
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolynuclear tantalum(V) coordination complexes: from dinuclear {Ta2O} to octanuclear {Ta8O12} oxo species connected through aryl monotopic carboxylate linkers†
Alejandro
Vieyra Huerta
 a,
Mahmud Beji
Ahmed
b,
Nadine
Essayem
b,
Laurent
Djakovitch
b,
Adel
Mesbah
b,
Thierry
Loiseau
a and
Sylvain
Duval
*a
a,
Mahmud Beji
Ahmed
b,
Nadine
Essayem
b,
Laurent
Djakovitch
b,
Adel
Mesbah
b,
Thierry
Loiseau
a and
Sylvain
Duval
*a
aUnité de Catalyse et Chimie du Solide (UCCS) – UMR CNRS 8181, Université de Lille, Centrale Lille, Université d'Artois, F-59000 Lille, France. E-mail: sylvain.duval@univ-lille.fr; Fax: +(33) 3 20 43 48 95; Tel: +(33) 3 20 43 40 13
bUniv. Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON, UMR5256, 69626, Villeurbanne, France
First published on 5th June 2025
Abstract
A series of monocarboxylic ligands (benzoic acid, 1-naphtoic acid, 2-naphtoic acid, antracene-9-carboxylic acid and 4′-methylbiphenyl-4-carboxylic acid) has been used to react upon mild heating with the pentavalent ethoxide tantalum(V) precursor Ta(OEt)5 in isopropanol solvent. Five new molecular coordination compounds were isolated from crystallization upon cooling of the reactant's mixtures at room temperature. The different systems crystallized in dimeric {Ta2O} (compound 1 and 2), tetrameric {Ta4O4} (compound 3 and 4) or octameric {Ta8O12} (compound 5) metal-oxo units stabilized by a combination of monocarboxylic ligands and isopropanolate solvent molecules. The five molecular samples were characterized in solid state by single-crystal and powder X-ray diffraction analyses, infrared spectroscopy, scanning electron microscopy (SEM) and thermogravimetric (TG) analysis. The chemical properties of compounds 1 & 5 have been tested for the catalytic conversion of dihydroxyacetone, showing Brønsted acidity.
Introduction
Tantalum and niobium are known to be two closely related transition metals belonging to the same column (group 5) of the periodic table and have attracted a lot of attention in the past decades owing to their acidic catalytic properties in different fields of applications; the used materials are usually inorganic such as oxides, hydroxides, phosphates or MOFs.1 They have gained significant attention in molecular chemistry due to their ability to form a wide variety of stable oxo-clusters. When decorated with organic ligands at the molecular level, they may exhibit tunable chemical properties that make them versatile in the construction of complex molecular architectures with various nuclearities ranging from dimers to hexadecamers for niobium and dimers to octanuclear species with tantalum.2,3 The organic ligands attached to the metal-oxo clusters play a crucial role in modulating the properties of these complexes and may influence solubility, stability and reactivity. Following previous work done in our laboratory with niobium(IV and V) cations,4–7 this work is focused on the coordination potentialities of the tantalum(V) cation with monocarboxylate linkers in the formation of polynuclear metal-oxo clusters.Surprisingly, with tantalum, relatively few examples of such molecular compounds have been described in the literature and they can be regrouped into two different categories. Indeed, these compounds are either full tantalum-oxo clusters or fall in the domain of organometallic chemistry with precursors possessing one or several tantalum–carbon bonds that are further used for coordination by oxo-donor ligands on the remaining free positions of the cationic centers. Most of the literature on organometallic tantalum compounds described the use of the well-known cyclopentadienyl Cp* ligand to form [Cp*TaCl4] precursors. Cp* hinders one face of the tantalum cation and the remaining labile chloride atoms can then be completely or partially substituted by various oxo-, N-, or C-donor ligands to form mainly mononuclear compounds.8–19 Following this route, some polynuclear entities were also obtained with the identification of the tri- and tetra-nuclear molecules [{TaCp*(OH)(OH2)}3(μ3-OH)(μ2-O)3](OThf)2, [{Cp*Ta}4(μ3-O)7](OH)2 and [{Cp*Ta}3(μ3-O)5Cl(H2O)2]Cl.20–22
Albeit being studied for several decades, these compounds do not present many applications other than acting as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond activators or as polymerization catalysts for methyl methacrylate synthesis.13,19 Few other examples were also reported using N-heterocyclic carbene as the ligand for tantalum complexation with catalytic properties for cyclic carbonate formation23,24 or incorporation of a carbon based electrophile to form a metal alkyl substrate or various compounds by additional reaction of the tantalum complex.25 It is worth mentioning that for catalytic purposes, some tantalum hydride compounds were also described and used for carbon dioxide reduction reactions.26,27 From a fundamental point of view, tantalum compounds presenting a unique coordination mode of dinitrogen were also described and further used for to react with propene or bromobenzyl.28
O bond activators or as polymerization catalysts for methyl methacrylate synthesis.13,19 Few other examples were also reported using N-heterocyclic carbene as the ligand for tantalum complexation with catalytic properties for cyclic carbonate formation23,24 or incorporation of a carbon based electrophile to form a metal alkyl substrate or various compounds by additional reaction of the tantalum complex.25 It is worth mentioning that for catalytic purposes, some tantalum hydride compounds were also described and used for carbon dioxide reduction reactions.26,27 From a fundamental point of view, tantalum compounds presenting a unique coordination mode of dinitrogen were also described and further used for to react with propene or bromobenzyl.28
In the approach based on classical coordination chemistry using oxo-donor ligands such as alkoxides or carboxylates, several molecular compounds were previously described by other research groups, sometimes with the addition of other hetero-ligands. Although the synthetic strategies and composition may diverge, the resulting products that were reported are constructed around oxo-tantalum species with nuclearities going from a simple cation to octanuclear oxo-clusters. For tantalum(V), most of the results turn around the synthesis of single tantalum complexes that were used for lots of applications. One may mention their use for NbV/TaV separation,29 the use of halide [MX5] compounds (M = NbV, TaV) for anhydride C–H activation reactions,30 deposition of thin films of Ta2O5 using the complex [Ta(NMe2)4(dmbl)] (dmbl = diterbutylmalonato)31 and, in a more fundamental approach, discovering the coordination potentialities of the Ta(V) cation.32–38
Several mixed-metal compounds containing tantalum were obtained using oxalate (C2O4)2− or salicylate (O2C–C6H4-2–OH)− ligands. In these systems, the additive metallic cation (Bi, Ni) is either directly incorporated in the polynuclear core to form a {BiTa4O4} or {Bi2Ta2O} oxo-cluster,39–41 or stand as a hybrid counter cation of [ML3]n+ type (with L = phenanthroline or bipyridine) compensating for the negative charge of [TaO(C2O4)]3− complexes in the work of Dubraja et al.42–46
The literature related to the topic we developed and described in the present contribution appears relatively sparse compared to the work presented above. Only a few examples of polynuclear tantalum-oxo species were described using only oxo-donor ligands. In these results, tantalum was isolated possessing various nuclearities with, for example, several dinuclear {Ta2O} and tetranuclear {Ta4O4} moieties and a few octanuclear {Ta8O12} cores. Most of the reactions described in the literature were performed starting from non-common precursors such as mixed chloride-dimethylsulfide [Ta2Cl6(SMe2)3] and tetraperoxo–guanidinium tantalum [(gu)3Ta(O2)4] or the more classical pentachloro tantalum TaCl5. They gave rise to the isolation of dimeric entities with various carboxylate ligands, for instance {TaIII2Cl5(piv)(SMe2)(THF)2} with piv = pivalate,47 {Ta2(O2)4(tart)2} with tart = tartrate,48 {Ta2Cl8(p-Cl-benz)} with p-Cl-benz = p-chlorobenzoate49 and {Ta2(lactate)4(O2)2O} with lactate50 or with some other ligands.51,52
Tetranuclear species like {Ta4(μ-O)4(μ-OBc)4(ONep)8}52 or {Ta4(μ-O)4Cl8(p-Mebenzoate)4}53 were also synthetized using more bulky carboxylate ligands (OBc = pivalate and p-Mebenzoate = para-methylbenzoate), eventually associated with alkoxide molecules (ONep = 2,2-dimethylpropan-1-olate) using standard solution synthesis. In these cases, the bulky nature of the ligands was reported as being the main driving force of the increase in nuclearity of the tantalum-oxo core. Another tetranuclear compound with applications in ring opening polymerization was described using the bifunctional (carboxylate and alcoholate) benzylic acid ligand.51
Finally, in the race to obtain large clusters, the first octanuclear tantalum compound {Ta8(μ-O)12(O2CNEt2)16} (O2CNEt2 = diethyl carbamate) was structurally characterized by Pampaloni et al. in 1998. It remains the only illustration in tantalum “hybrid” chemistry up to date.3
Herein we report on new results obtained by using five aromatic monocarboxylic acid ligands to investigate their reactivity towards the tantalum penta-ethoxide Ta(OEt)5 precursor as the tantalum(V) metal source and generating diverse Ta-centered nuclearities. Anthracene-9-carboxylic acid and 4′-methylbyphenyl-4-carboxylic acids (dimeric compounds 1 and 2), benzoic acid and 1-naphtoic acid (tetrameric compounds 3 and 4) and 2-naphtoic acid (octameric compound 5) were successfully used to form crystalline materials. A solvent approach was successfully used to ensure high crystalline yield of the octanuclear core 5. All the molecules were further analyzed by a combination of solid-state analyses (single crystal and powder X-ray diffraction, IR spectroscopy, TGA, optical binocular and SEM images). The acidity of the crystallized compounds 1 and 5 was tested for the conversion of hydroxyacetone (DHA) in water.
Experimental section
Reagents
The following reactants were used: tantalum(V) ethoxide complex (Ta(OEt)5, 99%, Thermo Fisher), benzoic acid (C7H6O2, 99.5%, Sigma Aldrich), 1-naphtoic acid (C11H8O2, 96%, Sigma Aldrich), 2-napthoic acid (C7H6O2, 98%, Sigma Aldrich), 4′-methylbiphenyl-4-carboxylic acid (C14H12O2, 96%, Sigma Aldrich), anthracene-9-carboxylic acid (C15H10O2, 99%, Sigma Aldrich), anhydrous isopropanol (C3H8O, 99.5%, Sigma Aldrich), anhydrous acetonitrile (C4H3N, 99.8%, Thermo Fisher). The chemical reactants were commercially available and used without any further purification.Syntheses
The water-sensitive tantalum(V) alkoxide precursor was used and manipulated in a glove box under argon to prevent any hydrolysis reaction of the tantalum(V) alkoxide into tantalum(V) oxide. Due to the high sensitivity of the tantalum ethoxide precursor with regard to moisture, all synthesis mixtures were prepared in a glovebox using dried solids and extra dried isopropanol (iPrOH) or acetonitrile (CH3CN) solvent. The dry solvents were purchased and used without any additional drying. After heating, the vials were removed from the oven and left to fast cooling at room temperature. After reaction, all the synthesis batches gave rise to the identification of five crystalline materials with single crystal solids of sufficient quality (Fig. S1†) for performing single crystal X-ray diffraction (SC-XRD) analyses.Compound 1 [(Ta2(μ-O)(OiPr)6)(C15H9O2)2]
A mixture of 3.5 μL (0.013 mmol) Ta(OEt)5 and 8.9 mg (0.040 mmol) of anthracene-9-carboxylic acid was placed in a 2 mL glass tube, sealed with a phenolic cap. The vial was heated at 100 °C for 5 days in an oven. Block transparent crystals appeared after 3 days upon cooling (Fig. S1†). Compound 1 was analyzed by optical and scanning electron microscopy showing large plate-shaped crystals up to 300 μm in size. The resulting crystals were filtered off, washed with ethanol and dried at room temperature. Crystallization yield was 88.3%Ta.Elemental analyses: C (49.1%), H (5.1%). Calc. C (51.1%), H (5.6%).
Compound 2 [(Ta2(μ-O)(OiPr)6)(C14H11O2)2]
A mixture of 3.5 μL (0.013 mmol) Ta(OEt)5 and 8.4 mg (0.040 mmol) of 4′-methylbiphenyl-4-carboxylic acid was placed in a 2 mL glass tube, sealed with a phenolic cap. The vial was heated at 100 °C for 5 days in an oven. Block transparent crystals appeared after 5 weeks upon cooling (Fig. S1†). Compound 2 was analyzed by optical microscopy showing parallelepiped-shape crystals up to 150 μm in size. The resulting crystals were filtered off, washed with ethanol and dried at room temperature. Crystallization yield was 12.8%Ta.Compound 3 [Ta4(μ-O)4(OiPr)8(C7H5O2)4]
A mixture of 3.5 μL (0.013 mmol) Ta(OEt)5 and 4.9 mg (0.040 mmol) of benzoic acid was placed in a 2 mL glass tube, sealed with a phenolic cap. The vial was heated at 100 °C for 5 days in an oven. Block transparent crystals appeared after 6 weeks (Fig. S1†). Compound 3 was analyzed by optical microscopy showing plate-shaped crystals up to 400 μm in size. The resulting yellowish crystals were filtered off, washed with ethanol and dried at room temperature. Crystallization yield was 17.3%Ta.Elemental analyses: C (35.2%), H (4.2%). Calc. C (35.8%), H (4.36%).
Compound 4 [Ta4(μ-O)4(OiPr)8(C11H7O2)4]
A mixture of 3.5 μL (0.013 mmol) Ta(OEt)5 and 6.9 mg (0.040 mmol) of 1-naphtoic acid was placed in a 2 mL glass tube, sealed with a phenolic cap. The vial was heated at 100 °C for 5 days in an oven. Block transparent crystals appeared after 1 day (Fig. S1†). Compound 4 was analyzed by optical microscopy showing square block crystals up to 150 μm size. The resulting yellowish crystals were filtered off, washed with ethanol and dried at room temperature. Crystallization yield was 68.3%Ta.Elemental analyses: C (42.2%), H (4.2%). Calc. C (42.0%), H (4.3%).
Compound 5 [(Ta8(μ-O)12(OiPr)8)(C11H7O2)8]
A mixture of 3.5 μL (0.013 mmol) Ta(OEt)5 and 6.9 mg (0.040 mmol) of 2-naphtoic acid was placed in a 2 mL glass tube, sealed with a phenolic cap. The vial was heated at 100 °C for 5 days in an oven. Rectangle transparent crystals appeared after 3 days (Fig. S1†). Compound 5 was analyzed by optical microscopy showing parallelepiped crystals up to 400 μm in size. The resulting yellowish crystals were filtered off, washed with ethanol and dried at room temperature. Crystallization yield was 75.3%Ta.Elemental analyses: C (38.3%), H (3.04%). Calc. C (38.6%), H (3.2%).
For analytical purposes, all the synthesis can be performed in higher quantity using a 12 mL vial increasing the precursor and solvent amount by five. The purity of compounds 1–5 was confirmed using powder X-ray diffraction analyses. Only compound 2 was systematically obtained with probably unreacted ligands as can be seen in the IR spectroscopy section where a νCO vibration is still observed at 1687 cm−1 after reaction in the compound mixture preventing the elemental analyses.
Single-crystal X-ray diffraction
Crystals of compounds 1–5 were selected under a polarizing optical microscope and all the crystals were mounted on MicroMount patented by MiTeGen, inserted into a goniometer base to perform single-crystal X-ray diffraction experiments. X-ray intensity data were collected on a Bruker DUO-APEX2 diffractometer using Mo-Kα (λ = 0.71073 Å) or Ag-Kα (λ = 0.56086 Å) radiation with an optical fiber as the collimator. Several sets of narrow data frames (10 s per frame) were collected at different values of θ for two initial values of ϕ and ω, respectively, using 0.3° increments of ϕ with ω scans.Data reduction was accomplished using SAINT V8.34a.54 The substantial redundancy in data allowed a semi-empirical absorption correction (SADABS V2014/5) to be applied, on the basis of multiple measurements of equivalent reflections.55 The structures were solved by direct methods, developed by successive difference Fourier syntheses, and refined by full-matrix least-squares on all F2 data using SHELX program suites,56 implemented in the OLEX2 graphical tool.57
The crystal data are given in Table 1. The ESI† is available in CIF format. CCDC numbers: 2446460 for 1, 2446461 for 2, 2446462 for 3, 2446463 for 4 and 2446464 for 5 contain the ESI† crystallographic data for this paper.
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Formula | C48H60Ta2O11 | C46H64Ta2O11 | C52H76Ta4O20 | C68H24Ta4O20 | C112H110Ta8O36 |
| Formula weight | 1174.86 | 1154.87 | 1744.92 | 1945.15 | 6961.23 |
| Temperature/K | 296 | 100 | 296 | 296 | 296 |
| Crystal type | Colorless plate | Colorless block | Colorless block | Colorless block | Colorless block |
| Crystal size/mm | 0.15 × 0.12 × 0.02 | 0.227 × 0.086 × 0.075 | 0.121 × 0.118 × 0.087 | 0.206 × 0.085 × 0.078 | 0.27 × 0.13 × 0.12 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/c | C2/c | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 22.0631(12) | 24.6357(5) | 11.9823(9) | 14.4485(8) | 11.3938(14) |
| b/Å | 10.1086(4) | 9.2183(2) | 18.5216(15) | 12.6909(7) | 23.187(4) |
| c/Å | 21.7781(11) | 21.2142(5) | 14.4260(12) | 20.2649(10) | 24.311(4) |
| α/° | 90 | 90 | 90 | 90 | 87.046(5) |
| β/° | 95.399(2) | 94.4971(9) | 95.784(3) | 102.8516(16) | 78.852(5) |
| γ° | 90 | 90 | 90 | 90 | 80.786(5) |
| Volume/Å3 | 4835.6(4) | 4802.90(18) | 3185.3(4) | 3622.8(3) | 6218.8(17) |
| Z, ρcalculated/g cm−3 | 4, 1.614 | 4, 1.597 | 2, 1.819 | 2, 1.783 | 1, 1.859 |
| μ/mm−1 | 2.465 | 4.608 | 3.717 | 3.275 | 3.806 |
| θ range/° | 2.711–20.551 | 1.926–26.382 | 2.065–23.638 | 2.526–21.982 | 0.674–20.702 |
| Limiting indices | −27 ≤ h ≤ 27 | −30 ≤ h ≤ 30 | −17 ≤ h ≤ 16 | −19 ≤ h ≤ 19 | −13 ≤ h ≤ 14 |
| −12 ≤ k ≤ 12 | −11 ≤ k ≤ 11 | −26 ≤ k ≤ 26 | −16 ≤ k ≤ 16 | −29 ≤ k ≤ 29 | |
| −27 ≤ l ≤ 27 | −26 ≤ l ≤ 26 | −20 ≤ l ≤ 19 | −26 ≤ l ≤ 27 | −30 ≤ l ≤ 30 | |
| Collected reflections | 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 322 322 |
61939 |
100994 |
116530 |
339380 |
| Unique reflections | 4952 [R(int) = 0.0777] | 4922 [R(int) = 0.0496] | 9720 [R(int) = 0.0407] | 8972 [R(int) = 0.0608] | 25543 [R(int) = 0.0725] |
| Parameters | 282 | 321 | 381 | 423 | 1343 |
| Goodness-of-fit on F2 | 1.092 | 1.082 | 1.166 | 1.220 | 1.116 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0281 | R 1 = 0.0346 | R 1 = 0.0262 | R 1 = 0.0279 | R 1 = 0.0766 |
| wR2 = 0.0544 | wR2 = 0.0661 | wR2 = 0.0461 | wR2 = 0.0531 | wR2 = 0.1469 | |
| R indices (all data) | R 1 = 0.0447 | R 1 = 0.0496 | R 1 = 0.0428 | R 1 = 0.0462 | R 1 = 0.1258 |
| wR2 = 0.0628 | wR2 = 0.0690 | wR2 = 0.0559 | wR2 = 0.0635 | wR2 = 0.1907 | |
| Largest diff. peak and hole/e Å−3 | 1.38 and −0.89 | 2.34 and −2.29 | 1.74 and −0.82 | 1.57 and −0.86 | 7.12 and −3.448 |
Catalytic reaction
1,3-Dihydroxyacetone dimer, >97%, purchased from Sigma-Aldrich was used to prepare 0.1 M solution in deionized water. 20 ml of this solution was placed into a glass tube (Radleys carousel) containing 150 mg of catalyst. The reaction was conducted at a temperature of 90 °C with continuous stirring at 400 rpm for 6 h. Samples were taken with a 1 mL syringe through the septum and analysed by high-performance liquid chromatography (HPLC) (Model Shimadzu Prominence) with a refractive index detector (RID) equipped with a COROGEL 107H column at 40 °C to quantify the products and the residual substrate, and acidified water (H2SO4 1.7 mM) was used as the mobile phase (0.6 mL min−1). Upon the completion of the reaction, the resultant solutions were subjected to centrifugation (10000 rpm, 15 min) to facilitate the separation of the catalyst from the solution.
Powder X-ray diffraction
X-ray powder diffraction was performed on a Bruker D8 Advance diffractometer (LynxEye detector) in a Bragg–Brentano θ–θ mode using Cu-Kα radiation. Each powder pattern was recorded within an angular range of 5–50° in 2θ, with steps of 0.02° and counting time of 0.5 s per step.Scanning electron microscopy
The SEM analyses of tantalum(V) coordination complexes 1–5 were performed on a Hitachi-S3400N microscope, equipped with a tungsten filament (accelerating voltage = 5 kV, secondary electron mode, working distance = 5.3 to 5.5 mm).Results and discussion
Structure description
Single crystal X-ray diffraction was performed on the five compounds to determine their structural arrangement. From this analysis, it appears that they crystallize to form three different structural assemblies. Compounds 1 and 2, synthesized with anthracene-9-carboxylic acid and 4′-methylbiphenyl-4-carboxylic acid, respectively, are obtained as tantalum dimers with a {Ta2O} core, compounds 3 and 4 with benzoic acid and 1-naphtoic acid crystallize as tetramers with {Ta4O4} moieties and compound 5 with 2-naphtoic acid is obtained as an octamer with a {Ta8O12} core.Compounds 1 and 2 of the general formula type [Ta2(μ2-O)(L)2(OiPr)6]
The two first compounds crystallize in a monoclinic C2/c space group with close cell parameters (see Table 1) and can be described the same way. The only difference lies in the change of the ligand used for the complexation of the tantalum centers. In both molecules, the asymmetric unit in the crystal structure is constructed from one independent tantalum center (Ta1), one monotopic ligand, three isopropanolate ligands coming from the solvent used in the synthesis and one oxygen atom bridging the two tantalum centers in the complete molecular architecture. The molecular system is a neutral compound composed of dimeric {Ta2O} moieties surrounded by a total of two carboxylate and six isopropanolate ligands (Fig. 1). Each tantalum center is six-fold coordinated with a distorted octahedral geometry in an oxygen environment. It is linked by three oxo groups from isopropanolate molecules, with Ta–OiPr bond distances varying from 1.859(4) Å to 1.877(3) Å for compound 1, and from 1.863(4) Å to 1.900(4) Å for compound 2; two oxo groups from carboxylate arms (Ta–O = 2.163(3) Å and Ta–O = 2.207(3) Å) for compound 1 and (Ta–O = 2.146(3) Å and Ta–O = 2.178(3) Å) for compound 2. Finally, the coordination sphere of both tantalum atoms is completed by one oxygen atom bridging the two centers together with Ta–O distances of 1.9133(12) Å for compound 1 and 1.9221(14) Å for compound 2. The Ta1–Ta1 distances in the molecular entity are about 3.657 Å and 3.677 Å for compounds 1 and 2, respectively. Interestingly, the two carboxylate functions from two distinct monotopic ligands (either anthracene-9-carboxylate or 4′-methylbiphenyl-4-carboxylate) linked two adjacent tantalum centers, in a syn–syn bidentate bridging fashion, adopting a cis position and causing a distortion of the dimeric {Ta2O} unit with a Ta1–O1–Ta1 angle of about 145.7°. | ||
| Fig. 1 Structural representation of compounds 1 and 2; (a) side view showing the “syn–syn” bridging coordination mode and (b) top view illustrating the “cis” coordination of both carboxylate ligands. | ||
Compounds 3 and 4 of the general formula type [Ta4(μ2-O)4(L)4(OiPr)8]
Both compounds crystalize in the monoclinic P21/n space group with different cell parameters (see Table 1). They can be both described as a neutral tetranuclear tantalum(V)-centered {Ta4O4} motif stabilized either by benzoate or 1-naphtoate ligands and further decorated by eight isopropanolate groups (Fig. 2). They are built up from two crystallographically inequivalent tantalum(V) atoms, Ta1 and Ta2, linked to each other via μ2-oxo groups, within a square-ring unit (Fig. 2). Conceptually, these tetranuclear blocks can be seen as the association of two dinuclear subunits as the one described for compounds 1 and 2 through the addition of two bridging μ2-oxo groups. It is worth noting that in this association, the two dimers are connected together in a sort of “head–tail” mode where two ligands are on the same plane and the two others are located above and under this plane (see Fig. 2b). Each tantalum center of complexes 3 and 4 is in a six-fold coordination oxo environment with a distorted octahedral geometry. The tantalum cations are surrounded by two μ2-oxo groups with Ta–O distances between 1.896(2) Å and 1.928(2) Å for compound 3 and between 1.896(3) Å and 1.939(3) Å in compound 4. Two oxygen atoms belong to isopropanolate groups and are bound to the tantalum with distances ranging from 1.863(3) Å to 1.899(2) Å for compound 3 and from 1.875(3) Å to 1.885(3) Å for compound 4. The coordination sphere is completed by the two carboxyl oxygen atoms belonging to the carboxylate ligand with Ta–O distances ranging from 2.141(3) Å to 2.175(3) Å for compound 3 and from 2.132(3) Å to 2.183(3) Å for compound 4. The Ta–O–Ta angles are about 144.9° and 149.5° for the concave and convex bending of compound 3 and about 144.7° and 150° for the concave and convex bending of compound 4. Within the tetramers, the Ta–Ta bond lengths are in the range 3.664–3.669 Å for complex 3 and about 3.667 Å to 3.674 Å for complex 4. In the lattice, the tetrameric units interact through σ–π stacking between the naphthoate ligands with distances of 2.876 Å and benzoate ligands with distances of 2.927 Å.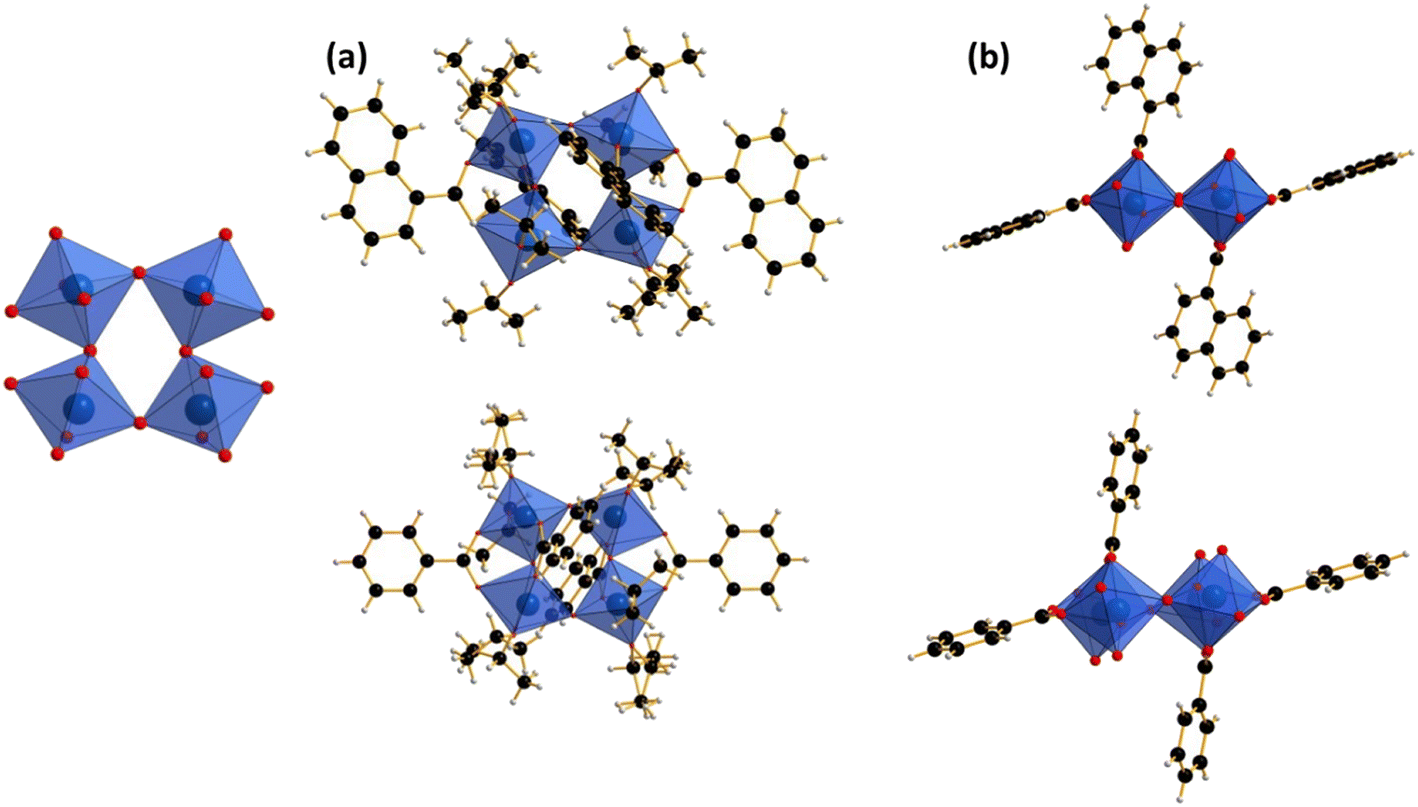 | ||
| Fig. 2 Structural representation of compounds 3 and 4; (a) illustration of the tetranuclear core coordination environment and (b) side view showing the relative position of the carboxylate ligands (isopropanolate groups have been removed for clarity). | ||
Compound 5 [Ta8(μ2-O)12(L)8(OiPr)8]
This system crystallizes in the triclinic P space group. The crystal structure of compound 5 is built up with eight tantalum centers, twelve bridging μ2-oxo groups, eight 2-naphtoate ligands and eight isopropanolate molecules. The cationic-oxo arrangement results in a cubic octanuclear core {Ta8O12}, where the tantalum atoms occupy the eight corners of a cube, and the bridging μ2-oxo groups are roughly located nearby the twelve edges (Fig. 3a). This octanuclear arrangement can be viewed as the condensation of two tantalum-centered tetranuclear blocks through four additional μ2-oxo groups, located perpendicularly to its square plane of Ta–O–Ta bonds. In the octamer, four crystallographic inequivalent tantalum atoms labelled Ta1, Ta2, Ta3 and Ta4 are present. All of them are six-fold coordinated with the same kind of distorted octahedral geometry as in the previous molecular species described in the manuscript. The coordination sphere of each tantalum center is composed of three μ2-oxo groups, two carboxyl oxygen atoms belonging to the 2-naphthoate ligand (in a bidentate syn–syn bridging mode between two adjacent Ta) and one remaining oxygen atom coming from one isopropanolate ligand (Fig. 3b). The Ta–Ooxo bond distances are in the range of 1.875(12)–1.913(12) Å, whereas the Ta–Onaphtoate bonds are longer and in the range of 2.113(14) Å to 2.177(12) Å. The last Ta–OiPr bond lengths (related to the isopropanolate) are observed in the range of 1.854(11) Å to 1.913(12) Å. The Ta–O–Ta angles in the octanuclear compound are all convex and range from 146.5° to 150°. All the Ta–Ta bond distances in the molecular species are in the range of 3.639 Å to 3.669 Å as previously observed in other dinuclear or tetranuclear compounds. The overall compound is also neutral. In each octanuclear species, the aromatic rings of the naphthoate ligands exhibit typical π–π interactions with average distances between 3.598 Å and 3.719 Å for the four pairs of ligands. Interestingly, the eight ligands are located on both sides of the cubic {Ta8O12} arrangement in the (b, c) plane. The ligands from two distinct octamers also interact with weaker π–π stacking interactions with distances ranging from 3.591 Å and 3.632 Å. This tantalum octanuclear block has only been observed once in a related complex [Ta8(O)12(O2CNEt2)16] stabilized by sixteen N,N-diethylcarbamato ligands acting either as monodentate or bidentate ligands and in a parent niobium compound described previously in our group.3,4
 | ||
| Fig. 3 Polyhedral representation of the molecular octanuclear coordination compound 5, [Ta8(μ-O)12(OiPr)8(C11H7O2)8]. (a) Nb-oxo core cluster and (b) top and side view of the complete octanuclear compound. | ||
Infrared spectroscopy and powder X-ray diffraction
All the compounds were characterized by IR spectroscopy and PXRD (see Fig. S2a–e, S3a and b† respectively). The powder X-ray diffraction analysis shows that most of the crystalline phases of the five compounds contained the compounds described in the structure section. In addition, all the compounds are stable out of the mother liquor as we observe a good agreement (either in thickness and in band positions) between the experimental and theoretical diagrams calculated from the SC-XRD analyses in Fig. S2.†The infrared measurements show that all the compounds present characteristic vibrations related to the organic part, either the carboxylate or the isopropanolate ligands. As shown in Table 2, the carbonyl group of the free carboxylic acid ligands (located around 1680 cm−1) disappears to the benefit of classical νasym(COO) and νsym(COO) vibrations that lie around 1575–1602 cm−1 and 1557–1564 cm−1 respectively. They clearly indicate the coordination of the ligands to the tantalum(V) centers through the carboxylate function.
O) vibrations (from the carboxylic acid function) and ν(COO) vibrations (from the carboxylate function) for complexes 1–5 (in cm−1)
|
ν(CO)acid |
ν asym(COO) | ν sym(COO) | ||
|---|---|---|---|---|
| Compound 1 | Anthracene-9-carboxylic acid | 1674 | — | — |
| Crystalline product | — | 1575 | 1557 | |
| Compound 2 | 4′-Methylbiphenyl-4-carboxylic acid | 1687 | — | — |
| Crystalline product | — | 1592 | 1564 | |
| Compound 3 | Benzoic acid | 1680 | — | — |
| Crystalline product | — | 1602 | 1562 | |
| Compound 4 | 1-Naphtoic acid | 1668 | — | — |
| Crystalline product | — | 1599 | 1563 | |
| Compound 5 | 2-Naphtoic acid | 1682 | — | — |
| Crystalline product | — | 1591 | 1560 |
We can also observe for compound 2, on both PXRD and IR characterizations, the presence of several diffraction peaks and vibrations probably related to unreacted ligands as we have a characteristic νCO vibration visible at 1687 cm−1 for the compound spectrum. For unknown reasons, modifying the ligand to tantalum ratio in several syntheses did not lead to the disappearance of the unreacted ligand.
Structure discussion
The utilization of the various monocarboxylic ligands mentioned in the present work for the coordination of tantalum(V) centers leads to the formation of several molecular species with nuclearities starting from a {Ta2O} dinuclear unit up to {Ta4O4} tetranuclear units and a cubic-like {Ta8O12} octanuclear core. As previously described, these polynuclear species are obtained by the addition of several μ2-oxo bridges allowing, in the case of the tetranuclear and octanuclear species, the condensation of the classical dinuclear motif already present in the tantalum ethoxide precursor. The oxygen atoms allowing the condensation process to occur are not present on the initial reaction scheme as the synthesis is performed with great care, in a glove box with controlled atmosphere, to exclude the presence dioxygen and water. As previously described in the work performed in our team on niobium oxo-species,5,6 the emergence of these bridging atoms comes from an esterification reaction occurring between the alcohol ligands of the precursor and the carboxylic acid leading to the formation of an ester and water. This reaction can be of course transposed using the tantalum precursor: 2× Ta–OEt + H2O → Ta–O–Ta + 2× Et–OH, and the water formed in the esterification reaction is the source of oxygen necessary for the occurrence of the poly-condensation reaction. The crystalline phases of the five systems described in this work are pure with the exception of compound 2 where, despite our efforts, the presence of supposedly unreacted ligands seems to be present concomitantly with the dimer [Ta2(μ2-O)(C14H11O2)2(iPrO)6] itself.It is worth noting that there is a new observation revealed in this work, regarding the formation of a polynuclear core of higher nuclearity. Indeed, the four compounds 1–4, including either dimeric or tetrameric tantalum oxo-cores, are thoroughly synthesized using dry isopropanol solvent. However, the octanuclear moiety (compound 5), stabilized with 2-naphtoate ligands, was systematically obtained in high crystalline yield (up to 75%) using a mixture of solvents corresponding to “diluting” isopropanol with acetonitrile, with the latter being a non-complexing species for this chemical system. The best crystallinity for SC-XRD purposes was obtained using a 9:1 molar ratio of the CH3CN/iPrOH mixture, but the increase of the isopropanolate ratio up to 20% prevented the formation of the Ta-centered octanuclear complex. This behavior was not described in our previous work using 2-naphtoate ligands associated with the niobium(V) cation as it was probably hidden by the synthetic procedure for obtaining the {Nb8O12} core by using a large excess of ligands (12 equivalents per niobium centers).4 In our case, only three equivalents of 2-naphtoate ligands were necessary for the formation of the compound. One may assume the significant role played by the nature of the solvent in such synthesis. Isopropanol is a complexing solvent and probably competes with the carboxylate ligands, thus preventing or limiting their coordination to the tantalum centers. Indeed, as can be seen in the three structural types, the alcoholate to carboxylate ligand ratio decreased from 3 to 2 down to 1 for the dimer, the tetramer and the octamer, respectively. Consequently, diminishing the isopropanol ratio in the solvent helps the coordination of the carboxylate ligands and the increase of the nuclearity without preventing the esterification reaction required for the oxolation condensation process (μ2-O group bridging two adjacent tantalum centers). As expected, performing the synthesis in pure acetonitrile solvent did not lead to the formation of any crystalline material, probably because of the too low amount of alcohol species in the reaction medium preventing the beginning of the oxolation condensation reaction.
Another interesting point is that the same strategy using the other ligands mentioned in this work did not lead to the formation of an octanuclear species considering the diverse synthesis attempts that were done. It indicates a probable second effect with the bulky character of the monotopic linkers due to the presence of several aromatic rings in their structures generating more hindrance than the 2-naphtoate ligand. Regarding this specific ligand it is possible that the π–π stacking interaction observed and described in the molecular structure of the octameric compound plays an important role in the stability and formation of such a crystalline molecular arrangement. In a purely speculative approach, with bulky ligands (anthracenoate, 4′-methylbiphenyl-4-carboxylic or 1-naphtoate) or the less bulky benzoate ligands, it may be more difficult to arrange the same π–π stacking interactions making the octanuclear cubic-like core more difficult to obtain in the appropriate solvent mixture.
Catalytic activity during the conversion of dihydroxyacetone (DHA)
The acido/basic properties of compounds 1 (dinuclear Ta2O core) and 5 (octanuclear Ta8O12 core) were tested in aqueous media via the conversion of dihydroxyacetone (DHA) at 90 °C. Such a reaction (Fig. S5†) is usually used to evaluate the Lewis/Brønsted acidity in water through the conversion of DHA that produces lactic acid (LA) or/and pyruvaldehyde (PA). In the presence of basic sites, it could show production of sugars (C6, such as glucose or fructose). Fig. 4 shows the evolution of the reaction as a function of the time observed using compounds 1 & 5, as well as the DHA conversion and yield of the identified resulting products. By considering the same DHA/catalyst ratio of the used complexes (150 mg), compound 1 shows higher kinetics for the conversion of DHA with 70% consumed in 6 hours compared to 47% in the case of compound 5. For both samples the observed products were PA, LA and sugars. The amount of each product obtained as a function of the DHA conversion can be seen in Fig. 4 (right), with about 50% pyruvaldehyde formation and 5–10% for lactic acid and C6 sugar formation (based on %C). | ||
| Fig. 4 DHA conversion (%) (left) and the identified resulting products (lactic acid (LA), pyruvaldehyde (PA) and C6 sugars) for each test (right). Compound 1 (dinuclear Ta2O core): lines; compound 5 (octanuclear Ta8O12 core): dash. | ||
As expected, both compounds 1 & 5 exhibit acidic behavior with mostly Brønsted acid owing to the high yield of pyruvaldehyde. The formation of lactic acid shows some Lewis sites in water alongside with C6 sugars in small amounts. Since the tantalum(V) atoms are octahedrally coordinated to six oxygens, the presence of mostly Brønsted acid sites agrees with the results already reported by Chen et al. in the case of the inorganic compounds Ta2O5·nH2O, for which a regular Ta-centered octahedral environment tends to exhibit Brønsted sites.58 The benefit of the use of coordination polymers stable in water resides in the low concentration of used metallic centers and the possibility to tune the acidity of the catalysts.
Conclusion
We have successfully obtained five molecular compounds 1–5 based on the complexation of the tantalum ethoxide Ta(OEt)5 precursor with monocarboxylate ligands in isopropanol solvent. Three nuclearities were observed in this reactivity panel, dimeric, tetrameric and octameric entities with {Ta2O}, {Ta4O4} and {Ta8O12} tantalum-oxo centers, respectively. All these molecules have been characterized with various solid-state techniques and appear to be stable out of the mother liquor with no visible degradation over several days. We observed the formation of the highest nuclearity (here {Ta8O12} brick) when mixing isopropanol with acetonitrile with a strict molar ratio of 1:9 (iPrOH:CH3CN). It showed the influence of the balance of the complexing strength of the solvent (here iPrOH vs. CH3CN) on the generation of higher nuclearity in the Ta-based complexes. This complexing behavior may be due to pKa differences between the iPrOH/iPrO− couple (17.1) and CH3CN/CH2CN− couple (25) making isopropanolate a much better complexing agent than acetonitrile. The geometry of these molecular moieties and the peculiar cubic-like octanuclear core could be interesting to use as building blocks for the formation of extended materials upon ligand substitution. We are currently investigating the ligand exchange possibilities in our research team. Preliminary catalytic tests (dihydroxyacetone conversion) on two selected compounds (1 & 5) indicated a Brønsted acid activity of compounds using Ta centers opening the way to multiple catalytic applications.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors would like to thank Mrs. Laurence Burylo, Mr. Rémi Lamblin and Mr. Philippe Devaux for their assistance with the synthesis, SEM images, and XRD powder pattern measurements (UCCS). The “Fonds Européen de Développement Régional (FEDER)”, “CNRS”, “Région Hauts de France” and “Ministère de l'Education Nationale de l'Enseignement Supérieur et de la Recherche” are acknowledged for the funding of X-ray diffractometers and ICP-AES apparatus from the Chevreul Institute platform. The catalytic tests were supported by the France 2030 framework under the PEPR DIADEM (DREAM-BIO, project ANR-23-PEXD-0007).References
- M. Lin, C. Mochizuki, B. An, Y. Inomata, T. Ishida, M. Haruta and T. Murayama, ACS Catal., 2020, 10, 9328–9335 CrossRef CAS.
- M. D. Korzyński, L. S. Xie and M. Dincă, Helv. Chim. Acta, 2020, 103, e2000186 CrossRef.
- P. B. Arimondo, F. Calderazzo, R. Hiemeyer, C. Maichle-Mössmer, F. Marchetti, G. Pampaloni and J. Strähle, Inorg. Chem., 1998, 37, 5507–5511 CrossRef CAS PubMed.
- D. Andriotou, S. Duval, C. Volkringer, X. Trivelli, W. E. Shepard and T. Loiseau, Chem. – Eur. J., 2022, 28, e202201464 CrossRef CAS PubMed.
- D. Andriotou, S. Duval, X. Trivelli, C. Volkringer and T. Loiseau, CrystEngComm, 2022, 24, 5938–5948 RSC.
- D. Andriotou, N. Henry, S. Duval, C. Volkringer, W. E. Shepard, F. Pourpoint and T. Loiseau, Eur. J. Inorg. Chem., 2023, 26, e202300432 CrossRef CAS.
- D. Andriotou, S. Duval, C. Volkringer, A. M. Arevalo-Lopez, P. Simon, H. Vezin and T. Loiseau, Inorg. Chem., 2022, 61, 15346–15358 CrossRef CAS PubMed.
- H. Tsurugi, T. Ohno, T. Yamagata and K. Mashima, Organometallics, 2006, 25, 3179–3189 CrossRef CAS.
- A. Conde, R. Fandos, A. Otero and A. Rodríguez, Organometallics, 2008, 27, 6090–6095 CrossRef CAS.
- V. C. Gibson and T. P. Kee, J. Chem. Soc., Chem. Commun., 1989, 656–657 RSC.
- R. Fandos, A. Otero, A. M. Rodríguez and S. Suizo, Organometallics, 2012, 31, 1849–1856 CrossRef CAS.
- C. Poulard, D. Perrey, G. Boni, E. Vigier, O. Blacque, M. M. Kubicki and C. Moïse, Eur. J. Inorg. Chem., 2003, 2003, 633–637 CrossRef.
- R. Fandos, C. Hernández, A. Otero, A. Rodríguez and M. J. Ruiz, Eur. J. Inorg. Chem., 2014, 2014, 6196–6204 CrossRef CAS.
- D. Kwon and M. D. Curtis, Organometallics, 1990, 9, 1–5 CrossRef CAS.
- O. Blacque, M. M. Kubicki, J.-C. Leblanc, A. Sadorge, P. Sauvageot and C. Moïse, J. Organomet. Chem., 2002, 656, 139–145 CrossRef CAS.
- A. S. Batsanov, A. V. Churakov, J. A. K. Howard, A. K. Hughes, A. L. Johnson, A. J. Kingsley, I. S. Neretin and K. Wade, J. Chem. Soc., Dalton Trans., 1999, 3867–3875 RSC.
- A. Conde, R. Fandos, A. Otero and A. Rodríguez, Organometallics, 2009, 28, 5505–5513 CrossRef CAS.
- M. Herberhold, A. Goller and W. Milius, Z. Anorg. Allg. Chem., 2003, 629, 1162–1168 CrossRef CAS.
- Y. Matsuo, K. Mashima and K. Tani, Angew. Chem., Int. Ed., 2001, 40, 960–962 CrossRef CAS PubMed.
- R. Fandos, C. Hernández, A. Otero, A. M. Rodríguez and M. J. Ruiz, ChemistrySelect, 2017, 2, 1871–1877 CrossRef CAS.
- V. C. Gibson, T. P. Kee and W. Clegg, J. Chem. Soc., Chem. Commun., 1990, 29 RSC.
- P. Jernakoff, C. D. M. De Bellefon, G. L. Geoffroy, A. L. Rheingold and S. J. Geib, Organometallics, 1987, 6, 1362–1364 CrossRef CAS.
- W. Zhenhong, Z. Wenbiao, L. Guangming, X. Feng, M. Yingxuan and C. Hu, J. Organomet. Chem., 2016, 808, 104–108 CrossRef.
- M. Bortoluzzi, E. Ferretti, F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2016, 45, 6939–6948 RSC.
- B. S. Bronk, J. D. Protasiewicz, L. E. Pence and S. J. Lippard, Organometallics, 1995, 14, 2177–2187 CrossRef CAS.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Inorg. Chem., 2013, 52, 1685–1687 CrossRef CAS PubMed.
- M. A. Rankin and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 10021–10023 CrossRef CAS PubMed.
- M. D. Fryzuk, S. A. Johnson, B. O. Patrick, A. Albinati, S. A. Mason and T. F. Koetzle, J. Am. Chem. Soc., 2001, 123, 3960–3973 CrossRef CAS PubMed.
- A. B. Weberg, S. Chaudhuri, T. Cheisson, C. Uruburo, E. Lapsheva, P. Pandey, M. R. Gau, P. J. Carroll, G. C. Schatz and E. J. Schelter, Chem. Sci., 2022, 13, 6796–6805 RSC.
- F. Marchetti, G. Pampaloni and S. Zacchini, Polyhedron, 2008, 27, 1969–1976 CrossRef CAS.
- M. Hellwig, A. Milanov, D. Barreca, J.-L. Deborde, R. Thomas, M. Winter, U. Kunze, R. A. Fischer and A. Devi, Chem. Mater., 2007, 19, 6077–6087 CrossRef CAS.
- P. D. Smith, S. M. Harben, R. L. Beddoes, M. Helliwell, D. Collison and C. D. Garner, J. Chem. Soc., Dalton Trans., 1997, 685–692 RSC.
- B. Perić, N. Brničević, M. Jurić, P. Planinić and D. Matković-Čalogović, Struct. Chem., 2009, 20, 933–941 CrossRef.
- P. B. Arimondo, F. Calderazzo, U. Englert, C. Maichle-Mössmer, G. Pampaloni and J. Strähle, J. Chem. Soc., Dalton Trans., 1996, 311–319 RSC.
- M. Hayatifar, F. Marchetti, G. Pampaloni and S. Zacchini, Inorg. Chem., 2013, 52, 4017–4025 CrossRef CAS PubMed.
- M. H. Chisholm, F. A. Cotton and M. W. Extine, Inorg. Chem., 1978, 17, 2000–2003 CrossRef CAS.
- M. H. Chisholm, L. S. Tan and J. C. Huffman, J. Am. Chem. Soc., 1982, 104, 4879–4884 CrossRef CAS.
- D. Bayot, B. Tinant, B. Mathieu, J. Declercq and M. Devillers, Eur. J. Inorg. Chem., 2003, 2003, 737–743 CrossRef.
- J. H. Thurston and K. H. Whitmire, Inorg. Chem., 2003, 42, 2014–2023 CrossRef CAS PubMed.
- V. Stavila, J. H. Thurston and K. H. Whitmire, Inorg. Chem., 2009, 48, 6945–6951 CrossRef CAS PubMed.
- J. H. Thurston and K. H. Whitmire, Inorg. Chem., 2002, 41, 4194–4205 CrossRef CAS PubMed.
- L. A. Dubraja, D. Matković-Čalogović and P. Planinić, CrystEngComm, 2015, 17, 2021–2029 RSC.
- L. Androš, M. Jurić, J. Popović, A. Šantić, P. Lazić, M. Benčina, M. Valant, N. Brničević and P. Planinić, Inorg. Chem., 2013, 52, 14299–14308 CrossRef PubMed.
- L. A. Dubraja, M. Jurić, W. Lafargue-Dit-Hauret, D. Pajić, A. Zorko, A. Ozarowski and X. Rocquefelte, New J. Chem., 2018, 42, 10912–10921 RSC.
- L. Androš, M. Jurić, J. Popović and P. Planinić, RSC Adv., 2014, 4, 37051 RSC.
- L. Androš, D. Matković-Čalogović and P. Planinić, CrystEngComm, 2013, 15, 533–543 RSC.
- F. A. Cotton, M. P. Diebold, S. A. Duraj and W. J. Roth, Polyhedron, 1985, 4, 1479–1484 CrossRef CAS.
- D. Bayot, B. Tinant and M. Devillers, Inorg. Chem., 2005, 44, 1554–1562 CrossRef CAS PubMed.
- D. A. Brown, M. G. H. Wallbridge, W.-S. Li and M. McPartlin, Polyhedron, 1994, 13, 2265–2270 CrossRef CAS.
- V. Petrykin, M. Kakihana, K. Yoshioka, S. Sasaki, Y. Ueda, K. Tomita, Y. Nakamura, M. Shiro and A. Kudo, Inorg. Chem., 2006, 45, 9251–9256 CrossRef CAS PubMed.
- X. Zhang, T. J. Prior and C. Redshaw, New J. Chem., 2022, 46, 14146–14154 RSC.
- T. J. Boyle, N. L. Andrews, T. M. Alam, M. A. Rodriguez, J. M. Santana and B. L. Scott, Polyhedron, 2002, 21, 2333–2345 CrossRef CAS.
- D. A. Brown, W. Errington and M. G. H. Wallbridge, J. Chem. Soc., Dalton Trans., 1993, 1163 RSC.
- Bruker Analytical X-ray Systems, Madison, WI, 2008 Search PubMed.
- G. M. Sheldrick, Brucker-Siemens Area detector Absorption and Other Correction, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- X. Chen, D. Huang, L. He, L. Zhang, Y. Ren, X. Chen, B. Yue and H. He, J. Phys. Chem. C, 2021, 125, 9330–9341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The data associated with the findings of this study can be found in the online version (optical microscopy photographs, powder XRD pattern, IR spectra, liquid 1H NMR and solid state 93Nb spectra). CCDC 2446460 for 1, 2446461 for 2, 2446462 for 3, 2446463 for 4, 2446464 for 5. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00444f |
| This journal is © The Royal Society of Chemistry 2025 |