabilized at the pH of precipitation (Fig. 5d). Further reduction leads to the growth of the BZS and propagation of the pH gradient further into the solution bulk. Upon charging, the generation of protons (e.g., by framework deprotonation) drives the dissolution of BZS. As long as the salt deposit is present, the pH change is delayed due to the buffering effect of the dissolution reaction (Fig. 5e and eqn (4)). After the complete dissolution of BZS, the pH decreases locally because no hydroxide deposit remains to neutralize protons (Fig. 5f).
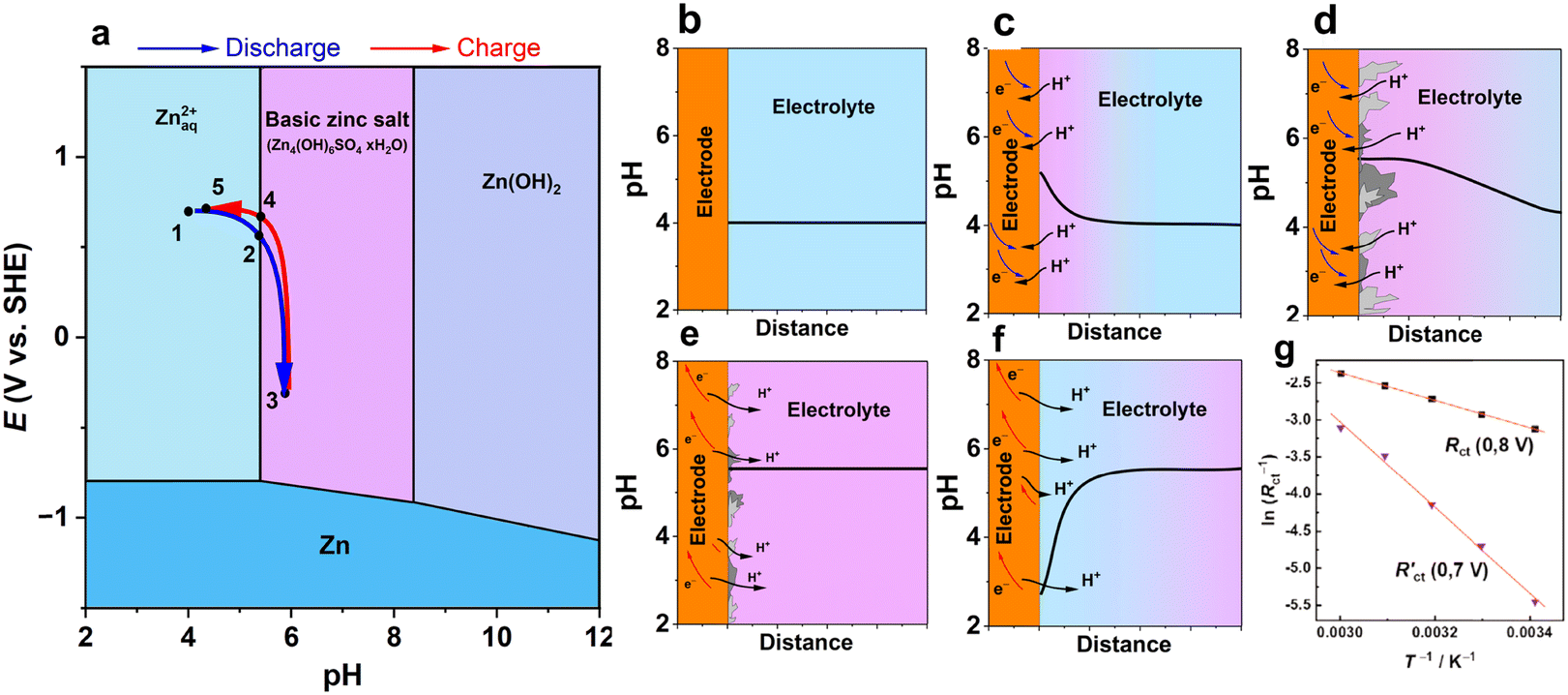 | ||
| Fig. 5 A schematic diagram of pH variation at the interface between the cathode and Zn2+ electrolyte. (a) Pourbaix diagram for Zn-water system built taking into consideration formation of basic zinc sulfate for c(ZnSO4) = 1 M. Blue and red arrows indicate potential-pH coordinates at a cathode while moving through the diagram upon discharge and charge for an H+-intercalating electrode; (b–f) represent pH variation at electrode | electrolyte interface at various states of discharge and formation/dissolution of BZS schematically plotted using systematization and observations of pH changes.58,65,100–102 (g) Arrhenius plot for charge transfer resistance before (0.8 V) and after (0.7 V) precipitation of basic triflate on the NaV3O8 cathode. Reproduced with permission from ref. 65. Copyright 2020, John Wiley & Sons. | ||
The described reversible scenario refers to an isolated electrode without the effect of the zinc anode, such as when recording pH changes in a beaker cell. However, depending on the experimental conditions, various scenarios can be observed, including an extreme pH drop due to BZS delamination.58,102 In addition, for small electrolyte volume cells, e.g., coin and pouch configurations, the continuous Zn-metal corrosion would increase the pH and result in pH changes near the cathode, especially after long conditioning. Thus, a scheme such as that depicted in Fig. 5 aids in understanding the changes on a qualitative level. For a full understanding of a particular system, rigorous calculations should be performed as demonstrated, for instance, for the Zn–MnO2 system.103
The precipitation of BZS on the electrode surface impacts the kinetics (Fig. 5g). The investigation of NaV3O8 in aqueous zinc triflate electrolyte indicated an increase in the interfacial charge-transfer resistance of 10–100 times upon the formation and growth of a basic triflate deposit.65 The corresponding activation energy for charge transfer associated with the grown interphase was measured to be 48 kJ mol−1, which is three times higher than that for charge transfer on the BZS “free” surface.65
The pH of precipitation depends on the solubility product constant and usually lies between 5 and 6 (Table 1). An exception is zinc acetate electrolyte, which deviates from the trend due to the basic properties of the acetate anion (hydrolysis).
| Basic salt | K s | Precipitation pH in 1 M Zn(II) salt | Examples of BZS precipitates on cathodes in zinc batteries | |
|---|---|---|---|---|
| Calculateda | Experimental | |||
| a Calculated in this review using Ks values and activity coefficients.125–127 | ||||
| Zn4(OH)6SO4·xH2O | 2.5 × 10−56 (ref. 88) | 5.31 | 5.47 (ref. 38), 5.74 (ref. 102) | MnO2,104–106 ZnMn2O4,107 NaV3O8,108,109 V2C MXene,110 AxV2O5,111,112 VO2113,114 |
| Zn5(OH)8Cl2·xH2O | 8.2 × 10−76 (ref. 115) | 5.04 | — | AxV2O5,116,117 Zn–air,118 VOx119 |
| Zn5(OH)8(NO3)2·xH2O | 1.11 × 10−76 (ref. 83) | 4.93 | — | — |
| Zn5(OH)8(CH3CO2)2·xH2O | — | — | ≈7 (ref. 120) | MnO2,106 Thionin,121 Zn-air122 |
| Zn hydroxy triflate | — | — | 5.2 (ref. 102), (2 M) | AxV3O8,65,100 Na3V2(PO4)2F3,86 NH4V4O10,123 VO2,124 MnO2,106 |
Most reports on basic salt precipitation cover the formation of Zn4(OH)6SO4·xH2O and basic triflate, and the different types of BZSs are summarized in Table 1. Most of the examples correspond to cathodes composed of vanadium and manganese oxides, which tend to accept H+ upon reduction. Thus, a question arises regarding the extent to which such materials can accommodate Zn2+ (and not H+) in the structure. This issue will be discussed in Section 3.
In summary, aqueous solutions of Zn(II) salts provide unique ionic equilibria. The acidity of Zn(H2O)62+ and the formation of BZSs correspond to a mildly acidic pH range, neither too low to cause strong acid etching of the materials nor too high to quickly passivate Zn, as occurs in alkaline electrolytes. More importantly, the formation of BZSs is fast and reversible. The hydrolysis of Zn2+ provides a stable supply of protons at the cathode during battery discharge while the deposited BZS neutralizes the protons during the charging process. At the same time, its deposition causes an increase in the surface impedance of both electrodes, which should always be considered whether studying the anode or cathode host.
2.2. Corrosion and deposition in Zn2+ aqueous solutions
According to the Zn–H2O–Xn− (Xn−: anion) Pourbaix diagram (Fig. 4b), zinc metal (Zn0) is thermodynamically unstable in contact with aqueous electrolyte at any pH. Depending on the pH, the products of the Zn reaction would be Zn2+, zinc basic salts, or zinc hydroxide:| Zn + 2H+ → Zn2+ + H2 | (6) |
| 3Zn + Znaq2+ + SO4(aq)2− + (6 + n)H2O → Zn4(OH)6SO4·nH2O + 3H2 | (7) |
| Zn + 2H2O → Zn(OH)2 + H2 | (8) |
At a longer timescale, the formation of zinc oxide is possible as a result of Zn(OH)2 aging:
| Zn(OH)2 → ZnO + H2O | (9) |
As a result, three important consequences for the use of zinc in Zn-metal batteries arise. First, prolonged contact of Zn with the electrolyte leads to a rise in pH from the initial value of 3–4 to 5–6, which is the pH of BZS precipitation (Table 1 and Fig. 4 and 6a). This finding implies that extended aging increases the pH of the electrolyte, which is higher than that of the fresh Zn2+ electrolyte, which spontaneously affects the surface state of the metallic Zn anode. Second, the abovementioned reactions (6)–(8) produce H2 gas, threatening the safety of cells (Fig. 6b). Unfortunately, the formed H2 bubbles adhere to the surface of the Zn anode, which in turn deteriorates cell performance caused by the uneven deposition of metallic Zn0 during reduction. Further accumulation of the H2 gas induces swelling or explosion of the cell. Third, the basic zinc salts and zinc hydroxide deposit as an inhomogeneous and loose film, inducing the formation of corrosion pits on the surface of the Zn anode.58,128 A loose hydroxide deposit does not prevent corrosion, and its inhomogeneous formation predisposes zinc to unequal deposition. Thus, the zinc surface gradually corrodes, increasing the surface impedance (Fig. 6c) while simultaneously leading to the accumulation of BZS deposits on the surface.128
 | ||
| Fig. 6 Zn corrosion and deposition in aqueous Zn-electrolyte. (a) Pourbaix diagram for Zn-water system built considering formation of Zn4(OH)6SO4·nH2O. (b) Hydrogen evolution during rest time in a pouch cell (3 M ZnSO4). Reproduced with permission from ref. 128. Copyright 2020, Elsevier. (c) Impedance spectra of Zn|Zn symmetrical cell by aging (3 M ZnSO4). Reproduced with permission from ref. 128. Copyright 2020, Elsevier. (d) Scanning electrochemical microscopy feedback images of local current upon Zn deposition. Reproduced with permission from ref. 129. Copyright 2023, John Wiley & Sons. (e) Dendrite formation upon Zn electrodeposition on bare Zn surface and moderate deposition (f) on the gold sputtered surface. Reproduced with permission from ref. 130. Copyright 2023, John Wiley & Sons. | ||
Reversible and homogeneous metal deposition is a key requirement for any battery with a metal electrode. Achieving these conditions has been a long-standing problem for zinc-metal batteries and the main obstacle to realizing commercial zinc-metal batteries. The main issue here is the formation of zinc dendrites, which can grow through a separator, causing an inner short-circuit and cell failure (Fig. 6d and e).
Although the dendrite problem is well known for non-aqueous batteries such as Li-metal batteries131,132 or Na-metal batteries,133,134 the presence of water significantly accelerates Zn dendrite formation through the aforementioned corrosion processes (reactions (6)–(9)). Corrosion during conditioning or a rest period creates an inhomogeneous surface. As a result, zinc is deposited at preferable sites, leading to inhomogeneous current distribution and a rough distribution of the grown metal (Fig. 6d and e). Upon subsequent oxidation, some of the grown zinc tips do not dissolve and act as new preferable sites for zinc deposition in the following cycles. The preferable deposition on tips, where the local potential stimulates faster metal growth, leads to dendrite growth and, after some time, to an inner short circuit. Furthermore, the freshly deposited zinc surface is also exposed to corrosion, which accelerates the rate of hydrogen evolution, dendrite formation, and, eventually, cell failure. These side reactions are sometimes referred to in the literature as the “vicious cycle”61 or “domino effect”.62 Investigation of zinc metal is interrelated with precise hydrogen quantification and understanding of pH changes, the hydrogen evolution reaction rate, and zinc deposition/dissolution. Therefore, further work is needed to minimize or eliminate hydrogen evolution and suppress dendrite growth.
The effect of pH and H2 evolution on zinc-metal electrochemistry and zinc-battery performance was considered in recent studies.98,135–141 Yang et al.142 investigated the relationship between hydrogen evolution and the current efficiency for Zn deposition. Fig. 7a shows the improvement in the average current efficiency (ACE) from 76% to 99% by increasing the current density from 0.033 to 20 mA cm−2 at a limited areal capacity of 1.0 mA h cm−2. Correspondingly, the hydrogen evolution reaction (HER) efficiency was reduced at higher current densities, resulting in a smooth Zn surface after zinc deposition.142
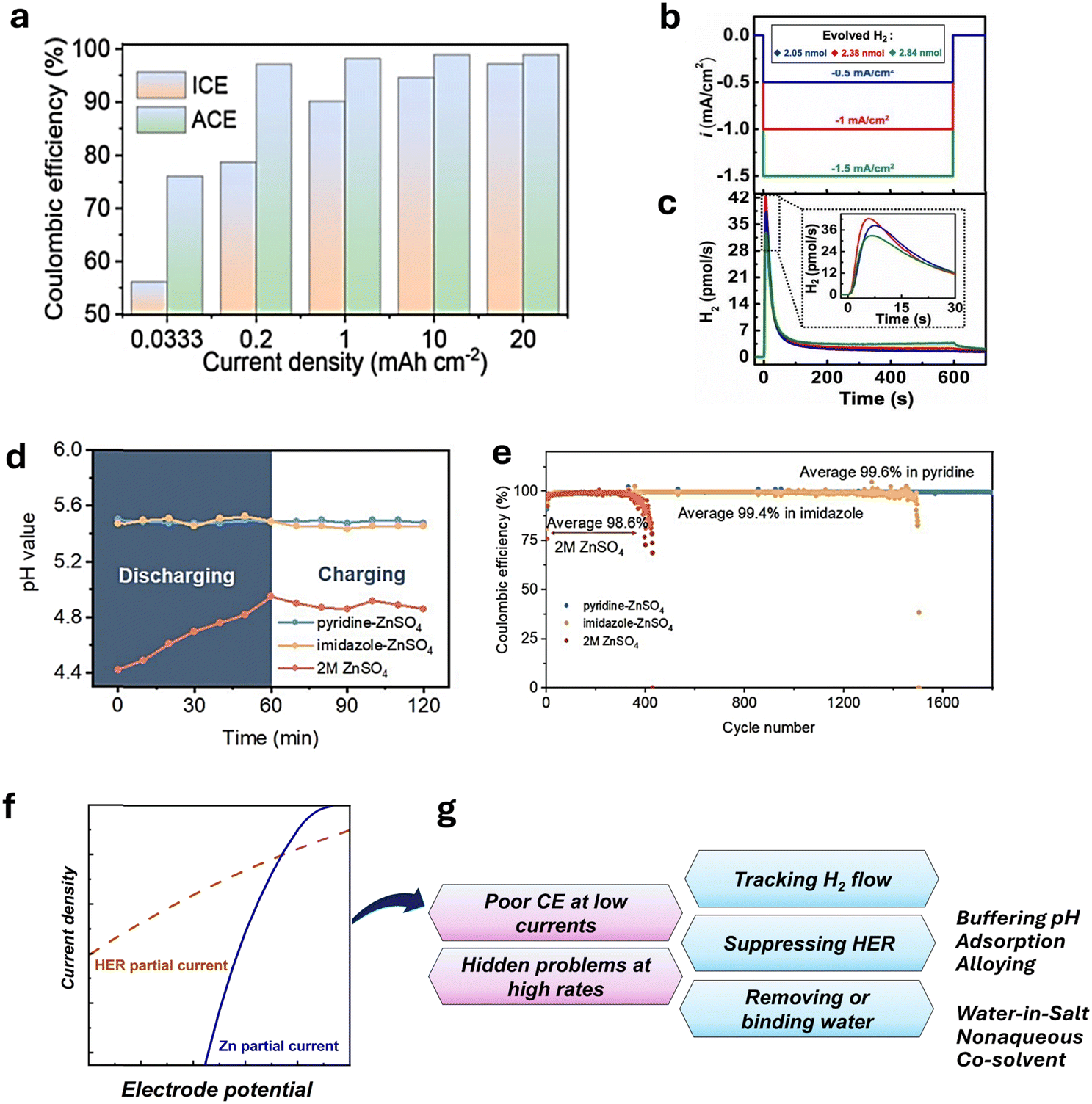 | ||
| Fig. 7 Efficiency of Zn deposition and HER. (a) Zn plating/stripping efficiency depending on current density. Reproduced from ref. 142 with permission from the Royal Society of Chemistry. Copyright 2024. (b) Galvanostatic Zn deposition and (c), corresponding hydrogen evolution reaction (HER) rate. Reproduced with permission from ref. 140. Copyright 2024, John Wiley & Sons. (d) pH in Zn anode proximity by time in buffered and blank 2 M ZnSO4 and (e), CE for Zn/Cu in 2 M buffered and blank 2 M ZnSO4 Reproduced with permission from ref. 139. Copyright 2023, John Wiley & Sons. (f) Schematic ratio of Zn deposition and HER partial currents. (g) Highlighted problems and issues of the Zn anode. | ||
In a recent study, Roy et al.140 utilized in situ electrochemical mass spectrometry (ECMS) for tracking hydrogen flux. As shown in Fig. 7b and c, an increase in the current density magnitude facilitates H2 evolution. In the set of experiments, current densities of −0.5 mA cm−2, −1.0 mA cm−2, and −1.5 mA cm−2 lead to the generation of 2.05, 2.38, and 2.84 nmol H2 over 600 s. At the same time, the HER efficiency dropped from 0.62% to 0.31%, indicating that zinc deposition intensifies faster than the HER rate under negative polarization. This finding agrees with the results of Yang et al.142 who reported a low CE at low rates (Fig. 7a). Additionally, the hydrogen evolution for 3 h of resting period resulted in 5.16 nmol H2, which cannot be neglected when compared with the gas generated during deposition.
The abovementioned studies strongly highlight the importance of zinc stability during long resting periods and its deposition efficiency under slow-charge conditions. One way to address the HER problem and control zinc deposition is to add pH buffer to the electrolyte, shifting and stabilizing the pH at more basic values than a pristine electrolyte. Such an approach has been used in various studies,136,139,143–145 often by adding amines to Zn2+ aqueous electrolyte. Fig. 7d shows the effect of pyridine and imidazole additives on pH stabilization during zinc deposition and the HER rate by pH upshifting.139 As a result, the corresponding coulombic efficiency (CE) and the cyclability of zinc were significantly enhanced by the effect of additives (Fig. 7e). It is worth mentioning that the ability of external buffers to upshift the pH is limited by the zinc salt used; namely, the resulting pH can be stabilized by forming BZS (Table 1) as the pH increases to the precipitation condition of BZS during the reaction. In addition, some functional molecule additives can suppress the HER through adsorption on the Zn surface or by altering the solvation structure.141 Hence, a critical and comprehensive evaluation of how strongly adsorption, solvation, or buffering can inhibit the HER is critical in designing additives for AZIB electrolytes. Tribbia et al.135 used an indium-based substrate to achieve dendrite-free zinc deposition. The measured hydrogen flux was found to be much lower for the indium substrate compared to that for the bare zinc, suggesting a correlation among hydrogen evolution, Zn deposition efficiency, and cyclability.
Although buffering, adsorptive additives, and alloying show promising results in enhancing Zn-metal performance,130,135,139,141 the slow H2 evolution and Zn corrosion appear to be inevitable in common water-based electrolytes.135,140,141 These findings imply that additives or surface-modification approaches can kinetically delay the HER, whereas, more importantly, the thermodynamic instability remains unresolved. Overcoming this issue is critical for practical-scale applications if AZIBs with high areal capacities (>2 mA h cm−2) are subjected to long resting periods (hours, days, and weeks).59 Switching the electrolyte to a nonaqueous system may be a solution to the problem; however, it is challenging to adopt nonaqueous Zn electrolytes due to their inferior salt solubilities, conductivities, and reaction kinetics, as considered in Fig. 3. More importantly, it should be considered that there are much fewer opportunities to find suitable cathode materials that warrant stable electrode performance in nonaqueous electrolyte.
Taking advantage of the benefits of both aqueous and nonaqueous systems, recent studies have explored the mixing of water and organic solvents over the last few years.35,36,146,147 In such systems, the water activity is suppressed by interaction with an organic co-solvent. The water hence cannot cause considerable corrosion and HER, but there is a sufficient amount of water to enable fast interfacial charge transfer for zinc ions. For example, a mixture of dimethyl carbonate, zinc triflate, and water enhances the current efficiency and cyclability, which eventually prevents the formation of BZSs during cycling.35 Nazar and colleagues36 demonstrated efficient Zn plating/stripping in water-sulfolane electrolytes at an areal capacity of 4 mA h cm−2 and long-term full-cell performance in combination with vanadium oxide and iodine-based positive electrodes. In addition, highly concentrated solutions or even water-in-salt electrolytes are suitable for enhancing the current efficiency for zinc deposition/stripping.148–150 Besides directly controlling water or proton activity, zinc deposition/stripping can be improved using various coatings, functional separators, and other approaches.61,66,67
Thus, there is no doubt that various approaches allow to manage the HER problem in zinc batteries in a certain way. Let's now consider how these approaches work on the micro level; for this, we need to consider kinetics, interfacial, and solvation structures.
As the HER on Zn is thermodynamically barely avoidable, even at low water content, most methods focus on slowing down the reaction kinetics to inhibit the process. The HER usually proceeds via Volmer–Heyrovsky or Volmer–Tafel mechanisms. In both mechanisms, the first step involves the reduction of a solvated proton, which generates a hydrogen adatom on the active surface site. In the Volmer–Heyrovsky mechanism, this is followed by the reduction of the second H+ at the same site. In contrast, the Volmer–Tafel mechanism involves the recombination of two adsorbed hydrogen adatoms, resulting in the formation of a hydrogen molecule (H2).151 During HER, hydrogen adsorption affects the reaction kinetics. Fig. 8a shows the dependency of HER exchange current density on metal–hydrogen bond energy.152 The plot has a volcano-shape following the Sabatier principle, which states that an intermediate in heterogeneous catalysis should be bound neither too weekly nor too strongly for facile reaction. In contrast to the catalysis field, zinc metal battery research should focus on HER inhibition. As seen in Fig. 8a, zinc lies in a low HER activity area, which, in fact, allows usage of mildly aqueous electrolytes. Following this principle, alloying or covering zinc electrodes with HER inhibiting metals would slow down hydrogen evolution. For instance, an amalgamation of zinc was shown to suppress the HER on the zinc surface153 as mercury is historically known for its extremely high HER overpotential. Alloys and coating with other metals that exhibit low affinity for hydrogen have been shown to reduce corrosion such as the indium,135 indium–gallium–zinc alloy,154 or lead coating.155 A recent study also shows promising behavior of zinc–bismuth alloy, while usage of nickel results in an opposite effect, as nickel enhances HER catalysis (Fig. 8b).156 Thus, the application of the electrocatalysis principles through zinc alloying appears to be promising in addressing aqueous zinc corrosion.
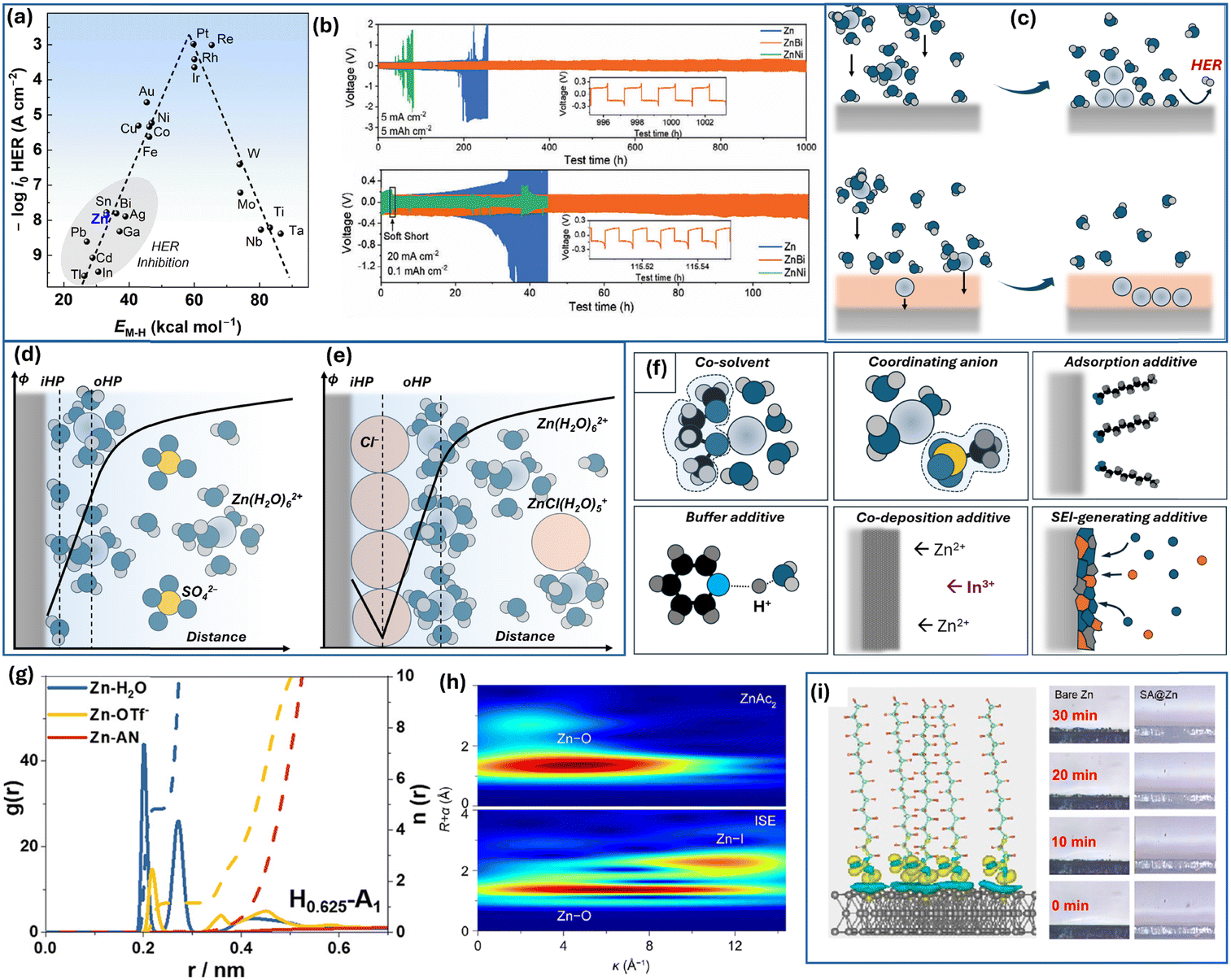 | ||
| Fig. 8 Interface of Zn electrode and Zn2+ solvation. (a) Volcano plot of HER activity on metal electrodes (data are from ref. 152). (b) Cyclic performance of Zn, Zn–Bi, and Zn–Ni alloys in symmetrical cells with Zn2+-electrolyte (Adapted with permission from ref. 156. Copyright 2024, John Wiley & Sons). (c) Schematic representation of “pre-desolvation” layer on the zinc surface. Scheme of the double layer structure for zinc electrode in contact with (d) zinc sulfate electrolyte and (e) zinc chloride electrolyte; based on ref. 157. (f) Various additives and modifications to electrolyte solution by their action principle. (g) The radial distribution function for zinc triflate in a H2O–acetonitrile mixture. Adapted with permission from ref. 158, Copyright 2022, Elsevier). (h) Wavelet transform images of the EXAFS spectra for Zn(OAc)2 and Zn(OAc)2 + NH4I mixed electrolyte (Adapted with permission from ref. 159, Copyright 2022, American Chemical Society). (i) Differential charge density for stearate adsorbed on the zinc surface and morphology of deposition at 10 mA cm−2 of bare and modified surface (Adapted with permission from ref. 160, Copyright 2023, American Chemical Society). | ||
Even if slowed down, the prolonged contact of zinc atoms with H2O molecules would lead to hydrogen evolution and corrosion. Moreover, upon deposition, hydrated zinc ions, Zn(H2O)62+, would release water molecules on fresh Zn surface upon desolvation.161 This, in combination with zinc ion depletion and negative polarization, would result in an enhanced HER rate. Thus, various approaches are applied to minimize local water concentration at the zinc interface or to remove direct Zn|H2O contact.
One way of preventing direct contact of zinc with H2O molecules is the design of a zinc-ion conductor layer, which accepts zinc ions upon desolvation and assists in their transport to the growing Zn interface. This process helps minimize the area of zinc exposure to the electrolyte and, ideally, promotes a uniform zinc ion flux across the electrode surface (see Fig. 8c). For instance, thin layers of ZnF2 have been shown to exhibit the desired ionic conductivity and transport properties.162,163 The ZnF2 layer, grown through HF-gas transport reaction by decomposition of NH4F, was demonstrated to have an interstitial diffusion mechanism allowing transport to the zinc surface.163 Having an open structure, metal hexacyanoferrates can act as suitable zinc ion transport layers.164–166 For example, a Zn3[Fe(CN)6]2 (ZnHCF)-modified separator can enhance cyclic stability.166 It is likely, that the ZnHCF-layer moderated the Zn2+ flux by increasing the zinc transference number. Various metal–organic frameworks and covalent frameworks are other open-channel structures suitable for such layer design.167–169 In recent work, a β-ketoenamine-linked fluorinated covalent organic framework was used to create a crystalline 2D layer on Zn.169 The layer contained 1D fluorinated nanochannels, which facilitated hopping for zinc ions while preventing water infiltration, as suggested by the observed lower HER currents and higher Tafel slope. Various polymer-based films were demonstrated to regulate Zn deposition.170–172 Interestingly, the immobilization of anions in the polyelectrolyte membrane allows increasing the Zn2+ transference number to 0.96, making the membrane layer almost exclusively Zn2+-conductive and enabling uniform Zn deposition.171
Overall, designing Zn2+-conductive layers is a viable approach to suppress corrosion and moderate deposition of zinc. It is crucial to analyze the mechanisms and estimate the ionic and electronic conductivity of these layers, as well as assess their structural integrity after prolonged cycling and susceptibility to cracks and zinc overgrowth.
Zinc deposition and water reduction are also sensitive to anion adsorption and double-layer structure. It is well-reported that ZnCl2 allows more stable Zn stripping–deposition than ZnSO4 or Zn(ClO4)2 and generates lower amount of passive films.173 Although it can be ascribed to the solvation shell structure as Cl− tends to substitute H2O in the zinc coordination environment at high concentration, chloride ions are known for specific adsorption. In a recent study by Lai et al., measurements of surface tension of a mercury drop in Zn(II) electrolytes and related analysis allowed recreating surface excesses of ions in the vicinity of the electrode.157 The chloride showed specific adsorption behavior with the potential of zero charge being negatively shifted and dependent on electrolyte concentration compared to ZnSO4 behavior. Chloride ions tend to occupy the inner Helmholtz plane, causing the attraction of Zn2+ (Fig. 8e). Thus, the interfacial vicinity is enriched with both Zn2+ and Cl− in 1 M ZnCl2 compared to 1 M ZnSO4 (Fig. 8d and e). Thus, such double layer structure and higher local concentration of Zn2+ likely contribute to better Zn2+/Zn kinetics.157
The deposition efficiency of zinc, the hydrogen evolution reaction, the double-layer structure, and the solvation shell of zinc ions are all closely interconnected. Consequently, various additives and electrolyte modifiers are actively being investigated (Fig. 8f). An effective approach involves the use of organic co-solvent additives. These co-solvents interact with water, modulating the hydrogen bonding network, weakening the bond between Zn2+ and H2O, or altering the Zn2+ solvation shell by incorporating co-solvent molecules into the coordination sphere.174 For instance, dimethoxyethane (DME) was shown to modify the solvation shell in a H2O–DME mixed electrolyte.175 Interestingly, the introduction of DME into the coordination environment also induced coordination between Zn2+ and CF3SO3−. This likely altered SEI composition and structure creating a ZnF2–ZnS-rich layer that contributed to more reversible behavior. Similar to DME, triethylene glycol (TEG) co-solvent acts as a bidentate chelating agent, reducing the number of H2O in the first coordination sphere from 6 to 3 and promoting the incorporation of triflate anion into the coordination shell.176 The SEI derived from this electrolyte was found to exhibit an organic–inorganic bilayer-like structure with a ZnF2-rich inner layer. Zhao et al. demonstrated that the acetonitrile co-solvent modifies the hydrogen bond network and promotes the substitution of water by triflate anion in the solvation shell (Fig. 8g).158 Notably, co-solvent does not necessarily participate in the first solvation shell. 2-Propanol, as a relatively weak ligand contributes to the outer solvation shell. Nevertheless, this modification facilitates stable zinc deposition with a preferential (101) orientation.177 A recent study demonstrated that the 2,2,2-trifluoroethanol (TFEA) co-solvent weakens the tip effect of zinc deposition and effectively suppresses the dendrite growth. While TFEA does not participate in the inner solvation shell, it has been shown to disrupt the hydrogen bonding network, facilitating the incorporation of triflate anions into the inner coordination shell.178 Overall, the co-solvent strategy appears to be one of the most promising approaches in ZIB development. In addition to its direct effects, such as reducing water activity and decreasing the number of water molecules in the Zn2+ coordination shell, co-solvents typically influence the SEI structure and, in many cases, mitigate the dissolution of cathode materials. Consequently, they contribute to enhanced cycling stability, functioning as multifunctional electrolyte components.
Similar to how the solvent affects the solvation shell, electrolyte can be modified by using different salts or anions. The effect of chloride on adsorption and coordination has been already mentioned above. Similarly, other halogen ions coordinate with zinc ions at high concentrations forming complexes such as ZnI(H2O)5+ in a mixed zinc acetate–ammonium iodide electrolyte (Fig. 8h).159 The addition of sodium tartrate as a dual-functional additive for absorption and solvation shell modification was shown to benefit (002) zinc growth and enable long-term stability.179 The elimination of “free” water and decrease in the amount of solvated water is often achieved by WiS electrolytes, which at the same time becomes less conductive and more viscous.148–150 To overcome this issue, a combination of cosmotropic and chaotropic anions could be effective in maintaining a sufficiently low water concentration while providing better conductivity and allowing long-term Zn deposition–stripping.180 The use of coordinating anions like acetate181 also allows stable cycling with zinc concentrations being relatively low compared to those of WiS electrolytes. In addition, the combination of Zn2+ ions and acetate (CH3COO−) ions in the electrolyte helps buffer the acidity of Zn2+ and manage the high viscosity of concentrated ZnCl2.182
Specific adsorption of some anions opens one more way to address Zn corrosion and side reactions. As verified by spectroscopic techniques and DFT calculations, the adsorption of dodecyl benzene sulfonate leads to an H2O-poor double-layer structure.183 It was also argued that such adsorbate contributed to the desolvation of zinc ions, which in turn allowed a long lifespan even in aqueous ZnSO4. The water-repelling adsorbate layers were created in different studies by utilizing trace amounts of perfluorooctanoic acid,184 phytic acid anions,185 amino acids,186 and fatty acids such as stearic acid160 (Fig. 8i). Thus, the adsorption of individual ions and solvated structures such as Zn(H2O)62+ or Zn(H2O)nCF3SO3− (ref. 187) as well as local hydrogen bond dynamics161 strongly affects the electrode kinetics of zinc deposition and HER.
Other electrolyte additives and modification strategies, including buffer,143–145 co-deposition/alloying ions,188–190 and functional species promoting in situ SEI layer formation,191–193 are of high importance for suppressing side reactions on zinc.
Overall, the study on additives holds much promise for zinc-ion and zinc-metal batteries. As evident from the examples above, rare additives affect only one parameter. More often, simultaneous changes in various properties are observed: a co-solvent changing both the solvation shell and the growing SEI, along with an anion capable of adsorbing on the zinc surface and coordinating with Zn2+ ions. Such multifunctionality introduces problems of “correlation vs. causation” in finding the key factors responsible for performance enhancement. Therefore, careful research of the electrolyte and interface structure must be performed by using a combination of experimental (Raman, FTIR, EXAFS, NMR, and XPS) and theoretical (molecular dynamics and DFT-calculations) methods.
Considering all these points, mildly acidic aqueous Zn2+ electrolytes have enabled the rapid growth of ZIB research but appear to be insufficient for long-term performance, especially with high, commercial-related current densities and extensive zinc utilization. Hence, further development requires the strict suppression of the side reactions and control of water activity. For this reason, the application of mixed electrolytes, water-in-salt electrolytes, and functional additives and coatings is promising. At the same time, research on Zn anodes and Zn deposition should be conducted critically, with an appropriate estimation of the deposition/stripping CE (e.g., by using the reservoir half-cell method194,195), without avoiding low current densities and resting times, and providing valid tracking of H2 evolution.
3. Protons, water, and cathodes
According to the literature survey, the main topics covered for AZIBs are the crystal structure, polymorphs, defects, and electrode compositions, whereas the role of water, pH, and protolytic reaction have often been overlooked in explaining the reaction mechanism of AZIBs. In addition, the electrode performance and synthesis-based studies usually do not highlight structural investigation or activation phenomena after multiple charge–discharge cycles. Recent studies have revealed that charge compensation by Zn2+ insertion and/or extraction into/out of the host structure is not the main reaction responsible for the reversible capacity of many host materials, although Zn2+ is the main component of the aqueous electrolyte. That is, many materials are unable to store Zn2+ at a high extent and instead serve as hosts for protons provided by Zn2+–H2O ionic equilibria. In addition, the high reactivity of water and its ability to accept and donate protons allows various dissolution, redeposition, and amorphization reactions, making AZIBs distinct from nonaqueous batteries.3.1. Scenarios of Zn2+ and H+ intercalation
Various types of active materials act as intercalation hosts when (dis)charged in aqueous zinc electrolytes (Fig. 9). The reversible X-ray diffraction (XRD) peak shift is often observed as a clear indicator of the intercalation reaction. As the aqueous zinc electrolyte can donate protons (reactions (2) and (5)), many materials tend to adopt H+ ions rather than Zn2+ ions when discharged or H+/Zn2+ co-intercalation can occur.11,18 BZS precipitation usually serves as an indicator of H+ intercalation and is typically observed for manganese-oxide- and vanadium-oxide-based materials as BZS peaks emerge and vanish in a cyclic manner (Fig. 9b). In addition, intercalation of Zn2+ can proceed in different ways, depending on the participation of solvated water in intercalation (Fig. 10a and b). The domination of a particular mechanism is strongly affected by the type and structure of the electrode materials.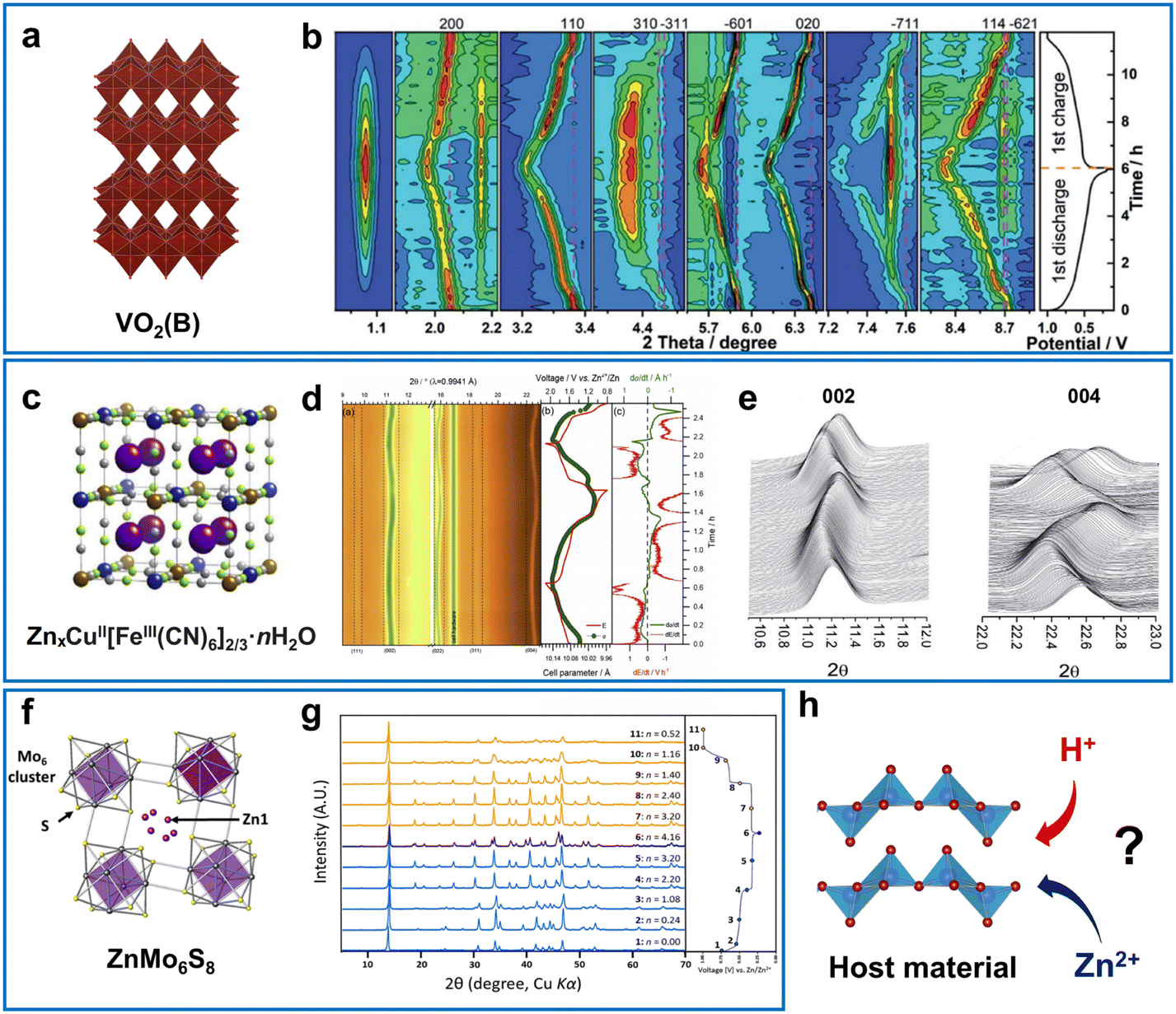 | ||
| Fig. 9 Examples of intercalation-type host materials in aqueous Zn-metal batteries. (a) Crystal structure and (b), operando XRD for VO2(B) in aqueous 1 M ZnSO4. Reproduced from ref. 196 with permission from the Royal Society of Chemistry. Copyright 2020. (c) Crystal structure and (d) and (e) operando XRD for copper hexacyanoferrate in aqueous 1 M ZnSO4 Reproduced with permission from ref. 197. Copyright 2017, Elsevier. (f) Crystal structure for ZnMo6S8 and (g), corresponding ex situ XRD after (dis)charges in 0.1 M ZnSO4. Adapted with permission from ref. 198. Copyright 2016, American Chemical Society. (h) The ambiguity of guest ions for aqueous ZIBs. | ||
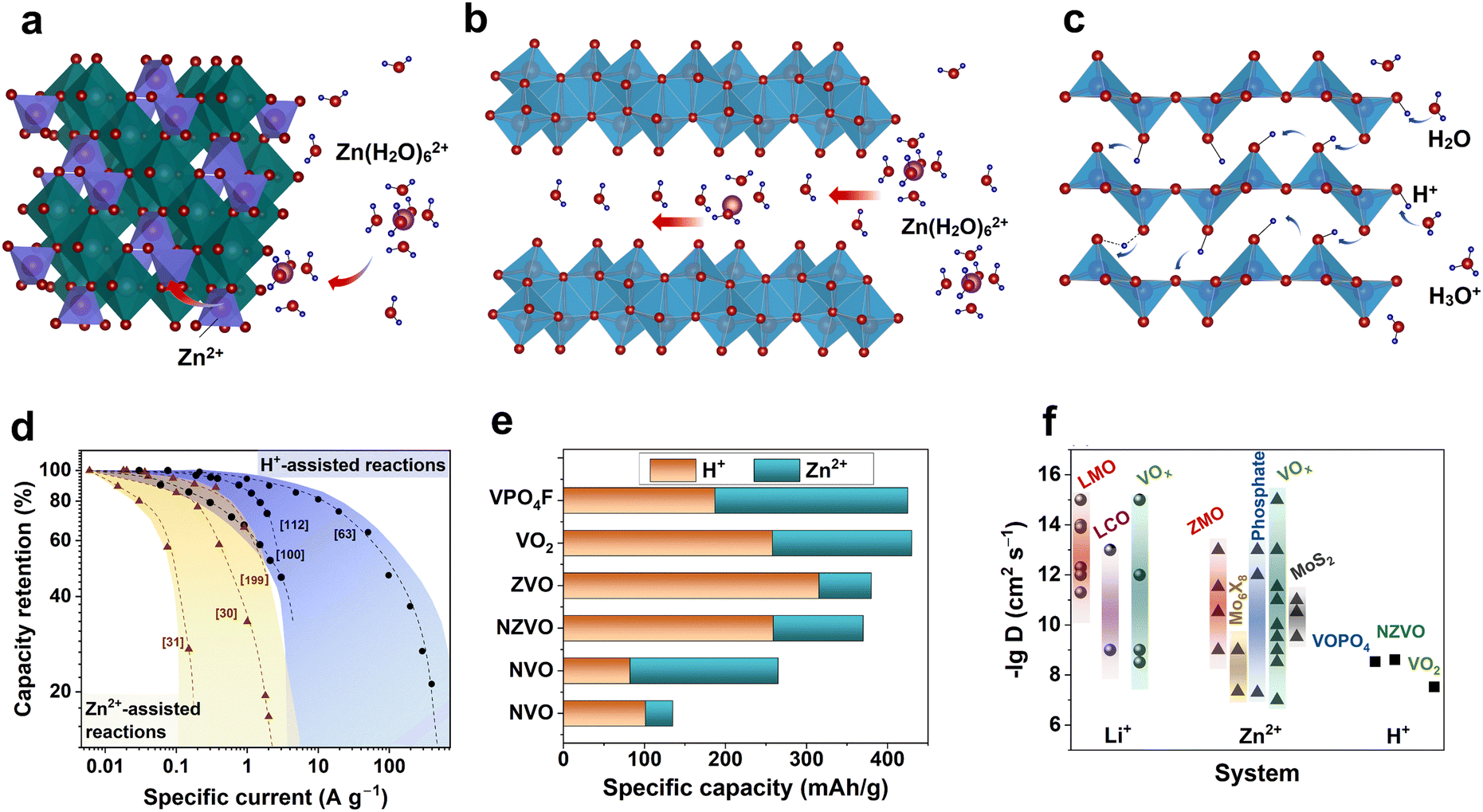 | ||
| Fig. 10 Intercalation of Zn2+ and H+ into host materials. Schematic illustrations of (a) Zn2+ intercalation accompanied by ion dehydration. (b) Zn2+ intercalation with participation of interlayered water acting as “charge screen”. (c) H+ insertion into metal oxide. (d) Rate capability of various systems relying on proton (Zn-metal aqueous battery) and zinc ion insertion (nonaqueous ZIBs).30,31,63,100,112,199 (e) Contribution of Zn2+ and H+ to specific capacity achieved from various materials.25,63,108,200–203 ZVO – zinc vanadium oxide, NZVO – sodium zinc vanadium oxide, NVO – sodium vanadium oxide. (f) Comparison of reported diffusion coefficients for Li+,204–214 Zn2+,107,215–234 and H+ (ref. 63, 235 and 236) for various cathode materials. LMO – lithium manganese oxide, LixMn2O4, LCO – lithium cobalt, LixCoO2, and ZMO – zinc manganese oxide, ZnxMn2O4. | ||
Molybdenum chalcogenides Mo6X8 (X = S, Se) known as Chevrel phases are well-known hosts for different ions due to their unique crystal structures. Their ability for electrochemical intercalation of zinc ions was demonstrated long before the resurgence of AZIB research.25 It is known that their electrochemistry usually includes the stepwise formation of ZnMo6X8 and Zn2Mo6X8 near 0.5 V and 0.3 V vs. Zn2+/Zn, respectively for Mo6S8.198,237 The limited specific capacity and low Zn intercalation potential have resulted in poor interest in practical applications; however, their unique structure makes them interesting materials for mechanistic studies.
Several studies have demonstrated that spinel oxides such as Zn–Al–Co30 and Zn–Ni–Mn–Co199 oxides serve as Zn intercalation cathodes in non-aqueous electrolytes. However, presumably due to the low current efficiency and the high potential of Co4+/Co3+ and Ni4+/Ni3+ redox, such compounds have not been examined in an aqueous environment. For aqueous systems, a highly stable battery based on Zn2+ intercalation has been reported for the cation-deficient ZnMn2O4 spinel.215 However, good capacity was observed in nanocomposites with carbon, indicating the importance of the material preparation method, composition, and vacancy character for effective zinc-intercalation kinetics.
Metal hexacyanoferrates (MHCFs), often referred to as Prussian blue analogues, usually have a cubic or rhombohedral open structure with channels suitable for accepting multivalent ions.238,239 Unlike metal oxides, the XRD patterns of MHCFs do not show feature of basic salt deposits (Fig. 9c–e), thus indicating the dominant Zn2+ insertion chemistry rather than H+ storage. The specific capacity of MHCFs is usually below 100 mA h g−1, activated by the iron redox, whereas the electrode performance is not so attractive due to the small capacity in Zn cells.238–243
Another class of materials with reported ability towards Zn2+ (de)intercalation is transition-metal phosphates, including NASICON-type vanadium phosphates.244 The reversible intercalation chemistry of Zn2+ has been reported for LiV2(PO4)3, which possesses high power density and high performance at low temperature.245 The ability for Zn storage is not limited to vanadium phosphates only; a recent study reported that Zn2Fe(PO4)2 undergoes reversible Zn2+ insertion/extraction in a nonaqueous electrolyte.246 However, notably, such compounds, especially V-containing ones, tend to decompose in a water environment, resulting in H+-affinitive vanadium oxides as decomposition products.247 Conventional Zn2+-ion intercalation is possible for various compounds mentioned above; however, it is not what generally allows facile kinetics, which makes research on aqueous ZIBs so popular. Proton insertion, the interaction of crystal water with zinc ions, and water co-intercalation are processes that enable high-rate performance and better kinetics in AZIBs.
However, introducing interlayer organic molecules, especially redox-active molecules with basic functional groups such as –NH2 results in high affinity for the H+-assisted redox process, and, hence, H+ rather than Zn2+ insertion for charge storage.
In addition to the vanadium oxide family, polyanionic phosphates can accept H+ instead of Zn2+. For the Na3V2(PO4)2F electrode, H+ works as a guest ion rather that Zn2+.86 Similarly, reduction also proceeded via the co-insertion of Zn2+ and H+ for VPO4F.202 This co-insertion reaction was also available in Na3V2(PO4)2O1.6F1.4 in a water-in-bisalt (WIBS) electrolyte.266 These studies indicate that H+ insertion occurs in V-containing frameworks among phosphate-based compounds and appears to be linked to the formation of V–O–H bonds.
Further understanding of the charge-storage mechanism in AZIBs requires identification of the inserted number of active ions and determination of how they contribute to the capacity. In other words, what amount of Zn2+ and H+ is inserted into the host materials and how can these amounts be quantified? Several groups have attempted to quantify the H+ and Zn2+ insertion contribution to the overall capacity (Fig. 10e).63,108,200–203 The studies were performed using vanadium-based oxide and phosphate materials. Usually, the Zn content is measured in discharged samples after washing the byproduct, whereas the remaining capacity is attributed to the inserted H+. In work on VOPO4F, in addition to conventional methods, a prompt-gamma neutron activation analysis was used as neutrons interact directly with a nucleus and can give the amount of inserted H+.202 According to the literature, the H+ quantity in most of the studied vanadium compounds is significant, exceeding that of the inserted Zn2+ ions. That allows saying that such examples are rather hybrid-ion than Zn-ion batteries.
The reported Zn2+ diffusion coefficients are distributed over a broad range from 10−15 to 10−7 cm2 s−1 (Fig. 10f). Note that the reported values are considerably higher than or comparable to those of Li+ in nonaqueous LIBs. The Zn2+ diffusion accompanied by a two-electron reaction presents such an unexpectedly fast response in AZIBs. Taking into account the mass and charge density of Zn2+, it is not possible to properly explain the reason for the fast diffusion of Zn2+ (as high as 10−7 cm2 s−1). Thus, it is most likely that such fast diffusivity can be attributed to the fast H+ diffusion as Zn2+ diffusion progresses rather slowly (∼10−11–∼10−12 cm2 s−1) as measured in nonaqueous ZIBs.267 Indeed, several studies have confirmed fast diffusion in the case of H+ intercalation, as shown in Fig. 10f. This tendency clearly supports the charge-compensation process by H+ and H+/Zn2+ co-insertion in many AZIB materials.
pH = −lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) γ[H+], γ[H+], | (10) |
| MOx + H+ + e− = HMOx | (11) |
 | (12) |
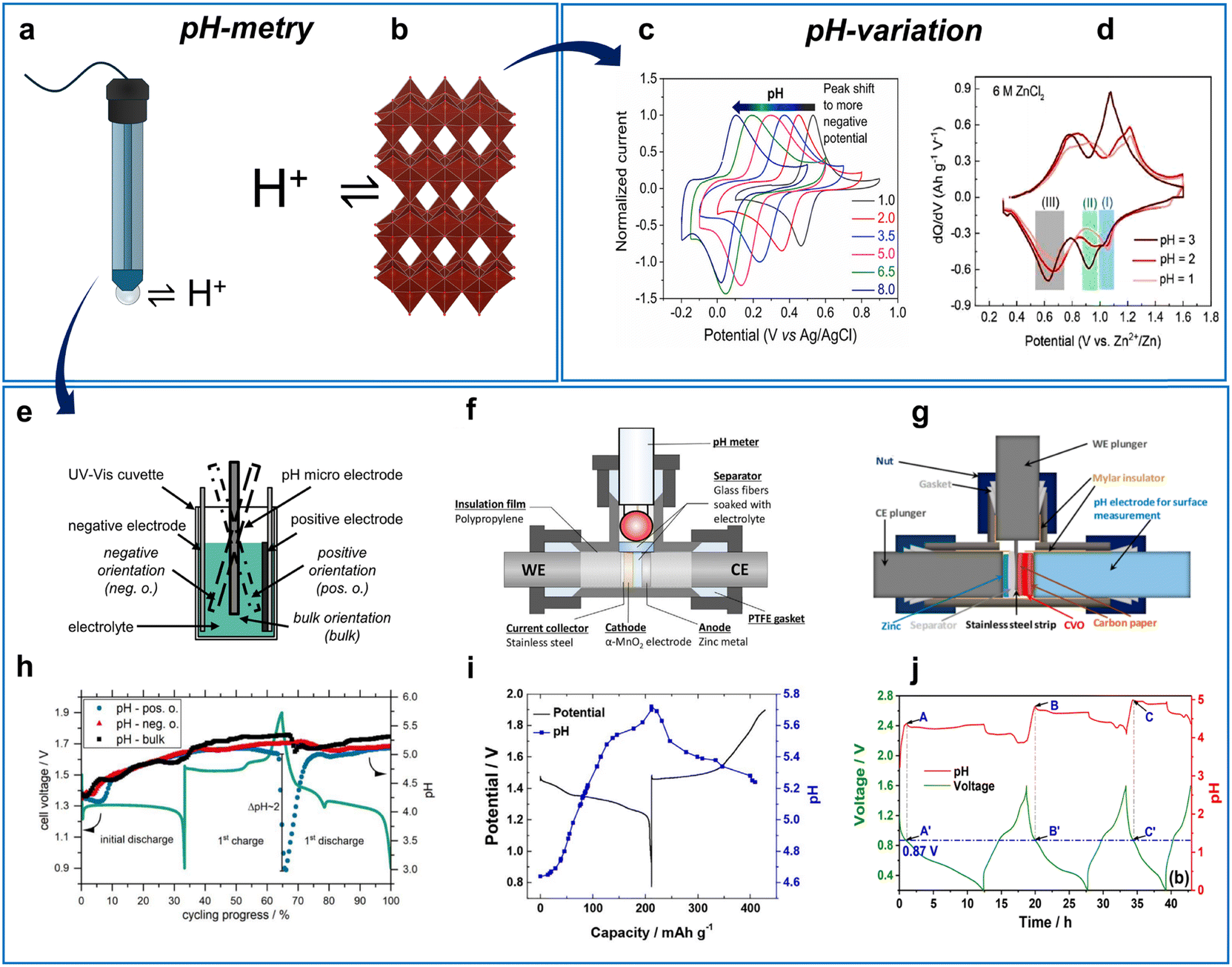 | ||
| Fig. 11 Study on H+ participation in AZIBs. (a) Schematic illustration of H+ exchange at pH electrode membrane and (b), host electrode. (c) pH-dependent voltammetry for H+ (de)insertion in polymers containing a quinone part. Reproduced with permission from ref. 269. Copyright 2021, Elsevier. (d) pH-dependent differential capacity plots for VOx in ZnCl2 solutions. Reproduced with permission from ref. 119. Copyright 2021, Elsevier. (e) Cuvette/beaker cell configuration for pH measurements. Reproduced with permission from ref. 270. Copyright 2020, IOP Publishing. (f), (g) T-shape cell configurations for pH measurements with different electrode arrangements. (f) Reproduced with permission from ref. 38. Copyright 2016, John Wiley & Sons. (g) Adapted with permission from ref. 101. Copyright 2020, American Chemical Society. (h) pH evolution in Zn|MnO2 cell shown in (e). Reproduced with permission from ref. 270. Copyright 2020, IOP Publishing. (i) pH evolution in Zn|MnO2 cell shown in (f). Reproduced with permission from ref. 38. Copyright 2016, John Wiley & Sons. (j) pH evolution in Zn|V2O5 cell shown in (g). Adapted with permission from ref. 101. Copyright 2020, American Chemical Society. | ||
A study on a Zn|VOx119 aqueous system demonstrated that the redox peaks shift negatively with the solution pH in accordance with H+ participation in the reaction (eqn (13) and Fig. 11d):
| VOx + aH+ + ae− → HaVOx | (13) |
In addition, pH variation of the electrolyte proved the H+ participation in the electrochemical reaction of MnO2 electrodes in AZIBs.45,271
More often for AZIBs, the H+ involvement is studied by operando pH monitoring, which can be realized using various cell designs (Fig. 11e–g). The simplest design includes a beaker cell or other big-volume cell design100,270,272–274 with an electrode placed close to one of the electrodes of interest (Fig. 11e and h). This method is quite simple and can be used in most laboratories as pH-sensitive electrodes and pH meters are readily available. Optionally, micro or macro electrodes can be used, and electrolyte convection may be introduced depending on the experimental design. One example is given in Fig. 11h, which shows the pH evolution upon charge–discharge for a Zn|MnO2 cell in a beaker-type cell.
To achieve conditions closer to those of a conventional battery protocol (i.e., a small amount of electrolyte, close-packed cell), several groups have used T-shape cells with a pH-sensitive electrode location either perpendicular to the cathode|anode (Fig. 11f and i)38 or on the active material side (Fig. 11g and j).101 Overall, the various designs of pH monitoring provide valuable data when investigating charge-storage mechanisms in AZIBs. The phase change of the active material, associated with proton-assisted reactions and dissolution–precipitation phenomena, can also be monitored by measuring the pH during prolonged electrochemical cycling tests.274
E-QCM (Electrochemical quartz crystal microbalance) is a powerful tool for distinguishing the charge carrier in an insertion-type electrode. The QCM principle is based on measuring the changes in frequency of a quartz crystal resonator, which reflects mass changes according to the Sauerbrey equation (Δf = −KΔm), where K is determined by the resonant frequency, density and shear modulus of quartz. In this regard, monitoring Δf during an electrochemical experiment on a metalized quartz-crystal electrode allows extracting mass changes (Fig. 12a). Ideally, the derivative of mass with respect to charge, dm/dq would allow the extraction of molar equivalent mass of the inserted ion, M/z. Hence, various scenarios emerge for a ZIB depending on the active material, electrolyte, and solvent used (Fig. 12b). The solo proton and Zn2+ insertion would lead to 1.0 and 32.7 g mole−1, respectively. However, intercalation of solvated species would result in higher values, e.g. 41.7 for Zn(H2O)2+. Furthermore, the pH-driven deposition of ZBS would lead to a severe mass increase depending on experimental conditions. As it is not a direct electrochemical process but rather one induced by pH change there is no universal theoretical mass increase; the experiment usually shows apparent M/z values ranging between a hundred and several thousand. As we describe below, all these scenarios and their simultaneous occurrence (e.g., co-insertion of two species) were reported in the literature.
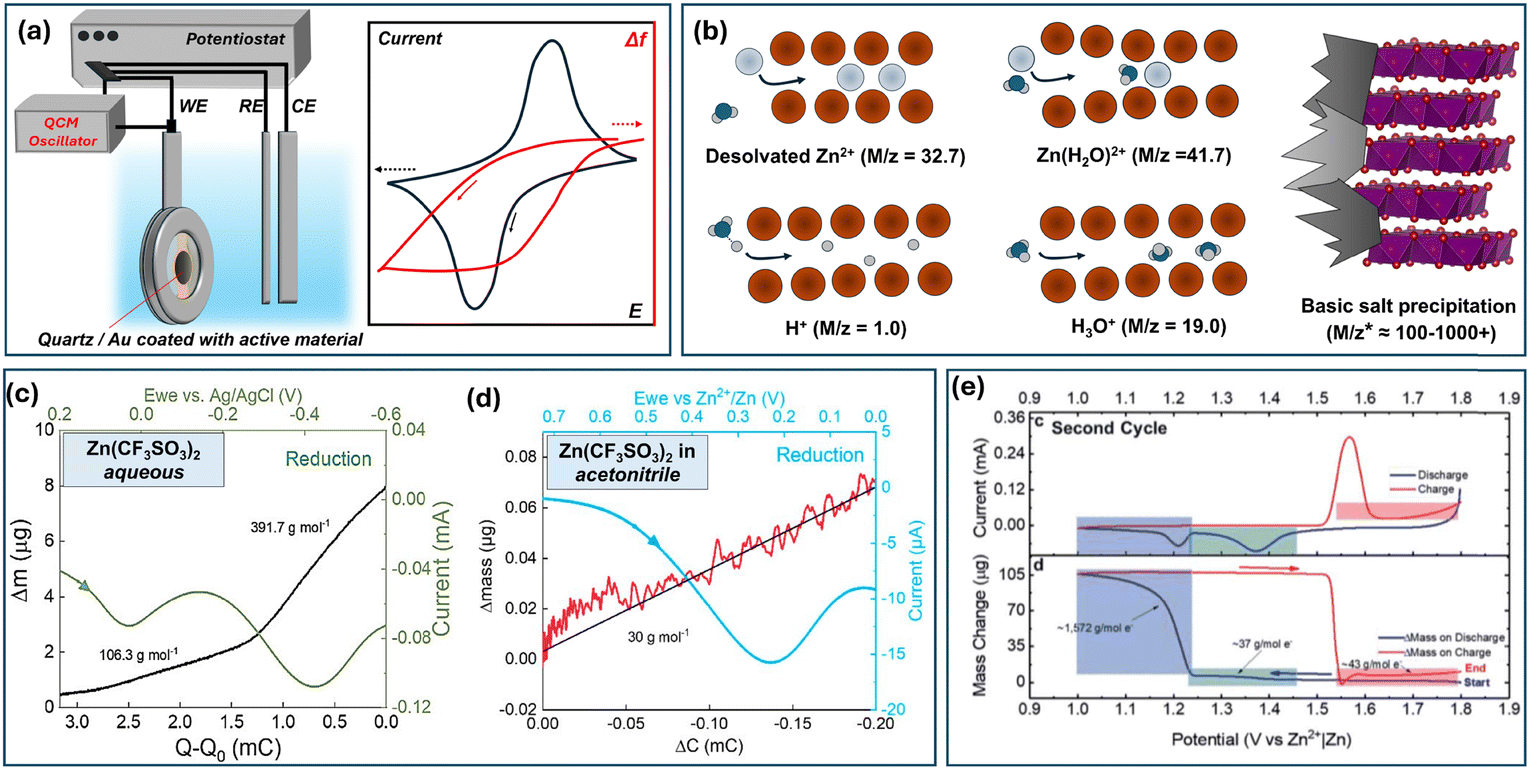 | ||
| Fig. 12 (a) Schema of an E-QCM experiment with a simple ion-insertion material. (b) Intercalation of various species reported in E-QCM ZIB experiments and ZBS precipitation as processes defining mass changes. Mass changes during reduction of Mn–V-based oxide in (c) aqueous and (d) non-aqueous Zn2+-electrolytes. Adapted with permission from ref. 275. Copyright 2025, John Wiley & Sons. (e) CV and mass change for a MnO2-based electrode during the 2nd cycle. Adapted with permission from ref. 276. Copyright 2021, Royal Society of Chemistry. | ||
E-QCM studies were performed for various vanadium oxide-based compounds.275,277,278 It was found that V2O5 undergoes H+ intercalation followed by precipitation of zinc hydroxy perchlorate Zn4(OH)7ClO4 upon discharge, and presumably Zn2+ and H+ co-insertion at lower potentials.277 In a recent work on a K–Mn–V-oxide (K1.92Mn0.54V2O5·H2O), the material was tested in both aqueous and nonaqueous Zn2+-electrolytes.275 The reduction in aqueous electrolyte led to a strong mass increase (≈100–400 g mol−1, Fig. 12c). Such a significant increase, especially at lower potentials, indicated the formation of zinc hydroxide triflate – the event caused by H+ intercalation or H+/Zn2+ intercalation. What's more interesting is that the experiment performed in acetonitrile-based electrolyte demonstrated values of 29–30 g mol−1 – which are quite close to the theoretical desolvated Zn2+ intercalation mass (Fig. 12d). Thus, Zn2+ was shown to intercalate into the oxide framework, but only in a limited amount – the resulting capacity was ≈60 mA h g−1 compared to over 300 mA h g−1 in aqueous electrolyte. Thus, again, the results support the limited ability of vanadium oxides to store Zn2+ ions and highlight the dominant contribution of H+ as a charge carrier when the electrolyte is an aqueous zinc salt providing protons through Zn2+/H2O equilibria (hydrolysis).
The most popular ZIB system, Zn–MnO2, was extensively studied using QCM over the last 4 years.276,279–282 Every work from this list reported the formation of basic zinc salts, which was evident from the severe mass increase, usually ranging from ≈100 g mol−1 to several thousand, both during the first and subsequent cycles. Depending on the cycle number and type of MnO2 used, the ZBS precipitation was caused by material dissolution276 or H+/H3O+ insertion.280,281 The ZBS precipitation was usually observed at the lower reduction peak at a curve inflection point (Fig. 12e). The charge led to its dissolution and subsequent deposition of MnO2, ZnMn2O4, and ZnMn3O7·nH2O as possible re-deposition products. What's interesting, little evidence was collected in such studies towards Zn2+ de/insertion, showing the dominant role of protonation and deposition/dissolution chemistry, at least for fresh and slightly cycled electrodes.
Besides vanadium and manganese oxides, various inorganic and organic compounds were studied. Investigation of Ti3C2 MXene revealed that the inserting ion may change depending on electrolyte concentration: 1 M aqueous ZnCl2 electrolyte demonstrated solely Zn2+ behavior while using 7 M ZnCl2 led to the co-intercalation of H3O+ species due to highly increased acidity of the concentrated electrolyte.283 Reports on layered TiS2 hosts showed a high sensitivity of the mechanism to the surface state.284,285 While TiS2 was demonstrated to accept hydrated Zn2+(H2O) species in the interlayer, the creation of a TiO2–TiS2 heterostructure promoted desolvation, leading to Zn2+ insertion.284 Another report on TiS2 demonstrated that exposing (011) facets favors the insertion of H3O+ instead of Zn(H2O)2+.285
Organic systems, mostly quinone-based were also studied by E-QCM techniques, highlighting Zn2+ or Zn2+/H+ associated charge storage mechanism.286–288 Interestingly, the poly(benzoquinonyl sulfide) behavior indicated that the triflate anion also participates in the reaction along with Zn2+ species.288 Such behavior should always be taken into account for polymer electrodes, which are much more prone to accepting anions and solvent molecules upon contact with electrolyte solutions and exhibit mixed cationic and anionic transport.289
Besides mass monitoring, more information can be gained using EQCM-D (Quartz Crystal Microbalance with Dissipation Monitoring), which helps investigate porous electrodes and viscoelastic properties.290,291 Overall, the E-QCM is a powerful technique for studying battery electrodes, which require careful analysis. Considering the inserting ions, the analysis should account for the possibility of solvated species (e.g., H3O+ and Zn(H2O)2+), anion de/insertion, and water dynamics, as various molecules can be inserted/extracted during an electrochemical process.
3.2. Water- and proton-assisted conversion reactions
Although the field of AZIB research has experienced tremendous growth in recent years, especially in the search for new materials and performance-oriented studies, understanding the reaction mechanisms remains challenging in many cases. This challenge largely stems from the numerous reactions with water, H+, and OH−, which often lead to phase changes or unwanted dissolution of the electrode materials. In this section, we discuss the associated structural changes of active materials in AZIBs. | ||
| Fig. 13 MnO2 polymorphs, their conversion products in aqueous ZIBs, and proposed mechanisms. (a) Most typical MnO2 polymorphs used for ZIB studies. (b) Zn–Mn–O compounds often found after Zn|MnO2 cycling. (c) Zinc hydroxide sulfate Zn4(OH)6SO4·xH2O, a typical discharge by-product. (d) pH changes upon MnO2 (dis)charge. Reproduced with permission from ref. 294. Copyright 2022, Elsevier. (e) Proposed Zn2+ (de)insertion mechanism. Reproduced with permission from ref. 37. Copyright 2011, John Wiley & Sons. (f) Proposed mixed mechanism. Adapted with permission from ref. 295. Copyright 2019, American Chemical Society. (g) Proposed dissolution-deposition mechanism. Reproduced with permission from ref. 296. Copyright 2023, John Wiley & Sons. | ||
Interest in AZIBs was retriggered by the study of α-MnO2 in a mildly acidic zinc electrolyte, for which the Zn|MnO2 cell using ZnSO4 electrolyte showed stable behavior over 100 cycles,37 although similar mildly acidic Zn|MnO2 cells were already demonstrated in 1986.24 First, it was argued that the reaction mechanism involves (de)introduction of Zn2+ from/into the MnO2 structure (Fig. 13e). Further studies confirmed the incorporation of zinc into γ-MnO2, revealing various discharge products such as spinel ZnMn2O4 or layered ZnyMnO2 (Fig. 13b).46 Other studies have proposed (de)insertion of Zn2+ ions as the main mechanism for charge storage in ZnMn2O4 spinels.215,297
Later, many studies rationalized that the reaction mechanism is much more complex than a simple (de)intercalation or (de)insertion of Zn2+.38,44,106,294,295,298,299 Many groups have observed the formation of basic zinc salts (for example, zinc hydroxide on discharged MnO2 electrodes) and its dissolution upon charging (Fig. 13c).38,44,106,294,295,298,299 Notably, this observation contradicts the abovementioned simple mechanisms because the formation of ZHS and the significant change in pH could not occur if the insertion of Zn2+ is the only electrochemical reaction. Therefore, MnO2-based electrodes consume protons during reduction and then release them upon oxidation. This behavior was measured directly by tracking the pH change during cell operation (Fig. 13d).38,270,294,300 Various reactions have been proposed to explain the proton consumption: the formation of MnOOH by H+,42,295,301 MnO2/Mn2+ deposition/dissolution,106,294,296,299 and a mixed insertion/deposition/dissolution mechanism.44,45,298,302–304
The simplest proton insertion implies the formation of a well-known MnOOH:
| MnO2 + H+ + e− → MnOOH | (14) |
This mechanism was verified by comprehensive studies using HRTEM, XRD, and 1H NMR analysis.42,305 Other studies have suggested that co-insertion of Zn2+ and H+ leads to the sequential formation of ZnMn2O4 and MnOOH depending on the depth of discharge (Fig. 13f).295,301 However, the formation of MnOOH was not detected, but the H+ insertion was assumed by the presence of zinc hydroxide sulfate.104 In addition, the ZnMn2O4 was found to be irreversibly accumulated on the MnO2 surface after prolonged cycling.104
The fading of capacity in many works was attributed to Mn2+-producing dissolution, particularly caused by disproportionation of manganese in the (+3) state; such as in
| ZnMn2O4 + 4H+ ⇌ Zn2+ + MnO2 + Mn2+ + 2H2O | (15) |
To suppress such disproportionation, the additive Mn2+ was introduced to the electrolyte, which enabled stable cycling behavior to be achieved in many works.294,299,306,307 The effect of the Mn2+ additive appeared to be more complex: Mn2+ is oxidized to form electrodeposited MnO2 on charge (which is a well-known reaction for industrially produced electrolytic manganese dioxide, EMD):
| Mn2+ + 2H2O ⇌ MnO2 + 4H+ + 2e− | (16) |
Other investigations on Zn|MnO2 cells with different polymorphs demonstrated that the charge storage is dominated by the dissolution–deposition reaction mechanism while (de)intercalation of Zn2+ and H+ has a minor contribution (Fig. 13g).296,299 The proposed dissolution–deposition mechanism included:
• Reductive dissolution of MnO2 (discharge):
| MnO2 + 4H+ + 2e− → Mn2+ + 2H2O | (17) |
| 4Zn2+ + 4H2O + SO42− + 6OH− → Zn4(OH)6SO4·4H2O | (18) |
| Mn2+ + 2H2O ⇌ MnO2 (birnessite) + 4H+ + 2e− | (19) |
| Zn4(OH)6SO4·4H2O + 6H+ = 4Zn2+ + 10H2O + SO42− | (20) |
Thus, pristine MnO2 material is mainly active during the initial cycles, whereas electrochemical oxidation of Mn2+ leads to layered birnessite-type MnO2, which is spontaneously deposited on the cathode surface. In principle, MnO2 can be directly electrodeposited in the cell without any initial manganese oxide.298 It was shown that BZS-based electrodes are available as a precursor to facilitate the in situ formation of MnO2-based electrodes by electrodeposition.298 The proposed mechanism included two steps during the charge–discharge process: Mn2+ ⇌ ZnMn2O4 ⇌ layered Zn-birnessite.
Operando X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) studies of α-MnO2 in three different electrolytes (ZnSO4, Zn(CF3SO3)2, and Zn(CH3COO2) demonstrated that the reaction mechanism in all three cases is dissolution–deposition rather than (de)intercalation of Zn2+.106 The reduction induces the formation of Mn2+ ions upon discharge, whereas a considerable fraction of MnO2 remained unreacted. On charge, the dissolved Mn2+ is oxidized, although the chemical state is not identical to that of the initial MnO2 structure but more likely transforms into a layered structure ZnMn3O7·3H2O. No considerable amount of Mn(III) compounds was detected, which may be the result of disproportionation to Mn2+ and Mn4+. Another study by the same group304 showed extensive investigation of the first cycles of charge–discharge for Zn/α-MnO2 using operando XRD, operando XAS, and ex situ Raman as well as other methods. They revealed a multi-stage dissolution–conversion charge-storage mechanism, including Zn2+ insertion and MnO2 dissolution/deposition reactions. Namely, the high-voltage plateau was attributed to the conversion of MnO2 to a poorly crystalline Zn–Mn–O compound with a locally layered structure, whereas the lower-voltage plateau was found to correspond to a proton-consuming MnO2 dissolution process responsible for BZS deposition.
A recent study on β-MnO2 using operando X-ray techniques (XRD, XAS, and X-ray nano-tomography) revealed several features of the dissolution–deposition mechanism (Fig. 14).47 It was shown that no new Mn-containing phase was formed during MnO2 reduction. The first discharge caused dissolution to Mn2+ and ZHS precipitation while maintaining the β-MnO2 active material (Fig. 14a). The Mn and Zn K-edge XAS results revealed the formation of amorphous Zn–Mn–O compounds such as chalcophanite (ZnMn3O7·3H2O) and spinel (ZnMn2O4) structures (Fig. 14b–d).47
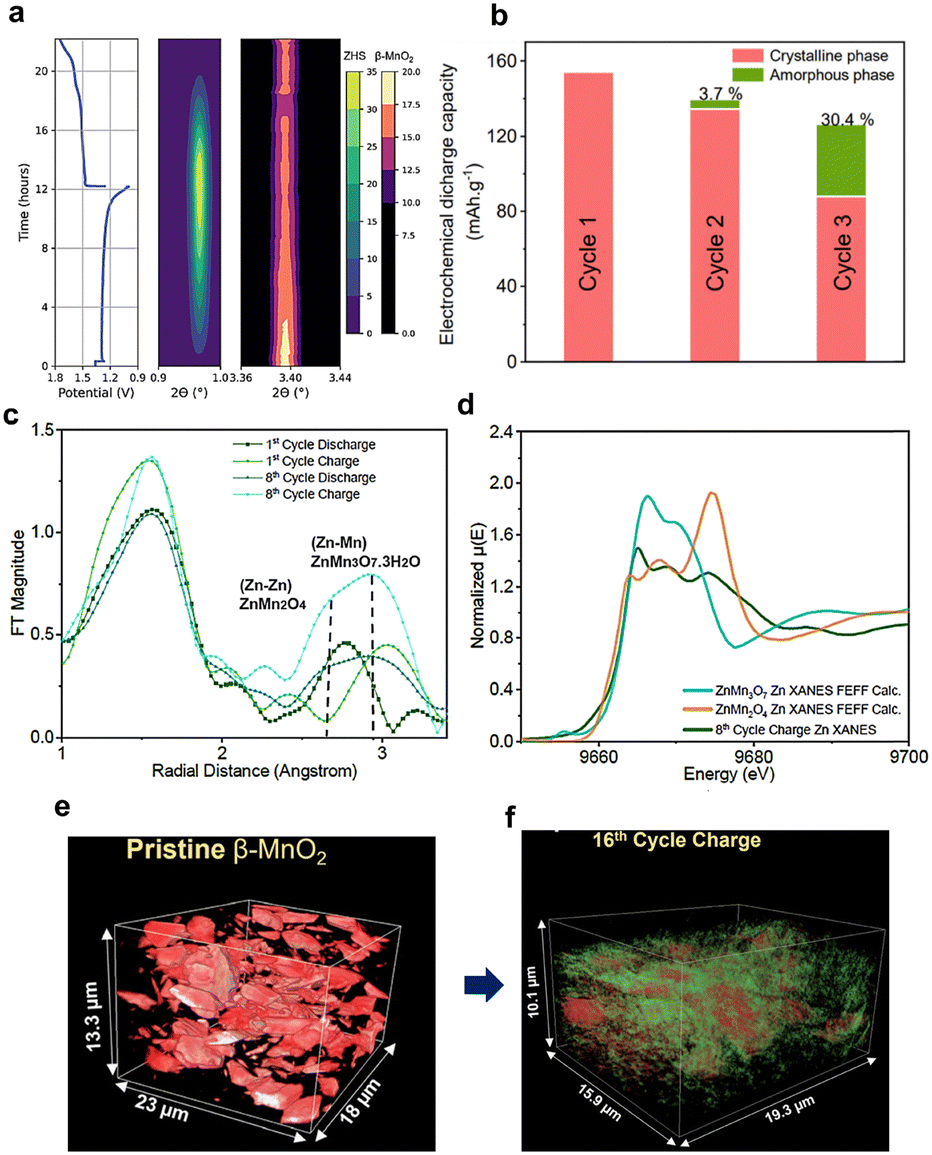 | ||
| Fig. 14 Operando XRD and ex situ XAS results of Zn|β-MnO2 cycling. (a) Discharge–charge profile and corresponding precipitation/dissolution of BZS and fading of MnO2 peak intensity. (b) Estimated relative capacity contributions by amorphous and crystalline phases. (c) EXAFS analysis (Zn) of charged and discharged states at the 1st and 8th cycles. (d) Comparison of experimental data (8th cycle) and calculated reference spectra for Zn K-edge. (e and f) X-ray synchrotron tomography showing amorphization of MnO2 after cycling. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. Copyright 2023. | ||
Amorphization was further supported by 3D synchrotron X-ray nano-tomography data after 16 cycles (Fig. 14e and f.). The results indicated that cyclability is not limited by the initial presence of MnO2 but is governed by the reversible formation and reaction of Zn–Mn–O compounds through the dissolution–deposition mechanism.
It is likely that the deposition–dissolution of MnO2 and other manganese phases is dependent on the solvation and adsorption of Mn2+ ion, that may explain higher capacity fluctuations in sulfate-based electrolyte than that of triflate-system.187
Electrolytic MnO2 (EMD) has been reported to be an active cathode material for AZIBs, which is enabled by multi-step reactions, including both Zn2+ (de)insertion and disproportionation/dissolution reactions.303 During discharge, EMD was found to form spinel-type ZnMn2O4 and tunnel-type ZnxMnO2; the latter was unstable at a certain state of discharge and disproportionated to Mn2+ ions accompanied by an increase in the pH of the electrolyte triggering the formation of BZS. Interestingly, Mn2+ in solution was detected only in the low-voltage region, which is contradictory to the above operando study in which Mn2+ dissolution was proposed for the entire potential range.106
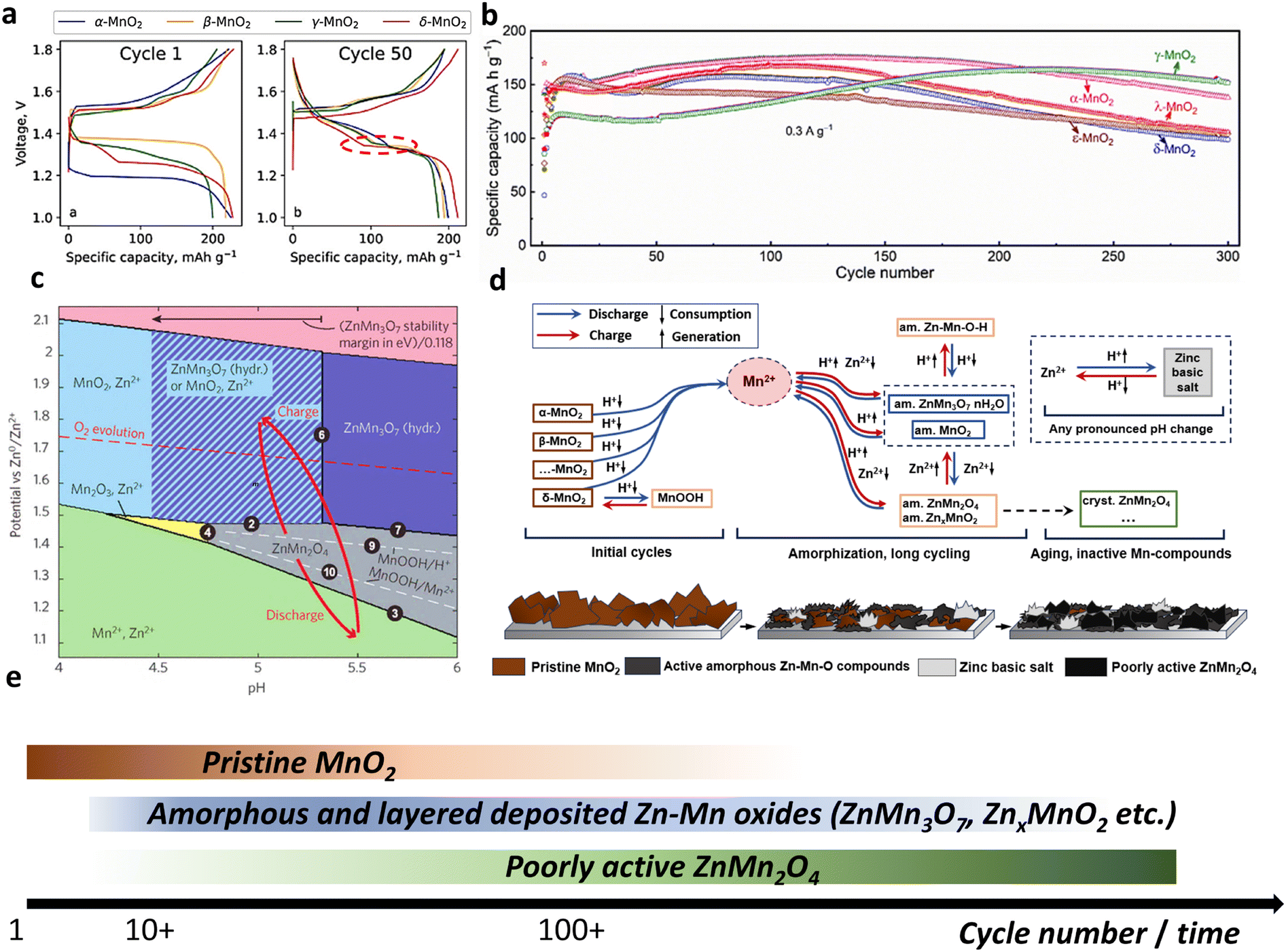 | ||
| Fig. 15 Behavior of various MnO2 polymorphs and generalized mechanism of Zn|MnO2 batteries. (a) Charge–discharge profiles for four different polymorphs at 1st and 50th cycles. Reproduced with permission from ref. 45. Copyright 2022, Elsevier. (b) Long-term cyclability of 5 different polymorphs over 300 cycles. Reproduced with permission from ref. 44. Copyright 2022, Elsevier. (c) Pourbaix diagram for Zn–Mn–O system according to ref. 302. Adapted with permission from ref. 302. Copyright 2022, American Chemical Society. (d) Generalized mechanism of Zn–MnO2 batteries. (e) Timescale of compounds stability in Zn–MnO2 cells. | ||
A recent study44 compared seven types of MnO2 polymorphs (α-, β-, γ-, ε-, δ-, λ-, and R-MnO2) over prolonged cycling. The initial discharge capacity showed dispersed data between 40 and 160 mA h g−1 for various polymorphs. Some cases exhibited an increase in capacity with cycling, whereas others resulted in fluctuating performance. Regardless of the types of MnO2, the resulting charge–discharge profiles became identical for the tested MnO2 polymorphs, with the capacity ranging between 120 and 180 mA h g−1 (Fig. 15b). This performance similarity was explained by Mn2+ dissolution caused by disproportionation followed by the formation of electrochemically active birnessite. The slow degradation was related to the formation of Mn3O4 and barely active ZnMn2O4 spinel during cycling.
Another study experimentally compared four types of MnO2 polymorphs (α, β, γ, and δ) in aqueous 1 M ZnSO4 electrolyte.45 The difference in the charge–discharge behavior was seen only at the first cycle with a single plateau for α- and β-MnO2 and two plateaus for γ- and δ-MnO2. Further cycling led to similar behavior and almost identical charge–discharge profiles (Fig. 15a). The specific capacities for all the electrodes were within the range of 200–230 mA h g−1, which is similar to the results of another comparative study.44 The extensively cycled electrode (200 cycles) clearly showed that ZnMn2O4 is one of the major reaction products, whereas unreacted initial phases and Mn2O3 were also detected as minor products. Moreover, Raman analysis of the cycled electrodes showed a very weak signal or the absence of the initial pristine materials and some broad peaks, suggesting an amorphous MnO2 deposit on the surface of the cathode. The strong dependence of anodic and cathodic processes on pH revealed the effect of H+ on charge and discharge, namely, Mn3+ disproportion, formation of BZS, and anodic oxidation of Mn2+, all of which are pH-dependent reactions. The similarity in the behavior of various polymorphs was attributed to the gradual dissolution and re-deposition of active materials, leading to a mixture of compounds (Zn4(OH)6SO4·xH2O, ZnMn2O4, Mn2O3, amorphous MnO2, etc.). Therefore, regardless of the starting polymorph materials, similar reactions triggered by the participation of H+ and the formation of similar compounds dominate the overall electrochemical behavior in a mildly acidic aqueous solution.
Recent work has rationalized the aforementioned reactions by building a refined Mn–Zn–H2O Pourbaix diagram, plotted using both experimental and theoretical data, with a Zn|MnO2 aqueous cell (Fig. 15c).302 Namely, the diagram predicts boundaries for Mn(IV)/Mn(III)/Mn(II) transitions depending on the pH. As MnO2 is reduced, thermodynamics governs the formation of the ZnMn2O4 spinel in the pH range of 5–6. An alternative form of trivalent manganese, MnOOH, was predicted to have a very narrow stability range. Further reduction of ZnMn2O4 resulted in pH-dependent dissolution, resulting in the release of aqueous Zn2+ and Mn2+ ions. As the pH shifted to higher values (>5) due to buffering of BZS, the more stable form of Mn(IV) became hydrated ZnMn3O7 (chalcophanite), not MnO2. As a consequence, Mn2+ or ZnMn2O4 is likely to be oxidized to form layered ZnMn3O7 upon charge. The researchers further validated their postulation by adjusting the pH of the electrolyte to 2.5, which did not result in the formation of chalcophanite, whereas a pH of 4.0 favored ZnMn3O7 deposition.302
To summarize the aforementioned operando,47,106,303,304 comparative,44–46 and theory-based studies,103,302 a general understanding of the mechanism for the aqueous Zn|MnO2 cells appears as follows. The MnO2 (polymorph) active materials dissolve to form Mn2+ upon the first discharge (reaction (17), Fig. 15d). This dissolution is supported by the consumption of protons and, therefore, leads to the formation of basic zinc salts (eqn (18)) almost immediately after the start of discharge.47,308,309 Although δ-MnO2 exhibits a slightly different behavior, accepting H+ to form MnOOH during the initial few cycles,305 it eventually follows the same tendency as other MnO2 polymorphs that show dissolution of Mn2+. Thereafter, during the subsequent charge, the Mn2+ in the electrolyte solution is oxidized to MnO2 (reaction (19)). However, the MnO2 formed after the charge is not the original MnO2 but amorphous MnO2 or amorphous layered zinc–manganese oxide. The low-crystalline deposit may vary depending on the conditions and is sometimes referred to as birnessite or, in later studies, as having a structure like layered ZnMn3O7·3H2O:47
| Zn2+ + 3Mn2+ + 10H2O → ZnMn3O7·3H2O + 14H+ + 6e− | (21) |
Some studies indicate that the deposit has a vernadite structure.310 Depending on the conditions, the low-crystalline deposit is sedimented on the surface of the cathode after several cycles (sometimes tens of cycles), depending on the operation condition of the cells (Fig. 15e).45,47,106 This activation typically results in two voltage plateaus. In the high-voltage region, for which the BZS is not present, (de)zincification is the main process to form amorphous oxides103,303,304 such as ZnxMnO2 or ZnMn2O4. As the active material transforms into the new active amorphous deposit, the resulting electrochemical performance is governed by the formed new phases as the new active material, such that the electrode performance becomes, in many cases, independent of the material properties of the original cathode active material. Furthermore, the lower voltage plateau is accompanied by BZS formation, and its deposition appears as an inflection point on the discharge curve (≈1.35 V).46,309,311 Concurrently with the growth of BZS, the Mn2+ concentration increases in the electrolyte,303,312 which is due to the proton-assisted dissolution of oxide phases to form Mn2+:
| ZnMn2O4 + 8H+ + 2e− → Zn2+ + 2Mn2+ + 4H2O | (22) |
Thus, the insertion of Zn2+/H+ at higher voltage and deposition/precipitation of Zn–Mn–O phases at the lower voltage plateau are responsible for the charge-storage mechanism of the cycle-induced new cathode active materials. This indicates that the cyclability is limited by the reversibility of the amorphous Zn–Mn–O phase.47 Eventually, the amorphous phase tends to transform into crystalline ZnMn2O4, which becomes thermodynamically stable as the pH shifts to higher values during cycling (Fig. 15c).302,313 Therefore, it is concluded that ZnMn2O4 accumulation is kinetically sluggish but thermodynamically favorable above pH 5, and the ZnMn2O4 spinel is a dominant “dead” form manganese compound, which impedes capacity retention and is associated with capacity decay.44,47,313,314
To summarize, the performance of Zn|MnO2 cells is determined by (1) the insertion of Zn2+/H+ into electrochemically deposited amorphous Mn–O or Zn–Mn–O compounds in the high-voltage region and (2) proton-assisted dissolution/deposition involving the Mn2+ process in the lower-voltage range. The service life of a positive electrode can be described in three areas: (1) amorphization of the original MnO2, (2) steady-state cycling, and (3) accumulation of electro-inactive products. Therefore, the electrolyte composition, pH, additives, and the substrate (current collector) appear to be more important for the design and optimization of Zn|MnO2 and addressing complex amorphization–deposition phenomena in AZIBs than the engineering of MnO2 crystalline and defect structures.
Although many metal vanadates are suitable for use as cathode materials for AZIBs, the actual redox-active phase may be different from the original material due to reactions involving an aqueous environment during the initial cycles. Recently, it was reported that oxidation of K2V3O8 up to 1.9 V vs. Zn2+/Zn led to the formation of amorphous vanadium oxide, presumably V2O5 (Fig. 16a).100 After that, a conversion reaction progressed during discharge, demonstrating a protonation-associated process: V2O5 + 4H+ + 4e ⇌ 2VOOH + H2O. Another type of V-based oxide, CaV4O9, was transformed into amorphous V2O5·nH2O after oxidation in a mildly acidic Zn2+ electrolyte.315 The related transformation resulted from the electrochemical displacement of Ca2+ ions by water molecules when the cell was charged to 1.6 V.
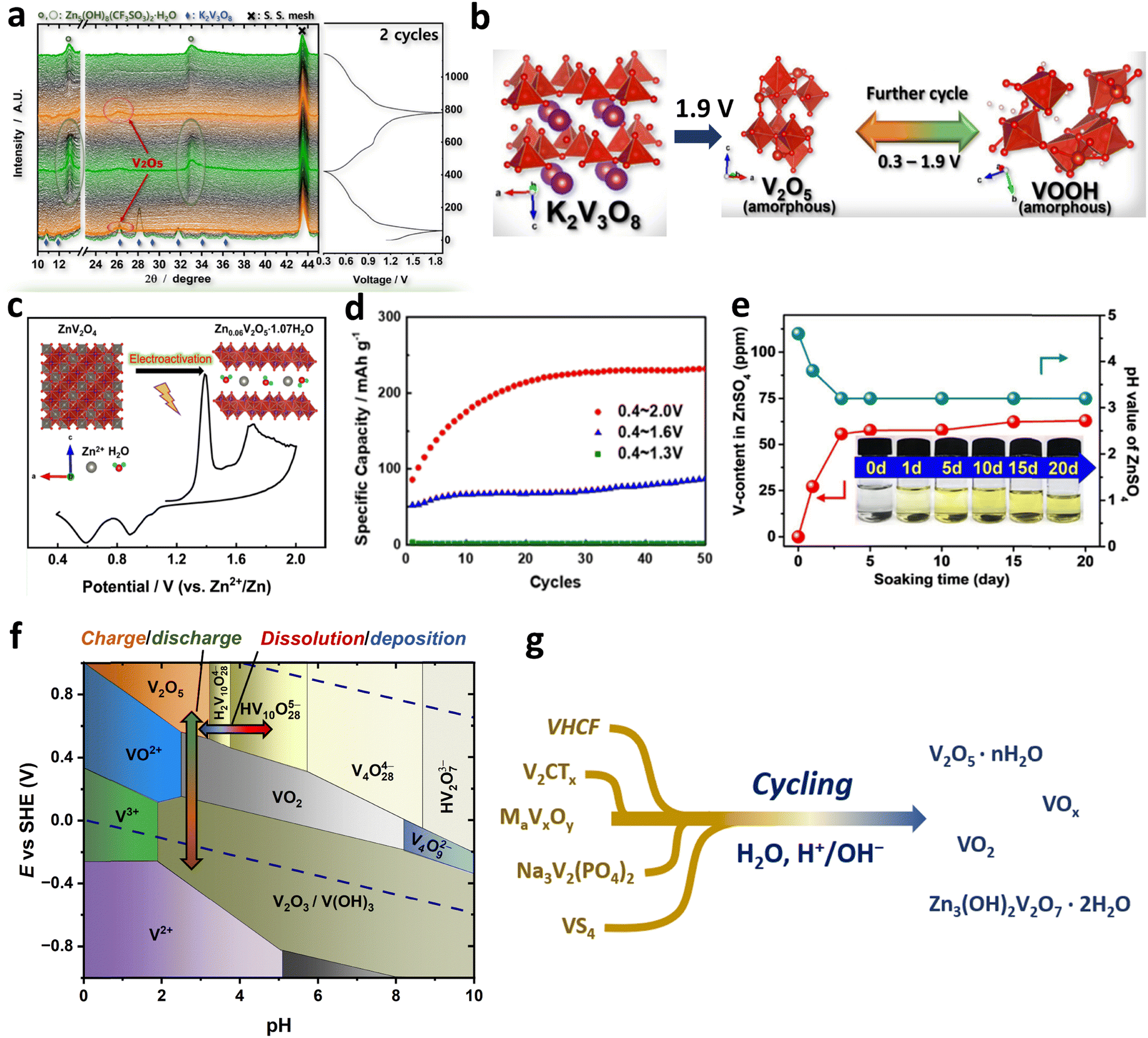 | ||
| Fig. 16 Transformations of vanadium-based electrodes in aqueous ZIBs. (a) Operando XRD of K2V3O8-based electrode showing conversion to amorphous V2O5 and (b), corresponding schematic representation of proton-associated V2O5/VOOH redox upon charge–discharge. Reproduced with permission from ref. 100. Copyright 2023, Elsevier. (c) Electrochemically-driven conversion of ZnV2O4 spinel to layered vanadium oxide, Zn-inserted V2O5·H2O, and (d) corresponding specific capacity increase associated with ZnV2O4/V2O5·H2O transformation. Adapted with permission from ref. 316. Copyright 2022, American Chemical Society. (e) Vanadium concentration and pH during long term α-V2O5 soaking in 2 M ZnSO4. Adapted with permission from ref. 317. Copyright 2021, American Chemical Society (f) Pourbaix diagram for vanadium species, c(V) = 0.1 M;318,319 arrows indicate charge–discharge processes of V2O5/VOx and dissolution/deposition phenomena triggered by pH change. (g) Examples of electrochemical transformation of vanadium-containing electrodes upon charge–discharge in aqueous electrolytes.247,274,315,316,320–322 | ||
This tendency, water-associated formation of V2O5·nH2O and other vanadium-based oxide materials, is not limited to metal vanadates. Investigation of NASCION-type Na3V2(PO4)3 in aqueous zinc triflate solution demonstrated the formation of V2O5, VO2, and Zn3V2O8 upon charge–discharge cycling.247 This eventually resulted in a decrease in the average charge and discharge voltage as the redox potential of VOx is lower than that of Na3V2(PO4)3. Interestingly, the use of a highly concentrated hybrid electrolyte with sodium ions, 10 M NaClO4 + 0.4 M Zn(CF3SO3)2, enabled not only stabilization but also suppressed the decomposition of the electrolyte.
Liu et al.320 demonstrated the formation of nano-sized vanadium oxide (VOx) during the electrochemical oxidation of V2C MXene. The VOx deposit grew on the top of the MXene material, and by controlling the potential, the MXene substrate could be preserved to serve as a 2D conductive layer for the grown nano VOx. Li et al.321 reported V2C MXene conversion to V2O5 by electrochemical cycling. The conversion eventually increased the capacity, with the phase transition being referred to as “activation”. Vanadium tetrasulfide (VS4) was recently found to convert into zinc pyrovanadate (Zn3(OH)2V2O7·2H2O) during the initial charge–discharge cycles.322 However, unlike the above examples of VOx formation, the conversion process was associated with an initial discharge involving the reaction with water molecules rather than the charging process. After several cycles, the resulting CV pattern represented two pairs of redox peaks at 0.6 and 1.0 V, which are typically observed for vanadium oxides and vanadate-based compounds.201,235,263–265
A spinel compound, ZnV2O4, was transformed into Zn-inserted V2O5·H2O upon electrochemical oxidation (Fig. 16c).316 Although the spinel phase was almost inactive, the newly formed vanadium oxide exhibited a prominent increase in capacity up to 230 mA h g−1 depending on the potential range used (Fig. 16d).
A phase change associated with water can result not only in a completely different type of material but also in the transformation of one oxide into another. The electrochemical oxidation of VO2 above 1.4 V (vs. Zn2+/Zn) was demonstrated to form V2O5·1.75H2O xerogel with a high specific capacity (610 mA h g−1 at 0.1 A g−1).323 The formed V2O5·1.75H2O possessed low crystallinity and exhibited a simultaneous H+ and Zn2+ (de)insertion mechanism. As in many previous cases, the transformation was accompanied by water consumption and protolytic reaction:
| 2VO2 + 2.75H2O → V2O5·1.75H2O + 2e− + 2H+ | (23) |
Another example is the transformation of α-V2O5 to hydrated V2O5·1.75H2O.317 The revealed mechanism of the transformation involved the partial dissolution of vanadium oxide to decavanadate [V10O26(OH)2]4− species in solution, which eventually transformed into Zn3(OH)2V2O7·2H2O and V2O5·1.75H2O. The dissolution progressed even without electrochemical treatment, mainly in the first 5 days (Fig. 16e):
| 5V2O5 + 3H2O → [V10O26(OH)2]4− + 4H+ | (24) |
The formation of VOx from solution can be used as a tool for electrode construction. Recently, highly disordered electrodeposited vanadium oxide was studied as a cathode material for AZIBs.324 Electrodeposition was performed using a VOSO4-containing solution in cyclic voltammetry mode, resulting in a highly stable VOx electrode. The deposit achieved a high areal capacity of 5 mA h cm−2 and remarkable stability over 5000 cycles.
In a recent study, vanadium hexacyanoferrate (VHCF) was revealed to undergo phase transformations to zinc hexacyanoferrate and amorphous vanadium oxide VOx.274 The intermediate of such transformations was also decavanadate:
| 10 VO2+ + 18H2O → V10O27(OH)5− + 35H+ + 10e− | (25) |
Combining all the above examples of phase transformations for vanadium-containing active materials, we can summarize the following common tendencies: (1) vanadium-containing electrodes tend to react with water to form vanadium oxides (V2O5·nH2O, VO2, VOx) or layered Zn3(OH)2V2O7·2H2O during cycling. Such reactions are usually initiated by extending the charge cutoff voltage (or, more precisely, extending the oxidation state of V to 5+ on charge). (2) The deposited vanadium oxide (VOx) often possesses an amorphous/nanosized nature, which makes it difficult to identify formed phases in terms of structure, composition, and chemical states. Usually, the associated reactions are accompanied by changes in pH because water, protons, and hydroxide ions participate in the reaction, which results in the formation of new oxo- and hydroxo-vanadium compounds. (3) Relatively closed-packed phases with vanadium atoms often tend to form open or layered structures. (4) The phase transformation usually progresses through dissolution–precipitation rather than direct solid-state phase transition. The most likely intermediates are decavanadates ions, as the most stable V5+ form in mildly acidic media.
The Pourbaix diagram is helpful in understanding such transformations (Fig. 16f) because vanadium tends to dissolve from many materials and form new compounds after reacting with water. This is especially valid for vanadium (+5) compounds: according to the diagram, V2O5 is not stable above pH 3.5, where V(+5) forms stable dissolved species. These species are mainly decavanadates because they are thermodynamically favorable for the typical pH of most Zn electrolyte solutions used (pH 3–6). Considering that many electrochemical reactions induce pH change, the cyclic pH variation may create conditions for multiple dissolution–precipitation cycles, eventually contributing to phase transformation and deposition of the V2O5·nH2O or some amorphous mixture of oxides. This provides a hint as to why so many completely different V-based materials, in turn, show an almost identical double CV peak shape after cycling, which does not belong to the original cathode material but to newly formed oxides (Fig. 16g). Such behavior is similar to the MnO2 mechanism when the originally different materials transform into similar mixtures with low-crystalline Zn–Mn–O phases.
Such transformations cannot always be defined as undesirable or favorable. In many cases, such “activation” is beneficial for electrochemical performance in terms of capacity and cyclability. In other cases, such as for phosphate compounds, such reactions lower the discharge voltage and degrade the stability and must be suppressed to achieve stable behavior.247
4. Generalized transformations of transition-metal active materials
As follows from the consideration of the electrochemistry of manganese oxides and vanadium-containing compounds in aqueous zinc electrolytes, many proton-activated reactions occur in an aqueous environment, including various precipitation–dissolution phenomena. Analysis of the available literature indicates that such phenomena are not limited to the aforementioned compounds but include other manganese materials such as phosphates or hexacyanoferrates and cobalt-and nickel-containing materials (Fig. 17a).325–332 For instance, as vanadium hexacyanoferrate (VHCF) transforms into VOx and ZnHCF upon cycling,274 manganese hexacyanoferrate particularly transforms in MnOx, as evident from the Raman signal accompanied by the appearance of MnO2-related features on discharge curves.332 Manganese phosphates327,331 have been shown to decompose during charge–discharge, producing a MnO2 layer on the electrode surface. Interestingly, cycling was used deliberately to create an appropriate surface oxide layer with enhanced performance in a recent work.331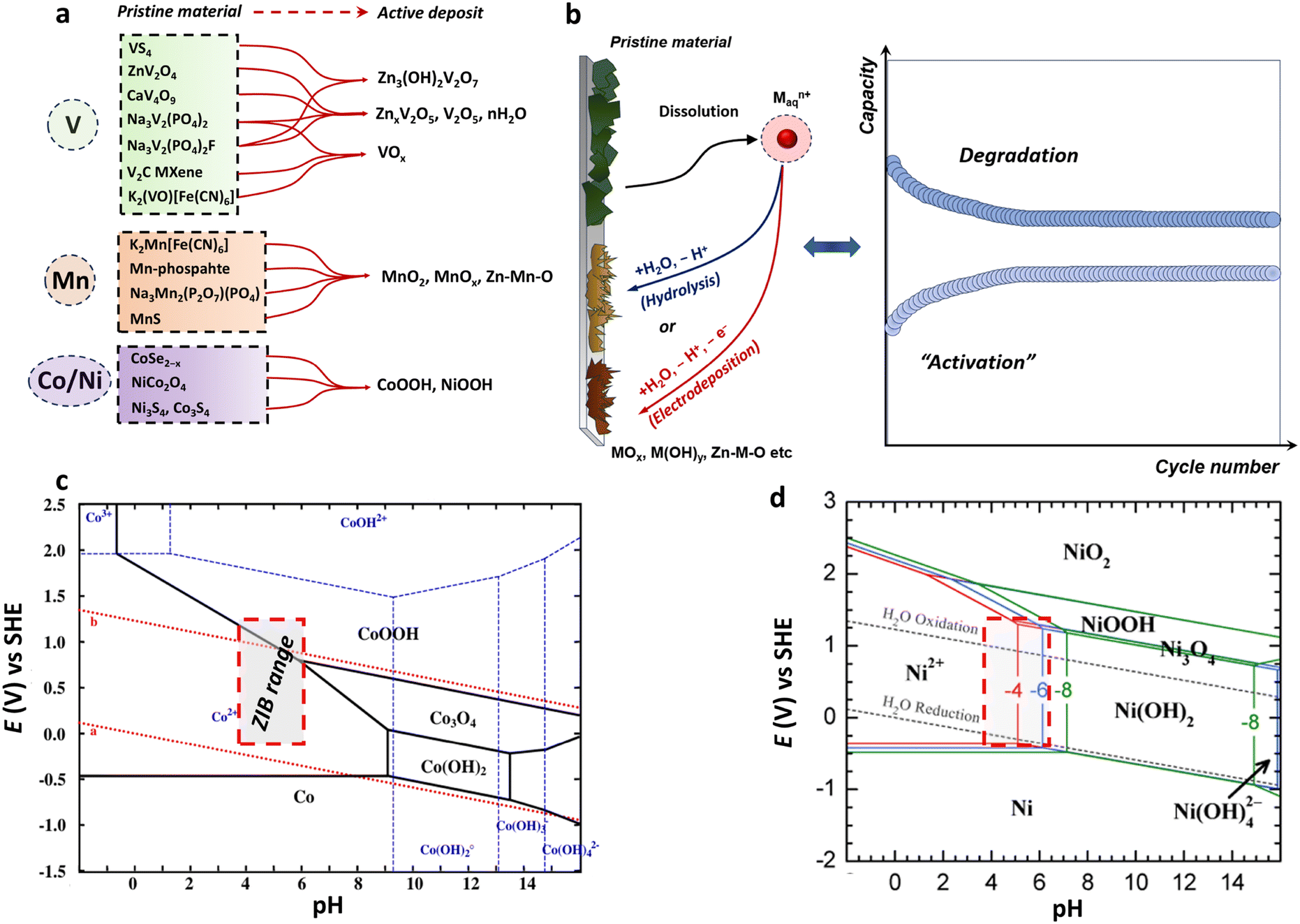 | ||
| Fig. 17 Transformations of transition metal-based materials in aqueous zinc batteries. (a) Examples of electrode material transformation into new electroactive compounds upon battery operation or prior to testing.247,274,315,316,320–322,325–331 (b) Generalized scheme of transformation as dissolution-deposition process and corresponding degradation or activation behavior. (c) Co-H2O Pourbaix diagram. Reproduced with permission from ref. 333. Copyright 2008, Elsevier. (d) Ni-H2O Pourbaix diagram. Adapted with permission from ref. 334. Copyright 2017, American Chemical Society. | ||
Cobalt and nickel compounds, mostly chalcogenides, have been extensively studied as electrodes in AZIBs and Zn hybrid batteries, often using alkaline electrolytes.325–330,335,336 Similar to MnOx and VOx generated from Mn- and V-containing materials, cobalt and nickel materials result in CoOOH/Co(OH)2 and NoOOH/Ni(OH)2 acting as redox-active materials for proton storage.328–330
Thus, there are obvious similarities in the behavior of many transition-metal-based materials in AZIBs caused by the transformation of the original into new redox-active species. The general transformation for most transition-metal-containing compounds consists of several steps (Fig. 17b). The transition-metal ion (Mn+) is leached due to the non-zero solubility of the active material into the solution or due to electrode polarization (oxidation or reduction). For V-ion compounds, the intermediate ions are likely decavanadates (V10O26(OH)24−/V10O27(OH)5−), whereas for Mn-, Co-, and Ni- containing materials, it would be Mn2+, Co2+, and Ni2+.
Then, metal ions interact with water, resulting in hydroxide or oxide formation. Depending on the potential, concentration, and pH, the deposition would proceed via a chemical or electrochemical route, generating a new oxide- or hydroxide-based compound, often in amorphous form. The chemical route may correspond to a hydrolysis reaction, e.g.:
| V10O27(OH)5− + 2H2O = 5V2O5 ↓ + 5OH− | (26) |
The electrochemical route would be electrochemical oxidation as for the deposition of MnO2, ZnMn2O4, or Co/Ni hydroxides:
| Co2+ + 2H2O = CoOOH + e− + 3H+ | (27) |
As such reactions include the participation of protons or hydroxide ions, the possibility and rate are highly dependent on pH, especially the local pH, which may vary during cycling. In addition, the dissolution–precipitation process depends on the solubility of the species, which, in turn, can be affected by the electrolyte concentration (e.g., by salting out in highly concentrated electrolytes). Thus, dissolution can be slowed down, and redeposition can occur directly on the surface of the active material, which is called “activation” (Fig. 17b). The case of dissolution without deposition on the current collector or carbon would lead to capacity decay. Therefore, it is of great importance to study the active material after cycling in case there is any evidence of “activation”, which, in combination with CV or voltage-shape changes, can be indicators of the phase-transformation phenomena.
In the simplest way, the deposit is formed according to the Pourbaix diagram. As seen for manganese (Fig. 15c), vanadium (Fig. 16f), cobalt (Fig. 17c), and nickel (Fig. 17d), the operation range of AZIBs overlaps with various oxide/hydroxide deposition regions. Hence, the new deposits start acting as electroactive materials, which are available only when they are deposited on the conductive part of the cell. The precise compositions of deposits may differ from the predicted M–H2O diagram because the presence of Zn2+ ions may be favorable for Zn-metal-oxide formation such as in the case of manganese-oxide phases. Nevertheless, the understanding of their formation through dissolution–precipitation routes appears to be general for most activation phenomena in the literature.
We suppose that the general driving force behind such activation–redeposition–amorphization phenomena is the thermodynamic stability of oxide- and hydroxide-based phases with strong M–O bonds compared to phosphates, hexacyanoferrates, and similar complex compounds. Water, which easily participates in many reactions and provides H+ due to ionic equilibria, acts as a reagent and a mediator of such transitions. We should note that a vast number of studies on activation phenomena lack analysis of extensively cycled electrodes and electrolytes, and hence, the redeposition and formation of oxides upon initial activation are extremely underreported in the literature and often are not properly studied in performance-oriented publications. The examples shown in Fig. 17a cover only a limited number of reported phenomena.
Another conclusion, which we draw from the considered literature, is the limited possibility for creating multiple redox centers in a single framework to boost zinc storage, such as an approach to increase the specific capacity of Prussian blue analogues by adding cobalt or vanadium atoms to the lattice. The specific capacity can indeed be increased via so-called activation;241 however, that increase is a result of phase separation274 due to proton- and water-assisted reactions in which at least two distinct phases and not two redox “sites” of one framework act as active materials. Thus, instead of zinc storage, proton storage occurs in the deposited oxide or hydroxide. It should be noted that the activation phenomena of various electrodes in various cases are not entirely detrimental for achieving high capacity and good cyclability. More detailed investigation of the cycled electrode is necessary to critically determine which processes represent “activation” and what is the real active material after many cycles. Because the discussed process requires water, it is of high interest to compare the behavior of AZIBs with that of their nonaqueous counterparts, where redeposition phenomena are prohibited or limited.
5. Key differences between aqueous and non-aqueous ZIBs
One of the main advantages of nonaqueous systems is the applicability of a broader electrochemical stability window, which is also available for ZIBs. Thus, many organic solvents allow high-voltage cathodes, and, more importantly, the stability of the Zn anode is secured by eliminating the parasitic HER. A comparison of the appearance of a Zn electrode in Zn(OTf)2/DMSO electrolyte and ZnSO4/H2O electrolyte after 100 cycles of deposition–stripping is shown in Fig. 18a and b.337 Multiple cycling (or long conditioning) of zinc metal in conventional aqueous zinc sulfate solution results in H2 evolution, and the DMSO-based electrolyte ensures stable behavior. Such an electrolyte was employed in Zn|δ-MnO2 batteries, showing good capacity retention for 1000 cycles.337 Although the HER on Zn is relatively slow, the resulting rate is critical for aqueous cell swelling and failure (Fig. 18c). Li et al.338 compared the behavior of K1.6Mn1.2Fe(CN)6 (MnHCF) in aqueous Zn(ClO4)2 and TEGDME (tetraethylene glycol dimethyl ether)-based electrolytes (Fig. 18d and e). The tetraglyme-based cell enabled evidently better cyclability and capacity retention, allowing thousands of cycles.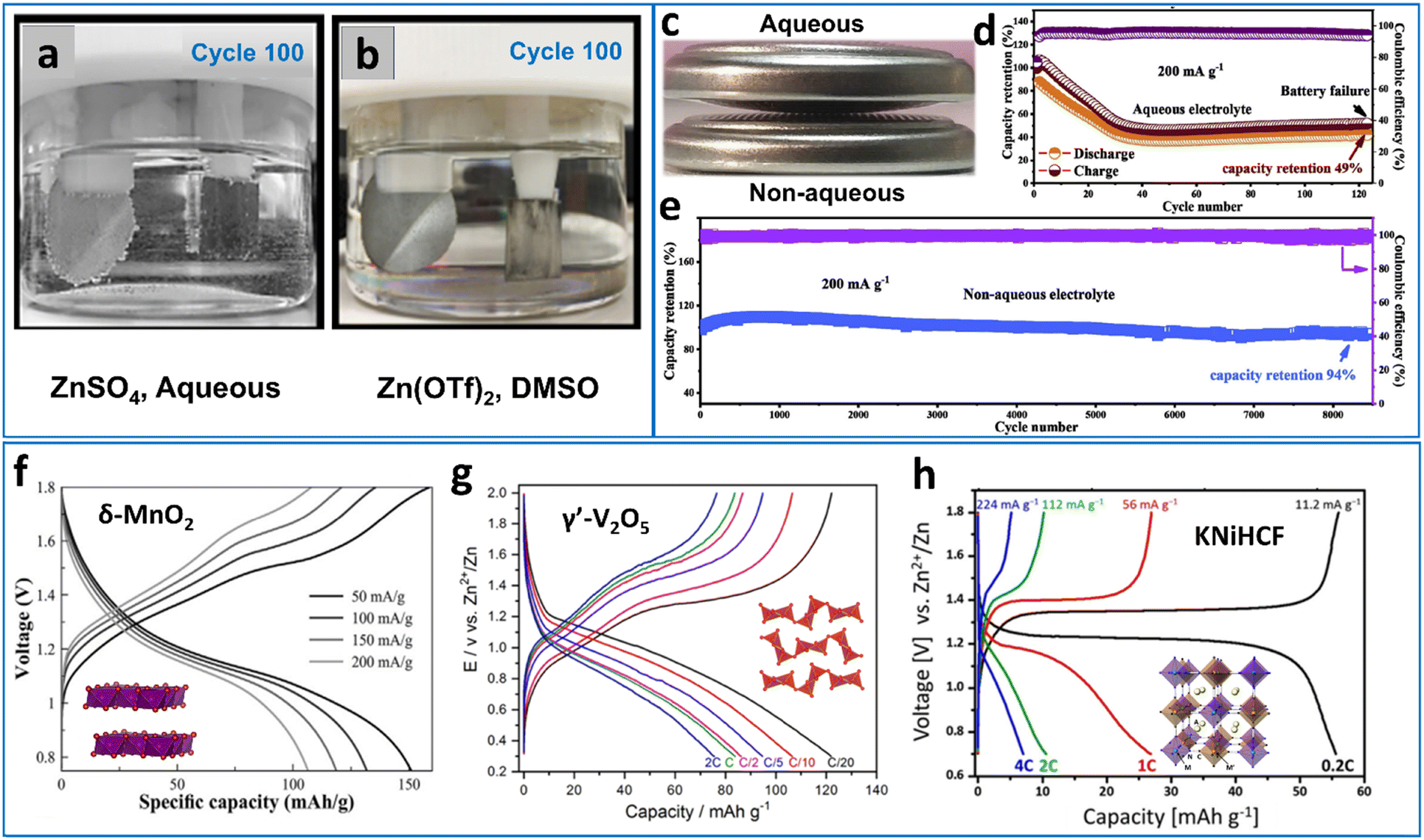 | ||
| Fig. 18 Non-aqueous ZIBs. Hydrogen evolution after 100 cycles for (a) 2 M ZnSO4/H2O and (b) 0.25 M Zn(OTf)2/DMSO. Reproduced with permission from ref. 337. Copyright 2021, Elsevier. (c) Zn|KMnFeHCF cells after cycling using Zn(ClO4)2 aqueous- and tetraglyme-based electrolyte and corresponding cyclability plots for (d), aqueous and (e), nonaqueous electrolyte. Reproduced with permission from ref. 338. Copyright 2020, Elsevier. Charge–discharge profiles for nonaqueous ZIBs using (f) δ-MnO2. Reproduced with permission from ref. 337. Copyright 2021, Elsevier. (g) γ′-V2O5 Adapted with permission from ref. 339. Copyright 2022, American Chemical Society. (h) KNiHCF Reproduced with permission from ref. 340. Copyright 2017, Elsevier. | ||
Regarding cathodes, non-aqueous electrolytes usually result in moderate capacities of approximately 50–150 mA h g−1 (Fig. 18f–h), which is far lower than the values obtained in aqueous systems. For instance, various studies on MnO231,337,341 revealed a smooth discharge profile without inflection, delivering a specific capacity in the range of 60–160 mA h g−1. This result is not surprising, as the abovementioned complex H2O-assisted dissolution–redeposition–amorphization phenomena are not available for nonaqueous Zn|MnO2 cells.
The difference between charge-storage ability using aqueous and nonaqueous electrolytes is much more strongly pronounced for vanadium oxides (Fig. 19a–c). For instance, Fig. 19a shows dis/charge profiles of the cathode material, δ-V2O5, in aqueous and acetonitrile-based electrolytes, with the capacity in the latter being only ≈25 mA h g−1vs. 300–350 mA h g−1 for the aqueous cell. Such results are similar to those of the abovementioned E-QCM study on nonaqueous electrolyte, where switching from H+ or H+/Zn2+ co-insertion to Zn2+ insertion in a nonaqueous system reduced the available capacity from ≈300 to 40 mA h g−1.275 Numerous vanadium oxides and metal vanadates are known to have capacities in the range of 300–450 mA h g−1 in AZIBs.99 However, there are only a few studies on nonaqueous ZIBs with vanadium oxides, with the capacity usually not exceeding ∼200 mA h g−1. To explain this gap, first, for general reasons, we must consider (a) kinetic limitations (interface charge transfer or solid-state diffusion) (Fig. 19d) or thermodynamic/structure limitation (the ability to store Zn2+ ions).
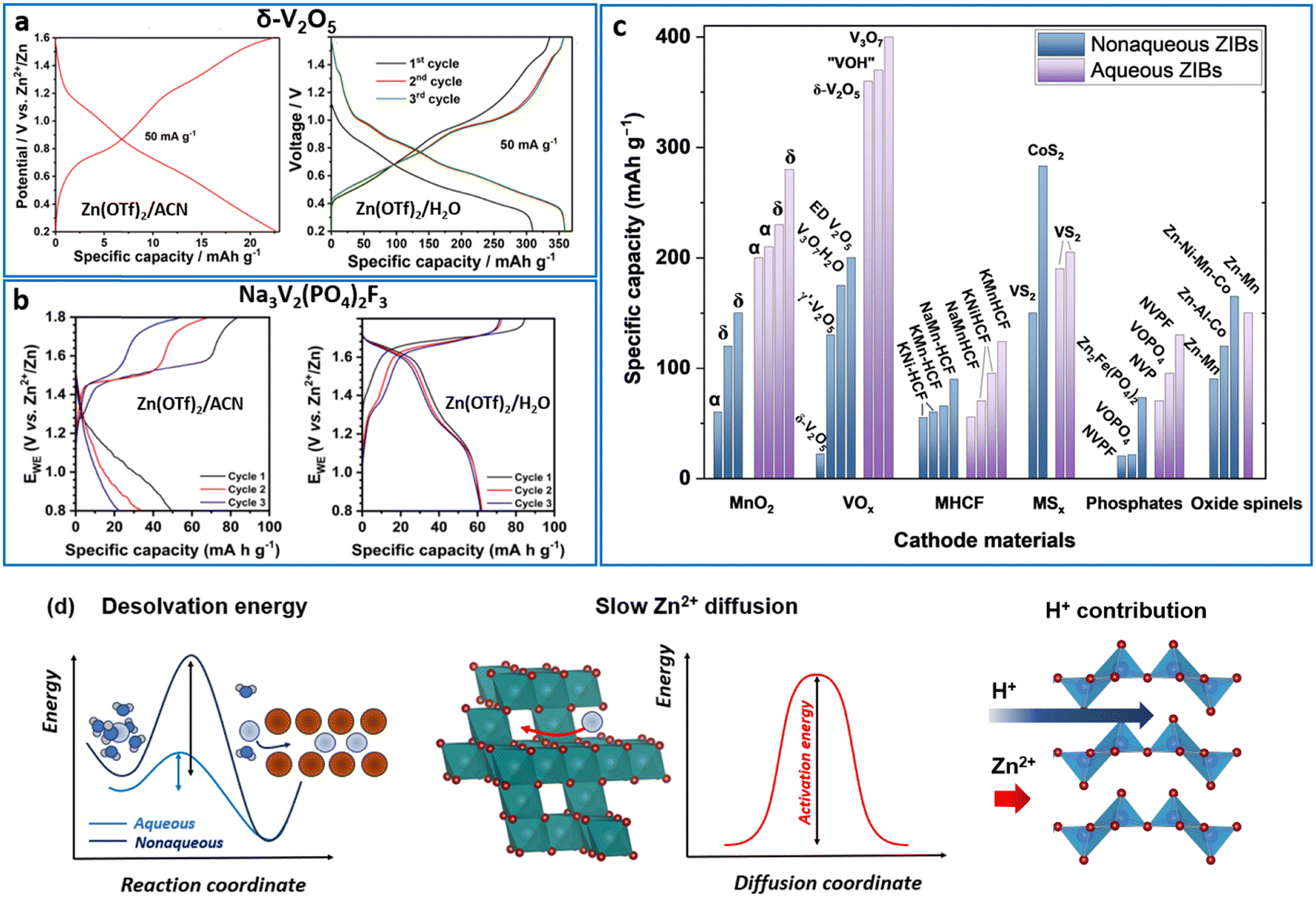 | ||
| Fig. 19 Aqueous vs. non-aqueous ZIBs. (a) Voltage profiles of δ-V2O5 in acetonitrile and water-based electrolytes. Adapted with permission from ref. 101. Copyright 2020, American Chemical Society. (b) Voltage profiles of Na3V2(PO4)2F3 in acetonitrile and water-based electrolytes. Adapted with permission from ref. 86. Copyright 2020, American Chemical Society. (c) Specific capacities achieved for different material families in nonaqueous and aqueous electrolytes.28–32,41,45,86,101,106,199,215,229,246,332,337–352 (d) Different reasons for inferior charge storage in nonaqueous ZIBs. | ||
Kundu et al.32 studied V3O7·H2O electrodes in both aqueous and nonaqueous electrolytes using operando XRD and electrochemical impedance spectroscopy (EIS) at various temperatures. Although they observed solid-solution-like changes in the XRD patterns for both electrolytes, the nonaqueous electrolyte allowed a capacity of only ∼60 mA h g−1, surprisingly lower than that obtained in the aqueous system (∼375 mA h g−1). A large difference in the activation energy of the interfacial charge transfer (19.47 kJ mol−1 in H2O vs. 75.34 kJ mol−1 in CH3CN) was proposed as evidence of the desolvation limitation at the interface which is the limiting factor for the nonaqueous Zn-ion storage.
In contrast, several studies have decisively shown that H+ (arising from Zn2+/H2O electrolyte) did contribute to the capacity along with Zn2+. For instance, Liu et al.101 comprehensively studied δ-V2O5 (Ca0.34V2O5) using XRD in both aqueous Zn(OTf)2, nonaqueous CH3CN-based Zn(OTf)2 electrolyte, and aqueous H2SO4 solution. Both Zn2+ and H+ were shown to be introduced via a solid-solution pathway. In addition, pH monitoring revealed Zn2+/H+ co-insertion behavior with a Zn2+/H+ exchange feature implying that at some potentials, Zn2+ intercalation can cause H+ deintercalation and vice versa. A recent study on pH monitoring in V2O5 has supported the co-insertion of H+ and Zn2+, where the H+ contribution strongly depends on the pH of the electrolyte.64 Additionally, a large number of AZIB articles showing high capacities of vanadium oxide and few reports on nonaqueous ZIBs with acceptable properties indirectly support the capacity being boosted by proton (co)insertion in AZIBs and moderate or limited ability to store Zn2+ ions in a VOx host for nonaqueous electrolytes (Fig. 19c).
Manganese- and vanadium-based oxide compounds are not the only material families that behave differently in nonaqueous media. Metal phosphates were found to be inferior in non-aqueous electrolytes, as reported for sodium vanadium fluoride phosphate and layered VOPO486,342 (Fig. 19b and c). Park and Manthiram86 found that the guest ion is H+ rather than Zn2+ in Na3V2(PO4)2F3, which explains the low capacity in nonaqueous cells, similar to the behavior of vanadium oxides. From this viewpoint, phosphate-based compounds need to be further investigated to clarify the reaction mechanism in nonaqueous ZIBs.
In contrast to metal oxides, metal hexacyanoferrates and metal sulfides demonstrate comparable charge-storage ability in both aqueous and nonaqueous solutions (Fig. 19c). As discussed earlier, metal hexacyanoferrates (MHCFs) have been actively studied in AZIBs with capacities reaching ∼60–100 mAh g−1.238–243 The use of nonaqueous electrolytes usually results in capacities of 50–90 mA h g−1,28,338,340,343 which are close to the values obtained in MHCF-based AZIBs. Such relatively low values are explained by the high molecular weight of the MHCF formula unit, rationalizing the delivery of capacity of 80–90 mA h g−1 provided the only Fe redox pair is active. Although some MHCFs were shown to overcome the one-electron limit in aqueous media by introducing Co353 or V241 in the HCF framework, the true reason for such activation is most likely associated with phase separation and formation of oxides due to reactions with water.274,332 As such reactions are prohibited in nonaqueous media, overcoming the theoretical limit of ∼90 mA h g−1 for nonaqueous MHCF-based ZIBs is hardly achievable.
Metal sulfides, in contrast to metal oxides, have demonstrated high and comparable capacities both in aqueous and organic-based electrolytes344,345,351,352 (Fig. 19c). Zhang et al.345 demonstrated a prominent capacity of 283 mA h g−1 for CoS2 in Zn(OTf)2/acetonitrile electrolyte. The capacity was achieved not by cobalt but by sulfide/disulfide anionic redox. No XRD peak shift was observed, whereas irreversible changes in the structure were detected from Zn2+ intercalation into the electrode material, likely leading to a poorly crystalline new phase. The other group used 0.5 M Zn(OTf)2/TMP electrolyte that enabled a long-lasting Zn|VS2 cell with a capacity of ∼140 mA h g−1.344 The structural analysis indicated irreversible changes occurring along with Zn2+ insertion/extraction, as evidenced by XRD. As the work was primarily focused on optimizing Zn electrode|electrolyte stability, no detailed information on the VS2 redox type was provided. The (de)intercalation behavior of Zn2+ was suggested in aqueous Zn|VS2 batteries by observing reversible shifts in XRD patterns and other analyses.352 The capacity did not greatly exceed the observed values in the nonaqueous system344 with minor or no proton contribution to the VS2/Zn2+ electrochemistry.
Finally, oxide spinel compounds have been reported as promising cathode materials for both aqueous and nonaqueous ZIBs both in terms of capacity and high operation voltage.30,199,215 This finding appears to be in disagreement with the degradation phenomena in the aqueous Zn|MnO2 system, for which the capacity of the cell fades concurrently with the formation of the ZnMn2O4 phase and Zn–Mn–O spinel that are poorly active. On the other hand, Zhang et al.215 used non-stoichiometric ZnMn2O4 spinel with Mn deficiency in the lattice in aqueous zinc triflate electrolyte. Interestingly, unlike for most MnO2 polymorphs, a double-plateau discharge profile, typical for dissolution–redeposition MnO2 phenomena, was not observed. The same electrode also demonstrated a moderate capacity (90 mA h g−1) in an acetonitrile-based electrolyte. Another group reported nonaqueous ZIBs using ZnAlxCo2−xO430 and ZnNixMnxCo2–2xO4199 spinels. Using ZnAlxCo2−xO4 allowed a capacity of 120 mA h g−1 at 0.1C in a Zn(OTf)2 solution in acetonitrile. The Zn–Ni–Mn–Co spinel was shown to have an even higher capacity, reaching ∼180 mA h g−1, enabled by Ni and Co redox pairs. Both systems demonstrated some issues with the CE being ∼90%–95% even after 100 cycles. An amine base additive, 200 ppm of 1,4-diazabicyclo [2.2.2]octane (DABCO), was used for Zn–Ni–Mn–Co spinel to make it feasible by suppressing electrolyte oxidation.199
However, despite the high potential and reasonable capacity in reported spinel work, this direction for nonaqueous ZIBs has not yet gained much attention and development. In addition to composition and defect management, all the aforementioned studies have addressed nanoparticle materials (12–20 nm for Zn–Mn, 10–25 nm for Zn–Al–Co, and 10–100 nm Zn–Ni–Mn–Co). The Zn–Al–Co and Zn–Ni–Mn–Co spinels were prepared by citric sol–gel technique, whereas the nano-sized ZnMn2O4/C spinel composite was obtained by ammonia-induced Zn2+, Mn2+ precipitation followed by 180 °C heating. Exploiting even 50-nm small ZnMn2O4 particles could achieve only ∼30 mA h g−1 because of the slow Zn2+ diffusion into the spinel structure. For spinel oxides to be active in nonaqueous ZIBs, several factors should be treated and optimized thoroughly: the use of an appropriate synthetic route to obtain nanomaterials with a high surface area and particle size not exceeding 30–50 nm to overcome slow divalent diffusion limitation; the use of composites with nanocarbon to mitigate conductivity and interparticle contact issues; and finding an appropriate electrolyte or electrolyte additive to suppress side reactions at high potentials.
In summary, various types of materials behave differently in aqueous and non-aqueous ZIBs (Fig. 19c and d). The oxide- and phosphate-based materials possess obviously inferior charge-storage ability towards Zn2+ compared to H+, which is the reason for the typically poor performance in nonaqueous electrolytes. In addition, kinetic reasons such as lower desolvation energy or higher diffusion rates boost high capacities in such frameworks in aqueous environments. Finally, the ability of water to accept or donate protons and dissolve various species enables deposition–dissolution reactions. In contrast, some metal sulfides, metal hexacyanoferrates, and spinels show similar capacities both in aqueous and non-aqueous electrolytes and demonstrate the ability to accept Zn2+; however, much more elaboration is required to improve the long-term electrode performance.
6. Conclusions and perspective
The role of water in AZIBs is much deeper and more complex than merely acting as a highly polar solvent. As has been intensively studied in the last few years, water is involved in many processes at the cathode and anode. Some of these processes are useful for achieving high performance of AZIBs, whereas others are harmful.The beneficial processes include capacity boosting by H+ intercalation or co-intercalation in vanadium-oxide electrodes and the high-capacity deposition–dissolution chemistry of MnO2-Mn2+, which is supported by Zn2+-H2O acid–base chemistry. From one point of view, Zn2+-H2O equilibria enable versatile ion-hybrid batteries with dual Zn2+/H+ roles. For this reason, aqueous zinc-ion batteries outperform their nonaqueous counterparts as well as Ca- and Mg-ion batteries, which are difficult to work with. However, the high water activity and low potential stability window are responsible for many undesirable reactions, such as zinc corrosion, which limits battery life and stimulates dendrite formation, dissolution of cathode materials, and amorphization.
Thus, there lies a question: which direction of ZIB development is beneficial in terms of battery performance and which direction of research is crucial for process understanding? For practice, one would desire to merge the high capacities and promising kinetics/power of AZIBs with the high stability and absence of various side reactions of non-aqueous ZIBs. Thus, various approaches to control the water activity354 may be used when selecting the appropriate electrolyte (Fig. 20).
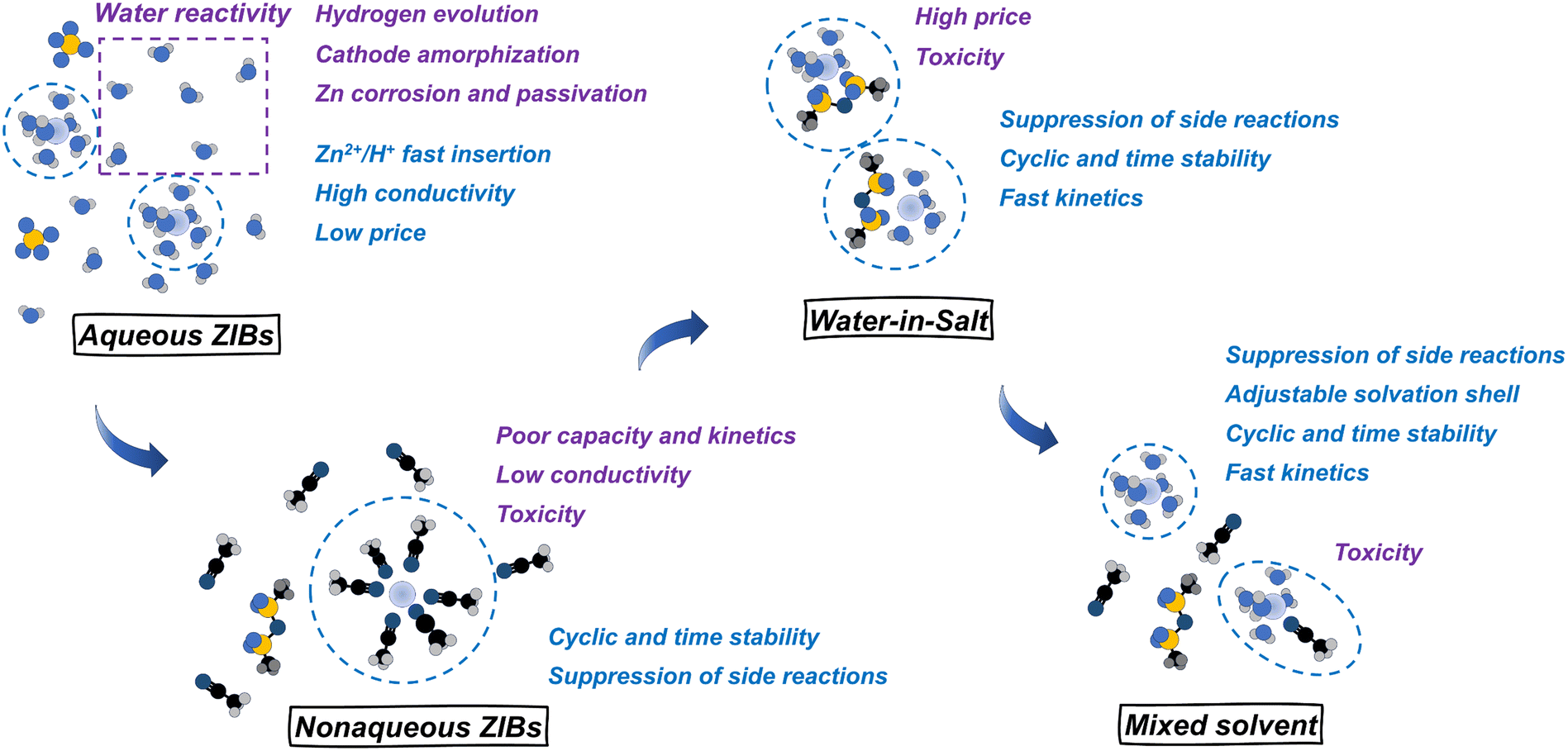 | ||
| Fig. 20 Research development on rechargeable Zn-ion batteries electrolytes. | ||
A well-known approach for decreasing the water activity and decelerating and suppressing side reactions is using highly concentrated water-in-salt electrolytes.355 Although such systems show promising performance, their scalability and applicability remain unclear as the salts used are usually expensive and the use of high concentrations raises price and toxicity issues. Another approach involves the use of mixed solvents, e.g., (H2O-PC); here, water enables hydration or some proton-donating properties beneficial for kinetics; however, its low concentration and interaction with an organic co-solvent decreases the activity and eliminates many harmful side reactions.35,36,146,147,174 The potential downside of such a system might be organic solvent toxicity but, eventually, it would depend on the solvent type used.
From the applied science view, we propose that the combined solvent approach holds the most promise, such as the use of organic-aqueous based mixed electrolytes to limit water activity and suppress corrosion and other undesired processes while benefiting from the H2O-driven capacity boost by H+-intercalation or H2O-solvated Zn2+ intercalation. At the same time, the mixed solvent-based electrolyte is inherently less expensive than WiS electrolyte due to the use of reasonable amounts of salts. From a fundamental perspective, studies on AZIBs require a broader use of research approaches and studies related to dissolved species and dissolution–deposition processes, particularly extensive use of UV-vis spectroscopy and operando pH monitoring. Due to the highly dynamic oxide–hydroxide chemistry, amorphization processes should be studied in detail, and comprehensive post-cycled electrode characterization is needed to identify the real active phase(s) and understand the cycling mechanism for prolonged cycling.
Author contributions
Yauhen Aniskevich: writing – original draft, visualization, validation, methodology, investigation, formal analysis, data curation, conceptualization. Seung-Taek Myung: writing – review & editing, validation, methodology, investigation, funding acquisition, data curation, conceptualization.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the International Research & Development Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT of Korea (NRF-2020R1A6A1A03043435, NRF-2022M3H4A1A04096478, NRF-2023R1A2C2003210, and NRF-2022H1D3A2A02092011).References
- Y. Liang, C. Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Q. Huang, D. Yu, Y. Liu, M. M. Titirici, Y. L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CAS.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 CAS.
- M. Walter, M. V. Kovalenko and K. V. Kravchyk, New J. Chem., 2020, 44, 1677–1683 CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CAS.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CAS.
- S. W. Kim, D. H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CAS.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Record, 2018, 18, 459–479 CAS.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- M. A. Schroeder, L. Ma, G. Pastel and K. Xu, Curr. Opin. Electrochem., 2021, 29, 100819 CrossRef CAS.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. W. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869 CrossRef.
- M. M. Huie, D. C. Bock, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, Coord. Chem. Rev., 2015, 287, 15–27 CrossRef CAS.
- M. E. Arroyo-De Dompablo, A. Ponrouch, P. Johansson and M. R. Palacín, Chem. Rev., 2020, 120, 6331–6357 CrossRef CAS PubMed.
- G. A. Elia, K. V. Kravchyk, M. V. Kovalenko, J. Chacón, A. Holland and R. G. A. Wills, J. Power Sources, 2021, 481, 228870 CrossRef CAS.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- Q. Li, J. Zhang, L. Zhong, F. Geng, Y. Tao, C. Geng, S. Li, B. Hu and Q. H. Yang, Adv. Energy Mater., 2022, 12, 2201734 CAS.
- Y. Aniskevich, J. H. Yu, J. Y. Kim, S. Komaba and S. T. Myung, Adv. Energy Mater., 2024, 14, 2304300 CAS.
- B. Yong, D. Ma, Y. Wang, H. Mi, C. He and P. Zhang, Adv. Energy Mater., 2020, 10, 2002354 CAS.
- Y. Shang and D. Kundu, Curr. Opin. Electrochem., 2022, 33, 100954 CAS.
- T. Yamamoto and T. Shoji, Inorg. Chim. Acta, 1986, 117, L27–L28 CAS.
- E. Gocke, W. Schramm, P. Dolscheid and R. Schöllhorn, J. Solid State Chem., 1987, 70, 71–81 CAS.
- N. S. Venkata Narayanan, B. V. Ashokraj and S. Sampath, J. Colloid Interface Sci., 2010, 342, 505–512 CAS.
- T. J. Simons, M. Salsamendi, P. C. Howlett, M. Forsyth, D. R. Macfarlane and C. Pozo-Gonzalo, ChemElectroChem, 2015, 2, 2071–2078 CAS.
- A. L. Lipson, S. D. Han, S. Kim, B. Pan, N. Sa, C. Liao, T. T. Fister, A. K. Burrell, J. T. Vaughey and B. J. Ingram, J. Power Sources, 2016, 325, 646–652 CAS.
- P. Senguttuvan, S. D. Han, S. Kim, A. L. Lipson, S. Tepavcevic, T. T. Fister, I. D. Bloom, A. K. Burrell and C. S. Johnson, Adv. Energy Mater., 2016, 6, 1600826 Search PubMed.
- C. Pan, R. G. Nuzzo and A. A. Gewirth, Chem. Mater., 2017, 29, 9351–9359 CrossRef CAS.
- S. D. Han, S. Kim, D. Li, V. Petkov, H. D. Yoo, P. J. Phillips, H. Wang, J. J. Kim, K. L. More, B. Key, R. F. Klie, J. Cabana, V. R. Stamenkovic, T. T. Fister, N. M. Markovic, A. K. Burrell, S. Tepavcevic and J. T. Vaughey, Chem. Mater., 2017, 29, 4874–4884 CrossRef CAS.
- D. Kundu, S. Hosseini Vajargah, L. Wan, B. Adams, D. Prendergast and L. F. Nazar, Energy Environ. Sci., 2018, 11, 881–892 RSC.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- J.-Q. Huang, X. Guo, X. Lin, Y. Zhu and B. Zhang, Research, 2019, 2019, 2635310 CAS.
- Y. Dong, L. Miao, G. Ma, S. Di, Y. Wang, L. Wang, J. Xu and N. Zhang, Chem. Sci., 2021, 12, 5843–5852 RSC.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., 2012, 124, 957–959 CrossRef.
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemSusChem, 2016, 9, 2948–2956 CrossRef CAS PubMed.
- R. Trócoli and F. L. Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- G. Li, Z. Yang, Y. Jiang, C. Jin, W. Huang, X. Ding and Y. Huang, Nano Energy, 2016, 25, 211–217 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- Y. Liao, H. C. Chen, C. Yang, R. Liu, Z. Peng, H. Cao and K. Wang, Energy Storage Mater., 2022, 44, 508–516 CrossRef.
- U. Siamionau, Y. Aniskevich, A. Mazanik, O. Kokits, G. Ragoisha, J. H. Jo, S. T. Myung and E. Streltsov, J. Power Sources, 2022, 523, 231023 CrossRef CAS.
- B. Sambandam, V. Mathew, S. Kim, S. Lee, S. Kim, J. Y. Hwang, H. J. Fan and J. Kim, Chem, 2022, 8, 924–946 CAS.
- V. R. Kankanallu, X. Zheng, D. Leschev, N. Zmich, C. Clark, C. H. Lin, H. Zhong, S. Ghose, A. M. Kiss, D. Nykypanchuk, E. Stavitski, E. S. Takeuchi, A. C. Marschilok, K. J. Takeuchi, J. Bai, M. Ge and Y. C. K. Chen-Wiegart, Energy Environ. Sci., 2023, 16, 2464–2482 CAS.
- H. Alt, H. Binder, G. Klempert, A. Köhling and G. Sandstede, J. Appl. Electrochem., 1972, 2, 193–200 CrossRef CAS.
- H. Alt, H. Binder, A. Köhling and G. Sandstede, Electrochim. Acta, 1972, 17, 873–887 CrossRef CAS.
- A. G. Macdiarmid, S.-L. Mu, N. L. D. Somasiri and W. Wu, Mol. Cryst. Liq. Cryst., 1985, 121, 187–190 CrossRef CAS.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Chem. Commun., 2009, 836–838 RSC.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Macromol. Chem. Phys., 2009, 210, 1989–1995 CrossRef CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- Z. Guo, Y. Uanyuan Ma, X. Dong, J. Ianhang Huang, Y. Onggang Wang and Y. Xia, Angew. Chem., 2018, 130, 11911–11915 CrossRef.
- Z. Song, L. Miao, H. Duan, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208821 CrossRef CAS PubMed.
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 Search PubMed.
- J. Cui, Z. Guo, J. Yi, X. Liu, K. Wu, P. Liang, Q. Li, Y. Liu, Y. Wang, Y. Xia and J. Zhang, ChemSusChem, 2020, 13, 2160–2185 CrossRef CAS PubMed.
- V. Verma, S. Kumar, W. Manalastas and M. Srinivasan, ACS Energy Lett., 2021, 6, 1773–1785 CrossRef CAS.
- X. Ji and L. F. Nazar, Nat. Sustainability, 2024, 7, 98–99 Search PubMed.
- M. Li, Z. Li, X. Wang, J. Meng, X. Liu, B. Wu, C. Han and L. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- C. Nie, G. Wang, D. Wang, M. Wang, X. Gao, Z. Bai, N. Wang, J. Yang, Z. Xing and S. Dou, Adv. Energy Mater., 2023, 13, 2300606 Search PubMed.
- A. Bayaguud, Y. Fu and C. Zhu, J. Energy Chem., 2022, 64, 246–262 CrossRef CAS.
- L. Wang, J. Yan, Y. Hong, Z. Yu, J. Chen and J. Zheng, Sci. Adv., 2023, 9, eadf4589 Search PubMed.
- C. Lee, Y. Hong, D. Kim, Y. Lim, J. W. Choi and S. Chung, Adv. Funct. Mater., 2023, 33, 2303763 CrossRef CAS.
- J. H. Jo, Y. Aniskevich, J. Kim, J. U. Choi, H. J. Kim, Y. H. Jung, D. Ahn, T. Y. Jeon, K. S. Lee, S. H. Song, H. Kim, G. Ragoisha, A. Mazanik, E. Streltsov and S. T. Myung, Adv. Energy Mater., 2020, 10, 2001595 CrossRef CAS.
- J. Shin, J. Lee, Y. Park and J. W. Choi, Chem. Sci., 2020, 11, 2028–2044 Search PubMed.
- X. Zhang, J. P. Hu, N. Fu, W. Bin Zhou, B. Liu, Q. Deng and X. W. Wu, InfoMat, 2022, 4, e12306 CAS.
- H. J. Kim, S. Kim, K. Heo, J.-H. Lim, H. Yashiro, S.-T. Myung, H. J. Kim, S. Kim, K. Heo, S.-T. Myung, J.-H. Lim and H. Yashiro, Adv. Energy Mater., 2023, 13, 2203189 CAS.
- V. Mathew, B. Sambandam, S. Kim, S. Kim, S. Park, S. Lee, M. H. Alfaruqi, V. Soundharrajan, S. Islam, D. Y. Putro, J. Y. Hwang, Y. K. Sun and J. Kim, ACS Energy Lett., 2020, 5, 2376–2400 CAS.
- J. O. Bockris, Z. Nagy and D. Drazic, J. Electrochem. Soc., 1973, 120, 30 CAS.
- M. Schlesinger and M. Paunovic, Modern Electroplating: Fifth Edition, John Wiley and Sons, 2010 Search PubMed.
- N. Sorour, W. Zhang, E. Ghali and G. Houlachi, Hydrometallurgy, 2017, 171, 320–332 CAS.
- Y. D. Gamburg and G. Zangari, Theory and Practice of Metal Electrodeposition, Springer, New York, 2011 Search PubMed.
- A. Agnew and R. Paterson, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 2885–2895 RSC.
- J. T. Hinatsu, V. D. Tran and F. R. Foulkes, J. Appl. Electrochem., 1992, 22, 215–223 CrossRef CAS.
- S. D. Han, N. N. Rajput, X. Qu, B. Pan, M. He, M. S. Ferrandon, C. Liao, K. A. Persson and A. K. Burrell, ACS Appl. Mater. Interfaces, 2016, 8, 3021–3031 CrossRef CAS PubMed.
- Y. Zhang and M. Muhammed, Hydrometallurgy, 2001, 60, 215–236 CrossRef CAS.
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, ACS Energy Lett., 2021, 6, 1015–1033 CrossRef CAS.
- C. Jana, G. Ohanessian and C. Clavaguéra, Theor. Chem. Acc., 2016, 135, 141 Search PubMed.
- P. Vanysek, CRC Hand Book Chem. Phys., 1993, 5–92 Search PubMed.
- A. Moezzi, A. McDonagh, A. Dowd and M. Cortie, Inorg. Chem., 2013, 52, 95–102 CrossRef CAS PubMed.
- S. Cousy, N. Gorodylova, L. Svoboda and J. Zelenka, Chem. Pap., 2017, 71, 2325–2334 CrossRef CAS.
- A. Moezzi, M. Cortie and A. M. McDonagh, Eur. J. Inorg. Chem., 2013, 1326–1335 CrossRef CAS.
- A. Moezzi, M. B. Cortie and A. M. McDonagh, Dalton Trans., 2013, 42, 14432–14437 RSC.
- C. S. Cordeiro, G. G. C. Arizaga, L. P. Ramos and F. Wypych, Catal. Commun., 2008, 9, 2140–2143 CrossRef CAS.
- M. J. Park and A. Manthiram, ACS Appl. Energy Mater., 2020, 3, 5015–5023 CrossRef CAS.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39, 107–114 CrossRef CAS.
- H. L. Clever, M. E. Derrick and S. A. Johnson, J. Phys. Chem. Ref. Data, 1992, 21, 941–1004 CrossRef CAS.
- X. Wang, J. Li, G. K. Das, S. Johanie, C. Vernon and R. Shaw, J. Solid State Chem., 2020, 290, 121483 CrossRef CAS.
- M. Ohnishi, I. Kusachi and S. Kobayashi, Can. Mineral., 2007, 45, 1511–1517 CrossRef CAS.
- F. C. Hawthorne and E. Sokolova, Can. Mineral., 2002, 40, 939–946 CrossRef CAS.
- L. A. Groat, Am. Mineral., 1996, 81, 238–243 CAS.
- A. Gordeeva, Y. J. Hsu, I. Z. Jenei, P. H. B. Brant Carvalho, S. I. Simak, O. Andersson and U. Haüssermann, ACS Omega, 2020, 5, 17617–17627 CAS.
- L. S. Germann, R. E. Dinnebier, X. Liu, Y. Dong and W. Li, Z. Anorg. Allg. Chem., 2016, 642, 255–259 CAS.
- N. Thomas and M. Rajamathi, J. Colloid Interface Sci., 2011, 362, 493–496 CAS.
- T. Biswick, W. Jones, A. Pacuła and E. Serwicka, J. Solid State Chem., 2006, 179, 49–55 CAS.
- A. Ziba, A. Pacuła, E. M. Serwicka and A. Drelinkiewicz, Fuel, 2010, 89, 1961–1972 Search PubMed.
- M. Liu, P. Wang, W. Zhang, H. He, G. He, S. Xu, L. Yao and T. S. Miller, Energy Storage Mater., 2024, 67, 103248 Search PubMed.
- J. Ding, H. Gao, D. Ji, K. Zhao, S. Wang and F. Cheng, J. Mater. Chem. A, 2021, 9, 5258–5275 CAS.
- H. J. Kim, J. H. Jo, J. Y. Kim, J. Jeong, J. H. Park, H. G. Jung, K. Y. Chung, M. G. Kim, N. Lee, K. S. Sohn, Y. Aniskevich, E. Streltsov and S. T. Myung, Energy Storage Mater., 2023, 55, 105–116 Search PubMed.
- X. Liu, H. Euchner, M. Zarrabeitia, X. Gao, G. A. Elia, A. Groß and S. Passerini, ACS Energy Lett., 2020, 5, 2979–2986 CrossRef CAS PubMed.
- P. Oberholzer, E. Tervoort, A. Bouzid, A. Pasquarello and D. Kundu, ACS Appl. Mater. Interfaces, 2019, 11, 674–682 CrossRef CAS PubMed.
- N. J. Herrmann, H. Euchner, A. Groß and B. Horstmann, Adv. Energy Mater., 2024, 14, 2302553 CrossRef CAS.
- X. Gao, H. Wu, W. Li, Y. Tian, Y. Zhang, H. Wu, L. Yang, G. Zou, H. Hou and X. Ji, Small, 2020, 16, 1905842 CrossRef CAS PubMed.
- Z. Tang, W. Chen, Z. Lyu and Q. Chen, Energy Mater. Adv., 2022, 2022, 9765710 Search PubMed.
- D. Wu, L. M. Housel, S. T. King, Z. R. Mansley, N. Sadique, Y. Zhu, L. Ma, S. N. Ehrlich, H. Zhong, E. S. Takeuchi, A. C. Marschilok, D. C. Bock, L. Wang and K. J. Takeuchi, J. Am. Chem. Soc., 2022, 144, 23405–23420 CrossRef CAS PubMed.
- S. Deng, Z. Tie, F. Yue, H. Cao, M. Yao and Z. Niu, Angew. Chem., Int. Ed., 2022, 134, e202115877 CrossRef.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- G. Singh, C. R. Tang, A. Nicoll, J. Torres, L. M. Housel, L. Wang, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, J. Phys. Chem. C, 2023, 127, 3940–3951 CrossRef CAS PubMed.
- C. Wang, S. Wei, S. Chen, D. Cao and L. Song, Small Methods, 2019, 3, 1900495 CrossRef CAS.
- M. Tamilselvan, T. V. M. Sreekanth, K. Yoo and J. Kim, J. Ind. Eng. Chem., 2021, 93, 176–185 CrossRef CAS.
- Y. Dong, M. Jia, Y. Wang, J. Xu, Y. Liu, L. Jiao and N. Zhang, ACS Appl. Energy Mater., 2020, 3, 11183–11192 CrossRef CAS.
- J. S. Park, J. H. Jo, Y. Aniskevich, A. Bakavets, G. Ragoisha, E. Streltsov, J. Kim and S. T. Myung, Chem. Mater., 2018, 30, 6777–6787 CrossRef CAS.
- Z. Li, S. Ganapathy, Y. Xu, Z. Zhou, M. Sarilar and M. Wagemaker, Adv. Energy Mater., 2019, 9, 1900237 Search PubMed.
- L. A. Hernandez-Alvarado, L. S. Hernandez, J. M. Miranda and O. Dominguez, Anti-Corros. Methods Mater., 2009, 56, 114–120 CAS.
- L. Zhang, I. A. Rodríguez-Pérez, H. Jiang, C. Zhang, D. P. Leonard, Q. Guo, W. Wang, S. Han, L. Wang and X. Ji, Adv. Funct. Mater., 2019, 29, 1902653 Search PubMed.
- L. Li, S. Liu, W. Liu, D. Ba, W. Liu, Q. Gui, Y. Chen, Z. Hu, Y. Li and J. Liu, Nanomicro Lett., 2021, 13, 34 Search PubMed.
- M. Cui, N. Ma, H. Lei, Y. Liu, W. Ling, S. Chen, J. Wang, H. Li, Z. Li, J. Fan and Y. Huang, Angew. Chem., Int. Ed., 2023, 62, e202303845 CrossRef CAS PubMed.
- Q. Pan, R. Dong, H. Lv, X. Sun, Y. Song and X. X. Liu, Chem. Eng. J., 2021, 419, 129491 CrossRef CAS.
- Q. Cui, K. Yu, N. Zhang and Z. Zhu, Appl. Surf. Sci., 2008, 254, 3517–3521 CrossRef CAS.
- P. Yu, J. Wang, X. Gan, Z. Guo, L. Huang and Z. Song, Batter Supercaps, 2023, 6, e202300010 CrossRef CAS.
- W. Sun, V. Küpers, F. Wang, P. Bieker and M. Winter, Angew. Chem., Int. Ed., 2022, 61, e202207353 CrossRef CAS PubMed.
- X. Liu, Q. Han, Q. Ma, Y. Wang and C. Liu, Small, 2022, 18, 2203327 CrossRef CAS PubMed.
- X. Liu, G. Xu, Q. Zhang, S. Huang, L. Li, X. Wei, J. Cao, L. Yang and P. K. Chu, J. Power Sources, 2020, 463, 228223 CrossRef CAS.
- R. A. Robinson and R. S. Jones, J. Am. Chem. Soc., 1936, 58, 959–961 CrossRef CAS.
- R. H. Stokes and B. J. Levien, J. Am. Chem. Soc., 1946, 68, 333–337 CrossRef CAS.
- Lutfullah, H. S. Dunsmore and R. Paterson, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 495–503 RSC.
- Z. Cai, Y. Ou, J. Wang, R. Xiao, L. Fu, Z. Yuan, R. Zhan and Y. Sun, Energy Storage Mater., 2020, 27, 205–211 CrossRef.
- J. Zhao, Z. Lv, S. Wang, Z. Chen, Z. Meng, G. Li, C. Guo, T. Liu and J. Hui, Small Methods, 2023, 7, 2300731 CAS.
- H. J. Kim, S. Kim, S. Kim, S. Kim, K. Heo, J. H. Lim, H. Yashiro, H. J. Shin, H. G. Jung, Y. M. Lee and S. T. Myung, Adv. Mater., 2024, 36, 2308592 CrossRef CAS PubMed.
- J. Xiao, Science, 2019, 366, 426–427 CAS.
- T. Foroozan, S. Sharifi-Asl and R. Shahbazian-Yassar, J. Power Sources, 2020, 461, 228135 CAS.
- Y. S. Hong, N. Li, H. Chen, P. Wang, W. L. Song and D. Fang, Energy Storage Mater., 2018, 11, 118–126 Search PubMed.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CAS.
- M. Tribbia, J. Glenneberg, G. Zampardi and F. La Mantia, Batter. Supercaps, 2022, 5, e202100381 CAS.
- Z. Zhao, P. Li, Z. Zhang, H. Zhang and G. Li, Chem. Eng. J., 2023, 454, 140435 CAS.
- L. N. Bengoa, P. Pary, P. R. Seré, M. S. Conconi and W. A. Egli, J. Mater. Eng. Perform., 2018, 27, 1103–1108 CAS.
- M. Zhang, H. Hua, P. Dai, Z. He, L. Han, P. Tang, J. Yang, P. Lin, Y. Zhang, D. Zhan, J. Chen, Y. Qiao, C. C. Li, J. Zhao and Y. Yang, Adv. Mater., 2023, 35, 2208630 CAS.
- Y. Lyu, J. A. Yuwono, P. Wang, Y. Wang, F. Yang, S. Liu, S. Zhang, B. Wang, K. Davey, J. Mao and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202303011 CrossRef CAS PubMed.
- K. Roy, A. Rana, J. N. Heil, B. M. Tackett and J. E. Dick, Angew. Chem., Int. Ed., 2024, 63, e202319010 CrossRef CAS PubMed.
- K. Roy, A. Rana, T. K. Ghosh, J. N. Heil, S. Roy, K. J. Vannoy, B. M. Tackett, M. Chen and J. E. Dick, Adv. Energy Mater., 2024, 14, 2303998 CrossRef CAS.
- H. Yang, Y. Yang, W. Yang, G. Wu and R. Zhu, Energy Environ. Sci., 2024, 17, 1975–1983 RSC.
- H. Li, L. Yang, S. Zhou, J. Li, Y. Chen, X. Meng, D. Xu, C. Han, H. Duan and A. Pan, Adv. Funct. Mater., 2024, 34, 2313859 CrossRef CAS.
- K. Ouyang, S. Chen, W. Ling, M. Cui, Q. Ma, K. Zhang, P. Zhang and Y. Huang, Angew. Chem., 2023, 135, e202311988 CrossRef.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang, J. Zhu, F. Guo, X. Gao, Z. Du, J. Chen, T. Wang, G. He and I. P. Parkin, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- F. Ming, Y. Zhu, G. Huang, A. H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- L. Miao, R. Wang, S. Di, Z. Qian, L. Zhang, W. Xin, M. Liu, Z. Zhu, S. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9678 CrossRef CAS PubMed.
- J. Zhao, Y. Li, X. Peng, S. Dong, J. Ma, G. Cui and L. Chen, Electrochem. Commun., 2016, 69, 6–10 CrossRef CAS.
- Z. Khan, D. Kumar and X. Crispin, Adv. Mater., 2023, 35, 2300369 CrossRef CAS PubMed.
- J. Wang, J. X. Tian, G. X. Liu, Z. Z. Shen and R. Wen, Small Methods, 2023, 7, 2300392 CrossRef CAS PubMed.
- S. Watzele, J. Fichtner, B. Garlyyev, J. N. Schwämmlein and A. S. Bandarenka, ACS Catal., 2018, 8, 9456–9462 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- C. Liu, Z. Luo, W. Deng, W. Wei, L. Chen, A. Pan, J. Ma, C. Wang, L. Zhu, L. Xie, X. Y. Cao, J. Hu, G. Zou, H. Hou and X. Ji, ACS Energy Lett., 2021, 6, 675–683 CrossRef CAS.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023, 35, 2300577 CrossRef CAS PubMed.
- L. Wang, S. Zhou, K. Yang, W. Huang, S. Ogata, L. Gao and X. Pu, Adv. Sci., 2024, 11, 2307667 CrossRef CAS PubMed.
- J. Lai, H. Zhang, K. Xu and F. Shi, J. Am. Chem. Soc., 2024, 146, 22257–22265 CrossRef CAS PubMed.
- H. Zhao, Q. Fu, X. Luo, X. Wu, S. Indris, M. Bauer, Y. Wang, H. Ehrenberg, M. Knapp and Y. Wei, Energy Storage Mater., 2022, 50, 464–472 CrossRef.
- Q. Zhang, Y. Ma, Y. Lu, Y. Ni, L. Lin, Z. Hao, Z. Yan, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 18435–18443 CrossRef CAS PubMed.
- M. Fu, H. Yu, S. Huang, Q. Li, B. Qu, L. Zhou, G. C. Kuang, Y. Chen and L. Chen, Nano Lett., 2023, 23, 3573–3581 CrossRef CAS PubMed.
- X. Yu, M. Chen, Z. Li, X. Tan, H. Zhang, J. Wang, Y. Tang, J. Xu, W. Yin, Y. Yang, D. Chao, F. Wang, Y. Zou, G. Feng, Y. Qiao, H. Zhou and S. G. Sun, J. Am. Chem. Soc., 2024, 146, 17103–17113 CAS.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, 2007406 CrossRef CAS PubMed.
- J. Han, H. Euchner, M. Kuenzel, S. M. Hosseini, A. Groß, A. Varzi and S. Passerini, ACS Energy Lett., 2021, 6, 3063–3071 CAS.
- M. Liu, W. Yuan, G. Ma, K. Qiu, X. Nie, Y. Liu, S. Shen and N. Zhang, Angew. Chem., 2023, 135, e202304444 CrossRef.
- Y. Liu, Y. Li, X. Huang, H. Cao, Q. Zheng, Y. Huo, J. Zhao, D. Lin, B. Xu, Y. Liu, Y. Li, X. Huang, H. Cao, Q. Zheng, Y. Huo, D. Lin, J. Zhao and B. Xu, Small, 2022, 18, 2203061 CAS.
- Y. Tan, D. Chen, T. Yao, Y. Zhang, C. Miao, H. Yang, Y. Wang, L. Li, V. Kotsiubynskyi, W. Han and L. Shen, Adv. Sci., 2024, 11, 2407410 CAS.
- Z. Cao, H. Zhang, B. Song, D. Xiong, S. Tao, W. Deng, J. Hu, H. Hou, G. Zou, X. Ji, Z. Cao, H. Zhang, D. Xiong, S. Tao, W. Deng, J. Hu, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2023, 33, 2300339 CAS.
- Y. Xiang, Y. Zhong, P. Tan, L. Zhou, G. Yin, H. Pan, X. Li, Y. Jiang, M. Xu and X. Zhang, Small, 2023, 19, 2302161 CAS.
- D. Lei, W. Shang, L. Cheng, W. Kaiser, P. Banerjee, S. Tu, O. Henrotte, J. Zhang, A. Gagliardi, J. Jinschek, E. Cortés, P. Müller-Buschbaum, A. S. Bandarenka, M. Z. Hussain and R. A. Fischer, Adv. Energy Mater., 2024, 14, 2403030 CrossRef CAS.
- B. Li, S. Liu, Y. Geng, C. Mao, L. Dai, L. Wang, S. C. Jun, B. Lu, Z. He and J. Zhou, Adv. Funct. Mater., 2024, 34, 2214033 CrossRef CAS.
- Y. He, R. Zhang, P. Zou, R. W. Chu, R. Lin, K. Xu and H. L. Xin, J. Am. Chem. Soc., 2025, 147, 6427–6438 CrossRef CAS PubMed.
- X. Cai, W. Tian, Z. Zhang, Y. Sun, L. Yang, H. Mu, C. Lian and H. Qiu, Adv. Mater., 2024, 36, 2307727 CrossRef CAS PubMed.
- G. Bergman, N. Bruchiel-Spanier, O. Bluman, N. Levi, S. Harpaz, F. Malchick, L. Wu, M. Sonoo, M. S. Chae, G. Wang, D. Mandler, D. Aurbach, Y. Zhang, N. Shpigel and D. Sharon, J. Mater. Chem. A, 2024, 12, 14456–14466 RSC.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- G. Ma, L. Miao, Y. Dong, W. Yuan, X. Nie, S. Di, Y. Wang, L. Wang and N. Zhang, Energy Storage Mater., 2022, 47, 203–210 CrossRef.
- D. Wang, R. Li, J. Dong, Z. Bai, N. Wang, S. X. Dou and J. Yang, Angew. Chem., Int. Ed., 2025, 64, e202414117 CrossRef CAS PubMed.
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed.
- S. Yang, G. Wu, J. Zhang, Y. Guo, K. Xue, Y. Zhang, Y. Zhu, T. Li, X. Zhang and L. Zhou, Adv. Sci., 2024, 11, 2403513 CrossRef CAS PubMed.
- J. Wan, R. Wang, Z. Liu, L. Zhang, F. Liang, T. Zhou, S. Zhang, L. Zhang, Q. Lu, C. Zhang and Z. Guo, ACS Nano, 2023, 17, 1610–1621 CrossRef CAS PubMed.
- I. Al Kathemi, Z. Slim, F. Igoa Saldaña, A. C. Dippel, P. Johansson, M. Odziomek and R. Bouchal, ACS Nano, 2025, 19, 6388–6398 CrossRef CAS PubMed.
- D. Gomez Vazquez, T. P. Pollard, J. Mars, J. M. Yoo, H. G. Steinrück, S. E. Bone, O. V. Safonova, M. F. Toney, O. Borodin and M. R. Lukatskaya, Energy Environ. Sci., 2023, 16, 1982–1991 RSC.
- R. Bouchal, I. Al Kathemi and M. Antonietti, Small, 2024, 20, 2309556 CrossRef CAS PubMed.
- X. Yang, Q. Zhou, S. Wei, X. Guo, P. J. Chimtali, W. Xu, S. Chen, Y. Cao, P. Zhang, K. Zhu, H. Shou, Y. Wang, X. Wu, C. Wang and L. Song, Small Methods, 2024, 8, 2301115 CrossRef CAS PubMed.
- F. Zhao, Z. Jing, X. Guo, J. Li, H. Dong, Y. Tan, L. Liu, Y. Zhou, R. Owen, P. R. Shearing, D. J. L. Brett, G. He and I. P. Parkin, Energy Storage Mater., 2022, 53, 638–645 CrossRef.
- Y. Chen, F. Gong, W. Deng, H. Zhang and X. Wang, Energy Storage Mater., 2023, 58, 20–29 Search PubMed.
- X. Fan, L. Chen, Y. Wang, X. Xu, X. Jiao, P. Zhou, Y. Liu, Z. Song and J. Zhou, Nanomicro Lett., 2024, 16, 270 CAS.
- H. Yang, D. Chen, R. Zhao, G. Li, H. Xu, L. Li, X. Liu, G. Li, D. Chao and W. Han, Energy Environ. Sci., 2023, 16, 2910–2923 CAS.
- V. Vanoppen, L. Zhang, E. J. Berg and X. Hou, ACS Mater. Lett., 2024, 6, 4881–4888 CAS.
- Q. Luo, K. Wang, D. Chen, K. Wang, Z. Sun, W. Xu, J. Li, Y. Wang, Q. Guo, J. Liao, Z. Deng, J. Hu and S. Xiong, Adv. Energy Mater., 2025, 2405169 CAS.
- T. A. Nigatu, H. K. Bezabh, S. K. Jiang, B. W. Taklu, Y. Nikodimos, S. C. Yang, S. H. Wu, W. N. Su, C. C. Yang and B. J. Hwang, Electrochim. Acta, 2023, 443, 141883 CAS.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X. Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CAS.
- D. Xie, Y. Sang, D. H. Wang, W. Y. Diao, F. Y. Tao, C. Liu, J. W. Wang, H. Z. Sun, J. P. Zhang and X. L. Wu, Angew. Chem., Int. Ed., 2023, 62, e202216934 CAS.
- L. Deng, X. Xie, W. Song, A. Pan, G. Cao, S. Liang and G. Fang, Chem. Eng. J., 2024, 488, 151104 CAS.
- D. Aurbach and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3529–3536 CAS.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CAS.
- Q. Pang, H. Zhao, R. Lian, Q. Fu, Y. Wei, A. Sarapulova, J. Sun, C. Wang, G. Chen and H. Ehrenberg, J. Mater. Chem. A, 2020, 8, 9567–9578 CAS.
- V. Renman, D. O. Ojwang, M. Valvo, C. P. Gómez, T. Gustafsson and G. Svensson, J. Power Sources, 2017, 369, 146–153 CAS.
- M. S. Chae, J. W. Heo, S. C. Lim and S. T. Hong, Inorg. Chem., 2016, 55, 3294–3301 CAS.
- C. Pan, R. Zhang, R. G. Nuzzo and A. A. Gewirth, Adv. Energy Mater., 2018, 8, 1800589 CrossRef.
- S. Zuo, J. Liu, W. He, S. Osman, Z. Liu, X. Xu, J. Shen, W. Jiang, J. Liu, Z. Zeng and M. Zhu, J. Phys. Chem. Lett., 2021, 12, 7076–7084 CrossRef CAS PubMed.
- X. Shan, S. Kim, A. M. M. Abeykoon, G. Kwon, D. Olds and X. Teng, ACS Appl. Mater. Interfaces, 2020, 12, 54627–54636 CrossRef CAS PubMed.
- F. Wang, L. E. Blanc, Q. Li, A. Faraone, X. Ji, H. H. Chen-Mayer, R. L. Paul, J. A. Dura, E. Hu, K. Xu, L. F. Nazar and C. Wang, Adv. Energy Mater., 2021, 11, 2102016 CrossRef CAS.
- L. Wang, K.-W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CAS.
- M. D. Chung, J. H. Seo, X. C. Zhang and A. M. Sastry, J. Electrochem. Soc., 2011, 158, A371 CAS.
- M. A. Kamenskii, S. N. Eliseeva, E. G. Tolstopjatova, A. I. Volkov, D. V. Zhuzhelskii and V. V. Kondratiev, Electrochim. Acta, 2019, 326, 134969 CrossRef CAS.
- S. Takai, K. Yoshioka, H. Iikura, M. Matsubayashi, T. Yao and T. Esaka, Solid. State Ion., 2014, 256, 93–96 CAS.
- S. B. Tang, M. O. Lai and L. Lu, Mater. Chem. Phys., 2008, 111, 149–153 CAS.
- E. Andrukaitis and I. Hill, J. Power Sources, 2004, 136, 290–295 CAS.
- N. Kuwata, M. Nakane, T. Miyazaki, K. Mitsuishi and J. Kawamura, Solid State Ion., 2018, 320, 266–271 CAS.
- G. Hasegawa, N. Kuwata, Y. Tanaka, T. Miyazaki, N. Ishigaki, K. Takada and J. Kawamura, Phys. Chem. Chem. Phys., 2021, 23, 2438–2448 CAS.
- P. M. Panchmatia, A. R. Armstrong, P. G. Bruce and M. S. Islam, Phys. Chem. Chem. Phys., 2014, 16, 21114–21118 CAS.
- F. D. A. Silva, F. Huguenin, S. M. De Lima and G. J. F. Demets, Inorg. Chem. Front., 2014, 1, 495–502 CAS.
- E. Potiron, A. Le Gal La Salle, A. Verbaere, Y. Piffard and D. Guyomard, Electrochim. Acta, 1999, 45, 197–214 CAS.
- J. Xie, K. Kohno, T. Matsumura, N. Imanishi, A. Hirano, Y. Takeda and O. Yamamoto, Electrochim. Acta, 2008, 54, 376–381 CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CAS.
- R. Si, S. Yi, H. Liu, F. Yu, W. Bao, C. Guo and J. Li, Chem. – Eur. J., 2023, 29, e202300409 CAS.
- C. Morando, P. Cofrancesco and C. Tealdi, Mater. Today Commun., 2020, 25, 101478 CAS.
- X. Yao, C. Li, R. Xiao, J. Li, H. Yang, J. Deng and M. S. Balogun, Small, 2022, 18, 2204534 CAS.
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao, X. S. Qu Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, P. Li, X. Qu, N. Zhang and L. Jiao, Adv. Mater., 2021, 33, 2007480 CAS.
- Z. Wu, C. Lu, F. Ye, L. Zhang, L. Jiang, Q. Liu, H. Dong, Z. Sun and L. Hu, Adv. Funct. Mater., 2021, 31, 2106816 Search PubMed.
- L. Hu, Z. Wu, C. Lu, F. Ye, Q. Liu and Z. Sun, Energy Environ. Sci., 2021, 14, 4095–4106 RSC.
- M. Z. A. Munshi, W. H. Smyrl and C. Schmidtke, Solid State Ion., 1991, 47, 265–270 CrossRef CAS.
- J. Lai, H. Zhu, X. Zhu, H. Koritala and Y. Wang, ACS Appl. Energy Mater., 2019, 2, 1988–1996 CrossRef CAS.
- W. Shi, B. Yin, Y. Yang, M. B. Sullivan, J. Wang, Y. W. Zhang, Z. G. Yu, W. S. V. Lee and J. Xue, ACS Nano, 2021, 15, 1273–1281 CrossRef CAS PubMed.
- W. Liang, D. Rao, T. Chen, R. Tang, J. Li and H. Jin, Angew. Chem., Int. Ed., 2022, 61, e202207779 CrossRef CAS PubMed.
- F. Ming, H. Liang, Y. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CAS.
- S. Deng, Z. Yuan, Z. Tie, C. Wang, L. Song and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 22002–22006 CAS.
- J. Zheng, C. Liu, M. Tian, X. Jia, E. P. Jahrman, G. T. Seidler, S. Zhang, Y. Liu, Y. Zhang, C. Meng and G. Cao, Nano Energy, 2020, 70, 104519 CAS.
- G. Yoo, B. R. Koo and G. H. An, Chem. Eng. J., 2022, 434, 134738 CAS.
- J. Chen, B. Xiao, C. Hu, H. Chen, J. Huang, D. Yan and S. Peng, ACS Appl. Mater. Interfaces, 2022, 14, 28760–28768 CrossRef CAS PubMed.
- C. Xia, J. Guo, P. E. Li, X. Zhang, N. Husam Alshareef, C. Xia, J. Guo, D. Li, X. X. Zhang and H. N. Alshareef, Angew. Chem., 2018, 130, 4007–4012 Search PubMed.
- L. F. Zhou, X. W. Gao, T. Du, H. Gong, L. Y. Liu and W. Bin Luo, J. Alloys Compd., 2022, 905, 163939 CAS.
- F. Gao, B. Mei, X. Xu, J. Ren, D. Zhao, Z. Zhang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CAS.
- L. Zhang, L. Miao, B. Zhang, J. Wang, J. Liu, Q. Tan, H. Wan and J. Jiang, J. Mater. Chem. A, 2020, 8, 1731–1740 CAS.
- S. H. Hwang, S. D. Seo and D. W. Kim, J. Mater. Chem. A, 2022, 10, 10638–10650 CAS.
- Y. Cheng, L. Luo, L. Zhong, J. Chen, B. Li, W. Wang, S. X. Mao, C. Wang, V. L. Sprenkle, G. Li and J. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13673–13677 CAS.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Sci. Rep., 2015, 5, 18263 CAS.
- G. Zampardi and F. L. Mantia, Curr. Opin. Electrochem., 2020, 21, 84–92 CAS.
- Y. Xue, X. Shen, H. Zhou, J. Cao, J. Pu, Z. Ji, L. Kong and A. Yuan, Chem. Eng. J., 2022, 448, 137657 CAS.
- Y. Zhang, Y. Wang, L. Lu, C. Sun and D. Y. W. Yu, J. Power Sources, 2021, 484, 229263 CAS.
- G. Ni, X. Xu, Z. Hao, W. Wang, C. Li, Y. Yang, C. Zhou, L. Qin, W. Chen, X. Yao and J. Cai, ACS Appl. Energy Mater., 2021, 4, 602–610 CrossRef CAS.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- X. Li, Z. Chen, Y. Yang, S. Liang, B. Lu and J. Zhou, Inorg. Chem. Front., 2022, 9, 3986–3998 RSC.
- F. Wang, E. Hu, W. Sun, T. Gao, X. Ji, X. Fan, F. Han, X. Q. Yang, K. Xu and C. Wang, Energy Environ. Sci., 2018, 11, 3168–3175 CAS.
- L. F. Zhou, X. W. Gao, T. Du, H. Gong, L. Y. Liu and W. Bin Luo, ACS Appl. Mater. Interfaces, 2022, 14, 8888–8895 CAS.
- W. Li, X. Jing, K. Jiang and D. Wang, ACS Appl. Energy Mater., 2021, 4, 2797–2807 CAS.
- N. Wang, C. Sun, X. Liao, Y. Yuan, H. Cheng, Q. Sun, B. Wang, X. Pan, K. Zhao, Q. Xu, X. Lu, J. Lu, N. Wang, H. Cheng, Q. Sun, K. Zhao, Q. Xu, X. Lu, C. Sun, X. Liao, X. Pan, Y. Yuan, J. Lu and B. Wang, Adv. Energy Mater., 2020, 10, 2002293 CAS.
- G. Shi, P. Zhao, P. Gao, Y. Xing and B. Shen, J. Energy Storage, 2024, 78, 110057 Search PubMed.
- M. Tian, C. Liu, J. Zheng, X. Jia, E. P. Jahrman, G. T. Seidler, D. Long, M. Atif, M. Alsalhi and G. Cao, Energy Storage Mater., 2020, 29, 9–16 Search PubMed.
- T. Lv, G. Zhu, S. Dong, Q. Kong, Y. Peng, S. Jiang, G. Zhang, Z. Yang, S. Yang, X. Dong, H. Pang and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202216089 CAS.
- Y. Tong, X. Li, S. Su, J. Li, J. Fang, B. Liang, J. Hou and M. Luo, J. Colloid Interface Sci., 2022, 606, 645–653 CAS.
- F. Zhang, X. Sun, M. Du, X. Zhang, W. Dong, Y. Sang, J. Wang, Y. Li, H. Liu and S. Wang, Energy Environ. Mater., 2021, 4, 620–630 CAS.
- Q. Zong, Y. Zhuang, C. Liu, Q. Kang, Y. Wu, J. Zhang, J. Wang, D. Tao, Q. Zhang and G. Cao, Adv. Energy Mater., 2023, 13, 2301480 CAS.
- S. Chen, K. Li, K. S. Hui and J. Zhang, Adv. Funct. Mater., 2020, 30, 2003890 CAS.
- M. Huang, Y. Mai, L. Zhao, X. Liang, Z. Fang and X. Jie, Electrochim. Acta, 2021, 388, 138624 CAS.
- H. Shuai, R. Liu, W. Li, X. Yang, H. Lu, Y. Gao, J. Xu and K. Huang, Adv. Energy Mater., 2023, 13, 2202992 CAS.
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Angew. Chem., Int. Ed., 2021, 60, 20286–20293 CAS.
- S. Li, C. Huang, L. Gao, Q. Shen, P. Li, X. Qu, L. Jiao and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202211478 CAS.
- H. Liu, J. G. Wang, W. Hua, Z. You, Z. Hou, J. Yang, C. Wei and F. Kang, Energy Storage Mater., 2021, 35, 731–738 Search PubMed.
- W. Liu, L. Dong, B. Jiang, Y. Huang, X. Wang, C. Xu, Z. Kang, J. Mou and F. Kang, Electrochim. Acta, 2019, 320, 134565 CAS.
- H. Yan, C. Yang, L. Zhao, J. Liu, P. Zhang and L. Gao, Electrochim. Acta, 2022, 429, 141003 CAS.
- H. Huang, X. Xia, J. Yun, C. Huang, D. Li, B. Chen, Z. Yang and W. Zhang, Energy Storage Mater., 2022, 52, 473–484 Search PubMed.
- F. Gong, Y. Feng, Y. H. Fang, Y. K. Hsu and Y. C. Chen, ACS Appl. Mater. Interfaces, 2022, 15, 18808–18818 Search PubMed.
- S. W. Kim, X. Shan, M. Abeykoon, G. Kwon, D. Olds and X. Teng, ACS Appl. Mater. Interfaces, 2021, 13, 25993–26000 CAS.
- Q. Ni, H. Jiang, S. Sandstrom, Y. Bai, H. Ren, X. Wu, Q. Guo, D. Yu, C. Wu and X. Ji, Adv. Funct. Mater., 2020, 30, 2003511 CrossRef CAS.
- M. J. Park, H. Y. Asl and A. Manthiram, Adv. Mater. Interfaces, 2021, 8, 2100878 CrossRef CAS.
- K. Malaie, F. Scholz, U. Schröder, H. Wulff and H. Kahlert, Chem. Phys. Chem., 2022, 23, e202200364 CrossRef CAS PubMed.
- L. Zhou, L. Liu, Z. Hao, Z. Yan, X. F. Yu, P. K. Chu, K. Zhang and J. Chen, Matter, 2021, 4, 1252–1273 CrossRef CAS.
- C. F. Bischoff, O. S. Fitz, J. Burns, M. Bauer, H. Gentischer, K. P. Birke, H.-M. Henning and D. Biro, J. Electrochem. Soc., 2020, 167, 020545 CrossRef CAS.
- O. Fitz, F. Wagner, J. Pross-Brakhage, M. Bauer, H. Gentischer, K. P. Birke and D. Biro, Energy Technol., 2023, 11, 2300723 CrossRef CAS.
- C. Li, X. Yun, Y. Chen, D. Lu, Z. Ma, S. Bai, G. Zhou, P. Xiao and C. Zheng, Chem. Eng. J., 2023, 477, 146901 CrossRef CAS.
- M. Chen, M. Yang, X. Han, J. Chen, P. Zhang and C. P. Wong, Adv. Mater., 2024, 36, 2304997 CrossRef CAS PubMed.
- O. Kokits, Y. Aniskevich, A. Mazanik, O. Yakimenko, G. Ragoisha, S. T. Myung and E. Streltsov, Energy Storage Mater., 2023, 63, 103017 CrossRef.
- S. Sariyer, N. M. Keppetipola, O. Sel and R. Demir-Cakan, ChemSusChem, 2025, e202402445 CrossRef PubMed.
- I. A. Rodríguez-Pérez, H. J. Chang, M. Fayette, B. M. Sivakumar, D. Choi, X. Li and D. Reed, J. Mater. Chem. A, 2021, 9, 20766–20775 RSC.
- C. Wang, J. Wang, S. Zhang, M. Li, F. Zeng, L. Tan, F. Liu, J. Wang, L. Huang, H. Lv, C. Zhi and C. Han, Adv. Energy Mater., 2023, 13, 2302683 CAS.
- Q. Guo, W. Li, X. Li, J. Zhang, D. Sabaghi, J. Zhang, B. Zhang, D. Li, J. Du, X. Chu, S. Chung, K. Cho, N. N. Nguyen, Z. Liao, Z. Zhang, X. Zhang, G. F. Schneider, T. Heine, M. Yu and X. Feng, Nat. Commun., 2024, 15, 2139 CrossRef CAS PubMed.
- A. O. Efremova, A. I. Volkov, E. G. Tolstopyatova and V. V. Kondratiev, J. Alloys Compd., 2022, 892, 162142 CAS.
- L. Y. Wu, Z. W. Li, Y. X. Xiang, W. Di Dong, X. D. Qi, Z. X. Ling, Y. H. Xu, H. Y. Wu, M. D. Levi, N. Shpigel and X. G. Zhang, Small, 2024, 20, 2404583 CAS.
- L. Y. Wu, Z. W. Li, Y. X. Xiang, W. Di Dong, H. Y. Wu, Y. H. Xu, Z. X. Ling, M. S. Chae, D. Sharon, N. Shpigel and X. G. Zhang, ACS Energy Lett., 2024, 9, 5801–5809 CAS.
- Y. Xu, G. Zhang, X. Wang, X. Li, J. Zhang, X. Wu, Y. Yuan, Y. Xi, X. Yang, M. Li, X. Pu, G. Cao, Z. Yang, B. Sun, J. Wang, H. Yang, W. Li, J. Zhang and X. Li, J. Colloid Interface Sci., 2024, 675, 1–13 CrossRef CAS PubMed.
- B. Gavriel, N. Shpigel, F. Malchik, G. Bergman, M. Turgeman, M. D. Levi and D. Aurbach, Energy Storage Mater., 2021, 38, 535–541 CrossRef.
- M. Chen, M. Zhou, Q. Wang, C. Xu, S. Wang, J. Ning, T. Wang, K. Wang and K. Jiang, Adv. Funct. Mater., 2025, 35, 2414032 CrossRef CAS.
- M. Chen, X. He, M. Zhou, J. Ning, Z. Zhang, S. Cao, T. Wang, K. Wang and K. Jiang, Adv. Energy Mater., 2024, 14, 2400724 CrossRef CAS.
- H. Cui, J. Zhu, R. Zhang, S. Yang, C. Li, Y. Wang, Y. Hou, Q. Li, G. Liang and C. Zhi, J. Am. Chem. Soc., 2024, 146, 15393–15402 CrossRef CAS PubMed.
- S. Yeşilot, Y. Solmaz, N. Kılıç, B. Unal, O. Sel and R. Demir-Cakan, ChemElectroChem, 2024, 11, e202400212 CrossRef.
- Y. Zhang, Y. Liang, H. Dong, X. Wang and Y. Yao, J. Electrochem. Soc., 2020, 167, 070558 CrossRef.
- G. Inzelt, Electrochim. Acta, 2000, 45, 3865–3876 CrossRef CAS.
- M. D. Levi, N. Shpigel, S. Sigalov, V. Dargel, L. Daikhin and D. Aurbach, Electrochim. Acta, 2017, 232, 271–284 CrossRef CAS.
- N. Shpigel, M. D. Levi and D. Aurbach, Energy Storage Mater., 2019, 21, 399–413 CrossRef.
- T. B. Reddy, Linden's Handbook of Batteries, McGraw-Hill Education, 4th edn, 2011 Search PubMed.
- K. Kordesh and M. Weissenbacher, J. Power Sources, 1994, 51, 61–78 CrossRef.
- H. Li, H. Yao, X. Sun, C. Sheng, W. Zhao, J. Wu, S. Chu, Z. Liu, S. Guo and H. Zhou, Chem. Eng. J., 2022, 446, 137205 CrossRef CAS.
- Y. Huang, J. Mou, W. Liu, X. Wang, L. Dong, F. Kang and C. Xu, Nanomicro Lett., 2019, 11, 49 CAS.
- Z. Li, Y. Li, X. Ren, Y. Zhao, Z. Ren, Z. Yao, W. Zhang, H. Xu, Z. Wang, N. Zhang, Y. Gu, X. Li, D. Zhu and J. Zou, Small, 2023, 19, 2301770 CrossRef CAS PubMed.
- L. Chen, Z. Yang, H. Qin, X. Zeng and J. Meng, J. Power Sources, 2019, 425, 162–169 CrossRef CAS.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 CAS.
- X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
- O. Fitz, C. Bischoff, M. Bauer, H. Gentischer, K. P. Birke, H. M. Henning and D. Biro, ChemElectroChem, 2021, 8, 3553–3566 CAS.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CAS.
- O. Rubel, T. N. T. Tran, S. Gourley, S. Anand, A. Van Bommel, B. D. Adams, D. G. Ivey and D. Higgins, J. Phys. Chem. C, 2022, 126, 10957–10967 CrossRef CAS.
- T. N. T. Tran, S. Jin, M. Cuisinier, B. D. Adams and D. G. Ivey, Sci. Rep., 2021, 11, 20777 CAS.
- D. Wu, S. T. King, N. Sadique, L. Ma, S. N. Ehrlich, S. Ghose, J. Bai, H. Zhong, S. Yan, D. C. Bock, E. S. Takeuchi, A. C. Marschilok, L. M. Housel, L. Wang and K. J. Takeuchi, J. Mater. Chem. A, 2023, 11, 16279–16292 CAS.
- O. Zhanadilov, H. J. Kim, A. Konarov, J. Jeong, J. H. Park, K. Y. Chung, Z. Bakenov, H. Yashiro and S. T. Myung, Energy Storage Mater., 2024, 67, 103283 Search PubMed.
- S. Islam, M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Kim, J. Jo, J. P. Baboo, D. T. Pham, D. Y. Putro, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2017, 5, 23299–23309 CAS.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 Search PubMed.
- D. Y. Putro, M. H. Alfaruqi, S. Islam, S. Kim, S. Park, S. Lee, J. Y. Hwang, Y. K. Sun and J. Kim, Electrochim. Acta, 2020, 345, 136189 CAS.
- S. Cui, D. Zhang, G. Zhang and Y. Gan, J. Mater. Chem. A, 2022, 10, 25620–25632 CAS.
- S. Cui, D. Zhang and Y. Gan, Adv. Energy Mater., 2024, 14, 2302655 CAS.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan, S.-J. Bao, H. Chen, F. Xiao, Q. Yang, S. Cai, M. Xu, S.-J. Bao, C. Dai and H. J. Fan, Adv. Mater., 2022, 34, 2109092 CAS.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- H. Yang, W. Zhou, D. Chen, J. Liu, Z. Yuan, M. Lu, L. Shen, V. Shulga, W. Han and D. Chao, Energy Environ. Sci., 2021, 15, 1106–1118 RSC.
- H. Yang, T. Zhang, D. Chen, Y. Tan, W. Zhou, L. Li, W. Li, G. Li, W. Han, H. J. Fan, D. Chao, H. Yang, D. Chen, Y. Tan, L. Li, G. Li, W. Han, T. Zhang, W. Zhou, W. Li, D. Chao and H. J. Fan, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- Y. Du, X. Wang, Y. Zhang, H. Zhang, J. Man, K. Liu and J. Sun, Chem. Eng. J., 2022, 434, 134642 CrossRef CAS.
- T. H. Wu, K. Y. Ni, B. T. Liu and S. H. Wang, ACS Appl. Energy Mater., 2022, 5, 10196–10206 CrossRef CAS.
- K. Zhu, T. Wu and K. Huang, Chem. Mater., 2021, 33, 4089–4098 CrossRef CAS.
- Y. Bo Hu, Y. Min Zhang, N. Nan XUE, P. Cheng Hu and L. Hong Zhang, Trans. Nonferrous Met. Soc. China, 2022, 32, 1290–1300 Search PubMed.
- I. Povar, O. Spinu, I. Zinicovscaia, B. Pintilie and S. Ubaldini, J. Electrochem. Sci. Eng., 2019, 9, 75–84 CrossRef CAS.
- Y. Liu, Y. Jiang, Z. Hu, J. Peng, W. Lai, D. Wu, S. Zuo, J. Zhang, B. Chen, Z. Dai, Y. Yang, Y. Huang, W. Zhang, W. Zhao, W. Zhang, L. Wang and S. Chou, Adv. Funct. Mater., 2021, 31, 2008033 CrossRef CAS.
- X. Li, M. Li, Q. Yang, H. Li, H. Xu, Z. Chai, K. Chen, Z. Liu, Z. Tang, L. Ma, Z. Huang, B. Dong, X. Yin, Q. Huang and C. Zhi, ACS Nano, 2020, 14, 541–551 CrossRef CAS PubMed.
- S. Gao, P. Ju, Z. Liu, L. Zhai, W. Liu, X. Zhang, Y. Zhou, C. Dong, F. Jiang and J. Sun, J. Energy Chem., 2022, 69, 356–362 CrossRef CAS.
- K. Zhu, T. Wu and K. Huang, Energy Storage Mater., 2021, 38, 473–481 CrossRef.
- Z. Wang, Y. Song, J. Wang, Y. Lin, J. Meng, W. Cui and X. X. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216290 CrossRef CAS PubMed.
- S. Shen, D. Ma, K. Ouyang, Y. Chen, M. Yang, Y. Wang, S. Sun, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2023, 33, 2304255 CrossRef CAS.
- S. Xu, S. Fan, W. Ma, J. Fan and G. Li, Inorg. Chem. Front., 2022, 9, 1481–1489 RSC.
- J. Li, Q. Kuang, G. Wang, Q. Lu, P. Jiang, Q. Fan, Y. Dong and Y. Zhao, Electrochim. Acta, 2023, 441, 141841 CAS.
- Y. Zeng, Z. Lai, Y. Han, H. Zhang, S. Xie, X. Lu, Y. X. Zeng, Z. Z. Lai, Y. Han, H. Z. Zhang, X. H. Lu and S. L. Xie, Adv. Mater., 2018, 30, 1802396 Search PubMed.
- M. Yin, H. Miao, J. Dang, B. Chen, J. Zou, G. Chen and H. Li, J. Power Sources, 2022, 545, 231902 CAS.
- Y. Tang, X. Li, H. Lv, D. Xie, W. Wang, C. Zhi and H. Li, Adv. Energy Mater., 2020, 10, 2000892 CrossRef CAS.
- Y. Zuo, T. Meng, H. Tian, L. Ling, H. Zhang, H. Zhang, X. Sun and S. Cai, ACS Nano, 2023, 17, 5600–5608 CrossRef CAS PubMed.
- G. Ni, Z. Hao, G. Zou, X. Xu, B. Hu, F. Cao and C. Zhou, Sustain, Energy Fuels, 2022, 6, 1353–1361 CAS.
- J. Chivot, L. Mendoza, C. Mansour, T. Pauporté and M. Cassir, Corros. Sci., 2008, 50, 62–69 CrossRef CAS.
- L. F. Huang, M. J. Hutchison, R. J. Santucci, J. R. Scully and J. M. Rondinelli, J. Phys. Chem. C, 2017, 121, 9782–9789 CrossRef CAS.
- J. He, X. Shi, Q. Liu, H. Wu, Y. Yu, X. Lu and Z. Yang, Small, 2024, 20, 2306258 CrossRef CAS PubMed.
- T. Fu, X. Tong, Y. Zhou, D. Wu, D. Xiong, S. Xu, L. Wang and P. K. Chu, J. Alloys Compd., 2022, 918, 165734 CrossRef CAS.
- W. Kao-ian, M. T. Nguyen, T. Yonezawa, R. Pornprasertsuk, J. Qin, S. Siwamogsatham and S. Kheawhom, Mater. Today Energy, 2021, 21, 100738 CrossRef CAS.
- Q. Li, K. Ma, G. Yang and C. Wang, Energy Storage Mater., 2020, 29, 246–253 CrossRef.
- A. Bhatia, J. Xu, J. P. Pereira-Ramos, G. Rousse and R. Baddour-Hadjean, Chem. Mater., 2022, 34, 1203–1212 CrossRef CAS.
- M. S. Chae, J. W. Heo, H. H. Kwak, H. Lee and S. T. Hong, J. Power Sources, 2017, 337, 204–211 CAS.
- R. D. Corpuz, L. M. De Juan-Corpuz, M. T. Nguyen, T. Yonezawa, H. L. Wu, A. Somwangthanaroj and S. Kheawhom, Int. J. Mol. Sci., 2020, 21, 3113 CrossRef CAS PubMed.
- V. Verma, S. Kumar, W. Manalastas, J. Zhao, R. Chua, S. Meng, P. Kidkhunthod and M. Srinivasan, ACS Appl. Energy Mater., 2019, 2, 8667–8674 CAS.
- T. Yimtrakarn, Y. C. Liao, A. S. MV, J. L. Chen, Y. C. Chuang, N. Lerkkasemsan and W. Kaveevivitchai, Mater. Today Commun., 2023, 34, 105231 CAS.
- A. Naveed, H. Yang, Y. Shao, J. Yang, N. Yanna, J. Liu, S. Shi, L. Zhang, A. Ye, B. He and J. Wang, Adv. Mater., 2019, 31, 1900668 Search PubMed.
- R. Zhang, C. Pan, R. G. Nuzzo and A. A. Gewirth, J. Phys. Chem. C, 2019, 123, 8740–8745 CAS.
- W. Wang, D. Liu, Y. Jiang, D. Zhang, X. Shen, S. Li, J. Liang and H. Xu, Chem. Eng. J., 2023, 463, 142309 CAS.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CAS.
- W. Li, C. Xu, X. Zhang, M. Xia, Z. Yang, H. Yan, H. Yu, L. Zhang, W. Shu and J. Shu, J. Electroanal. Chem., 2021, 881, 114968 CAS.
- Y. Xue, Y. Chen, X. Shen, A. Zhong, Z. Ji, J. Cheng, L. Kong and A. Yuan, J. Colloid Interface Sci., 2022, 609, 297–306 CAS.
- Z. Li, T. Liu, R. Meng, L. Gao, Y. Zou, P. Peng, Y. Shao and X. Liang, Energy Environ. Mater., 2021, 4, 111–116 CAS.
- H. Sun, L. Yang, E. Hu, M. Feng, C. Fan, W. Wang, H. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 40247–40256 CAS.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 Search PubMed.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. Antonio Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CAS.
- W. Yang, Y. Yang, H. Yang and H. Zhou, ACS Energy Lett., 2022, 7, 2515–2530 CAS.
- T. F. Burton, R. Jommongkol, Y. Zhu, S. Deebansok, K. Chitbankluai, J. Deng and O. Fontaine, Curr. Opin. Electrochem., 2022, 35, 101070 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |