
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow CO2 mass transfer promotes methanol and formaldehyde electrosynthesis on cobalt phthalocyanine†
Jie
Zhang
a,
Thi Ha My
Pham
b,
Shibo
Xi
c,
Liping
Zhong
 b,
David
Liem
a,
Futian
You
a,
Ben
Rowley
d,
Ramesha
Ganganahalli
e,
Federico
Calle-Vallejo
*fg and
Boon Siang
Yeo
*a
b,
David
Liem
a,
Futian
You
a,
Ben
Rowley
d,
Ramesha
Ganganahalli
e,
Federico
Calle-Vallejo
*fg and
Boon Siang
Yeo
*a
aDepartment of Chemistry, Faculty of Science, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmyeos@nus.edu.sg
bLaboratory of Materials for Renewable Energy (LMER), Institute of Chemical Sciences and Engineering (ISIC), Basic Science Faculty (SB), École Polytechnique Fedérale de Lausanne (EPFL) Valais/Wallis, Energypolis, Rue de l’Industrie 17, Sion CH-1951, Switzerland
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road Jurong Island, Singapore 627833, Republic of Singapore
dShell Global Solutions International B.V., Energy Transition Campus Amsterdam, Grasweg 31, 1031 HW, Amsterdam, The Netherlands
eShell India Markets Private Ltd Plot No. 7, Bengaluru Hardware Park, Mahadeva, Kodigehalli, Bangalore, North, 562149, India
fNano-Bio Spectroscopy Group and European Theoretical Spectroscopy Facility (ETSF), Department of Advanced Materials and Polymers: Physics, Chemistry and Technology, University of the Basque Country UPV/EHU, Avenida Tolosa 72, San Sebastián 20018, Spain. E-mail: federico.calle@ehu.es
gIKERBASQUE, Basque Foundation for Science, Plaza de Euskadi 5, Bilbao 48009, Spain
First published on 21st October 2024
Abstract
Cobalt phthalocyanine supported on carbon nanotubes (CoPcCNT) usually catalyzes the electroreduction of CO2 (CO2RR) to CO, although several reports have also indicated methanol formation. Herein, by analyzing the effects of CoPc loading and CO2 partial pressure on the CO2RR, we show that a lower rate of CO2 mass transfer to each CoPc favors methanol formation, while a higher rate of CO2 mass transfer favors CO evolution. The ratio of the production rates of methanol and CO is related to the average CO2 mass transfer rate by a power function with a negative exponent. Hence, methanol can only be formed when the supply of CO2 feed is low. This mass transfer effect is supported by supplementary experiments and computational modelling, which show that CO binding to CoPc is weaker than that of CO2, in agreement with previous studies. Consequently, *CO may only be reduced to methanol when the supply of CO2 is low and the dwelling time of CO is long. We further provide a quantitative guideline for the design of methanol-selective catalysts. At −0.86 V vs. RHE, enhanced CO mass transfer boosts the CORR to methanol with a faradaic efficiency up to 70% at a total current density of −19 mA cm−2. The production of formaldehyde, a reaction intermediate from CO reduction to methanol, is also boosted with a faradaic efficiency of up to 7%. We pinpoint CoPc containing Co(I) as the active CO2RR site.
Introduction
The electrochemical reduction of CO2 or CO (CO2RR or CORR) using renewable electricity is a promising method for producing green liquid fuels and chemical feedstocks.1–3 Extensive efforts have been dedicated in recent years to understanding the CO(2)RR process, with the aim of improving its selectivity and activity to value-added, highly reduced products, such as methanol, ethylene, and ethanol.4,5 Among these products, methanol has received relatively less attention, probably due to the lack of robust catalysts and clear routes for its electrocatalytic production.6,7 Indeed, while many materials have been reported to catalyze the CO2RR to methanol, only a few of them have been proven reproducible.6,8 Recently, the groups of Wang and Robert reported that cobalt phthalocyanine (CoPc) exhibits catalytic activity for the CO2RR to methanol.9–13 Yet, interestingly, alongside these reports, numerous other research groups have observed, using the same catalyst, that CO is the sole CO2RR product.14–17 Compared with CO, methanol has a higher volumetric energy density, can be easily stored and used directly as a fuel.18 Therefore, it is of commercial and scientific interest to understand in detail the mechanism of the CO2RR to methanol using CoPc catalysts.Wang and coworkers first identified the molecular dispersion of CoPc on a carbon nanotube support (CoPcCNT) as a factor to incline the selectivity of the CO2RR toward methanol.9,19 However, other studies using similar catalysts reported CO as the only product.20–22 This suggests that the molecular dispersion of CoPc may not govern methanol selectivity alone and other factors still need to be elucidated. Along these lines, Wang's group observed that the loading of CoPc on carbon nanotubes impacts the CO2RR and CORR selectivity to methanol.11,19 They also found that such selectivity depends on CO partial pressure.23 The curvature of the carbon nanotube support and the spin state of the cobalt center have further been associated with methanol production.24,25 More recently, Ren et al. showed that blending 10% CO2 into a CO feed gas can inhibit the CORR to methanol and attributed this inhibition to the stronger binding of CO2 on CoPc, as compared to CO on CoPc.13 McCrory and coworkers further revealed, on the basis of microkinetic analyses, that CO2 binds to CoPc with an effective equilibrium constant (11.1 atm−1) that is thrice that of CO binding (3.4 atm−1).26
Herein, we show that the average CO2 mass transfer rate to each CoPc active site is the factor that determines how CoPc loading and CO2 partial pressure affect the competing reduction of CO2 to methanol and CO. We further reveal a power function (R = 0.97ν−0.78) relating the ratio (R) of the production rates of methanol and CO to the average CO2 mass transfer rate (v). Computational modelling indicates that this relation is caused by the weaker adsorption of CO compared to CO2 on CoPc, such that only when the CO2 supply falls short, can the adsorbed *CO be further reduced to methanol. Further in situ Raman and cyclic voltammetry analyses show different behaviors of CoPc in CO2- and CO- saturated electrolytes, which are likely caused by the stronger adsorption of CO2 compared to CO on CoPc. The computational modeling together with the experimental results implies that the dwelling time of *CO on CoPc is critical in determining either its further conversion to methanol or its elution as a product. Additionally, DFT modelling based on the Sabatier principle provides a novel quantitative guideline for the design of methanol-selective catalysts, namely −0.2 eV < ΔGCO < 0.2 eV. Finally, we show that an increase in CO mass transfer to each CoPc molecule boosts the faradaic efficiency (FE) of methanol up to 70% and the FE of formaldehyde up to 7%. Our findings highlight the importance of CO2 mass transfer in tuning the product selectivity of the CO2RR on CoPcCNT to either C1 oxygenates or CO.
Results and discussion
Synthesis of molecularly dispersed CoPc on carbon nanotubes
We first anchored CoPc onto the surfaces of multiwalled carbon nanotubes (CNTs). The resulting materials are denoted as CoPcCNT_x%, where x% represents the loading of CoPc on the CNTs in weight percentages, from 0.2 to 7.0% (Fig. S6†). As the molecular dispersion of CoPc on CNTs is critical for the production of methanol,9 we characterized the CoPcCNT_x% extensively to confirm its successful synthesis. CoPcCNT_7.0%, which has the highest CoPc loading, is likely to contain CoPc aggregates, if these were formed.11 Hence, we used it here as a representative example for physical characterization.High resolution transmission electron microscopy (HRTEM) images show that CoPcCNT_7.0% and pristine CNTs have no discernible morphological differences (Fig. 1a and S7†). The inter-shell spacing of pristine CNTs was 0.35 nm (Fig. S7a†), which remained stable after calcination (Fig. S7b†) and when the CoPc was loaded onto the CNTs (Fig. 1a). No other phases indicative of CoPc were observed in the HRTEM images. Similarly, the fast Fourier transform patterns of the HRTEM images (Fig. S8†) only show the (002) and (100) crystalline planes of the CNTs, with no crystalline domains corresponding to CoPc.
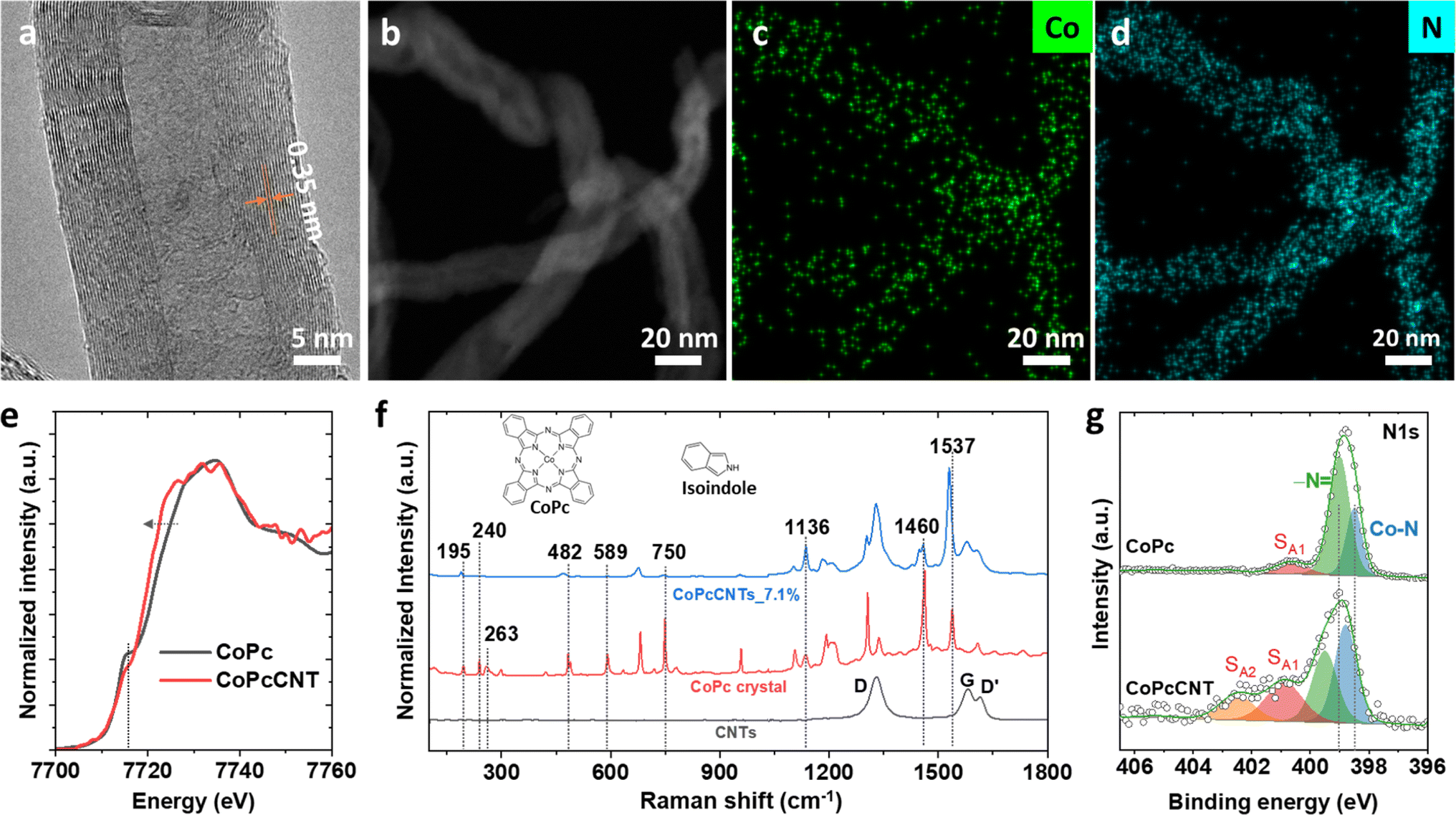 | ||
| Fig. 1 (a) HRTEM image, (b) scanning transmission electron microscopy image and the (c and d) corresponding elemental maps of Co and N of as-synthesized CoPcCNT_7.0%. (e) Co K-edge XANES spectra of CoPcCNT_7.0% and bulk CoPc crystals. (f) Raman spectra of CoPcCNT_7.0%, bulk CoPc crystals, and CNTs. (g) N 1s XPS spectra of bulk CoPc crystals and as-synthesized CoPcCNT_7.0%. | ||
Elemental mapping using energy dispersive spectroscopy (EDX), however, reveals a homogeneous distribution of cobalt and nitrogen atoms on the CNTs (Fig. 1b–d and S9†). Additionally, the Co K-edge X-ray absorption near edge structure (XANES) spectrum of CoPcCNT_7.0% exhibits a decrease in the intensity of its 1s → 4pz transition peak at 7716 eV and a red shift at the rising edge of its 1s → 4px,y transition at around 7723 eV, compared to that of bulk CoPc crystals (Fig. 1e).27,28 These changes indicate a distortion of the D4h planar symmetry of CoPc, due to interactions between CoPc and the CNTs.27,29
There are also differences between the Raman spectra of CoPcCNT_7.0% and bulk CoPc crystals (Fig. 1f and S10†). Specifically, the spectrum of CoPcCNT_7.0% shows smaller intensity ratios for several peaks against the peak of the pyrrole C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1537 cm−1. These peaks include those of the Co–N stretching at 240 cm−1, isoindole deformation at 589 cm−1, and macrocycle deformation at 750 cm−1.30 We further observed a red shift in the bands of the pyrrole CC stretching at 1537 cm−1 and the isoindole deformation at 482 and 195 cm−1. The N 1s X-ray photoelectron spectrum (XPS) of CoPcCNT_7.0% also exhibits a shift towards higher binding energy and an increase in the intensity of the shakeup satellites (SA) compared to that of the bulk CoPc crystals (Fig. 1g). The aforementioned changes in the Raman bands and N 1s XPS spectrum notably involve the nitrogen atoms of CoPc, which indicates that CoPc is likely to interact with the CNT surface via its nitrogen atoms.31–33 All in all, our characterization results indicate that CoPc is molecularly anchored to the CNT surface.
C stretching at 1537 cm−1. These peaks include those of the Co–N stretching at 240 cm−1, isoindole deformation at 589 cm−1, and macrocycle deformation at 750 cm−1.30 We further observed a red shift in the bands of the pyrrole CC stretching at 1537 cm−1 and the isoindole deformation at 482 and 195 cm−1. The N 1s X-ray photoelectron spectrum (XPS) of CoPcCNT_7.0% also exhibits a shift towards higher binding energy and an increase in the intensity of the shakeup satellites (SA) compared to that of the bulk CoPc crystals (Fig. 1g). The aforementioned changes in the Raman bands and N 1s XPS spectrum notably involve the nitrogen atoms of CoPc, which indicates that CoPc is likely to interact with the CNT surface via its nitrogen atoms.31–33 All in all, our characterization results indicate that CoPc is molecularly anchored to the CNT surface.
Co(I)Pc centers are catalytically active
The CO2RR activity of CoPcCNT_7.0% was evaluated in 0.1 M KHCO3 electrolyte saturated by 1 atm CO2 in an H-type cell (Fig. 2a, S11 and Table S1†). Each constant-potential electrolysis process was conducted for 15 min. H2 and CO products were quantified by online gas chromatography (GC), while methanol was identified by 1H nuclear magnetic resonance spectroscopy and quantified by headspace GC (Fig. S11a†). Formaldehyde (CH2O) was quantified by ultraviolet-visible spectroscopy. The product distribution as a function of applied potential is consistent with that in Wang et al.’s work.9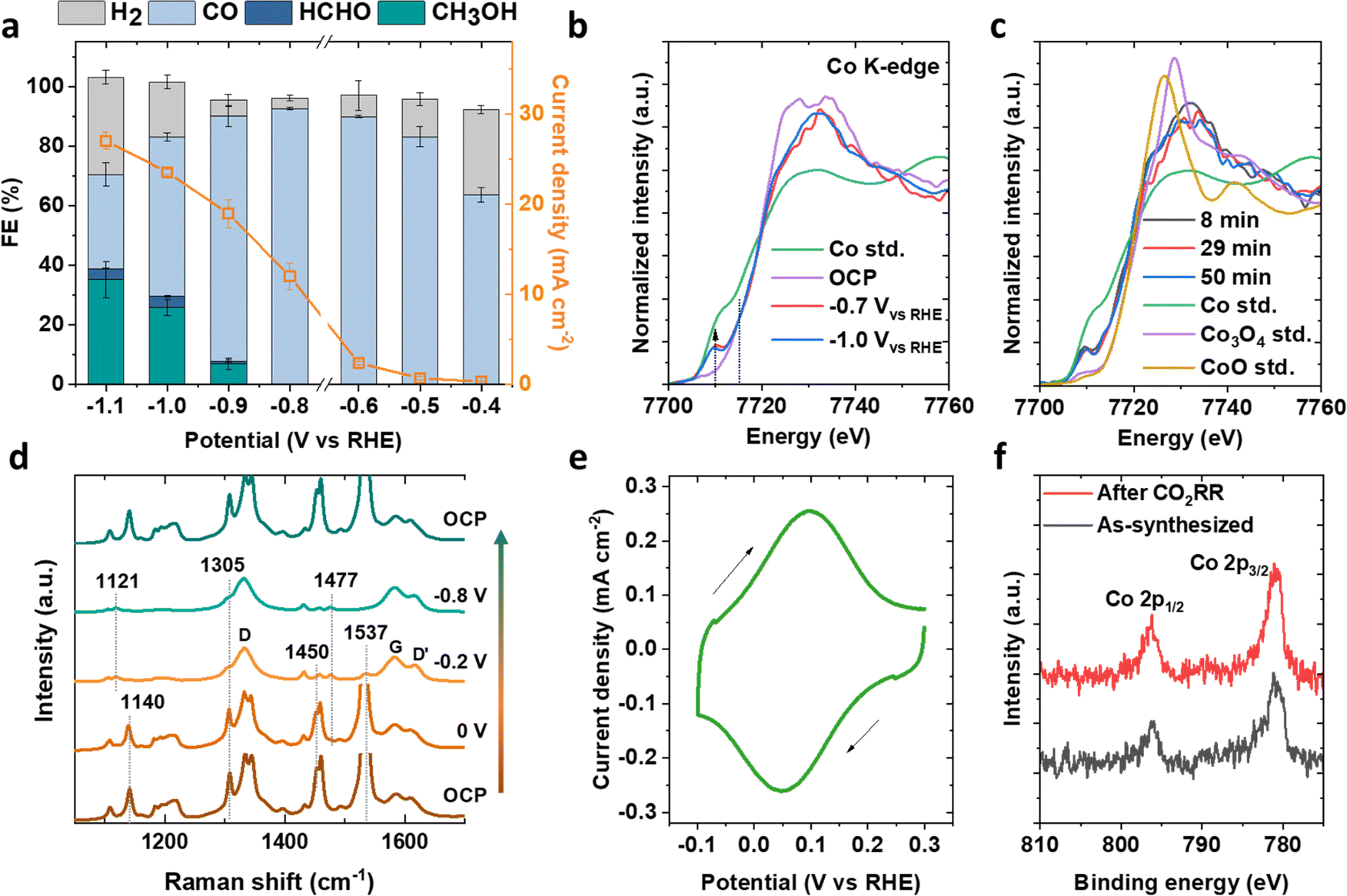 | ||
| Fig. 2 (a) Faradaic efficiencies of CO2RR products and the total current density as a function of the applied potential on CoPcCNT_7.0%. (b) In situ XANES spectra of CoPcCNT_7.0% at −0.7 and −1.0 V. OCP denotes open circuit potential and ‘Co std.’ denotes a Co metal standard. (c) Evolution of the in situ XANES spectra over reaction times at −1.0 V. Co std., Co3O4 std., and CoO std. are respectively the Co metal, Co3O4, and CoO standards. (d) In situ Raman spectra of CoPcCNT_7.0% measured at various potentials. The peak at 1537 cm−1 is truncated when its signal is too intense, so as to make the comparison clearer. (e) Cyclic voltammograms of CoPcCNT_7.0%. The scan rate is 50 mV s−1. (f) Co 2p XPS spectra of CoPcCNT_7.0% before and after its use for the 15 min CO2RR experiment at −1.0 V. CO2-saturated 0.1 M KHCO3 electrolyte was used for all the aforementioned experiments. | ||
From −0.4 to −0.8 V vs. the reversible hydrogen electrode (RHE; all potentials will hereafter be referenced to the RHE), CO was the sole CO2RR product. Methanol was observed with a FE of 7% at −0.9 V. CO2 as the carbon source for methanol was verified with 13CO2RR experiments (Fig. S11b†). As the potential changed from −0.9 to −1.1 V, the FE of methanol increased to 35%, accompanied by an increase in the FE of H2 from 6 to 33% and a decrease in the FE of CO from 82 to 32%. Formaldehyde was also detected with a FE of <4% (Fig. S11c and d†). This product has been recently identified as an intermediate of CO reduction to methanol.12 Overall, a minimum overpotential of ∼0.8–0.9 V is needed to reduce CO2 to methanol and formaldehyde, below which only CO is produced.
We conducted in situ XANES and Raman spectroscopy on CoPcCNT_7.0% during the CO2RR. A new peak at 7710 eV in the Co K-edge XANES evolved at representative potentials of −0.7 and −1.0 V, which were the potentials at which CO and methanol were formed. By comparison with the edge feature of a metallic cobalt standard (Fig. 2b), the oxidation state of Co in CoPc appeared to be reducing from +2 towards zero.19,34 However, the intensity of the newly formed peak remained fairly constant even after 50 min of electrolysis, suggesting that the metal in CoPc did not reduce to bulk metallic cobalt (Fig. 2c). Wang and coworkers recently assigned this peak to the Co 1s → 3d transition, asserting it as evidence for both the reduction of Co(II)Pc to Co(I)Pc and the binding of the CO intermediate to the Co center.19
The reduction of CoPc, as the potential changed from 0 to −0.2 V, was also indicated by changes in its Raman spectra (Fig. 2d). Specifically, its pyrrole breathing peak shifted from 1140 to 1121 cm−1, a pyrrole C–N peak at 1450 cm−1 and a pyrrole CC stretching peak at 1537 cm−1 faded, and a pyrrole ring stretching peak at 1477 cm−1 appeared. These changes persisted from −0.2 to −0.8 V. The peak shift from 1140 to 1121 cm−1 is induced by the reduction of Co(II)Pc to Co(I)Pc, as the Co atom is bound to the nitrogen atoms in the pyrrole ring.30,35 The decrease in the intensities of the bands at 1537 and 1477 cm−1 can be attributed to the change in the interaction between CoPc and CNTs, as the increased surface charge density of CNTs can weaken the molecule–substrate interaction.36 The Raman spectrum reverted to its original state upon switching the potential from −0.8 V to the open circuit potential, indicating a reversibility of the aforementioned changes. The observed reduction of CoPc is also consistent with its redox wave of Co(II)/Co(I) at 0.1 V in its cyclic voltammogram (CV, Fig. 2e).37 The Co 2p XPS spectrum of CoPcCNT_7.0% post-CO2RR closely aligns with that of the as-prepared one, notably lacking the characteristic satellite feature (around 804 eV) associated with cobalt oxides (Fig. 2f).38 The Co 2p3/2 binding energy of CoPcCNT_7.0% (780.9 eV) is also higher than that of metallic cobalt (778.3 eV).39 Additionally, the electron microscopy images of the catalyst post-CO2RR show no features corresponding to bulk metallic cobalt or cobalt oxide particles (Fig. S12 and S13†). Therefore, we assign the newly formed peak in the Co K-edge XANES spectrum to CoPc with Co in the +1 state, which we propose to be catalytically active for the CO2RR.
Low CO2 mass transfer positively impacts methanol selectivity
We separately investigated the effect of CoPc loading and the CO2 partial pressure on the electroreduction of CO2 to methanol on CoPc, and found that both factors impact the selectivity to methanol. Further analysis based on the effect of these two factors reveals an underlying relation: the average CO2 mass transfer rate (v) determines the ratio (R) of the production rates of methanol and CO. In fact, a power function (R = 0.97ν−0.78) fits the data from these two investigations with a high correlation coefficient of 0.948.In detail, we first evaluate CoPcCNT_x% samples with CoPc loading from 0.2 to 7.0% for the CO2RR at −1.0 V in an H-cell. Aqueous 0.1 M KHCO3 saturated with 1 atm CO2 was used as the electrolyte. CoPcCNT has rather poor stability when catalyzing the CO2RR to methanol due to the possible CoPc detachment and decomposition at a negative potential.9 We observed a small increase in the H2 FE (such as from 4.9 to 5.8% on CoPcCNT_1.9%) during 15 min of electrolysis, similar to what was reported in Wang's work.9 Hence, to minimize the effect of CoPcCNT instability on the performance assessment, we conducted the electrolysis for only 15 min (Fig. S14†).
The product distribution at various CoPc loadings is shown in Fig. 3a. As the CoPc loading on the CNTs increased from 0.2 to 7.0%, the FE of methanol increased from 2 to 26% (Fig. 3a and Table S2†). The FEs of formaldehyde and H2 also increased to 4 and 19%, respectively. Correspondingly, the FE of CO decreased from 99 to 53%. Electrolysis on bare CNTs produced only H2, indicating that the CNTs themselves are inactive for the CO2RR (Fig. 3a). We note that Wang and coworkers' recent study has also disclosed a positive correlation between FEmethanol and CoPc loading.19
 | ||
| Fig. 3 Faradaic efficiencies of CO2RR products and total current density as a function of the CoPc loading in the CoPcCNT catalysts at −1.0 V and 1 atm CO2. (b) CO2 conversion rate and average CO2 transfer rate to each CoPc as a function of the CoPc loading. (c) FE of CO2RR products and total current density as a function of the CO2 partial pressure (pCO2) on CoPcCNT_7.0% at −0.7 V (corrected based on the local pH). (d) Ratio of the production rates of CH3OH and CO versus pCO2 on CoPcCNT_0.5%, CoPcCNT_1.9%, and CoPcCNT_7.0%. (e) CO2 conversion rate and (f) average CO2 transfer rate to each CoPc as a function of pCO2 on CoPcCNT_0.5%, CoPcCNT_1.9%, and CoPcCNT_7.0%. (g) Ratio of the production rates of CH3OH and CO as a function of the average CO2 transfer rate to each CoPc obtained from four datasets. CoPcCNT_0.5%, CoPcCNT_1.9%, and CoPcCNT_7.0% denote the experiments where CO2 partial pressure is controlled with the results corresponding to the three curves in (3d), respectively. CoPcCNT_x% denotes the experiments where the CoPc loading is controlled with x% ranging from 0.2 to 7.0%, corresponding to the blue curve in (3b). ‘Fit’ denotes the black curve derived from the nonlinear curve fitting with the equation R = 0.97v−0.78, where R is the CH3OH/CO ratio and v is the CO2 transfer rate to each CoPc. ‘r’ denotes the correlation coefficient of the fit. All CO2RR experiments for (a–g) were conducted at −1.0 V in 0.1 M KHCO3 saturated with CO2 with the pressure noted in the graphs or in this caption. | ||
Further analysis reveals that the total CO2 conversion rate increased from ∼47 nmol s−1 cm−2 on CoPcCNT_0.2%, and plateaued at ∼77 nmol s−1 cm−2 when the CoPc loading exceeded 1% (Fig. 3b; See Section S1.2.7 for a sample calculation). The plateau indicates that the CO2 conversion rate is limited by CO2 mass transfer to the CoPc sites. Since the limiting CO2 mass transfer rate (77 nmol s−1 cm−2) to the electrode should be the same for all CoPcCNT_x% samples, an increase in CoPc loading from 0.2 to 7.0% will lower the average CO2 transfer rate to each CoPc molecular complex from 121 to 3 s−1. As no new catalytic species such as Co clusters were detected, we hypothesize that the CO2 mass transfer to each CoPc site must have played a significant role in its subsequent conversion to methanol or CO.
To evaluate our hypothesis on the effect of CO2 mass transfer on CO2RR selectivity, we controlled the CO2 mass transfer by varying the CO2 partial pressures (pCO2) on three catalysts, namely CoPcCNT_0.5%, CoPcCNT_1.9%, and CoPcCNT_7.0%, at around −0.7 V (corrected by the local pH, Table S6†). On all three catalysts, the FEmethanol and pCO2 exhibited non-monotonic correlations (Fig. 3c, S15 and Tables S3–S5†). It is also noteworthy that the maximum FEmethanol was not obtained at 1 atm, but at 0.1–0.5 atm of CO2. FECO, in contrast, increased with pCO2. It is further notable that while methanol could be generated at −0.8 V when the pCO2 is 0.2 atm, no methanol could be detected at 1 atm of CO2 (Fig. 2a and S16†). This phenomenon confirms that CO2 mass transfer indeed affects the production of methanol, but not in a monotonic way.
Further analysis of all three catalysts reveals a negative correlation between pCO2 and the ratio of the production rates (mole s−1 cm−2) of CH3OH and CO (Fig. 3d). The CH3OH/CO ratio also increased with CoPc loading. For instance, at a pCO2 of 0.3 atm, when the CoPc loading increased from 0.5 to 7.0%, the value of CH3OH/CO increased ∼7× from 0.2 to 1.6. These correlations imply the competitive formation of methanol and CO from the CO2RR, and this competition is modulated by both pCO2 and the CoPc loading. Specifically, we believe that the CO2 transfer rate to each CoPc directly influences the CH3OH/CO ratio.
To verify the validity of this conclusion, we first note that the CO2 conversion rate on all three catalysts is linearly related to pCO2 (Fig. 3e), which indicates that the reaction rate of the CO2RR is limited by CO2 mass transfer. This in turn suggests that the total CO2 transfer to the electrode has the same rate as CO2 conversion. The CO2 transfer rate to each CoPc complex can thus be calculated by dividing the CO2 conversion rate by the CoPc loading (Fig. 3f and Section S1.2.7). We then plotted the curves of the CH3OH/CO ratio (R) versus the CO2 transfer rate to each CoPc (v) in the same graph (Fig. 3g). Remarkably, the curves from four distinct datasets, either by controlling CoPc loading or pCO2, follow a consistent trend, and fit in a power-law function (R = 0.97v−0.78). This behavior reveals that the CO2 transfer rate to each CoPc controls the competition between methanol and CO formation. More specifically, CO2 may only be reduced to methanol when the supply of CO2 is low.
Mechanistic considerations and computational modelling
Thus far, we have identified CO, formaldehyde and methanol as CO2RR products on CoPcCNT (Fig. 2a). We have also electroreduced CO on CoPcCNT_7% and found that it can be converted to CH2O and CH3OH. CH2O itself can be reduced to methanol (Fig. S17a and Note S1†). On the other hand, the electrolysis of formate on CoPcCNT did not yield methanol (Fig. S17b†).10We performed density-functional theory (DFT) calculations to help connect these observations, elaborate a reaction pathway that complies with all of them, and provide simple catalyst design guidelines for future studies. In line with a previous study,13 we found that CoPc under negative potentials is reduced and contains a –H ligand on one of the N atoms (Fig. S18†). CoPc may also contain an extra electron under such conditions,13 but our calculations indicate that adding or removing it shifts the free energies of the intermediates on average by only 0.06 ± 0.03 eV and does not modify our conclusions (Fig. S19 and Table S7†). After inspecting each electrochemical step for possible bifurcations, the lowest-energy pathway for the CO2RR to methanol is: CO2 → *COOH → *CO → *CHO → *CH2O → *OCH3 → CH3OH, which agrees with previous studies (Fig. 4a and S19†).13,26
In Fig. 4b, we provide a free-energy diagram for the pathway in Fig. 4a on CoPc in which the equilibrium lines for CO (orange) and CH2O (blue) adsorption are shown. The potential-limiting step is the hydrogenation of *CH2O to *OCH3, and the thermodynamic potential to overcome it is −0.59 V. The first step, namely, the formation of *COOH requires a slightly lower potential (−0.50 V). Upon the second proton-electron transfer, *CO can either desorb as CO(g) or be further reduced to methanol via *CHO. This indicates that the extent of *CO adsorption/desorption is crucial in determining the selectivity of the CO2RR toward carbon monoxide or methanol. In the event that a *CO desorbs, the chances for it to re-adsorb onto an active site and hydrogenate to methanol will decrease if a species with a stronger adsorption energy is nearby. According to our calculations, such a species is likely CO2, as its adsorption energy is −0.14 eV, which is more negative than that of any other adsorbed species on CoPc, including *CO and *CH2O (Fig. 4b and Table S7†). In particular, the inset of Fig. 4b shows that CO2 adsorption is stronger than that of CO on CoPc at 0 V.
 | ||
| Fig. 4 (a) Side views of the lowest-energy electrochemical CO2RR intermediates to methanol on CoPc. Top views of the catalyst and the intermediates are provided in Fig. S18.† CO desorption and re-adsorption are possible upon the second proton-electron transfer, as indicated by the orange arrow. *CO and *CH2O appear in blue as CO(g) and CH2O(g) were shown in experiments to be reducible to methanol. (b) Free-energy diagram for CO2 electroreduction to methanol on CoPc at 0 V. The equilibrium adsorption/desorption lines for CO and CH2O are shown in orange and blue, respectively. For comparison, the free-energy landscape for the ideal catalyst is also provided. Inset: Adsorption energies of CO2 and CO on CoPc at 0 V. (c) Volcano plot for the CO2RR to methanol on FePc, CoPc and NiPc. Ag(211), Zn(0001) and Ag dendrites on Zn are also provided for comparison (AgZn).40 The datapoints are averages of the elementary steps on the phthalocyanines with and without extra electrons, and the average of two dendrites for AgZn. The grey area, where methanol production is enabled, corresponds to ΔGCO = 0 ± 0.2 eV. | ||
Several previous studies have reached a similar conclusion to ours that CO2 adsorption is stronger than that of CO on CoPc.13,26 In particular, the microkinetic analysis conducted by McCrory and coworkers indicated a 3.3-fold increase in the effective equilibrium binding constant for CO2-CoPc compared to CO-CoPc.26 However, it is worth noting that such an increase in the binding constant was supported by a calculated binding energy difference between CO2 and CO of only 0.03 eV (0.6 kcal mol−1). As DFT generally has an accuracy of no less than ±0.1 eV, even when hybrid functionals are used, the aforementioned agreement between the experiment and theory should be considered cautiously. The inaccuracies can be aggravated when gas-phase errors in e.g. CO2, CO, CH2O and CH3OH are not accounted for,41,42 and the impact can be observed not just in the adsorption energies but also in the predicted equilibrium and onset potentials. In this case, we used gas-phase energy corrections to ensure that (i) our DFT-predicted equilibrium potentials and reaction energies match the experimental ones (see Section S1.5 and Fig. S20†), and (ii) an accurate gas-phase reference is used for the adsorption/desorption energies in the free-energy diagrams. The qualitative and quantitative consequences of omitting gas-phase corrections are illustrated in the free-energy diagrams for CoPc and the ideal catalyst in Fig. S21.† Such considerations are still often overlooked in computational models and may severely limit their predictiveness.42
If *CO binding is to be strengthened to obtain more methanol, it is pertinent to ask by how much. To address this question and bearing in mind that previous studies found no liquid products on FePc and NiPc during the CO2RR,9,13 we modelled the pathway in Fig. 4a on these two phthalocyanines by means of DFT calculations and elaborated a Sabatier-type activity plot in Fig. 4c. The figure shows the potentials required by the various limiting steps (UL) of the phthalocyanines as a function of their free energies of adsorption of *CO. The phthalocyanine datapoints in Fig. 4c correspond to averages of charged and uncharged systems (Table S7†). We note that the narrow error bars indicate that the presence or absence of an extra electron at the metal-Pc sites does not change our conclusions in this case. This is also illustrated in the free-energy diagrams in Fig. S19 and in Table S7.†
Unlike the CO2RR volcano plot for the production of methane, which has a sharp summit,43 we observe here a relatively wide plateau where *CO hydrogenation is potential-limiting. The methanol activity of FePc is limited by this step, while that of NiPc is limited by *COOH formation. As mentioned before, CoPc is limited by the formation of *OCH3, but *COOH formation requires a similar potential (−0.59 vs. −0.50 V). The experimental inertness of NiPc can be explained by its weak binding of the reaction intermediates,9,13 which results in an average limiting potential of around −1.3 V. In turn, FePc is predicted to be active but experiments show otherwise,9 suggesting that *CO hydrogenation is kinetically impeded. This helps us formulate a hypothesis: active materials for the CO2RR to methanol ought to be found at around ΔGCO = 0 ± 0.2 eV, that is within the grey-colored zone in Fig. 4c. To the right of it, *CO and eventually *CH2O adsorption are too weak and the CO2RR only leads to CO(g). To the left of the grey zone, the plateau prevents an improvement of the limiting potential when *CO adsorption is strengthened, and *CO hydrogenation might be kinetically impeded, such that CO(g) will also be produced.
To test this hypothesis, we turn to our previous work, where Ag and Zn were shown to reduce CO2 mainly to CO, but Zn dendrites deposited on Ag foam were able to reduce CO2 to methanol with an experimental faradaic efficiency of 10.5%.40 We observe exactly this in Fig. 4c: Ag(211) and Zn(0001) bind *CO too weakly to be within the methanol production (grey) zone, while AgZn dendrites bind *CO strong enough to be within it. In perspective, our simple classification of the active sites in terms of *CO binding is similar to other classifications for CO2RR products44,45 and, certainly, its exciting predictions call for future experiments and calculations.
While we have no direct experimental evidence supporting the stronger CO2 adsorption on CoPc as compared to CO, our in situ Raman spectroscopy and CVs of CoPcCNT in CO2-, CO- and N2-saturated electrolytes indicate a different interaction of CO2 and CO with CoPc. At −0.2 V, the Raman bands of CoPc in CO2-saturated electrolyte were more attenuated than those in CO- and N2-saturated electrolytes (Fig. 5a and S22, and Note S2†). In the CVs of CoPcCNT_7.0%, the oxidation waves (A) and (B) at around 0.1 and 0.9 V, respectively, were suppressed in CO2-saturated electrolyte, relative to that in CO- and N2-saturated electrolytes (Fig. 5b and S23, and Note S3†). To directly show the competing binding of CO2 and CO and evaluate the dwelling time of *CO, more advanced electrochemical spectroscopic methods, such as in situ time-resolved infrared spectroscopy, should be used to quantify the binding strength of CO2 and CO on CoPc. These measurements may also reveal more microkinetic details and the physical model behind the power-law function as shown in Fig. 3g.
 | ||
| Fig. 5 (a) In situ Raman spectra of CoPcCNT_7.0% measured at −0.2 V in phosphate buffers (pH 7) saturated by N2, CO, or CO2. The 1537 cm−1 peak in the spectrum measured in N2-saturated electrolyte is truncated as its signal is too intense (see the full spectra in Fig. S22†). (b) Cyclic voltammograms of CoPcCNT_7.0% in 0.1 M KClO4 solution saturated by N2, CO, or CO2. The A and A′ peaks correspond to CoPc(II)/CoPc(I) and the C and C′ peaks to CoPc(II)/CoPc(III).37 The scan rate was 50 mV s−1. (c) Faradaic efficiencies of the CORR products and partial current density of methanol as a function of the limiting CO mass transfer to the electrode at −0.86 V. The electrolyte used was 0.1 M KHCO3 saturated by 1 atm of CO. | ||
On the basis of the above results, to enhance methanol production, it is necessary to moderately enhance the *CO adsorption energy of CoPc to prevent CO desorption, but still allow its hydrogenation. It is also necessary to reduce the competitive adsorption of CO2, so that *CO could be reduced to methanol. This can be controlled by decreasing the mass transfer of the CO2 reactant to the CoPcCNT catalysts as shown in Fig. 3.
Further directions for optimizing the selectivity of CoPc towards methanol can be obtained by comparing its free-energy diagram to that of the ideal catalyst. As shown in Fig. 4b, such a hypothetical catalyst has energy-symmetric electrochemical steps consecutively separated by 16.4 meV, which would allow it to operate with no energy losses at the equilibrium potential, in principle. A comparison of the two energy landscapes in Fig. 4b indicates that CoPc is relatively efficient for methanol production but is far from ideal. In particular, the first and fifth electrochemical steps are rather energy-intensive and the free energies of the adsorbates are visibly weaker than those of the ideal catalyst. For example, the ideal *COOH level with respect to CO2 should be at −0.02 eV, but it is 0.50 eV on CoPc.
Our results indicate that using CO as a feedstock is a more straightforward approach to boost the selectivity toward methanol, as no CO2 could then compete with CO for the active sites. Thus, we evaluated the CORR on CoPcCNT_7.0% under different mass transfer conditions (Fig. 5c, S1 and S2†). The selectivity and activity of methanol and formaldehyde production now positively correlate with the mass transfer of CO, unlike that of CO2. Faradaic efficiencies of 70% for methanol and 7% for formaldehyde could be achieved from the CORR at −0.86 V and at a total current density of −19 mA cm−2 in 0.1 M KHCO3 electrolyte. The stability of CoPcCNT for the CORR at −0.86 V in a flow cell was relatively poor, as evidenced by the increase in H2 FE from 20 to 36% and the decrease in the cathodic current density from around 13 to 10 mA cm−2 during a 2 h electrolysis (Fig. S24†). The instability of the CORR could be caused by the detachment of CoPc from the CNT support or by the decomposition of CoPc complexes, as observed in Wang's work.9 The catalyst stability could be improved by adding a functional group or ligand that enhances the anchoring of CoPc, or by optimizing the diameter of the CNT support.9,24,46 Moreover, unlike metal catalysts mounted in flow cells that often exhibit current densities larger than 100 mA cm−2,47 the CORR on CoPcCNT in a flow cell still exhibits a rather small current density of ∼13 mA cm−2. This is likely due to the easy wetting of CoPcCNT by the aqueous electrolyte that results in only a small fraction of CoPc active sites being accessible to CO.48 The microenvironment in the catalyst layer may be optimized to mitigate this issue and, hence, increase the current density.23,48
Overall, we have elucidated in this work that (1) Co(I)Pc is the likely catalytically active species under CO2 reduction conditions, and (2) the CO2 transfer rate to each CoPc together with the overpotential inclines the CO2RR toward either methanol or CO. The underlying reason is that CO2 mass transfer modulates the coverage of *CO2 and *CO. These findings, which align with those from several other recent studies,13,19,26 suggest two approaches to modulate the selectivity of methanol and CO from the CO2RR on CoPc: first, by adjusting the CoPc loading in the electrode, and second, by regulating the mass transfer of CO2. We note that similar mass-transfer effects have previously been reported on the copper-catalyzed CORR, showing that the competitive production of ethylene, acetate, and n-propanol may be correlated with the mass transfer of CO.49,50 However, the underlying mechanism still remains a matter of debate. Herein, our findings highlight the crucial role of adsorption intermediates from a new perspective, as on CoPc an excess or lack of CO2 around the active sites suffices to prevent or enable CO adsorption and hydrogenation.
Conclusions
In this work, by means of in situ XANES and in situ Raman spectroscopies, we determined that reduced CoPc with Co(I) centers are the active sites for the CO2RR. We found that the selectivity to methanol is positively correlated with the CoPc loading. The negative correlation between the ratio of the CH3OH/CO production rate and the CO2 partial pressure further led us to identify the CO2 transfer rate to each CoPc as a key factor for methanol production relative to CO. This rate is crucial in view of the competitive adsorption of CO2 and CO on CoPc, inferred from experiments and corroborated by DFT calculations. Inspired by this finding, we tuned the mass transfer of CO to each CoPc molecule, which enabled us to reach a 70% faradaic efficiency and a partial current density of 13 mA cm−2 for methanol production from the CORR. Computational modelling based on the Sabatier principle revealed that the CO2RR on CoPc can be enhanced by strengthening the binding of *CO under the condition that −0.2 eV < ΔGCO < 0.2 eV, and by making the free-energy diagram more energetically symmetric along the entire CO2RR catalytic pathway. This body of work provides valuable guidelines and inspiration for developing efficient metal complex-based catalysts, as well as for identifying the reaction conditions that optimize their activity and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Research Foundation of Singapore (Urban Solutions and Sustainability, Industry Alignment Fund (Pre-Positioning) Programme, A-0004543-00-00), and Shell Global Solutions International B.V. (A-0004543-01-00) for financial support of this project. FCV received financial support from grants PID2021-127957NB-I00 and TED2021-132550B-C21, which are funded by MCIN/AEI/10.13039/501100011033 and by the European Union. FCV also acknowledges financial support through grant IT1453-22 “Grupos Consolidados UPV/EHU del Gobierno Vasco” and thanks the Red Española de Supercomputación for computational resources (grants QHS-2023-2-0007 and QHS-2023-3-0006). The use of supercomputing facilities at SURFsara was sponsored by NWO Physical Sciences, with financial support by NWO. The authors thank Dr Jia Liu from the Department of Chemistry, National University of Singapore, for useful scientific discussions.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 Search PubMed.
- S. Sarp, S. Gonzalez Hernandez, C. Chen and S. W. Sheehan, Joule, 2021, 5, 59–76 Search PubMed.
- H. Choi, D.-K. Lee, M.-K. Han, G. Janani, S. Surendran, J. H. Kim, J. K. Kim, H. Cho and U. Sim, J. Electrochem. Soc., 2020, 167, 164503 Search PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 Search PubMed.
- Q. H. Low and B. S. Yeo, J. Electrochem. Energy Convers. Storage, 2020, 17, 040802 Search PubMed.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. F. de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, JPhys Energy, 2022, 4, 042003 Search PubMed.
- Y. Liu, F. Li, X. Zhang and X. Ji, Curr. Opin. Green Sustainable Chem., 2020, 23, 10–17 Search PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 Search PubMed.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 Search PubMed.
- Y. Wu, G. Hu, C. L. Rooney, G. W. Brudvig and H. Wang, ChemSusChem, 2020, 13, 6296–6299 Search PubMed.
- E. Boutin, A. Salamé, L. Merakeb, T. Chatterjee and M. Robert, Chem.–Eur. J., 2022, 28, e202200697 Search PubMed.
- X. Ren, J. Zhao, X. Li, J. Shao, B. Pan, A. Salamé, E. Boutin, T. Groizard, S. Wang, J. Ding, X. Zhang, W.-Y. Huang, W.-J. Zeng, C. Liu, Y. Li, S.-F. Hung, Y. Huang, M. Robert and B. Liu, Nat. Commun., 2023, 14, 1–10 Search PubMed.
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Chem. Soc. Rev., 2021, 50, 12985–13011 Search PubMed.
- Z. Yue, C. Ou, N. Ding, L. Tao, J. Zhao and J. Chen, ChemCatChem, 2020, 12, 6103–6130 Search PubMed.
- D. Grammatico, A. J. Bagnall, L. Riccardi, M. Fontecave, B.-L. Su and L. Billon, Angew. Chem., Int. Ed., 2022, 61, e202206399 Search PubMed.
- Q. Feng, Y. Sun, X. Gu and Z. Dong, Electrocatalysis, 2022, 13, 675–690 Search PubMed.
- S. Zhang, X. Jing, Y. Wang and F. Li, ChemNanoMat, 2021, 7, 728–736 Search PubMed.
- C. L. Rooney, M. Lyons, Y. Wu, G. Hu, M. Wang, C. Choi, Y. Gao, C.-W. Chang, G. W. Brudvig, Z. Feng and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202310623 Search PubMed.
- X. Wu, J. W. Sun, P. F. Liu, J. Y. Zhao, Y. Liu, L. Guo, S. Dai, H. G. Yang and H. Zhao, Adv. Funct. Mater., 2022, 32, 2107301 Search PubMed.
- H. Xu, H. Cai, L. Cui, L. Yu, R. Gao and C. Shi, Nano Res., 2023, 16, 3649–3657 Search PubMed.
- Q. Wu, D. Si, J. Liang, Y. Huang and R. Cao, Appl. Catal., B, 2023, 333, 122803 Search PubMed.
- J. Li, B. Shang, Y. Gao, S. Cheon, C. L. Rooney and H. Wang, Nat. Synth., 2023, 2, 1194–1201 Search PubMed.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 Search PubMed.
- J. Ding, Z. Wei, F. Li, J. Zhang, Q. Zhang, J. Zhou, W. Wang, Y. Liu, Z. Zhang, X. Su, R. Yang, W. Liu, C. Su, H. B. Yang, Y. Huang, Y. Zhai and B. Liu, Nat. Commun., 2023, 14, 6550 Search PubMed.
- L. Yao, K. E. Rivera-Cruz, P. M. Zimmerman, N. Singh and C. C. L. McCrory, ACS Catal., 2024, 14, 366–372 Search PubMed.
- Y. Liu, A. Deb, K. Y. Leung, W. Nie, W. S. Dean, J. E. Penner-Hahn and C. C. L. McCrory, Dalton Trans., 2020, 49, 16329–16339 Search PubMed.
- W. Hu, D. Wang, Q. Ma, B. J. Reinhart, X. Zhang and J. Huang, J. Photochem. Photobiol., 2022, 11, 100132 Search PubMed.
- N. Li, W. Lu, K. Pei and W. Chen, RSC Adv., 2015, 5, 9374–9380 Search PubMed.
- D. Masheder and K. P. J. Williams, J. Raman Spectrosc., 1987, 18, 391–398 Search PubMed.
- F. Evangelista, A. Ruocco, R. Gotter, A. Cossaro, L. Floreano, A. Morgante, F. Crispoldi, M. G. Betti and C. Mariani, J. Chem. Phys., 2009, 131, 174710 Search PubMed.
- B. J. Palys, G. J. Puppels, D. van den Ham and D. Feil, J. Electroanal. Chem., 1992, 326, 105–112 Search PubMed.
- B. J. Palys, D. M. W. van den Ham, W. Briels and D. Feil, J. Raman Spectrosc., 1995, 26, 63–76 Search PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 Search PubMed.
- S. Ren, E. W. Lees, C. Hunt, A. Jewlal, Y. Kim, Z. Zhang, B. A. W. Mowbray, A. G. Fink, L. Melo, E. R. Grant and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 4414–4420 Search PubMed.
- S. Jiang, Z. Chen, X. Chen, D. Nguyen, M. Mattei, G. Goubert and R. P. Van Duyne, J. Phys. Chem. C, 2019, 123, 9852–9859 Search PubMed.
- J. Zagal, M. Páez, A. A. Tanaka, J. R. dos Santos and C. A. Linkous, J. Electroanal. Chem., 1992, 339, 13–30 Search PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 Search PubMed.
- W. Luo and S. Zafeiratos, J. Phys. Chem. C, 2016, 120, 14130–14139 Search PubMed.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew. Chem., Int. Ed., 2019, 58, 2256–2260 Search PubMed.
- R. Urrego-Ortiz, S. Builes, F. Illas and F. Calle-Vallejo, EES Catal., 2024, 2, 157–179 Search PubMed.
- L. P. Granda-Marulanda, A. Rendón-Calle, S. Builes, F. Illas, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2020, 10, 6900–6907 Search PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 Search PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 Search PubMed.
- A. Bagger, Curr. Opin. Electrochem., 2023, 40, 101339 Search PubMed.
- S. C. Jesudass, S. Surendran, G. Janani, T.-H. Kim and U. Sim, Adv. Energy Mater., 2023, 13, 2301918 Search PubMed.
- J.-J. Lv, M. Jouny, W. Luc, W. Zhu, J.-J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 Search PubMed.
- S. Cheon, J. Li and H. Wang, J. Am. Chem. Soc., 2024, 146, 16348–16354 Search PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 Search PubMed.
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, X. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y.-M. Yiu, T.-K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Nature, 2023, 617, 724–729 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic details, supplementary figures, tables, and equations related to material characterization, pH simulation, computational modeling, and ICP, Raman, TEM, and EDS studies. See DOI: https://doi.org/10.1039/d4ta03531c |
| This journal is © The Royal Society of Chemistry 2024 |