
α-MnO2 catalysts with efficient ozone-catalyzed decomposition under high humidity conditions†
Jiafan
Ji
,
Qianqian
Yan
,
Yi
Chen
,
Gaosheng
Zhao
 ,
Bin
Jia
*,
Li
Xu
and
Ping
Cheng
*
,
Bin
Jia
*,
Li
Xu
and
Ping
Cheng
*
School of Environmental and Chemical Engineering, Shanghai University, Shanghai, 200444, China. E-mail: jiabin_bj@shu.edu.cn; Pingcheng@shu.edu.cn
First published on 7th November 2024
Abstract
Ground-level ozone pollution poses significant risks to ecosystems and human health and requires effective control measures. This study focused on the monolithic ozone degradation catalyst based on powdered α-MnO2 and comprehensively investigated its catalytic performance, moisture resistance, and stability. The monolithic catalyst achieved the optimal catalytic activity with an ozone conversion rate of 99% after being calcined at 400 °C for 3 hours. The detailed characterization of the catalyst properties at pH = 1, 4, and 7 revealed the adverse effects of residual acid ions on the catalyst activity. The catalyst at pH = 7 had more oxygen vacancies, which was related to the reduction of sulfate ion residues and the exposure of more active sites during the washing process. At pH = 7 and a space velocity of 900![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, the conversion rates of α-MnO2 to 18 ppm ozone reached 100% and 95% within 3 hours under 90% relative humidity and dry conditions, respectively. In addition, the monolithic catalyst exhibited significant moisture resistance and performed well in continuous alternating humidity cycle tests and sustained high humidity. It still maintained 90% ozone decomposition efficiency after 3 hours of testing under high humidity conditions. Meanwhile, the α-MnO2 monolithic catalyst showed excellent stability, with an ozone conversion rate exceeding 99% during the 50 – hour test period. These findings highlight the great potential of the α-MnO2 monolithic catalyst in ozone removal applications.
000 h−1, the conversion rates of α-MnO2 to 18 ppm ozone reached 100% and 95% within 3 hours under 90% relative humidity and dry conditions, respectively. In addition, the monolithic catalyst exhibited significant moisture resistance and performed well in continuous alternating humidity cycle tests and sustained high humidity. It still maintained 90% ozone decomposition efficiency after 3 hours of testing under high humidity conditions. Meanwhile, the α-MnO2 monolithic catalyst showed excellent stability, with an ozone conversion rate exceeding 99% during the 50 – hour test period. These findings highlight the great potential of the α-MnO2 monolithic catalyst in ozone removal applications.
Introduction
Ozone (O3) in the stratosphere can protect life on Earth from the harmful effects of ultraviolet radiation. However, near the ground, ozone is one of the most pervasive air pollutants, with an extremely high oxidizing capacity that is harmful to public health and the environment.1 Near-surface ozone is primarily formed through the photochemical cycle of precursor volatile organic compounds (VOCs) and nitrogen oxides (NOX).2 In addition, anthropogenic sources of ozone, such as ozone emissions from sewage treatment plants, hospitals, UV curing equipment, corona discharges, indoor appliances, copy machines, and disinfection products, also pose a significant threat to the environment.3,4 Increases in outdoor ozone concentrations can exacerbate indoor ozone concentrations due to indoor–outdoor air exchange. Ozone is a powerful oxidizing agent, and high levels of exposure can have serious effects on human health, including cardiovascular and respiratory disorders, and increased mortality rates.5 In addition, near-surface Ozone is one of the major causes of forest mortality and crop yield loss.6 The research has shown that even if ozone levels in indoor air are below the mandated standard concentration, it can still lead to the production of various oxidation products. These oxidation products include unsaturated hydrocarbons, VOCs, and secondary organic aerosols (SOA) formed through the reaction of ozone with VOCs. The secondary pollutants that result from ozone pose a greater threat to human health than ozone itself.7 Therefore, it is important to study techniques for removing ozone in order to protect the environment, promote human health, and support economic development.Catalytic decomposition is widely considered the most effective method for low-temperature ozone treatment in both academic and industrial settings. This preference is based on its high efficiency in decomposition and technical feasibility. The active components of catalytic materials include noble metals (e.g., Pt, Au, Pd, and Ag)8,9 and transition metal oxides (e.g., MnOX, Fe2O3, NiO, and CuO).10–15 Transition metal oxides, especially manganese oxides, have been extensively studied due to their cost-effectiveness and superior catalytic activity. In recent years, there has been a significant increase in research on amorphous and low-valent manganese oxides. Compared with the materials prepared by traditional methods, these materials have limited crystal growth, poor crystallinity, and more crystal defects. Moreover, a larger surface area and a lower valence state of manganese endow them with better ozone-degrading capabilities. Meanwhile, the mild preparation conditions and simple preparation methods make these materials more practical for production. Yang et al.16 prepared six types of MnO2 with crystal phases of α-, β-, γ-, ε-, λ-, and δ-MnO2, and studied their catalytic activity for ozone-assisted catalytic oxidation of toluene at room temperature. The order of research is: δ-MnO2 > α-MnO2 > ε-MnO2 > γ-MnO2 > λ-MnO2 > β-MnO2. Ji et al.17 used potassium permanganate to treat carbon nanotubes (CNTs) and produce amorphous MnO2/CNTs. The porous structure of carbon nanotubes enables efficient gas diffusion. Additionally, in the composite material, the electron transfer between manganese oxide and graphite carbon enhances its catalytic activity. Liu et al.18 used manganese acetate and potassium permanganate to react and produce amorphous MnOX with a mesoporous structure at room temperature. This material has a high ozone decomposition activity due to its large surface area and abundant oxygen vacancies. The calculation results of the apparent reaction rate constant suggest that the presence of abundant low-valent Mn2+ and Mn3+ species is more favorable for ozone degradation compared to Mn4+. The unsaturated coordination of surface atoms in amorphous metal oxides creates many active sites and oxygen vacancies on the surface, resulting in a large number of catalytic reaction sites.19
However, amorphous materials synthesized under mild conditions usually suffer from the problem that the preparation process is easily disturbed. It should be particularly noted that the rapid deactivation under humid conditions limits their wide range of applications. Compared with widely studied catalysts, there are relatively few reports on the modification of amorphous manganese oxide catalysts to improve ozone degradation activity and moisture resistance, and to make them suitable for practical engineering. Therefore, it is of great significance to develop monolithic amorphous ozone-catalytic materials that can operate efficiently and stably in high-humidity environments and to explore the mechanisms behind their moisture resistance. Metal sulfates, nitrates, and chlorides are commonly used as sources for synthesizing manganese oxides. Residual ions adsorbed on the catalyst surface often affect its performance. They may block the active sites of the catalyst or inhibit the effective adsorption of reactants, thus having a negative impact on the catalytic behavior.20–23
The impact of residual acidic anions on the catalytic performance in ozone decomposition is often overlooked. Currently, research on how they affect the properties of amorphous manganese oxide is rather limited.24 Generally, the powder form of highly active manganese oxide catalysts prepared in the laboratory restricts their molding capabilities and increases the risk of dust contamination, which limits their application under actual atmospheric conditions. In order to meet the requirements of a wide range of applications, it is necessary to conduct large-scale synthesis of catalysts and construct monolithic catalysts.
In this study, we synthesized amorphous α-MnO2 using a simple and gentle preparation method. The precursor material was washed with deionized water at various pH values to investigate the impact of washing degree on the moisture resistance and ozone decomposition performance of α-MnO2. The results of the ozone decomposition indicate that under humid conditions (RH = 90%), catalysts with pH values of 4 and 7 exhibit higher ozone removal efficiency compared to the catalyst with a pH value of 1. The excellent properties of amorphous α-MnO2 were explored using different techniques, and the reaction process of ozonolysis on the catalyst surface in the presence of water was proposed. The research results indicate that through optimizing experimental parameters, a large amount of powder catalysts suitable for large-scale production has been prepared. Meanwhile, the catalyst was loaded onto a cordierite support to prepare a monolithic catalyst with strong activity and moisture resistance. This study aims to deepen our understanding of catalyst deactivation under high humidity conditions and contribute to the development of efficient and stable ozonolysis catalysts for practical environmental applications.
Materials and methods
Synthesis of catalysts
The powdered amorphous α-MnO2 catalyst was prepared using the redox precipitation method.25 The preparation process involved adding 200 mL of 0.1 mol L−1 KMnO4 solution to 200 mL of 0.15 mol L−1 MnSO4·H2O solution, creating a suspension. The mixture was then stirred for 6 hours. The formation of a deep brown precipitate occurred, which was subsequently washed with deionized water until the pH of the supernatant reached 1, 4, and 7. The precipitate was dried in air at 70 °C for 12 hours and then calcined at a heating rate of 2 °C min−1 to 300 °C for 3 hours. The prepared samples are labeled according to their experimental pH values: pH = 1, pH = 4, and pH = 7.The α-MnO2 monolithic catalyst was prepared by impregnating cordierite honeycomb ceramics (size: L 150 mm × W 150 mm × H 30 mm) with a loading of approximately 20 g L−1. The specific method involved scaling up the above powder preparation by 20 times to obtain α-MnO2 on a pilot scale. The α-MnO2 was dried in the air at 70 °C for 12 hours. The α-MnO2 powder catalyst was mixed with deionized water and then subjected to ball milling. After milling, a mixture of aluminum sol and binder was added and stirred for 12 hours to obtain α-MnO2 slurry. The cordierite honeycomb ceramics carrier was immersed in a prepared slurry. After impregnation and coating, the monolithic catalyst α-MnO2/CHM was formed through different temperature calcinations (300, 400, and 500 °C) for 3 hours.
Catalyst characterization
The catalysts were characterized using XRD, SEM, EDS, FTIR, XPS, O2− and H2O-TPD, and H2-TPR techniques to analyze their structures and properties. Additional information about these characterizations can be found in the ESI.†Catalytic activity evaluation
The activity tests for the powder samples were conducted in a fixed-bed continuous-flow quartz reactor with a 4 mm inner diameter. The catalyst size was 40–60 mesh. The experiments were conducted at a room temperature of 26 °C with a gas flow rate of 1500 ml min−1. The activity of the catalysts at pH values of 1, 4, and 7 was measured using a sample of 50 mg under conditions of 90% relative humidity and a space velocity of 900000 h−1. The activity of various pH catalysts at different space velocities (300000, 450000, 900000, 1800000, and 3600000 h−1) was determined under dry conditions by varying the catalyst dosage (150 mg, 100 mg, 50 mg, 25 mg, and 12.5 mg).
The prepared samples were cut into cylindrical shapes with a diameter of 20 mm and a thickness of 30 mm to measure the catalytic performance of the monolithic catalyst for ozone. The objects were then inserted into the quartz tube perpendicular to the wall. The ozone concentration was kept at 15 ppm, with a gas flow rate of 5 L min−1. The gas used was prepared using a zero air generator (model: AADCO 737), and ozone was generated using zero air. Ozone was produced by irradiating zero air with a vacuum ultraviolet lamp (10 W, China). The laboratory-made temperature and humidity monitor was used to measure the relative humidity of the airflow. The ozone concentrations at the inlet and outlet were measured using an ozone analyzer (model EC9810, Ecotech, Australia). The rate of ozone conversion was determined using the following equation:
| O3 conversion = (Cin − Cout)/Cin × 100% |
Results and discussion
Ozone decomposition catalyst performance
The catalytic performance of the synthesized samples at different pH levels (1, 4, and 7) was investigated for ozone decomposition. The experiments were conducted at room temperature under both dry and high-humidity conditions. As shown in Fig. 1a, all three catalysts demonstrated comparable high activity in decomposing ozone in a dry stream. After 3 hours of reaction at a space velocity of 900000 h−1, the conversion of all samples to 18 ppm ozone removal was nearly 100%. Water vapor is known to inhibit the catalytic activity of ozone decomposition. Ozone usually exists in high-humidity environments. For example, a large amount of ozone is contained in the wastewater and exhaust gases produced during the water treatment process. Therefore, it is necessary to explore the performance of the catalyst under a high relative humidity. To replicate real-world conditions, the performance of the prepared catalysts was tested under a high relative humidity of 90%. The catalytic activity of the pH = 1 and 4 catalysts were significantly affected by water vapor, resulting in a significant decline in ozone conversion rates within 3 hours. The pH = 1 catalyst reached a conversion rate of 50%, while the pH = 4 catalyst reached 79%. However, the catalytic activity of the pH = 7 catalyst was minimally affected by high humidity. It maintained a high degradation rate even under these conditions, with the activity still reaching 95% after 3 hours of reaction. This suggests that the pH = 7 catalyst has a higher resistance to humidity. The results above show that relative humidity has a significant impact on the performance of ozone decomposition catalysts, and the degree of washing greatly affects the catalyst's resistance to humidity.
 | ||
| Fig. 1 (a) Ozone conversion on catalysts with pH values of 1, 4, and 7 under dry and humid conditions; (b) effect of space velocity on ozone conversion over catalysts with pH values of 1, 4, and 7 under dry conditions; (c) ozone conversion over catalysts with pH values of 1, 4, and 7 at different relative humidity levels; (d) ozone conversion over catalysts with a pH value of 7 at 55% relative humidity and a space velocity of 900000 h−1. | ||
The catalytic performance of the catalysts at pH levels of 1, 4, and 7 in ozone decomposition was further investigated. The activity was evaluated at different space velocities under dry conditions, and the ozone decomposition activity after 3 hours is shown in Fig. 1b. All three catalysts achieved 100% ozone conversion rates at space velocities of 300000 h−1 and 600000 h−1. When the space velocity increased to 1800000 h−1, the catalyst with a pH of 7 maintained a 100% ozone conversion rate, while the catalysts with pH values of 1 and 4 had decreased ozone conversion rates of 90% and 94% respectively. If the space velocity increased to 3600000 h−1, the ozone conversion rate on the pH = 7 catalyst remained at 99%. However, the ozone conversion rates on the pH = 1 and 4 catalysts decreased significantly. This suggests that the pH = 1 and 4 catalysts perform worse than the pH = 7 catalyst at high space velocity, indicating that the washing degree significantly affects ozone decomposition under drying conditions. The presence of residual sulfate ions has adverse effects on the moisture resistance and high space velocity performance of the catalyst.
Continuous alternating humidity cycling experiments were conducted to investigate the catalytic decomposition activity under different humidity conditions. As shown in Fig. 1c, when the humidity varies from 0% to 90%, the ozone catalytic activities of all three catalysts remain at approximately 100% under dry conditions. When the reaction conditions were changed to high humidity for a continuous 7-hour experiment, the catalyst with a pH of 7 still maintained a relatively high ozone decomposition efficiency, slightly decreasing from 100% to 90%. The ozone conversion rates on the catalysts with a pH of 1 and a pH of 4 decreased significantly, with the pH = 1 catalyst dropping to 50% and the pH = 4 catalyst dropping to 70%. When returning to dry conditions, the activities of all three catalysts were fully restored. This suggests that the deactivation caused by the competitive adsorption of H2O molecules on the active sites is reversible for the amorphous α-MnO2 catalyst. Furthermore, it is easier for the H2O molecules to be desorbed from the active sites under dry conditions. Additionally, the pH = 7 catalyst underwent long-term stability testing at 55% RH. As shown in Fig. 1d, the catalyst consistently achieved a high ozone removal rate (>90%) for over 40 hours of reaction. Experiments have demonstrated that the catalyst with a pH of 7, which has a higher washing degree and fewer residual acid ions, exhibits better moisture resistance and high space velocity resistance, and can better catalyze the degradation of ozone.
Crystallinity and microstructure of the catalysts
The morphology of the amorphous α-MnO2 catalyst was characterized using SEM, as shown in Fig. 2. The catalysts at pH levels 1, 4, and 7 were composed of nanoparticle aggregates without clear boundaries between particles. The diameter of the spherical units was around 150 nm, suggesting a similar morphology for the catalysts at different pH values. The pH = 1 catalyst has a rough surface with interlaced needle-shaped nanorods distributed on the particles. As the pH value increased, the number of nanorods on the catalyst surface decreased gradually, while the uniformity of particle distribution increased. The observation suggests that residual acid ions on the surface have an influence on the morphology of the catalyst. The washing process can remove inert inorganic components from the catalyst, which exposes more reactive Mn sites and enhances catalytic performance. | ||
| Fig. 2 Scanning electron microscopy (SEM) images of the catalysts (a and b) pH = 1, (c and d) pH = 4, and (e and f) pH = 7. | ||
XRD characterization was used to analyze the variations in the crystalline structure of the catalysts. The XRD patterns of the catalysts at pH 1, 4, and 7 are shown in Fig. 3a. All samples showed broad and weak peaks, suggesting a low level of crystallinity and a typical amorphous structure. The diffraction peaks in the patterns closely matched the standard JCPDS card. The peaks at 12.53°, 27.51°, 37.34°, 41.53°, 56.33°, and 65.85° corresponded to the crystal faces (110), (310), (211), (301), (600), and (002) of α-MnO2 (JCPDS number 44-0141).26 Low crystallinity can introduce more defects and increase the surface area of the material, which is beneficial for the adsorption and degradation of ozone molecules. The crystal structures of the samples before and after washing were essentially unchanged, suggesting that the impact of washing on the catalyst's crystalline phase can be neglected. In addition, significant changes in the XRD peaks were observed among samples with different calcination temperatures, suggesting that the calcination temperature has a significant impact on the crystal structure of amorphous α-MnO2 and improves the degree of crystallinity of the material (Fig. S1†). Fig. 3b presented the FT-IR spectrum, where the stretching modes of Mn–O and O–Mn–O in the MnOX matrix give rise to the low-frequency peaks at 400–800 cm−1.27 Compared to the pH = 1 and 4 catalysts, the peak intensities of the pH = 7 catalyst were located at 1650 cm−1 and 2900–3600 cm−1, corresponding to the surface hydroxyl vibration bands. The intensity of the band decreases, indicating a decrease in the number of surface hydroxyl groups. Previous studies have also shown that hydroxyl groups can block the active sites on the catalyst surface active sites and adversely affect ozone decomposition. Additionally, the interaction between surface hydroxyl groups and H2O molecules is stronger, creating a competitive relationship between water and ozone molecules. This interaction has an impact on the catalytic decomposition of ozone in humid conditions.28,29 The catalyst continuously adsorbs polar molecules, such as water molecules, from the feed gas due to the abundance of surface hydroxyl groups and their strong hydrophilicity. This may explain the excellent performance of the pH = 7 catalyst under high humidity conditions.
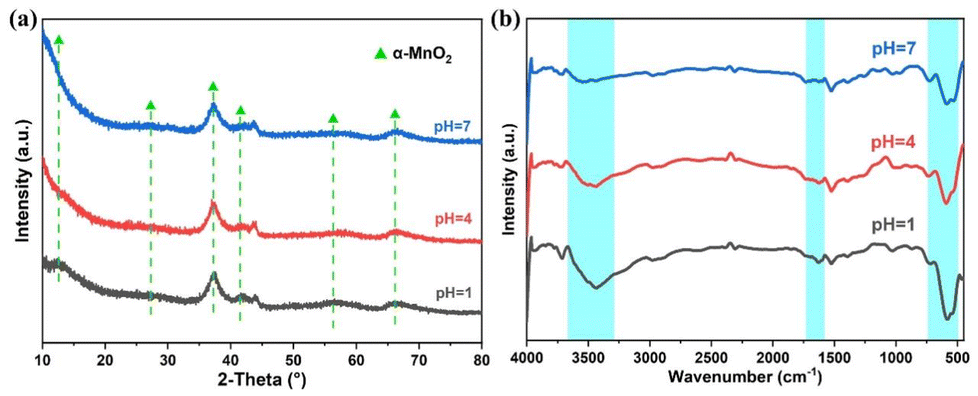 | ||
| Fig. 3 (a) XRD profiles and (b) FT-IR spectrum of pH = 1, 4, and 7 catalysts. | ||
Effects of acid ions on catalysts’ physical and chemical properties
To better understand the impact of residual acid ions on the catalyst, we analyzed the reducibility and oxygen migration rate of the samples using H2-TPR and O2-TPD, respectively. Fig. 4a shows the H2-TPR spectra of amorphous α-MnO2 catalysts at pH levels of 1, 4, and 7. The catalysts at pH 4 and 7 exhibited three main reduction peaks that were positioned similarly, suggesting comparable reducibility. The first reduction peak at low temperature is typically attributed to the reduction of Mn4+ to Mn3+, while the second reduction peak is associated with the reduction of Mn3+ to Mn2+.30 The catalyst with a pH of 1 showed two reduction peaks at temperatures of 305 °C and 435 °C, leading to a decrease in hydrogen consumption. The catalysts with pH values of 4 and 7 exhibited lower reduction temperatures and consumed more hydrogen compared to the catalyst with a pH value of 1. The reduction temperature is typically an indicator of the catalyst's reducibility, with lower temperatures suggesting higher reducibility. The high concentration of sulfate anion residues on the pH = 1 catalyst's surface hinders its redox behavior, resulting in a decrease in catalytic activity. Enhanced reducibility speeds up ozone decomposition and enhances catalytic activity.31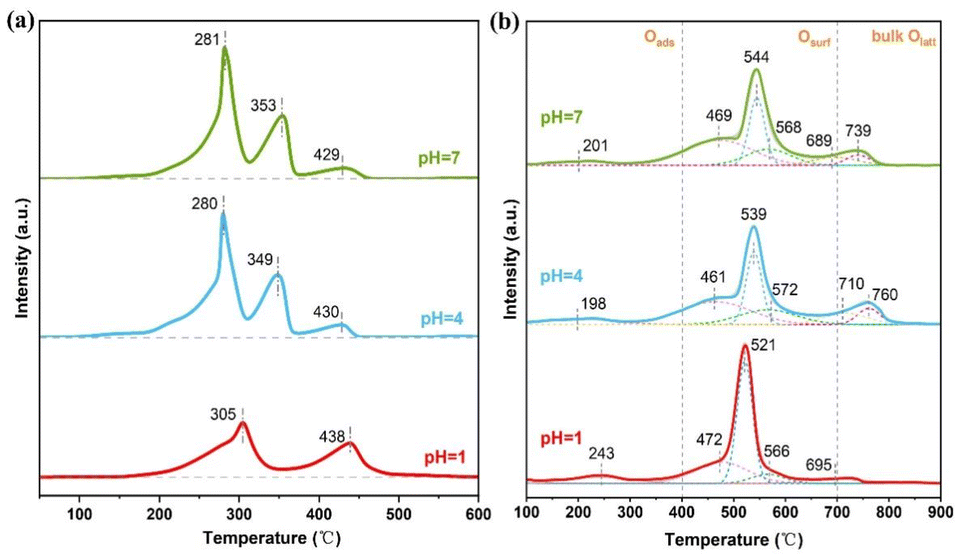 | ||
| Fig. 4 (a) H2-TPR spectra and (b) O2-TPD spectra for pH = 1, 4, and 7 catalysts. | ||
The results of the O2-TPD experiment can indicate the adsorption strength and the quantity of oxygen on the catalyst surface. The oxygen desorption processes of the three catalysts were examined using O2-TPD, as shown in Fig. 4b. The oxygen desorption curves of the MnO2 catalyst can be divided into three regions: a low-temperature region below 400 °C, a medium-temperature region of 400–700 °C, and a high-temperature region above 700 °C.32 Generally, peaks below 400 °C are attributed to the release of oxygen that is adsorbed on the surface. The peaks between 400 and 700 °C are caused by the release of surface lattice oxygen, while the peaks above 700 °C are attributed to the desorption of bulk lattice oxygen and the decomposition of the catalyst itself.33 The lower desorption peak temperature of the first peak indicates that the surface-adsorbed oxygen is released at lower temperatures, suggesting a weaker binding of oxygen species to the catalyst surface. This weaker binding is beneficial for ozone decomposition. According to the relevant reaction mechanism, oxygen vacancies play a crucial role in the ozone decomposition process. The release of surface-adsorbed oxygen and its binding to the catalyst surface can affect ozone decomposition. When oxygen is more easily released from the surface, it will promote the progress of the ozone decomposition reaction, thus enhancing the ozone decomposition effect.34 The catalysts at pH 4 and 7 showed minimal differences in the temperature of the initial desorption peak, suggesting that they had similar abilities to adsorb surface oxygen species. The desorption peak for the pH = 1 catalyst below 400 °C shifted to higher temperatures, suggesting a more difficult removal of surface oxygen molecules. This phenomenon hampers the effectiveness of ozone decomposition. The catalysts at pH 4 and 7 showed desorption of bulk lattice oxygen, indicating that the presence of residual acid ions hinders the presence of surface lattice oxygen species. It can be observed that the pH = 7 catalyst has a lower temperature for bulk lattice oxygen desorption. This suggests that as the washing degree improves, the bulk lattice oxygen becomes more unstable, leading to an increased oxygen migration and promoting the formation of oxygen vacancies.35,36 The presence of oxygen vacancies on the catalyst surface enhances the degradation of ozone. Thus, the catalyst with a pH of 7 showed the best performance in decomposing ozone.
The XPS analysis of amorphous α-MnO2 revealed the surface valence electron states, as showed in Fig. 5 and Table S1.† The Mn 2p3/2 spectrum of the α-MnO2 catalyst in Fig. 5a shows two distinct peaks at 642.0–643.0 eV and 644.0–645.0 eV, which correspond to Mn3+ and Mn4+ respectively.37 α-MnO2 possesses a specific crystal structure, and the internal atomic arrangement and coordination situation can influence the electronic state of Mn. In this study, there exist certain crystal defects in α-MnO2. The charge balance caused by the loss of lattice oxygen leads to the abundant presence of Mn3+.38 The cation-vacancy model assumes that the oxygen sublattice is complete and vacancies only occur on the manganese sublattice.39,40 The cation defects are manifested as the change of the oxidation state from Mn4+ to Mn3+ and the appearance of manganese cation vacancies. In the vicinity of the oxygen reduction reaction (ORR) region, when manganese dioxide is reduced to manganite, the Mn–O bonds in the MnO6 octahedron are forced to form a distorted Mn3+O6 octahedron (with four short and two long Mn–O bonds), and this change is attributed to the Jahn–Teller distortion and hydrogen bonding. During this process, the appearance of oxygen vacancies is associated with the formation of Mn3+, that is, the existence of oxygen vacancies may prompt the transformation from Mn4+ to Mn3+ and the structural distortion. Moreover, oxygen vacancies play a crucial role in catalytic reactions, such as adsorbing and activating reactant molecules.41,42 Therefore, the phenomenon of Mn3+ being dominant in this study may be an important factor for the good catalytic performance of α-MnO2. The XPS spectra of O 1s in Fig. 5b exhibited similar results. The peaks at ∼529.4 eV, ∼531.3 eV, and ∼533.0 eV were assigned to lattice oxygen (Olatt), surface-adsorbed oxygen (Oads), and surface-adsorbed water (H2Oads), respectively.43,44
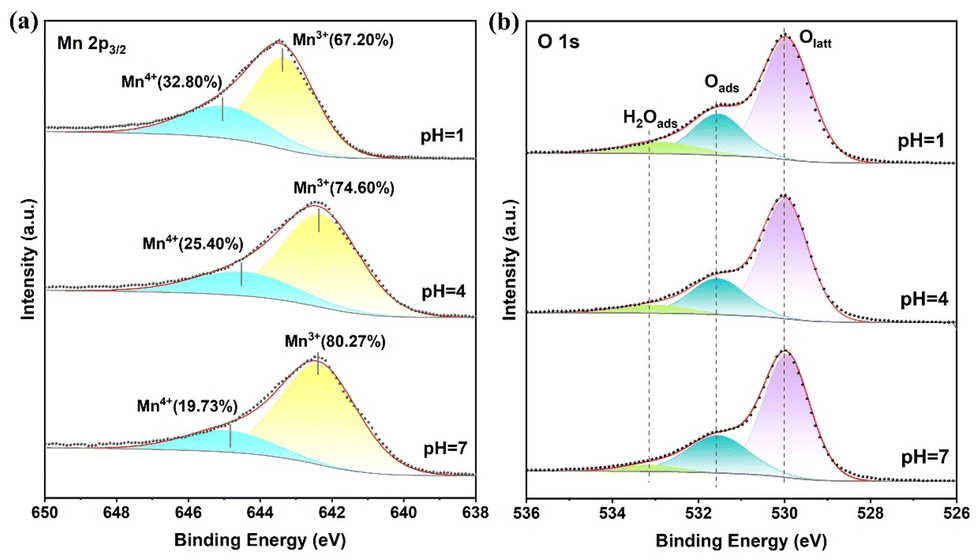 | ||
| Fig. 5 (a) Mn 2p and (b) O 1s XPS spectra of pH = 1, 4 and 7 catalysts. | ||
Oxygen tends to preferentially adsorb or activate on the oxygen vacancies of the MnO2 surface. Therefore, the oxygen adsorbed on the surface corresponds to the surface oxygen vacancies. The catalyst with a pH of 7 had a higher abundance of adsorbable oxygen, as shown in Table S1.† This suggests that there is a greater presence of surface oxygen vacancies. The S 2p spectra of different samples showed notable variations (Fig. S2†). The catalyst with a pH of 7 shows a distinct signal at 168.5 eV, indicating the presence of elemental sulfur.45
In this experiment, no sulfur-containing anions other than SO42− were introduced, so it can be determined that this is attributed to sulfate ions.46 Changes in pH affect the chemical environment and the state of a substance's chemical bonds. At different pH conditions, sulfur-containing compounds may undergo protonation or deprotonation, resulting in changes in their electronic structure that affect the central position of S 2p peaks. At the same time, changes in pH value may cause changes in intermolecular or intramolecular interactions. For example, at lower pH, surface hydroxyl groups may interact more with sulfates, causing the S 2p peak position to shift slightly.
According to Li et al., the desorption of SO42− from the catalyst surface was challenging, and surface sulfates only decomposed into SO2 and O2 at a temperature of 600 °C. The elemental S on the surfaces of the three catalysts primarily comes from SO42−. The relative intensities of the S 2p peaks followed the order of pH = 1 > pH = 4 > pH = 7. After treatment with deionized water, the S signal gradually disappeared, indicating a decrease in the surface SO42− content for the pH = 1, 4, and 7 catalysts. The amount of surface adsorbed water and S content on the catalysts increased as the pH value decreased. The presence of SO42− residue enhances the water adsorption capacity of the catalyst, suggesting that the residual SO42− on the catalyst surface affects its hydrophobicity.
Mechanism of residual sulfate ions’ impact on catalyst performance
The variances in water resistance among the catalysts can be better understood through H2O adsorption–desorption experiments conducted on the catalyst surfaces. The H2O-TPD spectra of the catalysts at pH 1, 4, and 7 are shown in Fig. 6. To gain a better understanding of the interaction between H2O and the catalyst, the H2O-TPD profiles were analyzed using Gaussian functions. The spectra can be divided into five peaks at temperatures below 120 °C, 120–200 °C, 200–350 °C, and above 350 °C. The peak below 120 °C corresponds to Type I water, which indicates water molecules located between the particles and not interacting with the catalyst surface.47 | ||
| Fig. 6 H2O-TPD spectra of catalysts: (a) pH = 1; (b) pH = 4; (c) pH = 7. | ||
The peak in the temperature range of 120–200 °C was identified as Type II water, which is believed to be caused by water molecules interacting with the catalyst surface through hydrogen bonding. The peak observed between 200–350 °C was identified as chemisorbed water (Type III water) on the surface of the catalyst. The peak above 350 °C was attributed to Type IV water, which is believed to be the dissociation of surface hydrophilic groups. The results showed that the pH = 7 catalyst had the least amount of Type III water. Weak water adsorption performance reduces the occupation of water molecules on the catalyst's active sites, promoting their desorption and preventing occupation of the reactive sites during the reaction. The catalyst with a pH of 7 showed greater resistance to moisture. The catalyst with a pH of 1 has a higher desorption temperature and adsorption capacity above 350 °C simultaneously, which is why it is prone to deactivation in a high-humidity environment. At the molecular level, acid ions such as sulfate on the catalyst surface may interact with water molecules through hydrogen bonds, thereby enhancing the adsorption of water molecules. At pH = 7, the number of these ions decreases, thereby weakening the adsorption capacity of water molecules on the catalyst surface.
By washing the catalyst surface, the remaining SO42− ions can be eliminated, reducing the catalyst's ability to adsorb water molecules and minimizing the competition between water and ozone molecules for adsorption. The pH = 7 catalyst demonstrated the best ozone catalytic degradation performance under humid conditions, compared to the pH = 1 and pH = 4 catalysts. According to the oxygen vacancy reaction mechanism,48 the activity of ozone decomposition is primarily determined by the number and nature of oxygen vacancies in the catalyst. Under humid conditions, water molecules fill the oxygen vacancies, gradually blocking the active sites and hindering further reactions. The α-MnO2 catalyst exhibited an amorphous structure, high specific surface area, and an abundance of oxygen vacancy active sites. Studies using XPS, O2-TPD, and H2-TPR techniques showed that the catalyst with a pH of 7, which had fewer residual sulfate ions, had more oxygen vacancies. Consequently, it exhibited excellent ozone degradation activity and high resistance to high space velocities in dry conditions. The H2O-TPD test showed that the catalyst with pH 7 was more hydrophobic than the catalyst with pH 1 and 4. This allowed it to have a higher adsorption capacity for gaseous ozone molecules, which was beneficial for decomposing ozone in humid conditions.
Based on the research analysis and experimental results, it was concluded that sulfate ions binding tightly to the catalyst's active sites were responsible for catalyst deactivation in humid conditions. The potential mechanism by which surface residual sulfate ions impact the catalyst's ozone decomposition process was proposed. The residual sulfate ions on the surface adsorb onto the oxygen vacancies of the catalyst, reducing the number of vacancies and limiting the access of reactive gases to the active sites. This resulted in reduced ozone decomposition performance and decreased resistance to high space velocities in dry conditions. As shown in Fig. 7, in humid conditions, sulfate ions have a stronger attraction to water molecules. This causes the active sites of the catalyst to be filled up rapidly, hindering the ongoing ozone decomposition reaction. This phenomenon leads to a gradual decline in the performance of the catalyst until it is deactivated.
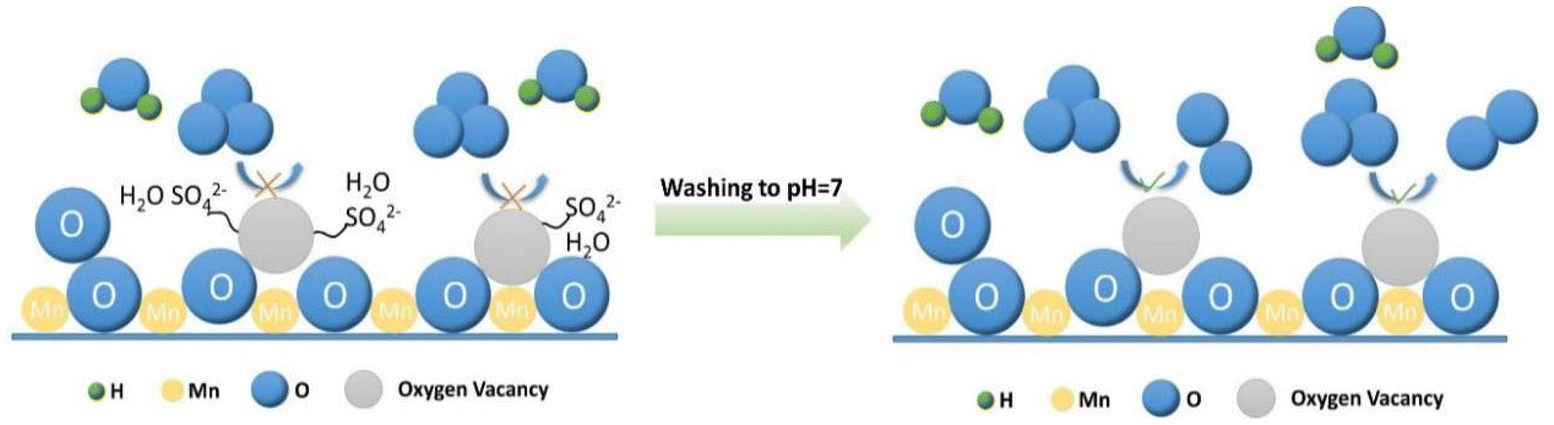 | ||
| Fig. 7 Effect of residual sulfate ions on catalyst degradation of ozone in humid conditions. | ||
The washing treatment removed residual sulfate ions from the catalyst surface, exposing more active sites.49 This enhanced the catalyst's adsorption capacity for gaseous ozone molecules, which was beneficial for ozone decomposition in humid conditions. Based on the aforementioned results and discussions, effectively removing residual sulfate ions is essential for enhancing the performance of amorphous α-MnO2 catalysts. Appropriate washing treatment improves the catalyst's resistance to high space velocity and humidity by effectively removing residues. This observation provides valuable insights for practical applications.
Monolithic catalyst
The geometric shape of the catalyst is essential for practical applications. Monolithic catalysts are a pioneering catalyst archetype that offer several advantageous attributes, including reduced bed pressure drop, improved mass and heat transfer efficiency, and increased mechanical robustness compared to conventional powdered catalysts.50,51 Generally, the preparation of a monolithic catalyst consists of several steps, including the preparation of catalytically active components, the production of an active slurry, and the impregnation or coating of the slurry onto a support material. Inspired by the efficient use of powdered materials in ozone decomposition, researchers have developed monolithic catalysts using impregnation and coating methods. These catalysts serve as a proof of concept for effectively removing high flux and low concentration ozone.The photographs and SEM images of the monolithic catalyst (ϕd = 20 mm, δ = 30 mm) after being calcined at 400 °C for 4 hours are shown in Fig. 8 and 9, respectively. The pristine cordierite blank support in Fig. 9a had a rough surface and was free of any foreign matter. Upon loading the catalyst, Fig. 9b and c showed that the α-MnO2 particles fully covered the monolith's surface, indicating a uniform dispersion on the support. This was further confirmed by the energy dispersive X-ray spectroscopy (EDS) elemental mapping images of a selected area of the α-MnO2 monolithic catalyst (Fig. S3†). It was observed that Mn, Al, and Mg were present on the catalyst surface as Mn oxide, Al oxide, and Mg oxide, respectively.
 | ||
| Fig. 8 (a) Blank support; (b) α-MnO2 monolithic catalyst; (c) blank support and α-MnO2 monolithic catalyst after cutting for activity testing. | ||
 | ||
| Fig. 9 SEM images of (a) blank support and (b and c) α-MnO2 monolithic catalyst. | ||
The XRD patterns of the α-MnO2 monolithic catalyst, the blank support, and the powder catalyst are shown in Fig. 10. The α-MnO2 monolithic catalyst showed similar peak positions to the blank support, indicating a spinel structure. The main components contributing to these peaks are a mixture of aluminum oxide, silicon oxide, and magnesium oxide.52 However, there were significant changes in the peak intensities, indicating a reduction in the crystallinity of the α-MnO2 monolithic catalysts. The poor crystallization and amorphous nature of α-MnO2 resulted in a reduction in the crystallinity of the monolithic catalysts. The findings confirmed the successful loading of amorphous α-MnO2 onto the support surface.
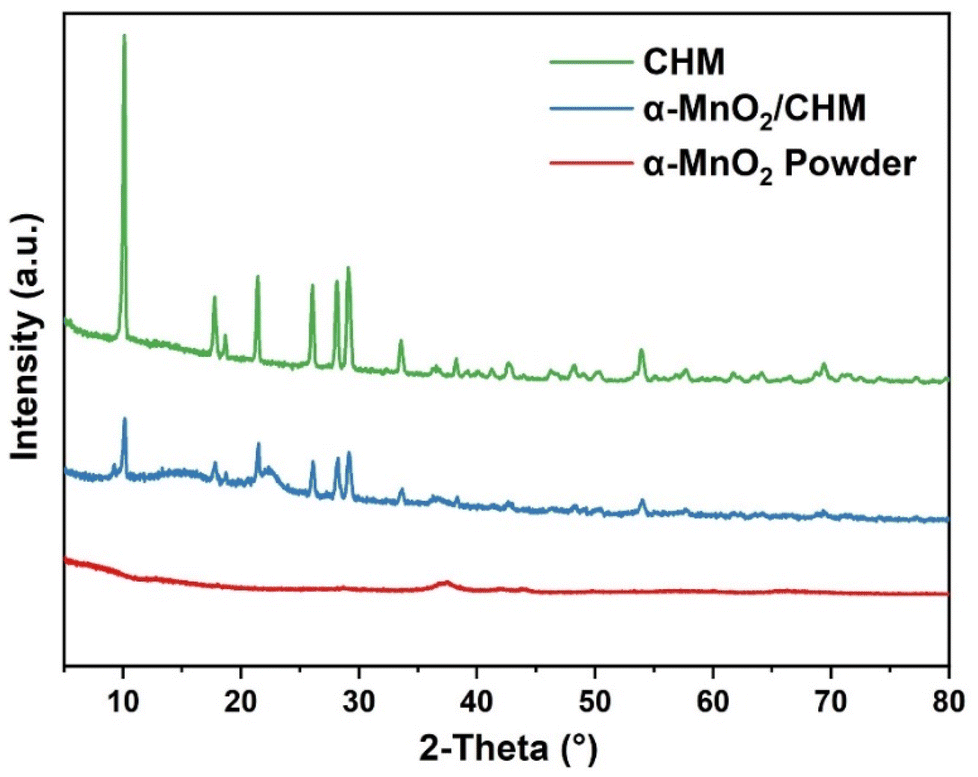 | ||
| Fig. 10 XRD patterns of blank support, α-MnO2 monolithic catalyst, and powder catalyst. | ||
The monolithic catalyst's activity was tested by exposing it to 15 ppm ozone. The performance of the catalyst was influenced by the duration and temperature of the calcination process. Fig. 11a shows the ozone catalytic degradation curves of the monolithic catalyst at various calcination temperatures. The figure shows that the highest catalytic activity was achieved after calcination at 400 °C, with a 99% ozone conversion rate maintained after 3 hours. Higher temperatures promote the decomposition and volatilization of organic compounds in the binder, leading to increased catalytic activity. Calcining amorphous MnO2 at different temperatures can effectively adjust the crystallinity of MnO2. After high-temperature calcination, MnO2 shows significantly lower activity due to fewer active sites and phase changes.29 As shown in Fig. S1,† the reduced activity observed in the product calcined at 500 °C can be attributed to changes in the crystal structure of the active component. The transformation from an amorphous configuration to a well-defined crystal structure weakened the bonding forces within the coating. When the calcination temperature reaches 500 °C, the surface structure of the catalyst is loose, the sheet structure is destroyed, and a large number of small sheet or rod-like structure materials are found, the pore size and pore volume increase, and the specific surface area decrease, resulting in the oxidation activity of the catalyst. As shown in Fig. 11b, the monolithic catalyst achieved complete ozone degradation under dry conditions after being calcined at 400 °C. Despite a decrease in catalyst activity under high humidity, its ozone decomposition efficiency remained at 90% after 3 hours of testing, indicating strong resistance to humidity.
 | ||
| Fig. 11 (a) Ozone catalytic activity of α-MnO2 monolithic catalysts at different calcination temperatures; (b and c) catalytic activity of α-MnO2 monolithic catalyst at different humidity. | ||
To further investigate the humidity resistance of the α-MnO2 monolithic catalyst, continuous alternating humidity cycling tests were conducted. When the humidity varies between 0% and 90%, the ozone catalytic activity of the α-MnO2 monolithic catalyst remains approximately 100% under dry conditions (Fig. 11c). Once the reaction conditions were changed to high humidity for a continuous 7-hour experiment, the efficiency of ozone decomposition on the pH = 7 catalyst decreased from 100% to 80%, indicating a significant decrease in activity. Additionally, when returning to low humidity conditions, the activity was fully restored. This demonstrates the exceptional humidity resistance of the α-MnO2 monolithic catalyst. In industrial applications, the stability of catalysts is crucial, particularly for maintaining ozone catalytic degradation activity over extended periods of continuous operation.53 The stability of the monolithic catalyst was evaluated under ambient temperature and pressure. The evaluation included a space velocity of 30000 h−1 and an ozone concentration of 15 ppm (Fig. S4†). During the 50-hour test, the catalyst maintained an ozone conversion rate of over 99% under constant space velocity and ozone concentration, demonstrating excellent stability. The results clearly highlight the significant potential of the monolithic catalyst in removing ozone, particularly in environments with high humidity.
Conclusions
In this study, three catalysts with different levels of sulfate ion residues were prepared using mild oxidative–reductive methods and different post-treatment washing procedures. The performance of these catalysts in ozone catalytic decomposition was evaluated. Based on powdered catalysts, monolithic catalysts with operational flexibility were prepared.Firstly, the crucial influence of calcination temperature on the catalytic activity of α-MnO2 catalysts was elucidated. The monolithic catalyst achieved the optimal catalytic activity after being calcined at 400 °C for 3 hours, with an ozone conversion rate of 99%. Higher temperatures led to a decrease in activity due to phase transitions and a reduction in active sites, highlighting the importance of carefully controlling the calcination process for catalyst optimization. Secondly, compared with the catalysts with pH values of 1 and 4, the catalyst with pH = 7 exhibited better ozone decomposition performance. Through characterization experiments, a mechanism was proposed to explain how the surface-residual sulfate anions affected the performance of the catalyst. It was found that the acidic anions on the catalyst surface decreased its hydrophobicity and covered the active sites, making it more prone to water molecule adsorption and competition with ozone molecules, ultimately reducing the performance of the catalyst under humid conditions. Meanwhile, the catalyst with pH = 7 had more oxygen vacancies, which was related to the reduction of residual sulfate ions and the exposure of active sites. Oxygen preferentially adsorbed on these vacancies, increasing the ability to decompose ozone. In addition, the monolithic catalyst also exhibited excellent moisture resistance. Under dry and humid conditions (with a relative humidity of 90% and a space velocity of 900000 h−1) and an ozone concentration of 18 ppm, the ozone removal rates of the catalyst with pH = 7 were 100% and 95% after 3 hours of reaction, respectively. In cycling tests, the α-MnO2 monolithic catalyst exhibited excellent stability during a 50-hour test period at a space velocity of 30000 h−1 and an ozone concentration of 15 ppm, with an ozone conversion rate exceeding 99%.
In summary, this study provides valuable insights into the efficacy and practical applications of α-MnO2 monolithic catalysts in ozone catalytic decomposition, considering factors such as calcination temperature, residual sulfate ions and pH value, oxygen vacancies, and moisture resistance.
Author contributions
Jiafan Ji (first author): methodology, investigation, formal analysis, writing – original draft; Qianqian Yan (Co-first author): methodology, investigation, data curation, writing – original draft; Yi Chen: conceptualization, visualization, investigation; Gaosheng Zhao: resources, supervision; Bin Jia (Co-corresponding author): conceptualization, resources, supervision, writing – review & editing; Li Xu: writing – review & editing; Ping Cheng (corresponding author): conceptualization, funding acquisition, resources, supervision, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 42277217, 41877374).References
- H. Z. Liu, J. F. Liu, Y. Liu, K. Yi, H. Z. Yang, S. L. Xiang, J. M. Ma and S. Tao, Atmos. Environ., 2021, 265, 10 Search PubMed.
- Y. Chen, M. Wang, Y. Yao, C. Zeng, W. Zhang, H. Yan, P. Gao, L. Fan and D. Ye, Sci. Total Environ., 2023, 869, 11 Search PubMed.
- B. W. Li, X. C. Zhao, X. H. Li, X. G. Y. Hu, L. T. Hu, D. Chen, M. D. An, Y. Yang, R. Feng, L. Y. Guo, P. N. Jiang, B. Yao, J. X. Hu and X. K. Fang, J. Cleaner Prod., 2023, 414, 9 Search PubMed.
- G. Viteri, A. Aranda, Y. D. de Mera, A. Rodríguez and D. Rodríguez, Sci. Total Environ., 2023, 858, 11 CrossRef PubMed.
- C. An, H. Li, Y. Ji, W. Chu, X. Yan and F. Chai, Sci. Total Environ., 2024, 921, 14 CrossRef PubMed.
- J. Zhang, H. Ran, Y. Guo, C. Xue, X. Liu, Y. Qu, Y. Sun, Q. Zhang, Y. Mu, Y. Chen, J. Wang and J. An, Sci. Total Environ., 2022, 803, 12 Search PubMed.
- H. L. Macintyre, C. Mitsakou, M. Vieno, M. R. Heal, C. Heaviside and K. S. Exley, Environ. Int., 2023, 178, 9 CrossRef PubMed.
- A. Lejeune, A. Cabrol, R. Lebullenger, A. Denicourt-Nowicki, A. Roucoux, A. Szymczyk, A. Couvert and P. F. Biard, ACS Sustainable Chem. Eng., 2020, 8, 2854–2864 CrossRef CAS.
- L. G. Tao, Z. Q. Zhang, P. J. Chen, G. F. Zhao, Y. Liu and Y. Lu, Appl. Surf. Sci., 2019, 481, 802–810 CrossRef CAS.
- S. Y. Gong, Z. Xie, W. M. Li, X. F. Wu, N. Han and Y. F. Chen, Appl. Catal., B, 2019, 241, 578–587 CrossRef CAS.
- Y. Ma, L. Wang, J. Ma, F. Liu, H. Einaga and H. He, Environ. Sci. Technol., 2022, 56, 9751–9761 CrossRef CAS PubMed.
- L. Yang, Y. Z. Li, Y. D. Sun, W. Wang and Z. P. Shao, Energy Environ. Mater., 2022, 5, 751–776 CrossRef CAS.
- T. Zumbar, I. Arcon, P. Djinovic, G. Aquilanti, G. Zerjav, A. Pintar, A. Ristic, G. Drazic, J. Volavsek, G. Mali, M. Popova, N. Zabukovec Logar and N. Novak Tusar, ACS Appl. Mater. Interfaces, 2023, 15, 28747–28762 CrossRef CAS PubMed.
- H. N. Ly, V. Parasuraman, H. Lee, M. Sheraz, A. Anus, W. R. Lee and S. Kim, Chemosphere, 2024, 351, 141261 CrossRef CAS PubMed.
- S. Y. Gong, A. Q. Wang, Y. Wang, H. D. Liu, N. Han and Y. F. Chen, ACS Appl. Nano Mater., 2020, 3, 597–607 CrossRef CAS.
- R. Yang, Z. Guo, L. Cai, R. Zhu, Y. Fan, Y. Zhang, P. Han, W. Zhang, X. Zhu, Q. Zhao, Z. Zhu, C. K. Chan and Z. Zeng, Small, 2021, 17, 11 Search PubMed.
- J. Ji, Y. Fang, L. S. He and H. B. Huang, Catal. Sci. Technol., 2019, 9, 4036–4046 RSC.
- S. L. Liu, J. Ji, Y. Yu and H. B. Huang, Catal. Sci. Technol., 2018, 8, 4264–4273 RSC.
- Y. H. Wang, Y. L. Sun, M. C. Gao, C. Z. Zhou, Y. J. Xin, G. S. Zhang, P. Xu and D. Ma, J. Cleaner Prod., 2022, 380, 15 Search PubMed.
- M. H. Li, F. F. Bi, Y. D. Xu, P. P. Hao, K. Xiang, Y. Zhang, S. Y. Chen, J. Guo, X. F. Guo and W. P. Ding, ACS Catal., 2019, 9, 11676–11684 CrossRef CAS.
- P. Wu, S. Q. Zhao, J. W. Yu, X. J. Jin, D. Q. Ye, S. H. Yang and Y. C. Qiu, ACS Appl. Mater. Interfaces, 2020, 12, 50566–50572 CrossRef CAS PubMed.
- C. L. Chen, H. J. Li, J. Z. Chen, D. Li, W. X. Chen, J. C. Dong, M. R. Sun and Y. J. Li, Energy Environ. Mater., 2023, 6, 8 Search PubMed.
- L. J. Zhong, D. Q. He, J. X. Xie, J. Zhong, Z. H. Kang, X. Yang, P. Y. Liu, Z. H. Sun, A. Mahmood, D. D. Wang, S. Y. Gan, Y. Bao and L. Niu, Chem. Eng. J., 2022, 435, 9 Search PubMed.
- R. Hu, G. Liu, H. Zhang, H. Xue, X. Wang and P. K. S. Lam, J. Cleaner Prod., 2020, 246, 11 CrossRef.
- J. Zeng, H. M. Xie, H. Y. Zhang, M. Huang, X. C. Liu, G. L. Zhou and Y. Jiang, Chemosphere, 2022, 291, 10 Search PubMed.
- F. Wang, Y. Y. Zheng, Q. H. Chen, Z. J. Yan, D. W. Lan, E. Lester and T. Wu, Coord. Chem. Rev., 2024, 500, 24 Search PubMed.
- D. L. Fang, B. C. Wu, A. Q. Mao, Y. Yan and C. H. Zheng, J. Alloys Compd., 2010, 507, 526–530 CrossRef CAS.
- R. R. Cao, L. X. Li and P. Y. Zhang, J. Hazard. Mater., 2021, 407, 10 CrossRef PubMed.
- Y. Yu, S. Liu, J. Ji and H. J. C. S. Huang, Catal. Sci. Technol., 2019, 9, 5090–5099 RSC.
- J. M. Shao, F. W. Lin, Z. H. Wang, P. X. Liu, H. R. Tang, Y. He and K. F. Cen, Appl. Catal., B, 2020, 266, 11 CrossRef.
- F. Liu, S. P. Rong, P. Y. Zhang and L. L. Gao, Appl. Catal., B, 2018, 235, 158–167 CrossRef CAS.
- S. L. Yang, H. C. Yang, J. Y. Yang, H. L. Qi, J. Kong, Z. Bo, X. D. Li, J. H. Yan, K. F. Cen and X. Tu, Chem. Eng. J., 2020, 402, 11 Search PubMed.
- W. Hong, T. L. Zhu, Y. Sun, H. N. Wang, X. Li and F. X. Shen, Environ. Sci. Technol., 2019, 53, 13332–13343 CrossRef PubMed.
- X. T. Li, J. Z. Ma, L. Yang, G. Z. He, C. B. Zhang, R. D. Zhang and H. He, Environ. Sci. Technol., 2018, 52, 12685–12696 CrossRef CAS PubMed.
- A. J. Wang, H. Zhao, Y. Wu, Q. Y. Zhang and C. Han, J. Environ. Sci., 2023, 131, 151–161 CrossRef CAS PubMed.
- Y. X. Zhao, Y. F. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2019, 31, 9 Search PubMed.
- N. Chubar, M. Szlachta and V. Gerda, ACS Appl. Mater. Interfaces, 2023, 15, 44572–44588 CrossRef CAS PubMed.
- J. Z. Ling, A. M. Gao, Y. L. Huang, F. Y. Yi, Q. Z. Li, G. Y. Wang, Y. Q. Liu and D. Shu, Chem. Eng. J., 2023, 452, 11 CrossRef.
- G. Cheng, S. L. Xie, B. Lan, X. Y. Zheng, F. Ye, M. Sun, X. H. Lu and L. Yu, J. Mater. Chem. A, 2016, 4, 16462–16468 RSC.
- K. Selvakumar, S. M. S. Kumar, R. Thangamuthu, K. Ganesan, P. Murugan, P. Rajput, S. N. Jha and D. Bhattacharyya, J. Phys. Chem. C, 2015, 119, 6604–6618 CrossRef CAS.
- H. Deng, C. B. Zhang, Y. Q. Lu, T. T. Pan and H. He, ACS Appl. Mater. Interfaces, 2023, 9362–9372, DOI:10.1021/acsami.2c21120.
- W. Liu, W. J. Xiang, X. Chen, Z. X. Song, C. X. Gao, N. Tsubaki and X. J. Zhang, Fuel, 2022, 312, 12 Search PubMed.
- L. Miao, J. L. Wang and P. Y. Zhang, Appl. Surf. Sci., 2019, 466, 441–453 CrossRef CAS.
- B. Lei, H. W. Zhang, W. W. Liu, Q. Zhao, Y. Wei, Y. Y. Lu, X. W. Yang, W. Q. Zhang, T. T. Xiao, J. L. Kong and W. P. Cai, Sens. Actuators, B, 2024, 401, 12 CrossRef.
- R. Hao, X. Wang, X. Zhao, M. Xu, Y. Zhao, X. Mao, B. Yuan, Y. Zhang and K. Gao, Chem. Eng. J., 2018, 333, 583–593 CrossRef CAS.
- D. J. Peng, Z. J. Zhou, Y. Zhou, Q. Guo, S. Y. Peng, X. W. Liu and M. H. Xu, Fuel, 2024, 363, 10 Search PubMed.
- C. Y. Ma, C. G. Yang, B. Wang, C. Chen, F. B. Wang, X. L. Yao and M. Y. Song, Appl. Catal., B, 2019, 254, 76–85 CrossRef CAS.
- Z. Wu, P. Y. Zhang, S. P. Rong and J. B. Jia, Appl. Catal., B, 2023, 335, 12 Search PubMed.
- X. Li, J. Ma, C. Zhang, R. Zhang and H. He, J. Environ. Sci., 2020, 91, 43–53 CrossRef CAS PubMed.
- Q. Peng, Y. J. Zhang, W. L. Zhong, K. Liu, J. J. Xing and X. K. Tang, J. Water Process Eng., 2023, 56, 12 Search PubMed.
- L. Kiewidt and J. Thöming, Chem. Eng. J., 2019, 359, 496–504 CrossRef CAS.
- N. Saheb, S. Lamara, F. Sahnoune and S. F. Hassan, Ceram. Int., 2022, 48, 23921–23930 CrossRef CAS.
- M. Namdari, C. S. Lee and F. Haghighat, Build. Environ., 2021, 187, 16 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01095g |
| This journal is © The Royal Society of Chemistry 2025 |