
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and reactivity of a triflimide-bridged bis(trimethylsilyl) cation†
Joshua H.
Daum‡
,
Nattamai
Bhuvanesh
and
Oleg V.
Ozerov
 *
*
Department of Chemistry, Texas A&M University, College Station, TX 77842, USA. E-mail: ozerov@chem.tamu.edu
First published on 2nd June 2025
Abstract
Preparation of a triflimide-bridged bis-silylium monocation [(Me3Si)2NTf2]+ has been accomplished as a [HCB11Cl11]− salt. Its structure has been analyzed and its potency in halide abstraction illustrated.
Silylium cations (R3Si+) are uniquely reactive Lewis acids with particular affinity for harder Lewis bases.1,2 They have been used in catalysis,3 especially in the context of C–F bond activation,4–6 but are also of special interest as powerful but gentle abstractors of halides and pseudohalides in the syntheses of otherwise difficult to access cations.2,3,7 Realization of the full potential of silylium cations in condensed phases requires the use of especially robust and weakly coordinating anions,8 in order to approach the reactivity of the “naked” silylium cation. Carborane anions have proven especially advantageous.9–11 A crystallographically characterized example of a true three-coordinate silylium cation has been reported.12 However, where the silylium reagent is intended to be used as a (pseudo)halide abstractor,1 a “naked” silylium cation is not necessary, nor is it necessary to use a reagent whose composition includes nothing besides the silylium cation and a weakly coordinating anion. Abstraction of a (pseudo)halide X− leads to the formation of R3Si–X, which can then form an adduct with the remaining R3Si+. If the formation of such a [R3Si–X–SiR3]+ adduct deactivates silylium and prevents the abstraction from happening, the reaction will not surmount 50% conversion even if base-free R3Si+ is initially used. The reports of isolation of [(Me3Si)2X][B(C6F5)4] (X = F, Cl, Br, I, OTf) by Schulz et al. served as guiding examples for our group.13,14 We have subsequently used the carborane version [(Me3Si)2OTf][HCB11Cl11] (and its analog [(Et3Si)2OTf][HCB11Cl11]) to abstract a (pseudo)halide from C–X bonds in organic and organometallic compounds, with the generation of reactive cations.15–17 Me3SiOTf binds Me3Si+ more strongly than does Me3SiH and so [(Me3Si)2OTf]+ is readily isolated in the reaction of Me3SiH with Ph3C+ in the presence of Me3SiOTf. [(Me3Si)2OTf][HCB11Cl11] is a solid, not an oil, and thus can be more effectively purified. The presence of the convenient 19F NMR reporter is also a plus, as it allows to monitor the release of free Me3SiOTf upon halide abstraction. Lastly, if the target (pseudo)halide for abstraction is not itself a triflate, the reaction ideally yields a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Me3SiX and Me3SiOTf – a ready 1H NMR spectroscopic verification of the stoichiometry of the desired reaction.
:1 mixture of Me3SiX and Me3SiOTf – a ready 1H NMR spectroscopic verification of the stoichiometry of the desired reaction.
The properties of triflate are sometimes compared with those of the triflimide anion NTf2− = [(F3CSO2)2N]−. Both HOTf and HNTf2 are considered superacids, and their relative strength depends on the medium.18 HNTf2 often outperforms HOTf in organic catalysis.19 Metal bis(triflimides) appear to be more Lewis acidic than the analogous metal triflates.20 Although the nitrogen in NTf2− is of course less electronegative than the oxygen in OTf−, the presence of two triflyl groups increases charge delocalization and steric encumbrance, which makes triflimide less of a nucleophile or a base, in at least some situations. With this in mind, we wished to access a triflimide analog of [(Me3Si)2OTf][HCB11Cl11] and evaluate its properties.
The reaction of [Ph3C][HCB11Cl11] with Me3SiH in the presence of either Me3SiNTf2 (prepared in situ from HNTf2 and allyltrimethylsilane)21 or HNTf2 proceeded smoothly and led to the isolation of [(Me3Si)2NTf2][HCB11Cl11] as a fine white crystalline solid in 99% yield (Scheme 1). In solution at ambient temperature, it displayed a single resonance for the cation in each of its 1H, 13C, 29Si, and 19F NMR spectra, in addition to the expected 1H and 13C NMR resonances for the [HCB11Cl11]− anion. The 19F NMR resonance for [(Me3Si)2NTf2][HCB11Cl11] (−75.8 ppm) is shifted downfield from the neutral Me3SiNTf2 (−78.2 ppm), similarly to the downfield shift of [(Me3Si)2OTf][HCB11Cl11] (−74.1 ppm) vs. Me3SiOTf (−78.2 ppm). The downfield shift upon coordination of a Me3Si+ is likely a consequence of the diminution of electron density on the fluorine atoms. The magnitude of this shift is smaller for NTf2 because of the presence of two CF3 groups instead of one in OTf, thus a lesser expected effect per fluorine. [(Me3Si)2NTf2][HCB11Cl11] is dramatically more soluble in non-polar solvents. Whereas [(Me3Si)2OTf][HCB11Cl11] displayed only sub-millimolar solubility in C6D6 and none in pentane, [(Me3Si)2NTf2][HCB11Cl11] appears to be freely soluble in benzene and even gives rise to ca. 3 mM concentration in pentane (NMR evidence, see ESI†).
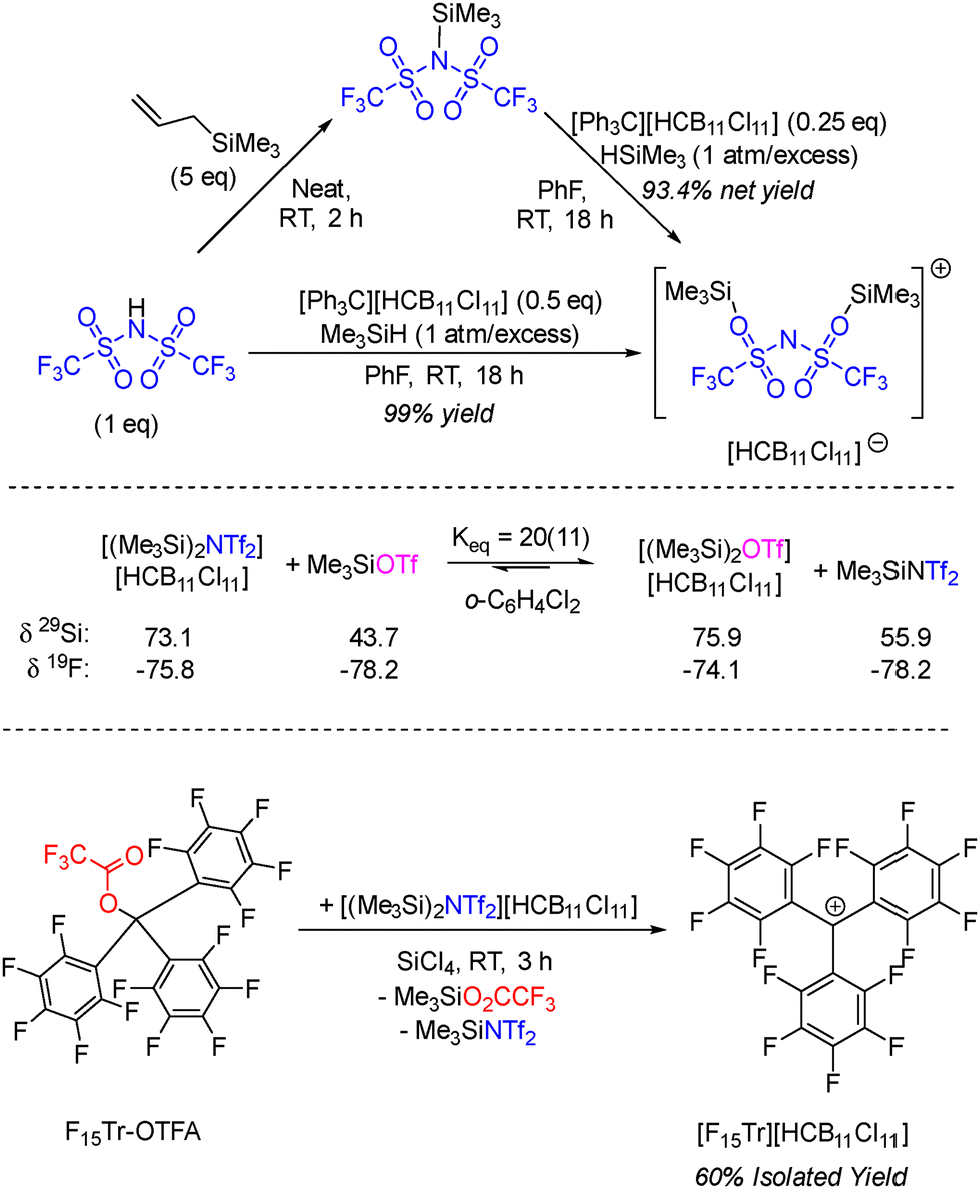 | ||
| Scheme 1 Synthesis of [(Me3Si)2NTf2][HCB11Cl11] (top), its reaction with Me3SiOTf with 29Si and 19F NMR chemical shifts given (middle), and its use in the synthesis of a perfluorotrityl cation (bottom). | ||
In order to evaluate the thermodynamic preference of the trimethylsilyl cation to bind to Me3SiOTf vs. Me3SiNTf2, [(Me3Si)2NTf2][HCB11Cl11] was treated with 0.95 equiv. Me3SiOTf in o-C6H4Cl2. The resultant homogenous mixture displayed only a single 29Si NMR resonance and two 19F NMR resonances (one for the NTf2 groups and the other for the OTf groups) at ambient temperature, indicating a fast equilibrium among [(Me3Si)2NTf2]+, [(Me3Si)2OTf]+, Me3SiNTf2, and Me3SiOTf. The equilibrium constant of ca. 20 favored [(Me3Si)2OTf]+. We also performed a van’t Hoff study in the 20–100 °C range in 1:9 mixture of C6D5CD3/o-C6H4Cl2. The Keq values were in the 1.5–2.5 range, corresponding to ΔH = 1.1 ± 0.4 kcal mol−1 and ΔS = 4.5 ± 1.1 cal (mol K)−1. These data show that [(Me3Si)2NTf2]+ is slightly less stabilized thermodynamically than [(Me3Si)2OTf]+. In other words, Me3Si+ displays a slight preference to bind to Me3SiOTf over Me3SiNTf2. Therefore, [(Me3Si)2NTf2]+ should function as a slightly more powerful (pseudo)halide abstractor.
Testing this notion, we used [(Me3Si)2NTf2][HCB11Cl11] to abstract a trifluoroacetate group from F15Tr-OTFA in a SiCl4 solution. This reaction resulted in a 60% isolated yield of [F15Tr][HCB11Cl11], comparable to that previously reported in a reaction using [(Me3Si)2OTf][HCB11Cl11].16 The data for the F15Tr+ are also consistent with the recent reports by Riedel et al. of its salts with other counteranions.22,23
The structure of [(Me3Si)2NTf2][HCB11Cl11] was determined by single-crystal X-ray crystallography (Fig. 1). The asymmetric unit was found to contain two independent units of [(Me3Si)2NTf2][HCB11Cl11]. The differences between the geometries of these two cations were not meaningful. There are no close contacts between the Si atoms and the carborane anions. The silicon atoms of the Me3Si groups are bound to the oxygens of the triflimide anion. In binding to Lewis acids, there are examples of triflimide utilizing its nitrogen, a single oxygen, or two oxygens.25,26 It appears to prefer to bind via the oxygen(s) to harder, more oxophilic Lewis acids.27 The Si–O(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) bond lengths of ca. 1.80–1.81 Å are comparable to those found in [(Me3Si)2OTf][B(C6F5)4] (1.81–1.82 Å),13 and Et3Si[CHB11Cl10OTf] (ca. 1.79 Å),28 and are slightly longer than those in [(Me3Si)3SO4][B(C6F5)4] (1.76–1.78 Å).29 The sums of C–Si–C angles in the four independent SiMe3 groups were found to be in the narrow 344°–346° range, similar to these known R3Si+–O(S) adducts. The two Me3Si groups are connected to the oxygens of the NTf2 fragment. The bonds between sulfur and the Si-bound oxygens are ca. 0.05 Å longer than the terminal S–O bonds. When compared to the parent HNTf230 and an “isolated” NTf2− anion such as in [C3(NPr2)3]NTf2,31 the N–S bond lengths of our silylated triflimide (ca. 1.56 Å) are more similar to the anionic NTf2− (ca. 1.56 Å) than neutral HNTf2 (ca. 1.64 Å). However, when comparing the S–N–S bond angle the opposite trend is seen with (Me3Si)2NTf2+ (123.8°) being more similar to HNTf2 (128.4°) than NTf2− (101.8°).
S) bond lengths of ca. 1.80–1.81 Å are comparable to those found in [(Me3Si)2OTf][B(C6F5)4] (1.81–1.82 Å),13 and Et3Si[CHB11Cl10OTf] (ca. 1.79 Å),28 and are slightly longer than those in [(Me3Si)3SO4][B(C6F5)4] (1.76–1.78 Å).29 The sums of C–Si–C angles in the four independent SiMe3 groups were found to be in the narrow 344°–346° range, similar to these known R3Si+–O(S) adducts. The two Me3Si groups are connected to the oxygens of the NTf2 fragment. The bonds between sulfur and the Si-bound oxygens are ca. 0.05 Å longer than the terminal S–O bonds. When compared to the parent HNTf230 and an “isolated” NTf2− anion such as in [C3(NPr2)3]NTf2,31 the N–S bond lengths of our silylated triflimide (ca. 1.56 Å) are more similar to the anionic NTf2− (ca. 1.56 Å) than neutral HNTf2 (ca. 1.64 Å). However, when comparing the S–N–S bond angle the opposite trend is seen with (Me3Si)2NTf2+ (123.8°) being more similar to HNTf2 (128.4°) than NTf2− (101.8°).
 | ||
| Fig. 1 POV-Ray rendition of the ORTEP24 drawing (50% thermal ellipsoids) showing one of the two independent [(Me3Si)2NTf2][HCB11Cl11] units with select atom labelling. Hydrogen atoms are omitted for clarity. Select bond distances (Å) and angles (°): Si1–O1, 1.818(4); Si2–O4, 1.802(4); S1–O1, 1.472(4); S2–O4, 1.479(4); S1–O2, 1.423(4); S2–O3, 1.414(5); N1–S1, 1.577(5); N1–S2, 1.562(5); Σ∢Si1–CH3, 345.2; Σ∢Si2–CH3, 344.3. | ||
In summary, we have been able to prepare an adduct of trimethylsilylium cation with trimethylsilyl triflimide in the form of the [(Me3Si)2NTf2][HCB11Cl11] salt. It appears that Me3Si+ binds Me3SiNTf2 slightly less strongly than Me3SiOTf. The new reagent provides another option for a silylium reagent for (pseudo)halide abstraction that also possesses increased solubility in solvents of low polarity.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [(Me3Si)2NTf2][HCB11Cl11] has been deposited at the Cambridge Structural Database under CCDC 2428563.We are thankful for the support of this research by the Office of Science of the US Department of Energy, Basic Energy Sciences (grant DE-SC0023280 to O. V. O.).
Conflicts of interest
There are no conflicts to declare.References
- C. A. Reed, Acc. Chem. Res., 1998, 31, 325–332 CrossRef CAS.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestrich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- J. C. L. Walker, H. F. T. Klare and M. Oestreich, Nat. Rev. Chem., 2020, 4, 54–62 CrossRef CAS.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- J. S. Siegel, O. Allemann, S. Duttwyler, P. Romanato and K. K. Baldridge, Science, 2011, 332, 574–577 CrossRef PubMed.
- B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS PubMed.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed.
- M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef PubMed.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS.
- L. Wang, Y. Jiang, S. Duttwyler, F. Lin and Y. Zhang, Coord. Chem. Rev., 2024, 516, 215974 CrossRef CAS.
- C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef CAS PubMed.
- K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed.
- A. Schulz, J. Thomas and A. Villinger, Chem. Commun., 2010, 46, 3696–3698 RSC.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2009, 48, 7444–7447 CrossRef CAS PubMed.
- O. Gunther, C.-I. Lee, E. Song, N. Bhuvanesh and O. V. Ozerov, Chem. Sci., 2022, 13, 4972–4976 RSC.
- W. Leong, A. R. Gogoi, T. Maity, C.-I. Lee, N. Bhuvanesh, O. Gutierrez and O. V. Ozerov, Angew. Chem., Int. Ed., 2025, e202422190 Search PubMed.
- J. Pell, Y. Zhu, R. Huacuja, D. E. Herbert, R. P. Hughes and O. V. Ozerov, Chem. Sci., 2017, 8, 3178–3186 RSC.
- B. Dhakal, L. Bohé and D. Crich, J. Org. Chem., 2017, 82, 9263–9269 CrossRef CAS PubMed.
- W. Zhao and J. Sun, Chem. Rev., 2018, 118, 10349–10392 CrossRef CAS PubMed.
- S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860–7888 CrossRef CAS PubMed.
- B. Rubial, A. Ballesteros and J. M. González, Eur. J. Org. Chem., 2022, e202200051 CrossRef CAS.
- K. F. Hoffmann, D. Battke, P. Golz, S. M. Rupf, M. Malischewski and S. Riedel, Angew. Chem., Int. Ed., 2022, 61, e202203777 CrossRef CAS PubMed.
- J. Schlögl, A. L. Brosius, A. N. Toraman, A. Wiesner, S. Steinhauer, C. Müller and S. Riedel, Angew. Chem., Int. Ed., 2025, 64, e202423857 CrossRef PubMed.
- J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- O. G. Polyakov, S. M. Ivanova, C. M. Gaudinski, S. M. Miller, O. P. Anderson and S. H. Strauss, Organometallics, 1999, 18, 3769–3771 CrossRef CAS.
- M. J. Earle, U. Hakala, B. J. McAuley, M. Nieuwenhuyzen, A. Ramania and K. R. Seddon, Chem. Commun., 2004, 1368–1369 RSC.
- M. Kawamura and S. Shimada, Inorg. Chim. Acta, 2007, 360, 2162–2168 CrossRef CAS.
- P. Press, B. J. McCulloch, W. Gu, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2015, 51, 14034–14037 RSC.
- K. Bläsing, R. Labbow, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2021, 60, 13798–13802 CrossRef PubMed.
- A. Haas, Ch Klare, P. Betz, J. Bruckmann, C. Krüger, Y.-H. Tsay and F. Aubke, Inorg. Chem., 1996, 35, 1918–1925 CrossRef CAS.
- J. Walst, R. Yunis, P. M. Bayley, D. R. MacFarlane, C. J. Ward, R. Wang and O. J. Curnow, RSC Adv., 2015, 5, 39565–39579 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, X-ray crystallographic data. CCDC 2428563. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01223f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |