
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the excited state proton transfer dynamics in N-oxide-based fluorophores: a keto–enol/enolate interplay to detect trace water in organic solvents†
Savita
 ,
Adarash Kumar
Shukla
and
Anupam
Bhattacharya
*
,
Adarash Kumar
Shukla
and
Anupam
Bhattacharya
*
Department of Chemistry, Birla Institute of Technology and Science-Pilani (Hyderabad Campus), Hyderabad-500078, India. E-mail: anupam@hyderabad.bits-pilani.ac.in; Tel: +91-40-66303522
First published on 22nd October 2024
Abstract
This work highlights the unique proton transfer ability observed in cyanoquinoxaline N-oxide-based fluorophores. A fluorophore HCQ was synthesized for this purpose, and a detailed study of its photophysical characteristics was undertaken. Preliminary structural characterization by NMR and single crystal XRD techniques indicated the possibility of a ground-state proton transfer (GSPT) reaction in the molecule. The absorbance and fluorescence spectroscopic studies further confirmed its sensitivity to the solvent environment and the possibility of the existence of three species enol (E)/enolate (RO−)/keto (K), with the predominance for a particular form based on the polarity of the solvent. Measurement of the fluorescence lifetime of the molecule allowed us to establish the role of protic solvents in the deprotonation of HCQ. In addition, the involvement of water in crystal packing and the significantly reduced lifetime of the molecule in water indicated the involvement of GSIPT and ESIPT processes. Based on the unique response of the molecule in the aqueous medium, its application for water detection in organic solvents was explored. HCQ demonstrated a water-induced fluorescence switch from its enol (E) form to the deprotonated (RO−)/keto (K) form, showcasing distinct spectral responses across various solvents with LOQ values in the range of 0.09–0.9%. The results were finally validated through Karl Fischer titration, showing similar outcomes.
Introduction
Excited state proton transfer dynamics, from salicylic acid to benzothiazoles and flavones, have been well explored and utilized for various applications.1N-Oxide-based systems, on the other hand, are versatile tools in various fields, including medicinal chemistry, where their utility as reactive intermediates and pharmaceutical agents is well recognized.2–6 The investigations on the excited state dynamics of these systems, particularly as fluorophores, remain largely unexplored. Given their significant biological activity, understanding their optical properties becomes crucial, as it can expand their applications as probes for bioimaging and material applications.7 The N-oxide group in these compounds acts as the proton acceptor site, whereas the presence of electron-donating/accepting groups on the scaffold increases the scope for spectral tuning (Scheme 1).8 In addition, introducing an amino/hydroxyl group in the neighbouring sites allows the possibility of intra/intermolecular proton transfer processes.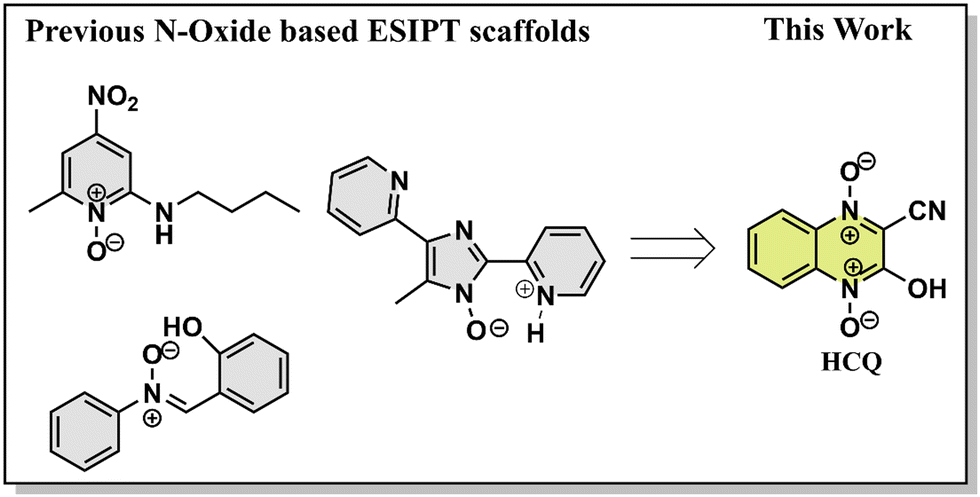 | ||
| Scheme 1 Structures of known fluorophores with the O/N–H⋯O−–N+ ESIPT site and fluorophore studied in this work. | ||
The excited state intramolecular proton transfer (ESIPT) is one of the most well-recognized mechanisms observed in the photochemical processes. ESIPT materials typically use π-delocalization/intramolecular hydrogen bonding for proton transfer.9 Different molecules with the abovementioned features are known to utilize the ESIPT route. However, research on proton transfer materials based on O/N–H⋯O−–N+ type intramolecular hydrogen bonding is limited. One of the initial reports on these systems was by Poór et al., who investigated the impact of an amino substituent in the ortho position on the excited-state dynamics. The molecule studied was 2-butylamino-6-methyl-4-nitropyridine N-oxide (2B6M), where intramolecular charge transfer triggered an intramolecular proton transfer.10 The same research group later determined the hydrogen bonding pattern in 2B6M using crystal structure analysis, and experimental/theoretical studies confirmed ESIPT in polar solvents like acetonitrile.11 Later, de Klerk et al. studied the same system and explored its behavior in nonpolar solvents. The study concluded that a monomeric ground state species dominates in the liquid state, emitting through two distinct pathways via internal proton-transfer processes from the normal and tautomeric excited states.12 A separate study on 2B6M revealed its unique fluorescence behavior in nonpolar solvents, where ratios and quantum yield of normal and tautomeric forms varied with excitation wavelength, challenging the Kasha–Vavilov rule. Transient absorption spectroscopy and fluorescence decay were used to identify multiple decay pathways, along with a detailed analysis of the impact of solvent polarity.13,14 The studies concluded that it might not be the S2 state but rather the high-energy vibrational mode of the S1 state that was involved. However, despite the abovementioned reports, a precise proton transfer mechanism remains elusive.15
Another series of rare ESIPT emitters are based on the 1-hydroxy-1H-imidazole scaffold. Shekhovtsov and coworkers demonstrated anti-Kasha (S2 → S0) emission behavior on (5-(4-fluorophenyl)-1-hydroxy-4-methyl-1H-imidazol-2-yl)(phenyl)methanone (HL).16 The study focussed on HL's proton transfer abilities, featuring an intramolecular N–O–H⋯N hydrogen bonding. The authors also demonstrated that the emission was excitation wavelength-dependent in CH3CN and the solid state, while in EtOH, the emission was relatively blue-shifted.17,18 The role of the π-conjugated quinoline-2-yl group in proton transfer processes and the influence of Zn2+ on the photochemistry and photophysics of an HL analog were also explored in another study.19 The same research group also explored the ESIPT feature of the HL core by increasing the number of donor N atoms and extending the π-system.20,21 Another compound that follows the ESIPT mechanism is α-(2-hydroxyphenyl)-N-phenylnitrone (Nit–OH). In this system, excited-state intramolecular proton transfer (ESIPT), twisted intramolecular charge transfer (TICT), and aggregation-induced emission in Nit–OH were investigated, which provide insights into the complex photophysical behavior of the molecule.22
Despite the wide scope for utility, there remains a gap in understanding the photophysics of these compounds. The current research work attempted to address this gap by investigating the optical characteristics of a substituted cyanoquinoxaline N-oxide (HCQ). The main objective of this study was to carry out a detailed study of its absorption and fluorescence processes across various solvent environments. Additionally, the enol/enolate/keto tautomerization phenomenon possible in the molecule was also studied by correlating fluorescence spectral features with the time-correlated single photon counting (TCSPC) data. Based on the photophysical studies, we also explored the application of HCQ.
Experimental section
Instruments
Nuclear magnetic resonance (NMR) spectroscopy: Both proton 1H and 13C NMR spectra were obtained using a Bruker AV NEO (400 MHz). HRMS data were collected using an Agilent 6546 LC/Q-TOF (Model AJS-ESI). Absorption and emission spectra were recorded using a UV-visible spectrophotometer Jasco V-650 and Horiba, Fl-3C, respectively. The time-correlated single-photon counting (TCSPC) technique was employed to determine time-resolved fluorescence measurements using Horiba DeltaFlex-01, Nano LEDs 405 L, 440 L, 510 L. Crystal data were procured using a single crystal XRD Rigaku Oxford XTALAB. The percentage of water in samples was measured using a Karl Fischer Titrator MA 101C (measuring range 10 ppm to 100% water contents). Mercury 4. 2. 0. Software was used for analyzing the crystal structure and intermolecular interactions.General information
All reagents obtained commercially were utilized as received. NMR spectra were recorded using a 400 MHz NMR instrument, with chemical shifts (δ) expressed in parts per million (ppm) and spin–spin coupling constants (J) in Hertz (Hz). Multiplicities were designated as s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Tetramethylsilane (TMS) was used as the internal standard.Theoretical calculations
The ground state (S0) structure of optimization and vibrational frequency calculations of HCQ were performed using the DFT B3LYP/6-311+G(d,p) and CPCM method.23–25 The calculated excited state transition energies were obtained using the TD DFT level using the same basis sets and functionals. All calculations were performed using the Gaussian 0926,27 suite program. The output files were analyzed using Chemcraft software.Synthesis
HCQ was prepared based on a previous literature report.28DBU (10 mmol) was introduced into a solution of 2H-benzo[d]imidazole 1-oxide (10 mmol) and ethyl 2-cyanoacetate (10 mmol) in DMF (15 mL) at 0 °C. Initially, a violet color developed in the mixture, which was left to stir for an additional 10 minutes under the same conditions. After the consumption of the starting material as indicated by TLC, the reaction was quenched by adding 50 mL of cold-water containing conc. HCl (1 mL). The precipitate thus obtained was filtered and washed with CH2Cl2 (50 mL) to yield pure HCQ.
Compound HCQ was obtained as a bright yellow solid. Melting point: 234.3–235.7 °C. (85.5% yield). 1H NMR (400 MHz, DMSO-d6): δ (ppm): 12.48 (s, broad, 1H), 8.2 (dd, 1H, J = 8.6 Hz, J = 0.8 Hz), 7.93 (t, 1H, J = 7.8 Hz, J = 1.2 Hz), 7.83 (dd,1H, J = 8.6 Hz, J = 0.8 Hz), 7.51 (t, 1H, J = 7.8 Hz, J = 1.2 Hz).13C NMR (101 MHz, DMSO-d6): δ 152.0, 136.0, 134.4, 130.5, 125.2, 120.3, 118.4, 114.5, 111.9. HRMS: m/z [M + H]+ Calculated: C9H5N3O3, 204.0409; found: 204.0387.
Results and discussion
Design of the probe and preliminary analysis
Our previous investigation showed variable responses of methyl and cyano substituents on the spectroscopic properties of cyanoquinoxaline N-oxide fluorophores29 (Fig. 1). While AMQ was non-emissive, ACQ emitted at λmax = 595 nm (577 nm in water). Based on the abovementioned study, it was envisaged that introducing a more electronegative heteroatom instead of nitrogen would enhance the acidity of the transferable hydrogen, thereby making the proton transfer process more feasible and possibly resulting in a considerable red shift in emission. The target molecule HCQ was initially synthesized, and subsequently, a detailed NMR analysis was carried out to understand the effect of solvent polarity on its structure.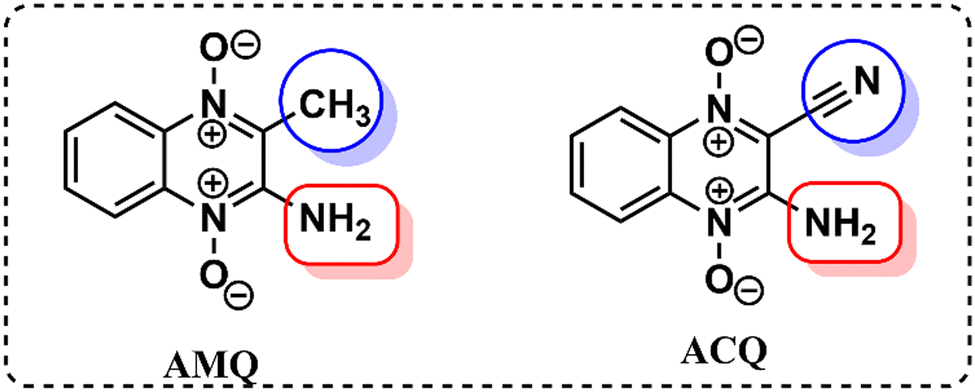 | ||
| Fig. 1 Molecular structures of AMQ and ACQ. | ||
In the 1H NMR spectra in DMSO-d6 and the D2O:DMSO-d6 mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), an upfield shift of peaks was observed, indicating the structural modification of the compound/formation of a new ground state species (Fig. 2(a)). However, the peak positions and peak patterns remained the same in D2O and CD3OD, suggesting the prevalence of the same species in aqueous and methanolic environments (Fig. 2(b)). Introducing one equivalent of triethylamine (TEA) to the NMR sample in DMSO-d6 revealed the generation of deprotonated species, plausibly existing in either enolate or keto form (Fig. 2(c)). However, the absence of a carbonyl peak in the 13C NMR spectrum indicated the predominance of enolate species (Fig. 2(d)). The 13C NMR spectra also showed an upfield shift for carbons at δ 136, 125.2, and 120.4 and a downfield shift for a peak at δ 114.5. In addition, a de-shielding of cyano carbon was observed with a shift in the δ value from 152 ppm to 154.8 ppm. The deshielding of cyano carbon plausibly arises due to the partial double bond character of the C–O− bond and its enhanced electron-withdrawing effect. Upon adding trifluoroacetic acid (TFA), a negligible influence on the 1H and 13C NMR spectrum was observed, confirming the existence of only enol form in DMSO in the absence/presence of acid.
:3), an upfield shift of peaks was observed, indicating the structural modification of the compound/formation of a new ground state species (Fig. 2(a)). However, the peak positions and peak patterns remained the same in D2O and CD3OD, suggesting the prevalence of the same species in aqueous and methanolic environments (Fig. 2(b)). Introducing one equivalent of triethylamine (TEA) to the NMR sample in DMSO-d6 revealed the generation of deprotonated species, plausibly existing in either enolate or keto form (Fig. 2(c)). However, the absence of a carbonyl peak in the 13C NMR spectrum indicated the predominance of enolate species (Fig. 2(d)). The 13C NMR spectra also showed an upfield shift for carbons at δ 136, 125.2, and 120.4 and a downfield shift for a peak at δ 114.5. In addition, a de-shielding of cyano carbon was observed with a shift in the δ value from 152 ppm to 154.8 ppm. The deshielding of cyano carbon plausibly arises due to the partial double bond character of the C–O− bond and its enhanced electron-withdrawing effect. Upon adding trifluoroacetic acid (TFA), a negligible influence on the 1H and 13C NMR spectrum was observed, confirming the existence of only enol form in DMSO in the absence/presence of acid.
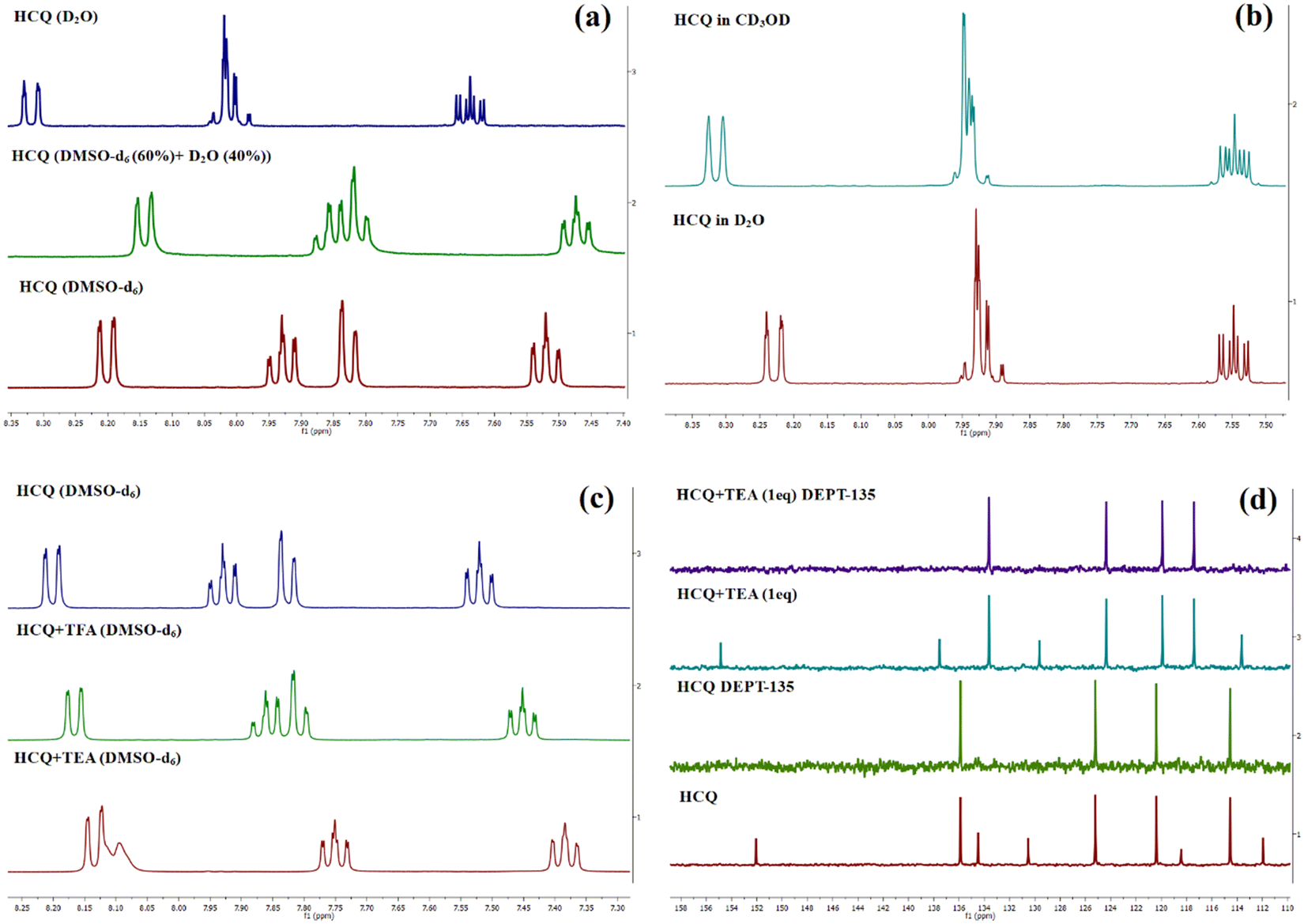 | ||
| Fig. 2 (a)1H NMR spectrum of HCQ in DMSO-d6, D2O, and DMSO-d6 (60%) + D2O (40%). (b) Comparison of similar splitting patterns in CD3OD and D2O. (c and d) Change in the 1H and 13C NMR spectrum of HCQ in the presence of triethyl amine (TEA) and trifluoroacetic acid (TFA). | ||
Crystal data for HCQ were also recorded (CCDC 2270859†). It revealed a monoclinic crystal system, with the C2/c group having a, b, and c values of 16.4937 (3) Å, 8.07760 (10) Å, and 14.6807 (3) Å, respectively. We also carried out a detailed structural analysis of ACQ, whose crystal structure was already reported in our previous study (CCDC 2194801†).29 A detailed structural analysis was necessary for this study to understand the effect of substituting nitrogen with oxygen in HCQ.
As shown by the crystal packing in ACQ (Fig. 3), the hydrogen bonds H3NA–N3 and H3NB–N3 both lie in the molecular plane and differ in bond length due to hydrogen bonding interaction with O1 and N4 of another molecule of ACQ (Fig. S34, ESI†). It was also validated by the dihedral angle values measured for N3–C5–C6–N1, O1–N2–C5–C6, O2–N1–C6–C5, and H3NB–N3–C5–C6 at −179.94°, −179.23°, 177.63° and −178.82° respectively (Table S11, ESI†) for ACQ. The dihedral angles for O1–N1–C9–C8 (178.43°), O3–N3–C8–C9 (−179.84°), and O2–C9–C8–N3 (178.64°) in HCQ also lie in a plane. However, in the same molecule, the measurements for H1–O1–N1–C9 (81.69°) and H1–O1–N1–C6 (−103.29°) confirm that the proton lies almost perpendicular to the plane and that the transfer of the proton takes place in the ground state. The elongated N1–O1 bond length of 1.3813 Å for HCQ further confirms the proton transfer because the typical O−–N+ bond is about 1.29 Å28 as observed for O2–N1 (1.2792 Å), N2–O1 (1.3088 Å) for ACQ and O3–N3 (1.2638 Å) for HCQ. The length of the C9–O2 bond (1.2259 Å) lies between a C–O (∼1.45 Å) and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼ 1.19 Å) bond, indicating that the C9–O2 bond attains a partial double bond character/bond length much closer to a carbonyl group (Table 1). The analysis shows the predominance of ground-state tautomerism in HCQ, in contrast to ACQ, which requires the presence of a base for the same. The observation in the case of HCQ is also substantiated by the observation from a previously reported N-oxide system with an ester substituent instead of –C
O (∼ 1.19 Å) bond, indicating that the C9–O2 bond attains a partial double bond character/bond length much closer to a carbonyl group (Table 1). The analysis shows the predominance of ground-state tautomerism in HCQ, in contrast to ACQ, which requires the presence of a base for the same. The observation in the case of HCQ is also substantiated by the observation from a previously reported N-oxide system with an ester substituent instead of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N.28 Besides, the crystal packing of HCQ also indicates the involvement of water molecules via hydrogen bonding and the concomitant proton transfer (Fig. S35, ESI†).
N.28 Besides, the crystal packing of HCQ also indicates the involvement of water molecules via hydrogen bonding and the concomitant proton transfer (Fig. S35, ESI†).
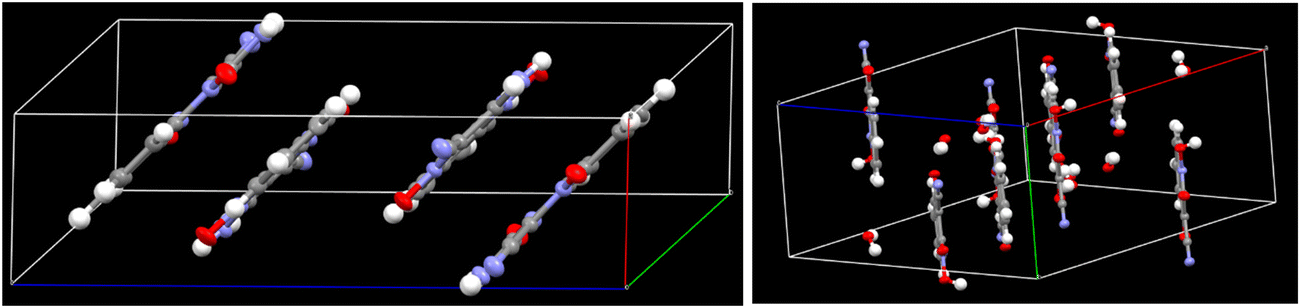 | ||
| Fig. 3 Crystal packing in ACQ (left) and HCQ (right). | ||
| ACQ | Bond length (Å) | HCQ | Bond length (Å) |
|---|---|---|---|
| O2–N1 | 1.2792 | O3–N3 | 1.2638 |
| N1–C6 | 1.3558 | N3–C8 | 1.3461 |
| C6–C5 | 1.4203 | C8–C9 | 1.4551 |
| C5–N2 | 1.3525 | N1–C9 | 1.3590 |
| N2–O1 | 1.3088 | N1–O1 | 1.3813 |
| C5–N3 | 1.3294 | C9–O2 | 1.2259 |
| H3NA–N3 | 0.8200 | O1–H1 | 0.8401 |
| H3NB–N3 | 0.8752 |
Absorption, emission, and excitation spectra of HCQ
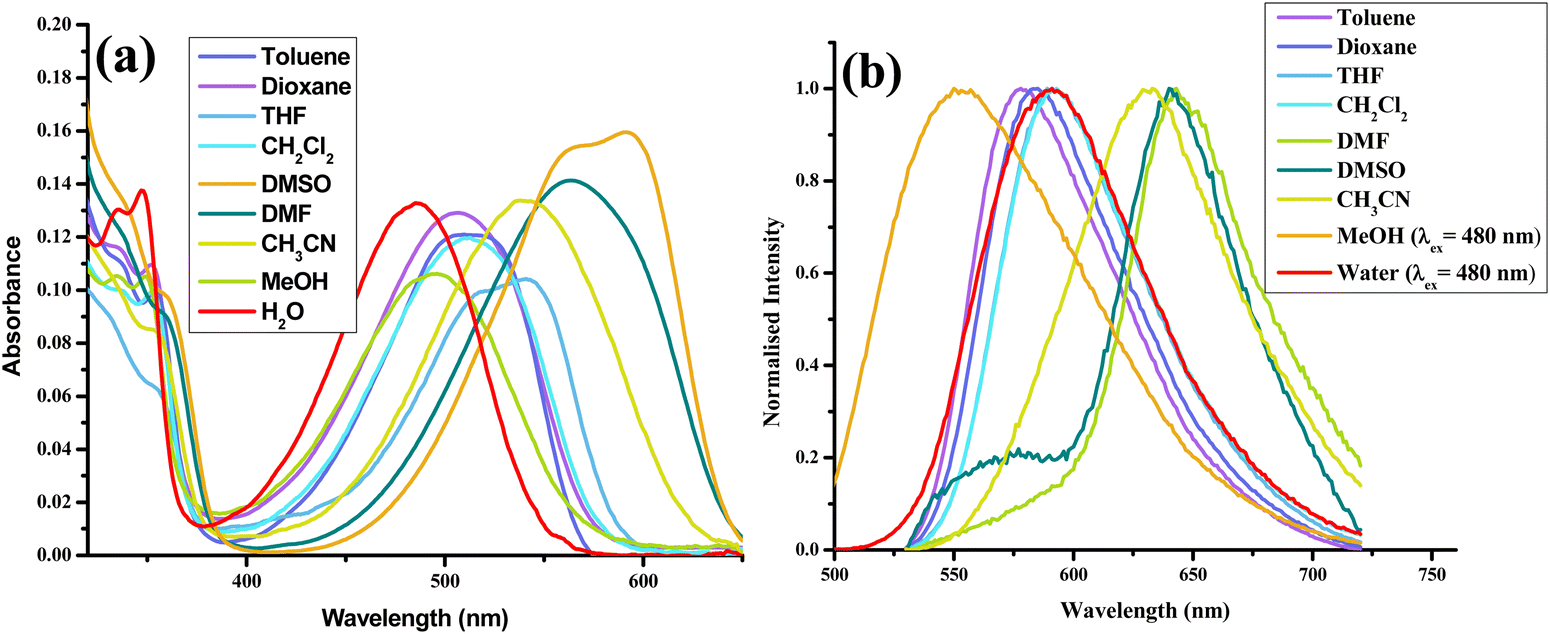
After thoroughly characterizing the probe, the basic photophysical properties of the system were explored. For a better understanding, solvatochromism data analysis was carried out by dividing the solvents into three categories: nonpolar (toluene, dioxane, THF, CH2Cl2), polar aprotic (CH3CN, DMF, DMSO) and polar protic (MeOH, H2O). The absorption spectra (Fig. 5(a)) of HCQ in different solvents indicate the possibility of the enol form (E), the deprotonated form (RO−), and the keto form (K). Similarly, the fluorescence spectrum (Fig. 5(b)) indicates the possibility of three species, which could be the excited enol (E*), an excited deprotonated form (RO−*), and the excited keto form (K*) (Fig. 4 and 6). | ||
| Fig. 4 Possible forms of HCQ in solution. | ||
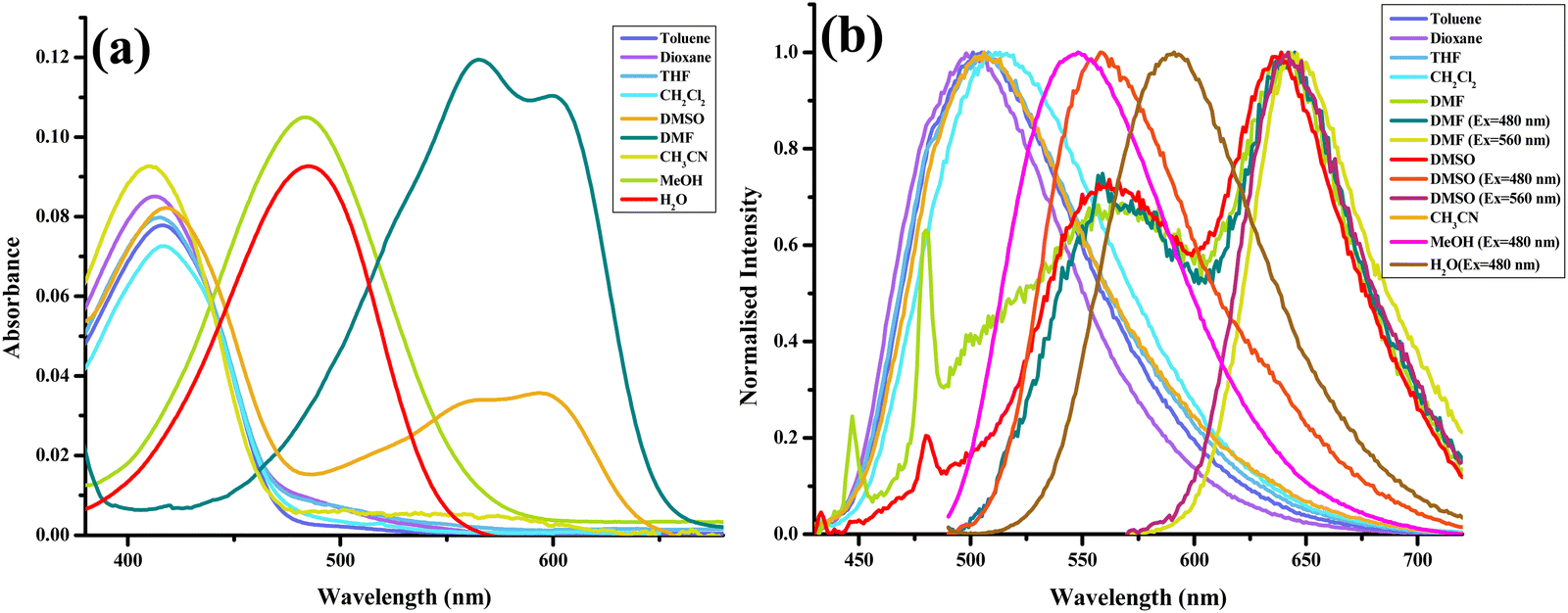 | ||
| Fig. 5 (a) Absorption spectra. (b) Normalised emission (λex = 420 nm) spectra of HCQ (10 μM) in solvents of varying polarities (note: arranged according to ET (30) with toluene-lowest to water-highest).30 | ||
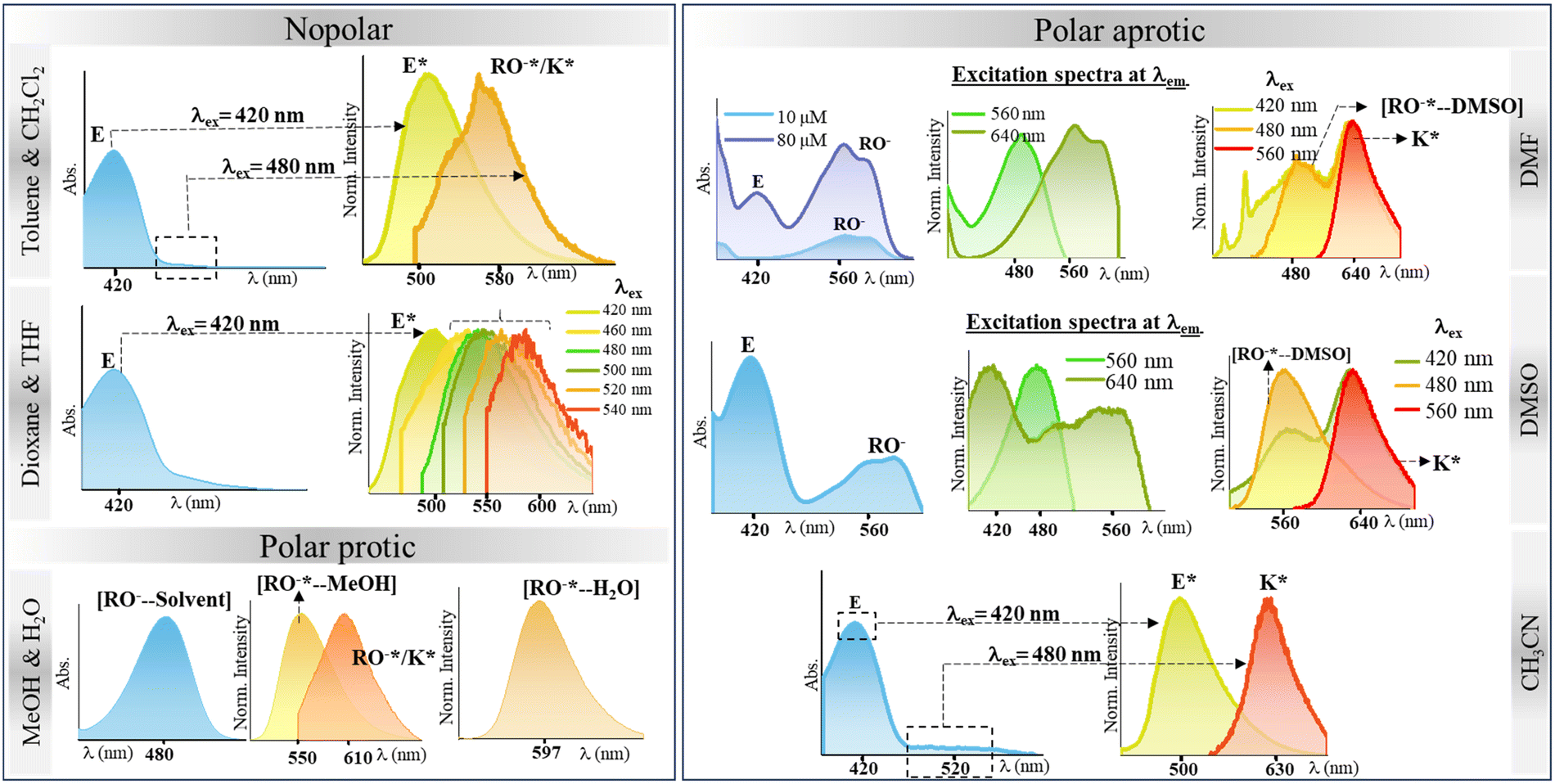 | ||
| Fig. 6 Overview of spectral responses of HCQ in solvents of varying polarities. | ||
Spectral features of the protonated and deprotonated forms
To gain further insights into the ground state and excited state forms in diverse solvents, solvatochromism (Fig. 7(a) and 8(a)) and solvatofluorochromism (Fig. 7(b) and 8(b)) of HCQ were studied under acidic (trifluoroacetic acid, TFA) and basic (triethylamine, TEA) conditions. In the presence of 100 μM TFA, in all solvents, the UV-visible spectrum showed a peak at ∼410 nm, initially observed only in non-polar solvents. This observation indicated that all the deprotonated/solvated forms can be converted to the E form in the presence of acid. Interestingly, for THF and dioxane, where a complex emission spectrum was initially observed, the presence of acid revealed a peak at ∼500 nm, which did not show any shift upon changing the excitation wavelength. Even the excitation spectrum overlapped with the absorbance spectrum of all solvents, demonstrating the prevalence of only one form in the ground state in an acidic environment. Noticeably, acid addition did not impact the emission wavelength of DMSO, DMF, MeOH, and water. The origin of such behavior could be the higher excited state acidity of the probe, which allows the proton to be transferred to either the acceptor N+–O− group/solvent in the presence of a high energy excitation wavelength of 420 nm.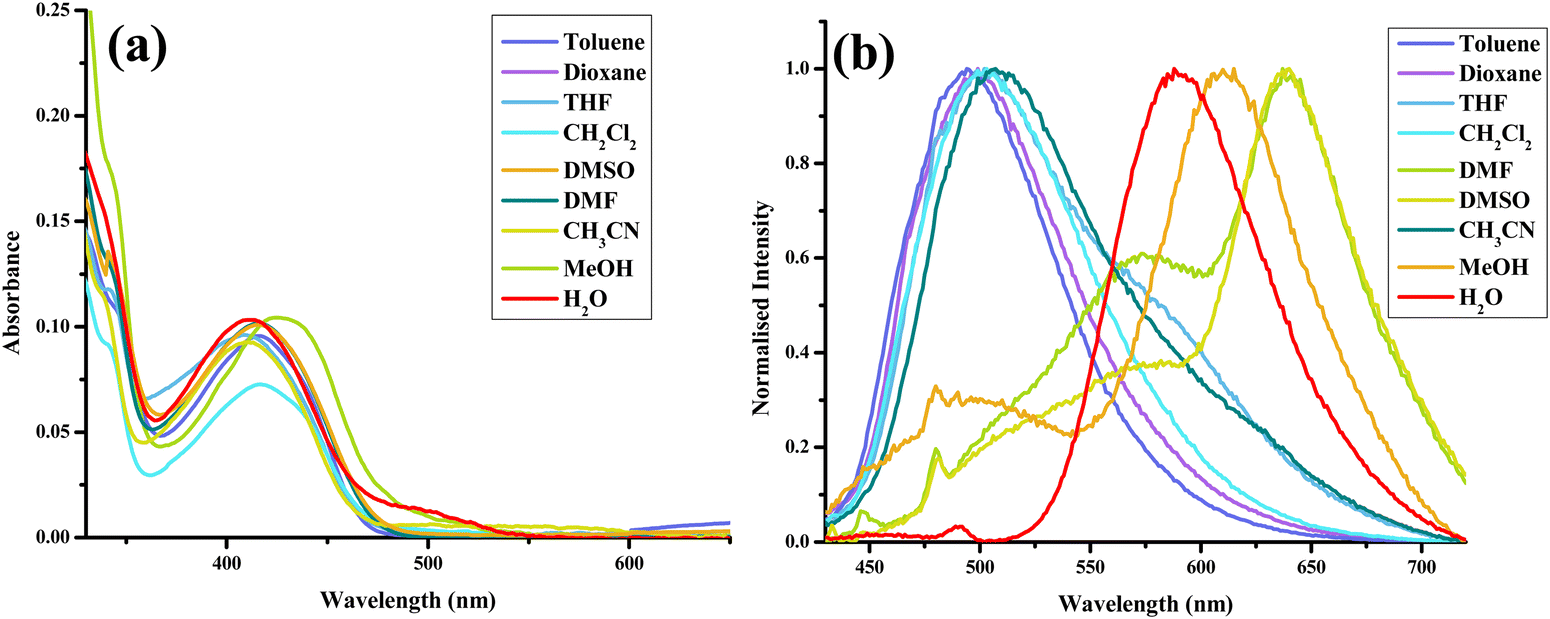 | ||
| Fig. 7 (a) Absorbance and (b) normalised emission spectra (λex = 420 nm) of HCQ (10 μM) with TFA (100 μM) in solvents of varying polarities. | ||
 | ||
| Fig. 8 (a) Absorbance and (b) normalised emission spectra (λex = 520 nm) of HCQ (10 μM) with TEA (100 μM) in solvents of varying polarities. (Note: λex for MeOH and water was kept at 480 nm since no changes in response to base were observed in the UV spectrum.) | ||
In the presence of TEA (100 μM), the disappearance of the peak at ∼410 nm and the concomitant increase in the absorption peak between 530 and 580 nm were observed for most solvents. This observation confirms that the 530–580 nm band in the absorbance spectrum of HCQ in DMSO, DMF, and CH3CN arises due to the deprotonation and that the E form and the RO−/K form exist in equilibrium in the ground state. However, no change in the absorbance signal was observed in water and MeOH. Similarly, in the excited state, the red-shifted emission band ranging from 578–643 nm appears at λex = 520 nm except for MeOH and water, where the emission spectrum remains unaltered in the presence of a base.
This study helped us to establish the prevalence of an enol E form and a deprotonated (RO−)/keto (K) form in the ground state of HCQ.
Fluorescence decay of HCQ
On completion of the preliminary photophysical studies, we focused on a detailed analysis of the lifetime for HCQ at different emission wavelengths. Fluorescence quantum yields (ϕF) and lifetimes (τ) were measured, followed by radiative (kr) and non-radiative rate constants (knr). These data, along with steady-state fluorescence information, are summarized in (Table S1, ESI†).Our initial study measured lifetimes at the blue and red end maxima of the emission spectra (Tables S2, S3 and Fig. S22, S23, ESI†). In addition, the obtained data were also compared with the lifetimes of the protonated and deprotonated species. The lifetime of the pure deprotonated form was obtained using a lower energy excitation laser source (510 nm) as it eliminates the interferences from all the species that emit below this excitation. It gave only one lifetime indicative of its keto/deprotonated state (Table S4 and Fig. S24, ESI†). For the protonated form, a certain percentage of the tautomeric isomer also persisted (Table S5 and Fig. S25, ESI†).
As observed in the fluorescence spectra of dioxane-HCQ and THF-HCQ conjugates, emission varied with excitation wavelength, which indicated the presence of multiple spectral components. In toluene and CH2Cl2, only two species/components were observed. At ∼500 nm, the lifetimes observed in these four solvents lie in the range of ∼0.45–0.55 ns. The lifetime obtained at red-end emission maxima for these solvents was inconsistent, possibly due to the HCQ–solvent interactions. However, upon deprotonation, the lifetimes were obtained in the ∼2.2–2.6 ns range.
Based on a large Stokes shift, the emission peak at λem ≈ 630–643 nm in CH3CN, DMSO, and DMF under basic conditions was attributed to the emission from RO−*/K* forms. This observation is comparable to the lifetime data (∼1.4–1.6 ns) obtained at the same wavelength for the solvent–HCQ ensemble without any acidic/basic additive. Additionally, the minor component observed only in DMSO-HCQ and DMF-HCQ combinations ranging from ∼1.4–1.6 ns showed an overall small amplitude (<11.7%). This indicated the presence of a small percentage of tautomeric form even at 560 nm and the prevalence of this species at 640 nm as a major component, as shown by the similar lifetime range. Apart from the E* and RO−*/K* forms, other components with lifetimes > 9 ns also exist in DMSO and DMF, plausibly arising due to the solvent–probe interactions. However, even upon protonation in DMF and DMSO, the persistence of a longer lifetime component of HCQ obtained at ∼640 nm was in line with the keto/deprotonated form. This observation shows the significant impact of proton transfer in an excited state. Interestingly, CH3CN, despite being a polar aprotic solvent, showed an intermediate behavior between nonpolar and polar aprotic solvents since the protonated form lifetime of ∼0.4 ns (>88.36%) lies close to that in non-polar solvents while the deprotonated state lifetime lies in the range of ∼1.4–1.6 ns which is similar to that in DMF and DMSO (Fig. 10).
Overall monitoring of lifetimes at the blue end of the spectrum with their emission at ∼500 nm in toluene, dioxane, THF, CH2Cl2, and CH3CN and ∼560 nm in DMSO and DMF also confirms the presence of two different species in these solvents. The scatter plot for solvents depicts that at ∼500 nm, the major component has a lifetime of ∼0.4 ns and ∼0.2 ns at ∼560 nm. While the red end spectrum (∼640 nm) lifetime of species generated in CH3CN, DMF, and DMSO is ∼1.4–1.6 ns and ∼2.4–2.6 ns at 570–590 nm in toluene, dioxane, THF, and CH2Cl2 which corresponds to the RO−*/K* form (Fig. 9).
 | ||
| Fig. 9 Scatter plot depicting the lifetime (ns) of the major component at respective λem in a pure solvent monitored at (a) blue end and (b) red end of the emission spectrum. | ||
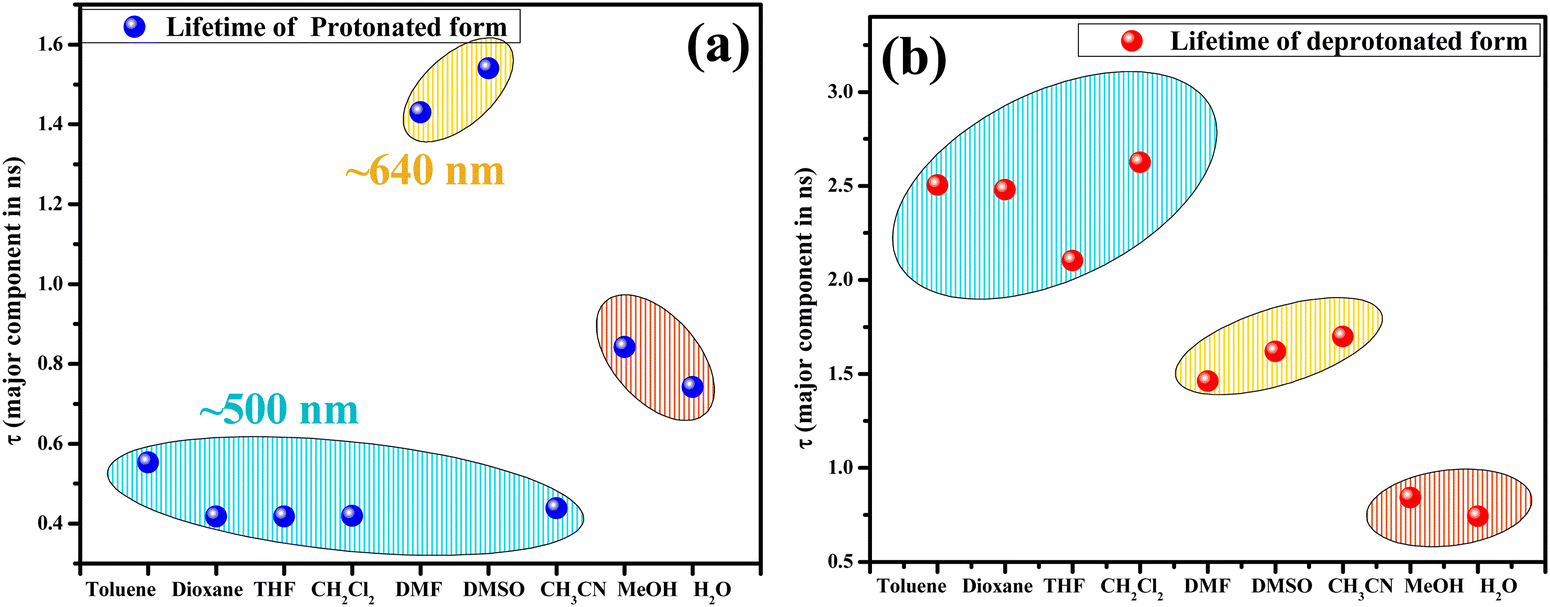 | ||
| Fig. 10 Scatter plot depicting the lifetime (ns) of the major component at respective λem for (a) the protonated and (b) the deprotonated form. [Note: In DMSO and DMF, the lifetime corresponding to λem 640 nm are shown for the protonated form because the maximum intensity was obtained at 640 nm. The 560 nm lifetimes are tabulated in Table S3, ESI†]. | ||
In water, the decay profile of HCQ exhibited a single exponential fit, indicating the prevalence of a single species irrespective of protonation or deprotonation at 597 nm. In MeOH, the decay lifetime revealed a tri-exponential decay at 610 nm, with a major component showing a lifetime of ∼0.84 ns. This value is comparable to that of water (∼0.7 ns), suggesting that the solvent environment in MeOH influences the excited state dynamics in a way that resembles that of water. Under acidic conditions, both MeOH–HCQ and water–HCQ conjugates exhibit a mono-exponential decay with comparable lifetimes. Shorter lifetimes in protic solvents at longer emission wavelengths can be attributed to the participation of the solvent molecules in the proton transfer process and the likely involvement of the ESIPT/ESPT mechanism.14 In order to substantiate the role of water molecules in the N-oxide proton transfer site, TD DFT B3LYP/6-311+G(d,p) calculations with excited state optimization of the [RO−—H2O] complex using a CPCM model in water were carried out. The oscillator strengths are tabulated in Table S12 (ESI†). The correlation between observed and predicted absorbance (505 nm) and emission spectra (599 nm) further substantiates a possible interaction between HCQ and water molecules [Fig. S39, ESI†].
Water detection
We decided to explore HCQ's potential for water detection by relying on its unique/stable photophysical response in the polar protic solvents against other solvents (Fig. 11). Despite the availability of traditional analytical techniques for assessing water content in organic solvents, there is a growing need to develop easily implementable methods for routine laboratory tasks and industrial applications.35 Various fluorescent probes are known for detecting trace water levels in organic solvents, from probes relying on mechanisms such as aggregation-induced emission/aggregation-caused quenching36–38 and hydrogen bonding to anti-Kasha emission.39,40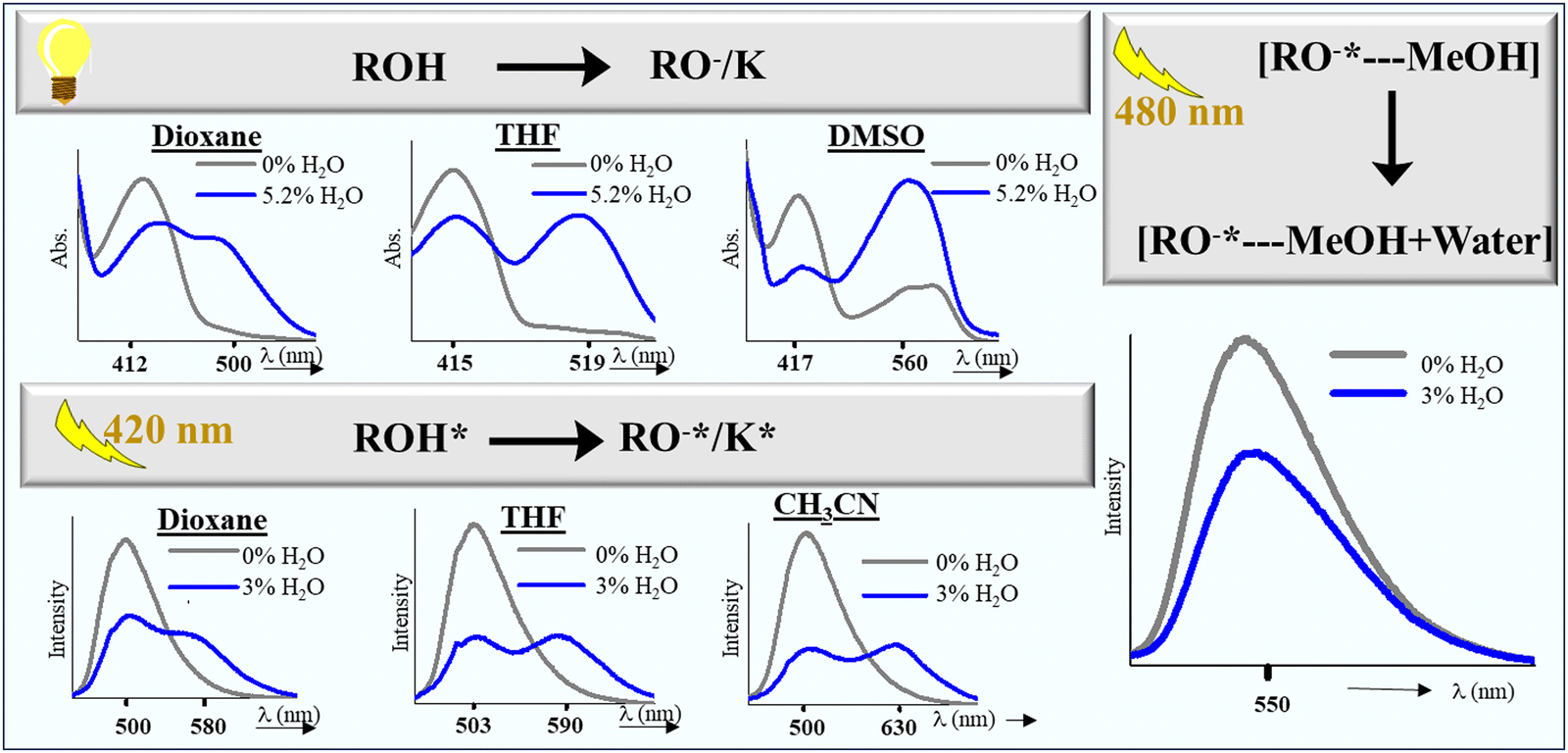 | ||
| Fig. 11 Overview of water detection using HCQ in different solvents. | ||
Our initial attempt started with dioxane and THF (Fig. S26–S29, ESI†). Herein, the addition of water in varying amounts in the dioxane–HCQ and THF–HCQ solution led to a decrease in the intensity of the absorption peaks at 412 nm and 415 nm, respectively, and a concomitant increase in the peaks at 500 nm and 519 nm, respectively. Due to the formation of multiple species in the excited state in dioxane and THF, these solutions were initially treated with 100 μM TFA before starting the fluorescence titration. It was done to generate a single system that can respond effectively to the variable water concentration. In the case of CH3CN, an inconsistent response was observed in the ground state signals upon titration with water (Fig. S31, ESI†). This forced us to conduct water analysis only via the fluorescence method by treatment with 100 μM TFA. Results included in the ESI for dioxane, THF, and acetonitrile reveal a consistent trend as the water content increased from 0–3%. The fluorescence intensity dropped at ∼500 nm, accompanied by a gradual red-shift in emission peak and an increase at 580 nm, 590 nm, and 630 nm, respectively. This variation in the turn-on fluorescence response stems from the solvent's variable polarities and solvation abilities. The limit of detection/quantitation (LOD/LOQ) of water for these solvents ranged from 0.001–0.08% and 0.09–0.9%, respectively (Table 2). Similar experiments with DMSO did not furnish any spectral responses towards water in the excited state; however, in the ground state, the peak at 412 nm decreased in intensity with a simultaneous increase in the intensity at 560 nm (Fig. S30, ESI†). Due to the prevalence of the deprotonated form in the ground state and inconsistent responses in the ground and excited state, water detection could not be carried out in DMF. In methanol, the water content was determined using the fluorescence route, where a decrease in the peak intensity at λmax 550 nm was observed, with a gradual increase in the water concentration (Fig. S32(a), ESI†). After completing the preliminary experiments, unknown samples were analyzed using the developed method. Volunteers were asked to prepare these samples in dry solvents with variable percentages of water (<3%) for further analysis. These samples were then recorded using fluorescence and UV spectrometers, and the value of the ratio of wavelengths as specified for each solvent was matched with the tabulated percentage range from the titration plot. To ensure the accuracy and reliability of the water content analysis, the same samples were also screened using a Karl Fischer titrator, further validating the obtained results (Tables S6–S10, ESI†).
| Solvent | Detection method | Shift in λ | LOD (%) | LOQ (%) | R 2 | Standard errora (±) | Water content (%) |
|---|---|---|---|---|---|---|---|
| [Note: For dioxane, THF, and CH3CN, 100 μM TFA was used along with HCQ only for fluorescence titration.]a Standard error denoted is from the slope of the calibration curve for three repeated experiments. | |||||||
| Dioxane | Absorbance | Bathochromic | 0.0483 | 0.4724 | 0.9972 | 0.0016 | 0–5.2 |
| Fluorescence | Bathochromic | 0.0864 | 0.9204 | 0.9964 | 0.0030 | 0–3 | |
| THF | Absorbance | Bathochromic | 0.0238 | 0.2588 | 0.9673 | 0.0072 | 0–5.2 |
| Fluorescence | Bathochromic | 0.0265 | 0.1773 | 0.9290 | 0.0226 | 0–3 | |
| CH3CN | Fluorescence | Bathochromic | 0.0522 | 0.3576 | 0.9423 | 0.0158 | 0–3 |
| DMSO | Absorbance | Bathochromic | 0.0015 | 0.0942 | 0.9871 | 0.0089 | 0–5.2 |
| MeOH | Fluorescence | Hypochromic | 0.2854 | 1.7093 | 0.9962 | 0.0020 | 0–3 |
The mechanism of detection
Two factors are mainly responsible for detecting water in the abovementioned solvents. In the solvents dioxane, THF, CH3CN, and DMSO that showed a bathochromic shift via absorption/fluorescence, the basis of water detection was an interplay of various excited state structures of HCQ. Water detection in methanol, on the other hand, relied on a hypochromic shift in peak intensity at λem = 550 nm (Fig. 11).Water detection via absorbance in dioxane, THF, and DMSO was possible due to the difference in their absorption maxima compared to water. Although observed even in CH3CN, this difference was only significant beyond a 10% water concentration (Fig. S33, ESI†). The absence of pronounced spectral changes in CH3CN can be attributed to its linear molecular structure, which allows the formation of a tighter solvent shell around the HCQ molecule, unlike dioxane, THF, and DMSO.
The detection of water in the dioxane–HCQ, THF–HCQ, and CH3CN–HCQ ensembles using the fluorescence technique relied on the previous analysis of the protonated states. In the presence of an acid, a single-emitting species was observed at λem = 500 nm, corresponding to the enol form as confirmed earlier. Due to the prevalence of a certain percentage of the keto form in these solvents at higher excitation, TFA was utilized to completely shift the equilibrium to the enol form (only for fluorescence titration in dioxane, THF, and CH3CN). Based on this, the excitation at 420 nm of the protonated form yielded emission peaks between 580 and 630 nm, which could be used to observe the effect of titration with water. Mechanistically, it is the water-assisted shifting of the equilibrium to the enolate/keto form.
The water detection method in methanol differs from that observed in other solvents. Although the ground state behavior of HCQ in methanol and water is comparable, the emission spectra differ by a wavelength of 47 nm (550 nm in MeOH and 597 nm in water). This disparity enabled water detection in methanol in the excited state using a λex of 480 nm.
Conclusion
The proton transfer capability of cyanoquinoxaline-N-oxide fluorophore HCQ has been demonstrated in this work. A detailed static/dynamic photophysical study of the probe in solvents of varying polarities, along with studies under acidic/basic conditions, disclosed the propensity of the probe to exist in variable structural entities. Owing to its stable behavior in the aqueous environment, the probe was also explored for detecting water in common organic solvents. It displayed advantages such as high sensitivity and rapid response. Given the simplicity of the probe structure, further efforts are undertaken in our laboratory to improve its sensitivity.Author contributions
Savita: conceptualisation, methodology, investigation, and writing – original draft. Adarash Kumar Shukla: methodology, investigation, and discussion. Anupam Bhattacharya: conceptualisation, resources, supervision, funding acquisition, and writing – review and editing.Data availability
Supporting data pertaining to this article have been included in the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
BITS-Pilani is acknowledged for the CDRF research grant (C1/23/138) and the Department of Science and Technology, Government of India, for the FIST grant (SR/FST/CS-I/2020/158). We thank Prof. Srikanta Dinda and Mr Apalla Reddy from the Department of Chemical Engineering, BITS-Pilani Hyderabad Campus, for providing access to the Karl Fischer instrument and for valuable discussions.References
- D. Ghosh, S. Batuta, S. Das, N. A. Begum and D. Mandal, Proton Transfer Dynamics of 4′-N,N-Dimethylamino-3-hydroxyflavone Observed in Hydrogen-Bonding Solvents and Aqueous Micelles, J. Phys. Chem. B, 2015, 119(17), 5650–5661 CrossRef CAS PubMed.
- S. Youssif, Recent trends in the chemistry of pyridine N-oxides, ARKIVOC, 2001, 1, 242–268 Search PubMed.
- J. d Klerk, I. H. van Stokkum, A. Szemik-Hojniak, I. Deperasinska, C. Gooijer, H. Zhang, W.-J. Buma and F. Ariese, Excited state processes of 2-butylamino-6-methyl-4-nitropyridine N-oxide in nonpolar solvents. A transient absorption spectroscopy study, J. Phys. Chem. A, 2010, 114(12), 4045–4050 CrossRef.
- S. Leyva-Ramos and A. Pedraza-Alvarez, Quinoxaline 1, 4-di-N-oxides: a review of the importance of their structure in the development of drugs against infectious diseases and cancer, Med. Chem. Res., 2021, 30(6), 1175–1184 CrossRef CAS.
- J. M. Turner and A. J. Messenger, Occurrence, biochemistry and physiology of phenazine pigment production, Adv. Microb. Physiol., 1986, 27, 211–275 CrossRef CAS PubMed.
- X. Shen and K. S. Gates, Enzyme-activated generation of reactive oxygen species from heterocyclic N-oxides under aerobic and anaerobic conditions and its relevance to hypoxia-selective prodrugs, Chem. Res. Toxicol., 2019, 32(3), 348–361 Search PubMed.
- G. Cheng, W. Sa, C. Cao, L. Guo, H. Hao, Z. Liu, X. Wang and Z. Yuan, Quinoxaline 1, 4-di-N-oxides: biological activities and mechanisms of actions, Front. Pharmacol., 2016, 7, 64 Search PubMed.
- Z. Ma, J. Li, K. Lin, M. Ramachandran, M. Li and Y. Li, Heterocyclic N-Oxides as Small-Molecule Fluorogenic Scaffolds: Rational Design and Applications of Their “On–Off” Fluorescence, Anal. Chem., 2020, 92(18), 12282–12289 CrossRef CAS.
- A. Shukla, V. T. N. Mai, V. V. Divya, C. H. Suresh, M. Paul, V. Karunakaran, S. K. M. McGregor, I. Allison, K. N. Narayanan Unni, A. Ajayaghosh, E. B. Namdas and S.-C. Lo, Amplified Spontaneous Emission from Zwitterionic Excited-State Intramolecular Proton Transfer, J. Am. Chem. Soc., 2022, 144(30), 13499–13510 CrossRef CAS PubMed.
- B. Poór, N. Michniewicz, M. Kállay, W. J. Buma, M. Kubinyi, A. Szemik-Hojniak, I. Deperasiñska, A. Puszko and H. Zhang, Femtosecond Studies of Charge-Transfer Mediated Proton Transfer in 2-Butylamino-6-methyl-4-nitropyridine N-Oxide, J. Phys. Chem. A, 2006, 110(22), 7086–7091 CrossRef.
- A. Szemik-Hojniak, I. Deperasiñska, L. Jerzykiewicz, P. Sobota, M. Hojniak, A. Puszko, N. Haraszkiewicz, G. van der Zwan and P. Jacques, Crystal Structure, Spectroscopic, and Theoretical Investigations of Excited-State Proton Transfer in the Doubly Hydrogen-Bonded Dimer of 2-Butylamino-6-methyl-4-nitropyridine N-Oxide, J. Phys. Chem. A, 2006, 110(37), 10690–10698 CrossRef CAS PubMed.
- J. S. de Klerk, A. Szemik-Hojniak, F. Ariese and C. Gooijer, Intramolecular Proton-Transfer Processes Starting at Higher Excited States: A Fluorescence Study on 2-Butylamino-6-methyl-4-nitropyridine N-Oxide in Nonpolar Solutions, J. Phys. Chem. A, 2007, 111(26), 5828–5832 CrossRef CAS PubMed.
- J. d Klerk, I. H. M. van Stokkum, A. Szemik-Hojniak, I. Deperasińska, C. Gooijer, H. Zhang, W.-J. Buma and F. Ariese, Excited State Processes of 2-Butylamino-6-methyl-4-nitropyridine N-oxide in Nonpolar Solvents. A Transient Absorption Spectroscopy Study, J. Phys. Chem. A, 2010, 114(12), 4045–4050 CrossRef PubMed.
- A. Szemik-Hojniak, Ł. Wiśniewski, I. Deperasińska, A. Makarewicz, L. Jerzykiewicz, A. Puszko, Y. Erez and D. Huppert, The impact of solvent polarity on intramolecular proton and electron transfer in 2-alkylamino-4-nitro-5-methyl pyridine N-oxides, Phys. Chem. Chem. Phys., 2012, 14(22), 8147–8159 RSC.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Breaking the Kasha Rule for More Efficient Photochemistry, Chem. Rev., 2017, 117(21), 13353–13381 CrossRef CAS PubMed.
- N. A. Shekhovtsov, E. B. Nikolaenkova, A. S. Berezin, V. F. Plyusnin, K. A. Vinogradova, D. Y. Naumov, N. V. Pervukhina, A. Y. Tikhonov and M. B. Bushuev, A 1-Hydroxy-1H-imidazole ESIPT Emitter Demonstrating anti-Kasha Fluorescence and Direct Excitation of a Tautomeric Form, ChemPlusChem, 2021, 86(10), 1436–1441 CrossRef CAS PubMed.
- N. A. Shekhovtsov, K. A. Vinogradova, S. N. Vorobyova, A. S. Berezin, V. F. Plyusnin, D. Y. Naumov, N. V. Pervukhina, E. B. Nikolaenkova, A. Y. Tikhonov and M. B. Bushuev, N-Hydroxy–N-oxide photoinduced tautomerization and excitation wavelength dependent luminescence of ESIPT-capable zinc(ii) complexes with a rationally designed 1-hydroxy-2,4-di(pyridin-2-yl)-1H-imidazole ESIPT-ligand, Dalton Trans., 2022, 51(25), 9818–9835 RSC.
- N. A. Shekhovtsov, A. A. Ryadun and M. B. Bushuev, Luminescence of a Zinc (II) Complex with a Protonated 1-Hydroxy-1H-imidazole ESIPT Ligand: Direct Excitation of a Tautomeric Form, ChemistrySelect, 2021, 6(44), 12346–12350 CrossRef CAS.
- N. A. Shekhovtsov, E. B. Nikolaenkova, A. S. Berezin, V. F. Plyusnin, K. A. Vinogradova, D. Y. Naumov, N. V. Pervukhina, A. Y. Tikhonov and M. B. Bushuev, Tuning ESIPT-coupled luminescence by expanding π-conjugation of a proton acceptor moiety in ESIPT-capable zinc (II) complexes with 1-hydroxy-1 H-imidazole-based ligands, Dalton Trans., 2022, 51(39), 15166–15188 RSC.
- N. A. Shekhovtsov, E. B. Nikolaenkova, S. N. Vorobyova, V. F. Plyusnin, K. A. Vinogradova, T. S. Sukhikh, A. Y. Tikhonov and M. B. Bushuev, Luminescence of ESIPT-capable zinc (II) complexes with a 1-hydroxy-1 H-imidazole-based ligand: exploring the impact of substitution in the proton-donating moiety, Dalton Trans., 2023, 52(23), 8114–8134 RSC.
- N. A. Shekhovtsov, E. B. Nikolaenkova, A. A. Ryadun, D. G. Samsonenko, A. Y. Tikhonov and M. B. Bushuev, ESIPT-Capable 4-(2-Hydroxyphenyl)-2-(Pyridin-2-yl)-1 H-Imidazoles with Single and Double Proton Transfer: Synthesis, Selective Reduction of the Imidazolic OH Group and Luminescence, Molecules, 2023, 28(4), 1793 CrossRef CAS PubMed.
- L. M. Carneiro, A. F. Keppler, F. F. Ferreira, P. Homem-de-Mello and F. H. Bartoloni, Mechanisms for the Deactivation of the Electronic Excited States of α-(2-Hydroxyphenyl)-N-phenylnitrone: From Intramolecular Proton and Charge Transfer to Structure Twisting and Aggregation, J. Phys. Chem. B, 2022, 126(38), 7373–7384 CrossRef CAS PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255(4–6), 327–335 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24(6), 669–681 CrossRef CAS PubMed.
- M. Cossi and V. Barone, Time-dependent density functional theory for molecules in liquid solutions, J. Chem. Phys., 2001, 115(10), 4708–4717 CrossRef CAS.
- M. Frisch; F. Clemente, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino and G. Zhe, Gaussian 09, revision a. 01, 2009, 20–44 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian09, R., 01, Gaussian Inc., Wallington, CT, 2016 Search PubMed.
- Y. Xu, F. Wu, Z. Yao and M. Z. S. Jiang, Synthesis of quinoxaline 1, 4-di-N-oxide analogues and crystal structure of 2-carbomethoxy-3-hydroxyquinoxaline-di-N-oxide, Molecules, 2011, 16(8), 6894–6901 CrossRef CAS PubMed.
- C. Savita, A. Nandikolla, A. K. Shukla, K. V. G. Chandra Sekhar and A. Bhattacharya, Visible light sensing of ions by a cyanoquinoxaline 1,4-dioxide-based probe and its applications, Dalton Trans., 2023, 52(13), 4103–4111 RSC.
- J. P. Cerón-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, Solvent polarity scales: determination of new ET(30) values for 84 organic solvents, J. Phys. Org. Chem., 2014, 27(6), 512–518 CrossRef.
- I. Kolthoff and T. Reddy, Acid-base strength in dimethyl sulfoxide, Inorg. Chem., 1962, 1(2), 189–194 CrossRef CAS.
- I. M. Kolthoff, M. K. Chantooni and H. Smagowski, Acid-base strength in N, N-dimethylformamide, Anal. Chem., 1970, 42(13), 1622–1628 CrossRef CAS.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Strengths of acids in acetonitrile, Eur. J. Org. Chem., 2021,(9), 1407–1419 CrossRef.
- R. Das, S. Mitra and S. Mukherjee, Proton transfer and anion formation in the ground and excited states of 4-methyl-2, 6-diformyl phenol in highly polar aprotic solvents, J. Photochem. Photobiol., A, 1993, 76(1–2), 33–38 CrossRef CAS.
- R. Oguchi, K. Yamaguchi and T. Shibamoto, Determination of Water Content in Common Organic Solvents by a Gas Chromatograph Equipped with a Megabore Fused-Silica Column and a Thermal Conductivity Detector, J. Chromatogr. Sci., 1988, 26(11), 588–590 CAS.
- H. S. Jung, P. Verwilst, W. Y. Kim and J. S. Kim, Fluorescent and colorimetric sensors for the detection of humidity or water content, Chem. Soc. Rev., 2016, 45(5), 1242–1256 RSC.
- F. Wu, L. Wang, H. Tang and D. Cao, Excited State Intramolecular Proton Transfer Plus Aggregation-Induced Emission-Based Diketopyrrolopyrrole Luminogen: Photophysical Properties and Simultaneously Discriminative Detection of Trace Water in Three Organic Solvents, Anal. Chem., 2019, 91(8), 5261–5269 CrossRef CAS PubMed.
- C. Zhang, X. Li, Z. Li, Y. Wang, J. Lu, L. Zhu and F. Zhang, Two-Stage Three-Dimensional Luminescent Sensing Strategy for Precisely Detecting a Wide Range of Water Content in Tetrahydrofuran, Anal. Chem., 2022, 94(19), 7004–7011 CrossRef CAS PubMed.
- H. Sun, S.-S. Sun, F.-F. Han, Y. Zhao, M.-D. Li, B.-X. Miao, J. Nie, R. Zhang and Z.-H. Ni, Water-stimuli-responsive dynamic fluorescent switch from Kasha's rule to anti-Kasha's rule based on a tetraphenylethene substituted Schiff base, Chem. Eng. J., 2021, 405, 127000 CrossRef CAS.
- Y. Zhou, G. Baryshnikov, X. Li, M. Zhu, H. Ågren and L. Zhu, Anti-Kasha's Rule Emissive Switching Induced by Intermolecular H-Bonding, Chem. Mater., 2018, 30(21), 8008–8016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2270859. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc02651a |
| This journal is © The Royal Society of Chemistry 2025 |