
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multilayered organosiloxane films with self-healing ability converted from block copolymer nanocomposites†
Yoshiaki
Miyamoto
a,
Takamichi
Matsuno
 abc and
Atsushi
Shimojima
*abc
abc and
Atsushi
Shimojima
*abc
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: shimojima@waseda.jp
bWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan
First published on 6th January 2025
Abstract
Self-healable, multilayered organosiloxane films were prepared via thermal conversion of lamellar organosiloxane films containing poly(ethylene oxide)–polydimethylsiloxane–poly(ethylene oxide) block copolymers. The incorporation of silanolate groups enabled crack healing through dynamic siloxane equilibration. The enhanced hardness and suppressed cyclic siloxane formation resulting from the multilayered structure exhibit potential for practical applications.
Polysiloxane materials are utilized in various fields, including optics, electronics, and aerospace, owing to their transparency, excellent thermal stability, weather resistance, and insulating properties.1–4 The dynamic nature of the Si–O–Si bonds, which impart self-healing properties, has recently gained considerable attention.3,5 In particular, polydimethylsiloxane (PDMS)-based elastomers with silanolate (Si–O−) end groups exhibit self-healing behavior through thermally activated equilibration among different siloxane chains.6,7 However, the formation and volatilization of low-molecular-weight cyclic siloxanes6 compromise the long-term stability of these materials and cause serious problems, such as contact failures in electronic components.5,8,9 Another significant issue is the enhancement of their mechanical properties, such as hardness, of the self-healing siloxane materials, particularly for their application in coatings.
The incorporation of inorganic nanofillers such as nanoparticles and nanosheets can generally improve the physical properties of polymer materials effectively. Nanocomposites of polymers and inorganic nanosheets have been reported to exhibit high mechanical strength, incombustibility, and gas-barrier properties.10,11 Furthermore, layer-by-layer-assembled nanocomposite films containing polymers (poly(vinyl alcohol)) and inorganic nanosheets (boron nitride) exhibit improved self-healing abilities owing to the in-plane anisotropic diffusion of the polymers.12 These features of multilayered nanocomposites greatly contribute to addressing the aforementioned issues of self-healing PDMS-based materials. However, one challenge is that stable linkages between PDMS and the nanosheet surface are required to prevent segregation due to the low surface energy of PDMS. Additionally, developing a more efficient approach for multilayered films than the conventional layer-by-layer assembly method12–14 is important from both scientific and practical viewpoints.
Herein, we report an efficient approach for fabricating multilayered films composed of organosiloxane and grafted PDMS layers through the thermal conversion of a lamellar silsesquioxane (O1.5Si–C2H4–SiO1.5)n thin film containing a poly(ethylene oxide)–PDMS–poly(ethylene oxide) block copolymer (PEO–PDMS–PEO) (Scheme 1). Lamellar thin films were prepared by an evaporation-induced self-assembly process starting with 1,2-bis(triethoxysilyl)ethane (BTSE), PEO–poly(propylene oxide) (PPO)–PEO triblock copolymer (Pluronic P123), and PEO–PDMS–PEO. By selectively removing the PEO and PPO blocks via calcination, we successfully obtained a multilayered siloxane film with a crack healing ability, exhibiting higher hardness and significantly lower volatilization of cyclic oligomers than previously reported self-healing PDMS-based elastomers.
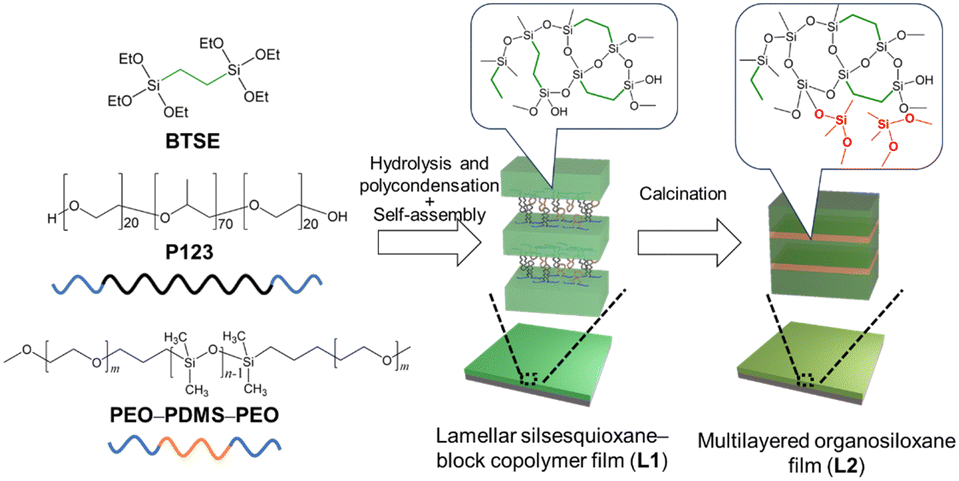 | ||
| Scheme 1 Preparation of multilayered organosiloxane films via the conversion of organosiloxane–block copolymer nanocomposites. | ||
PEO–PDMS–PEO (PEO27–PDMS36–PEO27) was synthesized according to the literature15 with some modifications (see ESI† for details). A transparent thin film with a lamellar structure (L1, Fig. 1a(i)) was prepared by spin-coating or drop-casting a precursor solution with a molar composition of BTSE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P123:PEO27–PDMS36–PEO27:EtOH:H2O:HCl = 1:0.015:0.015:15:20:0.008 onto silicon or glass substrates (see ESI† for details).
:P123:PEO27–PDMS36–PEO27:EtOH:H2O:HCl = 1:0.015:0.015:15:20:0.008 onto silicon or glass substrates (see ESI† for details).
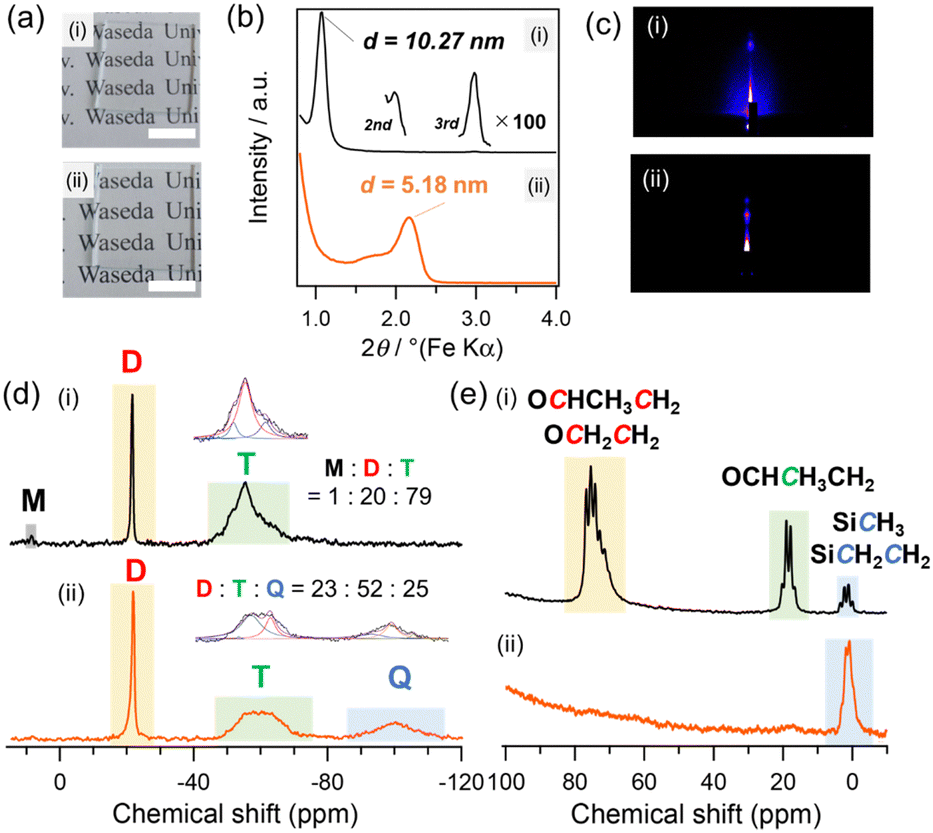 | ||
| Fig. 1 (a) Photographs of (i) L1 and (ii) L2 prepared on glass substrates (2 cm × 2 cm) by spin-coating. (b) XRD patterns and (c) GI-SAXS patterns of (i) L1 and (ii) L2 prepared on Si substrates by spin-coating. (d) 29Si MAS NMR and (e) 13C MAS NMR spectra of (i) L1 and (ii) L2 prepared by drop-casting the precursor solution on the substrates, followed by drying and pulverization. | ||
The X-ray diffraction (XRD) pattern of L1 (Fig. 1b(i)) shows a sharp peak at d spacing of 10.27 nm with second- and third-order diffractions. The grazing-incidence small-angle X-ray scattering (GI-SAXS) pattern (Fig. 1c(i)) shows a spot corresponding to the periodicity of the same d spacing along the direction perpendicular to the substrate surface, indicating the formation of a lamellar mesostructure oriented parallel to the substrate. The thickness of L1 was approximately 800 nm, as confirmed by scanning electron microscopy (SEM) analysis of the fractured surface (Fig. S1, ESI†). The control of the molar ratio of BTSE to block copolymers is crucial for the formation of the lamellar structure. For example, a two-dimensional hexagonal structure was formed with a molar ratio of BTSE:P123:PEO27–PDMS36–PEO27 = 1:0.01:0.01 (Fig. S2, ESI†).
The Fourier transform infrared (FT-IR) spectrum of L1 (Fig. S3, ESI†) shows absorption bands attributed to Si–O–Si (1000–1100 cm−1), C–O (1100–1200 cm−1), and Si–CH3 (1250 cm−1), suggesting the formation of a composite of siloxane and block copolymers. The solid-state 29Si magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectrum of L1 (Fig. 1d(i)) exhibits a sharp D2 signal (Dx: Me2Si(OSi)x(OH)2−x, –22 ppm) corresponding to the PDMS block and broad T1, T2, and T3 signals (Ty: CSi(OSi)y(OH)3−y, –51, –55, and –62 ppm, respectively) derived from BTSE. The D/T integral ratio was 20:79, which is consistent with the molar ratio of PEO27–PDMS36−PEO27/BTSE = 0.015 in the precursor solution. The solid-state 13C MAS NMR spectrum (Fig. 1e(i)) shows signals assigned to the PEO and PPO blocks (16–22 ppm and 67–78 ppm), along with the signals of the SiCH3 and SiC2H4Si groups (0–5 ppm) derived from PDMS and BTSE, respectively. These results indicate that L1 has a lamellar structure consisting of BTSE-derived silsesquioxane and block copolymer layers (P123 and PEO27–PDMS36–PEO27) (Scheme 1, middle).
PEO and PPO blocks were selectively removed from lamellar film L1 to produce a multilayered siloxane film composed of silsesquioxane and PDMS. The thermogravimetry–differential thermal analysis (TG–DTA) curves of L1 (Fig. S4, ESI†) show exothermic peaks with weight loss due to the combustion of PEO and PPO blocks at 130–250 °C.16 The SiC2H4Si groups in the silsesquioxane layers and SiCH3 groups of PDMS combusted at higher temperatures (>250 °C). Similar degradation behavior of the PEO blocks was also observed for PEO27–PDMS36–PEO27 (Fig. S5, ESI†). Based on these data, L1 was calcined in air at 170 °C for 4 h to remove the PEO and PPO blocks. The resulting calcined film (hereafter referred to as L2) was transparent and colorless, and no cracks were observed (Fig. 1a(ii)).
The FT-IR spectrum of L2 (Fig. S3(ii), ESI†) shows a decrease in the bands corresponding to the PEO and PPO blocks. The selective removal of the PEO and PPO blocks was confirmed from the 13C MAS NMR spectrum of L2 (Fig. 1e(ii)). The signals of the PEO and PPO blocks decreased significantly compared to those of the SiCH3 and SiC2H4Si groups. The 29Si MAS NMR analysis of L2 (Fig. 1d(ii)) confirmed that the D2 signal remained almost unchanged, whereas the integrated intensity of the T signals decreased slightly and a broad Q3 signal (Si(OSi)3OH, –100 ppm) appeared. This result indicates that the O3SiC2H4SiO3 units were partially cleaved to form SiO4 units, which may be due to the increase in the local temperature around the silsesquioxane layers upon the combustion of the PEO and PPO blocks of the block copolymers.
PDMS is known to degrade thermally through backbiting and chain transfer, resulting in evaporation of low-molecular-weight cyclic siloxanes.17,18 In the 29Si MAS NMR spectrum of L2, the signals of cyclic tetrasiloxane (−19 ppm)19 and cyclic trisiloxane (−9 ppm)19 were not observed (Fig. 1d(ii)). In addition, the integral ratio of the D units to total Si (D + T + Q) was almost unchanged from that of L1 (Fig. 1d). Therefore, the formation and evaporation of these cyclic siloxanes were negligible upon calcination at 170 °C. The decrease in the relative intensity of the 29Si NMR signal (9 ppm) corresponding to the terminal OSiMe2CH2– groups in PEO–PDMS–PEO (Fig. 1d) suggested that the PDMS blocks formed higher molecular-weight linear/cyclic siloxanes and/or linkages with the T and Q units.
The XRD pattern of L2 (Fig. 1b(ii)) shows a diffraction peak at d = 5.18 nm. The GI-SAXS pattern (Fig. 1c(ii)) shows a spot corresponding to an oriented lamellar structure with a reduced d spacing. These results indicate that a lamellar film consisting of partially decomposed silsesquioxane and PDMS layers was formed. The d spacing of L1 decreased to 4.37 nm after removal of the block copolymers (P123 and PEO–PPO–PEO) by solvent extraction (Fig. S6, ESI†). Assuming that the thickness of the silsesquioxane layers in L2 is 4 nm, the volume ratio of the silsesquioxane-based layers to the PDMS layers is approximately 4:1 (Scheme 1, right). No decrease in the d spacing of L2 was observed even after swelling in THF and subsequent drying (Fig. S7, ESI†). This result suggested that PDMS chains are partially grafted to the silsesquioxane layers. The volume ratio of PDMS can be varied using PEO–PDMS−PEO with different block lengths. A similar multilayered siloxane film prepared using PEO27–PDMS82−PEO27 exhibited a larger d spacing (7.11 nm) compared to that of L2 (Fig. S8, ESI†), which suggested the formation of thicker PDMS layers.
Several reports have shown that Si–O–Si bonds behave as dynamic bonds in the presence of nucleophiles such as silanolates and amines. These can induce the rearrangement of siloxane bonds by nucleophilic attack on other Si atoms or by reacting with residual silanol.5,20–22 Therefore, we introduced silanolate groups into L2 by immersing the film in a THF–water solution of KOH. The resulting film is referred to as L3. The X-ray photoelectron spectroscopy (XPS) depth profiles (Fig. S9, ESI†) of L3 revealed a uniform distribution of K on the surface for a depth of about 100 nm, which was not observed in L2. 29Si MAS NMR and 13C MAS NMR analyses confirmed that the silsesquioxane and PDMS moieties remained almost intact (Fig. S10 and S11, ESI†). The retention of the lamellar structure was confirmed by XRD and GI-SAXS (Fig. S12, ESI†). Cross-sectional scanning transmission electron microscopy (STEM) images of L3 cut vertically using focused ion beam milling revealed oriented layered patterns (Fig. S13, ESI†).
The healing of cracks, which were submicrometers in width and several micrometers in length, that were made on L3 was observed after treating the film at 80 °C and 40% relative humidity (RH) for 1 d (Fig. 2). Under these conditions, the layered structure of L3 was retained with a slight decrease in the d spacing (Fig. S14, ESI†). Cross-sectional TEM and STEM images and energy-dispersive X-ray spectroscopy mapping of the healed area suggested that the crack was filled with siloxane species with no layered structure (Fig. S15, ESI†).
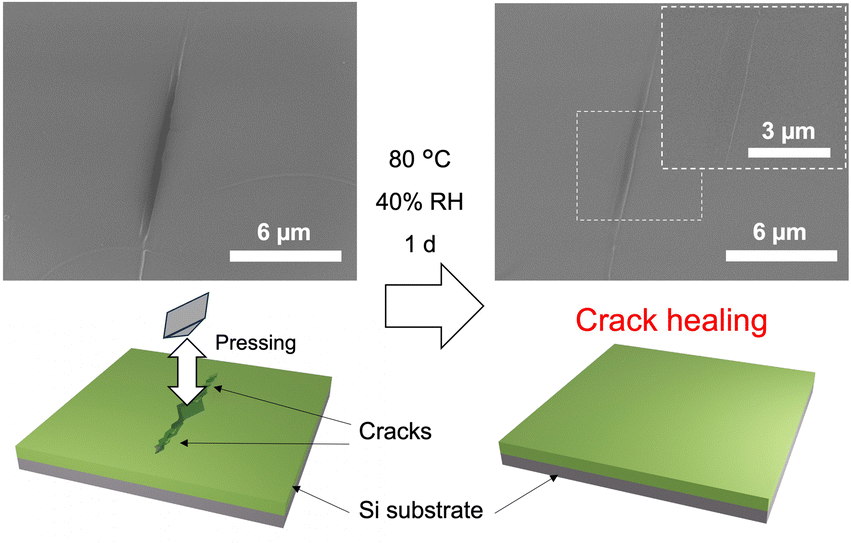 | ||
| Fig. 2 Crack healing behavior of L3 at 80 °C and 40% RH. The crack was created by pressing a diamond indenter on the film. The surface of the thin film before and after healing was observed by SEM. | ||
We confirmed that crack healing did not occur when L2 (without KOH) was treated under the similar conditions or when L3 was heated without increasing the humidity. It is reasonable to consider that the healing occurred via rearrangement of the siloxane networks mediated not only by potassium silanolate groups but also by silanol groups formed by partial hydrolysis of the Si–O–Si bonds.5 Healing was not observed for L3 at room temperature even at 90% RH, which is in contrast to room-temperature healing reported for PDMS elastomers with lower degrees of cross-linking.20 The heating and humid conditions required for the crack healing of L3 are likely due to the presence of a silsesquioxane layer with a high cross-linking density.
To verify the effects of the siloxane components on the healing behaviors, a multilayered film was prepared using tetraethoxysilane (TEOS) instead of BTSE under otherwise identical conditions. A lamellar thin film with d = 8.95 nm was formed by spin-coating. After calcination and introduction of KOH, the resulting layered silica–PDMS thin film exhibited a crack healing behavior (experimental details and Fig. S16, ESI†). These results suggest that the composition of the siloxane layers is tunable.
Our previous study showed that lamellar silica/silsesquioxane–surfactant composite films exhibit crack-healing abilities under humid conditions, even at room temperature.23,24 This healing mechanism is attributed to the lamellar swelling caused by water adsorption in the surfactant layers to induce crack closure, which fundamentally differs from the siloxane equilibration-induced healing of L3. The presence of quaternary ammonium cations or block copolymers between layers inevitably reduces their durability and mechanical strength. In contrast, the multilayered siloxane film L3 is an all-siloxane material that contains no interlayer organic molecules or polymers and exhibits excellent properties inherent to siloxanes, such as high thermal stability and weather resistance.
Nanoindentation tests revealed that L3 had a hardness of 1.50 GPa (Fig. S17, ESI†). This value is much higher than that of a conventional PDMS elastomer (49 MPa), which may be due to the presence of more rigid silsesquioxane layers. The value is also much higher than that of self-healing silsesquioxane–surfactant lamellar thin films that heal under humid conditions (261 MPa). In addition, the significant increase in hardness from L1 (112 MPa) can be attributed to the increased volume ratio of the silsesquioxane layers caused by the removal of the PEO and PPO moieties.
The TG–mass spectrometry (TG–MS) analysis was conducted to evaluate the release of low-molecular-weight cyclic siloxanes formed through siloxane equilibration (Fig. S18, ESI†). The self-healing PDMS elastomer exhibited degradation starting at 80 °C, and fragment ion peaks derived from cyclic hexamethyltrisiloxane (D3) and octamethyltetrasiloxane (D4) were detected up to 180 °C. Meanwhile, L3 exhibited relatively moderate weight loss at elevated temperatures. Importantly, no mass peaks derived from cyclic siloxanes were observed for L3, as the silsesquioxane layers likely blocked the release of volatile cyclic siloxanes from the interlayer space.
This material (L3) is promising as protective coatings because of its high hardness, crack-healing ability, suppressed cyclic siloxane volatilization, and transparency. The low-volatilization property also makes it suitable for electronic components that are sensitive to contamination risks. However, the processes involving KOH and H2O could pose challenges for practical applications. Further materials design is required to achieve crack healing under milder conditions.
In conclusion, a new class of self-healing siloxane materials composed of silsesquioxane and grafted PDMS layers was successfully prepared by the thermal conversion of a lamellar silsesquioxane film containing PEO–PDMS–PEO block copolymers. The multilayered structure of the material provides higher hardness and thermal stability than conventional self-healing PDMS-based materials. These findings contribute to the development of self-healing siloxane-based materials.
This work utilized research equipment JEOL JNM-ECX-400 (C1025), JEOL JNM-ECZ500R (C1028), BRUKER Dektak XT-S (C1049) ULVAC-PHI VersaProbe II (C1051), JEOL JEM-2100F (G1026), Hitachi S-5500 (G1028), JEOL JIB-4000 (G1033), Rigaku RINT Ultima IV (G1036), and Shimadzu HMV-2000 (G1064) shared in the MEXT Project for promoting the public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0440500024. The authors are grateful to Mr T. Goto (Material Characterization Central Laboratory, Waseda University) for the XPS analysis, Dr N. Hanzawa, Mr T. Akanuma, and Ms M. Itadani (Kagami Memorial Research Institute for Materials Science and Technology) for the STEM observation. We gratefully acknowledge Mr T. Hikino, Mr T. Miyake and Ms R. Wakino (Waseda University) for the useful discussions.
Yoshiaki Miyamoto: conceptualization, all experimental data collection, writing – original draft and editing. Takamichi Matsuno: analysis, writing – review & editing. Atsushi Shimojima: writing – review & editing, supervision.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26, 211–220 CrossRef CAS.
- B. Yi, S. Wang, C. Hou, X. Huang, J. Cui and X. Yao, Chem. Eng. J., 2021, 405, 127023 CrossRef CAS.
- H. Wang, X. Zhang, Y. Li and L.-W. Xu, Angew. Chem., Int. Ed., 2022, 134, e202210851 CrossRef.
- M. Suzuki, T. Hayashi, T. Hikino, M. Kishi, T. Matsuno, H. Wada, K. Kuroda and A. Shimojima, Adv. Sci., 2023, 10, 2303655 CrossRef CAS PubMed.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- X. Wu, X. Yang, X. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A., 2018, 6, 10184–10188 Search PubMed.
- X. Li, R. Yu, T. Zhao, Y. Zhang, X. Yang, X. Zhao and W. Huang, Eur. Polym. J., 2018, 108, 399–405 Search PubMed.
- M. Takahashi, in Organosilicon Chemistry III: From Molecules to Materials, ed. N. Auner, J. Weis, Wiley-VCH, Weinheim, 1997, pp. 555–565 Search PubMed.
- Y. Cui, S. Kumar, B. R. Kona and D. van Houcke, RSC Adv., 2015, 5, 63669–63690 RSC.
- J. Li, X. Liu, Y. Feng and J. Yin, Prog. Polym. Sci., 2022, 126, 101505 CrossRef CAS.
- X. Qi, L. Yang, J. Zhu, Y. Hou and M. Yang, ACS Nano, 2016, 10, 9434–9445 CrossRef CAS PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- Z. Zhang, J. Zeng, J. Groll and M. Matsusaki, Biomater. Sci., 2022, 10, 4077–4094 RSC.
- W. H. Li, X. Y. Zhang and J. B. Dai, Chin. Chem. Lett., 2008, 19, 1209–1211 CrossRef CAS.
- F. Kleitz, W. Schmidt and F. Schüth, Microporous Mesoporous Mater., 2003, 65, 1–29 CrossRef CAS.
- A. T. Wolf and A. Stammer, Polymers, 2024, 16, 2220 CrossRef CAS PubMed.
- B. Rupasinghe and J. C. Furgal, Polym. Int., 2022, 71, 521–531 CrossRef CAS.
- Y. Sato, R. Hayami and T. Gunji, J. Sol-Gel Sci. Technol., 2022, 104, 36–52 CrossRef CAS.
- W. Schmolke, N. Perner and S. Seiffert, Macromolecules, 2015, 48, 8781–8788 CrossRef CAS.
- T. Köhler, A. Gutacker and E. Mejía, Org. Chem. Front., 2020, 7, 4108–4120 RSC.
- M. O. Saed and E. M. Terentjev, ACS Macro Lett., 2020, 9, 749–755 CrossRef CAS PubMed.
- S. Itoh, S. Kodama, M. Kobayashi, S. Hara, H. Wada, K. Kuroda and A. Shimojima, ACS Nano, 2017, 11, 10289–10294 CrossRef CAS PubMed.
- S. Kodama, Y. Miyamoto, S. Itoh, T. Miyata, H. Wada, K. Kuroda and A. Shimojima, ACS Appl. Polym. Mater., 2021, 3, 4118–4126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05804f |
| This journal is © The Royal Society of Chemistry 2025 |