
Marcus inverted region in organic long-persistent luminescence host–guest systems designed from thermally activated delayed fluorescence molecules: a mechanistic study†
Yajie
Meng
ab,
Xi
Chen
 c,
Yingqi
Li
c,
Yunlong
Shang
b,
Yulin
Guo
c,
Yong
Wu
*c,
Haiyan
Wei
*c and
Jiawei
Xu
*abc
c,
Yingqi
Li
c,
Yunlong
Shang
b,
Yulin
Guo
c,
Yong
Wu
*c,
Haiyan
Wei
*c and
Jiawei
Xu
*abc
aFujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: jiawei.xu@chem.ox.ac.uk
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cMinistry-of-Education Key Laboratory of Numerical Simulation of Large-Scale Complex System (NSLSCS) and School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, Jiangsu 210023, P. R. China. E-mail: wuyong@njnu.edu.cn; weihaiyan@njnu.edu.cn
First published on 22nd October 2024
Abstract
Organic long-persistent luminescence (OLPL) systems have long been experimentally investigated. First reported by Ifor D. W. Samuel et al., OLPL can be observed upon doping thermally activated delayed fluorescence (TADF) molecules in host materials of PPT, TPBi and PMMA and is proposed to proceed via a two-photon mechanism. In this work, OLPL that occurs upon doping CzPhAP in PPT/TPBi systems with charge-separation features was theoretically investigated to understand the essence of transformation from TADF to OLPL and to provide insights for further research. Theoretical results of this study revealed that OLPL emission from CzPhAP:PPT/TPBi systems proceeded via a double-luminescence mechanism. The main S3 state emission peak (observed at 608 nm) was a mixture of TADF and OLPL. Moreover, large reorganization energy associated with converting S1 into a low-lying charge-separation S3 state enabled S3 fluorescence with a larger emission rate (7.92 × 107 s−1) compared to that of S1 fluorescence (3.94 × 107 s−1, shoulder peak observed at 584 nm). Thermodynamic equilibria between S1 and low-lying charge-separation states of S3 and T3 were constructed, which stimulated fast conversion among the S1, S3 and T3 states. Furthermore, our investigations indicated that when the reorganization energy of the ISC process is smaller than that of rISC, a larger ΔEST value is required to obtain krISC > kISC. Although a larger ΔEST value will make the ISC process deeply rooted in the Marcus inverted region, the ISC process will be strongly hindered and TADF emission could not be observed. Meanwhile, OLPL emission was enhanced with a larger ΔEST owing to frustrated charge recombination to the neutral T1 state and electron–hole dissociation could occur. More importantly, our study indicated that the essence of conversion of TADF into OLPL in CzPhAP:PPT/TPBi systems is due to a low-lying neutral T1 state.
1. Introduction
In ancient times, natural night-luminescent pearls were viewed as a precious heirloom and people believed that these pearls can keep corpses from decaying. However, in modern times, night-luminescent materials are no longer a noble secret and have gradually come into daily use. For example, the coat of paint on the hands of watches is one of the most common applications. Commercialized as glow-in-the-dark paints, long-persistent luminescence (LPL) materials have long been investigated and universally applied, stimulating the extensive research interests of materials scientists, chemists, physicists and biologists.1–8 Among them, doped metallic oxides,9–11 metal–organic frameworks12–16 and host–guest systems5,17–19 are three typical LPL materials. There are two major methods used to achieve the longer persistence of luminescence: doping with radioactive elements (such as radium) for continuous excitation in the dark and using materials with long-lived emission. As a typical example, MAl2O4 (M = Ca, Sr20,21 or Ba11,22,23) doped with europium and dysprosium was developed as a highly efficient LPL system. However, considering the requirement for RE elements and energy-consuming manufacturing processes, most pure inorganic LPL materials cannot meet the current principles of green chemistry and sustainable development.4 Organic compounds with a long-lived emission property have attracted considerable attention as alternatives to pure inorganic systems. In 1997, Ohkita et al. reported the first solid-state OLPL system of perylene:PnBMA (poly(n-butyl methacrylate)) with an after-glow reaching 200 s.21 In recent years, long-persistence has reached the thousand-second level for OLPL materials with an exciplex emission process, such as TMB (tetramethylbenzidine):PPT (2,8-bis(diphenyl-phosphoryl)dibenzo[b,d]thiophene), which was the first to reach the hour level,24m-MTDATA (4,4′,4′′-tris[phenyl(m-tolyl)amino]-triphenylamine):PPT,25 TMB:PBPO (poly(arylene ether phosphine oxide)),26 and carbazolyl units doped poly(vinyl alcohol) (PVA) films.27It is well known that the TADF process increases the fluorescence quantum yields via re-converting the T1 state to the S1 state, yet there is no clear evidence showing that molecular TADF emission may produce LPL. Inspired by TADF's breakthrough in efficiency, Ifor D. W. Samuel et al. designed a new TADF molecule CzPhAP and doped it into the conventional hosts PPT and TPBi and the polymer host PMMA.28 As reported, OLPL with a thousand-second level was observed with the three systems. Detailed spectroscopic research revealed that a combination of TADF small molecules and suitable hosts could bring great flexibility in the design of OLPL materials, making it possible to further decrease their cost and increase their efficiency. The OLPL mechanism of this novel system was reported to proceed via a two-photon mechanism, and experimental evidence showed that the OLPL duration increased with the application of a higher excitation density. However, the previous studies on OLPL were mainly based on spectroscopy and a theoretical study has not yet been carried out.
In this work, we studied CzPhAP-doped PPT and TPBi (CzPhAP:PPT, TPBi) (shown in Scheme 1) through density functional theory. According to our study, a different insight into the photophysical process is proposed to account for the experimental results, which is in contrast to the previous proposal discussed in the original work. First, our results revealed that the host material PPT plays the role of donor in this host–guest system, instead of the acceptor role mentioned in previous literatures, according to the inter-fragment charge-transfer (IFCT) analysis. Second, based on the non-radiative rates calculation proposed by Marcus–Levich–Jortner in their equation, out results showed that “OLPL” in this novel system (CzPhAP:PPT, TPBi) majorly resulted from a new kind of quasi-TADF process; whereas, traditional OLPL occurs via an electron–hole dissociation mechanism and charge-transfer excitation plays a negligible role. Through this work, a general computational scheme was constructed for investigating host–guest systems formed by the coupling of donor and acceptor molecules with OLPL or TADF properties.
 | ||
| Scheme 1 Guest molecule CzPhAP, where the AP moiety acts as an acceptor in intermolecular charge transfer, and the host molecules PPT and TPBi, where the DBT and PBi moieties act as donors in intermolecular charge transfer. | ||
2. Methodology
2.1. MD simulations
In order to obtain the reasonable stacking patterns of CzPhAP and PPT, six initial models were built by the Packmol program, corresponding to CzPhAP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPT ratios of 3:100, 1:100, 1:200, 1:300, 1:400, and 1:500, respectively. This ratio considers the high concentration of PPT in the experiment. The restrained electrostatic potential (RESP) atomic charges of CzPhAP and PPT were fitted by Multiwfn (development version 3.8).29
:PPT ratios of 3:100, 1:100, 1:200, 1:300, 1:400, and 1:500, respectively. This ratio considers the high concentration of PPT in the experiment. The restrained electrostatic potential (RESP) atomic charges of CzPhAP and PPT were fitted by Multiwfn (development version 3.8).29
Molecular dynamics (MD) simulations were performed using the GROMACS (version 2022.5) package30 and the topology file and forcefield parameters were created by Sobtop.31 The long-range electrostatic interactions were handled by the particle-mesh Ewald (PME) method and the cut-off value of the van der Waals (vdWs) interactions was set to 10 Å.32 After energy minimization, the six systems were heated from 0 K to 500 K in 1 ns simulations and then maintained for 3 ns at the NPT ensemble. Subsequently, the temperature of the six systems was dropped to 300 K and another 100 ns unrestrained MD simulations were performed at the NPT ensemble to analyze the stacking patterns of PPT and CzPhAP. Based on the MD results, the favoured stacking conformation was selected as AP and DBT moieties, and then used for the following quantum mechanical (QM) investigations.
2.2. QM computational details
All the geometric structures were optimized with the Gaussian16 program (Revision C.01),33 with the Minnesota hybrid functional with the M06-2X form34 and def2-SVP basis sets developed by Ahlrich and co-workers used.35 Grimme's third dispersion correction (D3) was adopted for better consideration of the intermolecular weak interactions.36 Frequency calculations at the same level were performed for all structures on steady points to ensure no imaginary frequency existed. Excited states were calculated under the framework of time-independent density functional theory (TD-DFT) except for the T1 states, which were calculated by the unrestricted Kohn–Sham method.The electronic coupling integrals (V) in non-adiabatic processes were calculated by the fragment charge-difference (FCD) method37,38 according to eqn (1), where ΔEFC stands for the vertical energy gap between the studied states. Elements of the donor–acceptor 2 × 2 charge-difference matrix Δq were defined using eqn (2), where ρmn stands for the transition density matrix between states m and n. For the diagonal elements Δq11 and Δq22, ρmn refers to the transition electronic density for the excited states. For the off-diagonal elements Δq12 and Δq21, eqn (3) was applied for symmetrizing. The integrals involved in the FCD calculation can be calculated as summations on the introduced basis set according to Slater–Condon's rule, and an approach for this can be found in the ESI.†
 | (1) |
 | (2) |
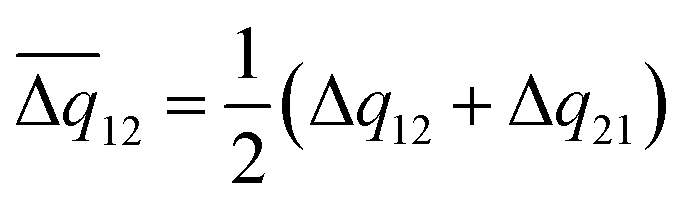 | (3) |
The spin–orbit coupling integrals (H) were calculated using the Dalton2020 program,39,40 with both single- and double-electron spin–orbit integrals precisely calculated under the CAM-B3LYP/def2-SVP level of theory.41 Wavefunction analysis was carried out using Multiwfn (development version 3.8),29 and visual molecular dynamics (VMD, version 1.9.3) was applied for better visualization of our results.42 More details and full results of the calculation works are available in the ESI.†
2.3. Theoretical backgrounds
Charge-separation states exist in many chemical systems.43 Consider an arbitrary donor–acceptor system D–A, where D refers to the donor and A refers to the acceptor. As pointed out by the electroneutrality principle,44,45 it is impossible to observe D+A−, where exactly one electron is transferred from D to A. Theoretically, we use the population number, which is commonly not an integer, to evaluate the fuzzy concept of the “number of electrons” evolved in a charge-transfer process.In an OLPL system, the charge-separation state is a key metastable intermediate.24 Generally, such a charge-separation state is a special charge-transfer state in which the obviously separation of electrons and holes can be observed. Conceptually, the population number change should be close to 1. Fig. 1 shows a potential energy surfaces diagram of the neutral state and charge-separation state with a conical intersection point. The charge-separation state can be reached via photoinduced or thermally activated charge transfer. On the opposite, charge recombination is usually realized by a non-radiative process. These concepts have been commonly acknowledged in the research on OLPL materials.24 Therefore, the charge-separation state is an excited state, if the neutral state is selected as the reference state. The main difficulty with this idea is that the charge-separation state of complicated systems is not the lowest excited state, especially for host–guest or highly conjugated systems. Moreover, ideal charge-separation states usually include a singlet state and a triplet state with a quite small energy gap. Therefore, in investigating OLPL systems, it is essential to refer to proper excited states as the charge-separation states.
 | ||
| Fig. 1 Charge transfer in the donor–acceptor system. | ||
In this framework, the rates of non-radiative processes are calculated by the Marcus–Levich–Jortner equation under the classical situation (eqn (4)), which gives a semi-empirical prediction of the rate of charge transfer between the donor and acceptor.
 | (4) |
 | (5) |
| Q2 = J·Q1 + ΔQ | (6) |
The Huang–Rhys factor Si is defined by eqn (7).
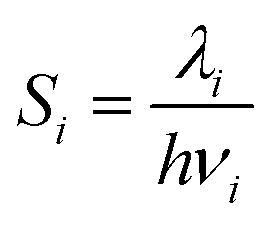 | (7) |
The Huang–Rhys factor reveals the strength of vibronic coupling.46–48 Non-radiative processes with large Huang–Rhys factors tend to occur easier. Apart from this, we may also gain insight into which normal vibrational mode makes a crucial contribution to the process.
Einstein's spontaneous emission rate formula (eqn (8)) was used to calculate the rates of the radiative processes including fluorescence and phosphorescence,49,50 where fij stands for the transition oscillator strength and ν stands for the transition energy in the form of a wavenumber (cm−1). Since phosphorescence is spin forbidden, spin–orbit coupling was considered to calculate the oscillator strength between the T1 state and S0 state.
 | (8) |
3. Results and discussion
3.1. Stacking conformation
Annealing MD simulations showed that the stacking conformation was favoured between the AP moiety of CzPhAP and the DBT moiety of PPT (see abbreviations in Scheme 1). Taking the MD system with 501 molecules (CzPhAP:PPT = 1:500) as an example (Fig. 2a), though the initial position and orientation of molecules were randomly generated, after annealing, the stacking conformation between the AP and DBT moieties was always observed, with a minimum distance of 3.51 Å between the mass centres of the AP and DBT moieties. Changing the concentration of the dopant did not affect obtaining such a conformation (Fig. S1–S5, ESI†). The statistics of the distance between the mass centres showed that such a conformation could be maintained during a notable length of simulation time. The mass centre distance led by the PPT molecule possessed the longest binding period with CzPhAP during the 100 ns MD simulation after annealing, as plotted in Fig. S1–S5 (ESI†). The minimum distance ranged between 2.8–5.1 Å, which is a suitable distance for π–π stacking interactions. This was also demonstrated by the reduced density gradient (RDG) analysis (Fig. 2b). At the same time, it has also been acknowledged in experimental designation that the AP moiety in CzPhAP usually acts as an electron acceptor while the DBT moiety in PPT acts as an electron donor. A D–A pair close in space is generally reasonable. Therefore, the following investigation based on quantum chemical calculations adopted the structure optimized from a stacking conformation between AP and DBT.
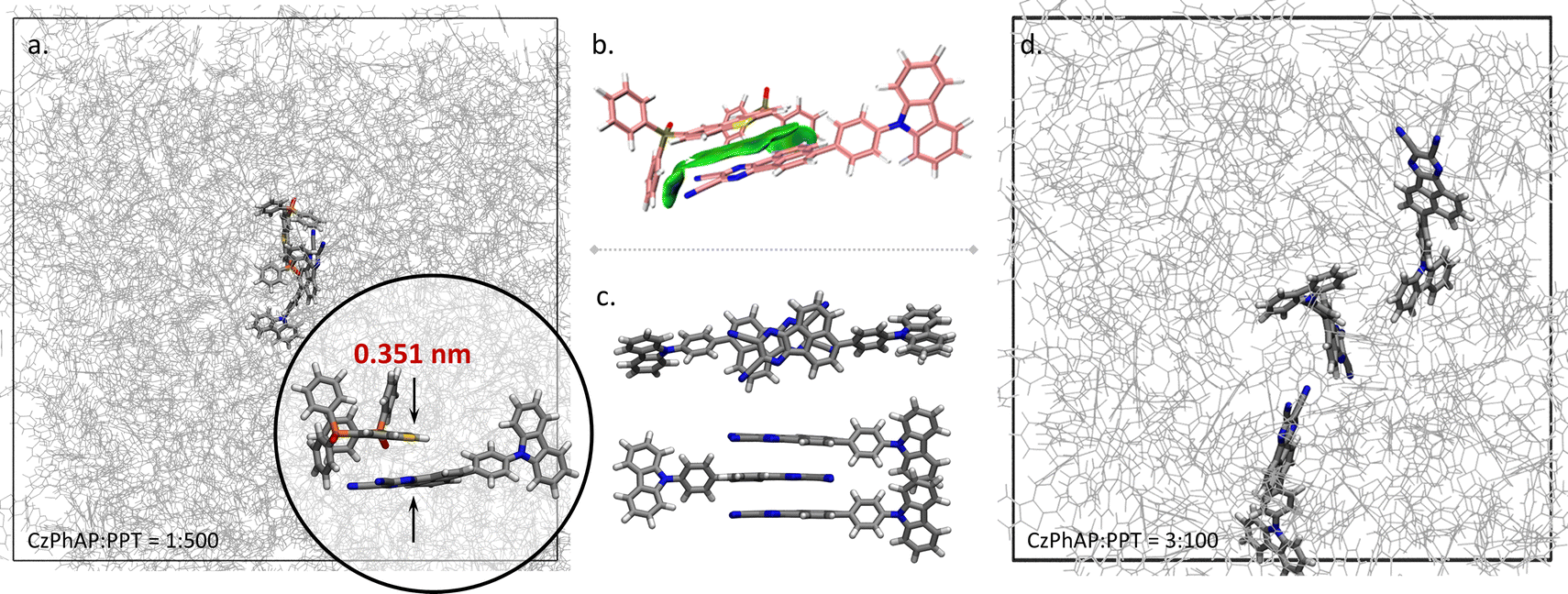 | ||
| Fig. 2 (a) MD simulation showing π–π stacking conformation between the DBT moiety of PPT and AP moiety of CzPhAP; (b) non-covalent interactions between DBT and AP revealed by reduced density gradient analysis (isovalue: 0.60); (c) Initial stacking conformation with multiple dopant molecules and (d) separated by host molecules after MD simulation. | ||
Considering that dopant clusters with more than one dopant molecule may influence performance, i.e. in this system, π–π stacking between AP and AP moieties may prevent that between AP and DBT moieties, the stacking behaviour in the model system with a high dopant concentration (CzPhAP:PPT = 3:100) was investigated as well. The AP–AP stacking conformation shown in Fig. 2c was used as the initial structure for the MD simulation, while after 100 ns MD simulations, these dopant molecules were largely separated by host molecules (Fig. 2d). These results demonstrated that the binding affinity between CzPhAP and PPT brought about by AP–DBT π–π stacking interactions plays a crucial role in the host–guest system investigated.
3.2. Charge-separation state
The electron–hole analysis and inter-fragment charge-transfer (IFCT) method51,52 were adopted to reveal the character of electron transfer in the excited states in the studied host–guest system. The distributions of electrons and holes in the CzPhAP:PPT host–guest system are shown in Fig. 3, where the electron area is coloured with green and the hole area is coloured with magenta. The lowest S1 state, which was reached via single photon excitation, showed charge transfer by a population number of 0.4575 from PPT to CzPhAP, as evaluated by the IFCT method. Thus, PPT molecules can be regarded as donors in this single photon excitation process. In contrast, the T1 state was associated with a negligible charge transfer by only the 0.0380 population number involved. Besides, the electron and hole areas were all distributed on the AP moiety of the CzPhAP molecule in the T1 state. Moreover, among the lowest 10 singlet excited states studied, only the S3 state showed the feature of charge separation. The IFCT population number of the S3 state reached the highest as 0.8418, being close to the ideal value of 1.0. Similarly, among the lowest 10 triplet excited states studied, the IFCT population number of the T3 state ranked the highest, with a calculated IFCT population number of 0.5634. Therefore, the T3 state can be viewed as a triplet charge-separation state. According to the isosurface distribution displayed in Fig. 3, the electron areas of both the S1 and S3 states were distributed mainly on AP moiety; however, the hole area of the S1 state was distributed on both the CzPhAP and PPT molecules, indicating that the S1 state was a mixture of through-space charge transfer (TSCT, PPT → CzPhAP) and through-bridge charge transfer (TBCT, within CzPhAP). | ||
| Fig. 3 Isosurface (isovalue: 0.0030 a.u.) of the electron–hole distribution and IFCT population change for different excited states of the CzPhAP:PPT host–guest system. Electron refers to the area in the excited state where electron density is increased compared with that of the ground state, while hole is the opposite, as evaluated by the population number. The detailed electron–hole distributions and IFCT results are listed in Tables S3 and S4 (ESI†). | ||
A similar conclusion was also reached for the host–guest system CzPhAP:TPBi. As shown in Fig. S6 (ESI†), both the higher S3 and T10 states showed a notable charge-transfer feature, with calculated IFCT population changes of 0.9253 and 0.7901, respectively. Whereas, the lowest excited states S1 and T1 did not show a charge-transfer character, with the calculated IFCT population change nearly zero. Moreover, it should also be pointed out that TPBi also acts as a donor molecule and the designed CzPhAP molecule plays the acceptor role in this host–guest system. Our conclusion is in contrast to the viewpoints in previous references, where PPT and TPBi molecules were claimed to be acceptors,24,25,53,54 which was based on HOMO–LUMO levels simply calculated by DFT, or wrongly took from redox potentials measured by cyclic voltammetry. The excited states reached by single photon excitation do not necessarily correspond to HOMO → LUMO excitation. Therefore, the optical gap and donor–acceptor roles cannot be determined via simply comparing the HOMO–LUMO levels of different molecules.55 As demonstrated by previous electron–hole analysis and inter-fragment charge-transfer (IFCT) analysis, the host–guest molecules should be viewed as a whole as the influence of the host–guest effect on the molecular optical or electronic properties cannot be ignored. However, in heterojunction structure systems, the properties of one bulk phase are not strongly influenced by another, which thus ensures materials can retain their major electronic–structural features. Therefore, the HOMO–LUMO levels of different materials determined in the bulk phase can be applied to predict their photochemical or electrochemical performance.
In the following, the transition density matrix (TDM) was calculated to analyze the transition feature in the host–guest systems CzPhAP:PPT and CzPhAP:TPBi. For a many-electron system, the real space function form of TDM is expressed as eqn (9.1), where, Φ0 and Ψexc represent the ground-state and excited-state wavefunctions, respectively. It can be further written as a three-dimensional real function in eqn (9.2) under the TDDFT framework, where ϕi and ϕa stand for occupied and unoccupied orbitals involved in a specific single excitation configuration function with the coefficient of wai.
 | (9.1) |
 | (9.2) |
By introducing a set of basis functions, the orbitals in eqn (9.2) can be expanded to eqn (10.1) and the TDM element is defined as eqn (10.2). Here, Cμi represents the coefficient of the basis function χμ in orbital ϕi and the double summation runs over every single excitation configuration function i → a.
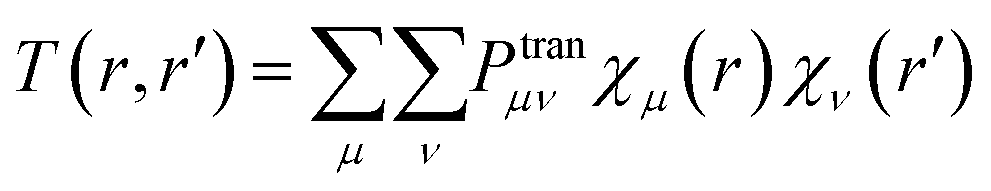 | (10.1) |
 | (10.2) |
The above definition extracts excitation information into each couple of basis functions. A more chemically meaningful expression is to condense the basis function TDM into atomic TDM, whose element corresponding to atoms A and B is given in eqn (11).
 | (11) |
Fig. 4 shows the atomic TDMs of the S1, S3, T1, and T3 states in the CzPhAP:PPT system, where the atom range is marked with PPT or CzPhAP. Elements in the diagonal area from the lower left to the upper right show a local-excitation (LE) character while elements in the non-diagonal area show a charge-transfer (CT) character. The S1 state of the CzPhAP:PPT system showed a strong PPT → CzPhAP CT character, while the CzPhAP → CzPhAP LE character also made a notable contribution. While in the S3 state, the bright area was majorly distributed within the PPT → CzPhAP CT area, indicating a stronger CT character in the S3 state, and thus it acts as a charge-separation state. This is consistent with the previous discussion. This also holds for other key excited states and moreover, the LE and CT characters showed an even starker contrast in the other three pairs of excited states. Therefore, in the studied system, the higher excited states acting as charge-separation states was confirmed, which is theoretically most relevant to OLPL. A similar conclusion also holds for the S3 and T10 states in the CzPhAP:TPBi system (as discussed in Fig. S7, ESI†). In the following sections, the photochemical mechanism in the CzPhAP:PPT system is discussed.
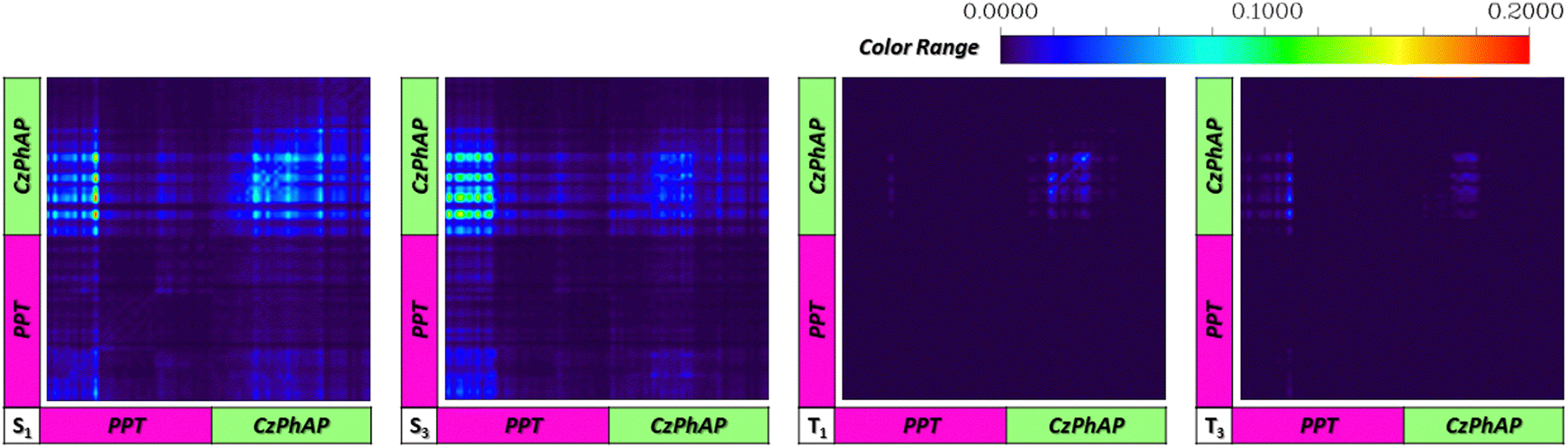 | ||
| Fig. 4 Atomic transition density matrices of key excited states in the form of heat maps for the CzPhAP:PPT host–guest system. | ||
3.3. Geometry and vibration
Previous theoretical works have pointed out that the geometric structure plays a significant role in non-radiative processes. As revealed by the Huang–Rhys factors calculated according to eqn (5)–(7), geometric reorganization during the (r)ISC process can be decomposed into the normal vibration modes, shown in Fig. 5a. Larger Huang–Rhys factors reveal a stronger vibronic coupling effect and indicate a faster non-radiative process.56–60 For S1–T1 processes, low-frequency normal vibration modes have larger Huang–Rhys factors, which are subjected to the rocking vibration modes of POPh2 groups in the PPT part (marked with white diamond in Fig. 5). This could also be revealed by the overlapping minimum points of both structures with the root-mean-square deviation (RMSD) minimized. Since the donor and acceptor molecules are not bound with chemical bonds, they are separated when evaluating the geometric change with the RMSD in order to avoid disturbing each other. | ||
| Fig. 5 (a) Huang–Rhys factor diagrams for S1–T1 and S3–T3 (r)ISC processes. (b) Root-mean-square deviation between S1–T1 and S3–T3 minimum structures. (c) Normal vibration modes with significant contributions to (r)ISC processes. | ||
As shown in Fig. 5b, the structural deformation of PPT was majorly contributed by the POPh2 groups. Thus, the RMSD value of the rigid central thiophene ring of PPT was first minimized, and then the total RMSD value was calculated to evaluate the structural deformation of PPT. The black framework (singlet state structure) overlapped with the red framework (triplet state structure) at the central aromatic rings in the PPT part, while showed a deviation at the lateral groups. In comparison of the S1 and T1 states for the CzPhAP part, the major deviation came from the Cz group. However, due to the micro Huang–Rhys factors associated with the related normal vibration modes, the non-radiative processes between the S1 and T1 state were not enhanced. In comparison between the structures of the S3 and T3 states, the deviation in the POPh2 group of the PPT part was much stronger, while the deviation in the CzPhAP part was negligible. This was the same with the two couples of structures where the structural deviation majorly originated from the lateral groups. Both the donor and acceptor parts contributed to structural deviation, notably in the S1–T1 couple; however, in great contrast, for the S3–T3 couple, the donor molecule accounted for most of the structural deviation, as revealed by the RMSD in Fig. 5b. Since both POPh2 and Cz groups are exposed to the surrounding environment, S1–T1 processes will be strongly affected by solvent molecules. A strong interaction with solvent molecules or a rigid environment (thin-layer for instance) make related processes harder to occur by hindering reorganization of the lateral groups.
Moreover, the deformation and bending modes of the PPT part (marked with black notations) had a greater impact in the S3–T3 related non-radiative processes. As shown in Fig. 5a, the deformation and bending modes of the PPT part were distributed in a wider region of wavenumber (700–1200 cm−1, but narrower in energy indeed), which was in contrast to the low-frequency region (50–100 cm−1) associated with the rocking modes. This is the most important feature that distinguishes the S1–T1 and S3–T3 processes. Although the rocking modes of the S3–T3 series had even larger Huang–Rhys factors compared with those of the S1–T1 series, they did not dominant the S3–T3 related non-radiative processes since the deformation and bending modes of the PPT part had comparatively larger Huang–Rhys factors, acting as the decisive strengths.
It is well known that the surrounding environment has a more important impact on the rocking mode compared with deformation and bending modes due to the steric hindrance. Hence, the S3–T3 related non-radiative processes are reliably ensured by normal vibration modes with higher Huang–Rhys factors. This is important since S3–T3 related non-radiative processes are crucial to thermochemical equilibrium between singlet and triplet charge-separation states (S3 and T3) and we discuss this further in the next section. Nevertheless, it is worth noting that for the normal vibration modes in the region of 700–1200 cm−1, although the large Huang–Rhys factors and reorganization energies indicated remarkable structure distortion in the related processes, they were not reliable for calculating the total reorganization energies of non-radiative processes. This is because in the calculation of the reorganization energies contributed by each normal vibration mode, harmonic oscillator approximation is applied to evaluate the vibration potential energy. It was obvious that the potential energy surfaces (PESs) of S3 and T3 presented notable differences and so eqn (5) is no longer applicable. Moreover, the reorganization energy decomposition and Huang–Rhys factors calculated under the harmonic oscillator approximation can still reveal the key normal vibration mode contribution to the non-radiative processes. But when calculating the total reorganization energy for rates prediction (Section 3.4), and to account for the reorganization energy induced by electronic structure change, the four-point method should be adopted instead of eqn (5).
Besides, the unchanged parts of the structures also play an important role in ensuring charge transfer and the photochemical properties. In the host–guest system CzPhAP:PPT, the donor and acceptor molecules are bound by intermolecular π–π stacking interactions, involving the dibenzothiophene (DBT) part of PPT and AP part of CzPhAP (Fig. 2b). The rigid aromatic ring structures ensure the π–π stacking structure act as a reliable TSCT tunnel. The related key normal vibration mode is a DBT ring deformation mode (1060.15 cm−1 in the S3 state and 1064.89 cm−1 in the T3 state, marked with “*”). Thus, the distance between π-conjugate systems does not increase obviously due to the within-plane deformation mode, and it can hardly influence the π–π stacking structure. As aforementioned, both S1–T1 and S3–T3 processes showed charge-transfer characters, and so an effective π–π stacking structure acting as a TSCT tunnel is crucial for good photochemical properties.
3.4. Photochemical mechanism
In the following sections, the detailed photochemical relaxation pathways and mechanism why the TADF molecule CzPhAP can display the OLPL property when doped in the host material PPT were studied based on Marcus’ theory.‡ | ||
| Fig. 6 Calculated UV-vis absorption spectra and electron (grey)–hole (white) distributions for CzPhAP and CzPhAP:PPT. | ||
| Process | Notation | λ/eV | ΔE/eV | H/cm−1a | k/s−1 |
|---|---|---|---|---|---|
| a Spin–orbit coupling matrices elements are listed in Table S5 (ESI). | |||||
| S1 → T1 | r 1 | 0.050 | −0.724 | 2.245 | 1.36 × 10−29 |
| S1 → T3 | r 2 | 0.107 | −0.015 | 1.165 | 4.95 × 108 |
| S3 → T1 | r 3 | 0.193 | −0.706 | 0.654 | 4.53 × 102 |
| S3 → T3 | r 4 | 0.162 | 0.003 | 1.774 | 3.91 × 108 |
| T1 → S1 | r −1 | 0.899 | 0.724 | 4.237 | 2.01 × 10−3 |
| T1 → S3 | r −3 | 0.104 | 0.706 | 4.171 | 2.66 × 10−17 |
| T3 → S1 | r −2 | 0.553 | 0.015 | 1.072 | 1.36 × 106 |
| T3 → S3 | r −4 | 0.095 | −0.003 | 1.110 | 4.36 × 108 |
We should also notice that since the potential energy surfaces (PESs) of the S1 and S3 states interact with each other, the minimum point reached by state-tracking the PES of the S3 state was even slightly lower in energy compared with the S1 state minima. This shows that partial charge recombination and relaxation bring extra stability for the charge-separation state. Since r±2 and k±1 were much larger than the rates for the following processes, we can consider both processes as fast equilibria reached between 1–1 order opposing reactions, with equilibrium constants of KISC2 = r2/r−2 = 2.75 × 10−3 and KIC1 = k1/k−1 = 2.32 × 10−2, respectively. Both equilibria hold the concentration of systems in the S1 state. With the T3 and S3 states being converted in the following processes and the S1 state produced by continuous excitation (since the excitation time was much longer than that needed for reaching fast equilibria, it could be viewed as continuous), the equilibria were pushed towards the direction of the products, within which S3 was the major product since k1 > r2.
Another equilibrium was then reached between S3 and T3 states (KISC4 = r4/r−4 = 0.897). The small energy gap (0.003 eV) between charge-separation states with different spin multiplicity enabled the Marcus inverted region to be impossible to be reached for both the ISC and rISC processes. This is different from (r)ISC processes between the S1 and T1 states. Such a difference was also consistent with the non-radiative coupling strength qualitatively revealed by the Huang–Rhys factors in Fig. 5a. Generally, the three fast equilibria discussed above ensure the concentration of excited systems as S1 > S3 > T3.
Previously, three OLPL mechanism have been reported: (1) two-photon mechanism proposed by Ohkita et al.,21,61,62 (2) charge-separation model based on CT excitation dissociation proposed by Kabe et al.,24,53 and (3) CT excitation dissociation based on spontaneous orientation polarization (SOP) proposed by Yamanaka et al.63 In previous charge-separation systems, the high-lying charge-separation state can be reconverted into a neutral state (usually the S1 state), and thus delayed luminescence emission at the S1 minimum would be observed, which is referred to as the OLPL mechanism (charge-separation model based on CT excitation dissociation). In those reported charge-separation OLPL mechanisms, the minimum points of the available charge-separation states are higher in energy than the neutral S1 state. Thus, charge-separation states can be continuously reconverted via non-radiative charge recombination processes, which obeys Kasha's rule and emits at the lowest singlet state. Vibronic coupling is significant for non-radiative processes in such a situation. From a more non-radiative viewpoint, the minimum energy crossing point (MECP)64,65 of neutral and charge-separation states is too high in energy to be reached. However, in the studied CzPhAP:PPT host–guest system, luminescence can also be emitted by the charge-separation state (S3).
In the studied CzPhAP:PPT host–guest system, for the S3 and T3 states, the RMSD calculation values of the donor molecule and DBT part of the acceptor molecule that acts as the hole were both very small (Fig. 5b). Hence, the two minimum points were quite close in nuclear coordinates. Therefore, MECP was strictly limited within the two minimum points and only a small energy barrier was needed to overcome this. Therefore, the related non-radiative processes could be forwarded by the thermal-driven force. This is why the harmonic oscillator approximation failed to give reasonable reorganization energies and Huang–Rhys factors. Meanwhile, a thermodynamic equilibrium was built between the two minimum points of the neutral and charge-separation states, as discussed in previous sections. The charge-separation state could be close to or even lower than that of the neutral S1 state. Furthermore, it resulted in double fluorescence emissions, which is anti-Kasha's rule. This new double-luminescence mechanism originated from the energy gap of the neutral and charge-separation states and is in great contrast to the previously reported mechanism.
As shown above, the CzPhAP:PPT host–guest system induced double luminescence. The S1 emission (observed at 584 nm) displayed a conventional OLPL peak generated by a charge-separation–recombination mechanism. This is because both the S3 and T3 states were charge-separation states. The S3 emission (observed at 608 nm) is more complicated. Since the S3 state can be reached from both the S1 and T3 states, a mixture of emissions from the two mechanisms can be generated. First, the new OLPL mechanism discussed above could be constructed via the S1 → S3 IC process, emitting directly via radiative charge recombination. Second, a quasi-TADF pathway could be constructed via the T3 → S3 ISC process. The difference between the “quasi-TADF” and TADF pathway is that the minimum point of the S3 state was slightly lower than that of the T3 state in this system. This photochemical mechanism also explains the influence of the quasi-TADF effort in the OLPL system.
However, in both mechanisms, the thermal barrier cannot be avoided and it still behaves as a thermally activated process. Moreover, the quasi-TADF mechanism demonstrated that TADF emission can not only be generated by the lowest couple of excited states (S1 and T1), but also can be generated from higher ones with a different excitation character. Table 3 also shows that the S3 emission had a larger oscillator strength and emission rate compared with the S1 emission, which well corresponded to the experimental spectrum showing that the main peak was longer in wavelength.
Apart from the fluorescence emission, phosphorescence emission also existed on the T1 state, albeit the emission strength was much weaker than fluorescence (Table 3). Compared with the neutral T1 state, the charge-separation T3 state was a high-lying excited state, with a calculated T3 → T1 IC rate of k−2 = 1.52 × 108. Therefore, the phosphorescence emission in the CzPhAP:PPT host–guest system still obeys Kasha's rule, which indicated that the high-lying state converted to the lowest triplet state via an IC process. These results were consistent with the experimental findings.
Fig. 7a gives the lnk–ΔEST curve of processes related to T1 state, where reorganization energies and coupling integrals are viewed as constants. With the experimental ΔEST value, ISC processes from both S1 and S3 states to T1 state will have notable rate constants, resulting in obvious phosphorescence emission. However, the theoretical ΔEST value (0.724 eV) reveals that the above two ISC processes are remarkably limited by Marcus inverted region (monotonic decreasing part of the curve). Only IC process from T3 state to T1 state is not strictly limited, with a rate constant k−2 = 1.52 × 108; however, ISC process from T3 state to S1 state (r−4 = 4.36 × 108) and low concentration of T3 state are extra limitations. This result indicates that that no obvious phosphorescence emission is observed in long-persistent luminescence, which is in consistence with the experiments.
 | ||
| Fig. 7 (a) The lnk–ΔEST curves of non-radiative processes related to the T1 state, with reorganization energies and coupling integrals viewed as constants. Energy of the S1 state was taken as the zero point. (b) Three conditions of possible crossing. | ||
As discussed above, the Marcus inverted region plays a significant role in non-radiative processes. Taking the CzPhAP:PPT host–guest system as an example, a larger ΔEST value is an advantage for OLPL; however, a smaller ΔEST value does not have a similar influence on TADF. In previous research, TADF systems were believed to have a small ΔEST value (usually < 0.10 eV) in order to enhance the rISC process from the T1 state to the S1 state. As shown in Fig. 7a, TADF emission induced by the rISC process from the T1 state to the S1 state could only be observed when the black dot line was higher than the black solid line (r1 < r−1). This condition can be satisfied within two regions.
First, when ΔEST > 0.36 eV, although the TADF condition is satisfied, TADF emission will be limited by the small ISC rate constant deeply located in the Marcus inverted region. In other words, the pre-process for rISC in the TADF mechanism is limited and a singlet system cannot be efficiently converted to a triplet system. Another region is where the T1 state is higher than the S1 state (ΔEST < 0), which is not displayed in Fig. 7a since it is unlikely to be realized in organic systems. Therefore, it is worth noting that a small ΔEST value is not always advantageous for TADF emission. Such a result is different from the viewpoint of previous research, where a small ΔEST value was always considered an advantage for TADF emission. Based on the above discussion, we can expand the old viewpoint for the better by taking the first-derivative of the lnk–ΔEST curve into consideration. According to the Marcus–Levich–Jortner equation (eqn (1)), this curve is a parabolic curve with a negative quadratic term coefficient (−1/4λkT, eqn (12)) and its first-derivative is eqn (13).
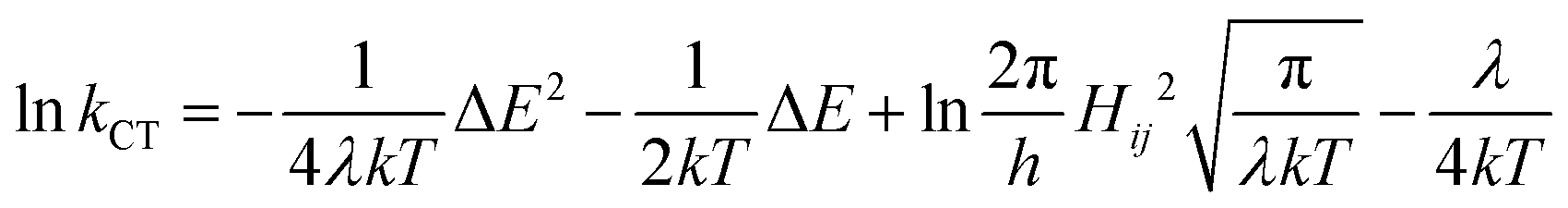 | (12) |
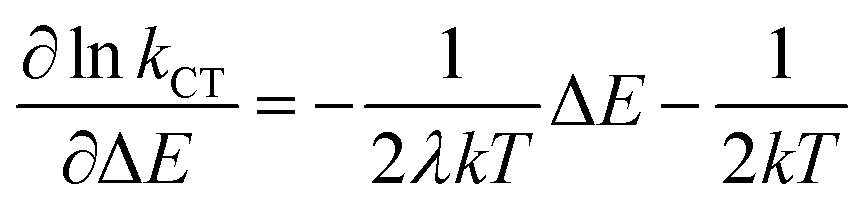 | (13) |
The absolute value of the quadratic term coefficient (1/4λkT) of the solid line is smaller than that of the dotted line for both the red and blue couples in Fig. 7a. The non-radiative process corresponding to the solid line has a larger reorganization energy compared with its reverse process. In such a circumstance, a larger ΔEST value will be beneficial for the TADF behaviour since the TADF region where the dotted line is higher than the solid line lies in the left side of the crossing point of two curves. This scheme can explain the observation in previous literature. Nevertheless, the crossing of two parabolic curves has three conditions (Fig. 7b). Only Condition III is allowed for relation eqn (12) (Fig. 7a can be an example) and we provide a simple proof for this conclusion in the appendix section (eqn (A1)–(A3)).
However, if the reorganization energy of the ISC process from the S1 to T1 process (0.050 eV) is smaller than that of the corresponding rISC process (0.899 eV), the smaller ΔEST value will not benefit the TADF behaviour and the T1 state cannot be effectively reconverted to the S1 state through the rISC process. When ΔEST value increases, although the rISC rate increases to be higher than the ISC rate, the Marcus inverted region limits both processes and conversion between the S1 and T1 states is almost banned, which also has no benefit for TADF emission. Conversely, this phenomenon may enhance OLPL emission by avoiding the loss of excited molecules on the neutral triplet state T1. This is because in the CzPhAP:PPT host–guest system, all non-radiative processes starting from the T1 state had quite low rates due to the low energy level of the T1 state and only phosphorescence emission or non-radiative decay to the ground state were possible pathways, which should be avoided in designing OLPL systems. Therefore, we suggest that a low-lying neutral T1 state is the key point for observation of the OLPL property upon a TADF molecule being doped in to host materials like PPT or TPBi.
Now we have noticed that the reorganization energy is an important influencing factor in OLPL emission, for a further step considering two minimum points connected by a couple of non-radiative processes, since the shift vector ΔQ is the same for both processes, the reorganization energies depend on the force constants of the normal vibration modes (k). Larger force constants correspond to larger gradients of PES, graphically behaving as a steeper PES. Therefore, the reorganization energies depend on whether the specific PES is steep or not. In experimental studies, reorganization energy can be examined by measuring Stokes shifts of absorption and the PL spectra, which provides a possible direction to adjust and improve the host–guest system. We may focus on the rational design of these two kinds of systems and relationship between them therefore.
4. Conclusions
In this work, we present a theoretical scheme for investigating the photochemical properties and mechanism of OLPL host–guest systems with charge-separation features. A recently reported OLPL system designed from the TADF molecule (CzPhAP:PPT/TPBi) was taken as an example. The rates of non-radiative processes and influence of the Marcus inverted region were analysed in detail. New insights into the rational design and adjustment of OLPL and TADF systems were provided.The host materials PPT and TPBi acted as donors in these novel systems, which is different from previous viewpoints reached by simply comparing the HOMO–LUMO gaps of the materials measured by cyclic voltammetry. The S1 and T1 states of both systems (CzPhAP:PPT and CzPhAP:TPBi) majorly showed local excitation, while the higher excited states S3, T3 for CzPhAP:PPT and S3, T10 for CzPhAP:TPBi displayed charge-transfer states. Based on this, the detailed photochemical mechanism of the example system CzPhAP:PPT demonstrated that the observed OLPL emission was a mixture of two emissions, corresponding to the steady-state PL spectrum with a main peak at 608 nm and a shoulder peak at 584 nm. This double-luminescence mechanism is different from the previously known three OLPL mechanisms.
In this double-luminescence mechanism, the singlet charge-separation state has a minimum point lower in energy and can directly luminate, which is an anti-Kasha's rule emission, corresponding to the main peak at 608 nm in the steady-state PL spectrum. Considering that the singlet and triplet charge-separation states form a thermodynamic equilibrium, the quasi-TADF emission resulting from the T3 → S3 rISC also contributed to this main peak notably. Emission at the shoulder peak was related to the traditional OLPL mechanism, in which the charge-separation state was reached by a further electron–hole dissociation of the charge-transfer excitation. With a lower emission rate (S1, kF1 = 3.94 × 107 s−1vs. S3, kF3 = 7.92 × 107 s−1), it behaved as a shoulder peak in the steady-state PL spectrum. These mechanisms can explain why the emission duration time did not display quadratic growth with respect to the increase I the excitation intensities. It also indicated that the CzPhAP:PPT system can be excited and show the OLPL property with a simple excitation source, where laser light is not necessary.
Moreover, it is worth noting that the traditional viewpoint that a smaller ΔEST value benefits TADF emission should be carefully examined. When the reorganization energy of the ISC process is larger than that of rISC, this conclusion can be accepted. However, when the reorganization energy of the ISC process is smaller than that of rISC, a larger ΔEST value is needed in order to get krISC > kISC. This will have the ISC process deeply rooted in the Marcus inverted region, which will hinder singlet-to-triplet conversion, resulting in no TADF emission. Conversely, for OLPL systems designed from TADF molecules like CzPhAP:PPT, OLPL emission will be enhanced since charge recombination of the charge-transfer excited-state S1 is hindered and further dissociation into charge-separation states can be enhanced. We believe this is the essence why TADF molecules can display an OLPL property when doped in host materials like PPT or TPBi. We also hope further experimental research can improve such novel systems based on our tentative mechanism study.
Author contributions
Yajie Meng and Xi Chen: data curation and investigation; Yingqi Li, Yunlong Shang and Yulin Guo: visualization and formal analysis; Yong Wu and Haiyan Wei: supervision and resources; Jiawei Xu: writing – original draft, review & editing.Data availability
Necessary data has been provided in supplemental information. Further details are available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Appendix
For non-radiative process with reorganization energy λ1, coupling integral V1 and energy difference ΔE, eqn (12) is written as eqn (A1). The corresponding reverse process (λ2, V2 and −ΔE) will be eqn (A2).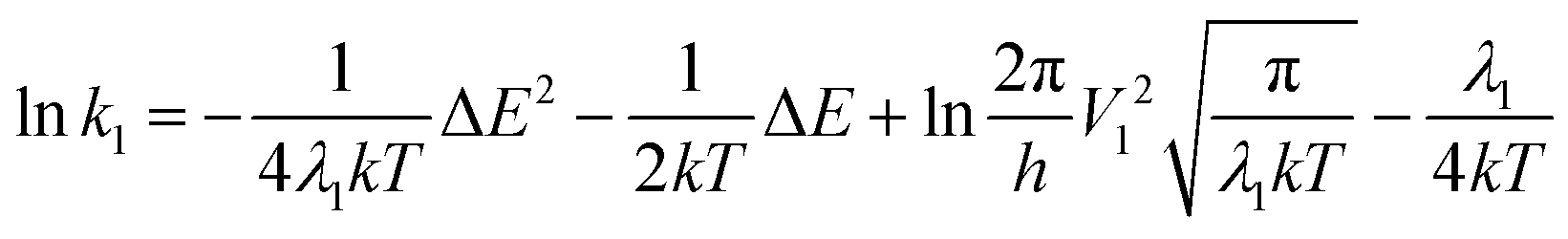 | (A1) |
 | (A2) |
To have crossing point, let ln k1 = lnk2 and apply the approximation that a couple of reverse processes have V1 ≈ V2 to get eqn (A3):
 | (A3) |
Assume that λ1 < λ2, coefficients and constant term of eqn (A3) have determined signs and eqn (A3) has one negative root and one positive root, which corresponds to Condition III. It is also the same case for λ1 > λ2.
Acknowledgements
We would like to acknowledge a project funded by Jiangsu Key Laboratory of Biofunctional Materials, and Ministry-of-Education Key Laboratory of Numerical Simulation of Large-Scale Complex System (NSLSCS) for their technical support.References
- G. Che, C. Liu, Q. Wang and Z. Xu, Chem. Lett., 2008, 37(2), 136–137 CrossRef CAS.
- C. E. Cui, G. Jiang, P. Huang, L. Wang and D. Liu, J. Lumin., 2014, 145, 665–668 CrossRef CAS.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27(40), 6195–6201 CrossRef CAS PubMed.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45(8), 2090–2136 RSC.
- Y. Hu, Y. Yuan, Y.-L. Shi, D. Li, Z.-Q. Jiang and L.-S. Liao, Adv. Funct. Mater., 2018, 28(32), 1802597 CrossRef.
- W. Jia, Q. Wang, H. Shi, Z. An and W. Huang, Chem. – Eur. J., 2020, 26(20), 4437–4448 CrossRef CAS.
- G. X. Xu, S. W. Zeng, B. T. Zhang, M. T. Swihart, K. T. Yong and P. N. Prasad, Chem. Rev., 2016, 116(19), 12234–12327 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. J. Wei, S. J. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118(4), 1770–1839 CrossRef CAS.
- P. F. Feng, Y. D. Wei, Y. N. Wang, J. C. Zhang, H. H. Li and Z. P. Ci, J. Am. Ceram. Soc., 2016, 99(7), 2368–2375 CrossRef CAS.
- X. Liu, X. X. Zhang and Z. F. Zhou, Mater. Res. Bull., 2016, 79, 84–89 CrossRef CAS.
- S. Das, J. Manam and S. K. Sharma, New J. Chem., 2017, 41(13), 5934–5941 RSC.
- X. G. Yang, X. Q. Lin, Y. B. Zhao, Y. S. Zhao and D. P. Yan, Angew. Chem., Int. Ed., 2017, 56(27), 7853–7857 CrossRef CAS.
- S. Yuan, Y. K. Deng and D. Sun, Chem. – Eur. J., 2014, 20(32), 10093–10098 CrossRef CAS.
- X. G. Yang and D. P. Yan, Chem. Sci., 2016, 7(7), 4519–4526 RSC.
- Y. Zhao, Y. J. Wang, N. Wang, P. Zheng, H. R. Fu, M. L. Han, L. F. Ma and L. Y. Wang, Inorg. Chem., 2019, 58(19), 12700–12706 CrossRef CAS.
- Z. Wang, C. Y. Zhu, Z. W. Wei, Y. N. Fan and M. Pan, Chem. Mater., 2020, 32(2), 841–848 CrossRef CAS.
- D. F. Li, F. F. Lu, J. Wang, W. D. Hu, X. M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140(5), 1916–1923 CrossRef CAS PubMed.
- T. Chatterjee and K. T. Wong, Adv. Opt. Mater., 2019, 7(1), 1800565 CrossRef.
- L. X. Xiao, Z. W. Wang, C. F. Zhang, X. Y. Xie, H. B. Ma, Q. Peng, Z. F. An, X. Y. Wang, Z. G. Shuai and M. Xiao, J. Phys. Chem. Lett., 2020, 11(9), 3582–3588 CrossRef CAS.
- T. Matsuzawa, Y. Aoki, N. Takeuchi and Y. Murayama, J. Electrochem. Soc., 1996, 143, 2670–2673 CrossRef CAS.
- H. Ohkita, W. Sakai, A. Tsuchida and M. Yamamoto, Bull. Chem. Soc. Jpn., 1997, 70, 2665–2670 CrossRef CAS.
- B. Cheng, L. Fang, Z. Zhang, Y. Xiao and S. Lei, J. Phys. Chem. C, 2011, 115(5), 1708–1713 CrossRef CAS.
- H. F. Brito, M. C. F. C. Felinto, J. Holsa, T. Laamanen, M. Lastusaari, M. Malkamaki, P. Novak, L. C. V. Rodrigues and R. Stefani, Opt. Mater. Express, 2012, 2(4), 420–431 CrossRef CAS.
- R. Kabe and C. Adachi, Nature, 2017, 550(7676), 384–387 CrossRef CAS PubMed.
- K. Jinnai, N. Nishimura, R. Kabe and C. Adachi, Chem. Lett., 2019, 48(3), 270–273 CrossRef CAS.
- Z. Lin, R. Kabe, N. Nishimura, K. Jinnai and C. Adachi, Adv. Mater., 2018, 30(45), 1803713 CrossRef.
- C. Y. Zhu, Z. Wang, J. T. Mo, Y. N. Fan and M. Pan, J. Mater. Chem. C, 2020, 8(29), 9916–9922 RSC.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32(45), e2003911 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4(3), 435–447 CrossRef CAS.
- T. Lu, Sobtop, (accessed 2024-01-01, 2024).
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Gaussian Inc., 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119(5), 525 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef.
- A. A. Voityuk and N. Rösch, J. Chem. Phys., 2002, 117(12), 5607–5616 CrossRef CAS.
- Z. Q. You, Y. C. Hung and C. P. Hsu, J. Phys. Chem. B, 2015, 119(24), 7480–7490 CrossRef CAS.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(3), 269–284 CAS.
- J. M. H. Olsen, S. Reine, O. Vahtras, E. Kjellgren, P. Reinholdt, K. O. Hjorth Dundas, X. Li, J. Cukras, M. Ringholm, E. D. Hedegård, R. Di Remigio, N. H. List, R. Faber, B. N. Cabral Tenorio, R. Bast, T. B. Pedersen, Z. Rinkevicius, S. P. A. Sauer, K. V. Mikkelsen, J. Kongsted, S. Coriani, K. Ruud, T. Helgaker, H. J. A. Jensen and P. Norman, J. Chem. Phys., 2020, 152(21), 214115 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- Y. Q. Hou, X. Zhang, K. P. Chen, D. Y. Liu, Z. J. Wang, Q. Y. Liu, J. Z. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7(39), 12048–12074 RSC.
- J. Xu, L. Deng, H. Zhang, H. Zhang, Y. Luo, X. Yan, J. Bao, M. Fang and Y. Wu, Chin. J. Inorg. Chem., 2020, 36(22), 2157–2168 CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85(22), 3533–3539 CrossRef CAS.
- M. F. Gelin, L. Chen, R. Borrelli and E. Thyrhaug, Chem. Phys., 2020, 528, 110495 CrossRef CAS.
- Y. Zhang, K. Wang, G. Jia, J. Li, H. Wang and Y. Tian, J. Alloys Compd., 2021, 860, 157914 CrossRef CAS.
- N. Christensson, H. F. Kauffmann, T. Pullerits and T. Mancal, J. Phys. Chem. B, 2012, 116(25), 7449–7454 CrossRef CAS.
- R. C. Hilborn, Am. J. Phys., 1982, 50(11), 982–986 CrossRef CAS.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Bredas, J. Phys. Chem. A, 2007, 111(42), 10490–10499 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- N. Nishimura, Z. S. Lin, K. Jinnai, R. Kabe and C. Adachi, Adv. Funct. Mater., 2020, 30(22), 2000795 CrossRef CAS.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32(45), e2003911 CrossRef.
- J.-L. Bredas, Mater. Horiz., 2014, 1(1), 17–19 RSC.
- N. Christensson, H. F. Kauffmann, T. Pullerits and T. Mančal, J. Phys. Chem. B, 2012, 116(25), 7449–7454 CrossRef CAS PubMed.
- M. F. Gelin, L. Chen, R. Borrelli and E. Thyrhaug, Chem. Phys., 2020, 528, 110495 CrossRef CAS.
- Y. Zhang, K. Wang, G. Jia, J. Li, H. Wang and Y. Tian, J. Alloys Compd., 2021, 860, 157914 CrossRef CAS.
- R. Zhu, X. Chen, N. Shu, Y. Shang, Y. Wang, P. Yang, Y. Tang, F. Wang and J. Xu, J. Phys. Chem. A, 2021, 125(47), 10144–10154 CrossRef CAS PubMed.
- Y. Shang, Z. Zhang, M. Huang, N. Shu, H. Luo, Q. Cao, B. Fan, Y. Han, M. Fang, Y. Wu and J. Xu, Phys. Chem. Chem. Phys., 2023, 25(47), 32666–32674 RSC.
- M. Yamamoto, H. Ohkita and S. Ito, Chin. J. Polym. Sci., 2001, 19(2), 129–134 CAS.
- M. Yamamoto, H. Ohkita, W. Sakai and A. Tsuchida, Synth. Met., 1996, 81, 301–304 CrossRef CAS.
- T. Yamanaka, H. Nakanotani and C. Adachi, Nat. Commun., 2019, 10, 5748 CrossRef CAS PubMed.
- J. Xu, J. Hao, C. Bu, Y. Meng, H. Xiao, M. Zhang and C. Li, J. Chem. Theory Comput., 2024, 20(9), 3590–3600 CrossRef CAS PubMed.
- C. M. Marian, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: supplemental figures and tables, molecular structures in xyz format and other computational results. See DOI: https://doi.org/10.1039/d4tc00805g |
| ‡ Note for non-theoretical readers: In the article, excited states S1, S3, T1 and T3 are all labelled according to the state energy at the ground state minima. However, as structure relaxing on the PES, states may go across conical intersections (CI), for example, going across the S1–S3 CI may lead to inversion of energy level (before: S1 < S3; after S3 < S1). In brief words, we use diabatic state notation for excited states in the article. |
| This journal is © The Royal Society of Chemistry 2025 |