
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClick-ready iridium(III) complexes as versatile bioimaging probes for bioorthogonal metabolic labeling†
Vincent
Rigolot‡
 a,
Clémence
Simon‡
a,
Aude
Bouchet
b,
Lucas
Lancel
a,
Veronica
Di Battista
c,
Dmitry
Karpov
de,
Boris
Vauzeilles
f,
Corentin
Spriet
ag,
Michel
Sliwa
bh,
Sylvain
Bohic
de,
Christophe
Biot
*a and
Cédric
Lion
*a
a,
Clémence
Simon‡
a,
Aude
Bouchet
b,
Lucas
Lancel
a,
Veronica
Di Battista
c,
Dmitry
Karpov
de,
Boris
Vauzeilles
f,
Corentin
Spriet
ag,
Michel
Sliwa
bh,
Sylvain
Bohic
de,
Christophe
Biot
*a and
Cédric
Lion
*a
aUniv. Lille, CNRS, UMR 8576 – UGSF – Unité de Glycobiologie Structurale et Fonctionnelle, Lille, France. E-mail: cedric.lion@univ-lille.fr
bUniv. Lille, CNRS, UMR 8516 – LASIRe – Laboratoire Avancé de Spectroscopie pour les Interactions la Réactivité et l’Environnement, Lille, France
cDTU Sustain, Technical University of Denmark, Building 115, Kgs., Lyngby, Denmark
dUniv. Grenoble Alpes, INSERM, UA 07 Synchrotron Radiation for Biomedicine (STROBE), Grenoble, ID16A, France
eNano-imaging beamline, European Synchrotron Radiation Facility, Grenoble, France
fChemical Biology Department, Univ. Paris-Saclay, CNRS, Institut de Chimie des Substances Naturelles, UPR 2301, 91198 Gif-sur-Yvette, France
gUniv. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, US 41 – UAR 2014 – PLBS, Lille, France
hLOB, CNRS, INSERM, Ecole Polytechnique, Institut Polytechnique de Paris, Palaiseau, France
First published on 3rd December 2024
Abstract
Herein, we report the synthesis, photophysical characterization and validation of iridium(III)–polypyridine complexes functionalized for click chemistry and bioorthogonal chemistry, as well as their versatile applications as probes in bioimaging studies exploiting metabolic labeling. The designed dyes are conjugated to chemical reporters in a specific manner within cells by CuAAC ligation and display attractive photophysical properties in the UV-visible range. They are indeed highly photostable and emit in the far-red to near-IR region with long lifetimes and large Stokes shifts. We demonstrate that they can be efficiently used to monitor nascent intracellular sialylated glycoconjugates in bioorthogonal MOE studies with a varied panel of optical and non-optical techniques, namely conventional UV-vis laser scanning confocal microscopy (for routine purposes), UV-vis time-resolved luminescence imaging (for specificity and facilitated multiplexing with nano-environment sensitivity), synchrotron radiation based X-ray fluorescence nanoimaging (for high resolution, elemental mapping and quantification in situ) and inductively coupled plasma mass spectrometry (for routine quantification on cell populations with high statistical confidence). The synthesized Ir(III) complexes were utilized in single labeling experiments, as well as in dual click-labeling experiments utilizing two distinct monosaccharide reporters relevant to the same metabolic pathway.
Introduction
Click and bioorthogonal chemistries empower researchers to interrogate biomolecules with high selectivity and biocompatibility, utilizing pericyclic mechanisms such as the copper-catalyzed azide–alkyne [3+2] cycloaddition (CuAAC),1 the strain-promoted azide–alkyne [3+2] cycloaddition (SPAAC),2 or the alkene-tetrazine inverse electronic-demand [4+2] Diels–Alder cycloaddition (IEDDA).3,4 Metabolic labeling approaches allow the introduction of bioorthogonal chemical handles into biomolecules of interest, enabling their covalent ligation with molecular dyes on or within living cells through such reactions. Metabolic oligosaccharide engineering (MOE), pioneered by the Reutter5 and Bertozzi6,7 groups, has garnered significant interest due to its pivotal role in studying glycosylation post-translational modifications (PTMs).8 Glycosylation PTMs indeed play a crucial role in countless biological processes that involve molecular recognition (e.g., cell–cell interactions, host–pathogen interactions, protein localization) and impact pathologies ranging from cancer to genetic disorders to viral and bacterial infections. Bioorthogonal chemistry is a field of chemical biology that has revolutionized the study of biological systems and continues to evolve as new reactions and chemical tools are developed and optimized.9In the context of bioimaging, the rapidly advancing field of bioorthogonal chemistry still requires improvements in key aspects that can perhaps be summarized in three words: resolution, versatility, and specificity. The first of these aspects can now be enhanced through super-resolution fluorescence imaging nanoscopy, which utilizes photonic techniques in the UV-vis range to achieve resolutions higher than those limited by the optical diffraction barrier (ca. 200–250 nm).10–12 Alternatively, improvements can be made by employing techniques involving dyes that emit at much shorter and energetic wavelengths than the UV-vis range (such as X-rays),13 or by utilizing non-optical techniques such as cryogenic electron microscopy (cryoEM).14 The lack of versatility is primarily due to the type of molecular probe used, which typically provides a single read-out but may not be suitable for correlative multimodal microscopy and multianalytical methods.15 Therefore, in many research studies, bioimaging must be supplemented with additional time-consuming bioanalytical techniques that may require cell lysis, the extraction/separation of biomolecule pools and/or the use of several distinct labeling methods (e.g., glycomic studies by LC-MS/MS). Finally, specificity is perhaps the main issue to tackle in many applications. Indeed, non-specific labeling may occur due to cross-reactivity of either reporters or probes with irrelevant off-target sites in the cell, or to non-covalent entrapment of unreacted dye molecules.16 This increases non-specific fluorescence background and diminishes the specific signal-to-noise ratio, thus hampering the experiment by decreasing its sensitivity. In some cases, it might even generate aberrant false positive signals and bias the results.17 If many efforts are constantly being made to improve bioorthogonal monosaccharide reporters and reactions in terms of specificity toward a given metabolic path, cell entry, level of incorporation, reporter group innocuity, stability or kinetics,18–24 the most promising advances also stem from the development of improved click-ready molecular probes. For example, near-IR dyes,25 long lifetime dyes,26 dyes that rely on Förster's resonance energy transfer (FRET) interactions or quenching27 or fluorogenic dyes28 can help circumventing autofluorescence and non-specific background issues.
In the search for new emissive probes to improve versatility, specificity and resolution in bioimaging studies, luminescent d6 transition metal complexes such as iridium(III)–polypyridines are very attractive candidates. Indeed, the physico-chemical properties of d6 metal ions and strong π-accepting ligands result in various charge transfer mechanisms that increase intersystem crossing from singlet to triplet excited states.29–31 Thus, using UV/vis excitation they often exhibit luminescence with long lifetimes (formally, T1 → S0 phosphorescence as the major signal contributor) and large Stokes shifts, both exploitable aspects to improve signal-to-noise ratio and unambiguously isolate specific signals in multiple labeling experiments. The true potential of Ir(III)-based luminophores is fully realized when their lifetimes in the range of hundreds of nanoseconds to microseconds are exploited through time-resolved luminescence imaging, as this technique enables the complete elimination of the sample's short-lived background fluorescence by employing time-gated detection. As a result, the sensitivity of the experiment and the specificity of the detected signal are significantly enhanced. Moreover, in emission lifetime imaging microscopy,32 signals originating from multiple dyes with overlapping emission spectra but differing lifetimes can be easily distinguished, thereby amplifying multiplexing capabilities. With such probes, it should be possible to utilize a single irradiation to simultaneously detect two dyes that have overlapping absorption spectra in either spectral mode or lifetime mode. In this regard, Ir(III)–polypyridine complexes present the advantage of easily modulating their photophysical parameters (absorption, emission and lifetime) when compared to other d6 metal ions such as Ru(II), Os(II) or Re(I). In addition, iridium is practically absent from living systems and presents emission rays that do not overlap with other endogenous elements, which enables elemental mapping by X-ray fluorescence (XRF).13 This offers an opportunity for nano-imaging with enhanced resolutions compared to optical diffraction limited confocal UV-vis microscopy, as well as straightforward quantification by XRF or by mass spectrometry.
Although iridium(III) luminophores have been attracting much attention in the bioimaging field in the past ten years, surprisingly such probes are scarcely used to label biomolecules with bioorthogonal click chemistry.33 Lo and co-workers, who pioneered the development of iridium sensors for bioconjugation on many application types,29 proposed the very first proof-of-concept for phosphorescent bioorthogonal reagents to detect azide-modified biomolecules at the cell surface, equipped with a dibenzocyclooctyne moiety for use with SPAAC labeling on living cells.34 Highly lipophilic probes entering the cell led to strong non-specific signal and high cytotoxicity, which the authors solved by appending a carboxylate group to prevent cell entry and visualize only cell–surface glycans. The same group also developed tetrazine-based probes with luminogenic behavior after IEDDA ligations.35–37 Focusing on spectral properties of the probes, these reports did not exploit their lifetime, and to our knowledge only two examples used time-resolved imaging so far. An iridium–alkynylbipyridine complex was described for photoluminescence lifetime imaging microscopy (PLIM) of protein pools after azido-homoalanine incorporation.38 In this article, the lifetime of the Ir-alkyne probe was significantly shortened upon formation of the triazole cycloadduct by CuAAC, thus allowing the authors to elegantly distinguish reacted probes from unreacted probes in single-labeling experiments. More recently, a FRET-based sensing approach involving SPAAC and IEDDA was found to enable detection of caspase-3 activity with PLIM.39 Importantly, we have found no report in the literature of clickable dyes for XRF nanoimaging and quantitative elemental mapping.
In this work, we report the synthesis, photophysical characterization and validation of click-ready azide- and alkyne-functionalized iridium(III)–polypyridine complexes, and we showcase their potential as versatile probes in studies exploiting bioorthogonal metabolic labeling. The designed heteroleptic Ir(III) complexes are of the type [Ir(C^N)2(N^N)]+[PF6]−, in which cyclometalating 2-phenylpyridine (ppy) is the C^N backbone ligand and a phenanthroline-based N^N ligand (phen) bears a spacer arm equipped with an azide or a terminal alkyne reactive moiety. These probes are specifically conjugated to chemical reporters inside cells by CuAAC ligation and display attractive photophysical properties in the UV-visible range (i.e., high photostability, large Stokes shift, long lifetime, emission in the far-red to near-IR region) and in the X-ray range. We demonstrate that they can be efficiently used to monitor nascent sialylated glycoconjugates in bioorthogonal MOE studies with a varied panel of optical and non-optical techniques (Fig. 1). The synthesized Ir(III) complexes can be detected with conventional laser scanning confocal microscopy (LSCM) for routine purposes, time-resolved photoluminescence lifetime imaging microscopy (PLIM) for facilitated multiplexing and high signal specificity in multicolor settings, synchrotron radiation based X-ray fluorescence nanoimaging (XRF-NI) for high resolution and quantitative elemental mapping, and inductively coupled plasma mass spectrometry (ICP-MS) for routine quantification on cell populations with statistical confidence. To our knowledge, this is the first time that XRF-NI is combined to bioorthogonal chemistry to detect metabolic reporters in cells, and such data can be used correlatively with photoluminescence.
 | ||
| Fig. 1 Chemical reporter strategy using unprotected N-acetylmannosamine (ManNAc) or N-acetylneuraminic acid (Neu5Ac) derivatives as chemical reporters and Ir(III) luminophores as molecular probes allows time-resolved PLIM bioimaging and X-ray fluorescence nanoimaging in addition routine confocal microscopy. The probes may also be used as mass tags in techniques such as ICP-MS. | ||
Results and discussion
Design and synthesis of click-ready iridium(III) luminophores
We envisaged bis-cyclometalated complexes of the [Ir(N∧C)2(N∧N)]+ structure family, as they are known to combine intense long-lifetime luminescence, good photostability and cell permeability while being more easily synthesized than their N6-coordinated counterparts. The auxiliary ligand can be modified as desired to fine-tune its photophysical parameters40 or to functionalize the complex for biological targeting41,42 or for specific reactivity, in our case click chemistry. We thus set out to synthesize Ir(III) complexes 3a–d with ppy as the main cyclometalating ligand, and a phenanthroline functionalized with amide-linked spacer arms and either azide or terminal alkyne reactive handles (Fig. 2), in order to label sialoglycoconjugates inside cells after metabolic incorporation of modified sialic acid precursors. As opposed to the two ratiometric or fluorogenic approaches cited above, our objective here was to generate robust, versatile large Stokes shift probes with long lifetime and photophysical properties that are not influenced by the bioorthogonal reaction used for labeling. Hence, the click-reactive moiety (azide or terminal alkyne) was not designed to be conjugated to the luminophore. Owing to our previous experience in intracellular sialylation studies, we considered the introduction of a small PEG spacer arm to improve solubility and allow enough flexibility for the azide/alkyne linker to react efficiently with the metabolically engineered glycans. We chose CuAAC as the bioorthogonal reaction for this study, as it remains, in our view, the most effective method for detecting intracellular glycans in fixed cells. The potential cytotoxicity of copper or of probes is not a concern here since the CuAAC reaction is performed after cell fixation and permeabilization. From our experience, CuAAC provides an optimal balance for labeling intracellular glycans, offering high specificity and efficiency.16 This reaction is well-known, extensively researched in bioorthogonal chemistry, and optimized for complex biological environments, offering a reliable and well-established protocol. CuAAC features fast reaction kinetics, high efficiency, and specificity, and uses azide and alkyne groups that are simple to synthesize, stable, and non-reactive with native biomolecules, making it ideal for fixed-cell applications where copper toxicity is not a concern. Although SPAAC avoids copper toxicity, its slower kinetics and bulkier probes often lead to higher background signals and lower signal-to-noise ratios due to hydrophobic trapping. IEDDA, while fast and copper-free, involves more complicated probe synthesis and bulkier reporter groups that are not yet fully optimized for MOE applications.8 | ||
| Fig. 2 (A) Synthesis of Ir(III) luminophores 3a–d. (i) 1,10-Phenanthrolin-5-amine, DCC, MeCN, rt, 20 h. 2a: 95%, 2b: 78%, 2c: 74%, 2d: 72%. (ii) [IrCl(ppy)2]2, CH2Cl2/MeOH, 45 to 55 °C, 5 to 15 h; (III) NH4PF6, rt, 15 nm. 3a: 93%, 3b: 54%, 3c: 88%, 3d: 99%. (B) Structure of sialylation chemical reporters. 4, 5 and 7 are derivatives of N-acetylmannosamine and enter the metabolic pathway at early stages in the cytosol (GNE/MNK) while SiaNAl 6 enters the pathway at the activation stage (CSS) in the nucleus. (C) CuAAC allows the ligation of probes into metabolically incorporated reporters within cells. | ||
The synthesis of photoluminescent complexes 3a–d and of chemical reporters N-azidoacetyl-D-mannosamine (ManNAz 4), N-pentynoyl-D-mannosamine (ManNAl 5) and N-pentynoylneuraminic acid (SiaNAl 6) is detailed in ESI.† In brief, carboxylic acids 1a–d were reacted with 1,10-phenanthrolin-5-amine and converted to the desired N^N bidentate auxiliary ligands 2a–d using dicyclohexylcarbodiimide (DCC) as activating agent in acetonitrile. 1a–d were added in a five-fold excess to compensate for the low nucleophilicity of the amine. Complexes [Ir(ppy)2(phen-Alk)]+[PF6]−3a, [Ir(ppy)2(phen-Az)]+[PF6]−3b, [Ir(ppy)2(phen-PEG2-Alk)]+[PF6]−3c and [Ir(ppy)2(phen-PEG2-Az)]+[PF6]−3d were obtained after reaction of two equivalents of N^N ligands 2a–d with one equivalent of the cyclometalated dimer [IrCl(ppy)2]2 in CH2Cl2/MeOH (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) followed by chloride anion exchange with ammonium hexafluorophosphate. Functionalized Ir(III) complexes 3a–d were then purified by silica column chromatography prior to characterization by 1H and 13C NMR, MALDI-ToF mass spectrometry and HPLC.
:1, v/v) followed by chloride anion exchange with ammonium hexafluorophosphate. Functionalized Ir(III) complexes 3a–d were then purified by silica column chromatography prior to characterization by 1H and 13C NMR, MALDI-ToF mass spectrometry and HPLC.
Photophysical characterization
The photophysical characterization of probes 3a–d was conducted in water and toluene in the presence or absence of oxygen, in order to measure their absorption/emission spectra and to determine their lifetimes and emission quantum yields.43 The steady state absorption spectra of all four probes in water exhibit identical characteristics (Fig. 3 and Fig. S1, ESI†) and are indicative of a mixed intraligand (ILCT) and metal–ligand charge transfer (MLCT) band. They cover a broad range from 250 nm to 500 nm, with an absorbance maximum at about 260 nm, and shoulders at 375 nm and at 460 nm. A broad emission spectrum (excitation at 375 nm) is measured covering the 500 to 800 nm range with a maximum at 615 nm (large Stokes shift of 240 nm/10407 cm−1).
 | ||
| Fig. 3 (A) Absorbance (dashed line) and normalized luminescence upon excitation at 375 nm (solid line) of probes 3a–d in water. (B) Emission decay at 375 nm excitation for 3c in water and toluene (non-degassed and degassed by bubbling argon), and in cells, versus emission decay of cell autofluorescence. Note: The decay in cells presents a fast component due to cell autofluorescence and a long component due to the Ir(III) probe. | ||
As the emission spectrum is usually assigned to a triplet excited state of 3MLCT character,44 we investigated whether the presence of oxygen might result in emission quenching. Therefore, the quantum yields (Φ) and lifetimes (τ) of 3a–d were measured in both aerobic and degassed conditions (Fig. S2 and S3, ESI†). The literature also documented the potential impact of polarity, leading us to determine these photophysical properties in toluene as well as in aqueous media. In water, the quantum yields (Φ) were measured at about 3–4% (Table 1), reasonable values corresponding to those previously described in the literature for similar structures (i.e. about 1–2%),35,36,45 and the emission lifetimes were revealed to be quite insensitive to oxygen. The emission decay τ in water is monoexponential with an average lifetime of ca. 70–80 ns. Interestingly, the lifetime in toluene was found to be longer and strongly dependent on the presence of oxygen. In degassed conditions, τ was indeed measured at ca. 700 ns, a 7-fold increase when compared to the value in aerobic conditions (ca. 100 ns). These results point out the sensitivity of the triplet excited state to polar environments as well as to oxygen. On average, probes 3c and 3d bearing a PEG3 spacer arm were found to have slightly larger τ values (ca. 15%) than probes equipped with a shorter, more hydrophobic alkyl spacer arm 3a and 3b.
| τ av H2O (ns) | τ av H2O degassed (ns) | τ av toluene (ns) | τ av toluene degassed | τ av in cells | Φ H2O (%) | |
|---|---|---|---|---|---|---|
| 3a | 68.8 ± 0.2 | 71.4 ± 0.3 | 98.7 ± 0.3 | 728.0 ± 0.6 | — | 3.8 |
| 3b | 67.8 ± 0.2 | 68.9 ± 0.3 | 88.7 ± 0.2 | 454.4 ± 0.7 | — | 3.4 |
| 3c | 81.9 ± 0.2 | 79.7 ± 0.3 | 98.9 ± 0.4 | 744.6 ± 0.5 | 369.5 ± 2.8 | 3.9 |
| 3d | 76.0 ± 0.2 | 74.2 ± 0.3 | 93.8 ± 0.3 | 696.2 ± 0.9 | 401.7 ± 1.2 | 4.0 |
In cell labeling experiments (Fig. S11–S13, ESI†), the obtained signal comprises two components: a fast lifetime (<5 ns) corresponding to cell autofluorescence, and a long lifetime corresponding to the Ir(III) complex. The long lifetime of 3c and 3d (ca. 400 ns) was consistently found to be longer in cells than in non-degassed solutions, in between the value obtained in solution for aerobic conditions and the value in degassed toluene. This τ value is homogeneous across the cell, regardless of the localization. The complex heterogeneous polarity microenvironments in cells may shield the triplet excited states from the effect of water thus leading to longer average lifetimes when compared to experiments in aqueous solutions, particularly when considering that the tagged N-glycoconjugates are majoritarily anchored in hydrophobic membranes. In addition, the fact that an anti-oxidant anti-fading mounting medium is used for imaging experiments might also aid in averting quenching and contribute to lengthening the lifetime of Ir(III) dyes.
Probe validation in vitro
We then investigated whether probes 3a–d could be efficiently conjugated by CuAAC to adequately functionalized monosaccharides in aqueous medium, in conditions relevant to bioorthogonal cellular assays.46 All four probes were reacted with a corresponding sugar reporter, namely either ManNAz 4 or ManNAl 5. The CuAAC ligations were monitored by HPLC equipped with UV and fluorescence detectors (Fig. S4 and S5, ESI†), and the formed cycloadducts were characterized by MALDI-ToF mass spectrometry of the isolated HPLC fractions (Fig. S6, ESI†). PEGylated probes 3c and 3d were quickly and fully converted to the desired cycloadducts as expected, indicating fast kinetics suitable for cell culture experiments. However, we observed that the reaction of probes 3a and 3b was significantly advanced but still incomplete after two hours, showing conversion yields of 94% and 85%, respectively (Fig. S4, ESI†). The reactivity assessed in aqueous solutions with a monosaccharide does not directly translate to the reactivity of probes reacting in the midst of cells, which are composed of a multitude of complex heterogeneous microenvironments with highly variable hydrophobic–hydrophilic balances, and the local concentration of reporter subunits is not uniform throughout the cell. However, this result hinted at a potentially lesser labelling efficiency for dyes 3a and 3b in subsequent biological experiments than for 3c and 3d.Metabolic labeling of sialoglycoconjugates in cells
The probes were then used to label intracellular sialylated glycoconjugates in HeLa cells through the MOE approach and subsequently visualized by laser scanning confocal microscopy. Sialic acids, a diverse group of monosaccharides, are found at the non-reducing ends of glycan chains of glycoproteins and glycolipids. Entering the sialic acid metabolic pathway, ManNAz 4 and ManNAl 5 are intracellularly converted in three steps to the corresponding azide- and alkyne-tagged sialic acids, respectively, then incorporated into nascent sialoconjugates in the Golgi apparatus after being activated as cytidine monophosphate nucleotide sugars in the nucleus. During their life cycle, the formed sialoconjugates are then generally exported to the cell membrane via the secretory pathway, and can be re-internalized during pinocytosis and recycled.46Whereas per-O-acetylated monosaccharides are commonly used for MOE in mammalian models, it has been shown that per-O-acetylation generates non-specific signal and false positives.21 It may also lead to metabolic perturbation due to partially acetylated forms, thus for reporters that are capable of entering cells by active transport such as ManNAc or Sia derivatives, we tend to use unprotected reporters in studies aimed at investigating intracellular metabolic paths.164 or 5 were metabolically incorporated in HeLa cells that were then fixed with paraformaldehyde and permeabilized with Triton X and imaged using confocal fluorescence microscope with 405 nm excitation. While brightness is lower, visible excitation wavelength was preferred to 375 nm one to avoid autofluorescence. A longpass was used to collect all the photon >500 nm. We would like to emphasize here the interest of bioorthogonal labelling when carried out in fixed cells, a method used in most articles but generally not advertised as deserved. Even if developing methods that implement bioorthogonal installation of the probe in vivo is of paramount importance, there are numerous applications and contexts ex vivo in which it is strategically and technically more advantageous to perform this step in cells that are fixed after the reporter incorporation. This is particularly true for intracellular bioimaging as we have argued in a previous review.16
Cells incubated with ManNAz 4 were reacted with alkyne-functionalized [Ir(ppy)2(phen-Alk)]+[PF6]−3a or [Ir(ppy)2(phen-PEG2-Alk)]+[PF6]−3c using the copper sulfate/ascorbate catalytic system in the presence of copper tetravalent ligand BTTAA, adapting our previous protocol, while cells incubated with ManNAl 5 were labeled with azide dyes [Ir(ppy)2(phen-Az)]+[PF6]−3b or [Ir(ppy)2(phen-PEG2-Az)]+[PF6]−3d. Iridium probe concentration, reporter incubation time and CuAAC parameters were optimized for maximum signal-to-noise ratio (Fig. S7–S9, ESI†).
Azide 3a and alkyne 3b at concentrations ranging from 1 to 100 μM did not lead to efficient marking of sialylated structures (Fig. 4). Although HPLC monitoring of the reaction in solution indicated a slower but decent rate, we had expected these probes to bind to sugar reporters in cells. An analogous Ir-alkyne complex with no spacer arm was indeed previously reported to bind to azide-tagged protein pools after incubating cells with 2 mM azidohomoalanine in methionine-depleted medium.38 However, in our case the non-specific background signal was consistently too high to detect any potential specific labeling. The short, rigid spacer arms of 3a and 3b combined to the hydrophobic nature of the complex core might not confer enough flexibility for the reactive handle to efficiently access reporter groups in the highly polar microenvironment of glycan chains. The issue might also stem from the low incorporation rate of monosaccharide reporters that leads to a lower concentration of tagged biomolecules in MOE than for residue-specific protein labeling.
 | ||
| Fig. 4 Bioorthogonal labeling of sialoglycoconjugates with Ir(III)-ppy complexes observed by laser scanning confocal microscopy. Cells were incubated with ManNAz 4 (top) or ManNAl 5 (bottom) and reacted with complementary probes: (A) TAMRA-N3 or TAMRA-Alk; (B) probes 3a–d. A negative control showing cells reacted with each probe after incubation of untagged N-acetylmannosamine (no chemical reporter) is shown in the top-right corner of each image. (C) ICP-MS analysis of cells reacted with 3c after incubation with ManNAc (control, grey) or ManNAz (reporter, red). Quantity of incorporated iridium is reported in femtogram per cell. Experiments were carried out as three biological replicates and two technical measures on each replicate. Scale bar = 30 μm. | ||
Conversely, probes 3c and 3d bearing a flexible PEG3 spacer arm capable of accepting hydrogen bonds provided images with efficient and highly specific labeling when compared to negative control conditions (Fig. 4). The obtained pattern is fully consistent with intracellular sialylation studies previously evidenced with small organic fluorophores such as click-reactive tetramethylrhodamines (TAMRA).46,47 Alkyne-functionalized probe 3c was selected for further studies.
ICP-MS quantitative analysis in cellulo
We went on to check whether these bioorthogonal probes could facilitate the quantification of reporters metabolized into cell glycoconjugates with ICP-MS. Given its low abundance in nature, iridium is characterized by a very low limit of detection (0.001 to 0.01 ppb), a clear benefit when considering that the rate of incorporation of monosaccharides in neo-formed glycoconjugates has been reported to be quite poor (ca. 5%). Cells were incubated with ManNAz 4 (or ManNAc as control) and labeled with 3c after cell fixation and permeabilization. A thorough washing procedure was optimized to eliminate the excess reactant as well as the pool of soluble metabolic intermediates prior to acidolysis. On average, the amount of iridium was found to be 7.3-fold higher in samples treated with ManNAz than in the control (Fig. 4C and Table S1, ESI†), illustrating the potential of such bulk quantitative analysis to complement photoluminescence imaging data.Ir(III) complexes photostability
The photobleaching of 3c was then assessed and compared to two dyes reputed for their photostability (Fig. 5). In cells incubated with either ManNAz 4 and labeled with either 3c, or with ManNAl 5 and labeled with Alexa Fluor 405-Azide, 108 × 108 μm squares (512 pixels × 512 pixels) corresponding to a single cell were continuously scanned with laser irradiation at 405 nm with a dwell time of 50 μs pixel−1 in the presence and absence of antifade reagent. 3c was revealed to be remarkably photostable and robust, with 85% of the mean photoluminescence intensity being retained after 80 seconds of exposure to the laser, time by which Alexa Fluor 405 was fully photobleached. Dye 3c excited at 405 nm even proved to be more photostable than Alexa Fluor 488 excited at the lower energetic wavelength 488 nm (Fig. S10, ESI†). Susceptibility to hydrolysis was also tested in PBS buffer over several weeks and no degradation was observed. Altogether the designed probes displayed consistent photophysical parameters independent of the bioorthogonal handle used, with large Stokes shift and long emission lifetime, and are chemically robust and highly photostable. They are thus ideal candidates for applications in multicolor time-gated bioimaging, whereby luminophores are distinguished using differing time ranges (Δt) of photon emission after an excitation pulse. | ||
| Fig. 5 Photostability of probe 3c compared to Alexa Fluor 405-Azide. The probes were linked to reporter units by CuAAC after metabolic incorporation of ManNAz 4 or SiaNAl 6, respectively, in HeLa cells for 48 h. (A) Mean photoluminescence (PL) intensity of confocal images (108 × 108 μm, 211 nm px size, 2 s per image). Three technical replicates were measured. (B) Confocal images of a single cell monitored during continuous data acquisition (35 × 35 μm, 211 nm px size). Excitation: 405 nm. Emission detection filter: bandpass 450/50 nm for AF405, longpass >600 nm for 3c. | ||
Time-resolved photoluminescence imaging microscopy
We then investigated Ir(III) complexes in lifetime imaging, using a single laser excitation pulse to distinguish two probes in time-gated experiments. Classical methods based on spectral properties acquire several images with different sets of excitation light and emission filters to separate probes which emit in different regions and/or can be excited at different wavelengths. In contrast, in time-gated emission methods, imaging of different marking patterns can be obtained in one experiment using a single excitation pulse, thus reducing photobleaching and any optical artefact that could occur changing the irradiation/detection conditions, improving temporal resolution, as well as enhancing multiplexing capabilities.Firstly, dual labeling experiments were conducted with 4/3c and DAPI. In this context, the two probes are not localized in the same region of the cell: at the incubation time used for 4, 3c (long lifetime) is conjugated upon bioorthogonal ligation to nascent sialoconjugates that are mainly localized in the Golgi apparatus, with a less intense secondary pool at the cell membrane. In contrast, DAPI (short lifetime, τ = 2 ns)48 is well-known to localize in the nucleus upon binding with the minor groove of DNA, thus providing a straightforward platform to validate signal specificity in single pulse time-gated experiments (Fig. 6). Images of labeled HeLa cells were acquired on an in-house designed microscope using a 405 nm picosecond pulsed laser excitation and a long-pass filter, to collect all photons with a wavelength superior to 500 nm with an avalanche photodiode-based single-photon counting detector. The experimental setup used for this “1-color/2-probes” approach and the negative control samples are shown in ESI† (Fig. S14). Time-gating was then applied to unmix emitted photons corresponding to DAPI (blue channel, photons arrival time Δt < 20 ns) and to sialoglycoconjugates (40 < Δt < 500 ns).
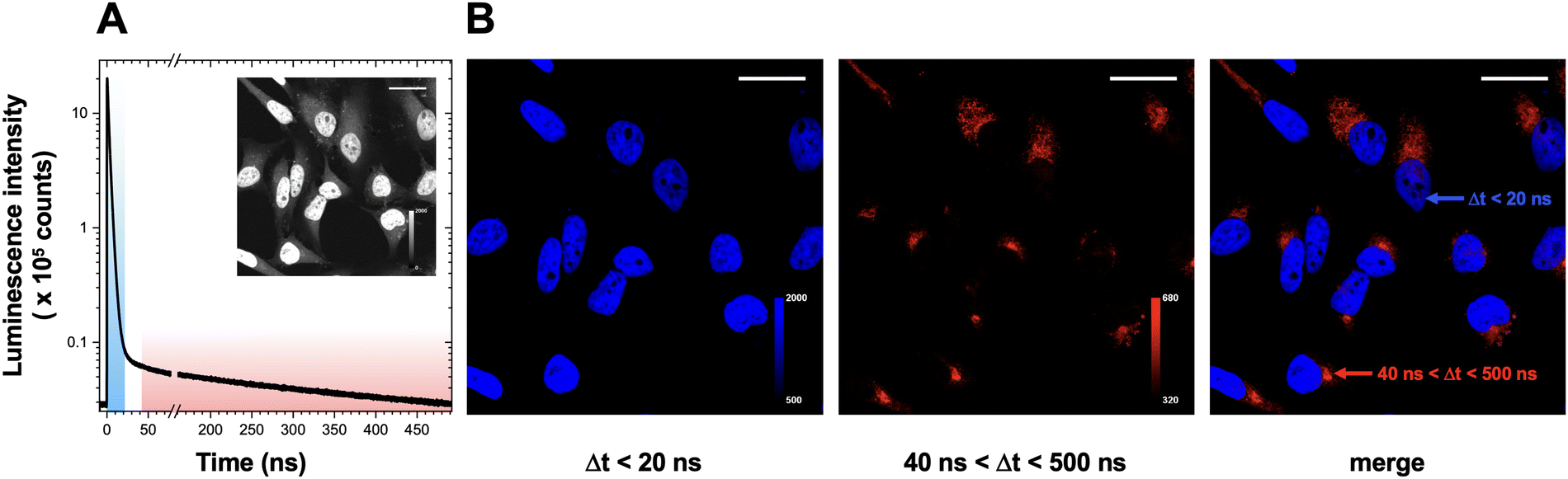 | ||
| Fig. 6 HeLa cells labeled with ManNAz 4/iridium complex 3c (sialoconjugates) and DAPI (nucleus). (A) Emission time decay and raw image with all collected photons used to generate time-gated channels. Excitation at 405 nm and detection with long pass filter >500 nm. (B) Time-gated images for early photon arrival times in blue (Δt < 20 ns, left), late photon arrival times in red (40 < Δt < 500 ns, middle), and merge of the two channels (right). The marking patterns are typical of DAPI and of sialylated glycoconjugates, respectively. Experiments were carried out as three biological replicates. Scale bar = 30 μm. | ||
These results clearly demonstrate the ability of such Ir(III) complexes to be exploited in multicolor imaging to generate highly specific channels. We then showed the full potential of probe 3c in dual labeling experiments exploiting two monosaccharide reporters of the same metabolic pathway. For that purpose, we used ManNAz 4 and SiaNAl 6, as they are indeed both incorporated as sialic acid units in glycoconjugates, but enter the metabolic network at two different points. SiaNAl 6 enters cells by pinocytosis and is directly activated in the nucleus as the sugar nucleotide donor cytidine 5′-monophospho N-pentynoylneuraminic (CMP-SiaNAl) acid prior to Golgi export and transfer onto a nascent glycoconjugate by a sialyltransferase. ManNAz 4 must follow a longer path as it penetrates cells through a yet unknown transport mechanism and undergoes three enzymatic conversions in the cytosol (phosphorylation to ManNAz-6-P, conversion to SiaNAz-9-P by condensation with phosphoenolpyruvate, then dephosphorylation to SiaNAz) before it can be transported to the nucleus and activated as CMP-SiaNAz.46 Therefore, the reporters are expected to be localized in the same regions of the cell, but with different kinetics of incorporation.
HeLa cells incubated with both reporters were submitted to two consecutive CuAAC labeling steps. ManNAz 4 was reacted with long-lifetime alkyne dye 3c, while SiaNAl 6 was ligated to short-lifetime Alexa Fluor 405-Azide. Cells were then imaged as described above (Fig. 7). Using similar time-gating analysis, we could get an image specific of Alexa Fluor 405 (green channel) and of 3c (red channel), with full signal specificity of each channel and with no need for fitting the time-dependent signal. To the best of our knowledge, this is the first time that both a sialic acid reporter and a N-acetylmannosamine reporter are used simultaneously in the same cell to generate dual color maps of intracellular sialylation. Partial co-localization of time-resolved signals was observed, but as expected some signals are not co-localized, thus evidencing differences in metabolism kinetics between the two reporters. This dual-labelling method could provide a useful imaging platform in the future for studying the early steps of sialylation pathways, or for investigating trafficking and recycling of sialoconjugates in pathologies such as congenital disorders of glycosylation (CDGs).
 | ||
| Fig. 7 Time-resolved imaging of HeLa cells dual labeled with SiaNAl 6/Alexa Fluor 405-Azide and with ManNAz 4/Ir(III) dye 3c. Top left: Time-gated channel showing photons emitted between 40 and 500 ns after the excitation pulse (specific of 3c). Top right: Time-gated channel showing photons emitted between 0 and 20 ns after the excitation pulse (specific of Alexa Fluor 405). Bottom left: Merge image showing both long (3c, red) and short (AF405, green) lifetime signals, overlayed with an image showing the raw full photon count (grey). The levels of the grey channel are not adjusted to the negative control in order to outline the cells. Bottom right: Correlation scatter plot of the red (3c) and green (AF405) channels. Excitation at 405 nm and detection with long pass filter >410 nm. Experiments were carried out as three biological replicates. | ||
Additional time-resolved experiments using a more conventional set-up using two excitation pulses at two different wavelengths to separately acquire the two channels were also successfully performed. In these experiments, ManNAz 4 and SiaNAl 6 were bioorthogonally labeled with 3c and AlexaFluor 488, respectively (Fig. S15, ESI†). The advantages of clickable Ir(III) probes in multiplexed settings are further showcased here: although the emission spectra of 3c and of AlexaFluor 488 partially overlap, as well as their absorption spectra, time-gating allows unambiguous distinction of the photon's origin (i.e., from which probe it was emitted), ensuring high confidence on the signal specificity of each channel.
X-ray fluorescence nanoimaging
While optical microscopy is the method of choice for imaging labeled biomolecules within the context of a living cell, X-ray fluorescence nanoimaging (XRF-NI) techniques developed at some synchrotron facilities provide a highly complementary subset of information. Indeed, XRF-NI now allows high-spatial resolution (30–50 nm) of the detected elements within a sub-cellular structure combined to a high degree of sensitivity, as shown for Zn in neuronal structures reaching a very low limit of detection of 130 atoms of zinc within a 40 nm pixel size.49 In addition, unlike most optical methods, XRF-NI provides absolute quantification through elemental composition mapping of the cell. Despite the low-throughput nature of the technique due to low temporal resolution, these benefits are great assets in the quest for quantifying intracellular metabolic probes that are expected to report locally for a very low number of molecules at the nanoscale.In our context, achieving optimal results entails the development of adequately designed click-reactive XRF sensitive probes such as 3c–d for quantitative subcellular mapping purposes. Iridium is indeed an ideal candidate for nanoscale XRF-NI, having no biological role and an extremely low abundance in living organisms (below 1 ng L−1 in serum for example).50 Thus, endogenous Ir is not detected by XRF within cells and does not induce any X-ray autofluorescence background. Furthermore, main X-ray fluorescence emission rays can be observed for Ir at 9.175 keV (Lα) and 10.708 keV (Lβ), a region nearly devoid of emission lines from biologically important elements observed in cells. This ensures a minimal background signal and provides clear and accurate results without interference from other cellular components. In our samples, only the Zn-Kβ X-ray emission line at 9.57 keV is situated close to the Ir-Lα emission line, while the Ir-Lβ line is in a spectral region completely free of other element contribution. Spectral fitting with a dedicated software allows the deconvolution of any potential spectral overlap as well as the removal of any background contributions, to get the net peak area for all elements detected. From these, a local concentration can be calculated for each pixel using the fundamental parameter method as already reported in similar XRF-NI studies (see ESI,† for details).13,51
To illustrate the capabilities of clickable Ir(III) dyes in XRF-NI, we explored their potential in distinguishing labeling patterns derived from two widely employed reporters for studying sialylation, namely per-O-acetylated Ac4ManNAz 7 and free-hydroxyl ManNAz 4. Whereas per-O-acetylated monosaccharides are commonly used in mammalian models, it has been shown that per-O-acetylation generates non-specific signal and false positives.21 It may also lead to metabolic perturbation due to partially acetylated forms, thus for reporters that are capable of entering cells by active transport such as ManNAc or Sia derivatives, we tend to prefer unprotected reporters in studies aimed at investigating intracellular metabolic paths.16 HeLa cells were grown for 48 h on graphene-coated silicon nitride membranes with either ManNAz 4, Ac4ManNAz 7 or ManNAc as control and submitted to CuAAC with 3c as described hereabove. The labeled cells were first observed by confocal fluorescence microscopy in PBS with an immersion objective to minimize any damages to the cells and/or the Si3N4 membranes. This allowed us to capture wide field images and to observe every cell on each membrane prior to XRF experiments at the synchrotron. Cells were kept under PBS at 4 °C until they were quickly rinsed with an ammonium acetate buffer, vitrified by plunge-freezing in liquid ethane and kept under liquid nitrogen before cryo-transfer for cryo-XRF nanoimaging.52 In the present case, the sample analysis at cryogenic temperature limits the impact of the intense focused X-ray beam and radiation damages on cells during acquisition of XRF data.53
The online transmitted light microscope allowed us to precisely select the cells of interest and generate a direct comparison between confocal UV/vis phosphorescence and XRF images (Fig. 8 and Fig. S17, ESI†).
 | ||
| Fig. 8 X-ray fluorescence nanoimaging (XRF-NI). (A) XRF elemental imaging of iridium (fire colorscale, 3c) and phosphorus (cyan, nucleus) in HeLa cell after 48 h ManNAz 4 incorporation shows 16-fold improved resolution compared to UV/vis confocal microscopy (50 nm px−1). (B) Comparison of iridium area density (ng mm−2) in XRF analyzed cells after incorporation of ManNAz 4 (orange), Ac4ManNAz 7 (purple) or ManNAc (grey, control) followed by CuAAC conjugation of 3c. Three replicates were measured. (C) Bimodal observation of HeLa cells after incorporation of ManNAz 4 (top) or Ac4ManNAz 7 (bottom) and CuAAC labeling with 3c. Cells were observed by laser scanning confocal microscopy (left, brightfield + 3c UV/vis photoluminescence) and analyzed with cryo-XRF-NI (middle). XRF-NI analysis allowed the creation of masks showing great correlation between UV/vis and XRF signal and improved sensitivity (right). Curves with SD are shown in Fig. S17 (ESI†), Negative control images are shown in ESI.† | ||
We observed a strong correlation between both experiments, with clear co-localization of the probes by UV/vis phosphorescence and the iridium XRF signal. While the UV/vis signal is fainter and blurrier, cryo-XRF allows much finer details to be observed, which is especially visible in the ManNAz condition where a punctiform pattern around the Golgi apparatus is revealed (Fig. 8A and C). A slightly different labeling profile is observed with the free-hydroxyl reporter ManNAz 4 compared to the peracetylated Ac4ManNAz 7, for which the pattern seems more diffuse and scattered, possibly due to partial non-specific incorporation of the reporter,18,21 or to a lower rate of incorporation at this incubation time point. XRF-NI experiments give access to the iridium surface mass expressed in ng mm−2 which, combined with surface measurements, allows us to precisely determine the quantity of iridium atoms and thus the number of probe molecules bound within the cells for each condition analyzed. In multimodal approaches, XRF-NI combined with UV-vis luminescence microscopy may thus be used to link the arbitrary luminescence units to actual quantitative values in future studies.
Interestingly, under the incubation conditions used here (48 hours for both sugars in HeLa cells), the surface density of the Ir(III) probe was found to be higher following incubation with 500 μM ManNAz compared to 50 μM Ac4ManNAz (Fig. 8B). This unexpected finding suggests that unprotected ManNAz holds significant potential as a sialylation reporter for studying the trafficking of intracellular sialylated glycoconjugates, as it minimizes much of the non-specific labeling typically observed with peracetylated reporters. Indeed, whereas incorporation of peracetylated reporters is usually thought to be more efficient than free-hydroxyl reporters because they enter cells by passive diffusion and require lower concentration in the culture medium, their incorporation is slower since they need to undergo full deacetylation through the action of non-specific esterases in order to release the free metabolite. Incomplete deacetylation might also hinder enzyme function or result in the incorporation of partially acetylated sialic acid analogs, potentially biasing downstream analyses, and the release of acetic acid may cause localized pH changes, which could impact cellular activity. Importantly, Chen and colleagues have also shown that per-O-acetylated sugars can non-enzymatically react with free cysteine residues in proteins,20 leading to off-target covalent binding and increased non-specific background signals. In our case, the actual metabolic incorporation of 7 in glycoconjugates was clearly less efficient than 4.
While detection of iridium probe 3c by ICP-MS allows bulk statistical analysis on a large pool of cells (Fig. 4C), its use in XRF-NI thus provides much finer analysis through iridium mapping as it retains subcellular spatial information with high resolution and sensitivity. Combined with photoluminescence techniques, such clickable organometallic complexes are thus versatile tools that may allow in-depth investigation of sialylation metabolism in pathological contexts.
Conclusions
We developed and validated click-ready Ir(III)–polypyridine complexes as robust bioimaging probes for metabolic labeling, offering compatibility with multimodal techniques. These tools displayed remarkable photophysical properties, and were shown to offer clear benefits terms of resolution, specificity or sensitivity using laser scanning confocal microscopy, time-resolved photoluminescence lifetime imaging, inductively coupled plasma mass spectrometry and X-ray fluorescence nanoimaging. They allowed dual imaging of two chemical reporters of the sialylation pathway with a single excitation pulse in time-gated experiments, which paves the way for future studies focusing on the kinetics and dynamics of sialoconjugate metabolism and trafficking in pathological vs. physiological contexts. They also led to high-resolution cryo XRF-NI elemental mapping of cells (40 nm) with quantification of iridium area density per pixel, providing an imaging platform for future studies comparing the incorporation of various reporters, or of a given reporter in different conditions. Such Ir(III) probes are thus amenable to multimodal correlative bioimaging studies, laying a foundation for further advances in glycosciences, but could also be used to monitor any other type of biomolecule accessible through bioorthogonal labeling. By addressing key challenges and opening new possibilities, such approaches are poised to play a pivotal role in advancing our understanding of complex biological processes, advocating for the continued integration of organometallic probes into the toolkit of chemical biologists.Author contributions
V. R., Cl. S., A. B., L. L., V. D. B., D. K., S. B. and C. L. performed the experiments and analyzed the data. V. R., C. L., L. L., M. S. and S. B. interpreted the results. C. L. wrote the manuscript. Cl. S. wrote the ESI.† V. R., C. L. and A. B. prepared figures. C. L., C. B., B. V. and M. S. conceptualized the project and acquired funding. C. L. and C. B. supervized the work. All authors revised the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Agence Nationale de la Recherche through the NEURAPROBE project (ANR-18-CE07-0042) (C. L., C. B., B. V., V. R.), and in part by the CNRS and the University of Lille. L. L. is a recipient from a research grant from ANR BIG_T-ReX (ANR-24-CE44-5061-01) (C. L., C. B., L. L., M. S., A. B.). Cl. S. was supported by the Human Frontier Science Program (LT000552/2020-L). We acknowledge the European Synchrotron Research Facility (ESRF) for granting beamtime through experiment LS-2975 at beamline ID16A.References
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- H. Kayser, R. Zeitler, C. Kannicht, D. Grunow, R. Nuck and W. Reutter, J. Biol. Chem., 1992, 267, 16934–16938 CrossRef CAS PubMed.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- M. Kufleitner, L. M. Haiber and V. Wittmann, Chem. Soc. Rev., 2023, 52, 510–535 RSC.
- S. S. Nguyen and J. A. Prescher, Nat. Rev. Chem., 2020, 4, 476–489 CrossRef CAS PubMed.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed.
- F. Balzarotti, Y. Eilers, K. C. Gwosch, A. H. Gynnå, V. Westphal, F. D. Stefani, J. Elf and S. W. Hell, Science, 2017, 355, 606–612 CrossRef CAS PubMed.
- M. Weber, H. von der Emde, M. Leutenegger, P. Gunkel, S. Sambandan, T. A. Khan, J. Keller-Findeisen, V. C. Cordes and S. W. Hell, Nat. Biotechnol., 2023, 41, 569–576 CrossRef CAS PubMed.
- J. J. Conesa, A. C. Carrasco, V. Rodríguez-Fanjul, Y. Yang, J. L. Carrascosa, P. Cloetens, E. Pereiro and A. M. Pizarro, Angew. Chem., Int. Ed., 2020, 59, 1270–1278 CrossRef CAS PubMed.
- J. Tan, X. Zhang, X. Wang, C. Xu, S. Chang, H. Wu, T. Wang, H. Liang, H. Gao, Y. Zhou and Y. Zhu, Cell, 2021, 184, 2665–2679.e19 CrossRef CAS PubMed.
- A. Walter, P. Paul-Gilloteaux, B. Plochberger, L. Sefc, P. Verkade, J. G. Mannheim, P. Slezak, A. Unterhuber, M. Marchetti-Deschmann, M. Ogris, K. Bühler, D. Fixler, S. H. Geyer, W. J. Weninger, M. Glösmann, S. Handschuh and T. Wanek, Front. Phys., 2020, 8, 47 CrossRef.
- V. Rigolot, C. Biot and C. Lion, Angew. Chem., Int. Ed., 2021, 60, 23084–23105 CrossRef CAS PubMed.
- R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS PubMed.
- K. N. Chuh, B. W. Zaro, F. Piller, V. Piller and M. R. Pratt, J. Am. Chem. Soc., 2014, 136, 12283–12295 CrossRef CAS PubMed.
- F. Doll, A. Buntz, A.-K. Späte, V. F. Schart, A. Timper, W. Schrimpf, C. R. Hauck, A. Zumbusch and V. Wittmann, Angew. Chem., Int. Ed., 2016, 128, 2303–2308 CrossRef.
- Y. Sun, S. Hong, R. Xie, R. Huang, R. Lei, B. Cheng, D.-E. Sun, Y. Du, C. M. Nycholat, J. C. Paulson and X. Chen, J. Am. Chem. Soc., 2018, 140, 3592–3602 CrossRef CAS PubMed.
- K. Qin, H. Zhang, Z. Zhao and X. Chen, J. Am. Chem. Soc., 2020, 142, 9382–9388 CrossRef CAS PubMed.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS PubMed.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, ACS Nano, 2020, 14, 4178–4187 CrossRef CAS PubMed.
- T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121, 6850–6914 CrossRef CAS PubMed.
- S. M. Usama, F. Inagaki, H. Kobayashi and M. J. Schnermann, J. Am. Chem. Soc., 2021, 143, 5674–5679 CrossRef CAS PubMed.
- L.-J. Cai, X. Wang, Q.-S. Zong, X.-P. Jiang and H.-Y. Hu, ChemPhotoChem, 2023, 7, e202300042 CrossRef CAS.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- S. Zeng, X. Liu, Y. S. Kafuti, H. Kim, J. Wang, X. Peng, H. Li and J. Yoon, Chem. Soc. Rev., 2023, 52, 5607–5651 RSC.
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265–287 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- L. Murphy, A. Congreve, L.-O. Pålsson and J. A. G. Williams, Chem. Commun., 2009, 8743–8745 Search PubMed.
- L. C.-C. Lee and K. K.-W. Lo, J. Am. Chem. Soc., 2022, 144, 14420–14440 CrossRef CAS PubMed.
- K. K.-W. Lo, B. T.-N. Chan, H.-W. Liu, K. Y. Zhang, S. P.-Y. Li and T. S.-M. Tang, Chem. Commun., 2013, 49, 4271–4273 RSC.
- S. P.-Y. Li, A. M.-H. Yip, H.-W. Liu and K. K.-W. Lo, Biomaterials, 2016, 103, 305–313 CrossRef CAS PubMed.
- T. S.-M. Tang, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2017, 53, 3299–3302 RSC.
- A. M.-H. Yip, C. K.-H. Lai, K. S.-M. Yiu and K. K.-W. Lo, Angew. Chem., Int. Ed., 2022, 61, e202116078 CrossRef CAS PubMed.
- J. Wang, J. Xue, Z. Yan, S. Zhang, J. Qiao and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14928–14932 CrossRef CAS PubMed.
- Q. Wu, K. Y. Zhang, P. Dai, H. Zhu, Y. Wang, L. Song, L. Wang, S. Liu, Q. Zhao and W. Huang, J. Am. Chem. Soc., 2020, 142, 1057–1064 CrossRef CAS PubMed.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- C. Liu, C. Yang, L. Lu, W. Wang, W. Tan, C.-H. Leung and D.-L. Ma, Chem. Commun., 2017, 53, 2822–2825 RSC.
- K. Vellaisamy, G. Li, C.-N. Ko, H.-J. Zhong, S. Fatima, H.-Y. Kwan, C.-Y. Wong, W.-J. Kwong, W. Tan, C.-H. Leung and D.-L. Ma, Chem. Sci., 2018, 9, 1119–1125 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- K. Y. Zhang, H.-W. Liu, T. T.-H. Fong, X.-G. Chen and K. K.-W. Lo, Inorg. Chem., 2010, 49, 5432–5443 CrossRef CAS PubMed.
- A. I. Solomatina, S.-H. Su, M. M. Lukina, V. V. Dudenkova, V. I. Shcheslavskiy, C.-H. Wu, P. S. Chelushkin, P.-T. Chou, I. O. Koshevoy and S. P. Tunik, RSC Adv., 2018, 8, 17224–17236 RSC.
- P. A. Gilormini, C. Lion, D. Vicogne, T. Levade, S. Potelle, C. Mariller, Y. Guérardel, C. Biot and F. Foulquier, Chem. Commun., 2016, 52, 2318–2321 RSC.
- P. A. Gilormini, C. Lion, D. Vicogne, Y. Guérardel, F. Foulquier and C. Biot, J. Inherited Metab. Dis., 2018, 41, 515–523 CrossRef CAS PubMed.
- A. K. Estandarte, S. Botchway, C. Lynch, M. Yusuf and I. Robinson, Sci. Rep., 2016, 6, 31417 CrossRef CAS PubMed.
- F. Domart, P. Cloetens, S. Roudeau, A. Carmona, E. Verdier, D. Choquet and R. Ortega, eLife, 2020, 9, e62334 CrossRef CAS PubMed.
- I. Rodushkin, E. Engström, A. Stenberg and D. C. Baxter, Anal. Bioanal. Chem., 2004, 380, 247–257 CrossRef CAS PubMed.
- F. Fus, Y. Yang, H. Z. S. Lee, S. Top, M. Carriere, A. Bouron, A. Pacureanu, J. C. da Silva, M. Salmain, A. Vessières, P. Cloetens, G. Jaouen and S. Bohic, Angew. Chem., Int. Ed., 2019, 58, 3461–3465 CrossRef CAS PubMed.
- C. Bissardon, S. Reymond, M. Salomé, L. André, S. Bayat, P. Cloetens and S. Bohic, J. Visualized Exp., 2019, 154, e60461 Search PubMed.
- C. Gramaccioni, Y. Yang, A. Pacureanu, N. Vigano, A. Procopio, P. Valenti, L. Rosa, F. Berlutti, S. Bohic and P. Cloetens, Anal. Chem., 2020, 92, 4814–4819 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00255e |
| ‡ V. R. and C. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |