
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpichaperomes: redefining chaperone biology and therapeutic strategies in complex diseases
Chiranjeevi
Pasala
 a,
Chander S.
Digwal
a,
Sahil
Sharma
a,
Shujuan
Wang
a,
Alessia
Bubula
a and
Gabriela
Chiosis
*ab
a,
Chander S.
Digwal
a,
Sahil
Sharma
a,
Shujuan
Wang
a,
Alessia
Bubula
a and
Gabriela
Chiosis
*ab
aChemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. E-mail: chiosisg@mskcc.org
bDepartment of Medicine, Division of Solid Tumors, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
First published on 19th March 2025
Abstract
The complexity of disease biology extends beyond mutations or overexpression, encompassing stress-induced mechanisms that reshape proteins into pathological assemblies. Epichaperomes, stable and disease-specific assemblies of chaperones and co-chaperones, exemplify this phenomenon. This review emphasizes the critical structural and functional distinctions between epichaperomes and canonical chaperones, highlighting their role in redefining therapeutic strategies. Epichaperomes arise under stress conditions through post-translational modifications that stabilize these assemblies, enabling them to act as scaffolding platforms that rewire protein–protein interaction networks and drive the pathological phenotypes of complex diseases such as cancer and neurodegeneration. Chemical biology has been instrumental in uncovering the unique nature of epichaperomes, with small molecules like PU-H71 elucidating their biology and demonstrating their therapeutic potential by dismantling pathological scaffolds and restoring normal protein–protein interaction networks. By targeting epichaperomes, we unlock the potential for network-level interventions and personalized medicine, offering transformative possibilities for diseases driven by protein–protein interaction network dysregulation.
 Chiranjeevi Pasala | Dr Chiranjeevi Pasala earned his PhD in Bioinformatics from SVIMS, Tirupati, where he identified therapeutic targets for multidrug-resistant Helicobacter pylori, designed novel lead compounds, and developed epitope-driven vaccines using computational approaches. Currently, he is a Research Scholar at Memorial Sloan Kettering Cancer Center, USA. His research focuses on the structural dynamics of epichaperome assemblies, integrating machine learning approaches to develop chemical toolsets for targeting epichaperomes. His work bridges computational and translational research, advancing the understanding and treatment of cellular stress-related disorders. |
 Chander S. Digwal | Dr Chander Singh Digwal received his BPharm in Pharmaceutical Sciences from Mohanlal Sukhadia University, Udaipur, and MS Pharm in Medicinal Chemistry from NIPER-Ahmedabad. He earned his PhD from NIPER-Hyderabad, where he specialized in small-molecule drug discovery. He is a Research Associate in Prof. Gabriela Chiosis’ lab at Memorial Sloan Kettering Cancer Center, focusing on the design and evaluation of chemical probes for epichaperomes. His research integrates medicinal chemistry and preclinical studies to advance diagnostic and therapeutic candidates targeting epichaperomes in cancer and neurodegenerative diseases. |
 Sahil Sharma | Dr Sahil Sharma earned his PhD in pharmaceutical chemistry from Guru Nanak Dev University, Amritsar, India, focusing on the design and synthesis of heterocyclic compounds with antitubulin and xanthine oxidase inhibitory properties for cancer and gout therapy. He is currently a Senior Research Scientist at Memorial Sloan Kettering Cancer Center in New York, USA. Dr Sharma has authored 40 research papers, 25 review articles, 6 book chapters, and holds 2 patents. His research focuses on developing small molecules targeting the epichaperome for the diagnosis and treatment of cancer and neurodegenerative diseases. |
 Shujuan Wang | Dr Shujuan Wang earned her bachelor's degree in biology from Henan University, China, followed by a master's degree in Analytical Chemistry from South China Normal University. She obtained her PhD in Analytical Chemistry from Hong Kong Baptist University. In 2024, she joined Prof. Gabriela Chiosis’ lab at Memorial Sloan Kettering Cancer Center as a Senior Research Scientist. Her research focuses on investigating protein conformation and protein–protein interaction interfaces in epichaperomes using crosslinking and quantitative mass spectrometry. |
 Gabriela Chiosis | Dr Gabriela Chiosis is a Member and Principal Investigator in the Chemical Biology Program at the Sloan Kettering Institute and a Professor at Weill Cornell Medical Center and Memorial Sloan Kettering Cancer Center. Her lab integrates chemical biology, systems biology, and translational research to define how internal and external stressors drive disease at the cellular, tissue, and organismal levels. These insights have led to first-in-class diagnostics and therapeutics, including epichaperome disruptors that have advanced to clinical evaluation in cancer and Alzheimer's disease. |
1. Introduction
In drug discovery, the structural understanding of proteins typically informs the design of chemical probes and therapeutics.1,2 However, the story of epichaperomes—distinctive, tightly bound hetero-oligomeric complexes of chaperones, co-chaperones, and other factors—unfolded in reverse. It was the discovery of a small molecule drug candidate—PU-H713–7—that illuminated the target and fundamentally changed our understanding of its complexity in disease.The small molecule PU-H71, also known as zelavespib in clinical settings,8 was initially discovered and developed as an heat shock protein 90 (HSP90) inhibitor.9 This compound emerged from medicinal chemistry efforts focused on a purine-scaffold molecule, PU3, which was optimized into PU-H71 through iterative chemical synthesis and rigorous testing both in vitro and in vivo.9–14 Based on its ability to bind the ATP-binding pocket of HSP90 in the N-terminal domain,15 PU-H71 was classified as a canonical HSP90 inhibitor and believed to exert its biological effects by disrupting the chaperone's folding activity.16
HSP90, often described as the guardian of the proteome, plays a critical role in maintaining cellular homeostasis by facilitating the folding, stabilization, and degradation of proteins.17,18 Under normal physiological conditions, HSP90 functions as a homodimer, with each protomer consisting of an N-terminal domain (NTD), a middle domain, and a C-terminal dimerization domain. The NTD contains a nucleotide-binding pocket where ATP binding and hydrolysis occur, driving the chaperone cycle. This cycle involves dynamic conformational changes,19 transitioning HSP90 from open to closed states as it folds client proteins.20–22 Co-chaperones participate at various stages of the HSP90 cycle, regulating its conformational states and fine-tuning the chaperone's activity.23 Post-translational modifications (PTMs) provide an additional layer of regulation,24,25 modulating co-chaperone and client protein binding, ATPase activity, and the overall conformational cycle (Fig. 1a).
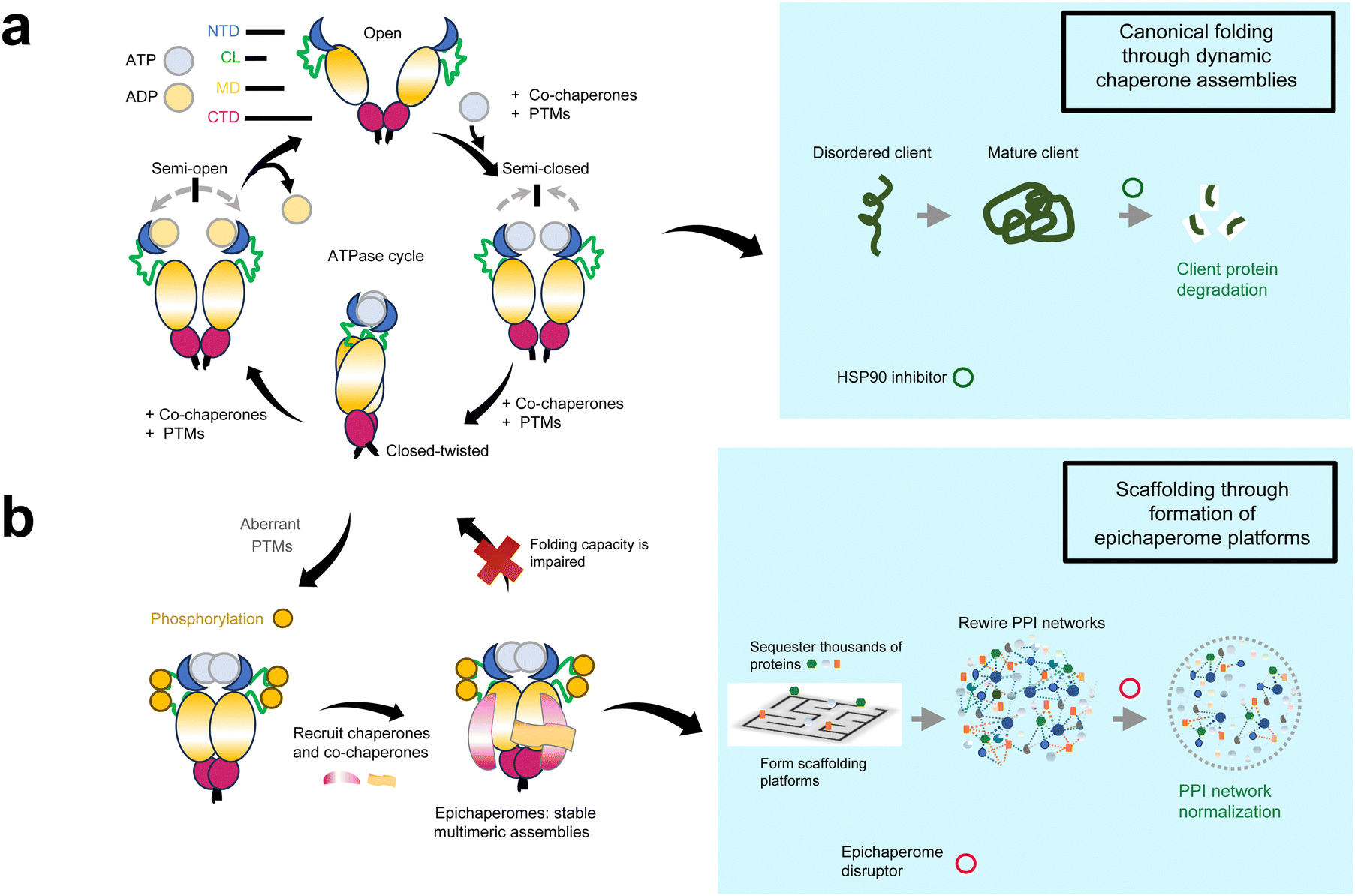 | ||
| Fig. 1 Distinct roles of HSP90 and epichaperomes in cellular proteostasis and disease pathogenesis. (a) HSP90, a critical molecular chaperone, functions as a homodimer composed of an N-terminal domain (NTD), a charged linker (CL), a middle domain (MD), and a C-terminal dimerization domain (CTD). Under normal physiological conditions, HSP90 undergoes a dynamic conformational cycle driven by ATP binding and hydrolysis in the NTD, enabling the folding, stabilization, and degradation of client proteins. Co-chaperones and post-translational modifications (PTMs) regulate this cycle, fine-tuning HSP90's activity and client protein interactions. Small molecules targeting the ATP-binding pocket of HSP90, such as early HSP90 inhibitors, block its folding activity, leading to destabilization of client proteins and their subsequent proteasomal degradation. This mechanism underpinned the initial rationale for HSP90 inhibition as a therapeutic strategy. (b) In disease settings, pathological post-translational modifications (PTMs) induce a conformational state of HSP90 that impairs its ability to transition through the ATPase cycle. This state favors the assembly of stable, long-lived hetero-oligomeric complexes known as epichaperomes. Epichaperomes act as scaffolding platforms, reorganizing protein–protein interaction (PPI) networks to sustain pathological phenotypes in diseases like cancer and neurodegeneration. Small molecules known as epichaperome disruptors, such as PU-H71, selectively bind to HSP90 when incorporated within epichaperomes, kinetically trapping and dismantling these pathological assemblies. By restoring normal PPI networks, epichaperome disruptors address the systems-level dysregulation underlying complex diseases. | ||
Within this framework, PU-H71 was initially believed to inhibit HSP90 by binding to its ATP-binding pocket, thereby blocking nucleotide binding and impeding the chaperone's folding activity. Given that many HSP90 clients are oncogenic kinases,26 PU-H71 was developed and introduced into clinical trials as an anti-cancer agent in 2011,27 specifically targeting tumors reliant on HSP90's folding activity. The rationale was that by inhibiting HSP90, PU-H71 would disrupt the folding of oncogenic kinases, leading to their degradation and consequently impairing their biological functions critical for tumor formation and maintenance.28
However, this client-centric view was challenged following a pivotal discovery in 2016,7 which revealed that the actual target of PU-H71 is a specific form of HSP90 integrated into epichaperomes. These epichaperomes, which include HSP90 as a key component, are structurally distinct from the canonical HSP90 complexes involved in protein folding.7,29,30 Rather than functioning as a protein-folding entity facilitating the maturation and stability of individual client proteins,22 the epichaperome acts as a scaffolding platform that orchestrates the reorganization of protein–protein interaction (PPI) networks,31–36 fundamentally altering cellular processes.7,31–33,37 This scaffolding function enables epichaperomes to sustain the pathological phenotypes of diseased cells by reorganizing PPI landscapes and stabilizing disease-specific PPI networks that sustain aberrant cellular behaviors,31–33,38–40 rather than by directly supporting the folding of client proteins (Fig. 1b). In disease settings where epichaperomes are present, they become a dominant driver of the disease phenotype through orchestrating the pathological reorganization of PPI networks.33 Notably, epichaperome formation is selective and context-dependent, with only a subset of HSP90 in the cell incorporated into these stable assemblies.7,31,32,37,41,42 This selective formation underscores why targeting epichaperomes specifically—not HSP90 more broadly—is a promising therapeutic strategy.
While PU-H71 has played a pivotal role in the discovery and characterization of epichaperomes, it is important to note that epichaperomes are not defined by their interaction with this or any specific chemical probe. Instead, their definition rests on distinct structural, compositional, and functional attributes which differentiates them from the canonical folding chaperone assemblies. This realization marked a paradigm shift in our understanding of chaperone biology in disease. What we have learned so far is that epichaperomes are not merely altered forms of canonical HSP90 complexes; they represent a distinct adaptation of the chaperone machinery to pathological stress. This review aims to explore how this discovery, driven by efforts to understand the biological activity of PU-H71, unraveled the role and significance of epichaperomes. It delves into the composition and function of epichaperomes, the molecular factors driving their formation, and their role in disease, specifically through remodeling PPI networks. To provide a foundation for this discussion, Table 1 provides an overview of the key distinctions between epichaperomes and canonical chaperones, including their structure, function, expression levels, and disease associations. This table offers a snapshot of their unique properties, which are explored in depth throughout this review. Lastly, this review also examines how the discovery of epichaperomes led to the development of various chemical probes and drug candidates designed to study, detect, monitor and target epichaperomes across different disease contexts, particularly in cancer and neurodegenerative diseases.
| Feature | Canonical chaperones | Epichaperomes |
|---|---|---|
| Structure | Dynamic, transient complexes that disassemble post-function | Stable, long-lived hetero-oligomeric assemblies |
| Core composition | Primarily HSP90/HSC70 or GRP94 with transient co-chaperones | HSP90, HSC70, GRP94 with tightly bound co-chaperones and factors |
| Assembly trigger | Normal cellular processes (e.g., folding, acute stress responses) | Chronic stress and aberrant PTMs (e.g., phosphorylation, glycosylation) |
| Function | Folding, stabilization, and degradation of client proteins | Scaffolding platforms that rewire protein–protein interaction networks |
| Expression levels | Ubiquitously expressed in all cells, with levels increasing under stress | Context-dependent; formed only in disease settings or highly proliferative/adaptive cells (e.g., pluripotent stem cells) |
| Client interactions | Specific, transient interactions with individual proteins | Sequesters and reprograms thousands of proteins |
| Role in disease | Generalized response to stress, protein folding maintenance | Drives pathological phenotypes by sustaining aberrant PPI networks |
| Response to PU-H71 | PU-H71 does not bind effectively, fast off-rate | PU-H71 kinetically traps and disrupts |
2. HSP90 vs. epichaperomes
The journey of PU-H71 began within the framework of the canonical understanding of HSP90 as a folding chaperone. Initially classified as a canonical HSP90 inhibitor, PU-H71 was believed to act by binding the ATP-binding pocket of HSP90, thereby inhibiting its folding activity and destabilizing its client proteins.9 Its activity in cancer was assumed to align with the prevailing view of HSP90 as a folding chaperone essential for the stability and activity of oncogenic kinases and other key cancer drivers.26,43 However, studies soon revealed a puzzling disconnect between this framework and the observed activity of PU-H71.5,7,44,45Specifically, tumor sensitivity to PU-H71 did not correlate with HSP90 expression levels or the induction of HSP70, both of which were thought to influence the efficacy of HSP90 inhibitors.7 Furthermore, sensitivity was independent of specific HSP90 clients. For instance, in breast cancers, estrogen receptor (ER)-positive tumors and HER2-positive tumors—tumor types characterized by the reliance of key oncogenic drivers such as ER and HER2 on HSP90 for their stability and activity—exhibited variable sensitivity to PU-H71.7 These findings suggested that neither the abundance of HSP90 nor its client proteins could fully explain tumor vulnerability to PU-H71.
A breakthrough occurred when Rodina et al.7 examined the complexes in which HSP90 participated in tumors that were sensitive and those that were insensitive to PU-H71. They examined over 100 cancer cell lines and primary tumor specimens spanning diverse tumor types.7 Using homogenates from tumors subjected to native-PAGE separation followed by immunoblotting with HSP90-specific antibodies, they observed distinct patterns: tumors insensitive to PU-H71 displayed primarily a broad band around 242 kDa, characteristic of canonical HSP90 complexes found in non-transformed cells and tissues.46 In contrast, tumors sensitive to PU-H71 exhibited additional high-molecular-weight species beyond the 242 kDa band.7 These species represented a structurally and functionally distinct form of HSP90-containing complexes: the epichaperomes (Fig. 2a).
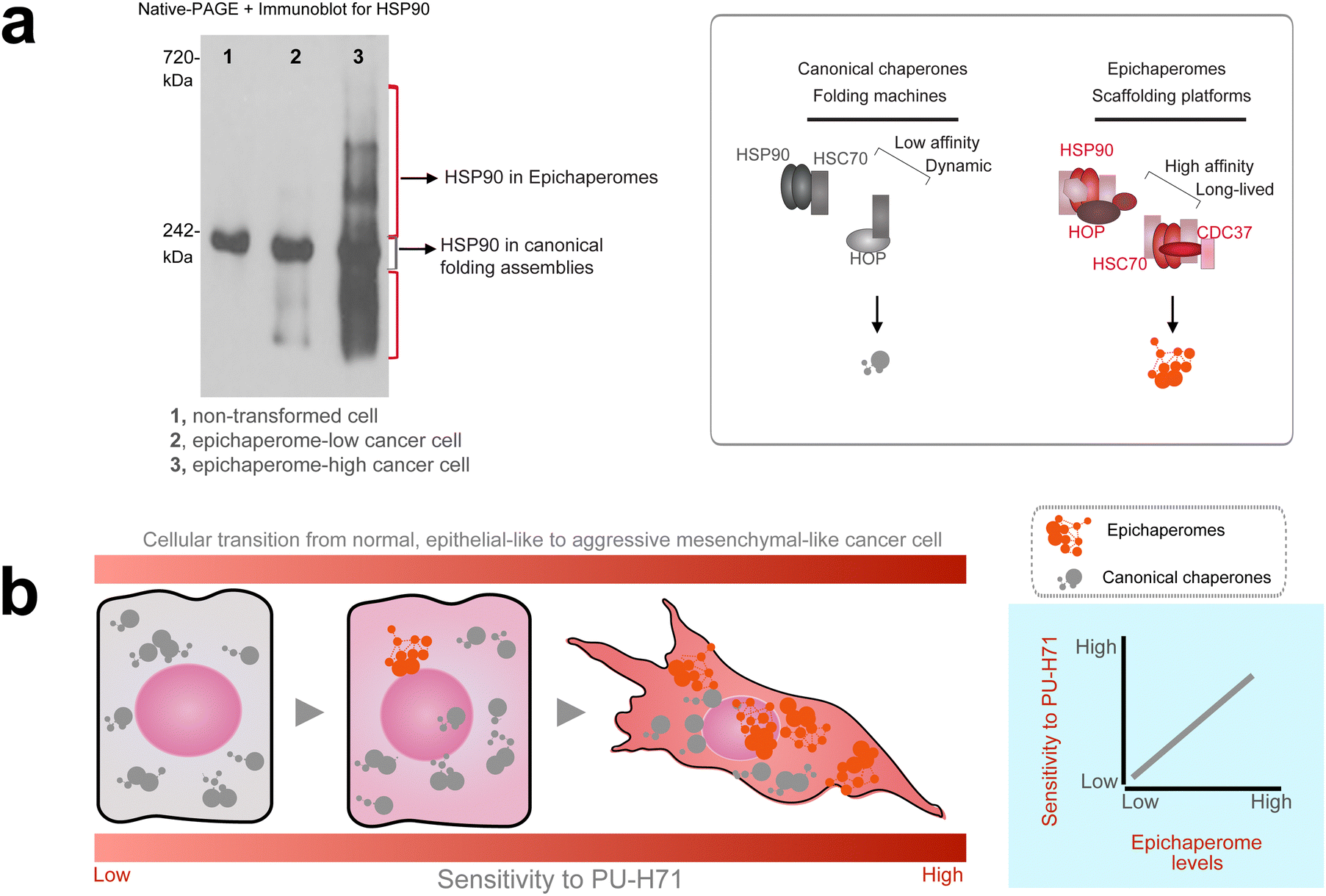 | ||
| Fig. 2 Epichaperome abundance is a key determinant of tumor aggressiveness and PU-H71 sensitivity. (a) Native-PAGE highlights the biochemical differences between canonical HSP90 complexes and epichaperomes. Canonical HSP90 complexes, which are transient and dynamic, disassemble under native-PAGE conditions, producing a single 242 kDa band. In contrast, epichaperomes, due to their stable and tightly bound assemblies, persist under native-PAGE as distinct high-molecular-weight bands. Heat shock protein 90, HSP90; heat shock cognate 70, HSC70; HSP-organizing protein, HOP. Adapted from Rodina et al. Nature Communications 2023. (b) Tumor sensitivity to PU-H71 correlates with epichaperome abundance. The schematic shows cells transitioning from a low-stress state with minimal epichaperomes and epithelial-like phenotypes to high-stress states characterized by abundant epichaperomes and aggressive mesenchymal-like behaviors. A graph illustrates that tumors with increased epichaperome levels exhibit greater sensitivity to PU-H71. Tumors with these aggressive behaviors—including uncontrolled proliferation, apoptosis inhibition, angiogenesis promotion, and therapy resistance—are marked by elevated epichaperome levels, which rewire PPI networks to sustain these pathological traits, making them more vulnerable to PU-H71 treatment. | ||
This difference in patterns stems from the inherent properties of canonical HSP90 complexes versus epichaperomes. Canonical HSP90 complexes are dynamic and transient, with weak and reversible interactions that disassemble under the conditions of native-PAGE analysis, leaving behind only the characteristic 242 kDa band. In contrast, epichaperomes are stable, long-lived assemblies composed of tightly bound chaperones, co-chaperones, and other factors.7,32,37,42,47,48 This stability enables epichaperomes to persist on native-PAGE, appearing as a set of high-molecular-weight bands that remain intact due to their structural rigidity and strong intermolecular interactions.
Further analysis revealed that tumor vulnerability to PU-H71, both in cellulo and in vivo, directly correlated with the abundance of these high-molecular-weight species.7,34–36,48 Tumors with higher levels of epichaperomes exhibited greater sensitivity to PU-H71, irrespective of tumor type, origin, genetic makeup, or HSP90 expression levels (Fig. 2b). Instead, baseline cellular stress and proliferative potential were common features of PU-H71-sensitive tumors, rather than the reliance on specific HSP90 client proteins.7,49,50 These findings suggest that epichaperome formation enables cancer cells to sustain survival and proliferation under high-stress conditions by reorganizing PPI networks to support their pathological needs. As a result, tumors with high levels of epichaperomes become uniquely vulnerable to epichaperome disruption.
These tumors shared aggressive disease behaviors that pose significant challenges for effective cancer treatment, including uncontrolled cell proliferation, inhibition of apoptosis, promotion of angiogenesis, enhanced metastasis, resistance to therapies, and immune evasion.7,34,35,49,50 This underscores the central role of epichaperomes in sustaining the pathological traits of aggressive cancers and highlight epichaperome formation as a mechanism of adaptation—or rather mal-adaptation—in high-stress conditions.
Importantly, unlike the ubiquitously expressed canonical HSP90 complexes found in all cells and tissues—whether normal or diseased—epichaperomes are uniquely present in disease contexts, such as cancer and neurodegeneration,7,29,30,32,38–40 and in limited cellular environments where robust proliferation and adaptive behavior are crucial, such as in the maintenance of pluripotent stem cells.7,30,32,36,41,48
In sum, these early studies in cancer position epichaperome formation as a maladaptive mechanism, allowing cancer cells to maintain their pathological phenotypes by reorganizing PPI networks to support stress adaptation. As a result, these tumors become uniquely vulnerable to epichaperome disruption, highlighting the therapeutic potential of targeting these assemblies to dismantle the pathological networks that sustain aggressive disease traits. Subsequent research, detailed in later sections, has expanded this understanding, implicating specialized adaptive roles for epichaperomes, including their involvement in maintaining key features of pluripotency—such as cellular adaptability and robust proliferation—through specialized protein network reconfigurations.30
3. Epichaperome disruption, not HSP90 inhibition, dictates activity
Why epichaperomes matter became further evident in cancer studies. While not all tumors harbor epichaperomes, in tumors where epichaperomes are prevalent, survival and signaling are critically reliant on these assemblies.7,31,34,50 Rodina et al. demonstrated this dependency by selectively downregulating HSP90 levels using siRNAs targeting both HSP90α and HSP90β paralogs in epichaperome-positive tumor models.7 By titrating siRNA amounts, they observed that even when 90% of HSP90 was depleted, as long as the remaining HSP90 was incorporated into epichaperomes, the cells remained viable, and signaling pathways, such as p-S6—a marker of translational activity and a driver of proliferative and survival signals in aggressive tumors51—remained active. Only when an inflection point was reached—where epichaperomes were completely disrupted, leaving only the 242 kDa band characteristic of folding HSP90 forms—did cell death occur, and signaling activity ceased.7 This finding underscores that in epichaperome-positive tumors, cell survival and aberrant signaling depend on epichaperomes rather than canonical folding HSP90 (Fig. 3a).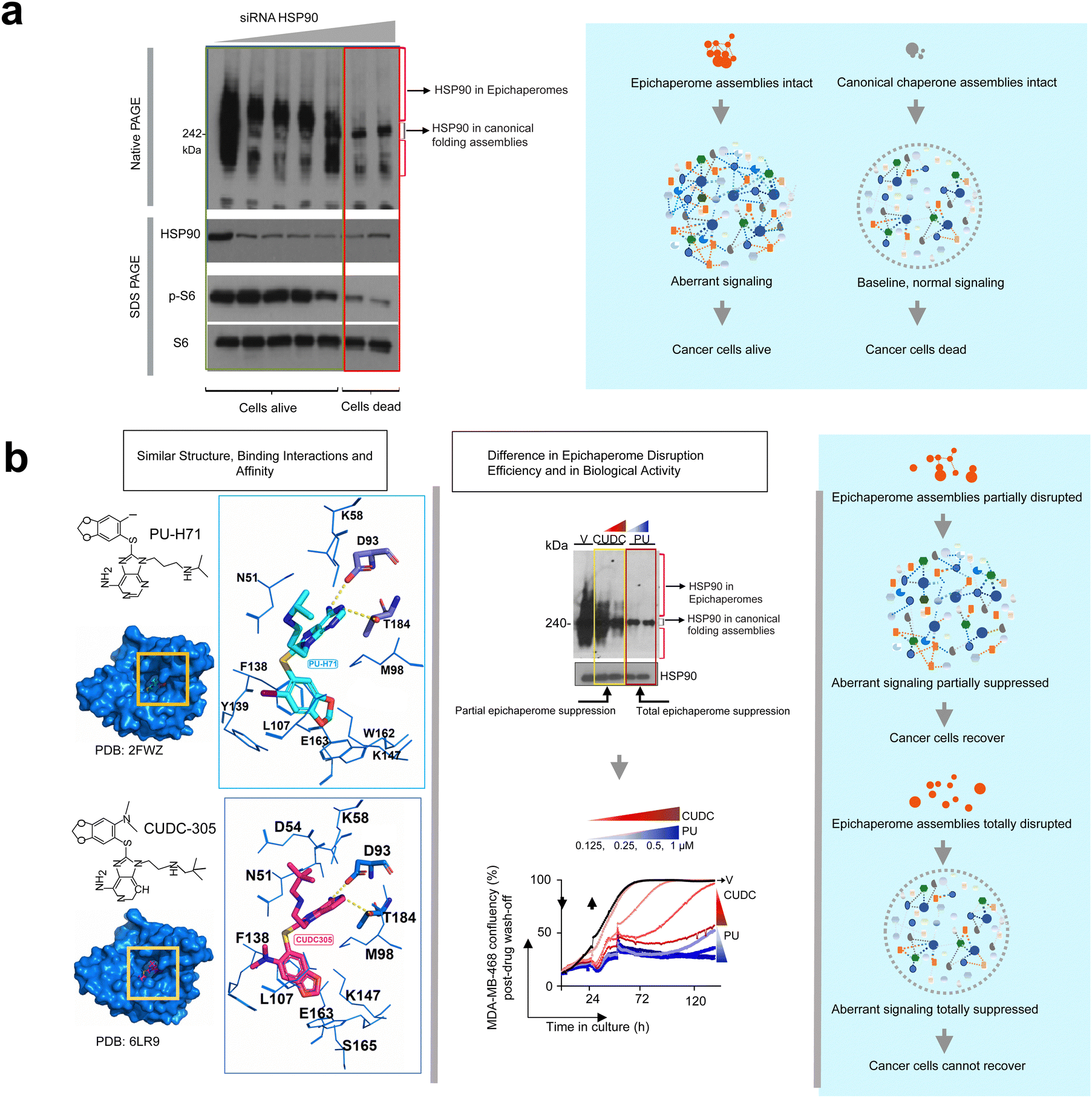 | ||
| Fig. 3 Epichaperome function, not canonical HSP90 activity, dictates tumor cell viability and aberrant signaling. (a) Epichaperome-positive tumor cells rely on epichaperome assemblies for survival and aberrant signaling. Selective downregulation of HSP90 levels using siRNAs targeting HSP90α and HSP90β paralogs demonstrates that as long as HSP90 is incorporated into epichaperomes, cells remain viable, and signaling pathways, such as p-S6, remain active. Cell death and signaling cessation occur only upon complete epichaperome disruption, correlating with the disappearance of high-molecular-weight species and the retention of only the 242 kDa band on native-PAGE, characteristic of folding HSP90. Adapted from Rodina et al. Nature 2016. (b) Epichaperome-targeting disruptors dismantle pathological epichaperome scaffolds, halting tumor regrowth. PU-H71 effectively disrupts epichaperomes, as evidenced by the loss of high-molecular-weight bands on native-PAGE, correlating with the inability of treated cells to regrow. In contrast, CUDC305, a compound with similar HSP90 binding affinity under equilibrium conditions, only partially disrupts epichaperomes, allowing residual high-molecular-weight species to persist and enabling cell recovery and regrowth. Adapted from Joshi et al. Communications Biology 2021. | ||
Further supporting this notion, where studies with PU-H71 and PU-AD (a CNS-permeable version of PU-H71 also called icapamespib in clinical settings)48,52 which demonstrated that the disruption of epichaperomes—rather than HSP90 inhibition—is the key driver of drug activity in epichaperome-positive tumors.32,36,37 These small molecules effectively dismantle epichaperomes, thereby abolishing their pathological scaffolding and restoring normal cellular processes.33 For example, Joshi et al.,37 compared PU-H71 with CUDC305—a structurally similar compound developed by Curis Inc.53 Both compounds bind HSP90 with similar binding affinity under equilibrium conditions, yet their abilities to disrupt epichaperomes differ significantly.37 In their study, Joshi et al.37 treated MDA-MB-468 cancer cells with high concentrations of PU-H71 or CUDC305 for 24 hours. After removing the drugs and washing the cells, they monitored cell regrowth over five days and assessed epichaperome levels using native-PAGE combined with immunoblotting against epichaperome components such as HSP90. The results showed that only PU-H71 completely disrupted epichaperomes, resulting in the absence of high-molecular-weight bands and the exclusive presence of the 242 kDa HSP90 band. This total epichaperome disassembly correlated with a lack of cell regrowth. In contrast, CUDC305-treated cells displayed partial epichaperome disruption, with residual high-molecular-weight bands still visible on the gel.37 These remaining epichaperomes allowed cells to recover and regrow, demonstrating a critical link between epichaperome persistence and cell survival (Fig. 3b).
Together, these studies illuminate the essential role of epichaperomes in maintaining disease-specific phenotypes in epichaperome-positive tumors and highlight the necessity of targeting these unique assemblies rather than simply focusing on HSP90 inhibition. These findings collectively establish that in epichaperome-positive tumors, the pathological role of epichaperomes—and not the folding function of HSP90—is the primary driver of disease phenotypes and thus the target of therapeutic intervention. This realization has catalyzed a wave of research aimed at unraveling the complexities of epichaperomes. What are the key components that constitute epichaperomes? What molecular factors drive their formation, and how do these assemblies remodel PPI networks to sustain disease-specific phenotypes? Addressing these questions is central to understanding the pathological roles of epichaperomes and their potential as therapeutic targets. The following sections explore these pivotal aspects in detail.
4. What are the epichaperome components?
Epichaperomes are context-dependent assemblies, with their composition varying according to the cellular environment and the specific stressors driving their formation.29,41 While these assemblies are nucleated on major chaperones, such as HSP90 and heat shock cognate 70 (HSC70), their precise components adapt dynamically to meet the demands of distinct stress conditions (Fig. 4).5,30–32,34,41,44,45,50,54,55 | ||
| Fig. 4 Context-dependent epichaperome assemblies and their role in rewiring cellular networks. Epichaperomes form context-specific assemblies whose composition varies depending on the cellular environment and stressors. These assemblies are nucleated on key chaperones, such as HSP90 and HSC70, and recruit additional components tailored to distinct pathological conditions. This figure illustrates the structural diversity of epichaperomes and their functional specificity in rewiring protein–protein interaction networks. By linking unique stress contexts to distinct network alterations, epichaperomes highlight their pivotal role in supporting disease-specific phenotypes. | ||
HSP90 and HSC70 serve as the core structural elements of epichaperomes across a variety of diseases and disorders, including cancer, Alzheimer's disease, Parkinson's disease and traumatic brain injury.7,31,32,37,41,42 These chaperones form a foundation for the recruitment of additional components tailored to the stress-specific cellular demands. For example, in dopaminergic neurons exposed to subtoxic concentrations of rotenone—a model of sporadic Parkinson's disease—HSP90 plays a pivotal role in recruiting HSP60.41 In this context, epichaperomes reorganize pathways related to dopamine synthesis, highlighting their role in mitigating mitochondrial dysfunction and other rotenone-induced stress effects. In contrast, in neurons stressed by a mutant form of parkin—a model of familial Parkinson's disease—HSC70 is the predominant chaperone incorporated into epichaperomes.41 Here, epichaperomes primarily regulate inflammatory pathways, demonstrating their ability to adjust their assembly and function to specific pathological conditions.
In highly proliferating and aggressive cancers, HSP90 and HSC70 work together within epichaperomes to remodel mitotic proteins and assemblies, facilitating more efficient mitosis.31 This role is crucial for sustaining the rapid division rates characteristic of these cancers and is exemplified by their involvement in reorganizing critical mitotic components. By ensuring robust mitotic function, epichaperomes contribute directly to the survival and proliferation of aggressive tumor cells.
Glucose regulated protein 94 (GRP94), a paralog of HSP90, is incorporated into epichaperomes in specific disease contexts through glycosylation at residue N62, shifting it from a folding chaperone to a stable epichaperome component.55–58 At the plasma membrane, GRP94-containing epichaperomes recruit signaling proteins, thereby enhancing signaling output in cancer by promoting overactivation of key oncogenic pathways, including ERK and NF-κB signaling.55,57 These epichaperomes also play key roles in immune modulation, inflammatory diseases, and viral infections,29,56,59–61 highlighting their versatile functions across diverse pathological contexts.
This adaptability underscores the modular and flexible nature of epichaperomes, enabling them to reorganize PPI networks in response to the unique demands of cellular stress. By nucleating on core chaperones and selectively incorporating components tailored to specific stress conditions, epichaperomes act as scaffolding platforms uniquely suited to pathological contexts. Understanding these context-dependent assemblies, along with the molecular factors that drive and enable their formation, is critical for deciphering their role in disease. In the next section, we examine how post-translational modifications (PTMs) on intrinsically disordered regions (IDRs) serve as key regulators of epichaperome assembly and function. IDRs are flexible regions within proteins that lack a fixed three-dimensional structure, making them highly adaptable in response to cellular stress and regulatory signals with PTMs located within these regions serving as critical regulators of protein conformation, acting as switches to direct structural transitions.62
5. PTMs on IDRs drive epichaperome assembly and function
Epichaperome formation represents a distinct reengineering of chaperone complexes, transforming canonical folding machinery into stable scaffolding platforms that reorganize PPI networks. A key driver of this transformation is the regulation of IDRs by PTMs.30,55 These modifications act as molecular switches, stabilizing specific conformations and influencing the stability of the formed assemblies, thereby enabling the incorporation of HSP90 and its paralog GRP94 into epichaperomes and defining their functional roles within these complexes (Fig. 5).30,55,57 | ||
| Fig. 5 Post-translational modifications (PTMs) on intrinsically disordered regions (IDRs) drive epichaperome formation. Phosphorylation at Ser226 and Ser255 within the intrinsically disordered charged linker of HSP90 stabilizes the linker structure, flipping it away to expose the middle domain (MD) of HSP90 and enhancing its interaction with co-chaperones such as HSC70. This phosphorylation induces a conformational switch that stabilizes a closed-like conformation of HSP90, impairing its ability to cycle through the conformational motions required for the ATPase cycle and disrupting its canonical folding activity. Instead this state stabilizes the epichaperome assembly by creating a microenvironment conducive to strong interactions with co-chaperones and other components. The resulting scaffolding platforms sequester proteins and rewire PPI networks that support aggressive tumor phenotypes. Adapted from Roychowdhury et al. Nature Communications 2024. | ||
A pivotal study identified phosphorylation of HSP90 at residues Ser226 and Ser255, located within the IDR of its charged linker, as a key determinant of epichaperome formation.30 This phosphorylation stabilizes a closed-like conformation of HSP90, enhancing its interactions with co-chaperones and creating a microenvironment conducive to epichaperome assembly.30 Locally, phosphorylation induces a conformational switch in the charged linker, flipping it into an “up” position that fully exposes the middle domain of HSP90, a critical interaction site for HSC70. Distally, these modifications stabilize the overall structure of the epichaperome, both by impairing HSP90's ability to refold denatured proteins and by reinforcing the stability of the formed complex.30
The functional significance of these modifications was demonstrated using HEK293 cells transfected with phosphomimetic (HSP90βS226E,S255E) and non-phosphorylatable (HSP90βS226A,S255A) mutants.30 Cells expressing the phosphomimetic mutant exhibited significantly higher levels of epichaperomes compared to wild-type or non-phosphorylatable mutants. Importantly, these cells also displayed enhanced signaling through pathways such as MEK, AKT, and mTOR, increased self-renewal and proliferation capacity, and phenotypic changes indicative of mesenchymal-like states.30
These findings directly link phosphorylation of HSP90 to epichaperome formation, stability, and the disease-specific functions mediated by these assemblies, while also highlighting the impact of phosphorylation on impairing HSP90's canonical folding activity.
For GRP94 (also called gp96 and endoplasmin), another critical chaperone incorporated into epichaperomes, N-glycosylation at residue Asn62 within an IDR was identified as a driver of epichaperome assembly.55,57 Under normal physiological conditions, GRP94 facilitates the folding of client proteins through transient interactions, primarily within the endoplasmic reticulum (ER).63 However, glycosylation at Asn62 disrupts this role, inducing a conformational shift that promotes its stable incorporation into epichaperomes.55,57
In the absence of Asn62 glycosylation, GRP94 transits between open and close conformations conducive to folding activity.64 When glycosylated, the ATP-lid is pulled into a more closed conformation, impairing folding activity and favoring stable interactions with other epichaperome components.57 This glycosylation event not only induces structural shifts but also stabilizes the formed epichaperome assembly, supporting its role in reorganizing PPI networks.55 At the plasma membrane, glycosylated GRP94 clusters oncogenic proteins into epichaperome platforms, driving the remodeling of cellular protein networks that support aggressive cancer phenotypes.55,56
This role was demonstrated in MDA-MB-468 breast cancer cells, which overexpress plasma membrane-localized EGFR, with downstream signaling via EGFR essential for promoting proliferation and survival in this cancer context.55,57 Using CRISPR-Cas9 to generate GRP94 mutants, researchers observed that cells expressing the N62Q mutant, which cannot undergo glycosylation, showed reduced epichaperome formation and a loss of plasma membrane EGFR localization and signaling. Conversely, cells expressing the N217A mutant, which represents the canonical folding form of GRP94, retained EGFR signaling. These findings directly link glycosylation of GRP94 to its pathological incorporation into epichaperomes, their stability, and the subsequent functional changes observed in cancer.
In sum, for both HSP90 and GRP94, PTMs on IDRs play a central role in regulating their conformation, stability, and assembly into epichaperomes. The flexibility of IDRs allows these regions to adopt multiple conformations, which are fine-tuned by PTMs acting as molecular switches. These PTMs not only stabilize specific conformations and reinforce the stability of the formed epichaperome assemblies but also impair the folding capacity of these chaperones. By disrupting the dynamic, transient nature of canonical folding chaperones, PTMs enable the transition to stable, long-lived scaffolding platforms. Through this mechanism, IDRs and their regulating PTMs mediate the structural shifts necessary for epichaperome formation, stabilize the complexes, and drive disease-specific PPI network remodeling. These findings highlight the dual role of PTMs as both structural regulators of epichaperome assembly and inhibitors of traditional chaperone folding activity, further emphasizing the unique properties of epichaperomes in pathological contexts.
This concept aligns with broader studies on chaperone biology, which emphasize the profound impact of PTMs on chaperone regulation. Due to the intrinsic need for chaperones to sample diverse conformational states during their functional cycles, PTMs serve as critical modulators that remodel chaperone structure and function with remarkable efficiency. This principle is encapsulated in the concept of the ‘chaperone code’, which describes how specific PTM patterns orchestrate chaperone activity, client interactions, and broader proteostasis networks.24,65,66 In this context, epichaperome formation represents a specialized instance of how PTM-driven regulation of chaperones can lead to the creation of stable, scaffolding platforms that fundamentally rewire PPI networks, underscoring the broader relevance of the chaperone code in both physiological and pathological settings.
6. What do epichaperomes do?
Epichaperomes are pivotal drivers of disease pathogenesis in cancer and neurodegenerative disorders.29,33,38,39 By sequestering thousands of proteins and reorganizing PPI networks, epichaperomes disrupt the conventional PPI landscape, leading to abnormal protein interactions that underlie dysfunctional cellular behaviors. These disruptions involve both the loss and gain of PPIs, fundamentally altering cellular functions in a context-dependent manner.29,33,38,39In Alzheimer's disease, epichaperomes significantly restructure PPIs within brain tissue, resulting in widespread disruptions of critical networks involved in synaptic plasticity, cell communication, protein translation, cell cycle regulation, axon guidance, and metabolic and inflammatory processes.29,32,39,67 Studies using post-mortem brain tissue from Alzheimer's disease patients revealed that during the transition from normal aging to Alzheimer's disease, epichaperomes drive the loss of connections for 942 proteins and the formation of 1191 new, abnormal interactions. Functional mapping of these dysfunctional PPIs revealed synaptic proteins as one of the most important class vulnerable to epichaperome formation. These synaptic proteins were linked to signaling pathways such as ‘signaling by second messenger’, ‘Gα(i) signaling’, ‘signaling by Rho GTPases’, ‘signaling by Wnt’, ‘response to elevated cytosolic Ca2+’, and ‘MAPK signaling’. Consequently, both short-term memories—dependent on post-translational modification of synaptic proteins through these signaling networks—and long-term memories—requiring synthesis of new proteins—are adversely affected by epichaperomes in Alzheimer's disease.29,32,39,67 Targeting epichaperomes with small molecules such as PU-H71 or PU-AD restored affected networks in cellular,32 organoid,68 or mouse models of Alzheimer's disease,32 normalizing synaptic protein connectivity and cognitive function. These findings underscore the therapeutic potential of targeting epichaperomes to achieve systems-level restoration of brain function, spanning cellular to connectome levels, in neurodegenerative diseases.
In cancer, epichaperomes sustain malignant phenotypes by sequestering proteins essential for the signaling, metabolic pathways, and immune regulation that define the aggressive behavior of cancer cells.7,29,38 For instance, in the highly proliferative MDA-MB-468 breast cancer cell line, epichaperomes sequestered 2481 proteins linked to processes critical for maintaining this aggressive phenotype.31 Among these were proteins indispensable for mitotic progression, particularly during the G2/M transition and anaphase.31 This subset included key regulators of spindle formation, spindle-assembly checkpoint signaling,69 centrosome regulation, and kinetochore-microtubule attachment. The rewiring of these mitotic PPI networks by epichaperomes was essential for the rapid and efficient division of these tumor cells.31 Notably, cells that entered mitosis in the presence of epichaperome disruptors were unable to progress through mitosis and ultimately died, underscoring the dependency of these cancer cells on epichaperome-driven mitotic rewiring.
In sum, epichaperomes are central orchestrators of disease phenotypes. By sequestering proteins and rewiring cellular networks, they underpin the functional imbalances critical to disease progression. Epichaperomes are highly organized scaffolding platforms that reconfigure cellular networks in response to disease-driven stress. These findings emphasize the potential of targeting epichaperomes to correct context-specific PPI network dysfunctions,33 offering a pathway to therapeutic interventions in diseases like AD and cancer.
7. Epichaperome-targeting small molecules
The discovery that major chaperones such as HSP90, GRP94, and HSC70 form the core of epichaperome assemblies in diseases like Alzheimer's disease, Parkinson's disease, traumatic brain injury, and cancer has driven the development of small molecules to selectively target these assemblies.31,36,48,58,70 Unlike conventional inhibitors, epichaperome disruptors specifically dismantle the stable scaffolding platforms formed by epichaperomes, addressing the disease-specific PPI networks they sustain (Fig. 6a).31,33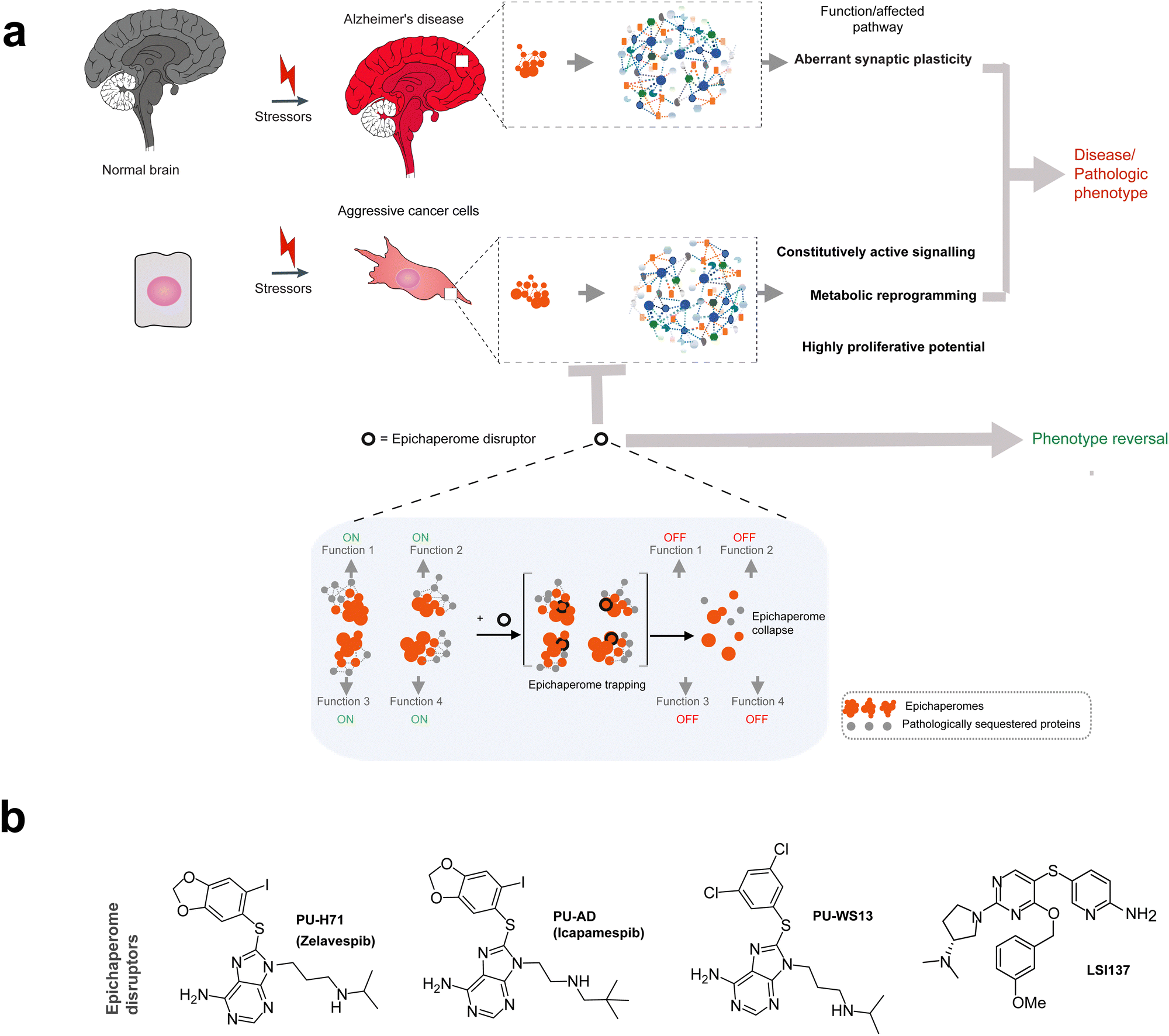 | ||
| Fig. 6 Epichaperome-targeting small molecules: mechanisms and therapeutic potential. (a) Epichaperome disruptors target the stable scaffolding platforms formed by epichaperomes, dismantling these disease-specific assemblies. By addressing the pathological protein–protein interaction (PPI) networks sustained by epichaperomes, disruptors restore normal cellular processes. Epichaperome disruptors represent a novel therapeutic class, transcending single-protein targeting by addressing the network-level dysregulation driven by pathological epichaperome scaffolds. These agents redefine the concept of molecular glues, uniquely combining stabilization and disassembly of disease-specific protein assemblies to restore cellular homeostasis (inset). (b) Chemical structure of PU-H71 and other epichaperome disruptors. PU-H71, the first-in-class epichaperome disruptor, kinetically traps epichaperomes by binding to HSP90 when it is incorporated within these assemblies. PU-AD (icapamespib) extends this mechanism to central nervous system (CNS) diseases, including Alzheimer's disease and glioblastoma, by targeting epichaperomes through HSP90. Similarly, LSI-137 acts on epichaperomes by targeting HSC70, whereas PU-WS13 disrupts epichaperomes by binding to GRP94. | ||
PU-H71, the first-in-class epichaperome disruptor, exemplifies this novel drug mechanism (Fig. 6b).7 By binding to HSP90 when it is incorporated within epichaperomes, PU-H71 kinetically traps these assemblies, inducing their disassembly and disrupting their pathological role.32,36,37,71 This action restores normal PPI networks by dismantling the epichaperomes, a process that affects the interactions of key constituent proteins, including HSP90 and HSC70, without broadly altering their overall expression levels.32,36,37,71
Pharmacokinetic and target occupancy studies conducted in vivo with PU-H71 have further illuminated its mode of action. The off-rate of PU-H71 from epichaperomes is significantly slower than expected from its tumor pharmacokinetic profile or from in vitro binding studies conducted under diluted conditions.36,71 Once bound to epichaperomes, PU-H71 becomes kinetically trapped, contributing to extended on-target residence times observed in clinical settings.35,36,72 For example, in epichaperome-positive tumors, PU-H71 exhibits residence times lasting days, compared to minutes or hours in epichaperome-negative tissues.35,36,72 This prolonged residence time is driven by the kinetics of epichaperome trapping and subsequent disassembly, rather than the drug's unbinding kinetics from its target.71
This mechanism aligns with and extends the concept of molecular glues—agents that stabilize specific protein interactions to achieve functional outcomes73—by adapting it to the unique context of epichaperomes. While molecular glues typically stabilize specific protein interactions, epichaperome disruptors go a step further by first stabilizing and then dismantling the pathological scaffolding platforms that define epichaperomes. This dual action enables targeted disruption of disease-specific assemblies and leads to systems-level restoration of cellular functions.33 The broader impact arises from the inherent role of epichaperomes themselves: by sequestering and restructuring thousands of proteins, epichaperomes reshape PPI networks to sustain disease-specific phenotypes. Disrupting epichaperomes not only dismantles these pathological scaffolds but also addresses the network-level dysregulation they drive, offering a therapeutic strategy that transcends single-protein targeting.
Building on the success of PU-H71, several other epichaperome-targeting molecules have been developed (Fig. 6b). PU-AD (icapamespib),32,48,52 which also targets epichaperomes via HSP90, has progressed to phase 2 trials for Alzheimer's disease and phase 1 trials for glioblastoma.29 PU-H71 (zelavespib) has moved to phase 1/phase 1b trials for various cancers, with activity in metastatic breast cancer35 and in myeloproliferative neoplasm transformed to refractory acute myeloid leukemia.34 LSI-137, which targets epichaperomes through HSC70,31 and PU-WS13, which disrupts epichaperomes by targeting GRP94,55,57,58 are both in preclinical development. These agents share the ability to selectively engage epichaperomes at disease sites for extended durations while rapidly dissociating from normal folding chaperone assemblies. This specificity, driven by the kinetics of epichaperome trapping and subsequent disassembly, underpins their therapeutic efficacy and favorable safety profiles.
By exploiting the pathological remodeling of chaperones into stable scaffolding platforms, epichaperome disruptors redefine the therapeutic potential of molecular glue-like agents. They address not just individual protein targets but also the broader network-level dysregulation that underlies diseases, providing a new paradigm for therapeutic intervention. Thus, while molecular glues traditionally stabilize protein interactions, epichaperome disruptors uniquely leverage this concept by first stabilizing epichaperome assemblies and then inducing their targeted disassembly, dismantling pathological scaffolds and restoring cellular homeostasis.
8. On the topic of selectivity and therapeutic index
With the above being said, why is that many of the HSP90 inhibitors that entered the clinic74,75 show selectivity for non-transformed cells vs. cancer cells, including CUDC305.53 If not all of these molecules act on epichaperomes, then why do they have cancer-cell selectivity? Is there a parallel mechanism for gaining therapeutic index?Before addressing the question of “selectivity,” it is important to clarify a key concept: small molecule binders of HSP90 that have entered clinic are likely not exclusively HSP90 inhibitors nor exclusively epichaperome disruptors. Instead, they may represent a “mixed bag” of activities, with each compound positioning itself somewhere along a continuum between a pure HSP90 inhibitor and a pure epichaperome disruptor.29,37 With this clarification in place, we now move to address the question of selectivity.
If we define “selectivity” by the proposed extended retention of such clinical HSP90 agents in tumors vs. normal tissues and plasma, this again would be true if such inhibitors do act, to some extent, on epichaperomes in a fashion similar to PU-H71 (i.e. become kinetically trapped). CUDC305, which interacts with epichaperomes but fails to disrupt them effectively,37 would likewise exhibit tumor retention, albeit to a lesser extent.53
If selectivity is defined by a compound's ability to spare normal cells, we argue that this feature is not solely a property of the compound but is largely influenced by the biological context of the target. Specifically, if normal cells predominantly contain canonical folding chaperones and lack epichaperomes, a crucial question arises: is inhibition of canonical folding HSP90 assemblies toxic to normal cells? Studies on HSP90 in yeast and normal cells suggest that inhibition of canonical HSP90 folding activity is not inherently toxic, at least under conditions of limited exposure.76,77
For example, studies by the Picard lab demonstrated that mouse NIH-3T3 fibroblast cells, which are considered normal except for their immortalization, and predominantly contain canonical folding chaperones and lack epichaperomes, are less affected by HSP90 inhibition under transient exposure conditions. These cells however exhibit heightened sensitivity to HSP90 agents when exposed to various stressors, including oncogenic Ras transformation and proteotoxic stress.77 This increased dependency was accompanied by metabolic shifts characteristic of a Warburg phenotype and a mesenchymal-like transition, both of which mirror the epichaperome-driven phenotypic changes observed in cancer cells.30
While not explicitly investigating epichaperomes, these findings align with our understanding that a variety of stressors can drive the formation of epichaperomes, rewiring PPI networks to sustain aggressive phenotypes (such as is the phenotype observed by Picard in the Ras-transformed NIH-3T3 cells), and in turn, rendering them uniquely vulnerable to HSP90-targeting agents.7 Thus, the heightened drug sensitivity in transformed cells may, in part, reflect their reliance on epichaperome scaffolding rather than canonical HSP90 folding activity, whereas the tolerance of non-transformed cells to drug exposure relies in their ability to overcome transient inhibition of canonical HSP90 folding activity.
This highlights a fundamental principle: inhibition of canonical HSP90 assemblies is not inherently toxic to normal cells unless exposure is prolonged.
Yeast models also reveal that the structural context of HSP90—whether in canonical folding assemblies or in epichaperome-like assemblies—plays a key role in determining inhibitor sensitivity. In particular, replacing endogenous yeast HSP90 with human HSP90β results in a dramatic increase in sensitivity to HSP90 inhibitors, despite comparable ATPase activity between the two proteins.78 This heightened drug sensitivity is likely due to the formation of more stable chaperone–cochaperone assemblies, modulated by the presence or absence of Sti1 (yeast HOP), and is reminiscent of epichaperome-like assemblies in mammalian cells.
Notably, yeast expressing only canonical chaperones remained viable upon HSP90 inhibition,78 reinforcing the concept that inhibition of dynamic, physiological chaperone assemblies is generally well tolerated, whereas stabilized, epichaperome-like chaperone complexes confer heightened drug sensitivity. Along these lines, the Lindquist lab79 also investigated the effects of transient HSP90 inhibition in yeast models. Their findings indicated that short-term inhibition led to persistent changes in mRNA translation without causing immediate toxicity, highlighting that transient disruption of HSP90 function does not necessarily compromise cell viability.
These observations combined imply that HSP90 inhibition may be tolerated in normal cells provided exposure is transient. However, chronic inhibition, which depends on how these agents are administered (i.e. dose and schedule, see discussion below), may overcome this tolerance.
With regards to why it matters where on the continuum between a pure HSP90 inhibitor and a pure epichaperome disruptor a compound places itself—the ‘HSP90 curve’ representing the spectrum of these mixed activities—one can draw an analogy to kinase inhibitors. These also span a spectrum of profiles, with some acting only on disease-associated kinase conformations, while others target both pathological and physiological kinase states.80–82 This distinction has direct implications for therapeutic index, as it strongly influences dosing schedules, on-target residence time, and off-target toxicities.
A key factor in therapeutic index is not just whether a compound binds its target but how effectively it modulates the pathological structure.83–87 In the context of epichaperome disruptors, agents that interact with epichaperomes but fail to efficiently dismantle them—such as CUDC305—may require higher or more frequent dosing to achieve therapeutic efficacy. This has several consequences:
(1) Increased systemic exposure—higher or more frequent dosing raises systemic drug levels, leading to greater potential for off-target toxicity.
(2) Incomplete target modulation—inefficient disruptors may not fully dismantle epichaperomes within a single dosing cycle, allowing pathological scaffolds to persist. This necessitates more sustained drug exposure, which could drive compensatory resistance mechanisms.
(3) Shorter on-target residence time—if a compound has fast dissociation kinetics from epichaperomes, it may fail to kinetically trap and dismantle these complexes. This means that higher concentrations are needed to maintain target engagement, further straining the therapeutic window.
(4) Cumulative toxicity—unlike agents which selectively dismantle epichaperomes with minimal impact on normal folding HSP90, weaker disruptors risk prolonged engagement with both epichaperomes and canonical chaperones, leading to off-target effects in normal tissues.
Thus, poor epichaperome disruptors will likely require higher systemic exposure and frequent dosing to sustain on-target effects, compromising selectivity and safety. In contrast, highly effective disruptors require lower doses and exhibit prolonged retention at the disease site due to kinetic trapping, enhancing their therapeutic index.
This underscores that therapeutic index is not solely a function of target binding but is also a function of sustained and efficient modulation of the pathological target. Therefore, where an HSP90-targeting agent falls on this continuum dictates not only its efficacy but also its safety and clinical viability.
9. On the topic of addiction—why knowing your target matters
Are epichaperomes the only pathological chaperone assemblies in tumors? While epichaperomes represent a distinct reengineering of chaperone networks, cancer cells also exhibit differential HSP90 conformational cycling and client dependency that extends beyond epichaperome formation.43,88,89 As a result, cancer cells may exhibit heightened chaperone addiction, which renders a greater fraction of HSP90 in conformations susceptible to inhibitor binding,90 independent of epichaperome formation. In this context, the activity of non-epichaperome-targeting HSP90 inhibitors may arise from exploiting this heightened dependency rather than from disrupting epichaperome scaffolds.28 Notably, if tumors exhibit elevated pools of chaperone-addicted HSP90, high-affinity binding to these chaperone forms, even in the absence of epichaperomes, may contribute to tumor accumulation of HSP90 inhibitors.91 Unlike epichaperome trapping, which leads to kinetic retention within tumor cells, this mechanism instead relies on the preferential engagement of these chaperones, leading to prolonged inhibitor residence times and selective accumulation in tumors. However, such an accumulation mechanism is distinct from epichaperome targeting and must be carefully considered when evaluating the pharmacokinetic and pharmacodynamic properties of HSP90 agents.Do all tumors harbor and depend on epichaperomes? Despite epichaperome presence in 60–70% of evaluated tumors,7 not all tumors harbor epichaperomes, yet they remain malignancies.7,30,31,48 These cancers may instead present and depend on such elevated pools of conformationally primed chaperones,92 reflecting a different form of chaperone dependence.93–96 For example, the ASPC1 pancreatic cancer cell line is tumorigenic, exhibits folding activity, and possesses oncogenic properties, yet does not inherently form or rely on epichaperomes.7 However, MYC overexpression is sufficient to induce epichaperome formation, shifting the tumor from a state of folding activity to one of epichaperome dependence and low folding capacity.7 This demonstrates that epichaperome formation is not an intrinsic tumor feature but a consequence of oncogenic stress—a tipping point where the chaperone network is fundamentally restructured.
Importantly, the higher the epichaperome levels in a cell, the more impaired its folding capacity.30 This loss of folding function persists despite the presence of high levels of HSP90 and other chaperones and co-chaperones, suggesting that epichaperome formation acts as a dominant-negative mechanism—sequestering chaperones, co-chaperones, and other proteins within the epichaperome structure, thereby reducing the pool of chaperones available for canonical folding functions.
Collectively, this transformation of cells from chaperone-dependence to epichaperome-addiction is not a gradual adaptation but a fundamental switch in tumor biology. Epichaperome formation does not simply modify existing chaperone function—it generates a distinct tumor state, characterized by a rewired proteome, altered stress response, and a pathological reliance on these supramolecular scaffolds for survival. This restructuring not only shifts tumor dependencies but also alters drug vulnerabilities, redefining how malignancies respond to therapeutic interventions.
Importantly, from a drug discovery perspective, these findings underscore a critical issue: the complexity of HSP90 in disease has been largely overlooked. Historically, drug development efforts treated HSP90 as a singular target, assuming that all cancer cells rely on a uniform HSP90-dependent mechanism. This is not the case. The biological context of HSP90—whether in dynamic folding complexes, epichaperomes, or other oncogenic conformational states—profoundly influences drug selectivity and therapeutic efficacy.
Given these insights, it is also critical to recognize that epichaperome detection serves as a functional biomarker for patient selection, allowing for the stratification of tumors based on their reliance on epichaperomes versus classical HSP90 chaperone activity. Positron emission tomography (PET) imaging with labeled epichaperome probes, for instance, has enabled the identification of tumors with high epichaperome burden in both preclinical models and patient samples, offering a non-invasive means to guide treatment decisions and predict therapeutic responses (see discussed below).
10. Chemical probes to dissect epichaperome biology
Chemical probes have been instrumental in elucidating the biology of epichaperomes, providing tools to both modulate and study these pathological assemblies in diverse biological contexts. Notably, chemical biology tools such as PU-H71 and its derivatives conceptually align with the activity-based probes (ABPs) described by Cravatt et al.,97–99 which highlight functionally relevant protein states in cellular contexts. While ABPs identify enzymatically active protein states through covalent attachment to active sites, PU-H71 distinguishes itself by targeting active supramolecular assemblies rather than enzymatic active sites.Epichaperome disruptors, such as PU-H71 and its derivatives, PU-WS13 and derivatives, and LSI-137 and derivatives, have been widely utilized to modulate epichaperomes in a variety of disease settings. These compounds enable researchers to investigate the role of epichaperomes in sustaining disease phenotypes.7,31,32,37,48,55,59,68,100 By selectively dismantling epichaperomes, these disruptors have revealed the critical contribution of epichaperomes to disease-specific PPI network dysregulation, offering valuable insights into their biology and therapeutic potential.
To visualize epichaperomes directly, click chemistry-based probes labeled with fluorescent dyes in situ, as well as directly fluorescently labeled probes, have been employed.6,7,31,101 These tools allow for the detection and quantification of epichaperomes within cells and tissues, providing spatial and temporal resolution to study their distribution and dynamics in both physiological and pathological settings. For example, PU-TCO, a tetrazine-clickable derivative of PU-H71, has been used to detect and quantify epichaperome levels in the mouse brain in a model of Parkinson's disease.101 In a recent study, this probe was also used in western blot analysis to detect epichaperomes, functioning similarly to antibodies in protein detection.30
Beyond visualization, solid-supported epichaperome probes have been developed to investigate the interactome of epichaperomes and dissect their biology at the systems level.7,31,32,37 One notable approach is the differential protein–protein interaction (dfPPI) method, which utilizes solid-supported epichaperome probes to capture and identify proteins sequestered by epichaperome assemblies (Fig. 7a).40 This method has enabled a comprehensive mapping of epichaperome-associated protein networks in cancer and neurodegenerative diseases, highlighting the extensive rewiring of cellular interactomes driven by epichaperomes. By revealing how epichaperomes reorganize PPI networks to sustain pathological phenotypes, the dfPPI approach has expanded our understanding of their systems-level impact.
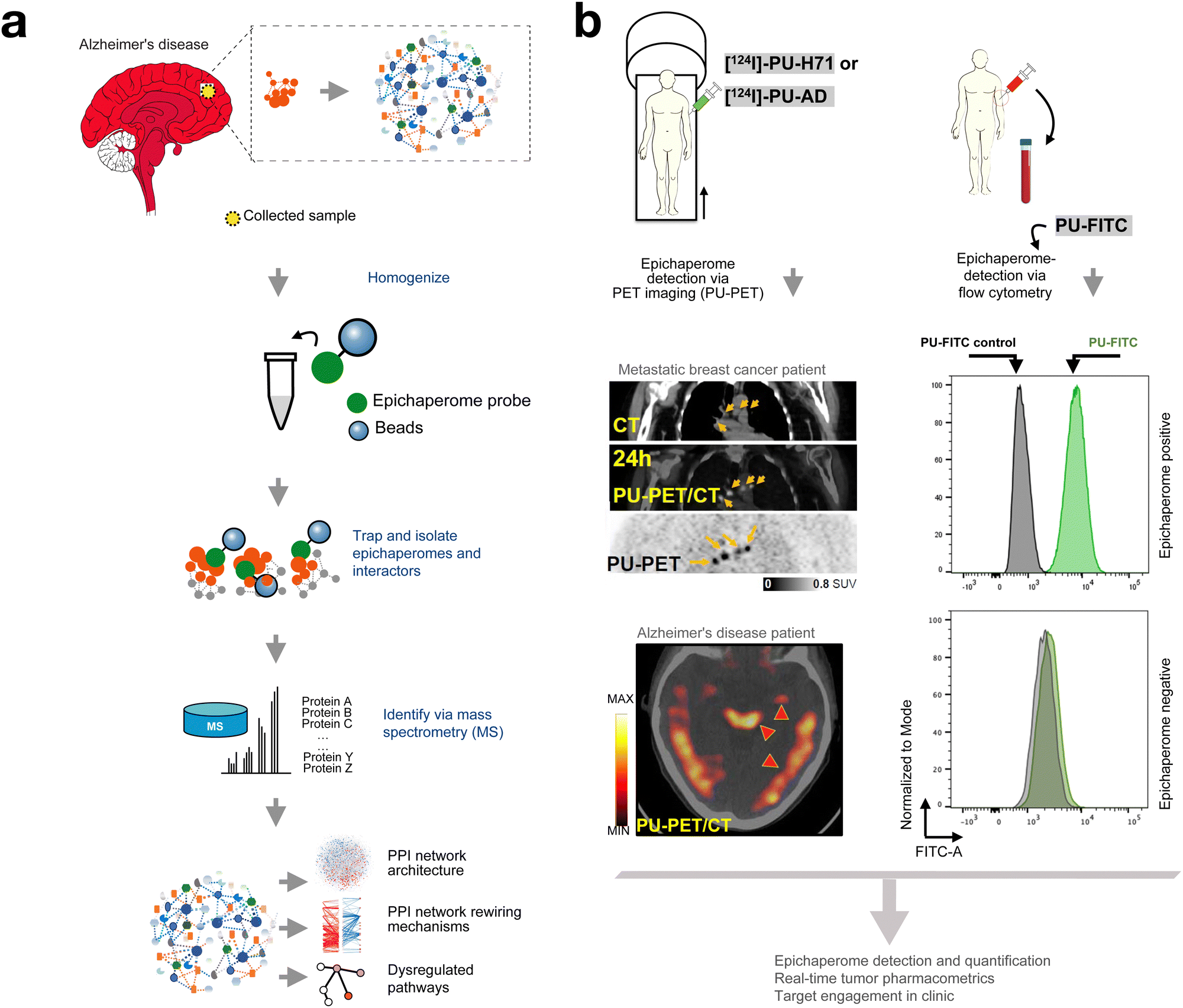 | ||
| Fig. 7 Chemical probes to study, detect, and quantify epichaperomes. (a) Tools for investigating epichaperome biology: differential protein–protein interaction (dfPPI) mapping leverages solid-supported epichaperome probes to dissect the interactomes of epichaperomes. By capturing and identifying proteins sequestered within epichaperome assemblies, dfPPI highlights the extensive rewiring of PPI networks driven by epichaperomes in disease contexts. This approach has provided critical insights into how epichaperomes reorganize PPI landscapes to sustain pathological phenotypes in diseases such as cancer and neurodegeneration. (b) Probes for detection, quantification, and pharmacometric analysis: epichaperome-targeting probes have advanced clinical and preclinical applications by enabling precise detection and quantification of epichaperomes. These radiolabeled probes have demonstrated tumor-specific retention times driven by the trapping mechanism within epichaperome assemblies. Fluorescently labeled probes, such as PU-FITC, are used to quantify epichaperome levels via flow cytometry in both patient samples and cell lines. Together, these imaging and detection tools underscore the pivotal role of chemical probes in advancing the understanding and clinical application of epichaperome-targeting therapies. Adapted from Rodina et al. Nature 2016 and Inda et al. Nature Communications 2020. | ||
Collectively, these chemical probes have not only illuminated the biology of epichaperomes but also underscored their potential as therapeutic targets. By modulating, visualizing, and characterizing epichaperomes, these tools have provided a robust platform for dissecting their function and exploring strategies to target them in complex diseases.
11. Probes in epichaperome research and therapy optimization
Radiolabeled and fluorescently labeled probes have proven invaluable in both preclinical and clinical settings for detecting epichaperomes, evaluating target engagement, and understanding pharmacokinetic (PK) and pharmacodynamic (PD) parameters.34,36,72,102Epichaperome levels serve as a biomarker for response to PU-H71 and other evaluated epichaperome disruptors, with higher epichaperome abundance correlating with greater tumor sensitivity to this therapy across all evaluated cancer types.7,31,37,45,48 The ability to quantify epichaperome levels is critical for identifying tumors most likely to benefit from targeted epichaperome disruption, emphasizing the translational significance of these probes.
In particular, 124I-labeled derivatives of PU-H71 (zelavespib) and PU-AD (icapamespib) have been used to detect and quantify epichaperomes in vivo through PET imaging (Fig. 7b).32,36,48 These agents are inherently suitable for radiolabeling due to the presence of an endogenous iodine atom, enabling the attachment of iodine-124, a PET-compatible radionuclide.
In preclinical mouse models and in clinical studies in cancer patients, PET imaging with 124I-labeled zelavespib or icapamespib allowed real-time individual-tumor PK measurements by co-injecting tracer amounts of radiolabeled agents with therapeutic doses of epichaperome drugs or alternatively, by injecting tracer amounts of the radiolabeled agent alone.35,36,48,71,72 These studies revealed the long residence time of zelavespib in tumors, with half-lives ranging from 24 to 100 hours. This prolonged retention is attributed to zelavespib's unique mechanism of interaction with epichaperomes, which involves an initial trapping phase, where the drug binds to HSP90 within the epichaperome assembly, followed by targeted disassembly of these pathological complexes.71
In addition to radiolabeled probes, fluorescently labeled compounds have been developed and utilized for the quantification of epichaperomes. For example, PU-FITC, a fluorescein-labeled derivative of PU-H71, has been employed in leukemia—both cell lines and patient samples—to quantify epichaperome levels via flow cytometry (Fig. 7b). This approach enables precise evaluation of epichaperome abundance within diverse cell populations, including blasts, lymphocytes, and granulocytes, facilitating patient selection for PU-H71 therapy.7,34,45,50,102 By quantifying epichaperome levels, PU-FITC played a critical role in identifying a relapsed-refractory AML patient who responded to PU-H71 therapy.34
PET imaging coupled with radiolabeled epichaperome probes not only enables the visualization of epichaperome presence but also provides critical insights into drug efficacy.36 In traditional drug development, plasma assays are commonly used to measure PK and guide dosing and schedule optimization.103 However, studies with PU-H71 and PU-AD revealed a disconnect between plasma PK and biological activity.36 Despite the rapid plasma clearance of these compounds—suggesting limited drug retention based on classical pharmacokinetic models—PET imaging demonstrated prolonged retention of these agents at their site of action—the tumor—driven by their unique mechanism of trapping within epichaperome assemblies.71 This tumor-specific retention correlated strongly with target occupancy and anti-tumor efficacy, as assessed by PET imaging.35,36,72 These findings underscore the limitations of plasma assays for evaluating epichaperome-targeting therapies and highlight the critical role of radiolabeled probes and PET imaging in optimizing dose and schedule selection by providing direct insights into tumor-specific drug retention and pharmacodynamic effects (Fig. 7b).35,36,72
These findings establish labeled epichaperome probes as powerful diagnostic and therapeutic tools, enabling precise, non-invasive assessments of drug–target interactions. The ability to detect epichaperomes, whether through imaging modalities like PET or flow cytometry using fluorescently labeled probes such as PU-FITC, defines a biomarker-driven approach to therapy, with epichaperome levels serving as a critical biomarker for both patient selection and therapeutic efficacy. By quantifying epichaperome abundance, these methods facilitate tailoring treatments to the unique molecular landscape of each patient. By leveraging these insights, epichaperome-targeted therapies hold the promise of achieving both precision and efficacy in treating diseases characterized by PPI network dysregulation.
12. Conclusions and future perspectives
The discovery of epichaperomes illuminated a new dimension of HSP90 biology, challenging the traditional view of this chaperone as merely a folding machine. It also clarified the mechanism of PU-H71, which targets HSP90 preferably when it is incorporated into epichaperomes. By binding to epichaperomes, PU-H71 disrupts their scaffolding function, restoring normal PPI networks and dismantling disease-specific cellular processes.33Despite these advancements, the development of epichaperome-targeting therapies has faced significant challenges, rooted in historical dogmas surrounding chaperone biology and the therapeutic targeting of HSP90. The “folding dogma,” which positions HSP90 primarily as a folding machine, has deeply influenced drug development. Under this framework, targeting HSP90's ATP-binding pocket was assumed to disrupt its folding activity, destabilizing client proteins and impairing their function. This rationale guided the early development of HSP90 inhibitors, including PU-H71. However, the discovery of epichaperomes revealed that this framework is insufficient to capture the complex biology of HSP90 in pathological contexts.
Unlike canonical folding chaperones, epichaperomes act as stable scaffolding platforms that rewire PPI networks to sustain disease phenotypes. This reengineering of chaperones into epichaperomes represents a distinct adaptation to cellular stress, driven by PTMs and context-specific demands. The conflation of epichaperomes with folding chaperones has hindered their recognition as a distinct therapeutic target, slowing the development of epichaperome disruptors. Similarly, classifying epichaperome disruptors as HSP90 inhibitors has obscured their unique mechanism of action. These compounds, including PU-H71, preferentially bind to HSP90 within epichaperomes, kinetically trapping and dismantling these pathological assemblies. This mechanism is fundamentally different from the inhibition of HSP90's ATPase activity or folding functions. Persisting with outdated classifications reinforces misconceptions that have impeded clinical progress.
Additionally, given the central role of epichaperomes in this field, it is crucial to clarify their definition. Epichaperomes are not defined by their ability to be pulled down by PU-H71 or related probes. Rather, they are stable, disease-specific assemblies of chaperones and co-chaperones that reorganize protein–protein interaction networks to sustain pathological phenotypes. While PU-H71 and related chemical probes are valuable tools for studying epichaperomes due to their preferential binding to these assemblies, the definition of epichaperomes is independent of such pulldown experiments. Their biochemical properties—including supramolecular assembly, stability, and unique functional roles—distinguish them from canonical chaperone complexes. We have elaborated on these structural and functional attributes throughout this review and continue to refine these definitions in our ongoing work.
It is imperative to move beyond these frameworks and embrace the robust evidence supporting epichaperomes as unique pathological entities and epichaperome disruptors as novel therapeutic agents. Epichaperomes are not merely altered chaperones, nor are epichaperome disruptors traditional HSP90 inhibitors. They represent a distinct target and therapeutic strategy, transcending the conventional focus on protein folding and inhibition. This perspective does not diminish the importance of protein folding and misfolding in disease but underscores the need to differentiate folding chaperones from epichaperomes. These are distinct entities that demand tailored approaches. Epichaperomes are central to the network-level dysregulation underlying diseases like cancer and neurodegenerative disorders, making them a promising target for therapeutic intervention.
From a drug development perspective, where a compound falls on the continuum between a pure HSP90 inhibitor and a pure epichaperome disruptor significantly influences its therapeutic index, selectivity, and clinical efficacy. Just as kinase inhibitors vary in targeting specific disease-associated conformations, HSP90-targeting agents must be assessed not only for their binding affinity but also for their ability to modulate pathological chaperone assemblies versus canonical folding chaperones. Thus, the next frontier in chaperone-targeted therapy is to design next-generation epichaperome disruptors that capitalize on the unique structural and functional properties of these assemblies, ensuring precise, context-dependent intervention. Understanding these mechanistic distinctions will be crucial for refining patient selection strategies, optimizing drug retention and efficacy, and ultimately, maximizing the therapeutic potential of HSP90-targeting agents.
Looking ahead, addressing these challenges will require a concerted effort to redefine the narrative within the scientific and clinical communities. Future research should focus on elucidating the molecular triggers and context-specific stressors driving epichaperome formation, while expanding the repertoire of chemical probes and disruptors to explore their roles across diverse diseases. Strategic efforts to incorporate biomarker-driven approaches, such as using PET imaging and labeled probes, will be critical for optimizing patient selection and therapy efficacy.
Are epichaperomes a driver or cause of disease? Current evidence strongly supports a causal role for epichaperomes, as their disruption leads to the reversion of pathological states to normal, pre-stressor conditions32,33,41,68—a hallmark of drivers rather than consequences.104 Data so far indicate that epichaperomes do not merely reflect underlying disease conditions but are key orchestrators of pathological phenotypes, with their formation marking a critical inflection point in disease progression.29 While these observations provide strong evidence for a causal role, definitive temporal causality requires further longitudinal studies. Ongoing research, including longitudinal analyses in preclinical models and patient-derived cellular systems, will be needed to clarify the precise timing of epichaperome formation and its impact on disease onset and progression.
More on this topic, a question raises on the potential role of epichaperomes in normal aging. Given that aging itself is a chronic stressor, it is conceivable that low-level epichaperome formation occurs in the aged brain, albeit at levels significantly lower than in Alzheimer's disease or other pathological conditions. This raises the intriguing possibility that such low-level assemblies could contribute to aspects of functional decline associated with aging, even in the absence of overt disease. If this hypothesis holds, a key question emerges: Could disrupting epichaperomes in aging tissues restore PPI networks to pre-stressor, more youthful configurations? Since epichaperome disruption in disease models reverses pathological PPI networks and restores cellular functions,33 it is plausible that targeting epichaperomes in aged individuals might similarly reset PPI networks, potentially mitigating certain aspects of functional decline. However, whether epichaperome disruption could reverse aging-associated phenotypes remains unknown, as this is an unexplored area of research. Future studies will be critical to determine whether epichaperome-targeting strategies could influence the aging process, potentially by restoring network homeostasis and cellular resilience.
A broader question in this context is on the normal roles of epichaperomes, if any exist. Do epichaperomes represent a normal process that goes awry during disease? This remains an important and evolving area of investigation. Current evidence suggests that epichaperome formation is not a common feature of normal physiology but rather represents a context-dependent cellular adaptation that emerges under conditions of chronic stress or in specific biological states. However, recent findings, including those in pluripotent stem cells,30 indicate that epichaperomes can also play specialized adaptive roles in non-pathological contexts, where they help sustain high cellular adaptability and robust proliferative capacity. In pluripotent stem cells, these specialized assemblies appear to support functional demands such as rapid proliferation and plasticity, which are essential for maintaining pluripotency. In this context, epichaperomes may provide a regulatory framework for orchestrating protein networks required for these physiological states.
This raises the intriguing analogy to conventional stress response pathways: while transient activation of stress responses is adaptive and protective, persistent or aberrant activation can lead to pathological outcomes, such as uncontrolled proliferation in cancer. Similarly, epichaperome formation may represent a mechanism that is co-opted or dysregulated in disease, transitioning from a specialized adaptive role in specific normal contexts to a maladaptive scaffolding system that supports disease progression.
With regard to mechanistic, structural, and drug discovery aspects, several open questions remain: an important open question is understanding the precise mechanistic pathway by which a limited number of PTMs, such as phosphorylation at S226 and S255 or a single glycosylation event, can stabilize complex epichaperome assemblies capable of sequestering thousands of proteins and restructuring extensive PPI networks. Additionally, the mechanistic underpinnings that differentiate HSP90 inhibitors from epichaperome disruptors remain to be fully investigated. Why does stabilization of the epichaperome by a small molecule lead to its “dismantling”? This seems counterintuitive and clearly not mechanistically satisfying yet. Another major outstanding question concerns how one would specifically synthesize a molecule with epichaperome activity versus one that lacks this activity. What are the structural properties required? What are the key contacts with HSP90? In other words, what lessons have been learned in how to make an epichaperome binder? These fundamental questions will guide future research and are critical for advancing our understanding of epichaperome biology and its therapeutic potential.
Epichaperome research exemplifies the power of chemical biology in uncovering novel disease mechanisms and developing targeted therapies. By disentangling epichaperomes from the folding dogma and recognizing their unique role in disease biology, the field is poised to accelerate the development of transformative, precision-targeted therapies. As our understanding deepens, epichaperome-targeted strategies have the potential to reshape the treatment landscape for complex diseases, offering new hope for more effective and personalized interventions.
Author contributions
C. P., C. S. D., S. S. and S. W.: writing original draft & editing, visualization. A. B.: writing original draft. G. C.: conceptualization, supervision, writing, visualization & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
Memorial Sloan Kettering Cancer Center (MSKCC) holds the intellectual rights to the epichaperome portfolio. G. C., C. S. D. and S. S. are inventors on the licensed intellectual property. All other authors declare no competing interests.Acknowledgements
This work was supported by the NIH (R01 CA172546, U01 AG032969, P01 CA186866, R56 AG061869, R01 AG067598, R01 AG074004, R01 AG072599, R56 AG072599, RF1 AG071805, P30 CA008748) and Mr William H. and Mrs Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of MSKCC (G. C.). S. S. would like to acknowledge funding support from BrightFocus Foundation (Award ID: A2022020F).References
- J. Wang, S. Yazdani, A. Han and M. Schapira, Drug Discovery Today, 2020, 25, 561–567 CAS
.
- J. P. Hughes, S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CAS
- M. Trendowski, Pharmacol. Res., 2015, 99, 202–216 CAS
- S. Saber, R. Abdelhady, M. A. Elhemely, E. A. Elmorsy, R. S. Hamad, M. A. Abdel-Reheim, A. F. El-Kott, M. A. AlShehri, K. Morsy, A. S. AlSheri and M. E. Youssef, Front. Pharmacol., 2024, 15, 1475998 CAS
- K. Moulick, J. H. Ahn, H. Zong, A. Rodina, L. Cerchietti, E. M. Gomes DaGama, E. Caldas-Lopes, K. Beebe, F. Perna, K. Hatzi, L. P. Vu, X. Zhao, D. Zatorska, T. Taldone, P. Smith-Jones, M. Alpaugh, S. S. Gross, N. Pillarsetty, T. Ku, J. S. Lewis, S. M. Larson, R. Levine, H. Erdjument-Bromage, M. L. Guzman, S. D. Nimer, A. Melnick, L. Neckers and G. Chiosis, Nat. Chem. Biol., 2011, 7, 818–826 CAS
- L. Shrestha, H. J. Patel and G. Chiosis, Cell Chem. Biol., 2016, 23, 158–172 CAS
- A. Rodina, T. Wang, P. Yan, E. D. Gomes, M. P. Dunphy, N. Pillarsetty, J. Koren, J. F. Gerecitano, T. Taldone, H. Zong, E. Caldas-Lopes, M. Alpaugh, A. Corben, M. Riolo, B. Beattie, C. Pressl, R. I. Peter, C. Xu, R. Trondl, H. J. Patel, F. Shimizu, A. Bolaender, C. Yang, P. Panchal, M. F. Farooq, S. Kishinevsky, S. Modi, O. Lin, F. Chu, S. Patil, H. Erdjument-Bromage, P. Zanzonico, C. Hudis, L. Studer, G. J. Roboz, E. Cesarman, L. Cerchietti, R. Levine, A. Melnick, S. M. Larson, J. S. Lewis, M. L. Guzman and G. Chiosis, Nature, 2016, 538, 397–401 CAS
- National Center for Biotechnology Information (2024). PubChem Compound Summary for CID 9549213, PU-H71. Retrieved December 9, 2024 from https://pubchem.ncbi.nlm.nih.gov/compound/Pu-h71.
- H. He, D. Zatorska, J. Kim, J. Aguirre, L. Llauger, Y. She, N. Wu, R. M. Immormino, D. T. Gewirth and G. Chiosis, J. Med. Chem., 2006, 49, 381–390 CAS
- M. Vilenchik, D. Solit, A. Basso, H. Huezo, B. Lucas, H. He, N. Rosen, C. Spampinato, P. Modrich and G. Chiosis, Chem. Biol., 2004, 11, 787–797 CAS
- G. Chiosis, M. N. Timaul, B. Lucas, P. N. Munster, F. F. Zheng, L. Sepp-Lorenzino and N. Rosen, Chem. Biol., 2001, 8, 289–299 CAS
- G. Chiosis, B. Lucas, A. Shtil, H. Huezo and N. Rosen, Bioorg. Med. Chem., 2002, 10, 3555–3564 CrossRef CAS PubMed
- L. Llauger, H. He, J. Kim, J. Aguirre, N. Rosen, U. Peters, P. Davies and G. Chiosis, J. Med. Chem., 2005, 48, 2892–2905 CAS
- B. Dymock, X. Barril, M. Beswick, A. Collier, N. Davies, M. Drysdale, A. Fink, C. Fromont, R. E. Hubbard, A. Massey, A. Surgenor and L. Wright, Bioorg. Med. Chem. Lett., 2004, 14, 325–328 CrossRef CAS PubMed
- R. M. Immormino, Y. Kang, G. Chiosis and D. T. Gewirth, J. Med. Chem., 2006, 49, 4953–4960 CrossRef CAS PubMed
- E. Caldas-Lopes, L. Cerchietti, J. H. Ahn, C. C. Clement, A. I. Robles, A. Rodina, K. Moulick, T. Taldone, A. Gozman, Y. Guo, N. Wu, E. de Stanchina, J. White, S. S. Gross, Y. Ma, L. Varticovski, A. Melnick and G. Chiosis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8368–8373 CrossRef CAS PubMed
- J. Li and J. Buchner, Biomed. J., 2013, 36, 106–117 CrossRef PubMed
- L. H. Pearl, Biopolymers, 2016, 105, 594–607 CrossRef CAS PubMed
- K. A. Krukenberg, T. O. Street, L. A. Lavery and D. A. Agard, Q. Rev. Biophys., 2011, 44, 229–255 CrossRef CAS PubMed
- L. H. Pearl and C. Prodromou, Annu. Rev. Biochem., 2006, 75, 271–294 CrossRef CAS PubMed
- M. Taipale, D. F. Jarosz and S. Lindquist, Nat. Rev. Mol. Cell Biol., 2010, 11, 515–528 Search PubMed
- F. H. Schopf, M. M. Biebl and J. Buchner, Nat. Rev. Mol. Cell Biol., 2017, 18, 345–360 CrossRef CAS PubMed
- M. E. Dean and J. L. Johnson, Cell Stress Chaperones, 2021, 26, 3–13 CrossRef CAS PubMed
- S. J. Backe, R. A. Sager, M. R. Woodford, A. M. Makedon and M. Mollapour, J. Biol. Chem., 2020, 295, 11099–11117 CrossRef CAS PubMed
- M. Mollapour and L. Neckers, Biochim. Biophys. Acta, 2012, 1823, 648–655 CrossRef CAS PubMed
- J. Trepel, M. Mollapour, G. Giaccone and L. Neckers, Nat. Rev. Cancer, 2010, 10, 537–549 CrossRef CAS PubMed
- J. F. Gerecitano, S. Modi, R. Rampal, A. E. Drilon, M. G. Fury, M. M. Gounder, J. J. Harding, D. M. Hyman, A. M. Varghese, M. H. Voss, F. O. France, T. Taldone, E. G. DaGama, M. Uddin, G. Chiosis, J. S. Lewis, S. K. Lyashchenko, S. M. Larson, C. Pressl and M. Dunphy, J. Clin. Oncol., 2015, 33, 2537 Search PubMed
- L. Neckers and P. Workman, Clin. Cancer Res., 2012, 18, 64–76 CAS
- G. Chiosis, C. S. Digwal, J. B. Trepel and L. Neckers, Nat. Rev. Mol. Cell Biol., 2023, 24, 797–815 CAS
- T. Roychowdhury, S. W. McNutt, C. Pasala, H. T. Nguyen, D. T. Thornton, S. Sharma, L. Botticelli, C. S. Digwal, S. Joshi, N. Yang, P. Panchal, S. Chakrabarty, S. Bay, V. Markov, C. Kwong, J. Lisanti, S. Y. Chung, S. D. Ginsberg, P. Yan, E. De Stanchina, A. Corben, S. Modi, M. L. Alpaugh, G. Colombo, H. Erdjument-Bromage, T. A. Neubert, R. J. Chalkley, P. R. Baker, A. L. Burlingame, A. Rodina, G. Chiosis and F. Chu, Nat. Commun., 2024, 15, 8912 CrossRef CAS PubMed
- A. Rodina, C. Xu, C. S. Digwal, S. Joshi, Y. Patel, A. R. Santhaseela, S. Bay, S. Merugu, A. Alam, P. Yan, C. Yang, T. Roychowdhury, P. Panchal, L. Shrestha, Y. Kang, S. Sharma, J. Almodovar, A. Corben, M. L. Alpaugh, S. Modi, M. L. Guzman, T. Fei, T. Taldone, S. D. Ginsberg, H. Erdjument-Bromage, T. A. Neubert, K. Manova-Todorova, M. B. Tsou, J. C. Young, T. Wang and G. Chiosis, Nat. Commun., 2023, 14, 3742 CrossRef CAS PubMed
- M. C. Inda, S. Joshi, T. Wang, A. Bolaender, S. Gandu, J. Koren Iii, A. Y. Che, T. Taldone, P. Yan, W. Sun, M. Uddin, P. Panchal, M. Riolo, S. Shah, A. Barlas, K. Xu, L. Y. L. Chan, A. Gruzinova, S. Kishinevsky, L. Studer, V. Fossati, S. A. Noggle, J. R. White, E. de Stanchina, S. Sequeira, K. H. Anthoney, J. W. Steele, K. Manova-Todorova, S. Patil, M. P. Dunphy, N. Pillarsetty, A. C. Pereira, H. Erdjument-Bromage, T. A. Neubert, A. Rodina, S. D. Ginsberg, N. De Marco Garcia, W. Luo and G. Chiosis, Nat. Commun., 2020, 11, 319 CAS
- S. D. Ginsberg, S. Sharma, L. Norton and G. Chiosis, Trends Pharmacol. Sci., 2023, 44, 20–33 CAS
- M. Sugita, D. C. Wilkes, R. Bareja, K. W. Eng, S. Nataraj, R. A. Jimenez-Flores, L. Yan, J. P. De Leon, J. A. Croyle, J. Kaner, S. Merugu, S. Sharma, T. Y. MacDonald, Z. Noorzad, P. Panchal, D. Pancirer, S. Cheng, J. Z. Xiang, L. Olson, K. Van Besien, D. S. Rickman, S. Mathew, W. Tam, M. A. Rubin, H. Beltran, A. Sboner, D. C. Hassane, G. Chiosis, O. Elemento, G. J. Roboz, J. M. Mosquera and M. L. Guzman, npj Precis. Oncol., 2021, 5, 44 CAS
- K. L. Jhaveri, C. H. Dos Anjos, T. Taldone, R. Wang, E. Comen, M. Fornier, J. F. Bromberg, W. Ma, S. Patil, A. Rodina, N. Pillarsetty, S. Duggan, S. Khoshi, N. Kadija, G. Chiosis, M. P. Dunphy and S. Modi, JCO Precis. Oncol., 2020, 4, 1414–1424 Search PubMed
- N. Pillarsetty, K. Jhaveri, T. Taldone, E. Caldas-Lopes, B. Punzalan, S. Joshi, A. Bolaender, M. M. Uddin, A. Rodina, P. Yan, A. Ku, T. Ku, S. K. Shah, S. Lyashchenko, E. Burnazi, T. Wang, N. Lecomte, Y. Janjigian, A. Younes, C. W. Batlevi, M. L. Guzman, G. J. Roboz, J. Koziorowski, P. Zanzonico, M. L. Alpaugh, A. Corben, S. Modi, L. Norton, S. M. Larson, J. S. Lewis, G. Chiosis, J. F. Gerecitano and M. P. S. Dunphy, Cancer Cell, 2019, 36(559–573), e557 Search PubMed
- S. Joshi, E. D. Gomes, T. Wang, A. Corben, T. Taldone, S. Gandu, C. Xu, S. Sharma, S. Buddaseth, P. Yan, L. Y. L. Chan, A. Gokce, V. K. Rajasekhar, L. Shrestha, P. Panchal, J. Almodovar, C. S. Digwal, A. Rodina, S. Merugu, N. Pillarsetty, V. Miclea, R. I. Peter, W. Wang, S. D. Ginsberg, L. Tang, M. Mattar, E. de Stanchina, K. H. Yu, M. Lowery, O. Grbovic-Huezo, E. M. O'Reilly, Y. Janjigian, J. H. Healey, W. R. Jarnagin, P. J. Allen, C. Sander, H. Erdjument-Bromage, T. A. Neubert, S. D. Leach and G. Chiosis, Commun. Biol., 2021, 4, 1333 CAS
- S. Joshi, T. Wang, T. L. S. Araujo, S. Sharma, J. L. Brodsky and G. Chiosis, Nat. Rev. Cancer, 2018, 18, 562–575 CrossRef CAS PubMed
- S. D. Ginsberg, S. Joshi, S. Sharma, G. Guzman, T. Wang, O. Arancio and G. Chiosis, J. Neurochem., 2021, 159, 958–979 CrossRef CAS PubMed
- S. Chakrabarty, S. Wang, T. Roychowdhury, S. D. Ginsberg and G. Chiosis, Curr. Opin. Struct. Biol., 2024, 88, 102886 CrossRef CAS PubMed
- S. Kishinevsky, T. Wang, A. Rodina, S. Y. Chung, C. Xu, J. Philip, T. Taldone, S. Joshi, M. L. Alpaugh, A. Bolaender, S. Gutbier, D. Sandhu, F. Fattahi, B. Zimmer, S. K. Shah, E. Chang, C. Inda, J. Koren, 3rd, N. G. Saurat, M. Leist, S. S. Gross, V. E. Seshan, C. Klein, M. J. Tomishima, H. Erdjument-Bromage, T. A. Neubert, R. C. Henrickson, G. Chiosis and L. Studer, Nat. Commun., 2018, 9, 4345 CrossRef PubMed
- S. E. Svirsky, Y. Li, J. Henchir, A. Rodina, S. W. Carlson, G. Chiosis and C. E. Dixon, Neurobiol. Dis., 2023, 188, 106331 CrossRef CAS PubMed
- L. Whitesell and S. L. Lindquist, Nat. Rev. Cancer, 2005, 5, 761–772 CrossRef CAS PubMed
- U. Nayar, P. Lu, R. L. Goldstein, J. Vider, G. Ballon, A. Rodina, T. Taldone, H. Erdjument-Bromage, M. Chomet, R. Blasberg, A. Melnick, L. Cerchietti, G. Chiosis, Y. L. Wang and E. Cesarman, Blood, 2013, 122, 2837–2847 CrossRef CAS PubMed
- H. Zong, A. Gozman, E. Caldas-Lopes, T. Taldone, E. Sturgill, S. Brennan, S. O. Ochiana, E. M. Gomes-DaGama, S. Sen, A. Rodina, J. Koren, 3rd, M. W. Becker, C. M. Rudin, A. Melnick, R. L. Levine, G. J. Roboz, S. D. Nimer, G. Chiosis and M. L. Guzman, Cell Rep., 2015, 13, 2159–2173 CrossRef CAS PubMed
- Y. Minami, H. Kawasaki, Y. Miyata, K. Suzuki and I. Yahara, J. Biol. Chem., 1991, 266, 10099–10103 CrossRef CAS PubMed
- T. Roychowdhury, A. R. Santhaseela, S. Sharma, P. Panchal, A. Rodina and G. Chiosis, Methods Mol. Biol., 2023, 2693, 175–191 CrossRef CAS PubMed
- A. Bolaender, D. Zatorska, H. He, S. Joshi, S. Sharma, C. S. Digwal, H. J. Patel, W. Sun, B. S. Imber, S. O. Ochiana, M. R. Patel, L. Shrestha, S. K. Shah, S. Wang, R. Karimov, H. Tao, P. D. Patel, A. R. Martin, P. Yan, P. Panchal, J. Almodovar, A. Corben, A. Rimner, S. D. Ginsberg, S. Lyashchenko, E. Burnazi, A. Ku, T. Kalidindi, S. G. Lee, M. Grkovski, B. J. Beattie, P. Zanzonico, J. S. Lewis, S. Larson, A. Rodina, N. Pillarsetty, V. Tabar, M. P. Dunphy, T. Taldone, F. Shimizu and G. Chiosis, Nat. Commun., 2021, 12, 4669 CrossRef CAS PubMed
- N. Kourtis, C. Lazaris, K. Hockemeyer, J. C. Balandrán, A. R. Jimenez, J. Mullenders, Y. Gong, T. Trimarchi, K. Bhatt, H. Hu, L. Shrestha, A. Ambesi-Impiombato, M. Kelliher, E. Paietta, G. Chiosis, M. L. Guzman, A. A. Ferrando, A. Tsirigos and I. Aifantis, Nat. Med., 2018, 24, 1157–1166 CrossRef CAS PubMed
- B. Z. Carter, P. Y. Mak, M. Muftuoglu, W. Tao, B. Ke, J. Pei, A. D. Bedoy, L. B. Ostermann, Y. Nishida, S. Isgandarova, M. Sobieski, N. Nguyen, R. T. Powell, M. Martinez-Moczygemba, C. Stephan, M. Basyal, N. Pemmaraju, S. Boettcher, B. L. Ebert, E. J. Shpall, B. Wallner, R. A. Morgan, G. I. Karras, U. M. Moll and M. Andreeff, Blood, 2023, 142, 1056–1070 CAS
- Y. Tang, J. Luo, Y. Zhou, H. Zang, Y. Yang, S. Liu, H. Zheng, J. Ma, S. Fan and Q. Wen, BMC Cancer, 2022, 22, 564 Search PubMed
- M. H. Silverman, S. Duggan, G. Bardelli, B. Sadler, C. Key, M. Medlock, L. Reynolds and B. Wallner, J. Prev. Alzheimer's Dis., 2022, 9, 635–645 CrossRef CAS PubMed
- R. Bao, C. J. Lai, H. Qu, D. Wang, L. Yin, B. Zifcak, R. Atoyan, J. Wang, M. Samson, J. Forrester, S. DellaRocca, G. X. Xu, X. Tao, H. X. Zhai, X. Cai and C. Qian, Clin. Cancer Res., 2009, 15, 4046–4057 CrossRef CAS PubMed
- M. N. Calvo-Vidal, N. Zamponi, J. Krumsiek, M. A. Stockslager, M. V. Revuelta, J. M. Phillip, R. Marullo, E. Tikhonova, N. Kotlov, J. Patel, S. N. Yang, L. Yang, T. Taldone, C. Thieblemont, J. P. Leonard, P. Martin, G. Inghirami, G. Chiosis, S. R. Manalis and L. Cerchietti, Cancer Res., 2021, 81, 5202–5216 CrossRef CAS PubMed
- P. Yan, H. J. Patel, S. Sharma, A. Corben, T. Wang, P. Panchal, C. Yang, W. Sun, T. L. Araujo, A. Rodina, S. Joshi, K. Robzyk, S. Gandu, J. R. White, E. de Stanchina, S. Modi, Y. Y. Janjigian, E. G. Hill, B. Liu, H. Erdjument-Bromage, T. A. Neubert, N. L. S. Que, Z. Li, D. T. Gewirth, T. Taldone and G. Chiosis, Cell Rep., 2020, 31, 107840 CrossRef CAS PubMed
- C. Pasala, S. Sharma, T. Roychowdhury, E. Moroni, G. Colombo and G. Chiosis, Biomolecules, 2024, 14, 282 CrossRef CAS PubMed
- M. Castelli, P. Yan, A. Rodina, C. S. Digwal, P. Panchal, G. Chiosis, E. Moroni and G. Colombo, Structure, 2023, 31(987–1004), e1008 Search PubMed
- A. Magni, C. Sciva, M. Castelli, C. S. Digwal, A. Rodina, S. Sharma, S. Ochiana, H. J. Patel, S. Shah, G. Chiosis, E. Moroni and G. Colombo, Chemistry, 2024, 30, e202401957 CAS
- K. Chaumonnot, S. Masson, H. Sikner, A. Bouchard, V. Baverel, P. S. Bellaye, B. Collin, C. Garrido and E. Kohli, Cell Death Dis., 2021, 12, 114 CAS
- A. Ratna, A. Lim, Z. Li, J. Argemi, R. Bataller, G. Chiosis and P. Mandrekar, Hepatol. Commun., 2021, 5, 1165–1182 CAS
- A. Pagetta, E. Tramentozzi, E. Tibaldi, L. Cendron, G. Zanotti, A. M. Brunati, M. Vitadello, L. Gorza and P. Finotti, PLoS One, 2014, 9, e86198 Search PubMed
- R. Trivedi and H. A. Nagarajaram, Int. J. Mol. Sci., 2022, 23, 14050 CAS
- M. Marzec, D. Eletto and Y. Argon, Biochim. Biophys. Acta, 2012, 1823, 774–787 CAS
- Y. S. Amankwah, Y. Fleifil, E. Unruh, P. Collins, Y. Wang, K. Vitou, A. Bates, I. Obaseki, M. Sugoor, J. P. Alao, R. M. McCarrick, D. T. Gewirth, I. D. Sahu, Z. Li, G. A. Lorigan and A. N. Kravats, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2309326121 CAS
- A. W. Truman, D. Bourboulia and M. Mollapour, J. Biol. Chem., 2021, 296, 100293 CAS
- S. J. Backe, R. A. Sager, J. A. Heritz, L. A. Wengert, K. A. Meluni, X. Aran-Guiu, B. Panaretou, M. R. Woodford, C. Prodromou, D. Bourboulia and M. Mollapour, Cell Rep., 2023, 42, 112807 CAS
- S. D. Ginsberg, T. A. Neubert, S. Sharma, C. S. Digwal, P. Yan, C. Timbus, T. Wang and G. Chiosis, FEBS J., 2022, 289, 2047–2066 CAS
- H. D. Rickner, L. Jiang, R. Hong, N. K. O’Neill, C. A. Mojica, B. J. Snyder, L. Zhang, D. Shaw, M. Medalla, B. Wolozin and C. S. Cheng, Nat. Commun., 2022, 13, 6275 CAS
- M. R. Woodford, A. W. Truman, D. M. Dunn, S. M. Jensen, R. Cotran, R. Bullard, M. Abouelleil, K. Beebe, D. Wolfgeher, S. Wierzbicki, D. E. Post, T. Caza, S. Tsutsumi, B. Panaretou, S. J. Kron, J. B. Trepel, S. Landas, C. Prodromou, O. Shapiro, W. G. Stetler-Stevenson, D. Bourboulia, L. Neckers, G. Bratslavsky and M. Mollapour, Cell Rep., 2016, 14, 872–884 CAS
-
C. S. Digwal, S. Sharma, A. R. Santhaseela, S. D. Ginsberg and G. Chiosis, Protein Homeostasis in Drug Discovery, 2022, pp. 1–26 DOI:10.1002/9781119774198.ch1
- S. Sharma, S. Joshi, T. Kalidindi, C. S. Digwal, P. Panchal, S. G. Lee, P. Zanzonico, N. Pillarsetty and G. Chiosis, Biomedicines, 2023, 11, 2599 CAS
- M. P. S. Dunphy, C. Pressl, N. Pillarsetty, M. Grkovski, S. Modi, K. Jhaveri, L. Norton, B. J. Beattie, P. B. Zanzonico, D. Zatorska, T. Taldone, S. O. Ochiana, M. M. Uddin, E. M. Burnazi, S. K. Lyashchenko, C. A. Hudis, J. Bromberg, H. M. Schoder, J. J. Fox, H. Zhang, G. Chiosis, J. S. Lewis and S. M. Larson, Clin. Cancer Res., 2020, 26, 5178–5187 CAS
- J. M. Sasso, R. Tenchov, D. Wang, L. S. Johnson, X. Wang and Q. A. Zhou, Biochemistry, 2023, 62, 601–623 CAS
- S. Rastogi, A. Joshi, N. Sato, S. Lee, M. J. Lee, J. B. Trepel and L. Neckers, Cell Stress Chaperones, 2024, 29, 519–539 CAS
- A. Yuno, M. J. Lee, S. Lee, Y. Tomita, D. Rekhtman, B. Moore and J. B. Trepel, Methods Mol. Biol., 2018, 1709, 423–441 CAS
- S. Maiti and D. Picard, Biomolecules, 2022, 12, 1166 CAS
- P. C. Echeverria, K. Bhattacharya, A. Joshi, T. Wang and D. Picard, PLoS One, 2019, 14, e0208287 CAS
- P. W. Piper, B. Panaretou, S. H. Millson, A. Truman, M. Mollapour, L. H. Pearl and C. Prodromou, Gene, 2003, 302, 165–170 CAS
- P. Tsvetkov, T. J. Eisen, S. U. Heinrich, Z. Brune, E. Hallacli, G. A. Newby, C. Kayatekin, D. Pincus and S. Lindquist, Cell Rep., 2020, 32, 108001 CrossRef CAS PubMed
- B. Wang, H. Wu, C. Hu, H. Wang, J. Liu, W. Wang and Q. Liu, Signal Transduction Targeted Ther., 2021, 6, 423 CrossRef CAS PubMed
- C. Temps, D. Lietha, E. R. Webb, X. F. Li, J. C. Dawson, M. Muir, K. G. Macleod, T. Valero, A. F. Munro, R. Contreras-Montoya, J. R. Luque-Ortega, C. Fraser, H. Beetham, C. Schoenherr, M. Lopalco, M. J. Arends, M. C. Frame, B. Z. Qian, V. G. Brunton, N. O. Carragher and A. Unciti-Broceta, Cancer Res., 2021, 81, 5438–5450 CrossRef CAS PubMed
- E. W. Lake, J. M. Muretta, A. R. Thompson, D. M. Rasmussen, A. Majumdar, E. B. Faber, E. F. Ruff, D. D. Thomas and N. M. Levinson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11894–E11903 CrossRef CAS PubMed
- P. J. Tonge, ACS Chem. Neurosci., 2018, 9, 29–39 CrossRef CAS PubMed
- S. Nunez, J. Venhorst and C. G. Kruse, Drug Discovery Today, 2012, 17, 10–22 CrossRef CAS PubMed
- D. V. Borisov and A. V. Veselovsky, Biomed. Khim., 2020, 66, 42–53 CAS
- R. A. Copeland, Expert Opin. Drug Discovery, 2010, 5, 305–310 CrossRef CAS PubMed
- A. C. Pan, D. W. Borhani, R. O. Dror and D. E. Shaw, Drug Discovery Today, 2013, 18, 667–673 CrossRef CAS PubMed
- P. Workman, F. Burrows, L. Neckers and N. Rosen, Ann. N. Y. Acad. Sci., 2007, 1113, 202–216 CAS
- D. Mahalingam, R. Swords, J. S. Carew, S. T. Nawrocki, K. Bhalla and F. J. Giles, Br. J. Cancer, 2009, 100, 1523–1529 CrossRef CAS PubMed
- A. Kamal, L. Thao, J. Sensintaffar, L. Zhang, M. F. Boehm, L. C. Fritz and F. J. Burrows, Nature, 2003, 425, 407–410 CrossRef CAS PubMed
- J. L. Eiseman, J. Lan, T. F. Lagattuta, D. R. Hamburger, E. Joseph, J. M. Covey and M. J. Egorin, Cancer Chemother. Pharmacol., 2005, 55, 21–32 CrossRef CAS PubMed
- A. Kamal, M. F. Boehm and F. J. Burrows, Trends Mol. Med., 2004, 10, 283–290 CAS
- J. Zhang, H. Li, Y. Liu, K. Zhao, S. Wei, E. T. Sugarman, L. Liu and G. Zhang, Cells, 2022, 11, 2778 CAS
- Z. N. Li and Y. Luo, Oncol. Rep., 2023, 49, 6 CAS
- W. F. Zuo, Q. Pang, X. Zhu, Q. Q. Yang, Q. Zhao, G. He, B. Han and W. Huang, J. Hematol. Oncol., 2024, 17, 81 Search PubMed
- C. A. Basset, E. Conway de Macario, L. G. Leone, A. J. L. Macario and A. Leone, J. Mol. Histol., 2023, 54, 105–118 CAS
- A. Saghatelian, N. Jessani, A. Joseph, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10000–10005 CAS
- A. E. Speers and B. F. Cravatt, Curr. Protoc. Chem. Biol., 2009, 1, 29–41 Search PubMed
- E. O. J. Porta and P. G. Steel, Curr. Res. Pharmacol. Drug Discovery, 2023, 5, 100164 Search PubMed
- L. Hinze, S. Schreek, A. Zeug, N. K. Ibrahim, B. Fehlhaber, L. Loxha, B. Cinar, E. Ponimaskin, J. Degar, C. McGuckin, G. Chiosis, C. Eckert, G. Cario, B. Bornhauser, J. P. Bourquin, M. Stanulla and A. Gutierrez, Mol. Cell, 2022, 82(2858–2870), e2858 Search PubMed
- S. Bay, C. S. Digwal, A. M. Rodilla Martín, S. Sharma, A. Stanisavljevic, A. Rodina, A. Attaran, T. Roychowdhury, K. Parikh, E. Toth, P. Panchal, E. Rosiek, C. Pasala, O. Arancio, P. E. Fraser, M. J. Alldred, M. A. M. Prado, S. D. Ginsberg and G. Chiosis, Biomedicines, 2024, 12, 1252 CAS
- S. Merugu, S. Sharma, J. Kaner, C. Digwal, M. Sugita, S. Joshi, T. Taldone, M. L. Guzman and G. Chiosis, Methods Enzymol., 2020, 639, 289–311 Search PubMed
- T. Tuntland, B. Ethell, T. Kosaka, F. Blasco, R. X. Zang, M. Jain, T. Gould and K. Hoffmaster, Front. Pharmacol., 2014, 5, 174 Search PubMed
- A. B. Hill, Proc. R. Soc. Med., 1965, 58, 295–300 CAS
| This journal is © The Royal Society of Chemistry 2025 |