
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging gut microbial glycoside hydrolase inhibitors
Mark E.
Kowalewski
 a and
Matthew R.
Redinbo
*abc
a and
Matthew R.
Redinbo
*abc
aDepartment of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, North Carolina, USA. E-mail: redinbo@unc.edu
bDepartment of Chemistry, University of North Carolina, Chapel Hill, North Carolina, USA
cDepartment of Microbiology and Immunology, and Integrative Program for Biological and Genome Sciences, University of North Carolina, Chapel Hill, North Carolina, USA
First published on 11th June 2025
Abstract
The human gut microbiota has been linked to numerous diseases through their metabolism of molecules in the gastrointestinal tract. Post-translational glycosylation is applied to many secreted proteins, including mucins and immunoglobulins, and glycosides are present in diet and generated by host metabolism systems. Thus, glycosides are key targets for degradation by gut microbial glycoside hydrolases (GHs). Indeed, diverse xenobiotic compounds, including therapeutics and dietary phytochemicals, along with endobiotics like neurotransmitters and hormones, are conjugated to monosaccharides making them substrates for GH enzymes. A range of GH inhibitors have been developed to study lysosomal storage diseases, treat viral infections, and to address type II diabetes. Recently, GH inhibitors have offered promising avenues for investigating gut microbial GHs and their influence on host health and disease. In this review we describe the growing classes of GH inhibitors and their applications in studying gut microbial GHs that target host-derived glycans and dietary and drug-xenobiotic molecules. We also review the use of GH-targeting activity-based probes to pinpoint specific proteins expressed by the gut microbiota that influence molecular and phenotypic outcomes. As we deepen our understanding of gut microbial GH function, we will further elucidate the roles played by the microbiota in host physiology and disease toward potential therapeutic interventions that target non-host factors in acute and chronic disorders.
Introduction
The human gut microbiota is composed of trillions of bacteria, encoding approximately 150 times more genetic information than the human genome.1,2 Gut microbiota composition has been associated to a variety of diseases including obesity,3 inflammatory bowel disease (IBD)4 and chronic kidney disease.5 The microbiota plays a key role in the metabolism of diet-derived compounds, hormones, neurotransmitters, host-glycans and drug–molecules.6–11 The metabolism of these molecules by the gut microbiota has implications in IBD,12 diabetes,13,14 drug efficacy and toxicity.8,15 Many of these substrates exist as glycoconjugates and glycoproteins, making them targets of carbohydrate active enzymes (CAZymes) produced by gut bacteria.16 A promising avenue for studying metabolism of these compounds is through CAZyme-targeting small-molecule inhibitors.The gut microbiota encodes a diverse suite of CAZymes, including glycoside hydrolases (GHs), sulfatases, carbohydrate esterases, polysaccharide lyases, and glycosyltransferases. The largest category of CAZymes are GHs, with nearly 200 families annotated according to sequence homology.16,17 GHs are active on glycosidic bonds, enabling the degradation of polysaccharides, oligosaccharides, sugar-conjugated small-molecules, and glycoproteins into monosaccharides to be used as an energy source for bacteria.16,18–20
Gut bacterial GHs are crucial in processing diet-derived plant polysaccharides and host-derived glycoproteins such as mucins and immunoglobulins, which are highly abundant in the gastrointestinal (GI) tract.21–25 The human genome lacks much of the machinery to degrade these glycans, relying on the gut microbiota to accomplish this chemistry. Given these substantial roles of gut microbial CAZymes in these processes (reviewed by Wardman et al.26), CAZyme inhibitors provide a valuable resource for uncovering the gut microbiota's role in human physiology and disease, and could serve as potential avenues for therapeutic intervention.
Due to the abundance of glycoconjugates and their importance in biology, numerous GH inhibitors have been discovered and developed. However, to date, the focus of GH inhibitors has generally been applied to host disease states like lysosomal storage diseases,27 type II diabetes,13,14 and viral infections,28 and has thus been outside the scope of the gut microbiota. This review examines small-molecule inhibitors and chemical probes that target GHs involved in O-glycan, N-glycan, dietary xenobiotic and drug xenobiotic metabolism in the gut microbiota.
Carbohydrate substrates for gut microbes
Mucin O-glycans
Mucin glycoproteins comprise the protective mucus layer that lines the GI tract, providing a barrier between the gut microbiota and intestinal epithelium. Mucins are highly glycosylated, with ∼80% of their total mass coming from the oligosaccharides that decorate its structure.29,30 The most abundant mucin in the gut is MUC2 and because of the high degree of glycosylation, MUC2 forms a gel-like matrix acting as a barrier separating the intestinal epithelium and intestinal microflora. The Ser- and Thr-linked O-glycans commonly contain the monosaccharides galactose, N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose, and sialic acid (Fig. 1).31 Additionally, the individual sugar moieties in these glycans can be decorated with a sulfate group. The chemical complexity of the glycosylations and sulfations requires a collection of gut microbial enzymes to achieve mucin oligosaccharide degradation. Several gut bacteria have been shown to metabolize mucin O-glycans, notably the well-studied Bacteroides thetaiotaomicron and Akkermansia muciniphila.32–34 Degradation of this protective layer has been associated with ulcerative colitis (UC),35–37 and understanding how inhibition of proteins responsible for specific steps in mucin O-glycan metabolism offers an avenue to more closely examine the processes that underpin mucin degradation by the gut microbiota. | ||
| Fig. 1 Substrates for glycoside hydrolases (GH) in the gastrointestinal (GI) tract. The range of carbohydrate substrates present in the GI tract including host-derived glycans (mucins & immunoglobulins), xenobiotic compounds (therapeutics and phytochemicals), and host-generated endobiotic compounds. The GH family responsible for removing the glycans are indicated, showing the diverse enzymes needed to degrade carbohydrate substrates. | ||
Host and dietary N-glycans
Non-barrier glycoproteins, like immunoglobulin A (IgA), are also abundant in the GI tract and are decorated by N-glycan oligosaccharides attached to Asn residues (Fig. 1).38–40N-Glycans are obtained through the diet, including α-mannan from yeast and N-glycans from plant-derived dietary fiber. N-Glycans usually contain the monosaccharides GlcNAc, fucose, mannose, sialic acid and galactose. Pathogenic microbes can utilize these glycans, providing a fitness advantage and has been implicated as a mechanism for pathogens to evade the immune system.41,42 Furthermore, IgA coating, or attachment to bacteria, is associated with the ability of certain commensal microbes to invade the colonic mucus layer.43 Inhibiting GHs involved in N-glycan degradation may provide mechanistic insights into how gut microbes use this to their advantage and potentially contribute to dysbiosis.Xenobiotics
The gut microbiome also plays essential roles in the metabolism of therapeutic xenobiotics.11 Zimmerman et al. analysed the ability of gut bacteria to metabolize 271 drugs, with the majority being chemically modified by at least one bacterial strain.11 An early well-known characterized example of drug metabolism by the gut microbiota is the chemotherapeutic irinotecan.8,44,45 The active form of irinotecan is conjugated to a glucuronic acid sugar, inactivating it and marking it for excretion through the GI tract (Fig. 1). GHs produced by gut microbes then remove the glucuronic acid and reactivate the drug, causing severe dose-limiting side effects.8 Small-molecule GH inhibitors have been shown to eliminate the gut toxicities of irinotecan,8,44,45 as well as numerous non-steroidal anti-inflammatory drugs and other therapeutics that damage the GI tract via a similar mechanism.46–50 Such reagents have helped to define the potential scope of therapeutically targeting the gut microbiota, with the goals of controlling drug toxicity and enhancing drug efficacy.Dietary glycans
The impact of dietary xenobiotics in the gut microbiota has recently emerged as a major research focus.10,51 Plant-based dietary compounds often exist as glucose conjugates, such as soy phytoestrogens (i.e. daidzin and genistin) and resveratrol (polydatin) (Fig. 1).10,52 Culp et al. demonstrated that these compounds can significantly alter gut microbial composition. Glycoside conjugates are generally less active on host systems than the aglycones created by gut microbial GHs.10 In plants, such glycosides are cloaked defence mechanisms against pathogens that are activated upon glycoside removal.10,52Recently, Kuziel et al. demonstrated the ability of gut microbes to metabolize dietary phenolic glycosides releasing active aglycones that influence host-health.53 Another class of diet-derived compounds are glucosinolates, which are abundant in cruciferous vegetables, like broccoli and cabbage.54 A recent paper highlights the ability of a commensal gut microbe, B. thetaiotaomicron, to release isothiocyanates conjugated to glucose, utilizing thio-specific GHs.55 These isothiocyanates have been implicated to possess anti-cancer properties.56
Dietary carbohydrates reach the GI tract and are exposed to gut microbes. Examples of dietary carbohydrates include cellulose and starches. Importantly, these molecules reach the GI tract largely intact, where they are then degraded by gut bacteria and fermented to produce short-chain fatty acids (SCFAs). Human milk oligosaccharides, and other dairy products, contain glycans that are not processed by human enzymes but are metabolized by gut microbial GHs. Indeed, the infant gut microbiota consists mostly of Bifidobacteria suggesting the potential in using such oligosaccharides as prebiotic compounds to promote certain species.57,58 Thus, inhibitors specific to relevant gut microbial GHs would help to define molecular mechanisms involved in diet, host and microbial factors.
Small-molecule CAZyme inhibitors
The complexity of the gut microbiota presents significant challenges for researchers seeking to define how these microbes influence human health. Small-molecule inhibitors provide generally actionable tools for understanding these intricate details and has broadly been reviewed by Woo et al.59 Thorough reviews of GH inhibitors have been presented recently by Asano et al.94 and Kim et al.;95 however, this is not the purpose of this review. Instead, here we focus on the applications of GH inhibitors in the context of the gut microbiota. Recent work has expanded the use of small-molecule inhibitors to investigate GHs in the gut microbiota.71 Host-targeting GH inhibitors can impact the function of the gut microbiota,96 and a recent study shows that common therapeutics influence endobiotic glycoconjugate availability by inhibiting gut microbial GHs.7 To date, most GH inhibitors have been substrate mimics, making selective targeting of microbial GHs difficult (Table 1). Indeed, the most prevalent class of GH inhibitors is derived from nitrogen-containing substrate analogues. Importantly, though, substrate-mimicking chemical probes have been developed and deployed to allow fluorescent labelling of specific GHs.97 These probes can also be conjugated to biotin to selectively enrich for GHs of interest, using streptavidin beads to increase sensitivity in proteomic studies.7| Enzyme | Substrate | Inhibitorsa | Significance | PDB | Ref. |
|---|---|---|---|---|---|
| a See relevant enzyme sections for names of inhibitor acronyms. | |||||
| α-Glucosidase | Carbohydrates, starch | Deoxynojirimycin | Type II diabetes | 3PHA (acarbose) | Tan et al.,60 Inouye et al.,61 Niwa et al.,62 Yoon et al.63 |
| Acarbose | 6C9X (voglibose) | ||||
| Miglitol | 6CA1 (miglitol) | ||||
| Voglibose | |||||
| Cyclophellitol-aziridine | |||||
| β-Glucosidase | Phytochemicals, plant polysaccharides | Isofagomine | Anti-inflammatory, anti-cancer, and antioxidant properties | 2J77 (nojirimcyin) | Street et al.,64 Saul et al.,65 Braun et al.,66 Gloster et al.,67 Beenakker et al.68 |
| Castanospermine | 5N6T (cyclophellitol) | ||||
| Conduritol B epoxide | |||||
| Cyclophellitol-aziridine | |||||
| β-Glucuronidase | Drug metabolites, endobiotic compounds | Saccharolactone | Drug toxicity, neurotransmitter and hormone levels | 8GEN (UNC10201652) | Wallace et al.,8 Simpson et al.,7 Pellock et al.,69 Jariwala et al.,70 Graboski et al.,71 Challa et al.,72 Roberts et al.,45 Boyland et al.,73 Rasmussen et al.,74 Awolade et al.,75 Pellock et al.76 |
| Uronic-noeurostegine | 8GEO (3-OH-desloratidine) | ||||
| Inhibitor 1 | 8GEQ (ceritinib) | ||||
| UNC10201652 | 3LPF (inhibitor1) | ||||
| UNC4917 | 6NZG (cyclophellitol) | ||||
| UNC10206581 | |||||
| TCH-3562 | |||||
| NCGC00253873 | |||||
| Ceritinib | |||||
| Desloratidine | |||||
| Cyclophellitol-aziridine | |||||
| β-Galactosidase | O-Glycans, N-glycans, GOS prebiotics, plant polysaccharides | PETG | Mucin-glycan and immunoglobulin-glycan degradation | 5A1A (PETG) | Bartesaghi et al.,77 Saur et al.,78 Huber et al.,79 De Bruyne et al.80 |
| Deoxygalactonojirimycin | 6TSH (nojirimycin) | ||||
| Cyclophellitol-aziridine | |||||
| α-Fucosidase | Host O-glycans and N-glycans | DFJ | Mucin-glycan and immunoglobulin-glycan degradation | 2EAC (DFJ) | Nagae et al.,81 Jiang et al.,82 Jiang et al.82 |
| Cyclophellitol-aziridine | 4WSK (cyclophellitol) | ||||
| α-Sialidase | O-Glycans, N-glycans | DANA | HMOs, developmental sialic acid, mucin degradation | 8AXI (DAN) | Shuoker et al.,83 Gut et al.,84 von Itzstein et al.,85 Kim et al.,86 Babu et al.87 |
| Zanamivir | 2YA7 (Zanamivir) | ||||
| Oseltamivir | |||||
| Peramivir | |||||
| β-Hexosamindase | O-Glycans, N-glycans | PUGNAc | Mucin-glycan and immunoglobulin-glycan degradation | 2VVS (PUGNAc) | Beer et al.,88 Knapp et al.,89 Macauley et al.90 |
| NAGT | |||||
| α-Mannosidase | Host N-glycans, alpha-mannans yeast cell-wall, plant N-glycans | Deoxymannojirimycin | Immunoglobulin-glycan degradation | 2WW0 (swainsonine) | Thompson et al.,91 Zhu et al.,92 Fuhrmann et al.93 |
| Swainsonine | 2WZS (mannoimidazole) | ||||
| Mannoimidazole | 2WVZ (kifunensine) | ||||
| Kifunensine | |||||
Broadly, GH inhibitors fall into several classes, most prominently including iminosugars (Fig. 2B), thiosugars (Fig. 2E), pyrrolidines (Fig. 2C), pyrrolizidines (Fig. 2D), fluorosugars (Fig. 2H), cyclophellitol-aziridines (Fig. 2G), and synthetic compounds97,98 (Fig. 2I, J and Table 1). Numerous structural studies have examined the binding modes of these inhibitors, offering a valuable resource for future inhibitor development (Fig. 3). Although a good portion of these compounds have been developed to treat lysosomal storage diseases, this review will discuss their potential in studying gut microbial CAZymes.
 | ||
| Fig. 2 (A) Chemical structures of monosaccharides, which are the targets for the glycoside hydrolases described in this review. Glycoside hydrolase inhibitor structural classes with representative chemical structures for each class: (B) afegostat, an iminosugar β-glucosidase inhibitor; (C) castanospermine, a pyrrolidine β-glucosidase inhibitor; (D) australine, a pyrrolizidine α-glucosidase inhibitor; (E) PETG, a thiosugar β-galactosidase inhibitor; (F) acarbose, a carbohydrate mimic α-glucosidase inhibitor (G) conduritol B epoxide, a cyclophellitol β-glucosidase inhibitor; (H) 6-azido-2,6-dideoxy-2-fluoro-β-D-galactosyl fluoride a fluorosugar β-galactosidase inhibitor and the synthetic β-glucuronidase inhibitors (I) inhibitor 1 and (J) UNC10201652. | ||
 | ||
| Fig. 3 Binding mode for (A) acarbose in complex with an α-glucosidase (3PHA), (B) PETG in complex with a β-galactosidase (6CVM), (C) deoxynojirimycin bound to an α-glucosidase (2JKE), (D) InhR1 in complex with a β-glucuronidase (5CZK), (E) UNC10206581 in complex with a β-glucuronidase (8UGT), (F) cyclophellitol in complex with a β-glucuronidase (6NZG), (G) fluorosugar in complex with an α-mannosidase (1QX1), (H) castanospermine bound to a β-glucosidase (2PWG). Inhibitors are coloured light orange and active site residues are coloured light blue. | ||
Glycoside hydrolases have traditionally been classified according to the two Koshland mechanisms,99,100 which utilize a catalytic nucleophile and acid/base residue to facilitate hydrolysis. These mechanisms differ in the stereochemistry of the anomeric carbon. Retaining GHs conserve the original configuration while inverting GHs reverse it. In the case of inverting GHs, the active site must accommodate a water nucleophile and therefore has a larger distance between the catalytic residues, providing unique active site architectures. These active site differences likely influence inhibitor binding and suggest mechanism-specific inhibitors could be developed for greater selectivity.
The mechanisms of GHs has been thoroughly reviewed previously by Davies and Henrisatt.101 Recently, a non-Koshland mechanism was discovered for bacterial GHs, utilizing anionic transition states, in contrast to cationic transition states observed in Koshland mechanisms.102 Notably the enzymes in this study show broad substrate promiscuity due to the ability for these GHs to accommodate both α- and β-anomers.102
These different mechanisms highlight the importance of diverse GH inhibitor classes. Current inhibitors may show preference for GHs of a certain catalytic mechanism over others. Finally, this discovery of non-Koshland GHs, particularly in the context of the gut microbiota, stresses the importance for new classes of GH inhibitors be discovered and to investigate the utility of current GH inhibitors against these non-Koshland GHs.
α-Glucosidase inhibitors
Glucose is an abundant monosaccharide in sucrose, maltooligosaccharides and starch. The first known α-glucosidase inhibitor, nojirimycin (Fig. 4A), was discovered in 1966 as an antibiotic produced by Streptomyces61 and then shown to be an α- and β-glucosidase inhibitor in 1970.62 A more stable form of this inhibitor, deoxynojirimycin (Fig. 4B), was identified in mulberry leaves and then isolated from Bacillus subtilis DSM704.106,107 Deoxynojirimycin served as a blueprint for subsequent inhibitor development, including those targeting GHs beyond glucosidases. Subsequent α-glucosidase inhibitors include acarbose (Fig. 2F), voglibose (Fig. 4C) and miglitol (Fig. 4D).108 They were developed to target host α-glucosidases to reduce carbohydrate degradation in the intestines to reduce free glucose levels as a therapeutic intervention for Type II diabetes.13,14 While originally focused on host enzymes, they also inhibit GHs in the gut microbiota.63 Tan et al. reported that a GH31 α-glucosidase from Blaubia obeum is inhibited by acarbose (Fig. 3A), voglibose and miglitol (Table 2).60 Notably, voglibose and acarbose have low absorption,109,110 and therefore remain mostly in the GI tract, where they can interact with microbial α-glucosidases. Furthermore, these inhibitors have been shown to impact the polysaccharide utilization loci employed by microbes in the Bacteroidota phylum, limiting their nutrient scavenging and thus impacting their growth through non-microbicidal means.105,111,112 Another study found increased short-chain fatty acid production, by-products of carbohydrate fermentation, in patients with Type II diabetes receiving acarbose, indicating that dietary fiber remains intact for fermentation by the gut microbiota.113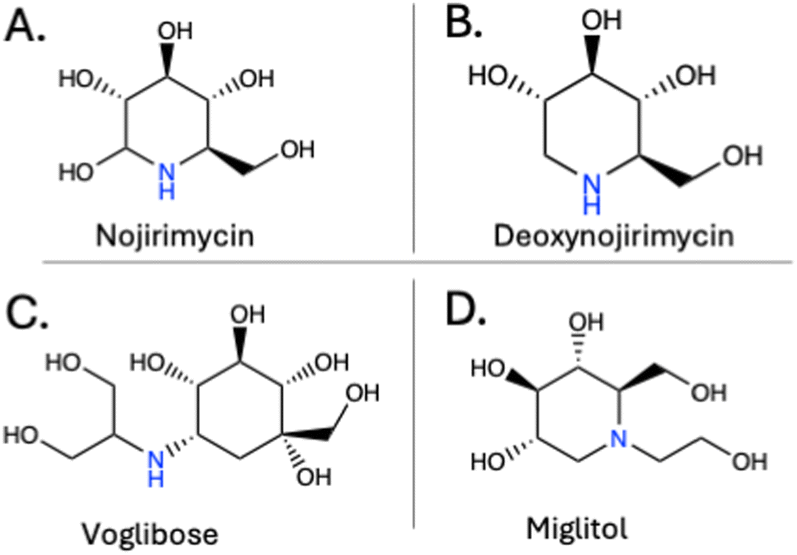 | ||
| Fig. 4 Chemical structures for representative α-glucosidase inhibitors (A) nojirimycin, (B) deoxynojirimycin, (C) voglibose, and (D) miglitol. | ||
| Compound | Microbial target | K i/IC50 | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| a IC50. b K i. | |||||
| Deoxynojirimycin | N/A (not available) | N/A | Competitive | In vivo studies | Li et al.103 |
| Voglibose | Blaubia obeum GH31 | 8.3 μMa | Competitive | In vivo studies | Tan et al.60 |
| Bae et al.104 | |||||
| Miglitol | Blaubia obeum GH31 | 32 μMa | Competitive | FDA approval for type II diabetes | Tan et al.60 |
| Acarbose | BoSusG | 2.2 μMa | Competitive | FDA approval for type II diabetes | Brown et al.105 |
| SusG | 68 μMa | ||||
| BoSusA | 123 nMb | ||||
| SusA | 95 nMb | ||||
| BoSusB | 69 nMb | ||||
| SusB | 54 nMb | ||||
| BoGH97D | 134 nMb | ||||
| BtGH97H | 162 nMb | ||||
Recent data have also indicated that the gut microbiota influences the effectiveness of the antidiabetic drug, acarbose.96,114,115 Work from the Donia Lab showed that gut microbes produce enzymes capable of deactivating acarbose via phosphorylation.96 Furthermore, Tian et al. found that increased Klebsiella grimontii abundance reduced efficacy in patients treated with acarbose and that acarbose efficacy is reduced in mice treated with K. grimontii.114K. grimontii encodes an acarbose-targeting glucosidase capable of degrading acarbose and reducing its inhibitory effects.114 Thus, using small molecular inhibitors to target α-glucosidases produced by gut bacteria alters microbial composition and reduces the level of glucose released from complex polysaccharides.
β-Glucosidase inhibitors
Plant phytochemicals are commonly conjugated to a glucose sugar that serves to inactivate the compound until its use is required. Many phytochemicals have antimicrobial properties, acting as a defence mechanism against pathogens, and are thus stored by plants as inactive glucoside conjugates.52 Plant β-glucosidases activate these reagents when a pathogen is detected. Phytochemicals are obtained by humans through diet and reach the GI tract where they are exposed to microbial β-glucosidases that have the capacity to remove the conjugating glucose, changing the activity of the ingested compound. Well studied phytochemicals include the soy-derived phytoestrogens, daidzein and genistein, which are of particular interest due to their activity on estrogen receptors,116 potentially interrupting estrogen signalling. Phytoestrogens are commonly found as inactive glucose conjugates, such as daidzin and genistin, making them targets for gut microbial β-glucosidases. The grape-derived phytochemical resveratrol is also commonly found in its glucoside conjugate, polydatin. Phytochemicals are implicated in host health due to their anti-cancer, anti-inflammatory, and antioxidant properties (Fig. 5A).117 Beyond their roles in activating glucose-conjugated small molecules, gut microbial β-glucosidases are essential proteins for degrading cellulose.118 | ||
| Fig. 5 (A) Daidzin activation pathway by gut microbial β-glucosidases, producing daidzein which has anti-inflammatory properties. (B) Diclofenac reactivation pathway by gut microbial β-glucuronidases resulting in intestinal ulcers. | ||
There are several β-glucosidase inhibitors: afegostat (isofagomine) (Fig. 2B), an iminosugar designed for treating Gaucher's disease, a lysosomal storage disorder;64 castanospermine (Fig. 2C), an alkaloid identified from Castanospermum australe seeds,65 and conduritol B epoxide (Fig. 2G), a mechanism-based covalent inhibitor that served as a prototype for covalent probe development.66 These inhibitors have seen limited application to the gut microbiota but have been examined as a therapeutic approach for Clostridium difficile infections (Table 3). Paparella et al. studied the ability of two β-glucosidase inhibitors to block the Clostridoides difficile toxins TcdA and TcdB.119 In this work, the β-glucosidase inhibitors isofagomine and noeuromycin reduced TcdA- and TcdB-mediated cell toxicities by blocking UDP binding to TcdA and TcdB glucosyltransferase domains (GTs) and preventing their activation of the host Rho GTPase.119 While this work focuses on C. difficile infection, future studies are needed to examine the ability of these inhibitors to potentially modulate microbial composition or alter phytochemical levels.
| Compound | Target | K i/IC50 | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| a IC50. b K i. | |||||
| Afegostat | β-Glucocerebrosidase | 5 nMa | Competitive | Clinical trials | Street et al.,64 Paparella et al.119 |
| TcdB | 4.8 μMb | ||||
| Castanospermine | Almond GH1 β-glucosidase | 10 μMb | Competitive | In vivo studies | Saul et al.,65 Paparella et al.119 |
| TcdB | 3.4 mMb | ||||
| Conduritol B epoxide | β-Glucocerebrosidase | 4.3 μMa | Covalent inhibitor | In vivo studies | Kuo et al.120 |
β-Glucuronidase inhibitors
Glucuronidation is a chemical modification that takes place primarily in the liver and gut epithelium and plays a central role in phase II metabolism. Glucuronidation most often inactivates compounds and typically marks them for excretion via the urine or bile.121 However, β-glucuronidases (GUS), produced by gut bacteria, can reverse this process in the GI tract by removing the glucuronide and reactivating the aglycone (Fig. 5B). The first reported GUS inhibitor, saccharolactone (Fig. 6A), acts as a substrate mimic,73 and other substrate mimics such as uronic-Noeurostegine (Fig. 6B)74 and iminosugars75 have also been reported. Because these compounds mimic the native substrate, they broadly inhibit GUS enzymes, targeting both host and bacterial GUS. The lethal lysosomal storage disease Sly Syndrome is caused by inactivating mutations of human GUS (hGUS). Thus, it has been considered important to employ GUS inhibitors that are selective for microbial GUS over hGUS. | ||
| Fig. 6 Chemical structures for representative β-glucuronidase inhibitors (A) saccharolactone, (B) noeurostegine, (C) TCH-3562, (D) UNC10206581, (E) UNC4917, (F) desloratidine, (G) ceritinib, (H) NCGC00253873, (I) vortioxetine, (J) amentoflavone, (K) scutellarein, and (L) quercetin. Reactive piperazine and piperadine moieties are highlighted in blue. | ||
Glucuronidation is a common metabolic step for drug compounds, inactivating them and marking them for excretion. A well-examined example is the glucuronidation of SN38, the active metabolite of irinotecan, a chemotherapy drug for colon and pancreatic cancer. Glucuronidated SN38-G is marked for excretion through the bile but when it reaches the gut lumen it is exposed to gut microbial GUS, that remove the glucuronic acid moiety. The reactivated antineoplastic drug SN38 moiety kills intestinal epithelial cells leading to severe, delayed and dose-limiting diarrhoea. The Redinbo Lab showed in 2010 that inhibiting gut microbial GUS alleviates GI toxicity caused by SN38 in a rodent model.8 This initial compound, Inhibitor 1, was identified through a high throughput screen and potently inhibits E. coli GUS (Fig. 2I and 3D).
In subsequent work, microbial GUS were found to be a structurally diverse family, with two loop (Loop 1, Loop 2) regions of varying length and some containing a flavin mononucleotide (FMN) binding site.76,122 These structural differences influence the substrate preferences of microbial GUS. Second generation GUS inhibitors, UNC10201652 (Fig. 2J and 3E) and UNC4917 (Fig. 6E), identified from the same high throughput screen that discovered Inhibitor 1, utilize a piperazine warhead that intercepts the catalytic cycle of the enzyme.69 The secondary piperazine amine forms a covalent bond with a glucuronic acid in the active site. Structural studies from the Redinbo Lab have identified a key motif that differentiates GH2 GUS from GH2 β-galactosidases.44 This NxK motif in GUS contacts the carboxyl group on the glucuronic acid and is responsible for its recognition (Fig. 7A). In contrast, GH2 β-galactosidases lack this motif (Fig. 7B). Similarly, other GHs have residues responsible for recognizing specific monosaccharides based on the orientation of hydroxyls. Such features could be exploited in future work to develop inhibitors that are selective for certain enzymes within a GH family. Recent work by Graboski et al. optimized the scaffold of UNC10201652 through an SAR campaign creating a potent FMN- and Loop-1 selective GUS inhibitor, UNC10206581.71 Additionally, the authors illustrate the utility of such inhibitors to target GUS extracted from faecal lysates and in bacterial cell culture. These inhibitors are promising due to their specificity for a structural category of GUS enzymes responsible for liberating small-molecule glucuronide conjugates. Furthermore, these inhibitors can serve as a prototype for developing similar mechanism-based inhibitors of other GHs.
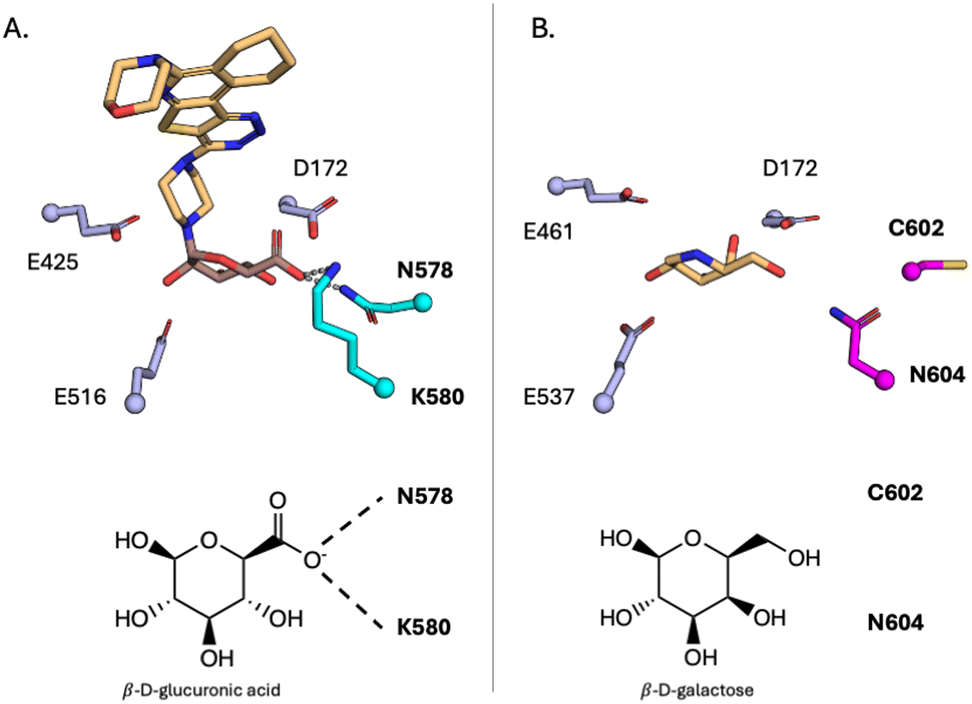 | ||
| Fig. 7 Active site differences for GH2 β-glucuronidases (GUS) and β-galactosidases (GAL). (A) Active site of a GH2 GUS (PDB 8GEN) bound to UNC10201652-glucuronide, with residues N578 and K580 highlighted in cyan. These residues recognize the carboxyl on glucuronic acid and differentiate enzymes within the GH2 family. (B) Active site of a GH2 GAL bound to deoxygalactonojirimycin (PDB 6TSH) with C602 and N604 highlighted in magenta. These residues differentiate the active sites of these proteins and can therefore be exploited to develop selective inhibitors. | ||
A wide range of human therapeutics reach the gut as inactive glucuronides, and several have been shown to be reactivated by gut microbial GUS including regorafenib,47 NSAIDs48–50,123,124 and the immunosuppressant mycophenolate.46 Modulation of GUS by small molecule inhibitors can alleviate toxicities associated with these therapeutics.69,73–76,121,122 The ubiquitous consumer product toxin triclosan is also glucuronidated and has been shown to reach the human gut and to cause GI toxicity and carcinogenesis.125 Blocking gut microbial GUS was recently found to prevent triclosan activation and to prevent colitis in a mouse model.126 New classes of E. coli GUS inhibitors identified through in silico screening efforts have recently been reported, providing a diverse collection of GUS targeting small-molecule inhibitors (Fig. 6 and Table 4).72,127,128 In all cases, the GUS inhibitors being developed are selective for microbial GUS over hGUS.8,44,45,69,71 Gut microbial GUS inhibitor results have helped to establish that selective compounds may be developed and deployed to modulate other intestinal symbiote GH proteins in future work.
| Compound | Microbial target | IC50 | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| Saccharolactone | C. perfringens | 6.2 μM | Competitive | In vitro | Bai et al.129 |
| E. coli | 28 μM | ||||
| S. pasteuri | 4.7 μM | ||||
| TCH-3562 | E. coli | 36 nM | Non-competitive | In vitro | Cheng et al.127 |
| Inhibitor 1 | E. coli | 283 nM | Competitive | In vivo | Wallace et al.8 |
| UNC10201652 | E. coli | 100 nM | Competitive | In vivo | Biernat et al.,136 Simpson et al.7 |
| E. eligens | 410 nM | ||||
| S. agalactiae | 133 nM | ||||
| C. perfringens | 26 nM | ||||
| UNC10206581 | E. coli | 29 nM | Competitive | In vitro | Graboski et al.71 |
| E. eligens | 88 nM | ||||
| UNC4917 | E. coli | 80 nM | Competitive | In vitro | Pellock et al.69 |
| Ceritinib | E. coli | 48 μM | Competitive | FDA approval for ALK–positive metastatic NSCLC | Simpson et al.7 |
| S. agalactiae | 12 μM | ||||
| C. perfringens | 3.4 μM | ||||
| E. eligens | 23 μM | ||||
| Gemmiger L1 | 45 μM | ||||
| Gemmiger FMN | 5.0 μM | ||||
| R. hominis 2 | 11 μM | ||||
| R. inulinvorans | 10 μM | ||||
| F. prausnitzii | 8.0 μM | ||||
| R. gnavus 3 | 98 μM | ||||
| Desloratidine | S. agalactiae | 81 μM | Competitive | FDA approved antihistamine | Simpson et al.7 |
| C. perfringens | 47 μM | ||||
| Gemmiger FMN | 38 μM | ||||
| R. hominis | 36 μM | ||||
| R. inulinvorans | 22 μM | ||||
| Vortioxetine | E. coli | 48 μM | Competitive | FDA approved antidepressant | Simpson et al.7 |
| S. agalactiae | 6.0 μM | ||||
| C. perfringens | 13 μM | ||||
| E. eligens | 8.0 μM | ||||
| Gemmiger L1 | 38 μM | ||||
| Gemmiger FMN | 31 μM | ||||
| R. hominis 2 | 10 μM | ||||
| R. inulinvorans | 0.3 μM | ||||
| F. prausnitzii | 8.0 μM | ||||
| NCGC00253873 | E. coli | 3.8 μM | Competitive | In vitro | Challa et al.72 |
| Scutellarein | E. coli | 5.8 μM | Competitive | Clinical trials | Weng et al.133 |
| Duan et al.137 | |||||
| Chen et al.138 | |||||
| Quercetin | E. coli | 21 μM | Competitive | Clinical trials | Weng et al.133 |
| Ferry et al.139 | |||||
| Amentoflavone | C. perfringens | 2.4 μM | Non-competitive | In vitro | Bai et al.129 |
| S. pasteuri | 2.9 μM | ||||
| E. coli | 3.4 μM |
Dietary flavonoids have been identified as microbial GUS inhibitors, highlighting the diverse compounds that inhibit microbial GUS.129,130 Flavonoid-based formulations have been of significant interest for alleviating chemotherapy-induced toxicity in clinical trials and other animal studies.131–133 Examples of flavonoids that inhibit microbial GUS are amentoflavone, scutellarein, and quercetin (Fig. 6J–L and Table 4). These compounds are ubiquitous in plants, and they highlight a key mechanism by which diet influences host physiology, potentially leading to avenues for alleviating chemotherapy-induced toxicity. Studies such as these suggest the potential for developing more potent GUS inhibitors based on these natural products.
While drug and xenobiotic inactivation has been a major focus of the gut GUS-mediated reactivation studies outlined above, a wide range of endobiotic hormones, neurotransmitters and other compounds also reach the mammalian GI tract as inactive glucuronides. A recent paper begins to highlight the intricate role of non-host GUS in modulating neurotransmitter and hormone levels.7 Simpson et al. showed that the glucuronides of serotonin, dopamine, estrone, estradiol and thyroxine are all substrates for specific sets of gut microbial GUS.7 They also demonstrated that commonly used medications at physiologically relevant concentrations can modulate GUS activity, reducing gut and systemic levels of serotonin. The drugs that appear capable of causing these effects contain either a piperazine or piperadine secondary amine that intercept the catalytic cycle of gut microbial GUS using the same mechanism employed by second-generation GUS inhibitors. These results indicate many drugs influence endobiotic homeostasis via gut microbial GUS enzymes, and may be capable of impacting other GHs, as well. Finally, several of the medications shown recently to inhibit gut microbial GUS are psychoactive drugs employed for diseases associated with serotonin and dopamine levels, suggesting that their efficacies may in part be due to effects on microbial as well as host factors.7,9,134,135 These data further support the conclusion that microbial symbiotes play important roles in host immunity, inflammation, neurological, and hormonal changes via mechanisms yet to be fully elucidated.
β-Galactosidase inhibitors
Many GH substrates in the GI tract contain galactose, including mucin glycans, galacto-oligosaccharides (GOS), N-glycans, and human milk oligosaccharides (HMOs). Several commensal gut microbes produce endo- and exo-β-galactosidases that are integral to mucin degradation.140 Potent β-galactosidases inhibitors include 1-deoxy-galactonojirimycin (Fig. 8A), galactosylamine (Fig. 8B),79 and 2-phenylethyl β-D-thiogalactoside (PETG) (Fig. 2E and 3B).80 To date, these inhibitors have been studied in the context of lysosomal storage disorders by targeting host enzymes.141 Interestingly, dietary compounds have been found to influence β-galactosidase activity. For example, caffeine and theophylline mildly inhibit E. coli β-galactosidase,142 suggesting dietary xenobiotics influence galactose liberation by gut microbes. | ||
| Fig. 8 Chemical structures for representative β-galactosidase inhibitors (A) 1-deoxy-galactonojirimycin, (B) galactosylamine, and (C) galactoisofagomine. | ||
A recent study from Zeng et al. demonstrated that a β-galactosidase from Lactobacillus vaginalis activates the soy phytoestrogen daidzein, which in turn reduced acetaminophen toxicity by inhibiting farnesyl diphosphate synthase, a ferroptosis pathway enzyme.143 The researchers show that the beneficial effects from daidzein were eliminated when mice were dosed with the β-galactosidase inhibitor D-Ribono-1,4-lactone, uncovering the importance of β-galactosidases in the availability of beneficial phytochemicals.143 This also indicates that some β-galactosidases can act on glucose-conjugated small molecules. Additionally, β-galactosidases are essential proteins for the degradation of GOS, and studies show that GOS increases the abundance of lactose degrading microbes β-galactosidase activity.144 The use of β-galactosidase inhibitors to study gut microbial processes to this point has been limited. However, these reagents offer valuable utility in future studies for uncovering the microbes and microbial factors that are responsible for the degradation key gut substrates (Table 5).
| Compound | Target | K i | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| 1-Deoxy-galactonojirimycin | S. pneumoniae BgaA | 34 μM | Competitive | In vitro | Singh et al.145 |
| Galactoisofagomine | S. pneumoniae BgaA | 25 nM | Competitive | In vitro | Singh et al.145 |
| Galactosylamine | E. coli β-galactosidase | 59 μM | Competitive | In vitro | Huber et al.79 |
| PETG | E. coli β-galactosidase | 8 μM | Competitive | In vitro | Hadd et al.146 |
α-Fucosidase inhibitors
Fucose is a common monosaccharide found in host-derived glycans, including mucins and immunoglobulins, both of which are abundant in the gut. Fucose is generally added via an α1,2 linkage to galactose or through an α1,3/4/6 linkage to GlcNAc and is commonly found at the termini of glycans. Two main GH families exhibit fucosidase activity: GH29 and GH95. The GH95 family is specific for α1,2 linkages while GH29 is divided into two subfamilies (GH29A and GH29B) based on substrate preference. Fucose is also a prevalent monosaccharide in HMOs, highlighting the importance of microbial α-fucosidases in infant development.147 The iminosugar fucosidase inhibitor, deoxyfuconojirimycin (DFJ) (Fig. 9A), was originally developed for treating the lysosomal storage disorder, fucosidosis.148 DFJ is a potent inhibitor for GH29 fucosidases and effectively inhibits the two human fucosidases, FUCA1 and FUCA2.149,150 More potent inhibitors of fucosidases were developed by modifying DFJ with various aglycone groups at the C1 position, leading to compound 2 (Fig. 9B) with a picomolar binding affinity for Corynebacterium sp. Fucosidase (Table 6).151 | ||
| Fig. 9 Chemical structures for representative α-fucosidase inhibitors (A) 1-deoxy-fuconojirimycin, and (B) compound 2. | ||
| Compound | Microbial target | K i/IC50 | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| a IC50. b K i. | |||||
| 1-Deoxy-fuconojirimycin | S. roseum fucosidase | 0.22 nMa | Competitive | In vitro | Bishnoi et al.153 Shuoker et al.83 |
| AmGH29A | 0.60 μMa | ||||
| AmGH29B | 1.5 μMa | ||||
| AmGH29C | 14 μMa | ||||
| AmGH29D | 7.3 μMa | ||||
| AmGH95A | 54 μMa | ||||
| AmGH95B | 24 μMa | ||||
| Compound 2 | Corynebacterium sp. fucosidase | 0.46 pMb | Competitive | In vitro | Chang et al.151 |
Recently Shuoker et al. showed that fucosidases are essential for the growth of Akkermansia muciniphila on a mucin substrate and the ability for this taxum to utilize mucin glycans is prevented when DFJ is present (Table 6).83 These data indicate that for A. muciniphila to fully utilize mucin glycans functionally active fucosidases are required to initiate this process.
Additionally, fucosidases are of great interest for HMO degradation, since these oligosaccharides are not metabolized by the infant-produced host factors but fermented in the GI tract by gut microbes.147 Indeed, the most prevalent HMO is 2′-fucosyllactose,152 emphasizing the importance of fucosidases in bacterial fermentation during infant development. Thus, fucosidase inhibitors may help to define how bacteria utilize mucin and HMO substrates via fucosidase enzymes.
α-Sialidase inhibitors
Sialic acid is a common moiety found at the termini of mucin glycans and HMOs.20 Like fucose, sialic acid removal facilitates the bulk degradation of the full oligosaccharide. Historically, many inhibitors of sialidases have been developed to combat viral infections.28 However, these inhibitors offer a promising avenue for unravelling the role of these enzymes in the gut microbiome. One such inhibitor is 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA) (Fig. 10A), a transition state analogue of sialic acid.154 This inhibitor served as the basis for future inhibitor development including, zanamivir (Fig. 10B),85 oseltamivir (Fig. 10C),86 peramivir (Fig. 10D)87 all of which have been developed to target influenza sialidases.28 | ||
| Fig. 10 Chemical structures for representative α-sialidase inhibitors (A) DANA, (B) zanamivir, (C) oseltamivir, and (D) peramivir. | ||
Sialidases have also been studied recently in the vaginal microbiota due to their ability to act on host mucin glycans.155 Pelayo and colleagues showed that prevalent bacterial sialidases in the vaginal microbiome are effectively inhibited by DANA and zanamivir (Table 7), indicating that these viral sialidase inhibitors can also target bacterial sialidases. Similar studies could be conducted focused on sialidases produced by the gut microbiota to probe their activities against mucin glycans and HMOs. Recently, A. muciniphila sialidases have been examined for their importance in mucin utilization, and it was found that that DANA inhibits this taxum's growth on porcine colonic mucus (Table 7).83 Thus, some bacteria rely on these proteins to access host glycans as a carbon source. Additionally, microbes that encode sialidases can release monosaccharides for other bacteria to utilize. Shuoker et al. describe in cross-feeding studies that A. muciniphila-released terminal sugars are the utilized by Clostridia bacteria to produce the short-chain fatty acid, butyrate.83
| Compound | Microbial target | IC50 | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| DANA | P. timonensis NanH1 | 56 μM | Competitive | In vivo | Pelayo et al.155 Shuoker et al.83 |
| P. timonensis NanH2 | 180 μM | Karhadikar et al.156 | |||
| P. Bivia NanH | 103 μM | ||||
| P. denticola NanH | 51 μM | ||||
| G. vaginalis NanH3 | 29 μM | ||||
| AmGH33A | 61 μM | ||||
| AmGH33B | 133 μM | ||||
| AmGH181 | 199 μM | ||||
| Zanamivir | P. timonensis NanH1 | 4200 μM | Competitive | FDA approval for influenza | Pelayo et al.155 |
| P. timonensis NanH2 | 34 μM |
β-Hexosaminidase inhibitors
N-Acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) are abundant monosaccharides found in mucins, N-glycans, and chitin. GlcNAc is one of the most abundant monosaccharides in mucins, and GalNAc is the first monosaccharide added in all O-glycans. β-Hexosamindases are produced by all forms of life and play a key role in the degradation of mucin glycans, immunoglobulin N-glycans, and other carbohydrates present in the GI tract. Inhibitors of β-hexosamindases have been developed with a focus on neurodegenerative lysosomal storage diseases, such as Tay-Sachs and Sandhoff diseases. Notable inhibitors include the phenyl carbamate inhibitor, PUGNAc (Fig. 11A),88 and 1,2-dideoxy-2′-methyl-D-glucopyranoso[2,1-D]-2′-thiazoline (NAGT) (Fig. 11B).89 | ||
| Fig. 11 Chemical structures for representative β-hexosaminidase inhibitors (A) PUGNAc, and (B) NAGT. | ||
The commensal microbe Bifidobacterium bifidum is a key player in HMO and mucin O-glycan degradation. A recent report identified an N-acetylglucosaminidase (GlcNAcase) specific for sulfated GlcNAc residues.157 Katoh et al. provided evidence for O-glycan degradation by this enzyme, along with a thorough structural analysis using X-ray crystallography. Furthermore, they synthesized two sulfo-GlcNACase inhibitors by adding a sulfate to PUGNAc and NAGT (Table 8). The authors then utilize NAGT-6S to inhibit O-glycan degradation by B. bifidum in culture.157 GlcNACase inhibitors have been the focus of groups developing novel antimicrobials for drug resistant pathogens like Staphylococcus aureus.158,159 Thus, novel GlcNAcase inhibitors could impact on gut microbiota composition along with advancing our understanding of gut microbial function.
α-Mannosidase inhibitors
Mannose is an abundant monosaccharide in N-glycans and is commonly found in the glycosylations that decorate immunoglobulins and the glycocalyx. Mannose is also abundant in yeast cell walls obtained through diet. A landmark study illustrated that a prevalent member of the gut microbiota, B. thetaiotaomicron, utilizes mannan glycans derived from yeast cell-walls.160 Additionally, other work has shown that mannosidases from Bacteroides species play key roles in the degradation of plant-derived N-glycans, further emphasizing their importance in dietary glycan metabolism.161 The first reported mannosidase inhibitor was 1-deoxymannojirimycin (Fig. 12A), an iminosugar,93 and others have subsequently been developed, including swainsonine (Fig. 12B), mannoimidazole (Fig. 12C) and kifunensine (Fig. 12D). | ||
| Fig. 12 Chemical structures for representative α-mannosidase inhibitors (A) 1-deoxymannojirimycin, (B) swainsonine, (C) mannoimidazole, and (D) kifunensine. | ||
Crystal structures of B. thetaiotaomicron mannosidases in complex with swainsonine, mannoimidazole and kifunensine have been reported and reveal conserved interactions.91,92 In this work from the Davies Lab, 22 mannosidases from B. thetaiotaomicron were examined and led to valuable information on substrate preferences and a validated roadmap for using mannosidase inhibitors in the context of the gut microbiome (Table 9).
| Compound | Microbial target | K i | Mechanism | Discovery phase | Ref. |
|---|---|---|---|---|---|
| Mannoimidazole | Bt3130 | 1.0 μM | Competitive | In vitro | Zhu et al.92 |
| Bt3965 | 0.4 μM | ||||
| Swainsonine | Bt2199 | 14 μM | Competitive | In vivo | Zhu et al.,92 Tulsiani et al.162 |
| Bt3990 | 5 μM | ||||
| Kifunensine | Bt2199 | 230 μM | Competitive | In vitro | Zhu et al.92 |
| Bt3990 | 140 μM | ||||
| 1-Deoxymannojirimycin | Bt2199 | 13 × 103 μM | Competitive | In vitro | Zhu et al.92 |
| Bt3990 | 12 × 103 μM |
Glycoside hydrolase activity-based probes
Activity-based probes (ABPs) have gained significant interest in examining gut microbial enzymes in recent years.163 ABPs are covalent inhibitors, capable of labelling proteins in complex mixtures like cell lysates or proteins extracted from faecal samples. ABPs are specifically designed to target the active site of the protein family of interest and often mimic the native substrate to allow for functional profiling of specific enzymes in a complex mixture. Several groups have used ABPs to fluorescently label proteins or enrich with biotin-conjugated probes for the enzyme of interest (Fig. 3F and 13). Many GH specific ABPs have been developed and have been reviewed recently by Artola et al.97 Here, we focus on uses of ABPs in studying the gut microbiome. A pioneering study was conducted by Bertozzi Lab in 2004.164 In this report, researchers conducted proteomic analyses of GHs in cell lysates164 using the fluorosugar, 6-azido-2,6-dideoxy-2-fluoro-β-D-galactosyl fluoride, to label β-galactosidase. Using an azide handle on this probe, researchers were able to label β-galactosidases with a FLAG tag for analysis by western blot.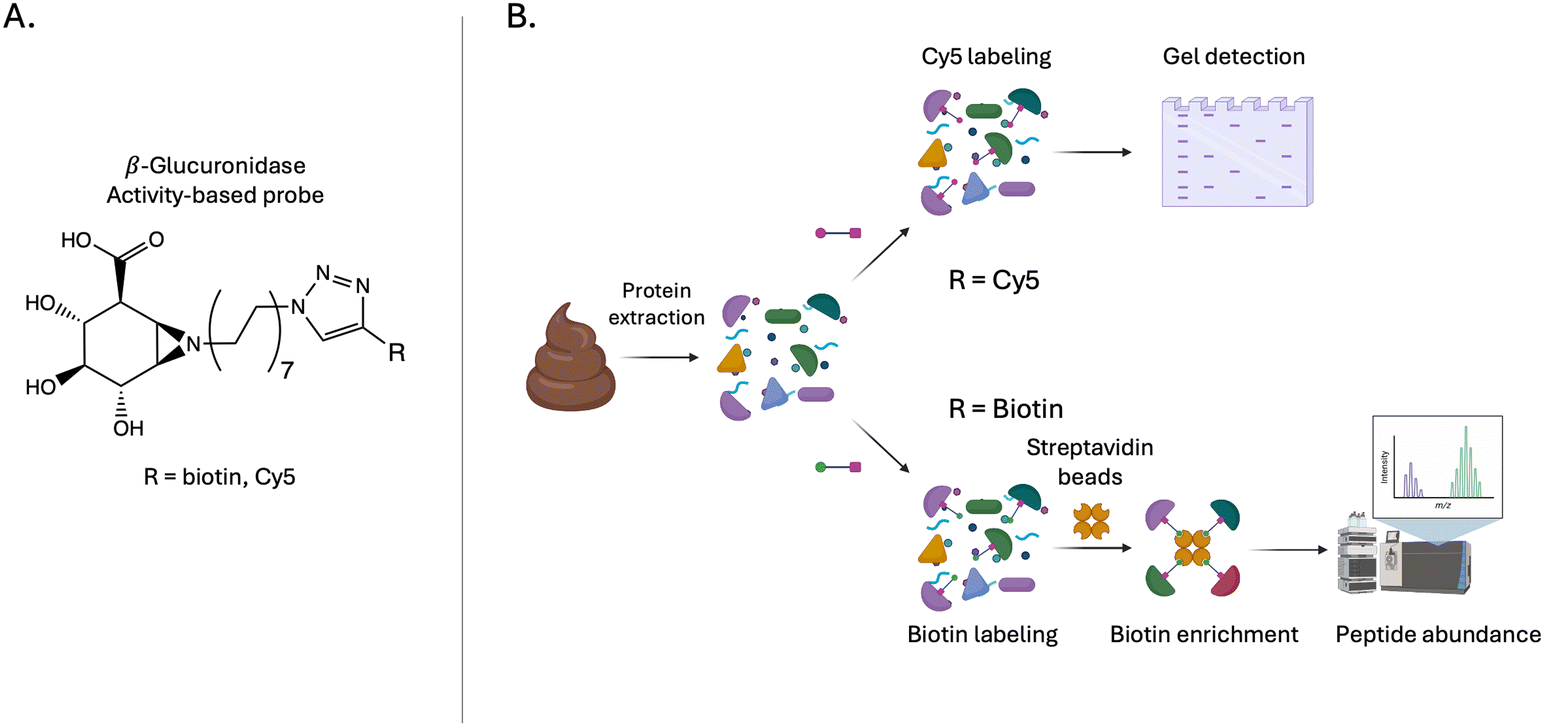 | ||
| Fig. 13 (A) Chemical structure of a β-glucuronidase activity-based probe. (B) Activity-based protein profiling pipeline, illustrating the ability to label specific GHs in complex biological samples. Probes contain a reactive warhead, mimicking the native substrate, linked to either a fluorophore for in-gel fluorescence detection or biotin for enrichment using streptavidin beads. These probes allow researchers to selectively identify and profile active GHs, enabling the study of GHs in complex biological samples. | ||
Another class of GH specific ABPs that has been successful in the past decade are the cyclophellitol-aziridine ABPs (Fig. 3F). This work has largely been accomplished by the Overkleeft Lab, with their initial progress reported in 2010 focused on targeting lysosomal glucocerebrosidase (GBA).165 Cyclophellitol-aziridine ABPs allowed for specific fluorescent labelling of GBA in vitro, in cultured cells and in vivo. Several other cyclophellitol derived ABPs have subsequently been created to target a range of GHs including fucosidases,82 glucosidases,68,166–169 glucuronidases,170 mannosidases171,172 and galactosidases,120,173 all which have utility in the context of the gut microbiome.
β-Glucuronidase ABPs
The first GH ABPs to be used in a gut microbiota context were the cyclophellitol-aziridine GUS ABPs.170 These compounds were developed in 2017 to target two hGUS orthologs, GUSB and HPSE, and were found to be effective in fluorescently labelling GUSB extracted from human spleen. They were then used to enrich for GUS in spleen lysates by attaching a biotin and performing a streptavidin pull down prior to proteomic profiling.170 Inspired by this work Jariwala et al. employed these probes to label GUS enzymes extracted from human faecal samples and selectively enriched for them in downstream proteomic analyses.70 This allowed the researchers to pinpoint a structural subset of GUS enzymes responsible for differential SN38 reactivation in ex vivo human samples. ABPs provide a powerful tool for identifying the proteins responsible for drug, toxin and endobiotic reactivation from complex human faecal mixtures. They have been applied to a variety of other substrates relating to GUS enzymes including triclosan,126 mycophenolate,46 and endogenous hormones and neurotransmitters.7 For triclosan, researchers found that a specific subset of the Loop 1 and FMN-binding gut microbial GUS enzymes drove the efficient reactivation of this toxin in the murine GI tract.126 This was validated by using a GUS inhibitor to block triclosan-induced colitis in a mouse model. Using the same APBB pipeline, researchers were able to pinpoint specific FMN-binding GUS proteins with a unique active site architecture were responsible for the reactivation of the immunosuppressant mycophenolate by human faecal sample extracts.46 Finally, in terms of endogenous compounds, a range of glucuronidated hormones and neurotransmitters were shown to be processed by specific sub-clades of gut microbial GUS proteins pinpointed from complex human faecal samples. These studies emphasize the influence of the Loop-1 and FMN-binding GUS classes in xenobiotic and endobiotic reactivation.7 In these examples, the GUS ABP was tethered to a biotin molecule allowing GUS enrichment using streptavidin-coated beads providing a robust signal in downstream mass spectrometry analyses for enzyme identification.α-Fucosidase ABPs
ABPs have similarly been turned to the study of α-fucosidases produced by gut microbes. The cyclophellitol fucosidase ABP labels bacterial fucosidases as well as fucosidases from human spleen, murine spleen, and murine kidney lysates.82 Furthermore, these probes have been used to identify fucosidase inhibitors through competition assays. Researchers challenged the ability of the ABP JJB256 to label recombinant FUCA1 by incubating with the high affinity fucosidase inhibitor, DFJ, showing that DFJ prevents labelling.82 In another study this probe was used to label GH29 α-fucosidases secreted by Bacteroides fragilis with a fluorophore for in-gel detection.174 This work highlights the importance of secreted fucosidases in B. fragilis-mediated invasion by the microbial pathogen Campylobacter jejuni. C. jejuni can utilize fucose liberated by B. fragilis and fucose utilizing strains of C. jejuni have been linked to increased virulence.175 This probe has also been used to fluorescently label α-fucosidases from mouse faecal extracts.176 α-Fucosidases were only detected when mice were fed 2′-fucosyllactose, demonstrating that diet influences gut microbial α-fucosidase abundance.176β-Glucosidase ABPs
Conduritol B epoxide was discovered as a covalent inhibitor of β-glucosidases in 197766 and cyclophellitol was discovered in 1990,177 laying the foundation for GH cyclophellitol probes. Breakthrough work in 2010 by the Overkleeft Lab described β-glucosidase-targeting cyclophellitol ABPs, allowing for the labelling of GBA, a retaining β-glucosidase in humans, both in vitro and in vivo.165 It was subsequently shown that these cyclophellitol ABPs are potent inhibitors of all 4 retaining β-glucosidases present in humans.165,166 In 2017, researchers developed a new class of cyclophellitol ABPs, carba-cyclophellitols, by substituting the oxygen for a carbon containing R-group substituents.68 Resultant ABPs were shown to be active against a GH1 β-glucosidase from Thermotoga maritima, indicating one of the utilities of these ABPs in studying bacterial GH1 β-glucosidases.β-Galactosidase ABPs
Cyclophellitol ABPs for β-galactosidase were developed in 2014 to examine human lysosomal β-galactosidases. More recently, these ABPs were shown to effectively label β-galactosidases in cell lysates and tissue extracts.120,173 These ABPs offer a strategy to study β-galactosidases produced by gut bacteria and how these enzymes are involved in mucin degradation and GOS catabolism.α-Mannosidase ABPs
In 2020, researchers reported α-mannosidase cyclophellitol ABPs and demonstrated the ability to label α-mannosidases in HEK293T cells expressing 5 human GH37 α-mannosidases and in murine tissue.172 These probes could be applied in studying degradation of N-glycans present in the GI tract as well as dietary polysaccharides containing mannose.Future directions
Numerous gut microbiota studies have emphasized the importance of glycan substrates on host-physiology and gut microbial composition.20,24 Several GH inhibitors have been developed to target host enzymes but can be employed as valuable tools for defining the actions of gut microbial enzymes on a range of host, dietary and microbial substrates. While some studies highlighted here interrogated gut microbial GH-mediated metabolism by using small molecule inhibitors, considerable work remains. Specifically, synthetically derived GH inhibitors, like GUS inhibitors, could allow for microbial-selective GH inhibition.To date, most GH inhibitors are substrate mimics targeting human proteins, and selectivity for microbial GHs remains elusive for the majority of GH families. Recent advances provide an avenue for overcoming these limitations. The synthetic bacterial GUS inhibitors have demonstrated selectivity for bacterial GUS. These GUS inhibitors, identified through high-throughput screening, selectively target bacterial GUS over mammalian GUS, and similar approaches can be applied to other GH families.8,69,71
Given these successes, future work should prioritize drug discovery efforts against bacterial GHs. Natural products and iminosugars have dominated the GH inhibitor landscape and expanding into synthetic and polyphenol-derived scaffolds offers the potential for greater selectivity and chemical diversity. Furthermore, many FDA-approved drugs have been found to modulate GH activity, highlighting a resource to guide GH inhibitor development.7 Specifically, some FDA approved drugs contain 6-member rings with a secondary amine, reminiscent of iminosugar inhibitors, and may be expected to impact GH function. Numerous studies have shown that dietary xenobiotics, like daidzein and genistein, inhibit GHs.130,133 Such studies implicate the potential for developing polyphenol-derived inhibitors, providing much needed chemical diversity for GH inhibitors.
Groups have successfully identified GUS inhibitors using in silico screening, which can readily be implemented for discovering inhibitors of other GH families.127 Advancements in machine learning and ultra-large-scale virtual screening now makes it feasible to evaluate billions of molecules in silico.178,179 These technologies could accelerate the discovery of GH inhibitors with greater selectivity. Furthermore, such approaches may enable to discovery of catalytic mechanism selective GH inhibitors, especially of interest due to the recent discovery of non-Koshland bacterial GHs.102 In addition to inhibitor discovery, there is increasing interest in leveraging microbiota-targeting inhibitors in personalized medicine approaches. Advances in omics technologies could allow for personalized manipulation of the gut microbiota to alleviate GI toxicity from therapeutics or reduce mucin-degrading GH activity associated with inflammation. Thus, future research is expected to consider these and other factors to fully understand the complex interplay between the gut microbiota, glycan metabolism and xenobiotic compounds.
Conflicts of interest
MRR is a founder of Symberix, Inc., and has received research funding from Lilly and Merck.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by a National Institutes of Health Training Grant T32 GM148376-01A1 to M. E. K. and R35 GM152079 to M. R. R.Notes and references
- E. A. Grice and J. A. Segre, Annu. Rev. Genomics Hum. Genet., 2012, 13, 151–170 CrossRef CAS PubMed.
- F. Bäckhed, R. E. Ley, J. L. Sonnenburg, D. A. Peterson and J. I. Gordon, Science, 2005, 307, 1915–1920 CrossRef PubMed.
- R. E. Ley, P. J. Turnbaugh, S. Klein and J. I. Gordon, Nature, 2006, 444, 1022–1023 CrossRef CAS PubMed.
- R. Caruso, B. C. Lo and G. Núñez, Nat. Rev. Immunol., 2020, 20, 411–426 CrossRef CAS PubMed.
- A. L. Graboski, M. E. Kowalewski, J. B. Simpson, X. Cao, M. Ha, J. Zhang, W. G. Walton, D. P. Flaherty and M. R. Redinbo, Cell Chem. Biol., 2023, 30, 1402–1413.e7 CrossRef CAS PubMed.
- R. K. Weersma, A. Zhernakova and J. Fu, Gut, 2020, 69, 1510–1519 CrossRef CAS PubMed.
- J. B. Simpson, M. E. Walker, J. J. Sekela, S. M. Ivey, P. B. Jariwala, C. M. Storch, M. E. Kowalewski, A. L. Graboski, A. D. Lietzan, W. G. Walton, K. A. Davis, E. W. Cloer, V. Borlandelli, Y.-C. Hsiao, L. R. Roberts, D. H. Perlman, X. Liang, H. S. Overkleeft, A. P. Bhatt, K. Lu and M. R. Redinbo, Cell Host Microbe, 2024, 32, 925–944.e10 CrossRef CAS PubMed.
- B. D. Wallace, H. Wang, K. T. Lane, J. E. Scott, J. Orans, J. S. Koo, M. Venkatesh, C. Jobin, L.-A. Yeh, S. Mani and M. R. Redinbo, Science, 2010, 330, 831–835 CrossRef CAS PubMed.
- S. M. Ervin, J. B. Simpson, M. E. Gibbs, B. C. Creekmore, L. Lim, W. G. Walton, R. Z. Gharaibeh and M. R. Redinbo, Biochemistry, 2020, 59, 3939–3950 CrossRef CAS PubMed.
- E. J. Culp, N. T. Nelson, A. A. Verdegaal and A. L. Goodman, Cell, 2024, 187, 6327–6345 CrossRef CAS PubMed.
- M. Zimmermann, M. Zimmermann-Kogadeeva, R. Wegmann and A. L. Goodman, Nature, 2019, 570, 462–467 CrossRef CAS PubMed.
- E. Bankole, E. Read, M. A. Curtis, J. F. Neves and J. A. Garnett, J. Clin. Med., 2021, 10, 1935 CrossRef CAS PubMed.
- H. Wu, E. Esteve, V. Tremaroli, M. T. Khan, R. Caesar, L. Mannerås-Holm, M. Ståhlman, L. M. Olsson, M. Serino, M. Planas-Fèlix, G. Xifra, J. M. Mercader, D. Torrents, R. Burcelin, W. Ricart, R. Perkins, J. M. Fernàndez-Real and F. Bäckhed, Nat. Med., 2017, 23, 850–858 CrossRef CAS PubMed.
- W. Wu, L. Liu, H. Zhu, Y. Sun, Y. Wu, H. Liao, Y. Gui, L. Li, L. Liu, F. Sun and H. Lin, FASEB J., 2019, 33, 12616–12629 CrossRef CAS PubMed.
- V. Maini Rekdal, E. N. Bess, J. E. Bisanz, P. J. Turnbaugh and E. P. Balskus, Science, 2019, 364, eaau6323 CrossRef PubMed.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- The CAZypedia Consortium, Glycobiology, 2018, 28, 3–8 CrossRef PubMed.
- K. A. Biernat, B. Li and M. R. Redinbo, mBio, 2018, 9, e02433-17 CrossRef PubMed.
- L. E. Tailford, E. H. Crost, D. Kavanaugh and N. Juge, Front. Genet., 2015, 6, 81 Search PubMed.
- A. S. Luis and G. C. Hansson, Cell Host Microbe, 2023, 31, 1087–1100 CrossRef CAS PubMed.
- M. He, X. Zhou and X. Wang, Signal Transduction Targeted Ther., 2024, 9, 1–33 CrossRef PubMed.
- Z. Roth, G. Yehezkel and I. Khalaila, Int. J. Carbohydr. Chem., 2012, 2012, 640923 Search PubMed.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef CAS PubMed.
- N. M. Koropatkin, E. A. Cameron and E. C. Martens, Nat. Rev. Microbiol., 2012, 10, 323–335 CrossRef CAS PubMed.
- A. S. Luis, C. Jin, G. V. Pereira, R. W. P. Glowacki, S. R. Gugel, S. Singh, D. P. Byrne, N. A. Pudlo, J. A. London, A. Baslé, M. Reihill, S. Oscarson, P. A. Eyers, M. Czjzek, G. Michel, T. Barbeyron, E. A. Yates, G. C. Hansson, N. G. Karlsson, A. Cartmell and E. C. Martens, Nature, 2021, 598, 332–337 CrossRef CAS PubMed.
- J. F. Wardman, R. K. Bains, P. Rahfeld and S. G. Withers, Nat. Rev. Microbiol., 2022, 20, 542–556 CrossRef CAS PubMed.
- T. D. Butters, R. A. Dwek and F. M. Platt, Glycobiology, 2005, 15, 43R–52R CrossRef CAS PubMed.
- M. von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS PubMed.
- M. E. V. Johansson, J. M. H. Larsson and G. C. Hansson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4659–4665 CrossRef CAS PubMed.
- J. M. H. Larsson, H. Karlsson, H. Sjovall and G. C. Hansson, Glycobiology, 2009, 19, 756–766 CrossRef PubMed.
- K. S. B. Bergstrom and L. Xia, Glycobiology, 2013, 23, 1026–1037 CrossRef CAS PubMed.
- S. Raimondi, E. Musmeci, F. Candeliere, A. Amaretti and M. Rossi, Sci. Rep., 2021, 11, 11094 CrossRef CAS PubMed.
- C. W. Png, S. K. Lindén, K. S. Gilshenan, E. G. Zoetendal, C. S. McSweeney, L. I. Sly, M. A. McGuckin and T. H. J. Florin, Off. J. Am. College Gastroenterol., 2010, 105, 2420 CrossRef CAS PubMed.
- C. R. Bakshani, T. O. Ojuri, B. Pilgaard, J. Holck, R. McInnes, R. P. Kozak, M. Zakhour, S. Çakaj, M. Kerouedan, E. Newton, D. N. Bolam and L. I. Crouch, Nat. Microbiol., 2025, 10, 585–598 CrossRef CAS PubMed.
- S. van der Post, K. S. Jabbar, G. Birchenough, L. Arike, N. Akhtar, H. Sjovall, M. E. V. Johansson and G. C. Hansson, Gut, 2019, 68, 2142–2151 CrossRef CAS PubMed.
- M. E. V. Johansson, J. K. Gustafsson, J. Holmén-Larsson, K. S. Jabbar, L. Xia, H. Xu, F. K. Ghishan, F. A. Carvalho, A. T. Gewirtz, H. Sjövall and G. C. Hansson, Gut, 2014, 63, 281–291 CrossRef CAS PubMed.
- A. Swidsinski, V. Loening-Baucke, F. Theissig, H. Engelhardt, S. Bengmark, S. Koch, H. Lochs and Y. Dörffel, Gut, 2007, 56, 343–350 CrossRef PubMed.
- C.-Y. Chung, N. I. Majewska, Q. Wang, J. T. Paul and M. J. Betenbaugh, Cell, 2017, 171, 258–258.e1 CrossRef CAS PubMed.
- L. I. Crouch, Essays Biochem., 2023, 67, 371–383 CAS.
- A. Mathias and B. Corthésy, Gut Microbes, 2011, 2, 287–293 CrossRef PubMed.
- Y. Cao, E. R. Rocha and C. J. Smith, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12901–12906 CrossRef CAS PubMed.
- M. Collin, EMBO J., 2001, 20, 3046–3055 CrossRef CAS PubMed.
- G. P. Donaldson, M. S. Ladinsky, K. B. Yu, J. G. Sanders, B. B. Yoo, W.-C. Chou, M. E. Conner, A. M. Earl, R. Knight, P. J. Bjorkman and S. K. Mazmanian, Science, 2018, 360, 795–800 CrossRef CAS PubMed.
- B. D. Wallace, A. B. Roberts, R. M. Pollet, J. D. Ingle, K. A. Biernat, S. J. Pellock, M. K. Venkatesh, L. Guthrie, S. K. O’Neal, S. J. Robinson, M. Dollinger, E. Figueroa, S. R. McShane, R. D. Cohen, J. Jin, S. V. Frye, W. C. Zamboni, C. Pepe-Ranney, S. Mani, L. Kelly and M. R. Redinbo, Chem. Biol., 2015, 22, 1238–1249 CrossRef CAS PubMed.
- A. B. Roberts, B. D. Wallace, M. K. Venkatesh, S. Mani and M. R. Redinbo, Mol. Pharmacol., 2013, 84, 208–217 CrossRef CAS PubMed.
- J. B. Simpson, J. J. Sekela, A. L. Graboski, V. B. Borlandelli, M. M. Bivins, N. K. Barker, A. A. Sorgen, A. L. Mordant, R. L. Johnson, A. P. Bhatt, A. A. Fodor, L. E. Herring, H. Overkleeft, J. R. Lee and M. R. Redinbo, Gut Microbes, 2022, 14, 2107289 CrossRef PubMed.
- S. M. Ervin, R. P. Hanley, L. Lim, W. G. Walton, K. H. Pearce, A. P. Bhatt, L. I. James and M. R. Redinbo, ACS Chem. Biol., 2019, 14, 2737–2744 CrossRef CAS PubMed.
- A. LoGuidice, B. D. Wallace, L. Bendel, M. R. Redinbo and U. A. Boelsterli, J. Pharmacol. Exp. Ther., 2012, 341, 447–454 CrossRef CAS PubMed.
- K. S. Saitta, C. Zhang, K. K. Lee, K. Fujimoto, M. R. Redinbo and U. A. Boelsterli, Xenobiotica, 2014, 44, 28–35 CrossRef CAS PubMed.
- A. P. Bhatt, D. B. Gunasekara, J. Speer, M. I. Reed, A. N. Peña, B. R. Midkiff, S. T. Magness, S. J. Bultman, N. L. Allbritton and M. R. Redinbo, ACS Infect. Dis., 2018, 4, 46–52 CrossRef CAS PubMed.
- M. Bae, C. Le, R. S. Mehta, X. Dong, L. M. Pieper, L. Ramirez, M. Alexander, S. Kiamehr, P. J. Turnbaugh, C. Huttenhower, A. T. Chan and E. P. Balskus, Cell Host Microbe, 2024, 32, 1887–1896.e8 CrossRef CAS PubMed.
- T. Khare, U. Anand, A. Dey, Y. G. Assaraf, Z.-S. Chen, Z. Liu and V. Kumar, Front. Pharmacol., 2021, 12, 720726 CrossRef CAS PubMed.
- G. A. Kuziel, G. L. Lozano, C. Simian, L. Li, J. Manion, E. Stephen-Victor, T. Chatila, M. Dong, J.-K. Weng and S. Rakoff-Nahoum, Cell, 2025, 188, 1967–1983.e22 CrossRef CAS PubMed.
- J. W. Fahey, A. T. Zalcmann and P. Talalay, Phytochemistry, 2001, 56, 5–51 CrossRef CAS PubMed.
- C. S. Liou, S. J. Sirk, C. A. C. Diaz, A. P. Klein, C. R. Fischer, S. K. Higginbottom, A. Erez, M. S. Donia, J. L. Sonnenburg and E. S. Sattely, Cell, 2020, 180, 717–728.e19 CrossRef CAS PubMed.
- I. Herr and M. W. Büchler, Cancer Treat. Rev., 2010, 36, 377–383 CrossRef CAS PubMed.
- P. Chaturvedi, C. D. Warren, C. R. Buescher, L. K. Pickering and D. S. Newburg, in Bioactive Components of Human Milk, ed. D. S. Newburg, Springer US, Boston, MA, 2001, vol. 501, pp. 315–323 Search PubMed.
- L. Bode, Nutr. Rev., 2009, 67, S183–S191 CrossRef PubMed.
- A. Y. M. Woo, M. A. Aguilar Ramos, R. Narayan, K. C. Richards-Corke, M. L. Wang, W. J. Sandoval-Espinola and E. P. Balskus, Nat. Rev. Chem., 2023, 7, 319–339 CrossRef CAS PubMed.
- K. Tan, C. Tesar, R. Wilton, R. P. Jedrzejczak and A. Joachimiak, Protein Sci., 2018, 27, 1498–1508 CrossRef CAS PubMed.
- S. Inouye, T. Tsuruoka and T. Nida, J. Antibiot., 1966, 19, 288–292 CAS.
- T. Niwa, S. Inouye, T. Tsuruoka, Y. Koaze and T. Niida, Agric. Biol. Chem., 1970, 34, 966–968 CrossRef CAS.
- S.-H. Yoon and J. F. Robyt, Carbohydr. Res., 2003, 338, 1969–1980 CrossRef CAS PubMed.
- R. A. Steet, S. Chung, B. Wustman, A. Powe, H. Do and S. A. Kornfeld, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13813–13818 CrossRef CAS PubMed.
- R. Saul, J. P. Chambers, R. J. Molyneux and A. D. Elbein, Arch. Biochem. Biophys., 1983, 221, 593–597 CrossRef CAS PubMed.
- H. Braun, G. Legler, J. Deshusses and G. Semenza, Biochim. Biophys. Acta, Enzymol., 1977, 483, 135–140 CrossRef CAS PubMed.
- T. M. Gloster, P. Meloncelli, R. V. Stick, D. Zechel, A. Vasella and G. J. Davies, J. Am. Chem. Soc., 2007, 129, 2345–2354 CrossRef CAS PubMed.
- T. J. M. Beenakker, D. P. A. Wander, W. A. Offen, M. Artola, L. Raich, M. J. Ferraz, K.-Y. Li, J. H. P. M. Houben, E. R. van Rijssel, T. Hansen, G. A. van der Marel, J. D. C. Codée, J. M. F. G. Aerts, C. Rovira, G. J. Davies and H. S. Overkleeft, J. Am. Chem. Soc., 2017, 139, 6534–6537 CrossRef CAS PubMed.
- S. J. Pellock, B. C. Creekmore, W. G. Walton, N. Mehta, K. A. Biernat, A. P. Cesmat, Y. Ariyarathna, Z. D. Dunn, B. Li, J. Jin, L. I. James and M. R. Redinbo, ACS Cent. Sci., 2018, 4, 868–879 CrossRef CAS PubMed.
- P. B. Jariwala, P. B. Jariwala, P. B. Jariwala, S. J. Pellock, S. J. Pellock, D. Goldfarb, D. Goldfarb, E. W. Cloer, E. W. Cloer, M. Artola, M. Artola, J. B. Simpson, J. B. Simpson, J. B. Simpson, A. P. Bhatt, A. P. Bhatt, W. G. Walton, W. G. Walton, L. R. Roberts, L. R. Roberts, M. B. Major, M. B. Major, G. J. Davies, G. J. Davies, H. S. Overkleeft, H. S. Overkleeft, M. R. Redinbo and M. R. Redinbo, ACS Chem. Biol., 2020, 15, 217–225 CrossRef CAS PubMed.
- A. L. Graboski, J. B. Simpson, S. J. Pellock, N. Mehta, B. C. Creekmore, Y. Ariyarathna, A. P. Bhatt, P. B. Jariwala, J. J. Sekela, M. E. Kowalewski, N. K. Barker, A. L. Mordant, V. B. Borlandelli, H. Overkleeft, L. E. Herring, J. Jin, L. I. James and M. R. Redinbo, RSC Chem. Biol., 2024, 5, 853–865 RSC.
- A. P. Challa, X. Hu, Y.-Q. Zhang, J. Hymes, B. D. Wallace, S. Karavadhi, H. Sun, S. Patnaik, M. D. Hall and M. Shen, J. Chem. Inf. Model., 2022, 62, 1783–1793 CrossRef CAS PubMed.
- E. Boyland, D. M. Wallace and D. C. Williams, Br. J. Cancer, 1957, 11, 578–589 CrossRef CAS PubMed.
- T. S. Rasmussen, H. Koldsø, S. Nakagawa, A. Kato, B. Schiøtt and H. H. Jensen, Org. Biomol. Chem., 2011, 9, 7807–7813 RSC.
- P. Awolade, N. Cele, N. Kerru, L. Gummidi, E. Oluwakemi and P. Singh, Eur. J. Med. Chem., 2020, 187, 111921 CrossRef CAS PubMed.
- S. J. Pellock, W. G. Walton, S. M. Ervin, D. Torres-Rivera, B. C. Creekmore, G. Bergan, Z. D. Dunn, B. Li, A. Tripathy and M. R. Redinbo, J. Mol. Biol., 2019, 431, 970–980 CrossRef CAS PubMed.
- A. Bartesaghi, A. Merk, S. Banerjee, D. Matthies, X. Wu, J. L. S. Milne and S. Subramaniam, Science, 2015, 348, 1147–1151 CrossRef CAS PubMed.
- M. Saur, M. J. Hartshorn, J. Dong, J. Reeks, G. Bunkoczi, H. Jhoti and P. A. Williams, Drug Discovery Today, 2020, 25, 485–490 CrossRef CAS PubMed.
- R. E. Huber and M. T. Gaunt, Can. J. Biochem., 1982, 60, 608–612 CrossRef CAS PubMed.
- C. K. De Bruyne and M. Yde, Carbohydr. Res., 1977, 56, 153–164 CrossRef CAS PubMed.
- M. Nagae, A. Tsuchiya, T. Katayama, K. Yamamoto, S. Wakatsuki and R. Kato, J. Biol. Chem., 2007, 282, 18497–18509 CrossRef CAS PubMed.
- J. Jiang, W. W. Kallemeijn, D. W. Wright, A. M. C. H. van den Nieuwendijk, V. Coco Rohde, E. Colomina Folch, H. van den Elst, B. I. Florea, S. Scheij, W. E. Donker-Koopman, M. Verhoek, N. Li, M. Schürmann, D. Mink, R. G. Boot, J. D. C. Codée, G. A. van der Marel, G. J. Davies, J. M. F. G. Aerts and H. S. Overkleeft, Chem. Sci., 2015, 6, 2782–2789 RSC.
- B. Shuoker, M. J. Pichler, C. Jin, H. Sakanaka, H. Wu, A. M. Gascueña, J. Liu, T. S. Nielsen, J. Holgersson, E. Nordberg Karlsson, N. Juge, S. Meier, J. P. Morth, N. G. Karlsson and M. Abou Hachem, Nat. Commun., 2023, 14, 1833 CrossRef CAS PubMed.
- H. Gut, G. Xu, G. L. Taylor and M. A. Walsh, J. Mol. Biol., 2011, 409, 496–503 CrossRef CAS PubMed.
- M. von Itzstein, W.-Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. Van Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron and C. R. Penn, Nature, 1993, 363, 418–423 CrossRef CAS PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 CrossRef CAS PubMed.
- Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482–3486 CrossRef CAS PubMed.
- D. Beer, J.-L. Maloisel, D. M. Rast and A. Vasella, Helv. Chim. Acta, 1990, 73, 1918–1922 CrossRef CAS.
- S. Knapp, D. Vocadlo, Z. Gao, B. Kirk, J. Lou and S. G. Withers, J. Am. Chem. Soc., 1996, 118, 6804–6805 CrossRef CAS.
- M. S. Macauley, A. K. Bubb, C. Martinez-Fleites, G. J. Davies and D. J. Vocadlo, J. Biol. Chem., 2008, 283, 34687–34695 CrossRef CAS PubMed.
- A. J. Thompson, R. J. Spears, Y. Zhu, M. D. L. Suits, S. J. Williams, H. J. Gilbert and G. J. Davies, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 394–404 CrossRef CAS PubMed.
- Y. Zhu, M. D. L. Suits, A. J. Thompson, S. Chavan, Z. Dinev, C. Dumon, N. Smith, K. W. Moremen, Y. Xiang, A. Siriwardena, S. J. Williams, H. J. Gilbert and G. J. Davies, Nat. Chem. Biol., 2010, 6, 125–132 CrossRef CAS PubMed.
- U. Fuhrmann, E. Bause, G. Legler and H. Ploegh, Nature, 1984, 307, 755–758 CrossRef CAS PubMed.
- N. Asano, Glycobiology, 2003, 13, 93R–104R CrossRef CAS PubMed.
- Y. Kim, H. Li, J. Choi, J. Boo, H. Jo, J. Young Hyun and I. Shin, Chem. Soc. Rev., 2023, 52, 7036–7070 RSC.
- J. Balaich, M. Estrella, G. Wu, P. D. Jeffrey, A. Biswas, L. Zhao, A. Korennykh and M. S. Donia, Nature, 2021, 600, 110–115 CrossRef CAS PubMed.
- M. Artola, J. M. F. G. Aerts, G. A. van der Marel, C. Rovira, J. D. C. Codée, G. J. Davies and H. S. Overkleeft, J. Am. Chem. Soc., 2024, 146, 24729–24741 CrossRef CAS PubMed.
- T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683–694 CrossRef CAS PubMed.
- J. Gebler, N. R. Gilkes, M. Claeyssens, D. B. Wilson, P. Béguin, W. W. Wakarchuk, D. G. Kilburn, R. C. Miller, R. A. Warren and S. G. Withers, J. Biol. Chem., 1992, 267, 12559–12561 CrossRef CAS PubMed.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS.
- G. Davies and B. Henrissat, Structure, 1995, 3, 853–859 CrossRef CAS PubMed.
- S. A. Nasseri, A. C. Lazarski, I. L. Lemmer, C. Y. Zhang, E. Brencher, H.-M. Chen, L. Sim, D. Panwar, L. Betschart, L. J. Worrall, H. Brumer, N. C. J. Strynadka and S. G. Withers, Nature, 2024, 631, 199–206 CrossRef CAS PubMed.
- Y.-G. Li, D.-F. Ji, S. Zhong, T.-B. Lin, Z.-Q. Lv, G.-Y. Hu and X. Wang, Sci. Rep., 2013, 3, 1377 CrossRef PubMed.
- J. Bae, J. Y. Lee, E. Shin, M. Lee, Y. Lee, B.-W. Lee, E. S. Kang and B.-S. Cha, Sci. Rep., 2022, 12, 13595 CrossRef CAS PubMed.
- H. A. Brown, A. L. Morris, N. A. Pudlo, A. E. Hopkins, E. C. Martens, J. L. Golob and N. M. Koropatkin, mBio, 2024, 15, e01506-24 CrossRef PubMed.
- M. Yagi, T. Kouno, Y. Aoyagi and H. Murai, Bull. Agric. Chem. Soc. Jpn., 1976, 50, 571–572 CAS.
- D. C. Stein, L. K. Kopec, R. E. Yasbin and F. E. Young, Appl. Environ. Microbiol., 1984, 48, 280–284 CrossRef CAS PubMed.
- T. M. Gloster and G. J. Davies, Org. Biomol. Chem., 2009, 8, 305–320 RSC.
- A. S. Dabhi, N. R. Bhatt and M. J. Shah, J. Clin. Diagn. Res., 2013, 7, 3023–3027 CAS.
- L. A. McIver, C. V. Preuss and J. Tripp, StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- A. D. Santilli, E. M. Dawson, K. J. Whitehead and D. C. Whitehead, ACS Chem. Biol., 2018, 13, 1165–1172 CrossRef CAS PubMed.
- A. D. Santilli, J. T. Russell, E. W. Triplett, K. J. Whitehead and D. C. Whitehead, MedChemComm, 2019, 10, 1875–1880 RSC.
- L. Zhao, F. Zhang, X. Ding, G. Wu, Y. Y. Lam, X. Wang, H. Fu, X. Xue, C. Lu, J. Ma, L. Yu, C. Xu, Z. Ren, Y. Xu, S. Xu, H. Shen, X. Zhu, Y. Shi, Q. Shen, W. Dong, R. Liu, Y. Ling, Y. Zeng, X. Wang, Q. Zhang, J. Wang, L. Wang, Y. Wu, B. Zeng, H. Wei, M. Zhang, Y. Peng and C. Zhang, Science, 2018, 359, 1151–1156 CrossRef CAS PubMed.
- J. Tian, C. Li, Z. Dong, Y. Yang, J. Xing, P. Yu, Y. Xin, F. Xu, L. Wang, Y. Mu, X. Guo, Q. Sun, G. Zhao, Y. Gu, G. Qin and W. Jiang, Nat. Metab., 2023, 5, 896–909 CrossRef CAS PubMed.
- X. Zhang, Z. Fang, C. Zhang, H. Xia, Z. Jie, X. Han, Y. Chen and L. Ji, Diabetes Ther., 2017, 8, 293–307 CrossRef CAS PubMed.
- S. O. Mueller, S. Simon, K. Chae, M. Metzler and K. S. Korach, Toxicol. Sci., 2004, 80, 14–25 CrossRef CAS PubMed.
- G. Catalkaya, K. Venema, L. Lucini, G. Rocchetti, D. Delmas, M. Daglia, A. De Filippis, H. Xiao, J. L. Quiles, J. Xiao and E. Capanoglu, Food Front., 2020, 1, 109–133 CrossRef.
- J. R. Ketudat Cairns and A. Esen, Cell. Mol. Life Sci., 2010, 67, 3389–3405 CrossRef CAS PubMed.
- A. S. Paparella, B. L. Aboulache, R. K. Harijan, K. S. Potts, P. C. Tyler and V. L. Schramm, Nat. Commun., 2021, 12, 6285 CrossRef CAS PubMed.
- C.-L. Kuo, Q. Su, A. M. C. H. van den Nieuwendijk, T. J. M. Beenakker, W. A. Offen, L. I. Willems, R. G. Boot, A. J. Sarris, A. R. A. Marques, J. D. C. Codée, G. A. van der Marel, B. I. Florea, G. J. Davies, H. S. Overkleeft and J. M. F. G. Aerts, Org. Biomol. Chem., 2023, 21, 7813–7820 RSC.
- G. Dutton, Glucuronidation of Drugs and Other Compounds, CRC Press, 2019 Search PubMed.
- R. M. Pollet, E. H. D’Agostino, W. G. Walton, Y. Xu, M. S. Little, K. A. Biernat, S. J. Pellock, L. M. Patterson, B. C. Creekmore, H. N. Isenberg, R. R. Bahethi, A. P. Bhatt, J. Liu, R. Z. Gharaibeh and M. R. Redinbo, Structure, 2017, 25, 967–977.e5 CrossRef CAS PubMed.
- S. T. K. Yauw, M. Arron, R. M. L. M. Lomme, P. van den Broek, R. Greupink, A. P. Bhatt, M. R. Redinbo and H. van Goor, Surg. Infect.:Sel. Antibiot. Ther., 2018, 19, 417–423 CrossRef PubMed.
- U. A. Boelsterli, M. R. Redinbo and K. S. Saitta, Toxicol. Sci., 2013, 131, 654–667 CrossRef CAS PubMed.
- H. Yang, W. Wang, K. A. Romano, M. Gu, K. Z. Sanidad, D. Kim, J. Yang, B. Schmidt, D. Panigrahy, R. Pei, D. A. Martin, E. I. Ozay, Y. Wang, M. Song, B. W. Bolling, H. Xiao, L. M. Minter, G.-Y. Yang, Z. Liu, F. E. Rey and G. Zhang, Sci. Transl. Med., 2018, 10, eaan4116 CrossRef PubMed.
- J. Zhang, M. E. Walker, K. Z. Sanidad, H. Zhang, Y. Liang, E. Zhao, K. Chacon-Vargas, V. Yeliseyev, J. Parsonnet and T. D. Haggerty, Nat. Commun., 2022, 13, 136 CrossRef CAS PubMed.
- T.-C. Cheng, K.-H. Chuang, S. R. Roffler, K.-W. Cheng, Y.-L. Leu, C.-H. Chuang, C.-C. Huang, C.-H. Kao, Y.-C. Hsieh, L.-S. Chang, T.-L. Cheng and C.-S. Chen, Sci. World J., 2015, 2015, 740815 CrossRef PubMed.
- K.-W. Cheng, C.-H. Tseng, C.-C. Tzeng, Y.-L. Leu, T.-C. Cheng, J.-Y. Wang, J.-M. Chang, Y.-C. Lu, C.-M. Cheng, I.-J. Chen, Y.-A. Cheng, Y.-L. Chen and T.-L. Cheng, Pharmacol. Res., 2019, 139, 41–49 CrossRef CAS PubMed.
- Y. Bai, L. Chen, P.-P. Wang, Y.-Q. Tang, D.-C. Wu, C.-L. Zhang, Q. Zhou, R. Yan and J. Hou, Food Funct., 2021, 12, 11190–11201 RSC.
- P. Wang, R. Wu, Y. Jia, P. Tang, B. Wei, Q. Zhang, V. Y.-F. Wang and R. Yan, Int. J. Biol. Macromol., 2022, 220, 1532–1544 CrossRef CAS PubMed.
- M. M. Kooshyar, P. M. Mozafari, M. Amirchaghmaghi, A. Pakfetrat, P. Karoos, M. R. Mohasel, H. Orafai and A. A. Azarian, J. Clin. Diagn. Res., 2017, 11, ZC46–ZC50 CAS.
- Z. Nozhat, S. Heydarzadeh, Z. Memariani and A. Ahmadi, Cancer Cell Int., 2021, 21, 574 CrossRef CAS PubMed.
- Z.-M. Weng, P. Wang, G.-B. Ge, Z.-R. Dai, D.-C. Wu, L.-W. Zou, T.-Y. Dou, T.-Y. Zhang, L. Yang and J. Hou, Food Chem. Toxicol., 2017, 109, 975–983 CrossRef CAS PubMed.
- M. E. Walker, J. B. Simpson and M. R. Redinbo, Curr. Opin. Struct. Biol., 2022, 75, 102416 CrossRef CAS PubMed.
- J. Li, H. Jia, X. Cai, H. Zhong, Q. Feng, S. Sunagawa, M. Arumugam, J. R. Kultima, E. Prifti, T. Nielsen, A. S. Juncker, C. Manichanh, B. Chen, W. Zhang, F. Levenez, J. Wang, X. Xu, L. Xiao, S. Liang, D. Zhang, Z. Zhang, W. Chen, H. Zhao, J. Y. Al-Aama, S. Edris, H. Yang, J. Wang, T. Hansen, H. B. Nielsen, S. Brunak, K. Kristiansen, F. Guarner, O. Pedersen, J. Doré, S. D. Ehrlich, P. Bork and J. Wang, Nat. Biotechnol., 2014, 32, 834–841 CrossRef CAS PubMed.
- K. A. Biernat, S. J. Pellock, A. P. Bhatt, M. M. Bivins, W. G. Walton, B. N. T. Tran, L. Wei, M. C. Snider, A. P. Cesmat, A. Tripathy, D. A. Erie and M. R. Redinbo, Sci. Rep., 2019, 9, 825 CrossRef PubMed.
- H. Duan, Z. Cheng, X. Geng, G. B. Rajah, J. Gao, Y. Guo, L. Cai, Y. Tong, F. Li, Q. Jiang, Z. Han and Y. Ding, Front. Neurol., 2024, 15, 1382365 CrossRef PubMed.
- Y. Chen, R. Chen, X. Wang, Y. Zhou, L. Hong, N. Xiong, J. Zhu, S. Ye and X. Tan, Trials, 2023, 24, 429 CrossRef CAS PubMed.
- D. R. Ferry, A. Smith, J. Malkhandi, D. W. Fyfe, P. G. deTakats, D. Anderson, J. Baker and D. J. Kerr, Clin. Cancer Res., 1996, 2, 659–668 CAS.
- L. I. Crouch, M. V. Liberato, P. A. Urbanowicz, A. Baslé, C. A. Lamb, C. J. Stewart, K. Cooke, M. Doona, S. Needham, R. R. Brady, J. E. Berrington, K. Madunic, M. Wuhrer, P. Chater, J. P. Pearson, R. Glowacki, E. C. Martens, F. Zhang, R. J. Linhardt, D. I. R. Spencer and D. N. Bolam, Nat. Commun., 2020, 11, 4017 CrossRef CAS PubMed.
- P. Weber, M. Thonhofer, S. Averill, G. J. Davies, A. G. Santana, P. Müller, S. A. Nasseri, W. A. Offen, B. M. Pabst, E. Paschke, M. Schalli, A. Torvisco, M. Tschernutter, C. Tysoe, W. Windischhofer, S. G. Withers, A. Wolfsgruber, T. M. Wrodnigg and A. E. Stütz, Molecules, 2020, 25, 4025 CrossRef CAS PubMed.
- J. Horne, E. Beddingfield, M. Knapp, S. Mitchell, L. Crawford, S. B. Mills, A. Wrist, S. Zhang and R. M. Summers, ACS Omega, 2020, 5, 32250–32255 CrossRef CAS PubMed.
- Y. Zeng, R. Wu, F. Wang, S. Li, L. Li, Y. Li, P. Qin, M. Wei, J. Yang, J. Wu, A. Chen, G. Ke, Z. Yan, H. Yang, Z. Chen, Z. Wang, W. Xiao, Y. Jiang, X. Chen, Z. Zeng, X. Zhao, P. Chen and S. Gong, Cell Host Microbe, 2023, 31, 766–780.e7 CrossRef CAS PubMed.
- M. A. Azcarate-Peril, A. J. Ritter, D. Savaiano, A. Monteagudo-Mera, C. Anderson, S. T. Magness and T. R. Klaenhammer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E367–E375 CrossRef CAS PubMed.
- A. K. Singh, B. Pluvinage, M. A. Higgins, A. B. Dalia, S. A. Woodiga, M. Flynn, A. R. Lloyd, J. N. Weiser, K. A. Stubbs, A. B. Boraston and S. J. King, PLoS Pathog., 2014, 10, e1004364 CrossRef PubMed.
- A. G. Hadd, D. E. Raymond, J. W. Halliwell, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1997, 69, 3407–3412 CrossRef CAS PubMed.
- M. L. A. De Leoz, K. M. Kalanetra, N. A. Bokulich, J. S. Strum, M. A. Underwood, J. B. German, D. A. Mills and C. B. Lebrilla, J. Proteome Res., 2015, 14, 491–502 CrossRef CAS PubMed.
- K. M. Stepien, E. Ciara and A. Jezela-Stanek, Genes, 2020, 11, 1383 CrossRef CAS PubMed.
- Z. Armstrong, R. W. Meek, L. Wu, J. N. Blaza and G. J. Davies, Structure, 2022, 30, 1443–1451.e5 CrossRef CAS PubMed.
- B. Winchester, C. Barker, S. Baines, G. S. Jacob, S. K. Namgoong and G. Fleet, Biochem. J., 1990, 265, 277–282 CrossRef CAS PubMed.
- C.-F. Chang, C.-W. Ho, C.-Y. Wu, T.-A. Chao, C.-H. Wong and C.-H. Lin, Chem. Biol., 2004, 11, 1301–1306 CrossRef CAS PubMed.
- B. Hegar, Y. Wibowo, R. W. Basrowi, R. G. Ranuh, S. M. Sudarmo, Z. Munasir, A. F. Atthiyah, A. D. Widodo, Supriatmo, M. Kadim, A. Suryawan, N. R. Diana, C. Manoppo and Y. Vandenplas, Pediatr. Gastroenterol. Hepatol. Nutr., 2019, 22, 330–340 CrossRef PubMed.
- R. Bishnoi, S. Mahajan and T. N. C. Ramya, Glycobiology, 2018, 28, 860–875 CrossRef CAS PubMed.
- P. Meindl and H. Tuppy, Hoppe Seylers Z. Physiol. Chem., 1969, 350, 1088–1092 CrossRef CAS PubMed.
- P. Pelayo, F. A. Hussain, C. A. Werlang, C. M. Wu, B. M. Woolston, C. M. Xiang, L. Rutt, M. T. France, J. Ravel, K. Ribbeck, D. S. Kwon and E. P. Balskus, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2400341121 CrossRef CAS PubMed.
- T. R. Karhadkar, D. Pilling, N. Cox and R. H. Gomer, Sci. Rep., 2017, 7, 15069 CrossRef PubMed.
- T. Katoh, C. Yamada, M. D. Wallace, A. Yoshida, A. Gotoh, M. Arai, T. Maeshibu, T. Kashima, A. Hagenbeek, M. N. Ojima, H. Takada, M. Sakanaka, H. Shimizu, K. Nishiyama, H. Ashida, J. Hirose, M. Suarez-Diez, M. Nishiyama, I. Kimura, K. A. Stubbs, S. Fushinobu and T. Katayama, Nat. Chem. Biol., 2023, 19, 778–789 CrossRef CAS PubMed.
- J. Sluga, T. Tomašič, M. Anderluh, M. H. Rambaher, G. Bajc, A. Sevšek, N. I. Martin, R. J. Pieters, M. Novič and K. Venko, Antibiotics, 2024, 13, 751 CrossRef CAS PubMed.
- R. Wheeler, R. D. Turner, R. G. Bailey, B. Salamaga, S. Mesnage, S. A. S. Mohamad, E. J. Hayhurst, M. Horsburgh, J. K. Hobbs and S. J. Foster, mBio, 2015, 6, e00660–e00615 CrossRef CAS PubMed.
- F. Cuskin, E. C. Lowe, M. J. Temple, Y. Zhu, E. A. Cameron, N. A. Pudlo, N. T. Porter, K. Urs, A. J. Thompson, A. Cartmell, A. Rogowski, B. S. Hamilton, R. Chen, T. J. Tolbert, K. Piens, D. Bracke, W. Vervecken, Z. Hakki, G. Speciale, J. L. Munōz-Munōz, A. Day, M. J. Peña, R. McLean, M. D. Suits, A. B. Boraston, T. Atherly, C. J. Ziemer, S. J. Williams, G. J. Davies, D. W. Abbott, E. C. Martens and H. J. Gilbert, Nature, 2015, 517, 165–169 CrossRef CAS PubMed.
- L. I. Crouch, P. A. Urbanowicz, A. Baslé, Z.-P. Cai, L. Liu, J. Voglmeir, J. M. Melo Diaz, S. T. Benedict, D. I. R. Spencer and D. N. Bolam, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2208168119 CrossRef CAS PubMed.
- D. R. P. Tulsiani and O. Touster, Arch. Biochem. Biophys., 1983, 224, 594–600 CrossRef CAS PubMed.
- L. Han and P. V. Chang, Curr. Opin. Chem. Biol., 2023, 76, 102351 CrossRef CAS PubMed.
- D. J. Vocadlo and C. R. Bertozzi, Angew. Chem., 2004, 116, 5452–5456 CrossRef.
- M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907–913 CrossRef CAS PubMed.
- W. W. Kallemeijn, M. D. Witte, T. M. Voorn-Brouwer, M. T. C. Walvoort, K.-Y. Li, J. D. C. Codée, G. A. van der Marel, R. G. Boot, H. S. Overkleeft and J. M. F. G. Aerts, J. Biol. Chem., 2014, 289, 35351–35362 CrossRef CAS PubMed.
- K.-Y. Li, J. Jiang, M. D. Witte, W. W. Kallemeijn, W. E. Donker-Koopman, R. G. Boot, J. M. F. G. Aerts, J. D. C. Codée, G. A. van der Marel and H. S. Overkleeft, Org. Biomol. Chem., 2014, 12, 7786–7791 RSC.
- Y. Radchenko, Q. Su, S. S. Schröder, L. van Gijlswijk, M. Artola, J. M. F. G. Aerts, J. D. C. Codée and H. S. Overkleeft, Chem. – Eur. J., 2024, 30, e202402988 CrossRef CAS PubMed.
- D. van der Gracht, R. J. Rowland, V. Roig-Zamboni, M. J. Ferraz, M. Louwerse, P. P. Geurink, J. M. F. G. Aerts, G. Sulzenbacher, G. J. Davies, H. S. Overkleeft and M. Artola, Chem. Sci., 2023, 14, 9136–9144 RSC.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C.-L. Kuo, M. Artola, W. Dai, C. van Elk, M. van Eijk, G. A. van der Marel, J. D. C. Codée, B. I. Florea, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 CrossRef CAS PubMed.
- N. G. S. McGregor, C.-L. Kuo, T. J. M. Beenakker, C.-S. Wong, W. A. Offen, Z. Armstrong, B. I. Florea, J. D. C. Codée, H. S. Overkleeft, J. M. F. G. Aerts and G. J. Davies, Org. Biomol. Chem., 2022, 20, 877–886 RSC.
- Z. Armstrong, C.-L. Kuo, D. Lahav, B. Liu, R. Johnson, T. J. M. Beenakker, C. de Boer, C.-S. Wong, E. R. van Rijssel, M. F. Debets, B. I. Florea, C. Hissink, R. G. Boot, P. P. Geurink, H. Ovaa, M. van der Stelt, G. M. van der Marel, J. D. C. Codée, J. M. F. G. Aerts, L. Wu, H. S. Overkleeft and G. J. Davies, J. Am. Chem. Soc., 2020, 142, 13021–13029 CrossRef CAS PubMed.
- L. I. Willems, T. J. M. Beenakker, B. Murray, B. Gagestein, H. van den Elst, E. R. van Rijssel, J. D. C. Codée, W. W. Kallemeijn, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2014, 6044–6056 CrossRef CAS.
- Y. M. C. A. Luijkx, N. M. C. Bleumink, J. Jiang, H. S. Overkleeft, M. M. S. M. Wösten, K. Strijbis and T. Wennekes, Cell. Microbiol., 2020, 22, e13252 CrossRef CAS PubMed.
- M. A. Javed, A. J. Grant, M. C. Bagnall, D. J. Maskell, D. G. Newell and G. Manning, Microbiology, 2010, 156, 1134–1143 CrossRef CAS PubMed.
- P. Paone, D. Latousakis, R. Terrasi, D. Vertommen, C. Jian, V. Borlandelli, F. Suriano, M. E. V. Johansson, A. Puel, C. Bouzin, N. M. Delzenne, A. Salonen, N. Juge, B. I. Florea, G. G. Muccioli, H. Overkleeft, M. Van Hul and P. D. Cani, Gut, 2024, 73, 1632–1649 CrossRef CAS PubMed.
- S. Atsumi, H. Iinuma, C. Nosaka and K. Umezawa, J. Antibiot., 1990, 43, 1579–1585 CrossRef CAS PubMed.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X.-P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran, F. Tong, N. Zvonok, M. K. Jain, O. Savych, D. S. Radchenko, S. P. Nikas, N. A. Petasis, Y. S. Moroz, B. L. Roth, A. Makriyannis and V. Katritch, Nature, 2022, 601, 452–459 CrossRef CAS PubMed.
- F. Gentile, J. C. Yaacoub, J. Gleave, M. Fernandez, A.-T. Ton, F. Ban, A. Stern and A. Cherkasov, Nat. Protoc., 2022, 17, 672–697 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |