
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective C(sp3)–H acetoxylation of phosphoramidites†
Anirban
Mondal‡
 ,
Vanda
Dašková‡
,
Xiaobing
Chen
,
Niklas O.
Thiel
,
Georgios
Alachouzos
and
Ben L.
Feringa
*
,
Vanda
Dašková‡
,
Xiaobing
Chen
,
Niklas O.
Thiel
,
Georgios
Alachouzos
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen Nijenborgh 3, 9747AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 7th April 2025
Abstract
Chiral phosphines are important ligands in asymmetric catalysis, yet their potential as directing groups for asymmetric C–H activation remains unexplored due to the oxidative nature of these reactions. We present a Pd-catalysed, P(III)-directed diastereoselective acetoxylation of phosphoramidites, with DFT calculations elucidating their unique reactivity and supporting the proposed reaction mechanism.
In the past decades, transition-metal-catalysed C–H functionalisation has established itself as one of the cornerstones of synthetic organic chemistry.1,2 Compared to the traditional approaches to install functional groups such as cross-coupling reactions, the activation and subsequent functionalisation of C–H bonds offers atom- and step-economic methodology as no installation of intermediate functional groups (e.g. halides, triflates, boron or metal-compounds) is required.2 Moreover, the recent advances in C–H functionalisation protocols have revolutionised synthetic organic chemistry in the context of natural product synthesis and development of biologically active molecules by enabling late stage functionalisation and rapid diversification of complex molecules.3 The direct C–H functionalisation faces two major challenges, namely the inert character of C–H bonds and the selectivity during replacement of one hydrogen atom among several similar ones.4 In particular, the chemo-, regio- and especially stereoselectivity of C–H functionalisation remains challenging even 40 years after its discovery, due to the difficulty associated with differentiating between multiple similar hydrogen atoms.5 Among the different C–H bonds to be modified,6 direct methods for an enantioselective C(sp3)–H functionalisation still remain underdeveloped.5b,7 Over the past decade, a number of elegant studies on the asymmetric C(sp3)–H activation have emerged.5b,8
The two main approaches involve the enantioselective methylene C–H activation of 2° C–H bonds9 and the C–H activation/desymmetrisation of 1° C–H bonds,10 pioneered by Yu and co-workers (Schemes 1a and b, respectively). In the latter case, the selective differentiation between six equivalent C–H bonds of an α-gem-dimethyl moiety is particularly challenging10 and its C–H functionalisation will be discussed in this work. A kinetic resolution, the preferential recognition of one enantiomer of a racemate by a chiral catalyst, is being reported in the context of the asymmetric C(sp3)–H functionalisation (Scheme 1c).11 Fascinated by the potential of the asymmetric C(sp3)–H functionalisation combined with our longstanding efforts exploring new classes of chiral phosphoramidite ligands in asymmetric transformations,12,13 we devised a strategy for the Pd-catalysed P(III)-directed diastereoselective β–C–H functionalisation of amines via an iPr desymmetrisation reaction (Scheme 1d). As depicted in Scheme 1d, we propose that this diastereoselective C–H functionalisation involves the generation of a five-membered cyclometalated species D14 bearing a newly formed carbon stereocenter distal to the reacting side via a concerted metalation deprotonation (CMD) mechanism.15 A desymmetrisation reaction of the identical Me-substituents at the iPr-moiety is expected to take place due to the stereodirecting effect of the chiral binaphthol moiety in close vicinity of the reacting center. Subsequently, the reductive elimination from the transient Pd(IV)-intermediate enables the carbon–heteroatom bond formation. Thereby, the phosphoramidites show three distinct key functions: (i) P(III) as directing group for selective C–H activation; (ii) chiral BINOL for stereocontrol; (iii) amine moiety for desymmetrisation. Nevertheless, there are few other synthetic challenges that we need to address: first, unreactive C(sp3)–H bonds are substantially less prone to C–H cleavage/C–H insertion due to their high bond dissociation energy.16 Second, both the regio-and stereoselectivity might be difficult to control. However, tuning the starting material including the chiral BINOL auxiliary might be a good solution to tackle this problem. Finally, the oxidation of P(III) to P(V) poses a major challenge under the oxidising conditions of the reaction. In contrast to numerous reports on elegant Pd-catalysed C(sp3)–H functionalisations controlled by synthetically versatile and oxidant-compatible directing groups containing O-, N- or S-atoms,2f,4,17 to the best of our knowledge, the use of the P(III) as the directing group for asymmetric C(sp3)–H activation has not been reported before. Very recently, two examples of the oxidant-compatible P(III)-directed Pd-catalysed transformations have been reported i.e. C(sp2)–H carbonylation18 and silylation of indoles.19 However, they have not been used in the functionalisation of C(sp3)–H bonds. Herein, we report a versatile Pd-promoted diastereoselective C(sp3)–H acetoxylation of phosphoramidites involving alkyl amine desymmetrisation providing valuable chiral amino alcohol precursors.
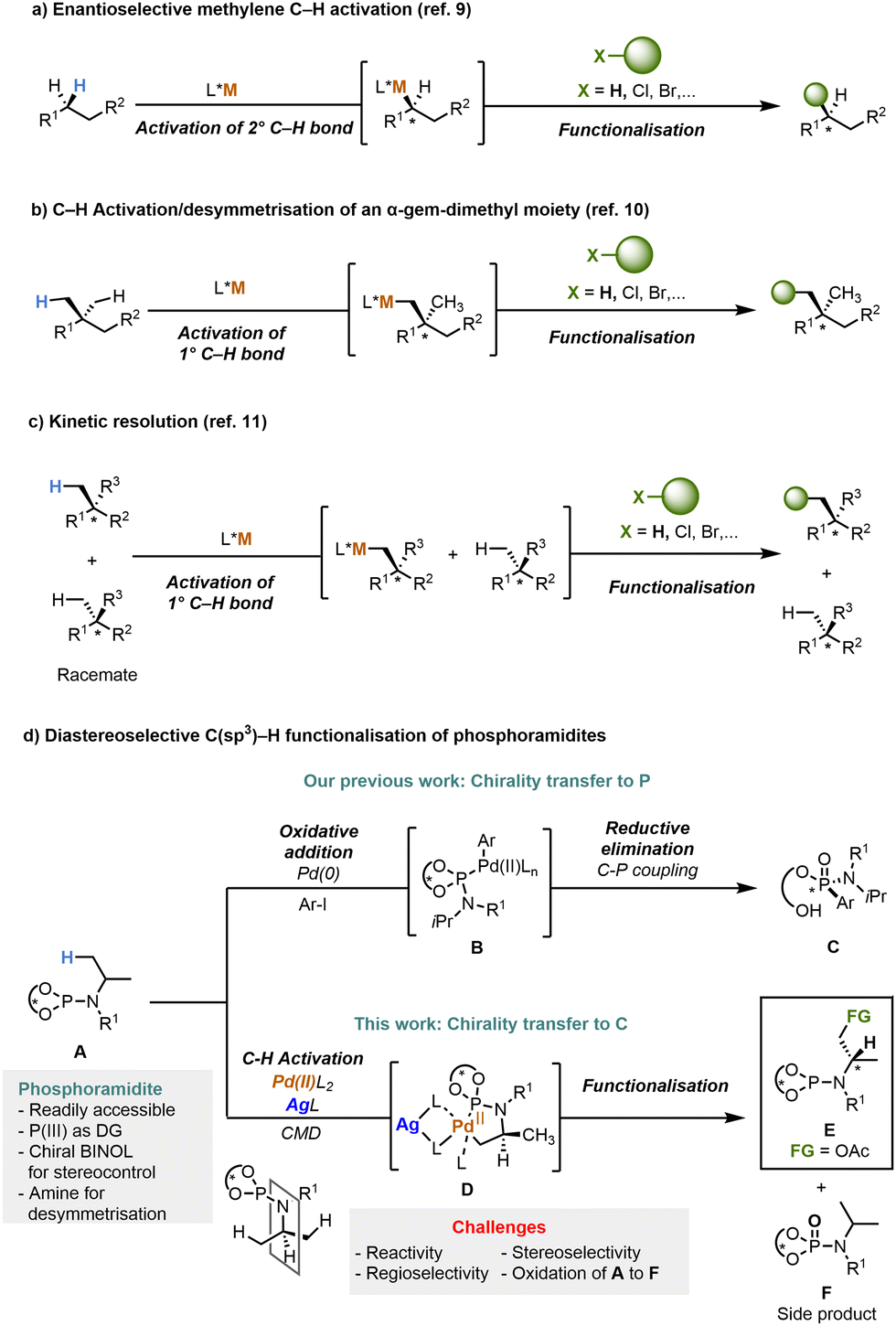 | ||
| Scheme 1 Approaches to asymmetric C(sp3)–H functionalisation via (a) enantioselective methylene C–H activation, (b) C–H activation/desymmetrisation and (c) kinetic resolution (d) proposed asymmetric C(sp3)–H functionalisation of phosphoramidites. | ||
To evaluate the proposed transformation shown in Scheme 1d the preliminary studies began with standard reaction conditions for C–H acetoxylation20 using phosphoramidite (R)-1a as substrate, namely PhI(OAc)2 (5.0 equiv.) and Pd(OAc)2 (10 mol%) in a deoxygenated AcOH/Ac2O-mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent at 100 °C. A comprehensive summary of the optimisation results is provided in Tables S1–S7 (see ESI†). It was found that the phosphoramidite (R)-1a mainly oxidised to (R)-1a-O and only a small amount of the product (R,R)-2a (<10% conversion, 88:12 d.r.) was formed (Table S1, entry 1, ESI†). This preliminary observation showed the potential of the chiral P(III) as a directing group for the C–H acetoxylation. However, the main challenge is the competing oxidation of the starting material to (R)-1a-O which does not undergo the desired transformation (as revealed by a control experiment using (R)-1a-O as the starting material, see ESI,† Table S7). To expedite C–H activation, we tested various silver salts, which are commonly used additives to enhance Pd-catalysed C–H activation in terms of selectivity and conversion.21,22 Gratifyingly, the C–H acetoxylation reaction of (R)-1a in the presence of AgOAc (2.0 equiv.) afforded (R,R)-2a with same level of d.r. (88:12), although with higher conversion (Table S2, entry 1, ESI†) which indicates that AgOAc accelerates the C–H activation step and, most importantly, hinders the early oxidation of the starting material. It's worth noting that no other Ag-salts were found to be as effective as AgOAc (see Table S2, ESI†). Next, various Pd-catalysts were evaluated in the acetoxylation of (R)-1a. No improved conversion was observed for the other Pd-complexes/salts tested and mainly oxidation of the directing group was detected (see Table S1, ESI†). However, an improved reactivity was found when an increased catalyst loading (15 mol%) was used, affording (R,R)-2a in 38% yield and 89:11 d.r. (Table S1, entry 7, ESI†). As solvent tuning for specific C–H functionalisations is an emerging topic of current research,23 the solvent influence was investigated next. The best result was achieved using a DCE/AcOH/Ac2O-mixture in 4:1:1 ratio yielding the product (R,R)-2a with much improved conversion and diastereoselectivity (44%, 92:8 d.r., Table S3, entry 9, ESI†). Finally, we shifted our attention towards the optimisation of both the stoichiometry between the PhI(OAc)2/AgOAc and temperature. It was found that optimal results were obtained using 15 mol% of Pd catalyst, 4.0 equiv. of hypervalent iodine reagent, 2.0 equiv. of AgOAc in deoxygenated DCE/AcOH/Ac2O (4:1:1) at 60 °C and a reaction time of 16 h (Table S5, entry 4, ESI†).
:1) as solvent at 100 °C. A comprehensive summary of the optimisation results is provided in Tables S1–S7 (see ESI†). It was found that the phosphoramidite (R)-1a mainly oxidised to (R)-1a-O and only a small amount of the product (R,R)-2a (<10% conversion, 88:12 d.r.) was formed (Table S1, entry 1, ESI†). This preliminary observation showed the potential of the chiral P(III) as a directing group for the C–H acetoxylation. However, the main challenge is the competing oxidation of the starting material to (R)-1a-O which does not undergo the desired transformation (as revealed by a control experiment using (R)-1a-O as the starting material, see ESI,† Table S7). To expedite C–H activation, we tested various silver salts, which are commonly used additives to enhance Pd-catalysed C–H activation in terms of selectivity and conversion.21,22 Gratifyingly, the C–H acetoxylation reaction of (R)-1a in the presence of AgOAc (2.0 equiv.) afforded (R,R)-2a with same level of d.r. (88:12), although with higher conversion (Table S2, entry 1, ESI†) which indicates that AgOAc accelerates the C–H activation step and, most importantly, hinders the early oxidation of the starting material. It's worth noting that no other Ag-salts were found to be as effective as AgOAc (see Table S2, ESI†). Next, various Pd-catalysts were evaluated in the acetoxylation of (R)-1a. No improved conversion was observed for the other Pd-complexes/salts tested and mainly oxidation of the directing group was detected (see Table S1, ESI†). However, an improved reactivity was found when an increased catalyst loading (15 mol%) was used, affording (R,R)-2a in 38% yield and 89:11 d.r. (Table S1, entry 7, ESI†). As solvent tuning for specific C–H functionalisations is an emerging topic of current research,23 the solvent influence was investigated next. The best result was achieved using a DCE/AcOH/Ac2O-mixture in 4:1:1 ratio yielding the product (R,R)-2a with much improved conversion and diastereoselectivity (44%, 92:8 d.r., Table S3, entry 9, ESI†). Finally, we shifted our attention towards the optimisation of both the stoichiometry between the PhI(OAc)2/AgOAc and temperature. It was found that optimal results were obtained using 15 mol% of Pd catalyst, 4.0 equiv. of hypervalent iodine reagent, 2.0 equiv. of AgOAc in deoxygenated DCE/AcOH/Ac2O (4:1:1) at 60 °C and a reaction time of 16 h (Table S5, entry 4, ESI†).
Next, the scope in terms of phosphoramidites containing 1°, 2° and 3° C–H bonds at the β-position were investigated (Scheme 2). Under the optimised reaction conditions, the standard acetoxylated substrate (R,R)-2a was obtained with a moderate isolated yield of 56% and excellent selectivity (97:3 d.r.). We were pleased to find that the C–H functionalisation was only taking place on the primary C–H bond, affording the desired products both with branched ((R,R)-2b-c) as well as cyclic aliphatic moieties ((R,R)-2d-f and (R,S)-2g) on the amine in low to moderate yields (22–51%), however, with excellent chemo-, regio- and diastereoselectivities (90:10–95:5 d.r.). The high level of diastereocontrol is attributed to: (i) the well-established preference for C–H functionalisation at the least hindered positions; and (ii) pre-arrangement of the C–H activation transition state by complexation of Ag.24 The absolute configuration of all products was predicted based on the known configuration of the BINOL moiety in the phosphoramidite and DFT analysis25 (Scheme 2). Additionally, the phenyl substituted substrate exhibited lower reactivity under the optimal reaction conditions ((R)-1j, Ph substituent), resulting in exclusive oxidation of the P(III)-directing group. Unexpectedly, the introduction of a bulkier tBu-group ((R)-1i) instead of Me or iPr had an adverse impact on the regioselectivity, leading to the formation of a mixture of multiple acetoxylation products. A preliminary mechanism of the C(sp3)–H acetoxylation is proposed based on previous C–H acetoxylation studies26 and density functional theory (DFT) calculations (obtained at the B3LYP/def2-SVP/PCM(DCM) level of theory) as shown in Scheme 3. The phosphoramidite (R)-1 first coordinates to Pd(OAc)2 and Ag(OAc) via the P(III) atom to afford the catalytic intermediate B′. The complexation of both Pd(OAc)2 and AgOAc is strongly exothermic (−33.7 kcal mol−1), and is highly stabilized by the AgOAc (by 12.9 kcal mol−1versus a B′ catalytic complex without the AgOAc, only coordinating the Pd(OAc)2). This species B′ then undergoes an intramolecular C–H activation through a concerted metalation deprotonation (CMD-type) mechanism15 to the five membered complex C. We envisioned that the cyclopalladated species C′ is formed with high diastereoselectivity by desymmetrisation resulting from transfer of axial (BINOL) to point chirality. Indeed, the diastereoselective C–H activation step was calculated to proceed via a stereochemically pre-arranged transition state involving both Pd and Ag, with a ΔG‡ of 37.5 kcal mol−1, and with an exothermic driving force of 7.8 kcal mol−1. Accordingly, the C–H activation transition state leading to the opposite diastereoisomer is proposed to be destabilized due to steric hindrance (see Scheme 3), and no viable transition state for the opposite diastereoisomer was found at the B3LYP/def-2SVP/PCM(DCM) level of theory. Intermediate C′ is then proposed to undergo an oxidative step by PhI(OAc)2 to form Pd(IV) species D′. A C–O reductive elimination step via an intermediate E′ follows, furnishing the desired chiral acetoxylated phosphoramidite product F′. Furthermore, both the acetoxylated product and the unreacted starting material undergo oxidation to form the final products (R,R)-227 and (R)-1-O, respectively. It's not clear from our current DFT studies whether the AgOAc is involved in the proposed steps beyond C′. The preliminary DFT investigations strongly support the cooperative effect of the Ag-salt (Scheme 3, intermediates B′ and C′, and the accompanying C–H activation transition state) under the tested reaction conditions. Similar to our DFT results, it has been previously reported that formation of bimetallic Pd–Ag complexes facilitates C–H cleavage by lowering the energy barrier of the transition states leading to the desired functionalized products.24
 | ||
| Scheme 2 Investigations on scope and limitations of the developed C(sp3)–H acetoxylation. aNMR yield. The stereochemistry of the acetoxylated products is assumed to be (R,R) based on DFT predictions, except for (R,S)-2g, which was confirmed by X-ray analysis. | ||
 | ||
| Scheme 3 A proposed catalytic cycle of the investigated C(sp3)–H acetoxylation of (R)-1 to (R,R)-2, which is supported by density functional theory calculations (see ESI†). | ||
It is important to note that the stereochemical organisation predicted by our DFT studies to lead to the (R,R)-2 stereochemistry could be overridden by bulkier phosphoramidite reactants, suggesting a degree of substrate-control is also at play here. Indeed, the obtained X-ray diffraction structure of product 2g bearing a bulky adamantane substituent shows that the (R,S)-2g stereochemistry was formed. We propose that in the formation of (R,R)-2, the steric bulk of BINOL causes C–H activation to proceed on the least sterically encumbered isopropyl functionality. However, in the formation of (R,S)-2g we anticipate that the adamantyl moiety, which cannot undergo this C–H functionalisation reaction, takes up the least sterically hindered space. Thereby in this specific case, the C–H activation may proceed via the more sterically encumbered N-substituent, forming the product with the opposite point chirality to that determined from the calculations. Further mechanistic studies are currently underway in our labs.
In summary, we have shown the first example of P(III) directed Pd-catalysed diastereoselective acetoxylation using readily available phosphoramidites. Remarkably, this study demonstrates the potential utility of P(III) as a directing group under highly oxidising conditions in the development of new asymmetric C–H functionalisation reactions. In particular, the critical role of the Ag-additive in preventing initial oxidation of directing group and preliminary DFT studies proposing a bimetallic intermediate lowering the C–H activation energy are noteworthy. The excellent stereocontrol provides a basis to access highly valuable chiral amino alcohol derivatives via asymmetric C–H activation.
We thank Dr Marta Castiñeira Reis for her contribution in the preliminary DFT calculations. Financial support from the Dutch Ministry of Education, Culture and Science (No. 024.001.035, to B. L. F), the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (No. 718.015.004 to A. N. M), the European Molecular Biology Organisation (EMBO LTF-232-2020, to GA), and China Scholarship Council (No. 202108330058 to X. C.). We are grateful to Charlotte Stindt for help with single crystal X-ray analysis, Renze Sneep for performing the HRMS measurements, and Dr Paco Visser for the helpful discussions.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for (R,S)-2g has been deposited at the CCDC under 2291901 and can be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437–2450 CAS; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 CAS; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CAS; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (e) J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2015, 55, 217–257 Search PubMed; (f) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 CAS; (g) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CAS; (h) J. A. Labinger, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS PubMed; (i) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed; (j) D.-S. Kim, W.-J. Park and C.-H. Yun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed; (k) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (l) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed; (m) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (n) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833–851 CrossRef CAS PubMed.
- (a) B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (c) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (d) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC; (e) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed; (f) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117(13), 8754–8786 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (c) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (d) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (f) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CAS.
- (a) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CAS; (b) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 Search PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CAS.
- F. Colobert and J. Wencel-Delord, C–H Activation for Asymmetric Synthesis, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed.
- A. P. Smalley, J. D. Cuthbertson and M. J. Gaunt, J. Am. Chem. Soc., 2017, 139, 1412–1415 CAS.
- For selected examples see: (a) S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 13, 4692–4695 CAS; (b) C. He, W. G. Whitehurst and M. Gaunt, Chem, 2019, 5, 1031–1058 CAS; (c) J. Pedroni, M. Boghi, T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 9064–9067 CAS; (d) P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nat. Chem., 2017, 9, 140–144 CAS; (e) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CAS; (f) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570–18572 CAS; (g) G. Chen, W. Gon, Z. Zhuang, M. S. Adra, J.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023–1027 CrossRef CAS PubMed.
- For selected examples see: (a) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed; (b) D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43, 5009–5031 RSC; (c) Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499–503 CrossRef CAS PubMed.
- (a) D. Katayev, M. Nakanishi, T. Burgi and E. P. Kündig, Chem. Sci., 2012, 3, 1422–1425 RSC; (b) L. Yang, R. Melot, M. Neuburger and O. Baudoin, Chem. Sci., 2017, 8, 1344–1349 RSC.
- (a) J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed; (b) B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS PubMed.
- (a) A. Mondal, N. O. Thiel, R. Dorel and B. L. Feringa, Nat. Catal., 2022, 5, 10–19 CrossRef CAS; (b) X.-B. Chen, D. Padín, C. N. Stindt and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e20230745 Search PubMed.
- C. A. Kiener, C. Shu, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14272–14273 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- B. Dong, J. Qian, M. Li, Z.-J. Wang, M. Wang, D. Wang, C. Yuan, Y. Han, Y. Zhao and Z. Shi, Sci. Adv., 2020, 6, eabd1378 CrossRef CAS PubMed.
- D. Wang, X. Chen, J. J. Wong, L. Jin, M. Li, Y. Zhao, K. N. Houk and Z. Shi, Nat. Commun., 2021, 12, 524 CrossRef CAS PubMed.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS PubMed.
- (a) O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046–4048 CrossRef CAS PubMed; (b) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- (a) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS PubMed; (b) K. L. Bay, Y.-F. Yang and K. N. Houk, J. Organomet. Chem., 2018, 864, 19–25 CrossRef CAS.
- C. Yu, J. Sanjosé-Orduna, F. W. Patureau and M. H. Pérez-Temprano, Chem. Soc. Rev., 2020, 49, 1643–1652 RSC.
- (a) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- The absolute configuration of all products was fixed as R,S determined by the crystals of (R,S)-2g.
- (a) C. S. Buettner, D. Willcox, B. G. N. Chappell and M. J. Gaunt, Chem. Sci., 2019, 10, 83–89 RSC; (b) A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109–3118 CrossRef CAS PubMed.
- The absolute configuration of the products in DFT calculations were fixed as R,R.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2291901. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00550g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |