
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-driven phase transformations on Mg3Ca(CO3)4 huntite carbonate†
David
Santamaría-Pérez
 *a,
Raquel
Chuliá-Jordán
b,
Benedito Donizeti
Botan-Neto
a,
Ganesh
Bera
a,
Julio
Pellicer-Porres
a,
Lkhamsuren
Bayarjargal
c,
Alberto
Otero-de-la-Roza
d and
Catalin
Popescu
e
*a,
Raquel
Chuliá-Jordán
b,
Benedito Donizeti
Botan-Neto
a,
Ganesh
Bera
a,
Julio
Pellicer-Porres
a,
Lkhamsuren
Bayarjargal
c,
Alberto
Otero-de-la-Roza
d and
Catalin
Popescu
e
aDepartamento de Física Aplicada-ICMUV, MALTA Consolider Team, Universitat de València, Valencia 46100, Spain. E-mail: David.Santamaria@uv.es
bDepartamento de Didáctica de las Ciencias Experimentales y Sociales, Universitat de Valencia, 46022, Valencia, Spain
cInstitute of Geosciences, Goethe University Frankfurt, Frankfurt 60438, Germany
dDepartamento de Química Física y Analítica, Facultad de Química, MALTA Consolider Team, Universidad de Oviedo, Oviedo 33006, Spain
eCELLS-ALBA Synchrotron Light Facility, Cerdanyola del Vallés, 08290, Barcelona, Spain
First published on 15th January 2025
Abstract
Magnesium and calcium carbonate minerals are significant reservoirs of Earth's carbon and understanding their behavior under different conditions is crucial for elucidating the mechanisms of deep carbon storage. Huntite, Mg3Ca(CO3)4, is one of the two stable calcium magnesium carbonate phases, together with dolomite. The distinctive cation coordination environment of Ca atoms compared to calcite-type and dolomite structures makes huntite a comparatively less dense phase. Here we examine the behavior of a polycrystalline natural huntite sample under room-temperature compression up to 38 GPa. Synchrotron X-ray diffraction and Raman spectroscopy experiments were carried out in a diamond-anvil cell using He as a highly hydrostatic pressure transmitting medium. XRD results suggest that the initial R32 huntite structure persists up to 21 GPa. The Raman experiment agrees with this result but also suggests the appearance of structural defects from 10 GPa on. Birch–Murnaghan equation of state parameters were fit to the pressure–volume huntite data resulting in zero-pressure volume V0 of 611.7(2) Å3, a bulk modulus B0 of 99.5(11) GPa and a pressure derivative of the bulk modulus of  . At 21 GPa, huntite transforms to another trigonal phase (R3), designated here as huntite II. This phase persists up to at least 38 GPa, the maximum pressure reached in this study. The major structural differences between huntite and the huntite-II phase involve the tilting of the [CO3] units with respect to the basal plane and a rotation, which cause a progressive change in the coordination number of the Ca atoms, from 6 to 9. DFT calculations complement the experimental data, providing new insights into the structural response to high-pressure conditions of this magnesium–calcium double carbonate mineral.
. At 21 GPa, huntite transforms to another trigonal phase (R3), designated here as huntite II. This phase persists up to at least 38 GPa, the maximum pressure reached in this study. The major structural differences between huntite and the huntite-II phase involve the tilting of the [CO3] units with respect to the basal plane and a rotation, which cause a progressive change in the coordination number of the Ca atoms, from 6 to 9. DFT calculations complement the experimental data, providing new insights into the structural response to high-pressure conditions of this magnesium–calcium double carbonate mineral.
Introduction
The Earth's deep carbon cycle plays a critical role in regulating the planet's climate, atmospheric composition, and overall geochemical processes.1–4 Central to this cycle are carbonate minerals, which act as significant reservoirs of carbon. Among these, magnesium (Mg) and calcium (Ca) carbonates are particularly important due to their abundance and stability in various geological settings. These minerals not only sequester carbon dioxide (CO2) from the atmosphere but also facilitate its long-term storage within the Earth's crust and mantle. Understanding the behavior of Mg and Ca carbonates under different conditions is crucial for elucidating the mechanisms of deep carbon storage and transport.Magnesite (MgCO3, space group (SG) R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c), calcite (CaCO3, SG. Rc) and dolomite (MgCa(CO3)2, SG. R) are trigonal stable phases at room conditions, where the Mg and Ca atoms are octahedrally coordinated by O atoms. Trigonal structures are also adopted by other carbonates with small divalent cations (Fe, Ni, Zn, Mn, Co or Cd).5–7 Aragonite, another CaCO3 polymorph, is an orthorhombic phase (SG. Pmcn) where the Ca atoms have 9 O neighbor atoms and this structure-type is adopted by carbonates with larger divalent cations (Sr, Ba or Pb).8,9 The Ca2+ cation size delimits the border of the (trigonal) calcite- to (orthorhombic) aragonite-type structures. The chemical stability of carbonates of divalent cations within the trigonal and orthorhombic groups therefore varies with cation radius, which is also directly related with its solubility behavior.10 Ca carbonates are at the center of the trigonal–orthorhombic structural discontinuity and their crystal chemistry, in particular, the local environment around the Ca atoms, is not yet well understood.
c), calcite (CaCO3, SG. Rc) and dolomite (MgCa(CO3)2, SG. R) are trigonal stable phases at room conditions, where the Mg and Ca atoms are octahedrally coordinated by O atoms. Trigonal structures are also adopted by other carbonates with small divalent cations (Fe, Ni, Zn, Mn, Co or Cd).5–7 Aragonite, another CaCO3 polymorph, is an orthorhombic phase (SG. Pmcn) where the Ca atoms have 9 O neighbor atoms and this structure-type is adopted by carbonates with larger divalent cations (Sr, Ba or Pb).8,9 The Ca2+ cation size delimits the border of the (trigonal) calcite- to (orthorhombic) aragonite-type structures. The chemical stability of carbonates of divalent cations within the trigonal and orthorhombic groups therefore varies with cation radius, which is also directly related with its solubility behavior.10 Ca carbonates are at the center of the trigonal–orthorhombic structural discontinuity and their crystal chemistry, in particular, the local environment around the Ca atoms, is not yet well understood.
Huntite Mg3Ca(CO3)4 mineral is produced in low-temperature surface or near surface environments by precipitation from magnesium-rich aqueous solutions or as a result of the interaction of such solutions with other carbonates.11–13 Its composition lies halfway between MgCO3 magnesite and MgCa(CO3)2 dolomite, and it is also a trigonal phase (SG. R32) but its structure consists of [CO3] carbonate units roughly perpendicular to the c axis, 3/4 of these carbonate groups having the same orientation (see Fig. 1). This means that, whereas the Mg atoms are octahedrally coordinated by oxygen atoms, the Ca atoms are at the center of trigonal prisms. This particular oxygen arrangement around the Ca atoms produces a relatively low-density structure, with a density of 2.875 g cm−3 at ambient conditions (similar to that of dolomite and smaller than the density average of dolomite and magnesite, ∼2.94 g cm−3).
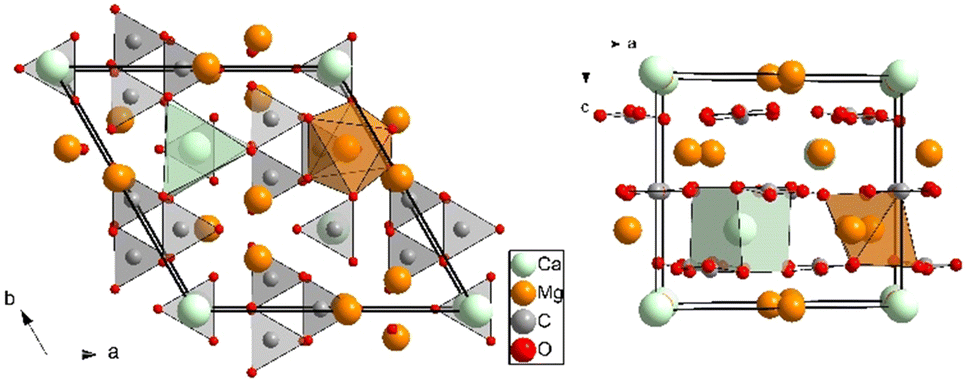 | ||
| Fig. 1 Projections along the c (left) and b axes (right) of the crystalline R32 Mg3Ca(CO3)4 huntite structure at room conditions. Thick black lines indicate the unit-cell. Light green, orange, light gray, and red spheres represent Ca, Mg, C, and O atoms, respectively. A [CaO6] trigonal prism and a [MgO6] octahedron are depicted to illustrate the different coordination sphere around the cation atoms. [CO3] trigonal planar units are also represented. | ||
Magnesium and calcium carbonates have been widely studied at high-pressure (HP) and high-temperature (HT) conditions. The structural behavior of the Mg and Ca end-members of the MgO–CaO–CO2 carbonate system under compression and heating differ significantly. MgCO3 magnesite is stable throughout a wide pressure and temperature range, with just one HP-HT phase transition reported above 115 GPa and 2200 K.14,15 CaCO3, on the other hand, undergoes numerous high pressure phase transitions below 30 GPa.16,17 Dolomite carbonate, MgCa(CO3)2, an intermediate composition between magnesite and calcite, has two room-temperature high-pressure polymorphs above 17 GPa and 35 GPa18,19 and high P,T phase at 46 GPa and 1800 K.19 The composition of Mg3Ca(CO3)4 huntite lies halfway between MgCO3 magnesite and MgCa(CO3)2 dolomite and it is reported to be structurally stable up to 12 GPa at room temperature.20 When heated below 6 GPa, huntite decomposes into CaCO3, MgO and CO2 at approximately 420 °C independently of pressure.20
Huntite's unique composition and structure provide valuable insights into the stability and behavior of mixed carbonates under various environmental conditions. In particular, the study of huntite upon compression will enhance our understanding of the coordination chemistry of the Ca atoms in carbonates and the stability fields of Mg and Ca carbonates. This work studies the structural and vibrational properties of huntite up to 30 and 38 GPa, respectively, extending more than 3 times the previously reported pressure range. Synchrotron powder X-ray diffraction (XRD) and Raman spectroscopy measurements using quasi hydrostatic pressure medium, reveal a phase transition and DFT calculations confirm the stability of the dense polymorph.
Experimental details
Naturally occurring huntite samples from the Tea Tree Gully in the South Australia State (Australia) were kindly provided by the Yale Peabody Museum (Specimen YPM MIN 035091). The chemical composition of the polycrystalline huntite sample was characterized by means of energy dispersive X-ray spectroscopy on a Philips XL30 scanning electron microscope. Only traces (<0.2 atom %) of Sr and Na were found besides the Mg, Ca, C, and O atoms present in the Mg3Ca(CO3)4 huntite composition. XRD measurements carried out at room conditions confirmed that the sample has the previously reported structure.Room temperature HP XRD experiments up to 30 GPa were carried out in a membrane-driven diamond-anvil cell (DAC) equipped with diamonds of 350 μm of culet diameter. The huntite sample was placed together with a ruby chip at the center of a 150 μm diameter 40 μm thick pressure chamber drilled in a preindented rhenium gasket. High-purity helium (He) gas was also loaded in the DAC at room temperature using the Sanchez Technologies gas loading apparatus. Helium is fluid up to 12 GPa at room temperature21,22 and acted as a quasi-hydrostatic medium in the pressure range of the study.23 Pressure was determined using the ruby fluorescence method.24In situ angle-dispersive powder X-ray diffraction measurements were carried out at MSPD beamline at ALBA-CELLS synchrotron using an X-ray wavelength of 0.4246 Å.25 This beamline provides an X-ray beam focused down to ∼20 × 20 μm2, and the diffracted signal was collected with a Rayonix CCD detector. The detector parameters were calibrated with LaB6 powder standard, and integration to conventional 2θ-intensity data was carried out with the Dioptas software.26 The indexing and refinement of the powder patterns were performed using the Unitcell,27 Powdercell28 and Fullprof29 program packages.
Raman spectra were measured in 2–3 GPa steps upon compression and decompression covering a range between ambient pressure and 38 GPa. A frequency-doubled 532.14 nm Nd:YAG Oxxius laser (LCX-532S) was focused on the sample with a spot size of 6 μm. Spectra were collected in backscattering geometry, using a grating spectrometer (Acton, SP-2356) equipped with a Pixis 256E charge coupled device (CCD) detector and a Mitutoyo microscope objective. The spectral resolution of the spectrometer is 3 cm−1.30,31 The laser power was set to 80 mW and spectra were collected for 300 s in a frequency window of 100–1300 cm−1, using a grating of 1800 grooves per mm.
Computational details
Density-functional theory (DFT) calculations were carried out in the huntite structure using Quantum ESPRESSO (QE), version 6.5.32 The projector augmented wave (PAW) method33 was used in combination with the B86bPBE-XDM functional34–38 implemented in QE. PAW datasets were obtained from the pslibrary39 for all atoms, with number of valence electrons equal to 10 (Ca), 4 (C), 10 (Mg), and 6 (O). The kinetic energy cuttoff for the plane-wave expansions of the wavefunction and the electron density were 100 Ry and 1000 Ry, respectively, and a uniform k-point grid of 2 × 2 × 2 was used for the huntite structure and doubled dolomite structures. The experimental zero-pressure geometry of huntite was relaxed at zero pressure and 50 GPa. All geometry relaxations were carried out with tight convergence parameters: 10−5 Ry in the energy and 10−4 Ry bohr−1 in the forces. The equilibrium volumes resulting from these initial relaxations were used to set up a uniformly spaced volume grid with 41 points spanning the corresponding pressure range. Constant-volume geometry relaxations were carried out at each of the 41 points.To probe the dynamical instability of the calculated structure, phonon frequency calculations were carried out with the density-functional perturbation theory method.40 Uniform reciprocal-space q-point grids with size 2 × 2 × 2 were used. As described below, our calculations predict that the R32 structure becomes dynamically unstable under pressure resulting in a broken-symmetry R3 structure, in agreement with experiment. For this reason, we calculated both the R32 structure, dynamically unstable at high pressure, and the R3 structure, which coincides with R32 at low pressure and is stable at high pressure.
The equation of state and the transition pressures were calculated with the gibbs2 code,41,42 using the data generated by Quantum ESPRESSO. To compare to the experimental data, Raman spectra were simulated at selected pressures in both the R32 and R3 phases. The QE equilibrium structures at a given pressure was relaxed using the crystal17 program,43 employing the pob-DZVP-rev2 double-zeta basis sets.44 Constant-volume geometry relaxations were carried out using the B3LYP-D345–47 functional with the same k-point grid as in the QE calculation. Raman intensities were then obtained using the coupled-perturbed Kohn–Sham method.48,49
Results and discussion
XRD measurements
The structural evolution of huntite, Mg3Ca(CO3)4, at high pressure and room temperature has been studied by means of in situ powder synchrotron X-ray diffraction measurements. The experiments were performed in hydrostatic conditions using He as pressure transmitting medium. Fig. 2 illustrates a selection of patterns at different pressures, which shows that there are no obvious changes in the XRD profiles except for the shift of the peaks to higher 2θ angles as a result of unit cell compression. High-pressure synchrotron powder XRD patterns present some texturing effects due to uneven crystal sizes (see the bottom of Fig. 2). Despite the fact that, in our case, this can cause small inaccuracies in the relative intensities of the diffraction maxima, we were able to perform full structural Rietveld refinements. The lattice parameters and the atomic coordinates at the lowest pressure (1.05 GPa) are consistent with those of the previously reported huntite structure12,13,20 (see Fig. S1 and Table S1 of ESI†). Raman spectroscopic results at room conditions, as we will discuss later, and DFT calculations also confirm that the low-pressure phase is the R32 huntite structure.12,13,20 | ||
| Fig. 2 Selected powder XRD patterns of huntite up to 29.5 GPa (λ = 0.4246 Å). All the patterns can be indexed with a trigonal unitcell and no significant peak intensity changes are observed in the studied pressure range. The diffraction signals corresponding to the rhenium of the gasket are marked with asterisks. The different colors denote the pressure ranges where indications of phase transitions were found either by discontinuities in lattice parameters dimensions or changes in the Raman spectra. The unrolled (or cake) image of the raw data from the CCD detector at 1.05 GPa and room temperature is given to illustrate the quality of our data. | ||
A short description of huntite needs to be done in order to understand its atomic arrangement and the phase transformations that could occur at high pressures. The lattice parameters of huntite are related to those of calcite (or magnesite) by ahunt ∼ 2 × acalc; chunt ∼ ccalc/2. The relationship to calcite arises from the ordering of the cations on the expanded cation layer, with a stoichiometric Mg/Ca ratio of 3/1.
Two crystallographically distinct carbonate groups are present in the structure: one parallel to the ab basal plane of the cell and three symmetry-related [CO3] slightly canted to the basal plane (6.4° tilt), the orientation of the former rotated 30° with respect to the others (see Fig. 1). In this atomic arrangement, Mg atoms are octahedrally coordinated by O atoms and Ca atoms are hexa-coordinated adopting a trigonal prism configuration, unlike the calcite, magnesite and dolomite structures, where all the cations are forming [MO6] octahedral units.
All the high-pressure XRD patterns can be indexed and fitted using a hexagonal unit cell. The experimental lattice parameters and unit cell volumes at different pressures are collected in Table 1 and their evolution as a function of pressure is represented in Fig. 3 and 4, respectively. As can be seen, the lattice parameters vary smoothly with increasing pressure up to 21 GPa, in reasonable agreement with theory (also depicted in Fig. 3 and 4), which supports the absence of first-order phase transitions in this pressure range.
| Pressure (GPa) | Space group | a axis (Å) | c axis (Å) | a/c ratio | Volume (Å3) |
|---|---|---|---|---|---|
| 1.05 | R32 | 9.4834(6) | 7.7712(8) | 1.2203(2) | 605.27(10) |
| 1.6 | R32 | 9.4731(6) | 7.7484(9) | 1.2226(2) | 602.19(11) |
| 2.35 | R32 | 9.4590(7) | 7.7185(11) | 1.2255(3) | 598.077(13) |
| 3.3 | R32 | 9.4418(8) | 7.6802(9) | 1.2294(2) | 592.95(12) |
| 4.3 | R32 | 9.4232(7) | 7.6382(11) | 1.2337(3) | 587.38(13) |
| 5.2 | R32 | 9.4092(4) | 7.6070(7) | 1.2369(2) | 583.25(8) |
| 6.45 | R32 | 9.3869(4) | 7.5609(6) | 1.2415(2) | 576.97(7) |
| 7.6 | R32 | 9.3686(4) | 7.5236(7) | 1.2452(2) | 571.89(8) |
| 8.5 | R32 | 9.3527(5) | 7.4909(7) | 1.2485(2) | 567.47(8) |
| 9.7 | R32 | 9.3349(5) | 7.4527(7) | 1.2526(2) | 562.43(8) |
| 11 | R32 | 9.3141(6) | 7.4083(7) | 1.2573(2) | 556.59(9) |
| 11.95 | R32 | 9.2995(8) | 7.3776(12) | 1.2605(3) | 552.55(14) |
| 13.4 | R32 | 9.2818(16) | 7.3391(19) | 1.2647(5) | 547.6(2) |
| 14.85 | R32 | 9.2634(7) | 7.2974(6) | 1.2694(2) | 542.30(9) |
| 16.25 | R32 | 9.2450(10) | 7.2558(9) | 1.2742(3) | 537.07(12) |
| 17.6 | R32 | 9.2284(9) | 7.2201(8) | 1.2782(3) | 532.512(11) |
| 18.95 | R32 | 9.2123(15) | 7.184(2) | 1.2824(6) | 528.0(3) |
| 20.0 | R32 | 9.1976(8) | 7.1561(7) | 1.2853(2) | 524.28(10) |
| 21.15 | R3 | 9.1841(15) | 7.1217(18) | 1.2896(5) | 520.2(2) |
| 22.2 | R3 | 9.1754(16) | 7.0936(18) | 1.2935(6) | 517.2(2) |
| 23.15 | R3 | 9.1643(6) | 7.0624(6) | 1.2976(2) | 513.67(8) |
| 24.25 | R3 | 9.1507(6) | 7.0255(8) | 1.3025(2) | 509.47(9) |
| 25.3 | R3 | 9.1380(8) | 6.9945(9) | 1.3065(3) | 505.82(11) |
| 26.35 | R3 | 9.1268(12) | 6.9655(13) | 1.3103(4) | 502.49(16) |
| 27.45 | R3 | 9.1157(10) | 6.9381(10) | 1.3139(3) | 499.29(13) |
| 28.45 | R3 | 9.1077(8) | 6.9151(8) | 1.3171(3) | 496.76(10) |
| 29.45 | R3 | 9.0971(11) | 6.8906(10) | 1.3202(4) | 493.85(13) |
 | ||
| Fig. 3 Evolution of the lattice parameters of Mg3Ca(CO3)4 huntite with pressure. Triangles and circles represent the dimensions of the a and c axes of the trigonal structure. Red and black symbols correspond to results of the present study and previous results from ref. 20, respectively. Solid and empty symbols correspond to upstroke and downstroke data, respectively. The evolution of the DFT calculated lattice parameters of both the initial R32 and the predicted R3 phases as a function of pressure are represented as orange and purple lines, respectively. | ||
 | ||
| Fig. 4 Unit-cell volume vs. pressure for Mg3Ca(CO3)4. Black squares and red circles represent experimental data from a previous study20 and the present study, respectively, and the dashed lines the fitted equation of state. Solid and empty symbols correspond to upstroke and downstroke data, respectively. The evolution of the DFT calculated unitcell volumes of both the initial R32 and the predicted R3 phases as a function of pressure are represented as orange and purple lines, respectively. | ||
The calculated volume is overestimated by 1.6% with respect to the experimental value at room conditions, as a consequence of the 0.7% overestimation of the a and b axis lengths. The a/c ratio is also affected by this small difference, being its 0.6% larger in the simulations but presenting a similar slope upon compression. The pressure evolution of the lattice parameters (Fig. 3) shows that the c axis is the most compressible. A linearization of the 3rd-order Birch–Murnaghan equation of state (through the substitution of the volume for the cube of a lattice parameter) was used to analyze the axial compression,50 providing evidence of the strong anisotropy in this compound. Thus, the bulk moduli values obtained from data up to 21 GPa are Ba = 151(3) GPa ( ) and Bc = 56.8(5) GPa (
) and Bc = 56.8(5) GPa ( ), for the a and c axes, respectively, which indicate the that the a axis is prominently more uncompressible and that the huntite bulk compressibility is mainly dominated by that of the c axis. The a/c ratio increases with pressure according to the expression a/c = 1.2163(2) + 0.00405(5)·P − 3.07(2)·P2 (see Fig. 5). This response to external pressure arises from the fact that the relatively incompressible [CO3] carbonate units are arranged roughly parallel to the ab plane and, therefore, the compressibility of the c axis can be directly attributable to that of [CaO6] trigonal prisms and [MgO6] octahedra. The unit cell volumes as a function of pressure can be explained with a third-order Birch–Murnaghan equation of state (BM-EoS)51 with a zero-pressure volume V0 = 611.7(2) Å3, a bulk modulus of B0 = 99.5(11) GPa, and a bulk modulus first-pressure derivative of
), for the a and c axes, respectively, which indicate the that the a axis is prominently more uncompressible and that the huntite bulk compressibility is mainly dominated by that of the c axis. The a/c ratio increases with pressure according to the expression a/c = 1.2163(2) + 0.00405(5)·P − 3.07(2)·P2 (see Fig. 5). This response to external pressure arises from the fact that the relatively incompressible [CO3] carbonate units are arranged roughly parallel to the ab plane and, therefore, the compressibility of the c axis can be directly attributable to that of [CaO6] trigonal prisms and [MgO6] octahedra. The unit cell volumes as a function of pressure can be explained with a third-order Birch–Murnaghan equation of state (BM-EoS)51 with a zero-pressure volume V0 = 611.7(2) Å3, a bulk modulus of B0 = 99.5(11) GPa, and a bulk modulus first-pressure derivative of  . These experimental results show a slightly larger compressibility than that reported using a less-hydrostatic pressure-transmitting medium (a mixture 4
. These experimental results show a slightly larger compressibility than that reported using a less-hydrostatic pressure-transmitting medium (a mixture 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol:ethanol) up to 12 GPa20 and compare well with those obtained from our DFT calculations below 10 GPa: V0 = 621.046 Å3, B0 = 99.55 GPa, and
:1 methanol:ethanol) up to 12 GPa20 and compare well with those obtained from our DFT calculations below 10 GPa: V0 = 621.046 Å3, B0 = 99.55 GPa, and  . The bulk modulus of Mg3Ca(CO3)4 huntite is slightly larger than that of MgCa(CO3)2 dolomite (B0 = 94 GPa)52 and it is only slightly smaller to that of MgCO3 magnesite (B0 = 103–108 GPa).53 Huntite's intermediate bulk modulus results from its mixed cation structure, where one fourth of calcium atoms and three-fourth magnesium atoms alternate within the crystal lattice. The trigonal prism coordination of the Ca atoms does not seem to have a significant effect on compressibility despite its higher polyhedral volume.
. The bulk modulus of Mg3Ca(CO3)4 huntite is slightly larger than that of MgCa(CO3)2 dolomite (B0 = 94 GPa)52 and it is only slightly smaller to that of MgCO3 magnesite (B0 = 103–108 GPa).53 Huntite's intermediate bulk modulus results from its mixed cation structure, where one fourth of calcium atoms and three-fourth magnesium atoms alternate within the crystal lattice. The trigonal prism coordination of the Ca atoms does not seem to have a significant effect on compressibility despite its higher polyhedral volume.
 | ||
| Fig. 5 Evolution of the a/c ratio of Mg3Ca(CO3)4 huntite with pressure. Black squares and red circles represent experimental data from a previous study20 and the present study, respectively. The dashed line is a 2nd-order polynomial fitting to the low-pressure phase. The evolution of the DFT calculated ratios of both the initial R32 and the predicted R3 phases as a function of pressure are represented as orange and purple lines, respectively. | ||
At approximately 21 GPa, a discontinuity in the evolution of the hexagonal lattice parameters starts to occur. Between this pressure and 25 GPa, the a/c ratio increases more pronouncedly as a consequence of a larger decrease of the c-axis, which suggests the existence of a progressive second-order phase transition between hexagonal crystal structures. Above 25 GPa, that sudden increase in slope is moderated and the a/c ratio increases smoothly according to the following linear expression: a/c = 1.2259(5) + 0.00321(17)·P. The intensities of the XRD peaks of the HP patterns show small changes upon compression, which points to small atomic displacements that could reduce the symmetry of the unit cell lattice. DFT calculations, as we will see later in detail, predict that the R32 structure is dynamically unstable above 8 GPa, and suggest a lower symmetry R3 at high pressures. Using the atomic coordinates given by the simulations for the HP R3 structure as a reference model, we performed a Rietveld refinement at 25.3 GPa, above the observed transition (see Fig. S2 and Tables S2, S3 in ESI†). The results show the atoms slightly rearrange as a consequence of the disappearance of the 2-fold axes, which entails both a rotation and a larger tilting of the [CO3] groups causing a distortion of the cation-centered polyhedra. In fact, Ca atoms can be considered as 9-fold coordinated by O atoms in the HP R3 phase (3 × 2.18 Å, 3 × 2.37 Å and 3 × 2.68 Å at 25.3 GPa), whereas in the low-pressure R32 phase Ca are hexacoordinated forming trigonal prisms (6 × 2.37 Å at 1.05 GPa). The new Ca-centered polyhedral are depicted in Fig. 6 (to be compared to Fig. 1). The phase transition is reversible upon decompression, as inferred by the unitcell volume and a/c ratio.
 | ||
| Fig. 6 Projections along the c (left) and b axes (right) of the crystalline high-pressure R3 Mg3Ca(CO3)4 huntite structure. Thick black lines indicate the unit-cell. Light green, orange, light gray, and red spheres represent Ca, Mg, C, and O atoms, respectively. A [CaO9] polyhedron and a [MgO6] octahedron are depicted to illustrate, in particular, the increase in the coordination sphere around the Ca atoms. [CO3] trigonal planar units are also represented and are significantly more tilted than in the initial huntite phase. | ||
Raman spectroscopy measurements
We performed Raman spectroscopy measurements of huntite up to 38 GPa also using He as hydrostatic pressure transmitting medium. In general, the spectra of carbonates can be divided into two regions.54,55 Those bands at frequencies above 600 cm−1 are mainly due to the internal motions of the molecular carbonate ion. Those below 600 cm−1 are generic lattice modes. Group theoretical analyses (point group D3) predict 26 Raman active modes ΓR = 7A1 + 19E.56,57Raman scattering spectra at some representative pressures are shown in Fig. 7 (top) and (bottom). The huntite sample from Tea Tree Gully, Australia, gave spectra with well-defined bands. A complete band assignment was made by Scheetz and White57 by carefully comparing IR and Raman measurements with the predictions of the factor-group calculation. Assignments can be summarized as follows:
 | ||
| Fig. 7 Room-temperature Raman spectra of Mg3Ca(CO3)4 at selected pressures on upstroke in the frequency regions 100–500 cm−1 (top) and 650–1300 cm−1 (bottom). The black and blue spectra correspond to the initial R32 huntite and the high-pressure R3 phase, respectively. Red spectra correspond to the pressure range were most of the bands can be assigned to the initial R32 huntite phase but small peaks and a band splitting in the [CO3] bending frequency region appear. Dashed lines show the appearance and evolution of new Raman bands. | ||
(i) There are two crystallographically distinct carbonate ions in the huntite structure. A unique [CO3] group which lies in the basal plane of the trigonal cell and has point symmetry D3. The remaining three [CO3] groups are arranged around the three-fold axis and are slightly tilted towards the basal plane. All three are crystallographically equivalent with site symmetry C2. The orientation of these two types of carbonate groups is different, one rotated 30° with respect to the other. Only one strong symmetric C–O stretching band appears at 1120 cm−1 in the Raman spectra at room and low pressures (below 3.9 GPa) due to the fact that the bands that arise from the two different crystallographic carbonate units and from combinations of the three symmetry-related equivalent [CO3] groups have similar frequencies.
Above 3.9 GPa, this band splits into two well-defined Raman bands due to the different pressure-induced vibrational frequency shifts of the C–O stretching bands caused by the change in its atomic environment. The evolution of the Raman band frequencies with increasing pressure is depicted in Fig. 8. In this figure it can be seen that the frequency difference between the two C–O stretching bands progressively and slowly increases upon compression. On the higher frequency region, at around 1460 cm−1, an antisymmetric C–O stretching mode appears. This band was observed at room conditions but, unfortunately, in static high pressure experiments the first-order Raman band of diamond anvils prevents access to the 1300–1500 cm−1 frequency range. The bands between 705 and 880 cm−1 correspond to [CO3] bending modes. At 11.5 GPa, the Raman mode of ∼880 cm−1 splits into two components and a new Raman band appear at 782 cm−1. Above 23 GPa, where the axial ratio discontinuity occurs, more changes occur in the vibrational spectra, appearing a new Raman band at 854 cm−1 and a triplet with well-defined bands between 725 and 830 cm−1.
 | ||
| Fig. 8 Experimental (symbols) and calculated (lines) pressure dependence of the Raman-active modes of Mg3Ca(CO3)4. Black and blue symbols represent bands of the initial R32 huntite and the high-pressure R3 phase, respectively. Red symbols correspond to the pressure range were most of the bands can be assigned to the initial R32 huntite phase but small peaks and a band splitting in the [CO3] bending frequency region appear. | ||
(ii) A detailed classification of the lattice modes, those below 420 cm−1, was done by Scheetz and White based on factor-group analysis.57 In Fig. 7 and 8 it can be seen the general blueshift of the bands of huntite with increasing pressure, which do not present significant broadening as expected by the crystallinity of the solid and the hydrostaticity provided by the pressure transmitting medium. Above 23 GPa, there are changes in the relative intensities of the modes in this low frequency region and the appearance of new modes like the one at 403 cm−1. These observations could be explained by the continuous change towards a higher coordination number of the Ca atoms induced by pressure, as revealed in XRD experiments and suggested in DFT calculations (see below).
The calculated Raman spectra are in reasonable agreement with those experimentally measured at low and high pressures, as shown in Fig. 9. The initial huntite spectrum was observed on pressure release, confirming the reversibility of the phase transitions.
 | ||
| Fig. 9 Comparisons between calculated (red lines) and experimental (black lines) Raman spectra of the low-pressure R32 (top) and the high-pressure R3 (bottom) phases of Mg3Ca(CO3)4 at 3.9 and 28.8 GPa. Calculated Raman spectra were used for the mode assignment of Raman frequencies in the experiment. | ||
Phase transitions
The behavior of the c/a ratio above 21 GPa, as characterized by XRD, as well as the changes observed in the Raman spectra, suggest the existence of a second order phase transition that takes place between 21 and 25 GPa. Although this is the main structural change observed, there are other subtleties which is worth commenting on.DFT calculations reveal that the initial R32 structure of Mg3Ca(CO3)4 is dynamically unstable above 8 GPa, the pressure where the first additional features start to appear in the Raman spectra. The dynamical instability of the system at this pressure is revealed by the existence of imaginary phonon frequencies at Gamma. Subsequent geometry relaxation of the structure at the same volume leads to the broken-symmetry R3 structure, which has been verified to be dynamically stable by performing further DFPT phonon frequency calculations. The high-pressure R3 structure is energetically more favorable above this pressure, as indicate the E–V and H–P graphs represented in Fig. 10.
 | ||
| Fig. 10 DFT energy as a function of the volume per Mg3Ca(CO3)4 formula unit for the initial R32 phase and the HP R3 phase. Inset: Enthalpy difference as a function of pressure, showing the stability with respect to R32 Mg3Ca(CO3)4 huntite. | ||
The question arises as to whether the changes observed in the Raman spectra at 10 GPa correspond to an additional phase transition or whether they are related to precursor effects to the transition to the R3 structure at 21 GPa. We have explored two possibilities.
Firstly, we envisaged a smooth transition from 10 GPa onwards, which would produce gradual changes on the Raman spectra as the distortion of the new phase increases. The twelve A2 modes of the huntite R32 phase are IR active but Raman forbidden. Space group correlations between the 32 and 3 point groups show that the A2 modes give rise to A modes in the R3 structure, which are both Raman and IR active. The Raman tensor of the new modes would increase in parallel to the distortion associated with the R3 phase, and for some modes it would not result in relevant intensities up to 21 GPa. We have plotted the calculated pressure dependence of the IR active modes in Fig. 8 as green lines. At first sight, at 10 GPa there are some of the new modes which are close enough to the calculated values to be assigned to IR modes, such as the one observed at approx. 865 cm−1. However, this line of reasoning cannot explain why there are some modes, such as the one appearing at around 400 cm−1 at 12 GPa, which disappears from 22 GPa on.
Another possibility is to consider a progressive appearance of structural defects starting at 10 GPa. The structural defects would relax the momentum conservation rule and allow Raman dispersion from phonons with wavevectors away from the Brillouin zone center. The most relevant vibrations involve wavevectors at edge points and flat bands, whose associated density of states is large. A similar reasoning was used in ref. 58 to explain Raman features appearing prior to the phase transition in the CuAlO2 delafossite. A relevant section of the huntite phonon dispersion curves is show in Fig. 11. States with a large density of states are highlighted with red dashed lines. There are three relevant contributions to the Raman scattering that could explain the progressive transformation of the Raman peak at 890 cm−1 into a triplet from 10 GPa on. Interestingly, the low energy side of the triplet is predicted to be soft (−0.8 cm−1 GPa−1), as it is actually observed in the experiment (−0.3 cm−1 GPa−1). Since the precursor defects do not significantly affect long-range order, this mechanism would be compatible with the XRD results, which show no evidence of the phase transition up to 21 GPa.
 | ||
| Fig. 11 Dispersion curves along a path in the Brillouin zone, including Γ(0 0 0), D(0 0 1/3), A(−1/2 1 1/3) and F(−1/2 1 0) points. Coordinates are given in fractional coordinates of the conventional (hexagonal) unit cell. Black and blue lines correspond to calculations at 7.6 and 11.1 GPa, respectively. Dashed red lines highlight states with a high density of states which may contribute to Raman dispersion. | ||
Conclusions
We report an experimental and theoretical work on the behavior of Mg3Ca(CO3)4 huntite under high pressure in which we have studied the stability of the initial R32 phase from a mechanical and dynamical perspective. Our experimental XRD and Raman spectroscopy results confirm that there is a R32 to R3 phase transition between 21 and 25 GPa, which entails small atomic displacements allowed by the symmetry break and a change in the coordination environment of the Ca atom. This atom goes from being hexacoordinated by O atoms and adopting a trigonal prism configuration in the initial huntite phase to an irregular 9-fold coordination in the high-pressure phase. DFT calculations also predict the dynamical instability of the initial R32 structure at high pressures and the stability of the R3 phase. The Raman study also reveals a pressure range (11.5–20.5 GPa) where significant changes in the Raman spectrum are observed, although most of the Raman peaks can still be assigned to the initial R32 huntite phase. The analysis of the phonon dispersion curves suggests that these changes could be related to structural defects that are precursors to the phase transition. DFT calculations reveal that the broken-symmetry R3 phase is dynamically stable in the 8–30 GPa pressure range. After decompression, the recovered sample is the initial huntite phase, showing the reversibility of the structural transition.Author contributions
Conceptualization, D. S. P., investigation, D. S. P., R. Ch. J., B. D. B. N., G. B., J. P. P., L. B. A. O. R., and C. P., formal analysis, R. Ch. J., D. S. P., and A. O. R., writing – original draft, D. S. P., writing – review & editing, all the authors.Data availability
Crystallographic data for huntite-II has been deposited at the CCDC database under number 2393118. The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Spanish Ministerio de Ciencia e Innovación and the Agencia Estatal de Investigación (MCIN/AEI/10.13039/501100011033) under projects PID2021-125518NB-I00 and RED2022-134388-T (cofinanced by EU FEDER funds) as well as by the Generalitat Valenciana under projects CIAICO/2021/241, CIGRIS/2022/202 and MFA/2022/007 (funded by the European Union—Next Generation EU). A. O. R. thanks the Principality of Asturias (FICYT), project AYUD/2021/51036 cofinanced by EU FEDER. The authors also thank Dr. Nicolescu and the Mineralogy and Meteoritic Department of the Yale Peabody Museum of Natural History for providing the mineral samples, and the MALTA Consolider supercomputing centre and Compute Canada for computational resources. These experiments were performed at the MSPD beamline at ALBA Synchrotron thanks to the 2023087689 beamtime Part of this study was performed while D. S. P. was carrying out two research stays in the Institute of Geosciences of the Goethe University Frankfurt funded by the Generalitat Valenciana CIBEST/2022/198 and the Spanish Ministerio de Ciencia e Innovación PRX22/00013 grants. The authors thankfully acknowledge the computer resources at MareNostrum5 and the technical support provided by BSC (RES-AECT-2024-2-0010).References
- J. F. Kasting, O. B. Toon and J. B. Pollack, Climate evolved on the terrestrial planets, Sci. Am., 1988, 258, 90–97 CrossRef CAS PubMed.
- R. Dasgupta, Ingassing, storage, and outgassing of terrestrial carbon through geologic time, Rev. Mineral. Geochem., 2013, 75, 183–229 CrossRef CAS.
- S. Poli, Carbon mobilized at shallow depths in subduction zones by carbonatitic liquids, Nat. Geosci., 2015, 8, 633 CrossRef CAS.
- F. E. Brenker, C. Vollmer, L. Vincze, B. Vekemans, A. Szymanski, I. Szaloki, L. Nasdala, W. Joswig and F. Kaminsky, Carbonates from the lower part of transition zone or even lower mantle, Earth Planet. Sci. Lett., 2007, 260, 1 CrossRef CAS.
- J. Zhang and R. J. Reeder, Comparative compressibilities of calcite-structure carbonates: Deviations from empirical relations, Am. Mineral., 1999, 84, 861–870 CrossRef CAS.
- R. Chulia-Jordan, D. Santamaria-Perez, A. Otero-de-la-Roza, J. Ruiz-Fuertes, T. Marqueño, O. Gomis, S. MacLeod and C. Popescu, Phase stability of natural Ni0.75Mg0.22Ca0.03CO3 gaspeite mineral at high pressure and temperature, J. Phys. Chem. C, 2020, 124, 19781–19792 CrossRef CAS.
- R. Chulia-Jordan, D. Santamaria-Perez, J. Ruiz-Fuertes, A. Otero-de-la-Roza and C. Popescu, Compressibility and phase stability of iron-rich ankerite, Minerals, 2021, 11, 607 CrossRef CAS.
- J. P. R. de Villiers, Crystal structures of aragonite, strontianite and witherite, Am. Mineral., 1971, 56, 758–766 CAS.
- K. Sahl, Verfeinerung der Kristallstruktur von Cerussit, PbCO3, Z. Kristallogr., 1974, 139, 215–222 CrossRef CAS.
- L. B. Railsback, Patterns in the compositions, properties, and geochemistry of carbonate minerals, Carbonates Evaporites, 1999, 14, 1–20 CrossRef CAS.
- G. T. Faust, Huntite, Mg3Ca(CO3)4, a new mineral, Am. Mineral., 1953, 38, 4–24 CAS.
- D. L. Graf and W. F. Bradley, The crystal structure of huntite, Mg3Ca(CO3)4, Acta Crystallogr., 1962, 15, 238–242 CrossRef CAS.
- W. A. Dollase and R. J. Reeder, Crystal structure refinement of huntite, CaMg3(CO3)4, with x-ray powder data, Am. Mineral., 1986, 71, 163–166 CAS.
- M. Isshiki, T. Irifune, K. Hirose, S. Ono, Y. Ohishi, T. Watanuki, E. Nishibori, M. Takata and M. Sakata, Stability of magnesite and its high-pressure form in the lowermost mantle, Nature, 2004, 427, 60–63 CrossRef CAS PubMed.
- J. Binck, L. Bayarjargal, S. S. Lobanov, W. Morgenroth, R. Luchitskaia, C. J. Pickard, V. Milman, K. Refson, D. B. Jochym, P. Byrne and B. Winkler, Phase stabilities of MgCO3 and MgCO3-II studied by Raman spectroscopy, X-ray diffraction and density functional theory calculations, Phys. Rev. Mater., 2020, 4, 055001 CrossRef CAS.
- K. Suito, J. Namba, T. Horikawa, Y. Taniguchi, N. Sakurai, M. Kobayashi, A. Onodera, O. Shimomura and T. Kikegawa, Phase relations of CaCO3 at high pressure and high temperature, Am. Mineral., 2001, 86, 997–1002 CrossRef CAS.
- M. Merlini, M. Hanfland and W. A. Crichton, CaCO3-III and CaCO3-IV, high-pressure polymorphs of calcite: Possible host structures for carbon in the Earth's mantle, Earth Planet. Sci. Lett., 2012, 333–334, 265–271 CrossRef CAS.
- M. Merlini, W. A. Crichton, M. Hanfland, M. Gemmi, H. Müller, I. Kupenko and L. Dubrovinsky, Structures of dolomite at ultrahigh pressure and their influence on the deep carbon cycle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13509–13514 CrossRef CAS PubMed.
- J. Binck, S. Chariton, M. Stekiel, L. Bayarjargal, W. Morgenroth, V. Milman, L. Dubrovinsky and B. Winkler, High-pressure, high-temperature phase stability of iron-poor dolomite and the structures of dolomite-IIIc and dolomite-V, Phys. Earth Planet. Inter., 2020, 299, 106403 CrossRef CAS.
- D. Santamaria-Perez, A. Otero-de-la-Roza, J. Ruiz-Fuertes, R. Chulia-Jordan, T. Marqueño, S. MacLeod and C. Popescu, Pressure and temperature effects on low-density Mg3Ca(CO3)4 huntite carbonate, J. Phys. Chem. C, 2020, 124, 1077–1087 CrossRef CAS.
- J. P. Pinceaux, J. P. Maury and J. M. Besson, Solidification of helium at room temperature under high pressure, J. Phys., Lett., 1979, 40, 307–308 CrossRef CAS.
- D. Santamaria-Perez, G. D. Mukherjee, B. Schwager and R. Boehler, High-pressure melting curve of helium and neon: Deviations from corresponding states theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 85, 214101 CrossRef.
- S. Klotz, J. C. Chervin, P. Munsch and G. Le Marchand, Hydrostatic limits of 11 pressure transmitting media, J. Phys. D: Appl. Phys., 2009, 42, 075413 CrossRef.
- K. K. Mao, J. Xu and P. M. Bell, Calibration of the Ruby Pressure Gauge to 800-Kbar under Quasi-Hydrostatic Conditions, J. Geophys. Res., [Solid Earth Planets], 1986, 91, 4673–4676 CrossRef.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, The new material science powder diffraction beamline at ALBA synchrotron, Powder Diffr., 2013, 28, 5360–5370 CrossRef.
- C. Prescher and V. B. Prakapenka, DIOPTAS: A Program for Reduction of Two-Dimensional X-Ray Diffraction Data and Data Exploration, High Press. Res., 2015, 35, 223–230 CrossRef CAS.
- T. J. B. Holland and S. A. T. Redfern, Unit cell refinement from powder diffraction data: the use of regression diagnostics, Mineral. Mag., 1997, 61, 65–77 CrossRef CAS.
- G. Nolze and W. Kraus, Powdercell 2.0 for Windows, Powder Diffr., 1998, 13, 256–259 Search PubMed.
- J. Rodriguez-Carvajal, Recent advances in magnetic-structure determination by neutron powder diffraction, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- L. Bayarjargal, C. J. Fruhner, N. Schrodt and B. Winkler, CaCO3 phase diagram studied with Raman spectroscopy at pressures up to 50 GPa and high temperatures and DFT modelling, Phys. Earth Planet. Inter., 2018, 281, 31 CrossRef CAS.
- B. D. Botan-Neto, D. Santamaria-Perez, L. Bayarjargal, E. Bykova, J. Gonzalez-Platas and A. Otero-de-la-Roza, Dense hydrated magnesium carbonate MgCO3·3H2O phases, Inorg. Chem., 2024, 63, 15762–15771 CrossRef CAS PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, C. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio Jr, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys. Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. D. Becke, On the large-gradient behavior of the density functional exchange energy, J. Chem. Phys., 1986, 85, 7184–7187 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. R. Johnson, The exchange-hole dipole moment dispersion model, in Non-covalent interactions in quantum chemistry and physics, ed. A. Otero-de-la-Roza and G. A. DiLabio, Elsevier, 2017, ch. 5, pp. 169–194 Search PubMed.
- A. D. Becke and E. R. Johnson, Exchange-hole dipole moment and the dispersion interaction revisited, J. Chem. Phys., 2007, 127, 154108 CrossRef PubMed.
- A. Otero-De-La-Roza and E. R. Johnson, van der Waals interactions in solids using the exchange-hole dipole moment model, J. Chem. Phys., 2012, 136, 174109 CrossRef CAS PubMed.
- A. Dal Corso, Pseudopotentials periodic table: From H to Pu, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef CAS.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozi, Phonons and related crystal properties from density-functional perturbation theory, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- A. Otero-De-La-Roza and V. Luaña, Gibbs2: A new version of the quasi-harmonic model code. I. Robust treatment of the static data, Comput. Phys. Commun., 2011, 182, 1708–1720 CrossRef CAS.
- A. Otero-De-La-Roza, D. Abbasi-Pérez and V. Luaña, Gibbs2: A new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation, Comput. Phys. Commun., 2011, 182, 2232–2248 CrossRef CAS.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. M. Maschio, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Quantum-mechanical condensed matter simulations with CRYSTAL, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- D. Vilela Oliveira, J. Laun, M. F. Peintinger and T. Bredow, BSSE-correction scheme for consistent Gaussian basis sets of double-and triple-zeta valence with polarization quality for solid-state calculations, J. Comput. Chem., 2019, 40, 2364–2376 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Ch Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- L. Maschio, B. Kirtman, M. Rérat, R. Orlando and R. Dovesi, Ab initio analytical Raman intensities for periodic systems through a coupled perturbed Hartree-Fock/Kohn-Sham method in an atomic orbital basis. I. Theory, J. Chem. Phys., 2013, 139, 164101 CrossRef PubMed.
- L. Maschio, B. Kirtman, M. Rérat, R. Orlando and R. Dovesi, Ab initio analytical Raman intensities for periodic systems through a coupled perturbed Hartree-Fock/Kohn-Sham method in an atomic orbital basis. II. Validation and comparison with experiments, J. Chem. Phys., 2013, 139, 164102 CrossRef PubMed.
- R. J. Angel, Equations of state, Rev. Mineral. Geochem., 2000, 41, 35–59 CrossRef.
- F. Birch, Finite elastic strain of cubic crystals, Phys. Rev., 1947, 71, 809 CrossRef CAS.
- N. L. Ross and R. J. Reeder, High-pressure structural study of dolomite and ankerite, Am. Mineral., 1992, 77, 412–421 CAS.
- J. Zhang, I. Martinez, F. Guyot, P. Gillet and S. K. Saxena, X-ray diffraction study of magnesite at high pressure and high temperature, Phys. Chem. Miner., 1997, 24, 122–130 CrossRef CAS.
- L. J. Bonales, V. Muñoz-Iglesias, D. Santamaria-Perez, M. Caceres, D. Fernandez-Remolar and O. Prieto-Ballesteros, Quantitative Raman spectroscopy as a tool to study the kinetics and formation mechanism of carbonates, Spectrochim. Acta, Part A, 2013, 116, 26–30 CrossRef CAS PubMed.
- D. Santamaria-Perez, J. Ruiz-Fuertes, M. Peña-Alvarez, R. Chulia-Jordan, T. Marqueño, D. Zimmer, V. Gutiérrez-Cano, S. MacLeod, E. Gregoryanz, C. Popescu, P. Rodríguez-Hernández and A. Muñoz, Post-tilleyite, a dense calcium silicate-carbonate phase, Sci. Rep., 2019, 9, 7898 CrossRef PubMed.
- E. Kroumova, M. I. Arroyo, J. M. Perez-Mato, A. Kirov, C. Capillas, S. Ivantchev and H. Wondratschek, Bilbao Crystallographic Server: Useful databases and tolos for phase-transition studies, Phase Transitions, 2003, 76, 155–170 CrossRef CAS.
- B. E. Scheetz and W. B. White, Vibrational spectra of the alkaline earth double carbonates, Am. Mineral., 1977, 62, 36–50 CAS.
- J. Pellicer-Porres, D. Martínez-García, A. Segura, P. Rodríguez-Hernández, A. Muñoz, J. C. C. Chervin, N. Garro and D. Kim, Pressure and Temperature Dependence of the Lattice Dynamics of CuAlO2 Investigated by Raman Scattering Experiments and Ab Initio Calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04200j |
| This journal is © the Owner Societies 2025 |