
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUranyl fluorescence in acidic solution: quenching effects by tetramethylammonium (TMA+)†
Thomas D.
Persinger
 *a,
Michael C.
Heaven
b and
Richard E.
Wilson
a
*a,
Michael C.
Heaven
b and
Richard E.
Wilson
a
aChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA. E-mail: tpersinger@anl.gov
bEmory University, Atlanta, Georgia 30322, USA
First published on 30th April 2025
Abstract
The quenching of uranyl luminescence by various cation species was studied in aqueous media at low pH. Solutions with different nitrate salts, held at constant uranyl nitrate, nitric acid, and ion concentration, were tested to examine the quenching effects of the cations from the nitrate salts. Alkali metal (Li+, Na+, Rb+) and quaternary ammonium cations (NH4+), (CH3)4N+ (TMA+), (C2H5)4N+ (TEA+) were investigated. Solutions containing TMA+ reduced the lifetime of uranyl fluorescence significantly more than the other cations. Uranyl emission spectra also showed that TMA+ increased the complex formation between uranyl and nitrate ions. Fluorescence decay lifetime measurements for most solutions yielded values between 1.4–1.9 μs at 20 °C, while 1.8 M TMA+ reduced the lifetime of uranyl fluorescence to 0.6 μs. Decay rate versus concentration data (Stern–Volmer plots) indicated a dynamic quenching process with increasing fluorescence decay rates at higher cation concentrations for Li+, TMA+, and TEA+. The temperature dependencies of the decay rates and the kinetics in D2O were also examined.
1. Introduction
Solution chemistry involving the uranyl ion (UO22+) and its interactions with the surrounding molecules in aqueous environments is important for research in nuclear waste management, environmental remediation, and heavy element separations.1 Speciation effects on UO22+ in solution is a multi-faceted problem that is still not completely understood. The complexation of UO22+ in nitric acid solution has been explored,2,3 where the low-pH environment results in the formation of UO22+–nitrate complexes. However, the equilibrium of these complexes in solution can be impacted by many factors. The introduction of additional molecules, both as electrolytes and coordinating ligands in solution leads to a variety of physical and chemical interactions with UO22+ that are difficult to predict. Photoexcitation of UO22+ populates a highly reactive excited state where it behaves as a strong oxidizer,4 leading to a variety of interactions with nearby molecules and providing alternative pathways for electronic relaxation. The quenching of UO22+ luminescence by some ions such as carbonate and chloride is efficient enough that even at low concentrations, they can make it difficult to determine uranyl concentrations in solution. Transient absorption spectroscopy (TAS) was used to understand the quenching mechanism of chloride and bromide. It was found that the halogen anions interacted with UO22+ through the equatorial plane and the uranyl solvent shell influenced the anion's ability to quench the luminescence.5 To effectively use uranyl luminescence for monitoring processes we must understand the mechanisms by which UO22+ interacts with other solute molecules, particularly cations that may facilitate the formation of ion-pairs or other ion–ion interactions.Tetramethylammonium (TMA+) has been shown to uniquely modify UO22+ speciation in solution, presumably through the formation of ion-pair complexes. Hydrolysis experiments of UO22+ can be challenging to perform because, under alkaline conditions, insoluble uranyl hydroxide salts begin to form. However, TMA+ was found to be an effective supporting electrolyte in solution as its presence inhibited the formation of insoluble uranyl salts.6 Clark et al. used this interaction between UO22+ and TMA+ to spectroscopically characterize larger uranyl hydroxide species (UO2(OH)n2−n (n = 4, 5)).7 They also discovered that this behavior was unique to TMA+ and that larger and smaller ammonium ions did not prevent uranyl salts from precipitating out of the solution. It appears that TMA+ participates in some interaction with UO22+ in solution, but the details are not yet known. The interaction with TMA+ has not been investigated in a low-pH solution, where the uranyl does not form hydroxides. Studies at low pH may provide new insights concerning the nature of the UO22+/TMA+ interaction and the possibility of ion association.
Time-resolved and steady-state fluorescence spectroscopy are powerful techniques that are used to investigate the interactions between UO22+ and the surrounding molecules in solution, facilitating the measurement of their thermodynamic formation constants and highlighting the impact of the local chemical environment on the UO22+ luminescence spectrum. Extensive studies have shown that UO22+ is subject to many types of quenching interactions, including electron transfer,5,8,9 hydrogen abstraction,10,11 self-quenching,12 and exciplex formation.2,13 These processes result in faster fluorescence decay rates for the excited UO22+ and studying how temperature and solute concentrations affect the rates can provide insight into the mechanism by which the UO22+ fluorescence is being quenched.14 In the present study, we explore the fluorescence quenching abilities of alkali metal and quaternary ammonium cations. Measurements were taken over a range of temperatures. Solvent effects were probed by comparing results obtained in H2O and D2O. Concentration dependences were determined to differentiate between static and dynamic quenching processes.
2. Experimental methods
2.1. Sample preparation
The UO2(NO3)2·6H2O used for these experiments was obtained from an Argonne 0.2 M stock solution prepared in 20 mM nitric acid. The NaNO3, LiNO3, RbNO3, (CH3)4NNO3, and (C2H5)4NNO3 salts (Fisher Chemicals), and the deuterated solutions D2O, DNO3 and (CD3)4NNO3 (Sigma Aldrich) were used to make 2 M stock solutions by dissolving the appropriate mass of nitrate salt in a 20 mM nitric acid solution to keep a consistent acid concentration. Varied salt concentrations for each sample were made by mixing the appropriate volumes of these stock solutions into 2 mL vials. For storage, the vials were sealed using parafilm. For optical measurements the samples were transferred via pipette into quartz fluorescence cuvettes.2.2. Instrumentation
All fluorescence experiments were carried out using an FLS1000 photoluminescence spectrometer (Edinburgh Instruments), equipped with an Opolette 355 (Opotek) OPO tunable pulsed laser source. Samples were placed inside fluorescence cuvettes and a variable-temperature stage was used to study fluorescence from 5–40 °C. Quenching behavior in the solution was investigated via lifetime measurements of uranyl transitions using the pulsed (10 ns) OPO source. Continuous stirring of the solution was used to avoid the effects of local heating caused by the absorption of the excitation laser pulses. The fluorescence was dispersed by a monochromator and the wavelength-selected light was collected using a photomultiplier tube (Hamamatsu, PMT-900) with a multichannel scaling (MCS) photon counting method. The signals were processed and plotted using the in-house Edinburgh program (Fluoracle). A Xenon lamp source was used in conjunction with excitation and emission monochromators to collect the excitation and emission spectra of the uranyl species.3. Results and analyses
3.1. Steady-state emission
The quenching properties of alkali metal and quaternary ammonium cations were explored using time-resolved laser fluorescence spectroscopy (TRLFS) and steady-state emission spectroscopy. At a concentration of 20 mM [UO22+] and 20 mM [HNO3] (pH = 1.7), hydroxide complexation with uranyl was avoided. The primary complexation with the uranyl aqua ion in solution was with the nitrate ion (NO3−), which formed uranyl nitrate complexes (UO2(NO3)n2−n) described by the following equations of equilibrium,3UO22+ + NO3− ↔ UO2NO3+, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β1 = −0.45 β1 = −0.45 | (1) |
| UO22+ + 2NO3− ↔ UO2(NO3), logβ2 = −1.4 | (2) |
The samples prepared in this study contained nitrate salt concentrations between 0–1.8 M. As nitrate concentration in the solution increased, the emission spectra broadened due to increasing uranyl nitrate complexation.3,15,16 The steady-state emission spectra in Fig. 1 show the UO22+ fluorescence from the lowest excited electronic state T1(3Δ) → S0(1Σ+). An excitation energy of 394 nm was used, which promoted UO22+ into the T2(3Φ) excited electronic states before quickly undergoing non-radiative decay into the lower T1(3Δ) state.9,17,18 The emission spectra show the ground state symmetric stretch vibrational progression. The spectra in Fig. 1a (left) show that the addition of all nitrate salts resulted in the broadening of emission bands due to complexation with nitrate. Uranyl nitrate complexation red-shifted the emission spectra, and the presence of multiple uranyl species caused the observed spectral broadening. Fig. 1b shows that the cations did not have a uniform effect on uranyl complexation. The replacement of Na+ with TMA+ resulted in greater complexation, which could be explained by TMA+ pushing the equilibrium of eqn (1) and (2) to the right. This shift in equilibrium could be due to ion interactions between UO22+ and TMA+.
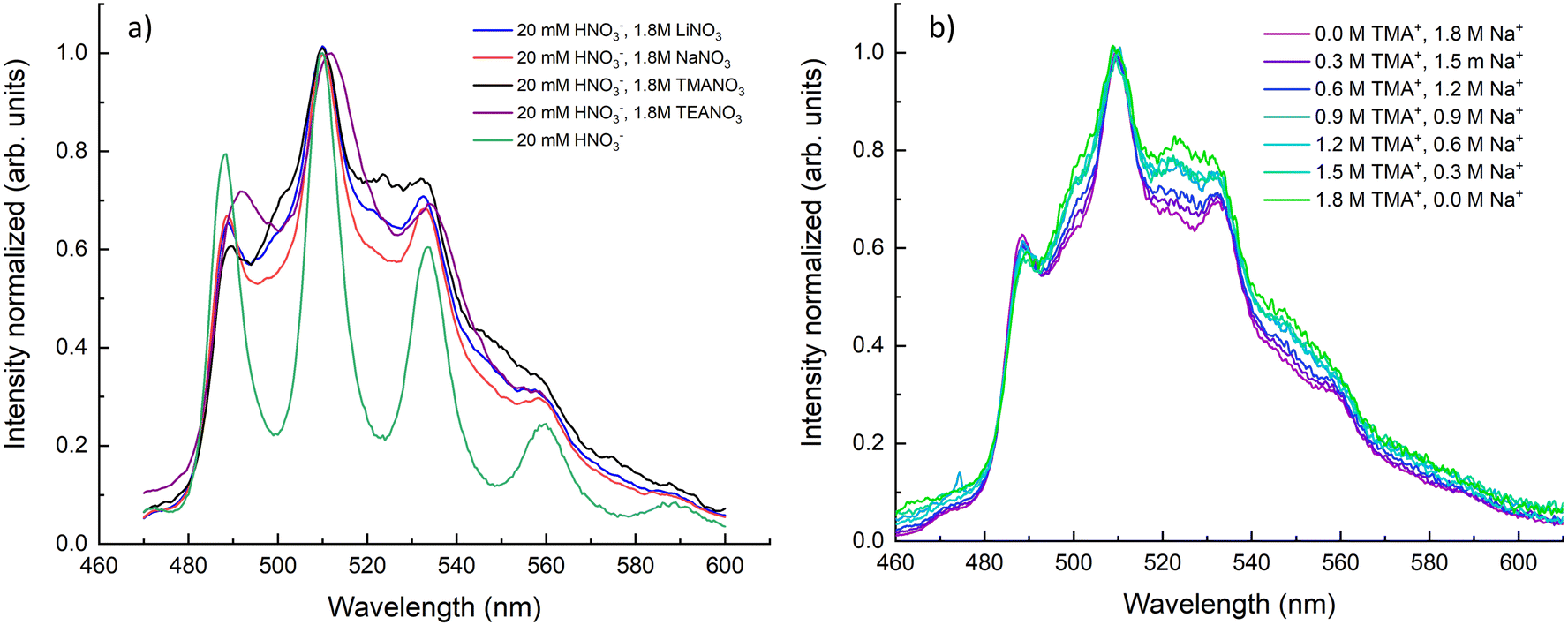 | ||
| Fig. 1 Steady-state emission spectra of uranyl from 460–600 nm. (a) The emission from solutions containing Li+, Na+, TMA+, and TEA+ nitrate salts are compared to uranyl nitrate in nitric acid. (b) Change in emission bands as NaNO3 is replaced with TMANO3. Excitation wavelength was 394 nm for all samples. | ||
3.2. Time-resolved laser fluorescence spectroscopy (TRLFS)
For the measurement conditions used in the present study, it is pertinent to consider the speciation of UO22+ in the presence of relatively high nitrate concentrations (up to 1.8 M). Moulin et al.3 analyzed uranyl emission spectra taken in low pH nitrate solutions to probe the speciation. For 1.8 M NO3−, their data show that the speciation would be 50% UO22+, 40% UO2+NO3, and 10% UO2(NO3)2 (see Fig. 2 of ref. 3). | ||
| Fig. 2 Time-resolved fluorescence curves for UO22+ in 1.8 M NaNO3 solution. The red and black curves correspond to fluorescence detection at 500 and 510 nm. The inset shows the difference between these curves. | ||
In the TRLFS experiments the control conditions (aqueous solution with 20 mM [UO22+] and 20 mM [HNO3]) yielded a fluorescence lifetime of 1.9 μs at 20 °C. The addition of 1.8 M Q+ (Q+ = Li+, Na+, Rb+) and 1.8 M NO3− brought the fluorescence lifetime of uranyl down to approximately 1.5 μs. This slight decrease in the fluorescence lifetime could be explained by weak dynamic quenching facilitated by the addition of cations and/or the contributions of fluorescence from UO22+(NO3−)n complexes. However, there is evidence that exponential regions of the fluorescence decay were not significantly influenced by the formation of nitrate complexes. Fig. 1 of ref. 3 shows that fluorescence detection at 510 nm favors the bare UO22+ ion while detection at 500 nm favors the complexes. Fig. 2 (present work) shows time-resolved fluorescence traces for a 1.8 M solution of NaNO3 recorded using detection of 510 and 500 nm light. Non-exponential kinetics were observed at short times, and the data for 510 nm detection have been shifted by 300 ns to superimpose the exponential decay regions. The inset in Fig. 2 shows the difference between the two fluorescence signals. From this plot it is apparent that the decay characteristics of the bare UO22+ ion and the nitrate complexes were closely similar.
As discussed below, many of the time-resolved fluorescence curves deviated from single exponential decay at the earliest times, but the longer-lived fluorescence was well represented by exponential decay. Fig. 3 shows a typical example, where the exponential decay portion of the curve corresponded to the fluorescence decay (T1(3Δ) → S0(1Σ+)) of the bare and complexed uranyl species. The analyses of the quenching kinetics used the single exponential decay regions of the curves to determine the decay rates.
 | ||
| Fig. 3 Semi-log plot of the time-resolved fluorescence from a uranyl solution that contained 1.8 M NaNO3 at 20 °C. | ||
The quaternary ammonium cation's ability to quench uranyl fluorescence was also studied. While NH4+ and TEA+ were found to be inefficient quenchers, the presence of TMA+ significantly reduced the lifetime. Fig. 4 shows the fluorescence decay curves from solutions containing 1.8 M of Q+(NO3)− (Q+ = Li+, Na+, Rb+, TMA+, TEA+). The presence of 1.8 M TMA+ in solution reduced the fluorescence lifetime to 580 ns, revealing TMA+ to be a markedly more effective quencher. Similar to the results observed by Clark et al.,7 smaller or larger ammonium cations were less effective quenching species (comparable to the alkali metal cations). These results indicate that TMA+ is involved in a unique interaction with UO22+. Hence, investigating how the presence of TMA+ affects uranyl fluorescence decay under changing conditions such as temperature and concentration may provide new insights concerning the TMA+ quenching mechanism and its ability to prevent hydroxide precipitation in alkaline solutions.
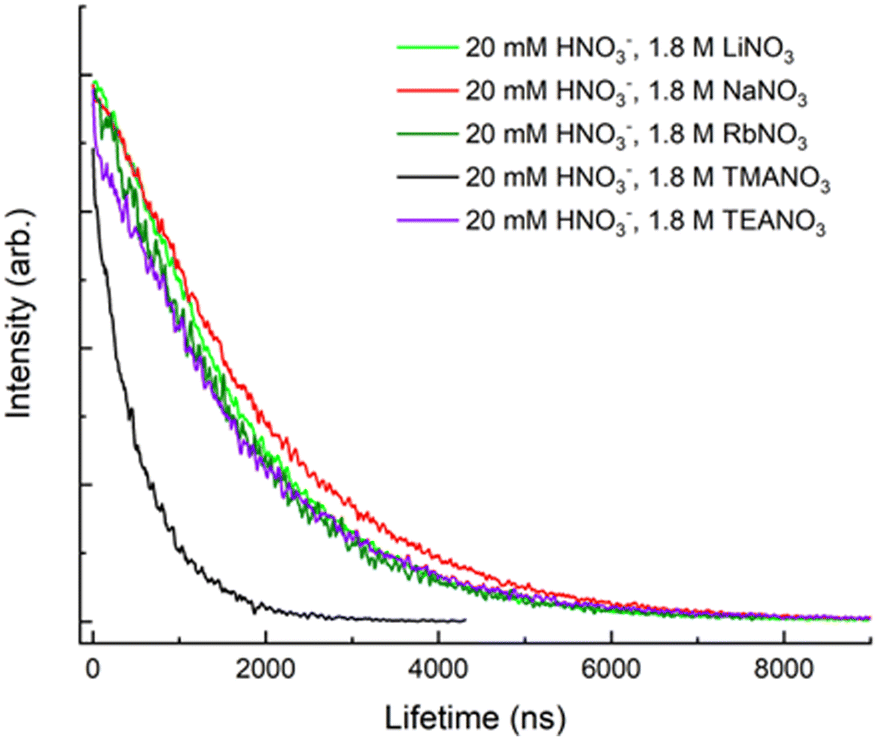 | ||
| Fig. 4 Fluorescence decay of uranyl at 510 nm, with an excitation wavelength of 394 nm and T = 20 °C. Each solution had a salt concentration of 1.8 M. | ||
3.3. Temperature-dependant measurements
To explore the quenching effects of the cations thermodynamically, the temperature dependence of the decay rates was measured using solutions containing 1.8 M of quenching ions. Fig. 5a shows the decay rates of UO22+ fluorescence in H2O and their dependence on temperature was consistent with the Arrhenius expression.2,19 The expression in the form: | (3) |
:1. This would be expected if quenching by TMA+ was dominant over the quenching by H2O/D2O.
 | ||
| Fig. 5 Fluorescence decay rates as a function of temperature. (a) Measurements were carried out in H2O with added nitrate salt concentrations of 1.8 M. (b) Measurements were carried out in D2O with added nitrate salt concentrations of 1.8 M. | ||
| Ion | ln(A)/H2O | E a/H2O, kJ Mol−1 | ln(A)/D2O | E a/D2O, kJ mol−1 |
|---|---|---|---|---|
| Li+ | 27.6 | 34.3 | 28.1 | 36.3 |
| Na+ | 30.2 | 41.3 | 30.2 | 42.2 |
| Rb+ | 30.2 | 41.0 | ||
| NH4+ | 30.7 | 42.4 | ||
| TMA+ | 24.9 | 25.8 | 24.3 | 24.8 |
| d-TMA+ | 24.6 | 25.9 | ||
| TEA+ | 26.2 | 31.0 |
3.4. Concentration-dependant measurements
The relationship between quencher concentration and uranyl fluorescence decay rate was used to investigate the quenching process between UO22+ and the cations. Experiments were carried out at a fixed temperature (20 °C) for quenching ion concentrations ranging from 0 to 1.8 M. Decay rate versus concentration data (referred to as Stern–Volmer plots in the following) were fitted using the expression:| Γ = Γ0 + k(Q+)[Q+] | (4) |
 | ||
| Fig. 6 Decay rate vs. Li+ concentration for uranyl fluorescence from H2O solutions. Measurements taken at 20 °C. | ||
The observable quenching rate constants measured in H2O and D2O are listed in Table 2. Due to the dominance of solvent quenching, we could not extract statistically significant quenching rate constants for Na+, Rb+ or NH4+. Quenching by Li+ was fast enough for determination of the rate constant, and the Stern–Volmer plot for TEA+ yielded a rate constant that was similar to that of Li+ (cf.Table 2). Given that NH4+ and TEA+ were found to be ineffective quenchers, it was surprising to discover that TMA+ was the most effective quencher examined in this study. Again, we saw that deuteration of the solvent had a smaller effect on the quenching rate for TMA+ as compared to the alkali metals. Li+ appears to quench by a dynamic process that involves the solvent, while TMA+ participates in quenching directly with UO22+.
| Ion | k(Q+)/104 dm3 mol−1 s−1 | k(Q+)/104 dm3 mol−1 s−1 |
|---|---|---|
| Solvent = H2O | Solvent = D2O | |
| a The quenching constant for the cation was not measurable. | ||
| Li+ | 11.7 | 5.0 |
| Na+ | ||
| Rb+ | ||
| NH4+ | ||
| TMA+ | 100 | 111 |
| TEA+ | 10.6 | |
The Stern–Volmer (S–V) plots for TMA+ and TEA+ also indicated that quenching was not purely collisional as their data deviated from a straight line. Fig. 7 shows S–V plots for TMA+ in H2O and D2O. The reason for this negative curvature is unclear, but we have assumed that the quenching rate constant can be defined by the linear coefficient of a quadratic fit to the S–V data. The rate constants determined using this method are listed in Table 2.
 | ||
| Fig. 7 Decay rate vs. TMA+ concentration for uranyl fluorescence from H2O and D2O solutions. Measurements taken at 20 °C. | ||
3.5. Non-exponential decay kinetics
The deviation from single exponential decay observed in solutions where the quenching was slow was another surprising detail of the decay kinetics. This effect was noted even for stock solutions that contained just 0.06 M nitrate, where the concentration of uranyl nitrate complexes would have been insignificant.Several steps were taken to ensure that the presence of a non-exponential decay component was not due to signal saturation of the PMT detector. When photon signals are counted using a MCS method, the dynamic range of fluorescence intensity for species with decay lifetimes <1 μs is low. To avoid saturation of the detector, emission intensities were kept low using neutral density filters. For additional verification, the fluorescence decay from a sample containing 20 mM [UO22+], 20 mM [HNO3] and 1.8 M [NaNO3] in H2O was measured using an analogue detection method with a far greater dynamic range. The non-exponential decay was observed in this alternative detection method, reproducing the temporal behavior obtained using MCS detection.
It is well established that, following excitation at short wavelengths (e.g., λ < 400 nm), UO22+ undergoes rapid non-radiative relaxation to the emitting state.15,16 Haubitz et al.9 assigned the state of UO22+ excited by light near 310 nm to T2(3Φg) which relaxes to the T1(3Δg) emitting state. For water-coordinated UO22+ they reported a non-radiative rate for transfer from T2(3Φg) to T1(3Δg) of 3.5 × 1011 s−1. Ghosh et al.18 followed the ultra-fast relaxation kinetics for UO22+ in nitric acid solutions using 400 nm excitation. They proposed that the 1Φg state was initially excited, with relaxation down to the emitting 3Δg state occurring on a picosecond timescale. The relaxation kinetics observed using ultrafast techniques occurred at rates that were too fast to be observed using the equipment available for the present study, and could not account for the observed deviations from single exponential decay.
We observed decay curves where there was a slower rate of decay at earlier times. This indicates that there is a minor, slow relaxation channel that feeds population to the emitting state. Consequently, the non-exponential decay curves have been simulated using the kinetic scheme shown in Fig. 8. We propose that there are overlapping electronic transitions excited at wavelengths near 394 nm. The dominant absorption populates excited states that are subject to rapid non-radiative relaxation. A weaker transition populates a state that has a lower rate of relaxation to the emitting state, resulting in a transfer that was slow enough to be observed. Fig. 8 shows an energy level scheme for this model, where state 3 represents excitation to T2(3Φg) (and other states that relax rapidly to T1(3Δg)). Γ31 is the rapid relaxation channel which is fast enough that we can consider this to be direct excitation of state 1. State 2 is the state (or group of states) with the slow relaxation rate. For this situation the time dependence of the emission signal is given by:
 | (5) |
 | ||
| Fig. 8 Proposed energy level scheme for the non-exponential fluorescence decay of uranyl. See text for details. | ||
4. Discussion
There have been several previous studies of the quenching of UO22+ fluorescence in the presence of metal cations. Moriyasu et al.20 noted that the alkali metal cations were closed-shell species that could not be expected to affect quenching by electron or energy transfer mechanisms. They reported that the ions Li+, Na+, K+, Rb+ and Cs+ had quenching rate constants that were less than an upper bound of 104 dm3 mol−1 s−1 at a temperature of 25 °C. The present results for Na+ and Rb+ are consistent with this observation, but the quenching rate constant for Li+ was found to be an order of magnitude greater than the upper bound of Moriyasu et al.20 Furthermore, it was possible to characterize the Li+ quenching rate in D2O, where the rate constant was approximately half the value measured in H2O. The Li+ measurements were repeated using several different LiNO3 solutions to check for the presence of impurities. These experiments yielded consistent results and no evidence of contaminants. It is difficult to rationalize why Li+ would be a more effective quencher than either Na+ or Rb+. This is clearly inconsistent with a heavy element effect. The marked decrease of the rate constant on switching from H2O to D2O indicates that the solvent is involved in the quenching mechanism. It is also of interest to note that the effective activation energy for quenching by Li+ is lower than that for quenching by H2O or D2O alone.Time-resolved fluorescence curves that deviated from single exponential decay were observed under conditions where the quenching rates were low. We speculate that this behavior is a consequence of the excitation of overlapping absorption bands. Quantitative analysis of the decay curves was achieved using a model where one of the initially excited states had a measurable rate for non-radiative relaxation into the emissive T1(3Δ) electronic state. There is no precedent for the slow relaxation rate needed for this kinetic model. We note, however, that many excited electronic states are accessible at an excitation energy of 25380 cm−1 (394 nm). This is evident from the many published discussions of the electronic states of UO22+, including the work of Denning,17 Ghosh,18 De Houwer and Gorller-Walrand,21 and Haubitz and co-workers.9 In a study of cryogenic matrix isolated UO2Cl2, Jin et al.22 observed seven electronically excited states of UO22+ at energies in the range of 20338 to 26000 cm−1. Decay channels with rates on the order of 1.5 × 106 s−1 were not observed as the fluorescence decay in the matrix was bi-exponential with lifetimes of 50(5) and 260(20) μs.
Quenching by the quaternary ammonium cations NH4+, TMA+ and TEA+ was also found to follow a non-monotonic dependence on the size of the cation. For these molecules, it might be expected that electronic to vibrational energy transfer would be an available deactivation mechanism. However, if this was a significant channel it would be probable that TEA+, having the highest density of vibrational states, would be the most efficient quencher. Hydrogen abstraction is another possible quenching mechanism for the quaternary ammonium cations, but this is not supported by the available data. Comparing the fluorescence decay rates for TMA+ and d-TMA+ in D2O, deuteration of TMA+ had a weak effect on the fluorescence decay rate, reducing the value at 20 °C from 1.36 × 106 s−1 to 1.17 × 106 s−1 (1.8 M solutions). This 4% change does not seem to be consistent with H/D atom transfer.
Conclusions
We have analyzed the emission spectra and time-resolved fluorescence decay of UO22+ in nitric acid and studied the quenching ability of various nitrate salts Q+(NO3)− (Q+ = Li+, Na+, Rb+, NH4+, TMA+, and TEA+) on uranyl. The addition of 1.8 M NO3− resulted in the broadening of emission spectra associated with an increase in uranyl nitrate complexation and the extent of complexation was dependent on the cation of the nitrate salt. In agreement with previous studies, we found that quenching of uranyl fluorescence by Na+ and Rb+ is negligibly slow under low pH conditions. Quenching by Li+ was found to be slow, but measurable in both H2O and D2O solutions. The solvent dependence of the Li+ quenching rate constant at 20 °C suggests that an interaction between Li+ and H2O/D2O is part of the dynamic quenching mechanism.Quenching of uranyl by the quaternary ammonium cations was found to be insignificant for NH4+, slow for TEA+ and relatively fast for TMA+ (by an order of magnitude as compared to TEA+). In this context, it is of interest to note that NH4+, TMA+ and TEA+ were previously investigated for their ability to inhibit the formation of insoluble uranyl hydroxide complexes under alkaline conditions. Clark et al.7 concluded that, “We have shown that monomeric uranyl hydroxides, UO2(OH)42− and UO2(OH)53−, can be stabilized from uranate formation using the Me4N+ (TMA+) cation, thereby allowing for a full characterization under highly alkaline conditions. TMA+ seems to be unique in this regard, as larger or smaller ammonium ions were unsuccessful at preventing precipitation.” Hence, it appears that there may be a link between the quenching behavior and the selective inhibition of uranyl precipitation in the presence of TMA+.
Author contributions
Thomas D. Persinger: conceptualization, investigation, formal analysis, visualization, and writing – original draft and review & editing. Michael C. Heaven: investigation, formal analysis, and writing – review and editing. Richard E. Wilson: conceptualization, funding acquisition, supervision and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted at ANL, operated by UChicago Argonne LLC for the United States Department of Energy and supported by the U.S. Department of Energy Office of Science, Office of Basic Energy Sciences, Chemical Sciences Geological and Biosciences Division, Heavy Element Chemistry program under Contract DE-AC02-06CH11357. Thanks to Srikanth Nayak for preparing the solutions that were used in this project.Notes and references
- Ion exchange and solvent extraction: a series of advances, ed. B. A. Moyer, 2010, vol. 19.
- M. D. Marcantonatos, Photochemistry and exciplex of the uranyl ion in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1980, 76(5), 1093–1115 RSC.
- C. Moulin, P. Decambox, P. Mauchien, D. Pouyat and L. Couston, Direct Uranium(VI) and Nitrate Determinations in Nuclear Reprocessing by Time-Resolved Laser-Induced Fluorescence, Anal. Chem., 1996, 68(18), 3204–3209 CrossRef CAS.
- A. B. Yusov and V. P. Shilov, Reduction of the photoexcited uranyl ion by water 1. U(IV) and H2O2 formation, Russ. Chem. Bull., 2000, 49(2), 285–290 CrossRef CAS.
- T. Haubitz, B. Drobot, S. Tsushima, R. Steudtner, T. Stumpf and M. U. Kumke, Quenching Mechanism of Uranyl(VI) by Chloride and Bromide in Aqueous and Non-Aqueous Solutions, J. Phys. Chem. A, 2021, 125(20), 4380–4389 CrossRef CAS PubMed.
- D. A. Palmer and C. Nguyen-Trung, Aqueous uranyl complexes. 3. Potentiometric measurements of the hydrolysis of uranyl(VI) ion at 25 °C, J. Solution Chem., 1995, 24(12), 1281–1291 CrossRef CAS.
- D. L. Clark, S. D. Conradson, R. J. Donohoe, D. W. Keogh, D. E. Morris, P. D. Palmer, R. D. Rogers and C. D. Tait, Chemical Speciation of the Uranyl Ion under Highly Alkaline Conditions. Synthesis, Structures, and Oxo Ligand Exchange Dynamics, Inorg. Chem., 1999, 38(7), 1456–1466 CrossRef CAS.
- M. D. Marcantonatos and M. Deschaux, Electron transfer and aqua cation-cation complex formation in the excited state of the uranyl ion, Chem. Phys. Lett., 1980, 76(2), 359–365 CrossRef CAS.
- T. Haubitz, S. Tsushima, R. Steudtner, B. Drobot, G. Geipel, T. Stumpf and M. U. Kumke, Ultrafast Transient Absorption Spectroscopy of UO22+ and [UO2Cl]+, J. Phys. Chem. A, 2018, 122(35), 6970–6977 CrossRef CAS PubMed.
- H. D. Burrows and S. J. Formosinho, Photochemical hydrogen abstractions as radiationless transitions. Part 3. Theoretical analysis of hydrogen abstraction by excited uranyl (UO22+) ion, J. Chem. Soc., Faraday Trans. 2, 1977, 73(2), 201–208 RSC.
- S. J. Formosinho, H. D. Burrows, M. da Graca Miguel, M. E. D. G. Azenha, I. M. Saraiva, A. C. D. N. Ribeiro, I. V. Khudyakov, R. G. Gasanov, M. Bolte and M. Sarakha, Deactivation processes of the lowest excited state of [UO2(H2O)5]2+ in aqueous solution, Photochem. Photobiol. Sci., 2003, 2(5), 569–575 CrossRef CAS PubMed.
- M. D. Marcantonatos, Dual luminescence of uranyl and self-quenching in aqueous acidic solution, Inorg. Chim. Acta, 1978, 26(1), 41–46 CrossRef CAS.
- M. D. Marcantonatos, The exciplex formation between uranyl species further evidenced by quenching data of the UO22+ luminescence, Inorg. Chim. Acta, 1977, 24(2), L37–L39 CrossRef CAS.
- R. Nagaishi, T. Kimura, J. Inagawa and Y. Kato, Isotope and temperature effects on photochemical reactions of uranyl ion in H2O-D2O mixtures, J. Alloys Compd., 1998, 271–273, 794–798 CrossRef CAS.
- C. P. Baird and T. J. Kemp, Luminescence, spectroscopy, lifetimes and quenching mechanisms of excited states of uranyl and other actinide ions, Prog. React. Kinet., 1997, 22(2), 87–139 CAS.
- H. D. Burrows and T. J. Kemp, Photochemistry of the uranyl ion, Chem. Soc. Rev., 1974, 3(2), 139–165 RSC.
- R. G. Denning, Electronic Structure and Bonding in Actinyl Ions and their Analogs, J. Phys. Chem. A, 2007, 111(20), 4125–4143 CrossRef CAS PubMed.
- R. Ghosh, J. A. Mondal, H. N. Ghosh and D. K. Palit, Ultrafast Dynamics of the Excited States of the Uranyl Ion in Solutions, J. Phys. Chem. A, 2010, 114(16), 5263–5270 CrossRef CAS PubMed.
- M. Moriyasu, Y. Yokoyama and S. Ikeda, Quenching of uranyl luminescence by water molecule, J. Inorg. Nucl. Chem., 1977, 39(12), 2211–2214 CrossRef CAS.
- M. Moriyasu, Y. Yokoyama and S. Ikeda, Quenching mechanisms of uranyl luminescence by metal ions, J. Inorg. Nucl. Chem., 1977, 39(12), 2205–2209 CrossRef CAS.
- S. De Houwer and C. Gorller-Walrand, Influence of complex formation on the electronic structure of uranyl, J. Alloys Compd., 2001, 323–324, 683–687 CrossRef CAS.
- J. Jin, R. Gondalia and M. C. Heaven, Electronic Spectroscopy of UO2Cl2 Isolated in Solid Ar, J. Phys. Chem., 2009, 113, 12734–12738 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: https://doi.org/10.1039/d5cc01299f |
| This journal is © the Owner Societies 2025 |