
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen exchange kinetics of BaGd0.3La0.7Co2O6−δ with exsolved Co3O4 nanoparticles in dry and humid atmospheres†
Jónína B.
Guðmundsdóttir
a,
Vincent
Thoréton
 a,
Bo
Jiang
a,
Anuj
Pokle
b,
Einar
Vøllestad
c,
Reidar
Haugsrud
a and
Jonathan M.
Polfus
*a
a,
Bo
Jiang
a,
Anuj
Pokle
b,
Einar
Vøllestad
c,
Reidar
Haugsrud
a and
Jonathan M.
Polfus
*a
aDepartment of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, PO Box 1033 Blindern, N-0315 Oslo, Norway. E-mail: jonathan.polfus@kjemi.uio.no
bDepartment of Physics, Centre for Materials Science and Nanotechnology, University of Oslo, PO Box 1033 Blindern, N-0315 Oslo, Norway
cDepartment of Sustainable Energy Technology, SINTEF Industry, Oslo, Norway
First published on 31st March 2025
Abstract
The oxygen exchange kinetics of BaGd0.3La0.7Co2O6−δ were investigated with pulsed isotope exchange (PIE) measurements in dry and humid atmospheres in the temperature range 400–600 °C in 0.005–0.21 bar O2. Synchrotron X-ray diffraction and scanning transmission electron microscopy (STEM) revealed exsolved Co3O4 nanoparticles dispersed on the surfaces that may contribute to the catalytic activity of the material towards the oxygen exchange reactions. The obtained oxygen exchange rate was 4.45 × 10−3 mol m−2 s−1 at 600 °C in 0.21 bar O2 with an activation energy of 0.76 eV. The rate determining step of the exchange reaction was determined to be dissociative adsorption of oxygen in both dry and humid atmospheres based on the individual rates of dissociative adsorption and incorporation, as well as pO2 dependencies of the oxygen exchange rate of around 1. The effect of water on the oxygen exchange rate was found to be dependent on the oxygen partial pressure, decreasing the rate at 0.21 bar O2 and 600 °C by a factor of approx. 2, while increasing the rate at 0.02 bar and 0.005 bar O2 by a similar amount. In situ thermogravimetric analysis was used to characterise the oxygen non-stoichiometry of the material throughout the oxygen exchange measurements.
1. Introduction
Proton ceramic electrolysis cells (PCEs) are promising electrochemical devices for efficient and clean production of hydrogen from steam using renewable electricity.1–4 Their operation at intermediate temperatures of 300–600 °C results in a lower thermodynamic cell voltage and enables utilization of waste heat for higher energy efficiency compared to water electrolysis cells, e.g., proton exchange membranes (PEM).5,6 The intermediate operating temperatures of PCEs also lead to improved electrode kinetics and reduced reliance on noble metal catalysts.7,8 Advantages of PCEs over solid oxide electrolysis cells (SOEC) – operating at higher temperatures – include their ability to produce dry pressurised hydrogen and presumably lower module costs and improved long-term stability.8–12The performance of PCEs is limited by the sluggish kinetics of the air/steam electrode (denoted positrode).11 The rate limiting reactions at the positrode are the oxygen reduction and evolution reactions (ORR/OER).13–15 These reactions involve several of the same steps as oxygen exchange, which thereby can be used to probe the catalytic activity towards the ORR and OER. Two main mechanisms for oxygen exchange have been defined. In the homoexchange mechanism, oxygen adsorbs dissociatively and exchange occurs between adsorbates on the surface.16–20 This exchange mechanism is typical for materials that are primarily electronic conductors with negligible oxide ion conductivity such as Sr-doped LaMnO3.21–23 In the heteroexchange mechanism, one or two oxygen atoms are incorporated into the surface after dissociative adsorption and the exchange occurs with oxygen from the bulk of the material.16–20 The heteroexchange mechanism is typical for mixed ionic electronic conductors such as La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF).21,24,25
Understanding the role of water on the oxygen exchange kinetics of positrodes in PCEs is essential as both species are involved in the electrochemical reaction under device operation. The presence of water and other co-adsorbates can lead to complex relationships with the oxygen exchange kinetics, depending on the type of exchange mechanism and the environmental conditions. On one hand, co-adsorbates such as H2O and CO2 may block catalytically active sites and reduce the exchange rate, unless the adsorption is not competitive (i.e., low surface coverage of adsorbates). A decrease in oxygen exchange rate in the presence of these co-adsorbates has been reported for LSCF, attributed to blocking of active sites.21,26 On the other hand, oxygen containing adsorbates may take part in the exchange reaction and contribute to the overall oxygen exchange, as shown for homoexchange on LSM, and heteroexchange on LSCF.26–28 In such cases, it is crucial to distinguish exchange from O2 involving oxygen reduction/evolution from other exchange reactions that do not represent the electrode redox processes.29 Notably, oxygen exchange between water and lattice oxygen proceeds at a faster rate and with lower activation energy compared to exchange from O2, as shown for (La0.8Sr0.2)0.95Cr0.5Fe0.5O3−δ and SrTiO3, since no electron transfer is required for chemisorption of water, i.e., oxygen reduction is only required for one of the molecules in the exchange reaction.30,31 Similarly, oxygen exchange has been shown to be significantly faster between lattice oxygen and CO2 compared to O2 on acceptor doped ZrO2 and Pd/Al2O3 catalysts.32–34 The presence of water was shown to increase the measured oxygen exchange rate by almost one order of magnitude on dense La0.6Sr0.4CoO3−δ (LSC) thin films at 400 °C,35 although the contribution from exchange with H2O was not quantified. A limited effect of humidity on the oxygen exchange coefficient was recently observed for SrTi0.65Fe0.35O3−δ.36 Finally, adsorbates can react to form secondary phases at the surface, induce cation segregation, or carry impurities such as silicon that are deposited on the surface, leading to degradation of the oxygen exchange rates as reported for LSC.35,37
Isotope exchange techniques are powerful methods for obtaining information about the oxygen exchange kinetics of metal oxides. Among these methods, pulsed isotope exchange (PIE) provides a quick way to gauge the oxygen surface exchange rate under different atmospheric conditions and temperatures.38 Parameters derived from isotope exchange experiments include the oxygen exchange rate and the contributions from dissociative adsorption and incorporation, as well as their activation energies.38
Double perovskite rare earth cobaltites LnBaCo2O5+δ (Ln = La, Pr, Nd, Sm, Gd, and Y) have been extensively studied as air electrode materials for solid oxide fuel cells.39 The oxygen exchange kinetics of PrBaCo2O5+δ and GdBaCo2O5+δ have been investigated with isotope exchange measurements. The obtained activation energies in 0.21 bar O2 were 0.8–1.0 eV for PrBaCo2O5+δ40–43 in the temperature range 300–850 °C and around 0.85 eV for GdBaCo2O5+δ42 in the temperature range 500–800 °C. Ba1−yGd0.8−xLa0.2+x+yCo2O6−δ (BGLCs) represents a group of double perovskites considered among the state-of-the-art positrodes for steam/water electrolysis over a wide temperature range (25–700 °C).44–46 Some compositions take up protons from water vapour and exhibit bulk protonic conductivity, making them particularly interesting for PCEs.47
In the present work, the oxygen exchange kinetics of BaGd0.3La0.7Co2O6−δ (BGLC37) were studied using pulsed isotope exchange in dry and humid atmospheres to further elucidate the role of steam on the materials performance as steam electrode. The oxygen non-stoichiometry and crystal structure of the material were quantified using thermogravimetry and synchrotron X-ray diffraction (XRD), respectively, to track the evolution of the material under the same conditions as the oxygen exchange measurements. Scanning transmission electron microscopy (STEM) and XRD revealed that the material contains exsolved Co3O4 nanoparticles dispersed on the surfaces that may contribute to the catalytic activity of the material towards the oxygen exchange reactions.48
2. Methods
2.1. Sample preparation
BaGd0.3La0.7Co2O6−δ (BGLC37) powder from spray pyrolysis (Marion Technologies, France) was uniaxially pressed into disks (25 mm diameter) and sintered at 1200 °C for 20 hours in ambient air to obtain dense samples (approx. 95% density). The sintered samples were crushed with a pestle and mortar, and sieved to acquire the particle size fractions 90–125 μm and 60–90 μm. A surface area of 0.10567 m2 g−1 for the 90–125 μm fraction was obtained with BET utilising a Belsorp mini II (Bel, Japan) with a pretreatment at 300 °C for three hours in vacuum.49,502.2. Synchrotron X-ray diffraction
Synchrotron X-ray diffraction experiments were conducted at the Swiss-Norwegian Beamline (SNBL) BM0151 at the European Synchrotron Radiation Facility (ESRF). A 0.5 mm diameter capillary filled with BGLC37 powder was rotated during measurements as the temperature increased from room temperature to 900 °C in air. An X-ray energy of 17.42 keV (λ = 0.72173 Å) was applied during the experiment and diffraction patterns were measured on a PILATUS@SNBL diffractometer. The 2D detector image was calibrated with a LaB6 standard and the in situ data were processed using the SNBL Toolbox and BUBBLE software.51 Rietveld refinements were performed using TOPAS v6 Academic software.522.3. Scanning transmission electron microscopy
High-angle annular dark field (HAADF) imaging and elemental mapping were conducted using STEM in combination with energy dispersive spectroscopy (EDS) using a Super-X detector. These analyses were performed on a Thermo Fisher Scientific Cs-corrected Titan G2 60–300 kV microscope, operated at an accelerating voltage of 200 kV. The BGLC37 sample was prepared by dispersing the crushed powder in ethanol using ultrasonic bath sonication to ensure uniform dispersion. A small amount of the dispersion was drop-cast onto a lacey carbon copper grid and allowed to dry.2.4. Electron probe microanalysis
Electron probe microanalysis (EPMA) was performed with a CAMECA SX100 electron microprobe on a sintered and polished BGLC37 disk sample. The microprobe was calibrated with cobalt metal, BaSO4, GdPO4 and LaPO4. The characteristic X-rays chosen for each of the elements were Ba Lα, Co Kα, La Lα and Gd Lβ. A focused beam was utilized with an energy of 15 kV and a beam current of 15 nA. The background was measured on each side of the chosen signals followed by a measurement of 20 s for each element.2.5. Pulsed isotope exchange
Pulsed isotope exchange measurements were performed in a custom setup with a gas supply line controlling both the carrier gas and the pulse, a packed bed reactor comprising a quartz tube inserted into a horizontal tube furnace, and a quadrupole mass spectrometer (Pfeiffer QME 220) connected to the exhaust line at the other end of the quartz tube. A powder sample of 43.7 mg of the 90–125 μm particle size fraction was placed in the quartz tube with an inner diameter of 2 mm. The powder bed (4 mm length) was held in place by quartz wool on both sides.The sample was pre-treated at 950 °C for 5 hours in a carrier gas with the desired pO2 and pH2O balanced with nitrogen at a flow rate of 50 mL s−1, before cooling to the first measurement temperature. Measurements were performed in 0.21, 0.02 and 0.005 bar O2 upon cooling from 600 °C to 200 °C. Measurements were performed isothermally at 25–50 °C intervals after a 30-minute equilibration time at each temperature and the heating and cooling rates were 3 °C per minute.
A pulse containing 18O (97% from Sigma Aldrich or 99% from Isotec) at the same pO2 as the carrier gas (balanced by 2% Ar and N2) was passed over the sample (pulse time 0.60 s), and the composition of the pulse was monitored at the outlet with the mass spectrometer. Gas containing 97% 18O was used for oxygen partial pressures of 0.21 bar, and 0.02 bar in dry conditions. Gas containing 99% 18O was used for oxygen partial pressures of 0.005 bar, and 0.02 bar O2 in humid conditions. The carrier gas was either bottle dry or bubbled through water to achieve a humidification of approximately 3% (0.03 bar), while the pulse was always bottle dry due to constraints in the setup. Thereby, the water content in the atmosphere while the pulse passes over the sample is not certain. Nevertheless, the measurements provide insight into the role of humidity on the oxygen exchange reactions. Blank measurements with only the quartz tube and quartz wool were performed to investigate possible exchange with the reaction chamber itself in a temperature range of 100–1100 °C. No exchange of oxygen with the reaction chamber was observed in the temperature range of interest (200–600 °C) with both dry and humid carrier gases (ESI†).
The PIE data was analysed according to a two-step model involving dissociative adsorption of O2 and incorporation of adsorbed oxygen into oxygen vacancies.38 The oxygen exchange reaction for heteroexchange can be written with Kröger–Vink notation as reversible adsorption, dissociation, and incorporation of oxygen into oxygen vacancies  :
:
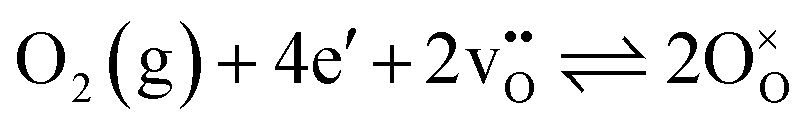 | (1) |
The oxygen exchange rate is calculated with the following equation:
 | (2) |
The oxygen exchange reaction can be split into two different reaction steps – dissociative adsorption and incorporation – and it is here not explicitly described in which step the oxygen reduction occurs:22,38
 | (3) |
| O* + vO ⇌ OO + * | (4) |
 | (5) |
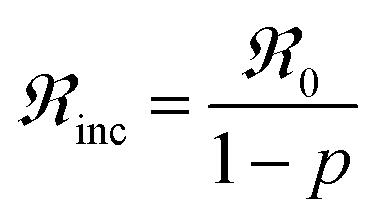 | (6) |
With this two-step model, the pO2 dependency of the oxygen exchange rate becomes 1 when dissociative adsorption is the rate limiting step and 0.5 when incorporation is the rate limiting step,22 as long as the materials properties do not change with pO2. Data analysis of the isotopologue fractions, extraction of reaction rates and activation energies were performed using a MATLAB script based on the aforementioned two-step model.53
2.6. Thermogravimetry
Thermogravimetry (TG) was performed using a Jupiter STA 449 F3 thermogravimetric analyser (Netzsch, Germany) with a 3.1 mL Al2O3 crucible. The TG measurements were performed for the 90–125 μm and 60–90 μm powder fractions, following the same pre-treatment and temperature profiles, and similar atmospheres to the PIE measurements. Blank measurements were performed with an empty crucible and the same atmospheres and temperature program. The mass change from the blank measurements was subtracted from the BGLC37 data to account for any instrumental background. All mass change after the correction with the blank measurements was attributed to the loss or uptake of oxygen. The absolute oxygen non-stoichiometry at the end of the 5-hour pre-treatments was separately determined by decomposition of BGLC37 in an atmosphere of 5% H2 in argon at 950 °C. The mass change was ascribed to oxygen loss due to reduction of cobalt to the metallic phase, i.e., assuming no change in oxidation states of the remaining cations.3. Results
3.1. Structure and exsolved Co3O4 nanoparticles
The structure of BaGd0.3La0.7Co2O6−δ has been reported to exhibit either tetragonal (P4/mmm)46 or orthorhombic (Pmmm)54 symmetry. The collected temperature dependent synchrotron X-ray diffraction patterns reveal no significant structural changes or phase transitions with increasing temperature (Fig. 1a and b). A minor Co3O4 phase (∼1 wt%) is identified at around 17° (Fig. 1b), with a crystallite size of about 35 nm that remained stable up to 800 °C (Fig. 1c). Rietveld refinements were first performed with the tetragonal P4/mmm space group for temperatures up to 900 °C. The lattice parameters increase linearly with increasing temperature, with a transition around 400 °C that may be ascribed to chemical expansion upon reduction (Fig. 1d).55 The weight fraction of the Co3O4 phase is given in Fig. 1e. Notably, the amount of the secondary Co3O4 phase remains essentially unchanged within the temperature range of the oxygen exchange measurements. Therefore, any contributions from the Co3O4 particles to the oxygen exchange kinetics can be expected to remain constant throughout the measurements. No significant improvement in the refinement quality (Rw) was obtained using the lower symmetry orthorhombic Pmmm space group (ESI†). Additionally, the lattice parameters and the amount of Co3O4 show similar trends for both P4/mmm and Pmmm phases as temperature increases. | ||
| Fig. 1 (a) Temperature-dependent synchrotron X-ray diffraction patterns of BGLC37 from room temperature to 900 °C. (b) Individual peaks of BGLC37 (P4/mmm) phase at (100), (002) and secondary phase Co3O4 at (311). (c) Grain size of Co3O4 estimation from the (311) peak calculated with the Scherrer equation. (d) Temperature-dependent lattice parameters of the primary P4/mmm phase from Rietveld refinements of synchrotron X-ray diffraction data. (e) Co3O4 weight percentage obtained from Rietveld refinements using a two-phase model comprising P4/mmm and a minor Co3O4 content of ∼1 wt%. | ||
To further investigate the nature of the Co3O4 phase, STEM analysis was performed on a sample from the crushed BGLC37 powder. STEM imaging revealed the presence of nanoparticles distributed across the surface of the sample, while some particles may also be embedded within the bulk (Fig. 2a and b). The size of the particles in these images appear to be somewhat smaller than the crystallite size of about 35 nm obtained from XRD (Fig. 1c). The elemental maps obtained through STEM-EDS confirmed the localized enrichment of cobalt in the particles protruding from the surface, consistent with Co3O4 (Fig. 2c–f).
 | ||
| Fig. 2 (a) and (b) high-angle annular dark field (HAADF) micrographs of the BGLC37 sample showing nanoparticles distributed over the surface. (c)–(f) Elemental maps of Ba (green), Gd (yellow), La (orange), and Co (pink), respectively, derived from micrograph (b), illustrating elemental distribution. | ||
The chemical composition of an as-sintered sample was investigated by electron probe microanalysis and the results are summarized in Table 1. The analysis includes contributions from the exsolved Co3O4 nanoparticles due to the comparatively large beam size and interaction volume. The obtained composition indicates minor cobalt deficiency, consistent with exsolution of Co3O4 not originating from excess cobalt or A-site deficiency in the BGLC material.
| Element | Mol fraction |
|---|---|
| Ba | 0.99 ± 0.12 |
| Gd | 0.32 ± 0.16 |
| La | 0.76 ± 0.08 |
| Co | 1.94 ± 0.12 |
| O | 5.51 ± 0.02 |
3.2. Oxygen exchange kinetics
 | ||
| Fig. 3 Oxygen isotopologue fractions as a function of temperature at 0.21 bar O2. The solid lines are fitted according to the two-step adsorption/incorporation model. | ||
Fig. 4 shows the oxygen exchange rates as a function of inverse temperature at different partial pressures of oxygen. The oxygen exchange rates exhibit Arrhenius behaviour and increase with oxygen partial pressure. The contributions from dissociative adsorption (ℜads) and incorporation (ℜinc) to the oxygen exchange rate (ℜ0) were calculated for a narrower temperature range (450–600 °C) where the data showed less scatter and consistent Arrhenius type behaviour (Fig. 4). The fastest process – in this case incorporation – generally exhibits the largest uncertainty. The oxygen exchange rate is limited by the dissociative adsorption of oxygen at all oxygen partial pressures, although to a lesser extent at 0.21 bar O2.
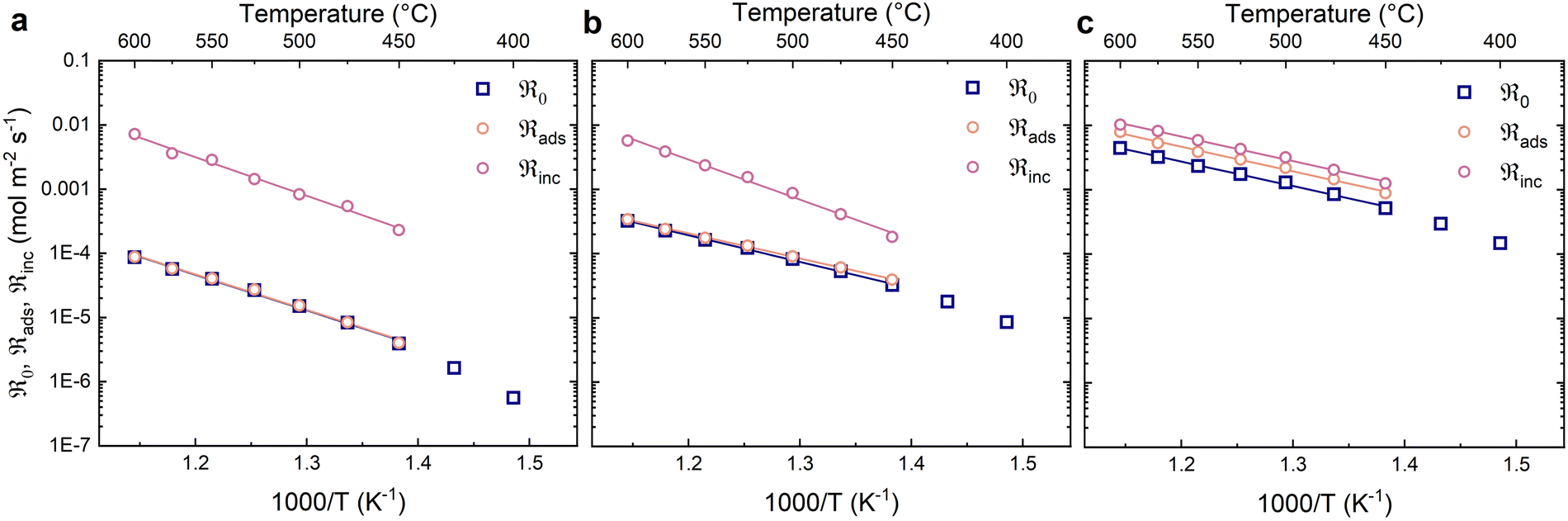 | ||
| Fig. 4 Oxygen exchange rate (ℜ0) and the contributions from dissociative adsorption (ℜads) and incorporation (ℜinc) at different oxygen partial pressures (dry carrier gas): (a) 0.005 bar (b) 0.02 bar (c) 0.21 bar. The solid lines are linear fits. | ||
The activation energies for the different reaction steps were obtained from the linear fits in Fig. 4 and are summarised in Table 2. The activation energies for dissociative adsorption (ℜads) are essentially the same as for the oxygen exchange rate (ℜ0). The activation energies of incorporation (ℜinc) are essentially the same as for the oxygen exchange at the highest oxygen partial pressure and slightly higher at the lower oxygen partial pressures.
| Atmosphere | Activation energies (eV) | |||
|---|---|---|---|---|
| pH2O (bar) | pO2 (bar) | ℜ 0 | ℜ ads | ℜ inc |
| 3E-5 | 0.005 | 1.10 ± 0.04 | 1.10 ± 0.04 | 1.20 ± 0.05 |
| 3E-5 | 0.02 | 0.82 ± 0.02 | 0.77 ± 0.01 | 1.24 ± 0.05 |
| 3E-5 | 0.21 | 0.76 ± 0.02 | 0.76 ± 0.02 | 0.76 ± 0.02 |
| 0.03 | 0.005 | 0.85 ± 0.02 | 0.87 ± 0.03 | — |
| 0.03 | 0.02 | 0.84 ± 0.02 | 0.90 ± 0.02 | — |
| 0.03 | 0.21 | 0.95 ± 0.04 | 1.10 ± 0.04 | 0.54 ± 0.05 |
Fig. 5 shows the oxygen exchange rate as a function of pO2 at 450–600 °C. The power-law dependence of the exchange rate is in the range of 1.0 to 1.3, decreasing slightly as the temperature increases. The pO2 dependency of the dissociative adsorption and incorporation rates are shown in Fig. 6. The rate of dissociative adsorption shows a pO2 dependency of around 1.2 to 1.4, and it increases with decreasing temperature. The incorporation rates show weaker dependencies on pO2 which appear to decrease with increasing temperature, and it is essentially zero at 600 °C. Overall, the pO2 dependencies are associated with some uncertainty as they are based on only three measurement points at each temperature.
 | ||
| Fig. 5 Oxygen exchange rate as a function of pO2 at 400–600 °C in dry carrier gas. The solid lines are linear fits. | ||
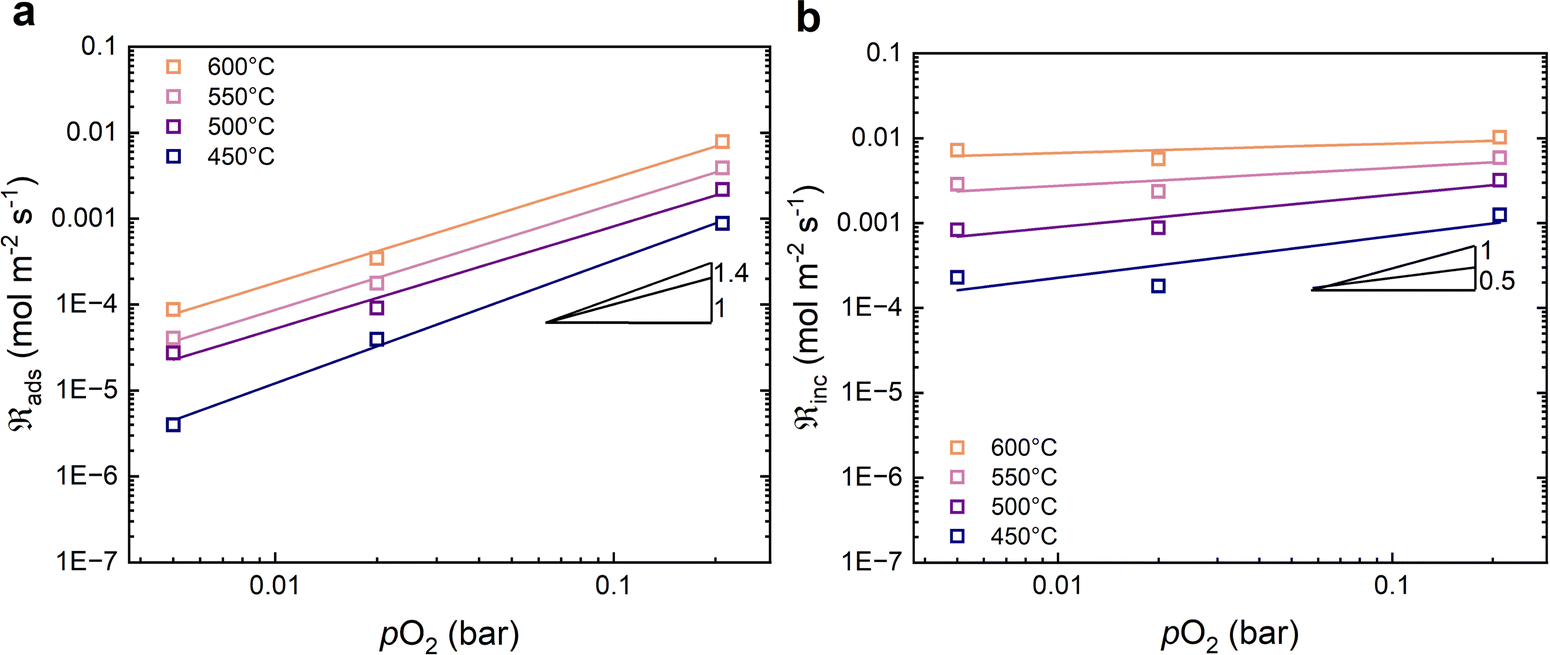 | ||
| Fig. 6 Oxygen partial pressure dependencies in dry carrier gas of (a) the dissociative adsorption rate, and (b) the incorporation rate. The solid lines are linear fits. | ||
 | ||
| Fig. 7 Oxygen exchange rate (ℜ0) and the contributions from dissociative adsorption (ℜads) and incorporation (ℜinc) at different oxygen partial pressures with humid carrier gas (0.03 bar H2O): (a) 0.005 bar (b) 0.02 bar (c) 0.21 bar. The solid lines are linear fits. | ||
The activation energies of the oxygen exchange processes are summarised in Table 2, showing a slight increase at 0.21 bar O2. The activation energies for dissociative adsorption are similar to the oxygen exchange rate at all measured oxygen partial pressures. The activation energies of incorporation exhibit the largest uncertainties and could not be reliably extracted from the data at 0.02 bar and 0.005 bar O2. The obtained activation energy for incorporation at 0.21 bar O2 was 0.54 eV, which is notably smaller than in dry carrier gas (0.76 eV). The pO2 dependencies of the oxygen exchange rate with a humid carrier gas exhibit a slight deviation from linearity as can be seen in Fig. 8. The pO2 dependency at the lower partial pressures is close to unity while the dependency at the highest pO2 is about 0.5.
 | ||
| Fig. 8 Oxygen exchange rate as a function of pO2 with humid carrier gas (0.03 bar H2O). | ||
It is noted that the obtained oxygen exchange rate in nominally dry atmosphere decreased after the measurements in humid atmospheres, e.g., the exchange rate at 500 °C and 0.21 bar O2 decreased from 1.30 × 10−3 mol m−2 s−1 to 1.06 × 10−3 mol m−2 s−1. This may be ascribed to changes to the material and/or experimental uncertainty between measurement series.
3.3. Oxygen non-stoichiometry
The oxygen non-stoichiometry of BGLC37 as measured by thermogravimetry is shown for the same temperature program as the PIE measurements in Fig. 9. The oxygen content of the material decreases with decreasing oxygen partial pressure and increasing temperature. The oxygen uptake is approximately proportional to the temperature from 600 °C to around 350 °C where it appears to start leveling off. The pO2 dependency of the oxygen non-stoichiometry is close to linear in dry atmospheres (Fig. 9b). When the atmosphere is humidified, the oxygen non-stoichiometry is essentially the same as in dry conditions at 0.21 bar and 0.02 bar O2, while it is slightly lower at 0.005 bar O2. This deviation leads to a more pronounced non-linearity in the oxygen non-stoichiometry as a function of pO2 in humid atmosphere (Fig. 9b). | ||
| Fig. 9 Oxygen non-stoichiometry of BGLC37 measured by TG with the same temperature program as for the PIE measurements as a function of (a) temperature and (b) pO2 in dry (open symbols) and humidified (0.03 bar H2O) atmospheres (filled symbols). The solid and dashed lines are guides to the eye. | ||
The nominal oxidation state of cobalt can be evaluated from the oxygen non-stoichiometry by assigning formal oxidation states to the cations, i.e., Ba2+, La3+ and Gd3+. Accordingly, the oxidation state of cobalt approaches Co3+ as the oxygen non-stochiomtry δ approaches 0.5 at the highest temperatures in 0.005 bar O2, while it is Co3.35+ for δ = 0.15 at 200 °C in 0.21 bar and 0.02 bar O2. The Co3O4 nanoparticles exhibit mixed valency of Co2+ and Co3+. Exsolution of the Co3O4 nanoparticles may therefore originate from partial reduction of cobalt at the higher temperatures during sintering in air at 1200 °C, similar to observations after mild reduction of PrBaCo2O6−δ in inert atmosphere at lower temperature.56
4. Discussion
In dry atmosphere the rate determining step of the oxygen exchange reaction has been identified as the dissociative adsorption of oxygen. This has been determined through the calculation of dissociative adsorption and incorporation rates (Fig. 4) as well as through the pO2 dependency of the oxygen exchange rates (Fig. 5). A rate determining step of dissociative adsorption has been reported for other mixed ionic electronic conducting materials such as LSCF.22 The activation energy of 0.76 eV for the oxygen exchange rate in 0.21 bar O2 is similar to what has been reported for other double perovskite cobaltites, e.g., 0.85 eV for GdBaCo2O5+δ42 and 0.8–1.0 eV for PrBaCo2O5+δ40–43 and other perovskite materials, e.g., SrTi0.5Fe0.5O3−δ (0.85 eV),57 and Sr1.95Fe1.4Ni0.1Mo0.5O6−δ (0.82 eV).58When water is introduced into the carrier gas, additional exchange reactions involving 16O from water can take place. Notably, the data showed no sign of substantial exchange between water and lattice oxygen based on analysis of masses 17 (16OH), 18 (H216O), 19 (18OH), and 20 (H218O) under the pulse (ESI†). Therefore, all oxygen exchange was ascribed to exchange between gas phase and lattice oxygen. This contrasts the findings reported by Sha et al.30 for (La0.8Sr0.2)0.95Cr0.5Fe0.5O3−δ where the activation energy decreased significantly when water (0.03 bar) was introduced into the oxygen gas (0.20 bar).
When comparing the oxygen exchange rates in dry and humid atmospheres, the effect of water in the carrier gas is pO2 dependent (Fig. 10). With the addition of water in the carrier gas, the oxygen exchange rate decreases in high pO2 and increases in low pO2, e.g., at 600 °C the rate decreases from 4.45 × 10−3 mol m−2 s−1 by a factor of approx. 2 in 0.21 bar O2, while it increases from 3.24 × 10−4 mol m−2 s−1 by the same amount in 0.02 bar O2. Water can adsorb and block active sites for the rate limiting dissociative adsorption step and thereby cause a decrease in the oxygen exchange rate. Furthermore, blocking of active sites by water or hydroxide species may predominate at higher pO2 (0.21 bar) due to the need for a larger number of adsorption sites. Blocking of active sites at 0.21 bar O2 may also explain the decrease is the pO2 dependency of the oxygen exchange rate towards 0.21 bar considering the non-linearity in the measured data (Fig. 8). The activation energy of the oxygen exchange rate in 0.21 bar O2 increased in humid atmosphere from 0.76 eV to 0.95 eV. However, these activation energies appear to become similar at the highest temperatures where the effect of water or hydroxide adsorbates may be less prominent. Otherwise, these differences may indicate a change in the reaction mechanism that is not distinguishable here due to the limitations of the method.22,59
 | ||
| Fig. 10 Comparison of the oxygen exchange rate of BGLC37 in dry and humid carrier gases (0.03 bar H2O). The solid lines are linear fits. | ||
The activation energies of the oxygen exchange rate were similar between dry and humid atmospheres in 0.02 bar O2 (Fig. 10). At this oxygen partial pressure, the presence of water had a minor impact on the oxygen non-stoichiometry in the temperature range of interest (Fig. 9). In contrast, the presence of water appears to increase the amount of oxygen deficiency in the material at 0.005 bar O2 (Fig. 9), which may be related to the increased oxygen exchange rate and the significant change in activation energy from 1.1 eV to 0.85 eV.
In comparison to other materials measured with PIE, BGLC37 shows a similar oxygen exchange rate in dry 0.21 bar O2 (Fig. 11). In humid atmospheres, the oxygen exchange rate decreases slightly but remains quite similar to the other materials in dry atmospheres. When the oxygen exchange coefficient (k) is compared between different materials and methods, BGLC37 shows a slightly higher oxygen exchange coefficient at lower temperatures than other materials measured with PIE, but the values become more similar as the temperature increases (Fig. 11b). The surface exchange coefficient for BGLC37 was calculated by dividing the oxygen exchange rate at each temperature by the oxygen concentration in the oxide (8.8741 × 104 m3 mol−1).38 The oxygen exchange coefficient for BSCF measured by chemical expansion relaxation is significantly higher than the other materials (2.9 × 10−3 cm s−1 at 700 °C).38,60
 | ||
| Fig. 11 (a) Comparison of oxygen exchange rate of BGLC37 (this work), SrTi0.5Fe0.5O3−δ (STF50),57 SrTi0.65Fe0.35O3−δ (STF35),57 PrBaCo2O5+δ (PBCO),40 Sr1.95Fe1.4Ni0.1Mo0.5O6−δ (SFNMO),58 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF),38 and La2NiO4+δ (LNO)38 at 0.21 bar O2 from PIE measurements. (b) Comparison of the oxygen exchange coefficient of BGLC37 (this work), PrBaCo2O5+δ (PBCO),43 La0.6Sr0.4Co2O3−δ (LSCO),61 and La2NiO4+δ (LNO)38 measured with varying methods. Squares: PIE, Circles: Isotope exchange depth profile. | ||
5. Conclusions
The oxygen exchange rate of BGLC37 containing exsolved Co3O4 nanoparticles was measured with pulsed isotope exchange as a function of temperature and oxygen partial pressure in dry and humid atmospheres. The obtained oxygen exchange rate was 4.45 × 10−3 mol m−2 s−1 at 600 °C in 0.21 bar O2. The activation energy of the exchange rate was 0.76 eV and 0.82 eV in 0.21 bar and 0.02 bar O2, respectively, while it increased to 1.1 eV at 0.005 bar O2. The rate determining step of the exchange reaction was determined to be dissociative adsorption of oxygen in both dry and humid atmospheres based on the individual rates of dissociative adsorption and incorporation, as well as a pO2 dependencies of the oxygen exchange rate of around 1.The effect of water on the oxygen exchange rate was found to be dependent on the oxygen partial pressure, decreasing the rate at 0.21 bar O2 and 600 °C by a factor of approx. 2, while increasing the rate at 0.02 bar and 0.005 bar O2 by a similar amount. These results were interpreted in terms of blocking of active sites on the surface by water or hydroxide species at 0.21 bar O2, and changes to the material due to increased oxygen non-stoichiometry at 0.005 bar O2. The presence of water led to an increased activation energy of 1.01 eV at 0.21 bar, while the activation energy remained similar at 0.02 bar O2 and decreased to 0.85 eV in 0.005 bar O2.
Data availability
Data from pulsed isotope exchange and thermogravimetry measurements are available at the figshare repository at https://doi.org/10.6084/m9.figshare.28061276. The synchrotron X-ray diffraction data is available at https://doi.org/10.15151/ESRF-ES-1550904430.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Research Council of Norway (RCN) for financial support under the FRIPRO program (project no. 315058) and for support to the Norwegian Center for Transmission Electron Microscopy (NORTEM) national infrastructure (project no. 197405). The authors further acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal number ma6015 and we would like to thank Chloe Fuller for assistance and support in using beamline BM01.References
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230–240 CAS.
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 CAS.
- D. Kim, K. T. Bae, K. J. Kim, H.-N. Im, S. Jang, S. Oh, S. W. Lee, T. H. Shin and K. T. Lee, ACS Energy Lett., 2022, 7, 2393–2400 CAS.
- Y. Wang, Y. Ling, B. Wang, G. Zhai, G. Yang, Z. Shao, R. Xiao and T. Li, Energy Environ. Sci., 2023, 16, 5721–5770 CAS.
- D. Medvedev, Int. J. Hydrogen Energy, 2019, 44, 26711–26740 CAS.
- F. Liu, H. Deng, D. Diercks, P. Kumar, M. H. A. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, L. Fang, D. Chen, B. Liu and C. Duan, Nat. Energy, 2023, 8, 1145–1157 CAS.
- M. M. Rashid, M. K. Al Mesfer, H. Naseem and M. Danish, Int. J. Eng. Adv. Technol., 2015, 4, 2249–8958 Search PubMed.
- L. Bi, S. Boulfrad and E. Traversa, Chem. Soc. Rev., 2014, 43, 8255–8270 CAS.
- Y. Wang, Y. Ling, B. Wang, G. Zhai, G. Yang, Z. Shao, R. Xiao and T. Li, Energy Environ. Sci., 2023, 16, 5721–5770 RSC.
- M. Choi, D. Kim, T. K. Lee, J. Lee, H. S. Yoo and W. Lee, Adv. Energy Mater., 2025, 15, 2400124 CrossRef CAS.
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang and Y. Zhang, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- Z. Luo, Y. Zhou, X. Hu, N. Kane, T. Li, W. Zhang, Z. Liu, Y. Ding, Y. Liu and M. Liu, Energy Environ. Sci., 2022, 15, 2992–3003 RSC.
- J. Jing, Z. Lei, Z. Zheng, H. Wang, P. Zhang, Z. Wang, H. Xu and Z. Yang, Int. J. Hydrogen Energy, 2023, 48, 9037–9045 CrossRef CAS.
- Y. Yi, R. Ran, W. Wang, W. Zhou and Z. Shao, Curr. Opin. Green Sustainable Chem., 2022, 38, 100711 CrossRef CAS.
- F. He, Y. Zhou, T. Hu, Y. Xu, M. Hou, F. Zhu, D. Liu, H. Zhang, K. Xu and M. Liu, Adv. Mater., 2023, 35, 2209469 CrossRef CAS PubMed.
- M. Den Otter, B. A. Boukamp and H. J. Bouwmeester, Solid State Ionics, 2001, 139, 89–94 Search PubMed.
- G. Boreskov and V. Muzykantov, Ann. N. Y. Acad. Sci., 1973, 213, 137–170 CrossRef CAS PubMed.
- K. Klier, J. Novakova and P. Jiru, J. Catal., 1963, 2, 479–484 CrossRef CAS.
- A. Ezin, V. Tsidilkovski and E. K. Kurumchin, Solid State Ionics, 1996, 84, 105–112 CrossRef CAS.
- V. Muzykantov, G. Boreskov and G. Panov, React. Kinet. Catal. Lett., 1974, 1, 315–319 CrossRef CAS.
- Y.-L. Huang, C. Pellegrinelli and E. D. Wachsman, ACS Catal., 2017, 7, 5766–5772 CAS.
- C. Kan, H. Kan, F. Van Assche, E. Armstrong and E. Wachsman, J. Electrochem. Soc., 2008, 155, B985 CrossRef CAS.
- Y.-L. Huang, C. Pellegrinelli and E. D. Wachsman, ACS Catal., 2016, 6, 6025–6032 CrossRef CAS.
- Y. L. Huang, C. Pellegrinelli and E. D. Wachsman, Angew. Chem., Int. Ed., 2016, 55, 15268–15271 CrossRef CAS PubMed.
- Y.-L. Huang, C. Pellegrinelli, A. Geller, S.-C. Liou, A. Jarry, L. Wang, Y. Yu, H. Bluhm, E. J. Crumlin, K. J. Gaskell, B. W. Eichhorn and E. D. Wachsman, Energy Environ. Sci., 2017, 10, 919–923 RSC.
- V. Thoreton, M. Niania and J. Kilner, Phys. Chem. Chem. Phys., 2021, 23, 2805–2811 RSC.
- Y.-L. Huang, C. Pellegrinelli, A. Geller, S.-C. Liou, A. Jarry, L. Wang, Y. Yu, H. Bluhm, E. J. Crumlin and K. J. Gaskell, Energy Environ. Sci., 2017, 10, 919–923 RSC.
- J. Yang, J. M. Polfus, Z. Li, H. L. Tuller and B. Yildiz, Chem. Mater., 2020, 32, 5483–5492 CrossRef CAS.
- A. Nenning, E. Navickas, H. Hutter and J. Fleig, J. Phys. Chem. Lett., 2016, 7, 2826–2831 CrossRef CAS PubMed.
- Z. Sha, E. Cali, Z. Shen, E. Ware, G. Kerherve and S. J. Skinner, Chem. Mater., 2021, 33, 8469–8476 CrossRef CAS.
- J. Kler and R. A. De Souza, J. Phys. Chem. Lett., 2022, 13, 4133–4138 CrossRef CAS PubMed.
- E. K. Kurumchin and M. V. Perfiliev, Solid State Ionics, 1990, 42, 129–133 CrossRef CAS.
- S. Ojala, N. Bion, S. Rijo Gomes, R. L. Keiski and D. Duprez, ChemCatChem, 2010, 2, 527–533 CrossRef CAS.
- A. Nau, R. Pointecouteau, M. Richard, T. Belin, F. Can, C. Comminges and N. Bion, Catal. Commun., 2023, 180, 106704 CrossRef CAS.
- J. H. Joo, R. Merkle and J. Maier, J. Power Sources, 2011, 196, 7495–7499 Search PubMed.
- D. M. Schwenkel, R. A. De Souza and G. F. Harrington, J. Mater. Chem. A, 2025, 13, 8541–8548 Search PubMed.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Solid State Ionics, 2012, 208, 43–51 Search PubMed.
- H. J. Bouwmeester, C. Song, J. Zhu, J. Yi, M. van Sint Annaland and B. A. Boukamp, Phys. Chem. Chem. Phys., 2009, 11, 9640–9643 Search PubMed.
- J.-H. Kim and A. Manthiram, J. Mater. Chem. A, 2015, 3, 24195–24210 Search PubMed.
- C.-Y. Yoo, B. A. Boukamp and H. J. Bouwmeester, Solid State Ionics, 2014, 262, 668–671 CrossRef CAS.
- M. Ananyev, V. Eremin, D. Tsvetkov, N. Porotnikova, A. Farlenkov, A. Y. Zuev, A. Fetisov and E. K. Kurumchin, Solid State Ionics, 2017, 304, 96–106 CrossRef CAS.
- A. C. Tomkiewicz, M. Meloni and S. McIntosh, Solid State Ionics, 2014, 260, 55–59 Search PubMed.
- M. Burriel, J. Pena-Martinez, R. J. Chater, S. Fearn, A. V. Berenov, S. J. Skinner and J. A. Kilner, Chem. Mater., 2012, 24, 613–621 CAS.
- R. Strandbakke, V. A. Cherepanov, A. Y. Zuev, D. S. Tsvetkov, C. Argirusis, G. Sourkouni, S. Prünte and T. Norby, Solid State Ionics, 2015, 278, 120–132 CAS.
- E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Nat. Mater., 2019, 18, 752–759 Search PubMed.
- J. Zhu, J. B. Guđmundsdóttir, R. Strandbakke, K. G. Both, T. Aarholt, P. A. Carvalho, M. H. Sørby, I. J. Jensen, M. N. Guzik and T. Norby, ACS Appl. Mater. Interfaces, 2021, 13, 20313–20325 Search PubMed.
- T. Miruszewski, R. Strandbakke, K. Dzierzgowski, I. Szpunar, A. Mielewczyk-Gryń, S. Wachowski and M. Gazda, J. Mater. Chem. A, 2024, 12, 13488–13497 CAS.
- C. Nicollet, C. Toparli, G. F. Harrington, T. Defferriere, B. Yildiz and H. L. Tuller, Nat. Catal., 2020, 3, 913–920 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CAS.
- P. Llewellyn, F. R. Reinoso, J. Rouqerol and N. Seaton, Characterization of porous solids VII: proceedings of the 7th International Symposium on the Characterization of Porous Solids (COPS-VII), Aix-en-Provence, France, 26-28 May 2005, Elsevier, 2006 Search PubMed.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- V. Thoreton, https://github.com/vthoreton/PIE_fit.
- R. Strandbakke, D. S. Wragg, M. H. Sørby, M. N. Guzik, A. E. Gunnæs, I. Szpunar, S. L. Wachowski, M. Balaguer, P. A. Carvalho, A. Mielewczyk-Gryń, J. M. Serra and T. Norby, Dalton Trans., 2022, 51, 18667–18677 Search PubMed.
- I. Szpunar, R. Strandbakke, M. H. Sørby, S. L. Wachowski, M. Balaguer, M. Tarach, J. M. Serra, A. Witkowska, E. Dzik and T. Norby, Materials, 2020, 13, 4044 CrossRef CAS PubMed.
- A. J. Carrillo, M. Balaguer, C. Solís, A. López-García, S. Haas, M. Fabuel, B. Delgado-Galicia, I. Rodriguez, E. Vøllestad, S. Wachowski, R. Strandbakke, T. Norby and J. M. Serra, J. Phys.: Energy, 2025, 7, 025007 CAS.
- C.-Y. Yoo and H. J. Bouwmeester, Phys. Chem. Chem. Phys., 2012, 14, 11759–11765 Search PubMed.
- N. Porotnikova, A. Khodimchuk, D. Zakharov, N. Bogdanovich and D. Osinkin, Appl. Surf. Sci., 2023, 613, 156015 CrossRef CAS.
- R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2002, 4, 4140–4148 CAS.
- M.-B. Choi, S.-Y. Jeon, H.-N. Im, E. Wachsman and S.-J. Song, J. Electrochem. Soc., 2011, 159, P23 Search PubMed.
- A. Berenov, A. Atkinson, J. Kilner, E. Bucher and W. Sitte, Solid State Ionics, 2010, 181, 819–826 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04791e |
| This journal is © the Owner Societies 2025 |