
Visible-light induced 1,2-dicarbofunctionalization of alkenes with quinoxalin-2(1H)-ones and malonic esters†
Yi
Wang
a,
Jia
Wang
a,
Chao
Chen
*b,
Lianhui
Song
c,
Zu-Li
Wang
e,
Wei
Wei
 *c and
Dong
Yi
*d
*c and
Dong
Yi
*d
aSchool of Pharmacy, Shaoyang University, Shaoyang 422000, P. R. China
bZhejiang Collaborative Innovation Center for Full-Process Monitoring and Green Governance of Emerging Contaminants, College of Biology and Environmental Engineering, Zhejiang Shuren University, Hangzhou 310015, China. E-mail: chencc@zjsru.edu.cn
cSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, P. R. China. E-mail: weiweiqfnu@163.com
dGreen Pharmaceutical Technology Key Laboratory of Luzhou City, School of Pharmacy, Southwest Medical University, Luzhou 646000, Sichuan, P. R. China. E-mail: yidong@swmu.edu.cn
eCollege of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, P. R. China
First published on 11th June 2025
Abstract
Visible-light photoredox catalyzed 1,2-arylalkylation of alkenes with quinoxalin-2(1H)-ones and malonic esters has been developed through direct C(sp2)–H/C(sp3)–H functionalization under mild conditions. A number of quinoxalin-2(1H)-one containing esters could be obtained in moderate to good yields in a step and atom-economic manner. This transformation proceeded through a radical process, which features the advantages of mild conditions, a clean energy source, a wide substrate scope, and favorable functional group compatibility.
Difunctionalization of alkenes is a practical and powerful protocol for rapidly synthesizing complex molecular structures through the introduction of two functional groups into one molecular skeleton.1,2 During the past decade, visible-light photoredox catalyzed difunctionalization of alkenes has been increasingly utilized in modern organic synthesis due to its advantages such as mild conditions, a clean energy source and sustainability.3–6 In this context, photocatalytic difunctionalization of alkenes with malonates has been elegantly developed for constructing substituted esters via direct cleavage of the C–H bond of malonates.7–10 For example, in 2022, Reiser reported visible-light-induced Cu(II)-catalyzed oxoalkylation of alkenes with malonates and dioxygen leading to an oxoalkylated product (Scheme 1a).7 In 2022, Deng described photoinduced fac-Ir(ppy)3 three-component carboarylation of alkenes with cyano-substituted pyridines and malonic esters (Scheme 1b).8 In 2024, Yi's group presented photoredox-catalyzed 1,2-dicarbofunctionalization of alkenes with aldehydes and malonates to access δ-hydroxy esters and δ-keto esters in the presence of K3PO4 (Scheme 1c).9 Very recently, Abel and Beletskaya reported photoredox-catalyzed bi-functionalization of alkenes with malonic esters and CO2 to prepare 3-arylpropane-1,1,3-tricarboxylic acid esters (Scheme 1d).10 Although significant advances have been achieved in these reactions, the development of new difunctionalization of alkenes with malonic esters and aromatic C–H feedstocks to construct more diverse functionalized esters in an atom-economic manner is still highly desirable.
 | ||
| Scheme 1 Photoinduced difunctionlization of alkenes with malonic esters. | ||
Quinoxalin-2(1H)-ones are highly valuable heteroaromatic structural motifs present in many natural products, biologically active compounds and pharmaceuticals.11 The direct C3(sp2)–H functionalization of quinoxalin-2(1H)-ones has attracted great interest from chemists owing to the special chemical and biological activities of C3-substituted quinoxalin-2(1H)-one derivatives.12–15 With our continued interest in photoredox catalyzed reactions,16 herein, we wish to report visible-light photoredox catalyzed 1,2-arylalkylation of alkenes with quinoxalin-2(1H)-ones and malonic esters via direct C(sp2)–H/C(sp3)–H functionalization at room temperature. The present transformations could be conducted in a step and atom-economic manner to access a series of quinoxalin-2(1H)-one containing esters in moderate to good yields (Scheme 1e).
At the outset of our investigations, the model reaction of styrene (1a), quinoxalin-2(1H)-one (2a), and diethyl malonate (3a) was employed to screen the reaction conditions (Table 1). As shown in entry 1, the desired product 4aa could be obtained in 33% yield when Rose Bengal (2 mol%) was used as a photocatalyst with the addition of Na2CO3 in DMSO. The screening of other photocatalysts such as Rhodamine 6G, Eosin Y, Rhodamine B, [Ir(dtbbpy)(ppy)2]PF6, Ir[dF(CF3)ppy]2(dtbbpy)PF6, 4CzIPN, Mes-Acr-ClO4, Ru(bpy)3Cl2·H2O, and [Ir(dtbbpy)(ppy)2]PF6 found that Ir[dF(CF3)ppy]2(dtbbpy)PF6 was the best one to provide product 4aa in 81% yield (entry 5). Then, the reactions performed in a number of solvents including DCE, DCM, CH3CN, 1,4-dioxane, DME, THF, and toluene were examined. No product or only a trace amount of product was observed in the above solvents (entries 11–17). Further investigations of various bases demonstrated that Na2CO3 was still the best base to promote this transformation (ESI†). Changing the loading of the photocatalyst did not improve the reaction efficiency (entries 18 and 19). No transformation was observed in the absence of photocatalysts (entry 20). Testing of other light sources showed that green light LEDs, white light LEDs and purple light LEDs would lead to low reaction efficiency (entries 21–23). In addition, the desired product was obtained in 76% yield when the model reaction was conducted in the presence of dioxygen (entry 24).
|
|
|||
|---|---|---|---|
| Entry | Photocatalysts (x mol%) | Solvent | Yieldb (%) |
| a Conditions: 1a (0.1 mmol), 2a (0.2 mmol), 3a (0.25 mmol), photocatalyst (1–5 mol%), Na2CO3 (2 equiv.), solvent (2 mL), 5 W blue LEDs, air, r.t., 2 h. b Isolated yields based on 1a. c Green light LEDs. d White light LEDs. e Purple light LEDs. f Under dioxygen. | |||
| 1 | Rose Bengal (2) | DMSO | 33 |
| 2 | Rhodamine 6G (2) | DMSO | 0 |
| 3 | Eosin Y (2) | DMSO | 26 |
| 4 | Rhodamine B (2) | DMSO | 40 |
| 5 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMSO | 81 |
| 6 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2) | DMSO | 65 |
| 7 | 4CzIPN (2) | DMSO | 56 |
| 8 | Mes-Acr-ClO4 (2) | DMSO | 59 |
| 9 | Ru(bpy)3Cl2·3H2O (2) | DMSO | 48 |
| 10 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMF | 15 |
| 11 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DCE | 0 |
| 12 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DCM | 0 |
| 13 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DME | 0 |
| 14 | [Ir(dtbbpy)(ppy)2]PF6 (2) | 1,4-Dioxane | 0 |
| 15 | [Ir(dtbbpy)(ppy)2]PF6 (2) | CH3CN | Trace |
| 16 | [Ir(dtbbpy)(ppy)2]PF6 (2) | THF | Trace |
| 17 | [Ir(dtbbpy)(ppy)2]PF6 (2) | Toluene | 0 |
| 18 | [Ir(dtbbpy)(ppy)2]PF6 (1) | DMSO | 75 |
| 19 | [Ir(dtbbpy)(ppy)2]PF6 (5) | DMSO | 76 |
| 20 | — | DMSO | 0 |
| 21 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMSO | 26c |
| 22 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMSO | 30d |
| 23 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMSO | 5e |
| 24 | [Ir(dtbbpy)(ppy)2]PF6 (2) | DMSO | 76f |
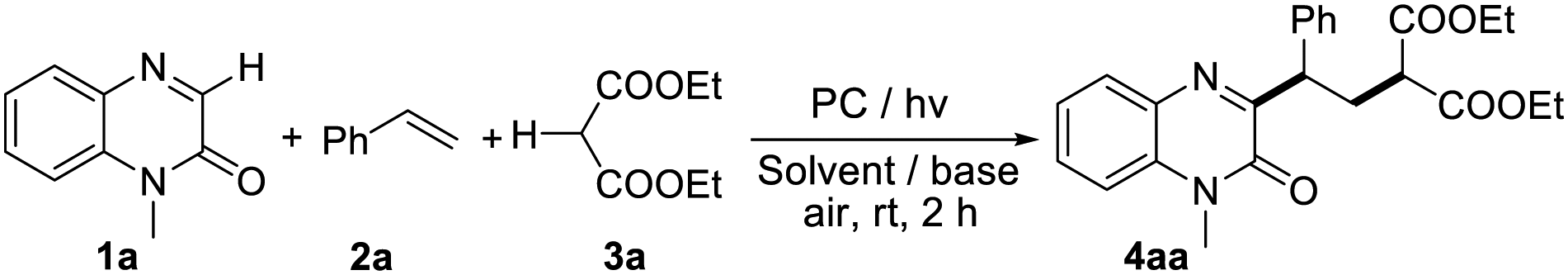
With the established reaction conditions in hand, we then investigated the scope of this 1,2-dicarbofunctionalization of alkenes with quinoxalin-2(1H)-ones and malonic esters (Table 2). A wide range of aromatic alkenes bearing both electron-donating groups and electron-withdrawing groups proved to be suitable for the reaction, and the corresponding products 4ab–4am were isolated in moderate to good yields. It is worth noting that sensitive functionalities such as halogen, ester, acetyloxy, and cyano groups were also tolerated in this procedure, highlighting the potential synthetic transformations of this protocol. Aromatic heterocyclic olefins such as 2-vinylthiophene and 2-vinylpyridine were suitable substrates to provide the corresponding products 4an and 4ao, albeit in relatively lower yields. 2-Naphthalene and internal olefin were also compatible with this procedure, and the corresponding products 4ap and 4aq were obtained in 70% and 69% yields, respectively. In addition, this protocol was also shown to be applicable to aliphatic olefins. When allylbenzene and 1-allylnaphthalene were employed as substrates, the desired products 4ar and 4as were obtained in good yields. Nevertheless, when 1,2-diaryl-substituted alkenes such as (E)-1,2-diphenylethene were employed in this reaction system, none of the desired products was observed.
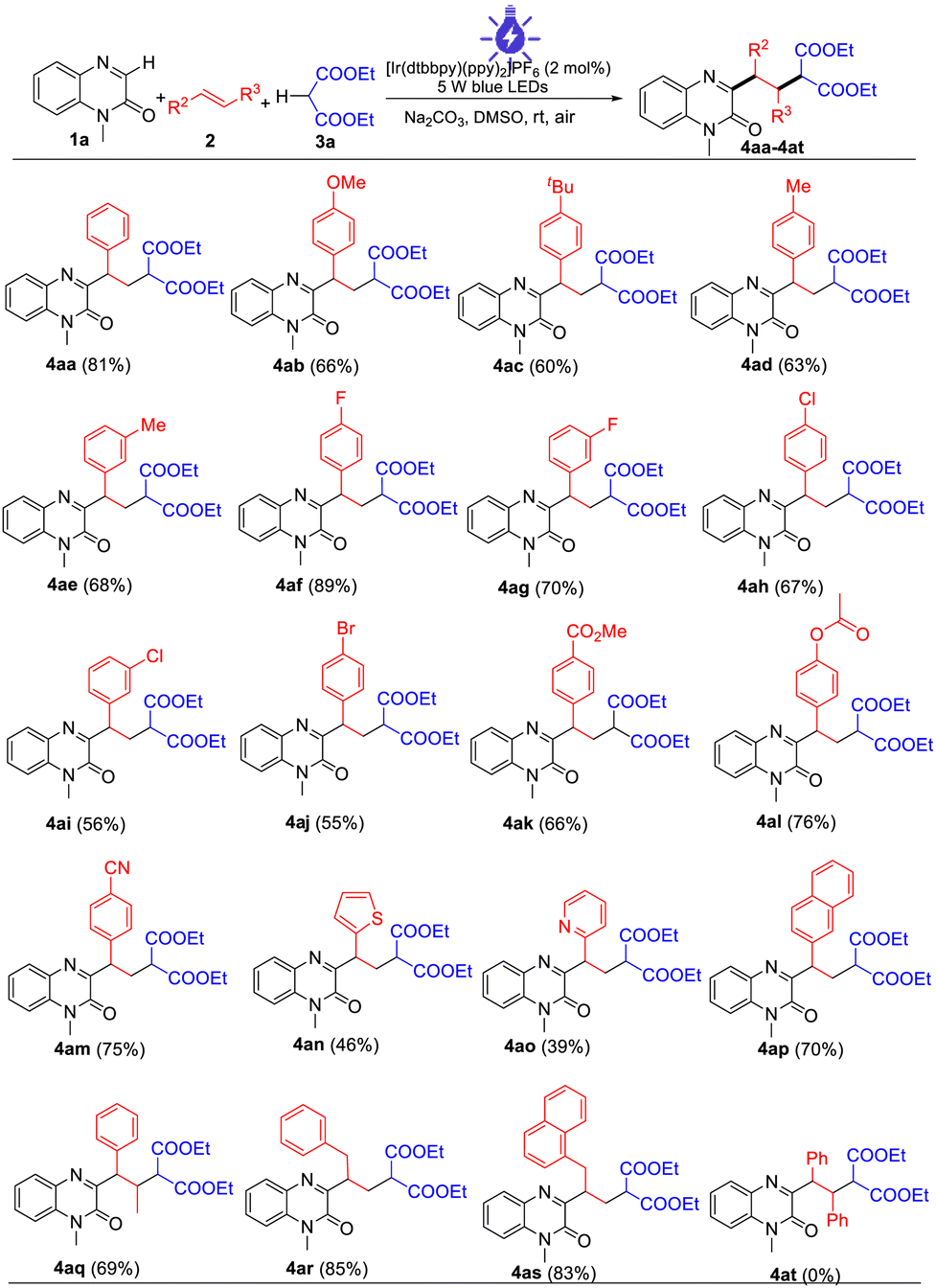
The capacity of the present multi-component reaction was also illustrated by the scope of quinoxalin-2(1H)-ones with malonic esters (Table 3). Quinoxalin-2(1H)-ones with substituents such as Me, MeO, and F on the benzene ring were compatible with this procedure, providing the desired products (4ba–4da) in moderate to high yields. Other N-substituted substrates such as N-ethyl, N-butyl, N-benzyl, and N-esteryl quinoxalin-2(1H)-ones also displayed good reactivity and provided the products 4ea–4ha in 73–82% yields. Notably, N-free protected quinoxalin-2(1H)-one also proceeded smoothly to afford the product 4ia in 51% yield. With respect to other malonic esters, in addition to diethyl malonate, dimethyl malonate, dibenzyl malonate, diisopropyl malonate, and di-tert-butylmalonate malonate were also suitable for the photocatalytic reactions, providing the corresponding 4ja–4ma in moderate to good yields. Unfortunately, the desired products were not detected when keto esters such as ethyl 3-oxo-3-phenylpropanoate and ethylacetoacetate were investigated in this reaction procedure.
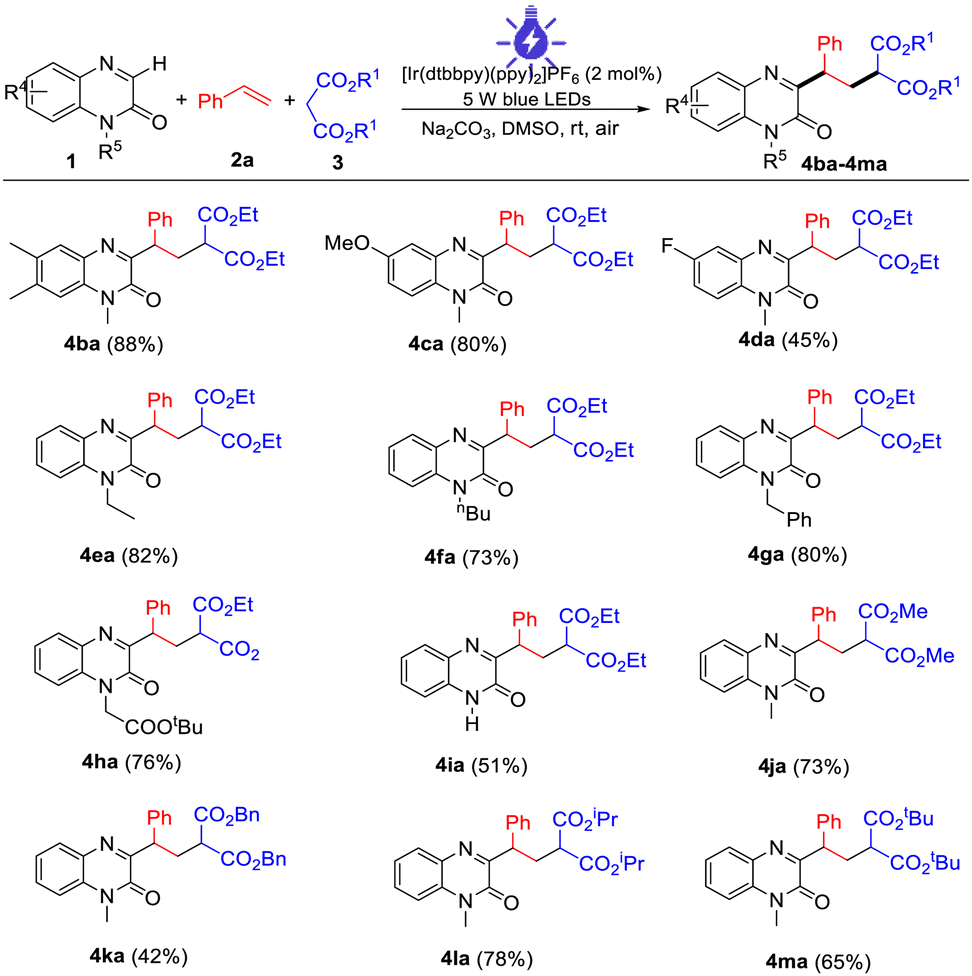
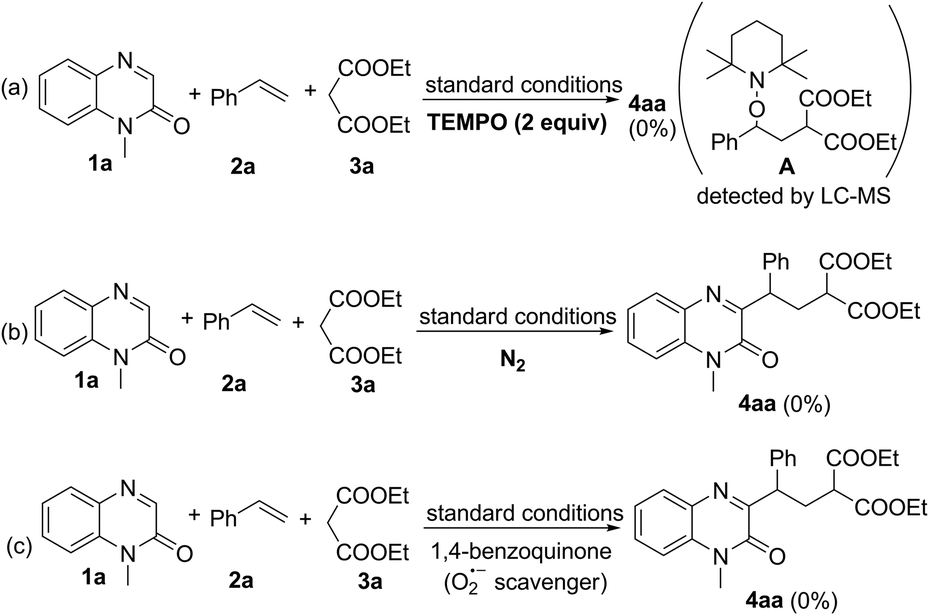
Next, several control experiments were performed to obtain some insight into the possible reaction mechanism (Scheme 2). First, when TEMPO (a radical scavenger) was added to the model reaction system, this photocatalytic multi-component reaction was completely suppressed and TEMPO-trapped complex A was observed by LC-MS. This result indicated that this reaction might involve the radical mechanism (Scheme 2a). Second, none of the desired products was observed when the model reaction was carried out under N2, suggesting that O2 in air is essential for promoting this transformation (Scheme 2b). Third, this transformation was also completely inhibited by the addition of 1,4-benzoquinone (a superoxide radical anion scavenger), indicating that O2˙− played a significant role in this reaction (Scheme 2c). Finally, the behavior of the visible light-excited step was further revealed by Stern–Volmer fluorescence quenching experiments. As a result, the emission intensity of the excited photocatalyst was obviously quenched with increasing concentration of the deprotonated enolate anion of malonate. Such a phenomenon was not observed between the excited photocatalyst and alkene 2a or malonate 3a under visible-light irradiation (Fig. 1). This result suggested that the interaction between the excited photocatalyst and the deprotonated enolate anion of malonate should exist in this reaction system (see the ESI†).
 | ||
| Scheme 2 Control experiments. | ||
 | ||
| Fig. 1 Stern–Volmer plots. I0 is the inherent fluorescence intensity of [Ir(dtbbpy)(ppy)2]PF6. I is the fluorescence intensity of [Ir(dtbbpy)(ppy)2]PF6 in the presence of Na+3a−, 2a or 3a. | ||
On the basis of the abovementioned results and literature,7–10,17 a possible mechanism was proposed as shown in Scheme 3. Under light irradiation, the photocatalyst IrIII complex was firstly excited to give the excited-state *IrIII, which can be readily reduced by the deprotonated enolate anion 5 affording the reduced IrII and malonate radical 6. Then, the IrII complex was oxidized by dioxygen (O2) to generate a superoxide radical anion (O2˙−), along with regeneration of the ground-state IrIII complex. Subsequently, the selective addition of malonate radical 6 to alkene 2 produced alkyl radical 7, which reacted with quinoxalin-2(1H)-one 1 to provide nitrogen radical intermediate 8. Finally, the deprotonation of the nitrogen radical 8 by the superoxide radical anion (O2˙−) afforded the desired product 4.
 | ||
| Scheme 3 Proposed mechanism. | ||
In conclusion, we have developed visible-light photoredox catalyzed 1,2-dicarbofunctionalization of alkenes with quinoxalin-2(1H)-ones and malonic esters at room temperature. Through this methodology, a series of quinoxalin-2(1H)-one containing esters were efficiently obtained in a step and atom-economic manner through direct C(sp2)–H/C(sp3)–H functionalization. This three-component reaction proceeded through a radical process, in which two carbon–carbon bonds were consecutively formed in a one pot procedure. The advantages of high atom economy, a clean energy source, mild conditions, and good functional group tolerance make this method attractive in synthetic chemistry.
This work was supported by the Hunan Provincial Natural Science Foundation of China (no. 2025JJ70231), the Scientific Research of Hunan Education Department (no. 24B0684), and the Collaborative Fund of Luzhou Government and Southwest Medical University (no. 2023LZXNYDJ019).
Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the ESI,† which include general synthetic experimental details, characterization data, and NMR spectra, respectively.Notes and references
- (a) M.-J. Luo, Q. Xiao and J.-H. Li, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC; (b) V. W. Bhoyare, A. G. Tathe, A. Das, C. C. Chintawar and N. T. Patil, Chem. Soc. Rev., 2021, 50, 10422–10450 RSC; (c) X. Chen, F. Xiao and W.-M. He, Org. Chem. Front., 2021, 8, 5206–5228 RSC; (d) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Bao, M. Tang, Z. Ye, Z. Yuan and G. Zhu, Chem. Commun., 2022, 58, 3847–3864 RSC; (b) C. Qu, Y. Lv, J. Huang, C. Ma, H. Yue, W. Wei and D. Yi, Green Chem., 2023, 25, 10678–10683 RSC; (c) Q. Zhao, Q. Zhang, X. Wu, L. Wang, K. Shen, Y. Hua, C. Gao, Y. Zhang, M. Peng and K. Zhao, Chin. Chem. Lett., 2025, 36, 110167 CrossRef; (d) K. Zheng, C. Chen, Y. Wang, H. Xu, K. Ge and C. Shen, Org. Lett., 2025, 27, 4747–4752 CrossRef CAS PubMed.
- (a) S. Gupta, A. Kundu, S. Ghosh, A. Chakraborty and A. Hajra, Green Chem., 2023, 25, 8459–8493 RSC; (b) T. Guan, J.-Y. Guo, Q. H. Zhang, X.-W. Xu, X.-Y. Yu, Y. Zhang and K. Zhao, Green Chem., 2022, 24, 6524–6530 RSC; (c) X.-M. Chen, L. Song, J. Pan, F. Zeng, Y. Xie, W. Wei and D. Yi, Chin. Chem. Lett., 2024, 35, 110112 CrossRef CAS.
- (a) H.-C. Li, G.-N. Li, H.-S. Wang, Y. Tan, X.-L. Chen and B. Yu, Org. Chem. Front., 2024, 11, 135–141 RSC; (b) Z. Wang, Q. Liu, R. Liu, Z. Ji, Y. Li, X. Zhao and W. Wei, Chin. Chem. Lett., 2022, 33, 1479–1482 CrossRef CAS; (c) Q.-W. Gui, F. Teng, Z.-C. Li, Z.-Y. Xiong, X.-F. Jin, H.-Y. Liu, Y.-W. Lin, Z. Cao and W.-M. He, Chin. Chem. Lett., 2021, 32, 1907–1910 CrossRef CAS.
- (a) H. Singh, R. K. Tak, D. P. Poudel and R. Giri, ACS Catal., 2024, 14, 6001–6008 CrossRef CAS PubMed; (b) Q. Feng, J. Hao, Y. Hu, R. Fu, W. Wei and D. Yi, Chin. Chem. Lett., 2025, 36, 110582 CrossRef CAS; (c) B. Sun, X.-L. Tang, X. Zhuang, L. Ling, P. Huang, J. Wang and C. Jin, Adv. Synth. Catal., 2023, 365, 1020–1026 CrossRef CAS.
- (a) J. Liu, S. Tang, S. Wang, M. Cao, J. Zhao, P. Zhang and P. Li, J. Org. Chem., 2022, 87, 9250–9258 CrossRef CAS PubMed; (b) H.-C. Li, M. Zhang, Q. Lv, K. Sun, X.-L. Chen, L. Qu and B. Yu, Chin. Chem. Lett., 2025, 36, 110579 CrossRef CAS.
- N. Katta, Q.-Q. Zhao, T. Mandal and O. Reiser, ACS Catal., 2022, 12, 14398–14407 CrossRef CAS PubMed.
- Y. Hong, M.-Y. Dong, D.-S. Li and H.-P. Deng, Org. Lett., 2022, 24, 7677–7684 CrossRef CAS PubMed.
- T. Li, W. Wang, M. Dong, Z. Zhang, S. Yu, Z. Chen, S. Wei and D. Yi, Chin. J. Chem., 2024, 42, 957–962 CrossRef CAS.
- A. D. Kharlamova, A. D. Averin, G. N. Bondarenko, A. S. Abel and I. P. Beletskaya, Adv. Synth. Catal., 2025, 367, e202400956 CrossRef CAS.
- (a) S. A. M. El-Hawash, N. S. Habib and M. A. Kassem, Arch. Pharm., 2006, 339, 564–571 CrossRef CAS PubMed; (b) R. Liu, Z. Huang, M. G. Murray, X. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747–5768 CrossRef CAS PubMed.
- (a) Q. Ke, G. Yan, J. Yu and X. Wu, Org. Biomol. Chem., 2019, 17, 5863–5881 RSC; (b) M. Zhang, G. Nan, X. Zhao and W. Wei, Chin. J. Org. Chem., 2022, 42, 4315–4322 CrossRef CAS; (c) W.-T. Ouyang, J. Jiang, Y.-F. Jiang, T. Li, Y.-Y. Liu, H.-T. Ji, L.-J. Ou and W.-M. He, Chin. Chem. Lett., 2024, 35, 110038 CrossRef CAS; (d) H.-Y. Song, J. Jiang, C. Wu, J.-C. Hou, Y.-H. Lu, K.-L. Wang, T.-B. Yang and W.-M. He, Green Chem., 2023, 25, 3292–3296 RSC.
- (a) N. Meng, Y. Lv, Q. Liu, R. Liu, X. Zhao and W. Wei, Chin. Chem. Lett., 2021, 32, 258–262 CrossRef CAS; (b) N. Zhou, R. Liu, C. Zhang, K. Wang, J. Feng, X. Zhao and K. Lu, Org. Lett., 2022, 24, 3576–3581 CrossRef CAS PubMed.
- (a) S. Singh, N. Dagar, G. Pal and S. R. Roy, Green Chem., 2022, 24, 8460–8465 RSC; (b) L. Wang, P. Bao, W. Liu, S. Liu, C. Hu, H. Yue, D. Yang and W. Wei, Chin. J. Org. Chem., 2018, 38, 3189–3196 CrossRef CAS; (c) Y.-D. Xu, Y.-M. Xing, H.-T. Ji, L.-J. Ou, W.-B. He, J. Peng, J.-S. Wang, J. Jiang and W.-M. He, J. Org. Chem., 2024, 89, 17701–17707 CrossRef CAS PubMed.
- (a) K.-L. Wang, H.-T. Ji, L.-J. Ou and W.-M. He, Eur. J. Org. Chem., 2023, e202300752 CrossRef CAS; (b) Q. Yang, B. Wang, M. Wu and Y.-Z. Lei, Molecules, 2023, 28, 2513 CrossRef CAS PubMed.
- (a) L. Song, C. Ma, J. Huang, Y. Lv, H. Yue, J. You, W. Wei and D. Yi, J. Org. Chem., 2024, 89, 10974–10986 CrossRef CAS PubMed; (b) Y. Lv, H. Ding, J. You, W. Wei and D. Yi, Chin. Chem. Lett., 2024, 35, 109107 CrossRef CAS.
- (a) S. Baś, Y. Yamashita and S. Kobayashi, ACS Catal., 2020, 10, 10546–10550 CrossRef; (b) M.-Y. Dong, C.-Y. Han, D.-S. Li, Y. Hong, F. Liu and H.-P. Deng, ACS Catal., 2022, 12, 9533–9539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02744f |
| This journal is © The Royal Society of Chemistry 2025 |