
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational design of gallium imides for methane activation†
Sylvester
Zhang
 a,
Ruofei
Cheng
a,
Peter H.
McBreen
b,
Chao-Jun
Li
a and
Rustam Z.
Khaliullin
*a
a,
Ruofei
Cheng
a,
Peter H.
McBreen
b,
Chao-Jun
Li
a and
Rustam Z.
Khaliullin
*a
aMcGill University, 801 Sherbrooke St W, Montréal, H3A 0B8, Canada. E-mail: rustam.khaliullin@mcgill.ca
bUniversité Laval, 2325 Rue de l'Université, Québec, G1V 0A6, Canada
First published on 1st July 2025
Abstract
In this work, computational modeling is used to modify NacNac gallium imide, which has recently been shown to cleave unactivated sp3 C–H bonds in organic substrates, with the aim of making it suitable for methane activation. Density functional theory predicts that the 123 kJ mol−1 methane activation barrier for the experimentally employed gallium imide can be reduced to 93 kJ mol−1 by changing substituents around the active gallium center. Furthermore, pre-straining the gallium imide reduces the reaction barrier to just 62 kJ mol−1. Dimerization of the gallium imides can be prevented with bulky groups that do not affect the reaction barrier. Several modified NacNac gallium imides are thus shown to be viable for the homogeneous activation of methane and higher alkanes.
Methane reserves are abundant on Earth and there are substantial economic and environmental benefits for converting methane to complex molecules instead of burning it as fuel. From the environmental perspective, using methane as fuel is not ideal because of the undesirable release of carbon dioxide into the atmosphere. In economics terms, products of methane conversion are more valuable than inert methane because they can be used in a variety of downstream chemical manufacturing processes. These considerations have driven extensive efforts to develop methane conversion reactions.1–7
Presently, the commercial processes of methane conversion, such as steam reforming or partial oxidation, are multi-step indirect procedures that produce synthesis gas that is then converted into high-value products, such as methanol, olefins, and parafins.3,5 Due to losses in each step, a direct conversion of methane is more desirable and has been studied extensively.8 Unfortunately, direct conversion of methane to value-added products is challenging because of the low chemical reactivity of methane. Direct methane activation usually involves a bulk catalyst, owing to the advantages of simply pumping methane gas onto solids that can handle high temperatures and pressures. Oxidative coupling of methane has mostly been reported for heterogenous catalysts.8 Methane dehydroaromatization, where methane is converted non-oxidatively into aromatic compounds, mainly benzene, has also been performed exclusively heterogeneously, using HZSM-5 and HMCM-5 zeolites containing molybdenum centers6 and more recently gallium nitride.9,10 However, homogeneous dehydroaromatization of higher alkanes has been demonstrated using a molecular iridium catalyst.11
Homogeneous methane activation reactions are of great interest because they offer several potential advantages over heterogeneous processes, including the stabilization of products through solvation effects, a variety of effective heat management strategies, lower reaction temperature, greater control of the reaction mechanism, and improved selectivity.12 The high reactivity of transition metal complexes has enabled considerable advances in homogeneous activation of methane in synthetic chemistry over the years, from the latter half of the 20th century onwards.1–3,8,13–15
Although the main-group activation of inert sp3 C–H bonds has not been nearly as well exploited as that on transition metals,8,11,12 it is more desirable because main group elements tend to be less toxic than late transition metals.
Methane activation has been reported using very strong bases in combination with a sterically hindered Lewis acid.16 Metal-free activation based on creating radicals has also been described.17 Since the introduction of frustrated Lewis pairs (FLPs),18 there has been extensive effort to develop FLPs for the C–H activation by exploiting the intermolecular or intramolecular combination of unquenched Lewis centers.16,18,19
There has also been progress in C–H bond activation using non-FLP molecules. Driess et al. showed that in situ generated iminosilanes could activate C–H bonds to yield an amide-silane.20 Braunschweig and coworkers described the cleavage of C–H bonds over a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, yielding aminoboranes.21 A phosphanyl-phosphogallene was also reported in 2020 to have FLP-like activity.22 Following a series of studies of C–H activation of polarizable or enolizable bonds on (NacNac)Ga = X (NacNac = [(ArNCMe)2 CH]−, Ar = 2,6-i-Pr2 –C6H3, X = O23 and X = PR24), Nikonov et al. used an in situ generated NacNac gallium imide, (X = NR) to cleave unactivated C–H bonds.25 In the latter work, gallium imide was formed by the reaction of an aromatic gallium molecule with an azide, expelling nitrogen gas in the process. This imide has been shown to react in situ with a wide range of C–H bonds on molecules including ketones, esters, and pyridine.25
N bond, yielding aminoboranes.21 A phosphanyl-phosphogallene was also reported in 2020 to have FLP-like activity.22 Following a series of studies of C–H activation of polarizable or enolizable bonds on (NacNac)Ga = X (NacNac = [(ArNCMe)2 CH]−, Ar = 2,6-i-Pr2 –C6H3, X = O23 and X = PR24), Nikonov et al. used an in situ generated NacNac gallium imide, (X = NR) to cleave unactivated C–H bonds.25 In the latter work, gallium imide was formed by the reaction of an aromatic gallium molecule with an azide, expelling nitrogen gas in the process. This imide has been shown to react in situ with a wide range of C–H bonds on molecules including ketones, esters, and pyridine.25
The aim of this study is to use computational modeling to design gallium imide derivatives capable of activating inert C–H bonds in methane. The choice of gallium imides is motivated by their success in cleaving unactivated sp3 bonds25 and by the structural similarity of their active center to the gallium nitride surface, which has been demonstrated to convert methane to benzene with high activity and selectivity in thermally-activated9 and photocatalytic10 processes.
Molecular models. Density functional theory (DFT) at the ωB97X-D/6-311++G(d,p) level of theory was used for molecular modeling as described in Methods (S1, ESI†). Energy profiles of the cleavage of inert C–H bonds in methane molecule were calculated for a series of nine substituted gallium imide structures shown in Fig. 1. Perfluorophenyl (C6F5) was used as an electron-withdrawing group (EWG), trimethylsilyl (TMS) as a σ electron donating group (EDG), while phenyl was considered as a neutral group. The gallium imide proposed by Nikonov et al.25 was represented by R1 = Ph and R2 = TMS.
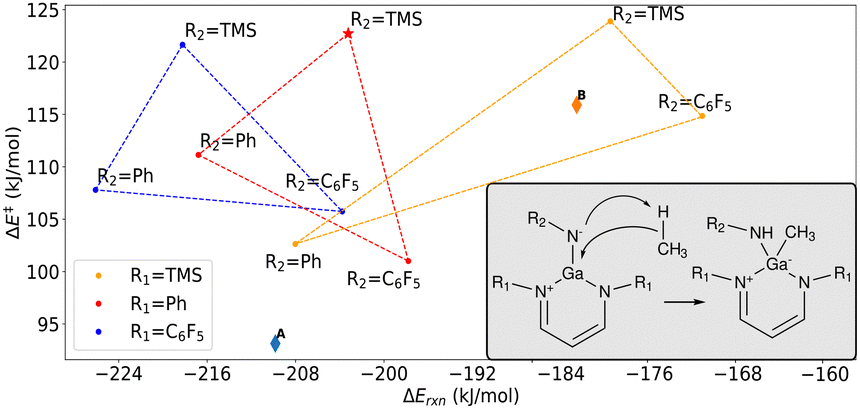 | ||
| Fig. 1 Computed ωB97X-D/6-311++G(d,p) reaction energies (ΔErxn) and activation energies (ΔE‡) for the C–H bond cleavage. R1 = TMS,Ph,C6F5 and R2 = TMS,Ph,C6F5 groups are shown in the inset, whereas structure A, and B are shown in Fig. 2. The red star labels the proxy to the imide proposed by Nikonov et al.25 | ||
Evaluation criteria. The energy barrier for the C–H cleavage (ΔE‡) was computed for all gallium imides in order to find the most reactive systems for nonpolar C–H bonds. In addition, we evaluated the overall reaction energy of the C–H activation (ΔErxn). An excessively exothermic reaction will preclude further chemical transformations of the activation products. These two considerations are typically mutually contradictory, as the Bell–Evans–Polanyi principle states that lower activation barriers for a given reaction correspond to more exothermic processes.
Finally, the stability of the gallium imides against spontaneous dimerization (ΔEdim) was estimated, as it is expected that the overcoordinated active centers in dimers cannot cleave C–H bonds.
Mechanism. We considered the nitrogen atom attached to R2 as the hydrogen acceptor (Fig. 1) because reacting on the R1 nitrogen was on the order of 100 kJ mol−1 less energetically favourable, and was liable to cause the imide molecule to fall apart. For all variants of the gallium imides, analysis of the intrinsic reaction coordinate starting from the transition state shows that C–H bond cleavage is a concerted reaction (Fig. 1): methane loses the hydrogen to the nitrogen while simultaneously forming a C–Ga bond.
Main trends and the best candidate. Fig. 1 shows the energy barrier and the reaction energy for methane activation. It is clear that no Bell–Evans–Polyani trend, or its inverse, is observed. Interestingly, placing TMS at R2 leads to elevated ΔE‡ in excess of 120 kJ mol−1 and replacing this TMS with phenyl or perfluorophenyl markedly reduces the barrier. It is also worth noting that phenyl at R2 makes the reaction more exothermic whilst perfluorophenyl has an opposite effect. For fixed R2, the reaction becomes increasingly exothermic as R1 goes from TMS to Ph to C6F5. However, there is no clear trend for the activation barriers when varying R1.
Thus, the calculations show that ΔE‡ can be lowered by 25 kJ mol−1 over the proxy to the previously reported gallium imide25 if Ph and C6F5 groups are used as R1 and R2, respectively. This reduction of ΔE‡ is substantial and will result in a ten-thousand-fold increase in the reaction rate at 300 K. Alternatively, using R1 = TMS and R2 = C6F5 makes the C–H activation process 30 kJ mol−1 less exothermic while also lowering the barrier by 10 kJ mol−1 and thus speeding up the cleavage by an order of magnitude.
Unfortunately, there are no structures in the set that can lower both the activation barrier and the absolute value of the reaction energy. Next, we explored modifications of the structure of the best candidate (R1 = Ph and R2 = C6F5) that can further improve the energetics of the C–H bond cleavage.
Energy decomposition analysis based on absolutely localized orbitals (ALMO EDA) suggests (S2, ESI†) that the most promising strategies to further improve the best candidate are to (a) increase the electron flow between the imide and methane and (b) straining the initial geometry so it more closely resembles the pyramidal geometry of the transition state.
Rational design to lower the activation energy. In order to increase the electron flow between the gallium imide center and methane, additional EWGs or EDGs were placed on the NacNac ligand in the reference structure. We hypothesized that adding the electron-withdrawing CHO group para to the gallium atom in the ring (A in Fig. 2) can increase the electron donation from CH3 to the gallium center. This hypothesis was only partially validated. The energy of charge transfer from the methane to imide increased, but only insignificantly (by ∼1 kJ mol−1). Instead it was the decrease in geometric distortion and frozen density interactions that reduced the activation energy by 7 kJ mol−1 (Fig. S2b, ESI†). Next, we substituted the Me groups of NacNac with NH2 (B in Fig. 2) with the goal of increasing the flow of electrons from the gallium center to methane. Unfortunately, the transition state barrier for structure B rose by 15 kJ mol−1 (Fig. S2b, ESI†). This unexpectedly high rise is not attributable to charge transfer between the imide and methane or from methane to the imide but rather to the increased penalty of distorting imide B, perhaps owing to the loss of aromaticity of the Ga-containing ring in the starting structure B in the reaction.
 | ||
| Fig. 2 Structures of gallium imides derived from the R1 = Ph, R2 = C6 F5 imide with the aim of (A) lowering the activation barriers by enhancing the electron flow from the methane to the imide, (B) increasing the electron donation from the imide to the methane, and (C) distorting the planar GaN3 active site. Structure (D) is structure (C) modified to increase steric hindrance against dimerization. | ||
Another strategy suggested by ALMO EDA is to distort the geometry of the gallium site. This is accomplished by tying R1 and R2 groups together as shown in structure C in Fig. 2. As a result, the flat GaN3 active center in the reference molecule becomes pyramidal in C (Fig. S3a, ESI†) and rotating the C6F4 group perpendicular to the NacNac plane breaks the π-conjugation of a nitrogen lone pair with the π-system of the Ga-containing aromatic ring. These structural changes reduce the C–H activation barrier by the dramatic 36 kJ mol−1 compared to the parent structure (Table 1). ALMO EDA also shows that the bulk of the improvement is from reducing the energy of the geometry distortion that is necessary to reach the transition state (Fig. S2b, ESI†). The distortion is accompanied by stronger charge transfer from methane to the active center, further stabilizing the transition state. This data indicates that straining gallium imide is a better strategy to lower the activation barrier of the reference imide than modifying its π-conjugated system through EDGs or EWGs.
| ΔE‡ kJ mol−1 | ΔErxn kJ mol−1 | ΔEdim kJ mol−1 | ΔG0dim kJ mol−1 | |
|---|---|---|---|---|
| Ref. | 100 | −197 | −224 | −127 |
| A | 93 | −210 | −272 | −112 |
| B | 115 | −183 | −148 | 1 |
| C | 73 | −260 | −588 | −477 |
| D | 62 | −251 | −105 | 7 |
Reaction energies. It is desirable that the overall reaction energy for the interaction between methane and gallium imide is only moderately negative, so that the product of the C–H bond cleavage can undergo further transformation instead of becoming trapped in the low-energy state. We found that using C6F5 groups as R1 makes the reaction more exothermic, which is undesirable, whilst using electron-donating TMS has the opposite effect (Fig. 1).
Fig. 1 and Table 1 show that the reference imide and its derivatives A, B, and C follow a perfect Bell–Evans–Polyani trend with the decrease (increase) in ΔE‡ being accompanied by a nearly linear decrease (increase) in ΔErxn. The highly exothermic nature of the reaction of CH4 with C is unsurprising in light of prior studies on the activation of C–H bonds by group 13–15 trisubstituted molecules.19,26
Further conversion of the CH4 activation product requires a strong acceptor of methyl and hydrogen. For example, the methylated imide A can react with carbon dioxide forming a gallium acetate complex (Fig. S5, ESI†). This reaction is exothermic releasing −48 kJ mol−1 and facilitates the incorporation of methane-derived fragments into downstream transformations.
Dimerization energies. Tri-coordinated gallium molecules tend to associate in solution forming dimers, trimers or oligomers, much like their group 13 analogues25,27–30 or to undergo secondary reactions.31 It is a common strategy to employ bulky, chemically inert substituents around the Lewis acid centers to provide kinetic stabilization and suppress undesired reactions, including dimerization.28,32,33 In this work, the same strategy is used to prevent the dimerization of gallium imides. Adding bulky peripheral substituents is not expected to affect the accessibility of the active gallium centers to small methane molecules.
Bulky groups such as TMS used in ref. 25 decrease the propensity of imides to dimerize (Fig. 3). The dimerization energies for all imides that have at least one bulky TMS group remain consistently within the realm of non-bonded interactions with |ΔEdim|<100 kJ mol−1. Such dimerization energies are much better compared to those for the other R1 and R2 substituents with |ΔEdim|>200 kJ mol−1 (Fig. 3). For example, phenyl and perfluorophenyl substituents in the reference imide (R1 = Ph, R2 = C6F5) are insufficiently large to block two active centers from approaching each other and forming two strong Ga–N bonds. This results in a strongly exothermic dimerization reaction for the reference imide with ΔEdim = −224 kJ mol−1 (Table 1). The reaction becomes significantly more exothermic for the derivative imide C with its highly exposed active center (ΔEdim = −558 kJ mol−1, Fig. S3, ESI† and Table 1).
 | ||
| Fig. 3 ωB97X-D/6-311++G(d,p) dimerization energies (ΔEdim) shown together with the C–H bond activation energy (ΔErxn) and barrier (ΔE‡). R1 and R2 groups are defined in Fig. 1, whereas structure A, B, and D are shown in Fig. 2. The proxy to the imide proposed by Nikonov et al. is R1 = Ph, R2 = TMS.25 | ||
To prevent strong interaction between two active sites in the dimer of C, bulky inert C6F5 groups were added, producing structure D (Fig. 2). This modification achieves its goal and reduces the dimerization energy to just −105 kJ mol−1 (Table 1), comparable with that of van der Waals interactions seen in TMS-containing imides, which include the structure used for C–H bond activation in experiments.25
While the dimerization of imide D remains exothermic, the dimerization entropy is negative. This results in a positive entropic contribution to the free energy that largely offsets the negative energy term. Therefore, the free energy of the dimerization of D is not expected to be very low (Table 1) and both dimers and monomers will exist in diluted solutions of D. Moreover, dimers of D can still cleave C–H bonds because their active centers retain the same structure as that of the monomers (Fig. S3d, ESI†). It should also be noted that bulky substituents in D do not affect the energetics of the methane activation compared to imide C (Table 1).
Final recommendations. Among the candidates studied, imide D emerges as the most promising for facile methane activation with its lowest ΔE‡ = 62 kJ mol−1 and active center protected from the dimerization, albeit with the second most exothermic ΔErxn. We suggest that imide D can be synthesized using an intramolecular version of a deazidification process (S4, ESI†). Another candidate is the R1 = TMS R2 = Ph imide, which has a low ΔE‡ (103 kJ mol−1), does not dimerize easily, with an even less exothermic ΔEdim than D, has a moderate ΔErxn (Fig. 3), and may also be synthesized in the manner suggested in ref. 25. An even lower barrier for imide A (ΔE‡ = 93 kJ mol−1) makes it suitable for methane activation if its gallium core is additionally protected with bulky groups against the dimerization, as demonstrated before.25 Given the potential for solvent activation on the Ga-N centers of imides, we recommend using inert solvents such as benzene or perfluorobenzene that do not have reactive alkyl C–H bonds and provide adequate methane solubility.
In summary, modified NacNac gallium imides with pre-strained reactive cores and suitable steric protection are promising for homogeneous methane activation.
This work was supported by the Fonds de recherche du Québec – Nature et technologies (FRQNT) through a Team Research Project grant 2021-PR-285170.
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including all optimized structures and their energies are available at Figshare at https://doi.org/10.6084/m9.figshare.28747208.References
- A. Shilov and A. Shteinman, Coord. Chem. Rev., 1977, 24, 97–143 CrossRef CAS.
- R. G. Bergman, Science, 1984, 223, 902–908 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Löffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340–343 CrossRef CAS PubMed.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- S. Ma, X. Guo, L. Zhao, S. Scott and X. Bao, J. Energy Chem., 2013, 22, 1–20 CrossRef CAS.
- S. V. Konnov, Pet. Chem., 2022, 62, 280–290 CrossRef CAS.
- F. Nkinahamira, R. Yang, R. Zhu, J. Zhang, Z. Ren, S. Sun, H. Xiong and Z. Zeng, Adv. Sci., 2022, 10, 2204566 CrossRef PubMed.
- L. Li, X. Mu, W. Liu, X. Kong, S. Fan, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 14106–14109 CrossRef CAS PubMed.
- L. Li, S. Fan, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2014, 136, 7793–7796 CrossRef CAS PubMed.
- R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167–171 CrossRef CAS PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809 RSC.
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS PubMed.
- S. Ahn, D. Sorsche, S. Berritt, M. R. Gau, D. J. Mindiola and M.-H. Baik, ACS Catal., 2018, 8, 10021–10031 CrossRef CAS.
- M. Uzelac, A. R. Kennedy and E. Hevia, Inorg. Chem., 2017, 56, 8615–8626 CrossRef CAS PubMed.
- G. dePetris, A. Troiani, M. Rosi, G. Angelini and O. Ursini, Chem. – Eur. J., 2009, 15, 4248–4252 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. Mahaut, A. Chardon, L. Mineur, G. Berionni and B. Champagne, ChemPhysChem, 2021, 22, 1958–1966 CrossRef CAS PubMed.
- K. Hansen, T. Szilvási, B. Blom and M. Driess, J. Am. Chem. Soc., 2014, 136, 14207–14214 CrossRef CAS PubMed.
- H. Braunschweig, W. C. Ewing, K. Geetharani and M. Schäfer, Angew. Chem., Int. Ed., 2015, 54, 1662–1665 CrossRef CAS PubMed.
- D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., 2020, 132, 21100–21104 CrossRef.
- A. Kassymbek, S. F. Vyboishchikov, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, Angew. Chem., Int. Ed., 2019, 58, 18102–18107 CrossRef CAS PubMed.
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784–6790 CrossRef CAS PubMed.
- A. Kassymbek, D. Spasyuk, A. Dmitrienko, M. Pilkington and G. I. Nikonov, Chem. Commun., 2022, 58, 6946–6949 RSC.
- D. Rodrigues Silva, L. de Azevedo Santos, M. P. Freitas, C. F. Guerra and T. A. Hamlin, Chem. – Asian J., 2020, 15, 4043–4054 CrossRef CAS PubMed.
- L. J. Murphy, K. N. Robertson, J. D. Masuda and J. A. Clyburne, N-Heterocyclic Carbenes, John Wiley and Sons, Ltd, 2014, ch. 15, pp. 427–498 Search PubMed.
- M. G. Gardiner and C. L. Raston, Coord. Chem. Rev., 1997, 166, 1–34 CrossRef CAS.
- P. L. Baxter, A. J. Downs, M. J. Goode, D. W. H. Rankin and H. E. Robertson, J. Chem. Soc., Dalton Trans., 1990, 2873–2881 RSC.
- A. Storr, J. Chem. Soc. A, 1968, 2605 RSC.
- S. Schulz, L. Häming, R. Herbst-Irmer, H. W. Roesky and G. M. Sheldrick, Angew. Chem., Int. Ed. Engl., 1994, 33, 969–970 CrossRef.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172–2174 CrossRef CAS.
- R. J. Wright, A. D. Phillips, T. L. Allen, W. H. Fink and P. P. Power, J. Am. Chem. Soc., 2003, 125, 1694–1695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02152a |
| This journal is © The Royal Society of Chemistry 2025 |