
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific in vivo protein SUMOylation via translational incorporation of a proximity-reactive pyrrolysine analogue†
Yuk Hei
Chan
 ,
Marianne M.
Lee
and
Michael K.
Chan
*
,
Marianne M.
Lee
and
Michael K.
Chan
*
School of Life Sciences & Center of Novel Biomaterials, The Chinese University of Hong Kong, Shatin, Hong Kong SAR. E-mail: michaelkcchan88@cuhk.edu.hk
First published on 28th January 2025
Abstract
Here, we present a novel strategy that integrates genetic-code expansion and proximity-induced crosslinking to achieve site-specific in vivo SUMOylation. This approach involves incorporating the unnatural amino acid 2-chloroacetyl-Nε-lysine (ClAcK) into the target protein using MmFAcKRS1, a previously reported pyrrolysyl-tRNA synthetase mutant that we have repurposed for ClAcK incorporation. Once incorporated, ClAcK can be specifically targeted to react with a cysteine engineered at the C-terminus of SUMO variants leading to a chemically SUMOylated protein. This reaction is proximity-induced, and preferentially promoted when the two reactive groups are in close spatial proximity. We therefore leverage the natural affinity of SUMO for SUMO-interacting motifs (SIMs) on target proteins to generate the targeted SUMO conjugation. Using this approach, site-specific SUMO-conjugates have been produced for two distinct proteins in cells, thus demonstrating its potential as a strategy for helping to dissect the role of SUMOylation in its native cellular context.
SUMOylation is a reversible post-translational modification that involves the covalent attachment of small ubiquitin-like modifiers (SUMOs) to the ε-nitrogen of lysine residues on target proteins.1 Mirroring the ubiquitination system, it employs a dedicated cascade of enzymes. The heterodimeric SUMO-E1 activating enzyme (SAE1/SAE2) initiates the process by activating SUMO and transferring it to the SUMO-E2 conjugating enzyme (Ubc9), which then attaches SUMO to the target protein with or without a specific SUMO-E3 ligase.1
SUMOylation is important in diverse cellular processes, regulating protein stability, localization, activity, and interactions with other macromolecules.2 It also plays essential roles in mediating cell proliferation and stress responses,3 and has been implicated in various human diseases, including neurodegenerative disorders,4 cancer,5,6 and cardiovascular diseases.7,8 In cancer cells, the SUMOylation cascade is frequently upregulated,5 and contributes to their proliferation by facilitating decatenation and adaptation to stress.5,6 Conversely, SUMOylation exhibits protective effects in synucleinopathies, with SUMOylated α-synuclein (αSyn) displaying an inhibitory effect on αSyn aggregation,9,10 suggesting a promising therapeutic strategy for these disorders.11
Despite its importance, studies of SUMOylation in vivo have been hampered by a lack of control over the specific lysine to be modified. Many substrates possess multiple lysines susceptible to SUMOylation, and thus blocking the dominant site can lead to compensatory modifications at secondary sites. This significantly complicates the production of site-specific SUMO-conjugates via enzymatic methods.
To address this challenge, multiple chemical approaches including disulfide exchange,12 Cu(I)-catalyzed azide–alkyne cycloaddition,13 thiol–ene reaction,14 and oxime ligation15 have been developed. While these methods can generate site-specific SUMOylated proteins, most require in vitro settings or are sensitive to reducing conditions within cells, rendering them unsuitable for studying SUMOylation in vivo. An approach to perform site-specific in vivo SUMOylation would be beneficial, as it would facilitate studies aimed at dissecting the in vivo consequences of SUMOylation at different positions on the same protein. A recent attempt to achieve this used genetic-code expansion and sortase.16 Through the genetic incorporation of an azido-protected glycylglycyl-lysine and sortase-mediated transpeptidation, site-specific SUMOylation was achieved in living cells.
Here we present an alternative method for in vivo site-specific SUMOylation. Our approach utilizes a proximity-induced crosslinking strategy that avoids the need for sortase co-expression. The key innovation lies in the genetic incorporation of 2-chloroacetyl-Nε-lysine (ClAcK; single letter code ‘X’), a non-canonical amino acid equipped with a 2-chloroacetyl group that can readily form thioether bonds with a proximal thiol moiety (Fig. 1). Previous research has successfully employed the SN2 reaction between cysteine and the 2-chloroacetyl group for affinity-guided peptide conjugation.17,18 The balanced electrophilicity of the 2-chloroacetyl group allows it to remain inert towards distant cysteines within the protein while efficiently reacting with the proximal cysteine. Furthermore, once incorporated in a peptide, its moderate electrophilicity minimizes unwanted reactions with free thiol-containing compounds, making the reaction suitable for conjugation within living cells.19 One potential concern, however, is the potential depletion of ClAcK due to the high concentrations (∼10 mM) of glutathione in E. coli20 and mammalian cells.21 Indeed, in vitro studies revealed that 2 mM ClAcK reacts with 10 mM reduced glutathione with a half-life of ∼3 hours at pH 7.4 (Fig. S1, ESI†). Despite this, as detailed below, we were able to successfully implement ClAcK for SUMO-conjugation in E. coli. Presumably the reservoir of extracellular ClAcK present in the growth medium is sufficient to replenish the cellular ClAcK levels at a concentration suitable for translational incorporation, making it feasible for use in cell-based studies.
 | ||
| Fig. 1 Genetic code expansion. L-Pyrrolysine (Pyl; O) and 2-chloroacetyl-Nε-lysine (ClAcK; X). | ||
For translational incorporation of ClAcK into proteins using the UAG codon, we employed the pyrrolysine translational machinery with a modified Methanosarcina mazei PylRS mutant, MmFAcKRS1, that was previously identified by Kobayashi et al. for the genetic incorporation of Nε-fluoroacetyllysine (FAcK).22 While FAcK can also react with proximity-confined thiols, its reactivity is limited, with the yield of intramolecular crosslinking in calmodulin estimated at only 14%.22 We hypothesized, however, that ClAcK with a more reactive chloroacetamide group,23 should result in enhanced crosslinking efficiencies while remaining mild enough to mediate the proximity-induced reaction needed for site-specificity.
Since ClAcK and FAcK differ only by one atom, our initial investigation focused on determining whether MmFAcKRS1 could accept ClAcK as a substrate. Using the mCherry reporter assay, where mCherry fluorescence reflects tRNA-synthetase charging efficiency, we confirmed that MmFAcKRS1 can indeed incorporate ClAcK into mCherry at the UAG codon in a specific and dose-dependent manner (Fig. 2a), while having low readthrough for endogenous amino acids (Fig. 2b). To assess the efficiency of ClAcK incorporation by MmFAcKRS1 relative to the benchmark (PylRS for BocK), we compared their readthrough efficiencies for these respective ncAAs. It was found that MmFAcKRS1 exhibited only slightly lower readthrough efficiency for ClAcK than PylRS for BocK (Fig. S2, ESI†).
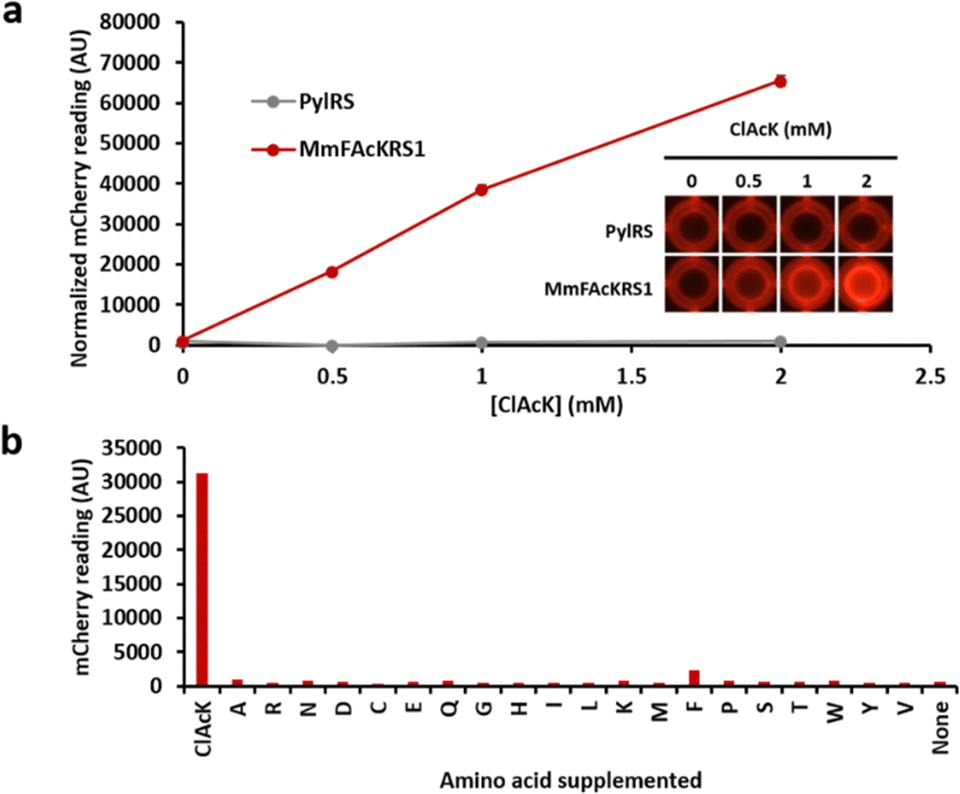 | ||
| Fig. 2 mCherry reporter assay. (a) Comparison of PylRS and MmFAcKRS1 readthrough of ClAcK. BL21(DE3) cells were transformed with a reporter plasmid containing either PylRS or MmFAcKRS1, along with PylT and an mCherry gene with Lys55 mutated to an amber stop codon. Transformants were grown in a 24-well plate in the presence of varying concentrations of [ClAcK] in M9 minimal medium. Normalized mCherry fluorescence was calculated by dividing the mCherry signal by the OD600. (b) Evaluation of background readthrough by MmFAcKRS1 using the 20 canonical amino acids. BL21(DE3) cells were transformed with the plasmid pDuet-PylT-MmFAcKRS1-KanR-K46X-mCherry-K55UAG and grown in M9 minimal medium supplemented with 2 mM of each of the 20 canonical amino acids. | ||
An initial assessment of the reactivity of the 2-chloroacetyl-handle with a thiol group via “proximity-induced reaction” within live cells was performed using E. coli arginine decarboxylase (EcADC) as a model system. EcADC naturally exists as a non-covalent tetramer, comprising two associated dimers, thus the generation of a covalent intermolecular crosslink between subunits should be easily confirmed by SDS-PAGE analysis. Based on the previously determined crystal structure,24 we engineered EcADC-K240X/D535C by introducing ClAcK at position 240, which is proximal to the mutated Cys residue on an adjacent monomer (Fig. S3a, ESI†). We then co-expressed the EcADC-K240X/D535C with PylT/MmFAcKRS1 in the presence of 2 mM ClAcK in E. coli (Fig. S3b, ESI†). SDS-PAGE confirmed the formation of the covalent dimer with a crosslinking efficiency of ∼83%, thereby demonstrating the robust reaction between ClAcK and Cys in vivo (Fig. S3c and d, ESI†).
We next investigated its application for site-specific in vivo SUMOylation. Studies show that nearly 90% of SUMO binders are also SUMOylation targets, with the SUMO-interacting motif often mediating the outcome of SUMOylation.25,26 Given the functional relationship between SUMO–SIM interaction and SUMOylation,27,28 we hypothesized that the SUMO–SIM interaction could be used to naturally position the C-terminus of SUMO close to native SUMOylation sites on the target proteins, enabling “proximity-induced” SUMO conjugation.
We first tested this idea with thymine-DNA glycosylase (TDG). To generate the SUMO–TDG conjugate, we engineered the C-terminus of human SUMO1 from its native sequence QTGG to QTC (SUMO(QTC)). This introduced a reactive Cys for a thioether linkage with the site-specifically incorporated ClAcK on TDG, while maintaining chain length as the native SUMOylation linkage via diglycine motif deletion (Fig. 3a). Subsequently, SUMO(QTC) was co-expressed with TDG-K330X-His6, which has ClAcK incorporated at the native SUMOylation site, in E. coli. The formation of the SUMO–TDG conjugate was confirmed by both mass spectrometry and western blotting analysis. LC-MS detected the intact protein with a mass corresponding to the combined mass of SUMO(QTC), TDG(112–360)-His6, and the linker (Fig. S4, ESI†), while LC-MS/MS further identified the SUMO C-terminal tryptic peptide with a modification corresponding to the mass of the 7-residue TDG tryptic peptide containing TDG-K330X (Fig. S5, ESI†). Western blotting analysis also detected the conjugate (Fig. 4a), and suggested a 15.7% SUMO-conjugation yield. This compares favourably to the 5–10% SUMO-conjugation yield reported for sortylation in mammalian cells.16
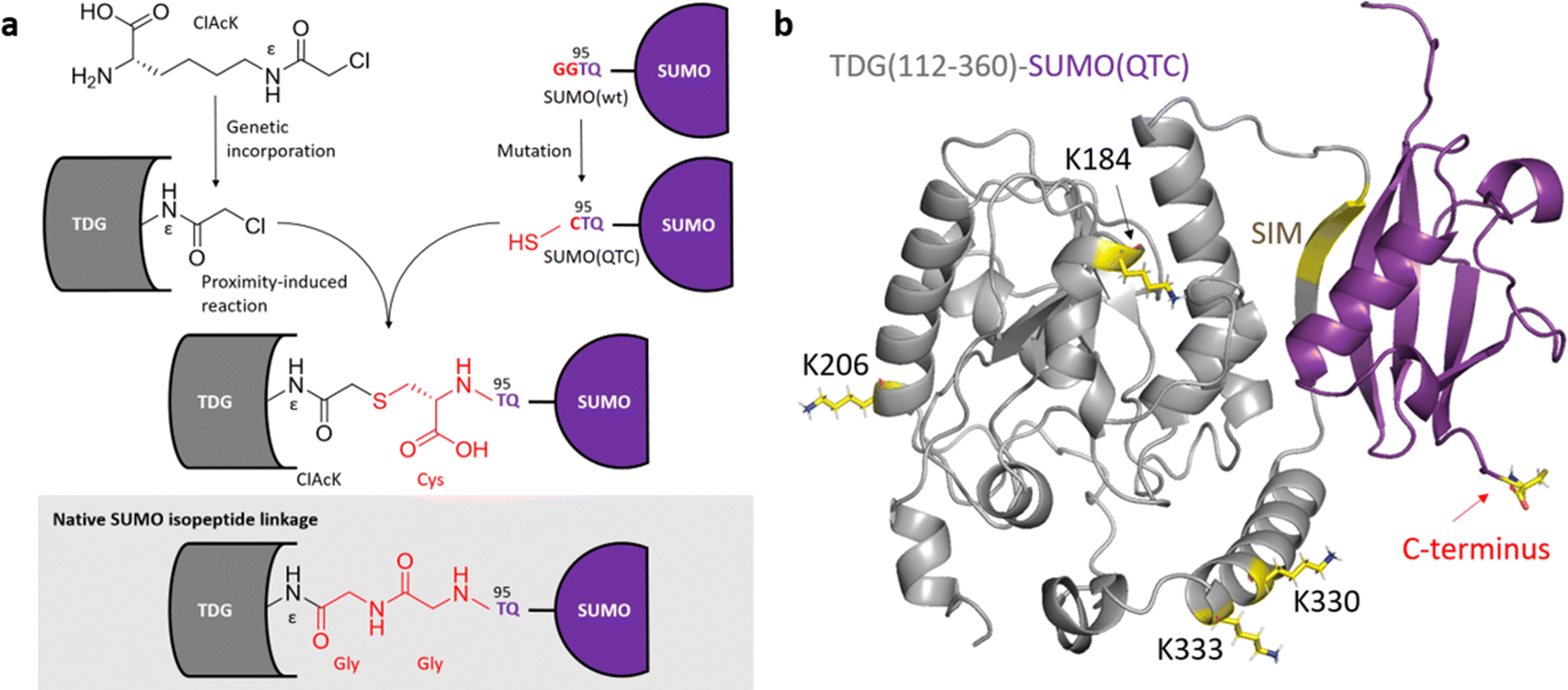 | ||
| Fig. 3 ClAcK-mediated SUMOylation. (a) ClAcK is genetically incorporated at a specific site within TDG using MmFAcKRS1. ClAcK-bearing TDG then reacts with a co-expressed SUMO variant, SUMO(QTC), harbouring a C-terminal cysteine through a proximity-induced SN2 reaction in cytosol upon expression. Notably, the linkage of the SUMO-conjugate generated by the Cys-ClAcK crosslink possesses identical chain length as the native SUMO isopeptide linkage, though it contains an additional carboxylate group from cysteine. (b) Illustration of SUMO(QTC) non-covalently bound to TDG(112–360). The model was generated using AlphaFold 2.2.0.29 | ||
 | ||
| Fig. 4 Western blotting analysis of ClAcK-mediated TDG SUMOylation with anti-His antibody. (a) Comparison of the conjugation yield between SUMO(QTC) and ΔN-SUMO(QTC) to TDG(112–360)-His6 with ClAcK installed at K330. (b) Comparison of the conjugation yield between TDG(112–360)-K330X-His6 and the SIM-defective TDG(112–360)-E310Q-K330X-His6 with ΔN-SUMO(QTC). (c) Conjugation of ΔN-SUMO(QTC) to TDG(112–360)-His6 with ClAcK installed at either the native SUMOylation site (K330) or the different alternate sites. The full gel images can be found in the “full gels and blots” section of the ESI.† | ||
Previous studies have shown that truncating the intrinsically-disordered N-terminus of SUMO (ΔN-SUMO) can improve its SIM-mediated binding affinity to its SUMOylation targets.11,30 To explore whether this tighter SUMO–SIM interaction could be used to enhance the conjugation efficiency, we designed a truncated SUMO1 variant, ΔN-SUMO(QTC), lacking the N-terminal 14 residues of SUMO1 and containing the C-terminal QTGG to QTC mutation and co-expressed it with TDG-K330X-His6 in E. coli. As expected, the conjugation yield increased from 15.7% to 34% (Fig. 4a), supporting the role of the SUMO–SIM interaction in directing the SUMO C-terminal thiol for “proximity-induced” crosslinking with ClAcK. To further validate this role, we introduced a known SIM-disrupting mutation (E310Q) into the TDG-K330X-His6 mutant.31 Co-expression of the SIM-defective mutant with ΔN-SUMO(QTC) resulted in markedly reduced amount of conjugated product (∼1.4%) on western blot (Fig. 4b and Fig. S6, ESI†). This confirms that the SUMO–SIM interaction is directly responsible for guiding the C-terminus of SUMO to K330.
Since the SUMO-conjugation efficiency in our method is heavily influenced by the SUMO–SIM interaction, we wondered if our approach could be extended for use in mapping the SUMOylation landscape. To explore this, we designed TDG mutants with ClAcK incorporated at alternate sites (Fig. 3b), including proximal (K333), intermediate (K184), and distal (K206) positions relative to the native SUMOylation site (K330). Notably, despite successful ClAcK incorporation, none of these mutants formed crosslinks with ΔN-SUMO(QTC) (Fig. 4c and Fig. S7–S9, ESI†). This suggests that our method is highly selective for the native SUMOylation site.
To better understand the differences between the SUMO–TDG conjugate generated by our method and the enzymatically SUMOylated TDG, we treated these conjugates with Ulp1 SUMO protease (Fig. S10, ESI†). While the enzymatically SUMOylated TDG can be completely deSUMOylated by Ulp1 treatment, the ClAcK-mediated conjugate remained intact, thus confirming the resistance of the thioether linkage to SUMO protease cleavage. Furthermore, we investigated the impact of ClAcK-mediated SUMOylation on the substrate binding activity of TDG. Similar to enzymatic SUMOylation,32,33 ClAcK-mediated SUMO conjugation at K330 of TDG resulted in a weakening of substrate binding (Fig. S11, ESI†).
To demonstrate the versatility of our in vivo ClAcK-mediated SUMOylation strategy, we extended its application to other SUMO substrates. Unlike TDG, which is ordered and has a single major SUMOylation site, α-synuclein (αSyn) is intrinsically disordered and possesses multiple native SUMOylation sites.9 K96 and K102 are the dominant sites, accounting for over 50% of SUMO–αSyn conjugates, while at least 9 of its remaining 13 lysines are reported as minor SUMOylation sites.9 It was therefore intriguing to test if our method could successfully conjugate SUMO to the various acceptor sites on αSyn.
Since αSyn is predominantly SUMOylated at K96 and K102, we first introduced ClAcK at these two positions. We co-expressed SUMO(QTC) with either αSyn-K96X or αSyn-K102X in E. coli and confirmed successful conjugation at both sites (Fig. 5 and Fig. S12, ESI†). To assess whether SUMO–SIM interaction likewise drives the conjugation in αSyn, we tested the conjugation efficiency of ΔN-SUMO(QTC), which has higher binding affinity to SIM, by co-expressing it with αSyn-K96X. Consistent with the results for TDG, ΔN-SUMO(QTC) produced a higher αSyn-K96X conjugation yield (∼48.5%) compared to SUMO(QTC) (∼22.5%) (Fig. S13, ESI†).
 | ||
| Fig. 5 ClAcK-mediated SUMOylation of αSyn at its two dominant native SUMOylation sites (K96 or K102). Boiled cell lysates of BL21(DE3) co-expressing SUMO(QTC) and different αSyn variants were analyzed by western blotting using either anti-αSyn or anti-SUMO antibodies. AcK (Nε-acetyl-lysine). Conjugation yield determined by ImageJ: αSyn-K96X, 27.6%; αSyn-K102X, 25%. The full gel images can be found in the “full gels and blots” section of the ESI.† | ||
Encouraged by these results, we expanded our investigation to other sites on αSyn. We selected four alternate sites for ClAcK incorporation (Fig. S14, ESI†): K10, a minor SUMOylation site on the N-terminus of αSyn; V52, a residue on SIM2 of αSyn;11 and A91, and G93, proximal sites with varying distances from the native SUMOylation site (K96). Each of these mutants was co-expressed with ΔN-SUMO(QTC) in E. coli and analysed by western blot using anti-αSyn.
As expected, ΔN-SUMO(QTC) was successfully conjugated to αSyn-K10X. However, contrary to our expectation, all other mutants modified at non-SUMOylation sites also exhibited robust crosslinking (Fig. S15–S17, ESI†). This unexpected promiscuity contradicts our observation with TDG, where SUMO(QTC) reacted solely with ClAcK at native SUMOylation sites. To reconcile these findings, we propose two possibilities: (1) the specificity of SUMO(QTC) for native SUMOylation sites is context dependent. For structurally disordered targets like αSyn, increased accessibility to alternate sites due to its conformational flexibility could lead to promiscuous conjugation; or (2) the observed non-native conjugation sites could represent cryptic SUMOylation targets. Mutation of these sites to lysine might reveal their latent potential for enzymatic SUMOylation.
To probe deeper into the concept of cryptic SUMOylation targets, we focused on site V52, owing to its further distance from the dominant native SUMOylation sites (K96 and K102). We engineered αSyn-V52K-K96R-K102R by replacing V52 with lysine and mutating the two dominant SUMOylation sites to arginine. Purified αSyn-V52K-K96R-K102R was then incubated with a complete enzymatic SUMOylation system (Fig. S18, ESI†). LC-MS/MS confirmed V52K as a genuine SUMOylation target (Fig. S19, ESI†). This suggests that the crosslink between ClAcK and SUMO(QTC) can accurately predict the positions of cryptic enzymatic SUMOylation sites.
To the best of our knowledge, the mutation of V52 to lysine in αSyn is the first reported instance of an engineered, enzymatic SUMOylation site. Interestingly, upon analysis with JASSA,34 the mutated sequence was found to exhibit low inherent potential for SUMOylation (Table S1, ESI†). Considering that both our crosslinking experiment (Fig. S15, ESI†) and molecular dynamic simulation (see ESI†) revealed that SUMO–SIM interaction favours positioning of the SUMO C-terminus near residue 52 of αSyn, we propose that its unexpected SUMOylation is primarily driven by SUMO–SIM interaction, similar to SIM-dependent SUMOylation.28
Conclusions
In summary, we describe a straightforward approach for generating site-specific SUMO-conjugates in cells by leveraging native SUMO–SIM interactions to precisely position SUMO for efficient crosslinking at the native SUMOylation sites. Using this approach, we demonstrate the successful conjugation of SUMO to two distinct proteins, thymine-DNA glycosylase and α-synuclein at their native SUMOylation sites. Furthermore, we have illustrated the ability of this method for mapping the SUMOylation landscape and for unveiling previously unknown cryptic SUMOylation sites.While these current studies are demonstrated in E. coli, our method for site-specific in vivo SUMOylation is likely adaptable to mammalian cells. Efficient reaction between the chloroacetyl handle and targeted cysteine has been demonstrated in the cytosol of human breast cancer cells.19 Additionally, the PylT/MmPylRS system for ClAcK incorporation has also been shown to be orthogonal in mammalian cells.35 We envision that translating our system to mammalian cells could enable in vivo SUMOylation studies with unprecedented spatial resolution.
Author contributions
Conceptualization, M. K. C.; methodology, Y. H. C.; investigation, Y. H. C.; writing – original draft, Y. H. C.; writing – review & editing, M. M. L. and M. K. C.; funding acquisition, M. K. C.; supervision, M. M. L. and M. K. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the CUHK Center of Novel Biomaterials, as well as AoE grant AoE/P-705-16 from the Hong Kong Research Grants Council and grant ITS/230/22 from the Innovation and Technology Commission Fund. We thank Professor Zhongping Yao, Drs Pui-Kin So and Tsz-Fung Wong for their assistance in obtaining mass spectrometry data.References
- R. Geiss-Friedlander and F. Melchior, Concepts in sumoylation: a decade on, Nat. Rev. Mol. Cell Biol., 2007, 8(12), 947–956 CrossRef CAS PubMed.
- A. B. Celen and U. Sahin, Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts, FEBS J., 2020, 287(15), 3110–3140 CrossRef CAS PubMed.
- M. L. Acuña, A. García-Morin, R. Orozco-Sepúlveda, C. Ontiveros, A. Flores, A. V. Diaz, I. Gutiérrez-Zubiate, A. R. Patil, L. A. Alvarado and S. Roy, Alternative splicing of the SUMO1/2/3 transcripts affects cellular SUMOylation and produces functionally distinct SUMO protein isoforms, Sci. Rep., 2023, 13(1), 2309 CrossRef PubMed.
- N. Mandel and N. Agarwal, Role of SUMOylation in neurodegenerative diseases, Cells, 2022, 11(21), 3395 CrossRef CAS PubMed.
- J.-S. Seeler and A. Dejean, SUMO and the robustness of cancer, Nat. Rev. Cancer, 2017, 17(3), 184–197 CrossRef CAS PubMed.
- J. S. Kroonen and A. C. Vertegaal, Targeting SUMO signaling to wrestle cancer, Trends Cancer, 2021, 7(6), 496–510 CrossRef CAS PubMed.
- C. Kho, A. Lee, D. Jeong, J. G. Oh, A. H. Chaanine, E. Kizana, W. J. Park and R. J. Hajjar, SUMO1-dependent modulation of SERCA2a in heart failure, Nature, 2011, 477(7366), 601–605 CrossRef CAS PubMed.
- Z. Cai, Z. Wang, R. Yuan, M. Cui, Y. Lao, Y. Wang, P. Nie, L. Shen, J. Yi and B. He, Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of β-catenin and regulation of its stability, EBioMedicine, 2021, 67, 103386 CrossRef CAS PubMed.
- P. Krumova, E. Meulmeester, M. Garrido, M. Tirard, H.-H. Hsiao, G. Bossis, H. Urlaub, M. Zweckstetter, S. Kügler and F. Melchior, Sumoylation inhibits α-synuclein aggregation and toxicity, J. Cell Biol., 2011, 194(1), 49–60 CrossRef CAS PubMed.
- T. Abeywardana and M. R. Pratt, Extent of inhibition of α-synuclein aggregation in vitro by SUMOylation is conjugation site-and SUMO isoform-selective, Biochemistry, 2015, 54(4), 959–961 CrossRef CAS PubMed.
- Z. Liang, H. Y. E. Chan, M. M. Lee and M. K. Chan, A SUMO1-derived peptide targeting SUMO-interacting motif inhibits α-Synuclein aggregation, Cell Chem. Biol., 2021, 28(2), 180–190.e6 CrossRef CAS PubMed.
- F. Meier, T. Abeywardana, A. Dhall, N. P. Marotta, J. Varkey, R. Langen, C. Chatterjee and M. R. Pratt, Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation, J. Am. Chem. Soc., 2012, 134(12), 5468–5471 CrossRef CAS PubMed.
- S. Eger, M. Scheffner, A. Marx and M. Rubini, Synthesis of defined ubiquitin dimers, J. Am. Chem. Soc., 2010, 132(46), 16337–16339 CrossRef CAS PubMed.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y.-C. Chen, Y. Ge and E. R. Strieter, Forging isopeptide bonds using thiol–ene chemistry: site-specific coupling of ubiquitin molecules for studying the activity of isopeptidases, J. Am. Chem. Soc., 2012, 134(16), 6916–6919 CrossRef CAS PubMed.
- M. Stanley and S. Virdee, Genetically directed production of recombinant, isosteric and nonhydrolysable ubiquitin conjugates, ChemBioChem, 2016, 17(15), 1472–1480 CrossRef CAS PubMed.
- M. Fottner, A.-D. Brunner, V. Bittl, D. Horn-Ghetko, A. Jussupow, V. R. Kaila, A. Bremm and K. Lang, Site-specific ubiquitylation and SUMOylation using genetic-code expansion and sortase, Nat. Chem. Biol., 2019, 15(3), 276–284 CrossRef CAS PubMed.
- J. Wang, Y. Yu and J. Xia, Short peptide tag for covalent protein labeling based on coiled coils, Bioconjugate Chem., 2014, 25(1), 178–187 CrossRef CAS PubMed.
- Y. Lu, F. Huang, J. Wang and J. Xia, Affinity-guided covalent conjugation reactions based on PDZ–peptide and SH3–peptide interactions, Bioconjugate Chem., 2014, 25(5), 989–999 CrossRef CAS PubMed.
- Y. Yu, Y. Nie, Q. Feng, J. Qu, R. Wang, L. Bian and J. Xia, Targeted covalent inhibition of Grb2–Sos1 interaction through proximity-induced conjugation in breast cancer cells, Mol. Pharmaceutics, 2017, 14(5), 1548–1557 CrossRef CAS PubMed.
- R. Fahey, W. Brown, W. Adams and M. Worsham, Occurrence of glutathione in bacteria, J. Bacteriol., 1978, 133(3), 1126–1129 CrossRef CAS PubMed.
- A. Pastore, G. Federici, E. Bertini and F. Piemonte, Analysis of glutathione: implication in redox and detoxification, Clin. Chim. Acta, 2003, 333(1), 19–39 CrossRef CAS PubMed.
- T. Kobayashi, C. Hoppmann, B. Yang and L. Wang, Using protein-confined proximity to determine chemical reactivity, J. Am. Chem. Soc., 2016, 138(45), 14832–14835 CrossRef CAS PubMed.
- N. Shindo, H. Fuchida, M. Sato, K. Watari, T. Shibata, K. Kuwata, C. Miura, K. Okamoto, Y. Hatsuyama, K. Tokunaga, S. Sakamoto, S. Morimoto, Y. Abe, M. Shiroishi, J. M. M. Caaveiro, T. Ueda, T. Tamura, N. Matsunaga, T. Nakao, S. Koyanagi, S. Ohdo, Y. Yamaguchi, I. Hamachi, M. Ono and A. Ojida, Selective and reversible modification of kinase cysteines with chlorofluoroacetamides, Nat. Chem. Biol., 2019, 15(3), 250–258 CrossRef CAS PubMed.
- F. Forouhar, S. Lew, J. Seetharaman, R. Xiao, T. B. Acton, G. T. Montelione and L. Tong, Structures of bacterial biosynthetic arginine decarboxylases, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66(Pt 12), 1562–1566 CrossRef CAS PubMed.
- J. Lascorz, J. Codina-Fabra, D. Reverter and J. Torres-Rosell, In SUMO–SIM interactions: From structure to biological functions, Seminars in Cell & Developmental Biology, Elsevier, 2022, pp. 193–202 Search PubMed.
- T. Yau, W. Sander, C. Eidson and A. Courey, SUMO interacting motifs: structure and function, Cells, 2021, 10, 2825 CrossRef CAS PubMed.
- J. Zhu, S. Zhu, C. M. Guzzo, N. A. Ellis, K. S. Sung, C. Y. Choi and M. J. Matunis, Small Ubiquitin-related Modifier (SUMO) Binding Determines Substrate Recognition and Paralog-selective SUMO Modification, J. Biol. Chem., 2008, 283(43), 29405–29415 CrossRef CAS PubMed.
- D.-Y. Lin, Y.-S. Huang, J.-C. Jeng, H.-Y. Kuo, C.-C. Chang, T.-T. Chao, C.-C. Ho, Y.-C. Chen, T.-P. Lin and H.-I. Fang, Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors, Mol. Cell, 2006, 24(3), 341–354 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- M. Lussier-Price, H. M. Wahba, X. H. Mascle, L. Cappadocia, V. Bourdeau, C. Gagnon, S. Igelmann, K. Sakaguchi, G. Ferbeyre and J. G. Omichinski, Zinc controls PML nuclear body formation through regulation of a paralog specific auto-inhibition in SUMO1, Nucleic Acids Res., 2022, 50(14), 8331–8348 CrossRef CAS PubMed.
- C. Smet-Nocca, J.-M. Wieruszeski, H. Léger, S. Eilebrecht and A. Benecke, SUMO-1 regulates the conformational dynamics of thymine-DNA glycosylase regulatory domain and competes with its DNA binding activity, BMC Biochem., 2011, 12, 1–15 CrossRef PubMed.
- D. Baba, N. Maita, J.-G. Jee, Y. Uchimura, H. Saitoh, K. Sugasawa, F. Hanaoka, H. Tochio, H. Hiroaki and M. Shirakawa, Crystal structure of thymine DNA glycosylase conjugated to SUMO-1, Nature, 2005, 435(7044), 979–982 CrossRef CAS PubMed.
- C. T. Coey, M. E. Fitzgerald, A. Maiti, K. H. Reiter, C. M. Guzzo, M. J. Matunis and A. C. Drohat, E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex, J. Biol. Chem., 2014, 289(22), 15810–15819 CrossRef CAS PubMed.
- G. Beauclair, A. Bridier-Nahmias, J.-F. Zagury, A. Saïb and A. Zamborlini, JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs, Bioinformatics, 2015, 31(21), 3483–3491 CrossRef CAS PubMed.
- P. R. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. H. Geierstanger and P. G. Schultz, A facile system for encoding unnatural amino acids in mammalian cells, Angew. Chem., Int. Ed., 2009, 48(22), 4052–4055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00135d |
| This journal is © The Royal Society of Chemistry 2025 |