
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative neuropeptide analysis by mass spectrometry: advancing methodologies for biological discovery
Angel Erbey
Ibarra
 a,
Wenxin
Wu
a,
Haoran
Zhang
a and
Lingjun
Li
*abcd
a,
Wenxin
Wu
a,
Haoran
Zhang
a and
Lingjun
Li
*abcd
aDepartment of Chemistry, University of Wisconsin–Madison, Madison, WI 53706, USA. E-mail: lingjun.li@wisc.edu
bSchool of Pharmacy, University of Wisconsin–Madison, 777 Highland Ave, Madison, WI 53705, USA. Fax: +1-608-262-5345; Tel: +1-608-265-8491
cLachman Institute for Pharmaceutical Development, School of Pharmacy, University of Wisconsin–Madison, 777 Highland Ave, Madison, WI 53705, USA
dWisconsin Center for NanoBioSystems, School of Pharmacy, University of Wisconsin–Madison, 777 Highland Ave, Madison, WI 53705, USA
First published on 12th June 2025
Abstract
Neuropeptides are critical endogenous signaling molecules involved in a wide range of biological processes, including neurotransmission, hormonal regulation, immune responses, and stress management. Despite their importance, the field of neuropeptide research has been historically hampered by significant technical challenges. These include their low abundance in biological systems, diverse and complex post-translational modifications, dynamic expression patterns, and susceptibility to degradation. As such, traditional proteomics approaches often fall short of accurately characterizing neuropeptides, underscoring the need for specialized methodologies to unlock their biological and translational potential. This review evaluates state-of-the-art quantitative mass spectrometry (MS)-based peptidomics, emphasizing their impact on neuropeptide analysis. We highlight how strategies in label-free and label-based quantitation, tandem MS acquisition, and mass spectrometry imaging provide unprecedented sensitivity and throughput for capturing the landscape of neuropeptides and their modifications. Importantly, the review bridges technological innovation with practical applications, highlighting how these approaches have been utilized to uncover novel neuropeptides and elucidate their roles in systems biology and disease pathways.
 Angel Erbey Ibarra | Angel Erbey Ibarra received his Bachelor of Arts degree in Chemistry and American Studies from Williams College. His undergraduate thesis research focused on characterizing the substrate specificities of phosphopantetheinyl transferases under the mentorship of Dr Amy Gehring. He then joined the doctoral program in the Department of Chemistry at the University of Wisconsin–Madison, where he utilizes powerful instrumentation, chemical biology tools, and bioinformatics to advance neuroscience in the Lingjun Li research group. His graduate work develops mass spectrometry-based tools to characterize neuropeptidergic signaling and glycoconjugate identification and characterization. |
 Wenxin Wu | Wenxin Wu received her Bachelor of Arts degree in Biochemistry and Molecular Biology from Boston University, where she began her research in targeted protein engineering under the mentorship of Dr Pinghua Liu. Her work focused on mechanistic enzymology and biocatalysis. She then joined the research group of Professor Lingjun Li at the University of Wisconsin–Madison, where she is currently pursuing her PhD in Chemistry. Her graduate work centers on the development of advanced high throughput mass spectrometry methods, including DDA-MS, DIA-MS and MALDI imaging, to explore neuropeptidomics, glycoproteomics, and small molecule analysis. |
 Haoran Zhang | Haoran Zhang received his Bachelor of Science degree in Chemistry from the University of Science and Technology of China, where he conducted research on nanomaterials and carbon-based aerogels under the mentorship of Professor Shuhong Yu. He went on to earn a Master of Engineering in Materials Science at the University of Wisconsin–Madison, where he deepened his expertise in nanotechnology and biomaterials. Building on this interdisciplinary foundation, Haoran joined the research group of Professor Lingjun Li in the Department of Chemistry at the University of Wisconsin–Madison, where he is currently pursuing his PhD. His research focuses on mass spectrometry-based multi-omics and the development of sustainable carbon nanofiber substrates for advanced imaging applications and enrichment strategies for biomarker discovery in Alzheimer's disease. |
 Lingjun Li | Dr Lingjun Li is a Vilas Distinguished Achievement Professor and the Charles Melbourne Johnson Distinguished Chair Professor of Pharmaceutical Sciences and Chemistry at the University of Wisconsin–Madison (UW-Madison). Dr Li received her PhD degree in Analytical Chemistry/Biomolecular Chemistry from the University of Illinois at Urbana-Champaign in 2000. She then did joint postdoctoral research at the Pacific Northwest National Laboratory and Brandeis University before joining the faculty at UW-Madison in December 2002. Dr Li's research interests include the development of novel mass spectrometry (MS)-based tools such as new isotopic and isobaric labeling strategies that enable hyperplexing for quantitative proteomics, peptidomics, and glycomics, and their applications in neuroscience and cancer research. She and her team also develop microscale separations, in vivo microdialysis and imaging MS tools for functional discovery of neuropeptides in model organisms and (glyco)protein biomarkers in neurodegenerative diseases with a strong focus on Alzheimer's disease. Her lab also explores novel use of ion mobility MS to address technical challenges in peptidomic research. Professor Li has established a highly productive research program and published more than 400 peer-reviewed research journal papers and has given more than 300 invited talks. She has successfully trained and graduated 71 PhDs and is currently training 27 PhD graduate students, 4 postdoctoral scientists, and 7 undergraduate students. She has been recognized with numerous awards, including ASMS Research Award, NSF CAREER Award, Sloan Fellowship, PittCon Achievement Award, and ASMS Biemann Medal, and was included on the Analytical Scientist Power List for 4 times (2016, 2019, 2021, 2024). Dr Li is currently serving as an Associate Editor for the Journal of the American Society for Mass Spectrometry (JASMS) and sitting on the Advisory Board for Mass Spectrometry Reviews and Analytical and Bioanalytical Chemistry. She served on the Board of Directors for the US Human Proteome Organization (US HUPO) 2015–2021 and currently chairs the Board of Directors for the Chinese American Society for Mass Spectrometry (CASMS). |
Introduction
Neuropeptides are essential regulators for numerous cellular and physiological processes, acting as key neurotransmitters and neuromodulators. They are synthesized in neurons primarily through mRNA translation on ribosomes and undergo extensive post-translational processing, including proteolytic cleavage from larger precursor proteins.1 These bioactive peptides, typically composed of 3 to 50 amino acids, range in molecular weight from 200 Da to 10 kDa. Their amino acid sequence and composition largely determine their function, while post-translational modifications (PTMs) further refine their physicochemical properties, influencing their solubility, receptor binding, and stability.2–4 This enables neuropeptides to participate in diverse neuromodulatory functions beyond being mere byproducts of protein degradation. Neuropeptides play critical roles in synaptic transmission, neuroinflammation, brain development, stress responses, pain perception, appetite regulation, and psychiatric behavior.5–11 Some also exhibit antimicrobial properties, contributing to neuroimmune interactions that help regulate inflammation and host defense mechanisms.12,13 The regulation of neuropeptide biosynthesis, processing, and secretion is critical for maintaining neuronal homeostasis, as dysregulation can lead to neuropsychiatric disorders, neurodegenerative diseases, and other pathological conditions.The interdependent functions of many neuropeptides collectively regulate diverse physiological and neural processes, making studies that focus on a single neuropeptide inadequate for capturing their full scope of influence. Neuropeptide co-transmission, along with classical neurotransmitters, further complicates the isolation of their specific contributions to neuronal signaling, while their rapid degradation, low in vivo concentrations, and diffusion in the extracellular space present additional challenges for detection and analysis.14,15 Neuropeptide analysis by mass spectrometry (MS)-based techniques, termed neuropeptidomics, has supplanted classical methods such as radioimmunoassays and Edman degradation due to its ability to handle complex samples while providing detailed chemical information.16 Therefore, MS-based techniques have become the premier method for neuropeptide analysis by providing invaluable information on peptide sequence, relative abundance, PTM characterization, and spatially resolved localization.
Neuropeptidomics involves the direct analysis of endogenous peptides without relying on enzymatic digestion and does not require prior knowledge of peptides present in a sample.17,18 These peptidomic methods stem from a branch of proteomics that similarly aims to characterize protein compositions. So-called “bottom-up” proteomics is based on the controlled proteolysis of proteins into sets of cleaved peptide fragments followed by liquid chromatography-tandem mass spectrometry analysis (LC-MS/MS).19–22 However, several neuropeptides are endogenously produced fragments derived from larger prohormones with more variable peptide lengths than tryptic fragments. Since trypsin digestion produces a highly complex mixture of peptides, these may mask observations of some other low-abundance short peptides.23 By forgoing the use of enzymatic proteolysis, native peptide sequences are preserved, allowing for the identification of physiological cleavage patterns without introducing artificial peptide fragments. This enables the detection of low-abundance short neuropeptides in their naturally processed, cleaved form.
Electrospray ionization (ESI) is a soft ionization technique that facilitates the transition into the gas phase with minimal degradation and is readily coupled to online reversed-phase LC separation, enabling high sensitivity and quantitative capabilities in peptide analysis.24,25 Peptides partially elute according to their size and hydrophobicity and the analyte is continuously sprayed into and measured by the mass analyzer. MS first measures the mass-to-charge ratios (m/z) of intact peptide “parent” ions in the gas phase (MS1). These precursor ions are then selected for further fragmentation (MS2 or MS/MS) to reveal characteristic peptide fragmentation patterns, allowing for peptide characterization and PTM differentiation.
In MS-based peptide analysis, fragment ions are categorized as a/x-, b/y-, and c/z-type, depending on which peptide backbone bond is broken, and peptide sequences are then determined from the MS2 fragment ion series following an initial MS1 selection.26,27 Commonly employed fragmentation strategies, such as high-energy collision-induced dissociation (HCD) and electron transfer dissociation (ETD) methods, produce MS2 spectra that contain critical fragment ion information for identifying peptides.28–30 In proteomics, these spectra are often searched against an in silico-generated library of theoretical spectra obtained from predicted proteins with predictable cleavage sites from enzymatic proteolysis. Tryptic peptide fragments thus serve as proxies for the protein they originated from, and quantification is performed by aggregating peptides into a single value at the protein level.31–33 Whereas bottom-up proteomics attempts to piece together tryptic peptide fragments to inform protein compositions and their isoforms, peptidomics forgoes the use of proteolytic enzymes to directly measure naturally occurring endogenous peptides. Specifically, quantitative peptidomics refers to the systematic characterization and quantification of endogenous peptides present in a biological sample through label-based and label-free methods for relative and absolute quantification.34,35
There is no one-size-fits-all approach for endogenous peptide quantitation by MS-based methods. Label-based quantitative techniques provide increased accuracy and reproducibility with high multiplexing capabilities. In contrast, label-free quantitation can accommodate large sample sets due to straightforward, cost-effective sample preparation and LC-MS/MS analysis. These approaches vary depending on whether the study is discovery-based (untargeted) or if specific peptides (targeted) are being explored. Further, peptides may exert their function based on their PTMs and cellular/tissue localization. Therefore, quantitative MS-based techniques are powerful and multifaceted approaches to probe peptide organization, structure, localization, PTMs, and overall distribution. By bridging technological innovations with practical applications, we aim to demonstrate how these techniques have been utilized to identify novel peptides, unravel their functions in complex systems, and uncover their relevance to clinical medicine.
General considerations for peptide analysis and quantitation
Peptidomic workflows can be divided into four stages: sample preparation, chromatographic separation, mass spectral acquisition, and data processing and analysis (Fig. 1). Each stage introduces variation that can affect quantitation downstream. Careful experimental design is necessary to reduce experimental variation to ensure optimal peptide coverage and quantification, which have been discussed previously for peptidomic and proteomic workflows in detail.17,35,36 Nevertheless, achieving reliable, high-quality quantitation by MS can be complicated due to biological factors underlying neuropeptide biology and challenges inherent to MS.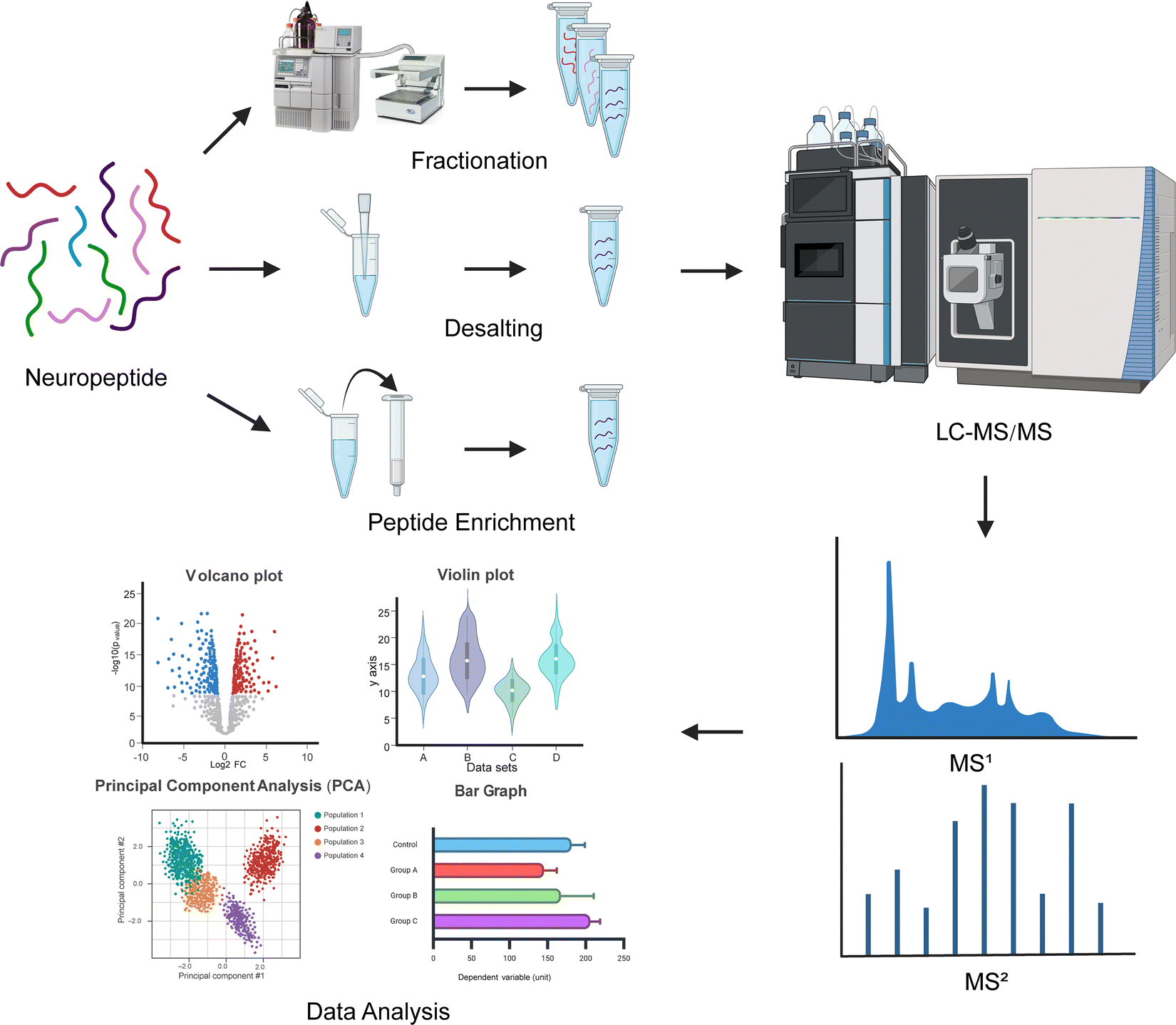 | ||
| Fig. 1 Experimental mass spectrometry-based neuropeptidomic workflow. Peptides are extracted from neural tissue or circulating biofluids and forego the use of enzymatic digestion, as commonly applied to bottom-up proteomic workflows. Neuropeptides are processed and cleaned for downstream LC-MS/MS analysis. MS1 precursor ions are then isolated and fragmented into smaller fragment ions (MS2), where different fragment ions correspond to specific peptide bonds. MS2 spectra are then analyzed via database searching and de novo sequencing for quantitative data analysis of neuropeptides. | ||
Sampling neuropeptides from neural tissue is difficult as these are often expressed in very low in vivo concentrations (pM–nM) and prone to proteolytic processing.37 The high sample complexity of neural tissue may introduce interference from high salt content and lipids. In addition, many neuropeptides require PTMs in order to become bioactive such as C-terminal amidation and N-terminal pyroglutamate formation, which cannot be predicted from genomic information.38 This results in dynamic levels of chemical diversity of neuropeptides that require analytical methods with high resolution and sensitivity.
Preventing degradation must begin at harvest to mitigate enzymatic proteolysis and preserve the in vivo peptide sequence and PTMs. Endogenous bioactive peptides are more susceptible to degradation in biological matrices than proteins, as various proteases and peptidases can readily target them.39–42 Their rapid turnover is essential for timely peptide signaling and regulation in order to maintain homeostasis and prevent prolonged bioactivity. Further, endogenous peptides generally lack tertiary structures and are shorter in length than larger folded proteins, making their cleavage sites accessible to proteolytic enzymes in these biological environments. The proteolytic background in the mass spectra can mask the true peptidome due to peptide artifacts resulting from proteolytic degradation. This susceptibility to degradation poses challenges for accurately detecting and quantifying endogenous peptides in peptidomics studies, as it often necessitates using protease inhibitors and rapid sample processing.42–47 Several techniques have been developed to minimize peptide degradation. These include protease inhibitor cocktails to inhibit a variety of endogenous peptidases and heat stabilization through tissue boiling and microwave irradiation.48–54 The concentration of endogenous peptides also presents a concern in quantitation capabilities. Their sparse cellular localization, tight regulation, and PTMs often result in a wide range of peptide concentrations, with some falling outside the linear dynamic range of high-resolution mass spectrometers.55,56
Although LC-MS/MS approaches are among the most sensitive and selective analytical techniques, matrix effects are a significant issue affecting peptide quantitation.57–59 Biological samples are inherently complex matrices possessing interferences from salts, lipids, abundant proteins, and metabolites. Matrix effects cause alterations in the ionization efficiencies of analytes and differences in LC retention time due to co-eluting compounds from the same sample.59–63 This can lead to signal suppression or enhancement that may result in dramatic differences in analytical performance. Several strategies have been proposed that mitigate matrix effects in quantitative MS-based analysis.57,58 Matrix interferences can be mitigated in peptidomics by employing isotopically labeled internal standards or offline peptide fractionation to reduce interference between peptides, enhance signal-to-noise ratios, and boost peptidome coverage.63–65 Acidified methanol (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:1 (v/v/v) MeOH:H2O:glacial acetic acid) is commonly used for neuropeptide extraction due to its ability to simultaneously precipitate proteins, inhibit protease activity, and solubilize peptides.66 Molecular-weight cutoff (MWCO) devices can help further concentrate neuropeptides while removing abundant, larger proteins and are featured in common neuropeptidomic workflows.67,68
:9:1 (v/v/v) MeOH:H2O:glacial acetic acid) is commonly used for neuropeptide extraction due to its ability to simultaneously precipitate proteins, inhibit protease activity, and solubilize peptides.66 Molecular-weight cutoff (MWCO) devices can help further concentrate neuropeptides while removing abundant, larger proteins and are featured in common neuropeptidomic workflows.67,68
Quantitation techniques
Neuropeptides can be abundantly expressed in tissues or synthesized at trace levels.69 MS-based methods can provide quantitative information on the abundance of expressed neuropeptides due to their broad dynamic range capabilities across several orders of magnitude.70–73 Mass spectrometry is inherently nonquantitative due to several factors that affect the relationship between analyte concentration and the signal intensity following the ionization process.74,75 The ionization efficiency varies significantly between endogenous peptides under similar analytical parameters, depending on their physicochemical properties, including size, charge, and polarity, meaning analytes at the same concentration can produce vastly different signals.76–78 Instrument factors such as variability in optics, ion transmission, and detector sensitivity introduce inconsistencies unrelated to the analyte concentration.79,80 Nonlinear detector response, particularly at high ion counts where signal saturation occurs, reduces reliability for highly abundant analytes.81 Additionally, fragmentation efficiency in tandem MS can vary between precursors, affecting the intensity of peptide fragment ions used for quantitation.82 Matrix effects further complicate quantitation, as other components in the sample can suppress or enhance ionization, skewing signal intensities.83,84 To overcome these challenges, quantitative MS for endogenous peptide analysis relies on techniques like isotopic labeling, internal standards, and carefully constructed quality control metrics to correct for variability and improve accuracy.85–87 Quantitation of endogenous peptides can be categorized as label-based or label-free method performed by either evaluating the area under the curve of the chromatogram (signal intensity) of precursor ions, the signal intensity of diagnostic reporter ions following peptide fragmentation (MS2) or counting the number of MS/MS spectra assigned to a peptide (Fig. 2).88,89 | ||
| Fig. 2 Overview of mass spectrometry-based strategies for neuropeptide quantification. Label-free quantification measures the intensity of peptide ions at the MS1-level over an elution time. The area under the curve correlates with peptide abundance. Stable isotopes can be incorporated into peptides through isotopic labeling or metabolic incorporation (e.g. SILAC). Labeled and unlabeled samples are mixed, and the ratio intensities of labeled to unlabeled peptides are measured. Isobaric labeling incorporates stable isotopes where labeled peptides are indistinguishable at the MS1-level. Upon fragmentation, reporter ions are generated, and their MS2 intensities are used for quantification, enabling multiplexing of samples. Data-dependent acquisition (DDA) and data-independent acquisition (DIA) are commonly employed for neuropeptide analysis, with DDA enabling targeted fragmentation of the most abundant precursor ions for detailed sequencing. DIA systematically fragments all ions within a mass range, providing comprehensive coverage and improved detection of low-abundance neuropeptides. | ||
Label-based quantification
Label-based methods incorporate stable isotopes in order to obtain quantitative ions at the MS1 or MS2 level. Stable isotopes enable accurate quantitation in LC-MS/MS by serving as internal standards that closely mimic the physicochemical behavior of natural peptides. Labeled peptides are chemically identical to their natural counterparts, ensuring they co-elute during chromatography and share similar ionization efficiency, while their distinct mass shift allows for spectral differentiation.90 By comparing the intensity ratio of labeled and unlabeled peptides, relative or absolute quantification is achieved, with calibration curves enabling precise measurements of peptide concentrations.Stable isotopes can be introduced through metabolic, enzymatic, or chemical methods. Metabolic labeling was considered the gold standard for quantitative proteomics as isotopically labeled amino acids can be introduced in vivo and metabolically encoded into the proteome.91–93 This early-stage labeling enhances quantitative accuracy and reduces variability from sample preparation or enzymatic proteolysis that can propagate downstream. SILAC, stable isotope labeling with amino acids in cell culture, is the most popular approach and typically uses 13C or 15N-labeled arginine and lysine in the culture media.93–96 Briefly, two isotopically distinct culture mediums are used, with one containing the light medium of the natural amino acids and the other heavy medium containing the stable isotope-labeled residues. Following a sufficient number of cell incorporations to complete labeling (≥95% labeling efficiency), the two cell populations are then pooled together in equal concentrations before injection into the mass spectrometer. It is demonstrated that SILAC bolsters reproducibility and quantification accuracy due to its ease of implementation and is especially powerful at probing intracellular signaling.97–101 With SILAC, quantitation is determined from the extracted ion chromatograms of peptide precursor ions from both the heavy and light peptides, where small mass differences are distinguishable at the MS1 level. Despite its benefits, SILAC is limited almost exclusively to cell cultures, and attempts at multiplexing may cause overlapping isotope clusters at the MS1 level, which increases spectral complexity. If deuterium is used, this may also alter chromatographic retention times. Nevertheless, though introduced over two decades ago, continuous innovations in SILAC technology such as pulsed SILAC, super SILAC, and spike-in SILAC, among others, are becoming amenable towards spatiotemporal analysis of the dynamic peptidome along with integration of select model organisms.101–104
Chemical and enzymatic labeling are typically done in vitro with samples from biological extracts such as biological fluids and tissue samples, including cell cultures. This type of chemical labeling can be performed with either isotopic or isobaric tags; however, isobaric tagging is more commonly used in quantitative peptidomic workflows and will be the focus of this discussion.105,106 In general, precursor ion-based quantification methods with SILAC or isotopic tags suffer from isotopic envelope interference, thus requiring a mass difference of >4 Da.107,108 Isotopic envelope interference occurs when overlapping signals from isotopic variants of different peptides distort the intensity of a target precursor ion, leading to inaccurate quantification. This is particularly problematic in complex samples where isotopic peaks from multiple peptides overlap in the same m/z range.109
Isobaric tags overcome challenges faced in MS1-based quantitation rising from isotope-induced spectral complexity by shifting quantitation to the MS2 level.82 These labels incorporate isotopically encoded tags with the same nominal mass addition and structure that generate reporter ions of differing masses upon MS2 fragmentation. Regardless of isotopic distribution along the tag, labeled peptides co-elute as a single composite peak during the precursor scan with a modest increase in spectra complexity at the MS2-level. This enables increased multiplexing capabilities as the MS1 spectral complexity does not increase with the number of samples.110
Isobaric tags such as tandem mass tags (TMT), isobaric tags for absolute and relative quantification (iTRAQ), and N,N-dimethyl leucine (DiLeu) follow a common structure motif consisting of a reactive group, a balancing group, and a reporter ion group (Fig. 3).111–113 The reactive group selectively reacts with specific functional groups or residues on peptides, typically targeting amine groups located on the N-terminus and the ε-amine in lysine. The balancing group functions as a mass normalization via encoded isotopes. The reporter ion group contains discrete isotopes for different ion channels to generate diagnostic ions with different masses for quantitation. This unique structural configuration allows for the synthesis of significantly higher-order multiplexed tags. For instance, whereas SILAC-based quantitation can modestly achieve up to 4-plex multiplexing, isobaric tags can readily achieve up to 8-plex with iTRAQ, 21-plex with DiLeu, or 35-plex with TMTpro.101,114–118 These amine-reactive tags, however, require at least one primary amine; therefore, endogenous peptides with PTMs altering the amino groups or those lacking a primary amine will require appropriate adjustments, such as using cysteine-reactive tags.119–121 Further, bolstering the multiplexing capacity of these tags can lead to decreased quantitative accuracy in the MS2 scan due to chemical noise, signal interference, false positives, and ratio distortion.110,122–124 Analytes with similar m/z values to the tagged peptide can be co-isolated during the initial MS1 scan; the isobaric reporter ion signal can be masked by the contaminating peptide, leading to distorted quantitation. Nevertheless, sample multiplexing enabled by isobaric tags at the MS2 level drastically reduces sample analysis time and significantly boosts throughput capacities without compromising the high-quality quantitation of endogenous peptides. Lastly, the additional steps in sample preparation and high-cost reagents are a significant limitation of label-based approaches, deterring the widespread use and adoption of these techniques when sampling low-abundance, precious peptides.
 | ||
| Fig. 3 Chemical structures of common isobaric tags used in quantitative peptidomics. Isobaric tags follow a common structure motif consisting of an amine-reactive group (green), a mass balancing group (black), and a reporter ion group (blue). (A) Tandem mass tags (TMT), (B) isobaric tag for relative and absolute quantification (iTRAQ), and (C) N,N-dimethyl leucine. | ||
Label-free quantification
Label-free quantification (LFQ) forgoes the use of stable-isotope labels, providing several experimental advantages, including large-scale cohort analysis, inexpensive and cleaner sample processing, and amenability to any sample matrix.125,126 LFQ analysis is performed by aligning multiple data sets using m/z and LC retention times and correlating peptide abundance to the number of MS/MS spectra or precursor signal intensities.127,128 A relatively straightforward method for LFQ analysis involves integrating the chromatographic peak areas for a given peptide at the MS1-level (MS1-AUC).129 The area under the curve of the extracted ion chromatogram (XIC), which measures the signal intensity of a peptide as it elutes from a chromatographic column, is observed to be linearly related to its concentration across very broad dynamic ranges.129,130 Therefore, by comparing the peak areas between samples, relative quantification of peptides can be achieved. Conversely, spectrum count approaches infer peptide quantity indirectly from the total number of MS/MS spectra acquired.127,131 The spectral counts of peptides are reproducibly observed to be linearly correlated with protein abundance over a linear dynamic range of over 2 orders of magnitude.127,132 For both strategies, high-quality LFQ necessitates narrow LC peak widths to enhance the signal-to-noise ratios of peptide ions while extending the dynamic range of peptide quantitation to account for low-abundance ions. While isotopic labeling methods can accommodate reasonably sized sample cohorts, the number of samples that can be quantitatively assayed is limited to the multiplexing capacity of the chemical tag. When combined with costly tagging reagents, LFQ methods offer flexible sample preparation steps, rendering them attractive options for large-scale analyses.133,134Common challenges in LFQ analysis stem from limitations of LC-MS/MS, referred to as batch effects, which are the systematic differences in measurements caused by technical factors.135 For example, run-to-run variability in peptide elution time differences due to difficulties obtaining stable chromatography complicates the alignment of RT and m/z values.136 Multiple sample injections into the same LC column can lead to chromatographic shifts where, even within the same sample, peptides may exhibit different responses across injections, resulting in variations in peak intensity and elution between runs.137 This fluctuation in reproducibility often compromises accuracy and precision. Isobaric peptides with higher precursor intensities can co-elute, making it difficult to identify, much less quantify, these peptides. Various normalization strategies have been proposed to mitigate systemic bias, employing novel algorithms and benchmarks to account for the variation introduced by batch and matrix effects.138–141
Data acquisition methods
When considering ways to achieve high-quality LC-MS/MS quantitation, the type of study must be delineated. Peptidomics research can be categorized into two groups: discovery-based (untargeted) and targeted. Discovery-based platforms aim to maximize overall peptide coverage and identification by exploring “all” peptides across a broad dynamic range – known, novel, or unexpected. These untargeted approaches ultimately generate hypotheses and focus on peptide screening. Here, only relative quantification can be achieved, yet this remains a powerful method for biomarker discovery and understanding cellular mechanisms in disease.142–144 Targeted platforms are hypothesis-driven and limit peptide coverage to fully exploit the analytical capabilities of high-resolution MS. Selected or multiple reaction monitoring (SRM) and parallel reaction monitoring (PRM) are employed as they enhance quantitative capacity by boosting sensitivity and reproducibility with rapid detection due to a predefined set of peptides.145–147Following LC separation, the MS detects the signal intensities of the ionized peptides and quickly acquires mass spectra. Two broad approaches to generating tandem MS spectra exist: data-dependent acquisition (DDA) and data-independent acquisition (DIA). DDA is the conventional approach for quantitative peptidomics; however, rapid advancements in DIA have also demonstrated robust and reproducible peptide quantitation.148 DDA selects the most abundant precursor ions, top N, for MS2 fragmentation. Recently fragmented precursor ions are excluded to enhance peptide coverage for low-abundance ions and prevent redundant fragmentation.149 However, DDA is inherently stochastic as ion selection in complex mixtures is nontrivial due to interfering artifacts obstructing peptide MS1 selection, MS scanning speeds, exclusion efficiency, and separation resolution.127 Yet, since only the top N precursors are selected, this typically results in MS/MS spectra with numerous data points, allowing for sensitive and accurate quantitation. Given the stochastic nature of DDA, run-to-run variability, and inherently low abundance of endogenous peptides, this may result in missing peptides lacking sufficient data points for sensitive and accurate quantification. On the other hand, DIA overcomes the stochastic nature and bias selection of DDA wherein MS/MS spectra are collected systemically and independently of precursor ion information.150,151 In DIA mode, the instrument accumulates and fragments all precursor ions within a user-defined m/z isolation window and proceeds iteratively, typically within a mass range of m/z 400–1200.152–154 In addition to its increased specificity and greater reproducibility, DIA can detect low-abundance peptides owing to its ability to analyze the whole mass range. Since biologically active endogenous peptides are typically present in the low mass range and may not produce enough of a response to surpass the signal threshold in DDA, DIA is ideally suited for global discovery-based experiments for increased coverage and relative quantification.88 For instance, DIA has made major advancements in large-scale de novo sequencing of neuropeptides possible with improved neuroptidome coverage.155,156 These have detected nearly twice as many neuropeptides than traditional data-dependent acquisition (DDA) techniques, many of which were detected for the first time.157–159 However, because DIA methods generate significantly more data compared to DDA and all MS1 precursor ions are selected for fragmentation, advanced deconvolution, and processing tools are necessary to reduce spectral complexity.160
In bottom-up proteomics approaches, which generate various peptide fragments from protein precursors, the sum of all peptide-spectrum matches (PSMs) derived from a protein can be used to quantify protein abundance.161 It is assumed that in DDA mode, the more abundant peptides selected for fragmentation produce a larger number of MS/MS spectra. Quantitative DDA approaches may face substantial missing data and values, a challenge that is amplified with larger sample sets.148,162 Therefore, accurate and sensitive quantification by spectral count requires large numbers of tandem spectra, whereby abundant peptides result in more PSM data points than less abundant proteins and may result in detector saturation, compromising quantitative accuracy.163 MS1-AUC quantifies peptides by leveraging precursor ion signals, making it suitable for peptides that may not generate sufficient fragmentation events. This approach enhances the accurate quantification of low-abundance peptides by integrating their signal across the entire chromatographic elution profile. As a result, peptidomics LFQ approaches commonly employ precursor ion-based integration strategies to capture the full elution profile of endogenous peptides.164,165
DDA and DIA are routinely used in untargeted discovery peptidomics as both acquisition modes aim to maximize the number of peptides that can be identified and quantified. Typically, discovery-based experiments aim to screen peptides across a broad dynamic range while measuring their relative abundance across multiple samples.166 By profiling the differential abundance of peptides between a control and experimental group, insights into how peptide regulation is altered on a global, comprehensive scale can be achieved.142,167 This hypothesis-generating approach is, therefore, suitable not only for biomarker discovery and validation but also for naturally allowing investigators to quantify unique peptide differences for downstream targeted analysis of their involvement in signaling pathways. Further, tuning acquisition parameters for low-abundance peptide ions has been demonstrated to boost their identification and quantity confidence.156,168
Although DDA and DIA are instrumental in discovery-based peptidomics for probing the global peptide landscape, targeted acquisition methods like MRM and PRM provide the precision needed for quantifying specific peptides with greater sensitivity and reproducibility. MRM leverages the mass filtering capabilities of a triple quadrupole mass spectrometer where specific precursor ions are selected and fragmented with transitions (precursor-to-fragment ion pairs) monitored for quantification.169,170 This approach is highly selective but requires detailed prior knowledge of target peptides, often requiring significant assay development. PRM, derived from MRM, uses high-resolution hybrid quadrupole-orbitraps or time-of-flight instrumentations to collect full MS/MS spectra for the targeted precursor ions, providing enhanced specificity by monitoring all fragment ions simultaneously.171
While relative quantification methods are commonly used to compare peptide abundances across samples and depend on the fold changes in peptide expression, there is a need to provide precise absolute concentration measurements of peptides. Absolute quantification, commonly referred to as AQUA, relies on spiking a known concentration of synthetic stable isotope-labeled peptide internal standards (SIL-IS) into samples.172 When combined with SRM or PRM, AQUA enables precise quantification of target peptides in complex matrices by detecting both the native peptide and SIL-IS. This technique is commonly applied for bottom-up proteomics, where quantitative measurements occur at the peptide level from proteolytic peptide fragments.173 This approach eliminates variability associated with inter-sample comparisons, making it particularly valuable for applications requiring high accuracy, such as biomarker validation and allergen quantification. Ultimately, while targeted methods bring substantial rigor to quantitation, they demand a high level of specificity in peptidomics due to the diversity of native peptides being studied.
Post-translational modifications
Post-translational modifications (PTMs) are covalent processes where various modifying groups are added to or replace the side chain of one or more amino acids within a peptide sequence. Common PTMs for endogenous peptides include but are not limited to glycosylation, phosphorylation, citrullination, C-terminal amidation, N-terminal acetylation, and pyroglutamation on glutamic acid and glutamine.174 PTMs function as critical mediators for cellular signaling by influencing peptides’ and proteins’ function, activity, stability, and interaction with other biomolecules. Often, endogenous peptides only become bioactive once modified, which allows them to dynamically regulate cellular processes, including hormonal signaling, immune response, and neuromodulation.11,175–178 As such, PTM processing of endogenous peptides opens avenues for targeted therapeutic intervention.16,179Mass spectrometry-based characterization of modified peptides accounts for diagnostic features imparted by PTMs, including mass shifts, fragmentation patterns, and chromatographic retention.180–182 For untargeted profiling, most software for peptide identification offer variable PTM and fixed PTM searching abilities, allowing researchers to identify the differentially modified peptides from the sample.183–185 Generally, PTMs affect the peptides' overall chemical and physical characteristics, which can be exploited for their specific enrichment and purification.186 Glycosylation, for example, involves the addition of carbohydrates onto certain amino acid residues, which increases peptide hydrophilicity.187 Hydrophilic interaction liquid chromatography (HILIC) can relatively enrich the hydrophilic glycopeptides from the unmodified hydrophobic peptides. After enrichment, the interfering spectral background signal is lower, resulting in cleaner spectra and a higher signal-to-noise ratio for the glycopeptides.188 Cui et al. have similarly employed electrostatic repulsion hydrophilic interaction chromatography (ERLIC) to achieve simultaneous enrichment of both glycosylated and phosphorylated peptides from the MM.1S cell lysate to elucidate PTM crosstalk.189 Citrullination, the deiminiation of arginine, imparts a very small shift (+0.984 Da) which can be readily masked by deamidation (+0.894 Da) or 13C isotopic peaks (+1.0033 Da).190 To enhance their detection, Shi et al. developed a biotin thiol tag-assisted MS method to enrich and derivatize the citrullinated peptides, discovering 691 citrullination sites from 432 proteins.191
Different fragmentation techniques can also characterize modified peptides and determine PTM site localization. Collision-induced dissociation (CID) and high-energy collisional dissociation (HCD) induce fragmentation by colliding peptide ions with inert gas molecules (e.g., nitrogen or helium) to predominantly cleave amide bonds along the peptide backbone (C–N), generating sequence-informative b- and y-ions that are ideally suited for de novo sequencing and identification.192,193 Electron transfer dissociation (ETD) involves transferring an electron to multiply charged peptide ions, resulting in sequence-independent radical-driven fragmentation along the NH–Cα to generate c- and z-ions.194,195 This type of fragmentation can better retain intact labile moieties such as glycans and phosphates than HCD.196,197 Both dissociation methods are complementary and can generate a wealth of sequence-specific information in the resulting tandem MS spectra.
For specific PTMs of interest, instead of an untargeted approach, a targeted approach combining both dissociation methods can be used for characterization and quantification. Among these, electron transfer/high-energy collision dissociation (EThcD) is a hybrid technique that produces a mixture of c/z and b/y ions for detailed annotation of peptides and their PTMs, especially for glycopeptides.198,199 For instance, Yu et al. have used a known list of oxonium ions to selectively trigger EThCD for glycopeptides and achieved the first global profile of O-glycopeptides in both mouse and human pancreatic islets, discovering that signaling peptides such as insulin and BigLEN are glycosylated.200 Riley et al. subsequently determined that HCD is the preferred method for N-glycopeptide analysis, whereas EThcD fragmentation is indispensable for site-specific O-glycopeptide analysis.201 Using these techniques, glycosylated neuropeptides and peptide hormones have been discovered in the nervous system of mammals and crustaceans.202,203 Recently, D-amino acid-containing peptides (DAACPs) have been discovered as unusual neuropeptide PTMs across different phyla, including chordates, arthropods, and mollusks.204 Unlike other PTMs, peptide L/D isomerization does not induce a mass shift readily discernable by traditional neuropeptidomic approaches, thus requiring extensive sample processing for their characterization and functional assays to test their physiological and behavioral implications.205–208 However, ion mobility-mass spectrometry (IM-MS) uniquely resolves isomeric species through gas-phase separation based on peptide ions’ mass, charge, and collisional cross-section values.209,210 In particular, site-specific identification of DAACPs can be achieved first by online reversed phase chromatography followed by ion mobility analysis of epimeric fragment ions.211 This pioneering strategy has since been adopted to screen for DAACPs in the central nervous system of both vertebrates and invertebrates.212–216
In quantitative bottom-up proteomics, it is common to aggregate peptide fragments into a single quantitative value for each protein-coding gene. On the other hand, in neuropeptidomics, quantitation techniques must be adapted to account for the diverse and often modified nature of endogenous peptides, which differ from the predictable tryptic peptides typically analyzed in bottom-up proteomics. Unlike tryptic peptides, which result from enzymatic digestion and are typically unmodified and within a narrow mass range, endogenous peptides can have a broader range of PTMs, non-tryptic cleavages, and variable lengths. The in vivo abundance of endogenous peptides and their modified counterparts can vary significantly, with many existing at very low levels, thus requiring careful optimization of instrument settings to minimize modified peptide signal suppression.159 Specifically, SRM and PRM in peptidomics require tailored transitions that account for these unique features, often necessitating extensive optimization of precursor and fragment ion selections for each peptide target.217,218
Mass spectrometry imaging
Mass spectrometry imaging (MSI) leverages the specificity and sensitivity of mass spectrometric techniques to map and image the spatial distributions of diverse biomolecules directly.219 Unlike other imaging techniques, such as immunohistochemistry (IHC), fluorescent tags, or radio-immunoassays, MSI requires no prior knowledge of the targeted class of molecules, and thousands of molecules can be imaged in a single MSI experiment, covering a mass range from 1 m/z to as high as 120000 m/z.220 Sample preparation is relatively simple, making them amenable to thin tissue sections. MSI of proteins was first introduced by Caprioli et al. in 1997 by utilizing matrix-assisted laser desorption/ionization (MALDI) technique, and it allows spatial mapping of all classes of molecules, including lipids, metabolites, peptides, and intact proteins or protein complexes.221 MALDI is another soft ionization routinely used for MSI experiments as it is much more tolerant of impurities while enabling fast data acquisition.222Fig. 4 shows an example of a typical MALDI-MSI workflow. Aside from MALDI, other ionization techniques for MSI have been employed, such as desorption/ionization (DESI), atmospheric pressure MALDI, surface-assisted laser desorption/ionization, and secondary ion MS (SIMS).223–226
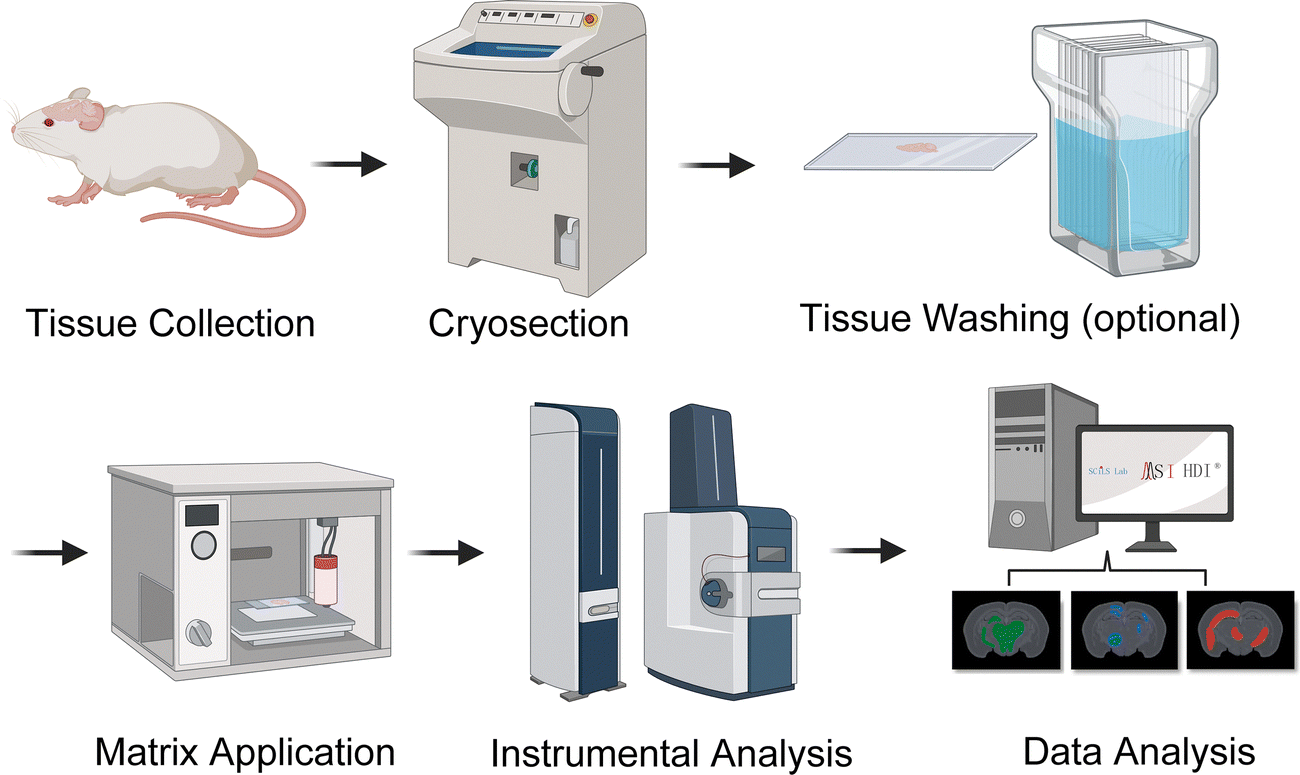 | ||
| Fig. 4 General overview of mass spectrometry imaging (MSI) workflows. Thin tissue sections are mounted on a plate and tissue washing can remove interfering ion signals. The tissue slices are then coated with or without a matrix, depending on the ionization technique employed. The produced ions are detected by a mass spectrometer, generating mass spectra at each spatial coordinate. MSI constructs mass spectrometric images corresponding to specific molecular weight values. This allows for the simultaneous imaging of many different molecules from a single tissue section. | ||
Before imaging, tissue sections must be properly prepared for the analyte of interest. Upon dissection, tissues need to be flash frozen or embedded into imaging matrices to preserve the tissue orientation and structure and prevent molecule degradation. Common embedding matrices include gelatin, carboxymethyl cellulose (CMC), paraffin, and optimal cutting temperature (OCT) compounds.227–229 Although paraffin and OCT compounds are common embedding reagents, both matrices are known to suppress analyte signals and need to be properly cleaned before MSI.229,230 Although tissue washing can remove interfering signals, it may also lead to diffusion, negatively affecting the analytes’ native localization. In these instances, fresh frozen tissue or gelatin and CMC embedding are recommended sample preparation methods for peptide imaging.231 Further, imaging endogenous and tryptic-digested peptides is particularly challenging due to their relatively low concentration compared to interfering lipid species. Lipids can be washed off tissues before imaging using organic solvents to remove these abundant molecular species to prevent their masking effect.232,233 Because neuropeptides are typically present in low abundance, several strategies have been developed, including removing formaldehyde-induced peptidyl crosslinks and employing alternative aqueous tissue washes.234,235 These have increased neuropeptide coverage while increasing their signal intensities by 5-fold.235,236
During imaging, mass spectra are generated for each defined pixel of the tissue slice, revealing the molecular composition and their relative abundance. The colored images from the experiment can reflect the abundance of specific ions across the whole tissue section, akin to an ion heat map. Similar to LC-MS/MS quantification techniques, both relative and absolute quantification can be achieved through MS imaging. In relative quantification, ion signals across tissues can be normalized using various imaging processing software methods, such as HDI, MSiReader, and SciLS lab software.237,238 The normalized intensity of the ions can then be used to compare tissue slices from different experimental conditions. Internal standards can also be used for relative and absolute quantification in MSI.239,240 Isotopically labeled standards are frequently used in MSI to correct for variations in ionization efficiency and matrix effects, enhancing the precision of quantitative analyses in complex samples. For instance, Bergman et al. integrated specific concentrations of multiple deuterated metabolites into the solvent stream for DESI imaging.241 This approach enhances the accuracy and reliability of quantification methods in mass spectrometry imaging experiments. In addition to internal standards, calibration curves are commonly employed in MSI for accurate quantification. These curves, as demonstrated by Jadoul et al., involve adding known concentrations of analytes to tissue homogenate, establishing a relationship between signal intensity and concentration.242 Further, comprehensive peptide identification and structural probing are crucial but difficult to achieve simultaneously. Using nanosecond photochemical reaction (nsPCR)-based click chemistry with 2-nitrobenzaldehyde (NBA), researchers developed a method to enhance neuropeptide identification and structural analysis by enabling on-demand removal of surrounding matrices, demonstrated with MALDI-MSI.243 This strategy achieves high labeling efficiencies (>90%) and facilitates neuropeptide visualization in complex tissues, with broad applicability for proteins up to 155 kDa across diverse samples and amine-containing metabolites.244 The 3D spatial peptidomics information provided by MSI can be integrated with microcomputed tomography for increased multiplexed mapping of neuropeptides in the brain microanatomy.245
Neuropeptidomic applications and challenges
Many MS-based approaches have been employed to profile and quantify the neuropeptidome of various animals. Nontargeted LFQ strategies are used extensively to study the endogenous peptidome change for several animal model systems across vertebrates and invertebrates, including mice, rats, crustaceans, nematodes, and insects.5,246,247 DeLaney et al. used a multifaceted approach to quantitatively investigate the neuropeptidome content in fed and unfed Cancer borealis tissue extracts by LFQ and spatially map neuropeptides of intact neural tissue.157 Wu et al. similarly conducted LFQ analysis across several regions of the brain on a rodent model of intense stress to discover novel neuropeptides involved in predator-stress-related behavior with implications for post-traumatic stress disorder.248 In total, 328 peptides were quantified from several key neuropeptide families, including ProSAAS, tachykinin, and opioid peptides. Probing the neuropeptidomes of early-branching lineages of Cnidaria, Porifera, and Ctenophora can offer insights into the evolutionary origins of peptidergic nervous systems.249,250 These findings suggest that animal peptidergic neurons first evolved from sensory-secretory cells, with neural function highly dependent on the complexity of neuropeptide-receptor interactions.251 Further, DIA and LFQ methods have emerged as powerful techniques to account for low-abundance peptides that may be unaccounted for in DDA analyses while reducing spectral complexity due to labeling and maintaining high degrees of reproducibility and coverage.252 For example, DeLaney et al. have used DIA-MS LFQ analysis for neuropeptide profiling for C. borealis hemolymph, successfully quantifying 217 neuropeptides compared to previous reports, 8 of which were documented to be significantly changed upon feeding for the first time.253,254As discussed above, peptidomics can be carried out using various quantification strategies. With untargeted LFQ, even scarce samples can be relatively quantified without the risk of sample loss, and the number of comparisons is not limited by the channels provided by the labeling strategies. For example, Anapindi et al. conducted discovery-based LFQ experiments to quantify over 1500 neuropeptides from a total of 200 LC-MS/MS runs, assessing their relationship with migraine and opioid-induced hyperalgesia (OIH) and discovering that pituitary adenylate cyclase-activating polypeptide mechanistically links chronic migraine with OIH.247 Because neuropeptides are commonly amidated, the authors accounted for this PTM by characterizing the peptide false discovery rate by adjusting false amidation assignments and normalizing the run-to-run variability from such extensive instrument time.255,256 Labeling methods can be used to minimize these batch effects and simultaneously screen multiple endogenous peptides. Sauer and Li leveraged the DiLeu isobaric tags to investigate how neuropeptide expression in neural tissues of Callinectes sapidus is affected by copper toxicity across different time points (1, 2, and 4 hours of copper exposure).257 Several neuropeptides from RFamide, allatostatin, orcokinin, and pigment-dispersing hormone families were significantly changed due to copper exposure. With DiLeu isobaric labeling, 4 conditions were analyzed in a single instrument run, enabling the time-course study to be done in an efficient and high-throughput way. Similarly, a 10-plex version of this tag was integrated with label-free techniques to assess the mouse hypothalamic neuropeptidome and proteome of germ-free and conventionally raised mice.258 Commercially available tagging reagents such as TMT and iTRAQ are commonly used for proteomics and tryptic-digested peptides; however, they have not been extensively used for endogenous peptidomics, although similar benefits provided by the isobaric labeling should be retained.111,259
Isotopic labeling strategies, including dimethyl labeling, SILAC, and 18O labeling, are also widely used to quantify endogenous peptides.94,260,261 Succinic anhydride and 4-trimethylammoniumbutyryl (TMAB) have been shown to label the mouse pituitary peptidome whereby deuterium incorporation generated channels with mass differences between the heavy and light labeled forms of 4 Da and 9 Da, respectively.262 Accordingly, TMAB reagents resulted in better separation and resolution than succinylated peptides and detected a broader range of peptides as the increased mass difference at the MS1-level minimized isotopic envelope overlap. Reductive dimethylation on the N-terminus and lysine residues with isotope-labeled formaldehyde is another strategy used for neuropeptide quantitation.263,264 These have been used to probe neuropeptide expression and localization across various environmental stressors in marine animals, including oxygen, salinity, and temperature fluctuations.265
Untargeted, profiling-based MS quantitation approaches provide a global understanding of the peptidome as these techniques require no prior knowledge of cellular content. Targeted quantitation techniques, such as SRM/MRM and PRM, require knowledge of specific peptides of interest to compare their expression level between control and experimental groups. These methods are desirable when analyzing bioactive peptide families. However, targeted approaches must account for dynamic peptide regulation, including proteolytic processing and PTMs. Informed from a previous discovery-based LFQ analysis to profile diet-induced neuropeptide changes, an extended SRM assay of 15 endogenous peptides from the Rattus norvegicus hypothalamus has been developed, discovering a significant upregulation of VGF-derived peptides as a result of a high caloric diet.266,267 Additionally, circulating peptide hormones are often present in low concentrations and require the high specificity of immunoassays, as matrix effects frequently hinder their detection from the complex composition of circulating body fluids.268 Neuropeptide Y (NPY), for example, is the most abundant neuropeptide in the brain and is prone to peptidolysis where NPY fragments are known to mediate distinct effects due to varying receptor affinities.269 The low concentration of NPY peptides, typically at picomolar concentrations, makes them difficult to quantify, even with techniques such as MRM in human plasma.270 However, combining the high affinity of monoclonal antibodies, using isotope-labeled analogs, and controlling for NPY chemical degradation makes it possible to assay the NPY family at subpicomolar concentrations.271 Similar targeted SRM-MS immunoassays have been developed for insulin-like growth factor 1 and parathyroid hormones.272–274
Recently, PRM techniques have garnered attention in neuropeptidomics for both relative and absolute quantification, as their mass accuracy and sensitivity rival SRM. Instead of measuring one transition at a time, all fragment ions of a selected precursor are measured in parallel.275 The renin-angiotensin system (RAS), a regulatory pathway controlling arterial blood pressure, produces angiotensin peptides by enzymatic cleavages from the prohormone angiotensinogen (AGT) with some peptides are short as five.276 Previous SRM-MS assays to study angiotensin peptides have yielded roughly 10 amol levels on the lower limit of detection for both relative and absolute quantitation.277–279 Angiotensin peptides in the brain, however, may be present at even lower concentrations and become trace-limited in regulatory regions with predicted concentrations from 400 zmol to 3 amol.280,281 To address these very low concentrations, PRM has been used to identify angiotensin peptides at very low limits of detection from 300 zmol to 5 amol using synthetic standards with roughly 10 amol sensitivity for quantification.282 PRM has also been used for targeted profiling experiments. For instance, Cockx et al. recently developed a discovery-driven targeted peptidomics strategy utilizing PRM to profile and quantify the neuropeptides of interest.283 With DDA LC-MS/MS LFQ analysis, they first generated a spectral library using 427 synthetic known and theoretical peptide sequences, resulting in a cumulative library of 510 peptide ions. By using RT scheduling for these low-abundance samples along with the sensitivity and specificity of PRM, they quantified 172 neuropeptides while functionally validating flp-7 and flp-11 as novel nictation regulating peptides in C. elegans. It is also possible to target the neuropeptide biosynthetic pathway directly to elucidate neuropeptide processing. For instance, affinity chromatography can be used to purify neuropeptides that are substrates of carboxypeptidase E (CPE), an enzyme commonly found in the final processing site for neuropeptide biosynthesis.284 This allows the analysis of peptides in genetically defined cell types with insights into peptide isoforms resulting from proteolytic cleavages and other PTMs with respect to CPE processing.285
Conclusion
Neuropeptide analysis has advanced significantly, driven by improvements in MS instrumentation, quantitative strategies, and data analysis workflows. The inherent challenges of neuropeptide analysis including low in vivo abundance, structural diversity, and complex functions have driven the development of both label-free (LFQ) and label-based strategies. While isotopic labeling methods such as dimethyl labeling and DiLeu tags improve quantitation and multiplexing, their additional processing steps can lead to sample loss, making DIA-based LFQ methods suitable for neuropeptidomic studies. Despite rapid progress, neuropeptidomics still lags behind proteomics regarding workflow standardization, enrichment techniques, and computational analysis. A major limitation is the high structural variability of neuropeptides, which necessitates the development of specialized MS methodologies tailored to specific neuropeptide families and their PTMs. Nonetheless, advances in hybrid MS fragmentation techniques and IM-MS have further improved neuropeptide and PTM analysis, particularly for glycopeptides and DAACPs. Additionally, enhanced spatial resolution in MSI is needed to better map neuropeptide distributions at the cellular level. IM-MS can be combined with MSI to probe neuropeptide spatial localization with enhanced sensitivity and specificity. Innovations in sample preparation and targeted techniques such as MRM and PRM have enabled the absolute quantification of neuropeptides, even in small-volume samples with significantly low limits of detection and quantification. These advancements are crucial for studying neuropeptide-mediated signaling in physiological and pathological contexts. Furthermore, integrating single-cell mass spectrometry with spatial peptidomics to resolve neuropeptide heterogeneity at the cellular level presents a promising approach.286 These innovations will further refine neuropeptide quantification, allow for more precise functional characterization, and ultimately enhance our understanding of neuropeptide dynamics in health and disease. By leveraging these advances, MS-based neuropeptidomics will continue to be instrumental in unraveling the complex neurochemical landscape of the brain.Author contributions
A. E. Ibarra: conceptualization, writing – original draft, review and editing, visualization. W. Wu: writing – original draft, review and editing, visualization. H. Zhang: writing – original draft, review and editing. L. Li: conceptualization, supervision, writing – review and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported in part by National Institutes of Health (NIH) through grants R01DK071801, R01NS029346, P41GM108538, and National Science Foundation (NSF) through the grant CHE-2108223. A. E. I. was supported in part by the NSF Graduate Research Fellowship Program under Grant No. DGE-2137424. Support was also provided by the Graduate School and the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin–Madison. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF. L. L. wishes to acknowledge additional support from NIH grants R01 AG078794, R01AG052324, S10OD028473, and S10OD025084, along with funding from the Vilas Distinguished Achievement Professorship, the Charles Melbourne Johnson Professorship, the Wisconsin Alumni Research Foundation, and the University of Wisconsin–Madison School of Pharmacy.References
- V. Hook, L. Funkelstein, D. Lu, S. Bark, J. Wegrzyn and S. R. Hwang, Annu. Rev. Pharmacol., 2008, 48, 393–423 CrossRef CAS PubMed.
- J. F. Rehfeld, L. Bardram, P. Cantor, J. Cerman, L. Hilsted, A. H. Johnsen, N. Mogensen and L. Odum, Acta Oncol., 1989, 28, 315–318 CrossRef CAS PubMed.
- M. Stawikowski and G. B. Fields, Curr. Protoc. Protein Sci., 2012, 69, 18.1.1–18.1.13 Search PubMed.
- M. L. Hennrich and A. C. Gavin, Sci. Signaling, 2015, 8, re5 CrossRef PubMed.
- L. E. Atkinson, Y. Liu, F. McKay, E. Vandewyer, C. Viau, A. Irvine, B. A. Rosa, Z. Li, Q. Liang and N. J. Marks, ACS Chem. Neurosci., 2021, 12, 3176–3188 CrossRef CAS PubMed.
- J. R. Schank, A. E. Ryabinin, W. J. Giardino, R. Ciccocioppo and M. Heilig, Neuron, 2012, 76, 192–208 CrossRef CAS PubMed.
- V. Kormos and B. Gaszner, Neuropeptides, 2013, 47, 401–419 CrossRef CAS PubMed.
- V. Cardoso, J. Chesne, H. Ribeiro, B. Garcia-Cassani, T. Carvalho, T. Bouchery, K. Shah, N. L. Barbosa-Morais, N. Harris and H. Veiga-Fernandes, Nature, 2017, 549, 277–281 CrossRef CAS PubMed.
- K. J. Blake, X. R. Jiang and I. M. Chiu, Trends Neurosci., 2019, 42, 537–551 CrossRef CAS PubMed.
- V. Neugebauer, M. Mazzitelli, B. Cragg, G. Ji, E. Navratilova and F. Porreca, Neuropharmacology, 2020, 170, 108052 CrossRef CAS PubMed.
- T. Zhang, D. Ai, P. Wei, Y. Xu, Z. Bi, F. Ma, F. Li, X. J. Chen, Z. Zhang, X. Zou, Z. Guo, Y. Zhao, J. L. Li, M. Ye, Z. Feng, X. Zhang, L. Zheng, J. Yu, C. Li, T. Tu, H. Zeng, J. Lei, H. Zhang, T. Hong, L. Zhang, B. Luo, Z. Li, C. Xing, C. Jia, L. Li, W. Sun and W. P. Ge, Nat. Neurosci., 2024, 27, 1103–1115 CrossRef CAS PubMed.
- E. Y. Lee, L. C. Chan, H. Wang, J. Lieng, M. Hung, Y. Srinivasan, J. Wang, J. A. Waschek, A. L. Ferguson, K. F. Lee, N. Y. Yount, M. R. Yeaman and G. C. L. Wong, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e1917623117 CrossRef CAS PubMed.
- X. Li, K. Chen, R. Liu, Z. Zheng and X. Hou, Front. Immunol., 2024, 15, 1496147 CrossRef CAS PubMed.
- M. P. Nusbaum, D. M. Blitz and E. Marder, Nat. Rev. Neurosci., 2017, 18, 389–403 CrossRef CAS PubMed.
- E. A. De La Toba, S. E. Bell, E. V. Romanova and J. V. Sweedler, Annu. Rev. Anal. Chem., 2022, 15, 83–106 CrossRef CAS PubMed.
- K. D. B. Anapindi, E. V. Romanova, J. W. Checco and J. V. Sweedler, Pharmacol. Rev., 2022, 74, 662–679 CrossRef CAS PubMed.
- R. Hellinger, A. Sigurdsson, W. Wu, E. V. Romanova, L. Li, J. V. Sweedler, R. D. Süssmuth and C. W. Gruber, Nat. Rev. Methods Primers, 2023, 3, 25 CrossRef CAS PubMed.
- C. S. Sauer, A. Phetsanthad, O. L. Riusech and L. Li, Expert Rev. Proteomics, 2021, 18, 607–621 CrossRef CAS PubMed.
- P. A. Rudnick, K. R. Clauser, L. E. Kilpatrick, D. V. Tchekhovskoi, P. Neta, N. Blonder, D. D. Billheimer, R. K. Blackman, D. M. Bunk, H. L. Cardasis, A. J. Ham, J. D. Jaffe, C. R. Kinsinger, M. Mesri, T. A. Neubert, B. Schilling, D. L. Tabb, T. J. Tegeler, L. Vega-Montoto, A. M. Variyath, M. Wang, P. Wang, J. R. Whiteaker, L. J. Zimmerman, S. A. Carr, S. J. Fisher, B. W. Gibson, A. G. Paulovich, F. E. Regnier, H. Rodriguez, C. Spiegelman, P. Tempst, D. C. Liebler and S. E. Stein, Mol. Cell. Proteomics, 2010, 9, 225–241 CrossRef CAS PubMed.
- A. L. Capriotti, C. Cavaliere, A. Cavazzini, F. Gasparrini, G. Pierri, S. Piovesana and A. Lagana, J. Chromatogr. A, 2017, 1498, 176–182 CrossRef CAS PubMed.
- J. Lenco, S. Jadeja, D. K. Naplekov, O. V. Krokhin, M. A. Khalikova, P. Chocholous, J. Urban, K. Broeckhoven, L. Novakova and F. Svec, J. Proteome Res., 2022, 21, 2846–2892 CrossRef CAS PubMed.
- Y. Jiang, D. A. B. Rex, D. Schuster, B. A. Neely, G. L. Rosano, N. Volkmar, A. Momenzadeh, T. M. Peters-Clarke, S. B. Egbert, S. Kreimer, E. H. Doud, O. M. Crook, A. K. Yadav, M. Vanuopadath, A. D. Hegeman, M. L. Mayta, A. G. Duboff, N. M. Riley, R. L. Moritz and J. G. Meyer, ACS Meas. Sci. Au, 2024, 4, 338–417 CrossRef CAS PubMed.
- Z. Z. Chen, J. Dufresne, P. Bowden, M. Miao and J. G. Marshall, ACS Omega, 2024, 9, 41343–41354 CrossRef CAS PubMed.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- P. Roepstorff and J. Fohlman, Biomed. Mass Spectrom., 1984, 11, 601 CrossRef CAS PubMed.
- H. Steen and M. Mann, Nat. Rev. Mol. Cell Biol., 2004, 5, 699–711 CrossRef CAS PubMed.
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2016, 537, 347–355 CrossRef CAS PubMed.
- D. F. Hunt, J. R. Yates, 3rd, J. Shabanowitz, S. Winston and C. R. Hauer, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6233–6237 CrossRef CAS PubMed.
- W. J. Kong, H. W. H. Hui, H. Peng and W. W. Bin Goh, Proteomics, 2022, 22, e2200092 CrossRef PubMed.
- D. L. Plubell, L. Kall, B. J. Webb-Robertson, L. M. Bramer, A. Ives, N. L. Kelleher, L. M. Smith, T. J. Montine, C. C. Wu and M. J. MacCoss, J. Proteome Res., 2022, 21, 891–898 CrossRef CAS PubMed.
- R. M. Miller and L. M. Smith, Analyst, 2023, 148, 475–486 RSC.
- R. E. Foreman, A. L. George, F. Reimann, F. M. Gribble and R. G. Kay, J. Proteome Res., 2021, 20, 3782–3797 CrossRef CAS PubMed.
- L. Fricker, Methods Mol. Biol., 2018, 1719, 121–140 CrossRef CAS PubMed.
- J. Peng, H. Zhang, H. Niu and R. Wu, TrAC, Trends Anal. Chem., 2020, 125, 115835 CrossRef CAS.
- A. Buchberger, Q. Yu and L. Li, Annu. Rev. Anal. Chem., 2015, 8, 485–509 CrossRef CAS PubMed.
- V. Hook, C. B. Lietz, S. Podvin, T. Cajka and O. Fiehn, J. Am. Soc. Mass Spectrom., 2018, 29, 807–816 CrossRef CAS PubMed.
- Y. G. Kim, A. M. Lone and A. Saghatelian, Nat. Protoc., 2013, 8, 1730–1742 CrossRef PubMed.
- H. Xu and D. Shields, J. Cell Biol., 1993, 122, 1169–1184 CrossRef CAS PubMed.
- A. J. Mitchell, A. M. Lone, A. D. Tinoco and A. Saghatelian, PLoS One, 2013, 8, e68638 CrossRef CAS PubMed.
- Y. G. Kim, A. M. Lone, W. M. Nolte and A. Saghatelian, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8523–8527 CrossRef CAS PubMed.
- L. Segerstrom, J. Gustavsson and I. Nylander, Biopreserv. Biobanking, 2016, 14, 172–179 CrossRef PubMed.
- A. Zissler, W. Stoiber, P. Steinbacher, J. Geissenberger, F. C. Monticelli and S. Pittner, Diagnostics, 2020, 10, 1014 CrossRef CAS PubMed.
- C. Stingl, M. Soderquist, O. Karlsson, M. Boren and T. M. Luider, J. Proteome Res., 2014, 13, 2807–2817 CrossRef CAS PubMed.
- E. Hartman, F. Forsberg, S. Kjellstrom, J. Petrlova, C. Luo, A. Scott, M. Puthia, J. Malmstrom and A. Schmidtchen, Nat. Commun., 2024, 15, 7128 CrossRef CAS PubMed.
- C. Lopez-Otin and C. M. Overall, Nat. Rev. Mol. Cell Biol., 2002, 3, 509–519 CrossRef CAS PubMed.
- J. Lindsay, M. Svensson, M. Boren, M. Faith, P. Andren, P. Svenningsson and K. Skold, Cancer Res., 2009, 69, 2613 Search PubMed.
- M. Svensson, M. Boren, K. Skold, M. Falth, B. Sjogren, M. Andersson, P. Svenningsson and P. E. Andren, J. Proteome Res., 2009, 8, 974–981 CrossRef CAS PubMed.
- N. Yang, K. D. B. Anapindi, E. V. Romanova, S. S. Rubakhin and J. V. Sweedler, Analyst, 2017, 142, 4476–4485 RSC.
- G. A. Dienel, J. Neurochem., 2021, 158, 1007–1031 CrossRef CAS PubMed.
- K. L. Rock, C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang and A. L. Goldberg, Cell, 1994, 78, 761–771 CrossRef CAS PubMed.
- A. Secher, C. D. Kelstrup, K. W. Conde-Frieboes, C. Pyke, K. Raun, B. S. Wulff and J. V. Olsen, Nat. Commun., 2016, 7, 11436 CrossRef CAS PubMed.
- E. Fridjonsdottir, A. Nilsson, L. D. Fricker and P. E. Andren, Methods Mol. Biol., 2024, 2758, 49–60 CrossRef PubMed.
- C. E. Bakalarski, J. E. Elias, J. Villen, W. Haas, S. A. Gerber, P. A. Everley and S. P. Gygi, J. Proteome Res., 2008, 7, 4756–4765 CrossRef CAS PubMed.
- H. Liu, L. Lam, L. Yan, B. Chi and P. K. Dasgupta, Anal. Chim. Acta, 2014, 850, 65–70 CrossRef CAS PubMed.
- P. J. Taylor, Clin. Biochem., 2005, 38, 328–334 CrossRef CAS PubMed.
- P. Panuwet, R. E. Hunter, Jr., P. E. D'Souza, X. Chen, S. A. Radford, J. R. Cohen, M. E. Marder, K. Kartavenka, P. B. Ryan and D. B. Barr, Crit. Rev. Anal. Chem., 2016, 46, 93–105 CrossRef CAS PubMed.
- R. King, R. Bonfiglio, C. Fernandez-Metzler, C. Miller-Stein and T. Olah, J. Am. Soc. Mass Spectrom., 2000, 11, 942–950 CrossRef CAS PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 1998, 70, 882–889 CrossRef CAS PubMed.
- C. Ghosh, C. P. Shinde and B. S. Chakraborty, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2012, 893–894, 193–200 CrossRef CAS PubMed.
- A. Nasiri, R. Jahani, S. Mokhtari, H. Yazdanpanah, B. Daraei, M. Faizi and F. Kobarfard, Analyst, 2021, 146, 6049–6063 RSC.
- T. S. Batth, C. Francavilla and J. V. Olsen, J. Proteome Res., 2014, 13, 6176–6186 CrossRef CAS PubMed.
- B. Manadas, V. M. Mendes, J. English and M. J. Dunn, Expert Rev. Proteomics, 2010, 7, 655–663 CrossRef CAS PubMed.
- R. M. Sturm, J. A. Dowell and L. Li, Methods Mol. Biol., 2010, 615, 217–226 CrossRef CAS PubMed.
- S. Podvin, Z. Jiang, B. Boyarko, L. A. Rossitto, A. O'Donoghue, R. A. Rissman and V. Hook, ACS Chem. Neurosci., 2022, 13, 1992–2005 CrossRef CAS PubMed.
- S. Mousavi, H. Qiu, F. I. Heinis, M. S. R. Abid, M. T. Andrews and J. W. Checco, ACS Chem. Neurosci., 2022, 13, 2888–2896 CrossRef CAS PubMed.
- A. F. Russo, Headache, 2017, 57(Suppl 2), 37–46 CrossRef PubMed.
- P. Yin, X. Hou, E. V. Romanova and J. V. Sweedler, Methods Mol. Biol., 2011, 789, 223–236 CrossRef CAS PubMed.
- T. R. T. Scoggins, J. T. Specker and B. M. Prentice, Analyst, 2024, 149, 2459–2468 RSC.
- R. A. Zubarev, Proteomics, 2013, 13, 723–726 CrossRef CAS PubMed.
- W. Bahureksa, T. Borch, R. B. Young, C. R. Weisbrod, G. T. Blakney and A. M. McKenna, Anal. Chem., 2022, 94, 11382–11389 CrossRef CAS PubMed.
- K. K. Murray, R. K. Boyd, M. N. Eberlin, G. J. Langley, L. Li and Y. Naito, Pure Appl. Chem., 2013, 85, 1515–1609 CrossRef CAS.
- S. Banerjee and S. Mazumdar, Int. J. Anal. Chem., 2012, 2012, 282574 Search PubMed.
- F. Jiang, Z. Lu, C. Zhang, J. Liu, J. Zhu, M. Huang and G. Zhong, J. Am. Soc. Mass Spectrom., 2022, 33, 1213–1220 CrossRef CAS PubMed.
- F. Jiang, Q. Liu, Q. Li, S. Zhang, X. Qu, J. Zhu, G. Zhong and M. Huang, Anal. Chem., 2020, 92, 7690–7698 CrossRef CAS PubMed.
- C. Z. Nie, H. Liu, X. H. Huang, D. Y. Zhou, X. S. Wang and L. Qin, Analyst, 2024, 149, 3140–3151 RSC.
- P. D. Piehowski, V. A. Petyuk, D. J. Orton, F. Xie, R. J. Moore, M. Ramirez-Restrepo, A. Engel, A. P. Lieberman, R. L. Albin, D. G. Camp, R. D. Smith and A. J. Myers, J. Proteome Res., 2013, 12, 2128–2137 CrossRef CAS PubMed.
- X. Zhou and Z. Ouyang, Rapid Commun. Mass Spectrom., 2016, 30(Suppl 1), 29–33 CrossRef CAS PubMed.
- A. Bilbao, B. C. Gibbons, G. W. Slysz, K. L. Crowell, M. E. Monroe, Y. M. Ibrahim, R. D. Smith, S. H. Payne and E. S. Baker, Int. J. Mass Spectrom., 2018, 427, 91–99 CrossRef CAS PubMed.
- M. K. Sivanich, T. J. Gu, D. N. Tabang and L. Li, Proteomics, 2022, 22, e2100256 CrossRef PubMed.
- H. Stahnke, S. Kittlaus, G. Kempe and L. Alder, Anal. Chem., 2012, 84, 1474–1482 CrossRef CAS PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS PubMed.
- K. A. Tsantilas, G. E. Merrihew, J. E. Robbins, R. S. Johnson, J. Park, D. L. Plubell, J. D. Canterbury, E. Huang, M. Riffle, V. Sharma, B. X. MacLean, J. Eckels, C. C. Wu, M. S. Bereman, S. E. Spencer, A. N. Hoofnagle and M. J. MacCoss, J. Proteome Res., 2024, 23, 4392–4408 CrossRef CAS PubMed.
- D. Bramwell, J. Proteomics, 2013, 95, 3–21 CrossRef CAS PubMed.
- F. Calderon-Celis, J. R. Encinar and A. Sanz-Medel, Mass Spectrom. Rev., 2018, 37, 715–737 CrossRef CAS PubMed.
- O. T. Schubert, H. L. Rost, B. C. Collins, G. Rosenberger and R. Aebersold, Nat. Protoc., 2017, 12, 1289–1294 CrossRef CAS PubMed.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007, 389, 1017–1031 CrossRef CAS PubMed.
- Z. Wang, P. K. Liu and L. Li, ACS Meas. Sci. Au, 2024, 4, 315–337 CrossRef CAS PubMed.
- Y. Oda, K. Huang, F. R. Cross, D. Cowburn and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6591–6596 CrossRef CAS PubMed.
- H. C. Harsha, H. Molina and A. Pandey, Nat. Protoc., 2008, 3, 505–516 CrossRef CAS PubMed.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell. Proteomics, 2002, 1, 376–386 CrossRef CAS PubMed.
- J. L. Hsu, S. Y. Huang, N. H. Chow and S. H. Chen, Anal. Chem., 2003, 75, 6843–6852 CrossRef CAS PubMed.
- M. Mann, Nat. Rev. Mol. Cell Biol., 2006, 7, 952–958 CrossRef CAS PubMed.
- X. L. Chen, S. S. Wei, Y. L. Ji, X. J. Guo and F. Q. Yang, Proteomics, 2015, 15, 3175–3192 CrossRef CAS PubMed.
- H. T. Lau, H. W. Suh, M. Golkowski and S. E. Ong, J. Proteome Res., 2014, 13, 4164–4174 CrossRef CAS PubMed.
- G. Pimienta, R. Chaerkady and A. Pandey, Methods Mol. Biol., 2009, 527, 107–116 CrossRef CAS PubMed.
- Z. Li, R. M. Adams, K. Chourey, G. B. Hurst, R. L. Hettich and C. Pan, J. Proteome Res., 2012, 11, 1582–1590 CrossRef CAS PubMed.
- D. Bourdetsky, C. E. Schmelzer and A. Admon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1591–E1599 CrossRef CAS PubMed.
- N. C. Beller and A. B. Hummon, Mol. Omics, 2022, 18, 579–590 RSC.
- V. Hung, N. D. Udeshi, S. S. Lam, K. H. Loh, K. J. Cox, K. Pedram, S. A. Carr and A. Y. Ting, Nat. Protoc., 2016, 11, 456–475 CrossRef CAS PubMed.
- D. A. Rothenberg, J. M. Taliaferro, S. M. Huber, T. J. Begley, P. C. Dedon and F. M. White, iScience, 2018, 9, 367–381 CrossRef CAS PubMed.
- H. W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328–1331 CrossRef CAS PubMed.
- K. Boonen, W. De Haes, J. Van Houtven, R. Verdonck, G. Baggerman, D. Valkenborg and L. Schoofs, Peptidomics: Methods and Strategies, 2018, vol. 1719, pp. 141–159 Search PubMed.
- C. S. Sauer and L. Li, Methods Enzymol., 2022, 663, 235–257 CAS.
- P. J. Boersema, T. T. Aye, T. A. van Veen, A. J. Heck and S. Mohammed, Proteomics, 2008, 8, 4624–4632 CrossRef CAS PubMed.
- C. Chang, J. Zhang, C. Xu, Y. Zhao, J. Ma, T. Chen, F. He, H. Xie and Y. Zhu, Anal. Chem., 2016, 88, 6844–6851 CrossRef CAS PubMed.
- J. Claesen, A. Rockwood, M. Gorshkov and D. Valkenborg, Mass Spectrom. Rev., 2023, 44, 22–42 CrossRef PubMed.
- N. Pappireddi, L. Martin and M. Wuhr, ChemBioChem, 2019, 20, 1210–1224 CrossRef CAS PubMed.
- A. Thompson, J. Schafer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann, R. Johnstone, A. K. Mohammed and C. Hamon, Anal. Chem., 2003, 75, 1895–1904 CrossRef CAS PubMed.
- P. L. Ross, Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski, S. Pillai, S. Dey, S. Daniels, S. Purkayastha, P. Juhasz, S. Martin, M. Bartlet-Jones, F. He, A. Jacobson and D. J. Pappin, Mol. Cell. Proteomics, 2004, 3, 1154–1169 CrossRef CAS PubMed.
- F. Xiang, H. Ye, R. B. Chen, Q. Fu and L. J. Li, Anal. Chem., 2010, 82, 2817–2825 CrossRef CAS PubMed.
- A. E. Merrill, A. S. Hebert, M. E. MacGilvray, C. M. Rose, D. J. Bailey, J. C. Bradley, W. W. Wood, M. El Masri, M. S. Westphall, A. P. Gasch and J. J. Coon, Mol. Cell. Proteomics, 2014, 13, 2503–2512 CrossRef CAS PubMed.
- G. Pottiez, J. Wiederin, H. S. Fox and P. Ciborowski, J. Proteome Res., 2012, 11, 3774–3781 CrossRef CAS PubMed.
- J. M. Li, Z. Y. Cai, R. D. Bomgarden, I. Pike, K. Kuhn, J. C. Rogers, T. M. Roberts, S. P. Gygi and J. A. Paulo, J. Proteome Res., 2021, 20, 2964–2972 CrossRef CAS PubMed.
- J. M. Li, J. G. Van Vranken, L. P. Vaites, D. K. Schweppe, E. L. Huttlin, C. Etienne, P. Nandhikonda, R. Viner, A. M. Robitaille, A. H. Thompson, K. Kuhn, I. Pike, R. D. Bomgarden, J. C. Rogers, S. P. Gygi and J. A. Paulo, Nat. Methods, 2020, 17, 399–439 CrossRef CAS PubMed.
- N. R. Zuniga, D. C. Frost, K. Kuhn, M. Shin, R. L. Whitehouse, T. Y. Wei, Y. C. He, S. L. Dawson, I. Pike, R. D. Bomgarden, S. P. Gygi and J. A. Paulo, J. Proteome Res., 2024, 23, 5153–5165 CrossRef CAS PubMed.
- P. Giron, L. Dayon, N. Turck, C. Hoogland and J. C. Sanchez, J. Proteome Res., 2011, 10, 249–258 CrossRef CAS PubMed.
- Z. Qu, F. Meng, R. D. Bomgarden, R. I. Viner, J. Li, J. C. Rogers, J. Cheng, C. M. Greenlief, J. Cui, D. B. Lubahn, G. Y. Sun and Z. Gu, J. Proteome Res., 2014, 13, 3200–3211 CrossRef CAS PubMed.
- M. Kuljanin, D. C. Mitchell, D. K. Schweppe, A. S. Gikandi, D. P. Nusinow, N. J. Bulloch, E. V. Vinogradova, D. L. Wilson, E. T. Kool, J. D. Mancias, B. F. Cravatt and S. P. Gygi, Nat. Biotechnol., 2021, 39, 630–641 CrossRef CAS PubMed.
- A. B. Arul and R. A. S. Robinson, Anal. Chem., 2019, 91, 178–189 CrossRef CAS PubMed.
- S. Y. Ow, M. Salim, J. Noirel, C. Evans, I. Rehman and P. C. Wright, J. Proteome Res., 2009, 8, 5347–5355 CrossRef CAS PubMed.
- A. Brenes, J. Hukelmann, D. Bensaddek and A. I. Lamond, Mol. Cell. Proteomics, 2019, 18, 1967–1980 CrossRef CAS PubMed.
- S. Nahnsen, C. Bielow, K. Reinert and O. Kohlbacher, Mol. Cell. Proteomics, 2013, 12, 549–556 CrossRef CAS PubMed.
- K. A. Neilson, N. A. Ali, S. Muralidharan, M. Mirzaei, M. Mariani, G. Assadourian, A. Lee, S. C. van Sluyter and P. A. Haynes, Proteomics, 2011, 11, 535–553 CrossRef CAS PubMed.
- H. Liu, R. G. Sadygov and J. R. Yates, 3rd, Anal. Chem., 2004, 76, 4193–4201 CrossRef CAS PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Mol. Cell. Proteomics, 2014, 13, 2513–2526 CrossRef CAS PubMed.
- D. Chelius and P. V. Bondarenko, J. Proteome Res., 2002, 1, 317–323 CrossRef CAS PubMed.
- W. Wang, H. Zhou, H. Lin, S. Roy, T. A. Shaler, L. R. Hill, S. Norton, P. Kumar, M. Anderle and C. H. Becker, Anal. Chem., 2003, 75, 4818–4826 CrossRef CAS PubMed.
- D. H. Lundgren, S. I. Hwang, L. Wu and D. K. Han, Expert Rev. Proteomics, 2010, 7, 39–53 CrossRef CAS PubMed.
- B. Zhang, N. C. VerBerkmoes, M. A. Langston, E. Uberbacher, R. L. Hettich and N. F. Samatova, J. Proteome Res., 2006, 5, 2909–2918 CrossRef CAS PubMed.
- X. Wang, S. Shen, S. S. Rasam and J. Qu, Mass Spectrom. Rev., 2019, 38, 461–482 CrossRef CAS PubMed.
- A. Medvedovici, E. Bacalum and V. David, Biomed. Chromatogr., 2018, 32, e4137 CrossRef PubMed.
- J. T. Leek, R. B. Scharpf, H. C. Bravo, D. Simcha, B. Langmead, W. E. Johnson, D. Geman, K. Baggerly and R. A. Irizarry, Nat. Rev. Genet., 2010, 11, 733–739 CrossRef CAS PubMed.
- X. Lai, L. Wang and F. A. Witzmann, Int. J. Proteomics, 2013, 2013, 756039 Search PubMed.
- M. R. Al Shweiki, S. Monchgesang, P. Majovsky, D. Thieme, D. Trutschel and W. Hoehenwarter, J. Proteome Res., 2017, 16, 1410–1424 CrossRef CAS PubMed.
- Y. V. Karpievitch, A. R. Dabney and R. D. Smith, BMC Bioinf., 2012, 13(Suppl 16), S5 CrossRef CAS PubMed.
- T. Valikangas, T. Suomi and L. L. Elo, Briefings Bioinf., 2018, 19, 1–11 CAS.
- N. M. Griffin, J. Yu, F. Long, P. Oh, S. Shore, Y. Li, J. A. Koziol and J. E. Schnitzer, Nat. Biotechnol., 2010, 28, 83–89 CrossRef CAS PubMed.
- N. W. Bateman, S. P. Goulding, N. J. Shulman, A. K. Gadok, K. K. Szumlinski, M. J. MacCoss and C. C. Wu, Mol. Cell. Proteomics, 2014, 13, 329–338 CrossRef CAS PubMed.
- E. S. Nakayasu, M. Gritsenko, P. D. Piehowski, Y. Gao, D. J. Orton, A. A. Schepmoes, T. L. Fillmore, B. I. Frohnert, M. Rewers, J. P. Krischer, C. Ansong, A. M. Suchy-Dicey, C. Evans-Molina, W. J. Qian, B. M. Webb-Robertson and T. O. Metz, Nat. Protoc., 2021, 16, 3737–3760 CrossRef CAS PubMed.
- E. V. Romanova and J. V. Sweedler, Trends Pharmacol. Sci., 2015, 36, 579–586 CrossRef CAS PubMed.
- R. Liu, Y. Huang, H. Xu, Y. Zheng, Y. Liu, S. Han, M. Yang, Y. Xie, K. Wang, J. A. Duan and L. Li, Anal. Chim. Acta, 2019, 1092, 32–41 CrossRef CAS PubMed.
- V. Lange, P. Picotti, B. Domon and R. Aebersold, Mol. Syst. Biol., 2008, 4, 222 CrossRef PubMed.
- P. Picotti and R. Aebersold, Nat. Methods, 2012, 9, 555–566 CrossRef CAS PubMed.
- A. Bourmaud, S. Gallien and B. Domon, Proteomics, 2016, 16, 2146–2159 CrossRef CAS PubMed.
- R. Bruderer, O. M. Bernhardt, T. Gandhi, S. M. Miladinovic, L. Y. Cheng, S. Messner, T. Ehrenberger, V. Zanotelli, Y. Butscheid, C. Escher, O. Vitek, O. Rinner and L. Reiter, Mol. Cell. Proteomics, 2015, 14, 1400–1410 CrossRef CAS PubMed.
- S. Kreimer, M. E. Belov, W. F. Danielson, L. I. Levitsky, M. V. Gorshkov, B. L. Karger and A. R. Ivanov, J. Proteome Res., 2016, 15, 3563–3573 CrossRef CAS PubMed.
- J. D. Venable, M. Q. Dong, J. Wohlschlegel, A. Dillin and J. R. Yates, Nat. Methods, 2004, 1, 39–45 CrossRef CAS PubMed.
- S. Purvine, J. T. Eppel, E. C. Yi and D. R. Goodlett, Proteomics, 2003, 3, 847–850 CrossRef CAS PubMed.
- L. C. Gillet, P. Navarro, S. Tate, H. Rost, N. Selevsek, L. Reiter, R. Bonner and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, O111.016717 CrossRef PubMed.
- H. L. Rost, G. Rosenberger, P. Navarro, L. Gillet, S. M. Miladinovic, O. T. Schubert, W. Wolski, B. C. Collins, J. Malmstrom, L. Malmstrom and R. Aebersold, Nat. Biotechnol., 2014, 32, 219–223 CrossRef PubMed.
- J. D. Egertson, A. Kuehn, G. E. Merrihew, N. W. Bateman, B. X. MacLean, Y. S. Ting, J. D. Canterbury, D. M. Marsh, M. Kellmann, V. Zabrouskov, C. C. Wu and M. J. MacCoss, Nat. Methods, 2013, 10, 744–746 CrossRef CAS PubMed.
- K. DeLaney and L. Li, Anal. Chem., 2019, 91, 5150–5158 CrossRef CAS PubMed.
- L. Fields, M. Ma, K. DeLaney, A. Phetsanthad and L. Li, Proteomics, 2024, 24, e2300285 CrossRef PubMed.
- K. DeLaney, M. Hu, T. Hellenbrand, P. S. Dickinson, M. P. Nusbaum and L. Li, ACS Chem. Neurosci., 2021, 12, 782–798 CrossRef CAS PubMed.
- K. DeLaney, M. Hu, W. Wu, M. P. Nusbaum and L. Li, Anal. Bioanal. Chem., 2022, 414, 533–543 CrossRef CAS PubMed.
- A. Phetsanthad, A. V. Carr, L. Fields and L. Li, J. Proteome Res., 2023, 22, 1510–1519 CrossRef CAS PubMed.
- F. Zhang, W. Ge, G. Ruan, X. Cai and T. Guo, Proteomics, 2020, 20, e1900276 CrossRef PubMed.
- T. S. Balbuena, D. R. Demartini and J. J. Thelen, Methods Mol. Biol., 2014, 1072, 171–183 CrossRef CAS PubMed.
- Z. Hamid, K. D. Zimmerman, H. Guillen-Ahlers, C. Li, P. Nathanielsz, L. A. Cox and M. Olivier, BMC Genomics, 2022, 23, 496 CrossRef CAS PubMed.
- W. M. Old, K. Meyer-Arendt, L. Aveline-Wolf, K. G. Pierce, A. Mendoza, J. R. Sevinsky, K. A. Resing and N. G. Ahn, Mol. Cell. Proteomics, 2005, 4, 1487–1502 CrossRef CAS PubMed.
- A. Latosinska, M. Frantzi and J. Siwy, Mass Spectrom. Rev., 2024, 43, 1195–1236 CrossRef CAS PubMed.
- W. Wu, L. Fields, K. DeLaney, A. R. Buchberger and L. Li, Methods Mol. Biol., 2024, 2758, 255–289 CrossRef PubMed.
- B. Domon and R. Aebersold, Nat. Biotechnol., 2010, 28, 710–721 CrossRef CAS PubMed.
- P. Patsali, P. Papasavva, S. Christou, M. Sitarou, M. N. Antoniou, C. W. Lederer and M. Kleanthous, Int. J. Mol. Sci., 2020, 21, 6671 CrossRef CAS PubMed.
- C. S. Sauer and L. Li, Chem. Res. Toxicol., 2021, 34, 1329–1336 Search PubMed.
- T. A. Addona, S. E. Abbatiello, B. Schilling, S. J. Skates, D. R. Mani, D. M. Bunk, C. H. Spiegelman, L. J. Zimmerman, A. J. Ham, H. Keshishian, S. C. Hall, S. Allen, R. K. Blackman, C. H. Borchers, C. Buck, H. L. Cardasis, M. P. Cusack, N. G. Dodder, B. W. Gibson, J. M. Held, T. Hiltke, A. Jackson, E. B. Johansen, C. R. Kinsinger, J. Li, M. Mesri, T. A. Neubert, R. K. Niles, T. C. Pulsipher, D. Ransohoff, H. Rodriguez, P. A. Rudnick, D. Smith, D. L. Tabb, T. J. Tegeler, A. M. Variyath, L. J. Vega-Montoto, A. Wahlander, S. Waldemarson, M. Wang, J. R. Whiteaker, L. Zhao, N. L. Anderson, S. J. Fisher, D. C. Liebler, A. G. Paulovich, F. E. Regnier, P. Tempst and S. A. Carr, Nat. Biotechnol., 2009, 27, 633–641 CrossRef CAS PubMed.
- L. Anderson and C. L. Hunter, Mol. Cell. Proteomics, 2006, 5, 573–588 CrossRef CAS PubMed.
- A. C. Peterson, J. D. Russell, D. J. Bailey, M. S. Westphall and J. J. Coon, Mol. Cell. Proteomics, 2012, 11, 1475–1488 CrossRef PubMed.
- S. A. Gerber, J. Rush, O. Stemman, M. W. Kirschner and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6940–6945 CrossRef CAS PubMed.
- D. S. Kirkpatrick, S. A. Gerber and S. P. Gygi, Methods, 2005, 35, 265–273 CrossRef CAS PubMed.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS PubMed.
- A. Kacen, A. Javitt, M. P. Kramer, D. Morgenstern, T. Tsaban, M. D. Shmueli, G. C. Teo, F. da Veiga Leprevost, E. Barnea, F. Yu, A. Admon, L. Eisenbach, Y. Samuels, O. Schueler-Furman, Y. Levin, A. I. Nesvizhskii and Y. Merbl, Nat. Biotechnol., 2023, 41, 239–251 CrossRef CAS PubMed.
- A. F. Bradbury, M. D. Finnie and D. G. Smyth, Nature, 1982, 298, 686–688 CrossRef CAS PubMed.
- T. A. Czyzyk, Y. Ning, M. S. Hsu, B. Peng, R. E. Mains, B. A. Eipper and J. E. Pintar, Dev. Biol., 2005, 287, 301–313 CrossRef CAS PubMed.
- A. L. Wiggenhorn, H. Z. Abuzaid, L. Coassolo, V. L. Li, J. T. Tanzo, W. Wei, X. Lyu, K. J. Svensson and J. Z. Long, Nat. Commun., 2023, 14, 8125 CrossRef CAS PubMed.
- D. A. Lovejoy, D. W. Hogg, T. L. Dodsworth, F. R. Jurado, C. C. Read, A. L. D'Aquila and D. Barsyte-Lovejoy, Front. Endocrinol., 2019, 10, 730 CrossRef PubMed.
- R. B. Kitata, W. K. Choong, C. F. Tsai, P. Y. Lin, B. S. Chen, Y. C. Chang, A. I. Nesvizhskii, T. Y. Sung and Y. J. Chen, Nat. Commun., 2021, 12, 2539 CrossRef CAS PubMed.
- D. J. Geiszler, D. A. Polasky, F. Yu and A. I. Nesvizhskii, Nat. Commun., 2023, 14, 4132 CrossRef CAS PubMed.
- T. Battellino, D. Yeung, H. Neustaeter, V. Spicer, K. Ogata, Y. Ishihama and O. V. Krokhin, J. Chromatogr. A, 2024, 1718, 464714 CrossRef CAS PubMed.
- B. Ma, K. Zhang, C. Hendrie, C. Liang, M. Li, A. Doherty-Kirby and G. Lajoie, Rapid Commun. Mass Spectrom., 2003, 17, 2337–2342 CrossRef CAS PubMed.
- A. T. Kong, F. V. Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, Nat. Methods, 2017, 14, 513–520 CrossRef CAS PubMed.
- P. Sinitcyn, H. Hamzeiy, F. Salinas Soto, D. Itzhak, F. McCarthy, C. Wichmann, M. Steger, U. Ohmayer, U. Distler, S. Kaspar-Schoenefeld, N. Prianichnikov, Ş. Yılmaz, J. D. Rudolph, S. Tenzer, Y. Perez-Riverol, N. Nagaraj, S. J. Humphrey and J. Cox, Nat. Biotechnol., 2021, 39, 1563–1573 Search PubMed.
- J. Brandi, R. Noberini, T. Bonaldi and D. Cecconi, J. Chromatogr. A, 2022, 1678, 463352 Search PubMed.
- M. Wuhrer, M. I. Catalina, A. M. Deelder and C. H. Hokke, J. Chromatogr. B, 2007, 849, 115–128 Search PubMed.
- B. Buszewski and S. Noga, Anal. Bioanal. Chem., 2012, 402, 231–247 CrossRef CAS PubMed.
- Y. Cui, K. Yang, D. N. Tabang, J. Huang, W. Tang and L. Li, J. Am. Soc. Mass Spectrom., 2019, 30, 2491–2501 CrossRef CAS PubMed.
- R. Vitorino, S. Guedes, C. Vitorino, R. Ferreira, F. Amado and J. E. Van Eyk, J. Proteome Res., 2021, 20, 38–48 CrossRef CAS PubMed.
- Y. Shi, Z. Li, B. Wang, X. Shi, H. Ye, D. G. Delafield, L. Lv, Z. Ye, Z. Chen, F. Ma and L. Li, Anal. Chem., 2022, 94, 17895–17903 CrossRef CAS PubMed.
- J. K. Eng, B. C. Searle, K. R. Clauser and D. L. Tabb, Mol. Cell. Proteomics, 2011, 10, R111 009522 CrossRef PubMed.
- J. V. Olsen, B. Macek, O. Lange, A. Makarov, S. Horning and M. Mann, Nat. Methods, 2007, 4, 709–712 CrossRef CAS PubMed.
- J. E. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed.
- N. M. Riley and J. J. Coon, Anal. Chem., 2018, 90, 40–64 CrossRef CAS PubMed.
- A. M. Mayampurath, Y. Wu, Z. M. Segu, Y. Mechref and H. Tang, Rapid Commun. Mass Spectrom., 2011, 25, 2007–2019 CrossRef CAS PubMed.
- N. M. Riley, A. S. Hebert, G. Durnberger, F. Stanek, K. Mechtler, M. S. Westphall and J. J. Coon, Anal. Chem., 2017, 89, 6367–6376 CrossRef CAS PubMed.
- A. Guthals and N. Bandeira, Mol. Cell. Proteomics, 2012, 11, 550–557 CrossRef CAS PubMed.
- Q. Yu, B. Wang, Z. Chen, G. Urabe, M. S. Glover, X. Shi, L.-W. Guo, K. C. Kent and L. Li, J. Am. Soc. Mass Spectrom., 2017, 28, 1751–1764 CrossRef CAS PubMed.
- Q. Yu, A. Canales, M. S. Glover, R. Das, X. Shi, Y. Liu, M. P. Keller, A. D. Attie and L. Li, Anal. Chem., 2017, 89, 9184–9191 CrossRef CAS PubMed.
- N. M. Riley, S. A. Malaker, M. D. Driessen and C. R. Bertozzi, J. Proteome Res., 2020, 19, 3286–3301 CrossRef CAS PubMed.
- T. D. Madsen, L. H. Hansen, J. Hintze, Z. Ye, S. Jebari, D. B. Andersen, H. J. Joshi, T. Ju, J. P. Goetze, C. Martin, M. M. Rosenkilde, J. J. Holst, R. E. Kuhre, C. K. Goth, S. Y. Vakhrushev and K. T. Schjoldager, Nat. Commun., 2020, 11, 4033 CrossRef CAS PubMed.
- Q. Cao, Q. Yu, Y. Liu, Z. Chen and L. Li, J. Proteome Res., 2020, 19, 634–643 CrossRef CAS PubMed.
- D. H. Mast, J. W. Checco and J. V. Sweedler, Biochim. Biophys. Acta, Proteins Proteomics, 2021, 1869, 140553 CrossRef CAS PubMed.
- I. Livnat, H. C. Tai, E. T. Jansson, L. Bai, E. V. Romanova, T. T. Chen, K. Yu, S. A. Chen, Y. Zhang, Z. Y. Wang, D. D. Liu, K. R. Weiss, J. Jing and J. V. Sweedler, Anal. Chem., 2016, 88, 11868–11876 CrossRef CAS PubMed.
- C. Y. Yang, K. Yu, Y. Wang, S. A. Chen, D. D. Liu, Z. Y. Wang, Y. N. Su, S. Z. Yang, T. T. Chen, I. Livnat, F. S. Vilim, E. C. Cropper, K. R. Weiss, J. V. Sweedler and J. Jing, PLoS One, 2016, 11, e0147335 CrossRef PubMed.
- J. W. Checco, G. Zhang, W. D. Yuan, Z. W. Le, J. Jing and J. V. Sweedler, J. Biol. Chem., 2018, 293, 16862–16873 CrossRef CAS PubMed.
- J. W. Checco, G. Zhang, W. D. Yuan, K. Yu, S. Y. Yin, R. H. Roberts-Galbraith, P. M. Yau, E. V. Romanova, J. Jing and J. V. Sweedler, ACS Chem. Biol., 2018, 13, 1343–1352 CrossRef CAS PubMed.
- G. Y. Li, D. G. Delafield and L. J. Li, TrAC-Trend Anal. Chem., 2020, 124, 115546 CrossRef CAS.
- D. G. Delafield, G. Y. Lu, C. J. Kaminsky and L. J. Li, TrAC-Trend Anal. Chem., 2022, 157, 116761 CrossRef CAS.
- C. Jia, C. B. Lietz, Q. Yu and L. Li, Anal. Chem., 2014, 86, 2972–2981 CrossRef CAS PubMed.
- K. Jeanne Dit Fouque, A. Garabedian, J. Porter, M. Baird, X. Pang, T. D. Williams, L. Li, A. Shvartsburg and F. Fernandez-Lima, Anal. Chem., 2017, 89, 11787–11794 CrossRef CAS PubMed.
- X. Pang, C. Jia, Z. Chen and L. Li, J. Am. Soc. Mass Spectrom., 2017, 28, 110–118 CrossRef CAS PubMed.
- G. Li, K. DeLaney and L. Li, Nat. Commun., 2019, 10, 5038 CrossRef PubMed.
- G. Li, C. K. Jeon, M. Ma, Y. Jia, Z. Zheng, D. G. Delafield, G. Lu, E. V. Romanova, J. V. Sweedler, B. T. Ruotolo and L. Li, Chem. Sci., 2023, 14, 5936–5944 RSC.
- S. Okyem, E. V. Romanova, H. C. Tai, J. W. Checco and J. V. Sweedler, Methods Mol. Biol., 2024, 2758, 227–240 CrossRef PubMed.
- V. Vidova and Z. Spacil, Anal. Chim. Acta, 2017, 964, 7–23 CrossRef CAS PubMed.
- E. Gianazza and C. Banfi, Expert Rev. Proteomics, 2018, 15, 477–502 CrossRef CAS PubMed.
- M. Stoeckli, P. Chaurand, D. E. Hallahan and R. M. Caprioli, Nat. Med., 2001, 7, 493–496 CrossRef CAS PubMed.
- L. A. McDonnell and R. M. Heeren, Mass Spectrom. Rev., 2007, 26, 606–643 CrossRef CAS PubMed.
- R. M. Caprioli, T. B. Farmer and J. Gile, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS PubMed.
- K. A. Dave, M. J. Headlam, T. P. Wallis and J. J. Gorman, Curr. Protoc. Protein Sci., 2011, 63, 16.13.1–16.13.21 Search PubMed.
- A. Venter, M. Nefliu and R. G. Cooks, TrAC, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS.
- M. Kompauer, S. Heiles and B. Spengler, Nat. Methods, 2017, 14, 90–96 CrossRef CAS PubMed.
- R. Arakawa and H. Kawasaki, Anal. Sci., 2010, 26, 1229–1240 CrossRef CAS PubMed.
- A. Benninghoven, F. Rudenauer and H. W. Werner, Surf. Interface Anal., 1987, 10, 435 CrossRef.
- S. Fan, B. Ung, E. P. Parrott and E. Pickwell-MacPherson, Phys. Med. Biol., 2015, 60, 2703 Search PubMed.
- R. Lemaire, A. Desmons, J. Tabet, R. Day, M. Salzet and I. Fournier, J. Proteome Res., 2007, 6, 1295–1305 CrossRef CAS PubMed.
- J. X. Truong, X. Spotbeen, J. White, J. V. Swinnen, L. M. Butler, M. F. Snel and P. J. Trim, Anal. Bioanal. Chem., 2021, 413, 2695–2708 CrossRef CAS PubMed.
- N. Q. Vu, A. R. Buchberger, J. Johnson and L. Li, Anal. Bioanal. Chem., 2021, 413, 2665–2673 Search PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Anal. Chem., 2018, 90, 240 Search PubMed.
- M. Andersson, M. R. Groseclose, A. Y. Deutch and R. M. Caprioli, Nat. Methods, 2008, 5, 101–108 CrossRef CAS PubMed.
- M. Pietrowska, M. Gawin, J. Polańska and P. Widłak, Proteomics, 2016, 16, 1670–1677 Search PubMed.
- D. K. Lee, S. S. Rubakhin, I. Kusmartseva, C. Wasserfall, M. A. Atkinson and J. V. Sweedler, Angew. Chem., Int. Ed., 2020, 59, 22584–22590 Search PubMed.
- N. Q. Vu, A. R. Buchberger, J. Johnson and L. Li, Anal. Bioanal. Chem., 2021, 413, 2665–2673 Search PubMed.
- A. R. Buchberger, N. Q. Vu, J. Johnson, K. DeLaney and L. Li, J. Am. Soc. Mass Spectrom., 2020, 31, 1058–1065 Search PubMed.
- E. Claude, E. A. Jones and S. D. Pringle, Imaging Mass Spectrometry: Methods and Protocols, 2017, pp. 65–75 Search PubMed.
- G. Robichaud, K. P. Garrard, J. A. Barry and D. C. Muddiman, J. Am. Soc. Mass Spectrom., 2013, 24, 718–721 Search PubMed.
- S. L. Koeniger, N. Talaty, Y. Luo, D. Ready, M. Voorbach, T. Seifert, S. Cepa, J. A. Fagerland, J. Bouska and W. Buck, Rapid Commun. Mass Spectrom., 2011, 25, 503–510 Search PubMed.
- S. R. Ellis, A. L. Bruinen and R. M. Heeren, Anal. Bioanal. Chem., 2014, 406, 1275–1289 Search PubMed.
- H.-M. Bergman, E. Lundin, M. Andersson and I. Lanekoff, Analyst, 2016, 141, 3686–3695 Search PubMed.
- L. Jadoul, R. Longuespée, A. Noël and E. De Pauw, Anal. Bioanal. Chem., 2015, 407, 2095–2106 Search PubMed.
- G. Li, F. Ma, Q. Cao, Z. Zheng, K. DeLaney, R. Liu and L. Li, Nat. Commun., 2019, 10, 4697 Search PubMed.
- G. Li, Y. Liu and L. Li, Methods Mol. Biol., 2022, 2437, 143–157 CrossRef CAS PubMed.
- B. Geier, E. Gil-Mansilla, Z. Liutkeviciute, R. Hellinger, J. Vanden Broeck, J. Oetjen, M. Liebeke and C. W. Gruber, PNAS Nexus, 2023, 2, pgad144 CrossRef PubMed.
- H. Zeng, Y. Qin, E. Du, Q. Wei, Y. Li, D. Huang, G. Wang, J. A. Veenstra, S. Li and N. Li, J. Proteome Res., 2021, 20, 1217–1228 CrossRef CAS PubMed.
- K. D. B. Anapindi, N. Yang, E. V. Romanova, S. S. Rubakhin, A. Tipton, I. Dripps, Z. Sheets, J. V. Sweedler and A. A. Pradhan, Mol. Cell. Proteomics, 2019, 18, 2447–2458 CrossRef PubMed.
- W. Wu, M. Ma, A. E. Ibarra, G. Lu, V. P. Bakshi and L. Li, J. Am. Soc. Mass Spectrom., 2023, 34, 1549–1558 CrossRef CAS PubMed.
- J. A. A. Resoles and E. T. Yu, Sci. Rep., 2025, 15, 7032 CrossRef CAS PubMed.
- E. Hayakawa, C. Guzman, O. Horiguchi, C. Kawano, A. Shiraishi, K. Mohri, M. F. Lin, R. Nakamura, R. Nakamura, E. Kawai, S. Komoto, K. Jokura, K. Shiba, S. Shigenobu, H. Satake, K. Inaba and H. Watanabe, Nat. Ecol. Evol., 2022, 6, 1438–1448 CrossRef PubMed.
- H. M. Jiang, Z. Yang, Y. Y. Xue, H. Y. Wang, S. Q. Guo, J. P. Xu, Y. D. Li, P. Fu, X. Y. Ding, K. Yu, W. J. Liu, G. Zhang, J. Wang, H. B. Zhou, A. J. Susswein and J. Jing, Sci. Rep., 2022, 12, 1213 CrossRef CAS PubMed.
- R. Bruderer, O. M. Bernhardt, T. Gandhi, Y. Xuan, J. Sondermann, M. Schmidt, D. Gomez-Varela and L. Reiter, Mol. Cell. Proteomics, 2017, 16, 2296–2309 CrossRef CAS PubMed.
- K. DeLaney, M. Hu, W. Wu, M. P. Nusbaum and L. Li, Anal. Bioanal. Chem., 2022, 414, 533–543 CrossRef PubMed.
- R. Chen, M. Ma, L. Hui, J. Zhang and L. Li, J. Am. Soc. Mass Spectrom., 2009, 20, 708–718 Search PubMed.
- A. Chawade, E. Alexandersson and F. Levander, J. Proteome Res., 2014, 13, 3114–3120 Search PubMed.
- K. D. B. Anapindi, E. V. Romanova, B. R. Southey and J. V. Sweedler, Talanta, 2018, 182, 456–463 CrossRef CAS PubMed.
- C. S. Sauer and L. Li, Methods Enzymol., 2022, 663, 235 CAS.
- R. Liu, P. Wei, C. Keller, N. S. Orefice, Y. Shi, Z. Li, J. Huang, Y. Cui, D. C. Frost and S. Han, Anal. Chem., 2020, 92, 14021–14030 Search PubMed.
- S. Wiese, K. A. Reidegeld, H. E. Meyer and B. Warscheid, Proteomics, 2007, 7, 340–350 Search PubMed.
- S. E. Ong and M. Mann, Nat. Protoc., 2006, 1, 2650–2660 CrossRef CAS PubMed.
- I. I. Stewart, T. Thomson and D. Figeys, Rapid Commun. Mass Spectrom., 2001, 15, 2456–2465 CrossRef CAS PubMed.
- F. Y. Che and L. D. Fricker, J. Mass Spectrom., 2005, 40, 238–249 CrossRef CAS PubMed.
- R. Chen, M. Xiao, A. Buchberger and L. Li, J. Proteome Res., 2014, 13, 5767–5776 CrossRef CAS PubMed.
- Y. Zhang, A. Buchberger, G. Muthuvel and L. Li, Proteomics, 2015, 15, 3969–3979 Search PubMed.
- A. R. Buchberger, C. S. Sauer, N. Q. Vu, K. DeLaney and L. Li, J. Proteome Res., 2020, 19, 1548–1555 Search PubMed.
- C. K. Frese, A. J. Boender, S. Mohammed, A. J. Heck, R. A. Adan and A. F. Altelaar, Anal. Chem., 2013, 85, 4594–4604 Search PubMed.
- T. Schmidlin, A. J. Boender, C. K. Frese, A. J. Heck, R. A. Adan and A. F. Altelaar, Anal. Chem., 2015, 87, 9966–9973 CrossRef CAS PubMed.
- J. A. Hutchinson, S. Burholt and I. W. Hamley, J. Pept. Sci., 2017, 23, 82–94 Search PubMed.
- Y. Zhang, C. Y. Liu, W. C. Chen, Y. C. Shi, C. M. Wang, S. Lin and H. F. He, Cell Biosci., 2021, 11, 151 CrossRef CAS PubMed.
- L. Xi, Y. Jin, E. A. Parker, P. Josh, A. Jones, G. Wijffels and M. L. Colgrave, Pept. Sci., 2012, 98, 357–366 Search PubMed.
- C. Vocat, M. Dunand, S. A. Hubers, N. Bourdillon, G. P. Millet, N. J. Brown, G. Wuerzner, E. Grouzmann and P. J. Eugster, Anal. Chem., 2020, 92, 859–866 CrossRef CAS PubMed.
- E. E. Chambers, M. E. Lame, J. Bardsley, S. Hannam, C. Legido-Quigley, N. Smith, K. J. Fountain, E. Collins and E. Thomas, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2013, 938, 96–104 Search PubMed.
- E. E. Niederkofler, D. A. Phillips, B. Krastins, V. Kulasingam, U. A. Kiernan, K. A. Tubbs, S. M. Peterman, A. Prakash, E. P. Diamandis, M. F. Lopez and D. Nedelkov, PLoS One, 2013, 8, e81125 Search PubMed.
- P. J. Eugster, H. Chtioui, A. Herren, M. Dunand, D. Cappelle, J. Bourquin, T. Buclin and E. Grouzmann, J. Pharm. Biomed. Anal., 2019, 166, 205–212 Search PubMed.
- M. van Bentum and M. Selbach, Mol. Cell. Proteomics, 2021, 20, 100165 Search PubMed.
- H. Lu, L. A. Cassis, C. W. Kooi and A. Daugherty, Hypertens. Res., 2016, 39, 492–500 Search PubMed.
- M. Olkowicz, A. Radulska, J. Suraj, A. Kij, M. Walczak, S. Chlopicki and R. T. Smolenski, J. Chromatogr. A, 2015, 1393, 37–46 Search PubMed.
- N. Pavo, G. Goliasch, R. Wurm, J. Novak, G. Strunk, M. Gyongyosi, M. Poglitsch, M. D. Saemann and M. Hulsmann, Clin. Chem., 2018, 64, 597–608 CrossRef CAS PubMed.
- L. Tan, Z. Yu, X. Zhou, D. Xing, X. Luo, R. Peng and Y. Tang, J. Chromatogr. A, 2015, 1411, 69–76 Search PubMed.
- T. W. Barber, J. A. Brockway and L. S. Higgins, Acta Neurol. Scand., 1970, 46, 85–92 Search PubMed.
- M. C. Chappell, Am. J. Physiol. Heart Circ. Physiol., 2016, 310, H137–H152 CrossRef PubMed.
- C. Lombard-Banek, Z. Yu, A. P. Swiercz, P. J. Marvar and P. Nemes, Anal. Bioanal. Chem., 2019, 411, 4661–4671 Search PubMed.
- B. Cockx, S. Van Bael, R. Boelen, E. Vandewyer, H. Yang, T. A. Le, J. J. Dalzell, I. Beets, C. Ludwig and J. Lee, Mol. Cell. Proteomics, 2023, 22, 100479 CrossRef CAS PubMed.
- L. D. Fricker, Methods Mol. Biol., 2024, 2758, 213–225 Search PubMed.
- L. D. Fricker, A. K. Tashima, A. K. Fakira, U. Hochgeschwender, W. C. Wetsel and L. A. Devi, Cell Chem. Biol., 2021, 28, 105–112 e104 Search PubMed.
- P. C. Chan-Andersen, E. V. Romanova, S. S. Rubakhin and J. V. Sweedler, J. Biol. Chem., 2022, 298, 102254 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |