
Boosting the oxygen vacancies in ZnO–Co3O4 composite by copper doping for bisphenol A abatement using persulfate†
Lei
Bai
 *ab,
Hui
Yan
a,
Jiao
Wang
c,
Qihang
Wu
a,
Guiling
Wang
ab,
Yi
Huang
c and
Bentian
Zhang
*ab
*ab,
Hui
Yan
a,
Jiao
Wang
c,
Qihang
Wu
a,
Guiling
Wang
ab,
Yi
Huang
c and
Bentian
Zhang
*ab
aCollege of Chemistry and Materials Engineering, Anhui Science and Technology University, Bengbu, Anhui 233030, China. E-mail: baileiwj2014@163.com; zhangbt@ahstu.edu.cn
bAnhui Province Quartzs and Purification and Photovoltaic Glass Engineering Research Center, Chuzhou, 233100, China
cCollege of Food Engineering, Anhui Science and Technology University, Fengyang, Anhui 233031, China
First published on 20th November 2024
Abstract
Through surface leaching and ion exchange, nest-like Cu–Co3O4 and Cu–ZnO–Co3O4 composites were obtained from solid ZIF-67 and hollow ZIF-8–ZIF-67, respectively. Their characterizations suggested that the ratio of oxygen vacancies/lattice oxygen in Cu–ZnO–Co3O4 was much higher (1.61) than that of Cu–Co3O4 (0.47). The Cu–ZnO–Co3O4 composite exhibited an ultra-high persulfate (PDS) activation ability and could degrade the target organic contaminant molecule bisphenol A (BPA) in six minutes under the reaction conditions of 20 mg L−1 BPA, 0.1 g L−1 PDS and 0.1 g L−1 Cu–ZnO–Co3O4, demonstrating a degradation rate constant (k) of 0.84 min−1, which was over 28-fold higher than that of the reference, Cu–Co3O4 (0.03 min−1). Notably, the robust reusability of the sample was also established based on three successive runs. The mechanistic study suggested that the influence of the active species derived from PDS followed the below order: 1O2 > HO˙ > O2˙− > SO4˙−. The robust catalytic activity of Cu–ZnO–Co3O4 could be ascribed to the rich oxygen vacancies, small size of active particles and the porous structure.
1. Introduction
Bisphenol A (BPA) is a kind of stable organic compound, which could cause serious effect on human body when exposed for ling time even under low concentration.1,2 BPA mainly leaches from improperly treated plastic products and enters the environment via wastewater, pollutants, surface water, and groundwater from plastic production industries.3,4 Stringent control and disposal of this compound are of great importance. Compared with the homogeneous catalytic degradation achieved by cobalt ions and iron ions, corresponding transition metal oxide catalysts are considered as promising catalysts for BPA elimination through advanced oxidation processes with persulfates due to several advantages, such as desirable activity and easy recovery.5–8 Among metal oxides, Co3O4 and its derived composites are well-established for their performance in the degradation of BPA under different conditions.9–13 However, previous works suggest that peroxymonosulfate (PMS) is a commonly used oxidation agent for BPA degradation in comparison with persulfate (PDS) in the presence of cobalt-oxide-based catalysts although PDS costs much less than PMS.14 This is mainly because of the poor PDS activation by Co3O4 due to the relatively higher O–O bond energy.15 Thus, the improvement of persulfate activation both from the catalyst development and method viewpoints has attracted special interest and attention, for instance, ferrite spinel and carbon-based composites as catalysts as well as activation by an electrochemical process and ultrasonic irradiation.16,17On the other hand, Lai et al. found that in the presence of PMS, the molecular structure of organic contaminants has a profound influence on the degradation efficiency.18 Besides, the ionization potential of pollutants greatly affects the degradation efficiency, and it has been shown that organic pollutants with ionization potential values lower than 9.0 can be degraded using PDS-based systems.19 Due to the lower cost and higher stability of PDS, the preparation of transition metal oxide catalysts with highly efficient PDS activation could be of great interest and importance for material science and wastewater treatment.
In this work, a hollow bimetallic zeolitic imidazolate framework (ZIF-8–ZIF-67) was employed as a precursor along with copper nitrate to generate the copper (Cu)-doped ZIF-8–ZIF-67. After calcination, a unique Cu–ZnO–Co3O4 composite with ultra-high PDS activation capability for BPA degradation was obtained. The catalytic experiments confirmed that the apparent degradation rate constant (k) was over 28- and 5-fold higher than those of the references Cu–Co3O4 and ZnO–Co3O4 without Cu doping, respectively. The control experiments and corresponding analysis suggested a composite mechanism for the oxidation reaction, involving non-radical and radical processes. The boosted catalytic activity of Cu–ZnO–Co3O4 towards BPA degradation with PDS could be ascribed to the rich oxygen vacancies, small active particles and the porous structure.
2. Experimental methods
2.1 Synthesis of samples
Solid ZIF-67 and hollow ZIF-8–ZIF-67 were synthesized according to the method described in previous work.20 Then, 120 mg ZIF-67 or ZIF-8–ZIF-67 were dispersed in 10 mL ethanol under ultrasonication. A 2 mL ethanol solution containing 24 mg Cu(NO3)2·3H2O was added to the above mixture and stirred for 6 h at 298 K. The solids were collected by centrifugation and dried at 333 K overnight before further use. Finally, the solids were calcined in the air at 603 K for 2 h with a heating rate of 1 K min−1. These samples were denoted as Cu–Co3O4 and Cu–ZnO–Co3O4, respectively. Moreover, Co3O4 and ZnO–Co3O4 were synthesized by the direct calcination of ZIF-67 and ZIF-8–ZIF-67. For Mn- and Ni-doped samples, the synthesis procedure was the same, except 0.1 mM of the corresponding nitrate was used.2.2 Characterization
X-Ray diffraction (XRD) was performed on an XRD-6100 X-ray diffractometer (XRD, SHIMADZU LIMITED Corp.) from 10° to 80° with a scanning rate of 10° min−1. Transmission electron microscopy (TEM) was performed using a JEOL JEM-2010. Field-emission scanning electron microscopy (FE-SEM) was performed on a Hitachi-4800. High-resolution transmission electron microscopy (HRTEM) and high-angle annular dark-field scanning-transmission electron microscopy (HAADF-STEM) were performed on JEOL 2100 operated at 200 kV. The X-ray photoelectron spectra (XPS) were obtained using a Thermo ESCALAB 250 instrument with a monochromatic Al Kα (hv = 1486.6 eV) X-ray source. The metal content was determined by inductively coupled plasma optical emission spectrometry (ICP-AES) analyses performed on an Agilent ICP-OES 720ES. Chronoamperometry and electrochemical impedance spectroscopy (EIS) were performed using a three-electrode system on a CHI 660E dual-channel electrochemical workstation. During the test, a platinum wire and a saturated calomel electrode (SCE) were used as the counter and reference electrodes, respectively. Ultraviolet-visible (UV-vis) absorption spectra of the solutions were recorded on a Shimadzu UV-2600 spectrophotometer. The electron paramagnetic resonance (EPR) measurements were carried out on a Bruker A300 spectrometer (Bruker, Germany) with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethyl-4-piperidone (TEMP) as the free radical scavengers. Total organic carbon (TOC) analysis was performed using a Shimadzu TOC-4200. Nitrogen adsorption–desorption isotherms were measured using a Micromeritics ASAP 2010 instrument. The samples were degassed for 4 h under a vacuum at 473 K before measurement. The BET method was used to calculate the surface areas. The pore size distributions were derived from the adsorption branches of the curves using the Barrett–Joyner–Halenda (BJH) method.2.3 Catalytic assays
Typically, in a 100 mL flask, 5 mg solid was dispersed in 50 mL 20 mg L−1 BPA solution under ultrasonic processing for 5 min. Then, the flask was put into a water bath at 298 K and stirred for 10 min to obtain the adsorption equilibrium. Later, 5 mg PDS was added into the solution under stirring. At a certain time, 1 mL reaction solution was drawn into a centrifuge tube containing 1 mL methanol. After centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min, BPA in the solution was analyzed by UV-vis spectroscopy or high-performance liquid chromatography.
000 rpm for 3 min, BPA in the solution was analyzed by UV-vis spectroscopy or high-performance liquid chromatography.
To determine the active species, ethanol (EtOH), tert-butanol (TBA), benzoquinone (BQ) and L-histidine (His) were selected to quench the sulfate radical anion (SO4˙−), hydroxyl radical (˙OH), superoxide anion radical (O2˙−) and single oxygen (1O2), respectively. To investigate the influence of pH, sodium hydroxide and sulfuric acid were used to adjust the pH of the solution. For evidencing the stability of Cu–ZnO–Co3O4 and in order to decrease the influence of the loss of catalyst during the recovering and washing, 10 mg solid and 5 mg PDS were used. After each run, the catalyst was recovered by centrifugation and washed once with distilled water before further use.
Chronoamperometry and electrochemical impedance spectroscopy (EIS) were carried out using a three-electrode system on a CHI 660E dual-channel electrochemical workstation, where a platinum wire and a saturated calomel electrode (SCE) were employed as the counter and reference electrodes, respectively. The working electrodes were prepared by dispersing 2 mg of the solids in a 0.5 mL mixture composed of 0.49 mL DMF and 0.01 mL Nafion (5 wt%) with ultrasonic treatment for 1 h. Then, 10 μL of each ink was dropped on a glass carbon electrode (5 mm diameter) and dried naturally. The EIS analysis of the prepared catalysts was performed in a 100 mM Na2SO4 + 5 mM K3Fe[(CN)6] solution. Chronoamperometry was performed in 30 mL of 0.5 M Na2SO4 by adding 1 mL of 20 mg L−1 BPA and 1 mL solution containing 5 mg PDS, respectively.
3. Results and discussion
First, the ZIF-67, Cu/ZIF-67, ZIF-8–ZIF-67 and Cu/ZIF-8–ZIF-67 solids were investigated by XRD. Compared with the pattern of ZIF-67, after interaction with copper ions, no obvious change was noticed, possibly due to the low amount and small size of the copper species. Similar phenomena were observed in the cases of ZIF-8–ZIF-67 and Cu/ZIF-8–ZIF-67. Besides, no peaks corresponding to cobalt hydroxide, copper hydroxide and zinc hydroxide were noticed, indicating that no obvious hydrolysis occurred under these conditions, as suggested by the XRD patterns in Fig. S1.† After calcination, ZIF-67 and Cu/ZIF-67 were successfully transformed to Co3O4-based solids. As shown in Fig. 1A, peaks arising from the (111), (220), (311), (400), (422), (511) and (440) crystal planes of Co3O4 (JCPDS no. 43-1003) were obtained both for Co3O4 and Cu–Co3O4. No peak of CuO, Cu2O or Cu was noticed in the Cu–Co3O4, which possibly resulted from the very small size or the incorporation of less amount of Cu into the Co3O4. The ICP-AES data evidenced that the content of Cu in Cu–Co3O4 was about 12.2 wt%. For ZnO–Co3O4 derived from ZIF-8–ZIF-67, in addition to the peaks of Co3O4, new peaks were seen (marked by stars) at 34.6°, 47.5°, 56.6°, 62.8° and 67.9° corresponding to the (002), (102), (311), (110), (103) and (112) planes of ZnO (JCPDS no. 36-1451), respectively. The broad peak located at about 20° was due to carbon, as observed in the previous work.20 In the XRD pattern of Cu-doped ZnO–Co3O4, no diffraction peaks corresponding to copper species were observed, and the Cu content was estimated to be 4.7 wt% by ICP-AES. A change in morphology during the transformation from ZIF-67 to Cu–ZnO–Co3O4 was observed by FE-SEM, as shown in Fig. 1B. ZIF-67 displayed a typical polyhedral structure, as shown in Fig. 1B-a. After ZIF-8 growth, the structure of ZIF-8–ZIF-67 was similar to ZIF-67 (Fig. 1B-b). However, after interaction with Cu2+, broken polyhedra were observed in Cu–ZIF-8–ZIF-67, indicating the ion exchange between Cu2+ and ZIF-8–ZIF-67 (Fig. 1B-c). After calcination, a porous nest-like sample was obtained, and the decomposition of organic ligands occurred during the thermal treatment (Fig. 1B-d). The other corresponding metal contents in the two solids were investigated by ICP-AES, and the data are listed in Table S1.† Clearly, the Cu–ZnO–Co3O4 composite had lower cobalt content compared with Cu–Co3O4. Besides, the BET (Fig. S2†) specific surface areas of Cu–ZnO–Co3O4 and Cu–Co3O4 were 47.1 and 24.6 m2 g−1, respectively. | ||
| Fig. 1 (A) XRD patterns of Co3O4 (a), Cu–Co3O4 (b), ZnO–Co3O4 (c) and Cu–ZnO–Co3O4 (d) as well as (B) SEM images of ZIF-67 (a), ZIF-8–ZIF-67 (b), Cu/ZIF-8–ZIF-67 (c) and Cu–ZnO–Co3O4 (d). | ||
The structure of Cu–ZnO–Co3O4 was further analyzed by TEM and HRTEM. As shown in Fig. 2A, a porous nest-like sample was observed, similar to that of ZnO–Co3O4 derived from ZIF-8–ZIF-67 shown in Fig. S3.† This observation confirmed that doping Cu2+ did not change the initial structure. The HRTEM image in Fig. 2B shows crystal distances measuring 0.243 and 0.248 nm, which can possibly be ascribed to the (311) crystal plane of Co3O4 (JCPDS no. 43-1003) and the (101) crystal plane of ZnO (JCPDS no. 36-1451), respectively. In addition, the diffraction peaks in the XRD pattern in Fig. 1A could be well-assigned to spinel Co3O4 and ZnO. No additional Cu phases were observed in the TEM and XRD analysis, suggesting that the Cu atoms were completely incorporated into the Co3O4 and ZnO lattice.21 The EDS spectrum evidenced the presence of Cu, Co and Zn, and on the other hand, the HADDF-STEM image together with the single element mapping and overlapping images (Fig. 2D–H) evidenced the homogeneous distribution of these metal elements. Moreover, for clarity, the O and N images are not presented, and their presence was confirmed by the subsequent XPS analysis.
 | ||
| Fig. 2 (A) TEM, (B) HRTEM, (C) HADDF-STEM image, (D) EDX image and the Co (E), Cu (F), Zn (G) element distribution as well as the overlapping image (H) of Cu–ZnO–Co3O4. | ||
The XPS analysis was performed to investigate the composition and chemical states of the elements in the composites. As shown in Fig. 3A, the peaks centered at 779.8 and 794.6 eV were ascribed to Co 2p3/2 and Co 2p1/2, respectively. A detailed analysis confirmed the presence of Co2+ and Co3+ in the composites, which is in accordance with the result from XRD. Moreover, the quantitative analysis revealed that the Co3+ content on the surface decreased from 0.504 in Cu–Co3O4 to 0.431 in Cu–ZnO–Co3O4 based on the estimated ratio of the area of Co3+ to the total area of Co2+ and Co3+. This suggests that the high content of Co2+ could facilitate the activation of PDS.22 The O 1s XPS profiles obtained for Cu–Co3O4 and Cu–ZnO–Co3O4 could be divided into three peaks at about 529 eV, 531 eV and 532 eV, which were assigned to the lattice oxygen (Olatt), oxygen vacancies (OVs) and adsorbed water molecules (Oads), respectively.23,24 By fitting the peaks, the amount of OVs was denoted by the ratio of OVs/Olatt peak area and it was found that the ratio of Cu–ZnO–Co3O4 (1.61) was much higher than that of Cu–Co3O4 (0.47). Clearly, the introduction of Cu ions significantly enhanced the oxygen vacancy in the Cu–ZnO–Co3O4. The above observation was further evidenced by the ESR spectra shown in Fig. 3C, which displays a symmetrical ESR signal with g = 2.003 (G = 3508), indicating that much more oxygen vacancies existed in Cu–ZnO–Co3O4 than in Cu–Co3O4. Moreover, in Fig. 3D, the peak at the binding energy of 934.9 eV can be attributed to Cu(II), while the peaks at 941.4 and 943.6 eV are the satellite peaks of Cu(II). The peak at binding energy of 933.6 eV may be ascribed to Cu(I) or Cu(0), since these two species were difficult to be deconvoluted in Cu 2p core level according to the previous work.25–27 However, taking into account of the experiment, the existence of Cu(0) was unlikely due to the calcination of the precursor in air. Residual nitrogen in the composites was also studied, as displayed in Fig. S4.† N 1s spectral deconvolution revealed peaks at 399.1, 400.1 and 400.9 eV for Cu–Co3O4 and at 398.1, 399.2 and 400.6 eV for Cu–ZnO–Co3O4. Despite the minor differences in the peak binding energy, previous works have indicated the presence of pyridinic N, pyrrolic N, and graphitic N or Co–Nx.28–31
 | ||
| Fig. 3 XPS spectra of Co 2p (A), O 1s (B), ESR spectra (C) and XPS spectrum of Cu 2p3/2 (D) of the samples. | ||
Subsequently, the solids were employed as catalysts for the degradation of BPA in the presence of PDS. As shown in Fig. 4A, a very low amount of BPA was eliminated by Co3O4 in the presence of PDS at a PDS-to-BPA molar ratio of 4.2:1. After the introduction of copper species, BPA elimination was promoted slightly to about 20% BPA in the presence of Cu–Co3O4 derived from Cu–ZIF-67, which was further evidenced by the k value of 0.03 min−1. As for ZnO–Co3O4, due to the small particle size of Co3O4, a desirable BPA degradation activity was observed, as suggested by 60% BPA elimination in six minutes with a k of 0.17 min−1. Notably, for Cu–ZnO–Co3O4, at the same time, nearly 100% BPA elimination and a significant increase in activity were observed, as proven by the k value of 0.84 min−1. Meanwhile, the TOC data suggested that after six minutes, the mineralization rate of BPA was 52%, that is, half of BPA was converted into harmless compounds. On the other hand, it was worth mentioning that the controlled experiment confirmed that copper oxide synthesized by calcination of copper nitrate was inactive for PDS activation and BPA degradation under the same condition. Moreover, it was confirmed that PDS was not able to degrade BPA without a catalyst and only about 8% of BPA in the solution was adsorbed on Cu–ZnO–Co3O4, demonstrating that the abatement of BPA was achieved by catalytic oxidation.
 | ||
| Fig. 4 BPA degradation in the presence of different catalysts, [0.1 g L−1 catalyst, 20 mg L−1 BPA, 0.1 g L−1 PDS] (A), under different additives [0.1 g L−1 catalyst, 20 mg L−1 BPA, 0.1 g L−1 PDS, additives: 0.15 M EtOH, 0.10 M TBA, 4.0 × 10−4 M BQ, 3.0 × 10−4 M His] (B), at different pH [0.1 g L−1 catalyst, 20 mg L−1 BPA, 0.1 g L−1 PDS] (C) with Cu–ZnO–Co3O4 and the reusability (D) of Cu–ZnO–Co3O4 for three successive runs [0.2 g L−1 catalyst, 20 mg L−1 BPA, 0.1 g L−1 PDS]. | ||
In order to know the active species responsible for BPA degradation, different additives were added to the reaction mixture to investigate their influence. As displayed in Fig. 4B, at the ethanol (EtOH)/PDS molar ratio of 250, the degradation BPA efficiency and k value were nearly not affected (Ct/C0 = 0.99 and 0.79 min−1, respectively). This observation is similar to the PDS consumption reported in the presence (26.5%) and absence (30.2%) of EtOH in a previous work.32 However, in the presence of TBA, these values decreased to 0.89 and 0.39 min−1, respectively. It is well-established in the literature that EtOH has high reactivity with both SO4˙− (k2 (EtOH, SO4˙−) = 1.6–7.7 × 107 L mol−1 s−1) and HO˙ (k2 (EtOH, HO˙) = 1.2–2.8 × 109 L mol−1 s−1), and the reaction rate constant of TBA with HO˙ (k2 (TBA, HO˙) = 3.8–7.6 × 108 L mol−1 s−1) is about 1000-fold higher than that with SO4˙− (k2 (TBA, SO4˙−) = 4.0–9.1 × 105 L mol−1 s−1).33 On the other hand, BQ and His were employed as scavengers of O2˙− and 1O2, respectively. The results in Fig. 4B suggest that the addition of a small amount of BQ and His led to a decrease in the k values to 0.46 and 0.32 min−1, respectively. The above data indicate that O2˙− and 1O2 also participated in the oxidation of BPA. Based on this observation, a composite mechanism including non-radical and radical pathways is proposed, and HO˙, O2˙−, and 1O2 play vital roles in BPA degradation rather than SO4˙−.
Furthermore, the influence of pH on the degradation efficiency was investigated at pH 3.2 and 11.6, respectively, as displayed in Fig. 4C. Reasonably, under the strong acid and basic conditions, the k values decreased to 0.39 and 0.06 min−1, respectively. Moreover, at pH 11.4, a higher electronic force could exist on the Cu–ZnO–Co3O4 surface compared with a pH 4.8, which is the zero potential point, as displayed in Fig. S5,† to repel the negative anions, leading to the obvious decrease in activity. At pH = 3.2, the decrease in catalytic activity is possibly due to the leaching of metal ions from the catalyst, as well as the hydrogen bond between H+ and O–O in PDS, which would inhibit the interaction between PDS and positively charged Cu–ZnO–Co3O4.34 The stability of the Cu–ZnO–Co3O4 was investigated by the catalytic tests for three successive runs and as shown in Fig. 4D, very slight decrease of the BPA removal content was noticed form 0.99 in the first run to 0.9 in the third run. Metal leaching in the solution was analyzed by ICP-AES, and 0.08 mg L−1 Cu2+, 0.22 mg L−1 Co2+ and 1.05 mg L−1 Zn2+ were found, indicating the robust stability of Cu–ZnO–Co3O4. To further evidence the advantage of this catalyst, some reported data were listed in Table S2.† It was suggested that under similar conditions, the present catalyst Cu–ZnO–Co3O4 demonstrated a much higher activity for BPA elimination in the presence of PDS. Finally, some control experiments for BPA degradation were performed using CuO, ZnO and Co3O4 derived from the nitrates calcined at the same temperature as Cu–ZnO–Co3O4, as well as with Cu–ZnO–Co3O4 washed in acid and base, respectively. The results presented in Fig. S6† suggest that CuO, ZnO and Co3O4 could not degrade BPA using PDS under the same conditions. Besides, the base- and acid-treated Cu–ZnO–Co3O4 samples showed k values of 0.66 and 0.06 min−1, respectively, suggesting that Cu–Co3O4 and the carbon species in the composite played vital roles in PDS activation and thus evidencing the synergistic effect of the structure and composition of the composite.
In order to confirm the active species generated by PDS and Cu–ZnO–Co3O4 during the reaction and verify the results of the control experiments, EPR measurements were performed, and the corresponding spectra are shown in Fig. 5A–C. In the absence of Cu–ZnO–Co3O4, no signal for the SO4˙−, HO˙, 1O2 and O2˙− was observed when only PDS was in the solution. After adding Cu–ZnO–Co3O4, signals corresponding to SO4˙− and HO˙ were clearly observed, as displayed in Fig. 5A. Notably, the intensity of HO˙ was much higher than that of SO4˙−, which is possible due to nucleophilic substitution, as reported in other systems.351O2 and O2˙− were also detected in the ESR experiment, as shown in Fig. 5B and C, in the presence of the as-obtained catalyst. In addition, nitroblue tetrazolium (NBT) was used as the probe for O2˙− during the reaction.36 The characteristic absorbance of NBT at about 260 nm was still observed for NBT–BPA–Cu–ZnO–Co3O4–PDS after adding PDS and stirring for 6 min, further confirming the participation of O2˙− in the degradation reaction, as shown in Fig. 5D. The results from the ESR and catalytic experiments collectively indicate that the PDS–Cu–ZnO–Co3O4 system involved a composite catalytic mechanism, and the HO˙, O2˙− and 1O2 species are the major reactive oxygen species involved in the degradation of BPA.
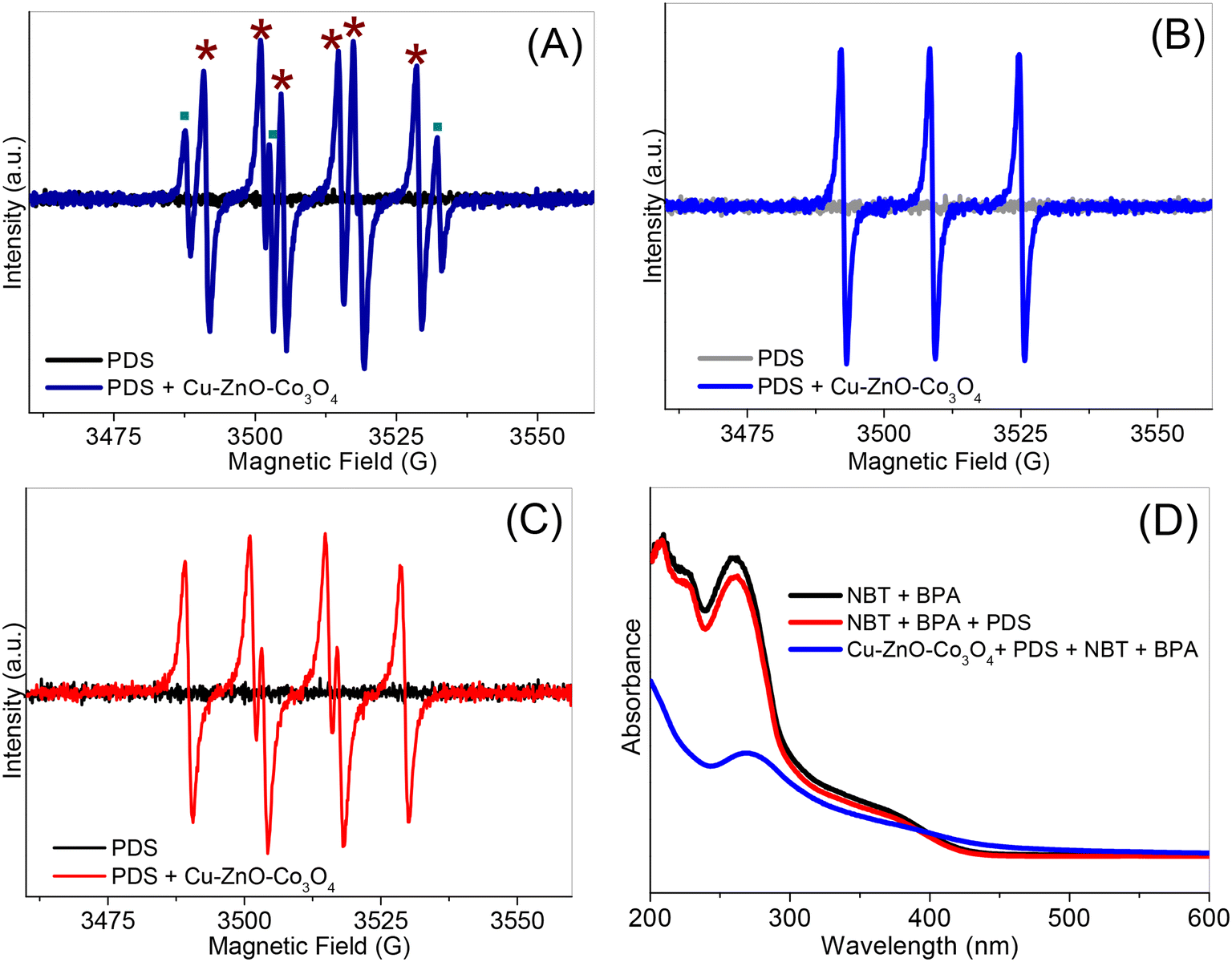 | ||
| Fig. 5 (A) ESR spectra of SO4˙− (■) and HO˙ (*), (B) 1O2, (C) O2˙− and (D) UV-vis spectra of the solutions under different conditions. | ||
Thus, according to the above observations and previous studies,34 the Co2+ and Cu+ species in Cu–ZnO–Co3O4 act as catalytic sites and activate the adsorbed S2O82− moiety to generate SO4˙−. Through nucleophilic substitution, HO˙ may also be generated from SO4˙− and water.37,38 In order to keep the charge balanced on the surface of the catalyst, the generated Co3+ would react with HO˙/SO4˙− and be reduced to Co2+. The oxygen vacancies (OV) in the catalyst will react with oxygen, Co3+, and electrons to produce O2˙− and 1O2. The above processes reduce Co3+ and Cu2+ to Co2+ and Cu+, respectively. Herein, the corresponding eqn (1)–(7) are presented below:
| S2O82− + Co2+ (Cu+) → Co3+ (Cu2+) + SO42− + SO4˙− | (1) |
| SO4˙− + H2O → HO˙ + HSO4− | (2) |
| Co3+ + S2O82− → Co2+ + S2O8˙− | (3) |
| Co2+ + HO˙/SO4˙− → Co3+ + OH−/SO42− | (4) |
| OV + O2 + e− → O2˙− | (5) |
| O2 + 4e− → 2Olatt2− | (6) |
| 2Olatt2− + Co3+ (Cu2+) → 1O2 + Co2+ (Cu+) + 3e− | (7) |
Electrochemical analysis was also employed to investigate the electron transfer process at the interface of the catalyst during PDS activation. It is known that EIS can be used to study the charge transfer and ion diffusion processes at the electrode surface.39 The EIS spectrum of Cu–ZnO–Co3O4 in Fig. 6A displays a smaller arc radius than that of Cu–Co3O4 in the high-frequency range, demonstrating lower electron transfer resistance at the interface of Cu–ZnO–Co3O4. Accordingly, the Cu–ZnO–Co3O4 composite is more favorable for electron tunneling during the PDS activation process. Furthermore, the chronoamperometry analysis was performed to study the detailed electron flow pathways, as presented in Fig. 6B. BPA and PDS were added in sequence to the solution. As shown, the addition of BPA resulted in the appearance of a much higher current density peak in Cu–ZnO–Co3O4 in comparison with Cu–Co3O4, suggesting that the transfer of electrons occurred from BPA to the working electrode coated with the catalysts, and the more intense peak demonstrates that the electron transfer from BPA to Cu–ZnO–Co3O4 was easier than to Cu–Co3O4. The subsequent addition of PDS resulted in an increase in negative current, which could be ascribed to instant electron movement from the electrochemical station to the working electrodes deposited with the catalysts. Thus, the change in current density is likely ascribed to the electron density redistribution between the BPA-catalysts and PDS when they come in contact.
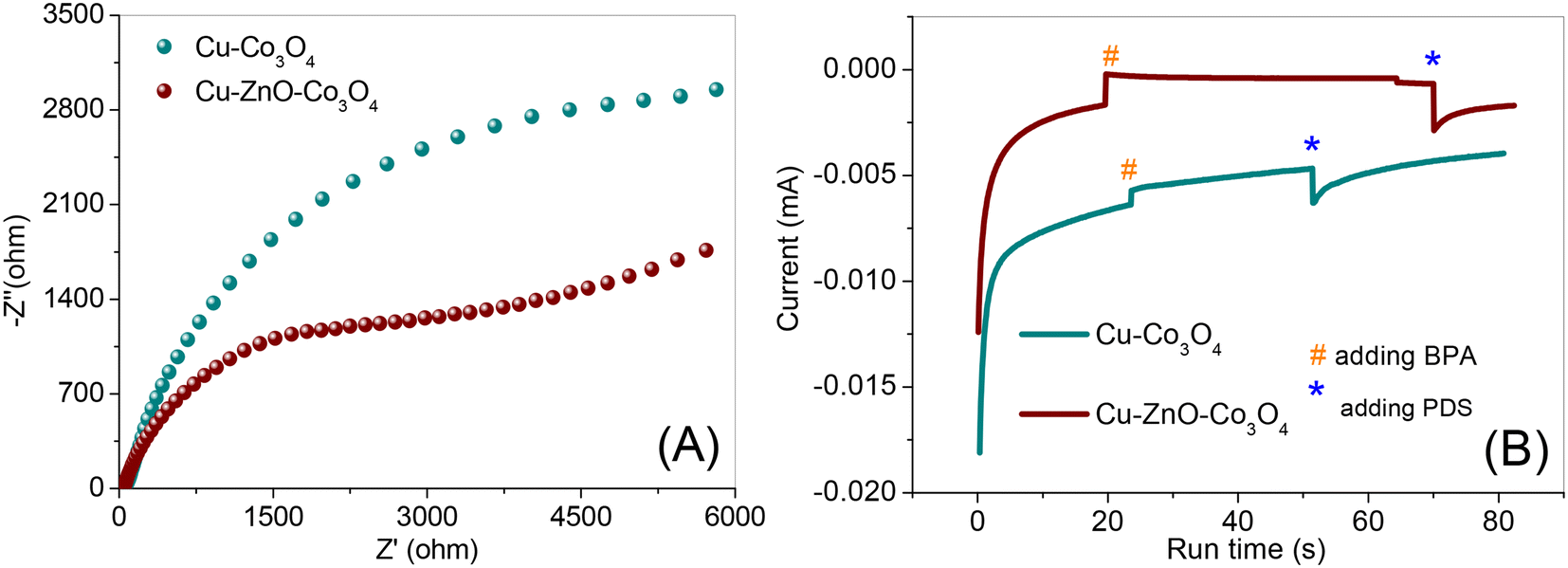 | ||
| Fig. 6 (A) EIS analysis of the prepared catalysts in a 100 mM Na2SO4 + 5 mM K3Fe[(CN)6] solution in the frequency range of 106–0.01 Hz. (B) Chronoamperometric analysis of the prepared catalysts in a 50 mM Na2SO4 solution, with a bias of 0.01 V. | ||
Furthermore, based on the intermediates identified by HPLC-MS analysis and the structures determined in previous works,40,41 the possible degradation pathways of BPA are proposed in Scheme S1 in the ESI.†
Finally, to evidence the practical application of this synthetic method of catalysts and Cu–ZnO–Co3O4 in BPA disposal under practical conditions, some extended experiments were performed. First, it was found that Cu and Ni doping could increase the performance of the corresponding catalysts compared with the references, however, the Mn doping resulted in the decrease of the activity as shown in Fig. S7A.† On the other hand, it was discovered that the BPA in the lake water from Longzi Lake in Bengbu City could not be eliminated by the Cu–ZnO–Co3O4 and PDS, which was possibly due to the existence of a large number of microorganisms and bacteria. Our experiment confirmed that the use of the same amount of PMS could degrade the BPA in 6 min (Fig. S7B†), which suggested a potential application of the present catalyst for water disposal in the nature.
4. Conclusion
Based on the interaction of hollow ZIF-8–ZIF-67 with copper nitrate, a nest-like Cu–ZnO–Co3O4 composite with a porous structure, small active species and abundant oxygen vacancies was synthesized after calcination. With a low consumption of PDS, based on a composite mechanism, including non-radical and radical path, the Cu–ZnO–Co3O4 displayed an ultra-high activity for BPA elimination with an apparent degradation constant of 0.84 min−1, where HO˙, O2˙− as well as 1O2 derived from PDS played vital roles for BPA degradation. The robust stability of Cu–ZnO–Co3O4 was also confirmed by cyclic tests. This work provides a novel strategy for the design and synthesis of robust catalysts from metal–organic frameworks for advanced oxidation reactions.Data availability
All relevant data are included within the manuscript and its ESI† files.Author contributions
Lei Bai: conceptualization, data curation, resources, supervision, writing – review & editing; Hui Yan: investigation, methodology, data curation; Jiao Wang: investigation, resources, supervision, methodology; Qihang Wu: investigation; Guiling Wang: formal analysis; Yi Huang: resources; Bentian Zhang: funding acquisition, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by Major Project of Natural Science Research Project (2023AH040278), Natural Science Research Project of Anhui Education Department (2023AH051873) and Anhui Province Applied Peak Cultivation Discipline (XK-XJGF005). Dr. B. T. Zhang thanks Talent-Introduction Program for Anhui Science and Technology University (HCYJ202105), Natural Science Research Project of Higher Education in Anhui Province (2023AH051849), Science and Technology Project of Bengbu City (2023gx05) and Yilin Environmental Protection Technology Wuxi Co., Ltd. (881406) for financial support.References
- M. A. Burgos-Aceves, H. G. Abo-Al-Ela and C. Faggio, J. Hazard. Mater., 2021, 404, 124114 CrossRef CAS.
- I. Ďurovcová, S. Kyzek, J. Fabová, J. Makuková, E. Gálová and A. Ševčovičová, Environ. Pollut., 2022, 306, 119346 CrossRef.
- N. G. Khan, J. Correia, D. Adiga, P. S. Rai, H. S. Dsouza, S. Chakrabarty and S. P. Kabekkodu, Environ. Sci. Pollut. Res., 2021, 28, 19643 CrossRef CAS.
- I. Ahmad, M. Kaur, D. Tyagi, T. B. Singh, G. Kaur, S. M. Afzal and M. Jauhar, Environ. Toxicol. Pharmacol., 2024, 108, 104467 CrossRef CAS PubMed.
- F. X. Wang, Z. C. Zhang and C. C. Wang, Chem. Eng. J., 2023, 459, 141538 CrossRef CAS.
- Y. Zhang, Y. Li and S. Tabassum, J. Water Process Eng., 2024, 61, 105286 CrossRef.
- J. Hou, X. He, S. Zhang, J. Yu, M. Feng and X. Li, Sci. Total Environ., 2021, 770, 145311 CrossRef CAS.
- L. Chen, T. Maqbool, C. Hou, W. Fu and X. Zhang, Sep. Purif. Technol., 2022, 281, 119882 CrossRef CAS.
- J. Wang, M. F. Wang, B. Leger, A. Ponchel and L. Bai, Inorg. Chem., 2023, 62, 12493 CrossRef CAS PubMed.
- J. Wang, X. Q. Huang and L. Bai, ACS Appl. Nano Mater., 2024, 7, 11900 CrossRef CAS.
- J. Wang, M. F. Wang, L. Fang and L. Bai, J. Sol-Gel Sci. Technol., 2023, 108, 448 CrossRef CAS.
- M. M. Wang, Y. K. Cui, H. Y. Cao, P. Wei, C. Chen, X. Y. Li, J. Xu and G. P. Sheng, Appl. Catal., B, 2021, 282, 119585 CrossRef CAS.
- M. Sun, S. N. Tang, Z. X. Chen, L. F. Zhai, Y. H. Xia and S. B. Wang, ACS ES&T Eng., 2024, 4, 1970 Search PubMed.
- Y. B. Ding, X. R. Wang, L. B. Fu, X. Q. Peng, C. Pan, Q. H. Mao, C. J. Wang and J. H. Yan, Sci. Total Environ., 2021, 765, 142794 CrossRef CAS PubMed.
- R. Tian, H. R. Dong, J. Chen, R. Li and Q. Q. Xie, Sep. Purif. Technol., 2020, 250, 117246 CrossRef CAS.
- (a) N. Olfatmehr, B. Kakavandi and S. M. Khezri, Sep. Purif. Technol., 2022, 281, 119882 CrossRef; (b) M. Golshan, N. Tian, G. Mamba and B. Kakavandi, Toxics, 2023, 11, 429 CrossRef CAS.
- (a) A. Son, J. Lee, C. Lee, K. Cho, J. Lee and S. W. Hong, Water Res., 2021, 191, 116803 CrossRef CAS PubMed; (b) B. Kakavandi, M. Z. Salmasi, M. Ahmadi, A. Naderi, P. Roccaro, J. Bedia, M. H. Firooz and R. R. Kalantary, J. Environ. Manage., 2023, 342, 118242 CrossRef CAS PubMed.
- Z. H. Xie, C. S. He, H. Y. Zhou, L. L. Li, Y. Liu, Y. Du, W. Liu, Y. Mu and B. Lai, Environ. Sci. Technol., 2022, 56, 8784–8795 CrossRef CAS PubMed.
- Y. Zhao, L. Yu, C. Song, Z. Chen, F. Meng and M. Song, Environ. Sci. Technol., 2022, 56, 10710 CrossRef CAS PubMed.
- L. Bai, J. Q. Zhang, J. X. He, H. X. Zheng and Q. Y. Yang, Inorg. Chem., 2021, 60, 13041 CrossRef CAS PubMed.
- Y. Tian, L. Cao and P. Qin, ChemCatChem, 2019, 11, 4420 CrossRef CAS.
- Y. F. Li, D. B. Wang, G. J. Yang, X. Z. Yuan, X. Q. Xu, X. R. Liu, Y. L. Wang, R. G. Guan, Q. Z. Fu and F. Chen, J. Taiwan Inst. Chem. Eng., 2019, 102, 259 CrossRef CAS.
- Q. H. Ma, F. Cui, J. J. Zhang, X. Qi and T. Y. Cui, Appl. Surf. Sci., 2022, 578, 152001 CrossRef CAS.
- L. L. Zhao, J. M. Zhang, Z. P. Zhang, T. Wei, J. Wang, J. Ma, Y. E. Ren and H. X. Zhang, J. Colloid Interface Sci., 2022, 623, 520 CrossRef CAS.
- T. Zhou, Y. Han, W. Xiang, C. Wang, X. H. Wu, J. Mao and M. J. Huang, Sci. Total Environ., 2022, 802, 149833 CrossRef CAS PubMed.
- J. Wang, M. Couillard and E. A. Baranova, ChemCatChem, 2023, 15, e202201514 CrossRef CAS.
- X. Y. Chen, C. Chen, Z. J. Zhang and D. H. Xie, Powder Technol., 2013, 246, 201 CrossRef CAS.
- S. Kundu, W. Xia, W. Busser, M. Becker, D. A. Schmidt, M. Havenith and M. Muhler, Phys. Chem. Chem. Phys., 2010, 12, 4351 RSC.
- J. Wang, Y. J. Cheng, H. D. Ye, Z. R. Li and L. Bai, J. Solid State Chem., 2024, 329, 124409 CrossRef CAS.
- S. J. Abdulrazzaq, Mol. Syst. Des. Eng., 2024, 9, 158 RSC.
- J. Liang and L. Fu, Appl. Surf. Sci., 2021, 563, 150335 CrossRef CAS.
- (a) T. S. Chen, J. H. Ma, Q. X. Zhang, Z. J. Xie, Y. Q. Zeng, R. B. Li, H. J. Liu, Y. Liu, W. Y. Lv and G. G. Liu, Sci. Total Environ., 2019, 690, 878 CrossRef CAS PubMed; (b) C. J. Liang, C.-F. Huang, N. Mohanty and R. M. Kurakalva, Chemosphere, 2008, 73, 1540 CrossRef CAS PubMed.
- J. Di, R. Jamakanga, Q. Chen, J. Y. Li, X. Gai, Y. Li, R. Q. Yang and Q. X. Ma, Sci. Total Environ., 2021, 784, 147258 CrossRef CAS.
- (a) Y. X. Liu, R. Luo, Y. Li, J. W. Qi, C. H. Wang, J. S. Li, X. Y. Sun and L. M. Wang, Chem. Eng. J., 2018, 347, 731 CrossRef CAS; (b) J. Deng, Y. S. Shao, N. Y. Gao, C. Q. Tan, S. Q. Zhou and X. H. Hu, J. Hazard. Mater., 2013, 262, 836 CrossRef CAS PubMed.
- L. Bai, Z. Y. Guan, S. J. Li, S. Q. Zhang, Q. X. Huang and Z. R. Li, Sep. Purif. Technol., 2021, 255, 117718 CrossRef CAS.
- V. Lakshmi Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155 CrossRef CAS PubMed.
- L. Wu, Q. Zhang, J. Hong, Z. Dong and J. Wang, Chemosphere, 2019, 221, 412 CrossRef CAS PubMed.
- S. Luo, Z. Wei, D. D. Dionysiou, R. Spinney, W. P. Hu, L. Chai, Z. H. Yang, T. T. Ye and R. Y. Xiao, Chem. Eng. J., 2017, 327, 1056 CrossRef CAS.
- M. M. Wang, L. J. Liu, J. R. Xi, Y. Ding, P. X. Liu, L. Mao, B. J. Ni, W. K. Wang and J. Xu, Chem. Eng. J., 2023, 45, 1138605 Search PubMed.
- H. Yin, F. B. Yao, Z. J. Pi, Y. Zhong, L. He, K. J. Hou, J. Fu, S. J. Chen, D. B. Wang, Z. Tao, X. M. Li and Q. Yang, J. Colloid Interface Sci., 2021, 586, 551 CrossRef CAS PubMed.
- Z. H. Diao, W. Qian, P. R. Guo, L. J. Kong and S. Y. Pu, Chem. Eng. J., 2018, 349, 683 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce00971a |
| This journal is © The Royal Society of Chemistry 2025 |