
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein-derived cofactors: chemical innovations expanding enzyme catalysis
Angelica
Graciano
 and
Aimin
Liu
*
and
Aimin
Liu
*
Department of Chemistry, The University of Texas at San Antonio, Texas 78249, USA. E-mail: Feradical@utsa.edu
First published on 28th March 2025
Abstract
Protein-derived cofactors, formed through posttranslational modification of a single amino acid or covalent crosslinking of amino acid side chains, represent a rapidly expanding class of catalytic moieties that redefine enzyme functionality. Once considered rare, these cofactors are recognized across all domains of life, with their repertoire growing from 17 to 38 types in two decades in our survey. Their biosynthesis proceeds via diverse pathways, including oxidation, metal-assisted rearrangements, and enzymatic modifications, yielding intricate motifs that underpin distinctive catalytic strategies. These cofactors span paramagnetic and non-radical states, including both mono-radical and crosslinked radical forms, sometimes accompanied by additional modifications. While their discovery has accelerated, mechanistic understanding lags, as conventional mutagenesis disrupts cofactor assembly. Emerging approaches, such as site-specific incorporation of non-canonical amino acids, now enable precise interrogation of cofactor biogenesis and function, offering a viable and increasingly rigorous means to gain mechanistic insights. Beyond redox chemistry and electron transfer, these cofactors confer enzymes with expanded functionalities. Recent studies have unveiled new paradigms, such as long-range remote catalysis and redox-regulated crosslinks as molecular switches. Advances in structural biology, mass spectrometry, and biophysical spectroscopy continue to elucidate their mechanisms. Moreover, synthetic biology and biomimetic chemistry are increasingly leveraging these natural designs to engineer enzyme-inspired catalysts. This review integrates recent advances in cofactor biogenesis, reactivity, metabolic regulation, and synthetic applications, highlighting the expanding chemical landscape and growing diversity of protein-derived cofactors and their far-reaching implications for enzymology, biocatalysis, and biotechnology.
 Angelica Graciano | Angelica Graciano is currently a Chemistry PhD student at the University of Texas at San Antonio (UTSA) under the supervision of Dr Aimin Liu. She obtained her BSc in Biochemistry at UTSA where her undergraduate research helped develop a deep interest in metalloenzymology. Her current research focuses on the elucidation of protein-derived cofactor biogenesis mechanisms through the incorporation of non-canonical amino acids, and characterization of metalloenzymes with functional roles in oxygen activation, amino acid metabolism, and carbohydrate metabolism. |
 Aimin Liu | Dr Aimin Liu specializes in amino acid chemistry, including metabolism and crosslinking. His research explores redox chemistries mediated by metalloproteins and free radicals, with conceptual contributions like biological charge resonance. He earned his PhD from Stockholm University after a BS from USTC. He held Royal Society scholarships and began his independent career in 2002 and is currently an endowed distinguished Professor of Chemistry and the Lutcher Brown Distinguished Chair in Biochemistry, and a member of the Academy of Distinguished Researchers at the University of Texas at San Antonio. He is an AAAS Fellow (Chemistry) and FRSC. |
1. Introduction
Enzymes, the workhorses of biological systems, rely on diverse strategies to catalyze an extraordinary range of chemical reactions. While genetically encoded amino acids provide the fundamental building blocks of proteins, nature often employs additional chemical entities known as cofactors to expand the catalytic repertoire of enzymes. These cofactors—comprising metal ions, exogenous organic molecules (such as vitamins and nucleotides), or complex structures of both endogenous and exogenous origins—reshape the catalytic machinery and modulate catalysis and reaction outcomes. Protein-derived cofactors are frequently encountered in oxidase, oxygenase, reductase, dehydrogenase, carboxylase, catalase, decarboxylase, lyases, hydratases, phosphotriesterase, racemase, sulfatase, synthetase etc. They are crucial for a vast array of biological processes, from challenging chemical transformations under mild conditions, metabolism for energy production, and biosynthesis to signal transduction and DNA replication. Typically, these cofactors bind non-covalently to proteins, forming functional holoenzymes.A fascinating class of cofactors is those “homemade cofactors”1 or “built-in cofactors”2 generated directly within proteins through covalent unidirectional posttranslational modifications (PTMs) of their constituent amino acid residues (Fig. 1). Among these PTMs, covalent crosslinking of amino acid side chains represents a remarkable strategy for creating specialized protein-derived cofactors. This intramolecular crosslinking, occurring within a single polypeptide chain, results in the formation of new covalent bonds (C–C, C–N, C–O, or C–S) that endow the protein with unique structural and chemical properties. This process is distinct from intermolecular crosslinking, which involves the formation of bonds between separate protein molecules and is often associated with protein aggregation or structural networks. This review focuses specifically on cofactors generated by intramolecular covalent crosslinking, excluding instances of intermolecular crosslinking.
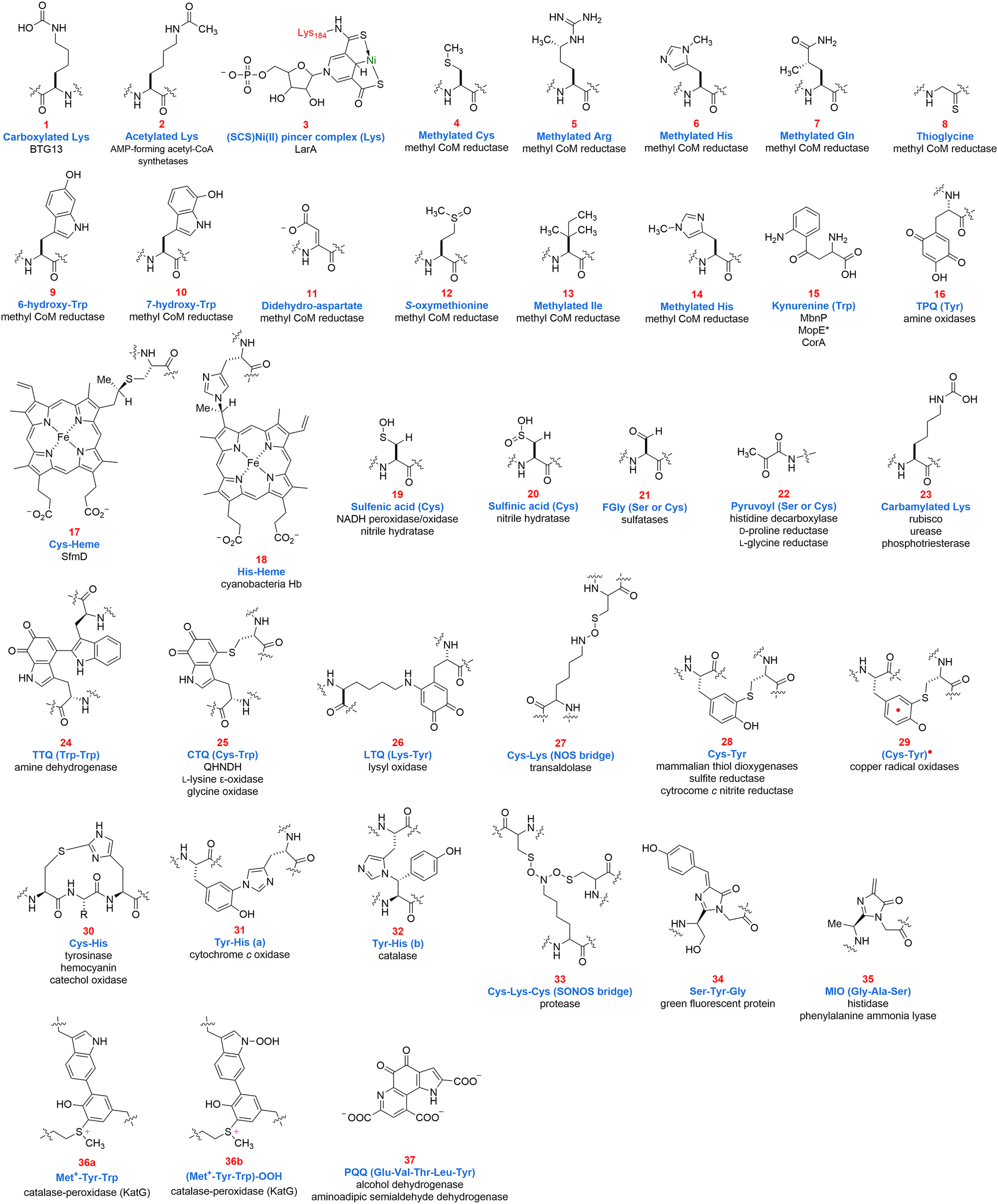 | ||
| Fig. 1 Thirty-eight structures of identified protein-derived cofactors and their corresponding systems from a total of 38 protein-derived cofactors. Throughout the text, these structures will be referenced by their assigned number. *The straight arrow in 3 indicates coordination. | ||
Formally known as protein-derived cofactors, these “homemade” intricate, atypical protein structural components are formed by chemical modification of a single amino acid side chain or by crosslinked side chains as a result of PTMs either through direct oxidation by metal/O2- or H2O2-derived intermediates or through outer sphere oxidation by highly oxidizing exogenous cofactors or auxiliary enzymes.3,4 These protein-derived cofactors often present significant challenges for prediction. Even advanced artificial intelligence (AI)-powered computational methods, such as the latest iterations of AlphaFold,5,6 currently lack the accuracy and reliability to consistently identify or predict these cofactors.5,7–10 Consequently, high-resolution structural studies, such as X-ray crystallography and cryo-electron microscopy (cryo-EM), remain essential for elucidating their precise structures and bonding arrangements. AlphaFold-guided molecular replacement for solving challenging crystal structures is evolving.11 Complementing these techniques, crosslinked peptide fragmentation (CLPF) mass spectrometry provides a powerful approach for identifying and validating the presence of novel crosslinks in proteins.12–16 Recent technological advances in rapid data collection at cryogenic temperatures to 13C-NMR investigations of 13C-labeled proteins and chemical modification protocols that can be integrated with both UV-visible and fluorescence spectroscopy offer crucial complementary information.17 These advances have facilitated the discovery of novel protein-derived cofactors. Emerging technologies, such as non-canonical natural and unnatural amino acid substitutions through genetic code expansion, have enabled precise interrogation of cofactor biogenesis and function. The resulting chemical and structural insights are vital for understanding the mechanisms underlying their biosynthesis and the precise role of these cofactors.
Approximately two decades ago, Okeley and van der Donk provided a foundational overview of protein-derived cofactors, identifying 17 distinct types and categorizing them based on their structural complexity.18 Since then, the field has witnessed significant growth, with the discovery and characterization of numerous new examples. Our current survey expands this list to 38 distinct types (Fig. 1), highlighting the rapid progress in this area. As emphasized in the work of Walsh and others, forming these crosslinked structures can significantly enhance a protein's structural variability by several orders of magnitude, enabling versatile functionalities in biological systems.4,18–24 This review builds upon the previous seminal work,18 focusing on protein-derived cofactors that have been characterized structurally and functionally within the past two decades. We will examine cofactors formed autocatalytically or enzymatically by other processing proteins, exploring how these modifications enhance existing protein functions, or add entirely new ones (Fig. 2).
 | ||
| Fig. 2 Overview of the paths that generate posttranslationally modified amino acids. Protein-derived cofactors are either enzymatically or autocatalytically formed and serve to enable a function or enhance an existing function. | ||
This review is organized as follows: First, we will discuss the various types of covalent crosslinks observed in protein-derived cofactors, categorized by the chemical nature of the bond formed and number of amino acid residues involved. While classifying these cofactors based on the type of bond formed during PTMs might seem intuitive, the frequent occurrence of multiple bond types within a single cofactor makes this approach less practical. Therefore, we maintain a classification based on the number of residues involved in the cofactor structure. Next, we will delve into the biosynthetic pathways leading to their formation, highlighting the enzymes and mechanisms involved. Subsequently, we will explore the diverse functional roles of these cofactors in biological systems, focusing on enzymatic catalysis and other key biological processes. Finally, we will briefly discuss emerging synthetic approaches to mimic these cofactors and their potential applications.
2. Protein cofactors derived from posttranslational modifications of single amino acids
A large subclass of protein-derived cofactors arises from PTMs of single amino acid residues within the polypeptide chain, totaling fifteen, as shown in Table 1. These modifications, encompassing a range of chemical transformations such as oxidation, reduction, carboxylation, and the formation of protein-derived unusual amino acids, generate unique structural motifs that play critical roles in modulating protein functions. Twelve genetically encoded amino acid residues—Asp, Arg, Cys, Gly, Gln, His, Ile, Lys, Met, Ser, Trp, and Tyr—have been identified as precursors to these modified cofactors (Table 1), highlighting the versatility of the proteinogenic amino acids. This section will explore these single amino acid-derived diverse cofactors, emphasizing their structural diversity, functional significance, and the biochemical pathways leading to their formation. We will divide this section into two subsections based on the final oxidation state of the final cofactor.| Source | Cofactor | New chemical bonds formed | Representative enzyme | Biogenesis | Function | Ref. |
|---|---|---|---|---|---|---|
| Arg | Methylated | C–C | Methyl-coenzyme M reductase | Enzymatic RCMT | Subunit interaction | 25 and 26 |
| Asp | Didehydroaspartate | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
Methyl-coenzyme M reductase | TBD | Tune enzyme structure | 27 |
| Cys | Pyruvoyl group | CO |
D-Proline reductase | Autocatalytic | Catalysis | 28 |
| Pyruvoyl group | CO |
L-Glycine reductase | Autocatalytic | Catalysis | 29 | |
| FGly | CO |
Human sulfatases | Enzymatic (SUMF1 FGE) | Catalysis | 30–32 | |
| Sulfenic acid | S–OH | NADH peroxidase/oxidase | Autocatalytic | Catalysis | 33 | |
| Nitrile hydratase | Autocatalytic | Catalysis | 34 and 35 | |||
| Sulfinic acid | S–OH | Nitrile hydratase | Autocatalytic | Catalysis | ||
| SO |
||||||
| Cys–Heme | C–S | 3-Methyl-L-tyrosine hydroxylase | Autocatalytic | Catalysis | 36 | |
| Methylated | C–S | Methyl-coenzyme M reductase | Enzymatic (MA4545) | Thermal stability, substrate binding | 25–27, 37–39 | |
| Cys˙ | Cys˙ | Class II ribonucleotide reductase | Autocatalytic | Regenerate AdoCbl cofactor | 40 | |
| Gln | Methylated | C–C | Methyl-coenzyme M reductase | Enzymatic (QCMT) | TBD | 25 and 39 |
| Gly | Thioglycine | CS |
Methyl-coenzyme M reductase | Enzymatic YcaO and TfuA* | Differing opinions based on catalysis or stability | 25, 37 and 38 |
| Gly˙ | Gly˙ | Class III ribonucleotide reductase | Enzymatic (AE) | Generate a transient protein radical, catalysis | 40 | |
| Gly˙ | Pyruvate formate lyase | Enzymatic (AE) | Generate a transient protein radical, catalysis | 40 and 41 | ||
| His | Methylated | C–N | Methyl-coenzyme M reductase | TBD | TBD | 25, 42 and 43 |
| His–heme | C–N | Cyanobacteria hemoglobin (PCC 6803 and 7002) | Autocatalytic | Catalysis | 44 | |
| Ile | Methylated | C–C | Methyl-coenzyme M reductase | TBD | TBD | 42 and 43 |
| Lys | Carboxylated | C–N | Questin oxidase (BTG13) | Autocatalytic | Catalysis | 45 |
| Carbamylated | C–N | Rubisco | Autocatalytic | Bridging ligand: Mg2+ | 46 | |
| Urease | Autocatalytic | Bridging ligand: Ni2+ | 47 | |||
| Phosphotriesterase | Autocatalytic | Bridging ligand: Zn2+ | 48 | |||
| Acetylated | C–N | Acetyl-CoA synthetase | Enzymatic | Regulates activity | 49 and 50 | |
| Ni-pincer-Lys | C–N | Lactate racemase | Enzymatic (LarB, LarC, and LarE) | Catalysis and/or nickel binding | 51 and 52 | |
| Met | Oxymethionine | SO |
Methyl-coenzyme M reductase | TBD | TBD | 42 and 43 |
| Ser | Pyruvoyl group | CO |
Histidine decarboxylase | Autocatalytic | Catalysis | 53 and 54 |
| FGly | CO |
Sulfatase | Enzymatic (AtsB) | Catalysis | 55 and 56 | |
| Trp | 7-Hydroxy-Trp | C–O | Methyl-coenzyme M reductase | TBD | TBD | 42 and 43 |
| 6-Hydroxy-Trp | C–O | Methyl-coenzyme M reductase | TBD | TBD | ||
| Kyn | CO |
Copper binding protein (MbnP) | Enzymatic (MbnH) | Copper binding | 57 | |
| CO |
Copper-binding protein (MopE*) | TBD | Copper binding | 58 | ||
| CO |
Copper-repressible protein (CorA) | TBD | Copper binding | 59 | ||
| Tyr | TPQ | CO |
Amine oxidases | Autocatalytic | Catalysis | 60 |
| Tyr˙ | Tyr˙ | Class I ribonucleotide reductase | Autocatalytic | Generate a transient protein radical | 40 | |
| Tyr˙ | Photosystem II | Autocatalytic | Catalysis | 61 | ||
| Tyr˙ | Prostaglandin H synthase | Autocatalytic | Catalysis | 62 |
2.1. Modified single amino acid cofactors
This subsection focuses on single amino acid residues that undergo PTMs to form non-radical cofactors. Several examples illustrate the diverse chemical transformations involved. | ||
| Fig. 3 Crystal structures showing novel lysine PTM's. (A) Iron-binding motif in BTG13 (yellow; PDB 7Y3W) is comprised of four histidine residues, a carboxylated lysine residue, and a water molecule. Thr299 and His58 from the second coordination sphere are shown due to their implications in regulation of Lys377 carboxylation (H-bonding shown in gray dashed lines). (B) Active site of LarA (orange; PDB 4YNS) showing the (SCS)Ni(II) pincer complex with Lys184 and His200 potentially playing roles in catalysis/Ni-binding (coordination shown in gray dashed lines). | ||
Mechanistic investigations into the C4a–C10 bond cleavage of anthraquinone catalyzed by BTG13, supported by computational studies, propose a stepwise mechanism initiated by hydrogen atom abstraction from C10 of the substrate by a ferric superoxide species, Fe(III)–O2˙−, generating a substrate radical–Fe(III)–OOH intermediate.63 Subsequent homolytic O–O bond cleavage and rebound of the distal oxygen to the substrate radical forms a high-valent Fe(IV)O species. Computational studies further suggest that Kcx377 facilitates the initial electron transfer from the iron center to dioxygen, promoting the formation of Fe(III)–O2˙−, the catalytically relevant species.63
A recently characterized protein, AcuA, functions as an acetyltransferase, transferring an acetyl group from either acetyl-CoA or acetyl phosphate to Lys549 on AcsA, thereby inactivating this synthetase.49 Interestingly, this process is reversible, and AcsA activity can be restored through the enzymatic deacetylation of Lys549.49 The presence of genetically encoded regulatory mechanisms—namely, an acetyltransferase and its corresponding deacetylase—highlights the critical need for AcsA regulation. Given the conservation of this lysine residue, it is speculated that other acetyl-CoA synthetases might regulate their activity similarly, based on metabolic conditions. These findings enhance our understanding of the AMP-forming acetyl-CoA synthetase in cellular metabolism and present potential therapeutic targets for interventions.
 | ||
| Fig. 4 Proposed biogenesis mechanism of (SCS)Ni(II) pincer complex. LarB is responsible for the formation of P2CMN from NaAD in a CO2/bicarbonate dependent reaction. Two LarE molecules are required for P2TMN formation. LarE activates P2CMN via adenylylation, covalently links the protein to P2CMN, and sacrifices a sulfur atom from its own cysteine residue to form the thioacid of P2CMN, a second LarE protomer repeats the set of reactions, where Dha is released as a product upon sulfur atom donation, forming P2TMN. LarC is suggested to be responsible for Ni insertion into P2TMN and transferring the cofactor to LarA (the straight arrow in the pincer indicates coordination). | ||
A comprehensive investigation of the cofactor biosynthetic pathway jointly by Hausinger and Hu has defined the functions of the three auxiliary proteins starting from a NAD+ precursor, nicotinic acid adenine dinucleotide (NaAD).52 LarB initiates cofactor assembly by carboxylating and hydrolyzing the pyridine ring of NaAD to yield AMP and pyridinium-3,5-biscarboxylic acid mononucleotide (P2CMN).69,70 Subsequent reactions with two LarE molecules, involving sacrificial desulfurization of Cys176, convert P2CMN to the thiocarboxylated intermediate, P2TMN.71–74 LarC purifies with bound nickel,52 and it is a CTP-dependent nickel-inserting cyclometallase playing the role in nickel insertion into P2TMN and subsequent transfer of the completed (SCS)Ni(II) cofactor to LarA (Fig. 3B).75
The identification of this unprecedented enzyme-generated pincer complex essential for catalyzing the racemization of lactate raises the possibility of similar cofactors in other biological systems. The broad distribution of LarB, LarC, and LarE homologs beyond organisms possessing LarA strongly suggests the potential for additional pincer-dependent enzymes.51,76
Further investigations of MCR homologs, including ethyl CoM reductase (ECR), MCR from Methanothermobacter marburgensis, and MCR from anaerobic methanotrophic archaea (ANME-1), have unveiled a diverse repertoire of novel PTMs. These include 6-hydroxy-tryptophan, 7-hydroxy-tryptophan, didehydro-aspartate, S-oxy-methionine, 3-methyl-isoleucine, and 2-N-methyl-histidine.42,43 Notably, didehydro-aspartate, was discovered in MCRs from M. marburgensis and Methanosarcina barkeri through peptide-sequencing and mass spectrometry and confirmed by a 2.15 Å resolution crystal structure.27 The formation of the double bond in didehydro-aspartate restricts the conformational space of the side chain, reducing the mobility of the carboxylate group and decreasing the residue's pKa. It is proposed that this modification fine-tunes enzyme structure to optimize substrate-binding and catalysis.27
The distribution of these modifications varies across different MCRs. For example, ANME-1 harbors 7-hydroxy-tryptophan and S-oxymethionine,80 while MCR from Methanotorris formicicus contains 6-hydroxy-tryptophan.81 3-Methyl-isoleucine and 2-N-methyl-histidine have been identified in the structure of Candidatus Ethanoperedens thermophilum MCR.82 The non-overlapping occurrence of 7-hydroxy-tryptophan and methylated arginine in ANME-1 suggests potential compensatory functions for these modifications.80 Similarly, 6-hydroxy-tryptophan may compensate for the absence of didehydro-aspartate in MCR.81 However, the precise roles and biosynthetic origins of many of these PTMs remain to be fully elucidated.
Significant progress has been made in understanding the biosynthesis of S-methyl-cysteine, 2-C-(S)-methyl-glutamine, 5-C-(S)-methyl-arginine, and thio-glycine.83 Gene knockout studies on the α subunit of MCR (McrA) have shown that the ycaO-tfuA locus is required for thio-glycine formation, where phylogenetic analyses showed their prevalence in methanogens.37 Thio-glycine, present in all examined MCRs, is implicated in stabilizing secondary protein structure near the active site, although its formation mechanism is still under investigation.37 A SAM-dependent methyltransferase, encoded by mcmA, is hypothesized to be responsible for the methyl-cysteine modification.38 Studies on MCR mutants lacking thio-glycine and methyl-cysteine showed significant growth defects, suggesting a synergistic interaction between these two modifications.38 While histidine methylation is conserved in all methanogens, the specific enzyme remains unidentified, although a SAM-dependent methyltransferase is suspected. It is hypothesized that this methylation positions the imidazole that coordinates coenzyme B, CoB.84
The methylation of arginine in MCR is catalyzed by the radical SAM methyltransferase MA4551 (Mmp10), also known as arginine C-methyltransferase (RCMT).26 RCMT utilizes methylcobalamin (MeCbl) as a cofactor.26,85 Booker et al. classified MaMmp10 (Mmp10 from Methanosarcina acetivorans) as a new member of subclass B in the radical SAM methyltransferase family, revealing a C-terminal cobalamin-binding domain.86 EPR studies have shown that a tyrosine residue coordinates the [4Fe–4S] cluster, enabling both radical and nucleophilic chemistry,85 similar to the initial steps of tyrosine cleavage by HydG of the radical SAM superfamily87,88 for cyanide production.89,90 The proposed mechanism involves SAM binding, assisted by tyrosine displacement in the [4Fe–4S] cluster. The 5′-deoxyadenosyl radical abstracts a hydrogen atom from arginine, initiating methyl transfer from MeCbl.85 The resulting Co(II) intermediate is likely reduced back to MeCbl by a ferredoxin (Fig. 5). Similarly, glutamine methylation in MCR is catalyzed by an enzyme of class B radical SAM methyltransferases, glutamine C-methyltransferase (QCMT), which utilizes a [4Fe–4S] cluster and MeCbl similarly. Studies have shown that QCMT transfers a methyl group from SAM to cobalamin(I), regenerating MeCbl, with 5′-deoxyadenosine and S-adenosyl-L-homocysteine as co-products.38,91 The similarities between QCMT and RCMT suggest analogous catalytic mechanisms.
 | ||
| Fig. 5 Proposed catalytic cycle of arginine C-methyltransferase (RCMT), also known as Mmp10, to produce 5-C-(S)-methylarginine, methionine, 5′-deoxyadenosine, and S-adenosyl-L-homocysteine.85 | ||
Intriguingly, the methylation of glutamine in MCR is also catalyzed by a class B radical SAM methyltransferase, termed glutamine C-methyltransferase (QCMT).39 Using the putative QCMT from Methanoculleus thermophilus, Layer et al. demonstrated glutamine methylation in an MCR-derived peptide substrate.39 QCMT possesses a single [4Fe–4S] cluster and enzyme-bound MeCbl cofactor utilized during catalysis. UV-vis spectra and HPLC analysis indicated that SAM was transferring a methyl group to cobalamin(I), regenerating MeCbl. A QCMT reaction with a peptide substrate revealed the presence of two co-products, 5′-deoxyadenosine and S-adenosyl-L-homocysteine.39 Due to the significant similarities in QCMT and RCMT, it is assumed that they catalyze similar reactions to methylate their corresponding residues.
 | ||
| Fig. 6 Active sites of copper-binding proteins containing kynurenine. (A) The copper ion in MbnPH (light green; PDB 7L6G) is coordinated in a tetrahedral geometry by a methionine, histidine, kynurenine, and a water molecule. (B) The copper ion in MopE* (turquoise; PDB 2VOV) is coordinated by two histidine residues, kynurenine and a water molecule. (C) CorA's copper ion (pink; PDB 4BZ4) is also coordinated by two histidine residues, kynurenine, and a water molecule. | ||
The TPQ cofactor was first identified in 1990 via biochemical studies and later confirmed by X-ray crystallography (Fig. 7A). Structural insights have been obtained from bacteria, plant, and mammal CuAOs, providing a comprehensive understanding of their catalytic mechanisms.111–117 Notably, the copper amine oxidase from Arthrobacter globiformis (AGAO) has been extensively studied to elucidate the stepwise formation of TPQ. Apo-AGAO crystals which feature an unmodified tyrosine residue Tyr382, were soaked in CuSO4 and exposed to an oxygen-saturated buffer to monitor TPQ (Fig. 8).118
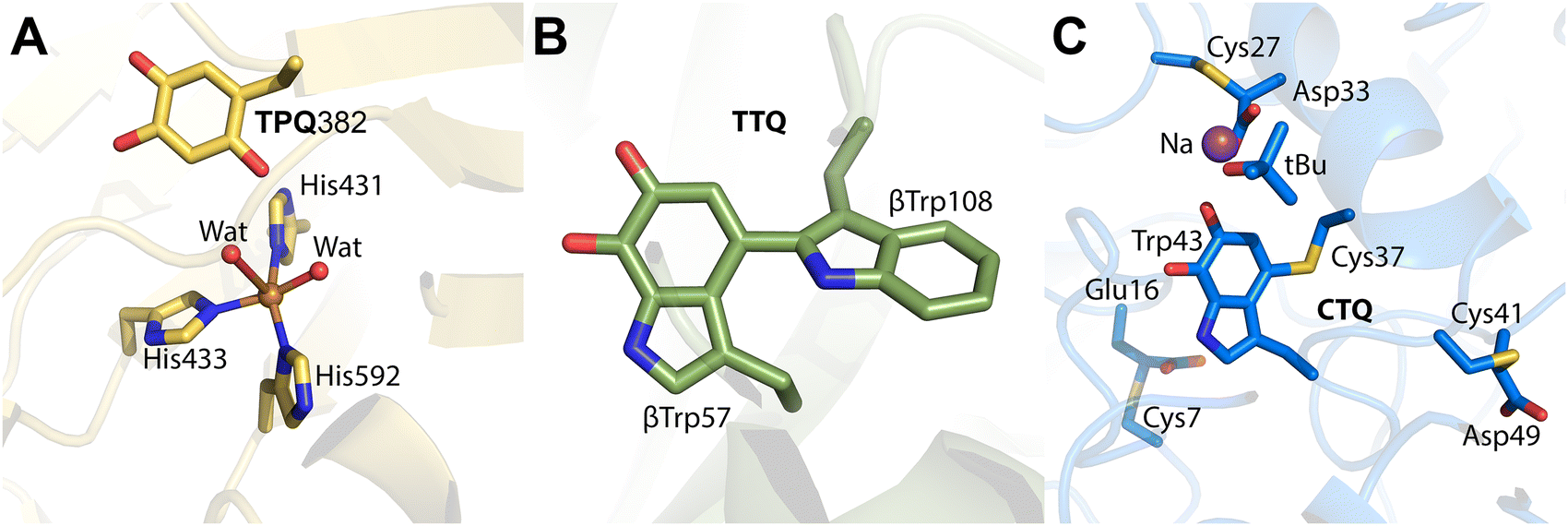 | ||
| Fig. 7 Crystal structures of cofactors in present in quinoproteins. (A) Amine oxidase of A. globiformis with mature TPQ cofactor (yellow; PDB 1IVX). The copper center is coordinated in a distorted square-pyramidal geometry by two water molecules and three histidine residues. (B) Wild-type MauG in complex with pre-MADH crystals aged 130 days, showing the crosslink between β-Trp57 and β-Trp108 (green; PDB 4FA1). (C) CTQ in the γ-subunit of QHNDH from Pseudomonas putida (blue; PDB 1JMX). Cys7, Cys27, and Cys41 of the γ-subunit are involved in thioether crosslinks with Asp or Glu residues that surround the CTQ cofactor. | ||
 | ||
| Fig. 8 Proposed mechanism of formation of TPQ in AGAO.118 Crystal structures for apo-AGAO, CuII-AGAO, DPQ, TPQred, and TPQox have been characterized and published with PDB codes 1AVK, 1IVU, 1IVV, 1IVW, and 1IVX, respectively. | ||
The first step in TPQ formation involves the binding of a copper(II) ion to the protein, positioning it approximately 2.5 Å from the hydroxyl group of Tyr382. Upon exposure to oxygen for 10 minutes, an O atom was detected near the C3 position of the tyrosine ring, with an occupancy of 0.5.118 This observation suggested the presence of a transient intermediate comprising both the unmodified tyrosine and its partially oxidized quinone form, referred to as dopaquinone (DPQ).118
Prolonged exposure to oxygen for 100 minutes revealed another intermediate lacking the characteristic 480 nm absorption peak of fully mature TPQ. This intermediate exhibited two oxygen atoms near the C2 and C5 positions of the tyrosine ring, consistent with the reduced form of TPQ (TPQred).118 Since mature TPQ contains oxygen atoms at the C2, C4, and C5 positions, the previously observed oxygen at C3 in DPQ likely corresponds to C5 in TPQred, implying a 180° rotation of the quinone ring during maturation.
The final step involves the oxidation of TPQred by a second molecule of molecular oxygen, yielding the fully mature oxidized form of TPQ (TPQox) and H2O2.118,119 This sequential mechanism highlights the intricate interplay between copper coordination, tyrosine oxidation, and ring conformational changes during TPQ biogenesis.
By contrast, LTQ biogenesis involves a crosslink between lysine and tyrosine residues, forming a distinct quinone cofactor, which will be further discussed in Section 3.3. Although less studied than TPQ, LTQ plays a similarly essential role in enabling CuAO catalysis and warrants further exploration to delineate its formation and function in enzymatic systems.
The catalytic mechanism of CuAOs has been studied, revealing a two-step process comprising a reductive half-reaction and an oxidative half-reaction (Fig. 9). In the reductive phase, the primary amine substrate is oxidized to an aldehyde, while TPQ is reduced. The oxidative half-reaction regenerates TPQ, releasing NH3 and H2O2 as byproducts.120
 | ||
| Fig. 9 Proposed catalytic mechanism of CuAOs. Steps 1–4 belong to the reductive half-reaction and the remaining steps are the oxidative half-reaction.120 TPQox, fully oxidized TPQ; TPQssb, TPQ substrate Schiff-base; TPQpsb, TPQ product Schiff-base; TPQamr, TPQ aminoresorcinol form; TPQsq, TPQ semiquinone radical, the “on-copper” catalytically active conformation, TPQ semiquinone form; TPQimq, TPQ iminoquinone form. | ||
A recent study provided new insights into the dynamic conformational changes during catalysis through in crystallo thermodynamic analysis of noncryocooled AGAO crystals.120 TPQ exists in two major conformations: the “off-copper” state, where it is distant from the copper(II) ion, and “on-copper” state, where it coordinates with the metal center and is catalytically active. During the reductive half-reaction, TPQ is reduced by the substrate to its aminoresorcinol form (TPQamr) which resides in the off-copper state. This form is in equilibrium with the on-copper semiquinone radical (TPQsq).120
Murakawa and colleagues ingeniously employed the humid air and glue-coating (HAG) method, commonly used in noncryogenic crystallography, to investigate the temperature-dependent behavior of TPQ. Using anaerobically substrate-soaked AGAO crystals, they demonstrate that increasing temperature facilitates the transition from TPQamr to TPQsq. This transition was further corroborated by single-crystal UV-vis spectroscopy, providing direct evidence of temperature-mediated conformational equilibrium.98 These findings emphasize the sophisticated interplay of cofactor conformation, substrate interactions, and environmental conditions in CuAO catalysis, offering deeper insights into the mechanistic versatility of protein-derived cofactors.
2.2. One oxidation state up: single amino acid residues harboring paramagnetic free radicals as cofactors
Free radicals on amino acid residues represent a unique class of protein-derived cofactors, serving as catalytic driving forces in diverse enzymatic systems. This section explores the biogenesis, stabilization, and functional roles of these radicals.This radical is formed during protein synthesis via an autocatalytic process involving a diiron cluster. Oxygen reacts with the diiron center to form a mixed-valence Fe(III)–O–Fe(IV) intermediate,127–129 which oxidizes a nearby tyrosine residue, yielding the tyrosyl radical while reducing the diiron cluster to the diferric state. The hydrophobic pocket surrounding the diiron center and the local electrostatic environment provide a protective environment that stabilizes the radical.130,131
The catalytic role of the tyrosyl radical becomes evident upon interaction with the R1 subunit. When R1 binds its substrate, a ribonucleotide diphosphate, the two subunits form a complex,132 enabling the transfer of oxidizing power from the R2 subunit's tyrosyl radical through a hydrogen-bonded network to the active site in R1. This generates a cysteine thiol radical that initiates substrate reduction.133,134 Following each turnover, the tyrosyl radical is regenerated, demonstrating its role as a true catalytic cofactor.135
Beyond RNRs, tyrosyl radicals as protein-derived cofactors play significant roles in other systems, such as photosystem II (PSII) and prostaglandin H synthase (PGHS).136 In PSII, two tyrosyl radicals, YZ˙ and YD˙, contribute distinct functions in water-splitting catalysis. YZ˙ mediates electron transfer to P680+ in the photosynthetic electron transport chain, rapidly reduced back to tyrosine and oxidized to the radical state. Conversely, YD˙ is exceptionally stable, persisting for minutes to hours, and is believed to assist in assembling and stabilizing the oxygen-evolving complex.137,138 Computational studies have revealed that YD˙'s stability is linked to a water-mediated proton transfer pathway within its hydrophobic environment, explaining its slower oxidation kinetics.139 PGHS is a heme-dependent bifunctional enzyme, a tyrosyl radical plays an active role in the cyclooxygenase activity that initiates prostaglandin biosynthesis.140–142 The radical is generated during catalysis and accumulates alongside product formation, signifying its central role in the reaction mechanism. These examples underscore the versatility and adaptability of tyrosyl radicals as catalytic cofactors in diverse enzymatic systems.
Other enzymes containing glycyl radicals include glycerol dehydratase, benzylsuccinate synthase, and hydroxyphenylacetate decarboxylase.153 Collectively termed glycyl radical enzymes (GREs), they share a common mechanism of radical transfer to cysteine residues for catalytic activity. Stabilization and precise transfer mechanisms of glycyl radicals remain active areas of research.
Thiyl radicals are essential to the catalytic mechanisms of RNRs, which play a central role in DNA synthesis. All RNR classes employ a transient thiyl radical to initiate nucleotide reduction, with variations in its generation mechanisms.144,156–163 To kinetically and spectroscopically study the elusive thiyl radical, researchers have utilized selenocysteine substitution.164 With its lower pKa and reduction potential, selenocysteine enhances radical stabilization, facilitating detection and advancing the understanding of thiyl radicals in enzymatic catalysis.165
Tryptophan radical cations are exemplified by cytochrome c peroxidase (CcP), a heme-dependent enzyme that catalyzes the oxidation of Fe(II)-cytochrome c to Fe(III)-cytochrome c while reducing H2O2 to water.166 The key intermediate is Trp191˙+,167 which facilitates long-range electron transfer to its substrate, cytochrome c.168–170 Structural and spectroscopic studies reveal that protein environments, such as solvent accessibility and proximal charges, critically influence the stability of Trp˙+.171 For example, the presence of a nearby cation-binding site in ascorbate peroxidase (APX) leads to the stabilization of a porphyrin π-cation radical instead of a Trp˙+.172,173 However, in Leishmania major CcP (LmP), a Trp˙+ radical forms despite the presence of a similar cation-binding site, suggesting that solvent environment differences may play a significant role.174 Recent computational studies have investigated the factors governing Trp˙+ stabilization, highlighting the roles of protein structure and solvent interactions.175 Despite substantial progress, questions remain about why CcP favors a Trp˙+ over a porphyrin radical and how this radical is reduced during catalysis.
3. Protein cofactors derived from the covalent crosslink of two amino acids
Covalent crosslinking between two amino acid residues forms a unique class of protein-derived cofactors, which play crucial roles in enhancing enzyme catalysis by creating specialized active sites with new redox centers. Among these cofactors, eight distinct types have been characterized to date, including Cys–His, Cys–Lys, Cys–Tyr, Cys–Trp, His–Tyr, Lys–Tyr, oxygenated Trp–Trp, and the radical form of (Cys–Tyr)˙ (Table 2). These crosslinked cofactors exhibit unique structural and functional properties, often arising through intricate biosynthetic pathways. This section examines these cofactors in detail, with a focus on their structural features, catalytic roles, and the underlying mechanisms of their formation.| Source | Cofactor | New chemical bonds formed | Representative enzyme | Biogenesis | Function | Ref. |
|---|---|---|---|---|---|---|
| Cys–His | Cys–His | C–S | Tyrosinase | Autocatalytic | TBD | 23 and 176–178 |
| Cys–His | Hemocyanin | Autocatalytic | TBD | 23, 179 and 180 | ||
| Cys–His | Catechol oxidase | Autocatalytic | TBD | 23 and 181 | ||
| Cys–Lys | NOS bridge | N–O–S | Transaldolase | Autocatalytic | Allosteric redox switch | 182 |
| Cys–Trp | CTQ | C–S | Quinohemoprotein amine dehydrogenase (QHNDH) | Enzymatic (QhpG, QhpA) | Catalysis | 183 |
| L-Lysine-ε-oxidase (LodA) | Enzymatic (LodB) | Catalysis | 184 | |||
| Glycine oxidase (GoxA) | Enzymatic (GoxB) | Catalysis | 185 | |||
| (Cys–Tyr)˙ | (Cys–Tyr)˙ | C–S | Galactose oxidase | Autocatalytic | Catalysis | 186 and 187 |
| Cys–Tyr | Cys–Tyr | C–S | Cysteine dioxygenase | Autocatalytic | Catalytic Amplifier | 188 |
| Cys–Tyr | Cysteamine dioxygenase | Autocatalytic | Catalytic Amplifier | 12 | ||
| Cys–Tyr | Copper radical oxidase (GlxA) | Autocatalytic | Catalysis | 189 | ||
| Cys–Tyr | Sulfite reductase (NirA) | Autocatalytic | Catalysis | 190 | ||
| Cys–Tyr | Putative zinc protease (BF4112) | Autocatalytic | Catalysis (putative) | 15 | ||
| Cys–Tyr | Cytochrome c nitrite reductase (TvNiR) | Autocatalytic | Catalysis | 191 | ||
| His–Tyr | His–Tyr (a) | aromatic C–N | Cytochrome c oxidase | Autocatalytic | Copper binding | 192–194 |
| His–Tyr (b) | Cβ–N | Catalase | TBD | TBD | 195 and 196 | |
| Lys–Tyr | LTQ | C–N | Lysyl oxidase | Autocatalytic | Catalysis | 110 |
| Trp–Trp | TTQ | C–C | Methylamine dehydrogenase (MADH) | Enzymatic (MauG) | Catalysis | 197–199 |
3.1. Tryptophan modification in amine dehydrogenases: tryptophan tryptophylquinone (TTQ)
The protein-derived cofactors accommodated by quinoproteins are among the most well-studied in terms of mechanisms of formation. Currently, five amino acid crosslinked cofactors with quinone moieties have been identified as redox centers in enzymes.22 These are:• Cysteine tryptophylquinone (CTQ, Fig. 1, 25) in quinohemoprotein amine dehydrogenase (QHNDH),60,110,183,197,200,201L-lysine ε-oxidase (LodA),202 and glycine oxidase (GoxA).185
• Lysine tyrosylquinone (LTQ, Fig. 1, 26) in lysyl oxidase.110
• Pyrroloquinoline quinone (PQQ, 37 in Fig. 1, 37)203 in alcohol dehydrogenases and aminoadipic 6-semialdehyde dehydrogenase.204,205
• 2,4,5-Trihydroxyphenylalanine quinone (TPQ, Fig. 1, 16) in amine oxidases.60
• Tryptophan tryptophylquinone (TTQ, Fig. 1, 24) in methylamine dehydrogenase.197–199
Among them, the biogenesis of TTQ is well studied. This quinone cofactor formed through the crosslinking and subsequent oxygenation of two tryptophan components (Fig. 1, 24). TTQ is the hallmark cofactor of methylamine dehydrogenase (MADH), an enzyme that catalyzes the oxidation of methylamine to formaldehyde and ammonia.197 TTQ serves as a prime example of how protein-derived cofactors confer remarkable catalytic capabilities, enhancing substrate specificity and turnover efficiency. The formation of TTQ involves a sophisticated posttranslational modification pathway, mediated by the diheme enzyme MauG.206,207
MauG must handle a large substrate with specificity. The biogenesis of TTQ is accomplished in a 119 kDa cofactor-free precursor protein, preMADH, containing two tryptophan residues at positions critical for cofactor assembly.93 MauG catalyzes the oxidative maturation of these residues into TTQ via a six-electron oxidation process (Fig. 10).97,102 The oxidant in this system, H2O2, interacts with the deeply buried hemes of MauG, initiating a cascade of high-energy intermediates.
 | ||
| Fig. 10 Proposed biogenesis mechanism of TTQ in MADH.96 TTQ maturation in MauG-preMADH crystals with slow release of H2O2 of the cryoprotectant illustrates TTQ formation, showing that the crosslink between the two Trp residues occurs prior to the addition of a second oxygen atom. The PDB codes for aged crystal structures are 4FA4, 4FA5, 4FA9, 4FAN, 4FAV, and 4FA1, in ascending order. | ||
Upon binding H2O2, the distal heme of MauG undergoes oxidation to form an FeO species coupled with a porphyrin radical. The oxidizing equivalent is then transferred to the proximal heme, generating a bis-Fe(IV) intermediate.98 This bis-Fe(IV) species is remarkably stable in the absence of preMADH, persisting for hours due to a biological charge resonance (CR) stabilization mechanism.99,100 This stabilization is characterized by a near-infrared (NIR) absorption band with maxima at 950–960 nm, a phenomenon rarely observed in biological systems. Unlike the short-lived ferryl intermediates that typically oxidize small-molecular substrates, the exceptional stability of bis-Fe(IV) in 43 kDa MauG enables the intermediate to oxidize a large substrate of 119 kDa preMADH on its specific two tryptophan residues (one is an already hydroxylated 7-OH-Trp) through long-range remote catalysis.96,101
The bis-Fe(IV) intermediate serves as the catalytic linchpin for oxidizing the tryptophan residues in preMADH.98 Despite the physical separation (∼40 Å) between MauG's catalytic center and the substrate tryptophans, the oxidation proceeds through a long-range electron transfer mechanism.102 This transfer is facilitated by a network of aromatic residues in MauG and involves a hole-hopping pathway via a surface-exposed tryptophan residue.101 The oxidative modification of preMADH occurs across an approximately 40 Å interface between the H2O2-binding site in MauG and the TTQ formation site in preMADH, producing a diradical of Trp˙ and (Trp-OH)˙ for crosslinking,96 exemplifying the efficiency of biological electron transfer mechanisms.
Each TTQ assembly cycle involves the consumption of one H2O2 molecule, repeating three times to deliver the six required oxidizing equivalents. During each cycle, MauG's bis-Fe(IV) intermediate oxidizes one target tryptophan residue in preMADH. The modifications include hydroxylation, crosslink formation, and further oxidation to achieve the mature TTQ structure.96 This cumulative oxidative process transforms preMADH into catalytically active MADH, equipped with a fully formed TTQ cofactor (Fig. 7B).
3.2. Cysteine tryptophylquinone (CTQ)
CTQ is a quinone cofactor derived from the crosslinking of cysteine and tryptophan residues (Fig. 1, 25).208 It has been identified in various enzymes, including quinohemoprotein amine dehydrogenase (QHNDH)183L-lysine ε-oxidase (LodA),202 and glycine oxidase (GoxA),185 where it plays a crucial role in the oxidation of primary amines or amino acids to their corresponding aldehydes or keto acids. These enzymatic reactions are significant in energy metabolism and catabolism, demonstrating the versatility and catalytic efficiency of CTQ-containing enzymes.QHNDH is a diheme enzyme that catalyzes the oxidation of primary amines to aldehydes, with energy generation as a byproduct. Its CTQ cofactor is formed during a multistep biosynthetic process involving several auxiliary proteins. The flavoprotein monooxygenase QhpG plays a central role in catalyzing the final oxidation step to form the mature CTQ (Fig. 7C) from the precursor peptide encoded by the qhpC gene, which forms the γ-subunit of QHNDH. However, this final step is preceded by sequential modifications orchestrated by QhpD and QhpE.209
QhpD mediates the formation of three Cys-to-Asp/Glu thioether bonds within the QhpC polypeptide, a step critical for stabilizing the cofactor's nascent structure. Structural studies revealed that QhpG preferentially binds to a QhpCD binary complex, facilitating efficient CTQ formation. This hierarchical assembly and multienzyme coordination are reminiscent of TTQ biogenesis in methylamine dehydrogenase (MADH), which similarly involves a diheme enzyme (MauG) to oxidize precursor tryptophan residues.
In L-lysine ε-oxidase (LodA) and glycine oxidase (GoxA), CTQ synthesis occurs via distinct pathways. Unlike the diheme protein-dependent biogenesis seen in QHNDH, LodA and GoxA rely on flavoproteins, LodB and GoxB, respectively, to generate their cofactors.184,185,202,210 These differences suggest unique evolutionary adaptations and mechanistic diversity among CTQ-bearing enzymes. Comparative studies have highlighted variations in the roles and structural contexts of CTQ within these systems, emphasizing the need for further research to delineate the precise biogenesis and catalytic mechanisms across different enzyme classes.
CTQ exemplifies the biochemical ingenuity of quinone cofactors in enzymology, showcasing the versatility of protein-derived modifications in facilitating challenging redox reactions. Its formation and function differ across enzyme families, illustrating how protein environments and accessory proteins adapt to specific biochemical needs. For a comprehensive overview of quinone cofactors, readers are directed to the detailed review by Klinman and Bonnot, which delves into the biogenesis and functionality of these remarkable molecular motifs.22 The formation and stabilization of these crosslinked quinone cofactors underscore the evolutionary creativity of enzymes in harnessing free radical chemistry and long-range electron transfer for catalysis. As research advances, further insights into the structural determinants and dynamic pathways governing CTQ and TTQ biogenesis will continue to inspire biomimetic and synthetic applications.
3.3. Lysine tyrosylquinone (LTQ)
The quinone moiety of LTQ is also derived from a tyrosine residue. However, in addition to the tyrosine modification, there is also a crosslink between the quinone tyrosine and the side chain nitrogen of a lysine residue (Fig. 1, 26). LTQ was first identified and characterized through the biochemical investigation of a peptide from bovine aorta lysyl oxidase (LOX), a protein responsible for the posttranslational modification of elastin and collagen in the metabolism of connective tissue.110 Reminiscent of TPQ, the formation of LTQ is also a copper and oxygen-dependent autocatalytic process, and is required for catalysis. Due to its membership in the CuAO family, it is assumed that the mechanism of formation of LTQ is similar to that of TPQ, but with the addition of the lysine nitrogen into the orthoquinone intermediate.4 A 2.4 Å resolution crystal structure of human lysyl oxidase-like 2 (hLOXL2) has a zinc ion occupying the copper-binding site, which prevented LTQ formation but the LTQ forming residues were observed 16.6 Å away from each other, suggesting that the published structure is in a precursor state to LTQ formation.211 The structure of the mature LOXL2 containing the LTQ cofactor is predicted through molecular modeling and simulations.212 However, a crystal or cryo-EM structure of LOX with a mature LTQ cofactor has yet to be determined experimentally.3.4. Cys–Lys crosslink with a NOS bridge in transaldolase
Transaldolase, a pivotal enzyme in the pentose phosphate pathway across all domains of life, has garnered significant attention as a potential drug target. Recent studies by Tittman and colleagues on the transaldolase enzyme of Neisseria gonnorhoeae (NgTAL) has revealed an unprecedented redox-regulated mechanism mediated by a unique protein-derived cofactor (Fig. 1, 27).182 This discovery underscores the enzyme's potential as a therapeutic target for combating gonorrhea, a sexually transmitted disease of global concern.The study demonstrates that NgTAL's enzymatic activity is modulated by redox conditions. Site-directed mutagenesis of its three cysteine residues revealed that none of these form disulfide bonds, yet the loss of redox regulation upon substitution of Cys38 pointed to its critical role. Structural characterization of the oxidized and reduced states of NgTAL provided further insights. In the reduced state, NgTAL exhibits catalytic activity, while in the oxidized state, activity is abolished. Crystallographic analyses of the oxidized form unveiled a novel covalent crosslink between Cys38 and Lys8, bridged by an additional oxygen atom—forming an NOS (nitrogen–oxygen–sulfur) bridge (Fig. 11B). This crosslink is absent in the reduced state (PDB 3CLM), wherein molecular oxygen is observed near Cys38, suggesting its role in initiating the oxidation that leads to the formation of the NOS bridge.182
 | ||
| Fig. 11 (A) General cofactor biogenesis mechanism of NOS/SONOS bridge.213 Crystal structures of NOS/SONOS redox switches. (B) Oxidized NgTAL, showing the NOS bridge formed by the crosslink of Lys8 and Cys38 (turquoise; PDB 6ZX4). (C) Mpro of SARS-CoV-2 in complex with inhibitor MPI8, possessing a SONOS bridge formed by the crosslink of Cys44, Lys61, and Cys22 (gray; PDB 7UUA). | ||
The NOS bridge distinguishes itself from other protein-derived cofactors by its reversibility, enabling it to function as an allosteric redox switch. Under oxidizing conditions, the formation of the NOS bridge disrupts enzymatic activity, whereas reducing conditions restore the enzyme's functionality by breaking the crosslink. This reversible mechanism provides an elegant means of modulating enzyme activity in response to cellular redox states.
Computational studies shed light on the plausible mechanism of NOS bridge formation. The favored pathway involves the oxidation of the cysteine thiol by reactive oxygen species (ROS) to generate sulfenic acid, while the lysine amine undergoes oxidation to form hydroxylamine or an amine oxide.214 A condensation reaction between sulfenic acid and hydroxylamine, accompanied by water elimination, results in the formation of the NOS bridge. This mechanism not only highlights the versatility of oxidative modifications in enzyme regulation but also adds to the growing repertoire of protein-derived cofactors with unique functional and structural properties. Similarly, cysteinesulfenic acid, Cys-(S)-OH, and derivatives stabilized by elements of protein structure as novel protein cofactors in enzyme catalysis and redox regulation have been appreciated.17,33–35
3.5. Cys–Tyr crosslinks
Cys–Tyr crosslinks are versatile protein-derived cofactors that differ in their functional roles depending on their structural context. While radical-containing Cys–Tyr cofactors are directly involved in catalysis as the catalytic driving force to provide an oxidizing equivalent to the bound substrate (Fig. 1, 29), radical-free Cys–Tyr crosslinks (Fig. 1, 28) often serve to amplify catalytic efficiency.12,215 These crosslinks are integral to the activity of a range of enzymes, including thiol dioxygenases and reductases, underscoring their importance in diverse biochemical processes.The Cys–Tyr crosslink was first observed in the mouse CDO crystal structure,188 and later in rat and human CDO structures (Fig. 12A).14,223 This crosslink is not essential for catalysis but enhances catalytic efficiency by approximately 20-fold. Initially, Tyr157 was believed to initiate cofactor formation;224 however, recent structural and computational studies indicate that Cys93 undergoes the initial oxidation.14,225 The protein-derived cofactor is “untouchable” by traditional site-directed mutagenesis, as such mutations disable cofactor synthesis.
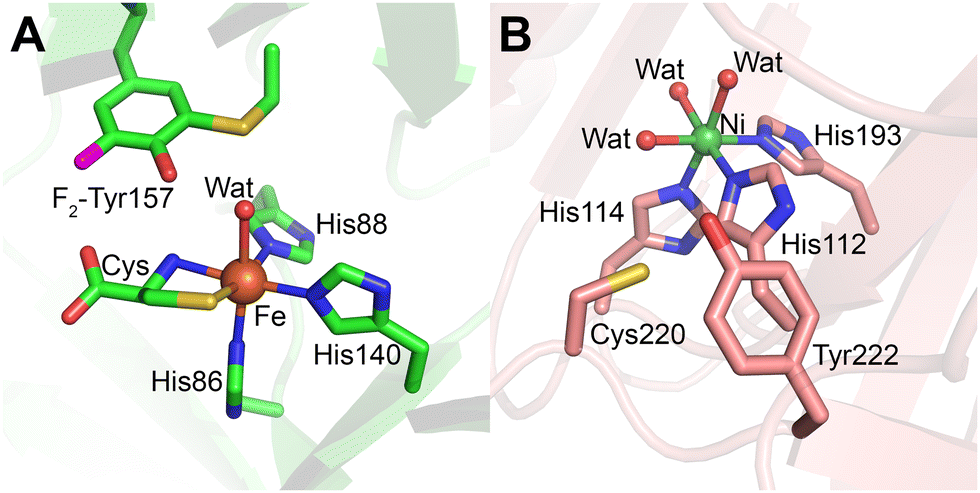 | ||
| Fig. 12 Cys–Tyr cofactor in mammalian thiol dioxygenases. (A) Cysteine-bound F2-Tyr157 hCDO (lime green; PDB 6BPV). (B) Cofactor-free Ni-ADO (salmon; PDB 7REI) is hexa-coordinated by three histidine residues, and three water molecules. Cofactor formation requires iron, cysteamine, and molecular oxygen therefore the crosslink is not expected in the Ni-bound crystal structure, but the crosslink has been detected via mass-spec in previous work. | ||
By introducing non-proteinogenic unnatural amino acids to specifically substitute the cofactor-bearing residues through genetic code expansion, the Cys–Tyr crosslink mechanism in human CDO has become better understood.14,225 Subsequent studies in human ADO12 and galactose oxidase13 have contributed to this understanding and showcased that this non-canonical amino acid substitution is a powerful approach to studying protein-derived crosslink cofactors. The ternary complex structure of a genetically modified variant with 3,5-difluoro-L-tyrosine 157 (F2-Tyr157) CDO bound to substrate cysteine and an O2 structural surrogate nitric oxide (˙NO) at 1.96 Å resolution revealed that ˙NO is equidistant (3.1 Å) from both Cys93 and Tyr157 residues, with F2-Tyr157 additionally hydrogen-bonded to the substrate's carboxyl group.225 This interaction suggests that Cys93 is the preferred site of initial oxidation.14,225
Mechanistically, substrate binding to the ferrous iron center generates an iron-bound superoxide radical, which oxidizes Cys93 to produce a thiyl radical. Tyr157 is subsequently oxidized by this thiyl radical, forming the Cys–Tyr crosslink (Fig. 13).225 CDO plays a central role in thiol metabolism with its substrate level fluctuating in cells in a large range depending on the metabolic states of fed, fasting, and starvation, it is proposed that the Cys–Tyr cofactor is formed after hundreds of turnovers to amplify the catalytic efficiency in the presence of excess substrate.226 This cofactor formation was first reported in 2006, establishing CDO as a model system for studying thiol dioxygenases.
 | ||
| Fig. 13 Proposed mechanism of formation of Cys–Tyr crosslink in CDO.225 Upon substrate binding to the Fe(II) center, an iron-bound superoxide radical is generated. The radical oxidizes Cys93, forming a thiyl radical and iron-bound hydroperoxide. The thiyl radical oxidizes Tyr157, generating the Cys–Tyr crosslink, and a transient-state radical species in Tyr157. C–H bond cleavage is driven by the formation of a ketone species after deprotonation of the hydroxyl group in the iron-bound hydroperoxide. | ||
In 2018, ADO was discovered to harbor a Cys–Tyr cofactor through mass spectrometry and 19F-NMR spectroscopy, representing a variation of this motif (Fig. 12B).12 Unlike CDO, ADO's cofactor residues, Cys220 and Tyr222, are located adjacent to each other in the protein sequence, allowing the crosslink to form without inducing significant structural changes. This proximity confers structural flexibility, enabling ADO to accept substrates of varying sizes, from small thiol metabolites like cysteamine to larger N-cysteine peptides. The presence of the Cys220-Tyr222 crosslink increases the catalytic rate of ADO by more than 10-fold12 through a concerted dioxygen transfer mechanism.227
Importantly, ADO exhibits dual functionality in thiol metabolism and oxygen sensing, leveraging its Cys–Tyr cofactor for catalytic enhancement only in thiol metabolism. The cofactor is not needed for oxygen sensing. Since the oxygenation of large protein substrates does not require a catalytic rate boost as cysteamine does during various metabolic states, the Cys–Tyr cofactor is not always synthesized in ADO.217,219 Additionally, Cys220 and Tyr222 sit in a dynamic loop that likely moves away from the catalytic iron center when the target residue of a protein substrate arrives.217 This trait makes the Cys–Tyr crosslink less frequently encountered in ADO and challenging to observe. It has been detected by protein mass spectrometry for the wild-type enzyme and shows approximately 50% occupancy for crosslinked and uncrosslinked forms in the F2-Tyr222 human ADO variant.12 Like LTQ,211 a crystal or cryo-EM structure of ADO with a mature Cys–Tyr cofactor remains unavailable to date, even though its uncrosslinked cofactorless structure is available for both human217 and mouse218 ADO proteins.
The unique properties of Cys–Tyr crosslinks, particularly their ability to amplify enzymatic efficiency and stabilize catalytic intermediates, make them a recurring motif in enzyme evolution. Continued exploration of these crosslinks promises to uncover new insights into their functional roles and mechanistic diversity in biological systems.
3.6. Stable (Cys–Tyr)˙ cofactor in copper-radical oxidases
The (Cys–Tyr)˙ cofactor represents a remarkable example of a stable yet catalytically potent crosslinked radical cofactor distinct from those derived from a single amino acid such as glycyl or tyrosyl radicals in RNR and PS II. It is a copper ligand in a superfamily of copper-radical oxidases oxidizing a variety of diverse carbohydrates.228,229 Permanently stabilized within the enzyme's active site, this tyrosyl radical (Tyr˙),186,230 covalently linked to a cysteine residue via a thioether bond,2,187 is akin to the tyrosyl radical in RNR. However, its reactivity is finely tuned to serve as the catalytic driving force for the specific substrates of its host enzyme. This duality of stability and reactivity distinguishes the (Cys–Tyr)˙ cofactor from the radical-free Cys–Tyr crosslinks discussed earlier.In copper-dependent galactose oxidase (GAO), the (Cys–Tyr)˙ cofactor is central to the enzyme's ability to catalyze the two-electron oxidation of a wide range of primary alcohols to their corresponding aldehydes, coupled with the reduction of molecular oxygen to hydrogen peroxide.2,229,231–233 This reaction is facilitated by the synergy between the (Cys–Tyr)˙ radical and the Cu(II) ion, a unique coupling that enables two-electron redox chemistry with a single copper center.229,231,233,234 The (Cys–Tyr)˙ radical is a metal ligand and permanently stabilized through antiferromagnetic coupling with the Cu(II) ion, making it EPR-silent, as confirmed by Whittaker and Whittaker using X-ray absorption near-edge spectroscopy (XANES).230,235 The antiferromagnetic coupling renders both paramagnetic centers, Cu(II), and the (Cys–Tyr)˙ ligand, spectroscopically silent in EPR experiments. However, the hidden Cu(II) center becomes EPR-active when the (Cys–Tyr)˙ radical is neutralized by a radical scavenger, hydroxyurea, and hidden again upon one-electron oxidation by K3Fe(CN)6.13
The radical cofactor is formed by the covalent linkage of Cys228 and Tyr272 through an autocatalytic, irreversible posttranslational modification.229,231 The crystal structure of GAO revealed that the sulfur atom of Cys228 forms a thioether bond with the Cε of Tyr272, a crosslink essential for catalytic function.187 This configuration supports the radical's stability while allowing it to participate in substrate oxidation.
Mutagenesis studies have reinforced the importance of the cofactor. Substitution of Cys228 with glycine (C228G) abolished cofactor formation and reduced catalytic efficiency by approximately 1000-fold, underscoring the critical role of the sulfur atom in stabilizing the Tyr˙ radical.236,237 Synthetic model studies have provided additional mechanistic insights, demonstrating that thioether substitutions lower the phenol's pKa and reduction potential, facilitating radical formation and stabilization.234
Recent advances in genetic code expansion have offered new perspectives on the cofactor's properties and formation.238 Substitution of Tyr272 with F2-Tyr retained cofactor formation but reduced catalytic efficiency to 12% of the wild-type enzyme. Structural studies of F2-Tyr GAO revealed the formation of a monofluorinated radical cofactor via C–F bond cleavage, illustrating how electronic modifications influence the radical's reactivity and stability (Fig. 14).13,239
 | ||
| Fig. 14 Active sites of GAO and F2-Tyr272 GAO showing the (Cys–Tyr)˙ cofactor. (A) Active site of GAO (purple; PDB 6XLT) shows the crosslink between Tyr272 and Cys228, with the copper ion coordinated in square pyramidal geometry by two histidines, Tyr495, and cofactor-bearing Tyr272. (B) Active site of F2-Tyr272 GAO (teal; PDB 6XLS) retaining a monofluorinated (Cys–Tyr)˙ cofactor through the oxidative C–F bond cleavage during autocatalytic cofactor formation.13 | ||
The proposed cofactor maturation mechanism involves a copper-bound superoxide radical oxidizing the thiol group of Cys228, forming a copper-bound hydroperoxide intermediate and a thiyl radical. This thiyl radical then oxidizes Tyr272 (or F2-Tyr272), resulting in the formation of the thioether bond and the mature (Cys–Tyr)˙ cofactor (Fig. 15).13,239
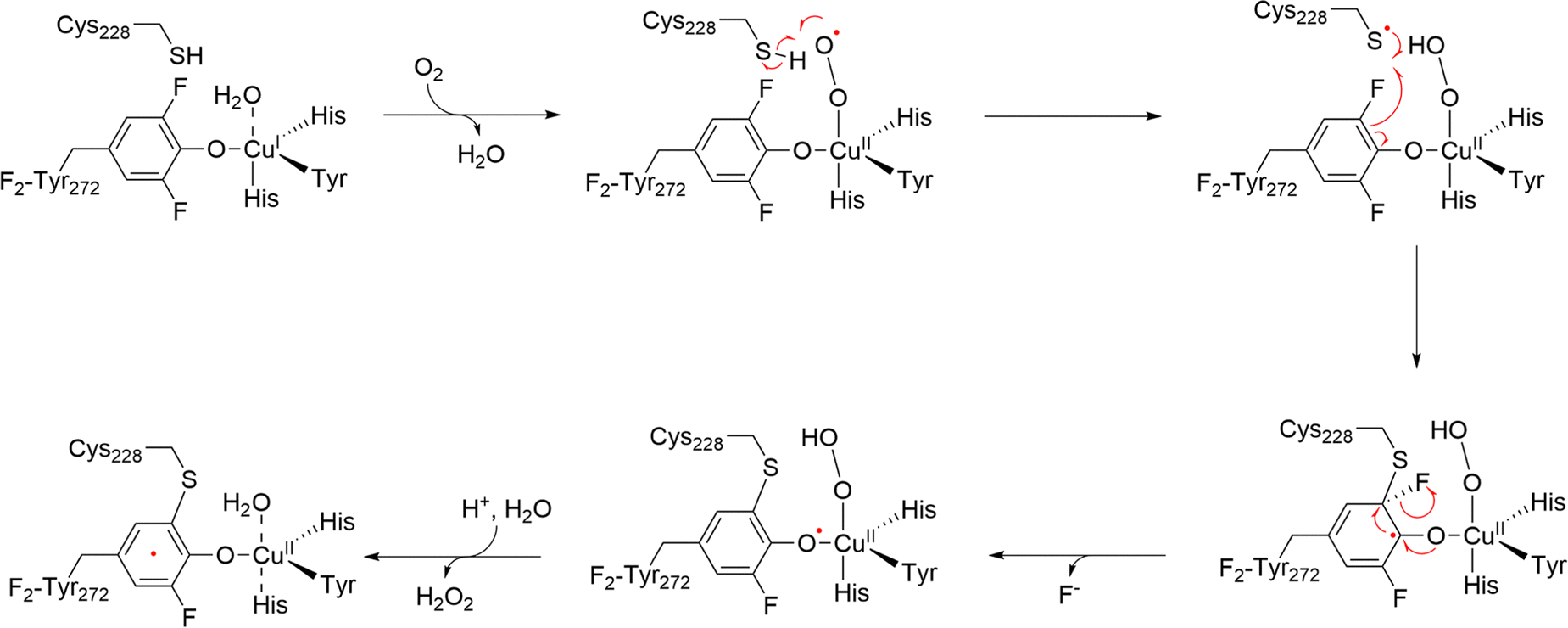 | ||
| Fig. 15 Proposed biogenesis mechanism of the (Cys–Tyr)˙ in F2-Tyr272 GAO. | ||
In addition to GAO, the (Cys–Tyr)˙ cofactor is found in glyoxal oxidase (GLOX) and other copper radical oxidases, where it drives diverse catalytic reactions tailored to their specific substrates.228,229,231 Its evolutionary significance and versatility underscore its importance as a powerful and highly specialized catalytic tool in enzymatic chemistry.
4. Protein cofactors derived from covalent crosslink of three or more amino acids
This section highlights protein-derived cofactors formed by the covalent crosslinking of three or more distinct amino acid residues. These trimeric or more crosslinks represent some of the most complex and functionally diverse modifications found in nature. By enhancing catalytic efficiency, stabilizing active sites, and introducing novel functionalities, these cofactors play critical roles in the biochemical processes of their host proteins. To date, four distinct trimeric crosslinked cofactors have been identified: Cys–Lys–Cys, Gly–Ala–Ser, Met–Tyr–Trp, and Ser–Tyr–Gly (Table 3). Additionally, the redox PQQ cofactor (Fig. 1, 37) is formed from five amino acid residues Glu–Val–Thr–Leu–Tyr with further posttranslational oxidations. Below, we examine their structure, function, and synthesis mechanisms.| Source | Cofactor | New chemical bonds formed | Representative enzyme | Biogenesis | Function | Ref. |
|---|---|---|---|---|---|---|
| Cys–Lys–Cys | SONOS bridge | S–O–N–O–S | Main protease | Autocatalytic | Allosteric redox switch | 213 and 240 |
| Gly–Ala–Ser | MIO | C–N | Histidase | Autocatalytic | Catalysis | 241 |
| MIO | C–N | Phenylalanine ammonia lyase | Autocatalytic | Catalysis | 242 | |
| MIO | C–N | Tyrosine ammonia lyase | Autocatalytic | Catalysis | 243 | |
| Met+–Tyr–Trp | M+YW | C–C, C–S | Catalase-peroxidase (KatG) | Autocatalytic | Enable catalase activity | 244 |
| Met+–Tyr–Trp–OOH | M+YW–OOH | C–C, C–S, C–O | Catalase-peroxidase (KatG) | Autocatalytic | Modulates catalase-peroxidase functions | 244 |
| Ser–Tyr–Gly | Ser–Tyr–Gly | C–N | Green fluorescent protein | Autocatalytic | Fluorescence emission | 245 |
| Glu–Val–Thr–Leu–Tyr | PQQ | C–C | Alcohol dehydrogenase, aminoadipic 6-semialdehyde dehydrogenase | Enzymatic: PqqE, PqqD, PqqF, PqqB, and PqqC | Catalysis | 203–205 |
| C–N | ||||||
| CO |
4.1. Cys–Lys–Cys crosslink with a SONOS bridge
The Cys–Lys–Cys cofactor represents a fascinating chemical feature involving a sulfur–oxygen–nitrogen–oxygen–sulfur (SONOS) bridge (Fig. 1, 33). Initially identified in the main protease (Mpro) of SARS-CoV-2, this structure has garnered significant attention due to its role in the pathogenesis and replication of the virus, making it a prime drug target in COVID-19 research.213,240Crystal soaking experiments revealed Y-shaped crosslinks between Cys22, Cys44, and Lys61 in Mpro (Fig. 11C). Computational studies suggest that the SONOS bridge forms through a series of oxidation reactions, likely mediated by reactive oxygen species (ROS).214 A thio-(hydro)peroxy acid intermediate has been identified as a plausible precursor to both NOS and SONOS bridges, with homolytic dissociation leading to NOS formation. However, the exact pathway for SONOS formation remains under investigation.
The regulatory role of SONOS and NOS bridges in Mpro exemplifies their functional versatility. The reversible formation and dissolution of these crosslinks appear to act as redox switches, modulating enzymatic activity in response to oxidative stress. Further structural and mechanistic studies are crucial for understanding the formation and potential therapeutic exploitation of these cofactors.
4.2. Ser–Tyr–Gly crosslink in green fluorescent protein
The Ser–Tyr–Gly crosslink forms the fluorescent chromophore at the core of green fluorescent protein (GFP), a widely used tool in molecular and cellular biology.245 GFP's chromophore is generated by the autocatalytic cyclization and oxidation of the Ser65 (or Thr65), Tyr66, and Gly67 residues within a tightly folded β-barrel structure (Fig. 1, 34).Unlike many cofactors, the formation of GFP's chromophore is spontaneous and requires no external enzymes. However, molecular oxygen is essential, as is precise protein folding to orient the residues for cyclization. The resultant chromophore exhibits a unique conjugated structure that absorbs blue light and emits green fluorescence (Fig. 16).
 | ||
| Fig. 16 Crystal structure of GFP cofactor formed by the cyclization and oxidation of Thr65, Tyr66, and Gly67 (pink; PDB 1EMA). | ||
Mechanistic parallels have been drawn between the GFP chromophore and the methylene imidazolone (MIO) cofactor in histidase enzymes,246 suggesting a shared evolutionary origin (Fig. 1, 35).21 Despite extensive use, the detailed mechanism of GFP chromophore biosynthesis remains elusive. Ongoing studies aim to elucidate these pathways, which may enable the engineering of fluorescent proteins with novel properties.
4.3. Met+–Tyr–Trp cofactor in catalase-peroxidase
In the catalase-peroxidase enzyme (KatG), the Met+–Tyr–Trp (MYW) cofactor is a trimeric crosslink critical for its bifunctional activity. KatG is a well-known enzyme, largely due to Mycobacterium tuberculosis KatG is an essential utility for activating the frontline antitubercular prodrug isoniazid (INH) and detoxifying reactive oxygen species such as H2O2, contributing to the pathogen's survival and virulence.247 This bifunctional enzyme has emerged as a promising target in tuberculosis drug development, with chemical modification of the MYW cofactor rendering the bacterium more susceptible to peroxide-mediated immune clearance and simultaneously enhancing peroxidase activity to improve INH activation.248The MYW cofactor, first characterized in Haloarcula marismortui KatG by X-crystallography, is autocatalytically formed during protein maturation.249 Spectroscopic studies suggest that an Fe(IV)O porphyrin cation radical oxidizes Met, Tyr, and Trp residues to generate the crosslinked structure.250,251 This cofactor endows KatG with an exceptional single-function catalase-like activity, synergistically enhancing H2O2 detoxification by several orders of magnitude while retaining its innate peroxidase functionality.251,252
It has been proven through EPR coupled with isotope labeling that MYW becomes a transient MYW˙ free radical after H2O2 binds to the adjacent heme and becomes activated, and a cofactor radical-based catalase mechanism is thus proposed.253 The MYW cofactor spares one of the oxidizing equivalents and located next to the Fe(IV)O, enabling strong coupling for 2e− oxidation chemistry against subsequent H2O2 for catalase activity. Otherwise, the oxidizing equivalents from H2O2 are directed outside the heme center of KatG via aromatic residues to support 1e− oxidation of organic substrates (i.e., peroxidase activity).252
Interestingly, two forms of the MYW cofactor exist in nature: MYW (catalase-active) and MYW-OOH (catalase-inhibitory) (Fig. 1, 36a and 36b).254–256 Studies of the solution state of as-isolated M. tuberculosis KatG indicate that the MYW-OOH cofactor can act as a molecular switch, toggling between active MYW and inactive MYW-OOH states depending on its environment conditions for the chemical structure of the trimeric cofactor, such as temperature and hydrogen peroxide levels.254 This dual functionality underscores the evolutionary significance of the MYW cofactor in balancing oxidative defense and metabolic processes.
4.4. Pyrroloquinoline quinone (PQQ) cofactor in dehydrogenases
The crosslinked protein cofactor PQQ (Fig. 1, 37) found in alcohol dehydrogenases and aminoadipic 6-semialdehyde dehydrogenase,203–205 possesses two remarkably unique features. First, it is the only known protein cofactor derived from more than three amino acid residues. Second, it is the only one that does not remain covalently bound to the parent protein. The synthesis of PQQ starts from a tyrosine residue and subsequently involves crosslinking and further chemical modifications of glutamate, valine, threonine, and leucine residues (Fig. 17).67,257 This complex process is facilitated by a series of auxiliary proteins: PqqE, PqqD, PqqF, PqqB, and PqqC. During the post-translational modifications (PTMs), this quinone cofactor loses the peptide connection but remains bound at the active site of the protein.203 | ||
| Fig. 17 Enzymatic synthesis PQQ consists of nine steps including crosslink and chemical modifications of five amino acid residues.67 | ||
5. Covalent crosslinks between heme and protein residues in metalloenzymes
Covalent linkages between heme and protein residues play crucial roles in the function and stability of heme-dependent enzymes. One well-known example is heme c, which is always covalently attached to two cysteine (Cys) residues of the associated protein. Its attachment occurs through thioether bonds formed between the vinyl side chains of the heme and the cysteine residues in the protein, creating a protein cofactor with the structure Cys–heme–Cys, which could be #38 if it were included in Fig. 1. While electron transfer is the primary function, heme c also plays other roles, such as in apoptosis and serving as a catalytic site in some enzymes. Due to its prevalence, heme c is not typically included in the protein-derived cofactor category.These crosslinks, formed between heme substituents (such as vinyl groups) and amino acid side chains (like cysteine or histidine), can significantly impact catalysis and heme retention. In addition to heme c, a recent discovery shows that SfmD, a monooxygenase involved in saframycin A biosynthesis, contains a mono-covalently linked Cys–Heme.36 SfmD possesses an autocatalytically generated thioether crosslink between the heme 4-vinyl group and Cys317 (Fig. 1, 17). This covalent bond is essential for catalytic activity, as demonstrated by the complete loss of activity upon mutation of Cys317 to serine or alanine. The observation of Cys–heme in the crystal structure of an oxygenase in SfmD is surprising, as all other characterized members of the same superfamily, known as heme-dependent aromatic oxygenase (HDAO), contain a heme b without crosslink and thioether bond attachment.258 This unique linkage enables dynamic rotation of the heme plane during substrate binding while maintaining heme retention at the active site. Such substantial heme center dynamics are warranted in this enzyme, as the heme has two axial histidine ligands; one of these dissociates during catalysis upon substrate binding and then re-coordinates to the heme iron at the end of the catalytic cycle (Fig. 18A). This dynamic coordination adds a significant trigger for enhancing the oxidation power of the heme. Given that this enzyme can also utilize L-Tyr as an alternative substrate, albeit with lower efficiency compared to its native substrate, 3-methyl-L-Tyr, this unusual heme cofactor with a single covalent crosslink to a cysteine residue and an additional mobile distal histidine ligand may be crucial for preventing misfire and L-Tyr depletion.
 | ||
| Fig. 18 Crosslinks to heme moieties in SfmD and GlbN. (A) SfmD has a unique Cys–heme crosslink at the 4-vinyl group used for catalysis, two conformations are shown. Initially, there is bis-His coordination and presence of the crosslink (left, navy blue; PDB 6VDQ), upon addition of 3-Me-L-Tyr, ascorbate, and oxygen, His274 disassociates (right, navy blue; PDB 6VE0).36 (B) GlbN has a His–heme crosslink at the 2-vinyl group also used for catalysis. Like SfmD, it presents bis-His coordination (left, green; PDB 4MAX).259 The right side shows the proposed biogenesis mechanism of the His–heme crosslink in GlbN where the 2-vinyl group is protonated at the Cβ position to yield a Cα carbocation. The carbocation then undergoes a nucleophilic attack by His117, producing the mature crosslink.260 | ||
The formation of this Cys–heme crosslink has been partially replicated in sperm whale myoglobin by introducing a cysteine residue near the heme 4-vinyl group under reducing conditions, further validating the feasibility of this type of modification.261 Another distinct type of covalent heme-protein crosslink involves a histidine residue and the heme group. This unprecedented histidine-heme bond was discovered in hemoglobins from the cyanobacteria Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002 (GlbN). In these proteins, the crosslink forms between the heme 2-vinyl group and a histidine residue (Fig. 1, 18). Site-directed mutagenesis and NMR studies have confirmed the functional role of this crosslink, particularly in catalysis.44,260 This discovery has spurred research into exploiting this type of linkage for protein engineering. Introducing a histidine residue near a heme vinyl group has been shown to improve heme retention. This strategy has been successfully applied to myoglobin and Chlamydomonas hemoglobin by mimicking the crosslink observed in cyanobacteria.262,263 Reminiscent of SfmD, GlbN also has a bis-His coordination with the heme and shows oxygenation activity. GlbN is capable of ˙NO dioxygenase activity, where the His–heme crosslink enables ˙NO reduction to HNO.264 The proposed biogenesis mechanism of this modification involves the nucleophilic attack of a neutral histidine residue to a Cα carbocation (Fig. 18B).260
6. Modern approaches revitalize crosslinked cofactor studies
While the discovery of protein-derived crosslinked cofactors initially sparked significant interest, the field has experienced limited progress in the past two decades. The inherent, irreversible nature of these crosslinks, forming only once per protein molecule, renders traditional mutagenesis approaches ineffective. This “untouchable” characteristic, coupled with the frequent absence of spectroscopic signatures, has hindered studies of their biogenesis and catalytic roles. Relying solely on mass spectrometry or structural determination for crosslink detection is impractical for routine analyses. Moreover, the stochastic, post-catalytic formation of these cofactors, occurring after an undefined number of catalytic cycles, further complicates mechanistic investigations. Consequently, the field has stagnated, demanding new strategies to unlock the secrets of crosslinked cofactor synthesis and function.Recently, an innovative approach employing site-specific non-canonical amino acid (ncAA) substitution has emerged as a powerful tool for reviving the crosslink cofactor field.12,14 By replacing cofactor-bearing residues with ncAAs, such as unnatural amino acid 3,5-difluoro-L-tyrosine, or L-3,4-dihydroxyphenylalanine (L-DOPA) from over 500 naturally occurring amino acids, the studies can maintain the essential chemical character of the target while introducing specific functional modifications. This method bypasses the limitations imposed by the irreversible crosslinks.
The ncAA-incorporation and site-specific cofactor substitution is an innovative, viable and rigorous approach to gain new insights into cofactor assembly and the catalytic properties of an enzyme. The ncAA incorporation strategy utilizes engineered tRNA/aminoacyl-tRNA-synthetase (aaRS) pairs and amber codon suppression to introduce desired amino acids during translation. The development of robust pEVOL vectors and commercially available ncAA systems has significantly enhanced this technology. This method consists of: (1) a plasmid that expresses aaRS pair which has been evolved to incorporate a specific ncAA, and (2) a plasmid of the gene of interest with a modified codon at the desired site (usually substituted with an amber stop codon) that will be recognized by the cognate charged tRNA (Fig. 19). This method allows for the ncAA to be incorporated using native protein translation machinery. After years of resilient research, the pEVOL vector was developed to enhance non-canonical amino acid incorporation in E. coli, with at least 34 plasmids coding for different ncAAs commercially available.265
 | ||
| Fig. 19 Cartoon diagram of genetic incorporation of non-canonical amino acids into proteins. | ||
Protein-derived cofactor studies greatly benefit from the ongoing development of aaRS/tRNACUA pairs that expand the toolbox to accurately and precisely probe mechanisms of formation, kinetics, and structural changes. Applications in bacterial RNR,266,267 thiol dioxygenases (CDO/ADO),12,14,225 and oxidases (GAO).13,238 These studies provided experimental evidence to support a new Cys–Tyr crosslink in ADO, direct evidence for the involvement of two tyrosine residues in radical propagation in RNR I, and evidence to support a concerted O–O transfer instead of a stepwise O-atom transfer proposed for CDO catalytic pathway, etc.
Continued development and expansion of ncAA incorporation techniques will be crucial for advancing our understanding of protein-derived cofactors. This approach offers a promising avenue for capturing catalytic intermediates, enabling novel reactions, and enhancing existing functions in a broad range of biological systems beyond those harboring crosslinked cofactors.268
7. Bridging synthetic applications and mechanistic understanding
Many of the protein-derived cofactors discussed are essential for catalytic activity, either directly or indirectly. This opens two major avenues of discovery: (1) synthesizing mechanistic probes to challenge a system and gain a mechanistic understanding of the cofactors for their role in catalysis, and (2) synthesizing inhibitors to prevent the formation of essential cofactors in undesired systems. Moreover, using synthetic models, either directly or through incorporation with proteins, has garnered attention, such as in the efforts led by Hayashi and colleagues.79,120,269–2727.1 Development of biomimetic systems
Quinones have applications in organic synthesis, catalysis, and industry due to their unique redox properties. They readily undergo reversible two-electron reductions, and their ability to present multiple oxidation states (quinone, semiquinone, and hydroquinone) allows for versatile reaction outcomes.273 Quinones resembling those present in the cofactors of CuAOs and other quinoproteins have been generated to investigate quinone-catalyzed oxidations of organic substrates and compare them to quinones with high reduction potentials.274 Using PQQ as a model, phenanthroline-derived quinones were synthesized and challenged with primary and secondary amines. The reaction products supported the hypothesis of a transamination mechanism.275 This mechanistic study focused on exploring how quinone moieties are reduced by their substrates. The quinones available in biological systems that operate at ambient conditions serve as a window to investigate how other naturally occurring quinones catalyze their designated reactions. These quinones are commonly seen in fungicides, allelochemicals, and siderophores.273Functional models of the SCS Ni(II) pincer complex found in LarA, also called the NPN cofactor, have been generated to study the cofactor's role in the catalytic mechanism.276–278 One of the previous models for the NPN cofactor contained thioamide groups at the C2 and C3 positions, and catalyzed alcohol dehydrogenation irreversibly with no racemization, opposite of the canonic mechanism of LarA.276 A further optimized model which replaced the thioamide groups with thiocarboxylates is more structurally similar to the NPN cofactor and displayed lactate racemization, serving as a better-suited mechanistic probe.277 Combined computational and experimental studies highlight two possible pathways, but the more favorable one involves the reversible proton-coupled hydride transfer to C1 of the NPN cofactor.277 These studies demonstrate the importance of using protein-derived cofactors as model systems when designing new metal-based catalysts and/or pincer ligands, in this case, for alcohol dehydrogenation reactions.276
GAO is a well-rounded enzyme that has gained a lot of attention due to its ability for C–H and, recently found, C–F bond functionalization under ambient conditions.13 A substantial amount of work has been done to generate GAO model compounds that are capable of C–sp3–H oxidations with equal or greater activity/selectivity.234,279–283 Many of these GAO biomimetic systems attempt to mimic the enzyme active site or the tyrosyl radical to catalyze the same chemistry. These systems are also incorporated into the use of biosensors given that galactose quantification is highly relevant in the dairy/fermentation industries. Methods using GAO as a biosensor date back to 1977,284 and continue to be optimized in the present day.285
7.2 Biocatalysis
Protein-derived cofactors have expanded the toolbox for enzymatic reactions, offering unique catalytic properties such as regio- and stereoselectivity. A prominent example is found in alcohol oxidation/dehydrogenation reactions. Copper-radical oxidases (CROs) such as galactose oxidase and glyoxal oxidases are a subset of enzymes that catalyze the two-electron oxidation of primary alcohols, concomitant with the reduction of molecular oxygen to hydrogen peroxide, without using external organic cofactors since they have a built-in Cys–Tyr radical cofactor that aids in catalysis.228 On the contrary, alcohol dehydrogenases (ADHs) and alcohol oxidases (AOXs) both require external cofactors that are either NAD+-based or FAD-based for catalysis.286 The broad substrate range accepted by CROs and their ability to only require molecular oxygen for catalysis has highlighted their potential applications as environmentally friendly biocatalysts for industrial, bioengineering, and biotechnological purposes.287,2888. Structural variations and function diversity
Protein-derived cofactors are remarkable products of posttranslational modifications that transform mundane amino acid residues into unique catalytic or structural moieties. These cofactors introduce novel functions such as redox switching, catalytic rate/efficiency amplification, adding a new function or converting a precursor protein to a functionally active enzyme, vastly expanding the functional repertoire of proteins. Even cofactors derived from identical amino acids can display distinct functions based on their position and conformations within their host enzymes. The evolution of these cofactors reflects nature's ability to optimize proteins for increasingly complex biological roles.A striking example of structural variation in a protein-derived cofactor is the two unique cofactor forms in KatG, MYW and MYW-OOH (Fig. 20). Previous crystallography studies identified an indole-N-linked hydroperoxyl group MYW-OOH cofactor, and recently this MYW-OOH form was extensively studied in solution and compared to the active MYW form.254 The MYW-OOH cofactor was first identified in crystal structures of KatG expressed at 25 °C, while the MYW form predominates in KatG expressed at 37 °C. Resonance Raman (rR), electron paramagnetic resonance (EPR), mass spectrometry, and X-ray crystallographic studies confirmed the reversible interconversion between these forms.
 | ||
| Fig. 20 Heme pocket of M. tuberculosis KatG with focus on MYW crosslink and His108, a catalytically relevant distal residue. (A) MYW form of KatG's cofactor that supports catalase activity (cyan; PDB 8W1X) shows a putative O2 molecule, presumably a decomposed product of the hydroperoxyl moiety. (B) Natural MYW-OOH form of KatG's cofactor that presents limited catalase activity (cyan; PDB 8W1W).254 | ||
Structural variation is also evident in the Lys–Cys–NOS cofactor of Neisseria gonorrhoeae transaldolase (NgTAL), which functions as a reversible redox switch. In its oxidized state, the cofactor contains a sulfur–oxygen–nitrogen–oxygen–sulfur (NOS) bridge, detectable via sulfur K-edge X-ray absorption spectroscopy (XAS).289 Upon reduction, the NOS bridge is absent, and NgTAL becomes catalytically active, reverting to inactivity over time as the oxidized form reforms. Unlike most protein-derived cofactors, which enhance catalytic efficiency, the NOS bridge decreases NgTAL's catalytic activity. Computational studies indicate that the high-energy nature of the NOS bridge supports its reversibility, allowing the enzyme to cycle between active and inactive states. These features make the NOS cofactor an intriguing target for studies in protein engineering and drug discovery, particularly for applications requiring redox-controlled enzymatic switches.
The structural plasticity of protein-derived cofactors highlights their adaptability in response to environmental or cellular cues. Variations in cofactor structure often directly influence enzymatic activity, substrate specificity, or regulatory mechanisms, underscoring the evolutionary ingenuity of these modifications. As seen with the MYW and NOS cofactors, structural variants may serve as molecular switches, toggling between states to regulate biological functions dynamically.
9. Summary and perspective
This review has explored the fascinating world of amino acid modifications and crosslinking in proteins, emphasizing the structural and functional diversity of protein-derived cofactors and their transformative role in enzymology. Originating from PTMs, these cofactors reveal a hidden layer of protein complexity, enabling enzymes to perform functions that extend beyond the capabilities of unmodified amino acid residues. In particular, the amino acid residue crosslinking process isn't just about molecular intricacies—it's about the creation of novel redox centers that significantly impact enzyme functionality. This burgeoning field has unveiled a new level of protein complexity, as evidenced by the discovery of protein-derived cofactors. Since the foundational work of Okeley and van der Donk two decades ago, which summarized 17 distinct cofactors, the field has expanded significantly, now encompassing 38 unique types (42 when free radical forms are counted), categorized into three major groups: single amino acid-derived cofactors, two-residue crosslinked cofactors, and trimeric crosslinked cofactors.A recurring theme in cofactor biogenesis is the involvement of metal centers. Protein-bound metals such as iron and copper often facilitate the complex chemical transformations necessary for cofactor formation, acting as catalytic mediators in autocatalytic or enzymatically driven pathways. While not all cofactors require metal centers, most characterized cases display the integral role of metalloproteins in enzymatic catalysis and the generation of protein-derived cofactors.
The discovery of these protein-derived cofactors underscores the versatility and ingenuity of nature's enzymatic toolbox. Beyond enhancing catalytic efficiency, protein-derived cofactors drive the evolution of novel biochemical pathways and regulatory mechanisms. Advances in structural biology, including X-ray crystallography, cryo-electron microscopy, and emerging biophysical techniques, have been instrumental in identifying and characterizing these cofactors, paving the way for future discoveries.
Despite these advances, significant challenges remain. The lack of predictive methods for identifying PTMs and the limited understanding of cofactor formation pathways highlight the need for further research. Protein-derived cofactors remain the most challenging aspect of protein structure–function relationships, for even the most advanced AI models. Research advances in this area will help generative AI grow stronger and more accurate in predicting protein structure and functions. Further studies will connect and integrate bioinformatics, structural biology, and metalloenzyme chemistry to elucidate the rules governing cofactor formation and function.
It is important to note that twenty-two genetically coded amino acids aren’t enough for catalysis. At least 38 protein-derived cofactors play critical roles in enhancing or even introducing new catalytic activities in enzymes. As the field progresses, the study of protein-derived cofactors promises to yield new insights into enzymology, protein evolution, novel catalytic functions, and bioengineering. These insights will deepen our understanding of fundamental biological processes, and offer innovative strategies for applications in biotechnology, drug discovery, and synthetic biology.
Data availability
All data supporting this study are included in the manuscript. All figures are original, created by the authors, and appropriately cited where external sources were used to support their illustrations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (NIH) under award numbers GM108988 and GM152982, the National Science Foundation (NSF) award under CHE-2204225, and the Welch Foundation grant AX-2110-20220331. A. L. acknowledges the generous support of the Lutcher Brown Endowment Fund.References
- L. Xie and W. A. Van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12863–12865 CrossRef CAS PubMed.
- N. Ito, Seibutsu Butsuri, 1992, 32, 300–305 CrossRef CAS.
- M. D. Swain and D. E. Benson, Proteins, 2005, 59, 64–71 CAS.
- V. L. Davidson, Biochemistry, 2007, 46, 5283–5292 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O’Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CAS.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- D. Chakravarty, M. Lee and L. L. Porter, Curr. Opin. Struct. Biol., 2025, 90, 102973 CAS (1–7).
- T. C. Terwilliger, D. Liebschner, T. I. Croll, C. J. Williams, A. J. McCoy, B. K. Poon, P. V. Afonine, R. D. Oeffner, J. S. Richardson, R. J. Read and P. D. Adams, Nat. Methods, 2024, 21, 110–116 CAS.
- H. Longin, N. Broeckaert, V. van Noort, R. Lavigne and H. Hendrix, Curr. Opin. Microbiol., 2024, 77, 102425 CAS (1-7).
- O. Carugo, Comput. Biol. Chem., 2024, 110, 108069 CAS (1-6).
- W. Wang, Z. Gong and W. A. Hendrickson, Acta Crystallogr., Sect. D:Struct. Biol., 2025, 81, 4–21 CAS.
- Y. Wang, W. P. Griffith, J. Li, T. Koto, D. J. Wherritt, E. Fritz and A. Liu, Angew. Chem., Int. Ed., 2018, 57, 8149–8153 CAS.
- J. Li, I. Davis, W. P. Griffith and A. Liu, J. Am. Chem. Soc., 2020, 142, 18753–18757 CrossRef CAS PubMed.
- J. Li, W. P. Griffith, I. Davis, I. Shin, J. Wang, F. Li, Y. Wang, D. J. Wherritt and A. Liu, Nat. Chem. Biol., 2018, 14, 853–860 CrossRef CAS PubMed.
- S. E. Hromada, A. M. Hilbrands, E. M. Wolf, J. L. Ross, T. R. Hegg, A. G. Roth, M. T. Hollowell, C. E. Anderson and D. E. Benson, J. Inorg. Biochem., 2017, 176, 168–174 CrossRef CAS PubMed.
- R. J. Martinie, P. I. Godakumbura, E. G. Porter, A. Divakaran, B. J. Burkhart, J. T. Wertz and D. E. Benson, Metallomics, 2012, 4(1037–1042), 1008 Search PubMed.
- A. Claiborne, T. C. Mallett, J. I. Yeh, J. Luba and D. Parsonage, Adv. Protein Chem., 2001, 58, 215–276 CrossRef CAS PubMed.
- N. M. Okeley and W. A. Van der Donk, Chem. Biol., 2000, 7, R159–R171 CrossRef CAS PubMed.
- C. T. Walsh, Posttranslational Modification of Proteins: Expanding Nature's Inventory, Roberts & Co, 2006, pp. 1–490 Search PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto Jr, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS PubMed.
- V. L. Davidson, Biochemistry, 2018, 57, 3115–3125 CrossRef CAS PubMed.
- J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- N. Fujieda, Biosci., Biotechnol., Biochem., 2020, 84, 445–454 CrossRef CAS PubMed.
- J. P. Klinman, Acc. Chem. Res., 2015, 48, 449–456 CrossRef CAS PubMed.
- U. Ermler, W. Grabarse, S. Shima, M. Goubeaud and R. K. Thauer, Science, 1997, 278, 1457–1462 CrossRef CAS PubMed.
- D. Deobald, L. Adrian, C. Schöne, M. Rother and G. Layer, Sci. Rep., 2018, 8, 7404 CrossRef PubMed (1-12).
- T. Wagner, J. Kahnt, U. Ermler and S. Shima, Angew. Chem., Int. Ed., 2016, 55, 10630–10633 CrossRef CAS PubMed.
- U. C. Kabisch, A. Grantzdorffer, A. Schierhorn, K. P. Rucknagel, J. R. Andreesen and A. Pich, J. Biol. Chem., 1999, 274, 8445–8454 CrossRef CAS PubMed.
- M. Wagner, D. Sonntag, R. Grimm, A. Pich, C. Eckerskorn, B. Sohling and J. R. Andreesen, Eur. J. Biochem., 1999, 260, 38–49 CrossRef CAS PubMed.
- B. Schmidt, T. Selmer, A. Ingendoh and K. von Figura, Cell, 1995, 82, 271–278 CrossRef CAS PubMed.
- M. P. Cosma, S. Pepe, I. Annunziata, R. F. Newbold, M. Grompe, G. Parenti and A. Ballabio, Cell, 2003, 113, 445–456 CrossRef CAS PubMed.
- T. Dierks, B. Schmidt, L. V. Borissenko, J. Peng, A. Preusser, M. Mariappan and K. von Figura, Cell, 2003, 113, 435–444 CrossRef CAS PubMed.
- J. I. Yeh, A. Claiborne and W. G. J. Hol, Biochemistry, 1996, 35, 9951–9957 Search PubMed.
- S. Nagashima, M. Nakasako, N. Dohmae, M. Tsujimura, K. Takio, M. Odaka, M. Yohda, N. Kamiya and I. Endo, Nat. Struct. Biol., 1998, 5, 347–351 CrossRef CAS PubMed.
- T. Murakami, M. Nojiri, H. Nakayama, N. Dohmae, K. Takio, M. Odaka, I. Endo, T. Nagamune and M. Yohda, Protein Sci., 2000, 9, 1024–1030 Search PubMed.
- I. Shin, I. Davis, K. Nieves-Merced, Y. Wang, S. McHardy and A. Liu, Chem. Sci., 2021, 12, 3984–3998 Search PubMed.
- D. D. Nayak, N. Mahanta, D. A. Mitchell and W. W. Metcalf, eLife, 2017, 6, e29218 CrossRef PubMed.
- D. D. Nayak, A. Liu, N. Agrawal, R. Rodriguez-Carerro, S.-H. Dong, D. A. Mitchell, S. K. Nair and W. W. Metcalf, PLoS Biol., 2020, 18, e3000507 CrossRef CAS PubMed (1-23).
- J. Gagsteiger, S. Jahn, L. Heidinger, L. Gericke, J. N. Andexer, T. Friedrich, C. Loenarz and G. Layer, Angew. Chem., Int. Ed., 2022, 61, e202204198 CrossRef CAS PubMed.
- J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed.
- A. F. Wagner, M. Frey, F. A. Neugebauer, W. Schäfer and J. Knappe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 996–1000 CrossRef CAS PubMed.
- A. Gendron and K. D. Allen, Front. Microbiol., 2022, 13, 867342 CrossRef PubMed (1-18).
- H. Chen, Q. Gan and C. Fan, Front. Microbiol., 2020, 11, 578356 CrossRef PubMed (1-7).
- B. C. Vu, A. D. Jones and J. T. Lecomte, J. Am. Chem. Soc., 2002, 124, 8544–8545 CrossRef CAS PubMed.
- X. Hou, H. Xu, Z. Deng, Y. Yan, Z. Yuan, X. Liu, Z. Su, S. Yang, Y. Zhang and Y. Rao, Angew. Chem., Int. Ed., 2022, 61, e202208772 CrossRef CAS PubMed.
- T. C. Taylor and I. Andersson, J. Mol. Biol., 1997, 265, 432–444 CrossRef CAS PubMed.
- E. Jabri, M. B. Carr, R. P. Hausinger and P. A. Karplus, Science, 1995, 268, 998–1004 CrossRef CAS PubMed.
- M. M. Benning, J. M. Kuo, F. M. Raushel and H. M. Holden, Biochemistry, 1995, 34, 7973–7978 CrossRef CAS PubMed.
- C. Qin, L. G. Graf, K. Striska, M. Janetzky, N. Geist, R. Specht, S. Schulze, G. J. Palm, B. Girbardt, B. Dörre, L. Berndt, S. Kemnitz, M. Doerr, U. T. Bornscheuer, M. Delcea and M. Lammers, Nat. Commun., 2024, 15, 6002 CrossRef CAS PubMed.
- B. Schwer, J. Bunkenborg, R. O. Verdin, J. S. Andersen and E. Verdin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10224–10229 CrossRef CAS PubMed.
- B. Desguin, T. Zhang, P. Soumillion, P. Hols, J. Hu and R. P. Hausinger, Science, 2015, 349, 66–69 CrossRef CAS PubMed.
- B. Desguin, P. Soumillion, P. Hols and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5598–5603 CrossRef CAS PubMed.
- W. D. Riley and E. E. Snell, Biochemistry, 1968, 7, 3520–3528 CrossRef CAS PubMed.
- P. A. Recsei and E. E. Snell, Biochemistry, 1970, 9, 1492–1497 CrossRef CAS PubMed.
- C. Miech, T. Dierks, T. Selmer, K. Von Figura and B. Schmidt, J. Biol. Chem., 1998, 273, 4835–4837 CrossRef CAS PubMed.
- C. Szameit, C. Miech, M. Balleininger, B. Schmidt, K. von Figura and T. Dierks, J. Biol. Chem., 1999, 274, 15375–15381 CrossRef CAS PubMed.
- A. C. Manesis, R. J. Jodts, B. M. Hoffman and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2100680118 CrossRef CAS PubMed.
- R. Helland, A. Fjellbirkeland, O. A. Karlsen, T. Ve, J. R. Lillehaug and H. B. Jensen, J. Biol. Chem., 2008, 283, 13897–13904 CrossRef CAS PubMed.
- K. A. Johnson, T. Ve, Ø. Larsen, R. B. Pedersen, J. R. Lillehaug, H. B. Jensen, R. Helland and O. A. Karlsen, PLoS One, 2014, 9, e87750 CrossRef PubMed.
- S. M. Janes, D. Mu, D. Wemmer, A. J. Smith, S. Kaur, D. Maltby, A. L. Burlingame and J. P. Klinman, Science, 1990, 248, 981–987 CrossRef CAS PubMed.
- B. A. Barry and G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7099–7103 CrossRef CAS PubMed.
- A.-l Tsai, R. J. Kulmacz and G. Palmer, J. Biol. Chem., 1995, 270, 10503–10508 CrossRef CAS PubMed.
- Z. Deng, H. Su, X. Hou, H. Xu, Z. Yuan, X. Sheng and Y. Rao, ACS Catal., 2024, 14, 797–811 CAS.
- M. Lammers, Front. Microbiol., 2021, 12, 757179 CrossRef PubMed.
- A. T. Blasl, S. Schulze, C. Qin, L. G. Graf, R. Vogt and M. Lammers, Biol. Chem., 2022, 403, 151–194 CrossRef CAS PubMed.
- J. G. Gardner, F. J. Grundy, T. M. Henkin and J. C. Escalante-Semerena, J. Bacteriol., 2006, 188, 5460–5468 CrossRef CAS PubMed.
- J. Nevarez, A. Turmo, J. Hu and R. P. Hausinger, ChemCatChem, 2020, 12, 4242–4254 CrossRef CAS PubMed.
- S. Gatreddi, S. Chatterjee, A. Turmo, J. Hu and R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2025, 1–16 Search PubMed.
- J. A. Rankin, S. Chatterjee, Z. Tariq, S. Lagishetty, B. Desguin, J. Hu and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106202118 CrossRef CAS PubMed.
- S. Chatterjee, J. L. Nevarez, J. A. Rankin, J. Hu and R. P. Hausinger, Biochemistry, 2023, 62, 3096–3104 CrossRef CAS PubMed.
- M. Fellner, B. Desguin, R. P. Hausinger and J. Hu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9074–9079 CrossRef CAS PubMed.
- M. Fellner, J. A. Rankin, B. Desguin, J. Hu and R. P. Hausinger, Biochemistry, 2018, 57, 5513–5523 CrossRef CAS PubMed.
- M. Fellner, K. G. Huizenga, R. P. Hausinger and J. Hu, Sci. Rep., 2020, 10, 5830 CrossRef CAS PubMed (1-9).
- S. Chatterjee, K. F. Parson, B. T. Ruotolo, J. McCracken, J. Hu and R. P. Hausinger, J. Biol. Chem., 2022, 298, 102131 CrossRef CAS PubMed (1-14).
- A. Turmo, J. Hu and R. P. Hausinger, Metallomics, 2022, 14, mfac014 CrossRef PubMed (1–8).
- B. Desguin, P. Goffin, E. Viaene, M. Kleerebezem, V. Martin-Diaconescu, M. J. Maroney, J.-P. Declercq, P. Soumillion and P. Hols, Nat. Commun., 2014, 5, 3615 CrossRef PubMed (1-12).
- R. E. Treviño and H. S. Shafaat, Curr. Opin. Chem. Biol., 2022, 67, 102110 CrossRef PubMed (1-8).
- V. Sanchez-Torres and T. K. Wood, Microb. Biotechnol., 2024, 17, e70000 CrossRef CAS PubMed (1-7).
- Y. Miyazaki, K. Oohora and T. Hayashi, Chem. Soc. Rev., 2022, 51, 1629–1639 RSC.
- S. Shima, M. Krueger, T. Weinert, U. Demmer, J. Kahnt, R. K. Thauer and U. Ermler, Nature, 2012, 481, 98–101 CrossRef CAS PubMed.
- T. Wagner, C.-E. Wegner, J. Kahnt, U. Ermler and S. Shima, J. Bacteriol., 2017, 199, e00197-17 CrossRef PubMed (1-15).
- C. J. Hahn, O. N. Lemaire, J. Kahnt, S. Engilberge, G. Wegener and T. Wagner, Science, 2021, 373, 118–121 CrossRef CAS PubMed.
- T. Selmer, J. Kahnt, M. Goubeaud, S. Shima, W. Grabarse, U. Ermler and R. K. Thauer, J. Biol. Chem., 2000, 275, 3755–3760 CrossRef CAS PubMed.
- W. Grabarse, F. Mahlert, S. Shima, R. K. Thauer and U. Ermler, J. Mol. Biol., 2000, 303, 329–344 CrossRef CAS PubMed.
- C. D. Fyfe, N. Bernardo-García, L. Fradale, S. Grimaldi, A. Guillot, C. Brewee, L. M. G. Chavas, P. Legrand, A. Benjdia and O. Berteau, Nature, 2022, 602, 336–342 CrossRef CAS PubMed.
- M. I. Radle, D. V. Miller, T. N. Laremore and S. J. Booker, J. Biol. Chem., 2019, 294, 11712–11725 CrossRef CAS PubMed.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS PubMed.
- S. J. Booker and T. L. Grove, F1000 Biol. Rep., 2010, 2, 52 Search PubMed.
- J. M. Kuchenreuther, W. K. Myers, T. A. Stich, S. J. George, Y. Nejatyjahromy, J. R. Swartz and R. D. Britt, Science, 2013, 342, 472–475 CrossRef CAS PubMed.
- J. Wang, R. P. Woldring, G. D. Román-Meléndez, A. M. McClain, B. R. Alzua and E. N. Marsh, ACS Chem. Biol., 2014, 9, 1929–1938 CrossRef CAS PubMed.
- Z. Lyu, N. Shao, C.-W. Chou, H. Shi, R. Patel, E. C. Duin and W. B. William, J. Bacteriol., 2020, 202, e00654-19 CrossRef PubMed (1-18).
- G. E. Kenney and A. C. Rosenzweig, J. Biol. Chem., 2018, 293, 4606–4615 CrossRef CAS PubMed.
- L. M. R. Jensen, R. Sanishvili, V. L. Davidson and C. M. Wilmot, Science, 2010, 327, 1392–1394 CAS.
- X. Li, R. Fu, A. Liu and V. L. Davidson, Biochemistry, 2008, 47, 2908–2912 CAS.
- M. Feng, L. M. R. Jensen, E. T. Yukl, X. Wei, A. Liu, C. M. Wilmot and V. L. Davidson, Biochemistry, 2012, 51, 1598–1606 CAS.
- E. T. Yukl, F. Liu, J. Krzystek, S. Shin, L. M. R. Jensen, V. L. Davidson, C. M. Wilmot and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4569–4573 Search PubMed.
- V. L. Davidson and A. Liu, Biochim. Biophys. Acta, 2012, 1824, 1299–1305 CAS.
- X. Li, R. Fu, S. Lee, C. Krebs, V. L. Davidson and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8597–8600 CAS.
- J. Geng, K. Dornevil, V. L. Davidson and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9639–9644 CrossRef CAS PubMed.
- J. Geng, I. Davis and A. Liu, Angew. Chem., Int. Ed., 2015, 54, 3692–3696 CrossRef CAS PubMed.
- N. A. Tarboush, L. M. R. Jensen, E. T. Yukl, J. Geng, A. Liu, C. M. Wilmot and V. L. Davidson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16956–16961 CrossRef PubMed.
- J. Geng, I. Davis, F. Liu and A. Liu, J. Biol. Inorg. Chem., 2014, 19, 1057–1067 CrossRef CAS PubMed.
- A. C. Manesis, J. W. Slater, K. Cantave, J. Martin Bollinger, Jr., C. Krebs and A. C. Rosenzweig, Biochemistry, 2023, 62, 1082–1092 CrossRef CAS PubMed.
- K. Rizzolo, S. E. Cohen, A. C. Weitz, M. M. López Muñoz, M. P. Hendrich, C. L. Drennan and S. J. Elliott, Nat. Commun., 2019, 10(1101), 1–10 CAS.
- T. W. Stone and L. G. Darlington, Nat. Rev. Drug Discovery, 2002, 1, 609–620 CrossRef CAS PubMed.
- R. Schwarcz, J. P. Bruno, P. J. Muchowski and H. Q. Wu, Nat. Rev. Neurosci., 2012, 13, 465–477 CrossRef CAS PubMed.
- A. Newton, L. McCann, L. Huo and A. Liu, Metabolites, 2023, 13, 500 CrossRef CAS PubMed (1-13).
- I. Davis and A. Liu, Expert Rev. Neurother., 2015, 15, 719–721 CrossRef CAS PubMed.
- E. M. Shepard and D. M. Dooley, J. Biol. Inorg. Chem., 2006, 11, 1039–1048 CrossRef CAS PubMed.
- S. X. Wang, M. Mure, K. F. Medzihradszky, A. L. Burlingame, D. E. Brown, D. M. Dooley, A. J. Smith, H. M. Kagan and J. P. Klinman, Science, 1996, 273, 1078–1084 CrossRef CAS PubMed.
- M. R. Parsons, M. A. Convery, C. M. Wilmot, K. D. Yadav, V. Blakeley, A. S. Corner, S. E. Phillips, M. J. McPherson and P. F. Knowles, Structure, 1995, 3, 1171–1184 CrossRef CAS PubMed.
- V. Kumar, D. M. Dooley, H. C. Freeman, J. M. Guss, I. Harvey, M. A. McGuirl, M. C. Wilce and V. M. Zubak, Structure, 1996, 4, 943–955 CrossRef CAS PubMed.
- L. Luhová, L. Slavík, I. Frébort, M. Sebela, L. Zajoncová and P. Peč, J. Enzyme Inhib., 1996, 10, 251–262 CrossRef PubMed.
- M. C. J. Wilce, D. M. Dooley, H. C. Freeman, J. M. Guss, H. Matsunami, W. S. McIntire, C. E. Ruggiero, K. Tanizawa and H. Yamaguchi, Biochemistry, 1997, 36, 16116–16133 CrossRef CAS PubMed.
- R. Li, J. P. Klinman and F. S. Mathews, Structure, 1998, 6, 293–307 CrossRef CAS PubMed.
- M. Lunelli, M. L. Di Paolo, M. Biadene, V. Calderone, R. Battistutta, M. Scarpa, A. Rigo and G. Zanotti, J. Mol. Biol., 2005, 346, 991–1004 CrossRef CAS PubMed.
- A. P. Duff, A. E. Cohen, P. J. Ellis, J. A. Kuchar, D. B. Langley, E. M. Shepard, D. M. Dooley, H. C. Freeman and J. M. Guss, Biochemistry, 2003, 42, 15148–15157 CrossRef CAS PubMed.
- M. Kim, T. Okajima, S. Kishishita, M. Yoshimura, A. Kawamori, K. Tanizawa and H. Yamaguchi, Nat. Struct. Biol., 2002, 9, 591–596 CAS.
- C. M. Wilmot, J. M. Murray, G. Alton, M. R. Parsons, M. A. Convery, V. Blakeley, A. S. Corner, M. M. Palcic, P. F. Knowles, M. J. McPherson and S. E. Phillips, Biochemistry, 1997, 36, 1608–1620 CrossRef CAS PubMed.
- T. Murakawa, S. Baba, Y. Kawano, H. Hayashi, T. Yano, T. Kumasaka, M. Yamamoto, K. Tanizawa and T. Okajima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 135–140 CrossRef CAS PubMed.
- A. Ehrenberg and P. Reichard, J. Biol. Chem., 1972, 247, 3485–3488 CAS.
- B. M. Sjöberg and P. Reichard, J. Biol. Chem., 1977, 252, 536–541 CrossRef.
- B. M. Sjöberg, P. Reichard, A. Gräslund and A. Ehrenberg, J. Biol. Chem., 1978, 253, 6863–6865 CrossRef.
- P. Reichard and A. Ehrenberg, Science, 1983, 221, 514–519 CAS.
- J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 13463–13472 CrossRef CAS PubMed.
- S. J. Booker, Biochim. Biophys. Acta, 2012, 1824, 1151–1153 CAS.
- J. M. Bollinger, Jr., D. E. Edmondson, B. H. Huynh, J. Filley, J. R. Norton and J. Stubbe, Science, 1991, 253, 292–298 Search PubMed.
- N. Ravi, J. M. Bollinger, Jr., B. H. Huynh, J. Stubbe and D. E. Edmondson, J. Am. Chem. Soc., 1994, 116, 8007–8014 CrossRef CAS.
- N. Mitić, M. D. Clay, L. Saleh, J. M. Bollinger and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 9049–9065 CrossRef PubMed.
- A. Liu, S. Pötsch, A. Davydov, A. L. Barra, H. Rubin and A. Gräslund, Biochemistry, 1998, 37, 16369–16377 CrossRef CAS PubMed.
- A. Liu, A.-L. Barra, H. Rubin, G. Lu and A. Gräslund, J. Am. Chem. Soc., 2000, 122, 1974–1978 CrossRef CAS.
- G. Kang, A. T. Taguchi, J. Stubbe and C. L. Drennan, Science, 2020, 368, 424–427 CrossRef CAS PubMed.
- B. M. Sjöberg, A. Gräslund and F. Eckstein, J. Biol. Chem., 1983, 258, 8060–8067 CrossRef.
- M. Ator, S. P. Salowe, J. Stubbe, M. H. Emptage and M. J. Robins, J. Am. Chem. Soc., 1984, 106, 1886–1887 CrossRef CAS.
- B. L. Greene, G. Kang, C. Cui, M. Bennati, D. G. Nocera, C. L. Drennan and J. Stubbe, Annu. Rev. Biochem., 2020, 89, 45–75 CrossRef CAS PubMed.
- R. P. Pesavento and W. A. Van Der Donk, Advances in Protein Chemistry, Academic Press, 2001, vol. 58, pp. 317–385 Search PubMed.
- R. J. Debus, B. A. Barry, I. Sithole, G. T. Babcock and L. McIntosh, Biochemistry, 1988, 27, 9071–9074 CrossRef CAS PubMed.
- S. Gerken, K. Brettel, E. Schlodder and H. T. Witt, FEBS Lett., 1988, 237, 69–75 CrossRef CAS.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7690–7695 CrossRef CAS PubMed.
- R. Dietz, W. Nastainczyk and H. H. Ruf, Eur. J. Biochem., 1988, 171, 321–328 CrossRef CAS PubMed.
- A. L. Tsai, G. Palmer and R. J. Kulmacz, J. Biol. Chem., 1992, 267, 17753–17759 CrossRef CAS PubMed.
- A.-L. Tsai and R. J. Kulmacz, Prostaglandins Other Lipid Mediators, 2000, 62, 231–254 CAS.
- V. Unkrig, F. A. Neugebauer and J. Knappe, Eur. J. Biochem., 1989, 184, 723–728 CAS.
- E. Mulliez, M. Fontecave, J. Gaillard and P. Reichard, J. Biol. Chem., 1993, 268, 2296–2299 CAS.
- A. Becker, K. Fritz-Wolf, W. Kabsch, J. Knappe, S. Schultz and A. F. Volker Wagner, Nat. Chem. Biol., 1999, 6, 969–975 CAS.
- D. T. Logan, J. Andersson, B. M. Sjöberg and P. Nordlund, Science, 1999, 283, 1499–1504 CAS.
- E. Torrents, G. Buist, A. Liu, R. Eliasson, J. Kok, I. Gibert, A. Gräslund and P. Reichard, J. Biol. Chem., 2000, 275, 2463–2471 CAS.
- J. Knappe and A. F. Wagner, Adv. Protein Chem., 2001, 58, 277–315 CAS.
- A. Liu and A. Gräslund, J. Biol. Chem., 2000, 275, 12367–12373 CAS.
- D. T. Logan, E. Mulliez, K. M. Larsson, S. Bodevin, M. Atta, P. E. Garnaud, B. M. Sjoberg and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3826–3831 CAS.
- M. Frey, M. Rothe, A. F. Wagner and J. Knappe, J. Biol. Chem., 1994, 269, 12432–12437 CAS.
- J. L. Vey, J. Yang, M. Li, W. E. Broderick, J. B. Broderick and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16137–16141 CAS.
- E. M. Shepard and J. B. Broderick, in Comprehensive Natural Products II, ed. H.-W. Liu and L. Mander, Elsevier, Oxford, 2010, pp. 625–661 Search PubMed.
- D. S. King and P. Reichard, Biochem. Biophys. Res. Commun., 1995, 206, 731–735 CAS.
- A. Becker and W. Kabsch, J. Biol. Chem., 2002, 277, 40036–40042 CAS.
- J. Stubbe, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2723–2724 CrossRef CAS PubMed.
- J. Stubbe and P. Riggs-Gelasco, Trends Biochem. Sci., 1998, 23, 438–443 CrossRef CAS PubMed.
- C. L. Atkin, L. Thelander, P. Reichard and G. Lang, J. Biol. Chem., 1973, 248, 7464–7472 CrossRef CAS PubMed.
- U. Griepenburg, G. Lassmann and G. Auling, Free Radical Res., 1996, 24, 473–481 CrossRef CAS PubMed.
- S. Licht, G. J. Gerfen and J. Stubbe, Science, 1996, 271, 477–481 CrossRef CAS PubMed.
- Y. Tamao and R. L. Blakley, Biochemistry, 1973, 12, 24–34 CrossRef CAS PubMed.
- W. H. Orme-Johnson, H. Beinert and R. L. Blakley, J. Biol. Chem., 1974, 249, 2338–2343 CAS.
- P. Reichard, Science, 1993, 260, 1773–1777 CAS.
- J. C. Cáceres, C. A. Bailey, K. Yokoyama and B. L. Greene, Methods Enzymol., 2022, 662, 119–141 Search PubMed.
- T. Nauser, S. Dockheer, R. Kissner and W. H. Koppenol, Biochemistry, 2006, 45, 6038–6043 CAS.
- T. Yonetani and H. Schleyer, J. Biol. Chem., 1966, 241, 3240–3243 CAS.
- M. Sivaraja, D. B. Goodin, M. Smith and B. M. Hoffman, Science, 1989, 245, 738–740 CrossRef CAS PubMed.
- T. M. Payne, E. F. Yee, B. Dzikovski and B. R. Crane, Biochemistry, 2016, 55, 4807–4822 CrossRef CAS PubMed.
- H. Mei, K. Wang, N. Peffer, G. Weatherly, D. S. Cohen, M. Miller, G. Pielak, B. Durham and F. Millett, Biochemistry, 1999, 38, 6846–6854 CrossRef CAS PubMed.
- K. Wang, H. Mei, L. Geren, M. A. Miller, A. Saunders, X. Wang, J. L. Waldner, G. J. Pielak, B. Durham and F. Millett, Biochemistry, 1996, 35, 15107–15119 CrossRef CAS PubMed.
- C. A. Bonagura, M. Sundaramoorthy, H. S. Pappa, W. R. Patterson and T. L. Poulos, Biochemistry, 1996, 35, 6107–6115 CrossRef CAS PubMed.
- W. R. Patterson and T. L. Poulos, Biochemistry, 1995, 34, 4331–4341 CrossRef CAS PubMed.
- W. R. Patterson, T. L. Poulos and D. B. Goodin, Biochemistry, 1995, 34, 4342–4345 CrossRef CAS PubMed.
- V. S. Jasion, J. A. Polanco, Y. T. Meharenna, H. Li and T. L. Poulos, J. Biol. Chem., 2011, 286, 24608–24615 CrossRef CAS PubMed.
- T. L. Poulos, J. S. Kim and V. C. Murarka, J. Biol. Inorg. Chem., 2022, 27, 229–237 CrossRef CAS PubMed.
- K. Lerch, J. Biol. Chem., 1982, 257, 6414–6419 CrossRef CAS PubMed.
- N. Fujieda, T. Ikeda, M. Murata, S. Yanagisawa, S. Aono, K. Ohkubo, S. Nagao, T. Ogura, S. Hirota, S. Fukuzumi, Y. Nakamura, Y. Hata and S. Itoh, J. Am. Chem. Soc., 2011, 133, 1180–1183 CrossRef CAS PubMed.
- N. Fujieda, K. Umakoshi, Y. Ochi, Y. Nishikawa, S. Yanagisawa, M. Kubo, G. Kurisu and S. Itoh, Angew. Chem., 2020, 132, 13487–13492 Search PubMed.
- C. Gielens, N. De Geest, X.-Q. Xin, B. Devreese, J. Van Beeumen and G. Preaux, Eur. J. Biochem., 1997, 248, 879–888 CAS.
- M. E. Cuff, K. I. Miller, K. E. Van Holde and W. A. Hendrickson, J. Mol. Biol., 1998, 278, 855–870 CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CAS.
- M. Wensien, F. R. von Pappenheim, L.-M. Funk, P. Kloskowski, U. Curth, U. Diederichsen, J. Uranga, J. Ye, P. Fang, K.-T. Pan, H. Urlaub, R. A. Mata, V. Sautner and K. Tittmann, Nature, 2021, 593, 460–464 CAS.
- A. Satoh, J.-K. Kim, I. Miyahara, B. Devreese, I. Vandenberghe, A. Hacisalihoglu, T. Okajima, S. I. Kuroda, O. Adachi, J. A. Duine, J. Van Beeumen, K. Tanizawa and K. Hirotsu, J. Biol. Chem., 2002, 277, 2830–2834 CAS.
- S. Okazaki, S. Nakano, D. Matsui, S. Akaji, K. Inagaki and Y. Asano, J. Biochem., 2013, 154, 233–236 CAS.
- A. Andreo-Vidal, K. J. Mamounis, E. Sehanobish, D. Avalos, J. C. Campillo-Brocal, A. Sanchez-Amat, E. T. Yukl and V. L. Davidson, Biochemistry, 2018, 57, 1155–1165 CrossRef CAS PubMed.
- M. M. Whittaker and J. W. Whittaker, J. Biol. Chem., 1990, 265, 9610–9613 CAS.
- N. Ito, S. E. V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Nature, 1991, 350, 87–90 CAS.
- J. G. McCoy, L. J. Bailey, E. Bitto, C. A. Bingman, D. J. Aceti, B. G. Fox and G. N. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3084–3089 CAS.
- A. K. Chaplin, M. L. Petrus, G. Mangiameli, M. A. Hough, D. A. Svistunenko, P. Nicholls, D. Claessen, E. Vijgenboom and J. A. Worrall, Biochem. J., 2015, 469, 433–444 CrossRef CAS PubMed.
- R. Schnell, T. Sandalova, U. Hellman, Y. Lindqvist and G. Schneider, J. Biol. Chem., 2005, 280, 27319–27328 CrossRef CAS PubMed.
- K. M. Polyakov, K. M. Boyko, T. V. Tikhonova, A. Slutsky, A. N. Antipov, R. A. Zvyagilskaya, A. N. Popov, G. P. Bourenkov, V. S. Lamzin and V. O. Popov, J. Mol. Biol., 2009, 389, 846–862 CrossRef CAS PubMed.
- C. Ostermeier, A. Harrenga, U. Ermler and H. Michel, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10547–10553 CrossRef CAS PubMed.
- M. A. Ehudin, L. Senft, A. Franke, I. Ivanović-Burmazović and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 10632–10643 CrossRef CAS PubMed.
- S. Yoshikawa, K. Shinzawa-itoh, R. Nakashima, R. Yaono, E. Yamashita, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki and T. Tsukihara, Science, 1998, 280, 1723–1729 CrossRef CAS PubMed.
- A. Buzy, V. Bracchi, R. Sterjiades, J. Chroboczek, P. Thibault, J. Gagnon, H. M. Jouve and G. Hudry-Clergeon, J. Protein Chem., 1995, 14, 59–72 CrossRef CAS PubMed.
- J. Bravo, I. Fita, J. C. Ferrer, W. Ens, A. Hillar, J. Switala and P. C. Loewen, Protein Sci., 1997, 6, 1016–1023 CrossRef CAS PubMed.
- W. S. McIntire, D. E. Wemmer, A. Chistoserdov and M. E. Lidstrom, Science, 1991, 252, 817–824 CrossRef CAS PubMed.
- L. Chen, F. S. Mathews, V. L. Davidson, E. G. Huizinga, F. M. D. Vellieux, J. A. Duine and W. G. J. Hol, FEBS Lett., 1991, 287, 163–166 CAS.
- L. Chen, R. C. Durley, F. S. Mathews and V. L. Davidson, Science, 1994, 264, 86–90 CrossRef CAS PubMed.
- S. Datta, Y. Mori, K. Takagi, K. Kawaguchi, Z.-W. Chen, T. Okajima, S. I. Kuroda, T. Ikeda, K. Kano, K. Tanizawa and F. S. Mathews, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14268–14273 CAS.
- J. G. Hauge, J. Biol. Chem., 1964, 239, 3630–3639 CAS.
- M. D. Chacón-Verdú, J. C. Campillo-Brocal, P. Lucas-Elío, V. L. Davidson and A. Sánchez-Amat, Biochim. Biophys. Acta, 2015, 1854, 1123–1131 Search PubMed.
- O. T. Magnusson, H. Toyama, M. Saeki, A. Rojas, J. C. Reed, R. C. Liddington, J. P. Klinman and R. Schwarzenbacher, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7913–7918 CAS.
- T. Kasahara and T. Kato, Nature, 2003, 422, 832 CAS.
- S. A. Salisbury, H. S. Forrest, W. B. Cruse and O. Kennard, Nature, 1979, 280, 843–844 CAS.
- Y. Wang, M. E. Graichen, A. Liu, A. R. Pearson, C. M. Wilmot and V. L. Davidson, Biochemistry, 2003, 42, 7318–7325 CAS.
- R. Fu, F. Liu, V. L. Davidson and A. Liu, Biochemistry, 2009, 48, 11603–11605 CAS.
- E. T. Yukl and V. L. Davidson, Arch. Biochem. Biophys., 2018, 654, 40–46 CAS.
- T. Oozeki, T. Nakai, K. Kozakai, K. Okamoto, S. I. Kuroda, K. Kobayashi, K. Tanizawa and T. Okajima, Nat. Commun., 2021, 12, 933 CAS.
- M. D. Chacón-Verdú, D. Gómez, F. Solano, P. Lucas-Elío and A. Sánchez-Amat, Appl. Microbiol. Biotechnol., 2014, 98, 2981–2989 CrossRef PubMed.
- X. Zhang, Q. Wang, J. Wu, J. Wang, Y. Shi and M. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3828–3833 CrossRef CAS PubMed.
- A. A. Meier, K. Kuczera and M. Mure, Int. J. Mol. Sci., 2022, 23, 13385 CrossRef CAS PubMed (1-13).
- F. Rabe von Pappenheim, M. Wensien, J. Ye, J. Uranga, I. Irisarri, J. de Vries, L.-M. Funk, R. A. Mata and K. Tittmann, Nat. Chem. Biol., 2022, 18, 368–375 CrossRef CAS PubMed.
- J. Ye, S. Bazzi, T. Fritz, K. Tittmann, R. A. Mata and J. Uranga, Angew. Chem., Int. Ed., 2023, 62, e202304163 CrossRef CAS PubMed.
- Y. Wang and A. Liu, Chem. Soc. Rev., 2020, 49, 4906–4925 RSC.
- N. Masson, T. P. Keeley, B. Giuntoli, M. D. White, M. L. Puerta, P. Perata, R. J. Hopkinson, E. Flashman, F. Licausi and P. J. Ratcliffe, Science, 2019, 365, 65–69 CrossRef CAS PubMed.
- Y. Wang, I. Shin, J. Li and A. Liu, J. Biol. Chem., 2021, 297, 101176 CrossRef CAS PubMed (1-10).
- R. L. Fernandez, L. D. Elmendorf, R. W. Smith, C. A. Bingman, B. G. Fox and T. C. Brunold, Biochemistry, 2021, 60, 3728–3737 CrossRef CAS PubMed.
- Y. Wang, I. Davis, Y. Chan, S. G. Naik, W. P. Griffith and A. Liu, J. Biol. Chem., 2020, 295, 11789–11802 CAS.
- J. E. Dominy, Jr., C. R. Simmons, L. L. Hirschberger, J. Hwang, R. M. Coloso and M. H. Stipanuk, J. Biol. Chem., 2007, 282, 25189–25198 Search PubMed.
- M. H. Stipanuk, J. Nutr., 2020, 150, 2494s–2505s Search PubMed.
- D. M. Gunawardana, K. C. Heathcote and E. Flashman, FEBS J., 2022, 289, 5426–5439 CAS.
- C. R. Simmons, Q. Liu, Q. Huang, Q. Hao, T. P. Begley, P. A. Karplus and M. H. Stipanuk, J. Biol. Chem., 2006, 281, 18723–18733 CAS.
- C. W. Njeri and H. R. Ellis, Arch. Biochem. Biophys., 2014, 558, 61–69 CAS.
- J. Li, T. Koto, I. Davis and A. Liu, Biochemistry, 2019, 58, 2218–2227 CAS.
- J. E. Dominy, J. Hwang, S. Guo, L. L. Hirschberger, S. Zhang and M. H. Stipanuk, J. Biol. Chem., 2008, 283, 12188–12201 CrossRef CAS PubMed.
- J. Li, R. Duan and A. Liu, J. Am. Chem. Soc., 2024, 146, 18292–18297 CrossRef CAS PubMed.
- D. T. Yin, S. Urresti, M. Lafond, E. M. Johnston, F. Derikvand, L. Ciano, J. G. Berrin, B. Henrissat, P. H. Walton, G. J. Davies and H. Brumer, Nat. Commun., 2015, 6, 10197 CrossRef CAS PubMed.
- J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2363 CrossRef CAS PubMed.
- K. Clark, J. E. Penner-Hahn, M. M. Whittaker and J. W. Whittaker, J. Am. Chem. Soc., 1990, 112, 6433–6434 CrossRef CAS.
- M. S. Rogers and D. M. Dooley, Adv. Protein Chem., 2001, 58, 387–436 CrossRef CAS PubMed.
- P. F. Knowles and N. Ito, Perspect. Bioinorg. Chem., 1993, 2, 207–244 CAS.
- M. J. McPherson, C. Stevens, A. J. Baron, Z. B. Ogel, K. Seneviratne, C. Wilmot, N. Ito, I. Brocklebank, S. E. V. Phillips and P. F. Knowles, Biochem. Soc. Trans., 1993, 21, 752–756 CrossRef CAS PubMed.
- Y. Wang, J. L. DuBois, B. Hedman, K. O. Hodgson and T. D. Stack, Science, 1998, 279, 537–540 CrossRef CAS PubMed.
- J. W. Whittaker and M. M. Whittaker, Pure & Appl. Chem., 1998, 70, 903–910 CrossRef CAS.
- A. J. Baron, C. Stevens, C. Wilmot, P. F. Knowles, S. E. V. Phillips and M. J. McPherson, Biochem. Soc. Trans., 1993, 21, 319S–319S CrossRef CAS PubMed.
- M. M. Whittaker, P. J. Kersten, D. Cullen and J. W. Whittaker, J. Biol. Chem., 1999, 274, 36226–36232 CrossRef CAS PubMed.
- Q. Zhou, M. Hu, W. Zhang, L. Jiang, S. Perrett, J. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 1203–1207 CrossRef CAS PubMed.
- R. E. Cowley, J. Cirera, M. F. Qayyum, D. Rokhsana, B. Hedman, K. O. Hodgson, D. M. Dooley and E. I. Solomon, J. Am. Chem. Soc., 2016, 138, 13219–13229 CrossRef CAS PubMed.
- K. S. Yang, L. R. Blankenship, S.-T. A. Kuo, Y. J. Sheng, P. Li, C. A. Fierke, D. H. Russell, X. Yan, S. Xu and W. R. Liu, ACS Chem. Biol., 2023, 18, 449–455 CrossRef CAS PubMed.
- T. F. Schwede, J. Retey and G. E. Schulz, Biochemistry, 1999, 38, 5355–5361 CrossRef CAS PubMed.
- B. Schuster and J. Retey, FEBS Lett., 1994, 349, 252–254 CrossRef CAS PubMed.
- K. T. Watts, B. N. Mijts, P. C. Lee, A. J. Manning and C. Schmidt-Dannert, Chem. Biol., 2006, 13, 1317–1326 CrossRef CAS PubMed.
- T. Bertrand, N. A. J. Eady, J. N. Jones, Jesmin, J. M. Nagy, B. Jamart-Gregoire, E. L. Raven and K. A. Brown, J. Biol. Chem., 2004, 279, 38991–38999 CAS.
- M. Ormö, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392–1395 CrossRef PubMed.
- B. Langer, M. Langer and J. Rétey, Adv. Protein Chem., 2001, 58, 175–214 CAS.
- T. Scior, I. Meneses Morales, S. J. Garcés Eisele, D. Domeyer and S. Laufer, Arch. Pharm., 2002, 335, 511–525 CAS.
- A. Liu, Crit. Rev. Biochem. Mol. Biol., 2024, 59, 434–446 Search PubMed.
- Y. Yamada, T. Fujiwara, T. Sato, N. Igarashi and N. Tanaka, Nat. Struct. Biol., 2002, 9, 691–695 CAS.
- R. A. Ghiladi, G. M. Knudsen, K. F. Medzihradszky and P. R. O. de Montellano, J. Biol. Chem., 2005, 280, 22651–22663 CAS.
- R. A. Ghiladi, K. F. Medzihradszky and P. R. Ortiz de Montellano, Biochemistry, 2005, 44, 15093–15105 CrossRef CAS PubMed.
- O. J. Njuma, I. Davis, E. N. Ndontsa, J. R. Krewall, A. Liu and D. C. Goodwin, J. Biol. Chem., 2017, 292, 18408–18421 CAS.
- X. Zhao, J. Suarez, A. Khajo, S. Yu, L. Metlitsky and R. S. Magliozzo, J. Am. Chem. Soc., 2010, 132, 8268–8269 CrossRef CAS PubMed.
- J. Li, R. Duan, E. S. Traore, R. C. Nguyen, I. Davis, W. P. Griffth, D. C. Goodwin, A. A. Jarzecki and A. Liu, Angew. Chem., Int. Ed., 2024, 63, e202407018 CrossRef CAS PubMed (1-10).
- P. C. Loewen, X. Carpena, P. Vidossich, I. Fita and C. Rovira, J. Am. Chem. Soc., 2014, 136, 7249–7252 CrossRef CAS PubMed.
- X. Carpena, S. Loprasert, S. Mongkolsuk, J. Switala, P. C. Loewen and I. Fita, J. Mol. Biol., 2003, 327, 475–489 CrossRef CAS PubMed.
- W. Zhu and J. P. Klinman, Curr. Opin. Chem. Biol., 2020, 59, 93–103 CrossRef CAS PubMed.
- I. Shin, Y. Wang and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e210656 Search PubMed (1-10).
- B. B. Wenke, J. T. Lecomte, A. Héroux and J. L. Schlessman, Proteins, 2014, 82, 528–534 CrossRef CAS PubMed.
- H. J. Nothnagel, M. R. Preimesberger, M. P. Pond, B. Y. Winer, E. M. Adney and J. T. Lecomte, J. Biol. Inorg. Chem., 2011, 16, 539–552 CrossRef CAS PubMed.
- H. M. Cheng, H. Yuan, X. J. Wang, J. K. Xu, S. Q. Gao, G. B. Wen, X. Tan and Y. W. Lin, J. Inorg. Biochem., 2018, 182, 141–149 CrossRef CAS PubMed.
- S. L. Rice, M. R. Preimesberger, E. A. Johnson and J. T. J. Lecomte, J. Inorg. Biochem., 2014, 141, 198–207 CrossRef CAS PubMed.
- S. Uppal, S. Salhotra, N. Mukhi, F. K. Zaidi, M. Seal, S. G. Dey, R. Bhat and S. Kundu, J. Biol. Chem., 2015, 290, 1979–1993 CrossRef CAS PubMed.
- M. R. Preimesberger, E. A. Johnson, D. B. Nye and J. T. J. Lecomte, J. Inorg. Biochem., 2017, 177, 171–182 CrossRef CAS PubMed.
- T. S. Young, I. Ahmad, J. A. Yin and P. G. Schultz, J. Mol. Biol., 2010, 395, 361–374 CrossRef CAS PubMed.
- E. C. Minnihan, M. R. Seyedsayamdost and J. Stubbe, Biochemistry, 2009, 48, 12125–12132 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, J. Xie, C. T. Y. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060–15071 CrossRef CAS PubMed.
- Z. Birch-Price, F. J. Hardy, T. M. Lister, A. R. Kohn and A. P. Green, Chem. Rev., 2024, 124, 8740–8786 CrossRef CAS PubMed.
- T. Hayashi, Generation of functionalized biomolecules using hemoprotein matrices with small protein cavities for incorporation of cofactors, In Coordination Chemistry in Protein Cages: Principles, Design, and Applications, ed T. Ueno and Y. Watanabe, John Wiley & Sons, Inc., 1st ed., 2013 Search PubMed.
- T. Matsuo and T. Hayashi, J. Porphyrins Phthalocyanines, 2009, 13, 1082–1089 CrossRef CAS.
- T. Hayashi, Y. Sano and A. Onoda, Isr. J. Chem., 2015, 55, 76–84 CrossRef CAS.
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945–954 CAS.
- M. Uchimiya and A. T. Stone, Chemosphere, 2009, 77, 451–458 CAS.
- A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CAS.
- T. S. Eckert and T. C. Bruice, J. Am. Chem. Soc., 1983, 105, 4431–4441 CAS.
- T. Xu, M. D. Wodrich, R. Scopelliti, C. Corminboeuf and X. Hu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1242–1245 CAS.
- R. Shi, M. D. Wodrich, H. J. Pan, F. F. Tirani and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 16869–16872 CAS.
- J. A. Rankin, R. C. Mauban, M. Fellner, B. Desguin, J. McCracken, J. Hu, S. A. Varganov and R. P. Hausinger, Biochemistry, 2018, 57, 3244–3251 CAS.
- S. Itoh, M. Taki and S. Fukuzumi, Coord. Chem. Rev., 2000, 198, 3–20 CAS.
- X. H. Liu, H. Y. Yu, J. Y. Huang, J. H. Su, C. Xue, X. T. Zhou, Y. R. He, Q. He, D. J. Xu, C. Xiong and H. B. Ji, Chem. Sci., 2022, 13, 9560–9568 CAS.
- P. Chaudhuri, M. Hess, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 1999, 38, 1095–1098 CrossRef CAS PubMed.
- E. saint-Aman, S. p Me′nage, J. L. Pierre, E. Defrancq and G. Gellon, New J. Chem., 1998, 22, 393–394 RSC.
- N. Kitajima, K. Whang, Y. Moro-oka, A. Uchida and Y. Sasada, J. Chem. Soc., Chem. Commun., 1986, 1504–1505 RSC.
- P. J. Taylor, E. Kmetec and J. M. Johnson, Anal. Chem., 1977, 49, 789–794 CrossRef CAS PubMed.
- C. Figueiredo, A. García-Ortega, T. Mandal, A. Lielpetere, F. Cervantes, D. Demurtas, E. Magner, F. J. Plou, W. Schuhmann, D. Leech, M. Pita and A. L. De Lacey, Electrochim. Acta, 2023, 472, 143438 CrossRef CAS.
- P. Goswami, S. S. Chinnadayyala, M. Chakraborty, A. K. Kumar and A. Kakoti, Appl. Microbiol. Biotechnol., 2013, 97, 4259–4275 CAS.
- K. Koschorreck, S. Alpdagtas and V. B. Urlacher, Eng. Microbiol., 2022, 2, 100037 CAS.
- Y. Mathieu, M. E. Cleveland and H. Brumer, ACS Catal., 2022, 12, 10264–10275 CAS.
- A. Tamhankar, M. Wensien, S. A. V. Jannuzzi, S. Chatterjee, B. Lassalle-Kaiser, K. Tittmann and S. DeBeer, J. Phys. Chem. Lett., 2024, 15, 4263–4267 CAS.
| This journal is © The Royal Society of Chemistry 2025 |