
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA point-of-need framework for illicit drug identification with high-resolution mass spectrometry†
Thomas P.
Forbes
 *a,
Elizabeth L.
Robinson
a,
Edward
Sisco
a and
Abigail
Koss
b
*a,
Elizabeth L.
Robinson
a,
Edward
Sisco
a and
Abigail
Koss
b
aNational Institute of Standards and Technology, Materials Measurement Science Division, Gaithersburg, Maryland 20899, USA. E-mail: thomas.forbes@nist.gov
bTOFWERK USA, Boulder, Colorado 80301, USA
First published on 18th March 2025
Abstract
The continually evolving drug landscape, with novel synthetic drugs and unique compositions, necessitates the need to advance technologies, data analysis methods, and data accessibility for compound detection and identification. Providing public health, first responder, and law enforcement communities with critical information in near real-time will aid emergency response and public awareness, and direct overdose prevention and interdiction efforts. A major component of this framework is the progression of accurate drug screening and preliminary identifications from a more rigid laboratory-based arrangement to an agile point-of-need paradigm. We investigated drug detection and identification of a field deployable ruggedized high-resolution time-of-flight mass spectrometer, employing both acetone-assisted vacuum ultraviolet (VUV) photoionization and dielectric barrier discharge ionization (DBDI) schemes. This preliminary fit-for-purpose exploration was conducted under laboratory conditions, considering ion sources not reliant on helium gas or external roughing pumps, building toward deployment in a mobile laboratory setting. The chromatography-free measurements enabled rapid analysis of neat drug solutions and multi-component mixtures. Characterization and optimization of system parameters demonstrated sensitive performance, with limits of detection in the tens to hundreds of picograms for a range of drug classes from multiple-component mixtures. The system's high mass resolution was calibrated with a polyethylene glycol calibrant, enabling accurate matching with spectral library entries. Integrating compound identification with the NIST DART-MS Forensics Database and NIST/NIJ DART-MS Data Interpretation Tool provided a solid foundation for transition to the point-of-need. The overarching framework seeks to support technology advancement and adoption, as well as the development of novel data analysis tools, processes, and management for public access and utilization.
Introduction
The development of measurement science and standards aimed at supporting the public health, first responder, law enforcement, and forensic science communities remains critical to combating the drug overdose epidemic.1 At the foundation of this support is the chemical analysis of samples, leading to accurate identification or classification of illicit drug composition. Advancements in analytical technology, novel data analysis algorithms, and growing chemical libraries and datasets are bolstering a move from laboratory-based analysis to on-site measurement.2,3 This shift enables near real-time feedback – in the form of chemical identification(s) – to first responders or harm reduction personnel, containing crucial information for individual and community safety.On-site detection and identification of illicit drugs is dominated by the use of color tests4 and lateral flow immunoassay (test strips),5 both supported by advancement in paper-based microfluidics;6 as well as spectroscopy-based techniques like Raman and Fourier transform infrared (FTIR) due to their low cost, ease of use, and portability.7 These techniques, however, are non-ideal for obtaining high-quality data on drug samples that are often complex mixtures. Color tests have been shown to frequently produce inconclusive or incorrect results, and color changes of novel psychoactive substances and new synthetic opioids are not well documented.8 Test strips provide information about a specific drug or class of drugs – presenting a narrow piece of information – and are prone to cross-reactivity.9 Spectroscopy techniques often cannot reliably detect minor components in a mixture. These minor components are critical to detect in drug mixtures as they often present the highest danger.
While the limited data that color tests, test strips, and spectroscopy-based techniques provide are useful, they do not allow law enforcement, harm reduction personnel, or people who use drugs with a way to fully understand the dangers present within a sample. Because of this, many agencies rely on sending samples to laboratories for additional, comprehensive analysis using a mass spectrometry-based approach. While this approach provides useful data for monitoring the illicit drug landscape, the time delays caused by shipping the sample and backlogs at laboratories render the information obsolete for the person who initially provided the sample. Over the past few years, efforts to close the time-gap on comprehensive drug product testing have been undertaken using rapid laboratory-based analyses,10 on-site analyses using laboratory-grade mass spectrometers,11 and mobile analyses12 using laboratory-grade mass spectrometers.
The sensitive and selective detection capabilities of mass spectrometry (MS) have made it a premier analytical technique for chemical identification from complex mixtures. Instrumentation advancements and miniaturization have yielded a range of fieldable mass spectrometers13–15 – from high-pressure ion traps16 to time-of-flight (TOF) analyzers.17,18 The evolution of portable mass spectrometers has coincided with the vast expansion in ambient and atmospheric pressure ionization techniques.19–21 The versatility of these ionization sources and simplicity of coupling with a variety of sample introduction avenues has enabled a plethora of adaptable fit-for-purpose front-end configurations22–24 that have been applied to innumerable applications.22,23,25–27
In response to the needs of the public health and forensic communities, and areas highlighted by the U.S. TRANQ Research Act28 and U.S. Office of National Drug Control Policy,29 we investigated the identification of illicit drugs with a rugged high-resolution TOF mass spectrometer. This investigation is an extension of the National Institute of Standards and Technology (NIST) rapid drug analysis and research (RaDAR) program.30,31 The RaDAR program works with local, state, and federal partners to monitor the chemical composition of the drug landscape. Here, characterization of sample introduction, ionization pathways, and MS performance was conducted for a 15-component drug mixture and nine (9) single-component drug solutions. The chromatography-free analysis employed the thermal desorption of extracted samples coupled with either dielectric barrier discharge ionization (DBDI) or acetone-assisted vacuum ultraviolet (VUV) photoionization, sources free from potential needs for helium gas or external pumping. Ionized samples were analyzed by a relatively compact transportable TOF mass spectrometer that has been designed, deployed, and demonstrated sensitive measurements for on-site, mobile, and field applications.18,32,33 Parametric and performance characterization was followed with an analysis of compound identification, exploiting related data tools – specifically, adapting analysis to the NIST DART-MS Forensics Database (an illicit drug centric MS library comprised of spectra collected with direct analysis in real time [DART] ionization) and NIST/NIJ DART-MS data interpretation tool (spectral matching algorithm). These studies lay the foundation toward rapid near real-time drug screening and preliminary identification at the point-of-need to aid in monitoring the drug landscape.
Methods
Materials
A standard mixture solution containing an array of fifteen drugs – each at 250 μg mL−1 in methanol – was purchased from Cayman Chemical (GC-MS Drug Standard Mixture 4, Ann Arbor, MI, USA)‡. The mixture was comprised of cathinones: α-pyrrolidinobutiophenone (α-PBP), α-pyrrolidinopentiophenone (α-PVP), ethylone (3,4-methylenedioxy-N-ethylcathinone or βk-MDEA), and butylone (β-keto-N-methylbenzodioxolylbutanamine or βk-MBDB); arylcyclohexylamines: tenocyclidine (TCP) and phencyclidine (PCP); stimulants: cocaine and methamphetamine; an opiate: heroin; synthetic opioids: furanyl fentanyl and furanyl fentanyl 3-furancarboxamide isomer; benzodiazepine: alprazolam; steroids: nandrolone and stanozolol; and a synthetic cannabinoid: 5-fluoro ADB (5-fluoro MDMB-PINACA). Single-component standards for methamphetamine, α-PBP, tenocyclidine, furanyl fentanyl, heroin, alprazolam, N,N-dimethylpentylone (N,N-DMP), fentanyl, and cocaine were also purchased from Cayman Chemical. Standard solutions were gravimetrically or volumetrically diluted in liquid chromatography (LC)-MS Chromasolv grade acetonitrile (Sigma-Aldrich, St. Louis, MO, USA) for further use. A selection of three (3) samples from the NIST RaDAR program were also analyzed for real-world demonstration. These samples were collected from used drug paraphernalia at harm reduction sites on the U.S. east coast and returned to NIST for extraction and analysis.10 Details of sample collection and preparation can be found in the literature.10 Briefly, meta-aramid wipes or cotton swabs were used to swipe the external surfaces of paraphernalia. Collection media (i.e., wipes and or swabs) were placed into coin envelopes and returned to NIST, where they were extracted in 1 mL acetonitrile and vortexed. Melting point capillary tubes were used as sampling substrates for dissolved drug solutions or solvent-extracted collections.Instrumentation
Analyte-laden sample substrates (e.g., melting point capillaries) were inserted into a GC interface heater (GC/SPME Module, Plasmion GmbH, Augsburg, Germany) operated at 250 °C unless noted (temperatures from 180 °C to 300 °C were investigated). Following thermal desorption, vaporized analytes were ionized by either an in-line dielectric barrier discharge ionization (DBDI) source (SICRIT [soft ionization by chemical reaction in transfer], Plasmion GmbH, Augsburg, Germany)34 or high-pressure acetone-adduct chemical ionization source (Aim reactor, TOFWERK AG, Thun, Switzerland).35 All hardware configurations were mounted on a Vocus S time-of-flight (TOF) mass spectrometer (TOFWERK). This class of instruments has been frequently deployed for mobile applications and come in a range of sizes, mass resolving power, and system requirements.18,32,33 The mass spectrometer was approximately 64 cm deep × 50 cm wide (62 cm with wheels) × 114 cm high, weighing approximately 120 kg and requiring standard 120 V (60 Hz) and 1 kW power. Custom mounting brackets are available for future deployment in a vehicle. The TOF mass spectrometer front-end hardware contained a sampling inlet separated by a pressure-reducing orifice from an ion-molecule reactor chamber. The ion-molecule reactor was followed by a series of RF-only quadrupole ion guides and focusing elements to accomplish differential pumping and interface with the mass analyzer. In the “Aim reactor” configuration, acetone chemical ionization reagent ions were introduced directly into the ion molecule reactor using a krypton vacuum ultraviolet (VUV) lamp (operated at 1600 V unless noted, with voltages from 900 V to 2000 V investigated) and reagent gas from a heated acetone permeation tube. In DBDI configuration, the DBDI source was mounted on the inlet, in front of the ion molecule reactor. The “Aim reactor” hardware (i.e., krypton lamp and permeation tube heater) remained in place for DBDI operation but were powered OFF. Details of the in-line DBDI source can be found in the literature.34,36–38 Briefly, a concentric electrode geometry generated a stable plasma for ionization between a thermal desorption unit and the MS inlet. Unless noted, the DBDI source was operated at 1400 V and 15 kHz, with the voltage range 1100 V to 1600 V investigated. Following thermal desorption and ionization, ions were focused and guided into an orthogonal reflectron TOF, impacting a multichannel plate detector.Safety considerations
Safety data sheet recommendations and standard practices were followed for storage and handling of hazardous materials (e.g., drug compounds). In this work, hazardous samples were already dissolved in solution. A portable fume extractor (containing carbon and HEPA filters) was used around the thermal desorber, and mass spectrometer pumps were appropriately vented through the building ventilation.Results and discussion
We explored the coupling of multiple sample introduction and ionization methods with a rugged TOF mass spectrometer to define critical parameters for on-site drug detection and identification. This investigation builds upon existing work of the NIST RaDAR program30,31 (Fig. 1), for which samples collected by local, state, or federal partners are shipped back to NIST for analysis. Here, we explore instrumentation for moving toward point-of-need drug identification, drug landscape monitoring, and associated public health applications (Fig. 1). For example, sample chemical composition could be provided at harm reduction sites or other points-of-need in near real-time, mapping the drug landscape. Bringing the laboratory to the point-of-need will remove the need for sample packaging and shipping (Step 2), a main contributor to the lengthy turn-around time. Similarly, ongoing and future avenues are considering removal of the solvent extraction step (Step 3) and directly analyzing wipes or paraphernalia-dipped glass capillaries. Here, we used a cause-and-effect analysis to aid in identifying critical process parameters (CPPs) impacting both measurement variability and compound identification confidence. An Ishikawa diagram laid out the major processes in compound identification – from sample collection to library matching – and established the main factors (and their relationships) impacting compound detection and identification (Fig. S1†). Critical process parameters for detailed investigation were chosen from this analysis and prior experience. | ||
| Fig. 1 Schematic representation of the NIST RaDAR workflow and overall reduction in process time by on-site analysis. Step 1: swipe sample collection from paraphernalia, 2: packaging of sample-laden wipes and shipping to NIST, 3: solvent extraction of wipes, 4: thermal desorption and ionization of samples, 5. TOF-MS analysis, and 6. Library matching and preliminary identification. | ||
Mass spectrometry detection CPPs
The process parameters investigated here were related to sample introduction, entrainment, ionization, transmission, and detection. The initial processes in mass spectrometric compound detection and identification revolve around sample introduction to the gas phase and analyte ionization (Fig. S1† – first two branches of the cause-and-effect diagram). | ||
| Fig. 2 Schematic representations of capillary-based thermal desorption (250 °C) coupled with (a) acetone-VUV ionization [ion-molecule reactor temp: 50 °C, perm tube flow: 150 cm3 min−1] or (b) DBDI ionization [15 kHz]. (c) and (d) Peak areas of select drugs from the 15-component mixture at 5 ng μL−1 (each compound) concentration as a function of (i) inlet pump flow (in percent speed [rpm] of pump) and (ii) ionization scheme (VUV or DBDI) voltage. Data points and uncertainty represented by average and standard deviation of 7–10 replicate measurements (schematics adapted from TOFWERK).35 | ||
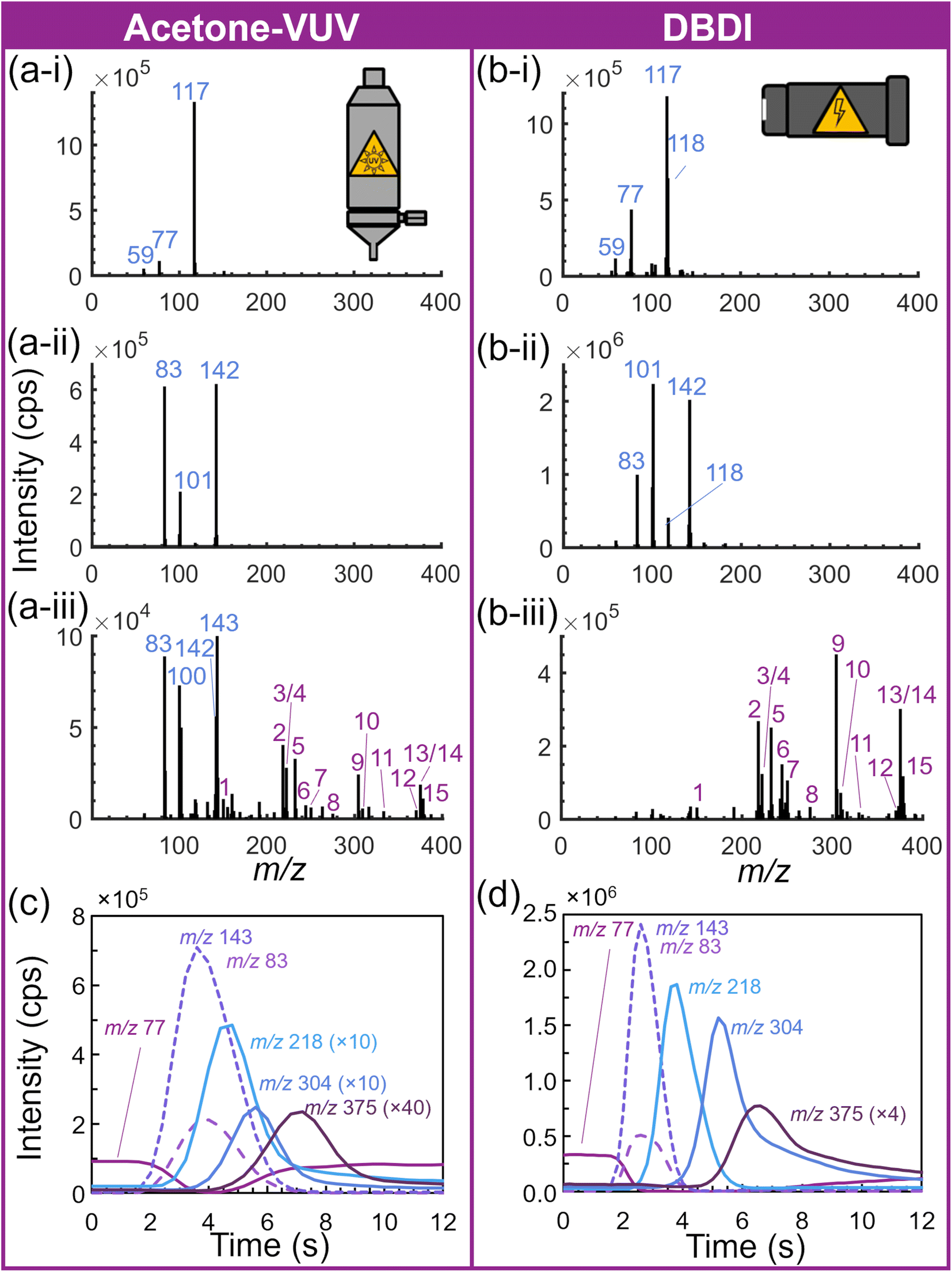 | ||
| Fig. 3 Mass spectra and extracted ion chronograms for (a) acetone-assisted VUV photoionization and (b) DBDI ionization configurations. Spectra display (i) background (ii) blank acetonitrile, and (iii) 15-component drug mixture [peak numbers 1–15 identified in Table 1] ion distributions. Blank acetonitrile spectra were background subtracted and the drug mixture spectra were blank acetonitrile subtracted. Extracted ion chronograms for the acetone water cluster (m/z 77), acetonitrile dimer (m/z 83) and cluster (m/z 143), α-PBP (m/z 218), cocaine (m/z 304), and furanyl fentanyl (m/z 375) from (c) acetone-VUV and (d) DBDI. | ||
During DBDI-based experiments, the acetone permeation tube heater and carrier gas flow were off (Fig. 2(b)), however, acetone related ions (e.g., protonated monomer, water adduct, and protonated dimer) were still observed (Fig. 3(b-i)). This was attributed to the volatility of acetone, its vapor pressure of ≈25 kPa at room temperature, and the vacuum suction from the reduced pressure in the ion-molecule reactor. To completely remove the acetone-related species from the system, the permeation tube and VUV lamp should be removed from the instrument and the lamp location capped. DBDI-generated hydronium clusters and excited gas species led to protonation of the analytes directly within the source (Fig. 2(b)), as well as charge exchange or proton transfer with downstream acetone vapor in the reaction region (detailed in ESI, eqn (S6)–(S13)†).37,38
The overall signal of the protonated acetone dimer base peak yielded similar intensity (i.e., counts per second, cps) across ionization schemes (Fig. 3(a-i) and (b-i)). However, using the differing inlet pump settings for drug samples of each configuration (Fig. 2) resulted in a total ion count (TIC) for the DBDI source approximately 20 to 50 times higher. This was attributed to the additional ambient air drawn into the thermal desorber, ion source, and mass spectrometer. The increased pull of ambient laboratory air also provided additional water molecules for adducting with the acetone and acetonitrile extraction solvent. Fig. 3(a-ii) and (b-ii) display the spectra of blank acetonitrile samples, exhibiting an acetonitrile dimer (m/z 83 [(C2H3N)2 + H]+), dimer water cluster (m/z 101 [(C2H3N)2 + H2O + H]+), and trimer water cluster (m/z 142 [(C2H3N)3 + H2O + H]+). The DBDI source yielded elevated signal of the acetonitrile species and an increase in the water clusters. The increased sampling of laboratory air was also corroborated by the observation of elevated m/z 118 in the background spectrum (Fig. 3(b-i) and (b-ii)). This ion was previously identified as diethylethanolamine ([C6H15NO + H]+), an HVAC corrosion inhibitor observed in the background of this specific laboratory of the Advanced Measurement Laboratory on the NIST Gaithersburg campus.
Both sources maintained similar analyte ionization, yielding protonated molecules of all compounds in the 15-drug mixture, with some minor solvent-based or dopant-based adduct formation. Fig. 3(a-iii) and (b-iii) display the background subtracted spectra of the 15-component mixture. Peak identifications were numbered as listed in Table 1. The chromatography-free nature of this platform prohibited the differentiation of the two sets of isomers in the mixture. In addition, slight differences in the temporal separation of species between the two ion source configurations were observed (Fig. 3(c) and (d)). For example, the acetone-VUV configuration yielded more significant overlap (in time) between the solvent peaks associated with the acetonitrile solvent and the ‘early’ drug mixture components (Fig. 3(c)). However, the solvent peaks were readily separable from the drug mixture when employing the DBDI configuration (Fig. 3(b-iii) and (d)). The combination of additional competitive ionization with the extraction (or dissolution) solvent (e.g., acetonitrile) and indications of poorer ionization efficiency, led to lower overall signal for the drug mixtures (i.e., fewer counts). The components of the drug mixture exhibited from 2× to 24× more counts from the DBDI source, with an overall average of approximately 10× more counts. Interestingly, with the differences in background, overall limits of detection were still similar.
| # | Compound | Theoretical [M + H]+m/z | Mixture ratio | Acetone-VUV LOD90 [95% upper CI] (ng μL−1) | DBDI LOD90 [95% upper CI] (ng μL−1) |
|---|---|---|---|---|---|
| a Compounds are isomers and undifferentiable in current implementation. b Stanozolol with acetone-VUV exhibited improved sensitivity for an acetone adduct, m/z 387.3012 [M + C3H7O]+. | |||||
| 1 | Methamphetamine | 150.1283 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 :15 |
0.08 [0.20] | 0.09 [0.26] |
| 2 | α-PBP | 218.1545 | 1:15 |
0.10 [0.34] | 0.16 [0.27] |
| 3 | Ethylonea | 222.1130 | 2:15 |
0.04 [0.10] | 0.07 [0.16] |
| 4 | Butylonea | ||||
| 5 | α-PVP | 232.1701 | 1:15 |
0.07 [0.20] | 0.06 [0.16] |
| 6 | PCP | 244.2065 | 1:15 |
0.08 [0.21] | 0.20 [0.36] |
| 7 | Tenocyclidine | 250.1629 | 1:15 |
0.48 [1.15] | 0.21 [0.38] |
| 8 | Nandrolone | 275.2011 | 1:15 |
0.45 [0.76] | 0.29 [0.57] |
| 9 | Cocaine | 304.1549 | 1:15 |
0.08 [0.21] | 0.23 [0.41] |
| 10 | Alprazolam | 309.0907 | 1:15 |
0.06 [0.13] | 0.07 [0.19] |
| 11 | Stanozolol | 329.2593 | 1:15 |
0.08 [0.19]b | 0.45 [0.75] |
| 12 | Heroin | 370.1654 | 1:15 |
0.24 [0.44] | 0.80 [1.49] |
| 13 | Furanyl fentanyl a | 375.2073 | 2:15 |
0.05 [0.11] | 0.04 [0.08] |
| 14 | Furanyl fentanyl 3-furancarboxamide isomera | ||||
| 15 | 5-Fluoro ADB | 378.2193 | 1:15 |
0.05 [0.11] | 0.07 [0.16] |
:15 mixture ratio relative to the 1:15 ratio the remaining compounds experienced.
The drug components of the mixture generally exhibited similar sensitivities across ionization schemes, demonstrating the dominance of the mass analyzer performance in overall sensitivity. Sensitivities generally fell in the tens to hundreds of picograms per microliter. Sample withdrawal by melting point capillary dipping was estimated on the order of a single microliter, yielding overall mass sensitivities of tens to hundreds of picograms. The detection of minor components in complex mixtures is critical for the analysis of drug samples and paraphernalia residues. Most compounds (from both ionization schemes) yielded linear response curves across the concentration range ([0, 0.1, 0.5, 1, 5, 10] ng μL−1) investigated for sensitivity calculations (Fig. S7 and S8†). Indications of competitive ionization – based on non-linearity in the response curve at higher concentrations – were observed for methamphetamine and the combined ethylone/butylone components with the DBDI configuration (Fig. S8†). Both signal suppression and signal enhancement matrix effects were observed from the analysis of mixtures. For example, with the DBDI configuration, methamphetamine exhibited signal suppression, while the cocaine peak areas were enhanced from analysis within the 15-component mixture (Fig. S9†). Matrix effects are a universal consideration for chromatography-free mass spectrometry and generally compound specific, based on properties such as proton affinity and ionization energy.51,52
Compound identification CPPs
To this point, our investigations largely considered process parameters impacting MS detection. We next focused on parameters and methods impacting compound identification. Our cause-and-effect analysis generally associated these factors with mass analysis, mass calibration, and library matching (Fig. S1† – final three branches). The high mass resolution afforded by the time-of-flight mass analyzer played a significant role in compound identification through library matching algorithms. | ||
| Fig. 4 Single-spectrum matching (i.e., low fragmentation) of (a) fentanyl [DBDI parameters: TD: 250 °C, DBDI voltage: 1400 V, DBDI frequency: 15 kHz, inlet pump: 25%] and (b) heroin [acetone-assisted VUV photoionization parameters: ion-molecule reactor temp: 50 °C, perm tube flow: 150 cm3 min−1, VUV voltage: 1600 V, inlet pump: 5%] using the DIT with NIST DART-MS Forensics Database library. DIT search results and match factors displayed in insets. | ||
| Compound | Theoretical [M + H]+ (m/z) | Acetone-VUV | DBDI | ||||
|---|---|---|---|---|---|---|---|
| Observed [M + H]+ | Δm/z (Da) | Match (RevMF) | Observed [M + H]+ | Δm/z (Da) | Match (RevMF) | ||
| a Tolerance window was opened to ±0.008 Da. | |||||||
| α-PBP | 218.1545 | 218.1518 | −0.0027 | 0.973 | 218.1560 | 0.0015 | 0.973 |
| Alprazolam | 309.0907 | 309.0876 | −0.0031 | 0.993 | 309.0884 | −0.0023 | 0.995 |
| Cocaine | 304.1549 | 304.1594 | 0.0045 | 0.985 | 304.1501 | −0.0048 | 0.995 |
| Fentanyl | 337.2280 | 337.2262 | −0.0018 | 0.993 | 337.2250 | −0.0030 | 0.992 |
| Furanyl fentanyl | 375.2073 | 375.2023 | −0.0050 | 0.981 | 375.2008 | −0.0065a | 0.985 |
| Heroin | 370.1654 | 370.1610 | −0.0044 | 0.332 | 370.1577 | −0.0077a | 0.325 |
| Methamphetamine | 150.1283 | 150.1275 | −0.0008 | 0.930 | 150.1282 | −0.0001 | 0.927 |
| N,N-Dimethylpentylone | 250.1443 | 250.1435 | −0.0008 | 0.961 | 250.1422 | −0.0021 | 0.971 |
| Tenocyclidine | 250.1629 | 250.1636 | 0.0007 | 0.540 | 250.1651 | 0.0022 | 0.442 |
Finally, a set of three samples from the NIST RaDAR program were analyzed and searched against the DIT and associated library. These samples were collected in March 2024 at various harm reduction sites and extracted upon receipt at NIST. Two of the samples (referred to as RaDAR samples #1 and #2) yielded high intensity signals and strong match scores (e.g., RevMF) for methamphetamine and cocaine, respectively (Fig. S12†). These findings were confirmed against previous analyses using DART-MS as outlined in the literature.10Fig. 5 displays the mass spectrum and identified compounds for the final RaDAR sample (#3), which exhibited several target peak matches. The mixture included fentanyl, fluorofentanyl, and the precursor 4-anilino-N-phenethylpiperidine (4-ANPP), all confirmed by previous analyses. The common diluent mannitol was also observed. Similar to the previous analysis, the poorer reverse match factor for mannitol (i.e., 0.533) was attributed to the numerous additional fragment peaks in the mannitol library spectrum (Fig. S13†).
 | ||
| Fig. 5 Low fragmentation DBDI-MS mass spectra of RaDAR sample #3 with matched compounds labeled. Insets display search results and match scores from the DIT. Parameters: TD: 250 °C, DBDI voltage: 1400 V, DBDI frequency: 15 kHz, inlet pump: 25%. | ||
The high-resolution time-of-flight mass analyzer used here enabled effective compound identification, predominately targeting the protonated molecule library matching of individual spectra. As interferences increase and the analysis of mixtures becomes more complex, this may be insufficient. The DIT and NIST Forensics MS library were created for matching a series of spectra at increasing fragmentation energies – specifically for systems without tandem MS capabilities. In these cases, isCID can be applied at a target location in the differentially pumped region, increasing the frequency and energy of collisions between incoming ions and remaining gas molecules. We conducted a preliminary investigation of isCID with the present system (Fig. S14†). The quadrupole ion guide configuration allowed for increasing voltages between the skimmer after the solid rod quadrupole and the second quadrupole (Fig. S15(a)†). Ions were driven by the voltage drop in this region. Low levels of fragmentation were achieved, sufficient to completely fragment the acetone dimer (or similar dopant adducts),56 but only partially fragment larger molecules such as cocaine (Fig. S14†). This level of fragmentation was insufficient to match the corresponding spectra of the DIT. To take full advantage of the DIT and existing libraries, an alternative quadrupole configuration was considered (Fig. S15(b)†). The solid rod quadrupole 1 was replaced with a short segmented quadrupole (SSQ) allowing a DC gradient to be generated and enabling a DC-only field to be applied across the skimmer-second quadrupole isCID region. The updated quadrupole configuration enabled sufficient isCID to fragment intact drugs (e.g., cocaine and fentanyl) to the extent observed in the library (Fig. S16†). Future work is aimed at optimizing the voltages necessary to match the NIST Forensics MS Library fragmentation levels more accurately.
Conclusions
In this work, we initiated development of a framework that will encompass technology characterization, advancement, and deployment pipelines, specifically targeting preliminary drug identification at the point-of-need for public health and forensic applications. Here, we focused on the analytical investigation of multiple ionization sources coupled with a rugged and transportable time-of-flight mass spectrometer. A range of parameters impacting MS detection were characterized, ultimately yielding sensitive detection (tens to hundreds of picograms) of numerous drug compounds from multicomponent mixtures (at 1:15 to 2:15 mass ratios). Mass calibration and library matching avenues were also examined, demonstrating compatibility with existing forensics-focused libraries (i.e., NIST DART-MS forensics database) and search algorithms (i.e., NIST/NIJ DART-MS data interpretation tool). The foundation presented here seeks to help build a framework to lower implementation barriers for new technologies and support community adoption.
Ongoing work is expanding the transportable TOF avenue into alternative ambient ionization sources such as DART and ASAP (atmospheric solids analysis probe). This initial work was focused on sources not requiring helium and operational without the need for an external rough pump (though modest signal improvement could be achieved with additional flow). However, DART configurations employing nitrogen gas (instead of helium) or using pulsed helium gas have been introduced. The need (or not) for additional gas tanks must be considered in mobile and fieldable applications. In addition, potential atmospheric conditions (e.g. temperature, humidity, wind currents) must be accounted for in sample introduction of ambient ion sources (e.g., DART). Ongoing development of a mobile laboratory related to this work will include a temperature-controlled work environment for sample analysis on-site. Investigation of alternative mass analyzers (e.g., single quadrupole and triple quadrupole) and fieldable analytical techniques (e.g., Fourier transform infrared spectroscopy and novel absorption spectroscopy instruments) is also underway for applicability to point-of-need drug analysis. Future work might also consider a thermal desorption ramp as a possible means of temporally differentiating isobars. In parallel with fit-for-purpose analytical investigations, rigorous workflow validation, implementation, and documentation are in progress for deployment within a mobile laboratory setting.
Associated content
Data availability
Raw data files, derived data files, extracted peak areas, and mass spectra are available on the NIST Public Data Repository: https://doi.org/10.18434/mds2-3591.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
AK was employed by TOFWERK, the manufacturer of the Vocus S mass spectrometer used here, during the completion of this study. Official contribution of the National Institute of Standards and Technology; not subject to copyright in the United States.References
- E. Sisco, Drug Detection, Analysis, and Monitoring Workshop Report, Special Publication (NIST SP), National Institute of Standards and Technology, Gaithersburg, MD, 2024 Search PubMed.
- B. W. Kammrath, P. E. Leary and J. A. Reffner, Forensic Applications of Portable Spectrometers, in Portable Spectroscopy and Spectrometry, 2021, pp. 125–147 Search PubMed.
- T. P. Forbes and R. Burks, Field-Deployable Devices, in Encyclopedia of Forensic Sciences, Third Edition, ed. M. M. Houck, Elsevier, 3rd edn, 2023, pp. 413–423 Search PubMed.
- M. Philp and S. Fu, A review of chemical ‘spot’ tests: A presumptive illicit drug identification technique, Drug Test. Anal., 2018, 10, 95–108 CrossRef CAS PubMed.
- E. Sisco, D. F. Nestadt, M. B. Bloom, K. E. Schneider, R. A. Elkasabany, S. Rouhani and S. G. Sherman, Understanding sensitivity and cross-reactivity of xylazine lateral flow immunoassay test strips for drug checking applications, Drug Test. Anal., 2024, 16, 942–947 CrossRef CAS PubMed.
- G. Musile, Y. Agard, L. Wang, E. F. De Palo, B. McCord and F. Tagliaro, Paper-based microfluidic devices: On-site tools for crime scene investigation, TrAC, Trends Anal. Chem., 2021, 143, 116406 CrossRef CAS.
- M. Hargreaves, Handheld Raman, SERS, and SORS, in Portable Spectroscopy and Spectrometry, 2021, pp. 347–376 Search PubMed.
- E. Sisco, A. Burns, E. Schneider, C. R. Miller IV and L. Bobka, Comparing two seized drug workflows for the analysis of synthetic cannabinoids, cathinones, and opioids, J. Forensic Sci., 2022, 67, 471–482 CrossRef CAS PubMed.
- S. E. Rodriguez-Cruz, Evaluating the sensitivity, stability, and cross-reactivity of commercial fentanyl immunoassay test strips, J. Forensic Sci., 2023, 68, 1555–1569 CrossRef CAS PubMed.
- M. G. Appley, E. L. Robinson, A. Thomson, E. Russell and E. Sisco, An analytical platform for near real-time drug landscape monitoring using paraphernalia residues, Forensic Chem., 2023, 34, 100489 CrossRef CAS.
- S. A. Borden, A. Saatchi, G. W. Vandergrift, J. Palaty, M. Lysyshyn and C. G. Gill, A new quantitative drug checking technology for harm reduction: Pilot study in Vancouver, Canada using paper spray mass spectrometry, Drug Alcohol Rev., 2022, 41, 410–418 CrossRef PubMed.
- H. West, J. Fitzgerald, K. Hopkins, E. Li, N. Clark, S. Tzanetis, S. L. Greene and G. E. Reid, Early Warning System for Illicit Drug Use at Large Public Events: Trace Residue Analysis of Discarded Drug Packaging Samples, J. Am. Soc. Mass Spectrom., 2021, 32, 2604–2614 CrossRef CAS PubMed.
- D. T. Snyder, C. J. Pulliam, Z. Ouyang and R. G. Cooks, Miniature and Fieldable Mass Spectrometers: Recent Advances, Anal. Chem., 2016, 88, 2–29 CrossRef CAS PubMed.
- H. M. Brown, T. J. McDaniel, P. W. Fedick and C. C. Mulligan, The current role of mass spectrometry in forensics and future prospects, Anal. Met., 2020, 12, 3974–3997 RSC.
- K. Evans-Nguyen, A. R. Stelmack, P. C. Clowser, J. M. Holtz and C. C. Mulligan, Fieldable Mass Spectrometry for Forensic Science, Homeland Security, and Defense Applications, Mass Spectrom. Rev., 2021, 40, 628–646 CrossRef PubMed.
- K. H. Blakeman and S. E. Miller, Development of High-Pressure Mass Spectrometry for Handheld and Benchtop Analyzers, in Portable Spectroscopy and Spectrometry, 2021, pp. 391–413 Search PubMed.
- C. J. Brais, J. O. Ibañez, A. J. Schwartz and S. J. Ray, Recent Advances in Instrumental Approaches to Time-of-Flight Mass Spectrometry, Mass Spectrom. Rev., 2021, 40, 647–669 Search PubMed.
- T. I. Yacovitch, B. M. Lerner, M. R. Canagaratna, C. Daube, R. M. Healy, J. M. Wang, E. C. Fortner, F. Majluf, M. S. Claflin, J. R. Roscioli, E. M. Lunny and S. C. Herndon, Mobile Laboratory Investigations of Industrial Point Source Emissions during the MOOSE Field Campaign, Atmosphere, 2023, 14, 1632 CrossRef CAS.
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernandez, Mass Spectrometry: Recent Advances in Direct Open Air Surface Sampling/Ionization, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- S. Rankin-Turner, P. Sears and L. M. Heaney, Applications of ambient ionization mass spectrometry in 2022: An annual review, Anal. Sci. Adv., 2023, 4, 133–153 CrossRef PubMed.
- C. L. Feider, A. Krieger, R. J. DeHoog and L. S. Eberlin, Ambient Ionization Mass Spectrometry: Recent Developments and Applications, Anal. Chem., 2019, 91, 4266–4290 CrossRef CAS PubMed.
- Z. E. Lawton, A. Traub, W. L. Fatigante, J. Mancias, A. E. O'Leary, S. E. Hall, J. R. Wieland, H. Oberacher, M. C. Gizzi and C. C. Mulligan, Analytical Validation of a Portable Mass Spectrometer Featuring Interchangeable, Ambient Ionization Sources for High Throughput Forensic Evidence Screening, J. Am. Soc. Mass Spectrom., 2017, 28, 1048–1059 CrossRef CAS PubMed.
- S. Mathias, M. Amerio-Cox, T. Jackson, D. Douce, A. Sage, P. Luke, R. Sleeman, C. Crean and P. Sears, Selectivity of Explosives Analysis with Ambient Ionization Single Quadrupole Mass Spectrometry: Implications for Trace Detection, J. Am. Soc. Mass Spectrom., 2024, 35, 50–61 CrossRef CAS PubMed.
- S. Mathias, M. Amerio-Cox, T. Jackson, D. Douce, B. McCullough, A. Sage, P. Luke, C. Crean and P. Sears, Performance Comparison of Ambient Ionization Techniques Using a Single Quadrupole Mass Spectrometer for the Analysis of Amino Acids, Drugs, and Explosives, J. Am. Soc. Mass Spectrom., 2024, 35, 2480–2489 CrossRef CAS PubMed.
- E. Sisco and T. P. Forbes, Forensic applications of DART-MS: A review of recent literature, Forensic Chem., 2021, 22, 100294 CrossRef CAS PubMed.
- M. d. M. Boronat Ena, D. A. Cowan and V. Abbate, Ambient ionization mass spectrometry applied to new psychoactive substance analysis, Mass Spectrom. Rev., 2023, 42, 3–34 CrossRef CAS PubMed.
- T. P. Forbes and E. Sisco, Recent advances in ambient mass spectrometry of trace explosives, Analyst, 2018, 143, 1948–1969 RSC.
- H.R.1734 - TRANQ Research Act of 2023, Public Law 118-23, (12/19/2023), 118th Congress, U.S. Government Publishing Office, https://www.congress.gov/bill/118th-congress/house-bill/1734/text.
- Biden-Harris Administration Designates Fentanyl Combined with Xylazine as an Emerging Threat to the United States. Office of National Drug Control Policy, U.S. White House Press Release, 2023. https://www.whitehouse.gov/ondcp/briefing-room/2023/04/12/biden-harris-administration-designates-fentanyl-combined-with-xylazine-as-an-emerging-threat-to-the-united-states/(accessed 2024, Sept. 4).
- E. Russell, E. Sisco, A. Thomson, J. Lopes, M. Rybak, M. Burnett, D. Heilman, M. G. Appley and R. M. Gladden, Rapid Analysis of Drugs: A Pilot Surveillance System To Detect Changes in the Illicit Drug Supply To Guide Timely Harm Reduction Responses—Eight Syringe Services Programs, Maryland, November 2021−August 2022, Morb. Mortal. Wkly. Rep., 2023, 72, 458–462 CrossRef PubMed.
- Rapid Drug Analysis and Research (RaDAR): Providing Near Real-Time Insight into the Illicit Drug Landscape. National Institute of Standards and Technology, https://www.nist.gov/programs-projects/rapid-drug-analysis-and-research-radar-providing-near-real-time-insight-illicit (accessed 2025, March 11).
- G. I. Gkatzelis, M. M. Coggon, B. C. McDonald, J. Peischl, J. B. Gilman, K. C. Aikin, M. A. Robinson, F. Canonaco, A. S. H. Prevot, M. Trainer and C. Warneke, Observations Confirm that Volatile Chemical Products Are a Major Source of Petrochemical Emissions in U.S. Cities, Environ. Sci. Technol., 2021, 55, 4332–4343 CrossRef CAS PubMed.
- D. Fillion, S. Perrier, M. Riva, C. George, F. Domine and R.-M. Couture, Emission of Volatile Organic Compounds to the Atmosphere from Photochemistry in Thermokarst Ponds in Subarctic Canada, ACS Earth Space Chem., 2024, 8, 563–574 CrossRef CAS.
- M. M. Nudnova, L. Zhu and R. Zenobi, Active capillary plasma source for ambient mass spectrometry, Rapid Commun. Mass Spectrom., 2012, 26, 1447–1452 CrossRef CAS PubMed.
- M. Riva, V. Pospisilova, C. Frege, S. Perrier, P. Bansal, S. Jorga, P. Sturm, J. Thornton, U. Rohner and F. Lopez-Hilfiker, Evaluation of a reduced pressure chemical ion reactor utilizing adduct ionization for the detection of gaseous organic and inorganic species, Atmos. Meas. Tech., 2024, 17, 5887–5901 CrossRef CAS.
- L. Gyr, F. D. Klute, J. Franzke and R. Zenobi, Characterization of a Nitrogen-Based Dielectric Barrier Discharge Ionization Source for Mass Spectrometry Reveals Factors Important for Soft Ionization, Anal. Chem., 2019, 91, 6865–6871 CrossRef CAS PubMed.
- M. Weber, J.-C. Wolf and C. Haisch, Effect of Dopants and Gas-Phase Composition on Ionization Behavior and Efficiency in Dielectric Barrier Discharge Ionization, J. Am. Soc. Mass Spectrom., 2023, 34, 538–549 CrossRef CAS PubMed.
- J.-C. Wolf, L. Gyr, M. F. Mirabelli, M. Schaer, P. Siegenthaler and R. Zenobi, A Radical-Mediated Pathway for the Formation of [M + H]+ in Dielectric Barrier Discharge Ionization, J. Am. Soc. Mass Spectrom., 2016, 27, 1468–1475 CrossRef CAS PubMed.
- J. L. Staymates, M. E. Staymates and J. Lawrence, The effect of reusing wipes for particle collection, Int. J. Ion Mobil. Spectrom., 2015, 19, 41–49 CrossRef PubMed.
- J. R. Verkouteren, J. L. Coleman, R. A. Fletcher, W. J. Smith, G. A. Klouda and G. Gillen, A method to determine collection efficiency of particles by swipe sampling, Meas. Sci. Technol., 2008, 19, 115101 CrossRef.
- D. Fisher, R. Zach, Y. Matana, P. Elia, S. Shustack, Y. Sharon and Y. Zeiri, Bomb swab: Can trace explosive particle sampling and detection be improved?, Talanta, 2017, 174, 92–99 CAS.
- T. P. Forbes, E. L. Robinson, E. Sisco and A. Koss, In-Line Thermal Desorption and Dielectric Barrier Discharge Ionization for Rapid Mass Spectrometry Detection of Explosives, Anal. Chem., 2024, 96, 13352–13357 CAS.
- E. Sisco and T. P. Forbes, Rapid detection of sugar alcohol precursors and corresponding nitrate ester explosives using direct analysis in real time mass spectrometry, Analyst, 2015, 140, 2785–2796 CAS.
- E. Sisco and T. P. Forbes, Direct analysis in real time mass spectrometry of potential by-products from homemade nitrate ester explosive synthesis, Talanta, 2016, 150, 177–183 CAS.
- E. Sisco, M. E. Staymates and T. P. Forbes, Optimization of confined direct analysis in real time mass spectrometry (DART-MS), Analyst, 2020, 145, 2743–2750 CAS.
- T. P. Forbes and S. T. Krauss, Confined DART-MS for Rapid Chemical Analysis of Electronic Cigarette Aerosols and Spiked Drugs, J. Am. Soc. Mass Spectrom., 2021, 32, 2274–2280 CAS.
- E. Sisco, T. P. Forbes, M. E. Staymates and G. Gillen, Rapid analysis of trace drugs and metabolites using a thermal desorption DART-MS configuration, Anal. Methods, 2016, 8, 6494–6499 RSC.
- S. Wang, W. Wang, H. Li, Y. Xing, K. Hou and H. Li, Rapid On-Site Detection of Illegal Drugs in Complex Matrix by Thermal Desorption Acetone-Assisted Photoionization Miniature Ion Trap Mass Spectrometer, Anal. Chem., 2019, 91, 3845–3851 CrossRef CAS PubMed.
- E. Sisco and A. S. Moorthy, NIST DART-MS Forensics Database (is-CID), National Institute of Standards and Technology, 2020, DOI:10.18434/mds2-2313.
- ASTM E2677 Limit of Detection Web Portal, 2014. https://www-s.nist.gov/loda/, (accessed 20162016).
- E. H. Denis, J. L. Bade, R. S. Renslow, K. A. Morrison, M. K. Nims, N. Govind and R. G. Ewing, Proton Affinity Values of Fentanyl and Fentanyl Analogues Pertinent to Ambient Ionization and Detection, J. Am. Soc. Mass Spectrom., 2022, 33, 482–490 CrossRef CAS PubMed.
- J. T. Shelley and G. M. Hieftje, Ionization matrix effects in plasma-based ambient mass spectrometry sources, J. Anal. At. Spectrom., 2010, 25, 345–350 RSC.
- A. S. Moorthy, S. S. Tennyson and E. Sisco, Updates to the Inverted Library Search Algorithm for Mixture Analysis, J. Am. Soc. Mass Spectrom., 2022, 33, 1260–1266 CrossRef CAS PubMed.
- E. Sisco, A. S. Moorthy, S. S. Tennyson and R. Corzo, NIST/NIJ DART-MS Data Interpretation Tool, National Institute of Standards and Technology, 2021, DOI:10.18434/mds2-2448.
- A. N. Couch, J. Sharp and J. T. Davidson, Assessing the effectiveness of the NIST DART-MS Forensics Database and Data Interpretation Tool for designer drug screening with alternative instrumentation, Int. J. Mass Spectrom., 2023, 483, 116964 CrossRef CAS.
- F. D. Lopez-Hilfiker, S. Iyer, C. Mohr, B. H. Lee, E. L. D'Ambro, T. Kurtén and J. A. Thornton, Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 2016, 9, 1505–1512 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental method details, ionization pathways, response curves, mass spectra, and figures as noted in the text. See DOI: https://doi.org/10.1039/d5an00082c |
| ‡ Certain commercial equipment, instruments, or materials are identified in this article in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose. |
| This journal is © The Royal Society of Chemistry 2025 |