
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new eco-friendly and highly emitting Mn-based hybrid perovskite toward high-performance green down-converted LEDs†
Asmae
Ben Abdelhadi
 ab,
Mario
Gutiérrez
a,
Boiko
Cohen
a,
Luis
Lezama
c,
Mohammed
Lachkar
b and
Abderrazzak
Douhal
*a
ab,
Mario
Gutiérrez
a,
Boiko
Cohen
a,
Luis
Lezama
c,
Mohammed
Lachkar
b and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, e INAMOL, Campus Tecnológico de Toledo, Universidad de Castilla-La Mancha (UCLM), Avenida Carlos III, S.N., 45071 Toledo, Spain. E-mail: abderrazzak.douhal@uclm.es
bEngineering Laboratory of Organometallic, Molecular Materials, and Environment (LIMOME), Faculty of Sciences, Sidi Mohamed Ben Abdellah University, 30000 Fez, Morocco
cDepartamento de Química Orgánica e Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, UPV/EHU, B Sarriena s/n, 48940 Leioa, Spain
First published on 27th November 2023
Abstract
As luminescent materials, lead-free zero-dimensional (0D) organic–inorganic metal halide perovskites have recently attracted special attention. Here, we have synthesized a novel lead-free manganese-based organic–inorganic hybrid halide (TPA)2MnBr4 where TPA = [(C3H7)3NH]+ (i.e., tri-n-propyl-ammonium). The single crystal X-ray diffraction (SCXRD) shows that (TPA)2MnBr4 crystallizes in the triclinic phase and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, with the lattice parameters of a = 9.5444(2) Å, b = 10.1056(2) Å and c = 15.1835(3) Å. The powder XRD (PXRD) pattern confirms the synthesized perovskite's high purity and crystalline nature. The crystalline structure of the melted sample at 72 °C and after cooling down to room temperature is similar to the not melted one, while the material shows high-temperature sustainability up to 180 °C. The EPR experiments confirm the tetrahedral environments of the Mn2+ ions. In this compound, the [MnBr4]2–tetrahedral units are isolated by large (TPA) organic cations, resulting in a unique 0D structure. This 0D perovskite emits a strong green light under UV excitation, with a maximum at 520 nm and a high photoluminescence quantum yield (PLQY) of 62% benefitting from the lowest d–d orbital transition of the Mn2+ ion in the tetrahedral crystal field and the long Mn–Mn distance, respectively. The photoluminescence lifetime of (TPA)2MnBr4 is 0.39 ms. Fluorescence lifetime imaging microscopy (FLIM) shows a uniform distribution of the emitting species with no apparent bulk or edge defects. A series of color-tunable down-converted light-emitting diodes (LEDs) were fabricated by using increasing amounts of (TPA)2MnBr4 as the phosphor layer, reflecting its high potential for application in LED technologies.
space group, with the lattice parameters of a = 9.5444(2) Å, b = 10.1056(2) Å and c = 15.1835(3) Å. The powder XRD (PXRD) pattern confirms the synthesized perovskite's high purity and crystalline nature. The crystalline structure of the melted sample at 72 °C and after cooling down to room temperature is similar to the not melted one, while the material shows high-temperature sustainability up to 180 °C. The EPR experiments confirm the tetrahedral environments of the Mn2+ ions. In this compound, the [MnBr4]2–tetrahedral units are isolated by large (TPA) organic cations, resulting in a unique 0D structure. This 0D perovskite emits a strong green light under UV excitation, with a maximum at 520 nm and a high photoluminescence quantum yield (PLQY) of 62% benefitting from the lowest d–d orbital transition of the Mn2+ ion in the tetrahedral crystal field and the long Mn–Mn distance, respectively. The photoluminescence lifetime of (TPA)2MnBr4 is 0.39 ms. Fluorescence lifetime imaging microscopy (FLIM) shows a uniform distribution of the emitting species with no apparent bulk or edge defects. A series of color-tunable down-converted light-emitting diodes (LEDs) were fabricated by using increasing amounts of (TPA)2MnBr4 as the phosphor layer, reflecting its high potential for application in LED technologies.
Introduction
During the last 2 decades, organic–inorganic hybrid lead (Pb) perovskites have attracted significant interest for their photoluminescence and electroluminescence properties such as large absorption coefficient, high charge carrier mobility, high photoluminescence quantum yield (PLQY), direct optical band gap and high defect tolerance.1–10 However, the high toxicity of Pb-based materials and their poor stability remain the major obstacles to their practical application.11,12 Therefore, an important way to overcome the toxicity issue is to replace Pb in perovskites with eco-friendly elements.13,14 Compounds based on divalent Mn(II) cations have long been considered promising candidates to replace Pb2+ in organic–inorganic perovskites.15,16 Manganese-based hybrid inorganic–organic materials have drawn significant attention not only in the fields of ferromagnetics17–19 and solar cells,20 but also for their good stability and high-performance emission with colors ranging from green to red depending on the coordination environment of Mn2+.21–24 The Mn2+ metal ion has a 3d5 electronic configuration and generally exhibits visible photoluminescence (PL) resulting from the 4T1-6A1 transition,25–27 which mainly depends on the crystal field surroundings of Mn2+ in the lattice. In some cases, the Mn2+ ions are tetrahedrally coordinated (weak crystal field), and each Mn2+ ion is surrounded by four ligands to form an independent [MnX4]2− tetrahedral unit, where the Mn–Mn distance is large enough to eliminate direct spin–spin coupling between adjacent Mn ions.28 In that case, the contributions from an individual Mn2+ ion dominate the optical transition, usually leading to green emission (500–550 nm) with a small full width at half maximum (FWHM) of 25–60 nm.29–31 For other cases, when the Mn2+ ions are octahedrally coordinated (a strong crystal field), each Mn2+ ion is surrounded by six ligands ([MnX6]4−), and the Mn–Mn distance is largely reduced due to the formation of edge-sharing infinite linear chains along one direction. Therefore, the octahedral coordination of Mn2+ ions (higher Mn–Mn coupling interaction) induces a shift in the emission spectrum to orange or deep red spectral regions, with a large FWHM of more than 60 nm.32–34 In some cases, it's has been found that the emission color of the Mn-based perovskites can be tuned from green to red during the phase transition from tetra-coordination to hexa-coordination of manganese(II) ions.35–37 Thus, Mn-hybrid perovskites show excellent gas sensing properties, X-ray scintillators, and good performance in terms of LEDs (light emitting diodes) with the advantages of low cost, environmental protection, and easy preparation.23,38–42The organic ligand is an important structural component of the Mn-based perovskites that affects the electronic band structures and the corresponding crystal field. Organic cations with a rigid structure could reduce the thermal vibrations and suppress nonradiative transitions thus increasing the PLQY.23 On the other hand, bulkier ligands in the correct molar ratio might allow tuning the optical properties of the resulting perovskites increasing the Mn–Mn separation. Although not as common, as an alternative strategy, smaller monovalent organic cations could also be used to obtain the required Mn–Mn distance to obtain highly efficient green-emitting Mn-based perovskites. Since they are monovalent, two such cations will be needed to charge balance the [MnBr4]2− perovskite tetrahedra, which should lead to a longer separation between the Mn centers.43 This approach has been used for example to produce green-emissive 0D Mn(II)-based perovskite of formula (LH)2MnX4 ((TEM)2MnBr4, TEM = triethylammonium) with a Mn–Mn distance of 8.85 Å and a PLQY of 50%.44 Another study has reported (DIPA)2MnBr4 (DIPA = diisopropylammonium) perovskites, with a similar distance between the [MnBr4]2− tetrahedron (8.85 Å) and a PLQY of 62.2%.45 Thus, it can be expected that the stereochemistry of small monovalent cation (both the DIPA and TEM provide a significant degree of steric hindrance) plays a fundamental role in the optoelectronic properties of the formed Mn-based perovskites. Notably, these studies did not provide information on the stability of these materials against moisture under ambient conditions. Therefore, small monovalent cations of the ligands with increased steric hindrance, like the tri-n-propylammonium cation, for example, could be used to improve the PLQY of the green emissive Mn-based organic–inorganic halide materials and will help evaluate the impact of tertiary ammonium cations on the long-term stability of Mn-based metal halide perovskites.
Here, we prepared, characterized, and studied the spectroscopy and photodynamical properties of a new Mn(II) based organic–inorganic hybrid perovskite material (TPA)2MnBr4 by using tri-n-propylammonium as the organic cation. The perovskite was obtained at a high yield using a slow evaporation method in two different solvents, with excellent reproducibility. It was fully characterized using a combination of single crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD), thermal analyses (TGA-DSC), electron-spin paramagnetic resonance (EPR), UV-Vis steady-state and time-resolved emission spectroscopy, and fluorescence confocal microscopy analyses. (TPA)2MnBr4 displays an intense green emission centered at 520 nm with a high quantum yield PLQY of 62% due to effective d–d transitions of tetrahedrally coordinated Mn2+. Its emission decays monoexponentially with a lifetime of 0.39 ms. Furthermore, a series of down-converted LED devices were fabricated using the (TPA)2MnBr4 powder as a green phosphor deposited on a blue (465 nm) LED chip, showing the possibility of controlling the emission color of the device with the amount of the Mn-based perovskite. Therefore, our work shows that (TPA)2MnBr4 has great potential to be the next-generation of hybrid organic–inorganic Mn halides for LED technologies and may also present great opportunities for its use in other optoelectronic applications.
Experimental section
The Experimental section is described in detail in the ESI,† Section 1. It includes the synthesis procedure of tri-n-propylammonium bromide salt (TPA), and the corresponding (TPA)2MnBr4 perovskite, both as a single crystal and as a crystalline powder (Fig. S1, ESI†). It also describes the fabrication of the LEDs and the techniques and experimental conditions used for the characterization of the perovskite.Results and discussion
Structural characterization
The SCXRD experiment was carried out at 150 K and shows that the synthesized perovskite possesses a typical zero-dimensional crystal structure, with a chemical formula of (TPA)2MnBr4. It crystallizes in the triclinic phase and P space group, with the lattice parameters of a = 9.5444(2) Å, b = 10.1056(2)Å, c = 15.1835(3) Å, α = 82.880(2)°, β = 73.734(2)°, γ = 83.119(2)° and Z = 2. The structure of a single unit of (TPA)2MnBr4 is shown in Scheme 1. Further details for the crystallographic parameters are provided in Tables S1–S3 (ESI†). The asymmetric unit of the compound (Scheme 1A) consists of two independent tri-n-propylammonium organic cations and one [MnBr4]2− anion as the inorganic part. The Mn2+ metal cation has a tetrahedral geometry composed of four bromide ions to form [MnBr4]2− anion units. The circumjacent space of [MnBr4]2− is filled with tri-n-propylammonium organic cations via hydrogen (H) bonding in opposite directions, forming a unique 0D structure. The geometry of [MnBr4]2− tetrahedrons can be regarded as a slightly distorted tetrahedral, with (Mn–Br) bond lengths varying from 2.5076(4) to 2.5408(4) Å, while the (Br–Mn–Br) angles vary from 106.654(14) to 109.781(14)° (Scheme 1B). These values of bond lengths and angles are in good agreement with those found in other reported [MnBr4]2− tetrahedrons.46–49 The crystal structure is stabilized by intermolecular H-bonding interactions between tri-n-propylammonium organic cations and [MnBr4]2− complex anions via an intricate network of C–H⋯Br and N–H⋯Br H-bonds into a 3-dimensional network that stabilizes the resulting 0D perovskite (Scheme 1C and Table S3, ESI†). The Mn–Mn distances in (TPA)2MnBr4 (approximately 9.5444(5) and 10.0066(5) (longest) Å (Fig. S2, ESI†)) are sufficiently large to eliminate the direct spin–spin coupling between Mn-ions in independent [MnBr4]2− tetrahedral units.41 Hence, it is expected that its optical transition will be dominated by an independent manganese ion, resulting in a green emission.
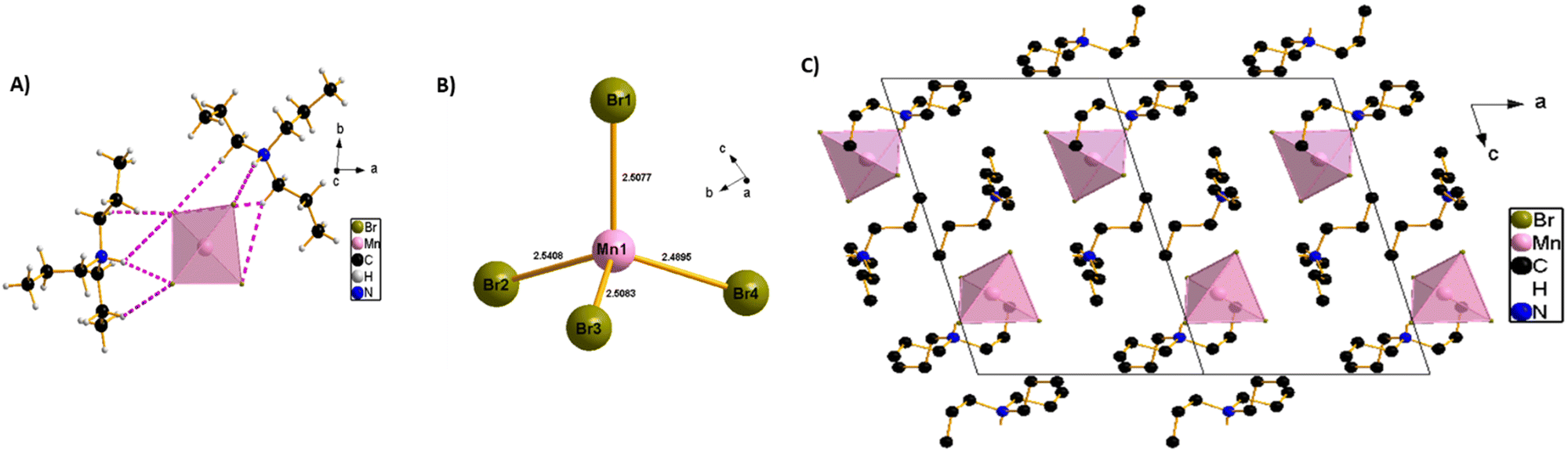 | ||
| Scheme 1 (A) Crystal structure of (TPA)2MnBr4 in the asymmetric part of the unit cell, the pink dashed line shows the H-bonds between tri-n-propylammonium organic cations and [MnBr4]2− tetrahedral units. (B) Mn–Br distances in the single [MnBr4]2− tetrahedral units. (C) The packing diagram of (TPA)2MnBr4 viewed from the b-direction at room temperature; all the H-atoms had been omitted for clarity. | ||
Powder X-ray diffraction (PXRD)
The crystalline structure of the as-synthesized perovskite was confirmed via powder X-ray diffraction (PXRD) analysis (Fig. 1A). The PXRD pattern of (TPA)2MnBr4 is consistent with the simulated one obtained from the single crystal with slightly varying intensities (Fig. 1A), most probably due to preferential orientations, demonstrating the high phase purity and the uniformity of the synthesized crystals.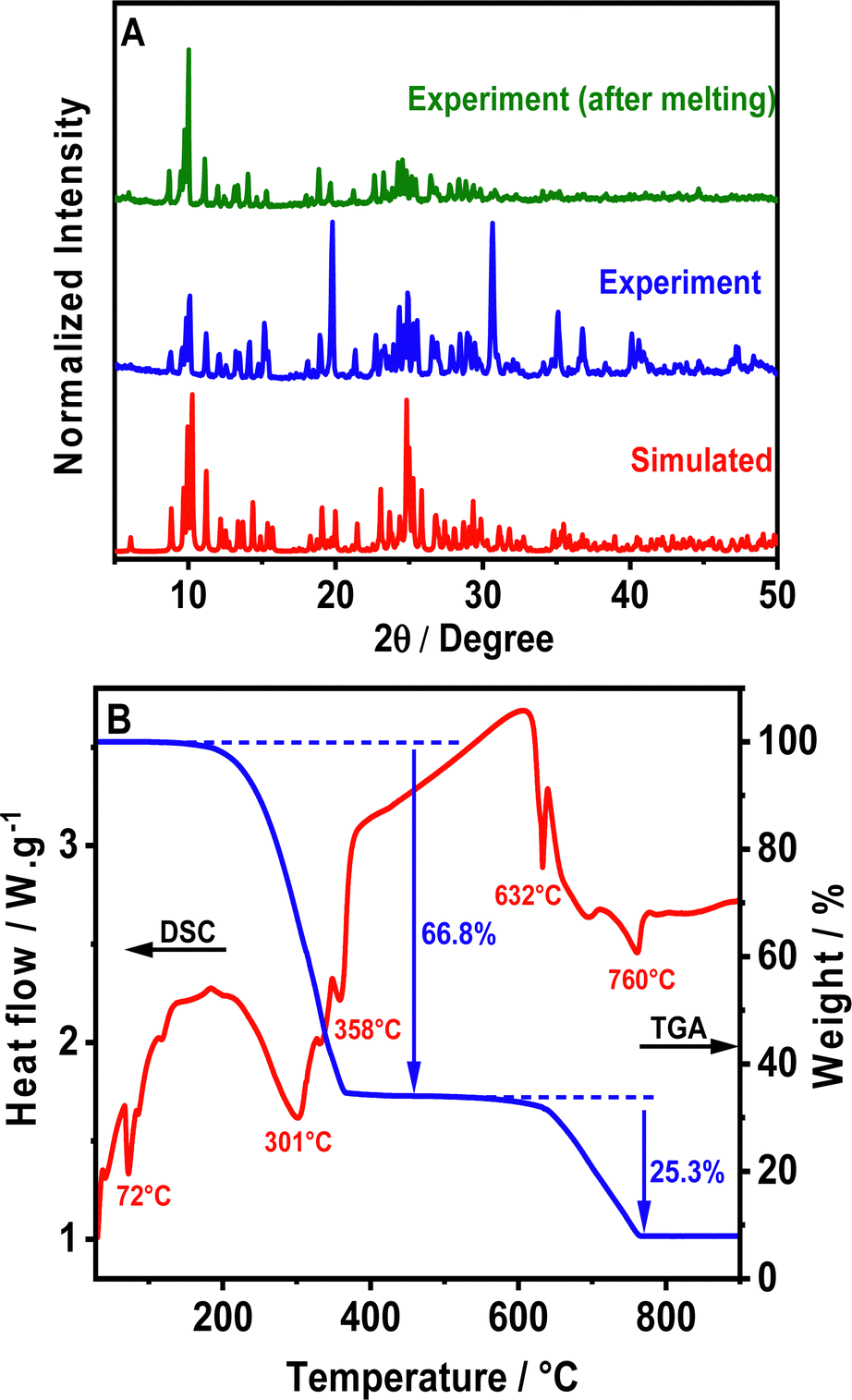 | ||
| Fig. 1 (A) PXRD patterns of the powder sample of (TPA)2MnBr4, before and after melting, as well as the simulated PXRD pattern from SCXRD. (B) TGA-DSC curves of (TPA)2MnBr4. | ||
Hirshfeld surface analysis
To gain more insights into the nature and robustness of interactions present in the crystalline arrangements of (TPA)2MnBr4, Fig. S3A (ESI†) shows the Hirshfeld surfaces (HS) mapped using dnorm. Clearly, the H–H intermolecular interactions provide the dominant contribution (69%) in the total surface of the molecular structure as it appears by the scattered points in the middle area of the fingerprint plot (Fig. S3B, ESI†). Furthermore, the H–Br/Br–H interactions, which appear as two pair sharp spikes in the fingerprint plot, have the second most dominant interaction contribution (30.7%). Due to the opposite partial electronic charge of the hydrogen and the bromine atoms, this kind of interaction is electrostatically favourable providing more robustness to the crystal. Notice that there are two different N–H⋯Br and C–H⋯Br H-bonds in the crystal structure (Fig. S3C and Table S3, ESI†) and the presence of these H-bonds exerts a large influence on the robustness of the packing in the crystalline arrangements. Finally, the Mn⋯Br intermolecular interaction (not within the same tetrahedron) is very weak, with only 0.3% contribution (Fig. S3D, ESI†).TGA-DSC analysis of the (TPA)2MnBr4 as-synthesized powder
Furthermore, we explored the thermal stability of (TPA)2MnBr4 through simultaneous thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements from room temperature to 900 °C (Fig. 1B). The results show that the compound remains stable up to 180 °C and above this temperature it starts to decompose in two main weightless steps. In addition to that, a sharp endothermic peak in the DSC signal is observed at 72 °C, reflecting the melting of (TPA)2MnBr4 powder, but without any signature of decomposition since the material is still green emissive upon cooling down to room temperature (Fig. S1, ESI†). Notice also that the PXRD at room temperature of a melted sample (heated to 75 °C) is very similar to the not melted one (Fig. 1A). This indicates that even after melting, the (TPA)2MnBr4 perovskite retains its initial crystalline structure and maintains its green emission. This result is similar to the one reported for (Bu4N)2[MnBr4] (Bu4N: tetrabutylammonium) in which the melting of the perovskite occurs at 60 °C.50On the other hand, the first weight loss occurring between 220 and 400 °C, is attributed to the decomposition of two (TPA)-Br moieties. The observed weight loss in this step is 66.8%, very close to the theoretical mass expected for this ligand in the material (67.3%). This decomposition process is accompanied by two endothermic peaks on the DSC curve (red curve), with the maximum being at 301 °C and 358 °C, respectively. The thermal decomposition mechanism could take place according to the following eqn (1):
| [(C3H7)3N]2MnBr4 → 2[(C3H7)3NBr] + MnBr2 | (1) |
To further investigate the possibility of an order–disorder-type structural transition, DSC measurement of (TPA)2MnBr4 from 200 to 300 K (Fig. S4, ESI†) was carried out. However, surprisingly, no thermal signals were observed in the DSC curves during two cycles of heating and cooling, which confirms the absence of any phase transition or thermal anomalies in the prepared Mn perovskite.
EPR spectroscopy characterization
EPR spectroscopy is a useful tool to get important information about the local coordination of high-spin Mn2+ ions in all types of material.53Fig. 2A shows the X-band EPR spectra of powder samples of (TPA)2MnBr4 measured at different temperatures from 100 to 5 K, while Fig. S5 (ESI†) exhibits the X-band EPR spectra carried out at room temperature. The observed spectra are very complexes, with several broad lines from zero field to 0.7 T, indicating that the zero-field splitting (ZFS) in this compound is larger than the microwave quantum energy, hv, at this frequency.54 Furthermore, the spectra do not change significantly when the temperature is lowered to 5 K, which means that the ground state does not change and that the magnetic interactions between the Mn2+ ions are negligible. Notably, we did not observe the manganese nuclear hyperfine splitting. We explain this behavior by the presence of dipolar interactions in solid samples that lead to a broadening of the lines and as a result the hyperfine structure is often missing.55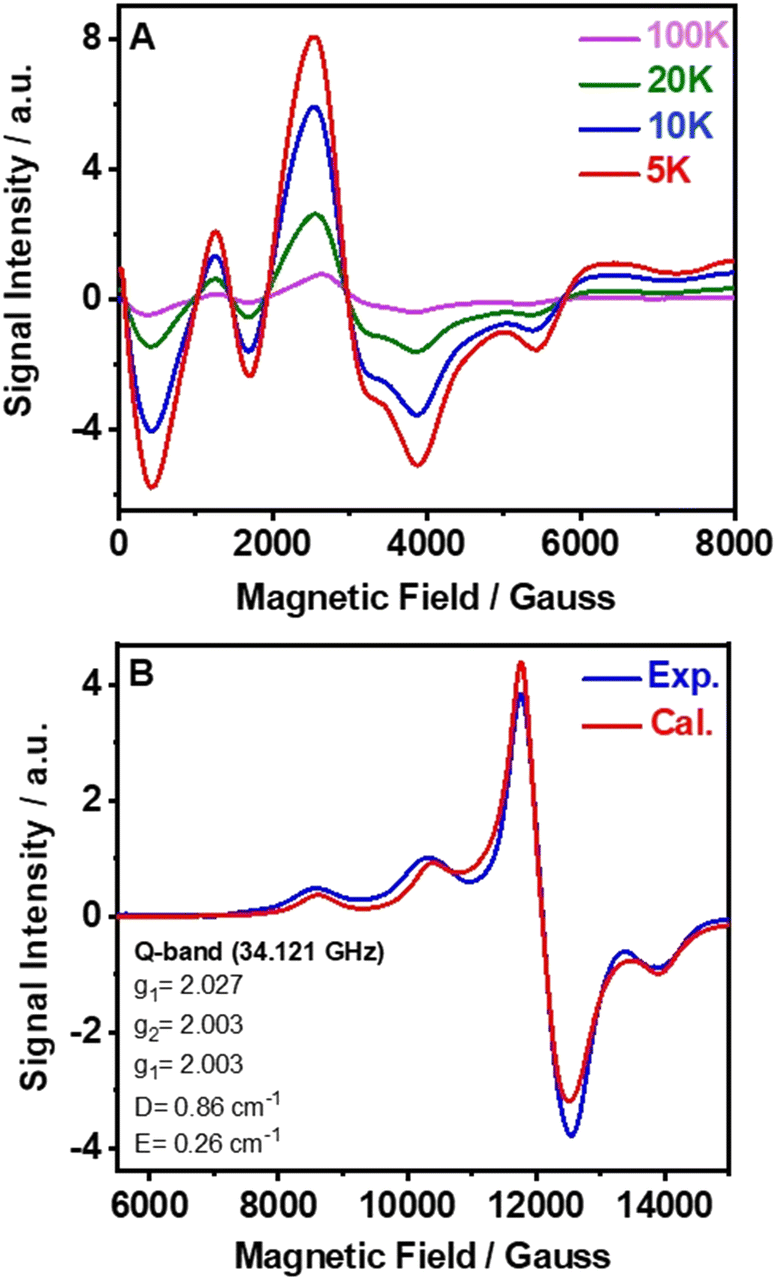 | ||
| Fig. 2 (A) Variation of the X-band electron-spin paramagnetic resonance (EPR) spectra of (TPA)2MnBr4 with temperature from 5 to 100 K. (B) Experimental and calculated Q-band room temperature spectra (see the text for details of the fit). | ||
The Q-band EPR spectrum recorded at room temperature has a much simpler appearance and a large signal intensity at g = 2 (Fig. 2B), indicating that the ZFS is smaller than the Zeeman energy at this frequency (34 GHz, 1.2 cm−1). This spectrum could be reasonably well fitted using the following spin-Hamiltonian (eqn (2)):
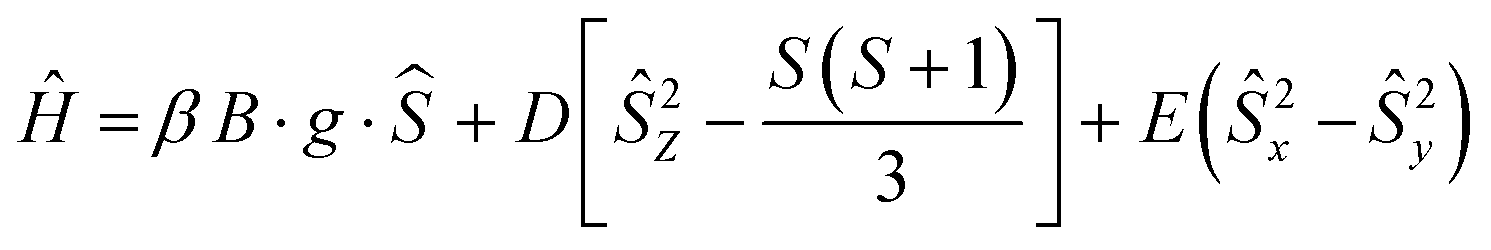 | (2) |
Photophysical characterization
To characterize the photophysical properties of (TPA)2MnBr4, we recorded the UV-vis absorption, excitation, and emission spectra (Fig. 3). As displayed in Fig. 3A, the diffuse reflectance (converted to Kubelka–Munk (KM) function) and the excitation spectra exhibit bands with multiple vibrational transitions in the UV and visible regions (G terms and D terms). In the UV region, the two most intense peaks are located at around 360 nm and 374 nm and correspond to the electronic transition from the 6A1 (6S) ground state of Mn2+ to 4E(D) and 4T2(D). On the other hand, the strong absorption bands at around 437 nm, 450 nm, and 467 nm (visible region) are attributed to the transition of Mn2+ from the 6A1 (6S) ground state to [4A1(G), 4E(G)], 4T2(G), and 4T1(G) levels, respectively. Besides, the positions of these peaks are consistent with the energy state splitting for tetrahedrally coordinated Mn2+.49,57–59 The excitation spectrum of (TPA)2MnBr4 (monitored at 520 nm) matches well with the diffuse reflectance one indicating a common origin of the ground state transitions.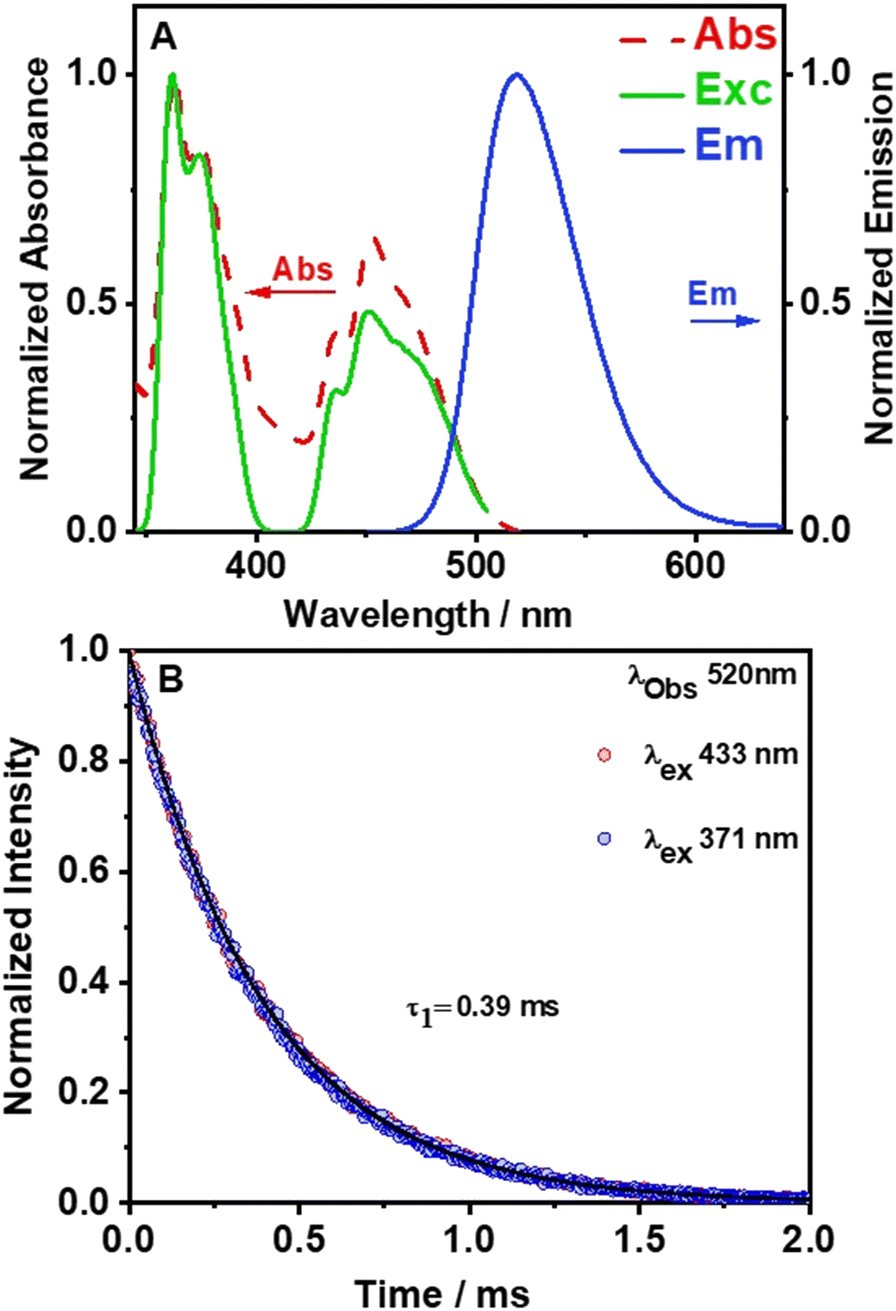 | ||
| Fig. 3 (A) Normalized to the maximum intensity of reflectance (converted to the K–M function), excitation (observation at 520 nm), and emission (excitation at 365 nm) spectra of the (TPA)2MnBr4 as-synthesized powder. (B) Emission decays of the (TPA)2MnBr4 as-synthesized powder following excitation at 371 nm (blue circles) and 433 nm (red circles). The solid lines represent the best monoexponential fit. | ||
The emission spectrum of (TPA)2MnBr4 (Fig. 3A) was recorded upon excitation at the most intense absorption wavelength (365 nm). It consists of a single and relative narrow band (FWHM = 54 nm, 2127 cm−1) with its maximum located at 520 nm (green color) and with a high PLQY of 62%. The strong green emission of this kind of Mn-perovskite is well known to be from the d–d (4T1 → 6A1) transition of Mn2+ ions with a tetrahedral coordination geometry in [MnBr4]2− units. This is in agreement with the results from the SCXRD experiment where the long distance between two adjacent Mn2+ (approximately 9.54(5) (shortest) and 10.01(5) (longest) Å) reflects the absence of or weak Mn–Mn coupling in the [MnBr4]2− tetrahedral units.48,60,61 The tetrahedral units are separated enough by tri-n-propylammonium in the crystal structure leading to a Jahn–Teller effect, which induces the emission corresponding with the d–d transition of the Mn ions. Furthermore, the emission spectrum does not shift with the excitation wavelength (Fig. S6, ESI†), demonstrating that the emission of the perovskite material has its origin in the d–d transition of the Mn ions. Previous studies have shown that the Mn–Mn distance is a highly dominant factor for the PLQY in this kind of organic–inorganic compounds.31,45,48,62 For example, in a systematic study of crystal structures and optical properties of several AmMnBr4-type perovskites (where m = 1 or 2, A = dimethylammonium, 3-methylpiperidinium, 3-aminomethylpiperidinium, heptamethylenimine, and trimethylphenylammonium) a direct correlation between the Mn–Mn distance and the PLQY was reported. The study demonstrated that higher PLQYs are associated with a longer Mn–Mn distance. The increase in the PLQY of these Mn-based compounds with the increase in the Mn–Mn distance was explained in terms of the reduction of the energy transfer processes (induced by coupling interactions such as dipole–dipole and spin–exchange interactions) between neighbouring Mn2+ ions.43 Fig. S7A (ESI†), based on Table S4 (ESI†) shows that the PLQY of Mn(II)-based perovskites increases with the Mn–Mn distance as a result of inhibition of the Mn–Mn quenching by coupling effects, and thus, the emission is dominated by the d–d transition of each individual Mn ion. It was shown that a small enhancement of PLQY could occur also by other factors such as the type of the organic linker, the crystallinity of the synthesized single crystals and their symmetry and the degree of distortion in [MnBr4]2−.48 The measured PLQY of 62%, which deviates from the ideal one (100%), suggests the presence of non-radiative processes that occur at shorter time scales.
As we previously discussed, the single crystal structure clearly shows the presence of H-bonds of CH⋯Br (2.91 and 3.12 Å) and NH⋯Br (2.36 and 2.39 Å). Thus, in addition to the weak Mn–Mn interaction and phonon (vibrations) channels for the non-radiative deactivation, upon electronic excitation to 4E(D), 4T2(D), [4A1(G), 4E(G)], 4T2(G), and 4T1(G) levels, the H-bond fluctuation in these states or in the emitting one, 4T1(G), should be an additional source for radiationless transitions which has been reported for many H-bonded systems.63–65 Notice also that the presence of bromide atoms in the Mn-tetrahedral configuration could also induce radiationless transitions due to the heavy-atom effect in the emission transition.66 Thus, we suggest that the weak Mn–Mn interaction, the H-bonds, the phonons, and the presence of bromide atoms around the Mn-emitter centre, all contribute to the decrease in the PLQY from 100% to 62%. Nevertheless, this is still a high value of PLQY, which encourages the use of this perovskite in lighting application as we will show in this contribution. Notice also that our experiments have been carried out at an ambient humidity of about 32%. Considering that the emission intensity in a down converter LED that was heated to 60 °C for 6 h increased by 13% (vide infra), we expect that the PLQY in a dry atmosphere should increase to at least 70–75%. Following Fig. S7 (ESI†) based on the published PLQY values of more than 30 tetrahedral Mn- and Br-based perovskites vs. Mn–Mn distance, the lowest expected value of the PLQY of our perovskite is about 50% while the highest one is about 90%.
To provide more insights into the photophysical properties of the hybrid Mn-perovskite (TPA)2MnBr4, the emission decay monitored at 520 nm was measured under two excitation wavelengths, 371 nm and 433 nm (Fig. 3B). The emission decay shows no dependency with either the observation wavelength nor the excitation one (Fig. S8, ESI†). The emission decays were accurately fitted using a single-exponential function with a time constant of 0.39 ms, indicating a single exciton recombination pathway. The long-lived emission lifetime value of this perovskite is due to the longer Mn–Mn distance (9.54 Å), which results in a lower energy transfer efficiency between the Mn2+ emitting centres, suggesting that the d–d orbital transition dominated the emission lifetime. Table S4 (ESI†) shows that previously reported organic–inorganic manganese-based perovskites where the Mn emissive centre is in a tetrahedral configuration, exhibit an average lifetime in the microsecond time scale.
Using the value of the emission lifetime and PLQY, the calculated non-radiative rate constant (knr) is 0.97 s−1. From the published PLQY and emission lifetime of tens of Mn(IV)- and Br-based perovskites (Table S4, ESI†), we calculated and plotted the change of knrvs. Mn–Mn shortest distance in the related single crystals (Fig. S7B and C, ESI†). The plot shows a significant dispersion of the data when considering all the data in Table S4 (ESI†). However, when the plot considers only the data for distances longer than 8.5 Å (Fig. S7D, ESI†), a clear trend is observed: a decrease in the knr value when the Mn–Mn distance increases. Notably, the Mn–Mn distance should be affected by the H-bond distances and forces of the CH⋯Br and NH⋯Br interactions (vide supra). Furthermore, an increase in the Mn–Mn distance will reduce the Mn⋯Mn coupling such as dipole–dipole and spin–exchange interactions between neighbouring Mn2+ centres. The value of knr obtained for (TPA)2MnBr4 correlates well with the reported data for other Mn- and Br-based perovskites with similar Mn–Mn distances.
To further evaluate the properties of (TPA)2MnBr4, we also studied the fluorescence lifetime images (FLIM), stability, anisotropy distribution, and spectra of several individual crystals (Fig. 4). The FLIM shows a uniform distribution of the emitting species with no apparent bulk or edge defect contributions (inset of Fig. 4A and Fig. S9, ESI†). We also tested the stability of the emission intensity over a longer irradiation time. The recorded emission trace maintains the same intensity for 5 min under constant laser irradiation. Notably, we did not observe blinking behavior, which suggests that under these experimental conditions, there is no significant contribution from trap states or structural defects (Fig. 4A). Fluorescence blinking in perovskite crystals is often associated with trap states.67 Therefore, the lack of blinking in the recorded emission traces indicates that no trap states are present in the studied crystals in agreement with the monoexponential fluorescence lifetime behavior described above. The high stability is further confirmed by the emission spectra collected before and after 10 min of constant laser irradiation (Fig. 4B). Furthermore, the emission spectra collected in the bulk of the crystal and on its edges (Fig. S10, ESI†) show similar positions (∼520 nm) and shapes with only small deviations in the intensity depending on the interrogated position. These spectra are also comparable to the steady-state emission one recorded for the powder samples, further supporting the homogeneous nature of this perovskite. Finally, we measured the anisotropy distribution of individual crystals. The obtained histograms are narrow and centred on values between −0.1 and 0 (inset of Fig. 4B and Fig. S11, ESI†). The narrow distribution agrees with the homogeneous nature of the crystals, while the value of the anisotropy suggests no preferential orientation of the emissive centres.
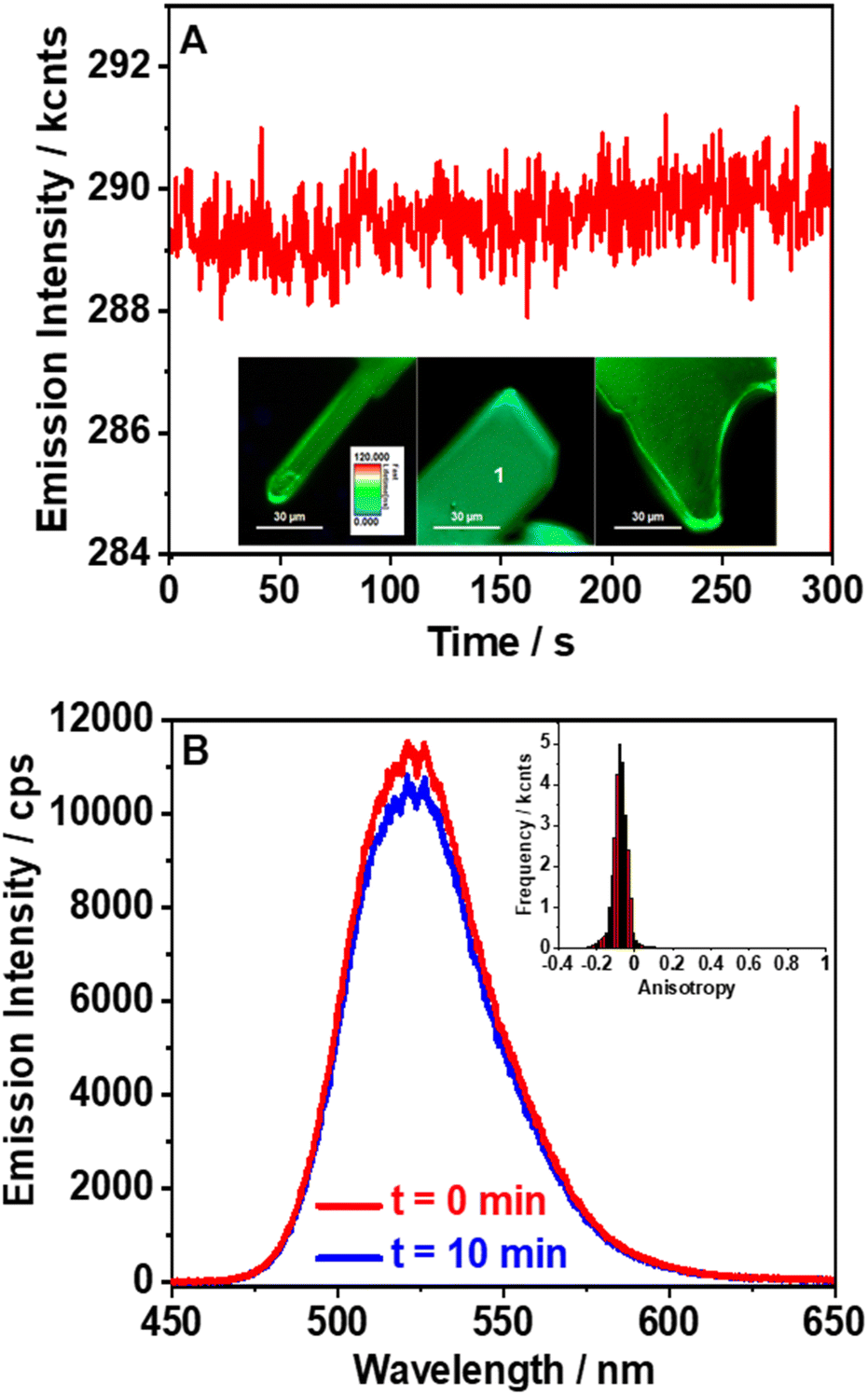 | ||
| Fig. 4 (A) Long-term emission intensity stability collected at point 1 for 5 min constant laser irradiation (inset: fluorescence lifetime imaging microscopy (FLIM) images of a single (TPA)2MnBr4 crystal). (B) Emission spectra collected at point 1 before and after 10 min continuous laser irradiation (inset: anisotropy distribution histogram for the isolated crystal in Panel A). The excitation wavelength was 390 nm. | ||
(TPA)2MnBr4 based down-converter LEDs
Next, based on the observed luminescence properties we employed (TPA)2MnBr4 for the construction of a series of down-converted LEDs. To this end, different amounts of (TPA)2MnBr4 were deposited on the surface of a blue (465 nm) LED chip (3.5 × 2.8 mm). Then, the 465 nm-LEDs were turned on to a voltage of 2.7 V (driving current of 18 mA), yielding different emission colours in line with of the amount of (TPA)2MnBr4 used in the preparation of the devices (Fig. 5A). The emission spectra of the LEDs show a combination of two narrow bands with intensity maxima at 465 and 520 nm, corresponding to the emission of the blue LED chip and the Mn-perovskite, respectively (Fig. 5B). The increment in the amount of perovskite (from 4 to 10 mg) produce a progressive increase in the emission band intensity with a the maximum at 520 nm, enabling the control of the final colour by adjusting the amount of perovskite deposited onto the LED surface. In this sense, the final colour of the LEDs was modulated from light blue, with a CIE coordinate of (0.14, 0.16), to a greenish colour (0.17, 0.38), as depicted in Fig. 5C. Note that it is important to reach a compromise between the amount of material used to tune the emission color of the down-converted LED while maintaining its luminance. For this reason, we have not deposited larger amounts than 10 mg, since the emission of the LED was partially absorbed/blocked by the material, diminishing the emission of the device.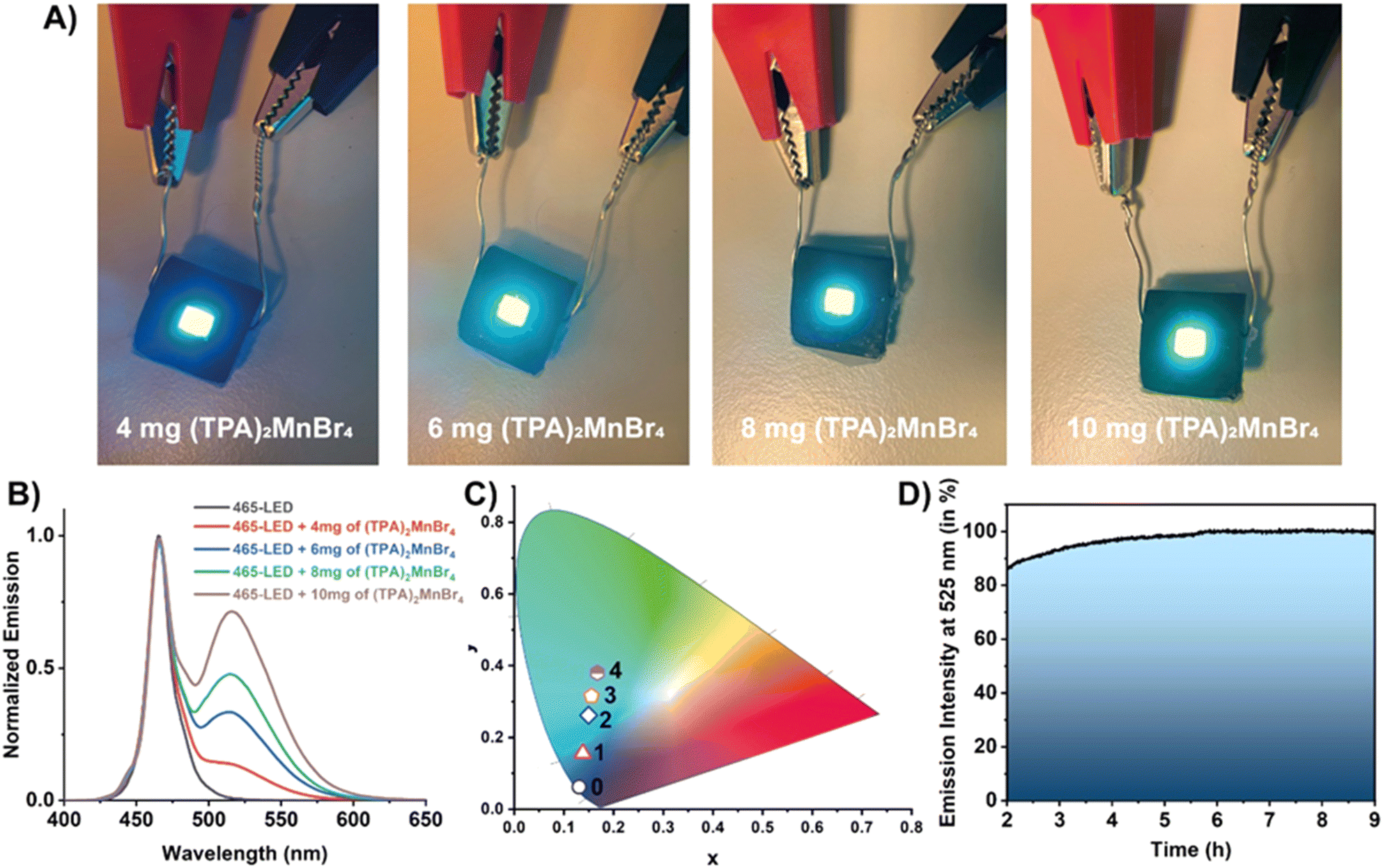 | ||
| Fig. 5 (A) Real photos of the down-converted LEDs fabricated by depositing a certain amount of (TPA)2MnBr4 onto a blue 465 nm-LED chip. (B) Emission spectra of the LEDs under a driving current of 18 mA (2.7 V). (C) CIE chromaticity coordinates of the LEDs fabricated by coating a 465 nm blue LED with different amounts of (TPA)2MnBr4 perovskite: 0 = blue 465 nm LED; 1 = 4 mg; 2 = 6 mg; 3 = 8 mg and 4 = 10 mg of (TPA)2MnBr4. (D) Representation of the emission intensity collected at 520 nm (corresponding to the emission of (TPA)2MnBr4) of the LED prepared with 10 mg of (TPA)2MnBr4 under working operation conditions for a total of 9 hours. | ||
The photostability of the material was also tested during 9 hours of continuous operation (Fig. 5D). We realised that the material requires an activation period (irradiation by the blue LED) of 2 h before stabilization. During this time, the material does not suffer any photodegradation, but some fluctuations in the emission intensity were observed. In particular, it shows a small fluctuation followed by an increase in the emission intensity. It is worth noting that the surface of this LED operating at 2.7 mV reaches a temperature of ∼40 °C. In general, higher temperatures usually produce a quenching of the emission intensity due to an increase in the molecular vibrations promoting no radiave transitions. However, we observed the opposite effect when the material is deposited onto the LED surface. Since the presence of water molecules might partially reduce the crystallinity of (TPA)2MnBr4, it is possible that the presence of ambient moisture could diminish the emission intensity of this material. However, (TPA)2MnBr4 shows low sensitivity to moisture over time as evidenced by the PLQY measured after 45 days of storage under ambient conditions, which remained almost unchanged (61% vs. 62%). This also supports that water molecules, under ambient conditions, interacting with this perovskite do not affect significantly its photobehavior. In this sense, we propose that while the optoelectronic properties of the material are not significantly affected, heat released by the LED favours the detachment of moisture adsorbed on the surface of the Mn-perovskite powder, therefore, enhancing its emission until reaching a thermal equilibrium. Once the LED is warmed up and the equilibrium in the perovskite is established, the emission of (TPA)2MnBr4 remains almost constant (with a slight increase in the emission over the first 6 hours) as shown in Fig. 5D. This observation is of utmost importance because of two reasons: (i) the Mn-based perovskite material is highly photostable under these experimental conditions, in agreement with the results from FLIM; and (ii) for the fabrication of a possible commercial LED device, it would be critical to consider an appropriate balance between the water molecules and the heat release by the LED. To this end, it would be paramount to firstly activate the material (i.e., heating for removing the surface-attached water molecules) followed by coating of the LED under controlled experimental conditions and appropriate encapsulation of the down-converted LED device to avoid possible deterioration and therefore, to increase its useful lifetime. Hence, these results indicate the promising properties of (TPA)2MnBr4 perovskite for use as an active phosphor layer for the fabrication of colour tunable LED devices.
Conclusion
In summary, we synthesized a new organic–inorganic 0D-manganese-based perovskite (TPA)2MnBr4 using a slow evaporation method, with a triclinic phase and P space group. The material exhibits high-temperature sustainability up to 180 °C, and the PXRD pattern after cooling down to room temperature of the melted sample at 72 °C is similar to the not melted one, indicating that the initial crystalline structure remains. (TPA)2MnBr4 displays an intense green emission centred at 520 nm with a high PLQY (62%), due to the effective d–d transitions of tetrahedrally coordinated Mn2+. It also exhibits a lifetime of 0.39 ms. The FLIM images for (TPA)2MnBr4 suggest a uniform distribution of the emitting species with no apparent bulk or edge defect contribution. Moreover, green-light LED devices were prepared based on (TPA)2MnBr4 crystals. We belive that our results will contribute to the advancement on novel low-cost, high-performance, and eco-friendly organic–inorganic manganese-based perovskites as green-light-emitting devices in the future.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by the following grants: grant PID2020-116519RB-I00 and TED2021-131650B-I00 funded by MCIN/AEI/10.13039/501100011033 and the European Union (EU); grant SBPLY/19/180501/000212 and SBPLY/21/180501/000108 funded by JCCM and the EU through “Fondo Europeo de Desarollo Regional” (FEDER); grant 2020-GRIN-28929 and 2022-GRIN-34325 funded by UCLM (FEDER). For her stay at the UCLM, A.B.A. thanks the grant from the Spanish Service for the Internationalization of Education (SEPIE), through the EU Erasmus+ key action program (2020-1-ES01-KA107-079868). M.G. thanks the EU for financial support through Fondo Social Europeo Plus (FSE+).References
- J. Sun, J. Wu, X. Tong, F. Lin, Y. Wang and Z. Wang, Adv. Sci., 2018, 5, 1700780 CrossRef.
- Z. Wangen and D. Yang, Small, 2017, 13, 1604153 CrossRef.
- Q. Chen, N. De Marco, Y. Yang, T.-B. Song, C.-C. Chen, H. Zhao, Z. Hong, H. Zhou and Y. Yang, Nano Today, 2015, 10, 355–396 CrossRef CAS.
- P. Xiao, Y. Yu, J. Cheng, Y. Chen, S. Yuan, J. Chen, J. Yuan and B. Liu, Nanomaterials, 2021, 11, 103 CrossRef CAS PubMed.
- Z. Ma, Z. Shi, D. Yang, F. Zhang, S. Li, L. Wang, D. Wu, Y. Zhang, G. Na, L. Zhang, X. Li, Y. Zhang and C. Shan, ACS Energy Lett., 2020, 5, 385–394 CrossRef CAS.
- T. He, L. Saisai, Y. Jiang, C. Qin, M. Cui, L. Qiao, H. Xu, J. Yang, R. Long, H. Wang and M. Yuan, Nat. Commun., 2020, 11, 1672 CrossRef CAS PubMed.
- H. Peng, S. Yao, Y. Guo, R. Zhi, X. Wang, F. Ge, Y. Tian, J. Wang and B. Zou, J. Phys. Chem. Lett., 2020, 11, 4703–4710 CrossRef CAS PubMed.
- K. X. Steirer, P. Schulz, G. Teeter, V. Stevanovic, M. Yang, K. Zhu and J. J. Berry, ACS Energy Lett., 2016, 1, 360–366 CrossRef CAS.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- W. Ning and F. Gao, Adv. Mater., 2019, 31, 1900326 CrossRef.
- Q. Fan, G. V. Biesold-McGee, J. Ma, Q. Xu, S. Pan, J. Peng and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 1030–1046 CrossRef CAS.
- I. Lahbib, R. Mohamed and W. Sta, J. Mol. Struct., 2016, 1120, 250–258 CrossRef CAS.
- J. Huang, T. Chang, R. Zeng, J. Yan, Q. Wei, W. Zhou, S. Cao and B. Zou, Adv. Opt. Mater., 2021, 9, 2002267 CrossRef CAS.
- T. Huang, H. Penga, Q. Wei, C. Peng, Y. Tian, S. Yao, X. Han and B. Zou, Nano Energy, 2021, 93, 106863 CrossRef.
- X. Bai, H. Zhong, B. Chen, C. Chen, J. Han, R. Zeng and B. Zou, J. Phys. Chem. C, 2018, 122, 3130–3137 CrossRef CAS.
- L. Septiany, D. Tulip, M. Chislov, J. Baas and G. R. Blake, Inorg. Chem., 2021, 60, 15151–15158 CrossRef CAS PubMed.
- S.-H. Park, I.-H. Oh, S. Park, Y. Park, J. H. Kim and Y.-D. Huh, Dalton Trans., 2012, 41, 1237–1242 RSC.
- Y. Sozen, S. Özen and H. Sahin, J. Magn. Magn. Mater., 2021, 531, 167845 CrossRef CAS.
- Z. Nie, J. Yin, H. Zhou, N. Chai, B. Chen, Y. Zhang, K. Qu, G. Shen, H. Ma, Y. Li, J. Zhao and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 28187–28193 CrossRef CAS.
- S. Balsamy, P. Natarajan, R. Vedalakshmi and S. Muralidharan, Inorg. Chem., 2014, 53, 6054–6059 CrossRef CAS.
- H.-Y. Ye, Q. Zhou, X. Niu, W.-Q. Liao, D.-W. Fu, Y. Zhang, Y.-M. You, J. Wang, Z.-N. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2015, 137, 13148–13154 CrossRef CAS.
- D. Liang, H. Xiao, W. Cai, S. Lu, S. Zhao, Z. Zang and L. Xie, Adv. Opt. Mater., 2023, 11, 2202997 CrossRef CAS.
- L.-J. Xu, C.-Z. Sun, H. Xiao, Y. Wu and Z.-N. Chen, Adv. Mater., 2017, 29, 1605739 CrossRef PubMed.
- Y. Zhang, W.-Q. Liao, D.-W. Fu, H.-Y. Ye, Z.-N. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2015, 137, 4928–4931 CrossRef CAS PubMed.
- T. Zhuang, Y.-M. Lin, H.-W. Lin, Y.-L. Guo, Z.-W. Li, K. Du, Z. Wang and X.-Y. Huang, Molecules, 2023, 28, 2380 CrossRef CAS PubMed.
- M. Li, J. Zhou, M. S. Molokeev, X. Jiang, Z. Lin, J. Zhao and Z. Xia, Inorg.Chem., 2019, 58, 13464–13470 CrossRef CAS PubMed.
- B. Su, G. Zhou, J. Huang, E. Song, A. Nag and Z. Xia, Laser Photonics Rev., 2021, 15, 2000334 CrossRef CAS.
- V. Morad, I. Cherniukh, L. Pöttschacher, Y. Shynkarenko, S. Yakunin and M. V. Kovalenko, Chem. Mater., 2019, 31, 10161–10169 CrossRef CAS.
- Y.-L. Wei, J. Jing, C. Shi, H.-Y. Ye, Z.-X. Wang and Y. Zhang, Inorg. Chem. Front., 2018, 5, 2615–2619 RSC.
- H.-M. Pan, Q.-L. Yang, X.-X. Xing, J.-P. Li, F.-L. Meng, X. Zhang, P.-C. Xiao, C.-Y. Yue and X.-W. Lei, Chem. Commun., 2021, 57, 6907–6910 RSC.
- Y. Zhang, W. Q. Liao, D. W. Fu, H. Y. Ye, C. M. Liu, Z. N. Chen and R. G. Xiong, Adv. Mater., 2015, 27, 3942–3946 CrossRef CAS.
- S. Wang, X. Han, T. Kou, Y. Zhou, Y. Liang, Z. Wu, J. Huang, T. Chang, C. Peng, Q. Wei and B. Zou, J. Mater. Chem. C, 2021, 9, 4895–4902 RSC.
- X.-F. Sun, P.-F. Li, W.-Q. Liao, Z. Wang, J. Gao, H.-Y. Ye and Y. Zhang, Inorg. Chem., 2017, 56, 12193–12198 CrossRef CAS.
- Y. Duan, Y. Ju, P. Xia, J. Qu, H. Shao, C. Wang, S. Xu and Y. Cui, Opt. Mater., 2022, 134, 113164 CrossRef CAS.
- K. Li, Y. Ye, W. Zhang, Y. Zhou, Y. Zhang, S. Lin, H. Lin, J. Ruan and C. Liu, Nano Res., 2022, 15, 9368–9376 CrossRef CAS.
- W. Gao, M. Leng, Z. Hu, J. Li, D. Li, H. Liu, L. Gao, G. Niu and J. Tang, Dalton Trans., 2020, 49, 5662–5668 RSC.
- G. Zhou, Q. Ren, M. S. Molokeev, Y. Zhou, J. Zhang and X.-M. Zhang, ACS Appl. Electron. Mater., 2021, 3, 4144–4150 CrossRef CAS.
- B. Li, Y. Xu, X. Zhang, K. Han, J. Jin and Z. Xia, Adv. Opt. Mater., 2022, 10, 2102793 CrossRef CAS.
- W. Shao, X. Wang, Z. Zhang, J. Huang, Z. Han, S. Pi, Q. Xu, X. Zhang, X. Xia and H. Liang, Adv. Opt. Mater., 2022, 10, 2102282 CrossRef CAS.
- S. Yan, W. Tian, H. Chen, K. Tang, T. Lin, G. Zhong, L. Qiu, X. Pan and W. Wang, Adv. Funct. Mater., 2021, 31, 2100855 CrossRef CAS.
- S. Chen, J. Gao, J. Chang, Y. Zhang and L. Feng, Sens. Actuators, B, 2019, 297, 126701 CrossRef CAS.
- L. Mao, P. Guo, S. Wang, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2020, 142, 13582–13589 CrossRef CAS.
- M.-H. Jung, Dalton Trans., 2023, 52, 3855–3868 RSC.
- C. Jiang, N. Zhong, C. Luo, H. Lin, Y. Zhang, H. Peng and C.-G. Duan, Chem. Commun., 2017, 53, 5954–5957 RSC.
- N. O. Giltzau and M. Köckerling, IUCrdata, 2020, 5, x200261 CrossRef CAS PubMed.
- L.-J. Xu, X. Lin, Q. He, M. Worku and B. Ma, Nat. Commun., 2020, 11, 4329 CrossRef CAS PubMed.
- N. N. Golovnev, M. A. Gerasimova, I. A. Ostapenko, A. O. Zolotov and M. S. Molokeev, J. Mol. Struct., 2023, 1277, 134851 CrossRef CAS.
- X.-W. Cai, Y.-Y. Zhao, H. Li, C.-P. Huang and Z. Zhou, J. Mol. Struct., 2018, 1161, 262–266 CrossRef CAS.
- A. Jana, S. Zhumagali, Q. Ba, A. S. Nissimagoudar and K. S. Kim, J. Mater. Chem. A, 2019, 7, 26504–26512 RSC.
- H. Fu, C. Jiang, J. Lao, C. Luo, H. Lin, H. Peng and C.-G. Duan, CrystEngComm, 2020, 22, 1436–1441 RSC.
- E. Styczeń, M. Gazda and D. Wyrzykowski, Thermochim. Acta, 2010, 503–504, 21–27 CrossRef.
- C. Duboc, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- D. M. L. Goodgame, H. E. Mkami, G. M. Smith, J. P. Zhao and E. J. L. McInnes, Dalton Trans., 2003, 34–35, 10.1039/B210407P.
- C. Duboc, M.-N. Collomb and F. Neese, Appl. Magn. Reson., 2010, 37, 229–245 CrossRef.
- J. Krzystek, A. Ozarowski and J. Telser, Coord. Chem. Rev., 2006, 250, 2308–2324 CrossRef CAS.
- Y. Rodríguez-Lazcano, L. Nataf and F. Rodríguez, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085115 CrossRef.
- T. Jiang, W. Ma, H. Zhang, Y. Tian, G. Lin, W. Xiao, X. yu, J. Qiu, X. Xu, Y. Yang and D. Ju, Adv. Funct. Mater., 2021, 31, 2009973 CrossRef CAS.
- W. Mao, J. Wang, X. Hu, B. Zhou, G. Zheng, S. Mo, S. Li, F. Long and Z. Zou, J. Saudi Chem. Soc., 2020, 24, 52–60 CrossRef CAS.
- Y. Cheng, H.-P. Ruan, Y. Peng, L. Li, Z. Xie, L. Liu, S. Zhang, H. Ye and Z.-B. Hu, Chin. Chem. Lett., 2023, 108554 CrossRef.
- H. Zhang, Y.-H. Tan, Y.-Z. Tang, X.-W. Fan, X.-L. Peng, R.-R. Han, Y.-K. Li and F.-X. Wang, Inorg. Chem., 2022, 61, 10454–10460 CrossRef CAS.
- S. Cao, C. Li, P. He, J. A. Lai, K. An, M. Zhou, P. Feng, M. Zhou and X. Tang, ACS Appl. Opt. Mater., 2023, 1, 623–632 CrossRef CAS.
- A. Douhal, F. Lahmani and A. H. Zewail, Chem. Phys., 1996, 207, 477–498 CrossRef CAS.
- T. Elsässer and H. Becker, Ultrafast hydrogen bonding dynamics and proton transfer processes in the condensed phase, SSBM, 2013.
- R. Ludwig, ChemPhysChem, 2007, 8, 2539 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books Sausalito, CA, 2010 Search PubMed.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2301462. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc03821a |
| This journal is © The Royal Society of Chemistry 2024 |