
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew salt-solvates of Mirabegron: a combined experimental and computational study†
Rafel
Prohens
 *a,
Rafael
Barbas
b,
Miquel
Barceló-Oliver
c and
Antonio
Frontera
*c
*a,
Rafael
Barbas
b,
Miquel
Barceló-Oliver
c and
Antonio
Frontera
*c
aLaboratory of Organic Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, Avda. Joan XXIII, 08028 Barcelona, Spain. E-mail: rafel_prohens@ub.edu
bUnitat de Polimorfisme i Calorimetria, Centres Científics i Tecnològics, Universitat de Barcelona, Baldiri Reixac 10, 08028 Barcelona, Spain
cDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Spain
First published on 1st April 2025
Abstract
A comprehensive combined virtual and experimental multicomponent solid-form screening has been carried out for Mirabegron, a therapeutic agent widely prescribed for interstitial cystitis and bladder pain syndrome. This systematic approach resulted in the identification and structural elucidation of three novel salt/solvate forms using single-crystal X-ray diffraction (SCXRD). These include two distinct mirabegron–orotate salts (1a and 1b) and a mirabegron–isobutyrate (acetonitrile solvate) salt (2). A detailed Density Functional Theory (DFT) computational analysis unveiled a diverse array of intermolecular interactions in these newly characterized forms, some of which were not observed in the elusive crystal structure of Mirabegron previously reported in the literature. These interactions, essential for understanding the stability and properties of the solid forms, have been meticulously evaluated both structurally and energetically. Advanced computational tools, including Quantum Theory of Atoms in Molecules (QTAIM), non-covalent interaction (NCIPlot) analysis, and Molecular Electrostatic Potential (MEP) surface mapping, were employed to provide in-depth insights into the nature and strength of these interactions, further enriching the understanding of Mirabegron's solid-state chemistry.
1. Introduction
Mirabegron (2-(2-amino-1,3-thiazol-4-yl)-N-[4-[2-[[(2R)-2-hydroxy-2-phenylethyl]amino]ethyl]phenyl]acetamide) is a drug commercialized under the trade names of Myrbetriq and Betmiga for the treatment of overactive bladder and urinary incontinence.1 In spite of receiving approval from the US Food and Drug Administration (FDA) in 2012 its solubility and physicochemical stability are still improvable issues. In this sense, its low bioavailability has motivated that solid-lipid nanoparticles,2 co-amorphous formulations3 or solid dispersions4 have been already explored in the literature. Two polymorphs, known as forms α and β,5 and an amorphous form,6 have been reported, and the crystal structure of form α has been determined recently by microcrystal electron diffraction.7 However, little is known about its potential to form multicomponent crystal forms, a well-known strategy for increasing water solubility.In this work, we have conducted a combined experimental/theoretical study to explore the multicomponent solid forms landscape from both a crystallographic and supramolecular points of view, which can open alternative approaches for others to develop improved formulations of this important Active Pharmaceutical Ingredient. Our work has led to the discovery of three new salt/solvate forms (see Scheme 1): two distinct mirabegron–orotate salts (1a and 1b) and a mirabegron–isobutyrate (acetonitrile solvate) salt (2), all characterized through single-crystal X-ray diffraction (SCXRD). To further understand these solid forms, we performed a detailed DFT computational analysis, which revealed a diverse array of intermolecular interactions absent in the recently reported crystal structure of Mirabegron. These interactions were explored structurally and energetically using QTAIM, NCIPlot, and MEP surface analyses, providing new insights into the molecular environment and stability of these forms.
 | ||
| Scheme 1 Structure of Mirabegron salts 1 and 2 reported herein. | ||
2. Materials and methods
2.1. Materials
Mirabegron form α was used as received by Interquim, S. A. (CCDC refcode: HOCHAM). Qualitative solubilities of mirabegron form α and of each coformer were determined experimentally before choosing the solvents used in the solid forms screening, (see ESI† for further details).2.2. Differential scanning calorimetry (DSC)
Differential scanning calorimetry analysis were carried out by means of a Mettler-Toledo DSC-822e calorimeter. Experimental conditions: aluminum crucibles of 40 μL volume, atmosphere of dry nitrogen with 50 mL min−1 flow rate, heating rates of 1 and 10 °C min−1. The calorimeter was calibrated with indium of 99.99% purity (m.p.: 156.8 °C, ΔH: 28.59 J g−1).2.3. Thermogravimetric analysis (TGA)
Thermogravimetric analysis were performed on a Mettler-Toledo TGA-851e thermobalance. Experimental conditions: alumina crucibles of 70 μL volume, atmosphere of dry nitrogen with 50 mL min−1 flow rate, heating rate of 10 °C min−1.2.4. X-ray powder diffraction analysis (XRPD)
X-ray powder diffraction patterns of the Oxyma-B were obtained on a PANalytical X'Pert PRO MPD diffractometer in transmission configuration using Cu Kα radiation (λ = 1.5418 Å) with a focusing elliptic mirror and a PIXcel detector working at a maximum detector's active length of 3.347°. Configuration of convergent beam with a focalizing mirror and a transmission geometry with flat sample sandwiched between low absorbing films measuring from 2 to 70° in 2θ, with a step size of 0.013° 2θ and a total measuring time of 8 to 30 minutes at room temperature (298 K).2.5. Multicomponent solid forms screening
Mirabegron has a thiazol group (pKα 4.5) and an amine group (pKα 8.0)8 in its structure. This makes expectable the formation of salts with acidic compounds. Thus, some carboxylic acids and aminoacids which are acceptable for human consumption were chosen and a comprehensive multicomponent solid forms screening was conducted following a general methodology we have previously reported9 and detailed below. The coformers tested in our screen were orotic acid, L-arginine, camphoric acid, sorbic acid, glycine, isobutyric acid, L-lysine and L-leucine.As a general procedure, three different experiments were conducted in the solid forms screening: liquid-assisted grinding (LAG), reaction crystallization (RC) and solvent-mediated transformation (SMT) experiments. LAG were conducted by grinding 20–30 mg of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of mirabegron and each coformer and one drop of selected solvents using a grinding mill (Retsch MM 2000). The mixtures were placed in 2 mL stainless steel containers, together with two stainless tungsten grinding balls (3 mm diameter). Experiments were conducted for 15–30 minutes, at 30 Hz mill frequency. Then, the resulting solids were collected without drying and immediately analyzed by XRPD. Evidences of new solid forms were detected by comparing XRPD patterns of all the known forms of mirabegron and the coformers against the resulting solids. RC experiments were conducted with a saturated solution of the most soluble component (mirabegron or the coformer) in selected solvents as follows: a small amount of the less soluble component was added to the saturated solution of the most soluble one until it did not dissolve anymore. Then, the mixture was stirred and the final solids filtered and analyzed by XRPD. Experiments by SMT were performed with suspensions of mirabegron and each selected coformer in different molar ratios. The mixtures were stirred and the resulting solids filtered and analyzed by XRPD.
:1 mixture of mirabegron and each coformer and one drop of selected solvents using a grinding mill (Retsch MM 2000). The mixtures were placed in 2 mL stainless steel containers, together with two stainless tungsten grinding balls (3 mm diameter). Experiments were conducted for 15–30 minutes, at 30 Hz mill frequency. Then, the resulting solids were collected without drying and immediately analyzed by XRPD. Evidences of new solid forms were detected by comparing XRPD patterns of all the known forms of mirabegron and the coformers against the resulting solids. RC experiments were conducted with a saturated solution of the most soluble component (mirabegron or the coformer) in selected solvents as follows: a small amount of the less soluble component was added to the saturated solution of the most soluble one until it did not dissolve anymore. Then, the mixture was stirred and the final solids filtered and analyzed by XRPD. Experiments by SMT were performed with suspensions of mirabegron and each selected coformer in different molar ratios. The mixtures were stirred and the resulting solids filtered and analyzed by XRPD.
2.6. Single X-ray crystallographic analysis
Single crystal X-ray diffraction intensity data of the mirabegron salts were collected using a D8 Venture system equipped with a multilayer monochromator and a Mo microfocus (λ = 0.71073 Å). Data reduction and cell refinements were performed using the Bruker APEX5 program.10 Scaling and absorption corrections were carried out using the SADABS program in all cases.10 Using Olex2,11 the structure was solved with the ShelXT structure solution program12 using intrinsic phasing and refined with the ShelXL refinement package12 using least squares minimization. All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2. Hydrogen atoms were generally inserted at calculated positions and refined as riders. Those from the protonation sites, were first located using a Fourier difference map and then replaced with idealized ones as riders. The structures were checked for higher symmetry with help of the program PLATON.13Both in 1a and 1b the terminal phenyl group and some more atoms from the chain, until the NH, present disorder and have been modelled over two complimentary positions. Regarding 2, the central phenyl ring, as well as de hydroxyl is disordered over two complimentary positions. Also in 2, the isobutyrate and the acetonitrile are also disorder over two positions.
In 1b, the isopropanol solvate could not be properly modelled so, a solvent mask was calculated using Olex2.11 35 electrons were found in a volume of 139 Å3 in 1 void per unit cell. This is consistent with the presence of 0.5[C3H8O] per formula unit, which account for 34 electrons per unit cell.
A summary of comparative crystallographic data is given in Table 1. The full key crystallographic information is given in ESI.† CCDC 2405158, 2405164 and 2405169 contain the supplementary crystallographic data for this paper.
| Crystal form | 1a | 1b | 2 |
|---|---|---|---|
| T (K) | 100 | 100 | 100 |
| System | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a (Å) | 10.6139(6) | 7.0067(9) | 7.9585(6) |
| b (Å) | 13.0164(11) | 11.6243(15) | 9.7595(8) |
| c (Å) | 13.2023(8) | 17.481(2) | 18.6559(16) |
| α (°) | 111.948(3) | 100.627(5) | 81.610(4) |
| β (°) | 95.785(2) | 97.557(5) | 88.461(4) |
| γ (°) | 114.007(2) | 98.458(5) | 83.493(3) |
| Vol (Å3) | 1474.99(18) | 1365.6(3) | 1424.2(2) |
| R (%) | 8.48 | 8.89 | 8.68 |
| CCDC | 2405169 | 2405158 | 2405164 |
2.7. Theoretical methods
The X-ray geometries were used to analyze the energetic features of each salt at the PBE0-D3/def2-TZVP14–16 level of theory. Only the positions of the H-atoms were optimized. The Gaussian-16 program17 has been used for the energetic calculations and generation of the wavefunctions. The QTAIM18 and NCIplot analyses19 have been performed using the AIMAll program at the same level of theory.20 For the NCIplot analysis, the following setting were used, S = 0.45, ρ cut-off = 0.04 a.u., color scale −0.035 ≤ sign(λ2)ρ ≤ −0.035 a.u. The H-bond energies were estimated using the values of the potential energy density and the methodology proposed in the literature.213. Results and discussion
3.1. Experimental
As a result of the solid forms screening, new multicomponent forms have been obtained with two coformers and different anhydrous and/or solvates of these new forms have been isolated and characterized by means of DSC, 1H-NMR, PXRD and TGA.For mirabegron and orotic acid salt, two different solvates have been obtained with (mirabegron:orotic acid:solvent) stoichiometries of (1:1:0.5) and (1:1:1) for ipOH and acetone respectively, both determined from SCXRD analysis.
The ipOH solvate crystallizes in the triclinic P space group with one mirabegron molecule, one orotic acid molecule and half molecule of isopropanol (50% disorder) in the asymmetric unit (Z′ = 1; Z = 2), Fig. 1.
 | ||
| Fig. 1 Ellipsoid representation (50% probability) for mirabegron:orotic acid ipOH solvate crystal structure. All hydrogens were omitted for clarity. | ||
The acetone solvate crystallizes in the triclinic P space group with one mirabegron molecule, one orotic acid and one acetone molecule in the asymmetric unit (Z′ = 1; Z = 2), Fig. 2.
 | ||
| Fig. 2 Ellipsoid representation (50% probability) for mirabegron:orotic acid acetone solvate crystal structure. All hydrogens were omitted for clarity. | ||
Also for mirabegron and orotic acid, a acetonitrile solvate has been obtained by reaction crystallization in acetonitrile (1:1:0.25 stoichiometry has been deduced according to 1H-NMR and TGA); a dioxane solvate has been obtained by reaction crystallization in dioxane (1:1:1 stoichiometry has been deduced according to 1H-NMR and TGA); and a hydrate has been obtained by slurry in H2O (a 1:1.5:1.5 stoichiometry has been deduced according to 1H-NMR and TGA analysis).
Moreover, for mirabegron and isobutyric acid salt, an anhydrous form has been obtained by slurry in acetonitrile (1:1 stoichiometry has been deduced from 1H-NMR); a THF solvate has been obtained by slurry in THF (a 1:1:0.5 stoichiometry has been deduced from 1H-NMR); and a acetonitrile solvate has been obtained by slow evaporation in acetonitrile (a 1:1:1 stoichiometry has been determined from SCXRD analysis). The acetonitrile solvate of the mirabegron/isobutyric acid salt crystallizes in the triclinic P space group with one mirabegron molecule, one isobutyric acid molecule and one acetonitrile molecule in the asymmetric unit (Z′ = 1; Z = 2), Fig. 3.
 | ||
| Fig. 3 Ellipsoid representation (50% probability) for mirabegron:isobutyric acid acetonitrile solvate crystal structure. All hydrogens were omitted for clarity. | ||
The characterization of all the forms is included in ESI.† The analysis of the crystal structures confirmed the proton transfer as expected since orotic acid has a pKα = 2.07 (ref. 22) and isobutyric acid has a pKα = 4.84.23
Since the only crystal structure reported for mirabegron is the anhydrous form α (CCDC refcode: HOCHAM) we analyzed this structure from a supramolecular chemistry point of view in order to determine what relevant interactions present in the neutral form are maintained or removed in our new salts. Mirabegron form α is a triclinic structure (P1) with two independent molecules in the asymmetric unit interacting through a strong array of hydrogen bonds of different nature. On the one hand, the thiazol moieties establish self-complementary R22(8) dimers while both amide⋯amide and alcohol⋯amine head-to-tail infinite chains are packed in a zig-zag motif, with no relevant or very weak aromatic interactions present in the structure (Fig. 4).
 | ||
| Fig. 4 Crystal cell of mirabegron form α (CCDC refcode: HOCHAM) and an schematic description of hydrogen bonds present in its structure. | ||
Interestingly, none of the mentioned supramolecular synthons are shown by any of the three new structures reported in this work. This is essentially due to the fact that on the one hand the proton transfer to the amine group precludes the formation of the alcohol⋯amine interaction and on the other hand the presence of the anionic contraction drives the formation of stronger charge assisted hydrogen bonds with both the thiazol and the amide groups. The study of these interactions is presented in the following sections.
3.2. DFT calculations
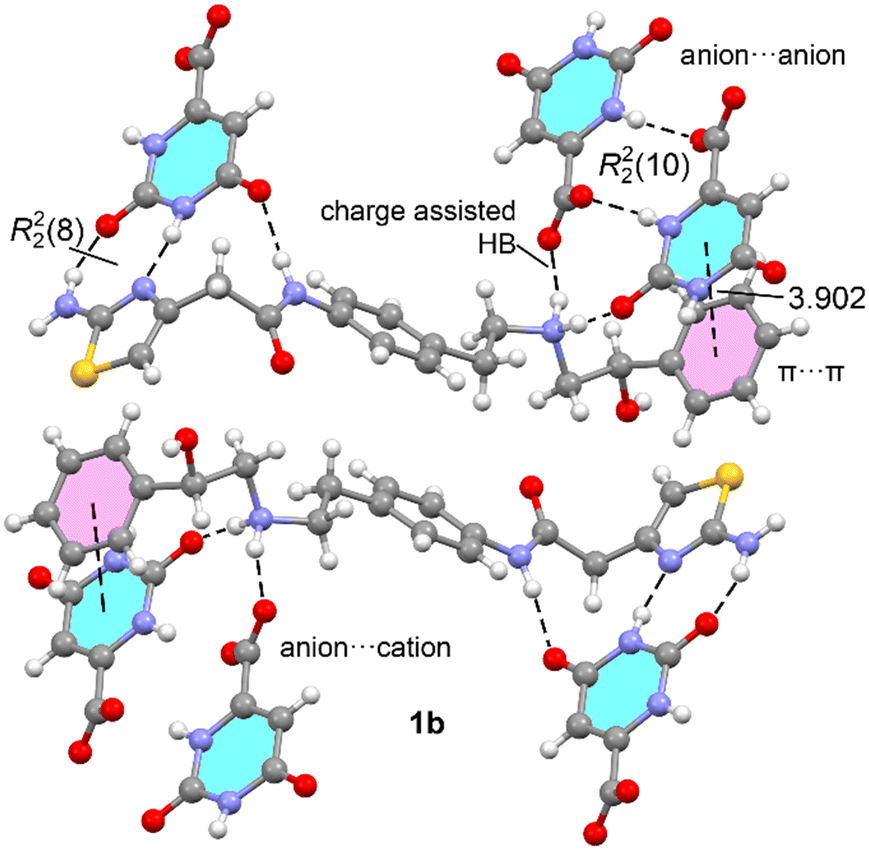
The DFT analysis investigates the hydrogen bonding patterns in the solid-state two structures of mirabegron–orotate (1a,b) and mirabegron–butyrate (2) co-crystals. As shown in Fig. 5–7, the orotate and isobutyrate anions interact with the ammonium group via the carboxylate, forming charge-assisted hydrogen bonds. Notably, in compound 1a,b, the orotate forms centrosymmetric homodimers through R22(8) synthons in 1a and R22(10) synthons, creating anion⋯anion hydrogen-bonded dimers. Additionally, the orotate interacts with the 2-(2-aminothiazol-4-yl)acetamide fragment through three hydrogen bonds, resulting in an anion⋯cation hydrogen-bonded dimer in both 1a and 1b salts. Different π-stacking interactions are observed in both salts. That is, in 1a it is between the orotate and the thiazole rings, with a centroid-to-centroid distance of 3.659 Å whilst in 1b it is between the ororate and the phenyl ring (see Fig. 5). | ||
| Fig. 5 Partial view of the X-ray solid structure of 1a. Distance in Å. | ||
 | ||
| Fig. 6 Partial view of the X-ray solid structure of 1b. Distance in Å. | ||
 | ||
| Fig. 7 Partial view of the X-ray solid structure of 2. Distances in Å. | ||
In compound 2, the anion plays a simpler role as a hydrogen bond acceptor, connecting the cations via a network of OH⋯O and N–H⋯O hydrogen bonds, including the formation of an R22(9) synthon between the protonated ammonium and the adjacent hydroxy group (see Fig. 7). Additionally, the central aromatic rings of mirabegron engage in π⋯π stacking interactions. The energetic characteristics of these interactions were calculated and compared. To further elucidate these interactions, a combination of QTAIM and NCIplot analyses was performed, effectively revealing noncovalent interactions in real space.
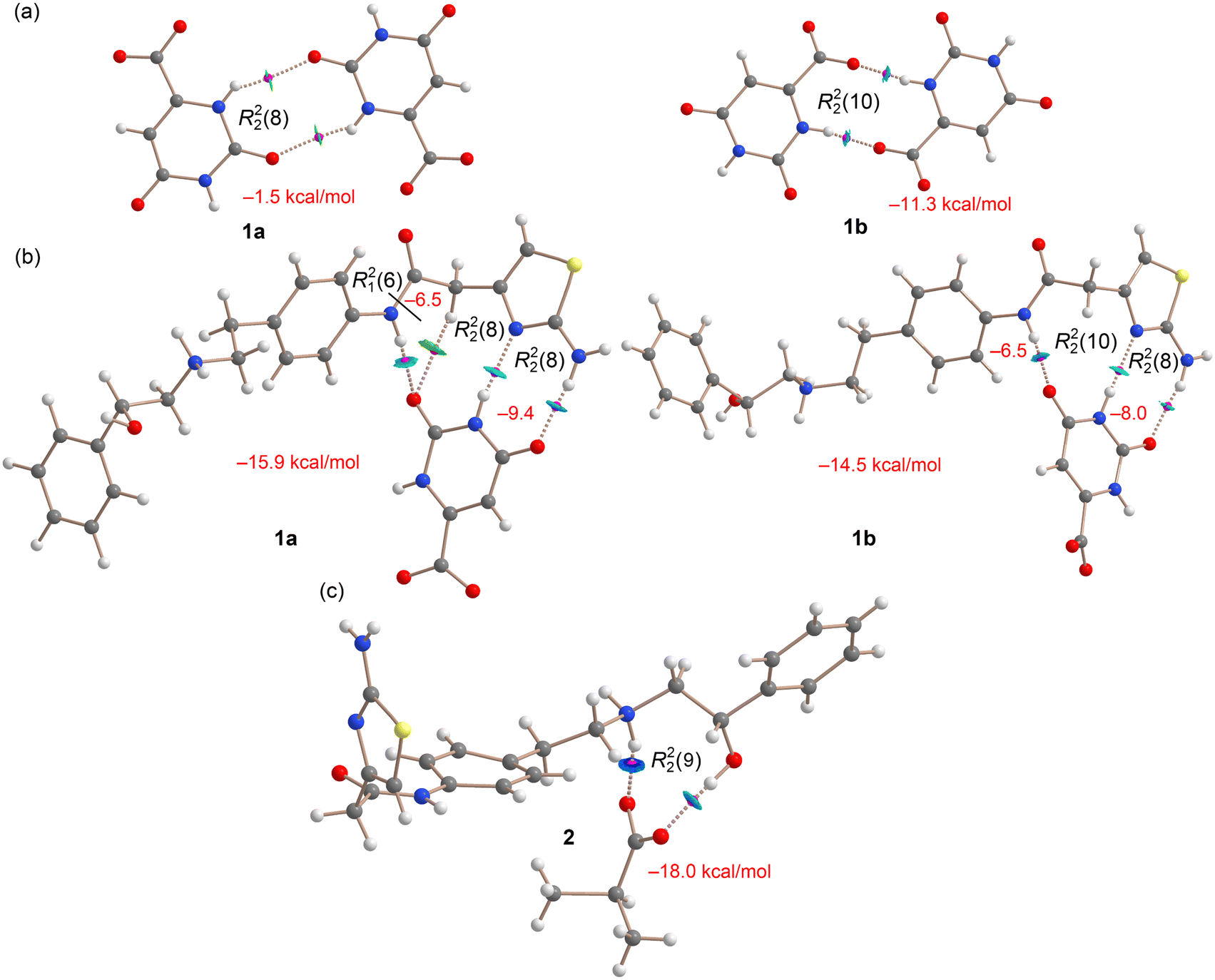
Fig. 8a and b shows the QTAIM analysis of the anion⋯anion and anion⋯cation hydrogen bonding dimers in compounds 1a,b. In both cases, the energies of the hydrogen bonds have been evaluated using the potential energy density at the bond critical point (BCP, magenta spheres in Fig. 8). This method is particularly useful for charged systems like ion pairs, as it eliminates the influence of pure Coulombic attraction or repulsion between fragments of opposite or the same charge, respectively. For the anion⋯anion dimers (Fig. 8a), the centrosymmetric R22(8) in 1a and R22(10) in 1b synthon is characterized by two BCPs and bond paths (dashed lines) connecting the oxygen and hydrogen atoms. Additionally, the interaction is further characterized by small reduced density gradient (RDG) isosurfaces, coincident with the location of the BCPs. In this manuscript, we have used blue color to denote strong interactions and green for weaker ones in the NCIplot color scheme. The small size and green color of the RDG isosurfaces that characterize the hydrogen bonds in the R22(8) anion⋯anion dimer correlate well with the low interaction energy calculated for this dimer in 1a (−1.5 kcal mol−1). In contrast, the R22(10) anion⋯anion dimer exhibits a significantly higher interaction energy (−11.3 kcal mol−1), consistent with the blue color of its RDG isosurfaces and the presence of a carboxylate group as the H-bond acceptor, which is a much stronger H-bond acceptor compared to a carbonyl group.
 | ||
| Fig. 8 Overlapped QTAIM and NCIplot analyses of the anion⋯anion (a) and anion⋯cation (b) dimers in mirabegron–orotate salt (1) and mirabegron–isobutyrate salt (2) anion⋯cation dimer (c). The energies of the synthons are given in kcal mol−1. | ||
In Fig. 8b, the QTAIM/NCIplot analyses of the cation⋯anion dimers are presented. For both 1a and 1b, the analysis identifies two NH⋯O and one NH⋯N hydrogen bonds, characterized by their corresponding BCPs, bond paths, and blue RDG isosurfaces, indicating their strong nature. Additionally, only for 1a the QTAIM/NCIplot analysis reveals the existence of an additional, much weaker CH⋯O interaction, as indicated by the green RDG isosurface. Therefore, the cation⋯anion dimer can be viewed as consisting of two fused R22(8) and one R21(6) synthons in 1a and two fused R22(8) and R22(10) synthons in 1b. It is observed that the R22(8) synthon in the anion⋯cation dimer in 1a (−9.4 kcal mol−1) is significantly stronger than the similar R22(8) synthon in the anion⋯anion dimer (−1.5 kcal mol−1), due to the shorter distances in the anion⋯cation dimer resulting from its ion-pair nature. The associated energy of the R21(6) synthon is −6.5 kcal mol−1 in 1a, identical to the NH(amidic)⋯O H-bond in 1b (−6.5 kcal mol−1). The total interaction energy of the dimers are −15.9 kcal mol−1 and −14.5 kcal mol−1 in 1a and 1b, respectively.
In Fig. 8c, the QTAIM/NCIplot analysis of the cation⋯anion dimer in compound 2 is depicted. The analysis reveals the presence of one NH⋯O and one OH⋯O hydrogen bond, each characterized by their corresponding BCPs and bond paths. The NH⋯O hydrogen bond is represented by a dark blue RDG isosurface, indicating its greater strength compared to the OH⋯O bond, which appears with a blue disk-shaped isosurface. Together, these hydrogen bonds form an R22(9) synthon with a binding energy of −18.0 kcal mol−1, which is notably stronger than the R22(8) synthons observed in compound 1. This increased strength is attributed to the shorter NH⋯O distance in the anion⋯cation dimer of compound 2, though the total interaction energy remains similar between both compounds.
The charge assisted H-bond and π-stacking interactions have been also analyzed. The QTAIM analysis of the tetrameric assemblies of 1a and 1b is shown in Fig. 9. For the charge-assisted hydrogen bonds between the ammonium and carboxylate groups in 1a, the QTAIM/NCIPlot analysis reveals a combination of a very strong NH⋯O interaction and a much weaker CH⋯O contact, resulting in a total interaction energy of −9.0 kcal mol−1. This value is very similar to that of the R22(8) synthon in the anion⋯cation dimer depicted in Fig. 8b, underscoring the exceptionally strong nature of the charge-assisted hydrogen bond. The analysis also identifies weak CH⋯S hydrogen bonds (−0.6 kcal mol−1) between the cationic coformers.
 | ||
| Fig. 9 Overlapped QTAIM and NCIplot analyses of the tetrameric assemblies observed in the mirabegron–orotate salts 1a (top) and 1b (bottom). The energies of the H-bonds are given in kcal mol−1. The ion-pair dimer used to estimate the π-stacking energy is depicted as a chemical diagram. | ||
The QTAIM/NCIPlot analysis confirms the presence of π-stacking interactions, as evidenced by two BCPs and bond paths interconnecting the orotate and thiazole rings. This interaction is further highlighted by an extended RDG isosurface that encompasses much of the π-systems. To quantify the contribution of the π-stacking interactions and compare them to the hydrogen bonds, we calculated the formation energy of the assembly as a dimer, using the hydrogen bond ion pair shown in Fig. 9 (chemical diagram) as the monomer.
In this monomer, the strong hydrogen bonds are pre-formed, so the dimerization energy reflects only the contributions from both π-stacking and weak CH⋯S contacts. The computed dimerization energy for 1a is −17.0 kcal mol−1. After isolating the contribution from CH⋯S contacts, this value reduces to −15.8 kcal mol−1. Thus, each π-stacking interaction can be estimated to have an energy of −7.9 kcal mol−1, which is weaker than the charge-assisted hydrogen bonds and the R22(8) cation⋯anion synthon, but stronger than the R21(6) synthon. This energetic analysis underscores the significance of π-stacking in the solid-state structure of the 1a mirabegron–orotate salt.
A similar analysis was conducted for 1b, revealing that the orotate establishes a π-stacking interaction with the phenyl ring of mirabegron while concurrently forming a charge-assisted H-bond (−5.2 kcal mol−1). In this form of the mirabegron–orotate salt, the thiazole rings of two mirabegron molecules also engage in π-stacking interactions, characterized by two BCPs, bond paths, and a green RDG isosurface spanning the entire space between the rings. The contribution of π-stacking in 1b was estimated using the same procedure as for 1a, where the dimerization energy of the salt was computed to yield the tetramer. The interaction energy of −5.0 kcal mol−1 corresponds to the energy of the thiazole⋯thiazole π-stacking, which is weaker than the orotate⋯thiazole stacking observed in 1a.
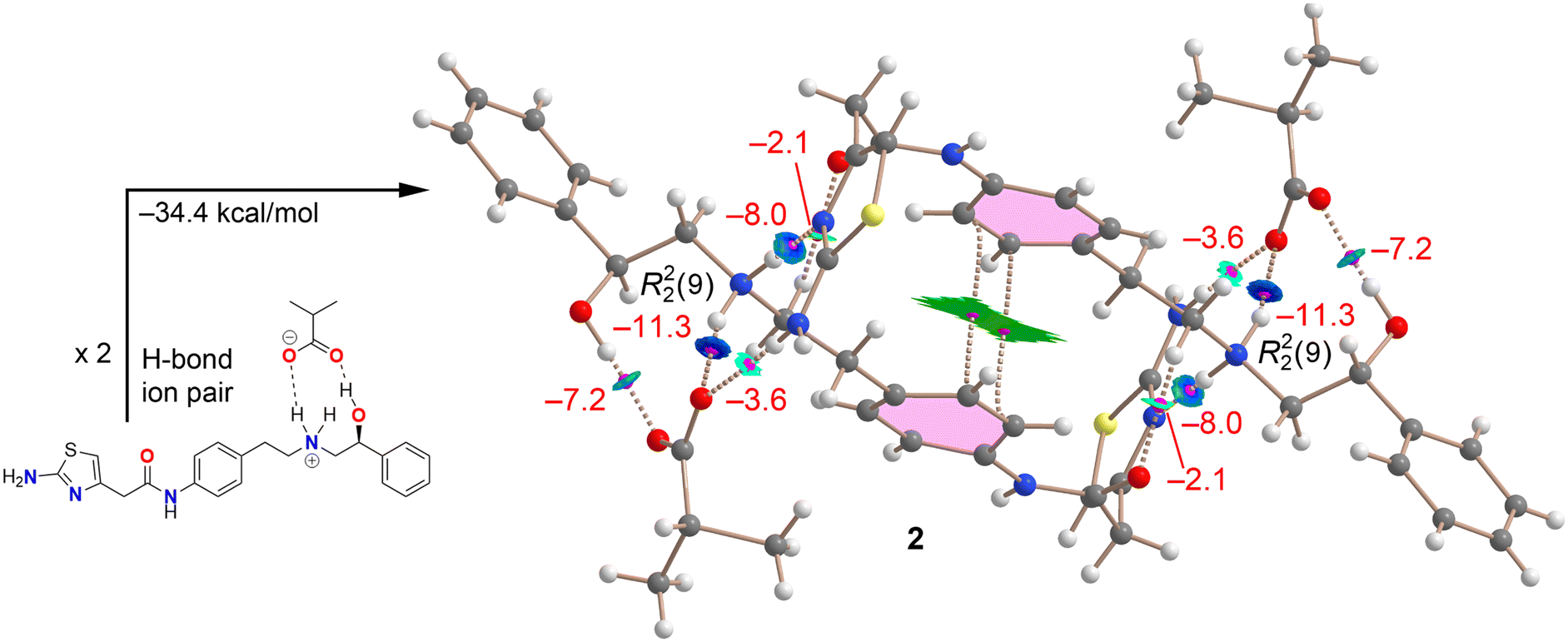
A similar study was conducted for compound 2, with the QTAIM analysis of the tetrameric assembly presented in Fig. 10. As previously described, the R22(9) synthon is notably strong due to the charge-assisted NH⋯O hydrogen bond. The analysis also uncovers additional hydrogen bonds, including a moderately strong NH⋯O H-bond (bluish RDG isosurface) between the anion and the NH2 group of the thiazole ring (−3.6 kcal mol−1), and a strong NH⋯N bond (dark blue RDG isosurface) between the ammonium group and the N-atom of the thiazole group (−8.0 kcal mol−1). Additionally, a weaker CH⋯O H-bond is formed between the amidic O-atom and the alkyl chain (−2.1 kcal mol−1). Altogether, this hydrogen bonding network contributes −64.4 kcal mol−1, highlighting the significant influence of this network in the X-ray solid state structure of compound 2.
 | ||
| Fig. 10 Overlapped QTAIM and NCIplot analyses of the tetrameric assembly observed in the mirabegron–butyrate (2) cocrystal. The energies of the H-bonds are given in kcal mol−1. The ion-pair dimer used to estimate the π-stacking energy is depicted as a chemical diagram. | ||
The QTAIM/NCIplot analysis also confirms the presence of π-stacking interactions, characterized by two BCPs and bond paths connecting the phenyl rings, along with an extended RDG isosurface encompassing the π-systems. As with compound 1, the dimerization energy was computed to estimate the contribution of the π-stacking interaction. Using the hydrogen bonded ion pair assembly depicted in Fig. 8 (bottom) as the monomer, the calculated dimerization energy for the assembly was −34.4 kcal mol−1. After subtracting the contributions from the additional NH⋯O, NH⋯N, and CH⋯O contacts, the π-stacking interaction energy was found to be −7.1 kcal mol−1, comparable to that in compound 1 (−7.9 kcal mol−1) and weaker than the charge-assisted hydrogen bonds.
These interactions have been compared with those observed in the only reported crystalline form of mirabegron (HOCHAM). As previously mentioned, the thiazol moieties form self-complementary R22(8) dimers, which have been analyzed using a combined QTAIM/NCIplot approach (see Fig. 11a). The QTAIM analysis reveals the presence of characteristic BCPs and bond paths interconnecting the H and N-atoms, forming an eight-membered supramolecular ring. The blue color of the RDG isosurfaces further supports the strong nature of this synthon. Notably, the computed formation energy of this dimer is −12.2 kcal mol−1, a significant value considering that the interaction occurs between two neutral mirabegron molecules. This synthon is stronger than the R22(8) synthons of 1a and 1b (vide supra) and similar to the R22(10) synthon of 1b (see Fig. 8a).
 | ||
| Fig. 11 Overlapped QTAIM and NCIplot analyses of the R22(8) synthon (a) and head-to-tail infinite chains (b) in dimers of mirabegron (HOCHAM). The energies of the synthons are given in kcal mol−1. | ||
In Fig. 11b, a similar analysis is presented for a dimer extracted from the infinite head-to-tail chain. The QTAIM/NCIplot analysis confirms the presence of hydrogen bonds between the amide groups, as well as between the amino and hydroxyl groups (OH⋯N). Notably, the OH⋯N hydrogen bond is stronger (−6.1 kcal mol−1) than the NH⋯O (amide⋯amide) hydrogen bond (−2.3 kcal mol−1), which aligns with its slightly shorter distance (2.01 Å vs. 2.11 Å) and the superior electron-donating ability of the secondary amine. Both hydrogen bonds also feature secondary CH⋯O contacts, where an aromatic CH bond acts as a hydrogen bond donor. The energies associated with these contacts are modest (approximately −1 kcal mol−1, see Fig. 11b). Additionally, the QTAIM/NCIplot analysis reveals a weak hydrogen bond between the aminothiazole moieties (NH⋯S), further contributing to the stabilization of the assembly. Overall, the total binding energy of the dimer is −12.0 kcal mol−1, comparable to that of the self-complementary R22(8) dimer.
4. Conclusions
The computational study of the new crystal structures of mirabegron salts with orotic acid and isobutyric acid has provided a comprehensive analysis of the various hydrogen bonding patterns and π-stacking interactions in the solid state of the mirabegron–orotate (1a, 1b) and mirabegron–isobutyrate (2) salts. Through the DFT analysis, we identified and characterized the significant noncovalent interactions, including charge-assisted NH⋯O hydrogen bonds, centrosymmetric R22(8) and R22(10) synthons in both anion⋯anion and anion⋯cation dimers, R22(9) synthons and π-stacking interactions. The QTAIM and NCIPlot analyses revealed the strengths and spatial characteristics of these interactions, confirming their roles in stabilizing the crystal structure. The charge-assisted NH⋯O hydrogen bonds were found to be particularly strong, with an energy of −9.0 kcal mol−1 in 1a −5.2 kcal mol−1 in 1b and −11.3 kcal mol−1 in 2, while the π-stacking interactions, estimated at −7.9 kcal mol−1 each in 1 and −7.1 kcal mol−1 in 2, also played a crucial role. These assemblies were compared to the neutral crystalline form of mirabegron, which also features energetically significant R22(8) dimers involving the aminothiazole rings. This detailed study is a new example that underscores the importance of combining non-covalent interactions of different strength in defining the supramolecular architecture of multicomponent assemblies and provides valuable insights for future research on similar systems.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the CCDC under CCDC numbers 2405169, 2405158 and 2405164, and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
The manuscript was collaboratively written by all authors, who have approved the final version. Rafel Prohens contributed to the study's conception, experimental work, and manuscript writing. Antonio Frontera contributed to the study's conception, computational work and manuscript writing. Rafael Barbas conducted experiments on crystal forms screening and characterization techniques. Miquel Barceló-Oliver analysed data on X-ray crystal structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MICIU/AEI of Spain (projects PID2020-115637GB-I00, PID2023-148453NB-I00 and PID2023-146632OB-I00 FEDER funds) for funding. We thank the CTI (UIB) for computational facilities and the CCiTUB for technical facilities. We also thank Interquim, S. A. (Barcelona, Spain) for providing Mirabegron. This article is dedicated to Dr. Mercè Font-Bardia on the occasion of her retirement. We would like to express our thanks to Dr. Font-Bardia for her continuing contribution in our research over many years.References
- C. R. Chapple, L. Cardozo, V. W. Nitti, E. Siddiqui and M. C. Michel, Neurourol. Urodyn., 2014, 33, 17–30 CrossRef CAS PubMed.
- P. Raut, M. Gambhire, D. Panchal and V. Gambhire, Pharm. Nanotechnol., 2021, 9, 120–129 CrossRef CAS PubMed.
- J.-H. An, C. Lim, A. N. Kiyonga, I. H. Chung, I. K. Lee, K. Mo, M. Park, W. Youn, W. R. Choi, Y.-G. Suh and K. Jung, Pharmaceutics, 2018, 10, 149–163 CAS.
- D. A. Patel and D. J. Patel, World J. Pharm. Pharm. Sci., 2018, 7, 973–991 CAS.
- S. Kawazoe, K. Sakamoto, Y. Awamura, T. Maruyama, T. Suzuki, K. Onda and T. Takasu, US7342117B2, 2008.
- V. Peddy and R. Boge, WO2012156998A2, 2012.
- J. Lin, J. Unge and T. Gonen, Adv. Sci., 2023, 10, 2304476–2304480 CrossRef PubMed.
- Y. Kasashima, K. Yoshihara, T. Yasuji, K. Sako, S. Uchida and N. Namiki, Chem. Pharm. Bull., 2016, 64, 1304–1309 CrossRef CAS PubMed.
- R. Barbas, A. Portell, C. A. Hunter, R. Prohens and A. Frontera, CrystEngComm, 2022, 24, 5105–5111 RSC.
- Bruker, APEX4, SAINT, and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–11 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CAS.
- AIMAll (Version 19.10.12), ed. T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CAS.
- E. P. Serjeant and B. Dempsey, Ionisation Constants of Organic Acids in Aqueous Solution, International Union of Pure and Applied Chemistry (IUPAC), IUPAC Chemical Data Series No. 23, Pergamon Press Inc., New York, New York, 1979 Search PubMed.
- G. Kortüm, W. Vogel and K. Andrussow, Dissociation Constants of Organic Acids in Aqueous Solution, International Union of Pure and Applied Chemistry, London, Butterworth, 1961 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cocrystal screening details and characterization of the new solid forms. CCDC 2405158, 2405164 and 2405169. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01228c |
| This journal is © The Royal Society of Chemistry 2025 |