
On the nature and interplay of Hg⋯O/S spodium bonding and O⋯S chalcogen bonding in one-dimensional phenylmercury(II) 3-alkoxycyclobutene-1,2-dione-4-thiolate coordination polymers†
Aparna
Kushwaha
 a,
Devyani
Srivastava
a,
Gabriele
Kociok-Köhn
b,
Sarfaraz
Ahmed
c,
Edward R. T.
Tiekink
*d and
Abhinav
Kumar
*a
a,
Devyani
Srivastava
a,
Gabriele
Kociok-Köhn
b,
Sarfaraz
Ahmed
c,
Edward R. T.
Tiekink
*d and
Abhinav
Kumar
*a
aDepartment of Chemistry, Faculty of Science, University of Lucknow, Lucknow 226 007, India. E-mail: abhinavmarshal@gmail.com
bPhysical Structure Characterization, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cDepartment of Pharmacognosy, College of Pharmacy, King Saud University, P.O. Box 2457, Riyadh 11451, Saudi Arabia
dDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Spain. E-mail: edward.tiekink@uib.es
First published on 19th November 2024
Abstract
Two one-dimensional coordination polymers, PhHg(3-alkoxycyclobutene-1,2-dione-4-thiolate) (alkoxy = OMe and OEt), have been synthesised, characterised spectroscopically, crystallographically and through computational chemistry techniques. Both species exhibit intermolecular Hg⋯O σ-hole spodium bonding as well as intramolecular Hg⋯O π-hole spodium bonding. In the ethoxy species there are complementary σ-hole O⋯S chalcogen bonds to the Hg⋯O σ-hole spodium bonding as well as orthogonal Hg⋯S π-hole spodium bonding. Each of the coordination polymers is connected into a supramolecular layer. For the methoxy derivative, the layer features π(cyclobutene)⋯π(phenyl) stacking and side-on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯π(cyclobutene)/anti-parallel carbonyl⋯carbonyl interactions. For the ethoxy species, Hg⋯S, offset π(cyclobutene)⋯π(cyclobutene) stacking and CO⋯π(cyclobutene) interactions occur within the layer. In each case, weak C–H⋯π interactions operate between the two-dimensional arrays. The nature of the electron transfer responsible for the spodium/chalcogen bonding has been established. In terms of QTAIM energies, those associated with Hg⋯O σ-hole spodium bonds were in the range of 17.5 to 23.3 kJ mol−1, which were less than the energies computed for the intramolecular Hg⋯O π-hole spodium bonds in each case (31.0 to 43.6 kJ mol−1). The lowest energies for the intermolecular Hg⋯O spodium bonding were computed for the ethoxy species, which correlates with the compensating Hg⋯S π-hole spodium bonding (2 × 11.0 kJ mol−1) and O⋯S chalcogen bonding (12.5 kJ mol−1). The electron transfer leading to the Hg⋯O/S spodium bonding involves donation of an oxygen- or sulphur-bound lone pair of electrons to the σ*(Hg–C) orbital, with the intramolecular Hg⋯O spodium bond involving two lone pair interactions. The O⋯S chalcogen bond is assigned to LP(O) → σ*(C–S).
O⋯π(cyclobutene)/anti-parallel carbonyl⋯carbonyl interactions. For the ethoxy species, Hg⋯S, offset π(cyclobutene)⋯π(cyclobutene) stacking and CO⋯π(cyclobutene) interactions occur within the layer. In each case, weak C–H⋯π interactions operate between the two-dimensional arrays. The nature of the electron transfer responsible for the spodium/chalcogen bonding has been established. In terms of QTAIM energies, those associated with Hg⋯O σ-hole spodium bonds were in the range of 17.5 to 23.3 kJ mol−1, which were less than the energies computed for the intramolecular Hg⋯O π-hole spodium bonds in each case (31.0 to 43.6 kJ mol−1). The lowest energies for the intermolecular Hg⋯O spodium bonding were computed for the ethoxy species, which correlates with the compensating Hg⋯S π-hole spodium bonding (2 × 11.0 kJ mol−1) and O⋯S chalcogen bonding (12.5 kJ mol−1). The electron transfer leading to the Hg⋯O/S spodium bonding involves donation of an oxygen- or sulphur-bound lone pair of electrons to the σ*(Hg–C) orbital, with the intramolecular Hg⋯O spodium bond involving two lone pair interactions. The O⋯S chalcogen bond is assigned to LP(O) → σ*(C–S).
1. Introduction
The design and development of tailored supramolecular architectures through varied intermolecular interactions is a prevalent theme in the chemical sciences.1 This arises as non-covalent interactions not only offer the opportunity to tune the desired framework but also can be exploited to tailor the desired photophysical properties of the resultant materials, including those containing mercury.2–4 However, the pivotal challenge still confronting crystal engineers is the rational assembly of the molecular components into three-dimensional arrays – how does the practitioner control the full gamut of intermolecular interactions which, despite being disparate, often contribute similar energies of stabilisation to the crystalline manifold?1–4Metal complexes with sulphur-rich ligands are very interesting from the viewpoint of their various properties like electrical conductivity, molecular magnetism, electrochemical characteristics, and optoelectronic properties and their applications in biological processes and medicinal chemistry.5 Of these, mono- and multi-functional dithiocarbamate ligands are of particular interest owing to their versatile coordination chemistry, material properties and industrial applications and have therefore been an active area of research for decades.6
Organomercury(II) compounds continue to attract significant attention owing to their importance in the preparation of other organometallic species, as intermediates in organic chemistry and their relevance to mercury detoxification.7 In this regard, organomercury(II) 1,1-dithiolates, including dithiocarbamates, are extremely versatile groups for the construction of supramolecular arrays, exhibit photoluminescence properties and can be used as dyes for solar energy harvesting.8 Previous crystallographic studies on phenylmercury(II) dithiocarbamates revealed that the bulkiness of the pendant group in the dithiocarbamate part of the compound plays an important role in the construction of the supramolecular architecture through Hg⋯S interactions;9 Alcock termed such contacts as “secondary-interactions”.10
Spodium bonding11 has recently emerged as a term to account for supramolecular association between any of the zinc triad elements, where the group 12 element functions as a Lewis acid interacting with electron-rich atoms, i.e. Lewis bases. These non-covalent interactions involve anti-bonding (zinc triad element)–ligand orbitals, which are markedly different from coordination (dative) bonds,11 and rely on the σ- and π-hole concepts of non-covalent bonding.12 There is increasing interest in evaluating these interactions for their nature and influence on molecular packing.13
Apart from 1,1-dithiolates, such as the pre-eminent dithiocarbamates, another class of thiolate ligands that has attracted interest are the thiosquarate ligands, i.e. the sulphur-substituted analogues of the squarate anion.14 The thiosquarate ligands have both soft sulphur and hard oxygen donor sites and have the capacity to coordinate soft and hard metal centres to generate heterobimetallic complexes.14 The substitution of only one oxygen centre with sulphur in the squarate anion reduces the bite parameter of this ligand in comparison to its 1,2-dithiolate derivative.15 The structural chemistry of monothiosquarates is relatively unexplored compared with their di-substituted analogues.14 The supramolecular frameworks and electrocatalytic properties of two new, tertiary phosphane-appended nickel(II) and copper(I) complexes were described, i.e. (LEt)2Ni(dppe) and (LEt)Cu(PPh3)2, where LEt is 3-ethoxycyclobutene-1,2-dione-4-thiolate and dppe is 1,1′-bis-(diphenylphosphino)ethane, being the first structurally characterised monothiosquarate derivatives.16a Even more recently, a series of complexes of the general formula (LEt)2M(dppf) (M = Ni, Zn and Cd, and dppf is 1,1′-bis-(diphenylphosphino)ferrocene) were described.16b With one exception, all complexes featured square-planar (Ni) or tetrahedral (Zn and Cd) centres with the monodentate LEt ligands being S-bound. The exceptional structure was found for the tetrahedral copper complex where the metal was S,O-chelated. Inspired by these encouraging results and in the quest of generating new supramolecular assemblies/architectures, two mono-anionic thiosquarate ligands, viz. 3-alkoxycyclobutene-1,2-dione-4-thiolates, i.e. [LMe]− and [LEt]−, having the same molecular framework but differing in the bulkiness of the pendant alkoxy groups, were reacted with phenylmercury(II) acetate, producing two new compounds, PhHgLMe and PhHgLEt, each of which self-associates in the solid state into a one-dimensional coordination polymer through interesting Hg⋯O spodium bonding. The present report evaluates the nature and importance of the various non-covalent interactions identified in the respective crystals, with an emphasis on the Hg⋯O/S spodium bonding and O⋯S chalcogen bonding as well as interactions involving the thiosquarate anions, through a computational chemistry study.
2. Results and discussion
2.1 Syntheses and spectroscopy
The phenylmercury compounds, PhHgLMe and PhHgLEt, were synthesised by reacting phenylmercury acetate in a stoichiometric ratio with sodium 3-alkoxycyclobutene-1,2-dione-4-thiolates generated in situ by the reaction of the respective thiol with NaSH (Scheme 1). The compounds are air-stable, soluble in halogenated solvents but insoluble in diethyl ether. The compounds were characterised by elemental analysis, 1H and 13C{1H} NMR and UV/vis spectroscopy; see the ESI,† Fig. S1–S5. Finally, their molecular and crystal structures were revealed through single-crystal X-ray diffraction analyses.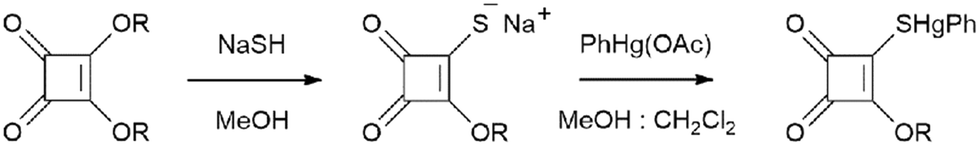 | ||
| Scheme 1 Synthetic route for the preparation of the phenylmercury(II) 3-alkoxycyclobutene-1,2-dione-4-thiolates: R = Me (PhHgLMe) and Et (PhHgLEt). | ||
1H NMR proved the purity and composition of the compounds by exhibiting the expected resonances and integration. For PhHgLMe, the resonances observed between δ 7.49–7.44 and a singlet at δ 4.51 justify the presence of phenyl and methoxy protons, respectively. Similarly, in PhHgLEt, the phenyl protons yielded resonances between δ 7.42 and 7.34, while the appearance of a quartet at δ 4.79 (OCH2) and a triplet at δ 1.45 (CH3) confirmed the presence of the ethoxy group. Owing to poor solubility, the 13C{1H} NMR spectrum of PhHgLMe did not exhibit all signals. A downfield signal at δ 207.2 is assigned to CS, whereas the signal δ 170.4 is assigned to ring-C-OMe, but those due to carbonyl-C nuclei were not observed. A multiplet in the region δ 137.6–128.4 is due to phenyl-C and the singlet at δ 65.9 is attributed to methoxy-C. More information is available in the 13C{1H} NMR spectrum of PhHgLEt, with the signal at δ 196.6 corresponding to CS and signals corresponding to the carbonyl-C (δ 184.5 and 182.2) and C-OEt (δ 169.4) nuclei were apparent. The resonances due to phenyl-C (δ 136.6–127.3) and ethoxy-C (δ 70.2 and 14.7) were also resolved.
The electronic absorption spectra were recorded in dichloromethane and presented very similar features. The bands appearing at ∼250 nm arise from intraligand π→π* charge transfer transitions of the phenyl ring.9c Another intense band, at ∼300 nm, is due to MLCT transitions between mercury and 3-alkoxycyclobutene-1,2-dione-4-thiolate ligands. Finally, weak bands at 370 and 351 nm, respectively, for PhHgLMe and PhHgLEt, arise due to the π→π* transition within the thiosquarate ring.
2.2 Crystallography
The crystal structures of PhHgLMe and PhHgLEt were determined but to relatively low resolution and with assumptions made to stabilise the refinement (see Experimental, X-ray crystallography). Nevertheless, the structures were determined unambiguously and confirm the molecular composition and supramolecular association of each compound.Two independent molecules comprise the crystallographic asymmetric unit of PhHgLMe. The molecular structure of the first independent molecule is shown in Fig. 1(a) and that for the second molecule in the ESI† (Fig. S6). The mercury atom in the first molecule exists in the expected linear geometry [C6–Hg1–S1 = 178.7(6)°] defined by thiolate-S [Hg1–S1 = 2.370(6) Å] and ipso-C(phenyl) [Hg1–C6 = 2.11(2) Å] atoms; the equivalent values for the Hg2 atom are equal within experimental error. Despite the equivalence in intramolecular geometric parameters, the independent molecules in PhHgLMe have rather distinct conformations. Thus, as readily seen in the overlay diagrams of Fig. 1(b), where it is apparent that the molecules are not quite superimposable in any residue except for the phenyl groups which are displaced but only marginally. The different conformations are readily quantified by the dihedral angles between the phenyl and the cyclobutene residues of 5.3(8)° and 10.1(8)°, respectively. Further, the RMS bond and angle fit between the two molecules were computed to be 0.0702 Å and 5.238°, respectively.17
 | ||
| Fig. 1 (a) Molecular structure of the first independent molecule in the crystal of PhHgLMe, showing the atom labelling scheme and displacement ellipsoids at the 35% probability level, and (b) overlay diagrams of the two independent molecules of PhHgLMe (red image: first independent molecule). The independent molecules have been superimposed so the cyclobutene (left-hand view) and phenyl residues are coincident. | ||
It is observed from Fig. 1(a) and S6† that one of the carbonyl-O atoms is oriented to be proximate to the mercury atom, the Hg1–S1–C1–O1 torsion angle is −3(1)°; the equivalent angle for the second independent molecule is 4.6(9)°. The intramolecular Hg1⋯O1 contact distance is 2.92(2) Å and this interaction closes a five-membered {⋯HgSC2O} synthon; the equivalent distance for the Hg2 atom is 2.86(2) Å. These separations may be compared to 3.07 Å, being the sum of the van der Waals radii.17 Intermolecular Hg⋯O contacts are also apparent in the molecular packing.
The individual molecules self-assemble via Hg⋯O(carbonyl) spodium bonding into one-dimensional coordination polymers with Hg1⋯O1i and Hg2⋯O5ii separations of 2.786(19) and 2.85(2) Å, respectively; Table 1 collates the geometric data characterising the discussed intermolecular contacts for both crystals. Each of the chains, illustrated in Fig. 2(a) and S7,† has a linear topology, is aligned along [1 1 0] and is close to flat (ESI† Fig. S7). The chains are parallel but offset with respect to each other and have the same pitch, i.e. 7.9830(9) Å. The supramolecular chains assemble into layers parallel to (1 1 0) (Fig. 2(b)), comprising alternating chains of independent molecules and feature π-stacking interactions between cyclobutene and phenyl rings derived from different independent molecules. In addition to the π⋯π stacking are side-on CO⋯π(cyclobutene) interactions whereby the respective carbonyl-O atom is projected to lie over the midpoint of the cyclobutene ring, again involving different independent molecules; intra- and intermolecular interactions involving related carbonyl residues are well documented.18 A closer inspection of the side-on CO⋯π(cyclobutene) interactions hints at a shorter O⋯C separation compared with the three other O⋯C interactions: O2⋯C14 = 3.22(2) Å (cf. range of O2⋯C = 3.27(2) to 3.36(3) Å) and O5⋯C3 = 2.90(3) Å (cf. range of O5⋯C = 3.06(3) to 3.39(3) Å). This leads to an alternate description of the supramolecular association between these residues, i.e. one based on an anti-parallel carbonyl⋯carbonyl interaction within a non-symmetric, four-membered {⋯CO⋯}2 synthon; see below for further discussion of this interaction. The supramolecular layers stack along the c-axis with the only obvious directional interaction between the layers being a weak methyl-C–H⋯π(phenyl) contact formed between symmetry-related Hg1-molecules (Table 1).
| A | Y | B | Y⋯B | A⋯B | A–Y⋯B | Symmetry operation |
|---|---|---|---|---|---|---|
| a Angle between the rings. b Slippage: 0.99 Å. c Slippage: 0.74 Å. d Slippage: 1.56 Å. e Slippage: 1.55 Å. f This interaction involves the second component of the disordered ethyl group. | ||||||
| PhHgL Me | ||||||
| C3 | O2 | Hg1 | 2.786(19) | 3.95(2) | 153(1) | ½ + x, ½ + y, z |
| C14 | O5 | Hg2 | 2.85(2) | 3.857(17) | 150(2) | −½ + x, −½ + y, z |
| Cg(C1–C4) | — | Cg(C17–C22) | — | 3.528(15) | 3a,b | x, −1 + y, z |
| Cg(C6–C11) | — | Cg(C12–C15) | — | 3.424(14) | 5a,c | x, −1 + y, z |
| C3 | O2 | Cg(C12–C15) | 3.16(2) | 3.568(19) | 98(1) | x, y, z |
| C14 | O5 | Cg(C1–C4) | 2.99(2) | 3.641(18) | 117(1) | x, y, z |
| C5 | H5b | Cg(C6–C11) | 2.93 | 3.66(3) | 133 | ½ + x, ½ − y, ½ + z |
| PhHgL Et | ||||||
| C3 | O2 | Hg | 2.992(6) | 4.064(8) | 147.2(5) | x, −1 + y, z |
| C3 | O2 | S1 | 2.928(5) | 4.064(8) | 165.3(5) | x, −1 + y, z |
| C1 | S1 | Hg | 3.334(2) | 4.305(7) | 113.2(2) | 1 − x, 2 − y, 1 − z |
| C1 | S1 | Hg | 3.337(2) | 4.302(7) | 113.0(2) | −x, 2 − y, 1 − z |
| Cg(C1–C4) | — | Cg(C1–C4) | — | 3.428(5) | 0a,d | −x, 1 − y, 1 − z |
| Cg(C1–C4) | — | Cg(C1–C4) | — | 3.431(5) | 0a,e | 1 − x, 1 − y, 1 − z |
| C2 | O1 | Cg(C1–C4) | 3.289(7) | 3.163(8) | 73.3(4) | −x, 1 − y, 1 − z |
| C2 | O1 | Cg(C1–C4) | 3.308(7) | 3.162(8) | 72.5(4) | 1 − x, 1 − y, 1 − z |
| C5af | H5a1 | Cg(C1a–C4a) | 2.99 | 3.89(2) | 151 | 1 − x, 2 − y, 1 − z |
 | ||
| Fig. 2 Molecular packing diagrams for PhHgLMe: (a) linear, supramolecular chain featuring Hg⋯O(carbonyl) spodium bonding and comprising the first independent molecule only, (b) supramolecular layer parallel to (1 1 0) (with all hydrogen atoms removed) and (c) unit cell contents viewed to highlight the stacking of layers shown in (b). The Hg⋯O, π⋯π, CO⋯π(cyclobutene) and methyl-C–H⋯π(phenyl) interactions are shown as black, violet, blue and orange dashed lines, respectively. | ||
The molecular structure of PhHgLEt is shown in Fig. 3 and presents essentially the same features as for the methoxy analogue: Hg–S1 = 2.3850(19) Å, Hg–C7 = 2.00(4) Å, C7–Hg–S1 = 177.6(10)° and Hg⋯O1 = 2.948(5) Å. Disorder in the molecular structure is evident (see the ESI,† Fig. S8). The mercury-bound phenyl ring is statistically disordered over two positions with the dihedral between the rings being nearly orthogonal at 87.8(13)°. The dihedral angles between each disordered phenyl ring and the cyclobutene ring are 43.4(10) (Fig. 3) and 44.5(10)°, respectively, indicating similar conformational relationships. Two orientations for the ethoxy group were also resolved. The statistically disordered ethyl groups are close to co-planar with the cyclobutene ring in each case [C1–C4–O3–C5, C5a torsion angles = 179(1) and 3(2)°, respectively] and best described as being anti to the thiolate-S1 atom, as shown in Fig. 3, or syn, ESI† Fig. S8.
 | ||
| Fig. 3 Molecular structure of PhHgLEt showing the atom labelling scheme and displacement ellipsoids at the 35% probability level. Only one orientation of the disordered components is illustrated. | ||
The molecular packing in the crystal of PhHgLEt bears some similarities to that in the crystal of PhHgLMe but exhibits significantly more complexity in the identified non-covalent interactions. Spodium bonding of the type Hg⋯O leads to a linear supramolecular chain along the b-axis (Fig. 4(a)). The spodium bonding involves the chemically equivalent carbonyl-O atom in each of the three one-dimensional coordination polymers. The pitch of the chain is marginally shorter, i.e. 7.8713(5) Å (cf. the Hg⋯Hg separations of 2 × 7.9830(9) Å in the chains of PhHgLMe). This difference may arise owing to the presence of a complimentary carbonyl-O⋯S(thiolate) chalcogen bond with a separation of 2.928(5) Å (cf. the sum of the respective van der Waals radii of 3.22 Å);17 the equivalent separations for PhHgLMe are 3.146(18) and 3.36(2) Å (see discussion below). Apart from the inclined disordered phenyl rings, the chains in PhHgLEt are flat. Running orthogonal to the one-dimensional coordination are a range of non-covalent interactions so a supramolecular layer in the ab-plane is apparent (Fig. 4(b)).
 | ||
| Fig. 4 Molecular packing diagrams for PhHgLEt: (a) linear, supramolecular chain featuring Hg⋯O(carbonyl) spodium bonding and carbonyl-O⋯S(thiolate) chalcogen bonding, (b) supramolecular layer parallel to (1 1 0) and (c) unit cell contents viewed in projection down the a-axis. The Hg⋯O, O⋯S, π⋯π, CO⋯π(cyclobutene) and Hg⋯S interactions are shown as black, yellow-red, violet, blue and yellow-orange dashed lines, respectively. | ||
Pivotal in the assembly of the layer is the participation of the cyclobutene residue. Thus, this ring self-associates on either side with centrosymmetrically related cyclobutene residues via π⋯π stacking interactions (Table 1). When viewed down the stack, there is an alternating sequence of overlapping C2–C3 atoms (see discussion below). Complementing the offset π⋯π interactions are carbonyl-CO⋯π interactions in which the C1-carbonyl bridges successive cyclobutene rings; there is no evidence for carbonyl⋯carbonyl interactions as found for PhHgLMe. Within the layer are close Hg⋯S contacts of 3.3342(19) and 3.3372(19) Å (cf. the sum of the van der Waals radii of 3.35 Å).17 These occur within two distinct centrosymmetric, four-membered {⋯HgS}2 synthons that assemble into an undulating tape (Fig. 4(b)); analogous short contacts are absent in the crystal of PhHgLMe. A recent review of organomercury(II) thiolate structures showed such synthons are frequently observed in their crystals, but not within the tapes highlighted in Fig. 4(b);9g however, concatenated {⋯HgS}2 synthons were observed within a square-wave arrangement in the crystal of MeHg[3-(2-hydroxybenzylideneamino)phenylthiolato].19a Tapes comprising connected {⋯HgS}2 synthons are sometimes observed in non-organomercury thiolates,13i with the closest analogue to that formed in the crystal of PhHgLEt being observed in the crystal of HgCl2(anhydro-5-mercapto-2,3-diphenyltetrazole).19b,c Further, a recent report highlighted that significant energies of stabilisation to the molecular packing can be contributed by Hg⋯S contacts at or longer than the sum of the conventional van der Waals radii.13k The supramolecular layers thus formed stack along the c-axis (Fig. 4(c)), with the closest interaction between them being a methylene-C–H⋯π(cyclobutene) contact involving an atom of the second orientation of the disordered component of the molecule (Table 1).
2.3 Hirshfeld surface analysis
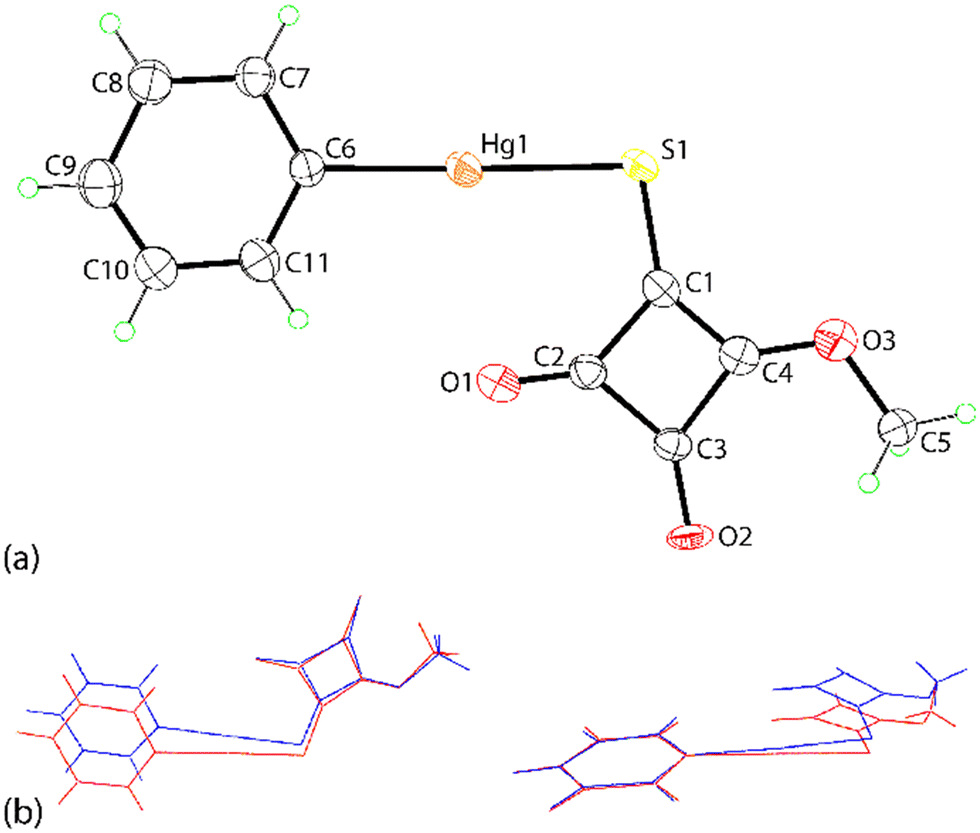
To gain additional insight into the intermolecular contacts in the respective crystals, Hirshfeld surface analyses, i.e. dnorm, shape-index, curvedness and two-dimensional fingerprint plots, were performed employing CrystalExplorer21 following established protocols.20a,bFor the first independent molecule of PhHgLMe, discernible red spots are noted about the mercury and carbonyl-O atoms in the dnorm plot (Fig. 5(a)), which are ascribed to the influence of the Hg⋯O spodium bonding and CO⋯π(cyclobutene)/anti-parallel carbonyl⋯carbonyl interactions, respectively. Fig. 5(b) highlights the concave region on the shape-index plot arising from the CO⋯π(cyclobutene) interactions, while the curvedness plot in Fig. 5(c) shows characteristic features, i.e. flat surfaces, arising from the π(cyclobutene)⋯π(phenyl) stacking. The equivalent Hirshfeld surfaces for the second independent molecule of PhHgLMe resemble closely those shown in Fig. 5 and are given in the ESI† (Fig. S9).
 | ||
| Fig. 5 Calculated Hirshfeld surfaces for the first independent molecule of PhHgLMe: (a) dnorm plot, (b) shape-index plot and (c) curvedness plot. | ||
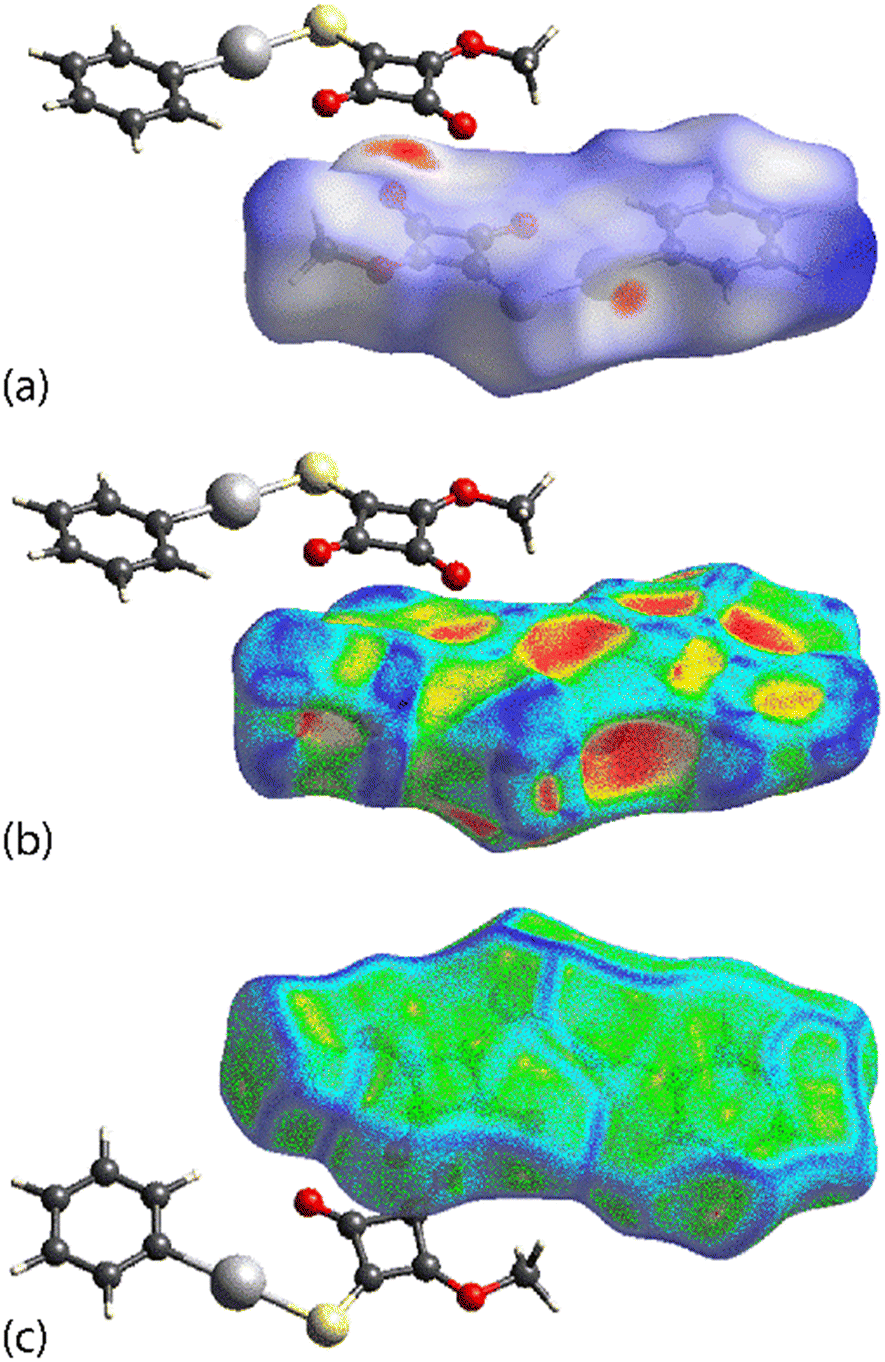
Distinctive Hirshfeld surfaces are calculated for PhHgLEt, i.e. corresponding to the disorder component illustrated in Fig. 3. As shown in Fig. 6(a), the dnorm plot features a prominent red region in the vicinity of the mercury, oxygen and sulphur atoms due to the formation of both Hg⋯O and Hg⋯S spodium bonding interactions. Further, red regions are evident near both carbonyl-O atoms in Fig. 6(b), with the feature about the O1 atom arising owing to its participation in the C2O1⋯π(cyclobutene) interaction, not present in the crystal of PhHgLMe, as well as the close C⋯C contacts within the stacked cyclobutene rings. The shape-index plot (Fig. 6(c)) and curvedness plot (Fig. 6(d)) highlight the concave and flat regions, respectively, arising from the offset interaction of two cyclobutene rings.
 | ||
| Fig. 6 Calculated Hirshfeld surfaces for PhHgLEt: (a and b) two orientations for the dnorm plots, (c) shape-index plot and (d) curvedness plot. | ||
Next, attention was directed towards the calculation of the percentage contributions to the Hirshfeld surfaces. For PhHgLMe, calculations were performed on the asymmetric unit and on the individual molecules; a compilation of surface contacts is given in the ESI† (Table S1). The individual molecules presented similar percentage values for each specific contact with the maximum differences being noted for H⋯H, i.e. 29.3 (cf. 30.5% for the Hg1- and Hg2-containing molecules, respectively), and O⋯H/H⋯O contacts, i.e. 17.3 and 16.6%, respectively, providing further evidence for the presence of two distinct molecules in the asymmetric unit;20c the remaining surface contributions differed by less than 1%. The importance of CH⋯HC interactions between alkanes has been highlighted in the literature.20d There are distinctive two-dimensional fingerprint plot profiles for PhHgLMe and PhHgLEt.
The most obvious difference in the percentage contributions to the Hirshfeld surfaces relates to the H⋯H contacts which increase from 31.1% to 40.6% for (overall) PhHgLMe and PhHgLEt, respectively, clearly related to the increased hydrogen content in PhHgLEt. The next most significant contributions to the Hirshfeld surface in PhHgLEt are O⋯H/H⋯O contacts at 14.7%, which is more significant than 12.3% in the crystal of PhHgLMe, followed by C⋯H/H⋯C contacts at 12.6% and 12.3%, respectively. There is an increase in C⋯O/O⋯C contacts passing from PhHgLMe to PhHgLEt, i.e. 3.5% to 8.6%, but a decrease in S⋯H/H⋯S (12.8% vs. 4.7%) and C⋯C contacts (7.2% vs. 3.7%), correlating with the presence of significant Hg⋯S/Hg⋯S contacts and reduced π-stacking in PhHgLEtvs.PhHgLMe.
Owing to the significant differences in the nature of the surface contacts between PhHgLMe and PhHgLEt, rather different two-dimensional fingerprint plots are apparent; ESI† Fig. S10 presents plots for the full and delineated fingerprint plots. The overall fingerprint plot for PhHgLMe has a shield-like shape in contrast to the elongated pattern seen for PhHgLEt (Fig. 7(b)). Of particular interest in the fingerprint plots are the features due to the spodium bonding. For PhHgLMe, two well-defined wings are apparent due to the Hg⋯O interactions (Fig. 7(a)), whereas two ticks terminating at values greater than the sum of the van der Waals radii are due to the long Hg⋯S separations (Fig. 7(a)).
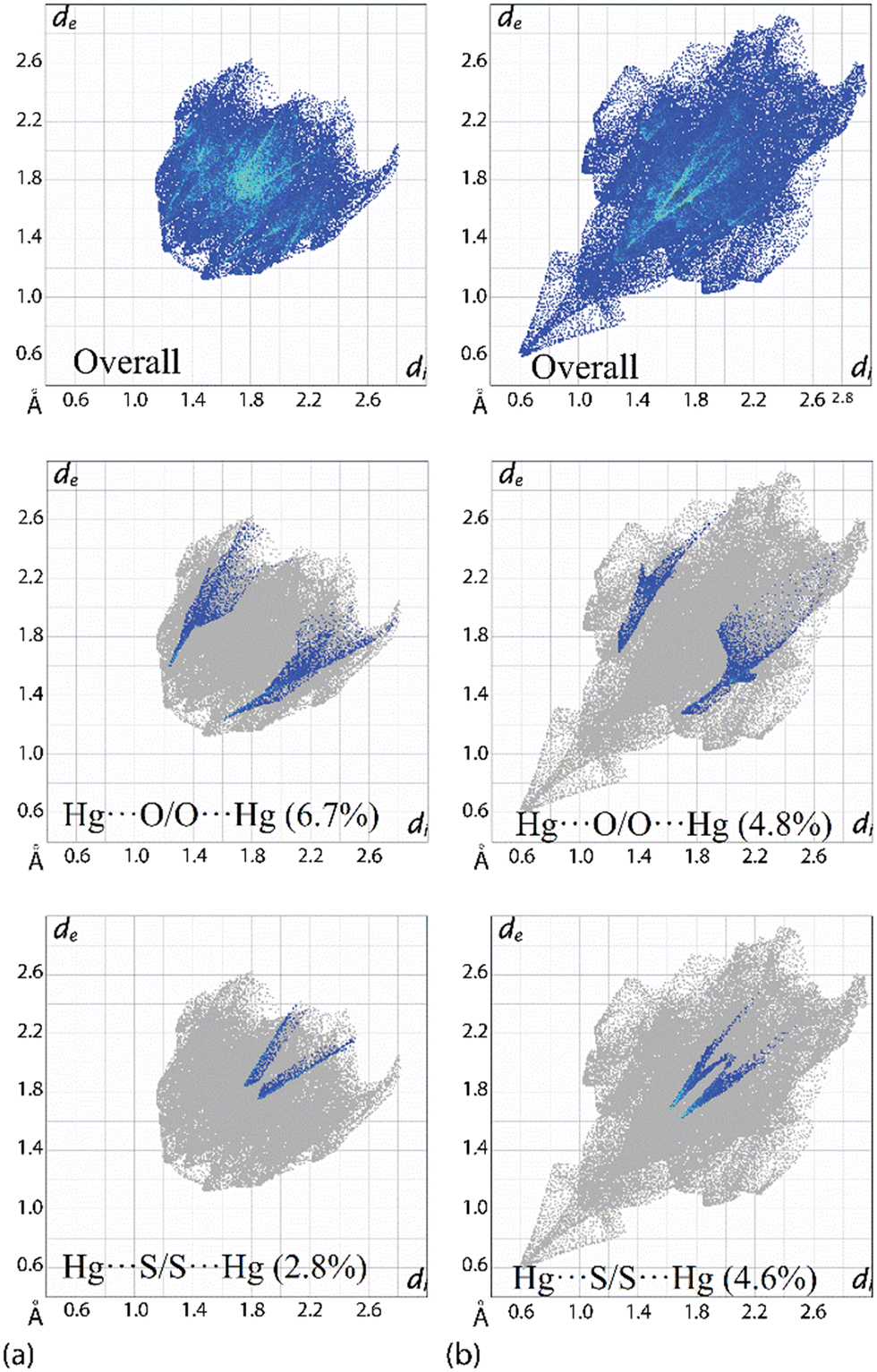 | ||
| Fig. 7 The overall two-dimensional fingerprint plots and those delineated into Hg⋯O/O⋯Hg and Hg⋯S/S⋯Hg contacts for (a) overall PhHgLMe and (b) PhHgLEt. The squid-like shape in the fingerprint plots for PhHgLEt arises from chemically irrelevant, impossibly short contacts between hydrogen atoms in the disordered residues. | ||
In the equivalent fingerprint plots for PhHgLEt, the wings in the delineated plot due to Hg⋯O/O⋯Hg contacts are less well-defined (cf.PhHgLMe), but the opposite is true for the delineated plot due to Hg⋯S/Hg⋯S surface contacts (Fig. 7(a) and (b)). These differences are reflected in the percentage contributions where Hg⋯O/O⋯Hg contacts are more prominent in PhHgLMe compared with PhHgLEt, i.e. 6.7% vs. 4.8%; the opposite trend pertains for the Hg⋯S/Hg⋯S contacts, i.e. 2.8% vs. 4.6%. The contribution to the calculated Hirshfeld surface by the O⋯S chalcogen bonding is 3.9% and is manifested by less distinct wings; cf. the contacts due to the Hg⋯O spodium bonding (ESI† Fig. S10).
2.4 Computational chemistry investigations
To further understand the nature of the non-covalent interactions observed in the crystals of PhHgLMe and PhHgLEt, several computational chemistry approaches were explored, namely molecular electrostatic potential (MEP) maps, non-covalent interaction (NCI) plots and quantum theory of atoms in molecules (QTAIM) investigations following procedures outlined in the experimental section. The calculations for PhHgLEt were performed on the molecule having the orientations shown in Fig. 3.The MEP map for the first independent molecule of PhHgLMe is shown in Fig. 8, with those for the second independent molecule of PhHgLMe and for PhHgLEt shown in ESI† Fig. S11 as these present very similar features; the MEP maps were calculated on the experimental structures in accord with literature precedents.21 The MEP minima are located on the carbonyl-O atoms, consistent with their involvement in intra- and intermolecular spodium bonding interactions. There are also minima located at the centre of the phenyl ring and at the sulphur atom. Positive electrostatic potential is evident at both mercury and at the centre of the cyclobutene ring. The opposite charges in the rings suggest that the π(cyclobutene)⋯π(phenyl) interactions in PhHgLMe have an electrostatic component which contributes to the stabilisation of the interaction, while the π(cyclobutene)⋯π(cyclobutene) interactions in PhHgLEt are likely to be predominantly dispersive in character.
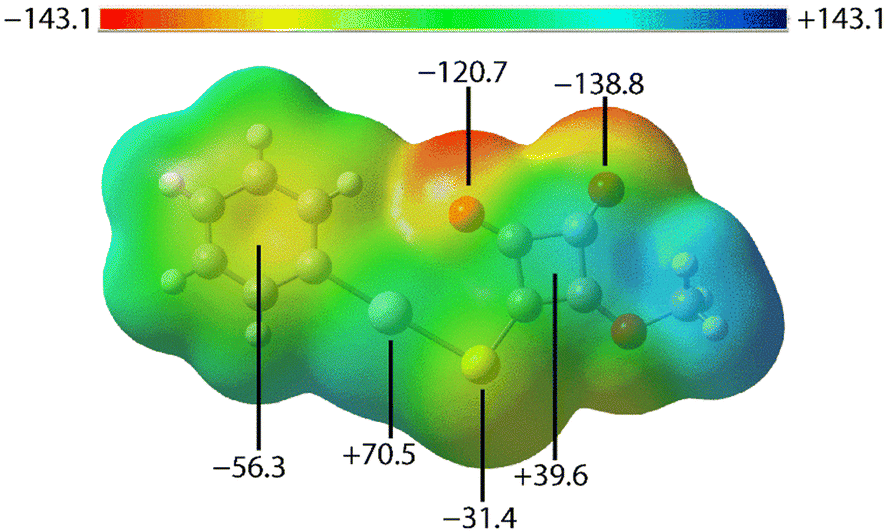 | ||
| Fig. 8 MEP surface plot calculated for the experimental geometry of PhHgLMe – the first independent molecule. The indicated energies are given in units of kJ mol−1. The isosurface for the MEP was 0.0004 a.u. and the plot was rendered between −0.054 and +0.054 a.u. | ||
The non-covalent interaction (NCI) index visualises non-covalent interactions qualitatively by mapping the non-covalent interaction zones in real space.22 This approach depicts the bonding characteristics using two scalar fields, the electron density (ρ) and the reduced density gradient (RDG) plots: these exhibited similar patterns, indicating the presence of a similar type of interactions in the crystals (see ESI† Fig. S12).
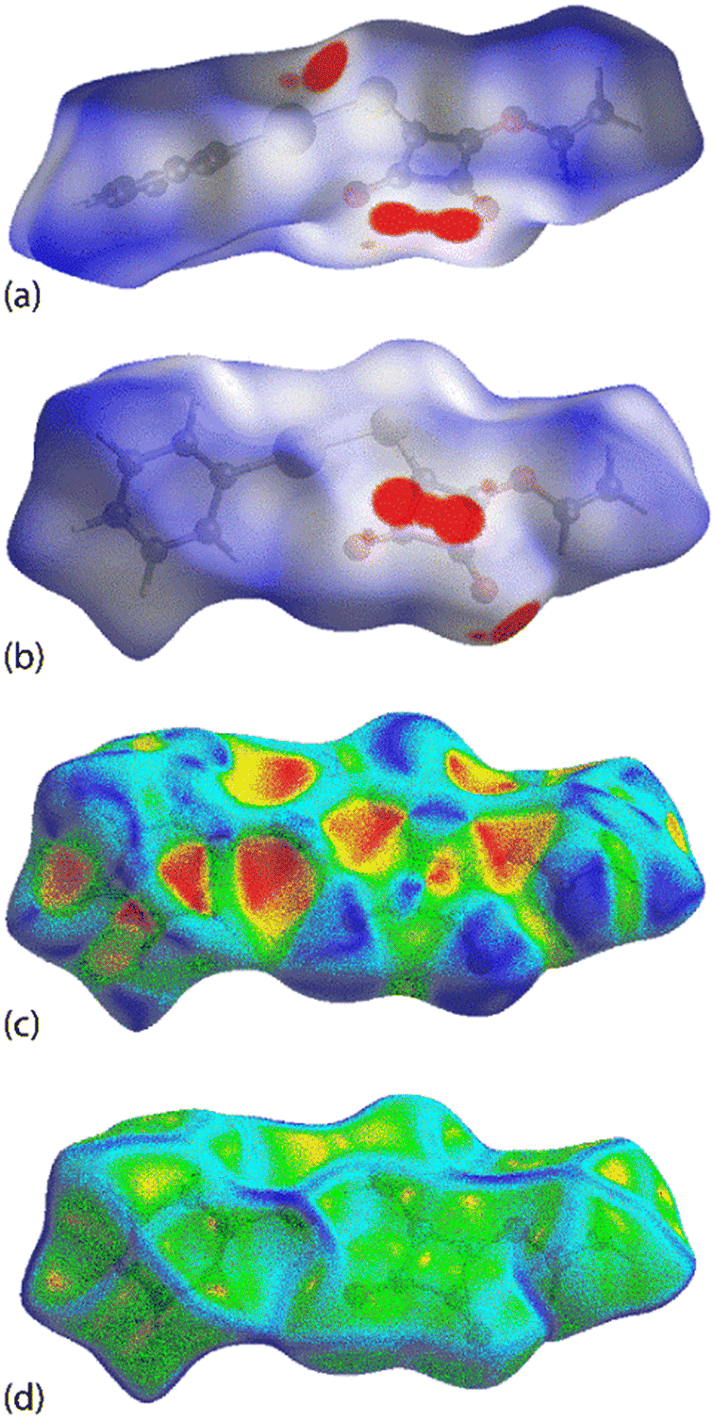
The complementary pair of π(cyclobutene)⋯π(phenyl) interactions occurring between the two independent molecules in PhHgLMe are clearly apparent from the NCI plot of Fig. 9(a). The CO⋯π(cyclobutene)/anti-bonding carbonyl⋯carbonyl interactions appear as significant green-red patches between the adjacent carbonyl oxygen atoms and cyclobutene rings, indicating an admixture of non-bonding steric clashes and van der Waals interactions between the respective atoms of interest, i.e. labelled (i) in Fig. 9(b). In the same region as (i) is a second green patch corresponding to the attractive intermolecular Hg⋯O spodium bonding; the intramolecular Hg⋯O spodium bonding is labelled with (ii) in Fig. 9(b) and very clearly discerned in Fig. 9(c) for the first independent molecule of PhHgLMe; see ESI† Fig. S13 for the equivalent image for the second independent molecule. The weak Hg⋯S spodium bonding is apparent in the region labelled with (iii) in Fig. 9(b).
 | ||
| Fig. 9 The non-covalent interaction (NCI) plots for PhHgLMe highlighting intermolecular (a) π(cyclobutene)⋯π(phenyl) interactions, (b) CO⋯π(cyclobutene)/anti-bonding carbonyl⋯carbonyl, (intramolecular) CO⋯Hg and Hg⋯S spodium bonding interactions indicated by (i), (ii) and (iii), respectively, and (c) intermolecular Hg⋯O spodium bonding within the supramolecular chain comprising the first independent molecule. Isovalue: 0.5 e per a.u.3. | ||
In the case of PhHgLEt, the NCI plots highlighting the less common interactions are shown in Fig. 10. Thus, the π(cyclobutene)⋯ π(cyclobutene) interactions are highlighted as extensive green patches, being indicative of van der Waals interactions operating between the carbon centres of the adjacent four-membered cyclobutene rings (Fig. 10(a)). The intra- and intermolecular Hg⋯O spodium bonding interactions are observed as green and blue regions, respectively, in Fig. 10(b). It is notable that the attractive region between the intermolecular Hg⋯O spodium bond extends to the region between the oxygen and sulphur atoms forming the chalcogen bond; in the equivalent NCI plots for the independent molecules of PhHgLMe, the attractive region between the equivalent oxygen and sulphur atoms are less pronounced (Fig. 9(c) and ESI† S13). Finally, the Hg⋯S spodium bonding interactions are clearly seen in Fig. 10(c).
 | ||
| Fig. 10 Non-covalent interaction (NCI) plots for PhHgLEt highlighting the (a) intermolecular π(cyclobutene)⋯π(cyclobutene) interactions, (b) intermolecular CO⋯π(cyclobutene), intra- and intermolecular CO⋯Hg spodium bonding, and intermolecular O⋯S chalcogen bonding interactions and (c) intermolecular Hg⋯S spodium bonding interactions. Isovalue: 0.5 e per a.u.3. | ||
Employing the BSSE-corrected wB97XD level of theory,23 successfully utilised for other mercury-containing systems,9f the non-covalent interaction energies were calculated for the three dimeric aggregates derived from the three independent coordination polymers featuring intermolecular Hg⋯O spodium bonding in the crystals of PhHgLMe and PhHgLEt. These energies were computed to be 24.5, 23.8 and 18.2 kJ mol−1, respectively; the latter value incorporates the contribution by the O⋯S chalcogen bonding within the coordination polymer. The reduced energy for the cooperative Hg⋯O spodium bonding and O⋯S chalcogen bonding in PhHgLEt correlates with the participation of the mercury atom in two Hg⋯S spodium bonding contacts, which provides a total interaction energy of 26.3 kJ mol−1. In PhHgLMe, the interaction energy between the two independent molecules featuring π(cyclobutene)⋯π(phenyl) and CO⋯π(cyclobutene) interactions is 16.1 kJ mol−1, while in PhHgLEt, the interaction energy for the pair of molecules featuring π(cyclobutene)⋯π(cyclobutene) interactions is 19.6 kJ mol−1.
The nature of the intra- and intermolecular interactions in PhHgLMe and PhHgLEt was also assessed by a quantum theory of atoms in molecules (QTAIM) analysis,24 a technique known to provide useful information on the nature of spodium bonding involving mercury;11,13k the listing of the topographical parameters defining the key bond critical points (BCPs) are listed in ESI† Table S2. There are BCPs apparent in the QTAIM analysis due to intra- and intermolecular Hg⋯O spodium bonding for each of the first (Fig. 11(a)) and second independent molecules comprising the asymmetric unit of PhHgLMe (ESI† Fig. S14). The same observation pertains to the crystal of PhHgLEt (Fig. 11(b)), but with the extra notable feature of a BCP between the carbonyl-O2 and translationally related thiolate-S1 atoms. It is noteworthy that equivalent BCPs are absent for PhHgLMe. Fig. 11(c) highlights the BCPs associated with the Hg⋯S spodium bonding in PhHgLEt.
 | ||
| Fig. 11 QTAIM analysis calculated at the wB97XD level of theory and 6-31G** and CEP-121G basis sets, and with BCPs represented as blue spheres and bond paths as dotted orange lines highlighting (a) Hg⋯O spodium bonding involving the first independent molecule of PhHgLMe, (b) Hg⋯O spodium bonding and O2⋯S1 chalcogen bonding in PhHgLEt and (c) Hg⋯S spodium bonding in PhHgLEt. | ||
The QTAIM associated with the anti-parallel carbonyl⋯carbonyl interactions in PhHgLMe is highlighted in Fig. 12(a). From this, only a BCP associated with the shorter C3⋯O5 interaction is apparent, supporting the notion that this region of the crystal is stabilised by a combination of CO⋯π(cyclobutene) and anti-parallel carbonyl⋯carbonyl interactions. The BCPs present within the stacked layers of cyclobutene rings are discerned in Fig. 12(b), consistent with offset π-stacking interactions between the cyclobutene rings.
 | ||
| Fig. 12 QTAIM analysis calculated at the wB97XD level of theory and 6-31G** and CEP-121G basis sets, and with BCPs represented as blue spheres and bond paths as dotted orange lines highlighting (a) one anti-parallel carbonyl⋯carbonyl interaction in PhHgLMe and (b) C⋯C contacts within the stacking of cyclobutene rings. | ||
From the QTAIM analysis, in contrast to the DFT study, estimations of the energies associated with the intramolecular Hg⋯O spodium bonds are possible. The energies associated with the intermolecular Hg⋯O spodium bonding approximate those determined from the DFT study (see above). For the first independent molecule of PhHgLMe, the energy associated with the intermolecular Hg⋯O spodium bond of 23.3 kJ mol−1 is less than the stabilisation provided by the intramolecular Hg⋯O spodium bond (15.4 + 15.6 = 31.0 kJ mol−1). The two energies for the intramolecular Hg⋯O spodium bond imply the participation of two oxygen-bound lone pairs in this interaction; see natural bond orbital (NBO) analysis below. For the second independent molecule of PhHgLMe, the respective energies are 19.1, 21.7 and 21.9 kJ mol−1, and for PhHgLEt, these are 14.4, 17.5 and 17.5 kJ mol−1, respectively. The two Hg⋯S spodium bonds in PhHgLEt each have an energy of 11.0 kJ mol−1, indicating that these provide less stabilisation to the molecular packing than the Hg⋯O spodium bonds. Further, in PhHgLEt, the energy of stabilisation provided by the O2⋯S1 chalcogen bond is computed to be 12.5 kJ mol−1.
The nature and relative strength of the Hg⋯O/S spodium bonds were further explored by calculating the Wiberg bond indices, Mayer bond orders and delocalisation indices; data are collated in ESI† Table S3. For the first independent molecule of PhHgLMe, the Wiberg bond indices, obtained from the natural population analysis, show that the intramolecular Hg⋯O spodium bond order (bond index = 0.077) is greater than the intermolecular Hg⋯O spodium bond order (bond index = 0.070), while the Mayer bond orders were relatively greater with values of 0.144 and 0.117, respectively; similar observations apply to the second independent molecule of PhHgLMe and to PhHgLEt.
Delocalisation index (DI) calculations provide insight into the number of electrons shared or exchanged between the interacting atoms. For PhHgLMe, the calculations indicate that DI(Hg, O) = 0.135 and 0.148 for the intra- and intermolecular Hg⋯O spodium bonds, respectively, suggesting appreciable delocalisation in the intermolecular spodium bond; the same applies to the second independent molecule of PhHgLMe. The DI values calculated for PhHgLEt are 0.140 and 0.095, respectively, indicating a reverse trend compared with PhHgLMe. The greater delocalisation of electron density in the intermolecular Hg⋯O spodium bonds in PhHgLMevs.PhHgLEt are consistent with the formation of the complementary O⋯S chalcogen bond and the additional Hg⋯S spodium bonds in the latter.
The final analysis to be conducted involved the calculation of the NBOs to analyse the nature of the electron transfer between the atoms involved in the Hg⋯O/S spodium bonding and O⋯S chalcogen bonding. The NBOs were calculated on the two molecule aggregates as for the QTAIM analysis. A listing of selected identified electron-transfer processes, their correlation with spodium/chalcogen bonding and associated energies is given in Table 2.
| Bond (symmetry operation) | Electron transfera | Designation | Energy (kJ mol−1) |
|---|---|---|---|
| a LP = lone pair. | |||
| PhHgL Me – first independent molecule | |||
| Hg1⋯O1 | LP(O) → σ*(Hg–C) | π-Spodium bond | 5.44 |
| Hg1i⋯O1i (i: ½ + x, ½ + y, z) | LP(O) → σ*(Hg–C) | π-Spodium bond | 6.36 |
| Hg1⋯O2i (i: ½ + x, ½ + y, z) | LP(O) → σ*(Hg–C) | σ-Spodium bond | 3.64 |
| PhHgL Me – second independent molecule | |||
| Hg2⋯O4 | LP(O) → σ*(Hg–C) | π-Spodium bond | 7.49 |
| Hg2i⋯O4i (i: −½ + x, −½ + y, z) | LP(O) → σ*(Hg–C) | π-Spodium bond | 8.75 |
| Hg2⋯O5i (i: −½ + x, −½ + y, z) | LP(O) → σ*(Hg–C) | σ-Spodium bond | 3.85 |
| PhHgL Et | |||
| Hg⋯O1 | LP(O) → σ*(Hg–C) | π-Spodium bond | 5.82 |
| Hgi⋯O1i (i: x, −1 + y, z) | LP(O) → σ*(Hg–C) | π-Spodium bond | 6.48 |
| Hg⋯O2i (i: x, −1 + y, z) | LP(O) → σ*(Hg–C) | σ-Spodium bond | 3.51 |
| Hg⋯S1ii (ii: 1 − x, 2 − y, 1 − z) | LP(S) → σ*(Hg–C) | π-Spodium bond | 0.46 |
| Hg⋯S1iii (iii: − x, 2 − y, 1 − z) | LP(S) → σ*(Hg–C) | π-Spodium bond | 0.46 |
| O2⋯S1i (i: x, −1 + y, z) | LP(O) → σ*(C–S) | σ-Chalcogen bond | 0.54 |
The NBOs calculated for the intra- and intermolecular Hg⋯O spodium bonding for the independent molecules of PhHgLMe (ESI† Fig. S15) and PhHgLEt (Fig. 13(a)) involve electron transfer from the lone pairs (LPs) of oxygen to the anti-bonding orbital of the Hg–C bond, i.e. LP(O) → σ*(Hg–C); this image clearly demonstrates the interaction of two distinct oxygen-bound lone pairs in the intramolecular Hg⋯O spodium bonding. With respect to the energies, while magnitudes vary, the same trends are apparent. Thus, the energies associated with the intramolecular electron transfer, e.g. 5.82 and 6.48 kJ mol−1 for PhHgLEt, are always greater than that for the intermolecular electron transfer, e.g. 3.51 kJ mol−1 for PhHgLEt.
 | ||
| Fig. 13 Illustrations of selected NBOs computed for PhHgLEt highlighting (a) intra- and intermolecular donor/acceptor interactions associated with the Hg⋯O spodium bonding, (b) intermolecular Hg⋯S spodium bonding and (c) intermolecular O⋯S chalcogen bonding. The second-order perturbation energies are indicated and have units of kJ mol−1. | ||
Fig. 13(b) shows the NBOs associated with one of the {⋯HgS}2 synthons arising from the intermolecular Hg⋯S spodium bonding with the electron transfer assigned to LP(S) → σ*(Hg–C). The energies associated with the electron transfer is approximately an order of magnitude less than for the electron transfer involved with the Hg⋯O spodium bonding, i.e. 0.46 kJ mol−1. The NBOs for the electron transfer leading to O⋯S chalcogen bonding are shown in Fig. 13(c). The electron transfer is assigned as LP(O) → σ*(C1–S1) and has an energy of 0.54 kJ mol−1, slightly greater than those associated with each Hg⋯S spodium bond.
3. Conclusions
Two essentially linear, one-dimensional coordination polymers have been characterised, i.e. PhHg(3-alkoxycyclobutene-1,2-dione-4-thiolate) (alkoxy = OMe and OEt). The nature and strength of the intramolecular Hg⋯O π-spodium bonds and intermolecular Hg⋯O σ-spodium bonds noted in both coordination polymers have been analysed. Also, for the ethoxy species, Hg⋯S π-spodium bonds and O⋯S σ-chalcogen bonds present in the crystal have been detailed. For the spodium bonding, the donor/acceptor interactions arise from the lone pair of electrons residing on oxygen or sulphur to an anti-bonding orbital of the Hg–C bond. For the chalcogen bonding, an oxygen bond lone pair is donated to an anti-bonding orbital of the C–C bond. The energies for the Hg⋯O σ-spodium bonds (17.5 to 23.3 kJ mol−1) were systematically greater than for the Hg⋯O π-spodium bonds (14.4 to 19.1 kJ mol−1), Hg⋯S π-hole spodium bonds (11.0 kJ mol−1) and O⋯S chalcogen bonds (12.5 kJ mol−1), pointing to a definitive structure-directing role for Hg⋯O σ-spodium bonding. It is likely that the subtle differences in the molecular packing arise from the greater electron-donating ability of the methoxy vs. ethoxy substituent.4. Experimental
4.1 General considerations
All synthetic procedures were carried out under a nitrogen atmosphere. All solvents used in the syntheses and measurements were distilled and dried prior to use using standard methods. The melting points were measured on an Ambassador melting point apparatus. Microanalyses were executed on a Model CE-440 CHN analyser. The 1H and 13C{1H} NMR spectra were collected in CDCl3 solution on a Bruker Avance III HD 300 MHz spectrometer. UV/vis spectra in dichloromethane solutions were recorded on a SPECORD 210 Plus BU spectrophotometer.4.2 Syntheses
To a methanol solution of sodium 3-methoxycyclobutene-1,2-dione-4-thiolate (0.042 g, 0.25 mmol)/sodium 3-ethoxycyclobutene-1,2-dione-4-thiolate (0.045 g, 0.25 mmol) was added a dichloromethane solution (10 ml) of PhHgO2CCH3 (0.084 g, 0.25 mmol) followed by stirring in an ice-bath for 4 h. Thereafter, the reaction mixtures were rotary evaporated and dissolved in a minimum amount of dichloromethane. To these solutions, diethyl ether was added to precipitate the desired compounds, i.e.PhHgLMe and PhHgLEt, respectively. The precipitates were filtered and washed twice with diethyl ether (2 × 10 ml) and dried in a desiccator.4.3 Characterisation data
PhHgL Me : light-yellow solid. Yield: 0.094 g, 90%. M. pt: 441 K. Anal. calc. for C11H8HgO3S: C, 31.39; H, 1.92; S, 7.62%. Found: C, 31.54; H, 2.04; S, 7.98%. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.49–7.44 (m, 5H, –C6H5), 4.51 (s, 3H, –OCH3). 13C{1H} NMR (CDCl3, 75.5 MHz, ppm): δ 207.2 (C–S), 170.4 (ring-C–O), 137.6–128.4 (C6H5), 65.9 (OCH3); the resonances due to (CO) were not resolved.
PhHgL
Et
: dark-yellow solid. Yield: 0.088 g, 81%. M. pt: 337 K. Anal. calc. for C12H10HgO3S: C, 33.14; H, 2.32; S, 7.37%. Found: C, 33.35; H, 2.41; S, 7.63%. 1H NMR (CDCl3, 300 MHz, ppm): δ 7.42–7.34 (m, 5H, C6H5), 4.79 (q, 2H, OCH2, J = 7.1 Hz), 1.45 (t, 3H, CH3, J = 3 Hz). 13C{1H} NMR (CDCl3, 75.5 MHz, ppm): δ 196.6 (C–S), 184.5, 182.2 (CO), 169.4 (ring-C–O), 136.6–127.3 (C6H5), 70.2 (OCH2), 14.7 (CH3).
4.4 X-ray crystallography
Intensity data for thin plates of PhHgLMe and PhHgLEt were collected at 150(2) K on a Rigaku SuperNova, Dual, EosS2 and on an Xcalibur, EosS2 single crystal diffractometer using monochromated Cu-Kα (λ = 1.54184 Å) and Mo-Kα radiation (λ = 0.71073 Å), respectively. Unit cell determination, data collection and data reduction were performed using the CrysAlis Pro software.25 The structures were solved with SHELXT26 and refined by a full-matrix least-squares procedure based on F2 (SHELXL-2017/1).27 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in their calculated positions and refined in the riding model approximation. A weighting scheme of the form w = 1/[σ2(Fo2) + (aP)2 + bP], where P = (Fo2 + 2Fc2/3), was introduced towards the conclusion of each refinement. The data sets were problematic owing to the presence of the heavy atom absorber mercury in the plate-like crystals.The sample of PhHgLMe was refined as an inversion twin in the ratio 0.67(4)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.33(4). The carbon and oxygen atoms were restrained to be nearly isotropic, and the phenyl ring was restrained to be approximately flat. The formally single and double bonds in the four-membered rings were restrained to 1.50 ± 0.01 and 1.38 ± 0.01 Å, respectively. The maximum and minimum residual electron density peaks of 1.13 and 2.00 e Å−3, respectively, were located 1.60 and 0.84 Å from the Hg1 atom. A comparative anisotropic refinement (employing the same restraints as for the model in Cc) in the centrosymmetric equivalent of Cc, i.e. C2/c with Z′ = 1, resulted in inferior results, e.g. R(obs. data) = 0.075, Rw(all data) = 0.236 and with maximum and minimum residual electron density peaks of 1.45 and 2.89 e Å−3, respectively.
:0.33(4). The carbon and oxygen atoms were restrained to be nearly isotropic, and the phenyl ring was restrained to be approximately flat. The formally single and double bonds in the four-membered rings were restrained to 1.50 ± 0.01 and 1.38 ± 0.01 Å, respectively. The maximum and minimum residual electron density peaks of 1.13 and 2.00 e Å−3, respectively, were located 1.60 and 0.84 Å from the Hg1 atom. A comparative anisotropic refinement (employing the same restraints as for the model in Cc) in the centrosymmetric equivalent of Cc, i.e. C2/c with Z′ = 1, resulted in inferior results, e.g. R(obs. data) = 0.075, Rw(all data) = 0.236 and with maximum and minimum residual electron density peaks of 1.45 and 2.89 e Å−3, respectively.
For PhHgLEt, each of the mercury-bound phenyl rings and oxygen-bound ethyl substituents were statistically disordered. The phenyl-carbon atoms were restrained to be nearly isotropic. The maximum and minimum residual electron density peaks of 1.18 and 0.80 e Å−3, respectively, were located 0.97 and 1.57 Å from the Hg atom.
The molecular structure diagrams were generated with ORTEP for Windows28 with 35% displacement ellipsoids, and the packing diagrams were drawn with DIAMOND.29 Additional data analysis was made with PLATON.17 Crystallographic data and refinement details are presented in Table 3.
| Compound | PhHgL Me | PhHgL Et |
|---|---|---|
| Formula | C11H8HgO3S | C12H10HgO3S |
| Molecular weight | 420.82 | 434.85 |
| Crystal size | 0.02 × 0.15 × 0.15 | 0.02 × 0.16 × 0.21 |
| Crystal system | Monoclinic | Triclinic |
| Space group | Cc |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 12.7721(3) | 6.1120(2) |
| b (Å) | 9.5804(2) | 7.8713(3) |
| c (Å) | 19.5867(4) | 14.3546(8) |
| α (°) | 90 | 101.423(4) |
| β (°) | 101.106(2) | 102.236(4) |
| γ (°) | 90 | 90.002(3) |
| V (Å3) | 2351.78(9) | 660.90(5) |
| Z, Z' | 8, 2 | 2, 1 |
| D x (g cm–3) | 2.377 | 2.185 |
| F(000) | 1552 | 404 |
| μ (mm–1) | 25.062 | 11.794 |
| No. reflections | 9204 | 10155 |
| No. unique reflections | 3832 | 2707 |
| No. reflections with I ≥ 2σ(I) | 3757 | 2393 |
| R (obs. data) | 0.040 | 0.040 |
| a and b in weighting scheme | 0.035, 21.797 | 0.030, 0 |
| R w (all data) | 0.101 | 0.075 |
| Max. and min. residual peaks (e Å−3) | 1.13; −2.00 | 1.18; −0.81 |
4.5 Computational details
The gas-phase molecular geometries of the monomer, dimers and pairs of dimer units of both organomercury(II) polymers were optimised using density functional theory (DFT) employing the wB97XD functional.23 For all atoms except Hg, the 6-31G** basis set was used, while for Hg, the CEP-121G basis set was employed. To calculate the interaction energies of the dimers held by non-covalent interactions, the interaction distances were fixed and the remaining degrees of freedom were relaxed during geometry optimisation. The stabilisation energies (ΔEdimer) for each dimeric entity were calculated using the formula ΔEdimer = Edimer − (2 × Emonomer), where Emonomer and Edimer are the energies of the monomer and dimer entities, respectively. Since the non-covalent interactions are considerably weaker than either ionic or covalent bonding, the basis set superposition error (BSSE) corrections were accounted for during the calculations employing the Boys–Bernardi scheme.30 All computations were performed using the Gaussian 09 revision B.01 program.31 The NBO calculations were conducted with NBO Version 3.1 as implemented in the Gaussian 09 suite of programs.32 The QTAIM analyses were performed using the AIMALL package (version 10.05.04).33a The NCI-RDG analysis was performed using the Multiwfn software33b and the VMD program was used to create 3D isosurfaces.33c Additionally, the coloured NCI plots were created using GNU plot.33d ESI† Table S4 comprises a listing of the coordinates for the calculated molecules and aggregates.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Aparna Kushwaha: syntheses and computational investigation. Devyani Srivastava: synthesis and spectroscopic studies. Gabriele Kociok-Köhn: X-ray data collection and refinement. Sarfaraz Ahmed: data analysis. Edward R. T. Tiekink: data analysis, writing – original draft. Abhinav Kumar: conceptualisation, supervision, data analysis, writing – original draft.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
AK acknowledges the Council of Science and Technology, Uttar Pradesh for sanctioning project ID 3701. The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R380), King Saud University, Riyadh, Saudi Arabia.References
- (a) D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen and S. L. Price, Innovation in crystal engineering, CrystEngComm, 2002, 4, 500–509, 10.1039/B207466B; (b) E. R. T. Tiekink, Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists, Coord. Chem. Rev., 2017, 345, 209–228, DOI:10.1016/j.ccr.2017.01.009; (c) A. V. Vologzhanina, Intermolecular interactions in functional crystalline materials: from data to knowledge, Crystals, 2019, 9, 478, DOI:10.3390/cryst9090478; (d) M. K. Corpinot and D.-K. Bučar, A practical guide to the design of molecular crystals, Cryst. Growth Des., 2019, 19, 1426–1453, DOI:10.1021/acs.cgd.8b00972; (e) D. Braga, Crystal engineering: from promise to delivery, Chem. Commun., 2023, 59, 14052–14062, 10.1039/d3cc04313d.
- (a) W. Yao, C. Ma, H. Ma, L. Fu, S. Lu, A. Lv, S. Cai, X. Hang, M. Singh, H. Shi, Z. An and W. Huang, Supramolecular organic frameworks with ultralong phosphorescence via breaking π-conjugated structures, Giant, 2020, 1, 100007, DOI:10.1016/j.giant.2020.100007; (b) J. D. Fernandes, M. D. Maximino, M. L. Braunger, M. S. Pereira, C. A. Olivati, C. J. Constantino and P. Alessio, Supramolecular architecture and electrical conductivity in organic semiconducting thin films, Phys. Chem. Chem. Phys., 2020, 22, 13554–13562, 10.1039/D0CP01293A; (c) J. Sautaux, F. Marx, I. Gunkel, C. Weder and S. Schretti, Mechanically robust supramolecular polymer co-assemblies, Nat. Commun., 2022, 13, 356, DOI:10.1038/s41467-022-28017-0; (d) G. Zhang, X. Li, G. Chen, Y. Zhang, M. Wei, X. Chen, B. Li, Y. Wu and L. Wu, Supramolecular framework membrane for precise sieving of small molecules, nanoparticles and proteins, Nat. Commun., 2023, 14, 975, DOI:10.1038/s41467-023-36684-w.
- (a) J. S. González-González, N. E. Magaña-Vergara, E. V. García-Báez, I. I. Padilla-Martínez, J. P. Mojica-Sánchez and F. J. Martínez-Martínez, Crystal structure and supramolecular architecture of antiallergic diphenylene diethyl dioxalamates, Crystals, 2020, 10, 1048, DOI:10.3390/cryst10111048; (b) Z.-T. Li, S.-B. Yu, Y. Liu, J. Tian and D.-W. Zhang, Cucurbituril homologues and derivatives: new opportunities in supramolecular chemistry, Acc. Chem. Res., 2022, 55, 2316–2325, DOI:10.1021/ar020254k; (c) M. Moradi, N. L. Lengweiler, C. E. Housecroft, L. G. Tulli, H. Stahlberg, T. A. Jung and P. Shahgaldian, Coordination-driven monolayer-to-bilayer transition in two-dimensional metal–organic networks, J. Phys. Chem. B, 2021, 125, 4204–4211, DOI:10.1021/acs.jpcb.1c01058; (d) C.-F. Ng, H.-F. Chow and T. C. W. Mak, Hydrogen-bonded supramolecular architecture by solvomorphism of hexahydroxytriptindane: from a rosette network to a rod structure, Cryst. Growth Des., 2022, 22, 3365–3375, DOI:10.1021/acs.cgd.2c00164.
- (a) T. S. B. Baul, S. Kundu, S. Mitra, H. Höpfl, E. R. T. Tiekink and A. Linden, The influence of counter ion and ligand methyl substitution on the solid-state structures and photophysical properties of mercury(II) complexes with (E)-N-(pyridin-2-ylmethylidene)arylamines, Dalton Trans., 2013, 42, 1905–1920, 10.1039/C2DT32283H; (b) Z.-Y. Ye, Z.-Y. Zhang, L.-H. Huo, Z.-P. Deng, X.-F. Zhang and S. Gao, Mercury supramolecular architectures constructed from flexible unsymmetrical bis(pyridyl) ligands: structural motif mediated by mercury-halide clusters, ligand conformations and non-covalent interactions, Polyhedron, 2016, 117, 338–351, DOI:10.1016/j.poly.2016.05.067; (c) A. Angeloski, A. Rawal, M. Bhadbhade, J. M. Hook, R. W. Schurko and A. M. McDonagh, An unusual mercury(II) diisopropyldithiocarbamate coordination polymer, Cryst. Growth Des., 2019, 19, 1125–1133, DOI:10.1021/acs.cgd.8b01619; (d) P. Kumar, S. Banerjee, A. Radha, T. Firdoos, S. C. Sahoo and S. K. Pandey, Role of non-covalent interactions in the supramolecular architectures of mercury(II) diphenyldithiophosphates: an experimental and theoretical investigation, New J. Chem., 2021, 45, 2249–2263, 10.1039/D0NJ05709F; (e) W. Wang, D. Dai, J. Wu, C. Wang, Y. Wang and Y. Yang, Recyclable supramolecular assembly-induced emission system for selective detection and efficient removal of mercury(II), Chem. – Eur. J., 2021, 27, 11879–11887, DOI:10.1002/chem.202101437; (f) F. M. Dolgushin and I. L. Eremenko, Structural chemistry of host–guest molecular architectures based on mercury-containing macrocycles, Russ. Chem. Rev., 2021, 90, 1493–1519, DOI:10.1070/RCR4998.
- (a) R. Eisenberg, Structural systematics of 1,1- and 1,2-dithiolato chelates, Prog. Inorg. Chem., 1970, 12, 295–369, DOI:10.1002/978047016613; (b) D. Cocouvanis, The chemistry of the dithioacid and 1,1-dithiolate complexes, Prog. Inorg. Chem., 1979, 26, 301–371, DOI:10.1002/978047016612; (c) E. R. T. Tiekink and G. Winter, Inorganic xanthates: a structural perspective, Rev. Inorg. Chem., 1992, 12, 183–302, DOI:10.1515/REVIC.1992.12.3-4.183; (d) I. Haiduc, D. B. Sowerby and S.-F. Lu, Stereochemical aspects of phosphor-1,1-dithiolato metal complexes (dithiophosphates, dithiophosphinates): Coordination patterns, molecular structures and supramolecular associations–I, Polyhedron, 1995, 14, 3389–3472, DOI:10.1016/0277-5387(95)00108-5; (e) W. Paw, S. D. Cummings, M. A. Mansour, W. B. Connick, D. K. Geiger and R. Eisenberg, Luminescent platinum complexes: tuning and using the excited state, Coord. Chem. Rev., 1998, 171, 125–150, DOI:10.1016/S0010-8545(98)90023-6; (f) N. Robertson and L. Cronin, Metal bis-1,2-dithiolene complexes in conducting or magnetic crystalline assemblies, Coord. Chem. Rev., 2002, 227, 93–127, DOI:10.1016/S0010-8545(01)00457-X; (g) A. Kobayashi, E. Fujiwara and H. Kobayashi, Single-component molecular metals with extended-ttf dithiolate ligands, Chem. Rev., 2004, 104, 5243–5264, DOI:10.1021/cr030656l; (h) E. R. T. Tiekink and I. Haiduc, Stereochemical aspects of metal xanthate complexes: Molecular structures and supramolecular self-assembly, Prog. Inorg. Chem., 2005, 54, 127–319, DOI:10.1002/0471725560.ch3.
- (a) A. M. Bond and R. L. Martin, Electrochemistry and redox behaviour of transition metal dithiocarbamates, Coord. Chem. Rev., 1984, 54, 23–98, DOI:10.1016/0010-8545(84)85017-1; (b) E. R. T. Tiekink, Molecular architecture and supramolecular association in the zinc-triad 1,1-dithiolates. Steric control as a design element in crystal engineering?, CrystEngComm, 2003, 5, 101–113, 10.1039/B301318A; (c) P. J. Heard, Main group dithiocarbamate complexes, Prog. Inorg. Chem., 2005, 53, 1–70, DOI:10.1002/0471725587.ch1; (d) G. Hogarth, Transition metal dithiocarbamates: 1978–2003, Prog. Inorg. Chem., 2005, 53, 71–561, DOI:10.1002/0471725587.ch2; (e) W. W. H. Wong, J. Cookson, E. A. L. Evans, E. J. L. McInnes, J. Wolowska, J. P. Maher, P. Bishop and P. D. Beer, Heteropolymetallic copper(II)–gold(III) dithiocarbamate [2]catenanes via magic ring synthesis, Chem. Commun., 2005, 2214–2216, 10.1039/B418453J; (f) J. D. E. T. Wilton-Ely, D. Solanki, E. R. Knight, K. B. Holt, A. L. Thompson and G. Hogarth, Multimetallic assemblies using piperazine-based dithiocarbamate building blocks, Inorg. Chem., 2008, 47, 9642–9653, DOI:10.1021/ic800398b; (g) E. R. T. Tiekink, Exploring the topological landscape exhibited by binary zinc-triad 1,1-dithiolates, Crystals, 2018, 8, 292, DOI:10.3390/cryst8070292; (h) E. R. T. Tiekink, On the coordination role of pyridyl-nitrogen in the structural chemistry of pyridyl-substituted dithiocarbamate ligands, Crystals, 2021, 11, 286, DOI:10.3390/cryst11030286; (i) S. M. Lee and E. R. T. Tiekink, A structural survey of poly-functional dithiocarbamate ligands and the aggregation patterns they sustain, Crystals, 2021, 11, 433, DOI:10.3390/cryst11040433; (j) J. C. Sarker and G. Hogarth, Dithiocarbamate complexes as single source precursors to nanoscale binary, ternary and quaternary metal sulfides, Chem. Rev., 2021, 121, 6057–6123, DOI:10.1021/acs.chemrev.0c01183; (k) C. I. Yeo, E. R. T. Tiekink and J. Chew, Insights into the antimicrobial potential of dithiocarbamate anions and metal-based species, Inorganics, 2021, 9, 48, DOI:10.3390/inorganics9060048.
- (a) C. Eaborn, A. Pidcock and B. R. Steele, Oxidative addition of triorganotin halides to platinum(0) complexes, J. Chem. Soc., Dalton Trans., 1976, 767–776, 10.1039/DT9760000767; (b) C. Eaborn, K. Kundu and A. Pidcock, Synthesis of platinum(II) alkyl and aryl complexes from K2[PtCl4] and tetraorganotin compounds in dimethyl sulphoxide, J. Chem. Soc., Dalton Trans., 1981, 933–938, 10.1039/DT9810000933; (c) A. F. M. J. van der Ploeg, G. van Koten and K. Vrieze, Aspects of transmetallation reactions of 2-Me2NCH2C6H4- and 2,6-(Me2NCH2)C6H3-Metal (Pd, Pt, Hg, Tl) complexes with metal carboxylates and low-valent metal (Pd, Pt) complexes, J. Organomet. Chem., 1981, 222, 155–174, DOI:10.1016/S0022-328X(00)89024-5; (d) G. K. Anderson, Preparation of chloride-bridged organopalladium(II) dimers and their role in the carbonylation of trans-[PdClR'(PR3)2], Organometallics, 1983, 2, 665–668, DOI:10.1021/om00077a015; (e) R. J. Cross and J. Gemmill, Stereospecific transfer reactions of phenyl and ethynyl groups between platinum(II) atoms, J. Chem. Soc., Dalton Trans., 1984, 205–209, 10.1039/DT9840000205; (f) E. Wehman, G. van Koten, J. T. B. H. Jastrzebski, H. Ossor and M. Pfeffer, Synthesis of cyclometallated naphthylpalladium complexes via transmetallation reactions of naphthylmercury with palladium species. Crystal structure determination of [{Pd(mxn)(O2CMe)}2], J. Chem. Soc., Dalton Trans., 1988, 2975–2981, 10.1039/DT9880002975.
- (a) R. C. Larock, Organomercury compounds in organic synthesis, Angew. Chem., Int. Ed. Engl., 1978, 17, 27–37, DOI:10.1002/anie.197800271; (b) J. G. Melnick and G. Parkin, Cleaving mercury-alkyl bonds: A functional model for mercury detoxification by MerB, Science, 2007, 317, 225–227, DOI:10.1126/science.
- (a) E. R. T. Tiekink, Phenylmercury dithiolates. The crystal and molecular structures of (C6H5Hg(S2COR)) (R = Me; iPr and (C6H5)Hg(S2CNEt2)), J. Organomet. Chem., 1987, 322, 1–10, DOI:10.1016/0022-328X(87)85018-0; (b) C. S. Lai and E. R. T. Tiekink, Supramolecular association in organomercury(II) 1,1-dithiolates. Complementarity between Hg⋯S and hydrogen bonding interactions in organomercury(II) 2-amino-cyclopent-1-ene-1-carbodithioates, CrystEngComm, 2003, 5, 253–261, 10.1039/B305363F; (c) N. Singh, A. Kumar, K. C. Molloy and M. F. Mahon, Syntheses, crystal and molecular structures, and properties of some new phenylmercury(II) dithiolate complexes, Dalton Trans., 2008, 4999–5007, 10.1039/B804635B; (d) N. Singh, A. Kumar, R. Prasad, K. C. Molloy and M. F. Mahon, Syntheses, crystal, photoluminescence and electrochemical investigation of some new phenylmercury(II) dithiocarbamate complexes involving ferrocene, Dalton Trans., 2010, 39, 2667–2675, 10.1039/B917871F; (e) V. Singh, R. Chauhan, A. Kumar, L. Bahadur and N. Singh, Efficient phenylmercury(II) methylferrocenyldithiocarbamate functionalized dye-sensitized solar cells, Dalton Trans., 2010, 39, 9779–9788, 10.1039/C0DT00575D; (f) V. Singh, A. Kumar, R. Prasad, G. Rajput, M. G. B. Drew and N. Singh, The interplay of secondary Hg⋯S, Hg⋯N and Hg⋯π bonding interactions in supramolecular structures of phenylmercury(II) dithiocarbamates, CrystEngComm, 2011, 13, 6817–6826, 10.1039/C1CE05678F; (g) E. R. T. Tiekink, Supramolecular association via Hg⋯S secondary-bonding interactions in crystals of organomercury(II) species: a survey of the Cambridge Structure Database, Crystals, 2023, 13, 385, DOI:10.3390/cryst13030385.
- N. W. Alcock, Secondary bonding to nonmetallic elements, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58, DOI:10.1016/S0065-2792(08)60016-3.
- A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Spodium bonds: noncovalent interactions involving Group 12 elements, Angew. Chem., Int. Ed., 2020, 59, 17482–17487, DOI:10.1002/anie.202007814.
- (a) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, Halogen bonding: the sigma-hole, J. Mol. Model., 2007, 13, 291–296, DOI:10.1007/s00894-006-0130-2; (b) P. Politzer, J. S. Murray, T. Clark and G. Resnati, The σ-hole revisited, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178, 10.1039/C7CP06793C; (c) J. S. Murray and P. Politzer, Interaction and polarization energy relationships in σ-hole and π-hole bonding, Crystals, 2020, 10, 76, DOI:10.3390/cryst10020076.
- (a) R. M. Gomila, A. Bauzá, T. J. Mooibroek and A. Frontera, Spodium bonding in five coordinated Zn(II): A new player in crystal engineering?, CrystEngComm, 2021, 23, 3084–3093, 10.1039/d1ce00221j; (b) H. S. Biswal, A. K. Sahu, A. Frontera and A. Bauzá, Spodium bonds in biological systems: expanding the role of Zn in protein structure and function, J. Chem. Inf. Model., 2021, 61, 3945–3954, DOI:10.1021/acs.jcim.1c00594R; (c) G. Ciancaleoni and L. Rocchigiani, Assessing the orbital contribution in the “spodium bond” by natural orbital for chemical valence−charge displacement analysis, Inorg. Chem., 2021, 60, 4683–4692, DOI:10.1021/acs.inorgchem.0c03650; (d) N. Liu, X. Xie and Q. Li, Chalcogen bond involving zinc(II)/cadmium(II) carbonate and its enhancement by spodium bond, Molecules, 2021, 26, 6443, DOI:10.3390/molecules26216443; (e) A. Singh, G. Kociok-Köhn, A. Dutta, A. Kumar and M. Muddassir, Diaminopyridine Hg(II)-based 1D supramolecular polymer: crystallographic and computational insights into spodium bonding, J. Solid State Chem., 2022, 315, 123517, DOI:10.1016/j.jssc.2022.123517; (f) N. Liu, Q. Li and S. Scheiner, Spodium and tetrel bonds involving Zn(II)/Cd(II) and their interplay, Chem. Phys., 2022, 556, 111470, DOI:10.1016/j.chemphys.2022.111470; (g) M. Gao, Q. Zhao, H. Yu, M. Fu and Q. Li, Insight into spodium–π bonding characteristics of the MX2⋯π (M = Zn, Cd and Hg; X = Cl, Br and I) complexes – a theoretical study, Molecules, 2022, 27, 2885, DOI:10.3390/molecules27092885; (h) A. V. Rozhkov, E. A. Katlenok, M. V. Zhmykhova, M. L. Kuznetsov, V. N. Khrustalev, K. I. Tugashov, N. A. Bokach and V. Y. Kukushkin, Spodium bonding to anticrown-Hg3 boosts phosphorescence of cyclometalated-PtII complexes, Inorg. Chem. Front., 2023, 10, 493–510, 10.1039/D2QI02047E; (i) Q. Wu, S. A. C. McDowell and Q. Li, Single-electron spodium bonds: substituent effects, Appl. Organomet. Chem., 2023, 37, e7052, DOI:10.1002/aoc.7052; (j) D. Majumdar, A. Frontera, S. Roy and D. Sutradhar, Experimental and theoretical survey of intramolecular spodium bonds/σ/π-holes and noncovalent interactions in trinuclear Zn(II)-salen type complex with OCN− ions: a holistic view in crystal engineering, ACS Omega, 2024, 9, 1786–1797, DOI:10.1021/acsomega.3c08422; (k) M. Gomila, A. Frontera and E. R. T. Tiekink, Supramolecular aggregation featuring Hg⋯S secondary-bonding interactions in crystals of mercury(II) species augmented by computational chemistry calculations, CrystEngComm, 2023, 25, 5262–5285, 10.1039/d3ce00590a; (l) R. M. Gomila, E. R. T. Tiekink and A. Frontera, A computational chemistry investigation of the influence of steric bulk of dithiocarbamato-bound organic substituents upon spodium bonding in three homoleptic mercury(II) bis(N,N-dialkyldithiocarbamato) compounds for alkyl = ethyl, isobutyl, and cyclohexyl, Inorganics, 2023, 11, 468, DOI:10.3390/inorganics11120468; (m) C. S. Onn, A. F. Hill and J. S. Ward, Spodium bonding in bis(alkynyl)mecurials, Chem. Commun., 2024, 60, 2552–2555, 10.1039/d3cc06027f.
- (a) D. Eggerding and R. West, Synthesis of the monothiosquarate and 1,2-dithiosquarate ions and their derivatives, J. Org. Chem., 1976, 41, 3904–3909, DOI:10.1021/jo00886a027; (b) R. Krause and R. Mattes, Dithioquadratato-komplexe von kupfer(ii) und kupfer(I). Darstellung und struktur von [CuII(en)2][CuII(C4O2S2)2] und [CuII(en)2(H2O)]2[CuI4(C4O2S2)4].2H2O, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1990, 45, 490–496 CrossRef CAS; (c) M. L. Calatayud, I. Castro, J. Sletten, J. Cano, F. Lloret, J. Faus, M. Julve, G. Seitz and K. Mann, Coordination modes of the 1,3-dithiosquarate (1,3-dtsq) ligand. Syntheses, crystal structures, and magnetic properties of [Ni(tren)(1,3-dtsq)(H2O)] and [Ni2(tren)2(1,3-dtsq)](ClO4)2 [tren = Tris(2-aminoethyl)amine], Inorg. Chem., 1996, 35, 2858–2865, DOI:10.1021/ic951393j; (d) I. Castro, M. L. Calatayud, J. Sletten, J. Cano, F. Lloret, M. Julve, G. Seitz and K. Mann, Ferro- and antiferromagnetic coupling through 1,n-dithiosquarate bridges (n = 2, 3) in copper(II) dimers. Syntheses, crystal structures, and magnetic properties of [Cu2(bpca)2(1,2-dtsq)(H2O)]·2H2O and [Cu2(bpca)2(1,3-dtsq)]·2H2O, Inorg. Chem., 1999, 38, 4680–4687, DOI:10.1021/ic9904384; (e) B. Wenzel, B. Wehse, U. Schilde and P. Strauch, 1,2-Dithioquadratato- und 1,2-dithiooxalatoindate(III), Z. Anorg. Allg. Chem., 2004, 630, 1469–1476, DOI:10.1002/zaac.200400143; (f) M. L. Calatayud, I. Castro, M. Julve and J. Sletten, Cu(II) and Cu(I) complexes with 1,2-dithiosquarate as a ligand; from molecular compounds to supramolecular network structures, J. Mol. Struct., 2008, 876, 328–338, DOI:10.1016/j.molstruc.2007.07.001; (g) P. Strauch, M. Neumann, A. Kelling and U. Schilde, Bis(1,2-dithiosquarato)nickelates(II): Synthesis, structure, EPR and thermal behavior, Acta Chim. Slov., 2015, 62, 288–296 CrossRef CAS PubMed.
- (a) D. Coucouvanis, F. J. Hollander, R. West and D. Eggerdling, Synthesis of 1,2-dithiosquarate salts and x-ray crystal structure of potassium bis(dithiosquarato)nickelate(II), J. Am. Chem. Soc., 1974, 96, 3006–3008, DOI:10.1021/ja00816a064; (b) D. Coucouvanis, D. G. Iiolah and F. J. Hollander, Metal dithiosquarates. II. Synthesis and characterization of bis(dithiosquarato) tetrahedral and planar complexes. Crystal and molecular structure of potassium bis(dithiosquarato)nickelate(II) dihydrate, Inorg. Chem., 1975, 14, 2657–2665, DOI:10.1021/ic50153a013.
- (a) D. Srivastava, A. Kushwaha, G. Kociok-Köhn, S. W. Gosavi, R. Chauhan, A. Kumar and M. Muddassir, The supramolecular frameworks and electrocatalytic properties of two new structurally diverse tertiary phosphane-appended nickel(II) and copper(I) thiosquarates, CrystEngComm, 2023, 25, 6822–6836, 10.1039/d3ce00817g; (b) A. Kushwaha, D. Srivastava, O. Prakash, G. Kociok-Köhn, S. W. Gosavi, R. Chauhan, M. Muddassir and A. Kumar, 1,1′-Bis-(diphenylphosphino)ferrocene appended d8- and d10-configuration based thiosquarates: the molecular and electronic configurational insights into their sensitization and co-sensitization properties for dye sensitized solar cells, Dalton Trans., 2024, 53, 6818–6829, 10.1039/D4DT00151F.
- A. Spek, checkCIF validation ALERTS: what they mean and how to respond, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 1–11, DOI:10.1107/S2056989019016244.
- (a) H. B. Bürgi, J. D. Dunitz and E. Shefter, Chemical reaction paths. IV. Aspects of O⋯C=O interactions in crystals, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1517–1527, DOI:10.1107/S0567740874005188; (b) F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Carbonyl−carbonyl interactions can be competitive with hydrogen bonds, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329, DOI:10.1107/S0108768198001463; (c) R. W. Newberry and R. T. Raines, The n→π* Interaction, Acc. Chem. Res., 2017, 50, 1838–1846, DOI:10.1021/acs.accounts.7b00121.
- (a) X. Almagro, W. Clegg, L. Cucurull-Sánchez, P. González-Duarte and M. Traveria, Schiff bases derived from mercury(II)–aminothiolate complexes as metalloligands for transition metals, J. Organomet. Chem., 2001, 623, 137–148, DOI:10.1016/S0022-328X(00)00672-0; (b) W. J. Kozarek and Q. Fernando, Crystal and molecular structure of C13H10N4S·HgCl2, the adduct of anhydro-5-mercapto2,3-diphenyltetrazolium hydroxide and mercury(II) chloride, Inorg. Chem., 1973, 12, 2129–2131, DOI:10.1021/ic50127a038; (c) R. E. Marsh and F. H. Herbstein, Some additional changes in space groups of published crystal structures, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 280–287, DOI:10.1107/S0108768183002402.
- (a) P. R. Spackman, M. J. Turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910; (b) S. L. Tan, M. M. Jotani and E. R. T. Tiekink, Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 308–318, DOI:10.1107/S2056989019001129; (c) M. M. Jotani, J. L. Wardell and E. R. T. Tiekink, Supramolecular association in the triclinic (Z′ = 1) and monoclinic (Z′ = 4) polymorphs of 4-(4-acetylphenyl)piperazin-1-ium 2-amino-4-nitrobenzoate, Z. Kristallogr. - Cryst. Mater., 2019, 234, 43–57, DOI:10.1515/zkri-2018-2101; (d) D. Danovich, S. Shaik, F. Neese, J. Echeverría, G. Aullón and S. Alvarez, Understanding the nature of the CH⋯HC interactions in alkanes, J. Chem. Theory Comput., 2013, 9, 1977–1991, DOI:10.1021/ct400070j.
- N. B. Topić, N. Bedeković, K. Lisac, V. Stilinović and D. Cinčić, Halogen-bonded cocrystals of 1,3,5-triiodo-2,4,6-trifluorobenzene and structural isomers of benzoylpyridine, Cryst. Growth Des., 2022, 22, 3981–3989, DOI:10.1021/acs.cgd.2c00382.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506, DOI:10.1021/ja100936w; (b) A. Otero-de-la-Roza and E. R. Johnson, A benchmark for non-covalent interactions in solids, J. Chem. Phys., 2012, 137, 054103, DOI:10.1063/1.4738961; (c) A. Otero-de-la-Roza and E. R. Johnson, Non-covalent interactions and thermochemistry using XDM-corrected hybrid and range-separated hybrid density functionals, J. Chem. Phys., 2013, 138, 204109, DOI:10.1063/1.4807330; (d) A. Otero-de-la-Roza, J. D. Mallory and E. R. Johnson, Metallophilic interactions from dispersion-corrected density-functional theory, J. Chem. Phys., 2014, 140, 18A504, DOI:10.1063/1.4862896.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/B810189B.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928, DOI:10.1021/cr00005a013.
- CrysAlis PRO, Rigaku Oxford Diffraction, Yarnton, Oxfordshire, England, 2022 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 1–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- L. J. Farrugia, WinGX and ORTEP for windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854, DOI:10.1107/S0021889812029111.
- K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2006 Search PubMed.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566, DOI:10.1080/00268977000101561.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, W. M. Wong, C. Gonzalez and J. A. Pople, Gaussian 09 revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) J. E. Carpenter and F. Weinhold, Analysis of the geometry of the hydroxymethyl radical by the different hybrids for different spins natural bond orbital procedure, J. Mol. Struct.: THEOCHEM, 1988, 139, 41–62, DOI:10.1016/0166-1280(88)80248-3; (b) J. P. Foster and F. Weinhold, Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102, 7211–7218, DOI:10.1021/ja00544a007; (c) A. E. Reed and F. Weinhold, Natural bond orbital analysis of near-Hartree-Fock water dimer, J. Chem. Phys., 1983, 78, 4066–4073, DOI:10.1063/1.445134; (d) A. E. Reed, R. B. Weinstock and F. Weinhold, Natural-population analysis, J. Chem. Phys., 1985, 83, 735–746, DOI:10.1063/1.449486; (e) A. E. Reed and F. Weinhold, Natural localized molecular orbitals, J. Chem. Phys., 1985, 83, 1736–1740, DOI:10.1063/1.449360; (f) A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926, DOI:10.1021/cr00088a005; (g) F. Weinhold and J. E. Carpenter, The structure of small molecules and ions, ed. R. Naaman and Z. Vager, Plenum, 1988, pp. 227–236, DOI:10.1007/978-1-4684-7424-4.
- (a) T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2019, (https://www.aim.tkgristmill.com); (b) T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885; (c) W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38, DOI:10.1016/0263-7855(96)00018-5; (d) T. Williams and C. Kelley, 1986–1993, 1998, 2004, 2007–2022, https://www.gnuplot.info.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra, electronic spectra, crystallographic images, two-dimensional fingerprint plots, listing of Hirshfeld surface contacts, MEP surface, RDG, NCI and NBO plots, topographical parameters for BCPs, a listing of Wiberg bond indices, Mayer bond orders, delocalisation indices and natural charges, and a listing of the coordinates for the calculated molecules and aggregates. CCDC 2375411 and 2375412 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00879k |
| This journal is © The Royal Society of Chemistry 2025 |