
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwisting of aryl groups affects thermal back reactivity of diarylbenzene photoswitches†
Oka
Fukata
a,
Daichi
Kitagawa
 *a,
Katsuya
Mutoh
b and
Seiya
Kobatake
*a
*a,
Katsuya
Mutoh
b and
Seiya
Kobatake
*a
aDepartment of Chemistry and Bioengineering, Graduate School of Engineering, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: kitagawa@omu.ac.jp; kobatake@omu.ac.jp
bDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 10th June 2025
Abstract
Herein, we demonstrated that the thermal back reactivity of diarylbenzene photoswitches depended on the degree of twisting of the aryl groups. Density functional theory calculations revealed that the π-conjugation length in the transition state was the key to determining the activation free energy.
Molecular photoswitches serve as important components of photochemistry in materials and life sciences.1–8 Among various photoswitches, diarylethene (DAE) derivatives are known as the representative photochemically reversible type (P-type) photoswitches that exhibit high thermal stability in both isomers and high durability.9,10 In the course of study on the photochromic reactivity of DAEs to date, it has been revealed that the thermal back reactivity from the closed-ring isomer to the open-ring isomer can be controlled by tuning the aromatic stabilization energy of the aryl groups, introducing bulky substituents at the reactive carbons, and introducing polar substituents at the lateral phenyl rings.11–15 Moreover, in 2019, our group developed diarylbenzenes (DABs) that are analogues of DAE with a benzene ring at the ethene bridge moiety.16 DABs function as thermally reversible type (T-type) photoswitches, and the thermal back reactivity of DABs can also be modulated by the three factors mentioned above.17–23 The change in the thermal back reactivity can be explained by the change in the energy difference between the open- and closed-ring isomers in the ground state. A larger energy difference corresponds to a lower activation energy barrier for the thermal back reaction, resulting in an accelerated thermal back reaction, which is called the Bell–Evans–Polanyi principle.24 Here, we investigated whether there were any other factors controlling the thermal back reactivity of DAB besides the above three factors.
Recently, the dihedral angle between two aromatic rings has been in the spotlight because it affects various physicochemical properties and reactivities.25–28 For instance, Risko and co-workers investigated the influence of the type of connected aromatic rings and the associated dihedral angles on the molecular energy landscape.29 Their study revealed four distinct patterns of energy variation, depending on the nature of the aromatic rings and the dihedral angle between them. Kim, Osuka, and co-workers reported that modulation of the dihedral angle between porphyrin units alters the extent of π-conjugation, significantly affecting the efficiency of two-photon absorption processes.30 Suganuma and co-workers reported that the photocyclization reactivity of inverse-type DAEs is affected by the dihedral angles in the aryl groups.31,32 These studies collectively highlight the effect of twisting the dihedral angles between aromatic units on inducing substantial changes in the physical and electronic properties of molecules, offering a powerful strategy for molecular design.
Inspired by these reports, in this study, to examine the effect of the dihedral angles in the aryl groups of DABs on thermal back reactivity, we focused on steric repulsion and designed novel compounds 2–7 with various alkyl groups at the lateral phenyl rings (Scheme 1). To estimate the dihedral angles of the open- and closed-ring isomers in the ground state for compounds 1–7, density functional theory (DFT) calculations were conducted. The dihedral angles for the open-ring isomers (φopen) 1o–7o were estimated to be 2.2°, 30°, 36°, 40°, 74°, 75°, and 82°, respectively, as shown in Fig. S1 (ESI†). The dihedral angles for the closed-ring isomers 1c–7c (φclosed) were also estimated to be 4.2°, 16°, 21°, 29°, 77°, 76°, and 83°, respectively, as shown in Fig. 1a and Fig. S2 (ESI†). These results indicate that by introducing different alkyl groups such as methyl, ethyl, isopropyl, and tert-butyl groups into the ortho position of the lateral phenyl ring, it is possible to prepare DABs with various dihedral angles in the aryl groups. Therefore, we synthesized 2o–7o using a procedure similar to that described in the literature.23 The details are described in the ESI.† The chemical structures were confirmed by 1H and 13C NMR spectroscopy, high-resolution mass spectrometry, and X-ray crystallographic analysis.
 | ||
| Scheme 1 Molecular structures of the diarylbenzene derivatives (1–7) investigated in this work. | ||
 | ||
| Fig. 1 (a) Optimized structure of 6c in the ground state using DFT calculations. (b) Absorption spectral change of 6 at 298 K in n-hexane: the open-ring isomer (black line) and the photostationary state upon irradiation with 254 nm light (blue line). (c) Absorption decay curves at λmax for 6c in n-hexane at various temperatures. (d) Eyring plot for the thermal back reaction of 6c. | ||
The photochromic behaviors of 1–7 in n-hexane were investigated. Fig. 1b shows the absorption spectral change of 6. 6o has an absorption maximum (λmax) of 249 nm in n-hexane. Upon irradiation with 254 nm light, the colorless solution turned blue, and an absorption maximum was observed at 580 nm. This spectral change is ascribed to photoisomerization from the open-ring isomer to the closed-ring isomer. After cessation of UV irradiation, the photogenerated colored closed-ring isomer 6c gradually underwent a thermal back reaction to the initial colorless open-ring isomer 6o, reducing the absorbance in the visible region. This indicates that 6o exhibits T-type photochromism, as in the case of 1o reported previously.212o–5o and 7o also exhibited similar absorption spectral changes, as shown in Fig. S3 (ESI†). The photophysical properties of 1–7 are summarized in Table 1. As the dihedral angle increases, the absorption maximum of both the open- and closed-ring isomers shift to shorter wavelengths (Fig. S4, ESI†). The reason is that the twisting of the phenyl ring reduces the effective π-conjugation length.
| Open-ring isomer | Closed-ring isomer | ΔH‡(exp)/kJ mol−1 | ΔS‡(exp)/J mol−1 | ΔG‡(exp)/kJ mol−1 | t 1/2/min | ||||
|---|---|---|---|---|---|---|---|---|---|
| φ open/° | λ max(exp)/nm | ε/M−1 cm−1 | φ closed/° | λ max(exp)/nm | |||||
| a Ref. 21. | |||||||||
| 1 | 2.2 | 312 | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
4.2 | 670 | 84.3 | −11.9 | 87.9 | 4.6 |
| 2 | 30 | 298 | 25300 |
16 | 647 | 85.6 | −9.3 | 88.4 | 5.7 |
| 3 | 36 | 289 | 20500 |
21 | 637 | 86.2 | −9.7 | 89.0 | 6.1 |
| 4 | 40 | 284 | 17200 |
29 | 629 | 85.5 | −14.5 | 89.8 | 10.2 |
| 5 | 74 | 266 | 14700 |
77 | 598 | 88.9 | −19.8 | 94.8 | 76.6 |
| 6 | 75 | 249 | 19000 |
76 | 580 | 93.3 | −12.7 | 97.1 | 193 |
| 7 | 82 | 248 | 18800 |
83 | 577 | 95.4 | −12.9 | 99.2 | 447 |
To quantitatively evaluate the thermal back reactivity of 2c–7c, the change in absorbance at various temperatures was measured, as shown in Fig. 1c and Fig. S5 (ESI†). The decay curve followed first-order kinetics, and the rate constants (k) of the thermal back reaction at various temperatures were determined (Fig. S6 and Tables S1–S6, ESI†). Fig. 1d and Fig. S7 (ESI†) show the Eyring plots for the thermal back reaction of 2c–7c. The experimental activation enthalpy (ΔH‡(exp)) and activation entropy (ΔS‡(exp)) in the thermal back reaction were determined from the slope and intercept, respectively. Using these values, the experimental activation free energy (ΔG‡(exp)) and half-life (t1/2) at 298 K were calculated. The results are summarized in Table 1. The values of ΔG‡(exp) for 2–7 were determined to be 88, 89, 90, 95, 97, and 99 kJ mol−1, respectively. The values of ΔG‡(exp) for 1–4 were almost the same, but those for 5–7 gradually increased in the order of 5, 6, and 7. The t1/2 values for 2–7 were also determined as 5.7, 6.1, 10.2, 76.6, 193, and 447 min, respectively. Again, the values of t1/2 for 1–4 were similar, but those for 5–7 were significantly longer in the order of 5, 6, and 7. These results demonstrate for the first time that the twisting of the aryl groups by introducing bulky alkyl groups on the phenyl ring reduces the thermal back reactivity.
To reveal the reason why the twisting of the aryl groups affects the thermal back reactivity, it would be desirable to experimentally determine the energy difference between the open and closed ring isomers. However, DABs are T-type compounds and closed-ring isomers cannot be isolated, making it difficult to accurately estimate the energy difference. Therefore, we adopted DFT calculations, and the values of the theoretical activation free energy (ΔG‡(calc)) at 298 K and the energy difference between the closed-ring isomer and the open-ring isomer (ΔG(calc)) were calculated. The results are summarized in Table S7 (ESI†). The values of ΔG‡(calc) were close to those of ΔG‡(exp), indicating that the DFT calculations reproduced the experimental results well. In addition, we checked the relationship between the ΔG(calc) and ΔG‡(calc). As mentioned in the introduction part, in the case of DAEs and DABs, the change in thermal back reactivity can be generally explained by the Bell–Evans–Polanyi principle.24 Namely, the smaller ΔG, the larger ΔG‡, resulting in the deceleration of the thermal back reaction (and vice versa). Therefore, there is usually a linear relationship between ΔG and ΔG‡. However, in this case, there is no correlation between the ΔG(calc) and ΔG‡(calc), as shown in Fig. S8 (ESI†). This indicates that the change in the thermal back reactivity cannot be simply explained by the Bell–Evans–Polanyi principle, and the twisting of the aryl groups greatly affects the energy level of the transition state compared with those of the open- and the closed-ring isomers.
To investigate the origin of the change in the energy level of the transition state, we focused on the dihedral angles in the transition states (φTS) of 1–7 and compared them with the dihedral angles of the closed-ring isomers. In addition, the molecular orbital distributions of the closed-ring isomer and the transition state at the highest occupied molecular orbital (HOMO) were visualized. The results are shown in Fig. 2a, b and Fig. S9, S10 (ESI†). For the transition state, only the molecular orbitals in the α spin state are shown because the molecular orbitals in the β spin state are mirror images of the molecular orbitals in the α spin state. The φTS of 1–7 were 1.8°, 19.2°, 19.9°, 32.5°, 48.9°, 57.5°, and 81.3°, respectively. There are no large differences in the dihedral angles between the transition state and the closed-ring isomer for 1–4 and 7, but there are large differences for 5 and 6. Moreover, in the transition state, all compounds have a greater spread of electron orbitals to the lateral phenyl ring. These results suggest that a longer π-conjugation length is preferable in the transition state. This is probably because the transition state is more delocalized in 6π electrocyclic reactions than in open- and closed-ring forms. Therefore, extending the π-conjugation further to the phenyl ring leads to stabilization. However, the π-conjugation length in the transition state tends to become shorter as the dihedral angles of the closed-ring isomer increase. This trend can be interpreted from the viewpoint of the steric repulsion between the thiazole and alkyl groups on the phenyl rings. In the case of 1–4, the alkyl groups are not very large, and the dihedral angles of the closed-ring isomer are already small. Therefore, no significant change in the molecular structure was observed in the transition state. For 5 and 6, the dihedral angles of the closed-ring isomers were large; thus, the molecular structure changed significantly to reduce the dihedral angles and extend the π-conjugation length for stabilization in the transition state. However, because of the steric repulsion, the dihedral angles in the transition state are larger than those in 1–4, resulting in a shorter π-conjugation length than in 1–4. On the other hand, although the dihedral angles of the closed-ring isomer of 7 are also large, the isopropyl groups on the phenyl rings are sterically very bulky, and steric repulsion occurs between the thiazole ring and the isopropyl groups. Therefore, no structural change can occur between the transition state and the closed-ring isomer, resulting in the largest dihedral angle and the shortest π-conjugation length in the transition state. The value of ΔG‡(exp) increases as the dihedral angles of the aryl groups in the transition state, as shown in Fig. 2c.
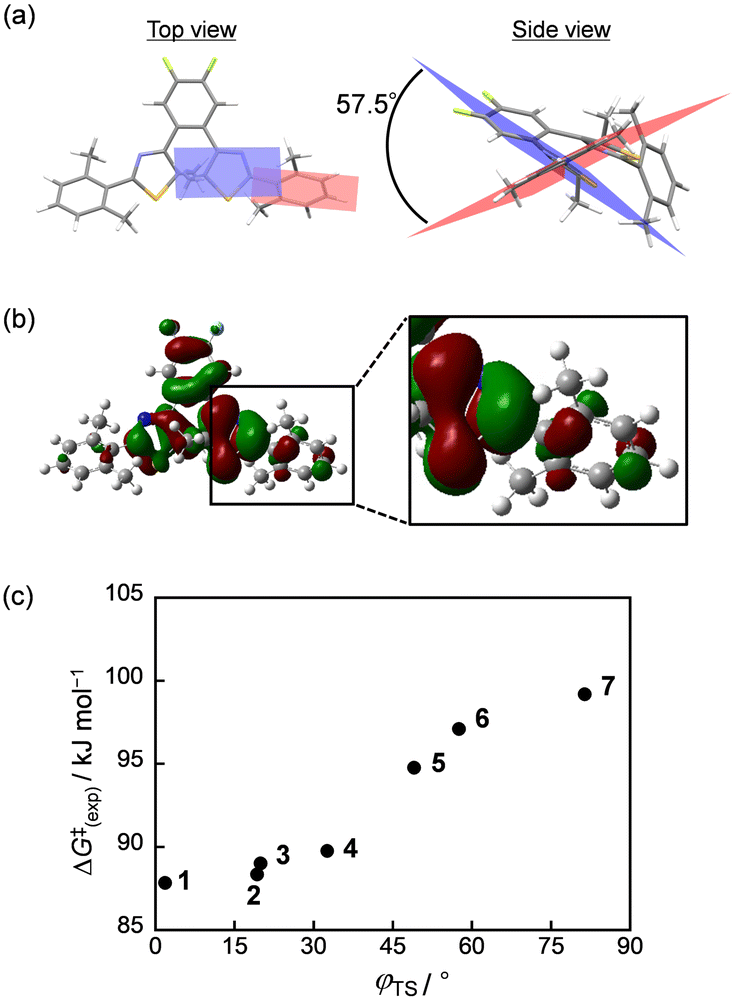 | ||
| Fig. 2 (a) Optimized structure and (b) molecular orbital distribution of 6 at the HOMO in the transition state using DFT calculations. (c) Relationship between the dihedral angles of aryl groups in the ground state and the experimental activation free energy. | ||
Summing up the results described above, we propose a mechanism for the change in thermal back reactivity induced by twisting of the aryl groups due to the introduction of bulky alkyl groups on the lateral phenyl ring. In the closed-ring isomer, the dihedral angles are set to eliminate the steric repulsion between the thiazole ring and the alkyl group on the lateral phenyl ring, but in the transition state, they likely become planer to extend the π-conjugation length for stabilization. However, the dihedral angles in the transition state also depend on the compounds due to steric repulsion between the thiazole ring and the alkyl group on the lateral phenyl ring, resulting in different π-conjugation lengths. This makes the energy of the transition state unstable, and ΔG‡ increases (Fig. 3). In particular, in compound 7, the large steric hindrance makes it impossible to reduce the dihedral angle in the transition state, resulting in an insufficient extension of the π-conjugation and a very low thermal back reactivity.
 | ||
| Fig. 3 Mechanism of the change in thermal back reactivity depending on the dihedral angles in the aryl groups. | ||
In conclusion, we demonstrated that the thermal back reactivity of DABs can be effectively tuned by controlling the dihedral angles in the aryl groups by introducing various bulky alkyl substituents. Both experimental and theoretical investigations revealed that the larger dihedral angles of the closed-ring isomers lead to an increase in the activation-free energy and prolongation of the thermal half-life. DFT calculations suggested that the destabilization of the transition state, caused by the inability to achieve planar geometries due to steric repulsion, results in the suppression of π-conjugation in the transition state, thereby elevating the energy barrier for thermal back reaction. This mechanism provides a clear explanation for the observed decrease in thermal back reactivity in DABs having highly twisted aryl groups. Overall, this study highlights a new structural strategy for controlling the thermal back reactivity of DABs by modulating aryl ring twisting, which may facilitate the design of advanced molecular switches with tunable kinetic properties.
This work was partly supported by JSPS KAKENHI (Grant Numbers JP23K26619, JP24K21794 (D. K.) and JP24K01458 (K. M.)), Iketani Science and Technology Foundation (D. K.), and JGC-S (Nikki-Saneyoshi) Scholarship Foundation (D. K.).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- M. Regehly, Y. Garmshausen, M. Reuter, N. F. Konig, E. Israel, D. P. Kelly, C. Y. Chou, K. Koch, B. Asfari and S. Hecht, Nature, 2020, 588, 620–624 CrossRef CAS PubMed.
- B. Shao, H. Fu and I. Aprahamian, Science, 2024, 385, 544–549 CrossRef CAS PubMed.
- S. Chakraborty, H. P. Q. Nguyen, J. Usuba, J. Y. Choi, Z. Sun, C. Raju, G. Sigelmann, Q. Qiu, S. Cho, S. M. Tenney, K. E. Shulenberger, K. Schmidt-Rohr, J. Park and G. G. D. Han, Chemistry, 2024, 10, 3309–3322 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2016, 138, 906–913 CrossRef CAS PubMed.
- T. Nakashima, K. Tsuchie, R. Kanazawa, R. Li, S. Iijima, O. Galangau, H. Nakagawa, K. Mutoh, Y. Kobayashi, J. Abe and T. Kawai, J. Am. Chem. Soc., 2015, 137, 7023–7026 CrossRef CAS PubMed.
- J. Wu, L. Kreimendahl, S. Tao, O. Anhalt and J. L. Greenfield, Chem. Sci., 2024, 15, 3872–3878 RSC.
- J. Wu and J. L. Greenfield, J. Am. Chem. Soc., 2024, 146, 20720–20727 CrossRef CAS PubMed.
- J. Wu, L. Kreimendahl and J. L. Greenfield, Angew. Chem., Int. Ed., 2025, 64, e202415464 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Irie, Diarylethene Molecular Photoswitches: Concepts and Functionalities, Wiley-VCH, Weinheim, 2021 Search PubMed.
- S. Nakamura and M. Irie, J. Org. Chem., 1988, 53, 6136–6138 CrossRef CAS.
- S. L. Gilat, S. H. Kawai and J. M. Lehn, Chem. – Eur. J., 1995, 1, 275–284 CrossRef CAS.
- S. Kobatake, K. Uchida, E. Tsuchida and M. Irie, Chem. Lett., 2000, 1340–1341 CrossRef CAS.
- V. Z. Shirinian, A. G. Lvov, M. M. Krayushkin, E. D. Lubuzh and B. V. Nabatov, J. Org. Chem., 2014, 79, 3440–3451 CrossRef CAS PubMed.
- G. Liu, S. Pu and R. Wang, Org. Lett., 2013, 15, 980–983 CrossRef CAS PubMed.
- D. Kitagawa, T. Nakahama, Y. Nakai and S. Kobatake, J. Mater. Chem. C, 2019, 7, 2865–2870 RSC.
- T. Nakahama, D. Kitagawa and S. Kobatake, J. Phys. Chem. C, 2019, 123, 31212–31218 CrossRef CAS.
- D. Kitagawa, N. Takahashi, T. Nakahama and S. Kobatake, Photochem. Photobiol. Sci., 2020, 19, 644–653 CrossRef CAS PubMed.
- S. Hamatani, D. Kitagawa, T. Nakahama and S. Kobatake, Tetrahedron Lett., 2020, 61, 151968 CrossRef CAS.
- R. Maegawa, D. Kitagawa, S. Hamatani and S. Kobatake, New J. Chem., 2021, 45, 18969–18975 RSC.
- S. Hamatani, D. Kitagawa and S. Kobatake, J. Phys. Chem. C, 2021, 125, 4588–4594 CrossRef CAS.
- S. Hamatani, D. Kitagawa, R. Maegawa and S. Kobatake, Mater. Adv., 2022, 3, 1280–1285 RSC.
- S. Hamatani, D. Kitagawa, T. Nakahama and S. Kobatake, Bull. Chem. Soc. Jpn., 2023, 96, 496–502 CrossRef CAS.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Part A: Structure and Mechanisms, Springer Science & Business Media, 2007 Search PubMed.
- D. H. Jeong, S. M. Jang, I.-W. Hwang, D. Kim, N. Yoshida and A. Osuka, J. Phys. Chem. A, 2002, 106, 11054–11063 CrossRef CAS.
- S. Cho, M.-C. Yoon, J. M. Lim, P. Kim, N. Aratani, Y. Nakamura, T. Ikeda, A. Osuka and D. Kim, J. Phys. Chem. B, 2009, 113, 10619–10627 CrossRef CAS PubMed.
- T. Mani and J. R. Miller, J. Phys. Chem. A, 2014, 118, 9451–9459 CrossRef CAS PubMed.
- B. Chandra Patra, R. Wan, C. E. Moore and Y. Wu, Chem. – Eur. J., 2025, 31, e202402535 CrossRef CAS PubMed.
- R. Duke, V. Bhat, A. Smith, S. Goodlett, S. Tretiak and C. Risko, Macromolecules, 2023, 56, 5259–5267 CrossRef CAS.
- T. K. Ahn, K. S. Kim, D. Y. Kim, S. B. Noh, N. Aratani, C. Ikeda, A. Osuka and D. Kim, J. Am. Chem. Soc., 2006, 128, 1700–1704 CrossRef CAS PubMed.
- M. Suganuma, D. Kitagawa, S. Hamatani, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, ChemPhotoChem, 2024, 8, e20230024 CrossRef.
- M. Suganuma, D. Kitagawa, S. Hamatani, H. Sotome, C. Mittelheisser, M. Sliwa, S. Ito, H. Miyasaka and S. Kobatake, J. Mater. Chem. C, 2025, 13, 5259–5267 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures of compounds used in this work and detailed experimental data. CCDC 2452875–2452880. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02172c |
| This journal is © The Royal Society of Chemistry 2025 |