DOI:
10.1039/D4CE01270D
(Paper)
CrystEngComm, 2025,
27, 1034-1043
Mixed-ligand uranyl ion complexes with two flexible, pyridinium-based dicarboxylate zwitterions and simple anionic dicarboxylates†
Received
17th December 2024
, Accepted 10th January 2025
First published on 13th January 2025
Abstract
Two new dizwitterionic dicarboxylates, E-bis(N-(2′-carboxylatoethyl)pyridinium-4-yl)ethene (L1) and E-bis(3-carboxylatopyridiniomethyl)ethene (L2) have been reacted with uranyl nitrate hexahydrate under solvo-hydrothermal conditions, in the presence of dianionic dicarboxylates, yielding a series of 7 complexes which have been characterized by their crystal structure and luminescence properties. Both [(UO2)2(L1)(1,2-pda)2]·2H2O (1) and [(UO2)2(L1)(1,4-pda)2]·H2O (2), where 1,2- and 1,4-pda2− are 1,2- and 1,4-phenylenediacetates, crystallize as monoperiodic coordination polymers, either two-stranded and ladder-like or sinuous and daisychain-like, respectively. [(UO2)2(L1)(t-1,4-chdc)2] (3), where t-1,4-chdc2− is trans-1,4-cyclohexanedicarboxylate, is a diperiodic assembly with the hcb topology. In situ formation of oxalate anions (ox2−) produces [(UO2)2(L2)(ox)(OH)2] (4), a diperiodic coordination polymer containing dihydroxo-bridged, dinuclear subunits. Simple chains are found in [(UO2)2(L2)(pht)2(H2O)2]·2H2O (5), where pht2− is phthalate, while [(UO2)2(L2)(ipht)2]·2H2O·2CH3CN (6), where ipht2− is isophthalate, is another hcb network. In all these cases, each dicarboxylate ligand connects two metal centres. Finally, [(UO2)2(L2)(t-1,4-chdc)2] (7) is a triperiodic framework with the unusual mog topology, in which t-1,4-chdc2− is either bis(κ2O,O′-chelating) or bis(μ2-κ1O:κ1O′-bridging). Bond valence calculations reveal no very significant difference in donor strength between the two types of ligands. The importance of weak interactions (hydrogen bonding, π–π stacking) is discussed. Only complex 5 is strongly emissive in the solid state, with a photoluminescence quantum yield of 19%, and 6 is weakly emissive (4%), while 1–3 and 7 are non-emissive. The spectra of 5 and 6 display the usual vibronic fine structure, the peak positions being dependent on the uranyl ion equatorial environment.
Introduction
In our previous investigations of the use of carboxylate zwitterions as ligands of interest for the design of uranyl ion coordination polymers, notably mixed-ligand species,1 the long-known tendency of the uranyl ion to form close-to-planar clusters and sheet-like coordination polymers2–5 was a factor seemingly limiting our success in obtaining triperiodic systems. However, di- or trizwitterionic polycarboxylates of large size, particularly those based on double or triple pyridinium cores, allowed isolation of original architectures, such as mixed-ligand discrete rings,6 cages,7 polycatenated8 and interpenetrated structures,6,9 as well as showing some potential for the formation of anion-encapsulating cavities.6,10 Flexibility allowing the ligands to adopt either convergent or divergent geometries for their coordination sites plays an essential part in the variety of structures which can be generated. In the ligands we have used previously, the two pyridinium rings were either linked to one another, flexibility being provided by the aliphatic chains bearing the carboxylate groups,7,8 or they were attached to a central aromatic ring through flexible linkages.6,8–11 We have now extended this work to two new dizwitterionic dicarboxylates presenting expected differences in conformational flexibility, which are also built on double pyridinium platforms and include a central linker containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond that introduces a measure of rigidity. In E-bis(N-(2′-carboxylatoethyl)pyridinium-4-yl)ethene (L1, Scheme 1(a)), the –(CH2)2–COO− terminal groups provide the required pliability. In contrast, E-bis(3-carboxylatopyridiniomethyl)ethene (L2, Scheme 1(a)) includes a longer –(CH2)–CHCH–(CH2)– central bridge, but the carboxylate groups are directly attached to the pyridinium rings. We have associated these ligands with various anionic dicarboxylates (Scheme 1(b)), 1,2- and 1,4-phenylenediacetates (1,2- and 1,4-pda2−), phthalate and isophthalate (pht2− and ipht2−), trans-1,4-cyclohexanedicarboxylate (t-1,4-chdc2−), and oxalate (ox2−) to synthesize a series of seven neutral complexes which span the complete periodicity range. In particular, the combination of the aliphatic dicarboxylate t-1,4-chdc2− with L2 has led to the isolation of a triperiodic framework.
C double bond that introduces a measure of rigidity. In E-bis(N-(2′-carboxylatoethyl)pyridinium-4-yl)ethene (L1, Scheme 1(a)), the –(CH2)2–COO− terminal groups provide the required pliability. In contrast, E-bis(3-carboxylatopyridiniomethyl)ethene (L2, Scheme 1(a)) includes a longer –(CH2)–CHCH–(CH2)– central bridge, but the carboxylate groups are directly attached to the pyridinium rings. We have associated these ligands with various anionic dicarboxylates (Scheme 1(b)), 1,2- and 1,4-phenylenediacetates (1,2- and 1,4-pda2−), phthalate and isophthalate (pht2− and ipht2−), trans-1,4-cyclohexanedicarboxylate (t-1,4-chdc2−), and oxalate (ox2−) to synthesize a series of seven neutral complexes which span the complete periodicity range. In particular, the combination of the aliphatic dicarboxylate t-1,4-chdc2− with L2 has led to the isolation of a triperiodic framework.
 |
| | Scheme 1 (a) The dizwitterionic dicarboxylates L1 and L2. (b) The dicarboxylate coligands employed. | |
Experimental
Synthesis
Caution! Uranium is a radioactive and chemically toxic element, and uranium-containing samples must be handled with suitable care and protection. Small quantities of reagents and solvents were employed to minimize any potential hazards arising both from the presence of uranium and the use of pressurized vessels for the syntheses.
[UO2(NO3)2(H2O)2]·4H2O (RP Normapur, 99%) was purchased from Prolabo; 1,2- and 1,4-phenylenediacetic acids (1,2- and 1,4-pdaH2), phthalic and isophthalic acids (phtH2 and iphtH2) were from Aldrich, and trans-1,4-cyclohexanedicarboxylic acid (t-1,4-chdcH2) was from Alfa-Aesar. For all syntheses of complexes, the solutions were placed in 10 mL tightly closed glass vessels (Pyrex culture tubes with SVL15 stoppers and Teflon-coated seals, provided by VWR) and heated at 140 °C in a sand bath (Harry Gestigkeit ST72). The crystals were grown in the hot, pressurized solutions and not as a result of a final return to ambient conditions.
L1H2Cl2.
The precursor to the ligand L1 was prepared by a minor modification of a literature method.12 A mixture of 1,2-di(4-pyridyl)ethylene (15 mmol) and acrylic acid (30 mL, 50-fold molar excess) was stirred in chloroform (20 mL) at ambient temperature for 2 days. After this time, acetone–HCl (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 15 mL) was added to the mixture, resulting in a light yellow precipitate that was subsequently collected by filtration. The product was washed with acetone and dried in the air (yield: 80%).
:1, 15 mL) was added to the mixture, resulting in a light yellow precipitate that was subsequently collected by filtration. The product was washed with acetone and dried in the air (yield: 80%).
L2H2Br2.
The precursor to the ligand L2 was prepared by a minor modification again of a literature method.13trans-1,4-Dibromo-2-butene (10 mmol) and ethyl nicotinate (50 mmol) were mixed in acetonitrile (50 mL) and heated at reflux for 2 days. After cooling to ambient temperature, the precipitate formed was collected by filtration, and dried in the air. It was then dissolved in 5% (w/v) HBr (50 mL) and heated at reflux for 5 hours. After evaporation under reduced pressure, the product was washed with acetone, resulting in a pale pink powder (yield: 41%).
[(UO2)2(L1)(1,2-pda)2]·2H2O (1).
L1H2Cl2 (20 mg, 0.05 mmol), 1,2-pdaH2 (20 mg, 0.10 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and N,N-dimethylacetamide (0.2 mL). Yellow crystals of complex 1 were obtained in low yield within two days.
[(UO2)2(L1)(1,4-pda)2]·H2O (2).
L1H2Cl2 (20 mg, 0.05 mmol), 1,4-pdaH2 (20 mg, 0.10 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 2 were obtained in low yield within three weeks.
[(UO2)2(L1)(t-1,4-chdc)2] (3).
L1H2Cl2 (20 mg, 0.05 mmol), t-1,4-chdcH2 (9 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 3 were obtained in low yield overnight.
[(UO2)2(L2)(ox)(OH)2] (4).
L2H2Br2 (24 mg, 0.05 mmol), CsI (26 mg, 0.10 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 4 were obtained in low yield within three days. The same complex was obtained with CsI replaced by 1,3-phenylenediacetic, camphoric, or pimelic acid.
[(UO2)2(L2)(pht)2(H2O)2]·2H2O (5).
L2H2Br2 (24 mg, 0.05 mmol), phtH2 (9 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 5 were obtained in low yield overnight.
[(UO2)2(L2)(ipht)2]·2H2O·2CH3CN (6).
L2H2Br2 (24 mg, 0.05 mmol), iphtH2 (9 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 6 were obtained in low yield overnight.
[(UO2)2(L2)(t-1,4-chdc)2] (7).
L2H2Br2 (24 mg, 0.05 mmol), t-1,4-chdcH2 (9 mg, 0.05 mmol), and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and N,N-dimethylacetamide (0.2 mL). Yellow crystals of complex 7 were obtained in low yield overnight.
Crystallography
Data collections were performed at 100(2) K on a Bruker D8 Quest diffractometer using an Incoatec Microfocus Source (IμS 3.0 Mo) and a PHOTON III area detector, and operated with APEX4.14 The data were processed with SAINT,15 and empirical absorption corrections were made with SADABS.16,17 The structures were solved by intrinsic phasing with SHELXT,18 and refined by full-matrix least-squares on F2 with SHELXL,19 using the ShelXle interface.20 When present, the hydrogen atoms bound to oxygen atoms were retrieved from residual electron density maps and they were refined with geometric restraints. All other hydrogen atoms in all compounds were introduced at calculated positions and treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom (1.5 for CH3). In 2, the water molecule is too close to its image by inversion and it has been given an occupancy factor of 0.5 accordingly. In 5, part of the zwitterionic ligand is disordered over two positions which have been refined with occupancy parameters constrained to sum to unity and restraints on bond lengths and displacement parameters; only part of the disorder of the aromatic ring could be resolved. For compounds 2, 3, and 6, the SQUEEZE21 software was used to subtract the contribution of disordered solvent molecules to the structure factors, the number of electrons added corresponding to approximately 2, 1 and 0.5 water molecules per formula unit, respectively. Crystal data and structure refinement parameters are given in Table 1. Drawings were made with ORTEP-3,22,23 and VESTA.24 The topological analyses were done with ToposPro.25
Table 1 Crystal data and structure refinement details
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| Chemical formula |
C38H38N2O18U2 |
C38H36N2O17U2 |
C34H38N2O16U2 |
C18H16N2O14U2 |
C32H30N2O20U2 |
C36H32N4O18U2 |
C32H34N2O16U2 |
|
M/g mol−1 |
1286.76 |
1268.75 |
1206.72 |
960.39 |
1238.64 |
1284.71 |
1178.67 |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P21/n |
P |
P |
P |
|
a/Å |
8.1663(3) |
11.2350(4) |
8.7832(4) |
9.7403(5) |
8.4515(3) |
8.3083(3) |
8.8565(2) |
|
b/Å |
10.5584(3) |
13.3313(5) |
9.7665(5) |
10.6942(5) |
10.5811(4) |
9.7524(3) |
9.0719(3) |
|
c/Å |
12.5335(4) |
14.3587(5) |
11.6623(6) |
10.9139(5) |
10.5850(3) |
13.3763(5) |
11.6001(3) |
|
α/° |
100.8155(12) |
90 |
76.391(2) |
90 |
75.8824(11) |
99.2243(17) |
73.5981(9) |
|
β/° |
91.1157(13) |
109.3383(16) |
83.846(2) |
96.3201(19) |
72.1083(12) |
100.6840(18) |
86.2684(9) |
|
γ/° |
111.4308(12) |
90 |
81.044(2) |
90 |
75.7896(14) |
108.6144(16) |
68.7753(9) |
|
V/Å3 |
983.50(6) |
2029.27(13) |
957.87(8) |
1129.93(9) |
858.55(5) |
980.89(6) |
832.72(4) |
|
Z
|
1 |
2 |
1 |
2 |
1 |
1 |
1 |
| Reflections collected |
47714 |
112330 |
67502 |
94664 |
45194 |
61474 |
53583 |
| Independent reflections |
5986 |
3855 |
3636 |
3448 |
5208 |
3717 |
5059 |
| Observed reflections [I > 2σ(I)] |
5596 |
3825 |
3536 |
3395 |
5005 |
3591 |
4890 |
|
R
int
|
0.045 |
0.048 |
0.051 |
0.046 |
0.040 |
0.044 |
0.048 |
| Parameters refined |
277 |
277 |
244 |
167 |
322 |
278 |
235 |
|
R
1
|
0.020 |
0.032 |
0.046 |
0.019 |
0.021 |
0.022 |
0.018 |
| wR2 |
0.052 |
0.075 |
0.119 |
0.047 |
0.052 |
0.057 |
0.043 |
|
S
|
1.058 |
1.349 |
1.231 |
1.126 |
1.114 |
1.118 |
1.118 |
| Δρmin/e Å−3 |
−1.16 |
−1.57 |
−2.41 |
−1.05 |
−2.51 |
−0.71 |
−1.33 |
| Δρmax/e Å−3 |
1.75 |
1.43 |
4.14 |
2.57 |
1.84 |
2.78 |
1.80 |
Luminescence measurements
Emission spectra were recorded on solid samples using an Edinburgh Instruments FS5 spectrofluorimeter equipped with a 150 W CW ozone-free xenon arc lamp, dual-grating excitation and emission monochromators (2.1 nm mm−1 dispersion; 1200 grooves per mm) and an R928P photomultiplier detector. The powdered compounds were pressed to the wall of a quartz tube, and the measurements were performed using the right-angle mode in the SC-05 cassette. An excitation wavelength of 420 nm was used in all cases and the emission was monitored from 450 to 640 nm. The quantum yield measurements were performed using a Hamamatsu Quantaurus C11347 absolute photoluminescence quantum yield spectrometer and exciting the samples between 300 and 400 nm.
Results and discussion
Syntheses
All complexes in the present series were obtained in rather low yield, an observation that may simply mean that they are of relatively high solubility in the reaction media used but also may be due to the instability of the zwitterionic ligands under the same conditions. That the latter may be the case, at least with L2, is indicated by the isolation of complex 4 in the presence of CsI, where the oxalate present could only originate from the zwitterion. It is possible that I2 formed by nitrate oxidation of iodide could facilitate oxalate formation but this seems to be excluded by the isolation of the same complex when CsI is replaced by various carboxylic acids. Once again, it is only possible to speculate as to the solution chemistry which may lead to the isolated crystals.
Crystal structures
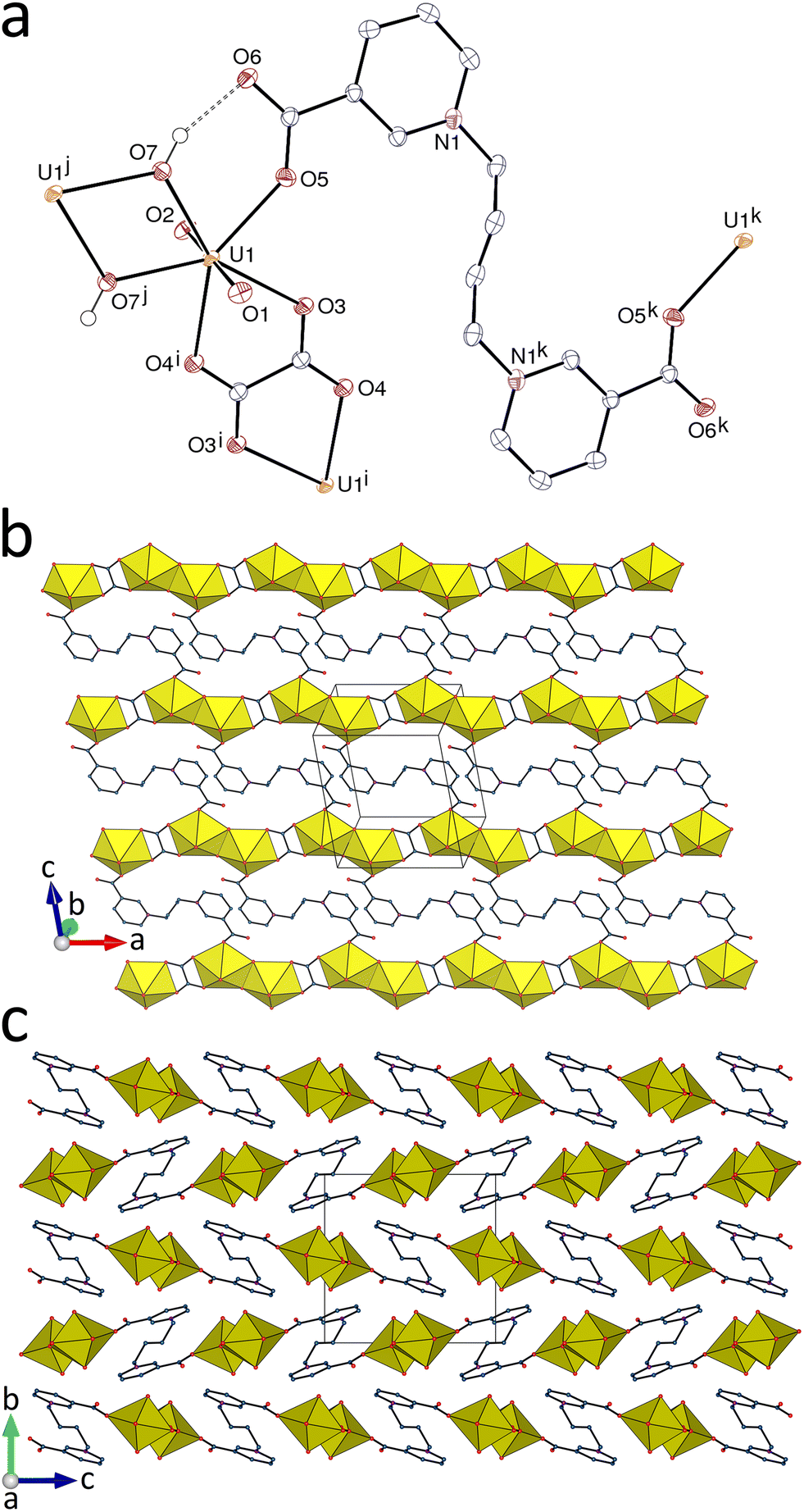
The uranyl cation in the complex [(UO2)2(L1)(1,2-pda)2]·2H2O (1) is κ2O,O′-chelated by one centrosymmetric L1 and two 1,2-pda2− ligands (Fig. 1), this being a mode relatively uncommon for a dizwitterionic ligand,1 which gives a hexagonal-bipyramidal uranium environment [U–O(oxo), 1.772(2) and 1.778(2) Å; U–O(carboxylato), 2.448(2)–2.5131(19) Å]. The two longest U–O bonds are those formed with L1, thus seemingly confirming the trend for zwitterionic carboxylates to be somewhat weaker donors than anionic carboxylates, as previously noted.1 Bond valence parameters (BVs)26 calculated with PLATON27 (Table 2) provide a convenient means of quantifying this effect. Both ligands have divergent donor groups, with the carboxylate groups of L1 divergently oriented approximately perpendicular to the bis(4-pyridyl)ethene plane, and the coordination polymer formed is monoperiodic and directed along [100]. The chains are double-stranded or ladderlike, with two UO2(1,2-pda) rows cross-linked by linear L1 ligands. The chains are arranged into layers parallel to (011) and they are interdigitated, with interlayer, parallel-displaced π–π interactions involving 1,2-pda2− and L1 [centroid⋯centroid distance, 3.9553(18) Å; dihedral angle, 11.43(15)°; slippage, 2.04 Å]. The Kitaigorodsky packing index (KPI, evaluated with PLATON) of 0.72 indicates a compact arrangement.
 |
| | Fig. 1 (a) View of complex 1 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = x − 1, y, z; j = x + 1, y, z; k = 1 − x, 2 − y, −z. (b) The two-stranded monoperiodic coordination polymer with uranium coordination polyhedra in yellow. (c) Packing with chains viewed end-on. | |
Table 2 Mean U–O bond lengths (Å) and bond valence parameters in complexes 1–7a
| Complex |
CNb |
U–Ooxo |
U–OAC (monodentate) |
U–OAC (chelating)c |
U–OZI (monodentate) |
U–OZI (chelating)c |
BVACd (monodentate) |
BVACd (chelating)c |
BVZId (monodentate) |
BVZId (chelating)c |
|
The esds on mean values measure the dispersion of individual values; no esd is given for single BV values.
CN, coordination number.
The term “chelating” refers here only to κ2O,O′-chelating species, not to those forming 5- or 7-membered chelate rings.
BVAC, mean bond valence parameter for anionic carboxylates, BVZI, mean bond valence parameter for zwitterionic carboxylates.
|
|
1
|
8 |
1.775(3) |
|
2.458(7) |
|
2.497(16) |
|
0.451(6) |
|
0.418(14) |
|
2
|
8 |
1.7785(15) |
|
2.46(3) |
|
2.47(4) |
|
0.45(3) |
|
0.44(4) |
|
3
|
8 |
1.776(9) |
|
2.46(2) |
|
2.460(4) |
|
0.452(18) |
|
0.449(4) |
|
4
|
7 |
1.789(4) |
2.469(5) |
|
2.339(2) |
|
0.441(4) |
|
0.567 |
|
|
5
|
7 |
1.774(4) |
2.368(15) |
|
2.368(2) |
|
0.538(16) |
|
0.536 |
|
|
6
|
8 |
1.7765(15) |
|
2.467(9) |
|
2.50(5) |
|
0.443(8) |
|
0.42(4) |
|
7
|
7 |
1.775(4) |
2.390(6) |
2.4373(6) |
2.3080(18) |
|
0.515(7) |
0.4690(10) |
0.604 |
|
Replacement of 1,2- by 1,4-pda2− gives the complex [(UO2)2(L1)(1,4-pda)2]·H2O (2) in which the uranium atom is also in a hexagonal-bipyramidal environment formed by one centrosymmetric, zwitterionic and two anionic ligands [U–O(oxo), 1.777(4) and 1.780(5) Å; U–O(carboxylato), 2.429(5)–2.516(4) Å] (Fig. 2). However, in contrast to 1, the ligand L1 has here both the longest and the shortest of the U–O bonds, resulting in very large standard deviations in mean bond length and BV values which make the difference between BVAC and BVZI insignificant (Table 2). Obviously, the variation of donor strength of the two ligands is sufficiently small to be masked by other small contributions due to weak interactions. The 1,4-pda2− ligand assumes a convergent shape, so that 22-membered, [UO2(1,4-pda)]2 rings are formed, which are further assembled by the divergent L1 ligands into a daisychain-like monoperiodic polymer directed along [201]. The same rings have been found in various other uranyl ion complexes of 1,4-pda2−,28–30 indicating that this may be the dominant factor determining the structure. Here, they are stabilized by the inclusion of a disordered water molecule involved in OH⋯O hydrogen bonds to the carboxylate oxygen atoms O4 and O5 pertaining to the dinuclear ring [O⋯O, 2.988(10) and 2.982(10) Å; O–H⋯O, 167(6) and 130(7)°], thus giving a hydrogen bonding ring with the graph set descriptor31 R22(13), and also in a CH⋯O interaction involving an L1 methylene group in a neighbouring chain. As seen when viewed down the a axis, these chains have a very sinuous shape and are tightly stacked (KPI, 0.69), with a possible interchain parallel-displaced π–π interaction involving 1,4-pda2− and L1 [centroid⋯centroid distance, 3.939(5) Å; dihedral angle, 17.2(4)°; slippage, 1.40 Å]. However, this interaction does not appear as exceeding dispersion on the Hirshfeld surface (HS),32,33 which does, however, provide evidence of CH⋯O hydrogen bonds between L1 and one uranyl oxo group, a factor which serves both to knit the polymer strands into a tridimensional array and to orient aromatic rings into proximity, possibly preventing formation of a simple sheet-like form.
 |
| | Fig. 2 (a) View of complex 2 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = −x, 1 − y, 1 − z; j = 2 − x, 1 − y, 2 − z. (b) Arrangement of monoperiodic coordination polymers parallel to (010). (c) Packing with chains viewed edge-on. | |
Tris-chelation of uranyl by one zwitterionic and two anionic ligands, all centrosymmetric, is also found in the complex [(UO2)2(L1)(t-1,4-chdc)2] (3), shown in Fig. 3 [U–O(oxo), 1.767(7) and 1.785(8) Å; U–O(carboxylato), 2.442(7)–2.495(6) Å]. The longest bond here is with atom O6 from t-1,4-chdc2−, the next two being those with L1, so that the mean BV values are not different (Table 2). The two centrosymmetric t-1,4-chdc2− ligands differ by the orientation of the carboxylate groups, which are both axial in the ligand containing O3 and O4 and equatorial in the case of O5 and O6 (the diequatorial conformation being generally more common in metal complexes34). In contrast to the monoperiodic coordination polymers found in 1 and 2, that formed here is diperiodic and parallel to (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 2). It has the {63} point symbol and the hcb topological type, a very common occurrence in complexes in which the uranyl ion is tris-chelated by three divergent dicarboxylate ligands. The hexanuclear cells are however very far from the regular hexagonal geometry, the L1 ligand having an S-shape. A methylene group of L1 is involved in a hydrogen bond with a uranyl oxo group, but there is no π–π interaction of L1, and the KPI of 0.67 indicates that only small solvent-accessible voids are present (see Experimental).
2). It has the {63} point symbol and the hcb topological type, a very common occurrence in complexes in which the uranyl ion is tris-chelated by three divergent dicarboxylate ligands. The hexanuclear cells are however very far from the regular hexagonal geometry, the L1 ligand having an S-shape. A methylene group of L1 is involved in a hydrogen bond with a uranyl oxo group, but there is no π–π interaction of L1, and the KPI of 0.67 indicates that only small solvent-accessible voids are present (see Experimental).
 |
| | Fig. 3 (a) View of compound 3 with displacement ellipsoids shown at the 40% probability level and hydrogen atoms omitted. Symmetry codes: i = 2 − x, 2 − y, −z; j = −x, 2 − y, 1 − z; k = −x, 1 − y, −z. (b) View of the hcb diperiodic coordination polymer. | |
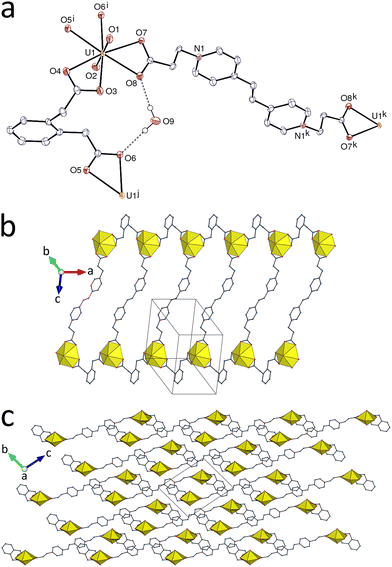
The complex [(UO2)2(L2)(ox)(OH)2] (4) was obtained in the presence of different additional reagents, CsI, 1,3-phenylenediacetic, camphoric, or pimelic acid (albeit always in extremely low yield), thus suggesting that oxalate is produced through oxidation of L2. Oxalate formation during solvothermal processes is frequently observed,35–40 and nitrate has been shown in several cases36,39 to be the oxidant involved. The uranyl cation in 4 is chelated by one centrosymmetric ox2− ligand forming a 5-membered ring, and it is also bound to one monodentate carboxylate group from the centrosymmetric ligand L2 and two hydroxide anions, the uranium environment being pentagonal-bipyramidal (Fig. 4) [U–O(oxo), 1.784(2) and 1.793(2) Å; U–O(carboxylato), 2.339(2)–2.473(2) Å; U–O(hydroxido), 2.279(2) and 2.348(2) Å]. The shortest U–O(carboxylato) bond here is that with L2 (Table 2), this being probably due to the constrained geometry of oxalate bonding. The hydroxo (instead of oxo) nature of O7 is confirmed by its overall bond valence parameter of 1.2. The L2 ligand assumes an S-shape and is divergent, both ligands being simple links. The double hydroxide bridges result in the formation of uranyl dimers with edge-sharing coordination polyhedra, these dimers being further assembled into linear chains running along the a axis by the oxalate links. These rows are cross-linked by the L2 ligands to give a diperiodic polymer parallel to (010). If the dimers are considered as 4-coordinated (4-c) nodes, the topological type is the usual sql. The hydroxide anion forms a strong hydrogen bond with the uncoordinated carboxylate atom O6 [O⋯O, 2.742(3) Å; O–H⋯O, 159(4)°], thus building an R11(6) ring. The L2 ligands are not involved in any π–π interaction, but two CH⋯O interactions involving both uranyl oxo groups are present, one with an aromatic CH group within the layers and the other with a methylene group in the adjoining layer [C⋯O, 3.105(4) and 3.411(4) Å; C–H⋯O, 130 and 156°]. In addition to CH⋯O(uranyl) hydrogen bonds, one UO⋯π(pyridinium) interaction involving a neighbouring layer is also apparent on the HS, as previously found in comparable systems.41 The KPI of 0.73 reveals no solvent-accessible space.
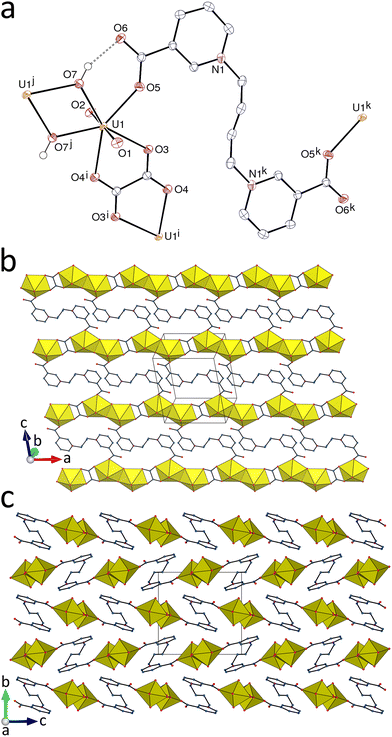 |
| | Fig. 4 (a) View of compound 4 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and the hydrogen bond is shown as a dashed line. Symmetry codes: i = 1 − x, 1 − y, −z; j = 2 − x, 1 − y, −z; k = 1 − x, 1 − y, 1 − z. (b) The diperiodic coordination polymer. (c) Packing with layers viewed edge-on. | |
Phthalate is a convergent ligand and, as expected, it forms a 7-membered chelate ring in [(UO2)2(L2)(pht)2(H2O)2]·2H2O (5), shown in Fig. 5. While one of its carboxylate groups is monodentate, the other is bridging in the syn/anti μ2-κ1O:κ1O′ mode. The uranium pentagonal-bipyramidal environment is completed by one monodentate carboxylate group from the extended, centrosymmetric L2, and one water molecule [U–O(oxo), 1.770(2) and 1.777(2) Å; U–O(carboxylato), 2.357(2)–2.389(2) Å; U–O(water), 2.450(2) Å]. The bond length with L2 is within the range of those with the anionic ligand (Table 2), so that no difference in donor strength is obvious here. The uranium atom is a 3-c node and both ligands are simple edges, and the coordination polymer formed is monoperiodic and directed along [10], with centrosymmetric dimers with double pht2− bridges being connected through L2 ligands. The packing (KPI, 0.75) involves a single parallel-displaced π–π interaction between pht2− and L2 [centroid⋯centroid distance, 3.969(10) Å; dihedral angle, 5.5(8)°; slippage, 2.06 Å].
 |
| | Fig. 5 (a) View of compound 5 with displacement ellipsoids shown at the 50% probability level. Only one position of the disordered parts is represented. Carbon-bound hydrogen atoms are omitted and the hydrogen bond is shown as a dashed line. Symmetry codes: i = 1 − x, 1 − y, −z; j = −x, 3 − y, −z. (b) Packing with chains viewed edge-on. (c) Packing with chains viewed end-on. | |
Replacing pht2− by its divergent positional isomer ipht2− gives the complex [(UO2)2(L2)(ipht)2]·2H2O·2CH3CN (6), in which the uranyl cation is tris(κ2O,O′-chelated) by one S-shaped L2 and two ipht2− ligands, all centrosymmetric, resulting in a hexagonal-bipyramidal uranium coordination polyhedron [U–O(oxo), 1.775(3) and 1.778(3) Å; U–O(carboxylato), 2.448(3)–2.549(3) Å] (Fig. 6). As L1 in complex 2, L2 is associated with both the shortest and longest equatorial bonds, with the consequence that, here also, no significant difference in donor strength between the two ligands is apparent (Table 2). With the uranium atom as a 3-c node and all ligands as edges, the diperiodic coordination polymer formed, parallel to (1![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), has the hcb topological type. The sheets are nearly planar and each elongated hexanuclear cell contains two acetonitrile molecules with their nitrogen atoms within the layer plane and involved in both OH⋯N (with water) and CH⋯O hydrogen bonds, thus forming links between layers. Due to the planar geometry of the sheets, the packing displays stacks of aromatic rings with parallel-displaced π–π interactions involving ipht2− and L2 [centroid⋯centroid distances, 3.690(2) and 3.768(2) Å; dihedral angles, 10.50(19) and 13.28(19)°; slippages, 1.23–2.08 Å], resulting in a compact arrangement (KPI, 0.72).
), has the hcb topological type. The sheets are nearly planar and each elongated hexanuclear cell contains two acetonitrile molecules with their nitrogen atoms within the layer plane and involved in both OH⋯N (with water) and CH⋯O hydrogen bonds, thus forming links between layers. Due to the planar geometry of the sheets, the packing displays stacks of aromatic rings with parallel-displaced π–π interactions involving ipht2− and L2 [centroid⋯centroid distances, 3.690(2) and 3.768(2) Å; dihedral angles, 10.50(19) and 13.28(19)°; slippages, 1.23–2.08 Å], resulting in a compact arrangement (KPI, 0.72).
 |
| | Fig. 6 (a) View of compound 6 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and the hydrogen bond is shown as a dashed line. Symmetry codes: i = 1 − x, 2 − y, −z; j = 2 − x, 1 − y, 1 − z; k = −x, −y, 1 − z. (b) View of the hcb diperiodic coordination polymer with included acetonitrile molecules. (c) Packing with layers viewed edge-on and solvent excluded. | |
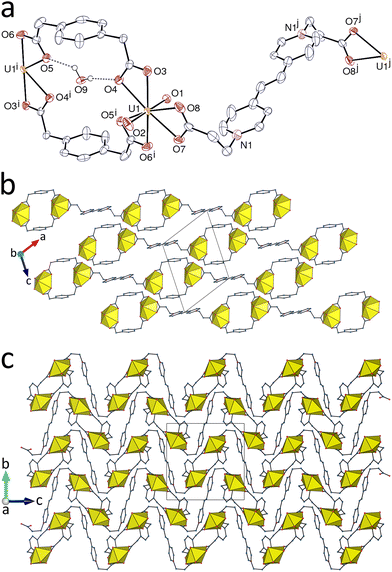
Using with L2 the same t-1,4-chdc2− ligand as in 3 results in the formation of the complex [(UO2)2(L2)(t-1,4-chdc)2] (7), represented in Fig. 7. The uranyl cation is κ2O,O′-chelated by one carboxylate group from an anionic ligand and bound in monodentate fashion to two more oxygen donors from two anionic ligands and one from L2, the environment being pentagonal-bipyramidal [U–O(oxo), 1.7711(18) and 1.7786(18) Å; U–O(carboxylato), 2.4367(18) and 2.4378(19) Å for the chelating group, 2.3080(18)–2.3964(17) Å for the monodentate groups]. In this case, as in 4 but without the constraints due to oxalate bonding geometry, the zwitterionic carboxylate appears to be the stronger donor (Table 2). All three ligands are centrosymmetric, the carboxylate groups of both t-1,4-chdc2− anions being in the equatorial position and L2 assuming a divergent, kinked conformation with the two carboxylatopyridinium groups in parallel, offset planes. While the chelating t-1,4-chdc2− ligand and L2 are simple edges, the bis(μ2-κ1O:κ1O′-bridging) anionic ligand is a 4-c node, as is also the metal centre. The triperiodic, 2-nodal net formed has the {4.64·8}2{42·62·82} point symbol and the unusual mog topological type.42,43 The same topology was previously found in a subunit formed in a uranyl ion complex with trans,trans-muconic acid, NiII cations forming however additional edges in this case.44 The framework in 7 contains neutral UO2(t-1,4-chdc) layers which are cross-linked by the L2 ligands, the latter being too far apart from one another for π–π interactions to be present. With a KPI of 0.74, the framework does not contain solvent-accessible voids.
 |
| | Fig. 7 (a) View of compound 7 with displacement ellipsoids shown at the 50% probability level and hydrogen atoms omitted. Symmetry codes: i = 1 − x, 1 − y, 1 − z; j = 2 − x, 1 − y, 2 − z; k = 2 − x, 1 − y, 1 − z; l = x + 1, y, z; m = −x − 1, 2 − y, 2 − z. (b and c) Two views of the mog triperiodic framework. (d) Nodal representation of the framework. U nodes, yellow; L2 edges, blue; t-1,4-chdc2− nodes and edges, red; same orientation as in (c). | |
In complexes 1–6, both zwitterionic and anionic dicarboxylates are simple edges (2-c), with both of them being bis(κ2O,O′-chelating) in 1–3 and 6. The zwitterionic ligand is bis(monodentate) in 4 and 5, i.e. in the two cases in which the anionic ligand is chelating but forms a larger, 5- or 7-membered chelate ring and thus limits the available equatorial space, resulting in pentagonal-bipyramidal uranium coordination. Only in compound 7 does one of the anionic ligands become a 4-c node, resulting in an increase in periodicity and formation of a triperiodic framework (with uranium in pentagonal-bipyramidal environment). Framework formation thus appears as a direct consequence of the bis(μ2-κ1O:κ1O′) bridging mode adopted by one of the t-1,4-chdc2− ligands in 7, weak interactions and ligand flexibility probably playing a very minor role. However, an interesting point is the difference between the structures of complexes 3 and 7, both containing t-1,4-chdc2−, in association with L1 or L2, respectively. The former is the usual hcb network often associated with uranyl tris(chelation), while the latter is of the rarer (4-c)2 2-nodal mog topology. A subtle influence of the difference in flexibility or donor strength between L1 and L2 cannot be ruled out, but is quite uncertain, as is a possible influence of the difference in organic cosolvent during the synthesis, acetonitrile for 3 and N,N-dimethylacetamide for 7.
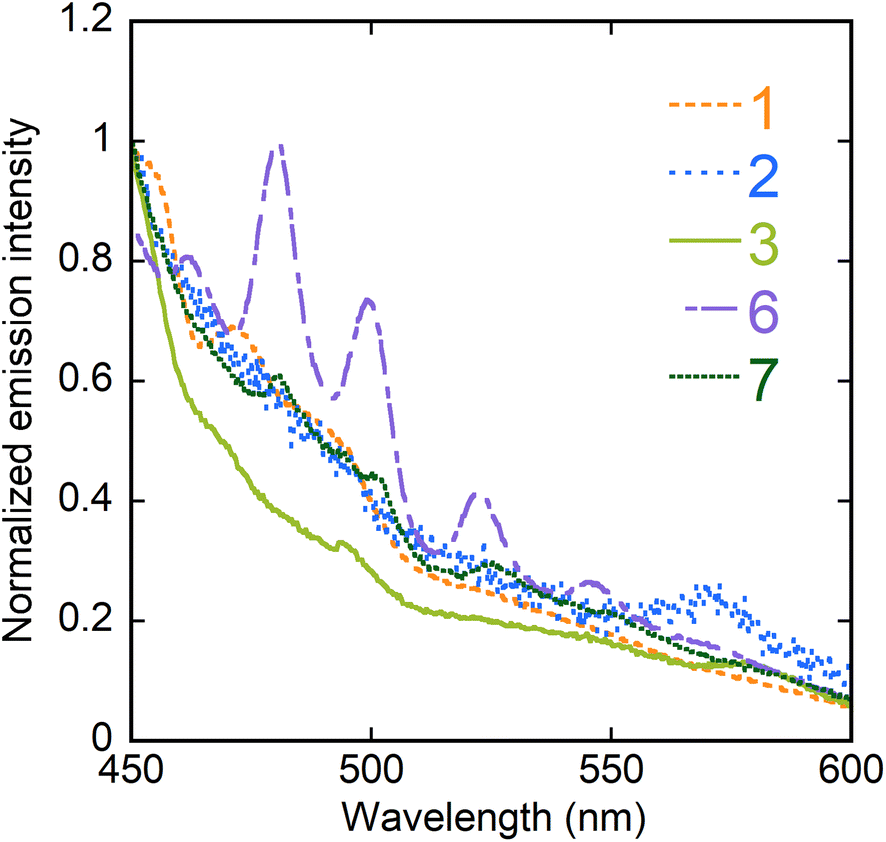
Luminescence properties
The photoluminescence quantum yields (PLQYs) have been measured and the emission spectra in the solid state under excitation at 420 nm have been recorded for all compounds but 4, which could not be obtained in sufficient quantity. Complexes 1–3 and 7 are non-emissive (PLQY ≤ 1%) and their spectra are essentially featureless, while complex 6 is only weakly emissive (PLQY, 4%) and shows five peaks at 461, 480, 499, 522, and 545 nm (Fig. 8). In contrast, complex 5 has a rather large PLQY of 19% and its spectrum displays six peaks at ∼482 (shoulder), 497, 518, 542, 567, and ∼594 nm (Fig. 9). Both spectra of 5 and 6 thus display the typical vibronic progression due to the S10 → S0ν (ν = 0–4) transitions of the uranyl ion,45,46 with the additional low intensity “hot-band” (S11 → S00) due to electron–phonon coupling, observed at the shortest wavelength.47 The positions of the peaks for 5 and 6 match those usual for uranyl ion complexes with equatorial O5 and O6 environments, respectively.48
 |
| | Fig. 8 Emission spectra of complexes 1–3, 6, and 7 in the crystalline state upon excitation at 420 nm. | |
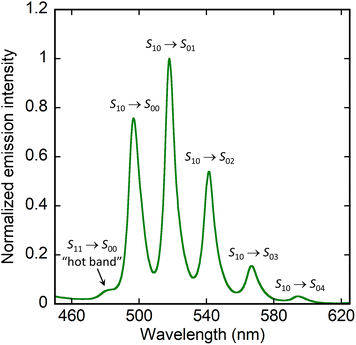 |
| | Fig. 9 Emission spectrum of complex 5 in the crystalline state upon excitation at 420 nm. | |
Conclusions
We have reported the synthesis and crystal structure of 7 mixed-ligand uranyl ion complexes involving a combination of zwitterionic and anionic dicarboxylates, as well as their luminescence properties in all but one case. The coordination polymers formed span the whole periodicity range, from chains, simple, daisychain or ladderlike in shape, to sheets and, in one case, a triperiodic framework. The connectivity of both types of ligands is generally low, of the 2-c, bis(monodentate) or bis(κ2O,O′-chelating) form, with only one instance of the 4-c, bis(μ2-κ1O:κ1O′) bridging mode in the triperiodic framework. The anticipated difference in flexibility between ligands L1 and L2 clearly does not have a major influence on the form of their uranyl ion complexes, with mono- and diperiodic polymers being dominant for both in the present series. A contrast in behaviour is seen in the case of the two complexes obtained with 1,4-chdc2− as a co-ligand, indicating perhaps that the aromatic co-ligands used in all other cases have a dominant effect due to aromatic⋯aromatic interactions, when present, coupled to differences in weak interactions of aromatic CH compared to aliphatic CH of 1,4-chdc2−.
Data availability
Full structural data for the seven complexes have been deposited with the Cambridge Crystallographic Data Centre under deposition numbers 2408741–2408747. Data in cif format may be obtained at https://www.ccdc.cam.ac.uk/structures.
Conflicts of interest
There are no conflicts of interest to declare.
References
- P. Thuéry and J. Harrowfield, Coord. Chem. Rev., 2024, 510, 215821 CrossRef.
- P. A. Giesting and P. C. Burns, Crystallogr. Rev., 2006, 12, 205 CrossRef CAS.
-
C. L. Cahill and L. A. Borkowski, in Structural Chemistry of Inorganic Actinide Compounds, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, Amsterdam, 2007, ch. 11 Search PubMed.
- T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS.
- S. Hickam and P. C. Burns, Struct. Bonding, 2017, 173, 121 CrossRef CAS.
- S. Kusumoto, Y. Atoini, S. Masuda, J. Y. Kim, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Inorg. Chem., 2022, 61, 15182 CrossRef CAS PubMed.
- S. Kusumoto, Y. Atoini, S. Masuda, Y. Koide, K. Chainok, Y. Kim, J. Harrowfield and P. Thuéry, Inorg. Chem., 2023, 62, 7803 CrossRef CAS PubMed.
- S. Kusumoto, Y. Atoini, Y. Koide, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, CrystEngComm, 2023, 25, 5748 RSC.
- S. Kusumoto, Y. Atoini, Y. Koide, K. Chainok, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Chem. Commun., 2023, 59, 10004 RSC.
- S. Kusumoto, Y. Atoini, Y. Koide, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, J. Inclusion Phenom. Macrocyclic Chem., 2024, 104, 209 CrossRef CAS.
- S. Kusumoto, Y. Atoini, S. Masuda, Y. Koide, J. Y. Kim, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, CrystEngComm, 2022, 24, 7833 RSC.
- C. Sun, G. Xu, X. M. Jiang, G. E. Wang, P. Y. Guo, M. S. Wang and G. C. Guo, J. Am. Chem. Soc., 2018, 140, 2805 CrossRef CAS PubMed.
- J. G. Mao, H. J. Zhang, J. Z. Ni, S. B. Wang and T. C. W. Mak, Polyhedron, 1999, 18, 1519 CrossRef CAS.
-
APEX4, ver. 2021.10–0, Bruker AXS, Madison, WI, 2021 Search PubMed.
-
SAINT, ver. 8.40A, Bruker Nano, Madison, WI, 2019 Search PubMed.
-
SADABS, ver. 2016/2, Bruker AXS, Madison, WI, 2016 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
-
M. N. Burnett and C. K. Johnson, ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, 1996 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2019, 58, 6550 CrossRef PubMed.
- P. Thuéry, Y. Atoini and J. Harrowfield, Cryst. Growth Des., 2019, 19, 6611 CrossRef.
- P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2020, 59, 2503 CrossRef PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer, University of Western Australia, 2012 Search PubMed.
- Y. C. Ou, R. M. Zhong and J. Z. Wu, Dalton Trans., 2022, 51, 2992 RSC.
- P. Thuéry, CrystEngComm, 2008, 10, 808 RSC.
- M. B. Andrews and C. L. Cahill, CrystEngComm, 2011, 13, 7068 RSC.
- H. H. Li, X. H. Zeng, H. Y. Wu, X. Jie, S. T. Zheng and Z. R. Chen, Cryst. Growth Des., 2015, 15, 10 CrossRef CAS.
- P. Thuéry, Y. Atoini and J. Harrowfield, Cryst. Growth Des., 2018, 18, 3167 CrossRef.
- D. E. Felton, T. A. Kohlgruber, Z. D. Tucker, E. M. Gulotty, B. L. Ashfeld and P. C. Burns, Cryst. Growth Des., 2023, 23, 8311 CrossRef CAS.
- Y. H. Lee, Y. Atoini, S. Kusumoto, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Polyhedron, 2024, 262, 117172 CrossRef.
- Y. Atoini, S. Kusumoto, Y. Koide, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Polyhedron, 2024, 250, 116848 CrossRef CAS.
- M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3 CrossRef.
- C. Y. Su, M. D. Smith, A. M. Goforth and H. C. zur Loye, Inorg. Chem., 2004, 43, 6881 CrossRef CAS PubMed.
- P. Thuéry and J. Harrowfield, Inorg. Chem., 2022, 61, 2790 CrossRef PubMed.
- A. Brachmann, G. Geipel, G. Bernhard and H. Nitsche, Radiochim. Acta, 2002, 90, 147 CrossRef CAS.
- M. Demnitz, S. Hilpmann, H. Lösch, F. Bok, R. Steudtner, M. Patzschke, T. Stumpf and N. Huittinen, Dalton Trans., 2020, 49, 7109 RSC.
- D. H. Chen, N. Vankova, G. Jha, X. Yu, Y. Wang, L. Lin, F. Kirschhöfer, R. Greifenstein, E. Redel, T. Heine and C. Wöll, Angew. Chem., Int. Ed., 2024, e202318559 CAS.
- P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 13464 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Youssef
Atoini
a,
Youssef
Atoini