
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origins of crystallisation-induced dual emission of terephthalic and isophthalic acid crystals†
Ljiljana
Stojanović
 *ab,
Michael
Dommett
c and
Rachel
Crespo-Otero
*d
*ab,
Michael
Dommett
c and
Rachel
Crespo-Otero
*d
aDepartment of Physics and Astronomy, University College London, London WC1E 6BT, UK. E-mail: ljiljana.stojanovic@stfc.ac.uk
bHartree Centre, Science and Technology Facilities Council, Daresbury Laboratory, Daresbury, WA4 4AB, UK
cSchool of Physical and Chemical Sciences, Queen Mary University of London, London, E1 4NS, UK
dDepartment of Chemistry, University College London, 2020 Gordon St., London WC1H 0AJ, UK. E-mail: r.crespo-otero@ucl.ac.uk
First published on 17th June 2025
Abstract
Metal-free organic crystals with room-temperature phosphorescence (RTP) present an innovative alternative to conventional inorganic materials for optoelectronic applications and sensing. Recently, substantial attention has been directed towards the design of new phosphorescent crystals through crystal engineering and functionalisation. In this paper, we investigate the excited-state deactivation mechanisms of two simple organic molecules: terephthalic acid (TPA) and isophthalic acid (IPA) using embedding models based on multiconfigurational MS-CASPT2 calculations. These molecules exhibit prompt and delayed fluorescence and RTP in the solid state. We explore intramolecular internal conversion pathways using high-level quantum chemistry methods in both solution and crystalline phases. We analyse deactivation mechanisms involving singlet and triplet states, quantifying direct and reverse intersystem crossing rates from the lowest triplet states, as well as fluorescence and phosphorescence rates. Additionally, our study examines singlet exciton transport in single crystals of TPA and IPA. Our findings clarify the mechanisms underlying the prompt and delayed fluorescence and RTP of crystalline TPA and IPA, revealing distinct differences in their deactivation processes. Notably, we explain the enhanced fluorescence and phosphorescence in IPA compared to TPA, emphasising how the positioning of the carboxylic group influences electronic delocalisation in excited states, (de)stabilising delocalised ππ* states along the reaction coordinate, thereby significantly impacting deactivation mechanisms.
1 Introduction
Room-temperature phosphorescence (RTP) has generated significant interest among experimental and computational scientists due to its wide range of applications, including its use in highly emissive materials for organic light-emitting diodes (OLEDs), anti-counterfeiting, photovoltaic devices, and bioimaging.1 Since efficient phosphorescence requires a substantial triplet population, these materials have traditionally been designed using inorganic and organometallic compounds that promote intersystem crossing (ISC) via strong spin–orbit coupling (SOC) and charge-transfer mechanisms. In contrast, organic molecules, due to their relatively small triplet populations and competing nonradiative pathways, typically exhibit phosphorescence only at cryogenic temperatures and under inert conditions in solution. However, recently RTP has been achieved in several purely organic crystals composed of molecules with relatively simple structures. These organic materials are significantly more cost-effective and environmentally friendly than their inorganic and organometallic counterparts.Efficient triplet formation can be achieved in RTP organic crystals in compounds where the presence of heteroatoms facilitates intersystem crossing rates between relevant singlet and triplet states.2 A second requirement for organic RTP is to establish a restrictive crystalline environment that suppresses large-amplitude molecular vibrations and reduces the rate of internal conversion to the ground state via conical intersections3 or vibrational wavefunction overlaps.4 Depending on the energy gaps and SOCs, RTP can compete with both prompt fluorescence (PF) and delayed fluorescence (DF).5 DF can occur through reverse intersystem crossing (rISC), which is crucial in thermally activated delayed fluorescence (TADF) chromophores.6
Two of the simplest organic crystals exhibiting RTP are the two isomers of benzenedicarboxylic acid in the crystalline phase: terephthalic acid (TPA) and isophthalic acid (IPA).7,8 These isomers differ in the relative positions of their substituents (Fig. 1). Transient photoluminescence spectroscopy of TPA and IPA in ethanol solution and disordered amorphous states reveals negligible luminescence quantum yields (below 0.6%). Upon crystallisation, both crystals exhibit dual emission, attributed to fluorescence and phosphorescence. The TPA crystal shows prompt deep blue emission at 388 nm with a lifetime of 〈t〉 = 0.53 ns and an emission efficiency of 8.4%. Additionally, a delayed fluorescence (DF) peak at 392 nm (slightly red-shifted compared to the prompt emission) and a shoulder at 511 nm, attributed to weak RTP, are observed. The experimental RTP lifetime is not determined on the millisecond timescale, suggesting longer lifetimes. The IPA crystal exhibits more efficient crystallisation-induced emission at 380 nm, with a quantum yield of 15.3%, attributed to prompt fluorescence. In addition to fluorescence, a long green afterglow lasting several seconds was observed, with distinct emission peaks at 384 nm and 506 nm, corresponding to DF and RTP, respectively. The measured lifetimes of prompt fluorescence, DF, and RTP in IPA are 1.81 ns, 9.6 ms, and 290 ms, respectively.7
 | ||
| Fig. 1 Structures of IPA and TPA molecules (upper panel). The crystal structures of of IPA and TPA crystals, with designated unit cells, are represented in two different perspectives in the bottom panels. | ||
Previous studies have investigated the mechanisms of fluorescence and phosphorescence in TPA and IPA.7,9 Shuai et al. found the crystalline phase induces a change of ordering of the lowest-lying singlet and triplet states of IPA and TPA in comparison with the vacuum where the S1 state is a nπ*, the higher ππ* state becomes the lowest according to the CASSCF(8,8)/CASPT2/AMBER level of theory. This change induces higher luminescence yields and larger intersystem crossing rates in the crystal.9 Gong et al. performed a similar optimisation of TPA and IPA in both the vacuum and crystalline phases based on QM/MM (TD-M06-2x/UFF) computations. They found that the energy gap between the closest-lying S1 and T2 states decreases while the spin–orbit coupling simultaneously increases in the crystal compared to the vacuum, leading to higher populations of the triplet state in the crystalline environment.7 Here, we evaluate the impact of using different levels of theory on predicting the ordering of states and consider the competition between various radiative and nonradiative mechanisms.
While some aspects of the luminescence of TPA and IPA have been explored both experimentally and computationally, the precise mechanisms of excited state relaxation in solution and the crystalline phase have yet to be fully understood. There are still many open questions about the competition between different nonradiative and radiative decay pathways and the origin of DF. Our study aims to fill this gap by systematically investigating the molecule-centred excited state relaxation pathways using high-level computational methods. In addition to these pathways, we also analyse the dimer-centred mechanisms and exciton transport mechanisms in both crystal environments. Our calculations provide critical insights into the excited state relaxation mechanisms of TPA and IPA, enhancing the current understanding of their behaviour in solution and crystalline phases.
2 Computational details
We evaluated the performance of several methods for predicting excited states in IPA and TPA across vacuum, solution, and crystal environments. First, we optimised the ground states using the DFT method with B3LYP, PBE0, and ωB97XD functionals, and then computed the vertical excitations of the first five singlet and triplet states with the time-dependent DFT (TD-DFT) method10–14 at the corresponding ground state minima. We also explored the resolution of identity coupled cluster method to the second order (RI-CC2)15–18 and the multi-state complete active space second-order perturbation theory19–21 ((MS-)CASPT2/CASSCF(14,11)) with the aug-cc-pVDZ basis set for describing the lowest-lying singlet and triplet states. For the RI-CC2 calculations, the ground state geometries were optimised at the CC2 level of theory, while for the CASPT2 computations, the ωB97XD/aug-cc-pVDZ ground state geometries were used. Solvent effects were considered by applying the polarisable continuum model (PCM) in the (TD-)DFT and CASPT2/CASSCF calculations, while the conductor-like screening model (COSMO) was used in the RI-CC2 computations. Ethanol was chosen as the solvent to facilitate comparison with available experimental results (dielectric permittivity εr = 24.55).The performance of the excited state methods for the prediction of vertical excitations in the crystal environment was evaluated using QM/MM techniques. To represent the crystal environment, we first refined the experimental crystal structures of TPA and IPA (retrieved from the Cambridge Crystallographic Database, with CCDC codes 1269122 for TPA22 and 1108748 for IPA23) by performing periodic boundary condition DFT calculations as implemented in Quantum Espresso.24 The PBE-D2 functional, including a dispersion correction, was employed with a plane-wave cutoff of 30 Ry and a Monkhorst–Pack k-point grid of (1 × 2 × 1), chosen based on the unit cell shape. The projector augmented wave (PAW) pseudopotential was used to model the nuclei and core electrons, while the valence electrons were treated explicitly.25,26 The optimisations were carried out by relaxing the structure within the unit cell while keeping the cell dimensions fixed at their experimental values. Clusters consisting of 123 TPA and 120 IPA molecules (2214 and 2160 atoms, respectively) were extracted from the optimised supercells. The QM/MM simulations included one or two central molecules in the QM region, with the surrounding molecules treated using MM. The QM region was relaxed, while the MM region remained fixed at its optimised lattice positions. For TD-DFT optimisations in the solid state, the ONIOM(QM:MM)method27,28 was applied, with the QM region treated at the (TD-)ωB97XD/6-311G(d,p) level of theory. For MS-CASPT2/CASSCF(14,11)/aug-cc-pVDZ calculations in the solid state, the QM/MM simulations were performed using the Molcas code for the electrostatic embedding QM calculations and the Tinker code for the MM part considering CM5 point charges. In the (TD-)DFT calculations, the MM region was described using the OPLS-AA force field, with ESP and CM5 charges derived from HF/3-21G calculations on a single molecule. The RI-CC2/aug-cc-pVDZ calculations were done only using electrostatic embedding with CM5 charges. While the calculations with TDDFT and CC2 methods were done using the geometries optimised with the same method, MS-CASPT2 calculations considered the ωB97XD/6-311G(d,p) ground-state structures. All (TD-)DFT computations were performed with the Gaussian 16 program,29 CASSCF and CASPT2 computations with the Molcas code,30 and RI-CC2 computations were performed with the Turbomole v7.0 code.31
To investigate the excited state relaxation mechanisms in solution and crystal phase, we optimised the excited states minima (S1, T1, T2) and S1–S0 and T1–S0 minimum energy crossing points in both environments. The excited states (S1, T1, and T2) were optimised at the CASSCF(14,11)/aug-cc-pVDZ level of theory because the TD-ωB97XD/6-311G(d,p) does not not predict correctly the types and ordering of all excited states of TPA and IPA. The S1–S0 minimum energy conical intersections (MECIs) optimisations were first carried out in solution and crystal phase at the SA-2-CASSCF(14,11)/6-31G(d) level of theory, using the branching plane update method32 as implemented in Molcas code. The obtained S1–S0 MECI geometries are reoptimised at the extended multistate CASPT2 level (XMS-CASPT2).33,34 The minimum energy crossing point between the ground and T1 states was optimised based on T1 states optimisation and S0–T1 energy gap criterion. In the solid state, the optimisations were done at the QM/MM level as described above.30 The active spaces comprised of twelve π orbitals and 2p-orbitals of carboxyl group oxygen atoms for IPA and TPA (Fig. S1, ESI†), were used in all optimisations and single point computations. The single-point CASPT2 computations were based on configuration state functions obtained at the SA-6-CASSCF(14,11)/aug-cc-pVDZ level.35 They were done without an IPEA shift and applying an imaginary shift of 0.1 a.u., which improves the convergence in the case of possible intruder states.36
Linearly interpolated pathways (LIIC) between S0 and S1–S0 MECI geometries in the solution and crystal were created and six lowest-lying singlet and triplet excited states were computed at the MS-6-CASPT2/CASSCF(14,11)/aug-cc-pVDZ level at the obtained geometries. The spin–orbit couplings (SOCs) between relevant singlet and triplet states were computed at the excited states minima in solution and crystal and along the LIIC pathway in the crystal, as implemented in the Molcas code. The method relies on the computation of matrix elements of one-electron spin–orbit part of the Hamiltonian in the atomic mean field approximation37,38 in the basis of the CASSCF wave functions. The SOCs between singlet and triplet pairs are obtained from the computed components corresponding to transitions between a singlet and three spin triplet components defined with quantum numbers ml ∈ {−1, 0, 1} as  .
.
Furthermore, to examine the effect of hyperfine interactions between singlet and triplet states on the excited-state deactivation mechanisms (in particular on the reverse intersystem crossing), we computed the hyperfine Hamiltonian matrix between excited states,39 as implemented in the Q-Chem program package.40 The matrix elements of the complete hyperfine Hamiltonian, including spin–spin dipole, Fermi contact, and orbital response term are evaluated in the basis of the TD-DFT excited states.39 The excited-states were computed with the ωB97XD functional and aug-cc-pVDZ basis set.
To investigate the dimer-centred relaxation mechanisms in the crystal phase, we optimised the S0 and S1 geometries of the dimers with the shortest centroid distances (π–π stacked dimers) applying the QM/QM′ approach as implemented in the fromage code.41,42 The QM (dimer) is represented by the algebraic diagrammatic construction ADC(2)43,44/aug-cc-pVDZ method and QM′ (environment) region was simulated at the second order (SCC-)DFTB method, employing the mio-1-1 set of Slater–Koster parameters parametrised for the tight-binding SCC-DFTB Hamiltonian.45,46 For the point charges in the environment, we used the ESP charges obtained at ωB97XD/6-31G(d) and PBE/6-31G(d) levels of theory, respectively. The DFTB calculations were performed with the DFTB+ program,45 and the ADC(2) computations with the Turbomole program. The lowest lying singlet and triplet states of the optimised dimers were computed at the ADC(2)/aug-cc-pVDZ level of theory.
We estimated the fluorescence and phosphorescence rates in vacuum and crystal applying Fermi's golden rule for the transition between initial (i) and final state (f) states:
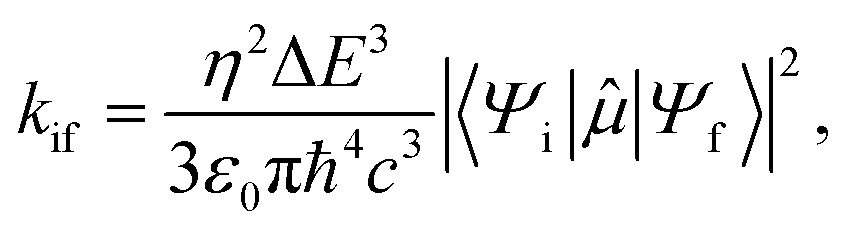 | (1) |
![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) is the electric transition dipole moment operator. The expectation values of this operator are computed between eigenstates of the Hamiltonian including the spin–orbit term as implemented in Molcas, allowing to estimate the transition dipole moments between the ground state and singlet and triplet excited states.
is the electric transition dipole moment operator. The expectation values of this operator are computed between eigenstates of the Hamiltonian including the spin–orbit term as implemented in Molcas, allowing to estimate the transition dipole moments between the ground state and singlet and triplet excited states.
The rates of the nonradiative electron transport (ET) processes (ISC, rISC and exciton transport) are estimated applying the Marcus semiclassical theory in the Condon approximations as
 | (2) |
 represents the vibrational density of states in the final state at the geometry of the initial state weighted by Franck–Condon factors in the limit of high temperatures.47
represents the vibrational density of states in the final state at the geometry of the initial state weighted by Franck–Condon factors in the limit of high temperatures.47
The reorganisation energy for (r)ISC was computed as λab ≈ ETb(RminSa) − ETb(RminTb) for the ISC and λab ≈ ESb(RminTa) − ESb(RminSb) for rISC (for clarification, for example ETb(RminSa) is the energy of the Tb state evaluated at the minimum of the Sa state). The Hab couplings correspond to the SOCs computed as described above. For the energy difference between the states (ΔEab) we used the difference between the energies of adiabatic states at the geometry at which the rates are computed (ΔEab = ΔEadab). The estimation of (r)ISC rates is done based on the MS-6-CASPT2/CASSCF(14,11)/aug-cc-pVDZ energies and CASSCF(14,11)/aug-cc-pVDZ spin–orbit couplings. We note that the energy gaps between singlet and triplet states correlate with the exchange interaction between those states, i.e. stronger exchange correlation induces larger gaps (ESI,† Section S2), and decreases the intersystem crossing rate. For this reason, an adequate treatment of electronic correlation using a high-level multiconfigurational methods, like CASPT2, is often necessary for the reliable prediction of ΔEST and consequently of the ISC rates.
In the case of exciton transport, the ΔEadab = 0 as the exciton hopping occurs between donor and acceptor molecules with identical geometries. The intramolecular reorganisation energy induced by the exciton transfer (λab) is computed considering one molecule (M1) going from the fully relaxed ground state S0 to the electronically excited state S1 and a neighboring molecule (M2) evolving in the opposite way as
 | (3) |
The exciton couplings (Hab = Jab) mediate the exciton transport process between the diabatic states (eqn (2)). The exciton transport rates are computed for the dimers with the centroid distances smaller than 10 Å, isolated from the optimised crystal structures. The Jab of isolated dimers were computed applying a diabatisation method based on the transition dipole moments of S1 states of isolated molecules and S1 and S2 states of dimers,48 as implemented in the fromage code.41 This method takes into account the short-range (exchange, orbital overlap, charge-transfer) and long-range Coulomb interactions. Both reorganisation energies for the exciton transport and exciton couplings are computed based on the TD-ωB97XD/6-311+G(d,p) excited states.
3 Results and discussion
3.1 Light absorption and emission
| B3LYP | PBE0 | ωB97XD | ωB97XD/PCM | RI-CC2/COSMO | CASPT2/PCM | Exp. | |
|---|---|---|---|---|---|---|---|
| a S5 states of IPA are predicted as nπ* at the B3LYP and PBE0 level. b T3–T5 states of TPA have different order at the B3LYP and PBE0 level. c Experimental excitation energies taken from ref. 7. | |||||||
| TPA | |||||||
| S1 (ππ*) | 4.52 (0.02) | 4.67 (0.03) | 4.84 (0.04) | 4.98 (0.03) | 4.56 (0.02) | 4.02 (0.04) | |
| S2 (nπ*) | 4.51 (0.00) | 4.67 (0.00) | 5.06 (0.00) | 5.30 (0.00) | 4.76 (0.00) | 4.80 (0.00) | |
| S3 (nπ*) | 4.57 (0.00) | 4.73 (0.00) | 5.11 (0.00) | 5.32 (0.00) | 4.80 (0.00) | 4.88 (0.00) | |
| S4 (ππ*) | 5.02 (0.43) | 5.16 (0.43) | 5.33 (0.41) | 5.52 (0.12) | 5.39 (0.55) | 4.92 (0.44) | 5.17c |
| S5 (ππ*) | 6.20 (0.00) | 6.44 (0.00) | 6.72 (0.15) | 6.01 (0.84) | 6.71 (0.14) | 6.16 (0.08) | 6.20c |
| T1 (ππ*) | 3.26 | 3.18 | 3.33 | 3.44 | 3.73 | 3.36 | |
| T2 (ππ*) | 3.68 | 3.75 | 3.94 | 4.20 | 4.24 | 3.97 | |
| T3 (ππ*)b | 4.46 | 4.46 | 4.62 | 4.48 | 4.78 | 4.07 | |
| T4 (nπ*)b | 4.17 | 4.27 | 4.65 | 4.89 | 5.63 | 4.69 | |
| T5 (nπ*)b | 4.23 | 4.33 | 4.70 | 4.90 | 5.82 | 4.76 | |
| IPA | |||||||
| S1 (ππ*) | 4.72 (0.02) | 4.87 (0.02) | 5.01 (0.02) | 4.79 (0.05) | 4.68 (0.01) | 3.94 (0.005) | |
| S2 (nπ*) | 4.77 (0.00) | 4.91 (0.00) | 5.21 (0.00) | 5.12 (0.00) | 4.92 (0.00) | 4.60 (0.00) | |
| S3 (nπ*) | 4.80 (0.00) | 4.94 (0.00) | 5.23 (0.00) | 5.18 (0.00) | 4.94 (0.00) | 4.61 (0.00) | |
| S4 (ππ*) | 5.32 (0.09) | 5.46 (0.09) | 5.60 (0.09) | 5.24 (0.50) | 5.70 (0.12) | 5.20 (0.09) | |
| S5 (ππ*) | 5.48a (0.00) | 5.76a (0.00) | 6.20 (0.75) | 6.74 (0.00) | 6.12 (0.84) | 5.34 (0.82) | 5.39c |
| T1 (ππ*) | 3.40 | 3.29 | 3.43 | 3.33 | 3.88 | 3.06 | |
| T2 (ππ*) | 3.99 | 4.05 | 4.22 | 3.91 | 4.45 | 3.64 | |
| T3 (ππ*) | 4.22 | 4.28 | 4.50 | 4.62 | 4.61 | 4.04 | |
| T4 (nπ*) | 4.39 | 4.47 | 4.78 | 4.72 | 4.68 | 4.33 | |
| T5 (nπ*) | 4.41 | 4.50 | 4.80 | 4.78 | 5.41 | 4.37 | |
| ωB97XD* | ωB97XD(CM5) | ωB97XD(ESP) | RI-CC2 | CASPT2 | Exp. | |
|---|---|---|---|---|---|---|
a S4 and S states of IPA show less nπ*/ππ* mixing according to the ωB97XD and RI-CC2 results, compared to the CASPT2 ones. They are predicted as ππ* at the ωB97XD level, and as nπ* and ππ* at the RI-CC2 level.
b S5 states of both IPA and TPA are predicted as nπ* at the RI-CC2 level.
c Photon energies used in the photoexcitation spectra for TPA and IPA crystals as reported in ref. 8. states of IPA show less nπ*/ππ* mixing according to the ωB97XD and RI-CC2 results, compared to the CASPT2 ones. They are predicted as ππ* at the ωB97XD level, and as nπ* and ππ* at the RI-CC2 level.
b S5 states of both IPA and TPA are predicted as nπ* at the RI-CC2 level.
c Photon energies used in the photoexcitation spectra for TPA and IPA crystals as reported in ref. 8.
|
||||||
| TPA | ||||||
| S1 (ππ*) | 4.82 (0.04) | 4.72 (0.04) | 4.69 (0.04) | 4.68 (0.03) | 3.97 (0.08) | 3.90c |
| S2 (nπ*) | 5.35 (0.37) | 5.23 (0.42) | 5.23 (0.42) | 5.39 (0.00) | 4.74 (0.29) | |
| S3 (nπ*) | 5.45 (0.00) | 5.46 (0.00) | 5.55 (0.00) | 5.41 (0.12) | 4.80 (0.00) | |
| S4 (ππ*) | 5.49 (0.04) | 5.50 (0.01) | 5.59 (0.01) | 5.53 (0.42) | 4.83 (0.07) | |
| S5 (ππ*) | 6.79 (0.00) | 6.68 (0.00) | 6.64 (0.00) | 7.02b (0.00) | 6.14 (0.38) | |
| T1 (ππ*) | 3.31 | 3.29 | 3.30 | 3.88 | 3.23 | |
| T2 (ππ*) | 3.86 | 3.81 | 3.77 | 4.32 | 3.75 | |
| T3 (ππ*) | 4.67 | 4.63 | 4.61 | 6.01 | 4.49 | |
| T4 (nπ*) | 5.04 | 5.07 | 5.17 | 4.95 | 4.75 | |
| T5 (nπ*) | 5.06 | 5.09 | 5.19 | 5.44 | 4.79 | |
| IPA | ||||||
| S1 (ππ*) | 5.08 (0.02) | 4.98 (0.02) | 4.96 (0.02) | 3.69 (0.00) | 3.93 (0.10) | 4.00c |
| S2 (nπ*) | 5.56 (0.00) | 5.53 (0.06) | 5.52 (0.07) | 4.13 (0.00) | 4.57 (0.00) | |
| S3 (ππ*) | 5.72 (0.08) | 5.56 (0.08) | 5.56 (0.01) | 4.17 (0.00) | 5.26 (0.00) | |
| S4 (ππ*/nπ*) | 5.60a (0.01) | 5.61a (0.02) | 5.74 (0.01) | 4.54 (0.00) | 5.72 (0.18) | |
| S5 (ππ*/nπ*) | 6.31a (0.71) | 6.10a (0.73) | 6.08 (0.71) | 4.66b (0.02) | 5.97 (0.00) | |
| T1 (ππ*) | 3.46 | 3.43 | 3.44 | 3.68 | 3.29 | |
| T2 (ππ*) | 4.27 | 4.19 | 4.20 | 4.00 | 3.85 | |
| T3 (ππ*) | 4.54 | 4.47 | 4.46 | 4.08 | 3.96 | |
| T4 (nπ*) | 4.99 | 4.89 | 4.88 | 4.15 | 4.47 | |
| T5 (nπ*) | 5.13 | 5.14 | 5.26 | 4.36 | 5.11 | |
We first compare the performance of three functionals, B3LYP, PBE0, and ωB97XD, for the prediction of excited states in vacuum. The bright S4 state energies in vacuum computed applying these functionals deviate from each other by less than 0.3 eV in both molecules, whereas the energies of other states are predicted to be higher at the TD-ωB97XD level. The calculations with TD-ωB97XD show that, when using the PCM model for ethanol, the energies are shifted by approximately 0.1–0.3 eV, except for the highest singlet state considered (S5), where the shift exceeds 0.5 eV. In Table 1, the vertical excitations obtained with RI-CC2 and MS-CASPT2 were calculated considering ethanol as the solvent, using the COSMO and PCM approaches, respectively. Comparing the excited state energies in the solution, we notice that the MS-CASPT2 S1 ππ* state has ∼1 eV and 0.6 eV lower energy compared to the TD-ωB97XD and RI-CC2 energies, respectively. The dark nπ* states (S2 and S3) energies are overestimated for ∼0.5 eV at the TD-ωB97XD level, whereas the RI-CC2 predicts their excitation energies closer to the MS-CASPT2 values.
The experimental absorption spectra in ethanol of TPA and IPA featured two bands,7 at 200 nm (6.20 eV) and 240 nm (5.17 eV) for TPA and at 210 nm (5.90 eV) and 230 nm (5.39 eV) for IPA. In the case of TPA, the MS-CASPT2/PCM reproduces well the first and second bright excitations at 4.92 eV and 6.16 eV. The TD-ωB97XD/PCM and RI-CC2/COSMO methods overestimate the energies of the bright singlet states and triplet excitations. The MS-CASPT2/PCM method reproduces well the first two bright excitations, the computed vertical excitation energies of bright S4 and S5 states are 5.20 eV and 5.34 eV in IPA, assignable to the first peak in the absorption spectrum with the maximum at 5.39 eV. The higher bright states contributing to the second absorption peak at 5.90 eV are not among the first five singlet states. These values are in very good agreement with the positions of the peak maxima (5.39 eV and 5.90 eV).
Table 3 shows the emission energies obtained after relaxation in S1. While the MS-CASPT2 emission energies are close to the experimental emission maxima, deviating by only 0.3–0.4 eV, the TD-ωB97XD energies are overestimated by more than 1 eV. In contrast to the results reported by Ma et al., we do not observe a change in the S1 state type from ππ* to nπ* upon crystallisation in the Franck–Condon region.9 According to our CASPT2 results, the S1 states of TPA and IPA in vacuum, solution, and crystal phases possess ππ* character, while the higher S2 and S3 states exhibit nπ* character. However, we found that the nπ* state becomes the lowest-lying state along the internal conversion coordinate, as will be discussed below (Section 3.2). These differences from previous works are attributed to the different active spaces employed. Our CASSCF/CASPT2 simulations considered a [14,11] active space (9π and 2n orbitals), while the smaller[8,8] active space used in the work by Ma et al. comprised 6π and 2n orbitals. This suggests that including a larger number of π orbitals in the active space enabled the identification of a lower eigenvalue of the Hamiltonian, corresponding to the ππ* state, whereas the energies of two quasi-degenerate nπ* states are predicted to be very close at both levels of theory (approximately 4.8 eV, ref. 9 and Table 2).
Based on this benchmark, we conclude that the MS-CASPT2 method with the selected active space ([14,11]) reproduces the absorption and emission properties of TPA and IPA well (Table 3). In Section 3.2.1, we will present the deactivation pathways connecting the ground state minimum with the optimised minimal energy conical intersections (S1–S0 MECI). These pathways were obtained applying the MS-CASPT2 method, which, apart from the good performance in description of absorption and emission, is suitable for the excited states simulations in the vicinity of the conical intersection, due to multireference character of the wave function.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–O hydrogen bonds between in-plane monomers. In the TPA crystal, each monomer is bonded with two CO⋯H–O hydrogen bonds with the average length of 1.53 Å, creating linear chains of hydrogen bonded molecules. In the case of IPA, the zig-zag molecular chains are formed by CO⋯H–O hydrogen bonds, with the average lengths of 1.66 Å. in both crystals, these chains create molecular planes with the distance of ∼3.4 Å, interacting with the adjacent planes through π⋯π interactions between molecules. The other O⋯H interactions between the molecules belonging to adjacent planes, between oxygen atoms from the CO groups and hydrogen atoms from the C–H and O–H groups, have substantially larger lengths (between 2.5 and 2.7 Å7) and are weaker compared to the in-plane hydrogen bonds and π⋯π interactions.
O⋯H–O hydrogen bonds between in-plane monomers. In the TPA crystal, each monomer is bonded with two CO⋯H–O hydrogen bonds with the average length of 1.53 Å, creating linear chains of hydrogen bonded molecules. In the case of IPA, the zig-zag molecular chains are formed by CO⋯H–O hydrogen bonds, with the average lengths of 1.66 Å. in both crystals, these chains create molecular planes with the distance of ∼3.4 Å, interacting with the adjacent planes through π⋯π interactions between molecules. The other O⋯H interactions between the molecules belonging to adjacent planes, between oxygen atoms from the CO groups and hydrogen atoms from the C–H and O–H groups, have substantially larger lengths (between 2.5 and 2.7 Å7) and are weaker compared to the in-plane hydrogen bonds and π⋯π interactions.
To get an insight into intermolecular interactions and exciton transport mechanisms in TPA and IPA crystals, we isolated dimers from an optimised supercell with centroid distances smaller than 10 Å, and computed the exciton couplings between the lowest-lying bright states of monomers at the TD-ωB97XD/6-311+G(d,p) level. At this level of theory, the S1 and S2 states of monomers at the optimised crystal structures are quasi-degenerate and the exciton couplings are very small. The exciton coupling are computed between the bright S2 (ππ*/nπ*) states, while the close-lying S1 (nπ*) are dark states. The S2 pairs of monomers combine in and form S3 and S4 states of isolated dimers. The dimers with the largest exciton couplings are the face-to-face slip-stacked dimers with the centroid distances ≈3.75 Å, interacting through π⋯π interactions between the π densities localised on phenyl rings and densities belonging to carboxylic groups (Fig. 2). Their interaction yields relatively weak couplings of 12 meV in both molecules. Other isolated dimers are mainly side-to-side dimers, including the dimers with the intermolecular CO⋯H–O bonds, and have negligible exciton couplings (<5 meV). The reorganisation energies for the exciton transport computed using eqn (3) is 382 meV for TPA and 277 meV for IPA. Upon reorganisation from the FC region to the excited state minima, the transition density changes from mixed ππ*/nπ* at optimised crystal geometry (Fig. 2) to pure ππ* state in TPA, whereas in IPA the S1 state has also a mixed ππ*/nπ* at its minimum (Fig. 6).
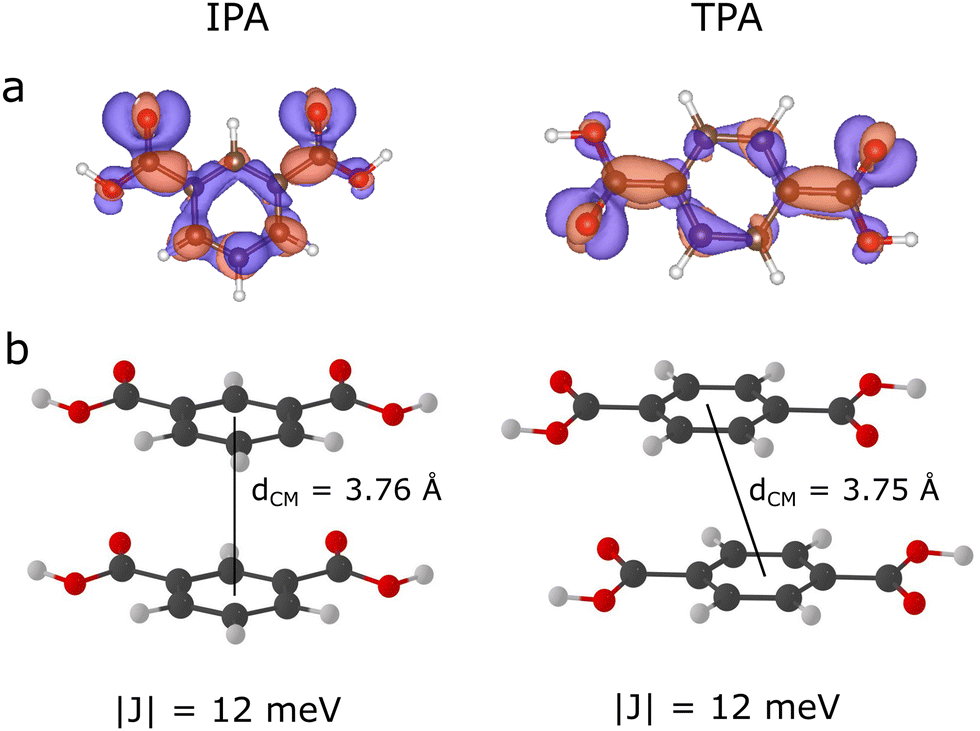 | ||
| Fig. 2 TD-ωB97X-D/6-311+G(d,p) transition densities of the S1 states of IPA and TPA computed at the optimised crystal geometry for an isolated molecule in vacuum (panel a). The isosurfaces corresponding to the 0.001 Å3 density isovalue are represented. Structures of IPA and TPA dimers with the largest exciton couplings are presented in panel b. The electronic couplings and centre of masses distances between monomers are designated. | ||
The exciton couplings are much smaller than the classical barrier for exciton hopping (λ/4), indicating that the S1 excitons mostly remain localised on a single site during exciton diffusion in crystals. The exciton diffusion mechanism can be represented as a series of hopping events between isolated molecules. In this context, the exciton hopping rates for this mechanism can be evaluated based on the Marcus theory (eqn (2)). Applying the computed exciton couplings and reorganisation energies for IPA and TPA, the estimated exciton hopping rates for these molecules are 2.7 × 1010 and 8.3 × 109 s−1, respectively. This will compete with the vibrational relaxation to S1 in the picosecond scale (rates around 1012 s−1) followed by other processes such as fluorescence and ISC. Due to a smaller reorganisation energy, the exciton hopping rate in IPA are ∼4 times larger than in TPA. We note that the thermal effects, which can significantly affect the exciton couplings and exciton diffusion rates and mechanism, are neglected here.
3.2 Excited state mechanisms
In this section, we analyse different mechanisms associated with the excited state relaxation of TPA and IPA. Our main focus is understanding the competition of different pathways in the solid state.O and CPh–CCarb bonds, i.e. the CO bond elongates by ∼0.25 Å, while the CPh–CCarb bond contracts by ∼0.15 Å with respect to the ground state values. Apart from this, the lone pair orbitals of oxygen atoms in carboxyl group rotate such that their axes lie in the COO-plane, and their interaction decreases their distance (by ∼0.2 Å) with respect to the standard distance. Similarly, ∠OC–O angle decreases to 80–90° from 120°.
 | ||
| Fig. 3 Structures of conical intersections of IPA and TPA optimised at XMS-CASPT2[14,11]/6-31G(d) level in vacuum and crystal environment. The geometries are represented in top and side views. The lengths of the C–O bonds, distances between O atoms, and ∠HOCO dihedral angles (α) in the carboxyl group are designated. | ||
In the S1–S0 MECI structures optimised in the solution, the ∠HOCO is close to 90°, while the rest of the molecule is planar. On the other hand, in the solid state, the more favourable CI structures involve the bending of the COO-plane with respect to the rest of the molecule, while the H-atom dihedral motion is less activated (∠HOCO is 60° in IPA and 53.5° in TPA). A similar type of MECI structure was found in a phenyl-derivative dicarboxylic acid.49
The XMS-CASPT2 method is an appropriate choice for the optimisation of conical intersections, due to the multireference character of the wave function near these intersections. We also optimised the S1–S0 MECI using less computationally expensive single-reference methods, TD-DFT and ADC(2). These methods predicted the minimal energy crossing structures involving benzene ring puckering, which are much higher in energy compared to the CASPT2-optimised structures. Additionally, we optimised the S1–S0 MECI of dimers bonded through CO⋯H–O hydrogen bonds at the TD-DFT and ADC(2) levels of theory to investigate whether internal conversion occurs through an intermolecular proton transfer mechanism. The obtained S1–S0 MECI structures involve geometry changes in only one of the molecules.
 | ||
| Fig. 4 CASPT2/SA-6-CASSCF(14,11)/aug-cc-pVDZ energies of the lowest-lying singlet and triplet states along the LIIC pathway connecting S0 and MECI geometries in the crystal in IPA (panel a) and TPA (panel b) in a diabatic representation. | ||
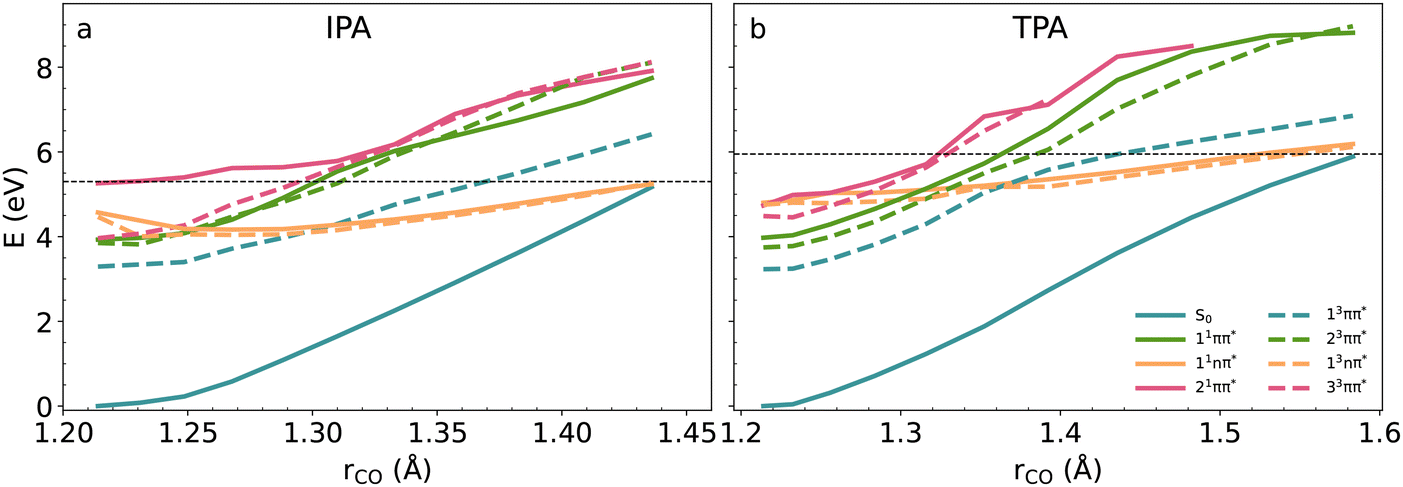
The S1–S0 MECI for IPA lies at ∼5.2 eV, ∼1.2 eV above the vertical excitation, and for TPA at ∼6.0 eV, ∼2 eV above the vertical excitation, and they are not classically accessible after the excitation to S1 state. The excitation of the second bright state of TPA at 4.74 eV (S2) would also not enable the internal conversion to the ground state. The excitation of high-energy bright states of IPA and TPA with energies close to 6 eV (∼200 nm), would enable internal conversion, but that would require the application of vacuum UV radiation, not used in the experiment.7 The crystal environment increases the energy barriers to the S1–S0 MECI with respect to solution in 0.4 eV for IPA and 0.8 eV for TPA. From the transition state theory, we can obtain that the fractions of nonradiative rates between the solid state and solution are in the order 10−7 for IPA and 10−14TPA respectively. This is in line with the restricted access to the conical intersection (RACI) model, where the crystal environment blocks the access to the conical intersections associated with the S1–S0 nonradiative transitions explaining the enhancement of solid-state luminescence for these systems.3,50,51 We should notice that for both S1–S0 MECI, T1 gets almost degenerate with S0 and S1 providing an additional nonradiative pathway to the ground state, which can be activated by providing larger excitation energies.
The excitation to the S1 state is followed by a relaxation to its minimum, where fluorescence and intersystem crossing can get activated. According to the PES and the spin–orbit couplings along the reaction coordinate (Fig. 4 and 5), the mechanisms involving triplet states are also possible. In the vicinity of the FC region, up to the (CO) distance of ∼1.3 Å, where the T1 and T2 states are close-lying to the S1, the S1/T1 and S1/T2 couplings have small values (below 5 cm−1), indicating the possibility for slow intersystem crossing. The S1/T1 and S1/T2 SOCs values significantly increase further along the IC coordinate for rCO > 1.3 Å, but in this region the S1 state energy is higher than the vertical excitation, and the probability for its population via tunneling is relatively small.
 | ||
| Fig. 5 Spin–orbit coupling matrix elements (SOC in cm−1) between relevant adiabatic singlet/triplet excited states computed along the linearly interpolated pathways for nonradiative relaxation for IPA (left) and TPA (right) in the crystal. The couplings between the states with large energy gaps are not shown. | ||
| k f (s−1) | k P (s−1) | k ISC (s−1) | k rISC (s−1) | |
|---|---|---|---|---|
| TPA | 9.9 × 107 | 0.5 | 9.1 × 106 (S1/T4) | 202 (T3/S2) |
| IPA | 1.7 × 108 | 9.9 | 1.8 × 1011 (S1/T1) | 523 (T2/S1) |
O bond stretching, in both systems the lowest-lying excited states in the FC region are the ππ* states. They cross the higher nπ* singlet and triplet states at rCO ∼ 1.3 Å in IPA and rCO ∼ 1.35 Å in TPA. For the larger CO lengths, the nπ* states become more stable (Fig. 5).
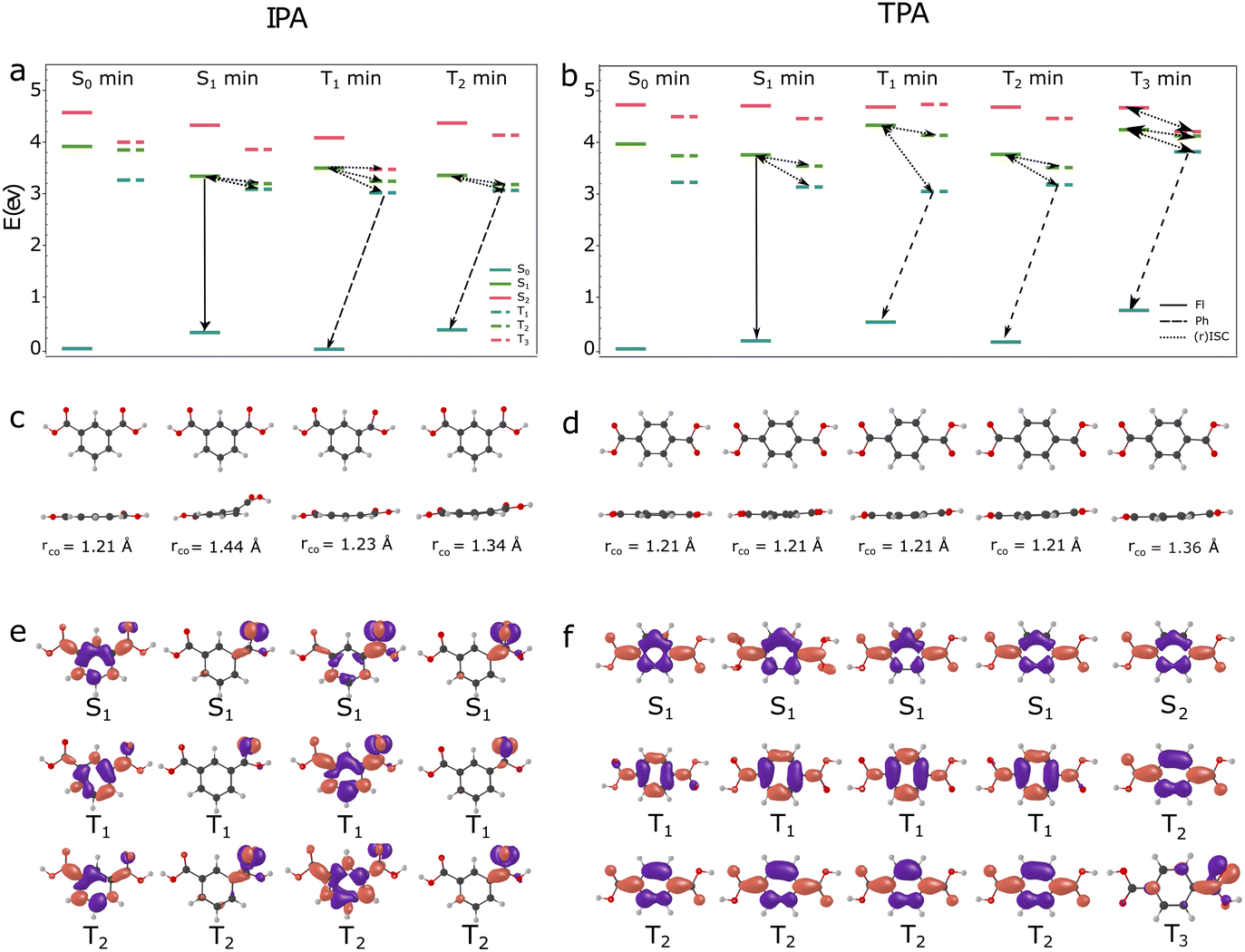 | ||
| Fig. 6 Energy levels of the lowest-lying singlet and triplet states of IPA (panel a) and TPA (panel b) in crystal computed at the S0, S1, T1, T2, and T3 (for TPA) states minima at the MS-CASPT2/CASSCF(14,11)/aug-cc-pVDZ level of theory. Possible fluorescence (full line), phosphorescence (dashed line), and (reversed) intersystem crossing (dotted line) transitions are designated. Optimised geometries of IPA (panel c) and TPA (panel d) molecules in S0, S1, T1, T2, and T3 states given in two perspectives. The lengths of the CO bond from the carboxylic group is given as well. The S1, T1, and T2 excited state MS-CASPT2 density differences in the crystal environment are represented for IPA (panel e) and TPA (panel f) at S0, S1, T1, and T2 minima. In the case of the excited states of TPA at the T3 minimum, the MS-CASPT2 density differences in the crystal environment are represented for S2, T2, and T3 states. | ||
The most striking difference between TPA and IPA is related to the nature of the most stable excited states. In IPA, the lowest-lying excited states are nπ* states at the S1 and T2, and mixed nπ* and ππ* states at the T1 geometry. Their geometries are non-planar, with signatures of distortions occurring at the conical intersection –CO bond stretching and out-of-plane bending of carboxyl group with respect to the phenyl ring. In TPA, the lowest-lying S1, T1, and T2 states are ππ* states. The geometries at their minima are planar structures and do not feature CO elongation.
3.2.4.1 Intersystem crossing. We explore possible intersystem crossing mechanisms from the S1 minima. In TPA, the energy gaps between S/T pairs (ΔE) at the S1 geometry are 0.62 eV for the S1/T1 and 0.22 eV for the S1/T2 pair. Since these states are ππ* states, the spin–orbit couplings between S1/T1 is small (∼4 cm−1) and ∼0 cm−1 for S1/T2 pair. The estimated ISC rates, based on the eqn (2), are negligible for the S1/T1 transition and zero ISC rate for the S1/T2 transition (Table 4, parameters used for the computations of all rates are given in the ESI†) at the S1 geometry. The ISC S1 (ππ*) and T1/T2 (ππ*) states are negligible along the reaction coordinate for the C
O lengths below 1.32 Å, as the gaps between states remain significant and spin–orbit couplings are close to zero. However, there is the possibility for the transition from the S1 ππ* state to the higher-lying nπ* triplet state along the reaction pathway. The S1 ππ* state crosses with the 13nπ* triplet state at rCO = 1.32 Å. The minimum of the 13nπ* state occurs at rCO ≈ 1.36 Å (Fig. 6), after the crossing between ππ* and nπ* states along the reaction coordinate (Fig. 4). At this geometry, the 13nπ* state (T3) is 0.2 eV above the S1 vertical excitation. The first two triplet states are ππ* states, whereas the first two singlet states are nπ* and ππ*, respectively. The calculated ISC rate for the S1/13nπ* transition at the minimum of the S1 state is 1.6 × 105 s−1. However, the spin–orbit couplings between these states increase along the reaction coordinate. The ISC rate computed applying the spin–orbit couplings in the vicinity of the crossing point between states (rCO ≈ 1.3 Å) is 9 × 105 s−1, indicating that a part of the population can be transferred to the 13nπ* state, and subsequently to the lower-lying T1 and T2 ππ* states.
In IPA, two triplet states are in the vicinity of the S1 state at its minimum (Fig. 6). The S1 geometry features significant CO stretching (rCO = 1.44 Å) and out-of-plane motions of carboxylic group (Fig. 6). As a consequence, the S1, T1, and T2 states are nπ* states at this geometry. The S1 and T2 states are mostly represented by a single nπ* configuration, while the T1 state is a linear combination of two nπ* configurations of opposite signs, inducing vanishing density around the CPh–CCarb bond, and the total transition density is localised on CO oxygen atom. Furthermore, the T1 transition density is rotated for π/2 around the CO bond with respect to the S1 density. The T2 density is also slightly rotated with respect to the S1 density. These differences induce significant spin–orbit couplings between S1/T1 and S1/T2 pairs (32.4 and 27.2 cm−1, respectively). Moreover, relatively small S–T gaps at this geometry (0.27 eV and 0.18 eV) enable significant ISC rates considerably faster than in TPA. The computed rates are 1.8 × 1011 s−1 for the S1/T1 transition and 9 × 106 s−1 for the S1/T2 transition (Table 4). Since the ISC rates to the T1 state are larger compared to the fluorescence rates from the S1 minimum (kf ≈ 1.7 × 108 s−1), a significant population transfer from the S1 to T1 state is expected. According to the reaction pathways (Fig. 4), the lowest-lying ππ* triplet states (T1 and T2 in the FC region) can be populated through internal conversion from the nπ* triplet state. This mechanism involves the planarisation of molecule and contraction of the CO bond.
In both systems, the population transferred to the lowest-lying triplet states could deactivate through phosphorescence, ISC to the ground state, and reversed ISC to the singlet states.5 According to the potential energy profiles along the reaction pathways, the ISC to the ground state is hindered in both systems, because the S0/T1 crossings, which are in the vicinity of the S1–S0 MECI (Fig. 6), are classically inaccessible following the S1 excitations.
3.2.4.2 Room temperature phosphorescence. Once the T1 states get populated, a possible relaxation mechanism is phosphorescence in both systems. Similar as fluorescence, the ultralong phosphorescence in IPA is more efficient compared to the TPA, with the computed rates of 9.92 s−1 and 0.5 s−1, respectively. In IPA, according to our results, phosphorescence occurs at 2.91 eV with a lifetime of ∼100 ms. The available experimental phosphorescence energies and lifetimes are in relatively good agreement with our results; RTP is observed at 2.45 eV (506 nm) with a lifetime of 290 ms by Gong et al.,7 and in the study by Kuno et al.8 at 2.51 eV (501 nm). Our computations predict an ultralong phosphorescence at 2.53 eV for TPA, in good agreement with the experimental weak phosphorescence signal at 2.43 eV (511 nm) by Gong et al.7 and at 2.40 eV (516 nm) by Kuno et al.8
3.2.4.3 Reverse intersystem crossing. To examine the deactivation pathways from the triplets populated from the singlet states through intersystem crossing, we optimised the geometries of the T1 and T2 states in both systems, and the T3 state in TPA, and computed the reverse ISC rates from the minima of the triplet states. Because these systems experimentally exhibit DF, there must be mechanisms that efficiently populate S1 from the triplet manifold. However, in both systems, population of singlet states from T1 seems unlikely due to the significant energy gaps. Nevertheless, we observe that when considering higher triplet states, such as T2 and T3, the rates for rISC can be one to two order of magnitudes faster than competing phosphorescence. Previous works highlight the relevant role of the higher-lying triplets in the rISC mechanism in organic systems.5,6,53–55 In cases where the energy gap between the T/S pair undergoing reverse ISC is substantial, intermediate triplet states can mediate this process. Vibronic interactions between closely spaced triplet states can promote internal conversion between them, effectively lowering the activation energy required for reverse ISC. Our hypothesis is that T2 in IPA and T3 in TPA lie very close in energy to T1 and T2, respectively, which should facilitate seamless population transfer between these states.
In TPA, the lowest T1 and T2 states at their minima are ππ* states. The computed rISC rates from the T1 and T2 state minima (the T2 state is the lowest triplet state at its minimum) to the S1 state are negligible (10−18 s−1) at the T1 minimum and zero at the (T2 minimum), as a consequence of negligible/zero SOCs between (ππ*) states. To explain the rISC mechanism, we take into account the higher-lying 13nπ* state. At the minimum of this state (T3), the lower-lying T1 and T2 states are ππ* states. The T3 state can get populated due to excess vibrational energy obtained following the S1/T3 ISC. At this geometry, there is a possibility for the rISC to the higher-lying S2 ππ* state (corresponding to the S1 state at the FC region) from the T3 (13nπ*) state, due to a significant SOC between the states (10 cm−1). The computed rISC rate is ∼200 s−1. The model used to compute the rISC rates (eqn (2)) does not take into account vibrational effects on spin–orbit interaction56 and vibronic coupling between triplet states,55 which could enhance the rISC rates. Another possible rISC mechanism at the T3 geometry includes the transition from the ππ* T1/T2 states to the close-lying S1 (nπ*) state. The T2/S1 rISC is expected to be efficient, since the ΔE is only ∼0.13 eV, while the T2/S1 SOC is 12.4 cm−1. Moreover, the SOCs increase substantially along the reaction coordinate, surpassing 40 cm−1 for the rCO > 1.38 Å (Fig. 5), which could further increase the rISC rates.
In IPA, the T1 state minium has a planar geometry with slightly elongated CO bond (rCO = 1.23 Å) compared to the ground state geometry (rCO = 1.21 Å) (Fig. 6). The S1 state corresponds to the nπ*, whereas lower-lying T1 and T2 states correspond to the mixed nπ*/ππ* states at this geometry. The SOCs between the S1 and T1 states are 3 cm−1. A significant S1–T1 gap at this geometry (0.71 eV), induces negligible rISC rate (Section S7 in the ESI†). The T2 state features CO stretching (rCO = 1.34 Å) and out-of-plane motion of –COOH group. Similar as at the S1 geometry, S1, T1, and T2 states are nπ* states at this geometry. We note that the T2 state is the second triplet state at its minimum. A significant SOC between S1/T1 pair (15.4 cm−1) and a relatively small energy gap (0.18 eV) induce more efficient rISC from T2 to the S1 state with the rate of 523 s−1, in comparison with the T1/S1 rISC. The T2/S1 rISC rates can increase along the stretching coordinate, as a results of an increase of the SOCs between them. Similar as for TPA, the T2/S1 SOCs significantly increase for rCO > 1.3 Å, reaching the values of 50 cm−1 (Fig. 5). Based on this analysis, we can conclude that a part of the population from the triplet states could be transferred back to the S1 state in both IPA and IPA due to spin–orbit interactions between states.
Previous studies suggest that apart from the spin–orbit interactions, the hyperfine interactions between singlet/triplet pairs could contribute to the rISC mechanism, in particular in intermolecular processes in multi-chromophore systems involving radical-pair charge transfer states.39,57 For the IPA crystal, Kuno et al.8 found that the phosphorescence decreases upon Zeeman splitting of the degenerate triplet state in an external magnetic field and CH-to-CD substitution, but increases in stronger magnetic fields. This was explained by a charge-transfer (CT) state delocalised over two molecules, with spins localised on each, forming nearly degenerate singlet and triplet radical-ion-pair states. Weak hyperfine couplings enable spin exchange between them, accounting for both the suppression (Zeeman effect) and enhancement (Δg mechanism) of phosphorescence. We have explored the effects of hyperfine interactions on intramolecular mechanisms. In the case of the excited states of TPA and IPA, the hyperfine couplings between the singlet and triplet states, calculated using the TD-ωB97XD method, are negligible—less than 10−14 meV (see ESI,† Section S9). Consequently, the influence of hyperfine interactions on singlet–triplet mixing can be considered negligible, suggesting that reverse intersystem crossing (rISC) will be primarily facilitated by spin–orbit interactions. Additionally, our calculations using electrostatic embedded RI-ADC(2)/aug-cc-pVDZ method for the lowest-lying singlet and triplet states (S1, S2, T1–T4) for the dimers of TPA and IPA (ESI,† Section S10), indicated these states are either localised or delocalised nπ* and ππ* states, without a significant radical-ion pair charge transfer character.
3.2.4.4 Delayed fluorescence. Our results show that the rISC, activated in both systems, enables the transfer of a fraction of the population from triplet manifold to the S1 states. A fraction of transferred population can undergo delayed fluorescence. In IPA, the delayed fluorescence is observed at 3.23 eV (slightly redshifted compared to the PF at 3.26 eV) with a lifetime of 9.6 ms.7 Similarly, in TPA a weak DF signal is recorded at 3.16 eV (392 nm), slightly redshifted compared to the prompt signal (3.20 eV) with the 0.16 ms lifetime. The computed rISC rates in both crystals (krISC = 523 s−1 for T2/S1 transition in IPA and krISC = 202 s−1 for T3/S2 transition in TPA) indicate that the delayed fluorescence lifetime is expected on the ms timescale, which is in line with the experiments despite we are not considering the times associated with the transitions between the different triplets.
Kuno et al. attributed the delayed fluorescence observed in IPA to a triple–triplet annihilation (TTA) mechanism, in which two triplet excitons on neighbouring molecules combine to form one S1 state and one S0 state, with emission arising from the former.8 To assess the thermodynamic feasibility of TTA, we evaluated the energy losses in both the singlet and triplet manifolds (ESI,† Section S11). Efficient TTA requires a small positive energy loss in the singlet states and a significantly negative loss in the triplet states.47 However, our calculations reveal that energy losses in the singlet manifold are large and positive (for both S1 and S2 excitations), and those in the triplet manifold are also large and positive (ESI,† Section S11). These results suggest that the TTA mechanism is energetically unfavourable in these crystals.
4. Conclusions
Motivated by distinct solid-state luminescence properties of two benzene dicarboxyl acid crystals, TPA and IPA, we have systematically explored their excited-state relaxation mechanisms in solution and crystal environments. Given the weak interactions between π–π stacked dimers, the exciton couplings in both molecules are very small compared to the reorganisation energies associated with exciton transport. As a result, excited-state processes are primarily localised, and exciton transport occurs predominantly through incoherent exciton hopping.We explore light-activated nonradiative decay mechanisms for both systems, considering multireference methods. We found the conical intersections geometries associated with the decay to the ground state, that involve mainly distortions of a carboxyl group –CO bond stretching, out-of-plane motions of the carboxyl group with respect to the phenyl ring, and out-of-plane motion of the H-atom with respect to the –COO plane of carboxyl group. In line with the restricted access to the conical intersection (RACI) mechanism, the conical intersections in the crystal environment are not classically accessible. We also explore the intersystem crossing pathways in both systems, enabling relatively efficient populations of triplet states. In general, ISC is more efficient in IPA due to larger SOCs associated with the nature of the excited states involved and smaller energy gaps. We have shown that in both systems, the reverse intersystem crossing could happen through intermediate triplet states, transferring back a part of the population to the singlet manifold, explaining experimentally observed delayed fluorescence. This phenomenon is more efficient in IPA, due to a larger SOC and a smaller energy gap between states involved in the transition. According to our calculations, TTA seems to be energetically unfavourable.
Our results show that for an efficient triplet states population and solid-state phosphorescence in organic crystals, apart from the hindrance of the internal conversion, the positioning of substituents in benzene dicarboxylic acids plays a significant role. In the case of para substitution, the electronic delocalisation over the entire molecule is stabilised in the low-lying singlet and triplet states, resulting in ππ* being the lowest in energy. With meta substitution, electronic delocalisation is less favorable, leading to the stabilisation of mixed nπ*/ππ* or pure nπ* at their respective minima. This electronic effect significantly influences deactivation pathways. Our results indicate that the fluorescence, phosphorescence, and triplet-state population thought intersystem crossing processes are more efficient in IPA, which is in line with experimental observations. We believe this effect can be generalised to other substituents involved in electronic delocalisation with the central ring, as well as to more complex conjugated systems. This result might be of interest for the design of highly efficient room-temperature phosphorescent materials.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This research was supported by the Leverhulme Trust (RPG-2019-122) and EPSRC (EP/R029385/1). We used the Queen Mary's Apocrita HPC facility, supported by QMUL Research-IT and UCL High Performance Computing Facilities Myriad@UCL and Kathleen@UCL. We also acknowledge the use ARCHER UK National Supercomputing Service (EP/X035859/1) via the Materials Chemistry Consortium and the Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/T022213/1). Funding by the UK Research and Innovation under the UK government's Horizon Europe funding guarantee (grant number EP/X020908/2) is acknowledged.Notes and references
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y. L. Wu, Chem. Sci., 2021, 12, 767–773 RSC.
- R. Crespo-Otero, Q. Li and L. Blancafort, Chem. – Asian J., 2019, 14, 700–714 CrossRef CAS PubMed.
- Y. Niu, Q. Peng and Z. Shuai, Sci. China, Ser. B: Chem., 2008, 51, 1153–1158 CrossRef CAS.
- A. Sidat, F. J. Hernández, L. Stojanović, A. J. Misquitta and R. Crespo-Otero, Phys. Chem. Chem. Phys., 2022, 24, 29437–29450 RSC.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989–15995 RSC.
- H. Ma, W. Shi, J. Ren, W. Li, Q. Peng and Z. Shuai, J. Phys. Chem. Lett., 2016, 7, 2893–2898 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- E. R. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef.
- C. V. Caillie and R. D. Amos, Chem. Phys. Lett., 1999, 308, 249–255 CrossRef.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- C. Hättig and A. Köhn, J. Chem. Phys., 2002, 117, 6939–6951 CrossRef.
- C. Hättig, J. Chem. Phys., 2003, 118, 7751–7761 CrossRef.
- A. Köhn and C. Hättig, J. Chem. Phys., 2003, 119, 5021–5036 CrossRef.
- B. O. Roos, A. J. Sadlej, P. Å. Malmqvist, K. Andersson and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef.
- K. Andersson, P. Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- S. Vancoillie, M. G. Delcey, R. Lindh, V. Vysotskiy, P.-Å. Malmqvist and V. Veryazov, J. Comput. Chem., 2013, 34, 1937–1948 CrossRef CAS PubMed.
- M. Bailey and C. J. Brown, Acta Crystallogr., 1967, 22, 387–391 CrossRef CAS.
- R. Alcala and S. Martnez-Carrera, Acta Crystallogr., Sect. B, 1972, 28, 1671–1677 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395–502 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Dapprich, I. Komaromi, K. Suzie Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 121 Search PubMed.
- L. W. Chung, W. M. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H. B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com.
- S. Maeda, K. Ohno and K. Morokuma, J. Chem. Theory Comput., 2010, 6, 1538–1545 CrossRef CAS PubMed.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- T. Shiozaki, W. Györffy, P. Celani and H.-J. Werner, J. Chem. Phys., 2011, 135, 081106 CrossRef PubMed.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- N. Forsberg and P. Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- P. Åke Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef.
- B. Schimmelpfennig, Amfi, an atomic mean-field spin–orbit integral program. Computer code, University of Stockholm, 1996 Search PubMed.
- S. R. May, C. Climent, Z. Tao, S. A. Vinogradov and J. E. Subotnik, J. Phys. Chem. A, 2023, 127, 3591–3597 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr., H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. Hanson-Heine, P. H. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. V. Voorhis, J. M. Herbert, A. I. Krylov, P. M. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- M. Rivera, M. Dommett and R. Crespo-Otero, J. Chem. Theory Comput., 2019, 15, 2504–2516 CrossRef CAS PubMed.
- M. Rivera, M. Dommett, A. Sidat, W. Rahim and R. Crespo-Otero, J. Comput. Chem., 2020, 41, 1045–1058 CrossRef CAS PubMed.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299 CrossRef CAS.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrica, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W.-Z. Yu and T. Frauenheim, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- F. J. Hernández and R. Crespo-Otero, Annu. Rev. Phys. Chem., 2023, 74, 547–571 CrossRef PubMed.
- J. Aragó and A. Troisi, Phys. Rev. Lett., 2015, 114, 1–5 CrossRef PubMed.
- Z. Li, C. Roscini, R. Núñez, F. Teixidor, C. Viñas, E. Ruiz and J. G. Planas, J. Mater. Chem. C, 2024, 12, 2101–2109 RSC.
- L. Stojanović and R. Crespo-Otero, ChemPhotoChem, 2019, 3, 907–915 CrossRef.
- L. Stojanovic and R. Crespo-Otero, Molecules, 2022, 27, 522 CrossRef CAS PubMed.
- L. Stojanović and R. Crespo-Otero, J. Phys. Chem. C, 2020, 124, 17752–17761 CrossRef.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- G. Herzberg and E. Teller, Z. Phys. Chem., 1933, 21B, 410–446 CrossRef.
- J. M. Dos Santos, D. Hall, B. Basumatary, M. Bryden, D. Chen, P. Choudhary, T. Comerford, E. Crovini, A. Danos, J. De, S. Diesing, M. Fatahi, M. Griffin, A. K. Gupta, H. Hafeez, L. Hämmerling, E. Hanover, J. Haug, T. Heil, D. Karthik, S. Kumar, O. Lee, H. Li, F. Lucas, C. F. R. Mackenzie, A. Mariko, T. Matulaitis, F. Millward, Y. Olivier, Q. Qi, I. D. W. Samuel, N. Sharma, C. Si, L. Spierling, P. Sudhakar, D. Sun, E. Tankelevičiutė, M. Duarte Tonet, J. Wang, T. Wang, S. Wu, Y. Xu, L. Zhang and E. Zysman-Colman, Chem. Rev., 2024, 124, 13736–14110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00603a |
| This journal is © the Owner Societies 2025 |