
Emission variation and spontaneous deformation of CsPbBr3 perovskite nanoplatelets at low concentrations†
Hui
Zhang
a,
Feifei
Cai
a,
Bo
Huang
*b,
Huichao
Zhang
 *b and
Shitong
Li
b
*b and
Shitong
Li
b
aSchool of Electronics and Information, Hangzhou Dianzi University, Hangzhou 310018, P. R. China
bInstitute of Carbon Neutrality and New Energy, School of Electronics and Information, Hangzhou Dianzi University, Hangzhou 310018, P. R. China. E-mail: hb@hdu.edu.cn; zhc@hdu.edu.cn
First published on 29th November 2023
Abstract
Due to the unique structural and optical anisotropy, colloidal quasi-two-dimensional nanoplatelets (NPLs) have been proposed as an important material platform for realizing quantum information technology. However, the optical and structural instability of emerging perovskite NPLs after rapid dilution is preventing such studies, along with an unknown intermediate evolution process. Herein, we investigate the photoluminescence evolution of CsPbBr3 NPLs during stepwise dilutions. Upon decreasing the concentration, a red shift of the emission peak can be observed in CsPbBr3 NPLs with different thicknesses and synthesis methods. The dilution ripening mechanism dominates this red-shifted photoluminescence, in which larger NPLs capture monomers when smaller NPLs are dissolved, and then the accumulated monomers increase the lateral size and thickness of large NPLs. Interestingly, the simple aging treatment can help improve the dilution stability of thicker NPLs of the hot-injection method. Moreover, it is confirmed that the dilution ripening effect can be suppressed by introducing PbBr2-saturated solution or exchanging pristine ligands with stronger binding ligands such as lecithin.
Introduction
As a quasi-two-dimensional material, colloidal semiconductor nanoplatelets (NPLs), with intrinsic attributes of structural and optical anisotropy, possess the potential for application in quantum information technology, low-threshold lasers, LEDs, and other fields.1–5 To evaluate whether an emerging colloidal NPL can be used in quantum information processing applications, a common practice is to measure the quantum optical properties of single NPLs.3,5 Therefore, the original solution of NPLs usually needs to be diluted by three (or four, or more) orders of magnitude to ensure that the space between adjacent NPLs in the spin-casted film is sufficient to avoid the influence of each other.6,7 If the NPLs change dramatically after dilution, it is impossible to determine whether the candidates possess appropriate quantum optical properties. In other words, the optical stability of the NPLs during dilution is crucial for subsequent quantum-light studies, as the detachment of surface ions and ligands or structural transformation can reconstruct the optical properties of samples as well as itself.8Recently, perovskite NPLs, especially CsPbBr3 NPLs, have attracted considerable research interest due to their strong quantum confinement effects and bright narrowband emission with the benefit of facile synthesis and solution-phase processing.9–12 Some synthesis strategies, such as hot injection and ligand-assisted reprecipitation methods, have been developed to obtain high-quality CsPbBr3 NPLs,9,11–13 while some applications such as LEDs and photocatalysis have also been extensively studied.14,15 Unlike conventional CdSe NPLs, only a few studies have investigated the optical properties of single perovskite NPLs likely due to their optical and structural instability in diluted solutions.8,16 Some studies have shown that the intrinsic ionic crystal nature and high concentration of vacancies in perovskite materials lead to instability of their crystal structure.17,18 Perovskite NPLs, with a thickness of only several atomic layers, are more likely to suffer from some structural modifications and optical property degeneration.19 Wang et al. found that under 1000-fold dilution, the emission of the sample shifted from blue to green, suggesting that pristine NPLs undergo a rapid aggregation process.20 These changes are different from perovskite materials of alternative shapes, such as nanocubes and nanorods.21–23 In another perovskite NPL study, the blue shift of the photoluminescence (PL) with dilution was interpreted as self-absorption suppression.24 Such mixed results mean that understanding the dilution evolution of perovskite NPLs remains a challenge. The large interval dilution method used in existing reports conceals much information on the optical properties and structural evolution of such intermediates of NPLs.20,24 Furthermore, dilution instability is also considered to offer potential pathways to creating innovative perovskite materials,21 but it prevents the use of perovskite NPLs in quantum information technology, becoming a key issue to be overcome.
Here, the optical evolution of CsPbBr3 NPLs with the dilution process is studied by a successive dilution method. The CsPbBr3 NPLs obtained by the hot injection method show significant red-shifted PL after dilution. A similar phenomenon can be observed in a sample synthesized by the ligand-assisted reprecipitation approach. We propose that the above red shift in the PL comes from a room-temperature ripening process of perovskite NPLs, in which the larger NPLs capture monomers from the smaller NPLs, and the monomers accumulate to increase the NPLs in lateral dimensions and thickness. Long-term placement of the 5 monolayer (ML) NPLs prepared by the hot injection method possibly benefits the improvement of NPLs’ surface structure, resisting changes caused by dilution. In addition, some methods to suppress the dilution ripening phenomena are investigated, such as the addition of a PbBr2-saturated solution and the ligand exchange with a strong binding one.
Results and discussion
It has been reported in the literature that the optical properties and morphology of perovskite NPLs may change due to environmental moisture, UV irradiation, and temperature.19,25–28 To minimize the influence of these factors on the study of NPL dilution, anhydrous hexane is used as the dilution solvent, and the NPL solution is usually sealed and stored at 4 °C. In addition, the irradiation time of the excitation light is shortened as much as possible during the measurement. Compared with the UV irradiation of dozens of minutes in the literature,25,27 our measurement time is only a few seconds, which is unlikely to have an impact on the NPLs. The purified NPLs dispersed in anhydrous hexane before the dilution experiment are considered as stock solution, and stepwise dilutions are taken with half-dilution of the previous one. In the following text, we use the dilution factor Dn to represent a dilution of n-fold.5 ML CsPbBr3 perovskite NPLs are obtained by using the hot injection method, which is denoted as 5 ML hot injection NPLs (h-NPLs). The details for the synthesis are described in the Experimental section. Fig. S1a (see the ESI†) shows the typical absorption and PL spectra of freshly prepared 5 ML h-NPLs. The first excitonic feature is located at 479 nm, and the narrow emission peak is near 486 nm. A typical PL decay curve of 5 ML h-NPLs is depicted in Fig. S1b (ESI†), which can be fitted by a bi-exponential function with an average lifetime of ∼7.1 ns, in agreement with the literature.13 To observe the emission evolution during the stepwise dilutions, we specially use a very dense stock solution of fresh 5 ML h-NPLs for dilution. The concentration of the stock solution is about 8.2 × 10−6 mmol mL−1 by inductively coupled plasma measurements. For ease of observation, a part of typical PL spectra with different dilution factors are plotted as shown in Fig. 1(a). We also record the evolving relationship between the dilution factor and the PL peak as well as emission full-width-half-maximum (FWHM) of the NPLs (Fig. 1(b)). With the increase in the dilution factor, the emission peak is gradually red shifted from 486 nm (at D0) to ∼500 nm (at D216). Further dilution induces a slight blue shift in PL, approximately 1 nm. Besides, an extra peak located at 463 nm starts to appear in PL at D214 (Fig. 1(a)), and it becomes more distinct after further dilution (at D218). By measuring the spectrum of anhydrous hexane alone, we observe that it has a characteristic PL peak at the same position (Fig. S2, ESI†). So, we consider this narrow peak comes from the emission (or scattering) of some impurity in anhydrous hexane. At a high concentration of NPLs, the luminescence of anhydrous hexane is inconspicuous. However, with the stepwise dilutions of the NPLs, fewer and fewer NPLs participate in the luminescence, and the characteristic peak of anhydrous hexane becomes apparent since we have to gradually increase the integration time. In addition to the influence of anhydrous hexane, more rounds of dilution (at D219, Fig. S3, ESI†) also make it difficult to accurately extract the spectral information of the NPL sample, so only 18 rounds of dilution treatment are performed on 5 ML h-NPLs. Such consideration also exists in the subsequent dilution measurement. The above redshift of the PL peak is observed in all the fresh 5 ML h-NPLs (Fig. S4a and b, ESI†). It should be noted that there are slight differences in the original PL and absorption spectra profile of NPLs due to the different synthesis batches and original concentrations of the samples.29 We also attempt to dilute the NPLs at large multiples (hundred-fold dilution in a single operation), which is often used in optical properties research of single colloidal nanocrystals.30,31 The PL obtained by applying this method shows an obvious redshift from the original 488 nm (D0) to 509 nm (D300) (Fig. S4c, ESI†), which is similar to the phenomenon in stepwise dilutions (Fig. S4b, ESI†). Likewise, the FWHM of the emission peak also changes significantly during the stepwise dilutions (Fig. 1(b)). It shows a slight decrease in the initial stage of dilution. Then, the trend reverses and the FWHM grows all the way towards ∼23 nm. According to the normalized PL spectra in Fig. S5 (ESI†), the PL component between 450 nm and 480 nm gradually reduces with increasing the dilution factor from D0 to D8. This indicates that many small-sized NPLs are dissolved, and then, the NPLs’ size distribution is focused, which leads to a more symmetric PL spectrum.32
 | ||
| Fig. 1 Characterization of fresh 5 ML h-NPLs during the stepwise dilutions. (a) A series of PL spectra upon dilution. (b) Evolution of PL peak wavelength and FWHM with increasing dilution. TEM images of NPLs (c) before (D0) and (d) after dilution experiment. Insets show the photographs of the corresponding samples under an ultraviolet lamp. | ||
To understand more about this PL shifting, we record transmission electron microscopy (TEM) of the fresh 5 ML h-NPLs during the stepwise dilutions. As shown in Fig. 1(c), the original NPLs have a regular morphology, with lateral dimensions of ∼13.1 nm and a thickness of ∼3.2 nm, which correspond to about five MLs.6,9,13 With the progress of dilution, the morphologic changes are observed in the sample. As shown in Fig. S6 (ESI†), some of the NPLs expand laterally, while others become smaller. Considering a very low content of the final sample after dilution that can’t be tested by TEM, we concentrate the diluted sample by volatilizing the solvent at room temperature. The TEM result is shown in Fig. 1(d). The size distribution becomes more inhomogeneous, which is mainly distributed from 5 nm to 20 nm, with a few reaching 50 nm. Additionally, the enhanced contrast indicates that the crystals also become distinctly thicker upon dilution, even somewhat like nanocubes.
To further investigate the underlying physical mechanism of the dilution process, we purposefully prepare the definite dual-emitting CsPbBr3 perovskite NPLs by the hot injection method. As shown in Fig. S7 (ESI†), the PL peaks are located at ∼451 nm and ∼477 nm by fitting the spectrum curve. These two peaks correspond to CsPbBr3 NPLs with 3 and 4 MLs, respectively.9 As the dilution factor increases to D28, both emission peaks are gradually red shifted to 461 nm and 490 nm, respectively (Fig. 2(a) and (b)). Fitting of the PL spectra using a sum of two Gaussian curves reveals an increase in the contribution of 4 ML NPLs and a decrease in a component of 3 ML NPLs upon dilution (Fig. 2(c)). This ultimately results in only the PL peak with longer wavelength being observed. Similar to the results from fresh 5 ML h-NPLs, a weak blue shift in the PL is also found at the end of the dilution (Fig. 2(b)). The FWHM of the two fitted spectra shows a somewhat narrowing trend with dilution (Fig. S8, ESI†), which decreased from 20 nm to 15 nm for 3 MLs NPLs and 28 nm to 26 nm for 4 MLs NPLs.
 | ||
| Fig. 2 PL results of fresh 3 (or 4) ML h-NPLs during the stepwise dilutions. (a) A series of PL spectra upon dilution. Evolution of (b) PL peak wavelength and (c) relative PL intensity with increasing dilution. | ||
Obviously, the above phenomena all show that with the progress of dilution, the PL peaks of NPLs with different thicknesses gradually red-shifted. In previous dilution studies, this red shift was attributed to a rapid NPL aggregation process,20 although the specific evolution information was not disclosed. It is not surprising that these NPLs have the potential to self-assemble into two different hierarchical structures, stacked columnar phases and large 2D sheets.9 A face-to-face merging of NPL stacks will result in a new PL peak or a jumping redshift process in emission (>10 nm), due to the one-dimensionally confined optical properties of NPLs.19,28 Clearly, it is not consistent with our experimental phenomenon, in which a single shift is less than 3 nm. The lateral crystallographic-oriented attachment of single plates is a possible explanation, but this kind of aggregation usually occurs after heating or aging.9,33 These driving factors are excluded from our experiment, and the lateral size of NPLs with dilution does not multiply manyfold as shown in Fig. S6 (ESI†). Therefore, the rapid NPL aggregation model cannot explain our experimental results well.
Considering that perovskite NPLs are easily prepared at room temperature, the above spectral changes naturally remind us that the dilution process may be similar to the ripening phenomenon observed in the preparation of traditional metal chalcogenide quantum dots (QDs), perovskite QDs, and metal nanoparticles.32,34,35 During dilution, the thinner (or small-sized laterally) NPLs gradually shrink/disappear, providing the monomers for the continued growth of the larger ones. This can be confirmed from the gradual reduction in the PL signal of the shorter wavelength components and the gradual red-shift of the PL peak, as observed in the above h-NPL surveys. Interestingly, a size focusing process occurs between D0 and D8 in samples with higher concentrations. The FWHM of 5 ML h-NPLs is decreased by ∼2 nm after three times dilution (Fig. 1(b)), which is credible evidence of size focusing as reported by past studies.32 Similar to the ripening process in QD synthesis, the size defocusing phenomenon of 5 ML h-NPLs also exists during further dilution, and thus their PL line width is increased by ∼7.1 nm, although there may be effects of defect states in this process.36 This is mainly related to monomer concentrations, which are depleted with dilution naturally, and the resulting increase in critical size triggers the size focusing/defocusing process, since the monomer concentration is inversely proportional to the critical size.32,34 Small NPLs below the critical size dissolve into the pool of monomers while some NPLs grow at a faster rate that allows them to catch up in lateral size (or thickness) to larger ones, resulting in focusing. During defocusing, more and more NPLs are below the critical size and dissolve, providing monomers for growing NPLs. In the mixed solution of 3 ML and 4 ML h-NPLs, the dilution ripening process is more obvious. The dissolution of 3 ML samples promotes the continuous growth of 4 ML h-NPLs, which can be inferred from the relative PL intensity changes and red spectral shifts, and eventually all of the 3 ML h-NPLs are almost consumed (Fig. 2).
A question that needs to be further discussed is the role of the thickness and lateral size of NPLs in the above process. In the formation of zinc blende CdSe NPLs, new growth is easier to occur at the narrow facets instead of the wide facets, inducing highly anisotropic morphology and precise atomic-scale thickness.37,38 Moreover, it is suggested that thinner NPLs are dissolved while new thicker NPLs are formed through laterally small nuclei during ripening.38 The CsPbBr3 perovskite NPLs studied here may also have a similar process, in which new nucleated islands are more easily formed on the narrow facets of the NPLs and then the islands gradually grow along the facets. Normally, this increasing lateral size of NPLs will cause red-shifted PL because of quantum confinement of the exciton in the lateral direction. It is worth noting that the total red shift in the PL due to ripening exceeds 10 nm and 14 nm for 5 ML h-NPLs and 13 nm for 4 ML h-NPLs. This seems to go beyond the effects of lateral growth. As a quasi-two-dimensional material, the thickness of NPLs dominates their optical properties, such as PL peak position, and the addition of a perovskite layer in the literature only results in a red shift of ∼10 nm for 4 ML and 5 ML NPLs.9,13 It is likely that the dilution ripening effect also leads to increasing thickness of NPLs, although they may need to overcome a greater barrier than lateral growth.38,39 In other words, NPLs can evolve into larger and thicker nanocrystals by diluting them, as shown by TEM results (Fig. 1(d)).
Besides the hot injection method, the reprecipitation protocol is often used to prepare CsPbBr3 perovskite NPLs.13,20 As an interesting attempt, we hope to observe whether a similar dilution ripening effect exists in CsPbBr3 NPLs synthesized in other schemes, which would help us to judge whether it is an intrinsic phenomenon. Hence, fresh 3 ML and 5 ML CsPbBr3 perovskite NPLs are prepared via the reprecipitation method, labeled as 3 ML r-NPLs and 5 ML r-NPLs, respectively. For 3 ML r-NPLs, the PL peak is red-shifted gradually from 461 nm to 478 nm with successive dilutions (Fig. 3(a) and (b)). The variation in FWHM is small, with a maximum deviation of only 3 nm compared with the initial value (Fig. S9, ESI†). 5 ML r-NPLs mainly display a red-shifted PL with successive dilutions, except for a slight blue shift (∼2 nm) at the initial stage (Fig. 3(a) and (c)). During the dilution process, the FWHM gradually decreases from ∼34 nm to ∼28 nm (Fig. S9, ESI†). Overall, the spectral evolution is similar to the above h-NPL results upon dilution, implying that preparation methods do not significantly affect the occurrence of the ripening process. In detail, the blue shift of the PL peak in 5 ML r-NPLs is different from that of 5 ML h-NPLs (or 3 ML, 4 ML h-NPLs) at the final stage. The latter may occur from the ion migration and removal in the sample after excessive dilution, resulting in degradation or lattice distortion of the crystal structure.30 The former is a ripening focusing process, because the onset of the PL spectra is constantly redshifted and their FWHM is decreasing.
 | ||
| Fig. 3 PL results of fresh 3 ML r-NPLs and 5 ML r-NPLs during the stepwise dilutions. (a) Evolution of PL peak wavelength with increasing dilution. A series of PL spectra upon dilution: (b) 3 ML r-NPLs, (c) 5 ML r-NPLs. | ||
In most application scenarios, the NPLs need to be stored for a period of time, which may cause some changes in their optical properties.20 Therefore, we next focus on the dilution measurements of aged NPLs. The NPLs are sealed and stored in a dark environment at 4 °C for several days. As depicted in Fig. 4(a), aged 5 ML h-NPLs exhibit a slight redshift (about 1.5 nm) of the PL peak and a small broadening (about 2 nm) of the FWHM compared with the fresh sample. Throughout the entire dilution process, the emission peak shift of the aged 5 ML h-NPLs is minimal, with a maximum deviation of approximately 3 nm compared with the initial PL (Fig. 4(b) and (c)). A slight increase in the FWHM is also observed in the stepwise dilutions, with a maximum increment of ∼2 nm (Fig. 4(b)). The total number of dilution rounds may be reduced due to the agglomeration and precipitation of some NPLs after aging.20,28 Compared with the result of fresh 5 ML h-NPLs, the variation in the PL peak and the FWHM is greatly decreased. This indicates that the spectral redshift level of 5 ML h-NPLs can be effectively suppressed in the stepwise dilutions when they are properly stored for some time.
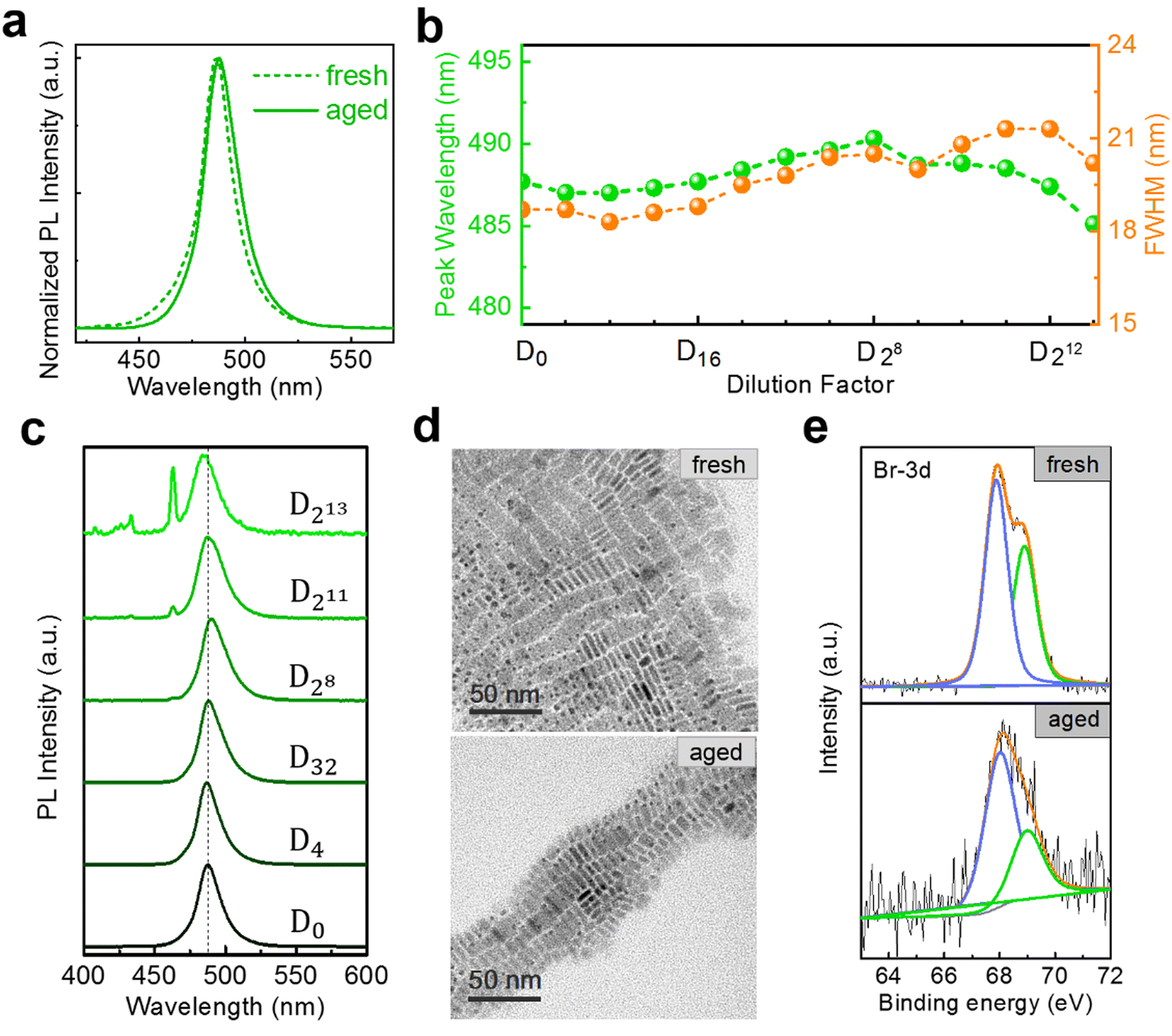 | ||
| Fig. 4 (a) Normalized PL spectra of fresh and aged 5 ML h-NPLs NPLs at D0. (b) Evolution of PL peak wavelength and FWHM in aged 5 ML h-NPLs during the stepwise dilutions. (c) A series of PL spectra of aged 5 ML h-NPLs upon dilution. (d) TEM images of fresh and aged 5 ML h-NPLs NPLs at D0. (e) XPS spectra corresponding to Br 3d of fresh and aged 5 ML h-NPLs. | ||
To further understand the above phenomenon, we compare the microstructure information of both fresh and aged samples. The TEM results indicate that the aged 5 ML h-NPLs exhibit almost identical morphology and size to those of the fresh sample (Fig. 4(d)). That is, the aged NPLs used in dilution tests do not contain spontaneous accumulation and deformation reported in the literature.20,28 This agrees well with the less spectral variation before and after aging, whereas that in the literature vary from blue to green.20,28 Meanwhile, X-ray photoelectron spectroscopy (XPS) spectra of the two samples are also recorded, and the results are shown in Fig. 4(e) and Fig. S10 (ESI†). Expect for Br 3d peaks, the shift of other peaks is inconspicuous with aging. The Br 3d peaks can be fitted into two peaks, corresponding to the surface and inner ions.40,41 Their binding energies shift from 68.9 eV and 67.9 eV to 69.0 eV and 68.0 eV, respectively, and the intensity ratio between the two peaks also changes from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 to 3:7. It can be speculated that some degree of surface rearrangement occurs upon aging, which improves the structural stability of NPLs. However this surface optimization is ineffective for diluting 5 ML h-NPLs at large multiples, which exhibit a considerable redshift in the PL after 3000-fold dilution (Fig. S11, ESI†). In addition, aging treatment fails to improve the spectral redshift of the 3 (or 4) ML h-NPLs, which exhibit a similar PL evolution upon stepwise dilutions (Fig. S12, ESI†). These two results elucidate that the spontaneous surface optimization effect of NPLs during aging is limited and unstable.
:6 to 3:7. It can be speculated that some degree of surface rearrangement occurs upon aging, which improves the structural stability of NPLs. However this surface optimization is ineffective for diluting 5 ML h-NPLs at large multiples, which exhibit a considerable redshift in the PL after 3000-fold dilution (Fig. S11, ESI†). In addition, aging treatment fails to improve the spectral redshift of the 3 (or 4) ML h-NPLs, which exhibit a similar PL evolution upon stepwise dilutions (Fig. S12, ESI†). These two results elucidate that the spontaneous surface optimization effect of NPLs during aging is limited and unstable.
As discussed above, the dilution ripening effect can greatly alter the optical properties of NPLs, making some experiments difficult to perform. So, it is necessary to find some methods to suppress this effect. Recently, a chemical post-treatment with PbBr2 solution was used to enhance the PL intensity and stability of NPLs by repairing their surface defects.13 The PbBr2-saturated toluene solution was used as a diluent to avoid dissolving and damaging the NPLs at low concentrations, despite lacking experimental details.8 Therefore, anhydrous hexane is saturated with PbBr2 before the next stepwise dilution tests here (see the Experimental section). As the PbBr2-saturated solution shows a brightly wide emission in the visible region (Fig. S13, ESI†), we have to remove it as background light when recording the PL signal of diluting NPLs. The PL peak maxima of fresh 5 ML h-NPLs in stepwise dilutions are plotted as shown in Fig. 5(a). They are nearly constant after the initial minor blueshift (∼3 nm). For comparison, we also offer the results of the same batch of NPLs upon dilution with pure anhydrous hexane. The NPL sample displays a redshifted emission, which shifts from 489 nm at D0 to 497nm at D213. It should be pointed out that, at D0, the sample in PbBr2-saturated solution shows a spectral redshift of ∼3.5 nm (∼18 meV) compared with that dispersed in anhydrous hexane, which is possibly due to the change of dielectric environment on the NPLs’ surface.13 Furthermore, the extra emission component at ∼480 nm, originating from thinner NPLs, is maintained throughout the dilution process of utilizing PbBr2-saturated solution (Fig. 5(b)). In contrast, this extra emission gradually weakens and becomes indistinguishable with the dilution of anhydrous hexane (Fig. 5(c)). This elucidates that the dilution ripening phenomenon can be effectively suppressed by using PbBr2-saturated solution as a diluting solvent, although slight spectral changes are inevitable. Dilution at large multiples also supports this viewpoint (Fig. S14, ESI†), due to the nearly constant PL profile and inconspicuous peak shift at D12000.
 | ||
| Fig. 5 PL results of fresh 5 ML h-NPLs diluted in different solvents: PbBr2-saturated solution and anhydrous hexane. (a) Evolution of PL peak wavelength during the stepwise dilutions. A series of typical PL spectra diluted in (b) PbBr2-saturated solution and (c) anhydrous hexane. | ||
In addition to the continuous surface passivation in the presence of excess PbBr2, the exchange of stronger binding ligands is a feasible manipulation to improve structural stability also. Oleic acid and oleylamine, used for preparing the NPLs, exhibit rather labile interactions with the NPL surface.20 As an example, lecithin has been successfully used to stabilize small-sized perovskite nanocrystals,42 inspiring us. Accordingly, we attempt to add lecithin into the fresh NPL stock solution to improve their dilution stability (see the Experimental section). The spectral results of dilution obtained after adding lecithin for 2 min are shown in Fig. 6(a), (b). As the same batch of NPLs, these results are compared with those without additional ligands (Fig. 5(a), orange broken line). The addition of lecithin postpones the overall evolution of the PL profile and peak maxima (Fig. 6(a), (b) and 5(a), (c)), despite arriving at a similar endpoint. At this point, the dilution ripening effect works at a slower rate. Unexpectedly, the NPLs with lecithin exceeding 1 h show wonderful dilution stability (Fig. 6(a) (green broken line), Fig. S15, ESI†). During the whole dilution procedure, the emission peak is mainly around 498 nm with a deviation of 1 nm. The dilution ripening process is inhibited by red-shifting of the PL peak from the original 489 nm to 499 nm at D0 (Fig. 6(c)). We suspect that this phenomenon is related to the changing of dielectric constant in NPLs due to the addition of lecithin. This operation should also be accompanied by the reorganization of the NPLs, due to the lack of an emission component located around 480 nm. Fortunately, the shape of the NPLs is maintained after adding lecithin (Fig. 6(d)), as shown by TEM results.
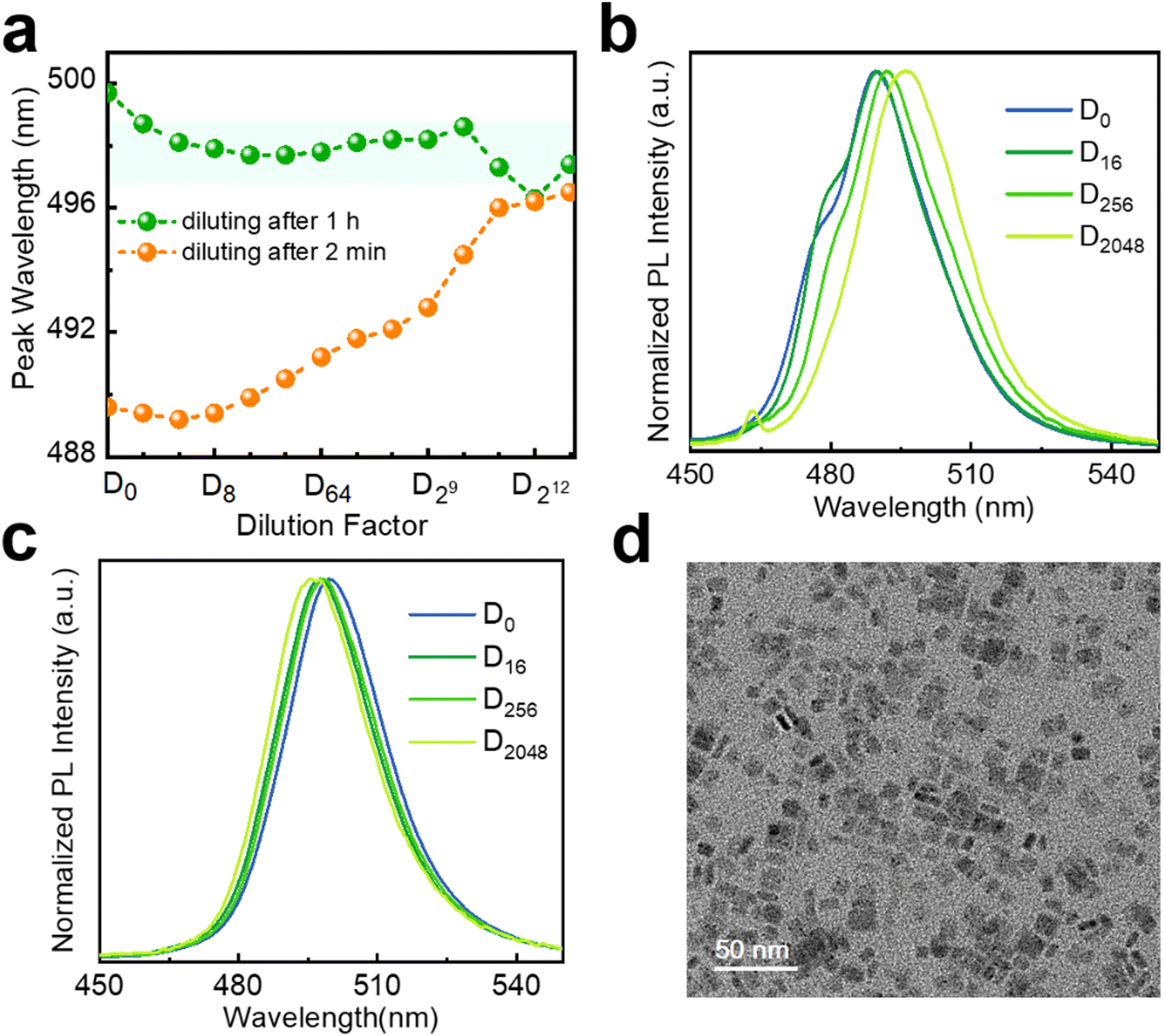 | ||
| Fig. 6 Stepwise dilution results of fresh 5 ML h-NPLs after adding lecithin to the stock solution for 2 min and 1 h. (a) Evolution of PL peak wavelength upon dilution. A series of typical PL spectra diluted after adding lecithin to the stock solution for (b) 2 min and (c) 1 h. (d) TEM image of 5 ML h-NPLs after adding lecithin to the stock solution for 1 h. | ||
A question arises: can the stabilization methods for perovskite nanocrystals be directly applied to enhance the dilution stability of NPLs? Wang et al. improved the dilution stability of CsPbBr3 NPLs using polysalt/PbBr2 ligands, and they also demonstrated that similar polysalt ligands can be used for perovskite nanocrystals as well.20,43,44 Here, we also tried two other ligands that can enhance the stability of perovskite nanocrystals (see the Experimental section).45,46 When using polymer zwitterions as a stabilizing ligand, we find a significant improvement in the dilution stability of NPLs after adding this ligand to the stock solution, with a minimal redshift in the PL spectrum (Fig. S16, ESI†). TEM image indicates that the addition of this ligand does not change the morphology of the NPLs (Fig. S17, ESI†). When using modular zwitterion-functionalized isopropyl methacrylate polymers as the stabilizing ligand, the PL spectrum is gradually blue-shifted throughout the dilution (Fig. S18, ESI†). At D0, the emission peak wavelength of the sample redshifts by ∼8 nm compared to the original spectrum. TEM result shows that the sample had transformed from NPLs to nanocrystals prior to the dilution experiment. These results indicate that the highly stable ligands applicable to perovskite nanocrystals may not be used to improve the dilution stability of perovskite NPLs. This could be attributed to the anisotropic morphology of NPLs, resulting in different requirements for surface ligands compared to nanocrystals. Although several strategies have been employed to improve the stability of diluted NPLs in our study, seeking more convenient methods is also necessary.
Conclusion
We investigate the dilution tests of CsPbBr3 NPLs with different thicknesses prepared by hot injection and ligand-assisted reprecipitation methods, respectively. The test results show that the gradually red-shifted spectra occur with an increase in the dilution factor, and it is not caused by the direct aggregation and fusion of NPLs according to TEM images. Hence, a dilution ripening mechanism is proposed to explain this phenomenon, in which smaller NPLs will gradually dissolve and release monomers during dilution to drive the growth of the larger NPLs. In a mixture solution of 3 ML and 4 ML NPLs, the ripening process is the most obvious, and the PL component corresponding to 3 ML NPLs decreases step by step while that of 4 ML NPLs is still prominent. Proper storage of NPLs is of great help in improving the dilution stability of 5 ML h-NPLs, but it is useless to the thinner samples. Furthermore, we explore two schemes to suppress the dilution ripening effect. When the PbBr2-saturated is used as the diluent, the PL peak of samples only has a minor blueshift, and the thinner NPLs can be well retained. A stronger binding ligand, such as lecithin, also helps improve the structural stability of the NPLs at low concentrations. An issue is that adding lecithin causes the NPLs to undergo a PL red shift at D0. In addition, we also find that stronger capping ligands suitable for cubic perovskite nanocrystals may not be used to improve the dilution stability of nanoplatelets. Therefore, the exploration of more suitable ligands or ligand exchange strategies for perovskite nanoplatelets will be the focus of further research.Experimental section
Materials
Cs2CO3 (99.9%, Aldrich), octadecene (ODE, 90%, Aldrich), oleic acid (OA, 90%, Aldrich), PbBr2 (99.999%, Aldrich), oleylamine (OLA, Aldrich, 70%), anhydrous hexane (95%, aladdin), anhydrous ethyl acetate (99.8%, aladdin), lecithin (98%, MACKLIN), toluene (sinopharm), and acetone (sinopharm). All chemicals were used as received.Hot injection method
000 rpm for 6 min. After centrifugation, the supernatant was discarded and the precipitates were redispersed in a small amount of anhydrous hexane forming a stable colloidal solution. The solution would suffer a second centrifugation (5200 rpm, 2 min) and the supernatant was collected for use or storage. In the dilution experiment, we mixed 100 μL of the purified sample with 2.9 mL of anhydrous hexane as D0 to minimize the effect of self-absorption.
Ligand-assisted reprecipitation method
Preparation of PbBr2-saturated solution
The PbBr2-saturated solution was prepared by dissolving 0.3 mmol PbBr2 in 30 mL of anhydrous hexane with the assistance of 300 μL each of oleic acid and oleylamine at 100 °C. In the corresponding dilution test, the CsPbBr3 NPLs were dispersed in a PbBr2-saturated solution according to the dilution factor.Ligand exchange with lecithin and others
The stock solution of lecithin (∼0.13 mol L−1) was prepared by dissolving 0.5 g of lecithin in 10 mL of anhydrous hexane at room temperature. Before the corresponding dilution test, 150 μL of lecithin stock solution was added to the purified CsPbBr3 NPL solution, and then the mixture was shaken for 1 min and stood for a period of time. During the dilution test, the mixture of NPLs and lecithin was dispersed in anhydrous hexane according to the dilution factor.Polymer zwitterions were prepared by a published procedure and dissolved in toluene (0.32 g mL−1).45 100 μL of polymer zwitterion solution was added to 100 μL purified CsPbBr3 NPLs which were dispersed in toluene, and then the mixture was shaken for 1 min before the dilution test. The anhydrous hexane was replaced by toluene in the dilution experiment.
Modular zwitterion-functionalized isopropyl methacrylate polymers were synthesized by using the Cohen's method.46 150 μL of polymer solution was mixed with the 150 μL of purified CsPbBr3 NPLs. The mixture was then sonicated for 2 min. The resulting NPL/polymer composites were precipitated out of the solution by adding an excess of hexanes and centrifuging at 3000 rpm for 3 min. This precipitate was then dispersed in 3 mL of anhydrous ethyl acetate. The solution was centrifugated again to remove the agglomerate. The supernatant was used as D0 for the stepwise dilution. Here, the anhydrous hexane was replaced by anhydrous ethyl acetate in the dilution experiment.
Characterization
Transmission electron microscopy (TEM) images of the NPLs were measured with a JEOL JEM-1400 Flash transmission electron microscope operated at an accelerating voltage of 120 kV. X-ray photoelectron spectroscopy (XPS) was recorded using an ESCALAB250Xi (Thermo Scientific) spectrometer with an Al Kα line X-ray source (1486.6 eV). The PL spectra were measured using a continuous wave diode laser at 405 nm as the excitation source and collecting the emitted light with a charged coupled device coupled to a spectrometer. The time-resolved PL decay curve was measured using a Hamamatsu's C5680 streak camera,4 and a 405 nm laser pulse was used as the excitation source. The absorption spectra were recorded by using a white light beam to transmit samples and collecting the transmitted light with a charged coupled device coupled to a spectrometer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (62274051 and 61605037), the Fundamental Research Funds for the Provincial Universities of Zhejiang (GK219909299001-001), and the Zhejiang Provincial Natural Science Foundation of China (LY20A040005 and LQ20F050007).References
- A. F. Vong, S. Irgen-Gioro, Y. Wu and E. A. Weiss, Nano Lett., 2021, 21, 10040–10046 CrossRef CAS PubMed.
- J. Yu and R. Chen, InfoMat, 2020, 2, 905–927 CrossRef CAS.
- Y. Huang, R. Su, Y. Wang, C. Zhu, J. Feng, J. Zhao, Z. Liu and Q. Xiong, Nano Lett., 2022, 22, 8274–8280 CrossRef CAS.
- B. Huang, Y. Huang, H. Zhang, X. Lu, X. Gao and S. Zhuang, ACS Appl. Mater. Interfaces, 2023, 15, 21354–21363 CrossRef CAS.
- A. Alfieri, S. B. Anantharaman, H. Zhang and D. Jariwala, Adv. Mater., 2023, 35, 2109621 CrossRef CAS.
- M. D. Tessier, C. Javaux, I. Maksimovic, V. Loriette and B. Dubertret, ACS Nano, 2012, 6, 6751–6758 CrossRef CAS.
- S. O. M. Hinterding, B. B. V. Salzmann, S. J. W. Vonk, D. Vanmaekelbergh, B. M. Weckhuysen, E. M. Hutter and F. T. Rabouw, ACS Nano, 2021, 15, 7216–7225 CrossRef CAS PubMed.
- A. Schmitz, F. Montanarella, L. L. Schaberg, M. Abdelbaky, M. V. Kovalenko and G. Bacher, Nano Lett., 2021, 21, 9085–9092 CrossRef CAS.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D. Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- C. Otero-Martinez, J. Ye, J. Sung, I. Pastoriza-Santos, J. Perez-Juste, Z. Xia, A. Rao, R. Hoye and L. Polavarapu, Adv. Mater., 2022, 34, 2107105 CrossRef CAS.
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Doblinger, K. Wang, R. Hoye, P. Muller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS.
- W. Shen, Y. Yu, W. Zhang, Y. Chen, J. Zhang, L. Yang, J. Feng, G. Cheng, L. Liu and S. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 5682–5691 CrossRef CAS.
- C. C. Lin, T. R. Liu, S. R. Lin, K. M. Boopathi, C. H. Chiang, W. Y. Tzeng, W. C. Chien, H. S. Hsu, C. W. Luo, H. Y. Tsai, H. A. Chen, P. C. Kuo, J. Shiue, J. W. Chiou, W. F. Pong, C. C. Chen and C. W. Chen, J. Am. Chem. Soc., 2022, 144, 15718–15726 CrossRef CAS.
- C. Huo, C. F. Fong, M. Amara, Y. Huang, B. Chen, H. Zhang, L. Guo, H. Li, W. Huang, C. Diederichs and Q. Xiong, Nano Lett., 2020, 20, 3673–3680 CrossRef CAS.
- M. Lai, A. Obliger, D. Lu, C. S. Kley, C. G. Bischak, Q. Kong, T. Lei, L. Dou, N. S. Ginsberg, D. T. Limmer and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11929–11934 CrossRef CAS PubMed.
- M. I. Bodnarchuk, S. C. Boehme, S. Ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef PubMed.
- Y. Wang, X. Li, S. Sreejith, F. Cao, Z. Wang, M. C. Stuparu, H. Zeng and H. Sun, Adv. Mater., 2016, 28, 10637–10643 CrossRef CAS.
- S. Wang, W. Wang, S. Donmez, Y. Xin and H. Mattoussi, Chem. Mater., 2022, 34, 4924–4936 CrossRef CAS.
- Y. Tong, F. Ehrat, W. Vanderlinden, C. Cardenas-Daw, J. K. Stolarczyk, L. Polavarapu and A. S. Urban, ACS Nano, 2016, 10, 10936–10944 CrossRef CAS PubMed.
- E. Fanizza, F. Cascella, D. Altamura, C. Giannini, A. Panniello, L. Triggiani, F. Panzarea, N. Depalo, R. Grisorio, G. P. Suranna, A. Agostiano, M. L. Curri and M. Striccoli, Nano Res., 2019, 12, 1155–1166 CrossRef CAS.
- S. K. Mehetor, H. Ghosh, B. Hudait, N. S. Karan, A. Paul, S. Baitalik and N. Pradhan, ACS Energy Lett., 2019, 4, 2353–2359 CrossRef CAS.
- F. Di Stasio, M. Imran, Q. A. Akkerman, M. Prato, L. Manna and R. Krahne, J. Phys. Chem. Lett., 2017, 8, 2725–2729 CrossRef CAS PubMed.
- J. Shamsi, P. Rastogi, V. Caligiuri, A. L. Abdelhady, D. Spirito, L. Manna and R. Krahne, ACS Nano, 2017, 11, 10206–10213 CrossRef CAS.
- S. K. Ha, C. M. Mauck and W. A. Tisdale, Chem. Mater., 2019, 31, 2486–2496 CrossRef CAS.
- V. Nagal, V. Kumar, R. Kumar, K. Singh, A. Khosla, R. Ahmad and A. K. Hafiz, ECS J. Solid State Sci. Technol., 2021, 10, 96002 CrossRef CAS.
- Z. Dang, B. Dhanabalan, A. Castelli, R. Dhall, K. C. Bustillo, D. Marchelli, D. Spirito, U. Petralanda, J. Shamsi, L. Manna, R. Krahne and M. P. Arciniegas, Nano Lett., 2020, 20, 1808–1818 CrossRef CAS.
- K. Antami, F. Bateni, M. Ramezani, C. E. Hauke, F. N. Castellano and M. Abolhasani, Adv. Funct. Mater., 2022, 32, 2108687 CrossRef CAS.
- H. Zhang, X. Fu, Y. Tang, H. Wang, C. Zhang, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, Nat. Commun., 2019, 10, 1088 CrossRef CAS PubMed.
- F. Hu, H. Zhang, C. Sun, C. Yin, B. Lv, C. Zhang, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, ACS Nano, 2015, 9, 12410–12416 CrossRef CAS PubMed.
- M. Koolyk, D. Amgar, S. Aharon and L. Etgar, Nanoscale, 2016, 8, 6403–6409 RSC.
- T. Le, S. Lee, H. Jo, G. Jeong, M. Chang and H. Yoon, J. Phys. Chem. Lett., 2021, 12, 5631–5638 CrossRef CAS.
- X. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef.
- Y. J. Yoon, Y. S. Shin, C. B. Park, J. G. Son, J. W. Kim, H. S. Kim, W. Lee, J. Heo, G. Kim and J. Y. Kim, Nanoscale, 2020, 12, 21695–22172 RSC.
- A. Riedinger, F. D. Ott, A. Mule, S. Mazzotti, P. N. Knüsel, S. J. P. Kress, F. Prins, S. C. Erwin and D. J. Norris, Nat. Mater., 2017, 16, 743–748 CrossRef CAS.
- F. D. Ott, A. Riedinger, D. R. Ochsenbein, P. N. Knüsel, S. C. Erwin, M. Mazzotti and D. J. Norris, Nano Lett., 2017, 17, 6870–6877 CrossRef CAS PubMed.
- P. N. Knüsel, A. Riedinger, A. A. Rossinelli, F. D. Ott, A. S. Mule and D. J. Norris, Chem. Mater., 2020, 32, 3312–3319 CrossRef.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- Y. Jing, K. Cao, B. Zhou, S. Geng, Y. Wen, B. Shan and R. Chen, Chem. Mater., 2020, 32, 10653–10662 CrossRef CAS.
- Q. A. Akkerman, T. Nguyen, S. C. Boehme, F. Montanarella, D. N. Dirin, P. Wechsler, F. Beiglbock, G. Raino, R. Erni, C. Katan, J. Even and M. V. Kovalenko, Science, 2022, 377, 1406–1412 CrossRef CAS PubMed.
- S. Wang, L. Du, S. Donmez, Y. Xin and H. Mattoussi, Nanoscale, 2021, 13, 16705–16718 RSC.
- S. Wang, L. Du, Z. Jin, Y. Xin and H. Mattoussi, J. Am. Chem. Soc., 2020, 142, 12669–12680 CrossRef CAS PubMed.
- H. Kim, N. Hight Huf, J. H. Kang, P. Bisnoff, S. Sundararajan, T. Thompson, M. Barnes, R. C. Hayward and T. Emrick, Angew. Chem., Int. Ed., 2020, 59, 10802–10806 CrossRef CAS.
- T. A. Cohen, Y. Huang, N. A. Bricker, C. S. Juhl, T. J. Milstein, J. D. MacKenzie, C. K. Luscombe and D. R. Gamelin, Chem. Mater., 2021, 33, 3779–3790 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02178e |
| This journal is © The Royal Society of Chemistry 2024 |