
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical lithiation reveals the structures of conjugated tetralithium dicarboxylates†
Kieran
Griffiths
 ab,
Chris
Cook
ab,
Valerie R.
Seymour
ab,
Ryan J.
Bragg
ac,
Nathan R.
Halcovitch
a and
John M.
Griffin
*ab
ab,
Chris
Cook
ab,
Valerie R.
Seymour
ab,
Ryan J.
Bragg
ac,
Nathan R.
Halcovitch
a and
John M.
Griffin
*ab
aDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: j.griffin@lancaster.ac.uk
bThe Faraday Institution, Quad One, Didcot, OX11 0RA, UK
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 3rd July 2025
Abstract
Conjugated metal dicarboxylates show promising properties as rechargeable battery anode materials; however, the structures of reduced (discharged) phases remain elusive. Here, we use chemical lithiation to solve the crystal structures of two tetralithium dicarboxylates for the first time. The structures are identical to those formed by electrochemical lithiation in coin cells. Solid-state NMR and DFT calculations show that there is complete mixing of Li cations upon intercalation into the structure.
The prevalence of rechargeable batteries in consumer devices is driving increasing interest in the development of sustainable battery chemistries. One family of materials that is receiving significant interest is conjugated lithium dicarboxylates (Li2DCs), which show considerable promise as anode materials in lithium-ion batteries.1–3 Li2DCs display high gravimetric capacities together with wide chemical diversity, which allows their properties to be tuned and optimised. Additionally, they can be derived from green and sustainable sources, such as waste plastic.4,5 However, organic anode materials tend to exhibit lower rate capabilities and cycle lives compared to more established anode materials such as graphite.
In 2009, Armand et al. showed that lithium benzenedicarboxylate (Li2BDC) can undergo reversible electrochemical lithiation to incorporate two additional Li+ ions per formula unit to form Li4BDC.6 This reaction occurred at 0.8 V vs. Li+/Li0 with a reversible capacity of 234 mA h g−1 after 50 cycles. Lithium intercalation is proposed to occur via reduction of the BDC ligand with rearrangement of the π electron distribution as shown in Scheme 1. Increasing the degree of conjugation can improve cycling stability; this has been attributed to increased electronic conductivity,7–9 although structural factors may also be relevant. This effect can be seen when comparing Li2BDC with lithium naphthalenedicarboxylate (Li2NDC), which also undergoes reduction via a similar mechanism and displays higher rate capability.7,10 However, despite the advances that have been made, there remains no clear consensus on the precise crystal structures of lithiated Li2DC phases. Knowledge of the locations of inserted Li ions, as well as any other structural rearrangements that take place upon reduction, will help to distinguish crystal structure and electronic structure effects to further optimise these materials.
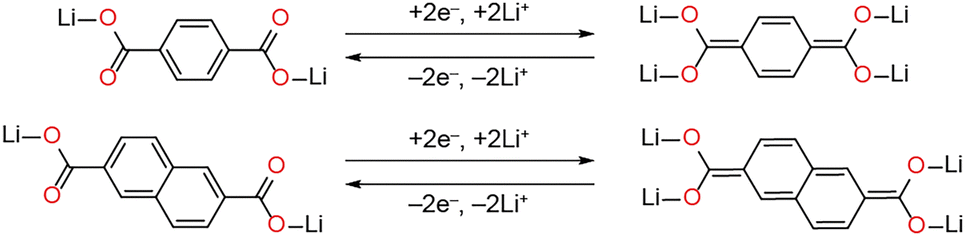 | ||
| Scheme 1 Reaction scheme for the reversible reductive lithiation of Li2BDC and Li2NDC. | ||
To gain insight into reduction mechanisms and the resulting structures, we prepared Li2BDC and Li2NDC as model dicarboxylate phases. Fig. 1a and b show the galvanostatic charge–discharge curves of Li2+xDC/Li(s) coin cells cycled at a rate of C/20 between 0.7 and 3.0 V. The first discharge capacities for Li2BDC and Li2NDC reach 425 mA h g−1 and 520 mA h g−1, respectively. These are higher than the theoretical capacities (Li2BDC – 255.1 mA h g−1; Li2NDC – 235.0 mA h g−1); however, additional capacity is expected due to contributions from carbon black,11,12 as well as irreversible solid–electrolyte interphase (SEI) formation in the first discharge cycle. For Li2BDC, a discharge capacity of 194 mA h g−1 is achieved in the second cycle and the accessible capacity continues to decrease to 163.6 mA h g−1 in the 5th cycle, which is lower than the previously reported value;6 this may be due to the unoptimised free-standing electrode used, as well as the non-uniform particle size of Li2BDC crystallites (Fig. S1, 0.6–25.0 μm, ESI†), where larger crystallites do not fully lithiate due to the low electronic conductivity. For Li2NDC, a capacity of 233.0 mA h g−1 is reached in the 5th discharge cycle, which is comparable to the theoretical capacity,7 although it is possible that the carbon additive and SEI could still contribute to the observed capacity at this point. The Li2NDC sample used had a smaller average crystallite size with a narrower range (Fig. S1, 0.9–9.0 μm, ESI†) and also higher intrinsic electronic conductivity.7
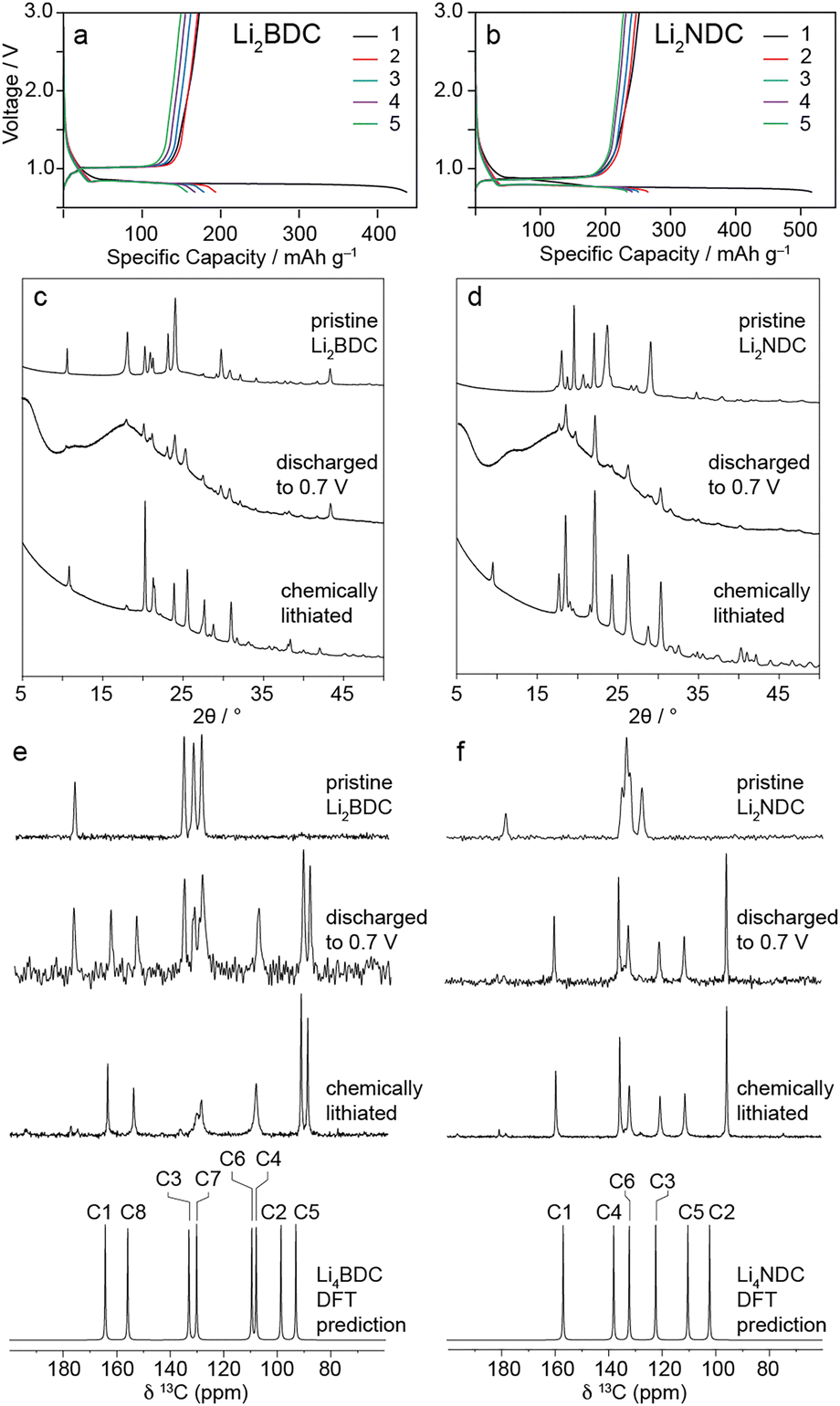 | ||
| Fig. 1 (a) and (b) Charge–discharge profiles of Li2DC electrodes cycled at C/20. (c) and (d) PXRD patterns of pristine, electrochemically discharged, and chemically lithiated Li2DCs. (e) and (f) 13C CPMAS NMR spectra of pristine, electrochemically discharged, and chemically lithiated Li2DCs, and DFT-predicted spectra for SCXRD Li4DC structures. | ||
PXRD patterns of the as-prepared electrodes (Fig. 1c and d) are characteristic of the pristine Li2DC phases. However, a broad background between 19° and 25° and discrete reflection at 17° are attributed to the carbon black additive and PTFE binder, respectively (Fig. S2, ESI†). The PXRD patterns of the discharged samples show an increased background due to the KaptonTM tape used to avoid air exposure (Fig. S2, ESI†). For the discharged Li2BDC anode, reflections of the pristine Li2BDC phase are still evident; however, additional reflections are observed at 25° and 27°. For discharged Li2NDC, pristine phase reflections are eliminated, and new reflections are observed between 24° and 31°. This indicates full conversion to a new phase, which is consistent with the capacity observed for this material. However, due to the low relative intensities, it was not possible to resolve or index the phase(s) present for either discharged sample.
The 13C CPMAS NMR spectrum of the discharged Li2BDC anode (Fig. 1e) shows residual resonances corresponding to the pristine phase, which is consistent with the observed capacity being lower than the theoretical value. The appearance of new resonances between 90.1 and 164.2 ppm indicates that a new phase has formed upon lithiation, which is consistent with the new PXRD reflections. No significant intensity is observed corresponding to SEI components or the carbon additive; this is likely due to the relatively low mass fraction of SEI and the absence of 1H in the carbon additive, which precludes magnetisation transfer in the CPMAS experiment. The 13C CPMAS NMR spectrum of the discharged Li2NDC anode (Fig. 1f) shows the pristine structure is no longer detected and instead six new resonances are observed between 95.4 and 159.4 ppm. This suggests that the close-to-theoretical capacity value observed for this material reflects a higher level of conversion to the lithiated phase. For both materials, charging to 3.0 V results in complete reversion to the pristine phase (Fig. S3, ESI†) for at least the first five cycles.
The PXRD and 13C CPMAS NMR data show that electrochemical lithiation and delithiation drive reversible conversion between the pristine and reduced phases. However, from these results, it is not possible to determine the structures of the reduced phases. An alternative approach for preparing reduced phases without additional electrode components is chemical lithiation. This involves adding solid Li2DC to a solution containing lithium ions and a redox mediator. Naphthalene has a reduction potential of 0.5 V vs. Li+/Li0, which is below the observed plateaus at 0.8 V for Li2BDC and Li2NDC. Therefore, in a suitable solvent, naphthalene should oxidise Li metal to form Li+ and simultaneously drive reduction and lithiation of Li2DCs.
Li metal and naphthalene were dissolved in THF under an argon atmosphere to prepare a solution with 3 M concentration. The Li2DC powders were soaked in this ethereal solution for 72 hours followed by washing with fresh THF and drying. 13C CPMAS NMR spectra of the recovered powders reveal almost complete conversion to reduced phases with the same chemical shifts as the electrochemically lithiated samples (Fig. 1e). Spectra of samples lithiated for shorter times show that the lithiation proceeds via a two-phase mechanism in both materials, with progressive loss of pristine phase resonances and simultaneous growth of lithiated phase resonances (Fig. S4, ESI†). Interestingly, chemical lithiation of Li2BDC appears to proceed more slowly, requiring 72 hours for complete conversion compared to 24 hours for Li2NDC. This mirrors the more sluggish electrochemical lithiation kinetics of Li2BDC observed in this work and other studies.7 EPR spectra of the lithiated samples show absorbances centred at g = 2.003 for Li4BDC and g = 2.004 for Li4NDC (Fig. S5, ESI†). These results agree with previous data indicating the formation of carbon-centred radicals.6,13 The radical concentration is low enough to permit the acquisition of undistorted NMR spectra; however, the 7Li T1 values for the pristine Li2DCs (Li2BDC, 191 s; Li2NDC, 264 s) decrease significantly after chemical lithiation (Li4BDC, 13 s; Li4NDC, 4 s). PXRD analysis of the chemically lithiated Li2DCs also confirms phase purity (Fig. 1c and d) and retention of crystallinity.
The retention of crystallinity and identification of single lithiated phases for Li2BDC and Li2NDC prompted us to investigate single crystals. Crystals of the pristine phases were grown from vapour diffusion of acetone into aqueous solutions of the Li2DCs. The structures were solved via single-crystal X-ray diffraction (SCXRD) analysis and the unit cell dimensions and structures agree well with literature structures (Tables S2 and S3; CCDC 664607 (Li2BDC); 722281 (Li2NDC), ESI†). Crystals were then soaked for 72 hours in the Li+/naphthalene solution. This resulted in block-shaped crystals with a dark-red hue (Fig. S8, ESI†). These were mounted in oil to prevent air exposure and SCXRD measurements were performed at 100 K. This gave two new structures with compositions Li4BDC and Li4NDC (Fig. 2); to our knowledge, these are the first experimental crystal structures reported for reduced forms of conjugated dicarboxylate materials. Crystallographic and structural refinement details are given in Tables S4 and S5 (ESI†). Unit cell parameters for both SCXRD structures agree well with Le Bail extracted unit cell parameters of the PXRD samples (Table S1, ESI†).
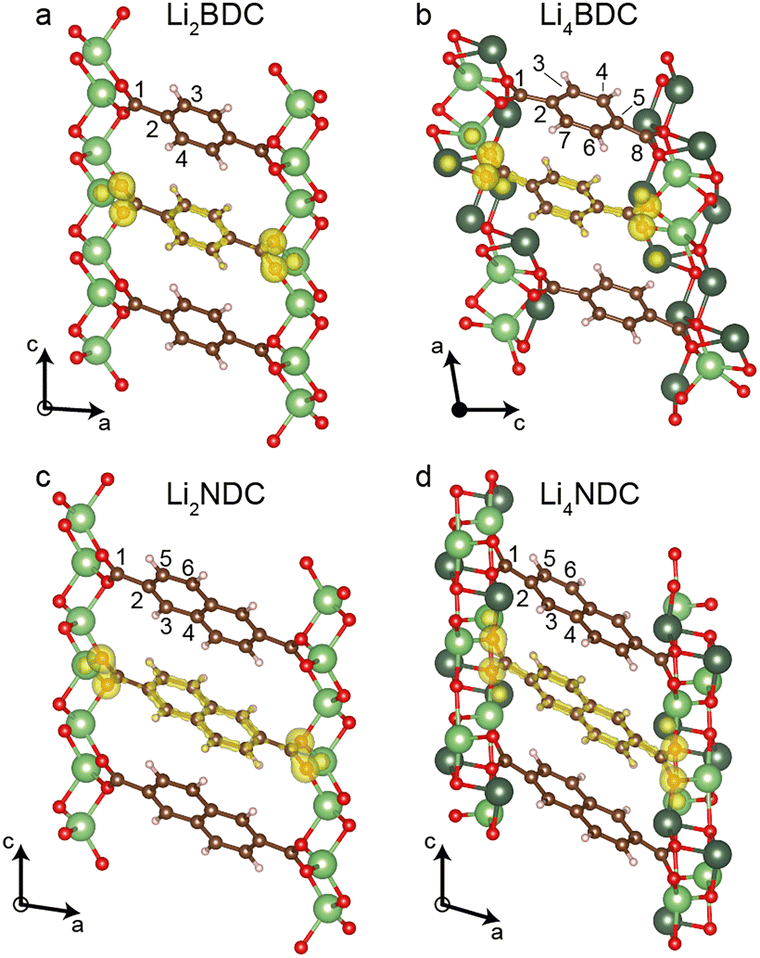 | ||
| Fig. 2 Crystal structures of (a) Li2BDC, (b) Li4BDC (c) Li2NDC and (d) Li4NDC. Three- and four-coordinate Li+ are shown in light and dark green, respectively. H, O and C atoms are shown in white, red and brown, respectively. DFT charge densities are superimposed in yellow. | ||
Li4BDC exhibits the monoclinic space group P21/n. Li sites Li1, Li3, and Li4 all have tri-planar coordination geometry, whereas Li2 retains the tetrahedral coordination geometry observed in the pristine phase (Fig. S9, ESI†). The carbon–carbon bond lengths C1–C2, C3–C4, C6–C7, and C5–C8 are contracted by 2.2–8.6% when compared to pristine Li2BDC (Table S6, ESI†). Each reduced BDC ligand has identical coordination geometry to surrounding Li+, and the Li4O4 dicarboxylate layers are separated by diagonally oriented reduced BDC linkers (Fig. S9, ESI†), with a herringbone-like stacking of the BDC linkers as seen in the pristine structure. However, due to differing chelation to Li+ at either end of the ligands, the structural symmetry is lowered and each carbon atom within each ligand is crystallographically distinct. Furthermore, each reduced ligand is bent out of plane with an angle of 7.4° between C2–C5–C8. Alternation in the bending direction between adjacent layers leads to an approximate doubling of the unit cell c axis (Fig. S9, ESI†).
Li4NDC exhibits the monoclinic space group P21/c. There are two distinct Li+ environments, Li1 – which has a highly distorted tri-planar coordination geometry, and Li2 – which has a distorted four-coordinate planar geometry (Fig. S10, ESI†). Carbon–carbon bond lengths C1–C2, C3–C4, and C5–C6 are also contracted by 0.7–7.4% compared to Li2NDC (Table S7, ESI†). The NDC ligands also display a herringbone-like stacking arrangement as seen in the pristine phase; however, in contrast to Li4BDC, the symmetry of the ligand is retained.
To rationalise the experimental 13C chemical shifts, periodic DFT calculations were performed on geometry-optimised crystal structures of the reduced phases (Fig. 1e and f, Fig. S11 and Tables S8, S9, ESI†). For Li4BDC, the calculated shifts are in good agreement with experiments, although the C4 and C6 resonances are unresolved in the experimental spectrum. For Li4NDC, the calculated shifts also show good agreement with experiments. DFT charge density plots (Fig. 2) also support the proposed reduction mechanism. In the pristine phases, the electronic charge is distributed approximately evenly around the rings within the ligand, and lower charge densities are observed on the carbonyl-quaternary C–C bonds. In the reduced phases, the charge density is concentrated between the CH–CH bonds and on the carbonyl-quaternary C–C bonds, reflecting the change in conjugation and loss of aromaticity of the rings.
The presented crystal structures of Li4BDC and Li4NDC unequivocally show that two Li+ cations intercalate per formula unit, and that the ligands are reduced via a two-electron process. The 13C CPMAS NMR data show that both chemical lithiation and electrochemical lithiation form the same structures, albeit with an incomplete reaction in the electrochemical method for Li2BDC. Within the lithiated structures, the herringbone-like ligand arrangements are retained from the Li2DC precursors. In a previous study of Li2NDC, it was proposed on the basis of PXRD refinement and DFT optimisation that the coordination environment and geometry of the Li2O4 dicarboxylate layer are completely retained, but additional Li cations intercalate in two locations between the NDC layers.8 However, the SCXRD structure determined here unambiguously shows only two Li+ cation environments, both of which are within the Li–O layer, and one of which has a tri-planar coordination geometry. DFT calculations show that the two previously proposed structures correspond to local energy minima, but our SCXRD structure is more stable than both of these by 0.07 and 0.20 eV per formula unit (Table S10, ESI†). For Li4BDC, there is a more substantial rearrangement of the Li–O layer, with only a single site retaining tetrahedral coordination and the more complex coordination mode and bent geometry of the reduced BDC ligand.
Static 7Li NMR spectra of the pristine phases (Fig. 3) show intense 7Li central transitions surrounded by quadrupolar-broadened satellite transition powder patterns. The reduced phases show broader satellite transition powder patterns; this is consistent with DFT calculations, which predict increased 7Li CQ values for the lithiated structures (Table S11, ESI†). For Li4BDC, DFT overestimates the powder pattern line width by approximately 25%. This may suggest partial averaging of the quadrupolar interaction due to Li dynamics within the structure.
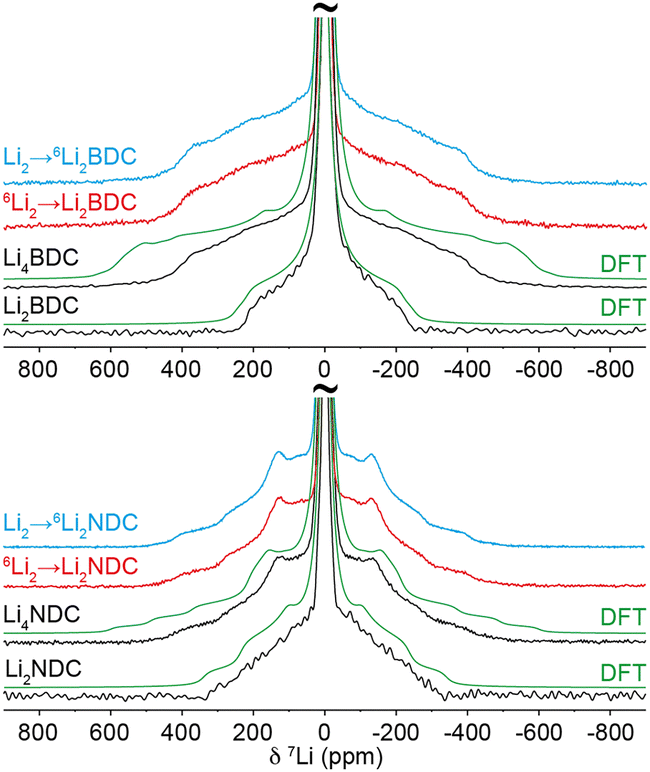 | ||
| Fig. 3 Experimental and calculated static 7Li NMR spectra of Li2DCs and Li4DCs. Calculated line shapes include 5 kHz line broadening. 6Li-enriched Li2DCs lithiated with natural abundance solution are denoted as Li2 → 6Li2DC; natural abundance Li2DCs lithiated with 6Li-enriched solution are denoted as 6Li2 → Li2DC. | ||
Given the substantial changes in Li ordering within the Li–O layers in both materials, it is natural to question on which crystallographic locations the inserted Li ions reside. It is important to note that, for the reduced phases, each observed powder pattern comprises the superposition of individual powder patterns from the four (Li4BDC) or two (Li4NDC) different crystallographic Li environments in each structure. Therefore, inserted Li+ can be potentially distinguished from those remaining from the pristine phase using 7Li isotope tracking and exploiting the large difference in natural abundance between 6Li (7.8%) and 7Li (92.2%). To test this, natural abundance pristine samples were chemically lithiated with isotopically enriched solutions containing 95% 6Li+. In addition, 6Li-enriched (95%) pristine samples were lithiated using natural abundance solutions. In this approach, if the inserted Li+ ions preferentially reside on particular crystallographic sites, the relative intensities of the 7Li line shape components corresponding to these sites should be reduced in natural-abundance samples lithiated with 6Li-enriched lithium and enhanced in 6Li-enriched samples lithiated with natural-abundance lithium. However, as shown in Fig. 3, the line shapes for all samples are identical, showing that 7Li ions are in fact distributed with equal probability across all sites within each structure. Therefore, there must be complete mixing of Li+ within the structure during lithiation. In addition, quantitative 7Li NMR measurements show that the 7Li population within the lithiated phase is always weighted towards the 7Li content of the ethereal solution and does not follow the 7Li content of the pristine phase (Tables S11, S12 and Fig. S12, ESI†). This indicates that, during insertion of Li ions into the structure, there is a global ion exchange between the solid structure and the solution.
Herein we have shown that chemical lithiation of model organic battery anode materials can enable full structure solution of lithiated phases. The combination of SCXRD and solid-state NMR unequivocally shows that chemical and electrochemical lithiation produce the same structures. Furthermore, 7Li NMR and DFT show that there is complete rearrangement of Li+ within the Li4O4 layer. Previously, the improved cycling efficiency of Li2NDC over Li2BDC has been attributed to only increased electronic conductivity of the former; however, the highlighted differences in the lithiated structures and complete rearrangement of the Li layers may allow for a re-examination of structure–property relationships in organic anode materials.
This work was funded by the Faraday Institution NEXGENNa project (FIRG018, K. G.; V. R. S.; J. M. G.), FutureCat project (FIRG017, C. C.), and EPSRC (EP/V05001X/1, R. J. B.; J. M. G.). Computational resources were provided by Lancaster University's High End Computing cluster.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
Crystallographic data have been deposited (2450774–2450777) at the CCDC. Additional details are available in the ESI,† and raw data underpinning this work are available at https://doi.org/10.17635/lancaster/researchdata/720.References
- M. Teusner, J. Mata, B. Johannessen, G. Stewart, S. Cadogan and N. Sharma, Mater. Adv., 2023, 4, 3224–3238 RSC.
- N. Ogihara, M. Hasegawa, H. Kumagai, R. Mikita and N. Nagasako, Nat. Commun., 2023, 14, 1472 CrossRef CAS PubMed.
- L. Wang, C. Mou, B. Wu, J. Xue and J. Li, Electrochim. Acta, 2016, 196, 118–124 CrossRef CAS.
- W. Walker, S. Grugeon, H. Vezin, S. Laruelle, M. Armand, J. M. Tarascon and F. Wudl, Electrochem. Commun., 2010, 12, 1348–1351 CrossRef CAS.
- D. Senthil Raja, C.-C. Pan, C.-W. Chen, Y.-H. Kang, J.-J. Chen and C.-H. Lin, Micropor. Mesopor. Mater., 2016, 231, 186–191 CrossRef CAS.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J.-M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- L. Fédèle, F. Sauvage, J. Bois, J.-M. Tarascon and M. Bécuwe, J. Electrochem. Soc., 2014, 161, A46–A52 CrossRef.
- N. Ogihara, T. Yasuda, Y. Kishida, T. Ohsuna, K. Miyamoto and N. Ohba, Angew. Chem., Int. Ed., 2014, 53, 11467–11472 CrossRef CAS PubMed.
- T. Yasuda and N. Ogihara, Chem. Commun., 2014, 50, 11565–11567 RSC.
- L. Fédèle, F. Sauvage, S. Gottis, C. Davoisne, E. Salager, J.-N. Chotard and M. Becuwe, Chem. Mater., 2017, 29, 546–554 CrossRef.
- K.-H. Nam, K. Hwa Chae, J.-H. Choi, K.-J. Jeon and C.-M. Park, Chem. Eng. J., 2021, 417, 129242 CrossRef CAS.
- J. Hu, S. Zhong and T. Yan, J. Power Sources, 2021, 508, 230342 CrossRef CAS.
- C. K. Borrowman, S. Zhou, T. E. Burrow and J. P. D. Abbatt, Phys. Chem. Chem. Phys., 2016, 18, 205–212 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC (2450774–2450777). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc03043a |
| This journal is © The Royal Society of Chemistry 2025 |