
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamics-informed neural networks and extensive data sets: key factors to accurate blind predictions of apparent pKa values in the euroSAMPL challenge†
Robert
Fraczkiewicz‡

Simulations Plus, Research Triangle Park, Inc., P.O. Box 12317, NC 27709, USA. E-mail: info@simulations-plus.com
First published on 8th April 2025
Abstract
Microscopic and macroscopic pKa values for 35 compounds selected by the organizers of euroSAMPL 1 challenge were blindly predicted with our thermodynamics-informed empirical S + pKa model (ranked submission 0x4cb7101f). Our results have received the first overall rank from the challenge organizers. We describe our methodology and discuss evaluation methods.
EuroSAMPL, the first European blind prediction challenge in the spirit of established SAMPL challenges ran from February 2024 until it concluded in June of that year.1 Participants were asked to predict ionization constants (pKa) of 35 newly synthesized drug-like compounds. The organizers made good effort to select compounds with only one deprotonation transition in the range of their experimental techniques (pH = 2–12).1 In addition, the single transition per compound in this pH range was confirmed by alternative experiments.2 Final prediction results are shown in Table 1. The organizers employed a simple null benchmark where all “predicted” pKa values were set to 7.0. The benchmark resulted in root mean square error (RMSE) of 2.444. Another benchmark was the organizers’ EC-RISM method achieving RMSE = 1.107. Based on these results, machine learning approaches tend to dominate methods based on quantum chemistry. In the previous SAMPL6 pKa competition it was a hybrid method (QM with COSMO-RS approach to solvation followed by a fit to experimental data, ID = “xvxzd”) that achieved the best RMSE = 0.68.3,4 The next two best methods were empirical. One of them, however, easily beat the “xvxzd” metric after retraining with more data.5
| Method1 | ID | RMSE “first” | MAE “first” | RMSE “best” | MAE “best” |
|---|---|---|---|---|---|
| Simulations plus, S + pKa5–7 | 0x4cb7101f | 0.529 | 0.379 | 0.529 | 0.379 |
| ORCA/DFT/DRACO/MM/ML | 0x4a6c0760 | 0.806 | 0.632 | 0.806 | 0.632 |
| RF/CDK/Jazzy | 0xc7960c21 | 1.207 | 0.812 | 1.207 | 0.812 |
| BIOVIA, COSMO-RS | 0x4b7b06e5 | 1.392 | 0.705 | 0.734 | 0.519 |
| QupKake8 | 0x216604d8 | 1.672 | 0.779 | 0.513 | 0.408 |
| Gaussian/DFT/SMD9 | 0x421c06f1 | 1.726 | 1.410 | 1.726 | 1.410 |
| ORCA/DFT/SMD | 0x4cb00786 | 2.123 | 1.757 | 2.123 | 1.757 |
| Gaussian/DFT/IEF-PCM | 0x3f2606c6 | 2.569 | 2.009 | 2.569 | 2.009 |
| Gaussian/uESE10 | 0x541007e2 | 5.280 | 3.375 | 3.422 | 2.231 |
| reference_EC-RISM 11 | 1.107 | 0.935 | 1.107 | 0.935 | |
| All pK a = 7 | 0xb8320bc2 | 2.444 | 2.142 | 2.444 | 2.142 |
Prior to discussing ranked results of all the participants, we must explain “first” and “best” – the two methods of matching predicted and observed pKa. Even though there was only one measured pKa per compound, some compounds were not monoprotic.
Therefore, methods with automatic detection of ionizable sites predicted multiple pKa for some compounds (see Table 2). For example, our S + pKa method5–7 predicted multiple pKa in the 2–12 range for seven compounds, albeit marginally. Fig. 1 shows one such example; others can be found in the ESI.† The natural question is which of the predicted pKa (i.e., which macroscopic deprotonation transition) should be matched against the one observed in the organizer's experiment? We have addressed this issue in our reference work:12 The only fair and objective method of matching a sequence of multiple predicted vs. a sequence of multiple observed values per compound is to form a pairing with minimal sum of absolute deviations while preserving the same order of both sequences. The latter is dictated by a simple physics of ionization: the order of pKa is descending as a function of the number of bound protons. After all, the more protons a compound has it is energetically more expensive to add another one. Such a method was employed by the organizers of SAMPL6 competition3 and it corresponds to the “best” matching in euroSAMPL. The “first” matching, although one may argue that best predictive algorithms should handle that part as well, was in our opinion subjective since each participant had to blindly guess, relying solely on their chemical intuition, which of the predicted pKa were measured in euroSAMPL experiments. Due to our vast experience in ionization chemistry, we have guessed each pairing correctly, but three participants were not so lucky. This is why their “first” and “best” results differ. In particular, developers of the empirical QupKake model8 mismatched just two of their guessed predictions resulting in high “first” RMSE while their “best” RMSE was much lower.
| SMILES | Name | Matched pKa | Other pKa |
|---|---|---|---|
o1cccc1-c1n(nc(c1)C(O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)C O)C |
euroSAMPL-01 | 3.5 | |
| OC1N(CC(CC1)c1ccccc1)CC(O)O |
euroSAMPL-02 | 3.49 | |
| O(C)c1ccc(cc1)-c1ncccc1 | euroSAMPL-03 | 5.13 | |
| n1cnc(N)cc1C(C)(C)C | euroSAMPL-04 | 6.54 | |
| OC(N)c1ccc(cc1)CN |
euroSAMPL-05 | 8.97 | |
| OC1N(c2c(C1)cc(cc2)C(O)O)C |
euroSAMPL-06 | 3.91 | |
| s1cc(nc1N)-c1ccc(cc1)C | euroSAMPL-07 | 4.68 | |
| Oc1ccc(cc1)C(O)NCc1ccccc1 |
euroSAMPL-08 | 8.91 | 11.65 |
| OC(O)c1ccc(N2CCN(CC2)C(O)C(C)(C)C)cc1 |
euroSAMPL-09 | 4.41 | 2.62 |
| Clc1cc(C(O)O)c(OC)cc1N |
euroSAMPL-10 | 4.84 | |
|
OC(O)c1nc2ncccc2cc1 |
euroSAMPL-11 | 3.45 | |
|
OC(O)c1cc(ncc1)NC(O)C |
euroSAMPL-12 | 3.59 | 11.96, 2.03 |
| o1cnnc1-c1ccc(O)cc1 | euroSAMPL-13 | 8.25 | |
| OC1C(Br)C(NCN1)C |
euroSAMPL-14 | 7.96 | |
| s1c2c(cccc2)c(O)c1C(O)C |
euroSAMPL-15 | 6.2 | |
| Oc1cc(NC(O)CC(C)C)ccc1 |
euroSAMPL-16 | 9.4 | |
| OC(O)c1cc(-n2cccc2)ccc1 |
euroSAMPL-17 | 3.83 | |
| s1c2NCNC(O)c2cc1CC |
euroSAMPL-18 | 9.46 | |
| OCc1n2c(nc1C)CCCC2 |
euroSAMPL-19 | 7.57 | |
| OC1N(c2c(cc(cc2)C(O)O)C1(C)C)C |
euroSAMPL-20 | 4.1 | |
|
OC(O)C1n2nc(cc2NC(C1)C1CC1)C(C)(C)C |
euroSAMPL-21 | 3.02 | |
| Oc1ccccc1C(O)N(C)C |
euroSAMPL-22 | 9.37 | |
|
OC(O)c1cnc(nc1C)CC |
euroSAMPL-23 | 3.49 | |
| O1CCN(CC1)c1cc(nc2c1cccc2)C | euroSAMPL-24 | 9.31 | |
| OC(N)CCn1c2cc(C)c(cc2nc1)C |
euroSAMPL-25 | 4.9 | 11.85 |
| Fc1cc2c(NCNC2O)cc1 |
euroSAMPL-26 | 9.51 | |
| OC(N)c1ccc(cc1)-c1ncccc1 |
euroSAMPL-27 | 3.91 | 11.88 |
| n1ccn(C)c1-c1cccnc1 | euroSAMPL-28 | 6.1 | 2.16 |
| OC(O)c1nnn(c1C1CC1)-c1ccc(cc1)C |
euroSAMPL-29 | 3.63 | |
| Oc1cc2c(cc1C(O)NCCO)cccc2 |
euroSAMPL-30 | 9.06 | 11.68 |
| O1CC(O)N(c2c1cccc2)CC(O)O |
euroSAMPL-31 | 3.35 | |
| o1nc(cc1-c1ccccc1)C(O)N1CCN(CC1)C |
euroSAMPL-32 | 7.27 | |
| s1cccc1-c1n2CCCc2nc1 | euroSAMPL-33 | 6.39 | |
| S(Cc1ccc(cc1)C(O)O)C |
euroSAMPL-34 | 4.02 | |
| n1cc(ccc1N)-c1ccccc1 | euroSAMPL-35 | 6.31 |
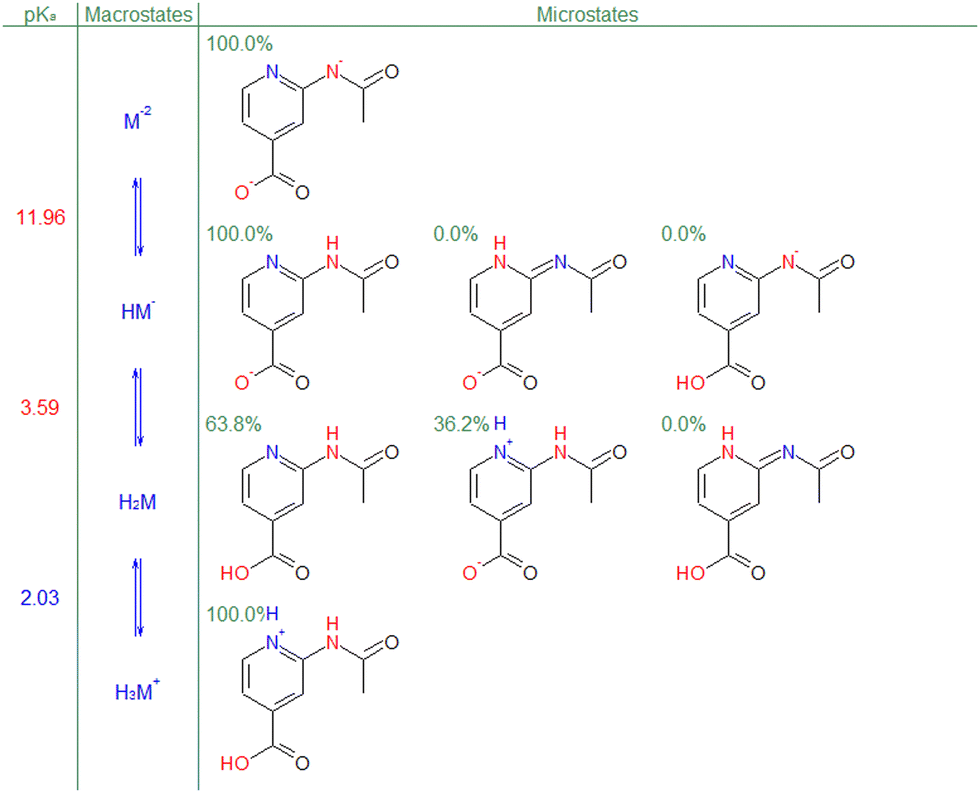 | ||
| Fig. 1 Multiple predicted pKa for compound euroSAMPL-12 with full resolution of ionization microstates (S + pKa method). The “0.0%” labels mean “<0.1%”. | ||
Our S + pKa method stands out as the best one in all “first” (MAE) categories outpacing the nearest participant by 0.3 log units. In the “best” category S + pKa's RMSE is higher than QupKake's RMSEs by 0.016 log units while its mean absolute error (MAE) is lower by 0.029 log units – this could be considered a tie. The observed vs. predicted plot for S + pKa is shown in Fig. 2 generated by the euroSAMPL organizers.
 | ||
| Fig. 2 Observed vs. predicted plot for the S + pKa predictions. Dashed line = identity line, solid line = trendline. | ||
Full details of the S + pKa method have been described elsewhere,5,6 but let us provide a short summary below. It belongs to a category of empirical, thermodynamics-informed machine learning methods. Its strength is in the exact, microscopic description of protic ionization. In fact, S + pKa predict all N × 2N−1 microconstants for a compound with N protonation sites, up to N = 20. The microconstants are calculated for each of the 2N microstates. For the calculation of each microconstant an artificial neural network ensemble (ANNE) is employed. Each of the component neural networks (ANN) is of the Multilayer Perceptron type with a single sigmoid hidden layer. Individual ANNs were obtained by training against different random splits of modelling data into actual training and verification subsets. An external test set was not used in this process.13 The ANNE uses atomic descriptors calculated for each ionization site and for each protonation state of other sites. Microequilibria theory12 is then used to calculate ionization macroconstants (a.k.a. apparent pKa) that can be matched against pKa obtained with standard experimental methods. S + pKa uses 10 ANNE trained for 10 types of ionizable groups (hydroxyacids, acidic amides, aromatic NH acids, thioacids, carboacids, amines, aromatic N bases, N-oxides, thiones, carbobases). In its latest incarnation S + pKa was trained against 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 810 measured apparent pKa obtained from public sources and industrial partnerships.5
810 measured apparent pKa obtained from public sources and industrial partnerships.5
It is interesting to ask to what degree the 35 challenge compounds represent the modern pharmaceutical chemistry space. In collaboration with our industrial partners, we have had a privilege to examine large sets of proprietary pharmaceutical and agrochemical compounds and their ionization patterns.5,6 Unequivocally, most of these compounds contains multiple ionizable groups. Moreover, a large portion of these multiprotic compounds exhibit complex ionization patterns in the following sense: Please refer to figures in the ESI† to convince yourself that microscopic deprotonation transitions of 31 challenge compounds are “simple”, i.e., dominated by a single microstate, taking 90% contribution as a cutoff value. In the same sense, deprotonations of the remaining 4 compounds (euroSAMPL-11, euroSAMPL-12, euroSAMPL-21, and euroSAMPL-23, indicated by a bold font in Table 2) have non-negligible contributions from other microstates, i.e., are “complex”. Fig. 1 illustrates that for euroSAMPL-12 the macroscopic pKa = 3.59 transition is dominated by two microstates contributing 63.8% and 38.2%, respectively. Our point is that predictive methods that do not take the microscopic thermodynamics of ionization into account and rely on single microstates will likely do better on “simple” cases. Fig. 3 illustrates this point: Neglect of the second dominant microstate in euroSAMPL-12 would present the 3.40 microconstant in lieu of the apparent pKa of 3.59.
 | ||
| Fig. 3 Predicted microconstants for the 3.59 transition in euroSAMPL-12 positioned next to the respective ionizable groups, acidic in red, basic in blue. | ||
In turn, methods with exact treatment of the ionization thermodynamics should perform well on both the “simple” and “complex” cases. Blind predictions on large sets of proprietary pharmaceutical and agrochemical compounds, mentioned above and conducted by our industrial partners prove this point for our S + pKa model.5,6 Unfortunately, euroSAMPL organizers have not revealed experimental pKa values for the 35 challenge compounds1 and we cannot present quantitative assessment of “complex” compound predictions across all participating methods. I hope the organizers will address this issue in their upcoming report. The good news is that five methods either included or probably included microstate analysis in their calculations (Table 3).
| Method1 | ID | Microstates? |
|---|---|---|
| Simulations plus, S + pKa5–7 | 0x4cb7101f | Yes |
| ORCA/DFT/DRACO/MM/ML | 0x4a6c0760 | Probably |
| RF/CDK/Jazzy | 0xc7960c21 | Unknown |
| BIOVIA, COSMO-RS | 0x4b7b06e5 | Yes |
| QupKake8 | 0x216604d8 | Yes |
| Gaussian/DFT/SMD9 | 0x421c06f1 | Unknown |
| ORCA/DFT/SMD | 0x4cb00786 | Unknown |
| Gaussian/DFT/IEF-PCM | 0x3f2606c6 | Yes |
| Gaussian/uESE10 | 0x541007e2 | Unknown |
Conclusions
Empirical and hybrid (quantum chemical + empirical post training) methods continue leading the pack of predictive accuracy of ionization constants. The pure ab initio approaches require massive computational resources seem to be less accurate, mainly due to significant difficulties in estimating solvation energies and entropies. On the other hand, the drawback of empirical methods is their reliance on massive amounts of experimental data.Future pKa competitions should use optimal matching algorithms instead of subjective guessing since it is only the former that evaluates the method in question, while the latter evaluates both the method and its developer or user. To make the competitions more meaningful at representing pharmaceutical chemistry their organizers should choose a sizable percentage of “complex” compounds.
Data availability
The S + pKa results are available in the accompanying ESI.† ADMET Predictor® is available at no charge to academic users.7 To reproduce results quoted in this communication turn on the detection of aliphatic –OH and aliphatic amides – these options are turned off by default to save processing time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has not received any external funding.Notes and references
- S. M. Kast The euroSAMPL pKa blind prediction challenge 2024. 2024. Available from: https://gitlab.tu-dortmund.de/kast_ccb/eurosampl/challenge [last accessed; 12/10/2024].
- S. M. Kast, Private communication, 2024 Search PubMed.
- M. Işık, A. S. Rustenburg and A. Rizzi, et al., Overview of the SAMPL6 pKa challenge: evaluating small molecule microscopic and macroscopic pKa predictions, J. Comput.-Aided Mol. Des., 2021, 35(2), 131–166, DOI:10.1007/s10822-020-00362-6.
- P. Pracht, R. Wilcken and A. Udvarhelyi, et al., High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge, J. Comput.-Aided Mol. Des., 2018, 32(10), 1139–1149, DOI:10.1007/s10822-018-0145-7.
- R. Fraczkiewicz, H. Quoc Nguyen and N. Wu, et al., Best of both worlds: An expansion of the state of the art pKa model with data from three industrial partners, Mol. Inf., 2024, 43(10), e202400088, DOI:10.1002/minf.202400088.
- R. Fraczkiewicz, M. Lobell and A. H. Göller, et al., Best of Both Worlds: Combining Pharma Data and State of the Art Modeling Technology To Improve in Silico pKa Prediction, J. Chem. Inf. Model., 2015, 55(2), 389–397, DOI:10.1021/ci500585w.
- ADMET Predictor(R), (Revision 11.0), Simulations Plus, Inc., Lancaster, CA, USA, 2023, https://www.simulations-plus.com.
- O. D. Abarbanel and G. R. Hutchison, QupKake: Integrating Machine Learning and Quantum Chemistry for Micro-pKa Predictions, J. Chem. Theory Comput., 2024, 20(15), 6946–6956, DOI:10.1021/acs.jctc.4c00328.
- J. Uranga, L. Hasecke and J. Proppe, et al., Theoretical Studies of the Acid–Base Equilibria in a Model Active Site of the Human 20S Proteasome, J. Chem. Inf. Model., 2021, 61(4), 1942–1953, DOI:10.1021/acs.jcim.0c01459.
- S. F. Vyboishchikov and A. A. Voityuk, Fast non-iterative calculation of solvation energies for water and non-aqueous solvents, J. Comput. Chem., 2021, 42(17), 1184–1194, DOI:10.1002/jcc.26531.
- N. Tielker, S. Güssregen and S. M. Kast, SAMPL7 physical property prediction from EC-RISM theory, J. Comput.-Aided Mol. Des., 2021, 35(8), 933–941, DOI:10.1007/s10822-021-00410-9.
- R. Fraczkiewicz, In silico Prediction of Ionization, Reference Module in Chemistry Molecular Sciences and Chemical Engineering, ed. J. Reedijk, Elsevier, 2013 DOI:10.1016/B0-08-045044-X/00143-7.
- ADMET Modeler(R), (Revision 11.0), Simulations Plus, Inc., Lancaster, CA, USA, 2023, https://www.simulations-plus.com.
Footnotes |
| † Electronic supplementary information (ESI) available: S + pKa predictions and microstates for all 35 compounds in PDF format. See DOI: https://doi.org/10.1039/d5cp00165j |
| ‡ Robert Fraczkiewicz has contributed to this work in its entirety. |
| This journal is © the Owner Societies 2025 |