
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceState-of-the-art and perspectives of hydrogen generation from waste plastics†
Feng
Niu
a,
Zeqi
Wu
a,
Da
Chen
 *a,
Yuexiang
Huang
a,
Vitaly V.
Ordomsky
b,
Andrei Y.
Khodakov
*b and
Kevin M.
Van Geem
*c
*a,
Yuexiang
Huang
a,
Vitaly V.
Ordomsky
b,
Andrei Y.
Khodakov
*b and
Kevin M.
Van Geem
*c
aCollege of Materials and Chemistry, China Jiliang University, Hangzhou 310018, Zhejiang, P. R. China
bUniversity of Lille, CNRS, Centrale Lille, University of Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, Lille, France. E-mail: andrei.khodakov@univ-lille.fr
cLaboratory for Chemical Technology (LCT), Ghent University, Technologiepark 125, B-9052 Ghent, Belgium
First published on 15th April 2025
Abstract
Waste plastic utilization and hydrogen production present significant economic and social challenges but also offer opportunities for research and innovation. This review provides a comprehensive analysis of the latest advancements and innovations in hydrogen generation coupled with waste plastic recycling. It explores various strategies, including pyrolysis, gasification, aqueous phase reforming, photoreforming, and electrocatalysis. Pyrolysis and gasification in combination with catalytic reforming or water gas-shift are currently the most feasible and scalable technologies for hydrogen generation from waste plastics, with pyrolysis operating in an oxygen-free environment and gasification in the presence of steam, though both require high energy inputs. Aqueous phase reforming operates at moderate temperatures and pressures, making it suitable for oxygenated plastics, but it faces challenges related to feedstock limitations, catalyst costs and deactivation. Photoreforming and electrocatalytic reforming are emerging, sustainable methods that use sunlight and electricity, respectively, to convert plastics into hydrogen. Still, they suffer from low efficiency, scalability issues, and limitations to specific plastic types like oxygenated polymers. The challenges and solutions to commercializing plastic-to-hydrogen technologies, drawing on global industrial case studies have been outlined. Maximizing hydrogen productivity and selectivity, minimizing energy consumption, and ensuring stable operation and scaleup of plastic recycling are crucial parameters for achieving commercial viability.
 Feng Niu | Feng Niu obtained his PhD degree in 2020 under the guidance of Prof. Andrei Khodakov and Prof. Vitaly Ordomsky in CNRS, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, Université de Lille, France. Then he spent 2 years at the Chinese University of Hong Kong, Shenzhen for his postdoctoral research. He is now an Associate Researcher in China Jiliang University, Hangzhou, China. His current research involves hydrogen energy and the photocatalytic organic synthesis. |
 Da Chen | Da Chen is currently a full professor at China Jiliang University, China. He received his MSc in 2004 from Zhejiang University and his PhD in 2008 from University of Science & Technology of China. He joined China Jiliang University in 2009. He spent 1 year at the Pennsylvania State University for his postdoctoral research in 2011, and spent 6 months as a visiting scholar at Loughborough University in 2019. His current research interests include (photo-)electrocatalysis, photoelectrochemistry, and energy conversion and storage. |
 Yuexiang Huang | Yuexiang Huang received his PhD degree in 1998 from Universiade de Aveiro. He did postdoctoral work in Shanghai Institute of Microsystem and Information Technology in 1999-2001. Then he established Tianjin Highland Energy Technology Development Co., Ltd. in China. He has over 20 years engineering experience in product development and national projects management in novel hydrogen storage materials and tanks for fuel cell system application and distributed hydrogen fueling stations. |
 Vitaly V. Ordomsky | Vitaly V. Ordomsky received his MSc (2006) and PhD (2009) degree from Moscow State University in the field of zeolite catalysis. He was a postdoctoral researcher in the Department of Chemical Engineering at the Eindhoven University in the Netherlands, working on biomass conversion. Afterwards, he received a permanent position as a researcher in 2013 at the CNRS (France) working in C1 chemistry. From 2016 to 2019 he was a CNRS researcher in the E2P2L laboratory in Shanghai. His main research field is the development of new metal-based nano-materials for sustainable catalysis. |
 Andrei Y. Khodakov | Andrei Y. Khodakov obtained his PhD degree in 1991 from Zelinsky Institute of Organic Chemistry of USSR Academy of Sciences. In 1992–1999, he worked as a postdoctoral researcher in academic and industrial laboratories in France, UK and USA. From 1999, he is a researcher of the National Centre for Scientific Research in France. Since 2017, he is a CNRS Senior Research Director in the UCCS-CNRS Laboratory in the University of Lille and Centrale Lille Institute. His major research topics address the chemical valorization of renewable and fossil feedstocks to fuels and chemicals using thermo and photocatalysis. |
 Kevin M. Van Geem | Kevin Van Geem (full professor) is a member of the Laboratory for Chemical Technology of Ghent University. His main interests are thermal reaction engineering, electrification, chemical recycling, kinetic modeling, scale-up, process design and transition from fossil to renewable resources. He is a former Fulbright Research Scholar of MIT and is the director of the Center of Sustainable chemistry and CTO of CAPTURE. Author of more than 300 scientific publications and managing director of spin-off company AVGI, Professor van Geem is currently the coordinator of 3 large-scale projects: ELECTRO (HORIZON Europe), e-cracker (ERC advanced grant), AVATAR (ERC proof of concept). |
1 Introduction
Renewable hydrogen has been considered the most promising alternative energy source, replacing traditional fossil fuels. Due to its abundance, high weight-based energy density, cleanliness, and non-toxicity, hydrogen is being widely applied in the fields of fuel-cell power generation,1 chemical synthesis,2 and heating supply.3 The global hydrogen demand reached 95 million metric tons (Mt) in 2022, which was almost 3% higher than that in 2021, and continuously increases.4 Hydrogen can be generated through state-of-the-art methods, such as electrolysis, thermochemical, photochemical, biochemical, and biological processes. Currently, 70 million tons of hydrogen are produced annually, with 75% derived from natural gas and 23% from coal, resulting in significant greenhouse gas emissions.Hydrogen can also be viewed as a sustainable energy carrier because it can be produced from renewable raw materials such as water, biomass, and waste. However, despite the numerous announced R&D programs and investments, the development of the green hydrogen economy has been slow. The industry's relatively mature and generally utilized techniques for high-yield hydrogen production remain reforming and partial oxidation of fossil fuels.5 Main drawbacks of these routes are high temperatures, high energy consumption, and significant carbon dioxide emissions. Moreover, hydrogen produced by methane steam reforming or partial oxidation without carbon capture, utilization, and storage (CCUS) is unsustainable and often called “grey hydrogen.” Sustainable “green hydrogen” of high purity is produced through water electrolysis using renewable electricity. However, due to its high cost, green hydrogen currently accounts for less than 2% of the hydrogen production market.6–8 Several emerging strategies for hydrogen production, such as photocatalytic water splitting,9 ammonia decomposition,10 and biomass conversion11 are still under active research and have a few limitations. For example, the solar-to-hydrogen (STH) efficiency of photocatalytic water splitting remains quite low (<10%). Ammonia decomposition is an energy-extensive strategy for hydrogen production, which is generally performed at 300–500 °C. Limited resources and complex composition of biomass and difficulties in purification makes it unsuitable for large-scale applications. The bioprocessing of plastics for hydrogen production is an emerging technology that integrates microbial and enzymatic pathways to degrade plastic waste and produce hydrogen. Sluggish reaction rates, instability of microbial and enzymatic components, and significant scalability hurdles impede the deployment of this technology. In addition, highly crystalline plastics like polyethylene and polypropylene, are resistant to microbial degradation. The increasing demand for sustainable hydrogen, particularly in the energy, chemistry, and transportation sectors, has created an urgent need for developing new technological solutions.
Plastics, also called synthetic polymers, are omnipresent in our economy and daily life due to their affordable price and convenient properties, such as strength, ductility, durability, and corrosion resistance. In many fields, plastics have replaced conventional materials like wood, metals, and ceramics. The most commonly used plastics are polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), polystyrene (PS), polyurethane (PU), polylactic acid (PLA) and polyethylene terephthalate (PET), which are widely applied in packaging, building industry, medical science, etc. Over the past five decades, global plastic production has continuously increased. Consequently, the amount of spent plastics and plastic waste is projected to almost triple by 2060 (Fig. 1(a)).12
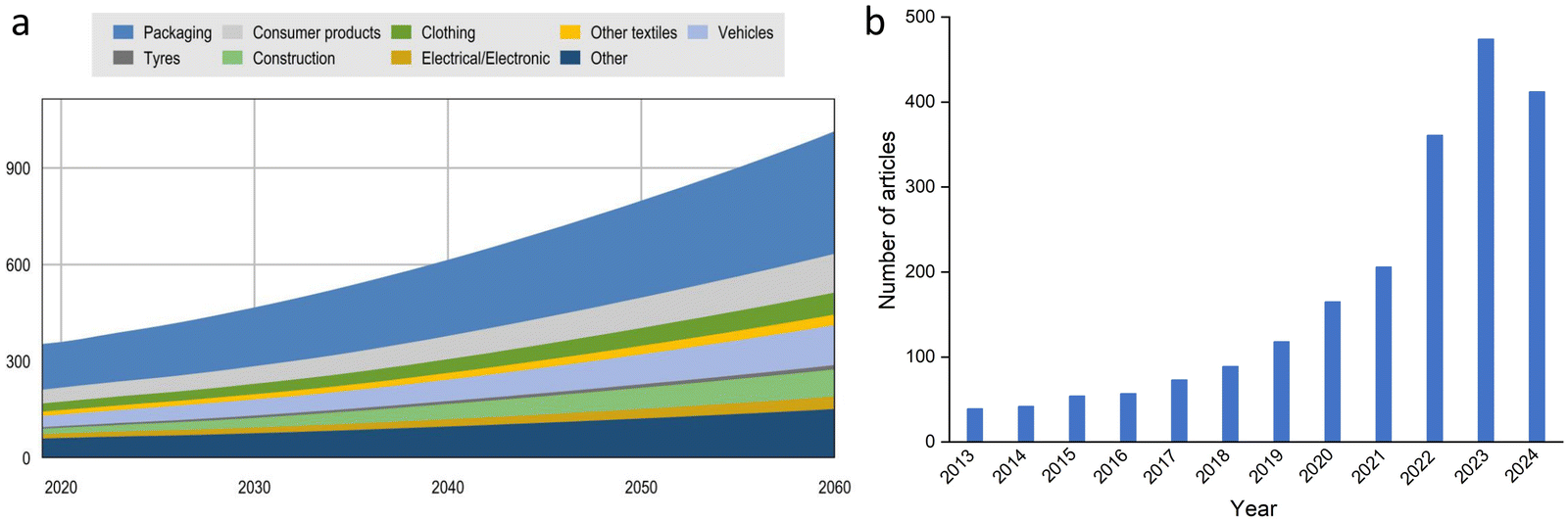 | ||
| Fig. 1 (a) Plastic waste use by application in million tons (Mt), Baseline scenario, Global Plastics Outlook: Policy Scenarios to 2060, https://www.oecd-ilibrary.org/ (b) Evolution of the number of publications dedicated to hydrogen and plastic recycling. Search results in Web of Science using “waste plastic” and “hydrogen” as search items (March 12, 2025). | ||
Microplastics,13 characterized as synthetic polymer particles measuring less than 5 mm in diameter, originate from primary microplastics, intentionally manufactured for use in products such as cosmetics and medical supplies, and from secondary microplastics, which result from the fragmentation of larger plastic debris through physical, chemical, or biological degradation processes. Their pervasive presence poses significant threats to both marine and terrestrial ecosystems. Studies estimate14 that 1.8–4.6% of the 275 million metric tons of global plastic waste generated in 2010 entered marine environments. Terrestrial microplastic pollution, conversely, is largely driven by wind-dispersed plastic debris from mismanaged waste that becomes airborne and infiltrates natural habitats. Over time, all plastic polymers released into these environments undergo gradual degradation into microplastics due to environmental stressors such as UV radiation, mechanical abrasion, and weathering. During the COVID-19 pandemic, more than eight million tons of medical plastic waste from personal protection equipment (PPE) have been generated globally, intensifying pressure on an already out-of-control global plastic waste problem.15 A recent survey of 84 shallow and deep coral ecosystems at 25 locations across the Pacific, Atlantic and Indian Ocean basins by Hudson Pinheiro and co-workers16 indicated that plastic and microplastic waste is even becoming an emerging threat to marine ecosystems. At the current rate, it is projected that by 2050, the ocean will have more plastic than fish when measured by weight.17
Growing environmental, economic, and social concerns have encouraged various plastic waste management strategies. Plastic recycling is essential for several environmental, economic, and social reasons, including reducing pollution, conserving fossil resources, lowering greenhouse gas emissions, and driving sustainable economic growth. The term “plastic recycling” covers not only the specific reprocessing, which converts plastic waste to new resources, but also the complete chain, which includes collection and sorting. The plastic waste represents an impure and diverse mix of different polymer materials. Among the methods for managing plastic waste, landfill disposal is the most economical and widely used. However, since plastic materials degrade very slowly, landfill disposal is not a sustainable solution due to the limited space in landfills and environmental consequences, such as vegetation degradation, groundwater, and air pollution.
Direct energy use of plastics can be an alternative to landfilling, such as heat or energy production through incineration. Due to the high energy density of plastic materials, this method produces electricity with high efficiency. Moreover, the volume of waste can be significantly reduced by about 90 to 95%. Unfortunately, toxic emissions produced during the incineration of plastic materials greatly hinder the use of this technology. Furthermore, incineration results in the loss of valuable resources that could serve as raw materials for the chemical industry. Over 4.9 billion tons of undegradable plastic products consumed and discarded are disposed of by direct landfilling and incineration. The number is predicted to be about 13 billion tons by 2050, which causes irreversible environmental detriment, including releasing toxic gases and infiltrating contaminants into soil, groundwater, and even deep oceans owing to their incredible stability and durability.18–20
Primary recycling of plastics, also known as re-extrusion, involves reintroducing plastic waste or mono-polymers during polymerization to generate products of similar quality to the original material. This type of reuse is only possible with semi-clean, i.e., uncontaminated, waste. Primary recycling can also include the re-extrusion of used plastics. However, this type of waste requires careful and systematic collection, particularly in plastic sorting.
On the other hand, secondary mechanical recycling, which typically involves the collection, sorting, washing, and granulation of waste, allows plastics to be used as raw materials in other recycling processes. This type of recycling is only viable for materials made from mono-polymer plastics, such as polyethylene, polypropylene, polystyrene, etc. As plastic waste becomes increasingly complex and contaminated, mechanical recycling becomes significantly more challenging. In solvent-based recycling, the solvent choice is specific to the target polymer, which makes this method ideal for selectively recycling components of complex, multicomponent products, such as multilayered plastics.
Finally, chemical recycling (tertiary recycling) allows plastic waste to be converted into shorter molecules, usually liquid or gas. These molecules can be used as raw materials to generate new chemical and plastic products. According to the type of bond in the main chain, the commonly used plastics can be divided into two groups: plastics with C–C backbone (PE, PP, PS, and PVC), and plastics with heteroatoms to form C–O or C–N backbone (PET, PU, and PLA). Generally, the products of waste plastics upcycling could differ depending on the plastics' characteristics and the methods used. For instance, PE, PP, and PS only contain C and H elements, which can be theoretically decomposed into pure hydrogen and carbon materials, like carbon nanofiber, graphene, and carbon nanotubes (CNTs) via different thermal catalytic processes under oxygen-free conditions to avoid massive CO2 emission. Although chemical recycling is costly and requires large amounts of used plastics to be economically viable, it is more tolerant of impurities than secondary recycling. There has been rising interest in efficient thermal catalytic upcycling of waste plastics into high value-added products, like gasoline,21 aromatic compounds,22 surfactant,23 diesel olefins,24 methane,25 syngas,26 and hydrogen.27 More recently, researchers are focusing more on producing hydrogen as the target product from different types of plastic waste upcycling through appropriate strategies because of the relatively high atomic H content (8–14 wt%) in common plastics,28 representing ideal hydrogen energy feedstocks.
Upcycling waste plastics into hydrogen and value-added products provides a promising, innovative route. The number of publications on hydrogen production from waste plastics has grown exponentially in the last decade (Fig. 1(b)). Upcycling of plastic wastes to hydrogen with an emphasis on catalyst design was reviewed in 2024 by Chen29et al. Although several review papers related to waste plastics upcycling to hydrogen have been published,30–34 few have focused so far on encompassing both innovative sustainable methods and conventional approaches, along with potential pathways to industrialization and economic feasibility.
Our review aims to give a comprehensive survey of hydrogen production from waste plastics upcycling. It features on the one hand, all available technologies and, on the other hand, economic and industrial aspects of hydrogen production from waste plastics (Fig. 2). Different types of plastics and strategies to deal with the produced hydrogen and carbon compounds are discussed. More comprehensive recent scientific strategies like aqueous phase and photothermal reforming are introduced and compared with conventional technologies. Additionally, detailed challenges and solutions are summarized for each strategy. More importantly, we introduce a growing number of advanced commercial plastic-to-hydrogen projects and plants under development by government agencies and energy companies around the world, as well as the evaluation of the economic feasibility of waste plastics to hydrogen pathways, driving breakthroughs in efficiency and providing opportunities for future commercialization. The circular economic feasibility analysis, alongside the challenges and insights on the prospects of hydrogen generation from waste plastics, are examined.
 | ||
| Fig. 2 Hydrogen production from waste plastics upcycling through different strategies. | ||
2 Strategies for hydrogen generation from waste plastic upcycling
2.1 Thermochemical upcycling
Today, about 96% of hydrogen in industry is derived from fossil fuels. The main process for hydrogen production is the steam-reforming of methane over nickel-based catalysts at high temperature and high pressure (700–1000 °C, 0.3–2.5 MPa), combined with subsequent water-gas-shift (WGS) reaction: CO + H2O → H2 + CO2. The final step requires the separation of hydrogen from carbon dioxide and removing residual carbon monoxide to obtain a high-purity product. Utilizing discarded plastics as raw materials to replace fossil resources while expanding the scope of thermochemical processes provides substantial environmental and sustainability benefits. Thermochemical upcycling13 is the most common technique to realize the plastics-to-hydrogen process. This chapter presents a summary of recent advancements in hydrogen production from waste plastic using thermochemical methods, such as pyrolysis and gasification, which are often combined with reforming processes (Tables S1 and S2, ESI†).2.1.1.1. Pyrolysis-catalytic reforming. In the conventional method for hydrogen production from waste plastics, pyrolysis is usually combined with catalytic reforming process and typically implemented using a tandem two-stage reactor system35 (Fig. 3(a)). The main pyrolysis-reforming strategy for hydrogen production is the operation under steam reforming conditions. The first-stage reactor is used for the thermal pyrolysis of plastics under an inert atmosphere. An inert carrier gas, typically N2 or Ar is generally introduced into the reactor at a constant rate, and the plastic is heated at 400–700 °C, and decomposed into gaseous, liquid and solid products (Fig. 3(b)).36 Temperatures above 500 °C tend to decrease the production of liquids and increase generation of gases. The second-stage reactor is designed for catalytic reforming of the generated gases and residues into hydrogen, carbon oxides and other by-products in the presence of a catalyst at 700–900 °C. The volatile gases are reformed over the catalyst surface, where carbon atoms are rearranged into graphitic structures. The metals in the reforming catalyst act as nucleation sites for CNT growth. The high-temperature gaseous products are then directed to a condenser, where hydrogen is separated from other by-products. In such a tandem system, water is sometimes introduced into the first-stage reactor with a dual purpose. First, steam serves as the inert carrier gas during the pyrolysis of plastics. Second, the introduced steam is utilized in the second-stage reactor for catalytic steam reforming. In practice, introducing a mixture of nitrogen and water into the system is primarily driven by the necessity to control the water content in the second-stage reactor. The introduction and optimization of suitable water vapor largely enhance hydrogen production efficiency. Carbon monoxide coproduced in waste plastic pyrolysis can also be used for hydrogen production via the water-gas-shift (WGS) reaction: CO + H2O → CO2 + H2. Thus, Alshareef et al.37 proposed a dedicated third-stage reactor for the WGS reaction, which provides a pathway for improving hydrogen yield while reducing the amount of undesired byproducts.
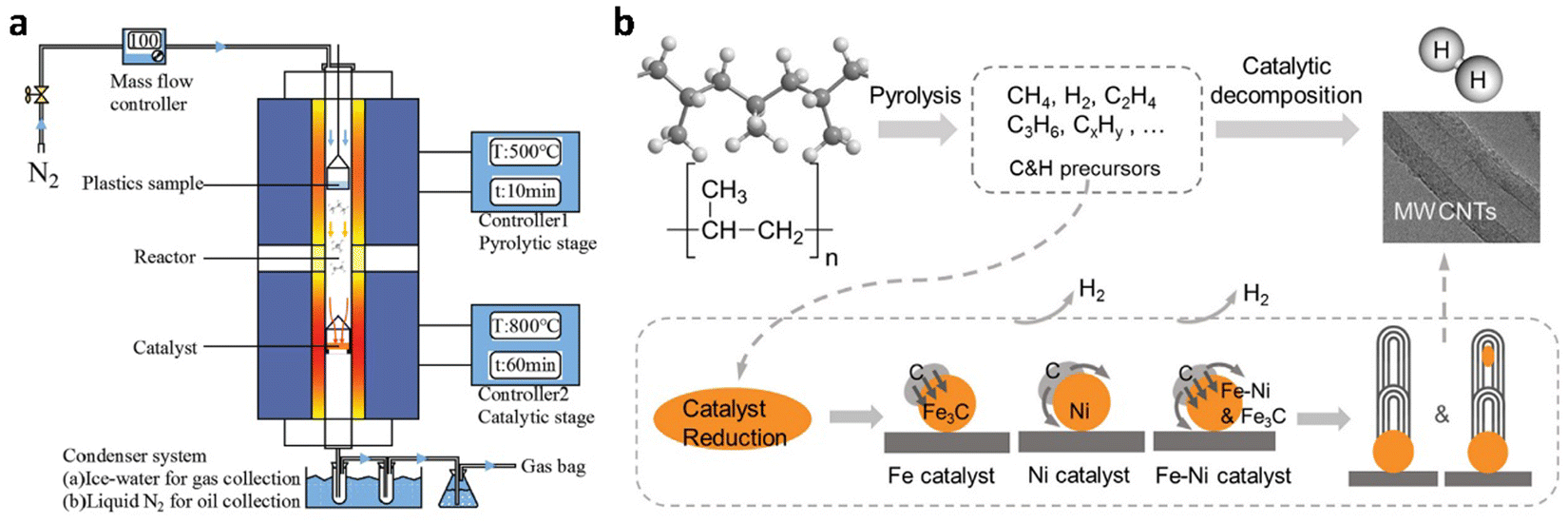 | ||
| Fig. 3 Schematic depiction of reaction apparatus and processes for conventional pyrolysis-catalytic reforming. (a) A tandem two-stage reactor system. Reproduced with permission.35 Copyright 2021, Elsevier. (b) Pyrolysis and catalytic decomposition of PP for H2 and CNTs production with Fe/Ni catalyst. Reproduced with permission.36 Copyright 2020, Elsevier. | ||
During the catalytic reforming, catalysts, feedstocks, and reaction conditions are crucial in determining the product composition. Nickel-based catalysts have been widely employed with excellent catalytic performance.38–46 Based on the investigations, the optimal Ni loading (15 wt%) and reaction temperature (700 °C) are needed for favorable performance. Higher Ni loading and reaction temperature would result in sintering, leading to a decrease in the thermal stability of the catalyst. In addition to the commonly used Al2O3 support, activated carbon has been explored recently due to its strong adsorption capacity for tar removal. Wang et al.38 conducted a study on hydrogen production from PP pyrolysis under different steam flow rates, including scenarios with no steam. In the absence of water, a significant amount of CNTs was generated. Lower water content enhanced the hydrogen conversion rate and gas yield from the raw material while simultaneously suppressing carbon deposition and improving the catalyst stability. When the water flow rate was increased to 2 mL h−1, the hydrogen conversion rose from 56.35% to 77.5%, resulting in a hydrogen yield of 112.8 mmol gplastic−1. As the water content continued to increase, there were only marginal variations in the yields of hydrogen and other gaseous byproducts, suggesting that the added water might not directly participate in the reaction. Producing CNTs during plastic pyrolysis could potentially increase hydrogen yield because the carbon is being captured into CNTs, which prevents it from forming heavier hydrocarbons or char, thereby releasing more hydrogen gas. Additionally, the conditions that favor CNT formation (like specific catalysts and higher temperatures) might also promote dehydrogenation reactions, further boosting hydrogen production.
Steam reforming, an inherently endothermic reaction, demands significant energy input. By introducing oxygen co-feeding, the energy needed for this step can be substantially reduced or even entirely offset.47,48 The volatile compounds generated during pyrolysis of waste plastics exhibit a complex composition, which accelerates catalyst deactivation through excessive coke deposition. Moreover, oxygen within the reforming reactor promotes the oxidation of these coke deposits on the catalyst surface, effectively improving the catalyst's durability and operational stability. The introduction of oxygen into the reforming reactor triggers partial oxidation of pyrolysis volatiles, resulting in lower hydrogen yields compared to conventional steam reforming systems.49
Polyolefin plastics have been proven to possess a significant hydrogen production potential. At the same time, hydrogen production capabilities are comparatively poor for other plastics, such as PVC and PS. This is not only due to the lower hydrogen content but also to the detrimental effect of Cl element in PVC and aromatics in PS on the catalyst stability during pyrolysis-catalytic reforming. Cl in PVC exhibits toxicity towards most metal catalysts, leading to catalyst deactivation and promoting polycyclic aromatic hydrocarbons, thereby enhancing coke formation and inhibiting the reforming reaction. Similarly, both PS and PET yield a substantial number of aromatic hydrocarbons upon pyrolysis, contributing to carbon deposition and decreasing hydrogen production efficiency. Jiang et al.39 comprehensively analyzed the hydrogen production performance from various plastics, including PET, PS, PP, PE, and PVC. In the case of PVC, the Cl element promoted the generation of polycyclic aromatic hydrocarbons. Among the plastics studied, PET exhibited the lowest hydrogen yield due to the production of oxygen-containing substances, which largely impeded hydrogen generation. Nabgan et al.42 conducted an in-depth study on the hydrogen production from PET pyrolysis. They employed a Ni–Pt/Ti–Al nano-sized catalyst to pyrolyze a mixture of phenol and PET. The obtained H2 yield over the fresh catalyst reached 75%, with liquid products primarily composed of phenol. However, after the long-term continuous pyrolysis for 6–8 days, a significant decline in catalytic performance was observed, attributed principally to pore blockage by carbon deposition on the Ti surface.
Acomb et al.50 explored the catalytic effects of Fe, Ni, Co, and Cu-based catalysts on the pyrolysis-catalytic reforming of LDPE for H2 production. In contrast to the Ni catalyst, the Fe catalyst demonstrated the highest yield of hydrogen and CNTs. Li et al.51 conducted an in-depth investigation of the performance of Fe2O3/Al2O3 catalyst in hydrogen production from HDPE (Fig. 4(a)). The hydrogen yield from the catalytic pyrolysis over Fe2O3/Al2O3 reached 50.53 mmol·gplastic−1, equivalent to over 70% of the H content in the plastic, with the generation of a substantial amount of CNTs. The catalytic performance of the fresh catalyst was significantly lower than that of the used counterpart. This is mainly attributed to forming the FeAl2O4 phase at the crystalline boundary of Fe2O3 and Al2O3 during the reaction, enhancing the cleavage of C–H bonds in hydrocarbon compounds. Liu et al.52 designed a monolithic multilayer stainless-steel mesh catalyst for hydrogen and CNT production from combined pyrolysis-catalysis upcycling of waste plastic (Fig. 4(b)). The fresh stainless-steel 316 (SS 316) catalyst was pretreated using acid etching followed by air calcination to increase the exposed surface sites. After modification, the yield of solid products significantly increased from 11.0 wt% to 47.6 wt%, and the filamentous carbon collected from the spent catalyst by simple ultrasound separation was mainly composed of MWCNTs. An H2 yield of 50 mmol gplastic−1 with 93.2% selectivity was achieved during the thermal catalytic conversion of HIPS with the pretreated SS 316 catalyst (Fig. 4(c)). When the single pyrolysis-catalysis test was completed, the spent SS 316 catalyst was recovered by ultrasound treatment to separate the produced CNTs from the catalyst. After removing CNTs, the catalyst was employed for the subsequent test without pretreatment. The hydrogen yield was stable, and the catalytic activity of the catalyst can be quickly recovered after simple recalcination for at least 10 cycles (Fig. 4(d)).
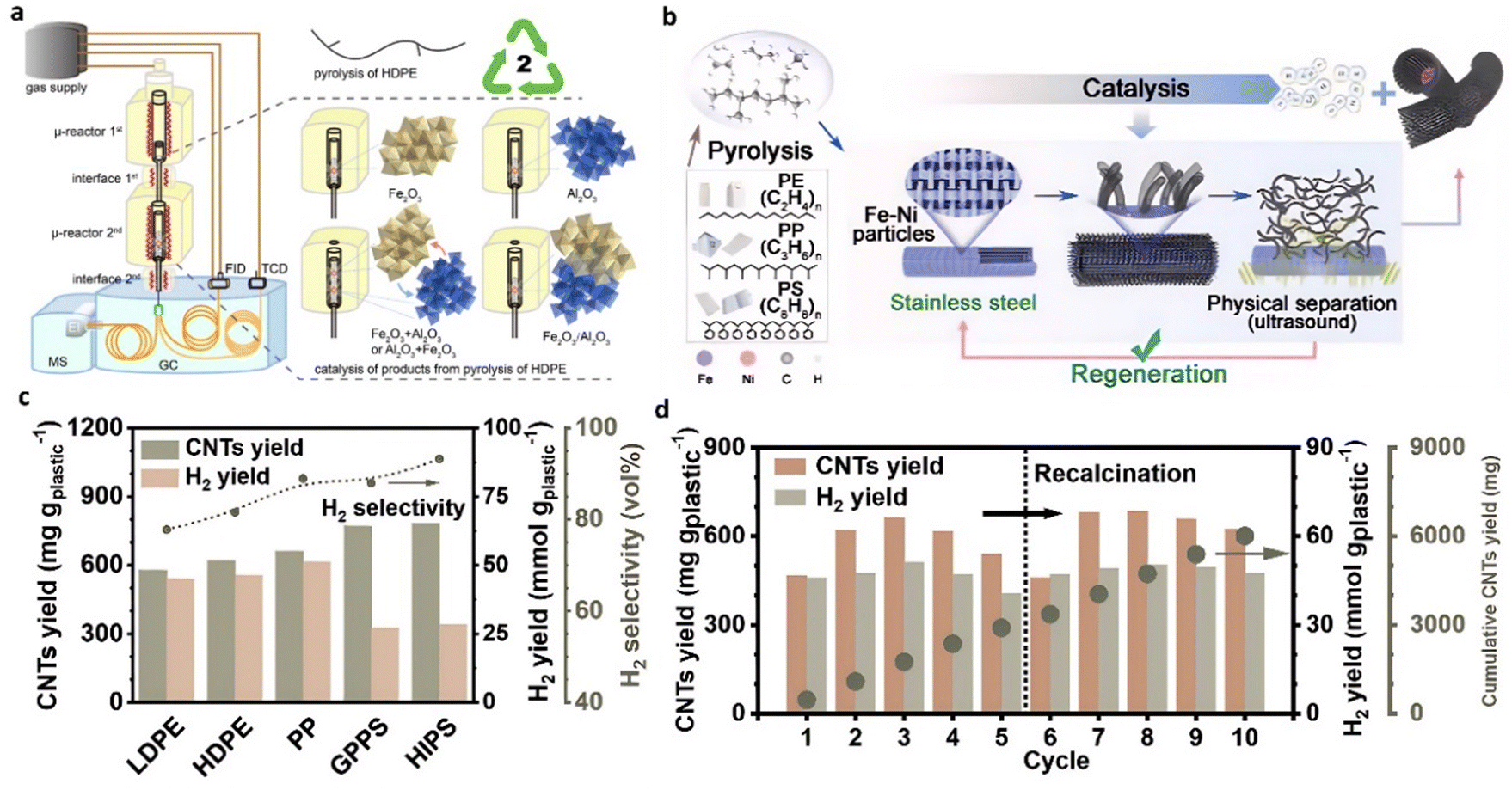 | ||
| Fig. 4 (a) Schematic diagram of the μ-reactor-GC/MS-FID-TCD system used for pyrolysis and catalytic upgrading of HDPE. Reproduced with permission.51 Copyright 2023, American Chemical Society. (b)–(d) Pyrolysis-catalysis of waste plastics to H2 and CNTs using a modified stainless-steel 316 catalyst. Reproduced with permission.52 Copyright 2023, Natl Acad Sciences. | ||
The successful implementation of pyrolysis-reforming processes at an industrial scale depends on the selection of the appropriate reactor design (Fig. 5). While many studies have been conducted in batch laboratory units, continuous operation is crucial for industrial applications. Continuous pyrolysis ensures a steady volatile stream, facilitating catalyst performance evaluation and better control of process conditions. This is also important for maintaining catalyst stability over time.
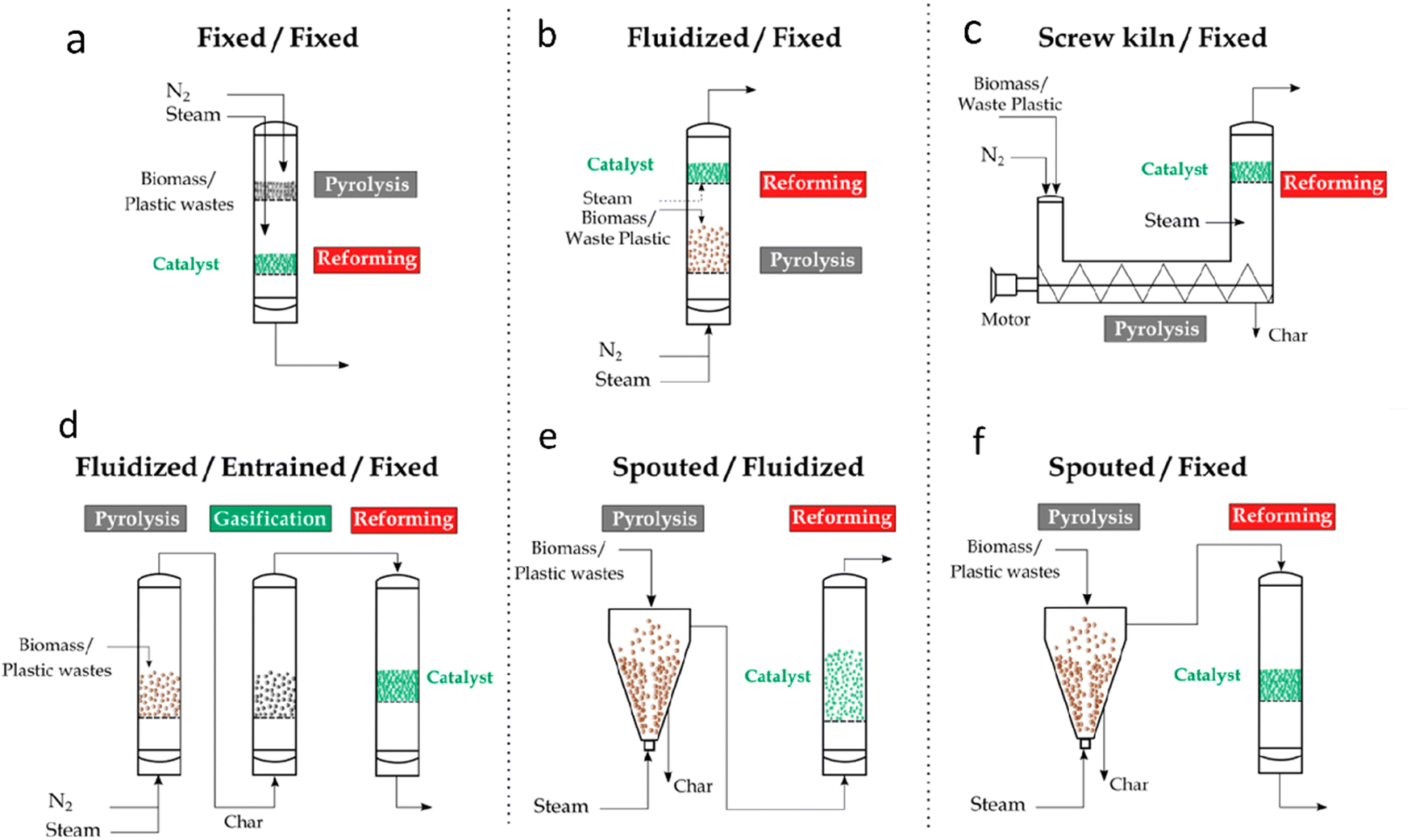 | ||
| Fig. 5 Reactor configurations for pyrolysis and in-line catalytic steam reforming of biomass and waste plastics: (a) fixed bed/fixed bed, (b) fluidized bed/fixed bed, (c) screw kiln/fixed bed, (d) fluidized bed/entrained flow/fixed bed, (e) spouted bed/fluidized bed, and (f) spouted bed/fixed bed. Reproduced with permission.32 Copyright 2021, American Chemical Society. | ||
While fixed bed reactors offer simplicity and cost-effectiveness, they are fraught with operational challenges, particularly rapid catalyst deactivation due to excessive coke formation. These limitations have driven researchers to explore alternative reactor designs that enhance efficiency and scalability. Fast pyrolysis reactors, such as fluidized and spouted beds, have emerged as more efficient alternatives to traditional fixed bed reactors. These reactors are designed for continuous feed operations and provide high heat and mass transfer rates, ensuring better gas–solid contact and bed isothermality. This efficiently converts plastic waste into volatiles with low char yields, thereby increasing the hydrogen production potential. Fast pyrolysis reactors have several advantages such as efficient operation, enhanced process control and easy catalyst regeneration. Fluidized bed53 and conical spouted bed reactors provide improved heat and mass transfer while allowing for shorter residence times. Fluidized bed reactors have however, limitations related to feed and particle size, with defluidization being a common issue, especially when dealing with plastic wastes. Spouted beds, particularly conical spouted bed reactors (CSBR), offer a solution by handling coarse solids with irregular textures due to their vigorous solid circulation movement. We recently introduced54 a vortex reactor for polystyrene pyrolysis. Its innovative design facilitates improved heat and mass transfer while reducing residence time, thereby minimizing secondary reactions.
Combining different reactor types can further optimize the pyrolysis-reforming process. For instance, the pyrolysis step in bubbling fluidized bed reactors can be combined with the reforming of volatiles in fixed or fluidized bed reactors. Similarly, spouted beds can be used in the pyrolysis step, followed by reforming in fixed or fluidized bed reactors. These combinations leverage the strengths of each reactor type while mitigating their weaknesses. Czernik55et al. have introduced a system comprising two fluidized beds, which demonstrated clear advantages for continuous plastic feed operations. More recently, Barbarias et al.45 developed a continuous process using a circulating spouted bed reactor (CSBR) combined with a fluidized bed reactor (FBR) for the valorization of high-density polyethylene (HDPE). This combination leveraged the high heat and mass transfer rates of the CSBR and the efficient performance of the FBR in volatile steam reforming, avoiding operational problems such as defluidization.
Pyrolysis-catalytic dry reforming (with CO2 instead of steam) is also a promising method for valorizing waste plastics. Saad et al.56 reported a dry reforming of various types of waste plastics (LDPE, HDPE, PS, PET, and PP) over a Ni–Co–Al catalyst using a two-stage pyrolysis reactor. The introduction of CO2 in the second stage markedly increased the dry reforming reaction and significantly improved the production of H2/CO with the highest yield of 154.7 mmolsyngas gplastic−1 for LDPE upcycling.
Rapid flash Joule heating (FJH) utilizes rapid current discharge in the presence of resistive feedstocks to achieve a super high temperature (≈3100 K) with a heating rate of up to 105 K s−1. Tour's group57 designed an FJH system for pyrolysis of polyolefins, polyesters, and mixed waste plastics into high yields of “flash H2” along with high-purity graphene as a value-added byproduct, without catalyst, solvent, or water additions (Fig. 6). Higher temperatures and faster heating rate resulted in a larger H2 yield from plastics through C–H bond homolytic fission and up to 47 mol H2 per kg of HDPE. More than 92% efficiency and 87% gas purity were obtained with the initial resistance of 6 Ohm. Based on life-cycle assessment (LCA), FJH of plastics generates <4 kg CO2 per 1 kg H2. Therefore, “flash H2” presents a new sustainable leading technology for H2 production from waste plastics compared with microwave-assisted pyrolysis, which exhibited high production costs due to using energy-intensive microwave and metal catalysts.
 | ||
| Fig. 6 (a) A schematic showing the typical flash Joule heating process used to convert waste plastic into flash H2. (b) A photo of the system to collect the gases evolved by FJH deconstruction of PE. (c) The relationship between initial sample resistance, H2 yield, and efficiency. Reproduced with permission.57 Copyright 2023, Wiley-VCH. | ||
In summary, the combined plastic pyrolysis-reforming is known for its ability to enable efficient hydrogen production. Achieving optimal results requires careful consideration of various factors, including reactors, temperature, feedstock composition, catalyst dosage, steam amount, etc. The right balance among these parameters is essential to maximize hydrogen efficiency. Introducing water into the process improves hydrogen productivity, but limits the formation of valuable by-products, such as CNTs. At the same time, a significant portion of C element is released in the form of CO2. The main challenges in combining pyrolysis and reforming for processing waste plastics are the low purity of hydrogen produced and the often-irreversible catalyst deactivation during the reforming stage. Pyrolysis is widely regarded as a well-established technology, with numerous operational commercial facilities already dedicated to processing both biomass and plastics.
2.1.1.2. Photo-assisted pyrolysis. Photothermal catalytic pyrolysis of waste plastics can potentially reduce the high energy consumption of thermopyrolysis. Recently, Luo et al.58 designed a photothermal catalytic pyrolysis system, which consists of an N2 feeding system, a solar simulator (0.2 to 1.5 MW m−2), a condenser unit, and a gas collecting station (Fig. 7(a)). In the presence of Ni–Ti–Al catalyst, the H2 yield from photothermal catalytic pyrolysis of LDPE was 34 mmol gplastic−1 with a jet fuel selectivity of 80%. Similarly, the plastic was first pyrolyzed to long-chain hydrocarbons at a high temperature (500 °C), which was reached using the solar simulator. These hydrocarbons were further catalytically fractured into hydrogen and small molecular weight hydrocarbons.59 The pyrolysis of LDPE was promoted by photo- and thermal synergistic effects (Fig. 7(c)). Nickel localized surface plasmon resonance (LSPR) also seems to enhance the reaction rates significantly.
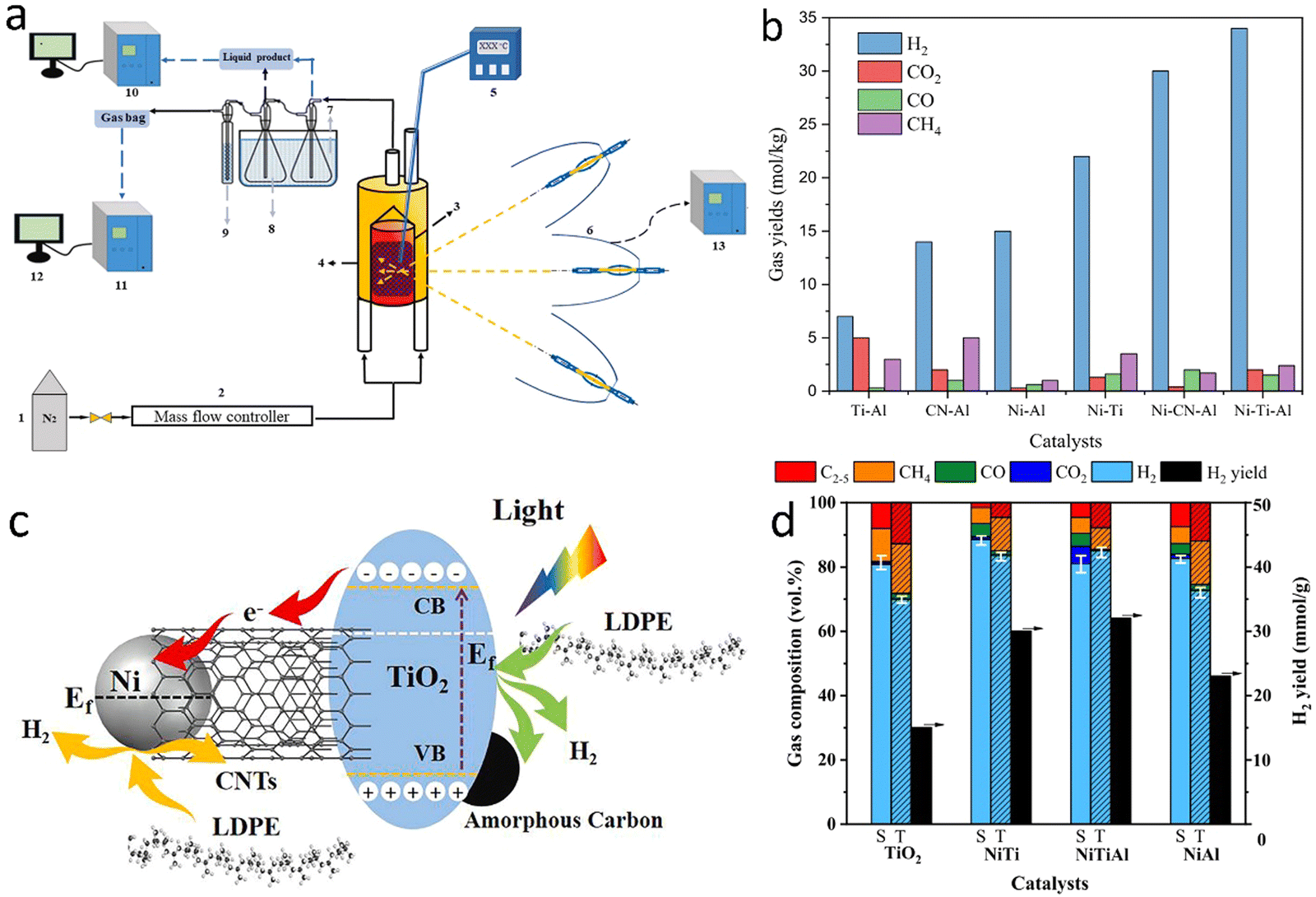 | ||
| Fig. 7 (a) The photothermal catalytic pyrolysis system. (b) Effects of catalyst on the gas yield of photothermal catalytic pyrolysis of LDPE. (a) and (b) Reproduced with permission.58 Copyright 2022, Elsevier. (c) Reaction mechanism for light-induced growth of CNTs and H2 production in photothermal catalytic pyrolysis of LDPE. (d) The comparison of the gas content for solar (S) and traditional (T) pyrolysis of LDPE at 500 °C. (c) and (d) Reproduced with permission.59 Copyright 2023, Elsevier. | ||
2.1.1.3. Plasma-assisted pyrolysis. In the early 1960s, non-thermal plasma-assisted catalytic reforming technology was developed and widely used in chemical engineering, particularly for solid waste disposal and air pollution control. This was due to the unique properties of this technology, such as operation in non-equilibrium conditions, high energy efficiency, and ability to initiate reactions that are otherwise kinetically and thermodynamically unfavorable at low temperatures (<250 °C).60–62 The non-thermal plasma is generated between two electrodes with a significant potential difference, which forms an intense electrical field where the gaseous substrates are ionized into high-energy electrons, ions, radicals, and excited species. The plasma temperature can exceed 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 °C, while the overall temperature of the gas system remains relatively low.
000 °C, while the overall temperature of the gas system remains relatively low.
To increase the hydrogen yield and decrease the energy consumption, Williams's group63 recently coupled a fixed bed pyrolysis reactor with a downstream dielectric barrier discharge (DBD) non-thermal plasma/catalytic steam reforming reactor for hydrogen production in the presence of Ni/MCM-41 catalyst (Fig. 8(a)). Specifically, the decomposed hydrocarbon volatiles derived from the pyrolysis reactor directly pass through the plasma reactor under a power input of 80 W for catalytic steam reforming (steam WHSV 2 g h−1 gcatalyst−1). A higher hydrogen yield of 18 mmol gplastic−1 for HDPE in the plasma-catalytic pyrolysis process was compared with that without plasma assistance. The pre-cracking of heavy intermediates produced by plastic waste pyrolysis by plasma significantly accelerated their diffusion within the catalyst pores and enhanced their catalytic conversion to hydrogen (Fig. 8(c)).64
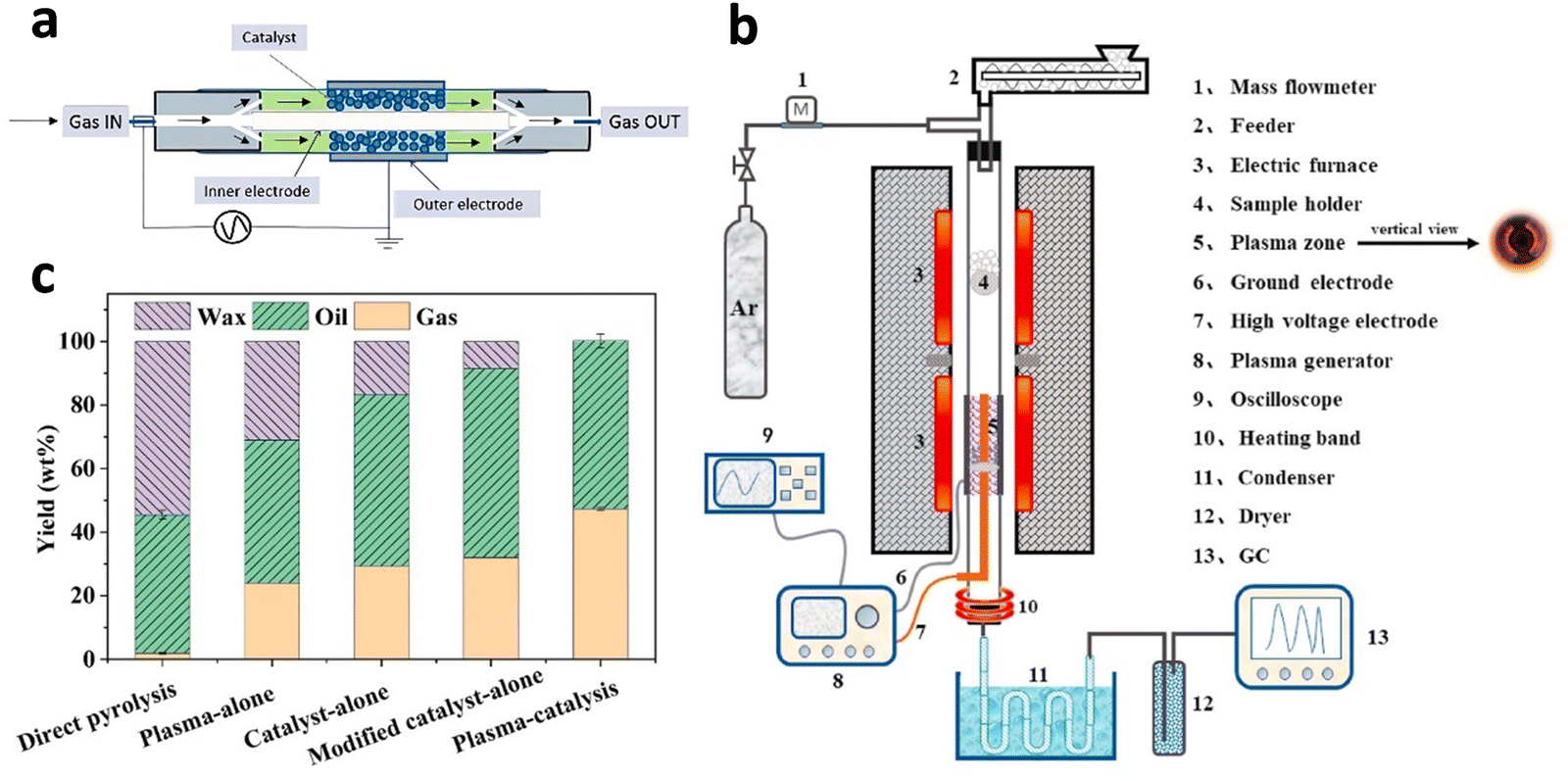 | ||
| Fig. 8 (a) Non-thermal plasma/catalytic reactor. Reproduced with permission.63 Copyright 2023, Elsevier. (b) A two-stage fixed bed system for plasma-catalytic pyrolysis. (c) Influence of plasma and catalyst on PP pyrolysis product distribution, gas and oil compositions. (b) and (c) Reproduced with permission.64 Copyright 2022, Elsevier. | ||
2.1.1.4. Microwave-assisted pyrolysis. Microwave assistance is widely applied in organic synthesis, material processing, and environmental applications, such as plastic waste treatment. Generally, the microwave-catalyzed pyrolysis of waste plastics into hydrogen can be performed either in a simple chamber with only a catalyst as a microwave-susceptor or in a multi-mode chamber with a catalyst and additional igniters. Edwards's group65 first reported a straightforward, simple, and rapid one-step process with microwave assistance for the catalytic deconstruction of various plastic feedstocks into hydrogen and multiwalled carbon nanotubes (MWCNTs) in the presence of a low-cost FeAlOx catalyst (Fig. 9(a)). The experiment was carried out on a homemade microwave reactor system consisting of a microwave generator with an operating frequency of 2450 MHz, a maximum output power of 2000 W, a purpose-built microwave cavity, and a controller (Fig. 9(b)). Throughout ∼90 s, an H2 yield of 55.6 mmol gplastic−1 with nearly 90% selectivity and MWCNTs productivity of 1.56 g gplastic−1 gcatalyst−1 were achieved from HDPE decomposition (Fig. 9(c)). Based on the experimental and computational studies, a tentative mechanistic model of the microwave-initiated solid–solid reaction using iron-based catalysts for hydrogen and MWCNTs production from waste plastic was proposed. Compared with conventional non-selective thermal heating, microwave electromagnetic energy is absorbed directly and preferentially by the microwave-absorbing FeAlOx catalyst and rapidly initiates the heating to a high temperature, while the plastics, which do not absorb microwave radiation, remain cold. This was verified by measuring the average temperature difference (>400 °C) of HDPE and FeAlOx catalyst under microwave irradiation. Such temperature gradient over the interface between the catalyst particles and the plastics causes the cleavage of C–H bonds, accelerating the desorption of produced H2 and minimizing the competing chemical side reactions. It was assumed that the formed Fe3C intermediate contributed to the subsequent formation of MWCNTs. This work was assessed by Lopez and Santamaria66 as a proof of concept at the laboratory scale. Several issues, however, relevant to the catalyst stability, MWCNTs separation should be solved before the scale-up of such a strategy.
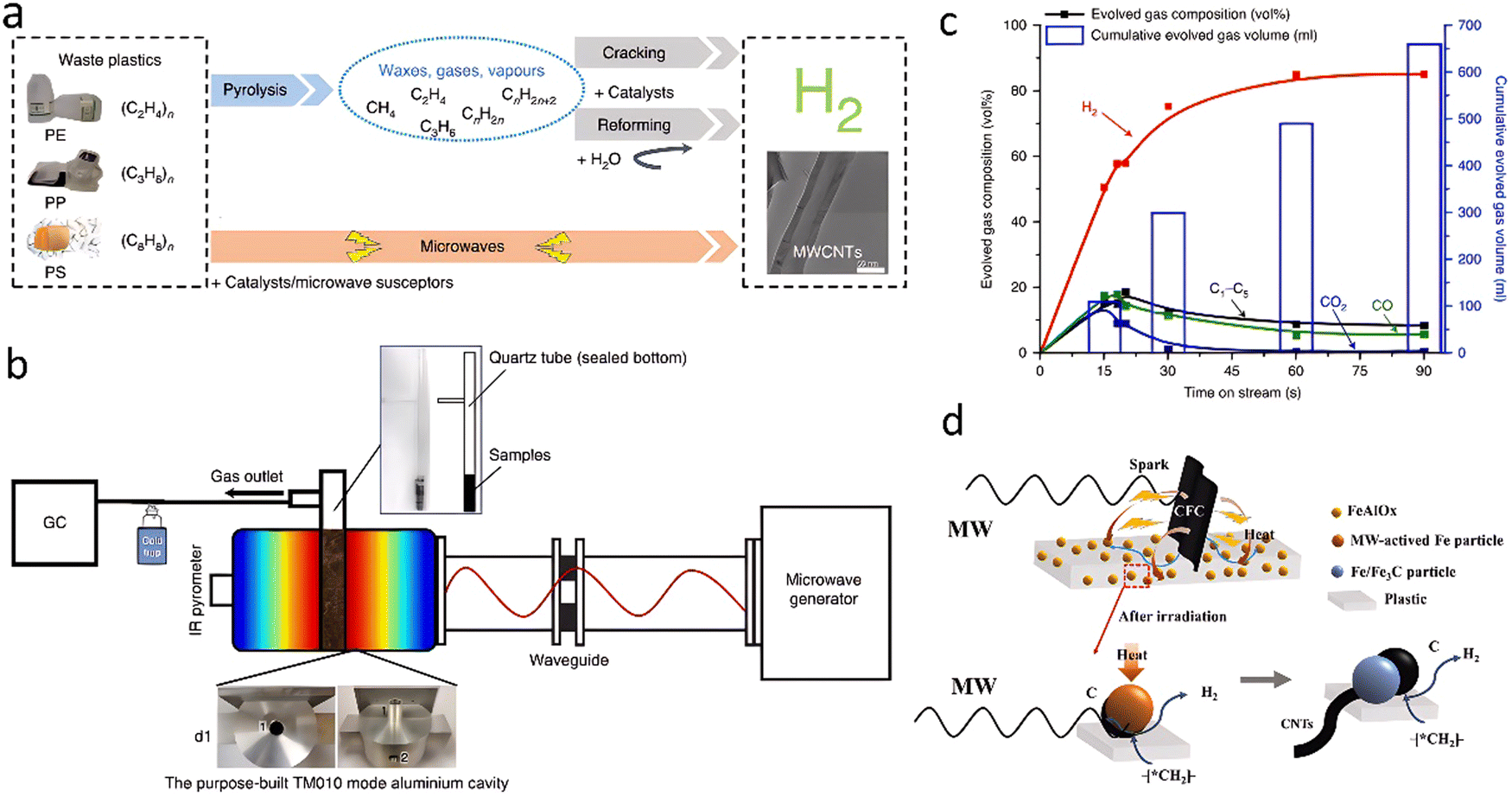 | ||
| Fig. 9 (a) The designed novel one-step microwave-initiated catalytic deconstruction of plastic waste to H2 and MWCNTs compared to the traditional two-step pyrolysis and gasification process. (b) The experimental set-up and reaction system configuration. (c) A time-on-stream analysis shows gas evolution as a function of the time of the microwave-initiated decomposition of HDPE. (a)–(c) Reproduced with permission.65 Copyright 2020, Springer Nature. (d) The mechanism of H2 production by microwave pyrolysis of PE with the assistance of carbon fiber cloth. Reproduced with permission.67 Copyright 2023, Elsevier. | ||
Catalyst optimization also allows the enhancement of hydrogen production from waste plastics through microwave-assisted catalytic pyrolysis.67–71 Li et al.68 developed the Co doped Fe–Al catalysts (Fe–Co–Al) showing the H2 yield up to 61.39 mmol gplastic−1 in the microwave-assisted catalytic deconstruction of PE. The productivity enhancement was mainly attributed to the easier formation of Fe3C.
To further improve the hydrogen production efficiency, a microwave-additive co-ignited cracking strategy is commonly applied for highly efficient conversion of waste plastics to hydrogen and high-quality CNTs. Zhang et al.67 induced carbon fiber cloth (CFC) as the microwave igniter in the FeAlOx@C catalyst. A hydrogen yield of 64.5 mmol gplastic−1 was achieved by cracking PE (Fig. 9(d)). As proposed, the CFC is responsible for absorbing the microwave, reaching a high temperature in a very short time under the mechanism of Joule heating and plasma arcing.
In summary, hydrogen generation through microwave-assisted catalytic deconstruction of waste plastics has advantages in terms of lower energy consumption and potential for reducing tar formation. At the same time, microwave pyrolysis of plastic waste faces significant scalability challenges when transitioning from laboratory to industrial use. Key obstacles include achieving uniform heating in large reactors due to variable dielectric properties of feedstocks and limited microwave penetration, which risks hot spots and inconsistent product quality. Feedstock variability in composition, moisture, and size complicates stable material flow and continuous feeding systems. Microwave generation demands costly equipment and optimized energy use to ensure economic viability. Maintaining consistent product quality is difficult due to uneven heating and feedstock diversity, alongside managing by-products like biochar and gases. Safety concerns, particularly preventing microwave leakage in high-power systems, add further complexity. Ongoing research aims to refine reactor design, process control, and energy efficiency to overcome these barriers and enable commercial adoption.
Catalysts play a crucial role in improving the efficiency of plastic gasification,74 primarily through two mechanisms: (a) tar cracking and (b) gas reforming. In the gasification process, two main types of catalysts are commonly employed: (a) mineral-based catalysts, such as dolomite and limestone, and (b) transition metal catalysts, including Ni and supported-Ni materials.
It is important to note that most of waste plastic gasification processes involve H2O and CO2, as the use of air as gasifying agent facilitates combustion and partial oxidation reactions, which yield CO, CO2, and H2O. The amount of hydrogen produced in air gasification is usually lower compared to steam gasification. In addition, the use of air results in the dilution of the produced syngas with nitrogen, decreasing its energetic and chemical value.
Higher ratio of steam to plastic waste ratio in steam gasification further increases the H2 and CO content, and the CO2, CH4, and C2–C3 hydrocarbon contents decrease due to intensification of water–gas shift and steam reforming.75 Steam gasification of plastic waste can be considered as a sequence of three steps76 (Fig. 10): drying, pyrolysis and reduction. All steps in steam gasification are endothermic.
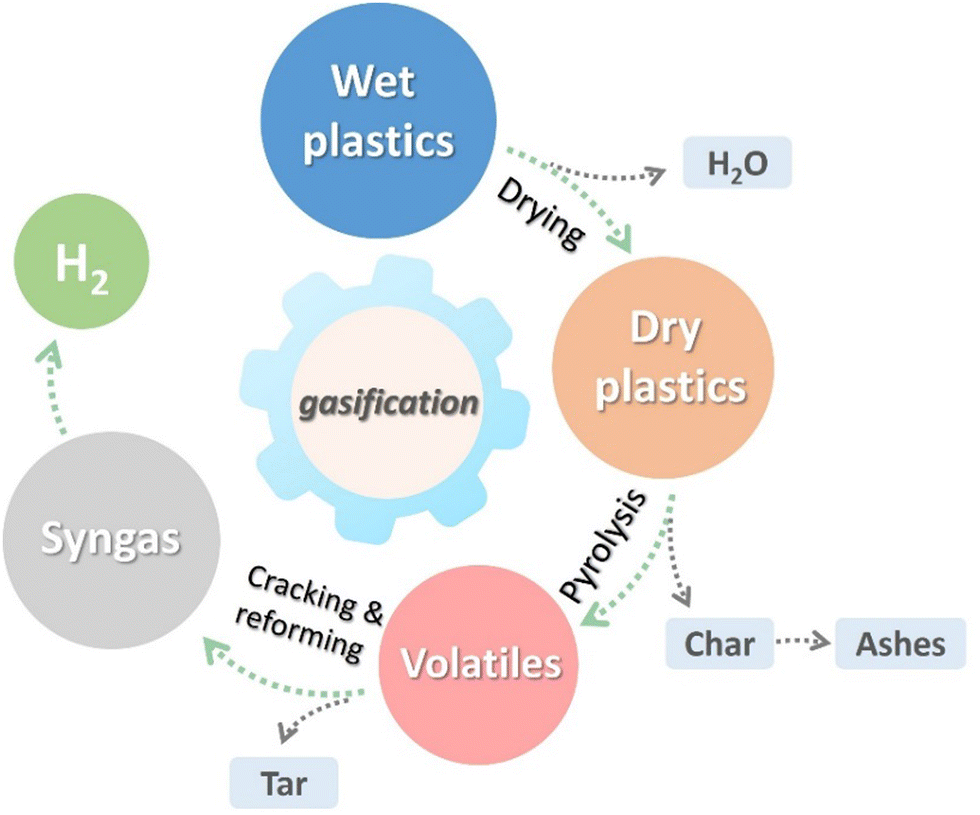 | ||
| Fig. 10 Main reactions and steps of plastic gasification process. | ||
Drying involves the vaporization of moisture present in plastics, with the energy required proportional to the moisture content. The pyrolysis step of gasification involves the thermochemical breakdown of the polymers, producing lower molecular weight molecules. Depending on the conditions, pyrolysis generates different proportions of solid, liquid, and gaseous products. Finally, during the reduction step, the gaseous, liquid, and solid products react to generate syngas.
The steam gasification of waste plastic can generate significant amounts of tar, potentially clogging pipelines and contaminating downstream equipment. While steam gasification produces more hydrogen than air gasification, the endothermic nature of the process requires significant energy input. The addition of a small amount of air and controlling the steam-to-oxygen molar ratio in steam gasification can be instrumental for smoother plant operation.77 Complementing extensive experimental trials, machine learning and numerical optimization have emerged as powerful tools for evaluating the influences of gasification parameters.78–80
Bai et al.81 introduced a novel approach by incorporating CO2 to modify the reaction environment for plastic gasification. Gasification efficiency was significantly enhanced in the mixed fluid compared to supercritical water alone, yielding higher amounts of H2 and CH4. Key factors such as reaction temperature, duration, and CO2 concentration positively influenced gasification. At 700 °C, the carbon conversion rate in the mixed CO2–water fluid was approximately 2.5 times higher than in supercritical water alone.
Gasification proceeds in combination with WGS or reforming, which converts syngas into H2 and carbon dioxide. Prominent advantages of hydrogen production via plastics gasification compared to pyrolysis are higher hydrogen yield, better feedstock flexibility, and greater scalability (Table S2, ESI†).
Generally, the gasification of waste plastic is similar to that of other feedstocks such as coal and biomass.13,82,83 However, high volatility, high thermal resistivity, sticky nature, and severe tar production from waste plastics largely limit their processing by conventional gasification technologies. For example, when waste plastic is fed into the reactor, it begins to melt and adhere to the walls of the feeding tube, which may block the pipe and hinder the feeding process. Thus, designing appropriate reactors for efficiently gasifying waste plastics into hydrogen is vital. Specifically, accelerating heat transfer rate, controlling the viscosity of plastics, and facilitating tar cracking should all be considered in the construction of gasifiers. Ruoppolo et al.84 showed that in the gasification of biomass and plastic mixtures, pelletization was an effective pre-treatment for improving the homogeneity of the fuel and resulted in hydrogen concentration increase up to 32% vol. Addition of plastics to biomass often increases hydrogen content in the syngas.85 There are numerous complexities regarding the kinetic modeling of plastic waste gasification relative to coal and biomass gasification.86
Presently, depending on the configuration, the reactors used for waste plastic gasification are fluidized bed reactors,84,85,87–96 conical spouted bed reactors,97,98 fixed bed reactors,99–103 plasma reactors104–106 and vortex reactors.86
A fluidized bed reactor (FBR) is a continuous flow reactor, which is widely used in gasifying various waste plastics. Based on the gas velocity conditions and processing step, FBR can be classified into a bubbling fluidized bed (Fig. 11(a)) and a circulating fluidized bed (Fig. 11(b)). For the bubbling fluidized bed, the fluidization agent is injected from the bottom part of the reactor along with additional sand to activate the feedstock at around 800–1000 °C with a low gas velocity (1–3 m s−1). Then, the produced syngas flows to the next stage through the gas outlet, leaving the particles in the bottom. The main difference for the circulating fluidized bed is the extra step in the reactor for separation and recirculation in the fluidized bed chamber with a higher gas velocity (3–10 m s−1). In general, the FBR provides good and stable hydrogen production performance, which can potentially be applied to large-scale power plants. Recently, Jeong and co-workers96 developed a two-stage gasifier consisting of a bubbling fluidized bed gasifier and a packed bed reactor filled with activated carbon, a tar removal additive for air gasification of 10 different types of plastics at ∼800 °C. The process consists of a feeding part, two reaction zones, a char-separating part, and a quenching part. A maximum hydrogen production content of 26 vol % was achieved by gasification of PP.
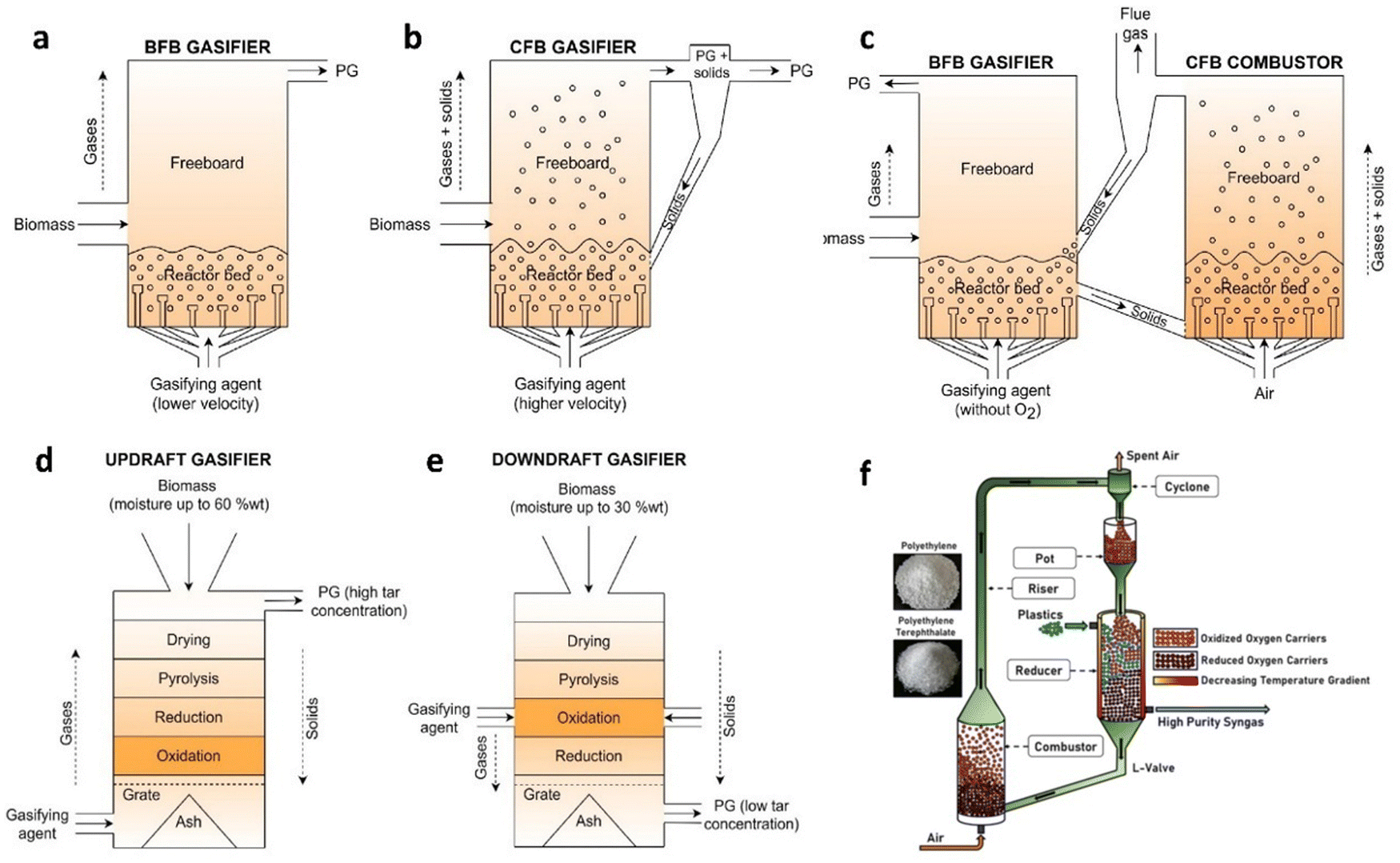 | ||
| Fig. 11 Different types of gasification reactors. Bubbling fluidized bed (a), circulating fluidized bed (b), dual fluidized beds (c), updraft fixed bed (d), and downdraft fixed bed (e). Reproduced with permission.99 Copyright 2021, Elsevier. (f) Scheme of chemical looping gasification process.107 Copyright 2024, Elsevier. | ||
Sorption-enhanced and chemical looping gasification are advanced approaches. In chemical looping gasification (Fig. 11(f)),107 a traditional gasification agent is replaced with an oxygen carrier for converting waste plastics into syngas. In the oxygen carrier, oxygen of transition metal oxides (e.g. iron and nickel oxides) is used as an oxygen donor. The process typically involves two interconnected reactors: a fuel reactor and an air reactor. In the fuel reactor, waste plastics are transformed into syngas while reducing the oxygen carrier. Then, in the air reactor, the reduced oxygen carrier is re-oxidized to replenish its lattice oxygen and remove any deposited coke. Additionally, the reduced oxygen carriers from the fuel reactor can act as catalysts, aiding in tar cracking. The heat balance is achieved by balancing the heat required by the endothermic reaction in the reducer with the heat from the exothermic re-oxidation of the reduced carrier in the combustor, potentially requiring no external heat input. Hydrogen productivity strongly depends on the operating conditions of chemical looping gasification, such as premixing, temperature, and heating rate.108
Dou109et al. proposed a fluidized-bed gasification (FBG) combined with sorption-enhanced steam reforming process (SERP) for continuous hydrogen production from waste plastic in the presence of Ni/NiAl2O4–CaO catalyst. Using high-pressure nitrogen, a riser facilitated the transport of catalyst particles between two fluidized bed reactors. Notably, the CaO adsorbent continuously removed CO2 generated during the WGS reaction, driving the WGS reaction forward, overcoming the equilibrium limitations, and ensuring higher yield and purity of H2. Moreover, at elevated temperatures, CaO can simultaneously eliminate gas pollutants such as HCl, which may be generated during PVC plastic pyrolysis. CaCO3 formed after CO2 absorption is subjected to high-temperature treatment in the regenerator section to release CO2 and regenerate CaO for the following reaction cycle. Ultimately, the combination of FBG and SERP processes has yielded approximately 88.4 vol% of high-purity hydrogen. This was achieved with an FBG temperature of 818 °C and a SERP temperature range of 706–583 °C.
The conical spouted bed reactor (CSBR, Fig. 11(f)) is considered an alternative to fluidized beds for waste plastics upcycling, which can avoid the defluidization process caused by melted plastic in FBR. The secondary reaction, like coke precursor formation, can be reduced due to the short residence time of the feedstocks. Moreover, CSBR has a lower pressure drop compared with FBR. Erkiaga et al.97 performed the steam gasification of PE in a bench-scale plant containing a CSBR for hydrogen production. To avoid the condensation of steam and tars, a CSBR is placed in a forced convection oven containing a high-efficiency cyclone and a sintered steel filter at 270 °C. Adjusting the steam/plastic mass ratio and gasification temperature allows an H2 concentration of 61.6% with minimum tar formation (9.6 g N m−3) over CSBR. The use of olivine and γ-Al2O3 instead of sand results in further moderate reduction in the tar formation.
The fixed bed gasifier (Fig. 11(d) and (e)) is the most basic and simplest gasifier for waste gasification through a slow-moving feedstock with a long residence time (∼900–1800 s). It includes updraft and downdraft configurations with the same feedstock feeding from the top of the gasifier and the gasifying agents from the side or bottom of the reactor, respectively. The reactor chamber is divided into drying, pyrolysis, gasification, and eventually oxidation zones. Fixed bed gasifier requires mechanically stable feedstocks of small particle size with low moisture content.
Plasma gasification is a novel technology used commercially as a waste-to-energy system that converts multiple waste streams into syngas using electrically ionized gas via plasma torches at a very high temperature (>1500 °C). It is categorized into plasma-assisted gasification and two-stage plasma gasification. Specifically, the plasma gas (Ar, N2, H2O, CO2, air, etc.) fed in the reaction acts as a reforming agent, cracking tar in the gas phase into lighter molecules and converting inorganic materials into vitrified slag without the pretreatment. Mallick et al.106 applied single-stage high-temperature plasma gasification for clean syngas production from acrylonitrile butadiene styrene (ABS) based computer keyboard plastic waste (CKPW) using CO2 as both plasma gas and gasifying agent. The experimental system consists of a feeding port, a plasma gasifier, a torch cooling system, a condenser, and a gas analysis system.110 Initially, the feedstock is fed through a hopper at the top of the reactor. Then, the plasma gas with controlled flowrate is purged through the center of the cathode in the specifically designed holes into the gasifier chamber. Finally, the produced syngas released at the top side of the reactor is cooled in a water condenser. Under the optimum process parameters (feed flow rates of feedstock and CO2 gas: 4 g min−1, 0.5 L min−1, and torch power of 1.12 kW), a higher percentage of H2 (30.16 vol%) and CO (46.09 vol%) were achieved. However, due to the high-temperature conditions, this plasma gasification technology has relatively high operational and maintenance costs, which restrict its large-scale application in commercialized hydrogen generation from waste plastic upcycling.
In oxygen or air gasification, the hydrogen production efficiency from waste plastic gasification is systematically determined by various factors like reaction temperature, the ratio of actual air used to stoichiometric air (equivalence ratio, ER), bed materials, etc. Mastellone et al.91 have investigated the effect of oxygen-enriched air during fluidized bed gasification of plastics. A higher hydrogen ratio in syngas production could be achieved, mainly due to the increased bed temperature. A similar result was explored by Zhao and co-workers.111 They showed that if the oxygen concentration increased by 5%, the hydrogen concentration in the produced gas would increase by about 14%. Higher temperatures favor endothermic reactions. Thus, the reaction temperature increase largely affects the main endothermic reactions like Boudouard reactions, carbon gasification, and the secondary cracking of tar. He et al.101 found that the H2 content almost doubled from 16.92% to 36.98% as the reactor temperature increased from 700 °C to 900 °C, which was due to the strong influence of temperature on the decomposition of CH4. Bed agglomeration and severe coke formation are common issues that must be solved during the two-stage gasification process. Jeong et al.96 investigated different types of bed materials in place of sand to reduce the tar inside the fluidized bed gasifier. Both natural olivine and calcined dolomite significantly decreased the tar content in produced gas.
In summary, plastic gasification is an emerging research area offering key advantages for managing contaminated or mixed plastic waste, as it eliminates the need for extensive sorting and can avoid catalyst use, which is prone to deactivation by impurities. This process, particularly steam gasification at 800–1500 °C, efficiently converts plastics into hydrogen-rich syngas, though its energy-intensive, endothermic nature necessitates supplementary heat sources. The resulting gas composition (H2, CO, etc.) depends on variables like plastic type, gasification agent (steam, CO2, oxygen, air), temperature, and pressure. However, challenges such as tar formation, the sticky nature of melted plastics, and thermal instability hinder conventional gasification. Innovations like dual-stage reactors, co-feeding with biomass/coal (despite reducing H2 content), and pilot-scale solutions such as conical spouted beds aim to address these issues, balancing efficiency with practical scalability. Optimizing energy demands, minimizing tar, and managing feedstock variability remain critical for advancing plastic gasification as a sustainable chemical recycling pathway.
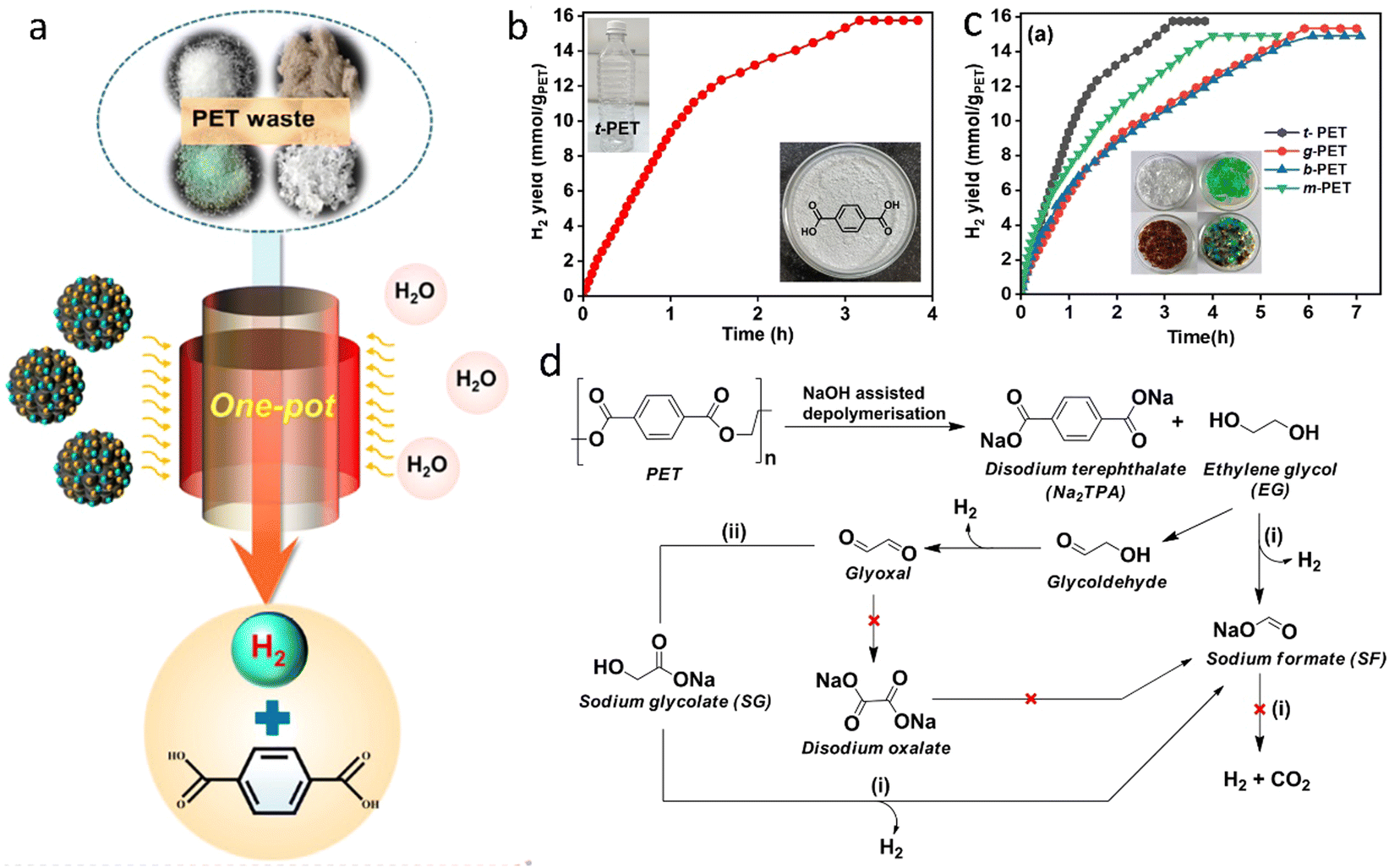 | ||
| Fig. 12 (a) One-pot H2 production from PET upcycling by aqueous phase reforming. Reproduced with permission.112 Copyright 2023, American Chemical Society. (b) Time course plot of H2 yield for the one-pot depolymerization of t-PET. (c) H2 yield for the depolymerization of different plastics. (d) The proposed reaction pathway. (b)–(d) Reproduced with permission.113 Copyright 2023, Wiley-VCH. | ||
In summary, APR presents distinct advantages compared to waste plastics pyrolysis and gasification, particularly in energy efficiency (lower temperature and moderate pressure), wet waste processing, which may remove soluble impurities (e.g. chlorine ions) and enhancement of water–gas shift (WGS) reaction, which allows producing H2 with negligible amounts of CO. At the same time, plastic waste should be dissolved in water to facilitate the reaction. APR is most efficient for hydrophilic oxygenated compoounds (e.g., glycerol, sugars) rather than hydrophobic non-oxygenated plastics like polyethylene or polypropylene. Preliminary liquefaction of waste plastics is required to increase the amount of hydrogen produced in APR. APR for plastic utilization is still in the early stages of development. More research is needed to optimize the pretreatment of waste plastics, to develop efficient catalysts, to improve reaction conditions, and to integrate the process with existing waste management systems.
2.2 Photoreforming
Unlike conventional thermochemical methods that require harsh conditions, photoreforming (PR) of waste plastics operates at room temperature and ambient pressure, enabling hydrogen production using sustainable solar energy. Semiconductor-based photocatalytic water splitting (H2O → H2 + 1/2O2; ΔG0 = +237 kJ mol−1) is considered a promising renewable and sustainable hydrogen production technology.114–118 However, the slower kinetics of water oxidation, also called oxygen evolution reaction (OER), largely restricts the H2 production efficiency. Numerous sacrificial reagents (S2−, SO32−, methanol, triethanolamine, etc.) have been used as hole scavengers for the collection of photogenerated holes. In recent years, a strategy of semiconductor-based dual-functional photocatalytic H2 generation coupled with waste plastics reforming/oxidation into high value-added chemicals has intrigued great interest.119–141 The overall reaction pathway involves two isolated half-reactions including hydrogen evolution reaction (HER) and selective oxidation of plastics in the presence of semiconductor-based photocatalysts with matched redox potentials.31 The photogenerated electrons (e−) in the conduction band (CB) of the catalyst reduce H+ to H2, while the holes (h+) in the valence band directly or indirectly with involvement of reactive oxygen species oxidize waste plastics into value-added oxygenates or ultimately to carbon oxides by reactive holes/oxygen species through a stepwise degradation process of C–C, C–O or C–N bonds (Fig. 13).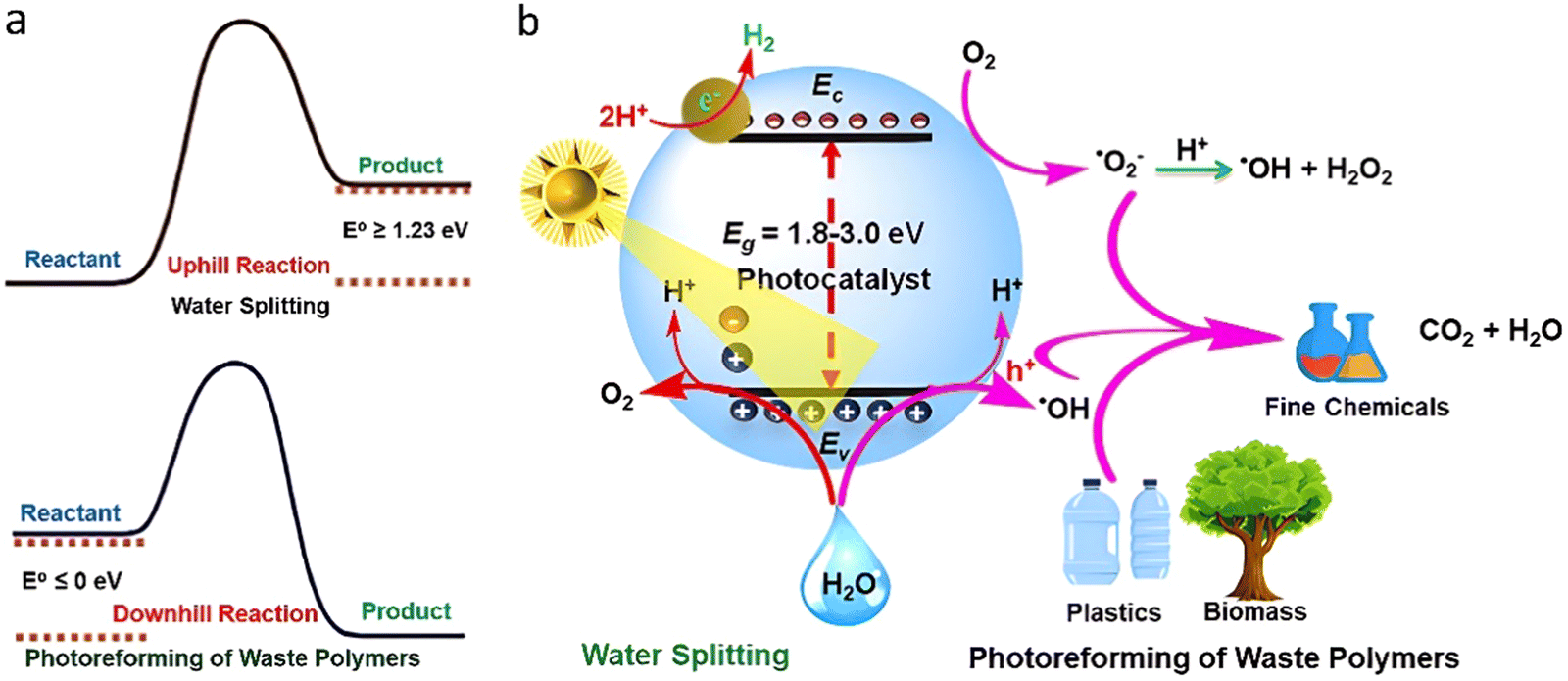 | ||
| Fig. 13 (a) Endothermic and exothermic water splitting and photoreforming of waste plastics. (b) Mechanistic pathways of waste plastics photoreforming. Reproduced with permission.31 Copyright 2023, American Chemical Society. | ||
Such a strategy has several advantages:
– PR of waste plastics shows a relatively lower thermodynamic barrier of ΔE0 = 0.001 V compared to overall water splitting (ΔE0 = 1.23 V). The waste plastics in the PR process act as sacrificial agents to reduce the recombination rate of e− and h+, which enhances the H2 productivity.
– Waste plastics can be upcycled into value-added fine oxygenates at room temperature in sunlight.
It was first reported by Kawai and Sakata142 that the chlorine-containing plastics, such as PVC, could be decomposed in water at room temperature under a 500 W Xe lamp irradiation by using Pt/TiO2 photocatalyst in a strong alkaline solution (5 M NaOH). H2 productivity increased much compared to the direct decomposition into H2 and O2. The PR process of waste plastics has been recently widely investigated by some leading research groups, including Reisner,137 Qiu,129 Xie,130 and Qiao122 using various catalytic systems. A summary of recent work on hydrogen production from waste plastic photoreforming is presented in Table S3 (ESI†).
Uekert et al.141 utilized a CdS/CdOx quantum dot (QD) to reform alkaline aqueous solution-treated polyesters with photocatalytic H2 production (Fig. 14(a)). Before the PR process, PLA was hydrolyzed to sodium lactate, while PET was hydrolyzed to terephthalate, ethylene glycol, and isophthalate. PU undergoes hydrolysis to 2,6-diaminotoluene and propylene glycol. When exposed to simulated solar light, these hydrolyzed compounds served electron donors and were oxidized into value-added smaller organics (pyruvate, formate, glycolate, ethanol, acetate, lactate, etc.). H2 was simultaneously produced from water with productivities of 64.3 ± 14.7, 3.42 ± 0.87, and 0.85 ± 0.28 mmolH2 gCdS−1 h−1 for PLA, PET and PU, respectively (Fig. 14(b)). PR of a PET water bottle was tested. A continuous H2 evolution with 4.13 ± 0.40 mmolH2 gCdS−1 h−1 and a conversion of 5.15 ± 0.72% was achieved in 6 days without catalyst deactivation. Thin Cd oxide/hydroxide shells (CdOx) of CdS QDs in aqueous NaOH, prevented photocorrosion. Considering the toxicity of Cd element, Uekert and co-workers140 developed an inexpensive and nontoxic carbon nitride/nickel phosphide (CNx|Ni2P) photocatalyst for PR of PET and PLA to H2 fuel under alkaline aqueous conditions (Fig. 14(c) and (d)). Although the H2 evolution rate was somewhat lower, the cost-efficiency and photostability make carbon nitride/nickel phosphide a sustainable catalyst for waste plastics upcycling.
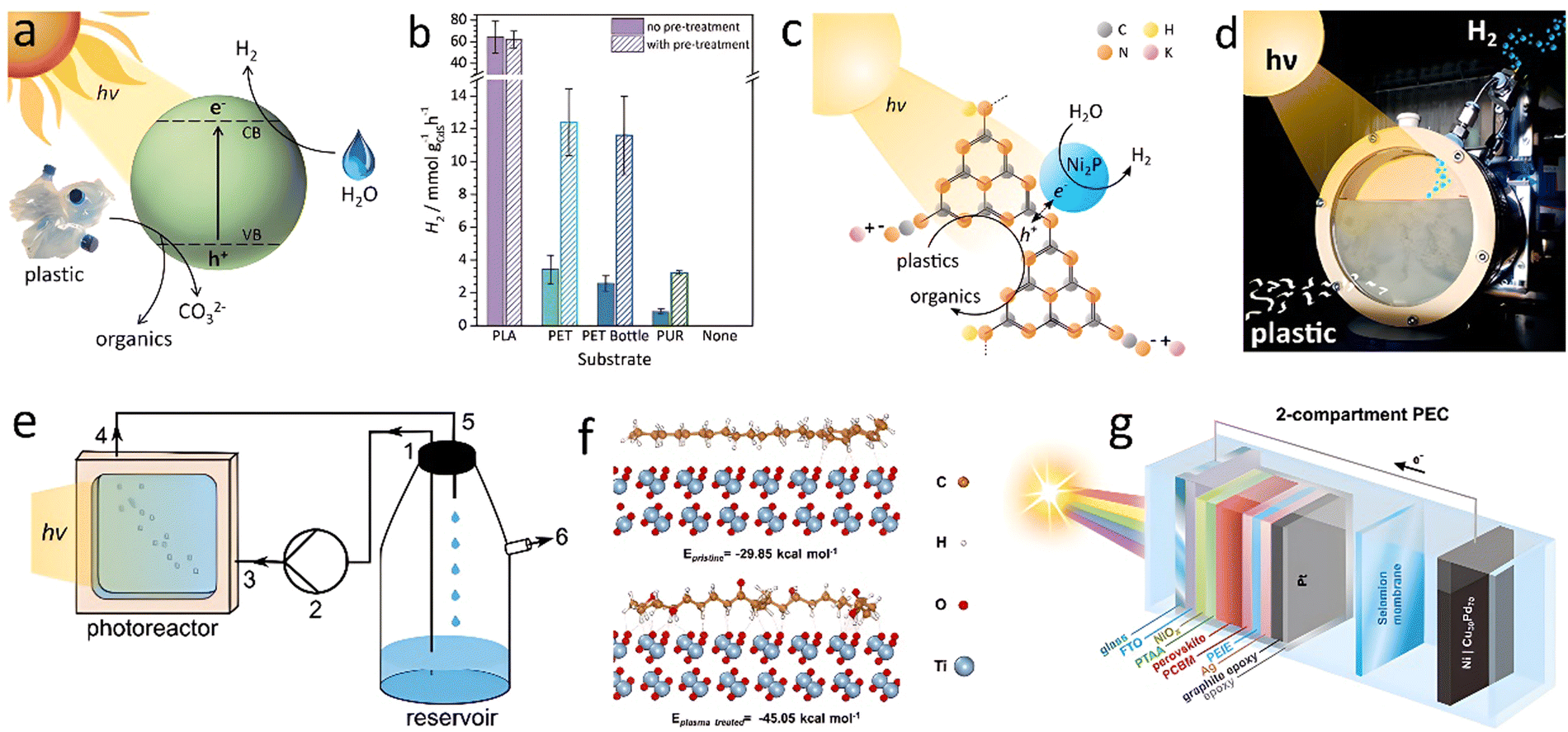 | ||
| Fig. 14 (a) Diagram of the photoreforming process with a CdS/CdOx QD photocatalyst in alkaline aqueous solution. (b) H2 yields for photoreforming of different plastics. Conditions: powdered plastics (50 mg mL−1 PLA, 25 mg mL−1 PET, PET bottle, or PUR) freshly prepared (no pre-treatment) or pre-treated in 10 M aq. NaOH (2 mL). (a) and (b) Reproduced with permission.141 Copyright 2018, Royal Society of Chemistry. (c) Schematic diagram of photoreforming using a CNx|Ni2P photocatalyst. (d) Photograph of the batch reactor. (c) and (d) Reproduced with permission.140 Copyright 2019, American Chemical Society. (e) Schematic diagram of large-scale panel experiments in a flow reactor. Reproduced with permission.138 Copyright 2020, Wiley-VCH. (f) The calculated interaction energies between PE and TiO2 surface before and after plasma treatment. Reproduced with permission.127 Copyright 2023, Wiley-VCH. (g) Overview of PEC waste reforming system in the two-compartment configuration. Reproduced with permission.143 Copyright 2022, Wiley-VCH. | ||
Using nitride/nickel phosphide in suspension posed several practical challenges, such as catalyst sedimentation, limited recyclability, and interference from plastic particles through competing light absorption and scattering. To address these drawbacks, the researchers developed a CNx|Ni2P catalyst immobilized on a textured glass surface, forming a 1 cm2 flat panel for PR of plastics. Such small photocatalyst panel under back irradiation generated 156 ± 15, 31 ± 3 and 15 ± 2 μmolH2 m−2 h−1 for PR of PET, α-cellulose and municipal solid waste (MSW), respectively. Scalability was further verified by developing a 25 cm2 panel for application in a custom-designed flow reactor (Fig. 14(e)), which consisted of a reservoir, peristaltic pump, and photoreactor. The panel showed an H2 yield of 21 μmolH2 m−2 h−1 for 5 days under “real-world” (seawater, low sunlight) conditions for PR of MSW. MOF-based materials were also reported as efficient photocatalysts for H2 generation due to their tailorable structure, high specific surface area, and strong adsorption capacity. Qin et al.132 decorated Ag2O into the pores of Fe-MOF to form an Ag2O/Fe-MOF heterojunction for converting PET into small hydrocarbon molecules and producing H2 with a rate of 1.9 mmol g−1 h−1.
High-entropy oxide (HEO) is another emerging material for photocatalysis. Recently, Hai and co-workers144 developed a high-entropy oxynitride (HEON) photocatalyst by nitrogen doping in a Ti–Zr–Hf–Nb–Ta-containing HEO, which leads to narrowing the bandgap (from 3.2 eV to 1.5 eV) and diminishes electron–hole recombination. Such HEON photocatalyst with distorted atomic-bond structures exhibits an H2 production rate of 1.63 mmol g−1 for photoreforming of PET in 4 h, which is 2 times higher than that of HEO. Necessity for plastic pretreatment in photoreforming of PET waste in harsh alkaline conditions (COH− = 5–10 M) results in the increasing burden of cost and photocatalytic devices. Therefore, Zhang's group145 proposed a tandem process for the photoreforming of waste PET plastics combining a binuclear zinc catalyst with an ultrasmall g-C3N4 nanosphere photocatalyst. An impressive H2 production rate of 2 mmol g−1 h−1 was achieved under mild conditions (COH− ≤ 0.1 M and T ≤ 60 °C), which was nearly 5-fold higher than that in strong alkali pretreatment system.
Most of the above works focused on the PR process of polar polyester-type plastics (PET, PLA, and PU). These plastics with lower bond energies of C–O and C–N can be hydrolyzed to corresponding monomer intermediates in strongly alkaline conditions. The monomer intermediates can be further converted into valuable products through subsequent PR process. However, non-polar polyolefin plastics like PE, PP, and PVC have rarely been investigated for H2 production using the PR strategy. Furthermore, the low water solubilities of the plastic particles hamper their dispersion in aqueous solution and decrease the interaction with catalysts, resulting in low H2 productivity. To address the challenges, Jiang et al.127 pretreated the PE plastic with non-thermal plasma to graft oxygenated groups (–OH, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CO) on the surface to form a high hydrophilicity polar polymer. After the plasma treatment for 30 min, the optimal photocatalytic H2 evolution rate for PE photoreforming by Pt/TiO2 catalyst reached 108.95 μmol g−1 h−1, 3.3 times higher than that of the pristine PE. The theoretical calculation showed that the interaction energies of PE with TiO2 were −29.85 kcal mol−1 and −45.04 kcal mol−1 for pristine PE and plasma-treated PE, respectively (Fig. 14(f)). Stronger interaction of treated PE with surface active sites of TiO2 seems to contribute to the enhanced reaction rate.
O, and CO) on the surface to form a high hydrophilicity polar polymer. After the plasma treatment for 30 min, the optimal photocatalytic H2 evolution rate for PE photoreforming by Pt/TiO2 catalyst reached 108.95 μmol g−1 h−1, 3.3 times higher than that of the pristine PE. The theoretical calculation showed that the interaction energies of PE with TiO2 were −29.85 kcal mol−1 and −45.04 kcal mol−1 for pristine PE and plasma-treated PE, respectively (Fig. 14(f)). Stronger interaction of treated PE with surface active sites of TiO2 seems to contribute to the enhanced reaction rate.
Reisner's group143 designed a perovskite-based photoelectrochemical (PEC) device (Cu30Pd70|perovskite|Pt) for hydrogen generation from a variety of waste feedstocks (Fig. 14(g)). The amounts of H2 evolved from PET powder and bottle under zero applied voltage were 737 and 705 μmol cm−2, respectively. The value-added glycolic acid formation rate was up to ≈130 μmol cm−2 h−1, 102–104 times higher than the conventional PR process.
In summary, photoreforming shows a potential for hydrogen generation coupled with the production of value-added chemicals from waste plastics. However, several issues should be addressed before commercialization: (1) development of efficient routes for pretreatment of waste plastics; (2) optimization of photocatalyst design; (3) scaling up and designing large-scale photoreactors; (4) investigations of photocatalytic reaction mechanisms; (5) establishment of standardized rules for evaluation of photocatalytic performance.
2.3 Electrocatalysis
Similar to photocatalytic water splitting, OER process is still the rate-limiting step in water electrolysis. In recent years, the thermodynamically favorable selective oxidation of organics has been widely applied in the anodic reaction for accelerating hydrogen production as well as achieving value-added chemicals. However, electrocatalysis is rarely reported for hydrogen production from waste plastics (Table 1). Based on the early work of Duan's group,146 ethylene glycol (EG) could be catalytically converted into formate through electrochemical oxidative cleavage of –C(OH)–C(OH)– bonds using a manganese-doped cobalt oxyhydroxide catalyst. More recently, Duan's group147 reported electrocatalytic upcycling of PET plastic to PTA and potassium diformate (KDF) coupled with H2 production in the presence of a bifunctional nickel-modified cobalt phosphide (CoNi0.25P) electrocatalyst in KOH electrolyte (Fig. 15(a)). A current density of 500 mA cm−2 at 1.8 V in a membrane-electrode assembly (MEA) reactor with >80% faradaic efficiency and selectivity to formate was achieved. Specifically, the electrocatalytic PET upcycling is mainly composed of PET hydrolysis (eqn (1)), electro-reforming of PET hydrolysate (eqn (2) and (3)), HER (eqn (4)), and product separation.| (PET)n + (2n − 1)H2O → nC8H6O4(PTA) + nC2H6O2(EG) | (1) |
| C2H6O2 + 2OH− → 2HCOO− + 3H2 | (2) |
| C2H6O2 + 8OH− → 2HCOO− + 6H2O + 6e− | (3) |
| 6H2O + 6e− → 3H2 + 6OH− | (4) |
| Catalyst | Plastic | Conditions | H2 yield (mmol gplastic−1) | Valued products | Ref. |
|---|---|---|---|---|---|
| Cu30Pd70 | PET | Cu30Pd70|perovskite|Pt PEC system, pretreated PET powder (1 M KOH), simulated solar light (AM 1.5G, 100 mW cm−2), 25 °C. | 22.1 | Glycolic acid | 143 |
| CuO NWs | PET | PV-EFR system, 0.1 M PET hydrolysate, cell voltage 1.9 V, simulated solar light (AM 1.5G, 100 mW cm−2), 25 °C. | 2.2 | Terephthalic acid, formic acid | 148 |
| Ni–Pi/α-Fe2O3 | PET | PEC system, 0.1 M PET hydrolysate, 300 W Xe lamp (AM 1.5 G, 100 mW cm−2), 25 °C. | 0.1 | Formate | 149 |
| Au/Ni(OH)2 | PET | 0.3 M EG (in 3 M KOH), 326.2 vs. 149.8 mA cm−2 at 1.15 V vs. RHE. | 6.0 | Glycolic acid | 150 |
| Ni3N/W5N4 | PET | PET lysate, 1 M KOH electrolyte, 1.33 V, 1 atm, 25 °C. | 0.5 | HCOOH | 151 |
| Co–Ni3N/CC | PET | 0.1 M EG, 1 M KOH electrolyte, 50 mA cm−2, 1.46 V | — | HCOOH | 152 |
| CoNi0.25P/NF | PET | 1 M KOH with EG, 500 mA cm−2, 1.8 V. | — | Terephthalate, formate | 147 |
| Pd–CuCo2O4 | PET | 1 M NaOH with EG, 600 mA cm−2, 1.15 V. | 0.2 | Glycolic acid | 153 |
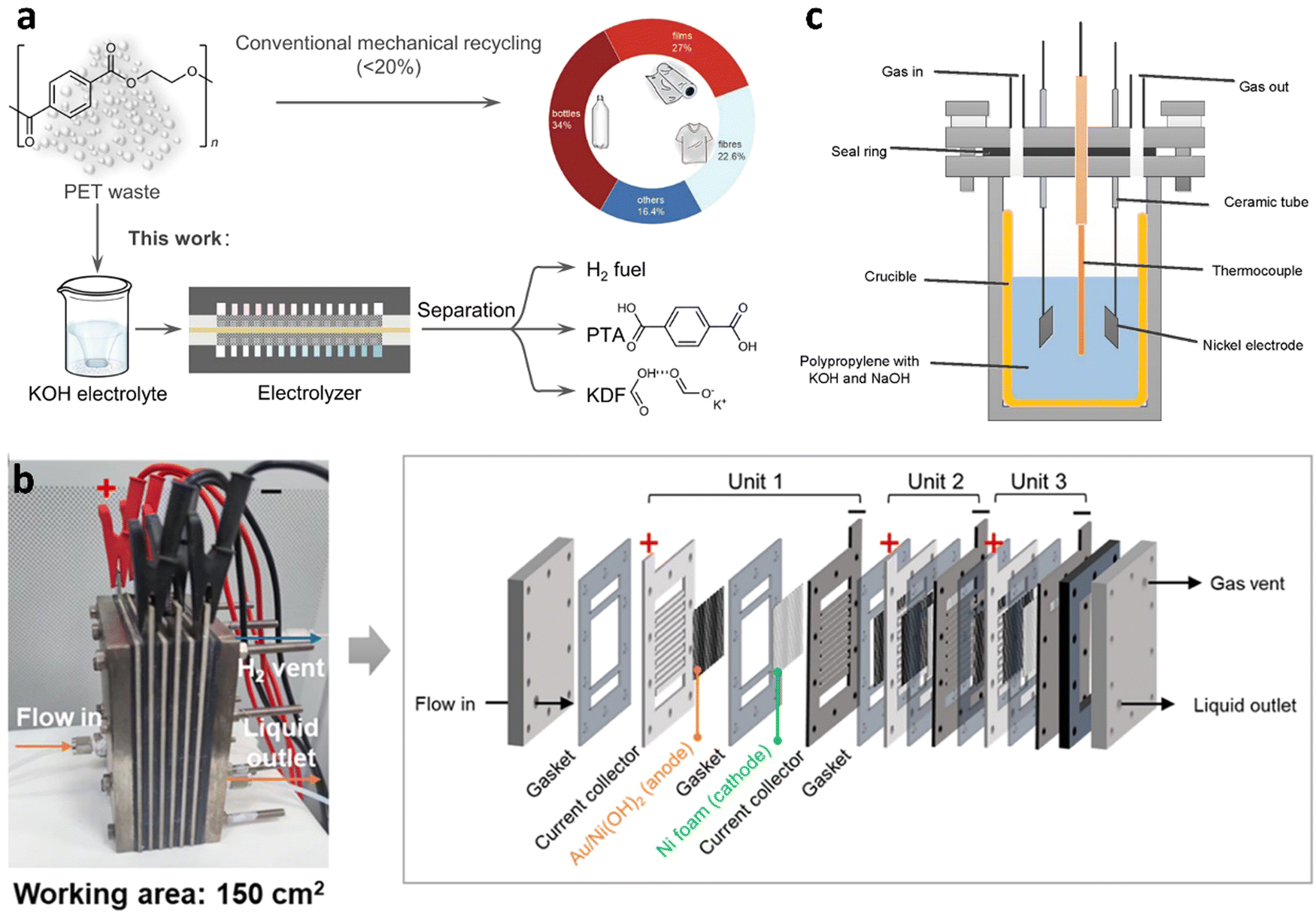 | ||
| Fig. 15 (a) Conventional and electrocatalytic routes for PET recycling to commodity chemicals and H2 fuel. Reproduced with permission.147 Copyright 2021, Springer Nature. (b) Photograph and schematic illustration of the stacked membrane-free flow electrolyzer. Reproduced with permission.150 Copyright 2023, American Chemical Society. (c) The solar thermo-coupled electrochemical set-up for indoor experiment. Reproduced with permission.154 Copyright 2020, Elsevier. | ||
The Raman spectroscopy and X-ray absorption near-edge structure (XANES) confirmed the presence of low-crystalline CoNi0.25Ox(OH)y species in the spent CoNi0.25P catalyst, which served as the real active site for electrocatalytic upcycling of PET. To evaluate the catalytic performance of electrooxidation of plastics in a more practical scenario, waste PET bottles and triglycerides in cooking oil were used for tests in a homemade membrane-free flow electrolyzer (Fig. 15(b)), using Au/Ni(OH)2 catalyst as the anode and Ni foam as the cathode.150 27.7 wt % yield of EG was firstly extracted from 70 g waste PET plastic bottles by 3 M KOH digestion. Then, EG was electrocatalytic upcycled into 13.7 g GA and 392.2 mmol of H2 (9.4 L) after a 23.3 h reaction. Jiang et al.154 designed a solar thermo-coupled electrochemical reactor (Fig. 15(c)) for plastics depolymerization to light fuel and hydrogen via solar-driven electrolysis coupled with pyrolysis. The mixture of the PP (2 g) and electrolyte (NaOH, 2.0 g and KOH, 2.8 g) was added into the reactor. The sunlight was irradiated to a solar heat concentrator and PV module to generate high-temperature heat and electricity. For the indoor experiment, the final volume of H2 was 72.5 ml, which was about 9.3 times higher than that produced by the pyrolysis at 400 °C. An outdoor experiment was conducted under optimal conditions (constant temperature and potential). The total conversion reached 66% at 350 °C, compared to 26% of the pyrolysis. Williams et al.155 developed another novel tandem thermo-electrochemical (elecATT) process for upcycling marine plastic wastes commingled with wet and salty seaweeds to high-purity H2 and high-value CNTs. The split-cell system consisted of the alkaline thermal treatment (ATT) and carbonate molten salt electroreduction. Plastic wastes were oxidized into carbonate salts of K and Li and valuable volatile gases. The produced gases were then further catalytically converted into H2 in the presence of Ni/ZrO2 catalyst. Subsequently, the molten carbonate salts (Li2CO3/K2CO3/LiOH) were continuously converted into carbon nanotubes via electrosplitting of carbonate ions with a nearly 100% Coulombic efficiency.
Direct seawater electrocatalysis presents a promising approach for large-scale green hydrogen production. However, high energy consumption and harmful chlorine corrosion largely impede its industrialization. Therefore, Liu et al.153 developed an energy-saving and chlorine-free H2 production system by coupling electrocatalytic seawater splitting and upcycling of PET waste into value-added glycolic acid (GA) over a Pd–CuCo2O4 synergistic catalyst, which exhibits high EG electrooxidation activity (600 mA cm−2 at 1.15 V vs. RHE), high FE (up to 96.1%) of value-added GA and 100 h stability at 1.6 A. Experimental and theoretical calculations reveal that the downshifted d-band center of Pd accelerates the desorption of GA to prevent over-oxidation. The strong adsorption of OH− on CuCo2O4 both enhanced EG electrooxidation and prevented chlorine corrosion by formating of a negative charge layer, which repels Cl− through the electrostatic repulsion effect.
In summary, waste plastics upcycling via electrocatalysis using renewable electricity with simultaneous production of value-added chemicals is a feasible technical route for hydrogen production coupled with value-added chemicals. The drawbacks of electrocatalysis for H2 generation from waste plastics are limitations to mostly oxygenated polymers, high energy consumption, the use of noble metals as catalysts, low efficiency, and scalability challenges.
3 Industrialization and economic feasibility
Compared with the traditional technologies of hydrogen production from fossil fuels by methane steam reforming/partial oxidation or water electrolysis, upcycling waste plastics presents a novel and promising approach for simultaneous waste management and sustainable hydrogen production. A growing number of government agencies, waste management, and energy companies worldwide are working to establish commercial-scale plastic-to-hydrogen plants. This “trash to treasure” technology, which transforms waste plastics into valuable hydrogen, is set for significant growth in the near future. Currently, there are approximately 17 industrial plastic-to-hydrogen projects either in the planning stages or under construction globally, mainly in Europe, the USA, Egypt, Korea, Japan, and China. Leading companies spearheading these efforts include Ways2H, SGH2 Energy, Waste2Tricity, Boson Energy, Powerhouse Energy, H2-Industries, and Dongfang Boiler (Table 2).| Country | Company | Technologies of plastic-to-hydrogen plants |
|---|---|---|
| UK | Waste2Tricity (W2T) | Early in 2020, a £1.25 m ($1.64 m) grant was invested in the first commercial plant to be built in northwest England with the partner PowerHouse Energy (PHE). It will be able to process 35 tons of plastic waste and produce up to 2 tons of H2 per day while generating 3.8 MW of electricity. |
| UK | Peel NRE | In 2022, Peel NRE received £20 m to build the second plastic-to-hydrogen plant in Rothesay Dock, which will produce 13500 tons of H2 for HGVs and buses. |
| Luxembourg | Boson Energy | Recently, Boson Energy and Siemens AG entered into an agreement to accelerate the green energy transition through waste-to-hydrogen (to-X) technology. The project aims to build over 300 plants and produce 1 million tons of H2 by 2030. |
| UK | Power House Energy | Powerhouse developed the first hydrogen generating plant at Peel Plastic Park using its proprietary Powerhouse DMG® technology. Forty tons of waste plastics could produce two tons of fuel cell-grade H2, which would generate 81 MWh of electric power. |
| Korea | SGH2 Energy | In 2022, SGH2 planned to build a plant in Ulsan by pyrolysis of recyclable waste papers using its SPEG technology. As estimated, 23–31 tons of carbon emission will be reduced per ton of H2. |
| USA | Ways2H | In 2021, Ways2H planned to build 40 plastic-to-hydrogen plants, each producing 0.5–1 tons of green H2 fuel per day. |
| Egypt | H2-Industries | H2-Industries announced plans to build a plastic-to-hydrogen plant in Egypt based on LOHC and CCUS technologies. The plant will process 4 million tons of plastics and produce 0.3 million tons of H2 per year. |
| China | Dongfang Boiler | In 2021, Dongfang Boiler signed a contract with the Chongqing government for waste power generation coupled with H2 generation, which is expected to be China's first demonstrative project of waste-to-hydrogen. |
| Korea | Hyundai Group | Recently, Hyundai Motor Group announced an “HTWO Grid” H2 solution. 2 factories of H2 generation from waste organics and plastics were built to produce over 30000 tons of H2 for fuel cell vehicles utilization. |
| Japan | JGC | In 2021, Japan Gasoline Company (JGC), Iwatani, and Toyota Tsusho announced a long-term hydrogen project based on EUP technology. They plan to construct a green waste-to-hydrogen plant until 2025. |
Early in 2019, the Waste2Tricity company was set up in northwestern England to convert unrecyclable plastics into hydrogen energy in a new UK clean energy hub. Millions of pounds were invested into the waste-to-power plant to destroy contaminated waste plastics and generate green hydrogen for vehicles and the national grid. The Powerhouse company also developed a hydrogen-generating plant using waste plastics based on its proprietary Powerhouse DMG® technology (Distributed Modular Power Generation)156 at the Peel Plastic Park in Protos, northwestern England, near Ellesmere Port. The Powerhouse's DMG® Technology converts the plastics from materials recycling facilities (MRFs) or plastic reprocessing plants to syngas, from which hydrogen is then extracted. Sustainably, a small portion of the syngas produced is used to run a thermal conversion chamber. The produced hydrogen is compressed on-site to be loaded onto tube trailers for onward transportation or piped to a nearby hydrogen fueling center. 40 tons of waste plastics could produce 2 tons of fuel cell grade hydrogen to generate 81 MWh of electric power integrated into the local grid. Based in Luxembourg, Boson Energy developed a plasma-assisted gasification process that converts waste plastics into hydrogen, CO2, and a molten slurry. This slurry can solidify into a glass-like material known as IMBY rock. The incomes from selling CO2 to industrial customers, stones to construction companies, and waste disposal fees paid by the local government sufficiently compensate for the cost of hydrogen production. They declared that 100 kg of carbon-negative hydrogen per ton of waste plastics can be realized, which needs 6 times less renewable electricity than electrolysis. Implementing this technology could transform 2 million tons of waste plastics disposed of in Europe yearly into 0.2 million tons of hydrogen, offering a cleaner and more sustainable solution. The SGH2 Energy company in Washington, DC, has planned to build a plant producing hydrogen by pyrolysis of recyclable waste papers using the innovative SPEG technology. The cost of hydrogen produced by this process is expected to be 5 to 7 times lower than that of other green hydrogen.
Over twenty years ago, Ebara and Ube Industries in Japan investigated a pressurized gasification technology for hydrogen production from waste plastics called the EUP process. In 2021, Japan Gasoline Company (JGC), Iwatani, and Toyota Tsusho announced a long-term hydrogen project based on the EUP technology. They plan to construct a green waste-to-hydrogen plant by 2025. The Chinese companies are also becoming the “forerunner” in the plastic-to-hydrogen technology. In 2021, Dongfang Boiler signed a contract with the Chongqing government about waste power generation coupled with hydrogen generation, which is expected to be the first demonstrative project of waste-to-hydrogen in China.
For the industrialization of waste plastics upcycling technologies, it is vital to systematically evaluate the economic feasibility of waste plastics to hydrogen pathway, which provides the reference for future improvement. Techno-economic analysis (TEA) is one of the most widely used tools to assess emerging technologies' economic and technical feasibility.157 Lan et al.158 initially conducted a TEA to evaluate the performance of gasifying waste plastics for hydrogen production and carbon capture and storage (CCS) based on the Aspen Plus simulation. In their work, the minimum hydrogen selling price (MHSP) was selected to assess the economic feasibility of the hydrogen plant by examining the impacts of varied steam/feedstock (S/F) ratio, feedstock costs, plant capacities, CCS adoption, and policy incentives. Apparently, the steam/feedstock ratio directly influences the hydrogen yield and further impacts the MHSP.
Feedstock cost is another important factor affecting economic feasibility. Lower-cost plastic feedstock and the large scale of plastic-to-hydrogen plants enhance the competitiveness of this process. An additional key factor impacting the cost of hydrogen derived from plastics is the availability of carbon credits provided by governments and regulatory agencies.
4 Conclusions and perspectives
In this review, multiple strategies such as pyrolysis, gasification, thermal reforming, photoreforming, aqueous phase reforming, and electrocatalytic reforming for hydrogen generation from waste plastics have been systematically explored in terms of reactor design, operating conditions, and catalyst development. In addition, potential industrialization and economic feasibility for the plastic-to-hydrogen route are discussed. The advantages and disadvantages of each technology are visually presented (Table 3). Pyrolysis and gasification are currently the most promising and scalable technologies for generating hydrogen from waste plastics. Both processes involve high-temperature treatment of plastics, with pyrolysis operating in the absence of oxygen and gasification utilizing principally steam as a gasifying agent. These methods are capable of processing mixed plastic waste and producing syngas (a mixture of hydrogen and carbon monoxide), which can be further converted and refined to extract hydrogen. However, they come with significant drawbacks, including high energy consumption and the production of carbon dioxide, which contributes to greenhouse gas emissions if not managed properly. Despite these challenges, their established infrastructure and ability to handle diverse feedstocks make them viable options for large-scale hydrogen production.| Parameter | Pyrolysis | Gasification | Photoreforming | Aqueous Phase reforming (APR) | Electrocatalytic reforming |
|---|---|---|---|---|---|
| Hydrogen yield from mixed plastics | Moderate to high <100 mmol g−1 | High <150 mmol g−1 | Low <10 mmol g−1 | Moderate <20 mmol g−1 | Low to moderate <20 mmol g−1 |
| Operating conditions | 400–700 °C, inert atmosphere | 800–1400 °C, gasifying agent (often steam) | Ambient temperature, sunlight | 110–250 °C, aqueous phase | Ambient to moderate temperature, electrochemical cell |
| Energy consumption | High | Very high | Low | Moderate | High |
| Catalyst | Optional | Optional | Required | Required | Required |
| Feedstock | Mixed plastics | Mixed plastics | Limited (e.g., PET) | Limited (oxygenated plastics) | Limited (oxygenated polymers) |
| Byproducts | Syngas, oil, char | Syngas, CO2 | CO2, organics | CO2, light hydrocarbons | CO2, organics |
| Scalability | High | High | Low | Moderate | Low |
| Technology maturity | Commercial | Commercial | Early research | Early research | Early research |
Aqueous phase reforming (APR) is another promising approach, particularly for oxygenated plastics. APR operates at moderate temperatures and pressures in an aqueous environment, using catalysts to convert plastic-derived compounds into hydrogen. While this method avoids the extreme conditions required by pyrolysis and gasification, it faces challenges related to catalyst costs, deactivation, and the need for pre-treatment, liquefaction of plastic feedstocks. Its applicability is limited to certain hydrophilic plastics, which reduces its versatility compared to other methods.
In contrast, photoreforming and electrocatalytic reforming represent emerging technologies that leverage renewable energy sources. Photoreforming utilizes sunlight and photocatalysts to break down plastics into hydrogen, offering a sustainable and low-energy approach. Similarly, electrocatalytic reforming employs electricity, ideally from renewable sources, to drive the electrochemical conversion of plastics into hydrogen. Both methods are environmentally friendly and operate under milder conditions compared to thermochemical processes. However, they are currently limited by low efficiency, slow reaction rates, and challenges in scaling up for industrial applications. Additionally, these technologies are often restricted to specific types of plastics, such as oxygenated polymers like polyethylene terephthalate (PET).
The choice of technology for hydrogen generation from waste plastics ultimately depends on several factors, including the type of plastic waste available, the desired hydrogen yield, the availability of energy resources, and economic considerations. Hydrogen productivity and selectivity, minimizing energy consumption, and ensuring stable operation of plastic recycling facilities are crucial parameters for achieving commercial viability. All plastic-to-hydrogen technologies should be subject to more detailed studies, challenges, and solutions before they are upscaled in the hydrogen market.
(1) Sorting and pretreatment of waste plastics should be explored. In contrast to the purified single plastics (PE, PP, etc.) used in the lab, “real-world” waste plastics generally represent more complex mixtures. Different types of plastics are mixed with other impurities such as chlorine, which would produce complex products and even cause the corrosion of the equipment and the poisoning of catalysts. Even in a plastic bottle, plasticizers, stabilizers, coatings, and other chemicals exist. So, sink float sorting, electrostatic sorting, and laser sorting should be applied before further plastic upcycling by suitable strategies. Some wet plastics with high moisture content need to be dried. Plastics with high stability can be pretreated by chemical or physical methods in advance to obtain corresponding monomers, which can be easily transferred into hydrogen and value-added chemicals.
(2) The large-scale upcycling reactors and systems based on carbon fixation and energy balance should be further optimized. Notably, the thermal pyrolysis of plastics is often accompanied by significant CO2 emissions. Therefore, efficient CCUS facilities should be coupled with the pyrolysis systems to accelerate “carbon neutrality.” The carbon char can be collected and processed for reuse in various applications, such as a soil amendment to enhance fertility (similar to biochar), a raw material for manufacturing activated carbon for filtration systems, or even as a precursor for producing carbon-based materials like graphene or carbon nanotubes. The quality of solid carbon products like CNTs and graphene for carbon fixation is expected to be improved via the catalyst designing process. Carbon dioxide has numerous applications: carbonating beverages, preserving food, enhancing oil extraction, producing construction materials, generating CO2-derived e-fuels, while also boosting plant growth in greenhouses. Carbon monoxide is a key feedstock in chemical synthesis (e.g., syngas for methanol, Fischer–Tropsch synthesis, acetic acid) and steel production, as well as in pharmaceuticals. Thus, the circular carbon economy in waste plastics should be optimized by using a suitable upcycling strategy. Thus, the circular carbon economy in waste plastics should be optimized by using a suitable upcycling strategy. To lower the cost of hydrogen production from plastics and enhance energy efficiency, the waste heat generated by certain exothermic reactions can be harnessed as an alternative energy source for upcycling plastics. Based on the properties of specific plastics, different strategies could be integrated for stepwise upcycling to obtain high H2 production efficiency.
(3) Developing efficient catalysts with low cost, high stability, and selectivity is necessary. Precise cleavage of C–C and C–H bonds during plastics pyrolysis by the active sites of catalysts plays a key role in hydrogen production with high selectivity. The catalyst with a high photothermal effect could be designed to achieve enough surface temperature for the catalytic pyrolysis process to improve the activity of photothermal catalytic pyrolysis. Moreover, catalysts with high stability are also required. During the thermal decomposition process, carbon deposition occurs easily on the catalyst's surface to block the active sites for further reaction. Also, the active metal sites are apt to be poisoned by S or Cl elements in the plastics, which largely decreases the catalyst's hydrogen production rate and lifetime.
(4) The reaction mechanism of hydrogen production from plastics upcycling is still unclear. Different advanced in situ characterization techniques integrated with computational calculations are vital for revealing the in-depth mechanism of how the H2 and other value-added products are formed. The comprehensive investigations of mechanism would further contribute to the development of reaction systems and optimization of catalyst design.
(5) The construction of plastic-to-hydrogen plants is still in the initial stage. More governmental policies and financial support are needed to establish enough plastic-to-hydrogen plants for real commercialization. Moreover, the solar energy-assisted decomposition of plastics coupled with hydrogen storage technology and fuel cell system provides a sustainable and novel pathway to produce electricity for community and household use. As Moritz Kuehnel from Swansea University said,159 “ultimately, maybe people could treat their own plastic waste in their gardens, similarly to compost, with a solar waste-reforming device. You put your plastic waste in it and get hydrogen to heat your house or fuel your car.”
(6) Hydrogen production from waste plastics utilizing solar energy is currently at a very early stage of development. The present H2 productivity from PR of plastics is generally in the μmol scale with low solar-to-hydrogen (STH) efficiency, far from scaling up and commercialization. The H2 generated by PR of plastics often mixes with oxidated organics, CO2, CH4, and other hydrocarbons and requires costly purification.
The potential of plastic waste as a long-term source for hydrogen production presents a dual narrative, one of immediate utility and another of future adaptability. Currently, the sheer volume of mismanaged plastic waste poses a significant environmental burden. Technologies like gasification and pyrolysis offer a promising avenue to mitigate this problem by converting this waste, including mixed and difficult-to-recycle plastics, into valuable hydrogen. Considering the technology maturity, on short notice, plastic waste pyrolysis is most likely to be implemented for hydrogen production.
However, the long-term viability hinges on the dynamic and evolving landscape of plastics, legislation and sustainability. The global push towards a more sustainable plastics future, characterized by increased adoption of bioplastics, enhanced mechanical recycling, and a general reduction in plastic consumption, could significantly alter the availability of traditional fossil-derived plastic waste. Consequently, for plastic-to-hydrogen to remain a sustainable solution, it must demonstrate adaptability. This entails developing processes capable of utilizing diverse feedstocks, including bioplastics, and seamlessly integrating with advanced recycling systems to process the residual, non-recyclable fractions.
Crucially, a truly sustainable approach necessitates integrating plastic-to-hydrogen within a broader circular economy framework. This involves prioritizing waste reduction and mechanical recycling, reserving plastic-to-hydrogen for the residual, non-recyclable waste, and ensuring the hydrogen production process itself adheres to stringent environmental standards. Therefore, while plastic waste offers a valuable resource for hydrogen production, particularly in addressing the current waste crisis, its long-term viability is contingent upon its ability to evolve alongside the changing plastics industry. To secure its place as a sustainable component of the future hydrogen economy, the technology must embrace adaptability, integrating diverse feedstocks and aligning with circular economy principles. New legislations are being implemented and they will play a crucial role on how the future of plastics recycling and use. Extensive collaboration of research institutions, universities, governments, and industries is hoped to accelerate advancements in hydrogen production from waste plastics, drive breakthroughs in efficiency, and pave the way for future commercialization.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the E-Cracker ERC Advanced Grant, OPTIMA ERC Consolidator Grant, FWVL Interreg OBIWAN project, National Natural Science Foundation of China (no. 22308343) and Zhejiang Provincial Natural Science Foundation of China (no. LQ24B030003).References
- K. Chaudhary, K. Bhardvaj and A. Chaudhary, Fuel, 2024, 358, 130090 CrossRef CAS.
- S. Q. Zhou, L. Shang, Y. X. Zhao, R. Shi, G. I. N. Waterhouse, Y. C. Huang, L. R. Zheng and T. R. Zhang, Adv. Mater., 2019, 31, 1900509 Search PubMed.
- J. J. Zhao, Z. K. Tu and S. H. Chan, Int. J. Hydrogen Energy, 2024, 78, 721–730 Search PubMed.
- D. Tang, G. L. Tan, G. W. Li, J. G. Liang, S. M. Ahmad, A. Bahadur, M. Humayun, H. Ullah, A. Khan and M. Bououdina, J. Energy Storage, 2023, 64, 107196 CrossRef.
- H. Barokh and M. Siavashi, Int. J. Hydrogen Energy, 2024, 83, 1294–1308 CrossRef CAS.
- H. Tüysüz, Acc. Chem. Res., 2024, 57, 558–567 Search PubMed.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 Search PubMed.
- D. Niblett, M. Delpisheh, S. Ramakrishnan and M. Mamlouk, J. Power Sources, 2024, 592, 233904 CrossRef CAS.
- X. P. Tao, Y. Zhao, S. Y. Wang, C. Li and R. G. Li, Chem. Soc. Rev., 2022, 51, 10120–10122 RSC.
- Y. G. Yuan, L. N. Zhou, H. Robatjazi, J. L. Bao, J. Y. Zhou, A. Bayles, L. Yuan, M. H. Lou, M. H. Lou, S. Khatiwada, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2022, 378, 889–893 CrossRef CAS PubMed.
- P. N. Ren, Z. Y. Gao, T. Montini, Z. T. Zhao, N. Ta, Y. K. Huang, N. C. Luo, E. Fonda, P. Fornasiero and F. Wang, Joule, 2023, 7, 1–17 CrossRef.
- Global Plastics Outlook: Policy Scenarios to 2060, https://www.oecd-ilibrary.org/.
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. M. Van Steenberge, D. R. D'hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. Lavender Law, Science, 2015, 347, 768 CrossRef CAS PubMed.
- Y. M. Peng, P. P. Wu, A. T. Schartup and Y. X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2111530118 Search PubMed.
- H. T. Pinheiro, C. MacDonald, R. G. Santos, R. Ali, A. Bobat, B. J. Cresswell, R. Francini-Filho, R. Freitas, G. F. Galbraith, P. Musembi, T. A. Phelps, J. P. Quimbayo, T. E. A. L. Quiros, B. Shepherd, P. V. Stefanoudis, S. Talma, J. B. Teixeira, L. C. Woodall and L. A. Rocha, Nature, 2023, 619, 311–316 CrossRef CAS PubMed.
- L. Magnier, R. Mugge and J. Schoormans, J. Cleaner Prod., 2019, 215, 84 CrossRef.
- N. Yan, Science, 2022, 378, 132–133 CrossRef CAS PubMed.
- J. Sills, Science, 2022, 378, 841–842 Search PubMed.
- S. Bhattacharjee, S. Linley and E. Reisner, Nat. Rev. Chem., 2024, 8, 87–105 CrossRef PubMed.
- Z. Y. Cen, X. Han, L. F. Lin, S. H. Yang, W. Y. Han, W. L. Wen, W. L. Yuan, M. H. Dong, Z. Y. Ma, F. Li, Y. B. Ke, J. C. Dong, J. Zhang, S. H. Liu, J. L. Li, Q. Li, N. N. Wu, J. F. Xiang, H. Wu, L. L. Cai, Y. B. Hou, Y. Q. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, P. Ferrer, D. C. Grinter, G. Held, Y. M. Liu and B. X. Han, Nat. Chem., 2024, 16, 871–880 CrossRef CAS PubMed.
- F. Zhang, M. H. Zeng, R. D. Yappert, J. K. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- Z. Xu, N. E. Munyaneza, Q. K. Zhang, M. Q. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. L. Liu, Science, 2023, 381, 666–671 Search PubMed.
- S. Yue, Z. Y. Zhao, T. Zhang, F. Li, K. W. Liu and S. H. Zhan, Angew. Chem., Int. Ed., 2024, 63, e202406795 CrossRef CAS PubMed.
- H. M. Dai and X. J. Gao, J. Cleaner Prod., 2024, 452, 142100 CrossRef CAS.
- W. C. Ma, C. Chu, P. Wang, Z. F. Guo, S. J. Lei, L. Zhong and G. Y. Chen, Adv. Sustainable Syst., 2020, 4, 2000026 Search PubMed.
- M. Lahafdoozian, H. Khoshkroudmansouri, S. H. Zein and A. A. Jalil, Int. J. Hydrogen Energy, 2024, 59, 465–479 CrossRef CAS.
- J. K. Lin, K. S. Hu, Y. T. Wang, W. J. Tian, T. Hall, X. G. Duan, H. Q. Sun, H. Y. Zhang, E. Cortés and S. B. Wang, Nat. Commun., 2024, 15, 8769 Search PubMed.
- Z. J. Chen, W. Wei, X. M. Chen, Y. W. Liu, Y. S. Shen and B. J. Ni, Renewable Sustainable Energy Rev., 2024, 195, 114333 CrossRef CAS.
- K. Zheng, Y. Wu, Z. X. Hu, S. M. Wang, X. C. Jiao, J. C. Zhu, Y. F. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- M. Ashraf, N. Ullah, I. Khan, W. Tremel, S. Ahmad and M. N. Tahir, Chem. Rev., 2023, 123, 4443–4509 CrossRef CAS PubMed.
- L. Santamaria, G. Lopez, E. Fernandez, M. Cortazar, A. Arregi, M. Olazar and J. Bilbao, Energy Fuels, 2021, 35, 17051–17084 CrossRef CAS PubMed.
- C. W. Xing, H. T. Cai, D. X. Kang and W. Sun, Adv. Energy Sustainability Res., 2023, 4, 2300015 CrossRef CAS.
- H. H. Shah, M. Amin, A. Iqbal, I. Nadeem, M. Kalin, A. M. Soomar and A. M. Galal, Front. Chem., 2023, 10, 960894 CrossRef PubMed.
- N. Cai, X. Q. Li, S. W. Xia, L. Sun, J. H. Hu, P. Bartocci, F. Fantozzi, P. T. Williams, H. P. Yang and H. P. Chen, Energy Convers. Manage., 2021, 229, 113794 CrossRef CAS.
- D. D. Yao and C. H. Wang, Appl. Energy, 2020, 265, 114819 CrossRef CAS.
- R. Alshareef, M. A. Nahil and P. T. Williams, Energy Fuels, 2023, 37, 3894–3907 CrossRef CAS PubMed.
- S. X. Wang, Y. Y. Zhang, R. Shan, J. Gu, T. L. Huhe, X. Ling, H. R. Yuan and Y. Chen, J. Cleaner Prod., 2022, 352, 131566 CrossRef CAS.
- Y. C. Jiang, X. L. Li, C. Li, L. J. Zhang, S. Zhang, B. Li, S. Wang and X. Hu, Renewable Energy, 2022, 200, 476–491 CrossRef CAS.
- D. D. Yao, H. Li, Y. J. Dai and C. H. Wang, Chem. Eng. J., 2021, 408, 127268 CrossRef CAS.
- R. X. Yang, S. L. Wu, K. H. Chuang and M. Y. Wey, Renewable Energy, 2020, 159, 10–22 Search PubMed.
- W. Nabgan, B. Nabgan, T. A. T. Abdullah, H. Alqaraghuli, N. Ngadi, A. A. Jalil, B. M. Othman, A. M. Ibrahim and T. J. Siang, Int. J. Hydrogen Energy, 2020, 45, 22817–22832 CrossRef CAS.
- D. D. Yao, Y. S. Zhang, P. T. Williams, H. P. Yang and H. P. Chen, Appl. Catal., B, 2018, 221, 584–597 Search PubMed.
- D. D. Yao, H. P. Yang, H. P. Chen and P. T. Williams, Appl. Catal., B, 2018, 239, 565–577 CrossRef CAS.
- I. Barbarias, G. Lopez, J. Alvarez, M. Artetxe, A. Arregi, J. Bilbao and M. Olazar, Chem. Eng. J., 2016, 296, 191–198 CrossRef CAS.
- C. F. Wu, M. A. Nahil, N. Miskolczi, J. Huang and P. T. Williams, Environ. Sci. Technol., 2014, 48, 819–826 CrossRef CAS PubMed.
- M. Cortazar, N. Gao, C. Quan, M. A. Suarez, G. Lopez, S. Orozco, L. Santamaria, M. Amutio and M. Olazar, Fuel Process. Technol., 2022, 225, 107044 CrossRef CAS.
- M. A. Suarez, K. Januszewicz, M. Cortazar, G. Lopez, L. Santamaria, M. Olazar, M. Artetxe and M. Amutio, Energy, 2024, 302, 131762 CrossRef CAS.
- G. Lopez, I. Garcia, A. Arregi, L. Santamaria, M. Amutio, M. Artetxe, J. Bilbao and M. Olazar, Energy Convers. Manage., 2020, 214, 112889 CrossRef CAS.
- J. C. Acomb, C. F. Wu and P. T. Williams, Appl. Catal., B, 2016, 180, 497–510 CrossRef CAS.
- S. J. Li, Y. Xue, Y. X. Lin, B. Wang and X. Gao, ACS Sustainable Chem. Eng., 2023, 11, 10108–10118 CrossRef CAS.
- Q. Y. Liu, D. Y. Jiang, H. Zhou, X. Z. Yuan, C. F. Wu, C. S. Hu, R. Luque, S. R. Wang, S. Chu, R. Xiao and H. Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305078120 CrossRef CAS PubMed.
- W. Kaminsky, Fuel Commun., 2021, 8, 100023 CrossRef.
- B. Goshayeshi, R. Kumar, Y. Wang, R. J. Varghese, S. Roy, B. Baruah, A. A. Lemonidou and K. M. Van Geem, J. Anal. Appl. Pyrolysis, 2025, 187, 107016 CrossRef CAS.
- S. Czernik and R. J. French, Energy Fuels, 2006, 20, 754–758 CrossRef CAS.
- J. M. Saad and P. T. Williams, Fuel Process. Technol., 2017, 156, 331–338 CrossRef CAS.
- K. M. Wyss, K. J. Silva, K. V. Bets, W. A. Algozeeb, C. Kittrell, C. H. Teng, C. H. Choi, W. Y. Chen, J. L. Beckham, B. I. Yakobson and J. M. Tour, Adv. Mater., 2023, 35, 2306763 CrossRef CAS PubMed.
- H. Luo, D. D. Yao, K. Zeng, J. Li, S. Yan, D. Zhong, J. H. Hu, H. P. Yang and H. P. Chen, Fuel Process. Technol., 2022, 230, 107205 CrossRef CAS.
- H. P. Yang, S. Yan, D. D. Yao, H. Luo, K. Zeng, J. Li, D. Zhong, H. Y. Xiao and H. P. Chen, Fuel, 2023, 333, 126496 CrossRef CAS.
- K. Stanley, S. Kelly and J. A. Sullivan, Appl. Catal., B, 2023, 328, 122533 CrossRef CAS.
- A. Bajpai, S. Mehta, K. Joshi and S. Kumar, Int. J. Hydrogen Energy, 2023, 48, 24328–24341 CrossRef CAS.
- S. Baig and B. Sajjadi, J. Energy Chem., 2024, 97, 265–301 CrossRef CAS.
- I. Aminu, M. A. Nahil and P. T. Williams, Catal. Today, 2023, 420, 114084 CrossRef CAS.
- H. Y. Xiao, J. Harding, S. S. Lei, W. Chen, S. W. Xia, N. Cai, X. Chen, J. H. Hu, Y. Q. Chen, X. H. Wang, X. Tu, H. P. Yang and H. P. Chen, J. Cleaner Prod., 2022, 350, 131467 CrossRef CAS.
- X. Y. Jie, W. S. Li, D. Slocombe, Y. G. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Z. Yao, H. AlMegren, S. Alshihri, J. Dilworth, J. Thomas, T. C. Xiao and P. Edwards, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- G. Lopez and L. Santamaria, Nat. Catal., 2020, 3, 861–862 CrossRef CAS.
- B. W. Zhang, H. Wang, Y. Y. Yang, Y. P. Zhou, B. Zhang and K. M. Huang, J. Environ. Chem. Eng., 2023, 11, 109710 CrossRef CAS.
- W. T. Li, K. Z. Qian, Z. X. Yang, X. X. Ding, W. M. Tian and D. Z. Chen, Appl. Catal., B, 2023, 327, 122451 CrossRef CAS.
- P. E. Ganza and B. J. Lee, Int. J. Hydrogen Energy, 2023, 48, 15037–15052 CrossRef CAS.
- L. S. Yao, B. K. Yi, X. Q. Zhao, W. L. Wang, Y. P. Mao, J. Sun and Z. L. Song, J. Anal. Appl. Pyrolysis, 2022, 165, 105577 CrossRef CAS.
- Q. Cao, H. C. Dai, J. H. He, C. L. Wang, C. Zhou, X. F. Cheng and J. M. Lu, Appl. Catal., B, 2022, 318, 121828 CrossRef CAS.
- S. Lee, S.-K. Yoo, J. Lee and J.-W. Park, J. Mater. Cycles Waste Manage., 2009, 11, 191–196 CrossRef CAS.
- S. Z. Li, I. C. Vela, M. Järvinen and M. Seemann, Waste Manage., 2021, 130, 117–126 CrossRef CAS PubMed.
- M. A. Bashir, Y. Soong, T. Ji, M. Gray, J. Weidman, F. Shi and P. Wang, Energy Adv., 2025, 4, 330–363, 10.1039/D4YA00292J.
- S. W. Han, D. Tokmurzin, J. J. Lee, S. J. Park, H. W. Ra, S. J. Yoon, T.-Y. Mun, S. M. Yoon, J. H. Moon, J. G. Lee, Y.-M. Kim, Y. W. Rhee and M. W. Seo, Fuel, 2022, 314, 123102 Search PubMed.
- G. Lopez, M. Artetxe, M. Amutio, J. Alvarez, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2018, 82, 576–596 CrossRef CAS.
- F. Parrillo, F. Ardolino, G. Calì, A. Pettinau, M. Materazzi, A. Sebastiani and U. Arena, Waste Manage., 2024, 183, 53–62 CrossRef CAS PubMed.
- A. Gharibi, R. Babazadeh and R. Hasanzadeh, Process Saf. Environ. Prot., 2024, 183, 46–58 CrossRef CAS.
- R. Hasanzadeh, M. Mojaver, T. Azdast and C. B. Park, Chem. Eng. J., 2022, 430, 132958 CrossRef CAS.
- M. Mojaver, R. Hasanzadeh, T. Azdast and C. B. Park, Chemosphere, 2022, 286, 131867 CrossRef CAS PubMed.
- B. Bai, J. L. Sun, Y. J. Wang, X. Y. Yu, W. H. Zhou and H. Jin, Chem. Eng. J., 2024, 502, 157847 CrossRef CAS.
- X. Lu, L. Cao, H. K. Wang, W. Peng, J. Xing, S. X. Wang, S. Y. Cai, B. Sheng, Q. Yang, C. P. Nielsen and M. B. McElroy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8206–8213 CrossRef CAS PubMed.
- A. Hasanoğlu, E. Faki, A. Seçer and Ş. T. Üzden, Fuel, 2023, 331, 125693 CrossRef.
- G. Ruoppolo, P. Ammendola, R. Chirone and F. Miccio, Waste Manage., 2012, 32, 724–732 CrossRef CAS PubMed.
- F. Pinto, C. Franco, R. N. André, M. Miranda, I. Gulyurtlu and I. Cabrita, Fuel, 2002, 81, 291–297 Search PubMed.
- S. Madanikashani, L. A. Vandewalle, S. De Meester, J. De Wilde and K. M. Van Geem, Materials, 2022, 15, 4215 CrossRef CAS PubMed.
- M. P. Aznar, M. A. Caballero, J. A. Sancho and E. Francés, Fuel Process. Technol., 2006, 87, 409–420 Search PubMed.
- R. Xiao, B. S. Jin, H. C. Zhou, Z. P. Zhong and M. Y. Zhang, Energy Convers. Manage., 2007, 48, 778–786 CrossRef CAS.
- U. Arena, L. Zaccariello and M. L. Mastellone, Waste Manage., 2009, 29, 783–791 Search PubMed.
- J. M. Toledo, M. P. Aznar and J. A. Sancho, Ind. Eng. Chem. Res., 2011, 50, 11815–11821 CrossRef CAS.
- M. L. Mastellone, L. Zaccariello, D. Santoro and U. Arena, Waste Manage., 2012, 32, 733–742 Search PubMed.
- M. H. Cho, T. Y. Mun and J. S. Kim, Energy, 2013, 58, 688–694 CrossRef CAS.
- S. Martínez-Lera, J. Torrico, J. Pallarés and A. Gil, Waste Manage., 2013, 33, 1640–1647 CrossRef PubMed.
- V. Wilk and H. Hofbauer, Fuel, 2013, 107, 787–799 CrossRef CAS.
- I. P. Lazzarotto, S. D. Ferreira, J. Junges, G. R. Bassanesi, C. Manera, D. Perondi and M. Godinho, Process Saf. Environ. Prot., 2020, 140, 60–67 CrossRef CAS.
- Y. S. Jeong, J. W. Kim, H. W. Ra, M. W. Seo, T. Y. Mun and J. S. Kim, ACS Sustainable Chem. Eng., 2022, 10, 4705–4716 CrossRef CAS.
- A. Erkiaga, G. Lopez, M. Amutio, J. Bilbao and M. Olazar, Fuel, 2013, 109, 461–469 CrossRef CAS.
- M. Arabiourrutia, G. Elordi, M. Olazar and J. Bilbao, Pyrolysis of Polyolefins in a Conical Spouted Bed Reactor: A Way to Obtain Valuable Products, 2017, ch. 12, p. 289 Search PubMed.
- D. T. Pio and L. A. C. Tarelho, Renewable Sustainable Energy Rev., 2021, 145, 111108 CrossRef CAS.
- A. Ponzio, S. Kalisz and W. Blasiak, Fuel Process. Technol., 2006, 87, 223–233 CrossRef CAS.
- M. Y. He, B. Xiao, Z. Q. Hu, S. M. Liu, X. J. Guo and S. Y. Luo, Int. J. Hydrogen Energy, 2009, 34, 1342–1348 Search PubMed.
- H. A. Baloch, T. H. Yang, R. D. Li, S. Nizamuddin, X. P. Kai and A. W. Bhutto, Clean Technol. Environ. Policy, 2016, 18, 1031–1042 CrossRef CAS.
- X. Y. Guo, Z. J. Song and W. Zhang, Waste Manag. Res., 2020, 38, 802–811 CrossRef CAS PubMed.
- H. Sekiguchi and T. Orimo, Thin Solid Films, 2004, 457, 44–47 CrossRef CAS.
- M. Hlina, M. Hrabovsky, T. Kavka and M. Konrad, Waste Manage, 2014, 34, 63–66 CrossRef CAS PubMed.
- R. Mallick and P. Vairakannu, J. Environ. Manage., 2023, 345, 118655 CrossRef CAS PubMed.
- E. Falascino, R. K. Joshi, S. Kumar, T. Jawdekar, I. K. Kudva, S. G. Shinde, Z. Cheng, A. Tong and L.-S. Fan, Appl. Energy Combust. Sci., 2024, 19, 100270 Search PubMed.
- B. J. Liu, C. F. You and H. M. Wang, Chem. Eng. J., 2024, 501, 157728 Search PubMed.
- B. L. Dou, K. Q. Wang, B. Jiang, Y. C. Song, C. Zhang, H. S. Chen and Y. J. Xu, Int. J. Hydrogen Energy, 2016, 41, 3803–3810 CrossRef CAS.
- A. R. Galaly, G. V. Oost and N. Dawood, ACS Omega, 2024, 9, 21174–21186 Search PubMed.
- J. Zhao, D. Xie, S. Z. Wang, R. Zhang, Z. Q. Wu, H. Y. Meng, L. Chen, T. Wang and Y. Guo, Int. J. Hydrogen Energy, 2021, 46, 18051–18063 CrossRef CAS.
- H. C. Su, Y. J. Hu, H. Y. Feng, L. J. Zhu and S. R. Wang, ACS Sustainable Chem. Eng., 2023, 11, 578–586 CrossRef CAS.
- A. Kumar, M. K. Awasthi, N. Sheet, T. A. Kharde and S. K. Singh, ChemCatChem, 2023, 15, e202300574 CrossRef CAS.
- H. Nishiyama, T. Yamada and M. Nakabayashi, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. J. Ma, Y. X. Xiao, P. Wang, Z. W. Ye, B. W. Zhou, K. Sun and Z. T. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- K. H. Chen, J. D. Xiao, J. J. M. Vequizo, T. Hisatomi, Y. W. Ma, M. Nakabayashi, T. Takata, A. Yamakata, N. Shibata and K. Domen, J. Am. Chem. Soc., 2023, 145, 3839–3843 CrossRef CAS PubMed.
- Y. J. Xiao, Y. Qi, X. L. Wang, X. Y. Wang, F. X. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- Y. W. Zhu, L. L. Wang, Y. T. Liu, L. H. Shao and X. N. Xia, Appl. Catal., B, 2019, 241, 483–490 CrossRef CAS.
- C. X. Zhu, J. Wang, J. B. Lv, Y. Q. Zhu, Q. X. Huang and C. Sun, Int. J. Hydrogen Energy, 2024, 51, 91–103 CrossRef CAS.
- R. Li, F. L. Wang, F. Lv, P. F. Wang, X. Guo, J. Feng, D. Li and Y. B. Chen, Int. J. Hydrogen Energy, 2024, 51, 406–414 CrossRef CAS.
- Y. Q. Zheng, P. Fan, R. J. Guo, X. H. Liu, X. T. Zhou, C. Xue and H. B. Ji, RSC Adv., 2023, 13, 12663–12669 Search PubMed.
- S. Zhang, H. B. Li, L. Wang, J. D. Liu, G. J. Liang, K. Davey, J. R. Ran and S. Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS PubMed.
- E. M. N. T. Edirisooriya, P. S. Senanayake, P. Xu and H. Y. Wang, J. Environ. Chem. Eng., 2023, 11, 111429 CrossRef.
- W. B. Qu, X. Y. Qi, G. X. Peng, M. C. Wang, L. X. Song, P. F. Du and J. Xiong, J. Mater. Chem. C, 2023, 11, 14359–14370 RSC.
- T. K. A. Nguyen, T. Trân-Phú, X. M. C. Ta, T. N. Truong, J. Leverett, R. Daiyan, R. Amal and A. Tricoli, Small Methods, 2023, 8, 2300427 Search PubMed.
- C. X. Liu, R. Shi, W. J. Ma, F. L. Liu and Y. Chen, Inorg. Chem. Front., 2023, 10, 4562–4568 RSC.
- Y. T. Jiang, J. J. Yu, H. Y. Zhang, L. F. Hong, J. J. Shao, B. W. Zhang, J. J. Yu and S. Chu, ChemSusChem, 2023, 16, e202300106 CrossRef CAS PubMed.
- E. M. N. T. Edirisooriya, P. S. Senanayake, H. B. Wang, M. R. Talipov, P. Xu and H. Y. Wang, J. Environ. Chem. Eng., 2023, 11, 109580 CrossRef CAS.
- M. M. Du, M. Y. Xing, W. F. Yuan, L. Zhang, T. Sun, T. Sheng, C. Y. Zhou and B. C. Qiu, Green Chem., 2023, 25, 9818–9825 RSC.
- J. Q. Xu, X. C. Jiao, K. Zheng, W. W. Shao, S. Zhu, X. D. Li, J. F. Zhu, Y. Pan, Y. F. Sun and Y. Xie, Natl. Sci. Rev., 2022, 9, nwac011 CrossRef CAS PubMed.
- J. Q. Yan, D. W. Sun and J. H. Huang, Chemosphere, 2022, 286, 131905 CrossRef CAS PubMed.
- J. B. Qin, Y. B. Dou, F. Y. Wu, Y. C. Yao, H. R. Andersen, C. Hélix-Nielsen, S. Y. Lim and W. J. Zhang, Appl. Catal., B, 2022, 319, 121940 CrossRef CAS.
- X. Q. Gong, F. X. Tong, F. H. Ma, Y. J. Zhang, P. Zhou, Z. Y. Wang, Y. Y. Liu, P. Wang, H. F. Cheng, Y. Dai, Z. K. Zheng and B. B. Huang, Appl. Catal., B, 2022, 307, 121143 CrossRef CAS.
- M. M. Du, Y. Zhang, S. L. Kang, X. Y. Guo, Y. X. Ma, M. Y. Xing, Y. Zhu, Y. Chai and B. C. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- B. Q. Cao, S. P. Wan, Y. N. Wang, H. W. Guo, M. Ou and Q. Zhong, J. Colloid Interface Sci., 2022, 605, 311–319 CrossRef CAS PubMed.
- H. Nagakawa and M. Nagata, ACS Appl. Mater. Interfaces, 2021, 13, 47511–47519 CrossRef CAS PubMed.
- S. Bhattacharjee, C. Z. Guo, E. Lam, J. M. Holstein, M. R. Pereira, C. M. Pichler, C. Pornrungroj, M. Rahaman, T. Uekert, F. Hollfelder and E. Reisner, J. Am. Chem. Soc., 2023, 145, 20355–20364 CrossRef CAS PubMed.
- T. Uekert, M. A. Bajada, T. Schubert, C. M. Pichler and E. Reisner, ChemSusChem, 2021, 14, 4190–4197 CrossRef CAS PubMed.
- Y. Q. Li, S. P. Wan, C. Lin, Y. J. Gao, Y. Lu, L. Y. Wang and K. Zhang, Sol. RRL, 2021, 5, 2000427 CrossRef CAS.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Kawai and T. Sakata, Chem. Lett., 1981, 81–84 CrossRef CAS.
- S. Bhattacharjee, V. Andrei, C. Pornrungroj, M. Rahaman, C. M. Pichler and E. Reisner, Adv. Funct. Mater., 2022, 32, 2109313 CrossRef CAS.
- H. T. N. Hai, T. T. Nguyen, M. Nishibori, T. Ishihara and K. Edalati, Appl. Catal., B, 2025, 365, 124968 CrossRef CAS.
- M. Li and S. B. Zhang, ACS Catal., 2024, 14, 2949–2958 CrossRef CAS.
- H. Zhou, Z. H. Li, S. M. Xu, L. L. Lu, M. Xu, K. Y. Ji, R. X. Ge, Y. F. Yan, L. Ma, X. G. Kong, L. R. Zheng and H. H. Duan, Angew. Chem., Int. Ed., 2021, 60, 8976–8982 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. H. Li, M. Xu, Y. Wang, R. X. Ge, X. G. Kong, L. R. Zheng and H. H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- T. Zhang, X. Li, J. Y. Wang, Y. F. Miao, T. F. Wang, X. F. Qian and Y. X. Zhao, J. Hazard. Mater., 2023, 450, 131054 CrossRef CAS PubMed.
- B. W. Zhang, H. Y. Zhang, Y. Y. Pan, J. J. Shao, X. T. Wang, Y. T. Jiang, X. Y. Xu and S. Chu, Chem. Eng. J., 2023, 462, 142247 CrossRef CAS.
- Y. F. Yan, H. Zhou, S. M. Xu, J. R. Yang, P. J. Hao, X. Cai, Y. Ren, M. Xu, X. G. Kong, M. F. Shao, Z. H. Li and H. H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- F. H. Ma, S. H. Wang, X. Q. Gong, X. L. Liu, Z. Y. Wang, P. Wang, Y. Y. Liu, H. F. Cheng, Y. Dai, Z. K. Zheng and B. B. Huang, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- X. Liu, Z. Y. Fang, D. K. Xiong, S. Q. Gong, Y. L. Niu, W. Chen and Z. F. Chen, Nano Res., 2023, 16, 4625–4633 CrossRef CAS.
- K. S. Liu, X. T. Gao, C.-X. Liu, R. Shi, E. C. M. Tse, F. L. Liu and Y. Chen, Adv. Energy Mater., 2024, 14, 2304065 CrossRef CAS.
- T. T. Jiang, X. L. Zhao, D. Gu, C. Yan, H. Jiang, H. J. Wu, B. H. Wang and X. R. Wang, Sol. Energy Mater. Sol. Cells, 2020, 204, 110208 CrossRef CAS.
- J. M. Williams, M. P. Nitzsche, L. Bromberg, Z. F. Qu, A. J. Moment, T. Alan Hatton and A. A. Park, Energy Environ. Sci., 2023, 16, 5805–5821 RSC.
- https://www.powerhouseenergy.co.uk/projects/plastics-to-hydrogen/ .
- R. W. Makepeace, A. Tabandeh, M. J. Hossain and M. Asaduz-Zaman, Int. J. Hydrogen Energy, 2024, 22, 1183–1192 CrossRef.
- K. Lan and Y. Yao, Commun. Earth Environ., 2022, 3, 300 CrossRef.
- https://www.chemistryworld.com/news/sunlight-converts-plastic-waste-to-hydrogen-fuel/3009467.article .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00604f |
| This journal is © The Royal Society of Chemistry 2025 |