
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing strain governs transmetalation behaviour at a tucked-in iron complex†
Connor S.
Durfy‡
 a,
Michelle
Huang‡
a,
Joseph A.
Zurakowski
ab,
Paul D.
Boyle
a and
Marcus W.
Drover
*a
a,
Michelle
Huang‡
a,
Joseph A.
Zurakowski
ab,
Paul D.
Boyle
a and
Marcus W.
Drover
*a
aDepartment of Chemistry, Western University, 1151 Richmond St, London, ON N8K 3G6, Canada. E-mail: marcus.drover@uwo.ca
bDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Ave, Windsor, ON N9B 3P4, Canada
First published on 7th January 2025
Abstract
Studies that independently investigate [M]–C transmetalation reactions using two different metals are uncommon and yet understanding this reactivity is important to unlocking new synthetic approaches and product classes. Here, we show that the strained [Fe]–C complex, [(η6-C5Me4-CH2)Fe(diphosphine)] undergoes transmetalation with rhodium(I) and iridium(I) diolefin salts, leading to rapid Fe–C(sp3) bond cleavage and M–C(sp3) (M = Rh or Ir) bond generation.
Carbon-element (C-E) bond forming reactions are key to accessing synthetic diversity.1–4 Contributions to the selective generation of C–C bonds, for example, were acknowledged with the 2010 Nobel prize in chemistry.5 These metal-mediated transformations have had a measurable impact on the fields of drug development and discovery. The Suzuki–Miyaura coupling reaction, for example, is one of the most utilized across medicinal chemistry.6 Such reactions proceed in the presence of a transition metal often via stepwise oxidative addition, transmetalation, and reductive elimination.7
Transmetalation can be used to forge reactive metal–carbon ([M]–C) bonds that can be later transferred or coupled via reductive elimination.8,9 Of steps associated with cross-coupling, a detailed mechanistic understanding of transmetalation is comparatively lagging and has consequently been the subject of numerous studies.10–15 Intimate knowledge surrounding preferred [M]–C generation routes helps to provide a general landscape for reaction optimization.16 Electronegativity trends and by-products can sometimes be used to predict transfer propensity and the reaction outcome.
As an example of a strained [Fe]–C bond, we recently reported the preparation of an Fe(II) tucked-in complex, [(η6-C5Me4-CH2)Fe(dnppe)] (dnppe = 1,2-bis(di-n-propylphosphino)ethane) (1).17,18 Despite the prevalence of related sandwich complexes, ([Cp/Cp*]2M; Cp = C5H5−; Cp* = C5Me5−), the reactivity of tucked-in compounds, especially those with late 3d elements, remains underexplored due to a size mismatch between the metal and L2X2-Cp* ring donor (compared to group 4 metals, for example).19,20 With 1 in hand, we wondered whether reaction with suitable metal sources would result in transmetalation, affording a programmable route towards heterometallic Cp*{Fe,M} compounds. This transformation would simultaneously enable a detailed study of Fe–C(sp3) bond cleavage and M–C(sp3) bond generation between two model organometallic molecules, helping to determine how ring strain impacts carbon-transfer chemistry, whilst informing the use of {[Fe]–C} compounds as transmetalation partners.
Herein, we investigate the transmetalation behaviour of 1 with Rh(I) and Ir(I) halides – metals known for application in a wide variety of carbon-element bond-forming cycles (Scheme 1). This reaction leads to rapid Fe–C(sp3) bond cleavage and the formation of new M–C(sp3) (M = Rh or Ir) bonds. This behavior is reversible: the addition of a diphosphine prompts regeneration of complex 1 and produces Cl–M(diphosphine). Intermolecular control reactions between [Cp*Fe(CH3)(diphosphine)] and Cl–MLn (Ln = diolefin or diphosphine) provide a differential outcome, resulting in clean formation of Fe–Cl and M–C species, pointing toward a heterometallic effect. These findings provide a clear example of Fe-to-Rh or – Ir hydrocarbyl transfer – and its reverse, differentiating inter- versus intramolecular transmetalation.
 | ||
| Scheme 1 (A) Concept: reversible metal-to-metal transmetalation; (B) present work: transmetalation at a bridged heterometallic. | ||
To begin, red C6D6 solutions of 117 were reacted with 0.5 equiv. of [Rh(nbd)Cl]2 (nbd = bicyclo[2.2.1]hepta-2,5-diene) or [M(COD)Cl]2 (M = Rh or Ir, COD = 1,5-cyclooctadiene), affording ring-opened μ-Cl heterometallic complexes [(η5-C5Me4-CH2-{M(Ln)})FeII(dnppe)(μ-Cl)] 2, 3, and 4, respectively (Scheme 2). These reactions proceeded similarly, with a notable colour change occurring immediately upon addition – yellow brown for 2/3 and burgundy for 4. The generation of 2–4 requires tucked-in ring-opening, giving new M–C(sp3) bonds via Fe-to-M alkyl transmetalation.
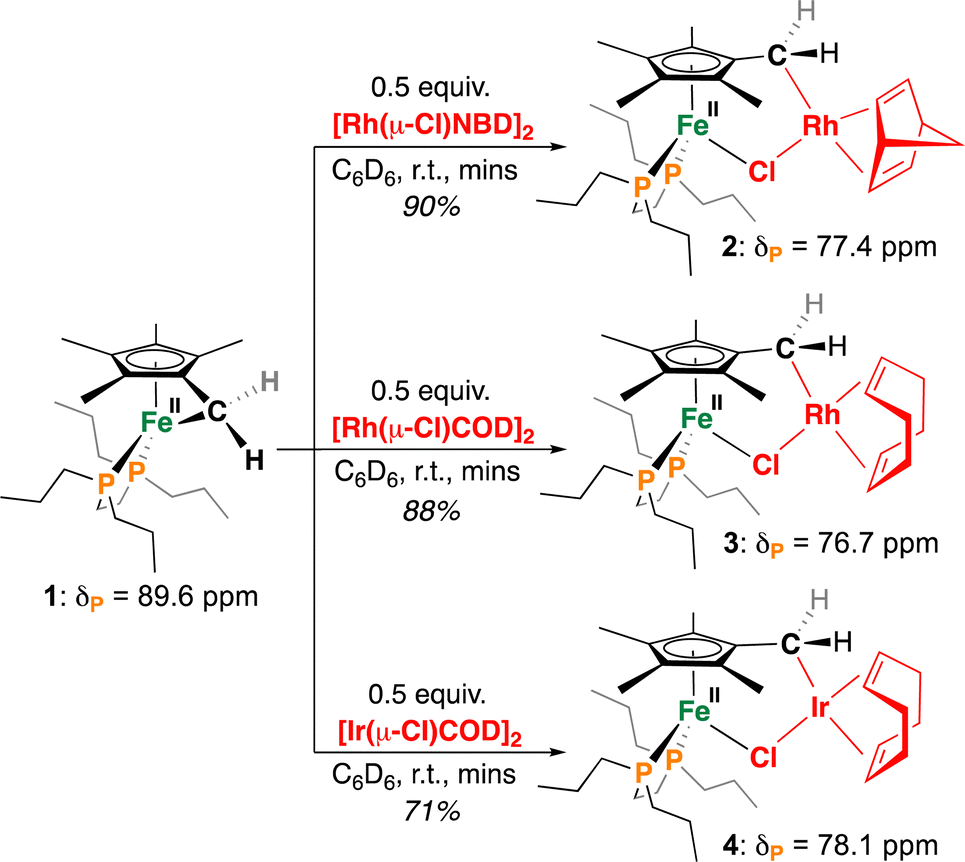 | ||
| Scheme 2 Reactions of 1 with M(I) (M = Rh, Ir) precursors to generate heterobimetallic complexes 2–4. | ||
The formation of these products is evidenced by a shift in their 31P{1H} NMR spectrum, from δP = 89.6 ppm (for 1)17 to 77.4, 76.7, and 78.1 ppm, for 2–4 respectively. These chemical shifts are similar to mononuclear [Cp*Fe(dnppe)Cl] (δP = 79.4 ppm)21 and suggest the formation of a dative Fe–Cl bond. Additionally supporting a μ-Cl bridge between Fe and Rh, the 31P{1H} NMR signature for 2 and 3 appears as a Rh-coupled doublet with JP–Rh = 3.1 and 2.7 Hz, respectively.
By 1H NMR spectroscopy, desymmetrization of the Cp*-Me protons proximal (2, 5-) and distal (3, 4-) to the [M]–CH2 (M = Rh or Ir) bond can be used as an additional means to support a Cp*-bound heterometallic with Δδ ∼ 1.40 ppm. We suggest that shielding of the proximal (2, 5-) methyl sites results from adjacent metalation of an electron-rich Rh or Ir center. This shielding effect is additionally observed in the Cp*-CH2-M(Ln) group, which shifts from δH = 2.74 ppm in 1 to 1.13 ppm in 2. Metallation of the Cp*-CH2-M(Ln) is further cemented by 13C{1H} and 1H–13C{1H} HSQC NMR spectroscopy, which for 2, displays a Rh-coupled doublet at δC = 27.5 ppm (1JC–Rh = 27.2 Hz).22 To the best of our knowledge, compounds 2-4 represent the only known Fe(II)/Rh(I) or Ir(I) complexes merged from a single Cp*-derived ligand scaffold.23 More broadly, this represents the first programmable route towards such heterometallics using a single tucked-in precursor, providing a vast space for future design.
The structures of 2–4 were confirmed by single-crystal X-ray diffraction analysis (Fig. 1). Each of the three compounds comprises a five-membered Fe–C–C–M–Cl (M = Rh or Ir) ring system having an envelope-type conformation, with the μ-Cl group occupying the endo-position. Across the series, the Fe–M distance increases from 3.816(1) Å (2) to 3.863(2) Å (3) to 3.875(1) Å (4) with most other inter-ring distances staying within ca. 0.02 Å of one another. This can be rationalized by the larger diolefin ligand (COD vs. nbd) for Rh(I) and the greater atomic radius of Ir cf., Rh on going from 3 to 4.
 | ||
| Fig. 1 Molecular structures of (A) 2, (B) 3, and (C) 4 with ellipsoids drawn at 40% probability. Hydrogen atoms except for those on C(1) are omitted for clarity. | ||
Given the inherent lability of metal-bound diolefin groups, we next became interested in the onwards functionalization chemistry of 2–4 with neutral L-type donor ligands, settling on diphosphines due to a strong drive for M–P bond formation and ease of monitoring by 31P{1H} NMR spectroscopy (Scheme 3A). Using the nbd precursor, 2, treatment with 1,2-bis(diphenylphosphino)ethane (dppe) at −78 °C immediately resulted in the formation of three new resonances by 31P{1H} NMR spectroscopy at δP = 79.2 for Fe-dnppe as well as 72.9 (dd, 1JRh–P = 234.7 Hz, 2JP–P = 26.7 Hz) and 47.0 (dd, 1JRh–P = 126.6 Hz, 2JP–P = 26.7 Hz) for the Rh-dppe component, attributed to the product 5, [(η5-C5Me4-CH2-{Rh(dppe)})FeII(dnppe)(μ-Cl)] (Scheme 3). A trans-influence of the bound Cp*CH2 group is borne out in a marked decrease in 1JRh–P coupling value from 234.7 (trans-Cl) to 126.6 Hz (trans-Cp*CH2).
 | ||
| Scheme 3 (A) Generation of complex 5 and onwards Rh-to-Fe–C(sp3) bond exchange; (B) the molecular structure of 5 with ellipsoids drawn at 40% probability. Hydrogen atoms are omitted except for those on C(1). | ||
Single crystals of 5 suitable for analysis by X-ray diffraction confirm a μ-Cl Fe–Cl–Rh(dppe) complex (Scheme 3B). Complex 5 maintains the longest distance between Fe and Rh (3.924(1) Å), lengthened by nearly 0.11 Å when compared to its nbd precursor 2. Of the heterobimetallic species generated, compound 5 has the shortest Fe–C2 (2.116(3) Å) and Fe–Cl (2.346(1) Å) bond lengths, suggesting the greatest “dissociation” from Rh. This point is further supported by a lengthened Rh–C1 bond of 2.142(3) for 5vs. 2.088(2) Å for 2, ascribed to a trans-influence of the newly installed phosphorus donor.
Solutions of 5 were found to be unstable at room-temperature, cleanly returning tucked-in complex 1 and mixtures of [Rh(dppe)(μ-Cl)]2 and [Rh(dppe)2]Cl (Scheme 3A).24 Given the preparative route used to access 2: reaction of 1 with 0.5 equiv. [Rh(nbd)(μ-Cl)]2, one might conclude that ring-opening or -closing (to return 1) is apparently dictated by group 9 metal ligand type i.e., Rh(diolefin) vs. Rh(diphosphine). This reaction is accelerated by the addition of excess dppe causing the formation of tucked-in complex 1 and [Rh(dppe)2]Cl, which precipitates from solution (Scheme 3A) – possibly via the unobserved intermediacy of five-coordinate complex 6. Examples of this elementary transformation, donor-induced transmetalation, where M = metal:
| [M1] − CR3 + [M2]+ + PR3 → {[M1] − PR3}+ + [M2] − CR3 |
To assess the favourability of this reaction the dissociation of 5 was modelled computationally. This reaction was found to be roughly thermoneutral (ΔG° = −0.3 kcal mol−1) having a positive value of ΔH° = 16.8 kcal mol−1 (see ESI†).
To explore the relationship between Fe–C bond strain and transmetalation outcome, the reactivity of the unstrained model, [Cp*FeII(dnppe)(CH3)] with [Rh(μ-Cl)(nbd)]2 was also tested. In this case, Fe–C(sp3) bond cleavage results to give [Cp*FeII(dnppe)(Cl)] by 31P{1H} NMR spectroscopy. By 1H NMR spectroscopy, the related Rh(I)–CH3 compound [Rh(nbd)(μ-CH3)]2 is not observed. However, the observation of CH4 is consistent with its implied intermediacy. Indeed, related reactions of [Rh(diolefin)(μ-Cl)]2 and CH3Li, to generate bridging alkyl Rh complexes, were reported as early as 1987 by Andersen and Muetterties.25,26 Unlike the Rh(I) alkyl diolefin compounds 2 and 3 presented here, [Rh(COD)(μ-CH3)]2 requires cryogenic preparation and storage. The authors note that the decomposition of this compound occurs at temperatures as low as 0 °C via elimination of CH4. Relatedly, they report that efforts to synthesize the nbd analogue, “[Rh(nbd)(μ-CH3)]2”, result in product decomposition via elimination of CH4 at −30 °C, thwarting isolation.
Expanding our study to the 5d congener Ir(I), treatment of 4 with dppe was pursued (Scheme 4A). For this reaction, however, Ir-COD coordination was maintained, as evidenced by a multiplet in the 1H NMR spectrum at δH = 3.48 ppm (4H) (and the absence of free COD). Moreover, the observation of two phosphorus resonances at δP = 79.8 and 27.1 ppm of integration 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 suggests the formation of a terminal [(η5-C5Me4-CH2-{Ir(dppe)(COD)})FeII(dnppe)(Cl)] (7) (Scheme 4A) cf., bridging chloride, where in the case of the latter, three distinct 31P NMR resonances would be expected due to local Cs-symmetry (as seen for 5). Speaking to COD lability, single-crystal X-ray diffraction analysis of 7 revealed the μ-Cl analogue of 5 – [(η5-C5Me4-CH2-{Ir(dppe)})FeII(dnppe)(μ-Cl)] (8). Chloride interaction (bridging or terminal) in 7, however, does not dictate group 9 metal loss, which generates the five-coordinate [Ir(dppe)(COD)(Cl)] (δP = 34.4 ppm) complex (along with 1) over time in solution. Consistent with the forward direction of this process, this reaction is irreversible – treatment of 1 with [Ir(dppe)(COD)(Cl)] does not result in 7.
:2 suggests the formation of a terminal [(η5-C5Me4-CH2-{Ir(dppe)(COD)})FeII(dnppe)(Cl)] (7) (Scheme 4A) cf., bridging chloride, where in the case of the latter, three distinct 31P NMR resonances would be expected due to local Cs-symmetry (as seen for 5). Speaking to COD lability, single-crystal X-ray diffraction analysis of 7 revealed the μ-Cl analogue of 5 – [(η5-C5Me4-CH2-{Ir(dppe)})FeII(dnppe)(μ-Cl)] (8). Chloride interaction (bridging or terminal) in 7, however, does not dictate group 9 metal loss, which generates the five-coordinate [Ir(dppe)(COD)(Cl)] (δP = 34.4 ppm) complex (along with 1) over time in solution. Consistent with the forward direction of this process, this reaction is irreversible – treatment of 1 with [Ir(dppe)(COD)(Cl)] does not result in 7.
 | ||
| Scheme 4 (A) Generation of complex 7 and onwards Ir-to-Fe–C(sp3) bond exchange. (B) An unstrained model undergoes clean transmetalation. | ||
As an intra- vs. intermolecular point of comparison, reactivity of the unstrained model, [Cp*FeII(dnppe)(CH3)] with [Ir(dppe)(COD)(Cl)] was also tested (Scheme 4B). In this case, the Fe–CH3 complex underwent clean methyl transfer to give [Ir(dppe)(COD)(CH3)]27 and [Cp*FeII(dnppe)(Cl)] as the only Fe-containing by-product. For the unstrained analogue, the intermolecular transfer of Fe–CH3 to Ir–Cl speaks to the drive for Ir–CH3 and Fe–Cl bond formation. For 5 and 7, however, this driving force is offset by the stability of 1, providing a reversal in the predicted outcome. This general reactivity trend additionally contrasts with that noted for the CpFe complex, [CpFe(CO)2I] (Cp = C5H5−) and Au–C bonds (another 5d-element), which results in Fe–C bond formation.28
A family of heterometallic Fe/M (M = Rh or Ir) complexes have been systematically prepared via an elementary transmetalation reaction between a strained Cp*Fe tucked-in complex 1 and group 9 diolefin salts. This work establishes routes for the systematic generation of such a compositionally distinct class of Cp*{Fe,M} compound and furthers our understanding of the factors that contribute to metal-to-metal transmetalation, an elementary reaction with direct implications for carbon-element bond formation using Fe.
The authors are grateful to Western University, the Council of Ontario Universities for a John C. Polanyi award to M. W. D., the Canadian Foundation for Innovation (LOF-212442), and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant, RGPIN-2020-04480 (M. W. D.), Discovery Launch Supplement, DGECR-2020-00183), and graduate award (CGS-D/NSERC Vanier to J. A. Z.) for funding. M. H. thanks the Inorganic Chemistry Exchange (ICE) program for the opportunity to conduct research at Western University.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2–5 and 8 have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2380939–2380943†).Conflicts of interest
There are no conflicts to declare.References
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- X. Chen, et al. , Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, et al. , Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- A. Biffis, et al. , Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- X. Yan and C. Xi, Coord. Chem. Rev., 2017, 350, 275–284 CrossRef CAS.
- S. C. Rasmussen, ChemTexts, 2021, 7, 1 CrossRef CAS.
- D. V. Partyka, Chem. Rev., 2011, 111, 1529–1595 CrossRef CAS PubMed.
- C. He, et al. , Angew. Chem., 2013, 125, 1567–1570 CrossRef.
- A. A. Thomas, et al. , J. Am. Chem. Soc., 2017, 139, 3805–3821 CrossRef CAS PubMed.
- A. A. Thomas and S. E. Denmark, Science, 2016, 352, 329–332 CrossRef CAS PubMed.
- M. J. Demchuk, et al. , Chem. Commun., 2022, 58, 68–71 RSC.
- M. L. Neidig, et al. , Acc. Chem. Res., 2019, 52, 140–150 CrossRef CAS PubMed.
- H. Kurosawa and A. Yamamoto, Fundamentals of Molecular Catalysis, Elsevier Science, Amsterdam, Netherlands, 2003 Search PubMed.
- J. A. Zurakowski and M. W. Drover, Chem. Commun., 2023, 59, 11349–11352 RSC.
- J. A. Zurakowski, et al. , Chem. Sci., 2024, 15, 10359–10365 RSC.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- (a) P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS; (b) T. Ostwald, et al. , Organometallics, 2019, 38, 829–843 CrossRef; (c) J. Pinkas, et al. , Dalton Trans., 2022, 51, 10198–10215 RSC.
- J. A. Zurakowski, et al. , Inorg. Chem., 2023, 62, 7053–7060 CrossRef CAS PubMed.
- S. Liu and G. S. Girolami, Organometallics, 2021, 40, 714–724 CrossRef CAS.
- T. Shima and Z. Hou, Organometallics, 2009, 28, 2244–2252 CrossRef CAS.
- A. Meißner, et al. , ChemPlusChem, 2015, 80, 169–180 CrossRef.
- M. A. Kulzick, et al. , J. Organomet. Chem., 1987, 336, 221–236 CrossRef CAS.
- M. A. Kulzick, et al. , J. Organomet. Chem., 1987, 333, 105–118 CrossRef CAS.
- M. R. Churchill and S. A. Bezman, Inorg. Chem., 1973, 12, 531–536 CrossRef CAS.
- A. S. K. Hashmi and L. Molinari, Organometallics, 2011, 30, 3457–3460 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, and computational methods. CCDC 2380939–2380943. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06176d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |