
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intramolecular phosphine-oxide stabilized germanium(IV) di-cation with enhanced Lewis acidity and catalytic applications†
Akanksha
Kumari
a,
Balakrishna
Peddi
a,
Cem. B.
Yildiz
 *b and
Moumita
Majumdar
*a
*b and
Moumita
Majumdar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune, Dr Homi Bhabha Road, Pashan, Pune-411008, India. E-mail: moumitam@iiserpune.ac.in
bDepartment of Basic Sciences, Faculty of Engineering, Architecture and Design, Bartin University, 74100 Bartin, Turkey
First published on 27th March 2025
Abstract
This work reports the intramolecular phosphine-oxide stabilized tetra-coordinated germanium(IV) di-cation on an acenaphthene platform, 1iPrPO. Computational study shows that the positive charges and the acceptor orbitals are localized on the Ge site. 1iPrPO is Lewis super acidic, capable of catalysing hydrodefluorination reaction. 1iPrPO also catalyses hydrosilylation of electron-deficient aldehydes.
Germanium compounds are less popular as Lewis acids compared to those of silicon1 analogues due to their inherent higher electronegativity. Contemporary research has shown an upsurge in developing Ge-based Lewis acid catalysts2 because of their relatively higher stability and thus handling ease. Prominent examples are the bis(perchlorocatecholato) germane and its derivatives3,4 exhibiting Lewis superacidity and high catalytic aptitude. Geometric constraint was introduced for Lewis acidity enhancement as observed in the anti-van’t Hoff-Le Bel reactivity of the calix[4]pyrrolato-germane.5 Significant increment in the Lewis acidity was noticed by installing positive charges on the Ge centre in the corrole-stabilized planar Ge(IV) mono-cation.6 The key factor behind the unusually high electrophilicity is the localization of the cationic charge on the Ge centre. There are a few more examples of germylium ions being used as Lewis acid catalysts.7–9
Very recently, our group has established the first example of a tetra-coordinated Ge(IV) di-cation as a Lewis acid catalyst (Fig. 1A).10 The catalytic proficiency of this intramolecular phosphine-stabilized Ge(IV) di-cation was compromised due to the dispersion of the di-positive charges. Therefore, we attempted to localize the acceptor orbitals on the Ge centre of the di-cationic system. A recent study has shown that phosphine oxide serves as an efficient donor towards the cationic sites without significantly hampering their electrophilicity (Fig. 1B and C).11–14 Inspired by these reports, in our continued efforts for the further advancement of Ge(IV) di-cationic Lewis acids, we have prepared the intramolecular phosphine oxide stabilized Ge(IV) di-cationic compound 1iPrPO (Fig. 1D), exhibiting an expanded catalytic portfolio. The detail is reported herein.
 | ||
| Fig. 1 (A) 1iPrP; (B) phosphine-oxide stabilized triaryl carbenium ion; (C) phosphine-oxide stabilized Sb(V) di-cation; and (D) 1iPrPO (this work). | ||
The intramolecular phosphine-oxide stabilized Ge(IV) di-cationic compound on an acenaphthene platform, 1iPrPO, was prepared by the simple oxidation of our previously reported compound 1iPr,10 with two equivalents of iodosobenzene in dichloromethane (Scheme 1). Colourless single crystals of 1iPrPO were grown from dichloromethane/pentane layering under room temperature conditions in 75% crystallization yield. Compound 1iPrPO was characterized in the solution state by multi-nuclear NMR spectroscopy in CD3CN (Fig. S1–S4, ESI†). The 31P{1H} NMR chemical shifts at +97.2 ppm (in CD3CN) and +97.6 (in CD2Cl2) were assigned to the phosphorus(V) centre present in 1iPrPO. The similar 31P{1H} NMR chemical shift values obtained invalidate the possible coordination of solvents at the Ge centre.10 The solid-state structure of 1iPrPO (Fig. 2 and Table S4, ESI†) shows a spirocyclic geometry with the Ge atom being shared between the two rings. The two triflate (CF3SO3 = OTf) counter anions are non-coordinating in nature (closest Ge–O(OTf) contact being 4.3 Å).15 The average Ge–C bond length in 1iPrPO (avg. 1.89 Å) is shorter than that observed in the case of 1iPr (avg. 1.92 Å).10 The average Ge–O bond length of 1.80 Å lies in the longer range of Ge–O covalent bonds for tetracoordinated Ge(IV) compounds.16 Correspondingly, the average P–O bond lengths (1.57 Å) exhibit partial double bond character.
 | ||
| Scheme 1 Lewis base binding and hydride and fluoride ion abstraction (see the ESI,† for detailed spectroscopic and experimental procedures of each reaction). | ||
 | ||
| Fig. 2 Molecular structure of 1iPrPO in the solid state (thermal ellipsoid 35%, H atoms, triflate counter anions and solvent molecules are omitted for clarity). Selected bond lengths [Å] Ge1–O1 = 1.803(2), Ge1–C1 = 1.895(3), P1–O1 = 1.575(2), Ge1–O2 = 1.797(3), Ge1–C2 = 1.891(3), P2–O2 = 1.570(2); selected bond angles [°]: O1–Ge1–C1 = 103.9(1), O2–Ge1–C2 = 104.1(1), O1–Ge1–O2 = 105.1(1), C1–Ge1–C2 = 121.9(1). | ||
The optimized geometry of 1iPrPO is in close agreement with the X-ray parameters (see ESI,† for the computational detail). The natural bond orbital (NBO) analyses of the optimized structure reveal donor–acceptor interactions between the lone pair orbitals on the oxygen atoms (from phosphine oxide) to the Ge-centred acceptor orbitals (Table S1, ESI†). The NBO frontier molecular orbitals depicted in Fig. 3A show that the acceptor orbitals are localized on the Ge centre, unlike our previously reported 1iPrP where the acceptor orbitals comprised Ge–C and Ge–P σ* orbitals.10 The Wiberg bond order (WBO) calculated for Ge–C (0.79) confirms the single bond between them, while the WBO for Ge–O (0.46) echoes the presence of comparatively weaker bonding interaction between them. The low value of WBO for P–O (0.81) reflects less than double bond character. The electrostatic potential map as shown in Fig. 3B reflects the concentration of the positive charges on the Ge centre in 1iPrPO, as opposed to the dispersed di-positive charges over the P–Ge–P framework found in 1iPrP. Overall, compared to our earlier report on 1iPr, our newly synthesized compound 1iPrPO has di-cationic charges and orbital vacancies localized6,17 on the Ge-centre.
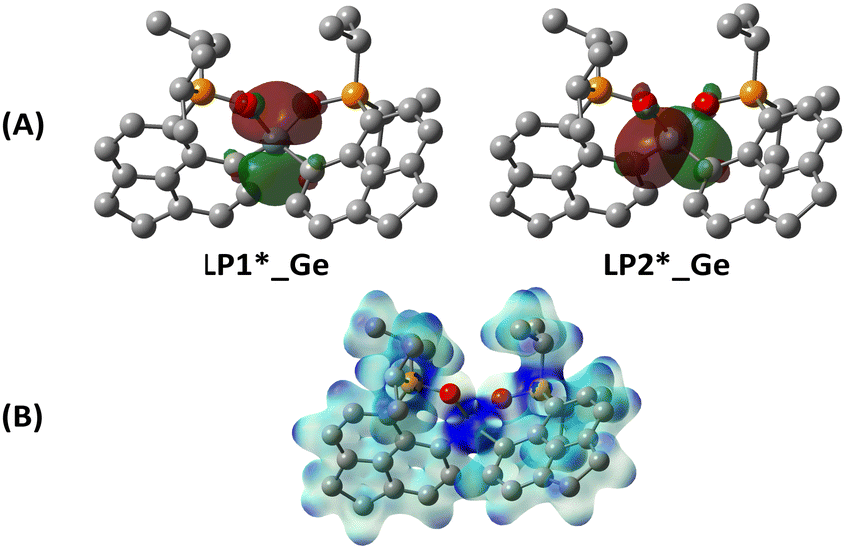 | ||
| Fig. 3 (A) Natural bond orbitals localized on the Ge center; (B) electrostatic potential map with the range of 0.05 (red)–0.40 (blue). | ||
The effective Lewis acidity18 of 1iPrPO following the Gutmann–Beckett (GB) method was investigated (see ESI,† for details). A Δδ31P value of 25.4 ppm was obtained from the addition of 0.2 equivalents of Et3PO to 1iPrPO in CD3CN (δ for free Et3PO in CD3CN is +49.7 ppm; for comparison Δδ31P for 1iPrP = 21.3 ppm) (Fig. S7–S9, ESI†). The formation of the mono-adduct 1iPrPO·Et3PO was confirmed from the 31P NMR study (δiPrPO at +75.57 ppm and δEt3PO at +75.08 ppm) (Fig. S10–S12, ESI†). The mono-adduct 1iPrPO·Et3PO remained in equilibrium with 1iPrPO, as revealed from the detailed NMR study (Fig. S12, ESI†). We were not successful in obtaining single crystals of the adduct 1iPrPO·Et3PO. Rather, single crystals of the adduct formed with 4-N,N′-dimethylaminopyridine (DMAP) 1iPrPO·DMAP (Scheme 1) were obtained. The molecular structure determined showed the coordination of the Lewis base leading to a distorted trigonal bipyramidal geometry at the Ge centre (Fig. S17, ESI†).
We have prepared 1iPrPO-F from the addition of one equivalent of KF/18-crown-6 to 1iPrPO (Scheme 1). The 31P{1H} NMR in CDCl3 showed a doublet at +77 ppm (3JP–F = 3.3 Hz) and a corresponding peak at −125.2 ppm in the 19F{1H} NMR spectrum (see ESI,† for detail). The molecular structure of 1iPrPO-F (Fig. 4A and Table S6, ESI†) exhibits a distorted trigonal bipyramidal geometry, analogous to 1iPrPO·DMAP. The Ge–F bond length is 1.746(2) Å, which is shorter compared to that found in the case of 1iPrP-F (1.785(2) Å).10 The Ge–O bond lengths have increased (avg. Ge–O = 1.97 Å) compared to those in 1iPrPO, with a corresponding decrease in the P–O bond lengths (avg. P–O = 1.53 Å). The calculated gas-phase fluoride ion affinity (FIA)19 at the Ge site of 1iPrPO gave a very high value of 865 kJ mol−1 (gas-phase FIA for reference SbF5 is 497 kJ mol−1) (Tables S2 and S3, see the ESI,† details). Incorporation of the acetonitrile (MeCN) solvated model decreased the calculated FIA value significantly to 142 kJ mol−1 (solvent corrected FIA for reference SbF5 is 315 kJ mol−1), respectively. This phenomenon of solvent damping is more pronounced with the cationic Lewis acids compared to the neutral ones.20,21 Nonetheless, 1iPrPO was found to be capable of abstracting a fluoride ion from AgSbF6 under heating conditions forming 1iPrPO-F (Scheme 1). 1iPrPO also abstracted fluoride from TBABF4 (TBA = n-tetrabutylammonium), which is a better fluoride ion donor, to give 1iPrPO-F under room temperature conditions. Thus, 1iPrPO proved to be Lewis superacidic22,23 under experimental conditions, despite low calculated values of FIA in solvated models.24
 | ||
| Fig. 4 Molecular structures of (A) 1iPrPO-F and (B) 1iPrPO-H in the solid state (thermal ellipsoids 35%, H atoms, triflate counter anions and solvent molecules are omitted for clarity). Selected bond lengths [Å] (a) Ge1–F1 = 1.746(2), Ge1–O1 = 1.971(2), P1–O1 = 1.524(2), Ge1–O2 = 1.990(2), P2–O2 = 1.525(2); (b) Ge1–H1 = 1.436(2), Ge1–O1 = 2.013(2), P1–O1 = 1.520(2), Ge1–O2 = 2.056(2), P2–O2 = 1.517(2). | ||
Gas-phase hydride ion affinity (HIA)19 calculation (Tables S2 and S3, see ESI† for detail) gave a very high value of 925 kJ mol−1, while incorporation of the solvated model gave low calculated values of 185 KJ mol−1 (MeCN). Notably, the gas phase HIA for reference B(C6F5)3 is 517 kJ mol−1 and the solvent corrected HIA is 244 kJ mol−1 for MeCN. Reaction of 1iPrPO with NaBH4 led to the formation of 1iPrPO-H (Scheme 1). The 31P{1H} NMR spectrum of 1iPrPO-H displayed a peak at +73.5 ppm. The molecular structure of 1iPrPO-H closely resembles that of 1iPrPO-F, possessing a Ge–H bond length of 1.436(2) Å (Fig. 4B and Table S7, ESI†). The inherently hydridophilic Ge center in 1iPrPO activated the Et3Si–H bond under room temperature reaction conditions to give 1iPrPO-H (Scheme 1).25,26 However, 1iPrPO does not abstract hydride from Ph3Si–H27 even upon heating.
Given the experimentally observed Lewis super acidic nature of 1iPrPO, we have explored the catalytic hydrodefluorination28 of an aliphatic C–F bond. With a catalyst loading of 5 mol% 1iPrPO and employing Ph3SiH as the hydride source, we have successfully achieved the hydrodefluorination of 1-adamantyl fluoride (76% conversion) in CD3CN upon heating overnight at 75 °C (Scheme 2A) (see ESI,† for detail). The in situ catalytic reaction mixture showed NMR signals at δ31P{1H} = +77.8 ppm and δ19F{1H} = −125.8 ppm corresponding to the formation of 1iPrPO-F, indicating the C–F bond activation over the Si–H bond activation (Fig. S40–S45, ESI†). Thus, we have proposed a catalytic pathway (Scheme S1, ESI†) involving the generation of the carbocation, which then abstracts hydride from Ph3SiH to form the alkane along with the regeneration of the catalyst 1iPrPO and Ph3SiF. We do not observe any Ge–H bond formation as an intermediate, discarding the Ph3Si–H bond activation pathway by 1iPrPO. This proposed mechanism is in line with that proposed by Müller et al. using the naphthalene-based digermyl hydronium borates as a catalyst.7 On the other hand, using Et3SiH as the hydride source for the same catalytic hydrodefluorination reaction (Scheme 2A) turned out to be less promising owing to the competing Si–H bond activation along with C–F bond activation by 1iPrPO. Only 40% conversion to the corresponding alkane was possible, the catalyst being transformed into 1iPrPO-H after 12 hours (Fig. S48–S51, ESI†). Notably, catalytic hydrodefluorination was unachievable using 1iPrP as the catalyst. As a matter of fact, there are very few reports on catalytic hydrodefluorination reactions achieved using molecular germanium as a catalyst.3,7,8,17 Our catalyst was ineffective for other C–F bonds. This lack of reactivity may be attributed to the stabilization of the Ge(IV) by the intramolecular P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O donors, thereby compromising the Lewis acidity at the Ge(IV) site.
O donors, thereby compromising the Lewis acidity at the Ge(IV) site.
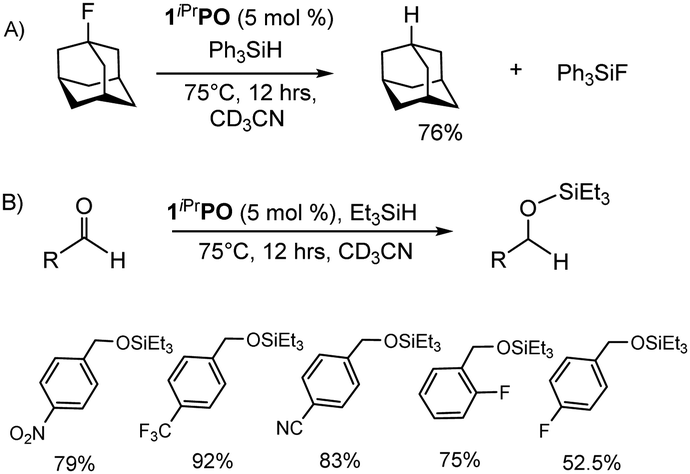 | ||
| Scheme 2 Catalytic applications of 1iPrPO: (A) hydrodefluorination of 1-adamantyl fluoride and (B) hydrosilylation of electron-deficient aromatic aldehydes. | ||
The hydridophilicity of 1iPrPO prompted us to explore the substrate scope for the catalytic hydrosilylation reactions (Scheme 2B) of aromatic aldehydes (see ESI,† for detail). In our earlier report, we have shown the catalytic hydrosilylation of p-methyl benzaldehyde using 1iPrP as a catalyst. However, catalytic hydrosilylation of electron-deficient aromatic aldehydes was not possible using 1iPrP. In this study, we were successful in converting a variety of electron-deficient aldehydes into the corresponding silyl ethers using 5 mol% of 1iPrPO under mild heating conditions (Scheme 2B).29
It was observed from the NMR study that the aldehydes did not bind to the Ge-centre of the catalyst. The NMR investigations of the catalytic reaction mixture showed the formation of a Ge–H bond (δ31P{1H} = +73.5 ppm) from Et3Si–H bond activation, corresponding to the formation of 1iPrPO-H as an intermediate (Fig. S53, see ESI,† for the NMRs). Therefore, the reaction is likely to proceed by Et3SiH bond activation by 1iPrPO followed by Si–H addition across the CO bond in the carbonyls (Scheme S2, see ESI†). A similar mechanism was observed in the case of 1iPrP as a catalyst. The 29Si{1H} NMR spectrum showed the formation of the hydrosilylated products (Fig. S59, ESI†). The formation of Et3SiOTf as a competent catalyst, though not detected in the NMR spectrum, cannot be completely overruled. The irreversible transformation of the catalyst to 1iPrPO-H was observed from in situ NMR study of the catalytic reaction mixture, which might be responsible for poor catalytic outcome in certain substrates (see ESI,† for detail).
In conclusion, we have successfully manipulated the frontier orbitals by treating 1iPrP with an oxidant to form the phosphine-oxide stabilized Ge(IV) di-cation 1iPrPO. Localization of the positive charges and acceptor orbitals on the Ge-centre has significantly enhanced the Lewis acidity. The well-exposed cationic Ge(IV) site is now capable of abstracting fluoride ions from a hexafluoroantimonate anion, thereby marking its Lewis super acidic nature. The hard and soft Lewis acidic nature of 1iPrPO have been manifested in catalytic hydrodefluorination of 1-adamantyl fluoride and hydrosilylation of electron-deficient aromatic aldehydes, which were unachievable with our previously reported 1iPrP. Thus, the localization of the cation charges on the Ge-centre has led to the expansion of the catalytic portfolio. Our group is engaged in investigating further catalytic applications of 1iPrPO, and extending this catalyst design strategy to other main-group Lewis acids.
M. M. thanks SERB India SPF/2022/000046 and CRG/2022/000673 for financial support. A. K. thanks UGC for the fellowship. We thank Prof. Deepak Chopra from the Indian Institute of Science Education and Research (IISER) Bhopal, India for his help in crystallography.
Data availability
Experimental details, spectroscopic data, X-ray diffraction analysis data, and computational data are available in the ESI.†Conflicts of interest
There is no conflict to declare.Notes and references
- H. Yamamoto and M. Oishi, Silicon(IV) Lewis Acids, Wiley-VCH, Weinheim Germany, 2000 Search PubMed.
- N. Mukherjee and M. Majumdar, J. Am. Chem. Soc., 2024, 146, 24209 CrossRef CAS PubMed.
- D. Roth, H. Wadepohl and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 20930 CrossRef CAS PubMed.
- A. T. Henry, T. P. L. Cosby, P. D. Boyle and K. M. Baines, Dalton Trans., 2021, 50, 15906 RSC.
- R. Yadav, P. Janβen, M. Schorpp and L. Greb, J. Am. Chem. Soc., 2023, 145, 177546 Search PubMed.
- H. Fang, H. Jing, A. Zhang, H. Ge, Z. Yao, P. J. Brothers and X. Fu, J. Am. Chem. Soc., 2016, 138, 7705 CrossRef CAS PubMed.
- N. Kordts, C. Borner, R. Panisch, W. Saak and T. Müller, Organometallics, 2014, 33, 1492 CrossRef CAS.
- A. Hayatifar, A. Borrego, D. Bosek, M. Czarnecki, G. Derocher, A. Kuplicki, E. Lytle, J. Padilla, C. Paroly, G. Tubay, J. Vyletel and C. S. Weinert, Chem. Commun., 2019, 55, 10852 RSC.
- M. Talavera, G. Meiβner, S. G. Rachor and T. Braun, Chem. Commun., 2020, 56, 4452 RSC.
- B. Peddi, S. Khan, R. G. Gonnade, C. B. Yildiz and M. Majumdar, Chem. Sci., 2023, 14, 13755 RSC.
- (a) J. E. Smith, H. Yang and F. P. Gabbaï, Organometallics, 2021, 40, 3886 CrossRef CAS; (b) Y.-H. Lo and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 10194 CrossRef CAS PubMed.
- E. D. Litle and F. P. Gabbai, Chem. Commun., 2024, 60, 690 RSC.
- M. Olaru, R. Kather, E. Hupf, E. Lork, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2018, 57, 5917 CrossRef CAS PubMed.
- W.-C. Liu and F. P. Gabbaï, Science, 2024, 385, 1184 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- S. E. Denmark, R. T. Jacobs, G. Dai-Ho and S. Wilson, Organometallics, 1990, 9, 3015 CrossRef CAS.
- D. Tanaka, A. Konishi and M. Yasuda, Chem. – Asian J., 2021, 16, 3118 CrossRef CAS PubMed.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, Chem. Phys. Chem., 2020, 21, 987 CrossRef CAS PubMed.
- A. Hermannsdorfer and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 23132 CrossRef CAS PubMed.
- D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845 CrossRef CAS PubMed.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. SantisoQuiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659 CrossRef PubMed.
- T. Thorwart and L. Greb, Lewis Superacids, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, Wiley, 2nd edn, 2021, pp 1–26 Search PubMed.
- P. Erdmann, M. Schmitt, L. M. Sigmund, F. Krämer, F. Breher and L. Greb, Angew. Chem., Int. Ed., 2024, 63, e202403356 CrossRef CAS PubMed.
- G. I. Nikonov, S. F. Vyboishchikov and O. G. Shirobokov, J. Am. Chem. Soc., 2012, 134, 5488 CrossRef CAS PubMed.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamaki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983 CrossRef CAS PubMed.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818 CrossRef CAS.
- G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546 CrossRef CAS PubMed.
- A. L. L. Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2416600, 2416604, 2416606 and 2416610. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00250h |
| This journal is © The Royal Society of Chemistry 2025 |