
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric total syntheses of immunosuppressive diterpenoids triptobenzenes N and R via a remote Csp3–H functionalization†‡
Nanda Kishore
Roy§
 a,
Ranjit
Murmu§
a,
Mintu
Munda
b,
Sovan
Niyogi
a and
Alakesh
Bisai
*ab
a,
Ranjit
Murmu§
a,
Mintu
Munda
b,
Sovan
Niyogi
a and
Alakesh
Bisai
*ab
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, 741246, West Bengal, India. E-mail: alakesh@iiserkol.ac.in
bDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal – 462 066, Madya Pradesh, India
First published on 17th June 2025
Abstract
The total syntheses of the naturally occurring diterpenoids triptobenzenes N (1c) and R (1d) have been accomplished via a late-stage δ-Csp3–H functionalization of an enone intermediate. The highly functionalized dienone intermediate 2 was utilized as a common scaffold for this study. An XRD analysis of compounds 8 and 4 confirmed the stereochemistry of the quaternary centers of the abietane core. Finally, a chemoselective ketal protection and reduction completed the first total syntheses of the immunosuppressive diterpenoids triptobenzenes N (1c) and R (1d).
The abietane diterpenoids (1a–d; Fig. 1) are architecturally complex secondary metabolites sharing a carbotricyclic core having a trans-decalin motif and are useful targets for drug discovery.1 In the modern era, scientists have been interested in synthesizing such diterpenoids not only because of their interesting structural scaffolds but also for their important bioprofiles.1 In 1999, Li et al. isolated diterpenoids named triptobenzenes L (1a) and N (1c) as well as 15 other diterpenoids from the TII extract of T. wilfordii.2 Later, in 2013, Song et al. isolated triptobenzene R (1d) from the roots of T. wilfordii, along with four more abietane diterpenoids.3 Structurally, both triptobenzenes N (1c) and R (1d) contain two quaternary stereogenic centers and a primary alcohol group in the ‘A’ ring. Triptobenzene N (1c) also contains an additional benzylic ketone functionality. Recent studies revealed significant pharmacological activities of the isolate of T. wilfordii, including antifertility,4anti-rheumatoid-arthritis5 and immunosuppressive6 activities.
 | ||
| Fig. 1 Naturally occurring abietane diterpenoids 1a–d. | ||
Structurally, abietane diterpenoids 1a–d share a common 6/6/6 carbotricyclic framework with three contiguous stereogenic centers [four for each of 1a and 1b] where a trans-decalin scaffold is embedded with an aromatic ring (Fig. 1). Importantly, two stereogenic centers feature challenging all-carbon quaternary centers situated at the 1 and 3 positions of the cyclohexane A ring.7
Despite important bioprofiles reported for selected congeners, there are only a few reports on asymmetric total syntheses of diterpenoids 1a–d. In 2018, Carter et al. reported on an L-proline–sulfonamide-catalyzed Yamada–Otani reaction and a Pummerer reaction as key strategies for synthesizing aromatic abietane diterpenoids.8 In 2023, Chou et al. reported a semi-synthetic route to aromatic abietanes starting from commercially available dehydroabietic acid.9 Recently, our group10 reported a catalytic asymmetric approach to diterpenoids 1a–b. However, no synthesis of immunosuppressive diterpenoids triptobenzenes N (1c) and R (1d) has yet been reported.
Metal-free activation of typically inert Csp3–H bonds is one of the most important research topics of modern organic synthesis. In this regard, in 2022, our group reported on functionalization of an aliphatic Csp3–H bond of an indolosesuquiterpene moiety for the total syntheses of naturally xiamycins C–F achieved by utilizing the pioneering work of Tahara et al.11a,b Herein, we report a highly regioselective remote Csp3–H activation for the first total syntheses of the abietane diterpenoids triptobenzenes N (1c) and R (1d). Our retrosynthetic approach is outlined in Scheme 1. It was envisioned that a collective total synthesis of naturally occurring abietane diterpenoids 1c–d could be accomplished from an advanced highly functionalized enone 2 (Scheme 1). Enone 2 could be made by carrying out a β-elimination of tricyclic bromo compound 3 followed by allylic oxidation. The secondary bromide 3 could be derived from the enone 4via a key formal Csp3–H functionalization,11a,b which in turn could be accessed from the methyl calistrisate 5 in two steps (Scheme 1).
 | ||
| Scheme 1 Our retrosynthetic analysis. | ||
Based on above hypothesis, we started our synthesis from abietic acid 6, a naturally occurring diterpenoid (Scheme 2). Compound 6 was converted to methyl calistrisate 5 following our reported procedure (Scheme 2).12a
 | ||
| Scheme 2 Synthesis of α-bromo ketone 8. | ||
Then, benzylic oxidation of 5 afforded product 7 in 78% yield. Next, we thought of accessing enone 4 following a two-step protocol, namely α-bromination of 7 with phenyl trimethyl ammonium tribromide (PTAB) followed by β-elimination. Although, a reaction of 7 with PTAB afforded, in 96% yield, α-bromo ketone 8, whose identity was unequivocally proved from an X-ray analysis [CCDC 2411032‡], we were unfortunately unable to access enone 4 from 8 under base-promoted β-elimination. In fact, the XRD analysis of 8 (as reported earlier by Clark et al.,12b CCDC 2411032‡) showed a syn orientation of β-H with the α-Br atom. Additionally, epimerization of α-Br was considered probably not possible due to the ‘α-H’ of keto 8 being stabilized through an H-bonding interaction with ‘O’ of the ester (Scheme 2). Gratifyingly, a one-step procedure using SeO2 under an elevated temperature afforded 4 in 82% yield.13 The structure of 4 was also confirmed from an XRD analysis (CCDC 2417922‡). A plausible mechanism for enone formation through intermediate 7a is shown in Scheme 3.
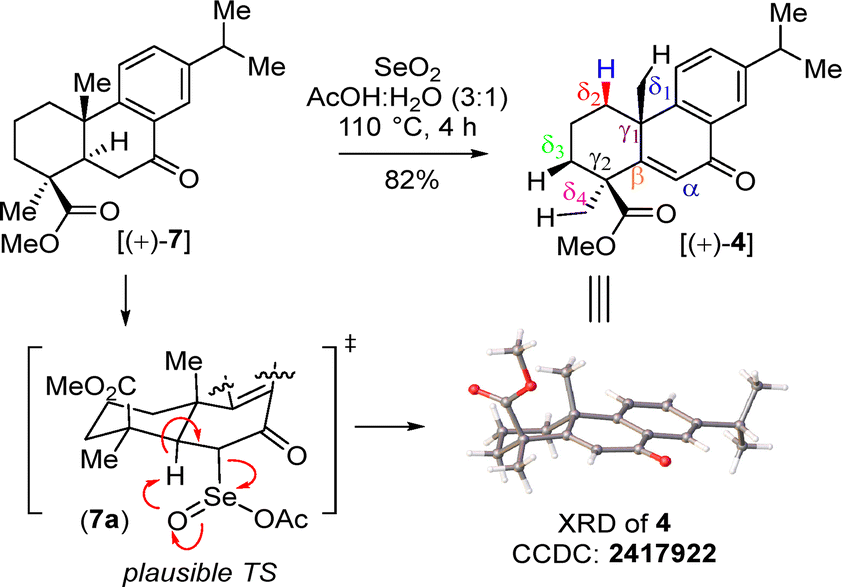 | ||
| Scheme 3 Synthesis of enone [(+)-4]. | ||
Having secured enone 4, we next looked into the key Csp3–H functionalization (Table 1). Towards this goal, we screened various conditions under metal-free formal C–H activation pathway affording Csp3–H halogenation. Initially, we tried several reactions using iodine with PhI(OAc)2 in the presence of light or under an elevated temperature. However, to our displeasure, these reactions led to a multitude of spots or decomposition of the mass balance. Initially, it was believed that the presence of angular methyl and axial ester at the 1,3-position of the A ring (Fig. 1) would create difficulties for the Csp3–H bond activation.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Br+ source | H+ source (cata.) | Temp (°C) | Time (h) | Yielda,b (%) |
| a Optimization reactions were carried out on 0.10 mmol of substrate. b Yields are reported for products obtained after column chromatography. c Starting materials at levels of 48–56% were recovered in addition to decomposition products. d Complex mixture of products. e DCE = 1,2-dichloroethane. | ||||||
| 1. | CH2Cl2 | Br2 (1.1 eq.) | HCl | 20 | 8 | —c |
| 2. | Ac2O | Br2 (1.2 eq.) | AcOH | 25 | 8 | —c |
| 3. | AcOH | NBS (1.2 eq.) | H2SO4 | 20 | 6 | —d |
| 4. | CH2Cl2 | NBS (1.1 eq.) | H2SO4 | 20 | 4 | 13 |
| 5. | CH2Cl2 | NBS (1.1 eq.) | H2SO4 | 25 | 8 | 17 |
| 6. | DCEe | NBS (1.1 eq.) | H2SO4 | 25 | 8 | 38 |
| 7. | Ac2O | NBS (1.1 eq.) | H2SO4 | 25 | 2 | 57 |
| 8. | Ac2O | NBS (1.1 eq.) | H2SO4 | 25 | 4 | 68 |
| 9. | Ac2O | NBS (1.1 eq.) | H2SO4 | 30 | 4 | 63 |
| 10. | Ac2O | NBS (1.1 eq.) | H2SO4 | 40 | 6 | 62 |

Thus, we thought of investigating our previously reported conditions on the formal Csp3–H functionalization (Table 1).11a,b We hence carried out a thorough screening of acids, solvents, electrophiles, and the best result was obtained using NBS as an electrophile in the presence of catalytic sulfuric acid in acetic anhydride solvent (Table 1). Following an exhaustive optimization, it was found that product 3 could be obtained in an isolated yield of 68% (entry 8, Table 1). Delightfully, secondary bromide 3 was obtained as a single diastereomer (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), suggesting a highly face-selective nature of this δ-Csp3–H functionalization.
:1), suggesting a highly face-selective nature of this δ-Csp3–H functionalization.
A proposed mechanism for the Csp3–H functionalization of 4 with NBS in the presence of catalytic sulfuric acid is illustrated in Scheme 3. According to this proposal, activation of the enone 4 with acetic anhydride leads to enol acetate carbocation intermediate 4bvia protonated species 4a (Scheme 3). Next, a syn-selective 1,2-migration of the angular methyl group occurs, forming olefin intermediate 4dvia a relatively stable benzylic carbocation 4c (Scheme 3). Subsequently, the formation of a bromonium ion intermediate 4e from the convex face is followed by another syn-selective 1,2-migration of the methyl group (see, intermediates 4f–g) ultimately resulting in compound 3 as a single diastereomer (Scheme 4).11b
 | ||
| Scheme 4 Plausible mechanism of Csp3–H bromination of [(+)-4]. | ||
After successful δ-Csp3-bromination, an E2-elimination of secondary bromide 3 was performed to achieve olefin 9 in 96% yield (Scheme 5 and Table 2).14
 | ||
| Scheme 5 Synthesis of di-enone [(+)-2]. | ||
Next, allylic oxidation of olefin 9 with SeO2 under an elevated temperature afforded allylic alcohol 10 in 84% yield as a single diastereomer (dr > 20:1). A plausible transition state for the allylic oxidation is depicted in Scheme 5. Due to steric hindrance created by the angular methyl group, SeO2 was believed to approach from the α-face of 9, forming a single diastereomer of 10. Next, DMP oxidation of 10 afforded diketo intermediate 2 in 97% yield (Scheme 5). Furthermore, hydrogenation of bis-enone derivative 2 in the presence of 10% (w/w) Pd/C and H2 (1 atm.) furnished 11 in 85% yield along with 12 as an over-reduced product (Scheme 6). During a longer period of hydrogenation, the benzylic enone could also co-ordinate with the Pd surface and stereoselectively transfer H2 onto the more exposed α-face of enone 2 (Scheme 6).
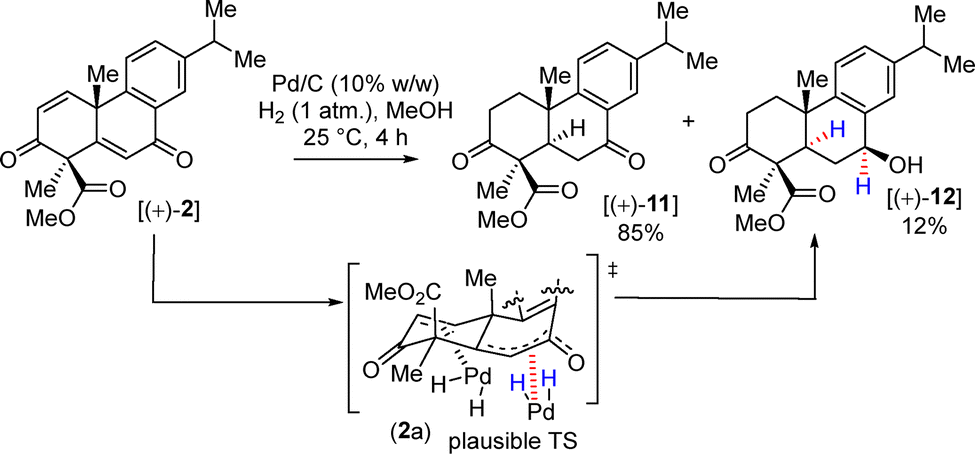 | ||
| Scheme 6 Synthesis of di-ketone [(+)-11]. | ||
Next, a highly diastereoselective reduction of diketone 11 with NaBH4 at −10 °C for 30 min furnished diol 13 in 98% yield with a dr > 20:1 (Scheme 7). With diol 13 secured, we then moved ahead with the synthesis of triptobenzene L (1a). Accordingly, chemoselective reduction of 13 using TFA/Et3SiH furnished the β-hydroxy ester 14 in 88% yield, which was followed by LiAlH4 reduction to complete the synthesis of triptobenzene L (1a) (Scheme 7). Along a similar line, a site-selective monoacetylation of triptobenzene L (1a) completed the total synthesis of nepetaefolin F (1b) (Scheme 7).
 | ||
| Scheme 7 Total syntheses of triptobenzene L [(+)-1a] and nepetaefolin F [(+)-1b]. | ||
Next, total syntheses of triptobenzene N (1c) and triptobenzene R (1d) were undertaken (Schemes 8 and 9). In this regard, bis-ketal protection of 11 using excess equivalents of ethylene glycol in the presence of catalytic p-TSA under refluxing toluene furnished bis-ketal 18a (Scheme 8).15 Next, LiAlH4 reduction of 18a in the same pot (see 18b) followed by deprotection of bis-ketal completed the first total synthesis of triptobenzene N (1c) (Scheme 8).
 | ||
| Scheme 8 Total synthesis of triptobenzene N [(−)-1c]. | ||
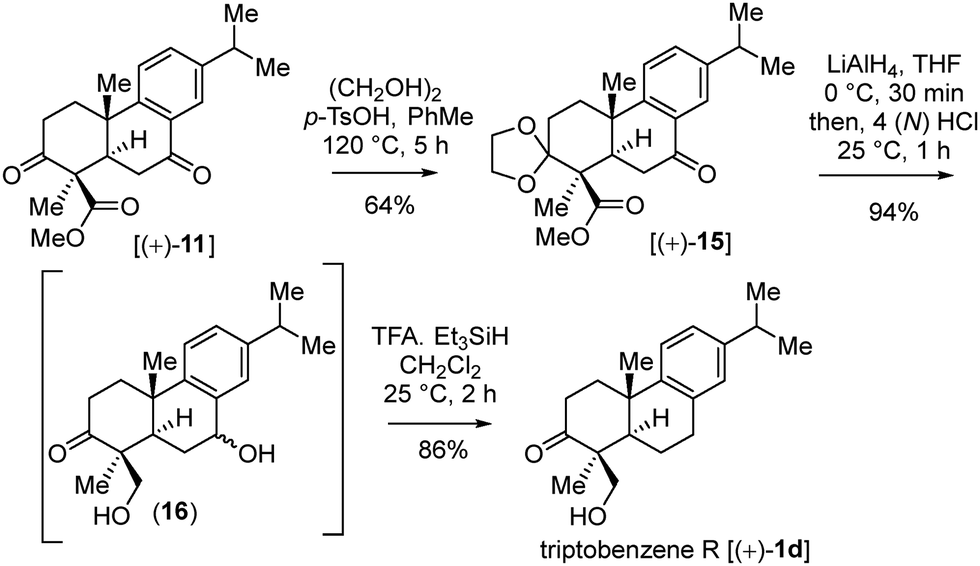 | ||
| Scheme 9 Total synthesis of triptobenzene R [(+)-1d]. | ||
Conversely, the chemoselective ketal protection in refluxing toluene afforded 15 in 64% yield (Scheme 9).16 Finally, LiAlH4 reduction of ester 15 followed by the ketal deprotection (see diol 16), and subsequent treatment with TFA/Et3SiH completed the first total synthesis of triptobenzene R (1d) (Scheme 9).
In conclusion, the first total syntheses of the immunosuppressive diterpenoids triptobenzenes N (1c) and R (1d) have been accomplished via a key Csp3–H functionalization. XRD analyses of enone 4 and α-bromoketone 8 indirectly confirmed the stereochemistry of the quaternary centers of these diterpenoids. Further application of this Csp3–H functionalization strategy in the context of total syntheses of other complex natural products is ongoing in our laboratory.
Financial support from the SERB [CRG/2023/000782] [SCP/2022/000486], STARS-MoE [STARS/2023/0753] and CSIR [02(0403)/21/EMR-II] is gratefully acknowledged. NKR thanks the UGC; RM thanks the CSIR for research fellowships. AB is a SERB-STAR Fellow and sincerely acknowledges the SERB [STR/2020/000061] for generous support.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the ESI.‡ CCDC 2411032 and 2417922 contain the crystallographic data for compounds 8 and 4 reported in this article.‡Notes and references
- H. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. Jia and D. Li, J. Nat. Prod., 1999, 62, 1522–1525 CrossRef CAS PubMed.
- C. G. Zhang, G. X. Chou, X. D. Mao, Q. S. Yang and J. L. Zhou, J. Nat. Prod., 2017, 80, 1742–1749 CrossRef CAS PubMed.
- J.-Y. Lia, Y. Peng, L.-Z. Lia, P.-Y. Gao, C. Gao, S.-X. Xia and S.-J. Song, Helv. Chim. Acta, 2013, 96, 313–319 CrossRef.
- J.-P. Bai and Y.-L. Shi, Contraception, 2002, 65, 441–445 CrossRef CAS.
- J. Cibere, Z. Deng, Y. Lin, R. Ou, Y. He, Z. Wang, A. Thorne, A. J. Lehman, I. K. Tsang and J. M. Esdaile, J. Rheumatol., 2003, 30, 465–467 Search PubMed.
- J. M. Fidler, G. Y. Ku, D. Piazza, R. Xu, R. Jin and Z. Chen, Transplantation, 2002, 74, 445–457 CrossRef CAS.
- J.-W. Yang and Y. Orihara, Tetrahedron, 2002, 58, 1265–1270 CrossRef CAS.
- X. Li and R. G. Carter, Org. Lett., 2018, 20, 5546–5549 CrossRef CAS PubMed.
- X.-D. Mao, T.-T. Du, Q. Gu Li. Yang, H.-L. Shi, R. Hong and G.-X. Chou, ACS Omega, 2023, 8, 14830–14840 CrossRef CAS.
- (a) R. Murmu, D. Jana, S. Noskar, M. Majhi and A. Bisai, J. Org. Chem., 2025, 90, 4973–4992 CrossRef CAS; (b) R. Murmu, S. Kundu, M. Maji, S. Pal, A. Mondal and A. Bisai, Chem. Commun., 2024, 60, 9737–9740 RSC.
- (a) H. Mizuno, T. Ohsawa and A. Tahara, Chem. Pharm. Bull., 1976, 24, 1527–1531 CrossRef CAS; (b) M. Munda, R. Nandi, V. R. Gavit, S. Kundu, S. Niyogi and A. Bisai, Chem. Sci., 2022, 13, 11666–11671 RSC.
- (a) M. Munda, D. Chatterjee, M. Majhi, S. Biswas, D. Pal and A. Bisai, RSC Adv., 2024, 14, 20420–20424 RSC; (b) J. F. Cutfield, T. N. Waters and G. R. Clark, J. Chem. Soc., Perkin Trans. 2, 1974, 150–157 RSC.
- M. A. El Had, J. J. Guardia, J. M. Ramos, M. Taourirte, R. Chahboun and E. Alvarez-Manzaneda, Org. Lett., 2018, 20, 5666–5670 CrossRef CAS PubMed.
- R. P. Robinson, L. Buckbinder, A. I. Haugeto, P. A. McNiff, M. L. Millham, M. R. Reese, J. F. Schaefer, Y. A. Abramov, J. Bordner, Y. A. Chantigny, E. F. Kleinman, E. R. Laird, B. P. Morgan, J. C. Murray, E. D. Salter, M. D. Wessel and S. A. Yocum, J. Med. Chem., 2009, 52, 1731–1743 CrossRef CAS PubMed.
- J.-L. Dong, L.-S.-H. Yu and J.-W. Xie, ACS Omega, 2018, 3, 4974–4985 CrossRef CAS.
- (a) F. Ono, H. Takenaka, T. Fujikawa, M. Mori and T. Sato, Synthesis, 2009, 1318–1322 CAS; (b) S. Kundu, D. Jana, N. Mandal, A. Mondal, R. Murmu, N. K. Roy, A. Datta and A. Bisai, Chem. Sci., 2024, 15, 14946–14953 RSC.
Footnotes |
| † This paper is dedicated respectfully to Prof. Deevi Basavaiah, University of Hyderabad, India, on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2411032 (8) and 2417922 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02354h |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |