
Rational design of bifunctional OER/ORR metal-free catalysts based on boron-doped graphene by strain engineering†
Jun
Zhou
a,
Yujia
Cheng
a,
Yuping
Ren
a,
Mo
Xiong
b,
Haozhen
Dou
c,
Yi
Jiang
c,
Luyuan
Wang
*a,
Chuangwei
Liu
 *ac and
Wenjun
Tang
*d
*ac and
Wenjun
Tang
*d
aSchool of Materials Science and Engineering, Northeastern University, Shenyang 110819, China. E-mail: wangluyuan20@gmail.com
bMOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, School of Physics, Xi’an Jiaotong University, Xi’an 710049, China
cDalian Institute of Chemical Physics, China Academy of Sciences, Dalian, 116023, China. E-mail: cwliu@dicp.ac.cn
dSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 610054, China. E-mail: wenjuntang9711@gmail.com
First published on 15th May 2025
Abstract
Designing high-activity and durable bifunctional catalysts for the oxygen reduction reaction and oxygen evolution reaction is crucial for advancing fuel cells. However, the development of electrocatalysts is usually based on transition metal materials in previous literature. Herein, we explore a group of single boron atoms doped on graphene with different N coordinations as promising bifunctional OER/ORR metal-free catalysts by density functional theory calculations. By constructing a volcano-shaped relationship between the OER/ORR activity and both ΔE*O and Bader charge (δ), the B–C2N structure is identified as a superior catalyst, with an overpotential of 0.38 V and 0.36 V for the OER and ORR, respectively. Although the thermodynamic barrier of the ORR is lower than that of the ORR in the same catalyst, the reaction rate of the ORR is only one-hundred-and-fiftieth that of the OER. Moreover, the catalytic activity is further enhanced in the presence of tensile strain, which can be attributed to the shift of the p-band center and charge accumulation.
Introduction
Extensive efforts to develop renewable energy technologies such as photovoltaics and wind turbines are driven by the ever-increasing detrimental effects of traditional fossil fuel energy on the environment.1,2 As a result, to handle the intermittent nature of renewable electricity, large-scale energy storage systems including fuel cells and electrolyzers must be developed as they offer much higher theoretical specific energies than state-of-the-art Li-ion batteries.3,4 The central reactions in these types of energy storage systems are the electrocatalytic oxygen reduction reaction (ORR) and oxygen evolution reaction (OER).5–7 As such, the key to achieving high efficiency lies in developing bifunctional electrocatalysts that can promote ORR and OER activity.8,9 Although precious metals, such as Pt/C, Ir/C, and RuO2, have been used as electrocatalysts, their high cost and poor bifunctionality hamper the commercialization of energy conversion and storage technologies.10,11 Tremendous efforts have been devoted to developing cost-effective alternatives with high electrocatalytic activity and stability.12,13 Over the years, various transition-metal-based (Fe, Co, Ni, etc.) sulfides, selenides, nitrides, carbides, phosphides, borides, oxides, hydro(oxy)oxides, and their alloys or complexes have been employed to drive fuel cells; however, corrosion of the metal is a major obstacle to electrolysis.14–16Carbon nanomaterials (e.g., carbon nanotubes and graphene) doped with heteroatoms offer unique advantages for intended catalysis due to their abundance and tunable properties.17,18 The accessible and tunable pore size with a high surface area of these carbon-based materials enables rapid diffusion of the reactants to active sites.19,20 Moreover, the introduction of suitable heteroatoms with different electronegativities into carbon materials can alter their behavior toward higher ORR and OER activity.21,22 The variation in the electrocatalytic activity of heteroatom-doped carbon nanomaterials is believed to be due to the different electronegativities of these doped elements, which perturb the π-orbital electrons in their graphitic domains.21,23 The different types of dopant elements in carbon nanomaterials result from varied sizes and electronegativities.24 Then, a series of single-atom electrocatalysts (SACs) with outstanding catalytic performance and durable stability, especially Pt, Fe, and Co, were investigated and applied to the ORR and OER.22,25–27 However, the poor selectivity and detrimental environmental impacts of the above SACs hindered their promising industrial application in fuel cells.28,29 A p-block element, boron atom (B), has been applied to various electrochemical reactions under ambient conditions, including the NRR,17,30 OER,31–33 CRR,34,35 HER,36 and ORR.37,38 We identified that the boron-based material has a similar catalytic activity to transition metal-based catalysts (TMs), but its activation mechanism is different from that of TMs in our previous works.17,34,36 A π-back bonding is formed during the catalytic process due to the B atom, an electron-deficient element, containing an empty orbital.17,39 In addition, we reported that the coordination environment, especially the different N species, can tune the inert small molecule activation ability of the catalytic site.40 The nitrogen coordination plays a pivotal role in tuning the electronic properties of boron by donating electron density. Particularly, the lower degree of nitrogen coordination may enhance the electron-donating ability of boron by limiting charge delocalization and constructing an electron-rich coordination site, which facilitates the adsorption and activation of reaction intermediates on the boron site.41
Herein, twelve promising structures were built by doping a single B atom onto 2D graphene with different N coordinations (B–Nx–C), involving pyridinic and pyrrolic nitrogen (denoted as “role”). For example, B–C1N1N2role indicates that one B atom is concerted by one C atom, one pyridinic N atom, and two pyrrolic N atoms. The OER and ORR catalytic performance on these potential models was evaluated by density functional theory (DFT) calculations. By comparing the thermodynamic barriers and scaling relationship results, a single boron atom with one pyridinic N and two carbon atoms (B–C2N) was selected as the best bifunctional catalyst in this work. Interestingly, the catalytic activity of pristine B–C2N was tuned by strain engineering, for which the overpotential of the promising catalyst was only 0.35 eV under 1% tensile strain. This exploration not only provides several descriptors for predicting the catalytic activity of metal-free catalysts but also offers valuable strategies for designing a new generation of materials with improved performance.
Results and discussion
Initially, the thermodynamic stability of the twelve potential SAC structures was investigated through the difference between the adsorption energy (Eads) of a single boron atom on the 2D N-doped graphene and the cohesive energy (Ecoh) of the boron atom, and the Ecoh is 5.81 eV.42,43 The optimized structure and adsorption energies are presented in Fig. 1 and Fig. S1 (ESI†), respectively. The B atom is coordinated with three atoms in the single vacancy (SV) of the structure, whereas the coordination environment of double-vacancy (DV) presents a more complicated state than the SV structure in Fig. 1. The results further indicate that the adsorption ability of the coordination atoms follows the order: pyrrolic nitrogen > carbon > pyridinic N atom. In other words, the pyrrolic nitrogen provides the strongest adsorption site for the B atom. The Bader charge transferred to the boron atom from the substrate further supports the above conclusion, as shown in Fig. S2 (ESI†). As we know, the difference between Eads and Ecoh is an important criterion for describing the thermodynamic stability of potential structures. Fig. S1 (ESI†) shows that the Eads values of all structures are larger than −6 eV, indicating a negative value difference. Then we can state that all the above models can be easily synthesized by experimental works. Indeed, Zettl identified that the experimental reaction temperature has a significant influence on the number and type of N-coordinated species.44 Therefore, the potential catalytic activities of the twelve promising structures are thoroughly investigated in the following section.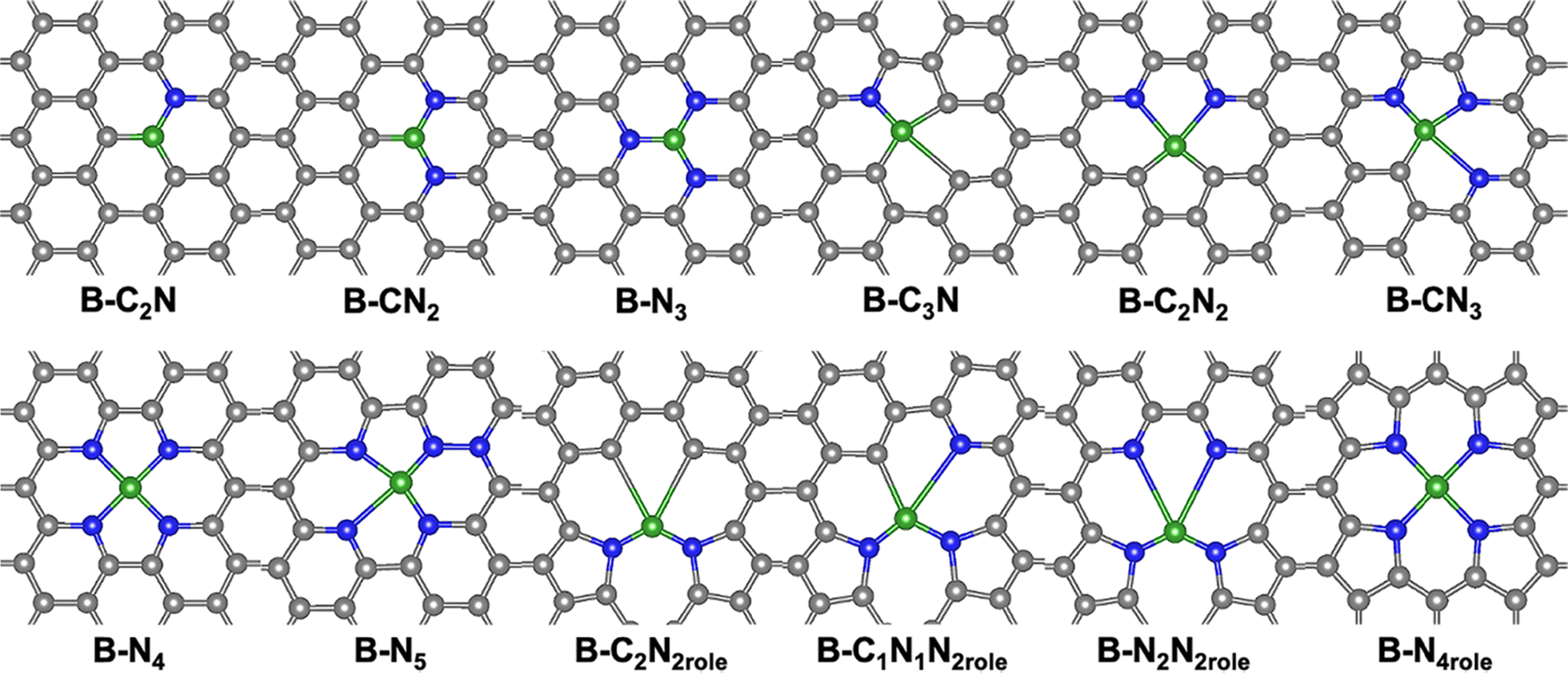 | ||
| Fig. 1 Optimized models of single B-doped 2D graphene with different nitrogen species. Black, blue, and green represent C, N, and B, respectively. | ||
During the oxygen evolution reaction (OER) process, activation of H2O on the 12 promising models was initially considered in this work. The optimized structures reveal that H2O is activated by the catalysts only via physical adsorption as shown in Fig. S3 (ESI†). Compared with a free water molecule, the bond angle of H2O is stretched from 104.50° to 105.58° to 107.32° after the adsorption process, indicating that H2O has been clearly activated in the initial step. Regarding the ORR procedure, the substrates also adsorb the oxygen molecule by the end-on and side-on models, as shown in Fig. S4 (ESI†). The substrate exhibits heavy distortion under the side-on configurations. The O–O bond lengths in this configuration range from 1.25 to 1.30 Å, which are significantly larger than the experimental length (1.21 Å), indicating that the substrates form strong interactions with *O2. From the adsorbed structure, it can be confirmed that the B atom is the main catalytic site for small molecule capture by chemisorption, rather than C. To further analyze the catalytic properties in the first steps, the charge density difference and Bader charge of two small molecules are presented in Fig. S5 and S6 (ESI†). The bond length change of H2O and O2 shows the same trend as the Bader charge vibration obtained by small molecules from B–Nx–C substrates. Interestingly, the activation degree of adsorbed species strongly depends on the electrons transferred from the catalytic site, and the results follow the order of C2N2role (2.40 |e|) > C3N (1.39 |e|) > CN3 (1.38 |e|) > N3 (1.31 |e|) in the O2 activation process. In other words, the B atom, having a lower degree of N-coordination and a relatively larger Bader charge value (Fig. S2, ESI†), provides more electrons to the species. The magnitude of adsorption energy also exhibits a powerful linear correlation with the N-coordination environment: the value increases with the decreasing number of N-coordinations in a similar substrate. Then the charge density difference of the two molecules is also shown in Fig. S5 and S6 (ESI†). The adsorbed species receive several electrons from the substrate, and the main catalytic site, the B atom, provides the most electrons during the activation step. Moreover, the charge density accumulation on O–B bonds and evident electron transfer around the B atom display a strong interaction between the B atom and the adsorbed species on B–Nx–C substrates. Compared to the TM-based catalysts, the relatively small charge density accumulation indicates that only the B-to-O π-back bonding is formed during the activation process due to the sp3 hybrid orbital of the boron atom. From the above results and analyses, it can be found that, with increasing N-coordination of the B atom in a similar substrate, the number of transferred electrons decreases during the catalytic process.
To systematically study and screen the promising bifunctional OER/ORR catalysts based on boron-doped graphene catalysts, two reactions with four consecutive elementary electronic steps were calculated, and the reaction intermediates and energy profiles on these surfaces are summarized in Fig. S7–S18 (ESI†). The maximum consumed energy (ΔGmax) step of each reaction was chosen as the criterion to evaluate the catalytic activity, and the step was named the thermodynamic rate-determining step (RDS) during the two processes. Nørskov et al. found that the adsorption energies of *OOH (ΔE*OOH) and *O (ΔE*O) have a linear scaling relationship with the energy of *OH (ΔE*OH).45 For example, the linear relationship between the Gibbs free energies of *OOH and *OH is ΔG*OOH = ΔG*OH + 3.2 ± 0.2 eV by the revised Perdew–Burke–Ernzerhof exchange–correlation functional on metal-based materials.46,47 Then the catalyst activity can be simply described by the adsorption free energies, and the scaling relationships between (ΔE*OOH), (ΔE*O), and (ΔE*OH) on the 12 potential models were established as shown in Fig. 2. As shown in Fig. 2a, the scaling relationship between ΔE*O and ΔE*OH is ΔE*OOH = 1.71ΔE*OH + 1.17 eV with an R2 value of 0.91 for all B–Nx–C substrates. The slope value (1.71) indicates that *OH and *O each have only one bond to the substrate surface, and the *O species is more stable than the *OH. In addition, the formal linear scaling relationship between ΔE*OOH and ΔE*OH is described by the equation: ΔE*OOH = 0.54ΔE*OH + 2.93 eV with an R2 value of 0.68 as shown in Fig. 2b. The intercept value (2.93 eV) has slightly different from that of metal-based materials (3.2 eV), as reported in Nørskov's work.46,48 This can be attributed to the PBE functional and the corrections of zero-point energies and entropy were considered in our work. The weaker correlation (0.68) can be attributed to the wrinkled 2D substrate when the *OOH adsorbed on the electron-deficient B site. A similar scaling relationship exists between the *O and *OOH in Fig. S19 (ESI†). These slopes further demonstrate that two adsorbed species have a single bond between the B atom, the main catalytic site, and the O atom. Moreover, the three constant intercepts indicate that the three species usually prefer a similar type of binding site.
 | ||
| Fig. 2 Scaling relationships of intermediate adsorption energies for (a) *O versus *OH and (b) *OOH versus *OH on the twelve potential structures. Activity volcano plots of ηOER (c) and ηORR (d) versus the free energies of *O (ΔE*O). Volcano plots of ηOER (e) and ηORR (f) as a function of the Bader charge (δ, in units of e) of a single boron atom. | ||
From the above linear scaling relationships between intermediates and the Sabatier principle, the adsorption energies can be used to predict the OER/ORR overpotential (η). Herein, several OER/ORR volcano models with ηOER and ηORR on the y-axis as a function of a series of descriptors, including ΔE*O, ΔE*OH, ΔE*OOH, and ΔE*O–ΔE*OOH, were established, which helps quickly identify the relationship between the catalytic performance and adsorption strength of intermediates. Then, eight volcano-shaped plots were constructed by using the as-obtained scaling relationships, as shown in Fig. 2 and Fig. S20–S22 (ESI†). These plots are intimately connected to the free energy variations for each reaction step in the whole ORR and OER. Comparison of the volcano plots versus the four descriptors, ΔE*O, ΔE*OH, ΔE*OOH, and ΔE*O–ΔE*OH, reveals a relatively excellent linear relationship on both sides of the volcano. Concerning the OER, the peak position of the volcano plots is located at ΔGmax = 1.61 eV (ηOER = 0.38 eV), corresponding to the adsorption energies of ΔE*O, ΔE*OH, ΔE*OOH, and ΔE*O–ΔE*OH, which are 2.15, 0.70, 3.64, and 1.45 eV, respectively. In simple terms, B–C2N displays a remarkable OER performance with ηOER = 0.38 eV, which is close to the overpotential of IrO2 (0.33 eV).49 In addition, the OER activity deteriorates when the adsorption energy values change. Interestingly, the catalytic performance correlates with the ΔE*O on both sides of the OER volcano plots. Under this condition, the stronger ΔE*O will increase the difficulty of *O to form the *OOH intermediate. Similar to the OER analysis, the B–C2N still provides the best ORR activity with ηORR = 0.36 eV, which is better than that of the benchmark catalyst (Pt(111), 0.43 eV).50 The ORR activity has a relatively linear scaling relationship with ΔE*O–ΔE*OH on the ascending side of Fig. S22b (ESI†), while ΔE*O displays a good linear relationship on the descending branch in Fig. 2d. In other words, the adsorption strength increases with a decrease of ΔE*O, but the strong adsorption will poison the catalytic site by blocking the formation of second water. Therefore, the promising adsorption energy of intermediates is very important for the design of bifunctional OER/ORR catalysts. From the above as-obtained scaling relationships and the volcano plots, we also need to understand the relationship between the boron chemical environment and bifunctional OER/ORR activity. As shown in Fig. S2, S5 and S300 (ESI†), the Bader charge (δ) is highly correlated with the intermediate adsorption. The ηOER and ηORRversus the δ values are summarized in Fig. 2e and f. The volcano plots display that the OER and ORR are activated when δ equals 0.51 |e|. The plot shows that optimal δ leads to a minimal energy requirement for the ORR and OER. As B → O2 electron transfer plays a key role in O2 adsorption and activation, more electrons in the boron site would increase the possibility for such electron injection and subsequent reduction. Combining the above analyses, the B-doped C2N substrate is the best bifunctional OER/ORR catalyst.
To further investigate the OER/ORR intrinsic activity on B-doped C2N, the full thermodynamic energy profiles of four electron transfer steps with calculated transition states (TSs) are summarized in Fig. 3a and b. For the OER process, the H2O is adsorbed on the B atom site by physisorption (Fig. S5, ESI†), and the slightly low Bader charge value also confirms the result. Two large energy fluctuations appear in this process, and the third evolution reaction (*O → *OOH, 0.38 eV) is the RDS based on thermodynamic performance. Interestingly, the maximum kinetic barrier (0.93 eV) also emerges in the same step. More importantly, the reaction rate depends on the energetic span, which emerges from the difference between the turnover frequency (TOF, s−1 site−1)-determining intermediate (TDI) and TOF-determining transition state (TDTS).51,52 According to the energetic span model, the TOF is calculated by 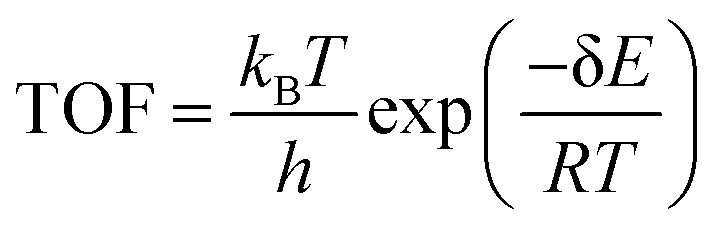 , where δE = GTDTS – GTDS. Essentially, TOF = 2.28 × 10−5 s−1 site−1 (1.04 eV) in the OER process, where the TDI and TDTS are *O and TS4, respectively. Fig. 3b shows that *O and TS2 are identified as the TDI and TDTS in the ORR process, respectively, and the TOF is equal to 1.50 × 10−7 s−1 site−1 (1.18 eV). Although the OER has a relatively larger kinetic barrier (0.93 eV, TS3), it exhibits a higher reaction rate under ambient conditions. In other words, the OER exhibits a narrower catalytic cycle than the ORR in this work. The intrinsic electronic properties of the B atom on all B–Nx–C catalysts were determined to investigate the different catalytic performance by using the projected density of states (PDOS), as shown in Fig. S23 (ESI†). Moreover, the PDOS of *O, *OH, and *OOH was calculated to investigate the electronic behavior of boron for intermediates (Fig. S24, ESI†). The antibonding π*-orbital of the O atom is well matched with the energy levels of the boron p-orbitals of the B atom, benefiting the occupation of the hybridized p–π*-orbitals for adsorption and activation of all small molecules. In catalyzing the following reactions, the σ2p state appears near the Fermi level, indicating that the adsorbed species can be further activated by the catalytic site. The AIMD and phonon spectrum calculations were applied to evaluate the stability of the B–C2N structure. Fig. 3c reveals that the geometric structure has slight distortion under 300 K, offering good structural stability. Moreover, no obvious vibration in the two plots further confirms that the B–C2N structure holds great stability. Afterward, density functional perturbation theory-based phonon calculations were employed to validate the stability of the B–C2N catalyst due to phonon methods that can predict variations in lattice thermal conductivity and identify the phonon scattering phenomenon. The absence of imaginary phonon frequencies in the profile shown in Fig. 3d further supports the excellent thermodynamic stability.
, where δE = GTDTS – GTDS. Essentially, TOF = 2.28 × 10−5 s−1 site−1 (1.04 eV) in the OER process, where the TDI and TDTS are *O and TS4, respectively. Fig. 3b shows that *O and TS2 are identified as the TDI and TDTS in the ORR process, respectively, and the TOF is equal to 1.50 × 10−7 s−1 site−1 (1.18 eV). Although the OER has a relatively larger kinetic barrier (0.93 eV, TS3), it exhibits a higher reaction rate under ambient conditions. In other words, the OER exhibits a narrower catalytic cycle than the ORR in this work. The intrinsic electronic properties of the B atom on all B–Nx–C catalysts were determined to investigate the different catalytic performance by using the projected density of states (PDOS), as shown in Fig. S23 (ESI†). Moreover, the PDOS of *O, *OH, and *OOH was calculated to investigate the electronic behavior of boron for intermediates (Fig. S24, ESI†). The antibonding π*-orbital of the O atom is well matched with the energy levels of the boron p-orbitals of the B atom, benefiting the occupation of the hybridized p–π*-orbitals for adsorption and activation of all small molecules. In catalyzing the following reactions, the σ2p state appears near the Fermi level, indicating that the adsorbed species can be further activated by the catalytic site. The AIMD and phonon spectrum calculations were applied to evaluate the stability of the B–C2N structure. Fig. 3c reveals that the geometric structure has slight distortion under 300 K, offering good structural stability. Moreover, no obvious vibration in the two plots further confirms that the B–C2N structure holds great stability. Afterward, density functional perturbation theory-based phonon calculations were employed to validate the stability of the B–C2N catalyst due to phonon methods that can predict variations in lattice thermal conductivity and identify the phonon scattering phenomenon. The absence of imaginary phonon frequencies in the profile shown in Fig. 3d further supports the excellent thermodynamic stability.
 | ||
| Fig. 3 OER (a) and ORR (b) free energy plots of the B-doped C2N substrate with four transition states. (c) Fluctuation of temperature and energy versus time (10 ps) in AIMD simulations. (d) Phonon dispersion spectra of the B-doped C2N potential structure. | ||
It is known that strain engineering can improve the catalytic activity by modulating the adsorption energy of intermediates because it can influence the electron distribution of the catalytic site.34,43,53 The bifunctional OER/ORR activity of B–C2N with biaxial strains from −5% to 5%, and the negative and positive values imply compressive and tensile strains, respectively. As shown in Fig. 4a and b, the adsorption energy of all intermediates decreases with increasing compressive strain. In other words, the adsorption capacity of the substrate is enhanced by compressive strain engineering, and the three adsorption energies of the three intermediates still maintain a good linear scaling relationship with applied tensile strain. Moreover, the bonding strength between *O and *OH and the B atom of B–C2N is weakened under tensile strain, decreasing the energy input to form the *OOH intermediate in the OER and further accelerating the desorption of H2O from the surface in the ORR. Interestingly, the balance is achieved under 1% tensile strain, offering the best bifunctional activity with both ηOER and ηORR values being 0.35 eV. In terms of electronic structures, compressive strain leads to stronger out-of-plane (π) states, which can not only strengthen OH adsorption, but also promote B-to-O π-back bonding, promoting subsequent electron transfer. Consequently, two contour plots of OER and ORR overpotentials as a function of ΔE*OH and ΔE*O, which are closely connected to the Gibbs free energy variations for each elementary reaction step in the OER and ORR, are shown in Fig. 4c and d. It is important to note that the two fitted lines pass through the triangular red-colored region, and excitingly several points are located in this region. In other words, the B–Nx–C structures are a group of potential two-dimensional materials with promising bifunctional OER/ORR catalytic activity. Furthermore, B–C2N with comprehensive strain is a group of potential electrocatalysts with excellent bifunctional OER/ORR activity.
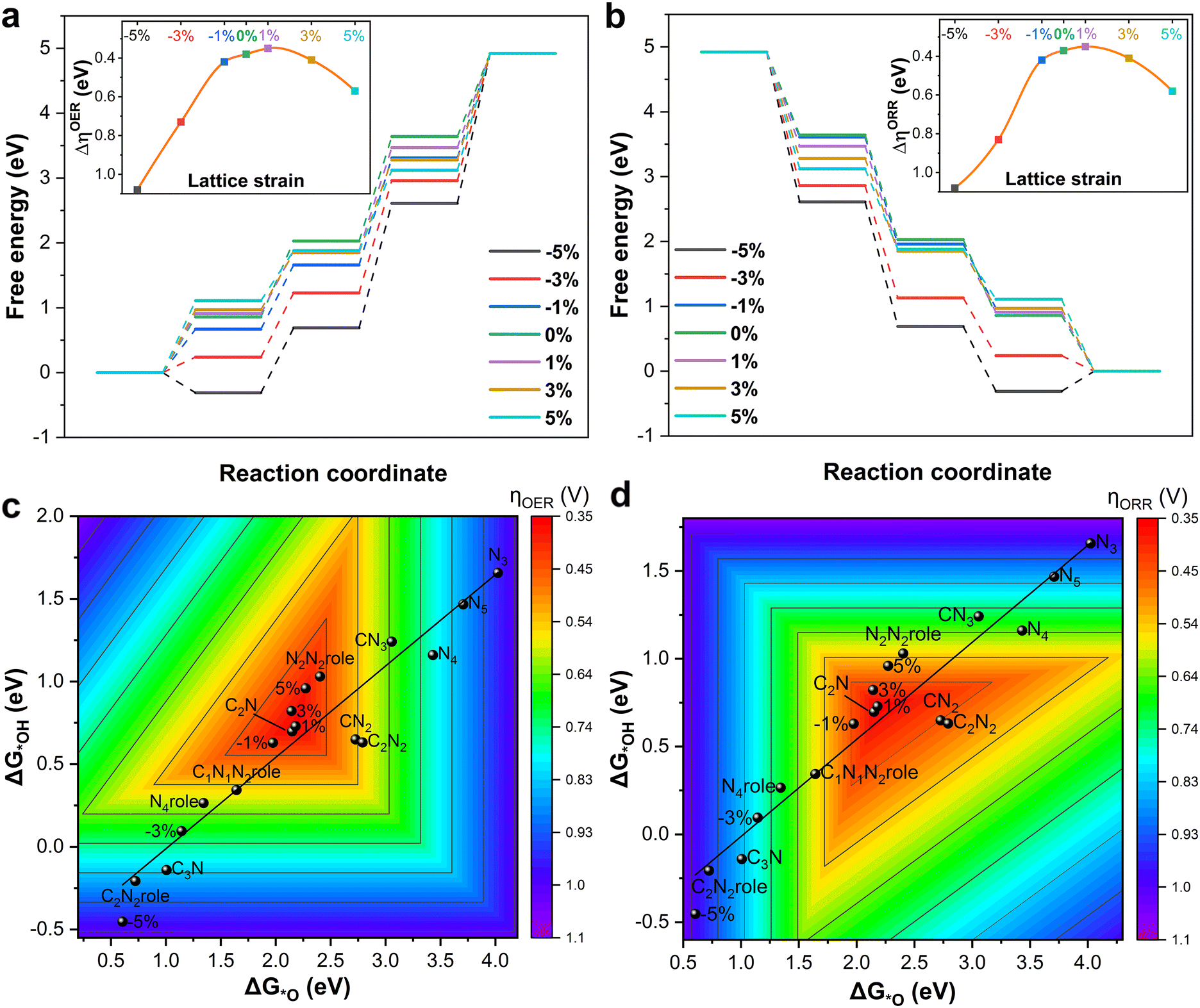 | ||
| Fig. 4 Gibbs free energy plot of OER (a) and ORR (b) performance on the B-doped C2N structure with different strains. The inset images are the corresponding ηORR and ηORRversus strain curves. Contour images of ηOER (c) and ηORR (d) as a function of ΔE*OH and ΔE*O constructed by the linear scaling relationship of B–Nx–C structures. | ||
Conclusions
In summary, the bifunctional OER/ORR catalytic activity of a single boron atom doped on graphene with different N coordinations was systematically investigated by density functional theory calculations in this work. Two outstanding catalysts, B–C2N and B–N2N2role, were screened using several descriptors, including ΔE*O, ΔE*OH, ΔE*O–ΔE*OH, and δ. Particularly, the B–C2N structure offers outstanding catalytic activity, and the overpotentials are 0.38 V and 0.36 V for the OER and ORR, respectively, and the promising catalyst exhibits remarkable stability under experimental conditions. Interestingly, the maximum TOF values of 2.28 × 10−5 s−1 site−1 and 1.50 × 10−7 s−1 site−1 were achieved for the OER and ORR, respectively. Although the thermodynamic barrier of the ORR is lower than that of the OER in the same catalyst, the kinetic reaction rate of the ORR is much slower than that of the OER. Moreover, the OER/ORR activity is further tuned by the application of strain, as the balance of intermediate adsorption energy is achieved by regulating the electronic accumulation at the boron site. The results indicate that the catalytic activity reaches its peak at 1% tensile strain, and the performance decreases gradually with further increase of tensile or compressive strain.Data availability
The data supporting this article have been included as part of the ESI.† And all conventional cells are from Materials Project (https://nextgen.materialsproject.org/).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate the financial support provided by the National Natural Science Foundation of China (grant no. 22309180 and 22473108) and the Foundation of National Key Technologies R&D Program of China (grant no. 2023YFB3810601).References
- C. Liu, Q. Wang, J. Guo, T. Vegge, P. Chen and H. A. Hansen, ACS Catal., 2022, 12, 4194–4202 CrossRef CAS.
- H. Guo, Z. Guo, K. Chu, W. Zong, H. Zhu, L. Zhang, C. Liu, T. Liu, J. Hofkens and F. Lai, Adv. Funct. Mater., 2023, 33, 2308229 CrossRef CAS.
- J. Li, J. Fleetwood, W. B. Hawley and W. Kays, Chem. Rev., 2022, 122, 903–956 Search PubMed.
- Z. Zeng, X. Shen, R. Cheng, O. Perez, N. Ouyang, Z. Fan, P. Lemoine, B. Raveau, E. Guilmeau and Y. Chen, Nat. Commun., 2024, 15, 3007 Search PubMed.
- D. Kan, D. Wang, Y. Cheng, R. Lian, B. Sun, K. Chen, W. Huo, Y. Wang, G. Chen and Y. Wei, ACS Appl. Mater. Interfaces, 2021, 13, 52508–52518 CrossRef CAS PubMed.
- B. Tang, Y. Zhou, Q. Ji, Z. Zhuang, L. Zhang, C. Wang, H. Hu, H. Wang, B. Mei, F. Song, S. Yang, B. M. Weckhuysen, H. Tan, D. Wang and W. Yan, Nat. Synth., 2024, 3, 878–890 CrossRef CAS.
- Y.-P. Deng, Y. Jiang, R. Liang, N. Chen, W. Chen, Z.-W. Yin, G. King, D. Su, X. Wang and Z. Chen, J. Am. Chem. Soc., 2023, 145, 20248–20260 Search PubMed.
- Y. Jiang, Y.-P. Deng, R. Liang, J. Fu, D. Luo, G. Liu, J. Li, Z. Zhang, Y. Hu and Z. Chen, Adv. Energy Mater., 2019, 9, 1900911 CrossRef.
- Y. Jiang, Y.-P. Deng, R. Liang, N. Chen, G. King, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 4783–4791 Search PubMed.
- K. J. Sawant, Z. Zeng and J. P. Greeley, Angew. Chem., Int. Ed., 2024, 63, e202312747 Search PubMed.
- X. Ping, Y. Liu, L. Zheng, Y. Song, L. Guo, S. Chen and Z. Wei, Nat. Commun., 2024, 15, 2501 Search PubMed.
- D. Kan, D. Wang, X. Zhang, R. Lian, J. Xu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 3097–3108 Search PubMed.
- D. Kan, R. Lian, D. Wang, X. Zhang, J. Xu, X. Gao, Y. Yu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 17065–17077 Search PubMed.
- Y. Jiang, Y.-P. Deng, R. Liang, J. Fu, R. Gao, D. Luo, Z. Bai, Y. Hu, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 5858 Search PubMed.
- Y. Han, Y. Xiong, C. Liu, H. Zhang, M. Zhao, W. Chen, W. Chen and W. Huang, J. Catal., 2021, 396, 351–359 CrossRef CAS.
- N. Karmodak, L. Bursi and O. Andreussi, J. Phys. Chem. Lett., 2022, 13, 58–65 Search PubMed.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 Search PubMed.
- C. Ehlert, A. Piras, J. Schleicher and G. Gryn’ova, J. Phys. Chem. Lett., 2023, 14, 476–480 Search PubMed.
- Y. Han, X. Ding, J. Han, Y. Fang, Z. Jin, W. Kong and C. Liu, Nanoscale, 2022, 14, 9893–9899 Search PubMed.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2021, 33, 2003075 Search PubMed.
- Q. Wu, H. Zou, X. Mao, J. He, Y. Shi, S. Chen, X. Yan, L. Wu, C. Lang, B. Zhang, L. Song, X. Wang, A. Du, Q. Li, Y. Jia, J. Chen and X. Yao, Nat. Commun., 2023, 14, 6275 CrossRef CAS PubMed.
- H. Sui, Q. Guo, M. Xiang, X. Kong, J. Zhang, S. Ding and Y. Su, J. Phys. Chem. Lett., 2024, 15, 8257–8264 CrossRef CAS PubMed.
- Y. Han, X. Mao, X. Yan, Q. Wu, Q. Fang, H. Yin, Y. Jia, Q. Li, X. Yao and A. Du, Chem. Commun., 2024, 60, 14216–14219 Search PubMed.
- L. Yan, Y. Mao, Y. Li, Q. Sha, K. Sun, P. Li, G. I. N. Waterhouse, Z. Wang, S. Tian and X. Sun, Angew. Chem., Int. Ed., 2025, 64, e202413179 Search PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 Search PubMed.
- J. Zhang, A. Yu, D. Li and C. Sun, Adv. Theory Simul., 2024, 7, 2400076 CrossRef CAS.
- K. Liu, J. Fu, T. Luo, G. Ni, H. Li, L. Zhu, Y. Wang, Z. Lin, Y. Sun, E. Cortés and M. Liu, J. Phys. Chem. Lett., 2023, 14, 3749–3756 CrossRef CAS PubMed.
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. Liu, D. Hao, J. Ye, S. Ye, F. Zhou, H. Xie, G. Qin, J. Xu, J. Liu, S. Li and C. Sun, Adv. Energy Mater., 2023, 13, 2204126 CrossRef CAS.
- Q. Liu, H. Zhao, M. Jiang, Q. Kang, W. Zhou, P. Wang and F. Zhou, J. Mater. Chem. A, 2020, 8, 13638–13645 RSC.
- Y. Cheng, Y. Tian, X. Fan, J. Liu and C. Yan, Electrochim. Acta, 2014, 143, 291–296 Search PubMed.
- O. L. Li, N. N. T. Pham, J. Kim, H. Choi, D. H. Lee, Y. Yang, W. Yao, Y.-R. Cho and S. G. Lee, Appl. Surf. Sci., 2020, 528, 146979 Search PubMed.
- C. Liu, T. Wang, D. Hao, Q. Li, S. Li and C. Sun, J. Mater. Sci. Technol., 2022, 110, 96–102 CrossRef CAS.
- G. Liu, X. Meng, H. Zhang, G. Zhao, H. Pang, T. Wang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2017, 56, 5570–5574 Search PubMed.
- C. Liu, Z. Dai, J. Zhang, Y. Jin, D. Li and C. Sun, J. Phys. Chem. C, 2018, 122, 19051–19055 CrossRef CAS.
- Z.-H. Sheng, H.-L. Gao, W.-J. Bao, F.-B. Wang and X.-H. Xia, J. Mater. Chem., 2012, 22, 390–395 Search PubMed.
- Z. Lei, H. Chen, M. Yang, D. Yang and H. Li, Appl. Surf. Sci., 2017, 426, 294–300 Search PubMed.
- S. H. Mir, S. Chakraborty, P. C. Jha, J. Wärnå, H. Soni, P. K. Jha and R. Ahuja, Appl. Phys. Lett., 2016, 109, 053903 CrossRef.
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J.-O. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Adv. Mater., 2022, 34, 2110653 CrossRef CAS PubMed.
- P. Jankowski, W. Wieczorek and P. Johansson, Phys. Chem. Chem. Phys., 2016, 18, 16274–16280 RSC.
- A. M. Halpern, J. Chem. Educ., 2012, 89, 592–597 Search PubMed.
- C. Liu, H. Zheng, T. Wang, Z. Guo, F. Zhu, H. Xie, G. Qin, H. Li and S. Li, J. Mater. Sci. Technol., 2023, 159, 244–250 Search PubMed.
- S. Turner, W. Yan, H. Long, A. J. Nelson, A. Baker, J. R. I. Lee, C. Carraro, M. A. Worsley, R. Maboudian and A. Zettl, J. Phys. Chem. C, 2018, 122, 20358–20365 Search PubMed.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 Search PubMed.
- R. Christensen, H. A. Hansen, C. F. Dickens, J. K. Nørskov and T. Vegge, J. Phys. Chem. C, 2016, 120, 24910–24916 Search PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. Feng, Z. Dong, Y. Ji and Y. Li, JACS Au, 2023, 3, 1131–1140 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 Search PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- N. Ouyang, C. Wang, Z. Zeng and Y. Chen, Appl. Phys. Lett., 2021, 119, 061902 Search PubMed.
- T. Wu, T. Vegge and H. A. Hansen, ACS Catal., 2019, 9, 4853–4861 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01345c |
| This journal is © the Owner Societies 2025 |