
Stationary phase effects in hydrophilic interaction liquid chromatographic separation of oligonucleotides†
Scott
Abernathy
 ,
Asif
Rayhan
and
Patrick A.
Limbach
*
,
Asif
Rayhan
and
Patrick A.
Limbach
*
Rieveschl Laboratories for Mass Spectrometry, Department of Chemistry, University of Cincinnati, PO Box 210172, Cincinnati, Ohio 45221-0172, USA. E-mail: Pat.Limbach@uc.edu; Fax: +1 (513) 556-9239; Tel: +1 (513) 558-0026
First published on 25th November 2024
Abstract
The use of liquid chromatography coupled with mass spectrometry (LC-MS) for the characterization of oligonucleotides and nucleic acids is a powerful analytical method. Recently, hydrophilic interaction chromatography (HILIC) has been proposed as a reasonable alternative to ion-pair reversed phase separations of oligonucleotides prior to MS. A wide variety of HILIC stationary phase surface chemistries are currently available. Although their selectivity can be considerably different, few studies have compared these chemistries for LC-MS analysis of oligonucleotides. We evaluated ten different HILIC column chemistries to understand their capabilities for separating a variety of oligonucleotides. In general, we found that most columns were ineffective at separating larger (n > 15-mer) oligonucleotides under the mobile phase and gradient conditions evaluated here. However, several stationary phases were found to be effective for separating smaller oligonucleotides such as endonuclease digestion products. Given that early eluting oligonucleotides were found to be compatible with standard electrospray ionization conditions, several different HILIC stationary phase options are available for LC-MS studies of smaller oligonucleotides including those generated in RNA modification mapping experiments.
Introduction
Oligonucleotide separation is extremely important for classification and analysis of both endogenous RNAs as well as synthetic RNAs used in drug development.1–3 Ion-pair reverse phase liquid chromatography (IP-RPLC) has been among the most common separation methods used for the on-line coupling of LC with mass spectrometry (MS).4–7 However, this approach has limitations including effects on instrumentation and detrimental impacts on the sensitivity as well as the selectivity of analyzing oligonucleotides in biological samples.8,9 These limitations have even led researchers to investigate using LC-MS in positive polarity to minimize workflow disruptions, especially on shared instruments.10Hydrophilic interaction liquid chromatography (HILIC) is an alternative to IP-RPLC that was also shown to be amenable to the separation of oligonucleotides in the late 1990s.5 However, it was not widely adapted due to limitations in sample amounts, selectivity, sensitivity, and general compatibility with MS. One of the first analytical methods demonstrating HILIC separation of oligonucleotides with MS analysis utilized inductively coupled plasma (ICP-MS) as this ionization technique is less prone to background effects from mobile phase constituents.11
The compatibility of HILIC with electrospray ionization for the on-line analysis of oligonucleotides has been examined by several groups.12,13 These initial studies were conducted as a comparison of LC-MS results between HILIC and IP-RPLC for the analysis of DNA-based oligomers and phosphorothioate oligonucleotides. While these studies demonstrated feasibility for creating HILIC-based LC-MS methods for oligonucleotides and related samples, these works also illustrated performance differences based on the type of HILIC column used. In the study by Lobue and colleagues,12 a polymer-based, dihydroxyl functionalized column was used. Not long after, a study by Demelenne and colleagues investigated a silica-based amide column.13 The amide column enabled smaller particle sizes to be used (1.7 μm vs. 5 μm for the dihydroxyl), which enhanced resolution while generating narrower peak widths for similar samples.13
More recently, additional studies into HILIC-MS performance for the analysis of therapeutic oligonucleotides have been reported.14,15 HILIC stationary phase composition has been found important for the separation of phosphorothioate diastereomers,15 which may possess higher-order structure. Moreover, column chemistry – including the column housing material – impacts oligonucleotide separations in both IP-RPLC16 and for HILIC.17 Although these studies are providing more insights into the importance of HILIC column chemistry on oligonucleotide separations, there remains a gap in defining what column chemistries are best for on-line LC-MS of non-therapeutic oligonucleotides of varying length and modification complexity.
Here we examined ten different HILIC columns to gain an understanding of how column chemistries may influence the separation of oligonucleotides, with a focus on shorter modified oligonucleotides and operational performance of LC-MS. The column functional groups included a range of polarities. To minimize the effects of varying mobile phase conditions, all analyses were conducted using fixed mobile phase and gradient conditions. A range of oligonucleotides, from DNA homopolymers to post-transcriptionally modified RNA oligomers, were examined. Similar to the recent report for phosphorothioate oligonucleotides,15 our findings suggest that there is no one “best” column chemistry for on-line HILIC-MS, rather the optimal stationary phase is determined by the sample characteristics. However, clear trends emerged relating to column polarity and effectiveness at oligonucleotide separation.
Experimental
Materials and reagents
DNA oligonucleotides consisting of polythymidylic acids (dT15, dT20, dT25, dT30, and dT35) were purchased as a premixed standard (MassPREP Oligonucleotide Standard) from Waters Corporation (Milford, MA). LC-MS grade ammonium acetate was purchased from MilliporeSigma (Burlington, MA). Triethylamine (TEA, HPLC grade) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, >99%) were purchased from MilliporeSigma (Burlington, MA). LC-MS grade acetic acid, methanol, and acetonitrile were purchased from Fisher Scientific (Waltham, MA). High-purity (18.2 MΩ cm−1) water was produced in house from a Barnstead Nanopure system (Thermo Scientific, Waltham, MA). Ribonuclease T1 was purchased from Worthington Biochemical (Lakewood, NJ), and Saccharomyces cerevisiae transfer RNA-Phe was purchased from MilliporeSigma (Burlington, MA). Invitrogen bacterial alkaline phosphatase (BAP) was purchased from Fisher Scientific (Waltham, MA).Table 1 list the columns used in this study along with their various properties, and the stationary phase chemistry is illustrated in ESI Fig. S1.† These columns were chosen to minimize variance in column particle size, length, and inner diameter, while providing significantly different bonded phases to explore the impact of column chemistry on performance.
| Manufacturer | Column name | Column size | Bonded phase | Particle size | Particle composition |
|---|---|---|---|---|---|
| Ace | ACE HILIC-N | 100 × 2.1 mm | Pentaol | 3 μm | Silica |
| HILICON | HILICON iHILIC-fusion | 100 × 2.1 mm | Zwitterionic | 3 μm | Silica |
| HILICON | HILICON iHILIC-fusion(+) | 100 × 2.1 mm | Zwitterionic | 3 μm | Silica |
| Shodex | HILICpak VN-50 | 150 × 2 mm | Diol | 5 μm | Polymer |
| Waters | Xbridge BEH HILIC | 150 × 2.1 mm | Silanol | 2.5 μm | Silica |
| Waters | Xbridge BEH amide | 150 × 2.1 mm | Amide | 2.5 μm | Silica |
| Waters | Torus DEA | 150 × 2.1 mm | Diethylamine | 2.5 μm | Silica |
| Waters | Torus Diol | 150 × 2.1 mm | Diol | 2.5 μm | Silica |
| Waters | Torus 2-pic | 150 × 2.1 mm | 2-Picolamine | 2.5 μm | Silica |
| Waters | Atlantis premier BEH Z-HILIC | 150 × 2.1 mm | Zwitterionic | 2.5 μm | Silica |
HPLC conditions
HILIC mobile phases were prepared from a stock solution of 50 mM aqueous ammonium acetate. This stock was diluted with acetonitrile in a ratio of 30/70 (stock/acetonitrile) and 70/30 (stock/acetonitrile) for mobile phase A (MPA) and mobile phase B (MPB), respectively. This resulted in a final concentration of 15 mM ammonium acetate in 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 water:acetonitrile for MPA and 35 mM ammonium acetate in 70:30 water:acetonitrile for MPB. For HILIC separations, the gradient started with a 5 min isocratic hold at 5% MPB followed by a linear gradient to 85% MPB at 40 min. The water percentage for this gradient starts at 32% aqueous and concludes at 64% aqueous. The run concluded with an isocratic hold for 5 minutes and a re-equilibration period of 15 min at 5% MPB. The flow rate was 220 μL min−1 and the column was thermostatted to 60 °C.
:70 water:acetonitrile for MPA and 35 mM ammonium acetate in 70:30 water:acetonitrile for MPB. For HILIC separations, the gradient started with a 5 min isocratic hold at 5% MPB followed by a linear gradient to 85% MPB at 40 min. The water percentage for this gradient starts at 32% aqueous and concludes at 64% aqueous. The run concluded with an isocratic hold for 5 minutes and a re-equilibration period of 15 min at 5% MPB. The flow rate was 220 μL min−1 and the column was thermostatted to 60 °C.
IP-RPLC mobile phases were prepared as previously described.18 Briefly, mobile phase A was 8 mM TEA and 200 mM HFIP in water and mobile phase B was 8 mM TEA and 200 mM HFIP in 50:50 water:methanol. For IP-RPLC, a 5 min isocratic hold of 5% MPB was followed by a ramp to 73% MPB at 40 min. Next, a flush of 100% MPB was performed for 5 min followed by 15 min re-equilibration at 5% MPB. The flow rate was 80 μL min−1 and the column was thermostatted to 60 °C.
LC separations with UV detection were conducted using a 1260 Infinity II (Agilent Technologies) ultra-high performance liquid chromatography (UHPLC) system. Detection was performed with an Infinity II diode array detector (DAD) (Agilent Technologies) monitoring at 260 nm and UV data was visualized with OpenLAB CDS ChemStation software Version 2.17.29 (Agilent Technologies).
LC-MS was conducted using a Dionex Ultimate 3000 (Thermo Scientific) UHPLC system connected in line with an LTQ-XL linear ion trap mass spectrometer (Thermo Fisher Scientific). The LTQ-XL was operated in negative polarity with a capillary temperature of 375 °C, spray voltage of 3.7 kV, capillary voltage of −30 V, and sheath gas, auxiliary gas, and sweep gas at 35, 20, and 20 arbitrary units, respectively. The data was acquired on a range of 600–2000 Da for the full scan followed by four data dependent scans triggered by the four most abundant precursors from the full scan event. The maximum injection was fixed at 250 ms with an AGC cutoff of 500 counts. Data was analyzed using Xcalibur v4.1 (Thermo Fisher Scientific). Extracted ion chromatograms (EICs) for the major components of the sample mixture were created using the summation of all generated charge states from 600 to 2000 m/z.
The Waters MassPREP oligonucleotide standard was diluted to 10 pmol μL−1 in MPA for both HILIC19 and IP-RPLC experiments and 4 μL were injected on column. The S. cerevisiae tRNA-Phe was hydrolyzed to 3′-terminal guanosine oligonucleotides using RNase T1. Enzymatic hydrolysis was carried out for 120 min at 37 °C. The sample was prepared in a solution of 110 mM ammonium acetate with 50 U of RNase T1 per 1 μg of tRNA.20 The hydrolysis reaction was taken to dryness and then reconstituted in a 50:50 mixture of MPA:MPB for LC-MS. In total, 4 μg were injected on column at a concentration of 1 μg μL−1. In cases where the enzymatic hydrolysis was carried out with bacterial alkaline phosphatase (BAP), the same digestion was followed as the normal RNase T1 digestion but with the addition of 0.01 U of BAP per 1 μg of RNA to cleave the 3′-phosphate to a 3′-hydroxyl. Chromatographic methods and mass spectral methods were consistent for the enzymatic digestions as they were for the Waters MassPREP oligonucleotide standard mix.
Results and discussion
Baseline for comparison of LC-MS analysis of oligonucleotides
The most common and effective method for the separation of oligonucleotides prior to on-line mass spectrometry analysis is ion-paired reverse phase liquid chromatography (IP-RPLC). As the goal of this work is to examine a variety of stationary phases for HILIC separation of oligonucleotides, we first established baseline IP-RPLC performance characteristics for subsequent HILIC comparisons. While HILIC separations have been compared to IP-RPLC in the past,13,18 here we sought to establish a standard set of gradient conditions that would then be applied consistently to all evaluated columns and could also be compared against the standard IP-RPLC results.The Waters MassPREP oligonucleotide standard, which is a commercial mixture of polythymidilic acids (dT15, dT20, dT25, dT30, dT35) and their various failure sequences, was used for creating this baseline data set. Failure sequences are truncated sequences arising from the synthesis process and can provide additional information related to the ability to separate n-mers from (n − 1)-mers. Fig. 1A contains a representative total ion chromatogram (TIC) obtained using IP-RPLC-MS for the analysis of 40 pmol of the polythymidilic acid mixture. These data were then used to extract the key chromatographic parameters (Table 2) to serve as a baseline for measuring the performance of various HILIC columns.
 | ||
| Fig. 1 Total ion chromatograms (TICs) showing separation of poly dTs by (A) IP-RPLC and (B) HILIC using a Waters BEH amide column. Inlays are combined extracted ion chromatograms (EIC) of the major components of this mixture. | ||
| Analyte | Retention factor (k′) | Resolution (R) | Peak capacity (Pc) | Symmetry (S) | w 1/2 (min) |
|---|---|---|---|---|---|
| IP-RPLC | |||||
| dT15 | 15.4 | — | 104 | 1.5 | 0.20 |
| dT20 | 18.4 | 15.1 | 138 | 2.0 | 0.15 |
| dT25 | 20.3 | 9.7 | 104 | 1.5 | 0.20 |
| dT30 | 21.7 | 6.3 | 115 | 3.0 | 0.18 |
| dT35 | 22.7 | 4.7 | 104 | 3.0 | 0.20 |
| HILIC (Waters BEH amide) | |||||
| dT15 | 14.9 | — | 95 | 1.6 | 0.22 |
| dT20 | 16.6 | 8.0 | 95 | 1.5 | 0.22 |
| dT25 | 17.9 | 4.9 | 75 | 1.3 | 0.28 |
| dT30 | 18.5 | 2.4 | 95 | 2.5 | 0.22 |
| dT35 | 19.5 | 4.4 | 80 | 12.5 | 0.26 |
For ease of comparison, we first optimized the HILIC mobile phase and gradient using the previously reported BEH amide column.13 We sought to keep the retention factors (k′) similar between the IP-RPLC and HILIC methods for the major sample components. Doing so allows for an evaluation of the other key chromatographic parameters, which included resolution (R), peak capacity (Pc), symmetry (S) and peak width at half-height (w1/2).
The retention factor (k′) was calculated from eqn (1):
 | (1) |
Column resolution (R) was calculated from eqn (2):
 | (2) |
Peak capacity (Pc), or the measure of the number of peaks that fit in an elution window, is determined from eqn (3) which has been adapted from literature:21
 | (3) |
Symmetry is calculated by measuring the ratio of the tail half of the peak to the front half of the peak by drawing a line from the apex of the peak to the baseline to designate the middle of the peak and the values were measured. Baseline measurements were chosen to ensure that differential ionization efficiency of the oligonucleotides would not affect these calculations.
Representative HILIC LC-MS results are shown in Fig. 1B and are consistent with those previously reported.13 Extracted ion chromatograms (EICs) for each of the major components were generated from both IP-RPLC and HILIC and are shown in the insets. The various figures of merit for IP-RPLC and HILIC with the BEH amide column are listed in Table 2 and were derived from the EIC data due to varying baseline and ESI responses seen in the total ion chromatograms. Despite the completely different retention mechanisms, the elution order for the poly dTs is the same with the shorter oligonucleotides eluting earliest and the longer oligonucleotides eluting last. The resolution for all analytes in both methods is also greater than 2, demonstrating sufficient separation of the major components. The symmetry values for both methods are three or less, with the HILIC method having slightly less tailing than the IP-RPLC method.
When comparing the mass spectral data and response, however, we observed differences between IP-RPLC-MS (Fig. 2A and B) and HILIC-MS (Fig. 2C and D) for the experimental conditions examined here. For example, the charge state distribution for dT15 obtained using IP-RPLC yields predominantly only the 3− charge state (Fig. 2A), whereas HILIC generates multiply charged ions from the 3− to 6− charge states (Fig. 2C). For dT35, which elutes at a much higher percentage of MPB, IP-RPLC yields a very broad and high charge state distribution (7− to 16−) (Fig. 2B) while HILIC generates only a few charge states (6− and 7−) (Fig. 2D).
 | ||
| Fig. 2 Mass spectral comparison of (A & B) ion-pair reverse phase chromatography and (C & D) HILIC chromatography. Charge states of the particular oligonucleotides generated by electrospray ionization are labeled. Mass spectrums for dT15 are on the left and dT35 are on the right. Both analytes were injected in 4 μL of their respective MPA at a concentration of 10 pmol L−1. | ||
These mass spectral results are consistent with both the role of water22,23 and mobile phase additives24–28 during electrospray ionization desolvation. With IP-RPLC, dT15 elutes at 74.5% water and dT35 elutes at 62.5% water. With HILIC, dT15 elutes at 54.4% water and dT35 elutes at 61.6% water. Higher ratios of water to organic can result in minimal charge state distributions. Shorter oligonucleotides elute in a lower ratio of water during HILIC (Fig. 2C) than for IP-RPLC (Fig. 2A). However, the relative amount of water is similar for both methods for the elution of dT35 suggesting the mass spectral differences (i.e., Fig. 2B vs. D) may be more driven by the mobile phase composition in IP-RPLC. It is also clear from these data that the ESI mass spectral results for dT35 in HILIC (Fig. 2D) are significantly impacted by salt adduction. Whether these salt adducts arise from the mobile phase components or are due to the lack of alkylamine displacement (as in IP-RPLC)29,30 remains to be determined.
Diol-based column chemistry
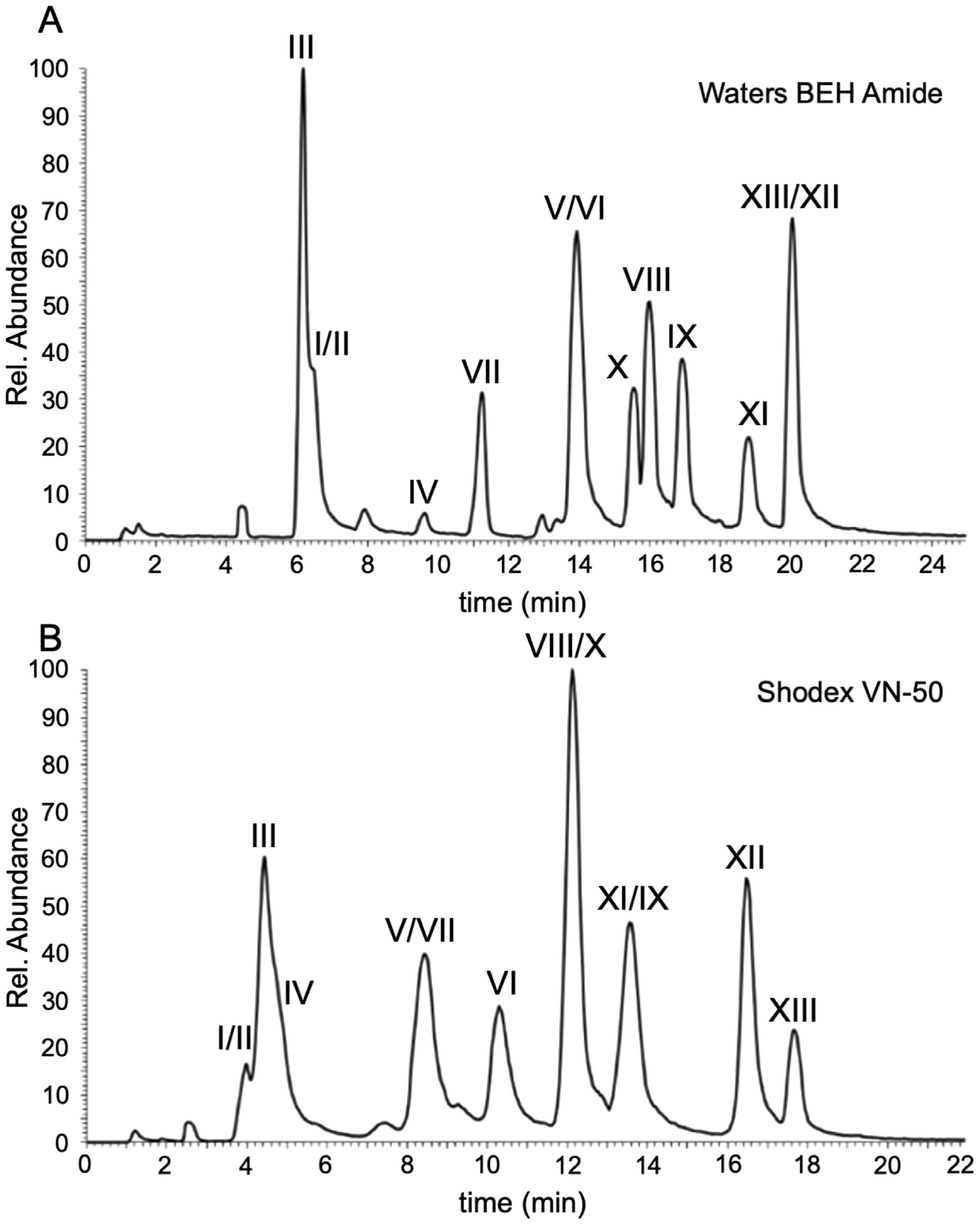
To facilitate further comparisons on the separation characteristics of all columns, the next set of studies were conducted using LC-UV rather than LC-MS. All data obtained were from single analyses as the interest here was focused on the differences amongst the stationary phases rather than the repeatability of each column. The BEH amide column served as our HILIC benchmark in this study because many of the other HILIC stationary phases can readily be evaluated against the amide functionality. The first HILIC comparisons were conducted using diol-bonded functionality that has been demonstrated to provide suitable separation of oligonucleotides.12 The Torus Diol contains similar functionality to the Shodex VN-50 previously used12 except that the Torus utilizes a silica-based particle whereas the Shodex is built on a polymer-based particle.The chromatograms for the separation and detection of the polythymidylic acid mixture by LC-UV using the Waters BEH amide, Shodex VN-50, and Waters Torus Diol are presented in Fig. 3. The mobile phase conditions were equivalent to those developed above for the initial comparison against IP-RPLC. The chromatographic figures of merit are listed in Table 3. Both the Waters BEH amide and Shodex VN-50 provided adequate resolution and peak capacity with the Shodex column exhibiting a smaller elution window for the polythymidylic acid mixture. As seen in Fig. 3C and Table 3, the Waters Torus Diol does not perform as well as the polymer-based diol of the Shodex. All chromatographic figures of merit, except for the retention factor, were worse for the Torus column including peak symmetry calculations, which could not be obtained due to the lack of baseline resolution among sample components.
 | ||
| Fig. 3 UV chromatograms of the columns that performed well in the separation of DNA thymidines. (A) Waters BEH Amide column. (B) Shodex VN-50 column. (C) Waters Torus Diol. | ||
| Analyte | Retention factor (k′) | Resolution (R) | Peak capacity (Pc) | Symmetry (S) | w 1/2 (min) |
|---|---|---|---|---|---|
| n.d. ∼not determined due to lack of baseline resolution. | |||||
| Waters BEH amide | |||||
| dT15 | 14.9 | — | 95 | 1.0 | 0.60 |
| dT20 | 16.5 | 3.3 | 95 | 1.5 | 0.50 |
| dT25 | 17.5 | 2.1 | 75 | 1.3 | 0.70 |
| dT30 | 18.2 | 1.0 | 95 | 1.5 | 1.00 |
| dT35 | 18.8 | 0.7 | 80 | 1.3 | 0.90 |
| Shodex VN-50 | |||||
| dT15 | 13.3 | — | 35 | 1.0 | 0.60 |
| dT20 | 14.6 | 2.2 | 42 | 1.5 | 0.50 |
| dT25 | 15.5 | 1.3 | 30 | 1.0 | 0.80 |
| dT30 | 16.2 | 0.7 | 22 | 0.8 | 1.10 |
| dT35 | 16.7 | 0.4 | 24 | 1.2 | 1.20 |
| Waters Torus Diol | |||||
| dT15 | 9.3 | — | 35 | n.d. | 0.60 |
| dT20 | 10.3 | 2.4 | 42 | n.d. | 0.50 |
| dT25 | 10.8 | 1.2 | 27 | n.d. | 0.80 |
| dT30 | 11.3 | 1.0 | 20 | n.d. | 1.10 |
| dT35 | 11.6 | 0.5 | 18 | n.d. | 1.20 |
Zwitterionic column chemistry
The amide and diol functionalities provided reasonable separations of the mixture of polythymidylic acids, although resolving capacity is limited for higher length oligonucleotides. Because HILIC retention mechanisms are largely partitioning of the analyte between the organic mobile phase and the aqueous layer surrounding each particle,31,32 we sought to determine whether other functional groups that are more polar than those investigated could be effective. By increasing the polarity of the stationary phase, our hypothesis was that the aqueous layer surrounding each particle would be larger and enable efficient separations of longer oligonucleotides.Three zwitterionic columns, iHILIC-fusion and iHILIC-fusion(+) both from Hilicon and the BEH Z-HILIC column from Waters, were chosen to try to maximize the aqueous layer of the particles (ESI Fig. S1†). The two columns from Hilicon are relatively similar in composition, with the latter having a larger positive charge density on its surface than the former. The Waters Z-HILIC column is also zwitterionic but is sulfobetaine functionalized and is oppositely polarized compared to the Hilicon columns; the positive charge is interior and the negative charge is exterior.
Representative chromatograms from these columns are shown in Fig. 4. Only the iHILIC-fusion column eluted the larger oligonucleotides (Fig. 4A). In contrast, the iHILIC-fusion(+) overly retained oligomers greater than n = 8, suggesting the zwitterionic functional group for this stationary phase retains the components too strongly.33 Additionally, as the positive charge of the zwitterionic pair gets stronger, the oligonucleotides become over-retained which is shown by the significant different in retention between the weaker positive charged iHILIC-fusion (Fig. 4A) and the stronger positive charged iHILIC-fusion(+) (Fig. 4B). Similar results to the iHILIC-fusion(+) were obtained using the BEH Z-HILIC column (Fig. 4C), which showed good separation in reasonable elution windows for dT10 and smaller but also appeared to be strongly retaining longer oligonucleotides.
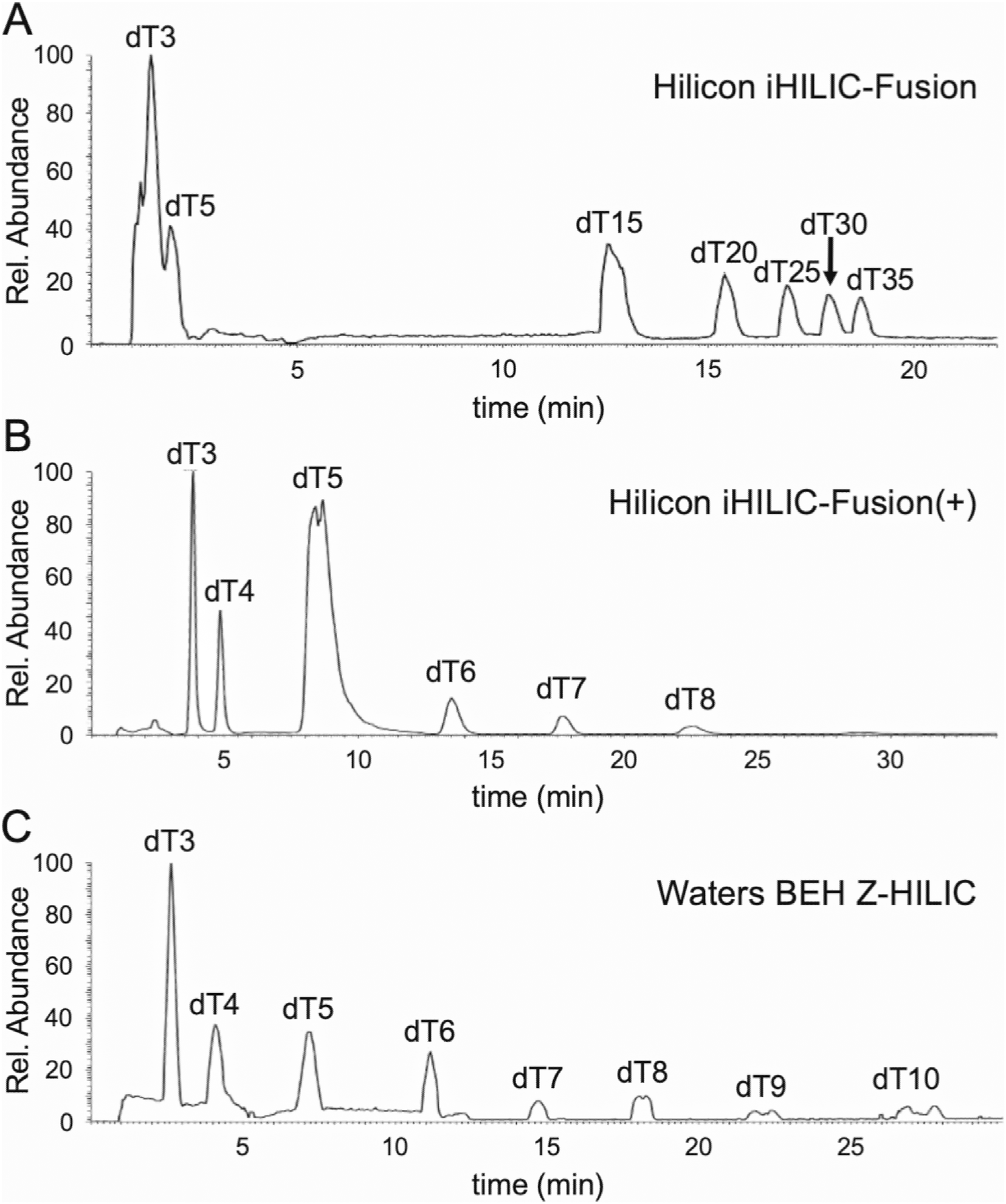 | ||
| Fig. 4 LC-MS total ion chromatograms (TICs) showing separation of poly dTs by (A) Hilicon iHILIC-fusion, (B) Hilicon iHILIC-fusion(+), and (C) Waters BEH Z-HILIC columns. In general, zwitterionic columns are less effective for the separation of longer oligonucleotides although they show some promise for smaller oligonucleotides. | ||
Alternative column chemistries
We next sought to examine the pentaol column ACE HILIC-N column, which should increase the polarity over diol and amide functional columns without adding any charge interactions. The Waters BEH HILIC column, which has only silanol functionality, was also investigated although silanol activity has been shown to be unpredictable even in well studied reverse phase conditions.34 Results from the ACE HILIC-N column (ESI Fig. S2A†) were similar to those found for the fusion(+) and BEH Z-HILIC columns with minimal elution of any oligonucleotides except small dT-mers. No separation of the individual oligomers was found for the BEH HILIC stationary phase, although there is some retention of sample components (ESI Fig. S2B†).The final two functionalities examined were the Waters Torus 2-pic and Torus DEA. Both columns should exhibit anion exchange behavior and will more likely be charged during separation. As before, strongly polar columns were ineffective at oligonucleotide separation with the Torus 2-pic (ESI Fig. S2C†) having better performance for small oligonucleotides than the Torus DEA (ESI Fig. S2D†). This is most likely due to the strong charge–charge interaction of the cationic amines of the stationary phases being too strongly retaining to the negatively charged phosphate backbones of oligonucleotides.
Beyond the BEH amide, both diol-based columns, and the iHILIC-fusion zwitterionic column, the other HILIC stationary phases generally over-retained the polythymidylic acid samples. Thus, it appears that HILIC columns of lower polarity are best for longer oligonucleotides. Moreover, the MS performance of these samples suggests that HILIC-MS of oligonucleotides should be optimized to elute oligonucleotides of interest at lower values for MPB, whereas achieving IP-RPLC quality results for later eluting oligonucleotides (i.e., longer oligonucleotides that are in a less aqueous environment in IP-RPLC) using HILIC will require adjustments to mobile phase conditions to enhance oligonucleotide desolvation. Such adjustments could include examination of alternative organic solvents as well as mobile phase additives for HILIC that might assist desolvation.
HILIC separation of shorter oligonucleotides
While the results above clearly demonstrate several HILIC column chemistries of higher polarity are ineffective for larger (n > 15-mer) oligonucleotides, there are a number of LC-MS applications that require separation of mixtures of smaller oligonucleotides. Further, noting that oligonucleotides eluting earlier in HILIC generate higher quality mass spectrometry data (cf.Fig. 2), we next investigated how these columns perform for the separation of a mixture of shorter length (n < 15-mer) modified oligoribonucleotides.The sample was generated by digesting S. cerevisiae tRNA-Phe with the endonuclease RNase T1. RNase T1 is a guanosine-specific ribonuclease that predictably cleaves RNA at the 3′-phosphodiester bond of all guanosines. Using the known sequence of yeast tRNA-Phe, the specific RNase T1 digestion products can be calculated including the 3′-terminus CACCA oligonucleotide (Table 4). The molecular weight and the electrospray series of the oligonucleotides that result from RNase T1 digestion are listed in ESI Table S1.† The expected digestion products range from dimers through the 12-mer (A[Cm]U[Gm]AA[yW]AΨ[m5C]UGp), where [Cm] is 2′-O-methylcytidine, [Gm] is 2′-O-methylguanosine, [yW] is wybutosine, Ψ is pseudouridine, and [m5C] is 5-methylcytidine.
| Yeast tRNAPhe RNase T1 digestion products | Mol wt. | DP-IDa | Waters BEH amide | Shodex VN-50 | Waters Torus Diol | HILICON iHILIC-fusion | Waters BEH Z-HILIC |
|---|---|---|---|---|---|---|---|
| a Digestion product ID. | |||||||
| CGp | 668.4 | I | 5.0 | 2.8 | 3.3 | 2.7 | 12.3 |
| UGp | 669.4 | II | 5.0 | 3.6 | 3.9 | 2.7 | 12.4 |
| AGp | 692.4 | III | 4.7 | 3.2 | 3.6 | 3 | 11.2 |
| DDGp | 979.6 | IV | 8.1 | 3.7 | 4.7 | 3.2 | 16.3 |
| [m5U]ΨCGp | 1294.8 | V | 12.4 | 7.2 | 7.0 | 5.8 | 21.7 |
| CCAGp | 1302.8 | VI | 12.9 | 9.1 | 8.9 | 6.4 | 23.3 |
| CACCA | 1512.0 | VII | 9.7 | 7.4 | 6.7 | 4.0 | 17.2 |
| CUCAGp | 1609.0 | VIII | 14.1 | 11 | 10.6 | 8.0 | 27.5 |
| AAUUCGp | 1939.2 | IX | 17.3 | 12.2 | 12.1 | 11.7 | 29.4 |
| AUUUA[m2G]p | 1954.2 | X | 14.5 | 10.9 | 10.5 | 9.2 | 27.7 |
| [m7G]UC[m5C]UGp | 1959.2 | XI | 15.4 | 12.5 | 12.1 | 9.8 | 30.9 |
| [m1A]UCCACAGp | 2586.6 | XII | 18.5 | 15.3 | 15.8 | 13.9 | 38.5 |
| A[Cm]U[Gm]AA[yW]AΨ[m5C]UGp | 4166.7 | XIII | 18.6 | 16.5 | 17.6 | 14.0 | — |
This mixture of tRNA-Phe RNase T1 digestion products was analyzed using the optimized HILIC gradient conditions identified earlier. All studies with these digestion products were conducted using LC-MS and LC-MS/MS to enable identification of individual digestion products. We first noted that, similar to their performance with polythymidylic acid mixtures, the Waters BEH amide and Shodex VN-50 (diol) columns could resolve the RNase T1 digestion products listed in Table 4. The total ion chromatograms for these separations are shown in Fig. 5. Both columns provided sufficient peak capacity in a reasonable elution window for analysis. The only discernable difference between these two columns was in the elution order of some of the digestion products as well as the ability of the Shodex column to resolve the two largest RNase T1 digestion products (XII and XIII), which was not possible using the BEH amide column.
 | ||
| Fig. 5 Total ion chromatogram (TIC) annotated with observed RNase T1 digestion product of S. cerevisiae tRNA-Phe obtained via LC-MS/MS data using the (A) Waters BEH amide and (B) Shodex VN-50. *Note, x-axis are not same to enhance clarity of chromatogram. Additional information on retention and analytical figures of merit is available in ESI Table S2.† | ||
Among the other columns available, we found that the Waters Torus Diol and the Hilicon iHILIC fusion columns also could separate RNase T1 digestion products, with retention generally correlated with the length of the RNase T1 digestion product. The total ion chromatograms are shown in Fig. 6A for the Waters Torus Diol and Fig. 6B for the Hilicon iHILIC fusion columns. Lastly, the Waters BEH Z-HILIC column also resolved the RNase T1 digestion products, and the corresponding total ion chromatogram is shown in Fig. 6C where all products are eluted and labelled except for the longest digestion product (i.e., XIII). The Torus Diol and iHILIC-fusion columns performed very similarly to the BEH amide and the Shodex VN-50 in retention times and elution window, but the BEH Z-HILIC (Fig. 6C) more strongly retains the longer digestion products leading to a larger elution window and peak broadening under the mobile phase and gradient conditions used here.
 | ||
| Fig. 6 Total ion chromatogram (TIC) annotated with observed RNase T1 digestion product of S. cerevisiae tRNA-Phe obtained via LC-MS/MS data using the (A) Waters Torus Diol (B) Hilicon iHILIC-fusion and (C) Waters BEH Z-HILIC columns. *Note, x-axis are not same to enhance clarity of chromatogram. Additional information on retention and analytical figures of merit is available in ESI Table S2.† | ||
The analytical figures of merit for separation of tRNA-Phe RNase T1 digestion products are listed in ESI Table S2† for all columns. When comparing these values, the five aforementioned columns had very similar peak capacities (Pc) and peak widths at half height (w1/2). Except for the Waters Torus Diol column, the columns showed good peak symmetry. The retention factor (k′) for these columns was also very similar, except for the Waters BEH Z-HILIC column, showing that the retaining power of these columns are relatively similar under the separation conditions investigated.
As Fig. 2 illustrates, HILIC-MS can potentially lead to a cleaner mass spectrum with a larger electrospray envelop for smaller oligonucleotides like dT15. In the tRNA-Phe digest with RNase T1, the digestion products were able to be characterized by mass spectrometry. Both the MS and MS/MS data are shown for an early eluting (11.2 min) oligonucleotide, CACCA, (ESI Fig. S3†), a middle eluting (16 min) oligonucleotide, CUCAGp, (ESI Fig. S4†) and a late eluting (20 min) oligonucleotide, A[Cm]U[Gm]AA[yW]AÑ°[m5C]UGp, (ESI Fig. S5†) taken from the LC-MS/MS analysis using the Waters BEH amide column. The charge states for these oligonucleotides are labelled in the mass spectra and the McLuckey fragmentation35 peaks are labelled in the fragment ion spectra. The quality of the MS and MS/MS data for these RNase T1 digestion products is generally good and enables accurate mass spectrometry-based sequencing of these oligonucleotides. The early and middle eluting oligonucleotides provided high quality MS/MS data while the latest eluting oligonucleotide (A[Cm]U[Gm]AA[yW]AÑ°[m5C]UGp) was of lower MS/MS quality. This is most likely due to the wybutosine modification, which is prone to base loss in electrospray conditions,36 rather than any LC-related conditions. Overall, these results find that there are a greater number of stationary phase options for smaller oligonucleotides, such as endonuclease digestion products, that are compatible with LC-MS/MS than might be available for larger oligonucleotides often examined for therapeutic uses.
Effect of 3′-phosphate on elution of RNase digestion products
To further understand the capabilities of these HILIC column chemistries for separating mixtures of endonuclease digestion products, we sought to determine whether the presence of the 3′-phosphate in the RNase T1 digestion products would significantly influence retention time and other chromatographic characteristics. A sample of S. cerevisiae tRNA-Phe was digested as before using RNase T1. It was additionally digested with bacterial alkaline phosphatase (BAP), which is an enzyme that will remove the 3′-terminal phosphate from each digestion product ending in 3′-Gp. Removing this terminal phosphate should make each oligonucleotide less polar and therefore reduce the retention time in HILIC.ESI Table S3† lists the retention factor for each RNase T1 digestion product after BAP treatment. Generally, as expected, removing the 3′-terminal phosphate shifts the retention time earlier for all digestion products. To provide a normalized comparison of the impact of the 3′-phosphate across the various columns investigated, a relative decrease in retention factor was calculated using eqn (4), where 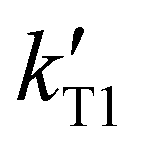 is the retention factor for the RNase T1 digestion and
is the retention factor for the RNase T1 digestion and  is the retention factor for the RNase T1 digestion with bacterial alkaline phosphatase.
is the retention factor for the RNase T1 digestion with bacterial alkaline phosphatase.
 | (4) |
The relative change for each digestion product across each column investigated is listed in Table 5. These values suggest that the terminal 3′-phosphate group of the oligonucleotide has an impact on retention time which decreases as the length of the oligonucleotide increases. This length dependency is not unexpected as the relative polarity increase due to the 3′-phosphate would lessen as the number of intermolecular phosphodiester bonds increase.
| Yeast tRNAPhe RNase T1 digestion products | Waters BEH amide | Shodex VN-50 | Waters Torus Diol | HILICON iHILIC-fusion | Waters BEH Z-HILIC |
|---|---|---|---|---|---|
| AG | 34% | — | 17% | 23% | 62% |
| DDG | 53% | — | 32% | 22% | 45% |
| [m5U]ΨCG | 44% | 38% | 34% | 41% | 30% |
| CCAG | 43% | 37% | 44% | 44% | 28% |
| CACCA | 9% | −7% | −7% | −20% | −4% |
| CUCAG | 29% | 18% | 40% | 45% | 30% |
| AAUUCG | 31% | 12% | 26% | 36% | 27% |
| AUUUA[m2G] | 39% | 4% | 32% | 53% | 31% |
| [m7G]UC[m5C]UG | 6% | 22% | 25% | 7% | 28% |
| [m1A]UCCACAG | 13% | 3% | 9% | 11% | 26% |
| A[Cm]U[Gm]AA[yW]AΨ[m5C]UG | 13% | 2% | 6% | 5% | — |
Of note, one digestion product, CACCA, arises from the 3′-terminal of tRNA-Phe. This digestion product is known to exist in the 3′-OH form in deacylated tRNAs, thus treatment with bacterial alkaline phosphatase should have a minimal effect on the retention of this oligonucleotide. As seen in Table 5, in most cases the variations are relatively minor (<10%) and often reveal a small increase in the retention factor after treatment with the phosphatase. Only the Hilicon iHILIC-fusion column exhibited a significant change (increase of 20%) in the retention factor of this digestion product for reasons that remain to be determined.
We also evaluated the variations in MS quality with these columns for digestion products with and without 3′-phosphate (ESI Fig. S6†). As an example the 12-mer, (A[Cm]U[Gm]AA[yW]AÑ°[m5C]UG), is shown for both the Waters BEH amide (ESI Fig. S6A and C†) column and the Shodex VN-50 (ESI Fig. S6B and D†) column. There is a stark difference in data quality when using RNase T1 with BAP, which produces much more complicated and poorer quality mass spectrum (ESI Fig. S6C and S6D†) than the spectra using solely RNase T1 for the digestion (ESI Fig. S6A and S6B†). These differences suggest that the current BAP digestion protocol increases the salt content of the sample, which may also be affecting overall sample retention. While BAP-treated RNase digestion products can provide some advantages during LC-MS/MS analyses, the current protocol should be optimized to reduce the salt content prior to implementation with HILIC-based separations.
Conclusions
Several HILIC column chemistries were compared for LC-MS compatibility using different oligonucleotide samples. Our results confirm previously reported data that less polar stationary phases such as diols, amides, and weakly zwitterionic such as sulfbetaine are the best for larger (n > 15-mer) oligonucleotides due to the strong interaction with the polar oligonucleotides without being overly retaining. Under the conditions examined here, the Waters BEH amide, the Shodex VN-50, the Waters Torus Diol, and the Hilicon iHILIC-fusion columns yielded appropriate chromatographic resolution and mass spectrometry sensitivity for a variety of different oligonucleotide samples. However, it is worth noting that the Waters BEH Z-HILIC column also performed well for smaller oligonucleotide digestion products suggesting some zwitterionic columns may have utility for these types of samples. Further investigation into the optimization of mobile phase and gradient conditions for these zwitterionic columns for smaller oligonucleotides seems warranted.The three columns that were found to be ineffective at both smaller oligonucleotides and the larger polythymidylic acid mixture under the specific conditions investigated here are the iHILIC-fusion(+), the Torus 2-pic, and the HILIC-N. It appears that the stronger positive charge of the zwitterion in iHILIC-fusion(+), the strongly anionic character of the Toros 2-pic, and the highly hydrophilic nature of the poly-ols in the HILIC-N, allow for very poor separation of longer oligonucleotides (15–35-mers) and similarly were outperformed on the smaller oligonucleotides resulting from the RNase T1 digestion.
Traditional LC-MS evaluation of new columns for oligonucleotide analysis has focused on larger oligonucleotides and oligonucleotide therapeutics.5,10,12,13,17 Such evaluations may miss important application areas. As found here, several HILIC columns that were less effective at separating larger oligonucleotides are shown to have utility for smaller oligonucleotides such as those generated during RNA modification mapping experiments. Additional studies to optimize the mobile phase and ESI conditions for the analysis of smaller oligonucleotides by these columns may yield even greater improvements that could prove valuable for future LC-MS studies.
Author contributions
Scott Abernathy: conceptualization, methodology, investigation, formal analysis validation, writing – original draft, writing – review & editing. Asif Rayhan: data curation, formal analysis. Patrick A. Limbach: conceptualization, resources, writing – review & editing, supervision, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support of this work was provided by the National Institutes of Health (GM058843). The generous support of the Rieveschl Eminent Scholar Endowment and the University of Cincinnati for these studies is also appreciated. Additionally the authors would like to thank Waters Corporation, especially Bill Warren and Christopher Romero, for generously supporting this research by sharing a variety of HILIC columns. The authors would lastly like to remember Peter A. Lobue (deceased) who helped brainstorm ideas for this work.References
- J. M. Sutton, G. J. Guimaraes, V. Annavarapu, W. D. van Dongen and M. G. Bartlett, J. Am. Soc. Mass Spectrom., 2020, 31, 1775–1782 CrossRef CAS PubMed.
- T. Suzuki and T. Suzuki, Methods Enzymol., 2007, 425, 231–239 CAS.
- S. C. Pomerantz, J. A. Kowalak and J. A. McCloskey, J. Am. Soc. Mass Spectrom., 1993, 4, 204–209 CrossRef CAS PubMed.
- A. Apffel, J. A. Chakel, S. Fischer, K. Lichtenwalter and W. S. Hancock, Anal. Chem., 1997, 69, 1320–1325 CrossRef CAS PubMed.
- A. Apffel, J. A. Chakel, S. Fischer, K. Lichtenwalter and W. S. Hancock, J. Chromatogr., A, 1997, 777, 3–21 CrossRef CAS.
- M. Gilar, K. J. Fountain, Y. Budman, U. D. Neue, K. R. Yardley, P. D. Rainville, R. J. Russell 2nd and J. C. Gebler, J. Chromatogr. A, 2002, 958, 167–182 CrossRef CAS PubMed.
- B. Chen and M. G. Bartlett, J. Chromatogr. A, 2013, 1288, 73–81 CrossRef CAS PubMed.
- S. Studzińska, R. Rola and B. Buszewski, J. Pharm. Biomed. Anal., 2017, 138, 146–152 CrossRef PubMed.
- F. Hagelskamp, K. Borland, J. Ramos, A. G. Hendrick, D. Fu and S. Kellner, Nucleic Acids Res., 2020, 48, e41–e41 CrossRef CAS PubMed.
- G. Weng, B. Sun, Z. Liu, F. Wang and Y. Pan, Anal. Bioanal. Chem., 2019, 411, 4167–4173 CrossRef CAS PubMed.
- R. N. Easter, K. K. Kröning, J. A. Caruso and P. A. Limbach, Analyst, 2010, 135, 2560–2565 RSC.
- P. A. Lobue, M. Jora, B. Addepalli and P. A. Limbach, J. Chromatogr. A, 2019, 1595, 39–48 CrossRef CAS PubMed.
- A. Demelenne, M.-J. Gou, G. Nys, C. Parulski, J. Crommen, A.-C. Servais and M. Fillet, J. Chromatogr., A, 2020, 1614, 460716 CrossRef CAS PubMed.
- H. Lardeux, V. D’'Atri and D. Guillarme, TrAC, Trends Anal. Chem., 2024, 176, 117758 CrossRef CAS.
- H. Lardeux, K. Stavenhagen, C. Paris, R. Dueholm, C. Kurek, L. De Maria, F. Gnerlich, T. Leek, W. Czechtizky, D. Guillarme and M. Jora, Anal. Chem., 2024, 96, 9994–10002 CrossRef CAS PubMed.
- G. J. Guimaraes, J. M. Sutton, M. Gilar, M. Donegan and M. G. Bartlett, J. Pharm. Biomed. Anal., 2022, 208, 114439 CrossRef CAS PubMed.
- H. Lardeux, A. Goyon, K. Zhang, J. M. Nguyen, M. A. Lauber, D. Guillarme and V. D'Atri, J. Chromatogr., A, 2022, 1677, 463324 CrossRef CAS PubMed.
- M. Jora, D. Corcoran, G. G. Parungao, P. A. Lobue, L. F. L. Oliveira, G. Stan, B. Addepalli and P. A. Limbach, Anal. Chem., 2022, 94, 13958–13967 CrossRef CAS PubMed.
- M. R. Taylor, J. Kawakami and D. V. McCalley, J. Chromatogr., A, 2023, 1700, 464006 CrossRef CAS PubMed.
- P. Thakur, S. Abernathy, P. A. Limbach and B. Addepalli, Methods Enzymol., 2021, 658, 1–24 CAS.
- U. D. Neue, J. Chromatogr., A, 2008, 1184, 107–130 CrossRef CAS PubMed.
- W. Clarke, in Mass Spectrometry for the Clinical Laboratory, ed. H. Nair and W. Clarke, Academic Press, San Diego, 2017, pp. 1–15, DOI:10.1016/B978-0-12-800871-3.00001-8.
- C. D. Daub and N. M. Cann, Anal. Chem., 2011, 83, 8372–8376 CrossRef CAS PubMed.
- B. Basiri, M. M. Murph and M. G. Bartlett, J. Am. Soc. Mass Spectrom., 2017, 28, 1647–1656 CrossRef CAS PubMed.
- B. Basiri, H. van Hattum, W. D. van Dongen, M. M. Murph and M. G. Bartlett, J. Am. Soc. Mass Spectrom., 2017, 28, 190–199 CrossRef CAS PubMed.
- M. Donegan, J. M. Nguyen and M. Gilar, J. Chromatogr., A, 2022, 1666, 462860 CrossRef CAS PubMed.
- G. J. Guimaraes, J. G. Saad, V. Annavarapu and M. G. Bartlett, J. Am. Soc. Mass Spectrom., 2023, 34, 2691–2699 CrossRef CAS PubMed.
- N. Li, N. M. El Zahar, J. G. Saad, E. R. E. van der Hage and M. G. Bartlett, J. Chromatogr., A, 2018, 1580, 110–119 CrossRef CAS PubMed.
- A. Liu, M. Cheng, Y. Zhou and P. Deng, Int. J. Mol. Sci., 2022, 23, 15474 CrossRef CAS PubMed.
- S. Studzinska, Talanta, 2018, 176, 329–343 CrossRef CAS PubMed.
- B. Buszewski and S. Noga, Anal. Bioanal. Chem., 2012, 402, 231–247 CrossRef CAS PubMed.
- S. M. Melnikov, A. Höltzel, A. Seidel-Morgenstern and U. Tallarek, Angew. Chem., Int. Ed., 2012, 51, 6251–6254 CrossRef CAS PubMed.
- M. Gilar, K. D. Berthelette and T. H. Walter, J. Sep. Sci., 2022, 45, 3264–3275 CrossRef CAS PubMed.
- S. Bocian and B. Buszewski, J. Sep. Sci., 2012, 35, 1191–1200 CrossRef CAS PubMed.
- S. A. McLuckey, G. J. Van Berkel and G. L. Glish, J. Am. Soc. Mass Spectrom., 1992, 3, 60–70 CrossRef CAS PubMed.
- T.-Y. Huang, J. Liu and S. A. McLuckey, J. Am. Soc. Mass Spectrom., 2010, 21, 890–898 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01155d |
| This journal is © The Royal Society of Chemistry 2025 |