
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(arylene piperidinium) terpolymer membranes with dual piperidinium cations and semi-fluoroalkyl pendants for anion exchange membrane water electrolyzers†
Vikrant
Yadav
 a,
Kenji
Miyatake
*abc,
Ahmed Mohamed
Ahmed Mahmoud
a,
Fanghua
Liu
a,
Fang
Xian
a,
Lin
Guo
a,
Chun Yik
Wong
a,
Toshio
Iwataki
b,
Makoto
Uchida
b and
Katsuyoshi
Kakinuma
ab
a,
Kenji
Miyatake
*abc,
Ahmed Mohamed
Ahmed Mahmoud
a,
Fanghua
Liu
a,
Fang
Xian
a,
Lin
Guo
a,
Chun Yik
Wong
a,
Toshio
Iwataki
b,
Makoto
Uchida
b and
Katsuyoshi
Kakinuma
ab
aClean Energy Research Center, University of Yamanashi, Kofu, 400-8510, Japan
bHydrogen and Fuel Cell Nanomaterials Center, University of Yamanashi, 4 Takeda, Kofu, Yamanashi 400-8510, Japan. E-mail: miyatake@yamanashi.ac.jp
cDepartment of Applied Chemistry, Waseda University, Tokyo 169-8555, Japan
First published on 30th August 2024
Abstract
To develop high-performance anion exchange membranes (AEMs), the effect of the length of semi-fluoroalkyl pendants on physical and electrochemical properties of the resulting AEMs was investigated. A series of 2,5-dichlorobenzene monomers differing in semi-fluoroalkyl pendant length were synthesized and copolymerized with 2,2-bis(4-chlorophenyl)hexafluoropropane and piperidine functionalized 2,7-dichlorofluorene. The terpolymers provided bendable membranes by solution casting. The membranes with comparable ion exchange capacity (IEC = ca. 1.85 meq g−1) showed similar water uptake, while the hydroxide ion conductivity increased by 24% on increasing the pendant chain from 7 to 11 carbons. The maximum ion conductivity of 112 mS cm−1 (at 80 °C) was achieved for the membrane formulated with the C11 pendant. Cx-QPip-n membranes showed good alkaline stability; in particular, C11-QPip-1.86 retained 75% of the original conductivity after 1056 h under harsh alkaline conditions (8 M KOH at 80 °C). An alkaline water electrolysis cell assembled with the C11-QPip-1.86 membrane and with a PGM-free anode catalyst (Ni0.8Co0.2O) showed good performance (1.0 A cm−2 at 1.64 V) with high voltage efficiency (75%). The cell was durable for 1000 h with minor voltage change (28 μV h−1) under constant current density (1.0 A cm−2) operation.
Introduction
With mounting concern over climate change with the use of fossil fuels, transition towards green hydrogen energy is one of the most promising solutions. Water electrolyzers (WEs) could produce green hydrogen from renewable energies and pave a way toward a carbon neutral society.1–4 Among several kinds of WEs, alkaline water electrolyzers (AWEs) and proton exchange membrane water electrolyzers (PEMWEs) are operable at low temperature (<100 °C) and have been commercialized.3 The use of concentrated alkaline solution (>ca. 5 M KOH) and low working current density, and use of noble metal catalysts (e.g., IrO2 and Pt) due to acidic environments are technical drawbacks for AWEs and PEMWEs, respectively.5,6As an emerging technique, anion exchange membrane water electrolyzers (AEMWEs) garnered considerable attention by combining the merits of AWEs and PEMWEs while mitigating most of the bottlenecks associated with them.7,8 AEMWEs are operable with diluted alkaline solution (<1 M KOH) using 3d transition metal catalysts (e.g., Ni, Co, Fe, etc.) at high current density (>2 A cm−2).9–11 Despite the promising potential, performance and durability of AEMWEs are limited by poor hydroxide ion conductivity and chemical instability of AEMs and needed to be improved at least to the level of PEMWEs. Overcoming the trade-off-relationship between the ion conductivity and mechanical stability is a challenge for AEMs.
To address the issues and meet the prerequisites of AEMs (high ion conductivity and chemical and mechanical stability) for AEMWEs, many efforts have been made in the last decade by tuning ion exchangeable species and/or the polymer main/side chain structure.11,12 Earlier reports confirmed that the polymer structures, cationic head groups and their position on the polymer backbone, and water absorbability influenced the ion conductivity and alkaline stability of the resulting AEMs and that the effect of these parameters was not independent but rather interrelated.13–16 For the polymer main chain, heteroatom linkages (in particular, aryl ether bonds) are vulnerable to hydroxide ion attack in alkaline media, leading to chain fragmentation.16–18 The aryl ether free polymer backbone is therefore preferable for chemically stable AEMs. A wide variety of cationic head groups with or without an alkyl spacer between the cationic unit and the main chain have been investigated, and it has been recognized that the placement of cationic head groups not on the main chain but on the alkyl side chain was effective in improving the ion conductivity as well as alkaline stability along with suppressing water absorption.19–24 We combined the advantages of the aryl ether free main chain and alkyl spacer side chain for several AEMs to achieve excellent hydroxide ion conductivity (>100 mS cm−1 at 80 °C) and alkaline stability (>1000 h in 8 M KOH at 80 °C).13,14,23,24 For example, Ozawa et al. prepared aryl ether linkage free terpolymers from 2,2-bis(4-chlorophenyl)hexafluoropropane, 1,6-bis(3-chlorophenyl)perfluorohexane, and 2,7-dichloro-9,9-bis[6′-(N,N-dimethylamino)hexyl]fluorene, and the resulting AEMs retained physical and mechanical integrity with no sign of backbone degradation (verified by nuclear magnetic resonance (NMR) spectroscopy) after a 1000 h harsh (8 M KOH at 80 °C) alkaline stability test.13 Compared to typical benzyltrimethylammonium groups in which methylene connected the head groups and main chain, alkyltrimethylammonium and alkyldimethylpiperidinium groups were much more alkaline stable.14,20–22 A study on small molecules revealed that benzyltrimethylammonium cations were unstable in alkaline media and that the stability was significantly improved by using a spacer between the aromatic and ammonium head groups.20 The results were also verified in polymeric systems.25,26 Recently, Mahmoud et al. reported that a 4-QPPAF-TMA membrane with N-hexyltrimethylammonium groups maintained 97% of its initial conductivity after a 1000 h test in 8 M KOH at 80 °C.14 Density functional theory (DFT) insights into the stability of N-hexyltrimethylammonium and heterocyclic quaternary ammonium groups disclosed that N,N-dimethylpiperidinium (DMP) was much more stable than N-hexyltrimethylammonium in all degradation pathways.21 We systematically studied the effect of substituents (with +I and −I effects) and position of the nitrogen atom in the piperidinium ring and found that the substituents significantly affected the water absorption and thus hydroxide ion conductivity. The position of nitrogen (1N- nitrogen attached to the side chain and N4- nitrogen at the terminal of the piperidinium ring) impacted water absorption as well as the alkaline stability. In particular, a membrane formulated with DMP cations with the nitrogen at the terminal was much more alkali tolerant than the membranes with nitrogen at the 1N position.23
The effect of fluorination on the aliphatic side chain was also investigated to strengthen the hydrophilic/hydrophobic differences between the head groups and the side chain and to promote development of microphase separated morphology of the membranes for improved ion conductivity.27–29 Xu et al.27 reported that replacing alkyl side chains (C5H8) with fluorinated ones (C5F8) in poly(biphenyl piperidinium) membranes enlarged hydrophilic clusters to ca. 5–6 nm from ca. 2 nm. At low IEC and thus low water uptake, poly(aryl piperidinium) membranes with a longer perfluoroalkyl side chain showed improved microphase separation and higher hydroxide ion conductivity.29 Nevertheless, formation of highly developed hydrophilic/hydrophobic phase separation seems still insufficient for poly(arylene)s partly because of the relatively short spacer length. Here, we adopted the semi-fluoroalkyl pendant strategy for our terpolymer-based AEMs aiming at better performance and stability. 1,3-Propane groups were inserted between the main chain phenylene and perfluoroalkyl groups for synthetic reasons. The effect of the semi-fluoroalkyl pendant length on physical and electrochemical properties of the resulting terpolymer membranes was investigated in detail. The optimized membrane was used in an AEMWE with an in-house developed transition metal-based anode catalyst (Ni0.8Co0.2O).30
Experimental section
Synthesis of hydrophobic and hydrophilic monomers
A series of semi-fluoroalkyl chain tethered 1,4-dichlorobenzene monomers (3a–c) were synthesized from the corresponding fluoroalkyl triphenyl phosphonium iodide salts and 2,5-dichlorobenzaldehyde (Scheme S1†). 2,2-Bis(4-chlorophenyl)hexafluoropropane (5) and 2,7-dichlorofluorene were synthesized following the procedure akin to that in our previous reports13,31 in 43% and 54% yields as a white crystalline solid and white powder, respectively. The di-tert-butyl-4,4′-((2,7-dichloro-9H-fluorene-9,9-diyl)bis(methylene))bis(piperidine-1-carboxylate) monomer (4b) was synthesized using 2,7-dichlorofluorene and tert-butyl 4-(iodomethyl)piperidine-1-carboxylate as the precursor (Scheme S2†).Synthesis of Cx-QPip-n terpolymers
Semi-fluoroalkyl chain tethered 1,4-dichlorobenzene (3a–c), di-tert-butyl-4,4′-((2,7-dichloro-9H-fluorene-9,9-diyl)bis(methylene))bis(piperidine-1-carboxylate) (4b), and 2,2-bis(4-chlorophenyl)hexafluoropropane (5) monomers were copolymerized via the Ni(0)-promoted coupling reaction. The ratio of monomers 5 and 3 was kept constant while the composition of monomer 4b was determined to afford the terpolymer with IEC = 2.10 meq g−1. A typical procedure for C11-Pip-2.10 as an example is as follows. To a previously dried 100 mL three-neck round bottomed flask fitted with a nitrogen inlet/outlet, dean stark trap, and reflux condenser, a mixture of monomers 5 (0.67 g, 1.80 mmol), 3c (0.22 g, 0.36 mmol), 4b (0.93 g, 1.47 mmol), and 2,2-bipyridyl (2.84 g, 18.18 mmol) was added under continuous N2 flow followed by the addition of DMAc (18 mL). After complete dissolution of the monomers at 80 °C, Ni(COD)2 (2.50 g, 9.09 mmol) was added quickly and the flask was sealed with a rubber septum. The polymerization reaction was allowed for 4 h at 80 °C under N2 flow. Then, the reaction mixture was diluted and poured into a large excess of 8 M HCl/MeOH. The obtained yellowish granular precipitate was washed with conc. hydrochloric acid (three-fold), water (two-fold), 0.1 M K2CO3 aq. (two-fold), and water (two-fold) followed by vacuum drying at 60 °C for 12 h. A simultaneous deprotection reaction was performed during washing of the terpolymer with hydrochloric acid.The resulting precursor terpolymers with different semi-fluoroalkyl side chains were quaternized using dimethyl sulfate and denoted as Cx-QPip-n, where x and n represent the number of carbon atoms in the semi-fluoroalkyl side chain and titrated IEC of the copolymer, respectively (Scheme 1). The quaternization reaction of the C11-Pip-n precursor terpolymer was performed in two steps: 1.25 g (3.64 mmol) of the copolymer was dissolved in 10 mL of DMAc followed by the addition of 4.59 g (36.40 mmol) of Me2SO4. The reaction was allowed for 48 h at 50 °C under a N2 atmosphere. Thereafter, the mixture was poured into water and the precipitate was washed with water (three-fold) followed by vacuum drying at 60 °C. To ensure full quaternization, a similar process was repeated with 0.97 g (7.00 mmol) of K2CO3 as the quaternization promoter.
 | ||
| Scheme 1 Schematic for the synthesis of Cx-QPip-n terpolymers. | ||
Preparation of Cx-QPip-n membranes
The solution casting method was adopted to obtain transparent and flexible Cx-QPip-n membranes. The Cx-QPip-n terpolymer was dissolved in DMAc, and the resulting homogeneous solution was filtered through a 0.45 μm membrane filter and degassed. The degassed solution was cast onto a glass plate and dried at 50 °C for 15 h. The membranes were treated with 1 M KOH aq. for 24 h at 80 °C in a closed PTFE container, washed with degassed DI water until a neutral pH was achieved, and then stored in degassed DI water. The thickness of the membranes was 45 ± 5 μm.Membrane electrode assembly (MEA) preparation
The cathode catalyst ink was prepared using a commercial carbon-black supported platinum catalyst (Pt/CB: TEC10E50E, Tanaka Kikinzoku Kogyo, K. K.), methanol, and water by zirconia ball milling for 30 min. To this slurry 5 wt% solution of QPAF-4 binder31 (IEC = 1.50 meq g−1) in methanol was added and ball-milled for another 30 min. The weight ratio of the binder to the catalyst support was 0.6. The resulting ink was directly sprayed onto Toray TGP H-120 carbon paper by the pulse-swirl-spray (PSS) technique (PSS, Nordson Co., Ltd) to prepare a gas diffusion electrode (GDE). The anode catalyst ink was prepared using the in-house developed Ni0.8Co0.2O catalyst,30 methanol, and water by zirconia ball milling for 30 min. A 5 wt% solution of QPAF-4 binder (IEC = 1.50 meq g−1) in methanol was added to the resultant slurry followed by ball milling for another 30 min. The weight ratio of the binder to the catalyst was 0.15. The catalyst ink was sprayed onto one side of the C11-QPip-1.86 membrane (50 μm thick) using a PSS machine to obtain a catalyst coated membrane (CCM). The electrode area was 1 cm2, and the amount of catalyst loaded on the cathode (Pt) and anode (Ni0.8Co0.2O catalyst) sides was 1.0 and 2.0 mg cm−2, respectively. The MEA was fabricated from the GDE and CCM. On both anode and cathode sides, EPDM gaskets (300 μm thick) and a Ni separator with straight flow channels were fixed. Gold-plated copper based current collectors were used and the electrolysis cell was sealed by applying a pressure of 8.5 kgf cm−2.Water electrolysis cell evaluation
The water electrolysis cell was operated at 80 °C and 1 M KOH aq. was circulated on both anodic and cathodic sides at a flow rate of 10 mL min−1. The electrolysis cell was preconditioned by sweeping the current density from 0 to 1.0 A cm−2 twice, prior to the performance evaluation.Results and discussion
Synthesis and characterization of monomers and polymers
A series of semi-fluoroalkyl chain tethered 1,4-dichlorobenzene monomers were synthesized from the corresponding semi-fluoroalkyl triphenyl phosphonium salts and 2,5-dichlorobenzaldehyde. Initial attempts to introduce a semi-fluoroalkyl chain on 2,5-dichlorobenzaldehyde were carried out using potassium tert-butoxide following the classical Wittig reaction. The reactions using potassium tert-butoxide even at low temperatures (0 °C and −30 °C) were unsuccessful due to instability of the ylide formed under the strong basic conditions which resulted in liberation of fluoride ions (confirmed by using 19F NMR spectra). The high temperature Wittig reaction using a mild base, K2CO3 under phase-transfer conditions (Scheme S1†) yielded the corresponding alkene (E/Z) intermediates in moderate to high yields. The chemical structures of the synthesized starting materials, intermediates and target monomers were well characterized by means of 1H, 19F, and 31P NMR spectroscopy (Fig. S1–S3†).The introduction of quaternary ammonium cations via flexible alkyl spacers was proven to be less susceptible to all alkali degradation pathways because of the high steric hindrance and electron density around the β-protons.32 Moreover, in such systems, ion conductivity is expected to be benefited by the high local mobility of cations. Previous study on small molecules revealed that alkyl spacer tethered heterocyclic quaternary ammonium cations were alkaline stable similar to the n-hexyltrimethylammonium cation and that the N,N-dimethylpiperidinium (DMP) cation was much more stable.21,23 In this work, two tert-butyloxycarbonyl protected piperidine groups were introduced via short spacers on the highly reactive 9H-position of 2,7-dichlorofluorene following the SN2 alkylation reaction (Scheme S2†). The nitrogen atom on the 4-position of the piperidine ring compared to the 1-(N) position could be advantageous in terms of high alkaline stability because all the β-protons are present in the ring to limit rotation of β-methyl and retention of the positive spacer effect. The precursor and target monomer were characterized by using 1H and 13C NMR spectra (Fig. S4 and S5†) wherein signals were assignable to the corresponding structures.
A series of precursor Cx-Pip-n terpolymers with different semi-fluoroalkyl pendant chains and target IECs were synthesized by using Ni(0)-promoted Colon's coupling of monomers 3a–c, 4b and 5. The precursor terpolymers were obtained in high yields (89–98%) (Table 1) with good solvent solubility. The tert-butoxycarbonyl (BOC) groups were removed during the purification process of the precursor polymers with hydrochloric acid, as confirmed by 1H NMR spectra. Fig. S6† displays the 1H and 19F NMR spectra of aryl ether linkage free Cx-Pip-n terpolymers, wherein all the characteristic peaks were well assigned, and the integral ratios were in accordance with the terpolymer composition. The composition (m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n:o) calculated from the integral of the aliphatic signals (δ = 0.58–1.14, δ = 1.40–1.82, and δ = 2.80–2.92) to that of the aromatic region signals (δ = 7.3–8.0) is tabulated in Table 1. The GPC data for the Cx-Pip-n precursor terpolymers were not available because of the interaction of –NH groups with our GPC columns. The intrinsic viscosity (η) of the terpolymers in DMAc at 25 °C ranged between 0.36 and 0.77 dL g−1, indicating moderate to high molecular weight.
:n:o) calculated from the integral of the aliphatic signals (δ = 0.58–1.14, δ = 1.40–1.82, and δ = 2.80–2.92) to that of the aromatic region signals (δ = 7.3–8.0) is tabulated in Table 1. The GPC data for the Cx-Pip-n precursor terpolymers were not available because of the interaction of –NH groups with our GPC columns. The intrinsic viscosity (η) of the terpolymers in DMAc at 25 °C ranged between 0.36 and 0.77 dL g−1, indicating moderate to high molecular weight.
| Sample | Compositiona | Compositionb | Yieldc (%) | IEC (meq g−1) | η (dL g−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M | n | o | m | n | o | Target | NMRd | Titrated | |||
| a Calculated from the feed monomer ratio (molar ratio). b Determined from 1H NMR spectra of Cx-Pip-n precursor terpolymers. c Precursor copolymer yield. d Calculated from 1H NMR spectra of Cx-QPip-n. e Measured by extrapolating ηred and ηinh to zero concentration. | |||||||||||
| C7-QPip-1.82 | 1.00 | 0.20 | 0.74 | 1.00 | 0.18 | 0.66 | 92 | 2.10 | 1.92 | 1.82 ± 0.04 | 0.38 |
| C9-QPip-1.85 | 1.00 | 0.20 | 0.78 | 1.00 | 0.19 | 0.74 | 89 | 2.10 | 2.06 | 1.85 ± 0.02 | 0.36 |
| C11-QPip-1.86 | 1.00 | 0.20 | 0.82 | 1.00 | 0.19 | 0.74 | 98 | 2.10 | 2.01 | 1.86 ± 0.02 | 0.76 |
| C11-QPip-2.29 | 1.00 | 0.20 | 1.07 | 1.00 | 0.21 | 0.99 | 94 | 2.40 | 2.30 | 2.29 ± 0.02 | 0.46 |
| C11-QPip-2.87 | 1.00 | 0.20 | 1.90 | 1.00 | 0.19 | 1.76 | 95 | 3.00 | 2.94 | 2.87 ± 0.01 | 0.77 |
The quaternization reaction was performed using dimethyl sulfate in DMAc solvent under mild reaction conditions (at 50 °C for 48 h). Cx-QPip-n terpolymers were obtained in reasonable yields (82–93%) and were soluble in both high (DMAc, DMSO, and NMP) and low boiling solvents (MeOH) but insoluble in less polar solvents such as chloroform. The pendant semi-fluoroalkyl chain was responsible for providing Cx-QPip-n terpolymers with solvent solubility especially in low boiling polar solvents. The chemical structure of quaternized terpolymers was analyzed by means of 1H and 19F NMR spectroscopy (Fig. 1). The chemical shifts for arylene protons were observed between 7.3 and 8.2 ppm, and signals originated from the piperidinium ring and methyl protons appeared between 2.7 and 3.2 ppm, which both indicate successful and quantitative quaternization of the precursor terpolymers. This was further verified by the fact that IEC values calculated from the terpolymer composition were comparable to those determined by using 1H NMR spectra (Table 1). The titrated IECtit (measured by Mohr titration) was slightly lower than the IECNMR (Table 1), possibly because the pendant semi-fluoroalkyl chain created a highly hydrophobic environment around the piperidinium cations which could have hindered access of water and ionic species to some of the quaternary nitrogen atoms. Henceforth, we refer to the titrated IEC values. The 19F NMR spectra did not show any significant changes after the quaternization reaction compared to those of the precursor terpolymers.
 | ||
| Fig. 1 1H (left) and 19F (right) NMR spectra of Cx-QPip-n copolymers tethered with different semi-fluoroalkyl chains recorded in DMSO-d6 at 80 °C. | ||
Morphology of membranes
The phase-separated morphology of the membranes was analyzed by using TEM images of tetrachloroplatinate stained samples. TEM micrographs of Cx-QPip-n membranes are presented in Fig. 2, wherein dark areas represent ionic domains while bright areas are associated with non-ionic domains. A well-developed hydrophilic-hydrophobic microphase separated morphology was observed for all membranes. For detailed investigation, hydrophilic and hydrophobic domain sizes were measured and plotted as a function of IEC as shown in Fig. 3a and b and the histogram of hydrophilic–hydrophobic domain distribution fitted with normal distribution is displayed in Fig. S7.† The mean hydrophilic domain size for C7-QPip-1.82, C9-QPip-1.85, C11-QPip-1.86, C11-QPip-2.29, and C11-QPip-2.87 membranes was 1.21 ± 0.33, 1.33 ± 0.42, 1.44 ± 0.41, 1.40 ± 0.32, and 1.42 ± 0.35 nm, respectively, averaged from 100 spots. The results indicate that the hydrophilic domain size was not dependent on the IEC but more likely related to the pendant chain length. The mean hydrophilic domain size is replotted as a function of the number of carbon atoms in the semi-fluoroalkyl chain as shown in Fig. S8.† The hydrophilic domain size for membranes with comparable IECs exhibited an approximate linearly relationship to the number of carbon atoms in the semi-fluoroalkyl chain. | ||
| Fig. 2 TEM images of Cx-QPip-n membrane samples stained with tetrachloroplatinate ions. | ||
 | ||
| Fig. 3 Hydrophilic (a) and hydrophobic (b) domain size for [PtCl4]−2 ions stained Cx-QPip-n membrane samples measured from the TEM images and averaged by 100 spots (domain size expressed in mean diameter with standard deviations). | ||
The hydrophobic domain size decreased on increasing the IEC; the mean hydrophobic domain size of C11-QPip-1.86, C11-QPip-2.29, and C11-QPip-2.87 membranes was 3.57 ± 0.79, 3.08 ± 0.66, and 2.77 ± 0.62 nm, respectively, averaged from 100 spots. The hydrophobic domain size decreased with increasing IEC for C11-Qpip-n membranes, probably because of a relatively low hydrophobic comonomer ratio (Table 1). The hydrophobic domain size is also replotted as a function of the number of carbon atoms in the semi-fluoroalkyl pendant as shown in Fig. S8,† where a nearly linear relationship was observed for membranes with comparable IECs, similar to the hydrophilic domain size. These results suggest that the highly hydrophobic semi-fluoroalkyl groups contributed to the development of the phase-separated morphology.
The humidity dependent SAXS pattern of Cx-QPip-n membranes in the chloride counter anion form was obtained at 40 °C (Fig. S9†). The SAXS pattern of C7-QPip-1.82 and C9-Qpip-1.85 membranes was similar and did not show any ionomer peaks. For the C11-QPip-1.86 membrane, a wide peak developed at q = ca. 0.9–1.2 nm−1 corresponding to a d-spacing of 5.2–7.0 nm. A minor (C11-QPip-1.86) or no (C7-QPip-1.82 and C9-QPip-1.85) ionomer peak was possibly due to the bulky nature of the short spacer tethered piperidinium cation which might have restricted the local mobility and thereby hindered the development of homogeneous morphology and efficient ion clustering. Since these peaks were not sensitive to humidity, the morphology must be related to the hydrophobic clusters.
Water uptake, swelling behavior, and hydroxide ion conductivity
Water absorbability is a crucial property to realize high hydroxide ion conductivity for AEMs, since water promotes ionic dissociation and develops ion transporting hydrophilic pathways across the membranes. However, excess water absorption undermines the conductivity by diluting charge carriers. In addition, mechanical properties are often compromised at high water uptake accompanied by high dimensional changes and swelling. To evaluate the effect of the chemical structure on swelling behavior of the membrane samples, the water uptake and swelling ratio were compared among the membranes formulated with different semi-fluoroalkyl pendants at comparable IECs. The membrane samples with different semi-fluoroalkyl pendants showed comparable water uptake (Fig. 4a), indicating that water absorbability more likely relies on IEC than the semi-fluoroalkyl pendant length. This is further verified by replotting the water uptake as a function of the semi-fluoroalkyl length and composition of the hydrophilic component in the terpolymers (Fig. S10a and b†). The effect of semi-fluoroalkyl side chains on water uptake at higher temperature (80 °C) was further investigated under humidified conditions (Fig. S11†). The water absorption of Cx-QPip-n membranes was lower than that in liquid water. A recent report by Xue et al. on poly(p-terphenyl dimethylpiperidinium) membranes also demonstrated a similar phenomenon of water absorption in liquid water and under humidified conditions.33 The water uptake of Cx-QPip-n membranes with different semi-fluoroalkyl pendants was very similar at each humidity investigated. These results further emphasize strong reliance of water absorption in Cx-QPip-n membranes on IEC rather than the semi-fluoroalkyl pendant length. In general, water uptake increased with increase in IEC. C11-QPip-n membranes followed this tendency and the water uptake increased from 57% to 133% with an increase in IEC from 1.86 to 2.87 meq g−1. The level of hydration in AEMs is typically illustrated by the number of water molecules absorbed per functional group (hydration number, λ) which is critical for ion conduction and alkaline stability. Similar to water uptake values, λ increased approximately linearly with IEC (Fig. 4a and b). Earlier findings demonstrated that higher λ benefited the alkaline stability of AEMs significantly.34–36 High alkaline stability for Cx-QPip-n membranes could be expected since λ values for Cx-QPip-n membranes ranged between 17 and 26. | ||
| Fig. 4 Water uptake (a), water molecules adsorbed per unit piperidinium cation (λ) (b), swelling ratio (c), and hydroxide conductivity at 30 °C (d) as a function of IEC for Cx-QPip-n membranes. | ||
Swelling behavior of Cx-QPip-n membranes at 30 °C was analyzed in liquid water. The volumetric swelling ratio of Cx-QPip-n membranes with comparable IEC (and thus comparable water uptake) was in the order of C7-QPip-1.82 (35.5 ± 0.5%) > C9-QPip-1.85 (32.2 ± 2.0%) > C11-QPip-1.86 (28.0 ± 1.3%) (Fig. 4c), indicating that the swelling was influenced by the semi-fluoroalkyl pendant length. The volumetric swelling ratio is replotted as a function of the number of carbon atoms in the semi-fluoroalkyl pendant in Fig. S12.† The results indicate that the interpolymer interactions would be strengthened with lengthening of the semi-fluoroalkyl pendant chain which suppressed swelling of the membranes. The swelling of C11-QPip-n membranes increased with IEC and showed an approximate linear relationship with IEC similar to water uptake (Fig. 4a and c).
The hydroxide ion conductivity of Cx-QPip-n membranes at 30 °C as a function of their IECs is displayed in Fig. 4d. Among Cx-QPip-n membranes with comparable IECs (ca. 1.85 meq g−1) and nearly the same water uptake, the C11-QPip-1.86 membrane showed the highest hydroxide ion conductivity of 53 mS cm−1, followed by C9-QPip-1.85 (45 mS cm−1) and C7-QPip-1.82 (40 mS cm−1), indicating that the lengthening of the semi-fluoroalkyl pendant improved the conductivity. The conductivity is replotted as a function of the number of carbon atoms in the semi-fluoroalkyl pendant and an approximate linear relationship was observed (Fig. S13†). Wu et al. also reported that the lengthening of perfluoroalkyl side chains significantly improved hydroxide ion conductivity, dimension stability, and microphase separation in FPAP membranes.29 A rather large hydrophilic domains size as a result of improved microphase separation for the C11-QPip-1.86 membrane tied for the high hydroxide conductivity.
For deeper understanding of the effect of the pendant length on the ion conductivity, the hydroxide ion diffusion coefficient (Dσ) was calculated for the Cx-QPip-n membranes. The normalized diffusion coefficient (Dσ/D0, where Dσ and D0 represent the diffusion coefficient of hydroxide ions in the membranes and in infinite dilution, respectively) is plotted as a function of λ in Fig. 5. In general, membranes with a higher level of hydration (λ) exhibit more facile ion transport and thus, greater Dσ/Do values. C11-QPip-n membranes followed this general trend and exhibited an approximately linear relationship with λ. The C11-QPip-1.86 membrane showed a relatively high Dσ/D0 value compared to the other Cx-QPip-n membranes at comparable IEC, demonstrating more facile hydroxide ion mobility in this membrane owing to the longer pendant semi-fluoroalkyl chain which might have facilitated the formation of more continuous, interconnected water-rich ion conducting channels. We assume that immiscibility of the main chain tethered with a long hydrophobic semi-fluoroalkyl chain and short spacer tethered hydrophilic piperidinium groups created nanoscale ionic domains and that these ionic domains might have facilitated and enhanced the mobility of the hydroxide ion in the membrane with water molecules. There were some earlier reports that demonstrated that immiscibility of hydrophobic main chain/grafted aliphatic chains with hydrophilic segments benefitted hydroxide ion conductivity by driving the formation of nanoscale ionic domains.37–39
 | ||
| Fig. 5 Volumetric IEC (IECvol) and normalized diffusion coefficient (Dσ/D0) as a function of water molecules adsorbed per unit piperidinium cation (λ). | ||
The volumetric IEC (IECvol) (molar concentration of the piperidinium cation per unit volume of the wet membrane; meq cm−3) as a function of λ is also displayed in Fig. 5. Generally, high bulk IEC membranes exhibit high water uptake and IECvol increases with increase in bulk IEC as long as membranes do not absorb excess water. C11-QPip-2.29 and C11-QPip-2.87 membranes with significantly high bulk IEC showed relatively low IECvol compared to the C11-QPip-1.86 membrane, indicating that their high water uptake and volumetric swelling apparently diluted the molar concentration of the piperidinium cations. For the membranes with comparable bulk IEC and nearly the same water uptake, IECvol followed a similar trend with λ to Dσ/D0. The hydroxide ion conductivity for the comparable IEC membranes (ca. 1.85 meq g−1) was well-correlated with the IECvol. For detailed investigation, the hydroxide ion conductivity is replotted as a function of IECvol in Fig. S14a.† The Cx-QPip-n membrane with comparable IEC (ca. 1.85 meq g−1) showed an approximately linear relationship of hydroxide ion conductivity and Dσ/D0 with IECvol (Fig. S14a and b†). The hydroxide ion conductivity and Dσ/D0 for C11-QPip-n membranes were not well-correlated with IECvol. C11-QPip-2.29 and C11-QPip-2.87 membranes showed significantly higher conductivity than the C11-QPip-1.86 membrane despite their low IECVol. C11-QPip-2.29 and C11-QPip-2.87 membranes exhibited similar IECvol (1.24 meq cm−3) to C7-QPip-1.82 and C9-QPip-1.85 membranes but their hydroxide ion conductivities were significantly high, suggesting that the hydroxide ion conductivity was more relevant to ion diffusion rather than IECvol.
Hydroxide ion conductivity increased with an increase in temperature and all membranes showed an approximate Arrhenius-type temperature dependence of the conductivity from 30 °C to 80 °C (Fig. 6). The highest conductivity of 112 mS cm−1 was realized for the C11-QPip-1.86 membrane at 80 °C. Allushi et al. reported that the DMP functionalized membrane labelled PDPF-DMP-1.8 showed 99 mS cm−1 at 80 °C (IEC = 1.80 meq g−1).40 Chu et al. reported that poly(phenylene oxide)-based PPO-DMP-40 membranes with an IEC of 1.98 meq g−1 showed a hydroxide ion conductivity of 71.8 mS cm−1 at 80 °C.41 In the present study, pendant semi-fluoroalkyl chains contributed to higher ion conductivity for membranes with moderate IEC and water uptake. As expected, the conductivity increased with an increase in IEC and 146 mS cm−1 (at 80 °C) was achieved for the C11-QPip-2.87 membrane at IEC = 2.87 meq g−1. Though the C11-QPip-2.87 membrane was the most conductive among all the membranes investigated, its dimensional changes were significantly high (Fig. 4c). The activation energy (Ea) for hydroxide ion conduction was calculated from the slope of the linear lines. The obtained values were comparable and ranged between 12.07 and 13.65 kJ mol−1, which implies that all membranes shared the same water-mediated hydroxide ion conduction mechanism.
 | ||
| Fig. 6 Arrhenius-type thermal dependence of the ion conductivity on temperature for C7-QPip-1.82, C9-QPip-1.85, and C11-QPip-n membranes. | ||
Mechanical properties
The viscoelastic properties (storage modulus (E′), loss modulus (E′′), and tanδ (E′′/E′)) were evaluated as a function of temperature and relative humidity at 60% RH and 80 °C, respectively (Fig. 7). The membrane samples in chloride counter anion forms were used for analysis to avoid carbonation in the samples. In relative humidity dependent analysis, despite comparable IECs (ca. 1.85 meq g−1), Cx-QPip-n membranes exhibited different storage moduli at 0% RH, and E′ decreased on increasing the humidity. At ca. 80 °C and ∼0% RH, the E′ values for C7-QPip-1.82, C9-QPip-1.85, and C11-QPip-1.86 were 1.25, 1.48, and 1.72 GPa, respectively while those at ca. 80 °C and 90% RH were 0.50, 0.55, and 0.57 GPa, respectively. The results indicate that the viscoelastic properties were strongly dependent on the pendant semi-fluoroalkyl chain length. E′ and E′′ for Cx-QPip-n membranes at comparable IECs were plotted as a function of the semi-fluoroalkyl pendant length in Fig. S15.† Both storage and loss moduli increased with an increase in pendant length and exhibited a linear relationship with the pendant length at ca. 80 °C and 0% RH. Similarly, the membranes with different IECs formulated with the same semi-fluoroalkyl pendant showed comparable storage and loss moduli at ca. 80 °C and 90% RH. Regarding the temperature dependence, a slight decrease in E′ was observed while none of the membranes showed glass transition behavior in the tanδ curves.
 | ||
| Fig. 7 DMA curves of Cx-QPip-n membranes as a function of temperature at 60% RH (left) and relative humidity at 80 °C (right). | ||
The tensile tests were performed at 80 °C and 60% RH for the membranes in the chloride counter anion form (Fig. 8). Generally, membranes with higher IEC exhibit reduced tensile strength with a larger elongation at break. In the present study, however, the IEC values had small impact on the tensile properties of C11-QPip-n membranes. The membranes with comparable IECs (ca. 1.85 meq g−1) showed significantly different tensile strength and elongation at break. Relatively high tensile strength and elongation at break were observed for the membranes with a longer semi-fluoroalkyl chain and the effect was more pronounced when the chain lengthened to C11. The tensile strength and elongation at break increased to 29.1 MPa and 124% from 21.4 MPa and 50%, respectively, when the semi-fluoroalkyl chain lengthened to C11 from C7. Significantly high values for tensile strength and elongation at break for the C11-QPip-1.86 membrane must be the result of increased entanglement of the longer semi-fluoroalkyl pendant in the polymer matrix. This was verified by replotting tensile strength and elongation at break as a function of the semi-fluoroalkyl pendant length in Fig. S16.† The tensile strength and elongation at break increased with pendant length up to C9 and a sharp jump was observed when the chain was lengthened to C11. Overall, taking water uptake, dimensional changes, and mechanical properties into consideration, the C11-QPip-1.86 membrane exhibited the most-balanced properties among the membranes discussed here.
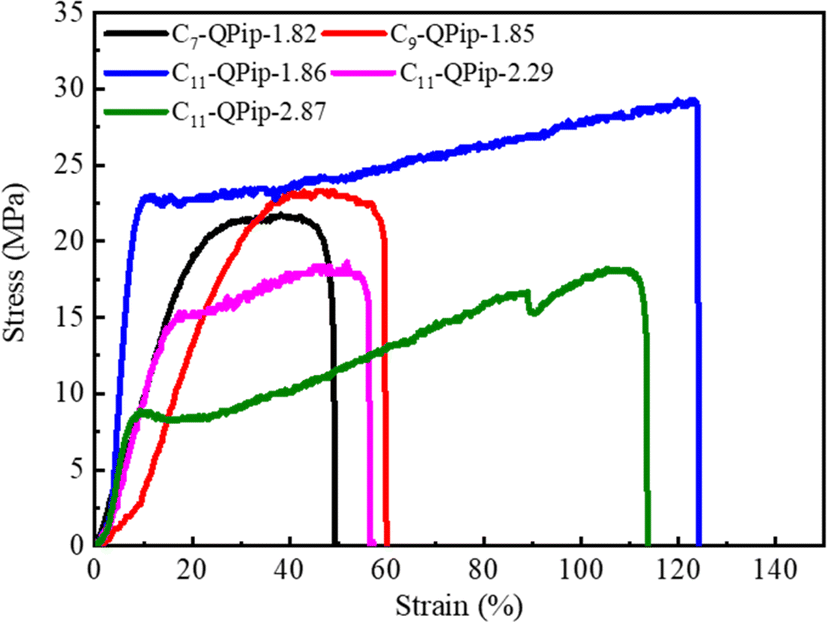 | ||
| Fig. 8 Stress versus strain curves for C7-QPip-1.82, C9-QPip-1.85, and C11-QPip-n membranes in the chloride counter anion form at 60% RH and 80 °C. | ||
Alkaline stability
The ex situ alkaline stability of Cx-QPip-n membranes was assessed in 8 M KOH at 80 °C, and the change in hydroxide ion conductivity with time was monitored at 30 and 80 °C (Fig. 9 and S17†). Over a test period of 1056 h, the remaining conductivity (at 30 °C) was in the order of C11-QPip-1.86 (75%; 39 mS cm−1) ≅ C11-QPip-2.29 (74%; 47 mS cm−1) > C9-QPip-1.85 (69%; 31 mS cm−1) > C7-QPip-1.82 (64%; 26 mS cm−1), indicating that the longer semi-fluoroalkyl pendants contributed slightly to better alkaline stability of the membranes. Slightly more pronounced hydrophobic–hydrophilic microphase separation with a longer pendant would be accountable. Zhu et al.42 also claimed that the development of hydrophobic–hydrophilic phase separation improved alkaline stability significantly. The C11-QPip-2.87 membrane with higher IEC survived up to 400 h; however, further testing was not continued because of excessive swelling. | ||
| Fig. 9 Change in hydroxide ion conductivity at 30 °C for Cx-QPip-n membranes during the alkaline stability test in 8 M KOH at 80 °C. | ||
The structural changes in the post-test membranes were scrutinized by using 1H NMR and 19F NMR spectra. From the conductivity loss, the degradation of the piperidinium cations was anticipated. In the 1H NMR spectra (Fig. 10 left), there were no changes observed in the aromatic region indicating alkaline stability of the main chain. Instead, new signals appeared at 4.0–5.2 ppm in which the intensity of the peaks of δ = 4.08, δ = 4.35, and δ = 5.17 ppm was nearly identical, suggesting the formation of vinyl groups via ring-opening Hofmann elimination in the piperidinium cations. Other new signals were also detected at δ = ∼2.2 and δ = ∼2.7 (overlapped with the signals of proton 7), suggesting the formation of tertiary amine groups either by nucleophilic substitution at α-carbon or by Hofmann β-elimination in the piperidinium cations. This was further verified by the fact that the peaks 7 and 8 became smaller and peaks 5 and 6 became larger compared to the peaks of the aromatic protons. For more quantitative analysis, IEC values were estimated from the 1H NMR spectra and Mohr titration method. The total ionic loss from both degradation pathways was estimated by comparing changes in integrals of the peaks 7, 8, and 8′ for pristine and post-test samples to those of the aromatic proton peaks. The total ionic loss estimated by 1H NMR for C7-QPip-1.82, C9-QPip-1.85, C11-QPip-1.86, and C11-QPip-2.29 was 29%, 31%, 28%, and 32%, respectively. This was further verified by the fact that IEC values for post-test samples calculated from the Mohr titration method were comparable to those determined by using 1H NMR spectra (Fig. S18†). Despite the loss of the ionic groups, the post-test membranes remained soluble in both high boiling (DMAc and DMSO) and low boiling (methanol) polar solvents.
Apart from the degradation of the piperidinium cations, the alkyl spacer (protons 1–3 (Fig. 1)) between the aryl moiety and fluoroalkyl chain underwent HF elimination (dehydrofluorination reaction). In the post-test 1H and 19F NMR spectra, new peaks appeared at δ = 5.3 ppm and δ = −116 ppm assignable to –CH = CF– groups. Notably, the semi-fluoroalkyl chain was not removed with the HF elimination reaction as confirmed with the 19F NMR spectra (Fig. 10 right).
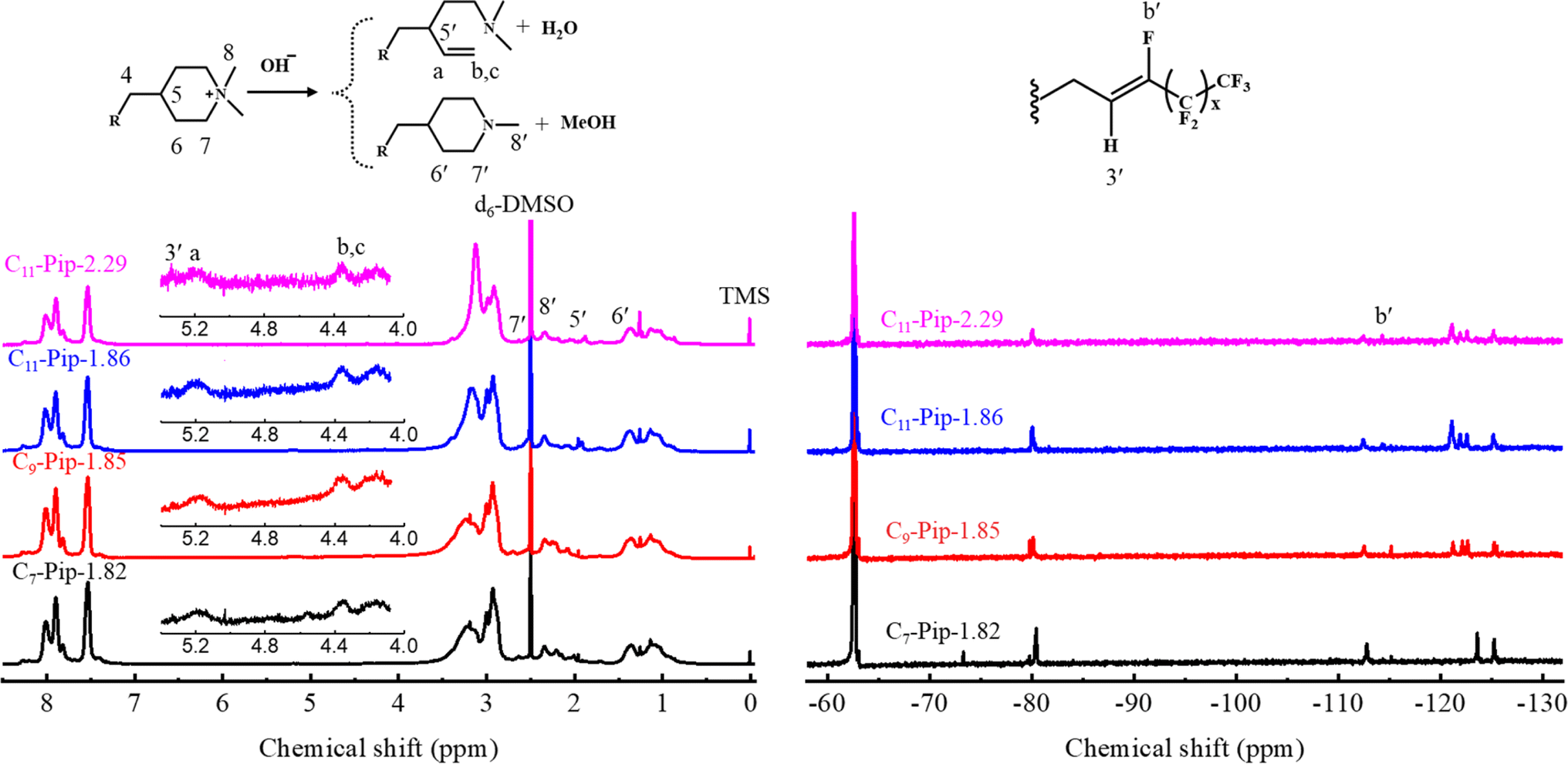 | ||
| Fig. 10 1H (left) and 19F (right) NMR spectra of Cx-QPip-n membranes in the chloride counter anion form after alkali-treatment for 1056 h in 8 M KOH at 80 °C. | ||
The post alkaline stability test morphology was examined for C7-QPip-1.82, C9-QPip-1.85, and C11-QPip-1.86 membranes through TEM images (Fig. S19†) wherein hydrophilic/hydrophobic phase separation remained for all samples. The mean hydrophilic domain size was only slightly decreased to 0.78 ± 0.19, 0.98 ± 0.17, and 1.11 ± 0.21 nm for C7-QPip-1.82, C9-QPip-1.85, and C11-QPip-1.86 membranes, respectively. The morphological data are in fair agreement with the changes in hydroxide ion conductivity and IEC (NMR and titration).
The post alkaline stability test mechanical properties were compared with those of the pristine samples (Fig. S20†). The post-test membranes showed smaller elongation at break due to smaller water absorbability accompanied by the ionic loss. The tensile strength was found to be comparable or slightly higher except for the C7-QPip-1.82 membrane, supporting the alkaline stability of the main chain.
Alkaline water electrolysis cell performance
Taking mechanical and alkaline stabilities and hydroxide ion conductivity into account, the C11-QPip-1.86 membrane (50 μm thick) was considered as the polymer electrolyte for alkaline water electrolysis cell performance evaluation. In-house developed Ni0.8Co0.2O30 and Pt/C catalysts were used for the anode and cathode, respectively, and QPAF-4 (ref. 31) (IEC = 1.50 meq g−1) was used as the ionomer for both electrodes which was also synthesized in our laboratory. Alkaline water electrolysis cell performance and ohmic resistance are displayed in Fig. 11(a). The onset voltage was 1.43 V, which was close to that in our earlier reports using the same catalyst layers23,43 and different membranes and that obtained by the rotating disk electrode for the same Ni0.8Co0.2O catalyst.30 The cell voltage increased with the increase in the current density to 1.64 V at 1.0 A cm−2 and to 1.77 V at 2.0 A cm−2. The voltage efficiency at 1.0 A cm−2 was 75% based on the theoretical voltage (1.23 V) for the electrochemical water splitting reaction. The ohmic resistance was nearly constant from 0 to 2.0 A cm−2. The average resistance was 0.086 Ω cm2 which was higher than that (0.045 Ω cm2) calculated from the conductivity (112 mS cm−1 at 80 °C) and the thickness of the C11-QPip-1.86 membrane, indicating that the interfacial resistance was included. The AEMWE performance of the C11-QPip-1.86 membrane (1.64 V at 1.0 A cm−2) was similar to that of our previously reported AEMWEs using a 37 μm thick QBM-2.7 membrane (1.62 V at 1.0 A cm−2)23 or 57 μm thick xQPAF-C3-VB (1.67 V at 1.0 A cm−2)43 membranes. The anode (Ni0.8Co0.2O) and cathode (Pt) catalysts, ionomer (QPAF-4), and MEA preparation procedure (including PTL and CCM) were the same for these three cells. The IEC of the QPAF-4 binder was 1.50 meq g−1 for the cells with xQPAF-C3-VB and C11-QPip-1.86 membranes but 2.00 meq g−1 for the cell with the QBM-2.7 membrane for better compatibility. The cell performance of the C11-QPip-1.86 membrane is compared with that in some of the recent reports (at 1.0 A cm−2 with 1 M KOH feed at 80 °C, Pt/C cathode catalyst, Table S1†). The voltage of the C11-QPip-1.86 cell was the lowest among the cells using ca. 50 μm thick membranes, and even comparable to or lower than that of the cells using a thinner (25 μm thick) membrane, partly because of lower cell resistance owing to higher ion conductivity of the C11-QPip-1.86 membrane. | ||
| Fig. 11 Water electrolysis cell performance and ohmic resistance (a) and durability at a constant current density of 1.0 A cm−2 (b) for the AEMWE cell assembled with the C11-QPip-1.86 membrane with 1 M KOH using Ni0.8Co0.2O (anode) and Pt/C (cathode) catalysts at 80 °C. | ||
The in situ durability of the AEMWE cell was assessed at a constant current density of 1.0 A cm−2 and 80 °C as displayed in Fig. 11(b). The cell potential increased from 1.62 V to 1.74 V during the initial 200 h presumably due to structural changes in the surface oxides of the OER catalyst.22,29 In the following 1000 h, the cell voltage was maintained at 1.74–1.77 V with a minor voltage decay of 28 μV h−1. Promising durability of the AEMWE cell was ascribed to the membrane stability as cell resistance increased only slightly to 0.12 Ω cm2 from 0.08 Ω cm2 during 1000 h operation.
Conclusions
We have designed and synthesized poly(arylene piperidinium) terpolymers with pendant semi-fluoroalkyl chains to evaluate the effect of the length of semi-fluoroalkyl pendants on physical and electrochemical properties of the resulting AEMs. The semi-fluoroalkyl pendant lengthening strategy not only lowered the swelling but also promoted hydrophobic–hydrophilic microphase separation in AEMs, leading to ion conductivity enhancement. Moreover, tensile strength, elongation at break, and storage modulus increased by 26, 60, and 27%, respectively, when the pendant chain lengthened to C11 from C7. The good solvent solubility of Cx-QPip-n terpolymers in methanol at room temperature is another advantage of the terpolymers with longer semi-fluoroalkyl pendants. Under very harsh alkaline conditions (in 8 M KOH at 80 °C for 1056 h), Cx-QPip-n membranes retained 64 to 75% of their initial conductivity and ionic loss was attributed to both Hofmann β-elimination and nucleophilic substitution. A high voltage efficiency of 75% and performance (1.64 V at 1.0 A cm−2) with a PGM-free anode catalyst (Ni0.8Co0.2O) were realized in an alkaline water electrolysis cell assembled with the C11-QPip-1.86 membrane. The electrolysis cell exhibited promising in situ durability at 1.0 A cm−2 and 80 °C for 1000 h with a voltage decay of 28 μV h−1.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Vikrant Yadav: investigation and writing. Kenji Miyatake: supervision, writing, reviewing and editing, funding acquisition, and project administration. Ahmed Mohamed Ahmed Mahmoud: reviewing. Fanghua Liu: reviewing. Fang Xian: reviewing. Lin Guo: reviewing. Chun Yik Wong: reviewing. Toshio Iwataki: investigation. Makoto Uchida: reviewing. Katsuyoshi Kakinuma: investigation and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the New Energy and Industrial Technology Development Organization (NEDO), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, through Grants-in-Aid for Scientific Research (22F22035, 23KF0037, and 23H02058), MEXT Program: Data Creation and Utilization Type Material Research and Development Project (JPMXP1122712807), JST (GteX JPMJGX23H2), and JKA promotion funds from KEIRIN RACE. VY and CYW are thankful for JSPS postdoctoral fellowships for research in Japan.References
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- D. Hua, J. Huang, E. Fabbri, M. Rafique and B. Song, ChemElectroChem, 2023, 10, e202200999 CrossRef CAS.
- N. DU, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- A. Khataee, A. Shirole, P. Jannasch, A. Krüger and A. Cornell, J. Mater. Chem. A, 2022, 10, 16061–16070 RSC.
- C. Hu, J. Y. Lee, Y. J. Lee, S. H. Kim, H. Hwang, K. S. Yoon, C. Park, S. Y. Lee and Y. M. Lee, Next Energy, 2023, 1, 100044 CrossRef.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. D. Noto, L. Elbaz, D. R. Dekel and F. Jaouen, ChemSusChem, 2022, 15, e202200027 CrossRef CAS PubMed.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhuo, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- L. Xiao, S. Zhang, J. Pan, C. X. Yang, M. L. He, L. Zhuang and J. T. Lu, Energy Environ. Sci., 2012, 5, 7869–7871 RSC.
- M. S. Cha, E. J. Park, S. Kim, S. H. Han, S. H. Shin, S. H. Yang, T. H. Kim, D. M. Yu, S. Y. So, Y. T. Hong, S. J. Yoon, S. G. Oh, S. Y. Kang, O. H. Kim, H. S. Park, B. Bae, Y. E. Sung, Y. H. Cho and J. Y. Lee, Energy Environ. Sci., 2020, 13, 3633–3645 RSC.
- M. Mandal, ChemElectroChem, 2020, 8, 36–45 CrossRef.
- C. Liu, Z. Geng, X. Wang, W. Liu, Y. Wang, Q. Xia, W. Li, L. Jin and C. Zhang, J. Energy Chem., 2024, 90, 348–369 CrossRef CAS.
- Y. Ozawa, Y. Shirase, K. Otsuji and K. Miyatake, Mol. Syst. Des. Eng., 2022, 7, 798–808 RSC.
- A. M. A. Mahmoud and K. Miyatake, ACS Appl. Polym. Mater., 2023, 5, 2243–2253 CrossRef.
- P. M. Bakvand and P. Jannasch, J. Membr. Sci., 2023, 668, 121229 CrossRef.
- E. J. Park, P. Jannasch, K. Miyatake, C. Bae, K. Noonan, C. Fujimoto, S. Holdcroft, J. R. Varcoe, D. Henkensmeier, M. D. Guiver and Y. S. Kim, Chem. Soc. Rev., 2024, 53, 5704–5780 RSC.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52(9), 2745–2755 CrossRef CAS PubMed.
- S. Haj-Bsoul, J. R. Varcoe and D. R. Dekel, J. Electroanal. Chem., 2022, 908, 116112 CrossRef CAS.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS.
- M. G. Marino and K. D. Kruer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- K. Zhang, W. Yu, L. Wu and T. Xu, J. Membr. Sci., 2023, 678, 121672 CrossRef CAS.
- J. Sharma and V. Kulshrestha, Alkaline Anion Exchange Membranes for Fuel Cells: From Tailored Materials to Novel Applications, 2024, pp. 241–284 Search PubMed.
- Y. Ozawa, T. Iwataki, M. Uchida, K. Kakinuma and K. Miyatake, J. Mater. Chem. A, 2023, 11, 19925–19935 RSC.
- T. Kimura, A. Matsumoto, J. Inukai and K. Miyatake, ACS Appl. Energy Mater., 2020, 3(1), 469–477 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- H. S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 RSC.
- G. Xu, J. Pan, X. Zuo, J. Jin, J. Zhang, P. Fang, Q. Zhang, Z. Sun and F. Yan, Adv. Funct. Mater., 2023, 33, 2302364 CrossRef CAS.
- J. Huang, Z. Yu, J. Tang, P. Wang, X. Zhang, J. Wang and X. Lei, Fuel, 2024, 357, 129686 CrossRef CAS.
- X. Wu, N. Chen, C. Hu, H. A. Klok, Y. M. Lee and X. Hu, Adv. Mater., 2023, 35, 2210432 CrossRef CAS PubMed.
- G. Shi, T. Tano, D. A. Tryk, M. Yamaguchi, A. Iiyama, M. Uchida, K. Iida, C. Arata, S. Watanabe and K. Kakinuma, ACS Catal., 2022, 12(22), 14209–14219 CrossRef CAS.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- J. Xue, J. C. Couglin, K. Yassin, T. Huang, H. Jiang, J. Zhang, Y. Yin, D. R. Dekel and M. D. Guiver, Joule, 2024, 8, 1457–1477 CrossRef CAS.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29(10), 4425–4431 CrossRef CAS.
- S. Willdorf-Cohen, A. Zhegur-Khais, J. Ponce- González, S. Bsoul-Haj, J. R. Varcoe, C. E. Diesendruck and D. R. Dekel, ACS Appl. Energy Mater., 2023, 6(2), 1085–1092 CrossRef CAS PubMed.
- K. Yassin, I. G. Rasin, S. Brandon and D. R. Dekel, J. Membr. Sci., 2024, 690, 122164 CrossRef CAS.
- N. Li, Y. Leng, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124–10133 CrossRef CAS PubMed.
- N. Li, T. Yan, Z. Li, T. Thomas-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888–7892 RSC.
- C. X. Lin, X. L. Huang, D. Guo, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2016, 4, 13938–13948 RSC.
- A. Allushi, P. M. Bakvand and P. Jannasch, Macromolecules, 2023, 56, 1165–1176 CrossRef CAS.
- X. Chu, L. Liu, Y. Huang, M. D. Guiver and N. Li, J. Membr. Sci., 2019, 578, 239–250 CrossRef CAS.
- L. Zhu, T. J. Zimudzi, Y. Wang, X. Yu, J. Pan, J. Han, D. I. Kushner, L. Zhuang and M. A. Hickner, Macromolecules, 2017, 50(6), 2329–2337 CrossRef CAS.
- A. M. A. Mahmoud, K. Miyatake, F. Liu, V. Yadav, L. Guo, C. Y. Wong, T. Iwataki, K. Kakinuma and M. Uchida, Adv. Energy Sustainability Res., 2024, 5, 2300236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03630a |
| This journal is © The Royal Society of Chemistry 2024 |