
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient reverse intersystem crossing process exploiting non-bonding states in an inverted singlet–triplet gap system†
Hwon
Kim
 and
Seung Kyu
Min
*
and
Seung Kyu
Min
*
Department of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulju-gun, Ulsan 44919, Republic of Korea. E-mail: skmin@unist.ac.kr
First published on 24th March 2025
Abstract
Reverse intersystem crossing (rISC) is an essential process in organic light-emitting diodes to populate singlet excited states from non-emissive triplet states. A small or negative singlet–triplet energy gap and a large spin–orbit coupling between low-lying singlet and triplet states are key requirements to enhance the rISC rate. Here, we present a molecular design exploiting the n–π* excited state to maximize the efficacy of the rISC process for efficient light emitters using thermodynamic and kinetic calculations validated with high-level quantum chemical methods. Heptazine-based molecules with carbonyl groups attached are shown to possess a reasonable singlet energy gap for blue-light emission with the energy level of the n–π* triplet state modulated by addition of electron withdrawing or donating groups to achieve the optimal energy level ordering of T(π–π*) > T(n–π*) > S1, leading to enhanced spin–orbit coupling between the lowest triplet and singlet states with an inverted energy gap.
1 Introduction
Reverse intersystem crossing (rISC) is a process involving transition from a state with higher spin multiplicity to a state with lower spin multiplicity.1–4 This is the key process for organic light-emitting diodes (OLEDs) utilizing thermally activated delayed fluorescence (TADF)5–7 due to the production of singlet states from non-emissive triplet states, increasing the electricity-to-light conversion efficiency. The rate of rISC is thus an important criterion in evaluating the efficacy of potential materials for optoelectronics.8–13 The two main strategies that are in use to facilitate rISC are (i) lowering the singlet–triplet gap (ΔEST)14–18 and (ii) aiming for S1 and T1 states with different characters19–23 to allow strong spin–orbit coupling (SOC).Finding molecules with small ΔEST that facilitates the rISC is non-trivial as the exchange energy puts the S1 state above the T1 state in the single reference description of the many-body electronic wavefunction. Molecules possessing the desired energy level usually show excited states with charge transfer (CT) character, with small overlap between the donor and acceptor orbitals resulting in low exchange energy. Recent works have also shed light on molecules with negative ΔEST,17,24–34 which makes rISC thermodynamically favored and thus are recognized as promising candidates for OLED materials. From a kinetic point of view, the main challenge is the weak SOC between the lowest singlet and triplet states. As OLED materials usually lack heavy atoms that strengthen the SOC, the rISC rate is expected to be slow even when energetically feasible. Moreover, states with the same electronic configuration have vanishing SOC between them because the change in spin angular momentum without the change in orbital angular momentum violates the conservation of momentum, known as the El-Sayed rule.35 Search for systems that have S1 and T1 states with different characters has thus been a popular means of achieving appreciable SOC, with locally excited (LE) configurations19–23 mixing with CT type configurations in either the lowest excited singlet or triplet state.
A family of molecules derived from phenalene possessing negative ΔEST that emanates from the small overlap of orbitals and coupling of the doubly excited configuration with the lowest singlet excited state17 have emerged as prospective emitters, owing to the appeal of the thermodynamic stability of the S1 state. The work conducted by De Silva17 demonstrated that using correlated electronic structure methods that take doubly excited states into consideration, such as equation of motion coupled cluster singles and doubles (EOM-CCSD) and algebraic diagrammatic correction (ADC(2)), is crucial for predicting the correct ΔEST, inspiring fruitful high-throughput computational studies in pursuit of discovering such inverted ΔEST (IST) emitters.24,28,36 The experimental verification of the inverted gap26 has also motivated further development and investigation of IST emitters. The computational studies to optimize the properties of IST molecules are mainly focused on finding IST molecules with appreciable oscillator strength,24 as the symmetry of the IST molecules derived from phenalene leads to weak transition dipoles. Direct kinetic assessment30,37 has also been performed to shed light on the importance of vibronic coupling in enhancing the transition dipole and the higher excited states that can become intermediates to enhance the spin–orbit coupling (SOC). Enhancing the weak SOC of singlet and triplet states sharing the same configuration is particularly not straightforward for the planar phenalene derivatives, as the mixing of LE and CT characters to increase the SOC is not an option without perpendicular moieties bonded to the molecule and adding branches to introduce low-lying excited states with CT character may bring the ΔEST to a positive value.24,38 Moreover, molecules with perpendicular donor–acceptor geometry have a propensity to exhibit broad emission due to high reorganization energy, which is undesirable for optoelectronic devices.39
Recently, molecules that bear carbonyl groups have been introduced as promising TADF emitters that show enhanced SOC from the non-bonding electrons.40,41 Non-bonding electrons in the carbonyl group are naturally in a perpendicular orientation from the π electrons along the molecular plane and thus allow finite SOC.42 Even though attaching carbonyl groups to IST emitters has been tried in previous studies, the focus was mainly on their effect on the ΔEST and the oscillator strength, rather than on the nature of the excited states and the SOC strength.24 The work of Pollice et al.24 has shown that the ΔEST of IST emitters can be kept negative with reasonable robustness, and thus investigating the effect of non-bonding electrons on the SOC and the electronic state manifold of IST emitters is a worthwhile research topic to optimize the properties of IST molecules for optoelectronic applications.
In this work, we propose a molecular design that can satisfy both IST and appreciable SOC based on heptazine derivatives with the carbonyl group attached to give low-lying n–π* states. While the HOMO and LUMO show delocalized π and π* characters, respectively, the non-bonding orbital of the carbonyl group (HOMO−1) is perpendicular to the molecular plane of heptazine, as shown in Fig. 1a. The key strategy is to adjust the relative energy of the T(n–π*) state to S1 and T(π–π*) states to maximize the efficacy of the T(n–π*) state by functional group engineering. We propose the desired energy level of the T(n–π*) state to be situated between those of the T(π–π*) state and the S1 state, with the S1 state as the state with the lower energy. As shown in Fig. 1b, having a lower-lying T(π–π*) state can result in internal conversion (IC) down to the T(π–π*) state from T(n–π*), thereby becoming unable to utilize the stronger SOC between the S1 and T(n–π*) states and behaving in the same manner as the conventional IST emitters. The T(n–π*) state with a lower energy than S1 means loss of the IST properties of the molecule, needing thermal activation (TA) to the S1 state. To achieve the optimal energetic ordering S1 < T(n–π*) < T(π–π*), the T(n–π*) energy can be tuned by attaching functional groups to the carbonyl branch, which modulates the energy of the non-bonding orbital and therefore the energy of the T(n–π*) state, as is suggested in Fig. 1c. As addition of an electron donating group (EDG) increases the energy of the n-orbital, the energy of the T(n–π*) state decreases, and vice versa upon addition of an electron withdrawing group (EWG).
 | ||
| Fig. 1 Schematic representation of utilizing n–π* states to facilitate rISC in IST molecules. (a) The non-bonding orbital and the π/π* orbitals of an IST molecule with the carbonyl group attached. (b) The outcome of rISC according to the energy of the T(n–π*) state, with the target energy of T(n–π*) between those of S1 and T(π–π*). (c) Scheme showing the tuning of the energy of T(n–π*) through attachment of electron donating or electron withdrawing functional groups. The potential energy surface of the T(n–π*) state is twisted and displaced with respect to the π–π* states, which show Duschinsky rotation and shift of relaxed geometry. | ||
2 Computational details
The molecules studied are shown in Fig. 2, with heptazine (1) and its derivatives having branches containing the carbonyl group attached (2 to 4). 2 has an acetyl group attached, 3 has a cyano group attached to 2 to tune the energy of the n–π* state by adding an electron-withdrawing group, and 4 has an amino group attached to 2 to tune the energy using an electron-donating group. Heptazine was chosen among IST molecules due to its small size that minimizes the computational cost and its S1 energy that coincides with the blue light region (∼2.7 eV). | ||
| Fig. 2 Chemical structures of the molecules studied. | ||
To analyze the capability of the molecules 2 to 4 to be used in OLEDs, the relaxed geometries of S0, S1, T(π–π*), and T(n–π*) states were obtained to examine the electronic state with the lowest adiabatic energy. The geometries of all four molecules in Fig. 2 were optimized using the B3LYP functional with the 6-31G(d) basis using Gaussian16 software.43 The optimized geometires for S1 states were obtained using time-depedent density functional theory (TDDFT) and Tamm–Dancoff approximation (TDA) methods. However, for triplet states, as previous studies report that TDDFT can have larger errors for triplet states compared to Tamm–Dancoff approximation (TDA) or unrestricted DFT (UDFT),44,45 three geometries, one from each of the TDDFT, TDA, and UDFT methods, were obtained for the T(π–π*) and T(n–π*) states. We confirmed that the optimized geometries of all four molecules showed real frequencies for all modes for each state of interest.
As a highly correlated electronic structure method is desirable to predict accurate ΔEST, each of the optimized geometries was subject to single point calculation using EOM-CCSD in the cc-pVDZ basis set to calculate the energy of the low-lying electronic states including S(n–π*) at the relaxed geometries of each electronic state of interest. The geometry that gives the lowest energy from the single point calculations was considered as the best guess for the relaxed geometry of each excited state (see ESI† for the corresponding geometries). The energies of each geometry obtained with each level of theory are listed in Tables S1–S4 (ESI†). The oscillator strengths of molecules 1–4 were obtained at the respective relaxed S1 geometries to examine the fluorescence intensity. To compare the rate of rISC and ISC involving the two triplet states, the SOC between the triplet states and the S1 state was computed at the relaxed geometries of the respective states. The SOC and non-adiabatic coupling (NAC) were also calculated from the EOM-CCSD calculations, with mean-field treatment of the two-electron interaction part of the SOC. To analyze the effects of geometry fluctuation due to finite temperature on the oscillator strength and the SOC, the displaced S1 geometries of the molecules 2–4 were obtained with Wigner sampling using the S1 state normal modes at 300 K, sampling 80 geometries for each molecule, with the oscillator strength and SOC at each geometry obtained using the EOM-CCSD method. All EOM-CCSD calculations were performed using Q-Chem 6.0 software.46
To assess the kinetics, the ISC and rISC rate between the singlet and the triplet excited states and the internal conversion (IC) rate between the T(π–π*) state and the T(n–π*) state were computed to compare the rate of rISC of two triplet states and verify that the internal conversion between the triplet states can effectively populate the state with stronger spin–orbit coupling. As previous works have shown that the difference in the relaxed geometry and normal modes of electronic states plays a pivotal role in determining the transition rate,47–49 the effect of Duschinsky rotation and displacement of normal modes should be taken into account correctly. The rate of the ISC/rISC process was thus calculated using the equation based on the golden rule50
 | (1) |
 .50 For ISC, the SOC is the sum of the SOC of the three levels:51
.50 For ISC, the SOC is the sum of the SOC of the three levels:51 . See computational details in the ESI† for more details regarding the computation of the integral in eqn (1).
. See computational details in the ESI† for more details regarding the computation of the integral in eqn (1).
The IC rate between the triplet states was also calculated based on the golden rule rate with consideration of the change in geometry and normal modes, according to the formalism of Miyazaki et al.,52 as
 | (2) |
 | (3) |
While the SOC strength of ISC and rISC between the S1 and the T(n–π*) state was simply taken as the value computed at the relaxed geometries of the initial states, the SOC strength of transition between S1 and the T(π–π*) was averaged according to the Boltzmann population of the initial state at its relaxed geometry and the relaxed geometry of the final state to take into account the low reorganization energy and considerable change in SOC upon reorganization. For example, the S1 → T(π–π*) SOC was computed to be
 | (4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Geom is the SOC strength computed at the relaxed geometry of the S1/T(π–π*) state. The T(π–π*) → S1 SOC was also computed in the same manner with the reorganization energy changed to that of the T(π–π*) state. The golden rule rates of the ISC/rISC were calculated using the correlation function formalism of Ianconescu et al., which is expressed in eqn (1).50,53–55
Geom is the SOC strength computed at the relaxed geometry of the S1/T(π–π*) state. The T(π–π*) → S1 SOC was also computed in the same manner with the reorganization energy changed to that of the T(π–π*) state. The golden rule rates of the ISC/rISC were calculated using the correlation function formalism of Ianconescu et al., which is expressed in eqn (1).50,53–55
3 Results and discussion
The energy diagrams of molecules 1–4 are shown in Fig. 3. The numerical values of the energies are listed in Tables S5–S8 (ESI†), showing agreement of the vertical excitation energies of the S1 and T1 states of molecule 1 with the theoretical best estimate using the CC3 approach56 with the energies within ∼0.1 eV. The adiabatic energies show slightly larger errors with 0.16 eV for the S1 state and 0.15 eV for the T1 state compared to the CC3 approach. Fig. S1–S4 (ESI†) show the dominant excitations corresponding to S1, T(π–π*), T(n–π*), and S(n–π*) states. The calculated energies show the promising prospects of utilizing the T(n–π*) state in rISC by showing that (i) T(n–π*) can be the lowest triplet excited state with energy close to the blue light region, (ii) the lowest singlet state is still the S(π–π*) state to achieve stronger spin–orbit coupling with the T(n–π*) state and (iii) the energy level of T(n–π*) can be tuned with functional groups with relatively little effects on (π–π*) state energies. In contrast to the negative energy gap of S1 and T(π–π*) states, the S(n–π*) state lies considerably higher in energy than the T(n–π*) state due to higher exchange energy between the non-bonding orbital and the π* orbital. Inclusion of the S(n–π*) state into the dynamics would lead to a more complete picture of the kinetics of our molecules, but optimization of the geometry of the S(n–π*) state at the DFT level did not agree with the single point calculation at the EOM-CCSD level of theory as the energy of the S(n–π*) state at the optimized geometry was higher than the energy of the S(n–π*) state at the relaxed geometry of the T(n–π*) state. To obtain a guess about the transition rates involving the S(n–π*) state, we optimized the S(n–π*) state with the EOM-CCSD/6-31G* level of theory to compute the adiabatic energy of the S(n–π*) state and referenced to a work55 that uses the theory we use to compute the ISC/rISC rate of systems. The adiabatic energies of the S(n–π*) states for molecules 2–4 are listed in Table S9 (ESI†). | ||
| Fig. 3 Energies of the electronic states of interest plotted at each of the relaxed geometries calculated at the EOM-CCSD/cc-pVDZ level of theory. | ||
The computed electronic energies give deeper insights into exploiting the n–π* state in rISC, showing that the addition of the carbonyl group does alter the π–π* state energies to an appreciable degree and may result in less negative ΔEST, as in the case of 3, and that reorganization to the T(n–π*) states’ minimum energy geometry incurs a significant change in the electronic state energies and thus the quantitative amount of reorganization is also a variable to be controlled to tune the relative adiabatic energies. The change in energy with respect to the geometry is especially prominent for 4, for which the energy of the T(n–π*) state stabilizes by ∼0.8 eV. The tuning of energy levels using functional groups is yet reliable, with 3 showing higher T(n–π*) energy and 4 showing lower T(n–π*) energy relative to 2 and exhibiting all three possibilities of energetic alignment shown in Fig. 1b. Molecules 2 and 4 have T(n–π*) as the triplet state with the lowest adiabatic energy, which means the T(n–π*) state is thermally more favored when the triplet excited state is produced upon electric excitation. The relative adiabatic energies of 2 resemble the desired energetic alignment with the adiabatic energy of T(n–π*) lying between those of S1 and T(π–π*), thus making the transition from the lowest triplet state to the lowest singlet excited state both kinetically and thermodynamically favorable.
The oscillator strengths computed at the relaxed geometries are shown in Table S10 (ESI†) and the histograms of oscillator strengths at thermally excited geometry sampled from Wigner sampling are shown in Fig. S5 (ESI†). The oscillator strengths at the relaxed geometry show that due to the loss of symmetry upon addition of the carbonyl group, the oscillator strength of the S1 → S0 state transition also increases, albeit to an order of 0.0001. The oscillator strength can occasionally reach 0.01 with thermal excitation and thus the molecules can still be used as prototypes when designing IST emitters with an enhanced rISC rate. It should be noted that the sampling shows that 4 is prone to dissociation, thus calling for the need to design emitters with stability put into consideration.57 The oscillator strength can be further improved by an additional modification of the molecule, as heptazine derivatives were shown to exhibit oscillator strength on the order of 0.01 in the work conducted by Aizawa et al.26
The |HISC/rISCSO| at the relaxed geometry of the initial states and ΔEadST between the S1 state and the triplet states are shown in Table 1. The SOC with S1 is shown to be at least an order of magnitude stronger for T(n–π*) than for T(π–π*), which results in a more than 100 times larger SOC factor for the rISC rate in eqn (1) after taking the square value. It is noteworthy that SOC strength between the S1 and T(π–π*) states gets significantly altered according to the geometry with the SOC strength computed at the relaxed geometry of one state one to two orders of magnitude stronger than the SOC strength computed at the relaxed geometry of the other state. This change in SOC can be attributed to the rotation of the bond between the heptazine molecule and the carbonyl group, which breaks the planar symmetry and mixes the non-bonding orbitals of the oxygen with the HOMO and LUMO of pre-dominantly π character, thus indicating that the carbonyl group facilitates the ISC by lending its SOC strength to transition between states of π–π* character. The dihedral angles of the heptazine plane and the plane made by the carbonyl group for the relaxed S1 and T(π–π*) geometries of each molecule are listed in Table S11 (ESI†), measured using the atoms shown in Fig. S6 (ESI†). The SOC strengths from Wigner sampled geometries also show that the SOC can vary appreciably with thermal fluctuation of the geometry, but the difference in the SOC between S1 and T(π–π*) and the SOC between S1 and T(n–π*) still remains significant. The histograms of the SOC of molecules 2–4 are shown in Fig. S7 (ESI†).
| T(π–π*) | |||||
|---|---|---|---|---|---|
| Molecule | |HISCSO|a | |HrISCSO|b | |HISCSO|effc | |HrISCSO|effc | ΔEadST |
a Calculated at the relaxed S1 state geometry, scaled down by a factor of 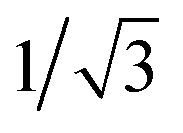 for a direct comparison with |HrISCSO|.
b Calculated at the relaxed T(π–π*) state geometry.
c Effective SOC from thermal average. See computational details.
d Calculated at the relaxed T(n–π*) state geometry. for a direct comparison with |HrISCSO|.
b Calculated at the relaxed T(π–π*) state geometry.
c Effective SOC from thermal average. See computational details.
d Calculated at the relaxed T(n–π*) state geometry.
|
|||||
| 1 | 1.62 × 10−4 | 1.40 × 10−4 | 1.53 × 10−4 | 1.49 × 10−4 | −1.92 × 10−1 |
| 2 | 2.08 × 10−1 | 5.19 × 10−3 | 1.53 × 10−1 | 3.67 × 10−2 | −3.05 × 10−2 |
| 3 | 7.98 × 10−3 | 1.41 × 10−2 | 9.66 × 10−3 | 1.18 × 10−2 | 1.03 × 10−2 |
| 4 | 3.14 × 10−2 | 6.55 × 10−3 | 2.81 × 10−2 | 1.42 × 10−2 | −3.84 × 10−2 |
The ISC, rISC, and the IC rates are shown in Table 2. The rates show that the energetic alignment of the T(n–π*) state placed with the T(π–π*) above and the S1 below is indeed advantageous for rISC, with IC transferring the population from T(π–π*) to T(n–π*) and large SOC facilitating the rISC from the T(n–π*) to the S1. The theoretical rISC rate from the T(n–π*) state in 3 also benefits from the strong SOC, but it also relaxes rapidly to the T(π–π*) state, which has a relatively slow rISC rate. 4 shows virtually negligible IC and ISC both to and from the T(n–π*) state. The kinetics of 4 is heavily affected by the large difference in the geometry of the T(n–π*) state from those of the states of (π–π*) nature, which leads to a rapid decay of the ρ factor in eqn (1) and (2). The decay of the integrand of eqn (1) and the trace of the ρkl term in eqn (2) are shown in Fig. S8–S10 (ESI†). As the rate calculation is based on harmonic approximation, the extremely slow rate of transition to and from the T(n–π*) state of 4 is expected to be an underestimation stemming from the large difference in geometry. The ISC and rISC rates involving the T(n–π*) state in all molecules thus have smaller integral values compared to the transitions involving the T(π–π*) state, thereby not achieving transition rates proportional to the much stronger SOC. The dynamics between the S1 and T(π–π*) states is also noteworthy in that despite the lower energy of the S1 state of 2, the ISC from the singlet to the triplet state is faster by an order of magnitude, which can be attributed to the stronger SOC strength at the S1 geometry given by the breaking of planar symmetry. The rates involving transition to S(n–π*) are not expected to be substantial enough to bring nontrivial changes to the overall kinetics, as for 2 and 3 the S(n–π*) state lies ∼0.4 eV higher in energy than either of the triplet states, which is an energy gap that resulted in the rISC rate on order of 102 s−1 in a recent study.55 The S(n–π*) state of 4 lies low enough for rISC from T(π–π*) to be energetically viable, but the large displacement between the states of (π–π*) character and the states of (n–π*) character is expected to hinder the transition, as how the transitions to and from T(n–π*) show negligible transition rates. The rISC from the T(n–π*) state to the S(n–π*) state is highly endothermic at ∼0.27 eV, which also results in the rISC rate on the order of 103 s−1.55
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|

|
6.37 × 10−1 | 3.46 × 106 | 1.61 × 105 | 4.30 × 105 |
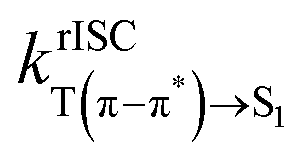
|
6.50 | 4.78 × 105 | 2.62 × 104 | 3.59 × 104 |

|
— | 1.66 × 106 | 1.43 × 103 | 4.60 × 10−3 |

|
— | 6.63 × 106 | 1.35 × 106 | 0.38 |
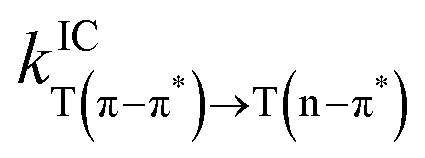
|
— | 1.32 × 109 | 8.27 × 104 | 0.54 |

|
— | 1.05 × 109 | 2.97 × 109 | 19.82 |
The difference in the geometry of the singlet and triplet states involved in ISC/rISC is shown in the plots of the Duschinsky matrices and displacement vectors involved in the ISC/rISC processes shown in Fig. 4a, and the rotation angles of the Duschinsky matrix are shown in Fig. 4b. The normal modes with the largest displacement vectors for each transition are shown in Fig. S11 (ESI†). The rotation angles are calculated as described in the ESI.† As expected from the different electronic structures, the T(n–π*) state has considerably larger relative displacements and more rotation angles are near the vicinity of π/4 with respect to S1 compared to the T(π–π*) state for all molecules, with especially large displacements for 4. The Duschinsky matrix and the displacement vectors between the triplet states involved in IC are shown in Fig. S12 (ESI†), again showing that the large change in geometry inhibits transitions to and from the T(n–π*) state.
 | ||
Fig. 4 (a) Plots of the absolute value of the Duschinsky matrix and displacement vectors between the S1 state and the triplet states for the molecules studied. (b) Histogram of the angle of rotation of the Duschinsky matrix, grouped into eight bins within the range 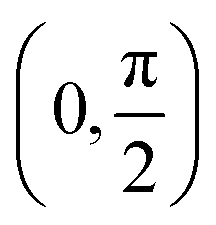 . . | ||
The steady state population for S1, T(π–π*), and T(n–π*) of molecules 2–4 can be readily obtained using the rates calculated, and thus the propensity of each molecules to populate the emissive singlet state can be quantified. 2 shows the most promising prospect as expected, with 42% populating the S1 at the steady state, and T(π–π*) and T(n–π*) taking 27% and 31%, respectively. Since T(π–π*) and T(n–π*) have similar energy levels, the IC rates between them are on the same order, which results in almost 1:1 population. The high population of the S1 state can be attributed to it being the lowest in energy and active participation of the T(n–π*) state in drawing population from the T(π–π*) state that subsequently undergoes rISC, counteracting the rather rapid ISC back to the T(π–π*) state. 3 shows the case when T(n–π*) is too high in energy to participate in the dynamics and the ΔEST inversion is broken to favor population of the triplet state, with 11% of the population in the S1 state and 89% in the T(π–π*) state and negligible population of the T(n–π*) state. The dynamics of 4 shows the situation when the T(n–π*) state is the lowest in energy but becomes inaccessible due to high reorganization energy and the SOC from the singlet state to the triplet state is stronger due to the breaking of planar symmetry, with 92% of the population in the T(π–π*) state and 8% in the S1 state, with virtually no population of the T(n–π*) state.
4 Conclusion
In this work, we show a new design strategy of OLED emitters that not only have negative ΔEST but also utilize the change in orbital angular momentum between the n–π* state and the π–π* state to overcome the weak SOC of previously discussed IST emitters. The computations on both energetics and kinetics provide encouraging prospects for development of carbonyl attached IST emitters by showing that (i) the T(n–π*) state energy is comparable to the T(π–π*) energy and can be populated using electronic excitation, (ii) the energy can be tuned using a relatively intuitive method of attaching functional groups that either donate or withdraw electrons, and (iii) the SOC of T(n–π*) → S1 transition is indeed stronger than that seen in conventional IST emitters. To make better use of the T(n–π*) state, means to control the geometric factors must be investigated, as the degree of displacement of relaxed geometry in the T(n–π*) state and the break of planar symmetry in either the S1 or T(π–π*) state are found to be especially impactful on the ISC/rISC dynamics. The large difference in geometries of states with (n–π*) character and states with (π–π*) character can become a major roadblock by (i) inhibiting the IC from T(π–π*) even in the case of T(n–π*) being the lower energy triplet state and (ii) inhibiting the rISC to S1. The break of planar symmetry can have synergistic effects with the non-bonding orbital as the contribution of non-bonding orbitals to the π and π* orbitals can also increase the rISC rate from the T(π–π*) to the S1 state. To better take into account the dependence of electronic state energies and couplings on the geometries, a molecular dynamics simulation to monitor the change in geometry at finite temperature and confirm the stability of the compound can be a promising topic for future studies. As satisfying the criteria of negative ΔEST, the right energy level of the T(n–π*) state, and moderate reorganization are not straightforward, a high-throughput study may help in discovering optimal molecules for rISC. Further investigations into optimizing the change in geometry and normal modes may lead to the discovery of numerous emitters for which the rISC happens in a prompt timescale and is thermodynamically driven.Author contributions
Conceptualization: Hwon Kim and Seung Kyu Min; data curation: Hwon Kim; formal analysis: Hwon Kim; funding acquisition: Seung Kyu Min; investigation: Hwon Kim and Seung Kyu Min; methodology: Hwon Kim and Seung Kyu Min; project administration: Hwon Kim and Seung Kyu Min; resources: Seung Kyu Min; software: Hwon Kim; supervision: Seung Kyu Min; validation: Hwon Kim; visualization: Hwon Kim and Seung Kyu Min; and writing – Hwon Kim and Seung Kyu Min.Data availability
All data generated in this study are available from the corresponding author upon request. The codes used for the rate calculation are available at: https://github.com/hkimaf/Nonbonding_ISC_calculation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (Ministry of Science and ICT(MSIT)) (NRF-2023M3K5A1094813, RS-2023-00257666, and RS-2024-00455131).Notes and references
- J. Larkin, W. Donaldson, T. Foster and R. Knox, Chem. Phys., 1999, 244, 319–330 CAS.
- T. Huang, X. Song, M. Cai, D. Zhang and L. Duan, Mater. Today Energy, 2021, 21, 100705 CAS.
- S. Gorgon, K. Lv, J. Grüne, B. H. Drummond, W. K. Myers, G. Londi, G. Ricci, D. Valverde, C. Tonnelé and P. Murto, et al. , Nature, 2023, 620, 538–544 CAS.
- L. Lv, Y. Zhang and Z. Ning, RSC Adv., 2024, 14, 23987–23999 CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CAS.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. G. Iii, J. Am. Chem. Soc., 2020, 142, 8074–8079 Search PubMed.
- A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 Search PubMed.
- K. R. Naveen, P. Palanisamy, M. Y. Chae and J. H. Kwon, Chem. Commun., 2023, 59, 3685–3702 RSC.
- H. S. Kim, J. Y. Lee, S. Shin, W. Jeong, S. H. Lee, S. Kim, J. Lee, M. C. Suh and S. Yoo, Adv. Funct. Mater., 2021, 31, 2104646 CrossRef CAS.
- Q. Ou, Y. Shao and Z. Shuai, J. Am. Chem. Soc., 2021, 143, 17786–17792 CrossRef CAS PubMed.
- J. Ochi, Y. Yamasaki, K. Tanaka, Y. Kondo, K. Isayama, S. Oda, M. Kondo and T. Hatakeyama, Nat. Commun., 2024, 15, 2361 CrossRef CAS.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, C.-C. Ren-Wu, H.-W. Lin and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 27026–27034 CAS.
- X. Chen, S. Bagnich, R. Pollice, B. Li, Y. Zhu, R. Saxena, Y. Yin, W. Zhu, A. Aspuru-Guzik and E. Zysman-Colman, et al. , Adv. Opt. Mater., 2024, 12, 2301784 CrossRef CAS.
- P. L. Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- P. de Silva, J. Phys. Chem. Lett., 2019, 10, 5674–5679 CrossRef CAS PubMed.
- L. Sun, W. Hua, Y. Liu, G. Tian, M. Chen, M. Chen, F. Yang, S. Wang, X. Zhang and Y. Luo, et al. , Angew. Chem., 2019, 131, 11433–11438 CrossRef.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- P. de Silva, C. A. Kim, T. Zhu and T. Van Voorhis, Chem. Mater., 2019, 31, 6995–7006 CrossRef CAS.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. B. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- R. Pollice, P. Friederich, C. Lavigne, G. dos Passos Gomes and A. Aspuru-Guzik, Matter, 2021, 4, 1654–1682 CrossRef CAS.
- Y. Lee, J. Kim, S. Lee, E. Sim and J.-I. Hong, Chem. Eng. J., 2023, 476, 146659 CrossRef CAS.
- N. Aizawa, Y.-J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K.-I. Nakayama, S. Maeda, F. Araoka and D. Miyajima, Nature, 2022, 609, 502–506 CrossRef CAS PubMed.
- J. Ehrmaier, E. J. Rabe, S. R. Pristash, K. L. Corp, C. W. Schlenker, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2019, 123, 8099–8108 CAS.
- O. H. Omar, X. Xie, A. Troisi and D. Padula, J. Am. Chem. Soc., 2023, 145, 19790–19799 CrossRef CAS PubMed.
- M. Bedogni, D. Giavazzi, F. Di Maiolo and A. Painelli, J. Chem. Theory Comput., 2023, 20, 902–913 Search PubMed.
- D. Valverde, C. T. Ser, G. Ricci, K. Jorner, R. Pollice, A. Aspuru-Guzik and Y. Olivier, ACS Appl. Mater. Interfaces, 2024, 16, 66991–67001 CAS.
- J. Terence Blaskovits, M. H. Garner and C. Corminboeuf, Angew. Chem., Int. Ed., 2023, 62, e202218156 CrossRef CAS PubMed.
- A. Actis, M. Melchionna, G. Filippini, P. Fornasiero, M. Prato, M. Chiesa and E. Salvadori, Angew. Chem., Int. Ed., 2023, 62, e202313540 CAS.
- R. Pollice, B. Ding and A. Aspuru-Guzik, Matter, 2024, 7, 1161–1186 CAS.
- G. Ricci, J.-C. Sancho-García and Y. Olivier, J. Mater. Chem. C, 2022, 10, 12680–12698 RSC.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- K. Jorner, R. Pollice, C. Lavigne and A. Aspuru-Guzik, J. Phys. Chem. A, 2024, 128, 2445–2456 CrossRef CAS PubMed.
- F. Dinkelbach, M. Bracker, M. Kleinschmidt and C. M. Marian, J. Phys. Chem. A, 2021, 125, 10044–10051 CrossRef CAS PubMed.
- H. Kim, G. D. Scholes and S. K. Min, Phys. Chem. Chem. Phys., 2024, 26, 5508–5516 RSC.
- Y. Yu, F. Liu, X. Meng, L. Ding, L. Liao and Z. Jiang, Chem. – Eur. J., 2023, 29, e202202628 CrossRef CAS PubMed.
- S. Wu, L. Zhang, J. Wang, A. K. Gupta, I. D. Samuel and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202305182 CrossRef CAS PubMed.
- Y.-C. Cheng, X. Tang, K. Wang, X. Xiong, X.-C. Fan, S. Luo, R. Walia, Y. Xie, T. Zhang and D. Zhang, et al. , Nat. Commun., 2024, 15, 731 CrossRef CAS PubMed.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- M. J. Peach, M. J. Williamson and D. J. Tozer, J. Chem. Theory Comput., 2011, 7, 3578–3585 CrossRef CAS PubMed.
- Y. Guo, L. Zhang and Z. Qu, Molecules, 2023, 28, 3257 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng and X. Feng, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- P. Karak and S. Chakrabarti, Phys. Chem. Chem. Phys., 2020, 22, 24399–24409 RSC.
- L. Paul, T. Moitra, K. Ruud and S. Chakrabarti, J. Phys. Chem. Lett., 2019, 10, 369–374 CrossRef CAS PubMed.
- P. Karak, P. Manna, A. Banerjee, K. Ruud and S. Chakrabarti, J. Phys. Chem. Lett., 2024, 15, 7603–7609 CrossRef CAS PubMed.
- I. Kim, S. O. Jeon, D. Jeong, H. Choi, W.-J. Son, D. Kim, Y. M. Rhee and H. S. Lee, J. Chem. Theory Comput., 2019, 16, 621–632 Search PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- K. Miyazaki and N. Ananth, J. Chem. Phys., 2022, 156, 044111 CAS.
- R. Ianconescu and E. Pollak, J. Phys. Chem. A, 2004, 108, 7778–7784 CAS.
- Y. Niu, Q. Peng, C. Deng, X. Gao and Z. Shuai, J. Phys. Chem. A, 2010, 114, 7817–7831 CAS.
- B. K. Min, D. Kim, D. Kim and Y. M. Rhee, Bull. Korean Chem. Soc., 2023, 44, 989–1003 CAS.
- P.-F. Loos, F. Lipparini and D. Jacquemin, J. Phys. Chem. Lett., 2023, 14, 11069–11075 CAS.
- N. Lin, J. Qiao, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2014, 118, 7569–7578 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00166h |
| This journal is © the Owner Societies 2025 |